 Angelina A. Kislichkina1*
Angelina A. Kislichkina1* Angelika A. Sizova1
Angelika A. Sizova1 Yury P. Skryabin2
Yury P. Skryabin2 Svetlana V. Dentovskaya3
Svetlana V. Dentovskaya3 Andrey P. Anisimov3*
Andrey P. Anisimov3*- 1Department of Culture Collection, State Research Center for Applied Microbiology and Biotechnology, Obolensk, Russia
- 2Department of Molecular Microbiology, State Research Center for Applied Microbiology and Biotechnology, Obolensk, Russia
- 3Laboratory for Plague Microbiology, Especially Dangerous Infections Department, State Research Center for Applied Microbiology and Biotechnology, Obolensk, Russia
16S rRNA genes sequencing has been used for routine species identification and phylogenetic studies of bacteria. However, the high sequence similarity between some species and heterogeneity within copies at the intragenomic level could be a limiting factor of discriminatory ability. In this study, we aimed to compare 16S rRNA genes sequences and genome-based analysis (core SNPs and ANI) for identification of non-pathogenic Yersinia. We used complete and draft genomes of 373 Yersinia strains from the NCBI Genome database. The taxonomic affiliations of 34 genomes based on core SNPs and the ANI results did not match those specified in the GenBank database (NCBI). The intragenic homology of the 16S rRNA gene copies exceeded 99.5% in complete genomes, but above 50% of genomes have four or more variants of the 16S rRNA gene. Among 327 draft genomes of non-pathogenic Yersinia, 11% did not have a full-length 16S rRNA gene. Most of draft genomes has one copy of gene and it is not possible to define the intragenomic heterogenicity. The average homology of 16S rRNA gene was 98.76%, and the maximum variability was 2.85%. The low degree of genetic heterogenicity of the gene (0.36%) was determined in group Y. pekkanenii/Y. proxima/Y. aldovae/Y. intermedia/Y. kristensenii/Y. rochesterensis. The identical gene sequences were found in the genomes of the Y. intermedia and Y. rochesterensis strains identified using ANI and core SNPs analyses. The phylogenetic tree based on 16S rRNA genes differed from the tree based on core SNPs of the genomes and did not represent phylogenetic relationship between the Yersinia species. These findings will help to fill the data gaps in genome characteristics of deficiently studied non-pathogenic Yersinia.
Introduction
The genus Yersinia, a member of the family Yersiniaceae, is currently composed of 26 species, including three human pathogens: the causative agent of plague, Yersinia pestis, and enteropathogenic Yersinia enterocolitica and Yersinia pseudotuberculosis (Mares et al., 2021; Le Guern et al., 2020). By reason of their medical significance, they have been well characterized, and data about their ecology, epidemiology, and molecular mechanisms of pathogenicity are available in many publications (Atkinson and Williams, 2016; Reuter et al., 2014). Other species of Yersinia are considered non-pathogenic for humans because they have not been shown to be associated with disease manifestation (Chen et al., 2010; Sulakvelidze, 2000). Nevertheless, the taxonomy of the genus Yersinia is evolving dynamically, and several novel species were recognized during WGS (Whole Genome Sequencing) investigations (Nguyen et al., 2020a; Nguyen et al., 2020b; Cunningham et al., 2019; Savin et al., 2019). Unlike the three human pathogens of Yersinia, other species have been less studied because most studies have focused on characterizing these Homo sapiens pathogens. As a result, our knowledge about non-pathogenic related species is very limited. Bacterial genome studies have shown that many pathogens can be separated from environmental, commensal, or zoonotic populations of microorganisms (Achtman et al., 1999; van Baarlen et al., 2007; Van Ert et al., 2007). The comprehensive studying of not clinically significant microorganisms is essential for understanding the evolution, ecology, virulence, and distribution of bacteria. The first step in studying of microorganism is properly to identify the species. Correct identification of clinical isolates is necessary for selecting optimal treatment strategies and determining the scope of public health measures.
16S rRNA sequencing has been used for decades for routine identification of bacterial isolates (Mignard and Flandrois, 2006). The advantage of the 16S rRNA gene over other genes is its presence in all known species of bacteria and archaea, as well as the existence of highly conserved regions, which made it possible to create universal primers suitable for ribotyping prokaryotes (Clarridge, 2004). This gene has become a widely used target for taxonomic and evolutionary studies of bacteria after the implementation of automatic genetic analyzers and the development of public databases containing a lot of nucleotide sequences of 16S rRNA genes (Cole et al., 2014; Caporaso et al., 2012). The 16S rRNA gene is approximately 1,500 bp long, and all known microorganisms have at least one copy of this gene. The nine hypervariable regions (V1–V9) and the conservative sequences separating them can be distinguished in the nucleotide sequence of the 16S rRNA gene (Abellan-Schneyder et al., 2021). Although 16S rRNA gene sequencing is widely used for microbe-species identification using WGS platforms, this method has several limitations and disadvantages (Johnson et al., 2019; Gonzalez et al., 2019; Muhamad Rizal et al., 2020). Results of identification could be unreliable in the case of using unsuitable primers, inadequate bioinformatic software, or outdated reference databases (Park and Won, 2018; Tatusova et al., 2015; Hsieh et al., 2022; Edgar, 2018). Among the factors limiting the discriminatory ability of this method are the high homology of the nucleotide sequences of this gene between several related genera and/or species and intragenic heterogenicity, i.e., polymorphisms between copies of 16S rRNA in the genome (Srinivasan et al., 2015; Rodriguez-R et al., 2018). In such cases, it is necessary to use additional genes or other methods to determine the species of bacteria.
The aim of our work was the evaluation of using 16S rRNA genes sequences and genome-based analysis for identification of non-pathogenic Yersinia. Complete genomes and WGS data from the NCBI Genome database were used in this study. We analyzed copies of the 16S rRNA gene in whole genomes to determine intragenic heterogenicity. The methods of the genome analysis such as core SNPs and ANI were used for species identification of Yersinia.
Materials and methods
Sample collection, DNA extraction, and whole-genome sequencing
A total of 33 non-pathogenic Yersinia strains were used in this study (Table 1). These strains are stored in the microorganism collection of the State Research Center for Applied Microbiology and Biotechnology (SRCAMB, Obolensk, Russia). Bacteria were originally collected as Yersinia enterocolitica-like, and were confirmed using microscopic examinations and biochemical identification tests. Before whole-genome sequencing, their species identifications were updated by matrix assisted laser desorption ionization (MALDI) Biotyper (Bruker, Germany).
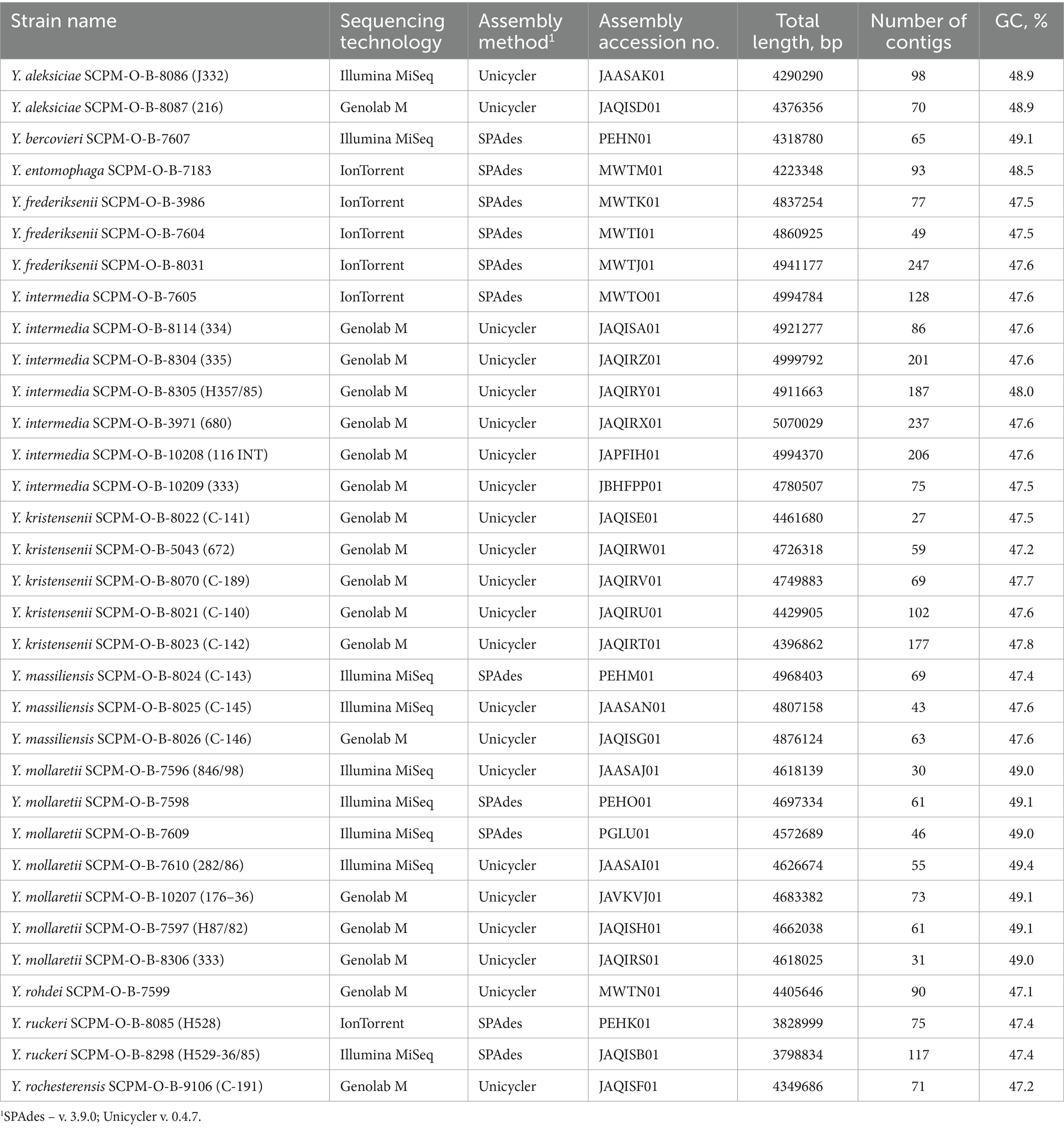
Table 1. Data for whole genome-sequenced Yersinia strains.
Bacteria were grown at 28°C on nutrient medium 1 (SRCAMB, Obolensk, Russia). DNA from each strain was extracted using the DNA minikit (BIOFACT Co., Ltd., Korea) following the manufacturer’s instructions. DNA quality was assessed using a Qubit 3 Fluorometer with the QubitTM dsDNA HS Assay Kit (Invitrogen, USA). Whole genome sequencing was performed in 2017, 2020 and 2022 using the Torrent PGM platform (Life Technologies, USA), Illumina MiSeq instrument (Illumina, USA), MGISeq-2000 (MGI Tech Co., China), and Genolab M (GeneMind Biosciences, China).
For sequencing on the Torrent PGM platform, the Ion 318 chip kit and 400-bp chemistry were used (Life Technologies, USA); on platform Illumina MiSeq - the Nextera DNA Library Preparation Kit and MiSeq Reagent Kits v3 (Illumina, USA); on the Genolab M - the library preparation kit SG GM (Raissol Bio, Sesana, Russia) and GenoLab M Sequencing Set V 1.0, FCM 300 cycles (GeneMind Biosciences, China); on MGISeq-2000 - MGIEasy FS DNA Library Prep Kit and MGI-Seq 2000RS High-throughput sequencing kit PE200 (MGI Tech Co., China).
The raw reads were de novo assembled using assemblers SPAdes v. 3.9.0 and Unicycler v. 0.4.7 with default settings, which included primary filtering and quality control (Bankevich et al., 2012; Wick et al., 2017). The draft genomes were deposited in GenBank database. Annotation was carried out by NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v. 5.3. Information on the assembly accession number in NCBI Genome database, total length, number of contigs and GC percentage is shown in Table 1.
Bacterial genomes
The genomes of 33 non-pathogenic Yersinia strains performed in this study and all complete and draft genomes of non-pathogenic Yersinia downloaded at NCBI Genome (September, 2022) were included in the investigation. Finally, the genomes of 368 strains of non-pathogenic Yersinia (Y. aldovae – 11, Y. aleksiciae – 11, Y. alsatica – 10, Y. artesiana – 4, Y. bercovieri – 17, Y. canariae – 3, Y. entomophaga – 2, Y frederiksenii – 37, Y. hibernica – 2, Y. intermedia – 41, Y. kristensenii – 38, Y. massiliensis – 15, Y. mollaretii – 27, Y. nurmii – 1, Y. pekkanenii – 2, Y. proxima – 10; Y. rochesterensis – 8; Y. rohdei – 12; Y. ruckeri – 99; Y. similis – 9, Y. thracica – 4, Y. vastinensis – 5), as well as genomes of Y. enterocolitica 8081, Y. enterocolitica subsp. palearctica Y11, Y. pseudotuberculosis IP 32953, Y. pestis CO92, and Y. wautersii WP-931201 were studied. The data are available in the NCBI Genome database, and accession numbers are provided in Supplementary Table S1.
Phylogenetic analysis of Yersinia genomes based on core SNPs
The core SNPs were determined using the Snippy 4.6.0 software with default settings.1 Visualization of the phylogenetic trees were performed using the Neighbor joining algorithm FigTree v. 1.4.42 and SplitsTree43 using NJ method.
Determination of ANI
Average nucleotide identity (ANI) values were determined using the FastANI software with default settings (Jain et al., 2018). Statistical calculations were performed using MS Office Excel.
Analysis of 16S rRNA gene in Yersinia genomes
The 16S rRNA gene searches were performed using BLAST. For phylogenetic analysis only full-length genes were selected from the genomes. The alignment was performed using MEGA11 with the ClustalW algorithm (Tamura et al., 2021) with default settings. The phylogenetic tree was constructed using the Neighbor joining algorithm in MEGA11 software.
Results
Diversity of copies 16S rRNA gene in whole genomes
The copies of 16S rRNA genes in complete genomes from the GenBank database (NCBI) were compared to evaluate the intragenomic heterogenicity of the 16S rRNA genes of Yersinia. Note that the complete genomes of Y. wautersii, Y. vastinensis, Y. artesiana, Y. proxima, Y. pekkanenii, Y. thracica, and Y. nurmii are not available from the NCBI database. Only one or a few complete genomes are available for other Yersinia species. Among the non-pathogenic Yersinia species for humans and other warm-blooded animals, Y. ruckeri is most represented among sequenced genomes because it is a cause of a serious septicemic bacterial disease in salmonid fish.
The complete genomes of Yersinia, their accession numbers, platforms on which WGS was performed, and assembly software are provided in Supplementary Table S2. The genomes of Yersinia species contain seven copies of the 16S rRNA gene, except for Y. pestis, which contains six copies per genome. The analysis of 16S rRNA gene copies in the complete genomes of Yersinia is shown in Table 2.
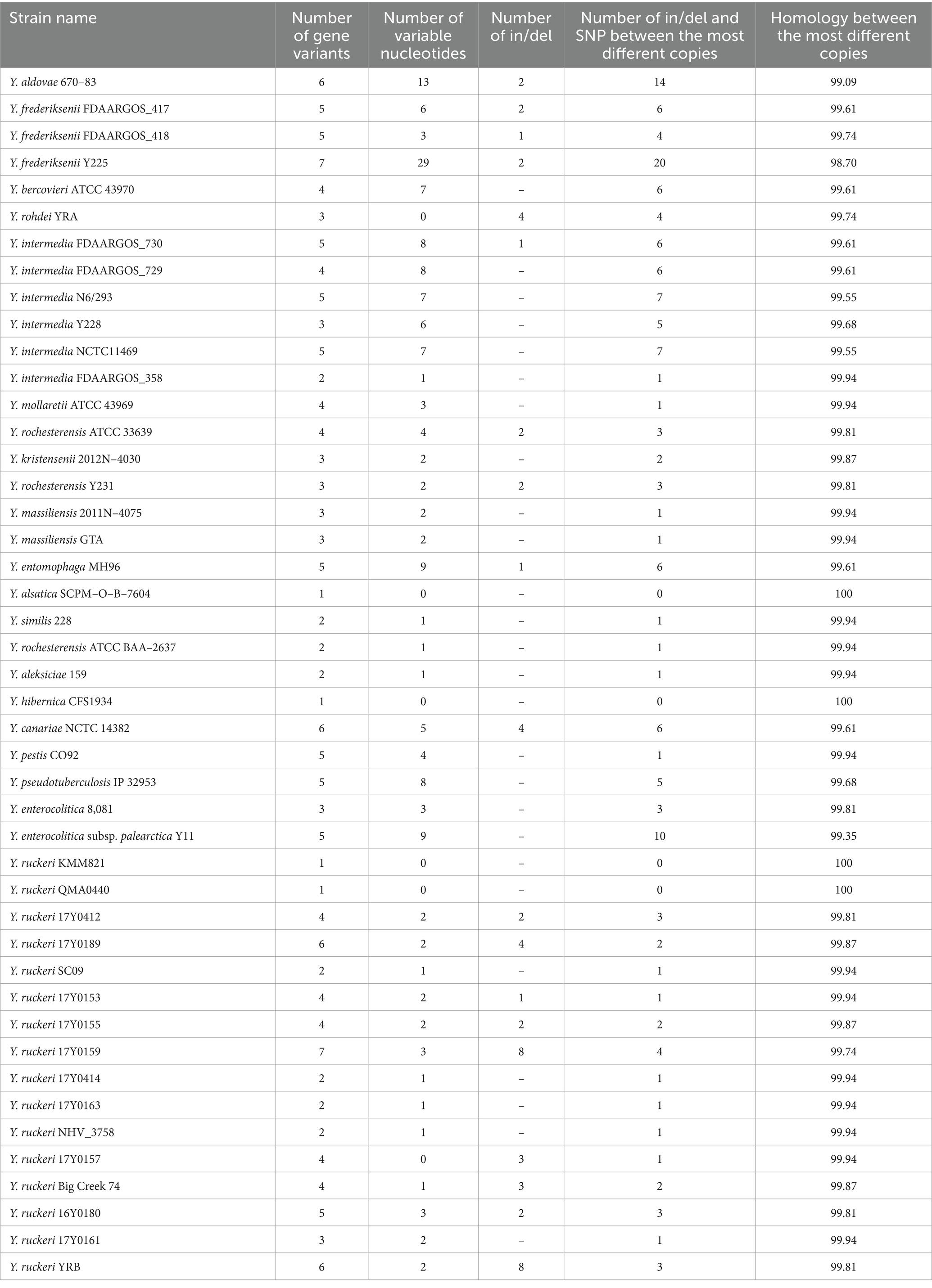
Table 2. Analysis of 16S rRNA gene copies in complete genomes of Yersinia.
Among investigated Yersinia were observed different number of 16S rRNA gene copy variants in the complete genomes. In two genomes (Y. frederiksenii Y225 and Y. ruckeri 17Y0159) of each copy of gene was unique. Six, five and four variants of 16S rRNA gene had 4, 10 and 9 genomes, respectively. Two and three variants of 16S rRNA gene had 8 genomes each. Interestingly, only four genomes (Y. alsatica SCPM-O-B-7604, Y. hibernica CFS1934, Y. ruckeri KMM821, Y. ruckeri QMA0440) among the complete sequences contained only one 16S rRNA gene variant. The nucleotide differences are more common than the formation of insertions or deletions. Ins/del were detected in 19 of 45 complete genomes of Yersinia. Number of in/del and SNP between the most different copies were varied, but in 25 genomes it was ≤3. More than 10 mismatches between the most different copies were observed in genome Y. frederiksenii Y225 (20) and in Y. aldovae 670–83 (14). In the rest genomes, number of in/del and SNP was ≥4 and ≤ 10 between the most different copies.
Pairwise comparison of gene copy sequences in one genome defined homology exceeding 99% for all strains, except for Y. frederiksenii Y225. The mismatch of 16S rRNA copies of this genome is presented in Figure 1. The total nucleotide mismatches of 31 points were detected, 29 SNPs and two insertion/deletion (ins/del). 26 points are in variable regions that are commonly used for species and genus identification; more than half of the mismatches (20) were in regions V1-V4, which are the most variable, and sequencing of these gene regions is most often used for routine identification of microorganisms. Each 16S rRNA copy in the Y. frederiksenii Y225 genome is unique. The maximum number (20 SNPs and ins/del) of differences between copies of the gene was also defined in this genome.
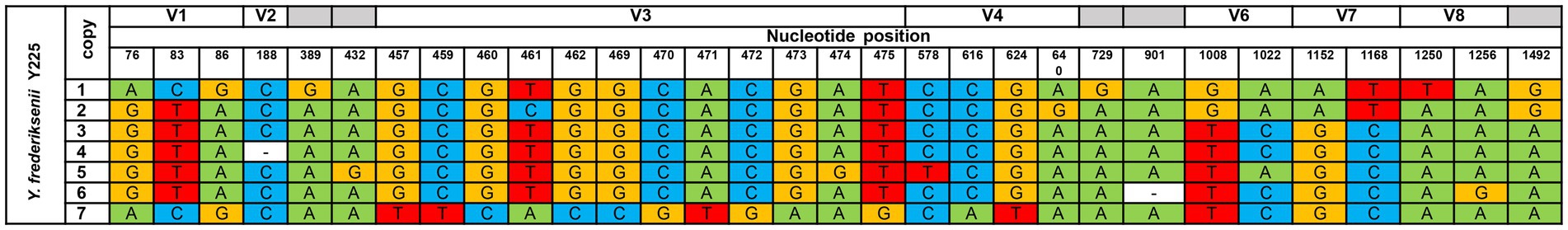
Figure 1. Comparison of the copies 16S rRNA gene Y. frederiksenii Y225. Coordinates of copies in chromosome (CP009364): 1 – 1663173–64703 bp; 2 – 3873144–3874674 bp; 3 – 3178807–3180337 bp; 4 – 2408072–2409601 bp; 5 – 3499799–3501329 bp; 6 – 3139875–3141404 bp; 7 – 3333051–3334581 bp. V1, V2, V3, V4, V6, V7, V8 – variable regions. Nucleotide positions were determined according to the E. coli gene nomenclature (Church et al., 2020).
Cluster analysis based on 16S rRNA gene sequence
For the analysis, only full-length genes were selected. From the complete and draft genomes, up to 10 scaffolds were taken of all copies of the gene. In most of the draft genomes one copy of the 16S rRNA gene was present; two full-length copies were identified in only four draft genomes. The full-length assembled 16S rRNA gene was absent in 36 drafts (9%). The total number of 16S rRNA genes (644 sequences) was aligned using MEGA11 (ClustalW algorithm). Each species of the genus Yersinia was represented by at least one copy of the 16S rRNA gene. Pairwise comparisons of the 16S rRNA genes are provided in Supplementary Table S3. A phylogenetic tree was constructed using the Neighbor joining algorithm. The expanded phylogenetic tree is presented in Supplementary Figure S1. The branches composed of identical genes or genes with some nucleotide differences were compressed (Supplementary Figure S2). As a result, the sequences of 16S rRNA genes formed six large clades, namely – 1a, 1b, 2a, 2b, 3a, and 3b, on the phylogenetic tree.
Clade 1a comprises two branches, the first includes all sequences of Y. ruckeri 16S rRNA and, the second contains part of the Y. kristensenii 16S rRNA gene sequences. The average homology rates inside the branches is 99.94 and 99.91%, respectively. Group 1b comprises of three branches. The first branch included all 16S rRNA sequences of Y. bercovieri and Y. aleksiciae, with an average homology of 99.66%. The second branch comprises all 16S rRNA genes of Y. mollaretii, with an average homology of 99.93%. The third branch includes the 16S rRNA sequences of several species: Y. pekkanenii, Y. proxima, Y. aldovae, Y. intermedia, Y. kristensenii, and Y. rochesterensis, with an average 16S rRNA homology of 99.64%. A few strains of Y. intermedia and Y. rochesterensis have the identical gene sequences.
Clade 2a included all 16S rRNA sequences of Y. massiliensis, and the average homology was 99.85%. The clade 2b are formed by three branches. The first branch included 16S rRNA sequences of Y. similis, Y. pseudotuberculosis, Y. pestis, and Y. wautersii, with an average homology of 99.76%. The second branch contains 16S rRNA sequences of Y. alsatica, with an average homology of 99.71%. The third branch includes Y. frederiksenii 16S rRNA sequences; the average gene homology was 99.91%.
Clade 3a contains two branches (Y. entomophaga/Y. nurmii and Y. vastinensis/Y. frederiksenii) and a separate 16S rRNA sequence from the complete genome of Y. frederiksenii Y225. Average homology of the group – 99.5%.
Clade 3b includes the 16S rRNA of Y. rohdei/Y. thracica, Y. canariae, Y. artesiana, Y. enterocolitica, and Y. hibernica/Y. kristensenii, with separate located sequences of Y. rohdei 68/02 and Y. rohdei 3,343, as well as the 16S rRNA gene of Y. aldovae IP07632. The average 16S rRNA homology in clade 3b was 99.05%.
Analysis of the 16S rRNA genes revealed identical sequences in some genomes of strains related to different species, according to NCBI data. The results are listed in Table 3.

Table 3. Sequences of identical 16S rRNA genes in the genomes of different Yersinia species.
Identification and phylogenetic analysis of Yersinia genomes based on core SNPs
The determination of core SNPs in 368 genomes of non-pathogenic Yersinia and genomes of Y. enterocolitica subsp. enterocolitica 8081, Y. enterocolitica subsp. palearctica Y11, Y. pseudotuberculosis IP 32953, Y. pestis CO92, and Y. wautersii WP-931201 was performed using Snippy 4.6.0 software (Snippy, 2015). A phylogenetic tree based on the 9,494 core SNPs was built using neighbor joining algorithm in the FigTree 1.4.3 software (FigTree 1.4.3, 2017) (Figure 2). The detailed phylogenetic tree is presented in Supplementary Figure S3.
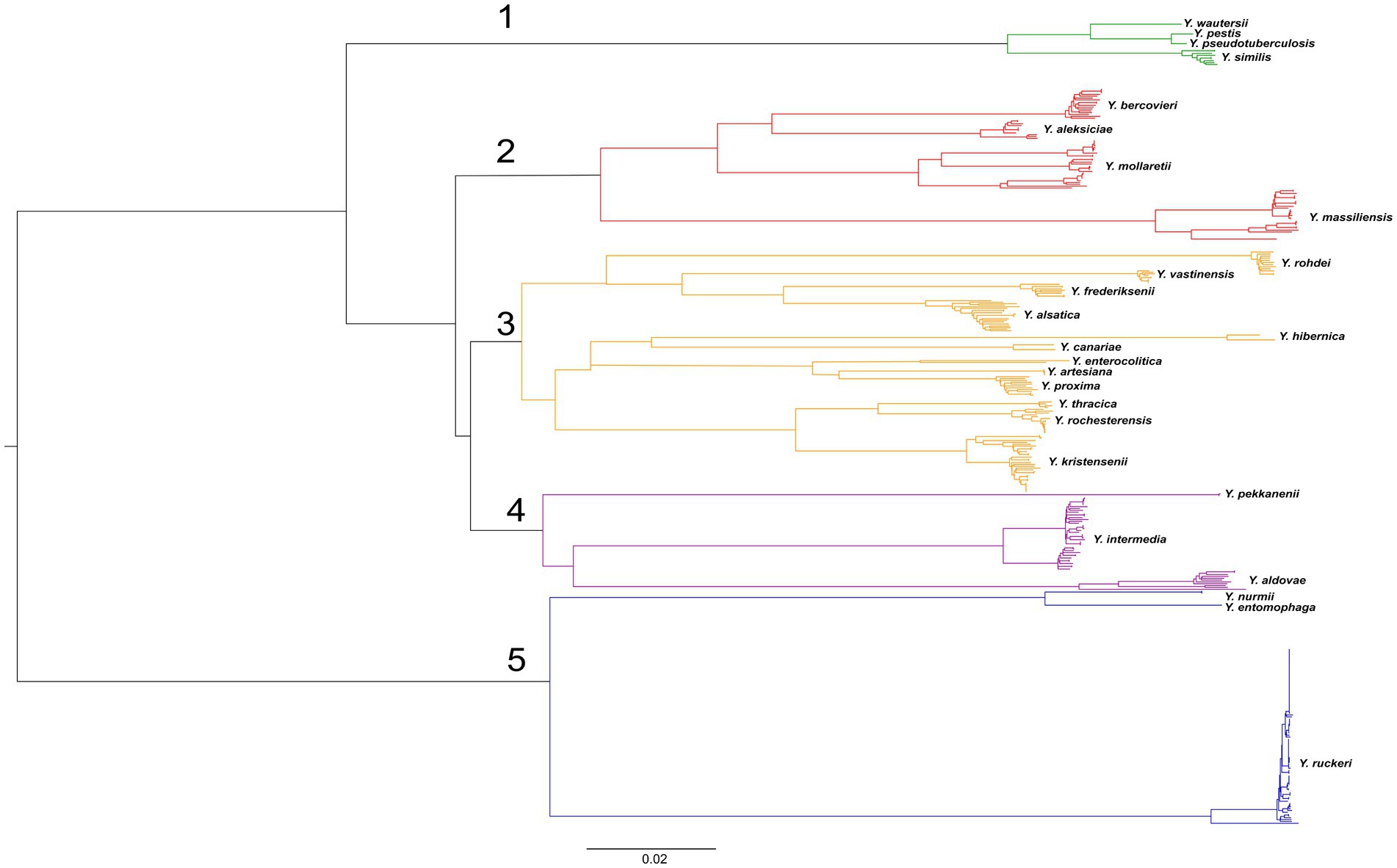
Figure 2. Phylogenetic tree of Yersinia genomes. A phylogenetic tree was conducted using the neighbor-joining (NJ) algorithm based on 9,494 core SNPs generated by the Snippy 4.6.0 software. This tree was rooted using midpoint option. The groups of genomes observed in the tree were identified according to the species names of the type strains. The scale bar shows the expected number of substitutions per site. Bar, 0.02 substitutions per nucleotide position.
The genomes formed five clades on the tree. The first clade includes the genomes of the closely related species Y. pseudotuberculosis, Y. pestis, Y. similis, and Y. wautersii. This clade can be reliably distinguished from other Yersinia species. The second clade consists of the species Y. aleksiciae, Y. bercovieri, Y. mollaretii, and the neighboring species Y. massiliensis on long, distant branches. The third clade can be distinguished on two branches.
The first included species Y. frederiksenii, Y. vastinensis, Y. alsatica, and Y. rohdei. The second branch comprises two groups: Y. enterocolitica together with its related species Y. proxima, Y. artesiana, and Y. canariae, and the closely related species Y. kristensenii, Y. thracica, and Y. rochesterensis. The fourth clade comprises the separate branches of Y. pekkanenii, Y. aldovae, and Y. intermedia. The fifth clade significantly outlies the other clades and consists of two branches: the first is represented by genomes of Y. ruckeri, and the second composes of genomes of related species Y. nurmii and Y. entomophaga.
The species relationship of most genomes of the genus Yersinia was matched with those specified in the GenBank database (NCBI), but taxonomic inconsistencies in 34 bacterial genomes were determined (Table 4). The genomes of 25 Yersinia strains were deposited as Y. frederiksenii, but according to the results of the analysis of core SNPs, 12 were assigned as Y. alsatica, nine as Y. massiliensis, four as Y. vastinensis, and one as Y. rochesterensis. Six of the Y. kristensenii genomes were identified as Y. rochesterensis and one as Y. hibernica. One Y. intermedia genome was located between Y. proxima genomes in the phylogenetic tree, and another was located between Y. massiliensis genomes.
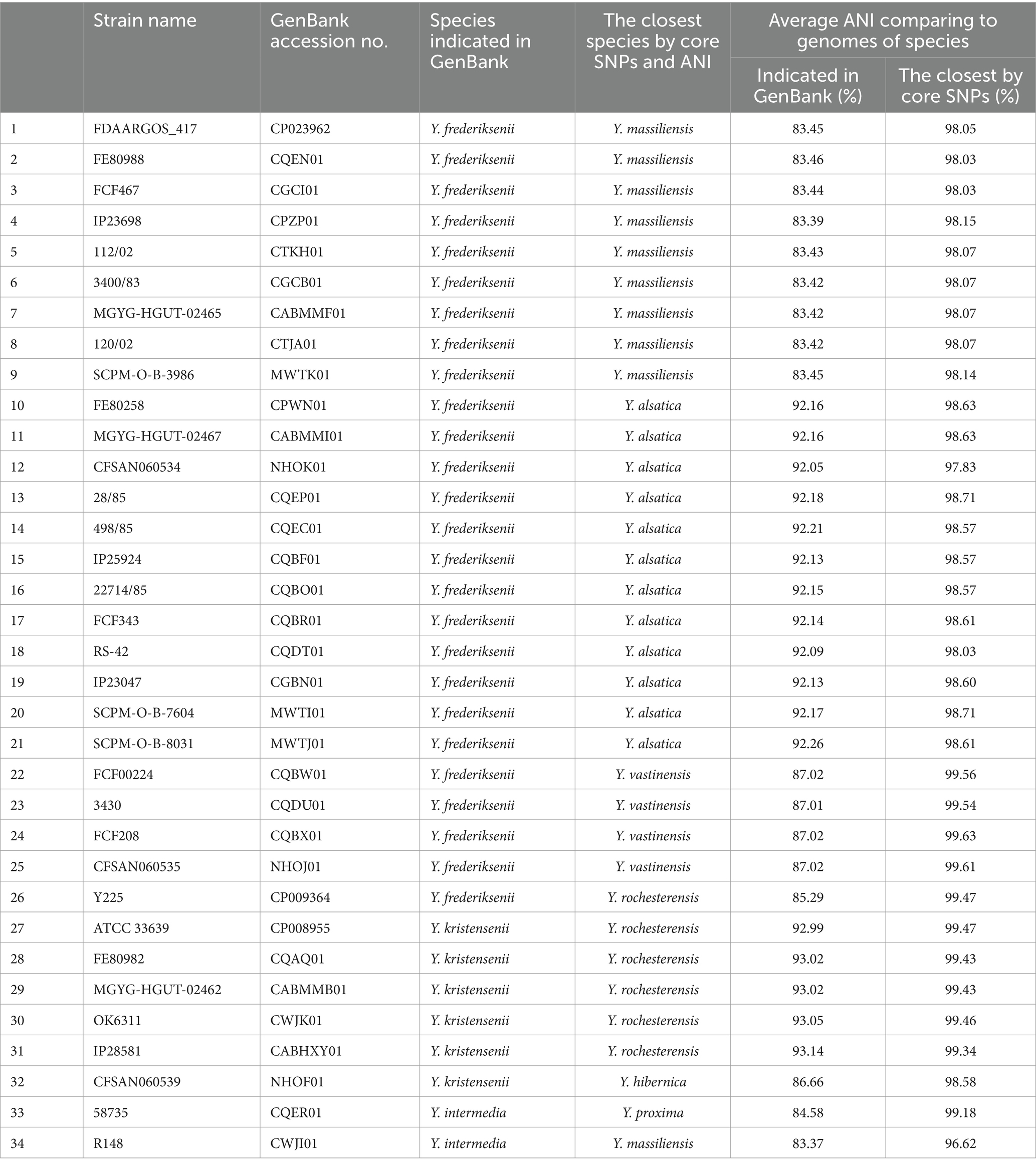
Table 4. Identification of mismatched Yersinia genomes.
Identification of Yersinia genomes based on ANI
ANI was calculated pairwise for each of the 373 genomes (Supplementary Table S4).
The genomes were grouped according to an ANI value of 95–96% which is the bacterial species threshold (Riesco and Trujillo, 2024). The species identified in of the same 34 bacterial genomes did not match those specified in the GenBank database (NCBI). The taxonomic affiliations of these genomes based on core SNPs were determined according to the ANI results. The mean ANI value for each of the 34 genomes relative to the genomes of the species indicated in the NCBI database and the genomes of the species identified by core SNPs are shown in the Table 4.
The average ANI value for all genomes of each species was determined in relation to the genomes of strains belonging to other species (Figure 3). Generally, Yersinia can be divided into three groups according to ANI results. The first group consisted of Y. ruckeri, Y. nurmii, and Y. entomophaga, with the lowest ANI (80.27–81.90%) in relation to other Yersinia species. The ANI values between Y. entomophaga and Y. nurmii is very close to the species threshold (94.71%).
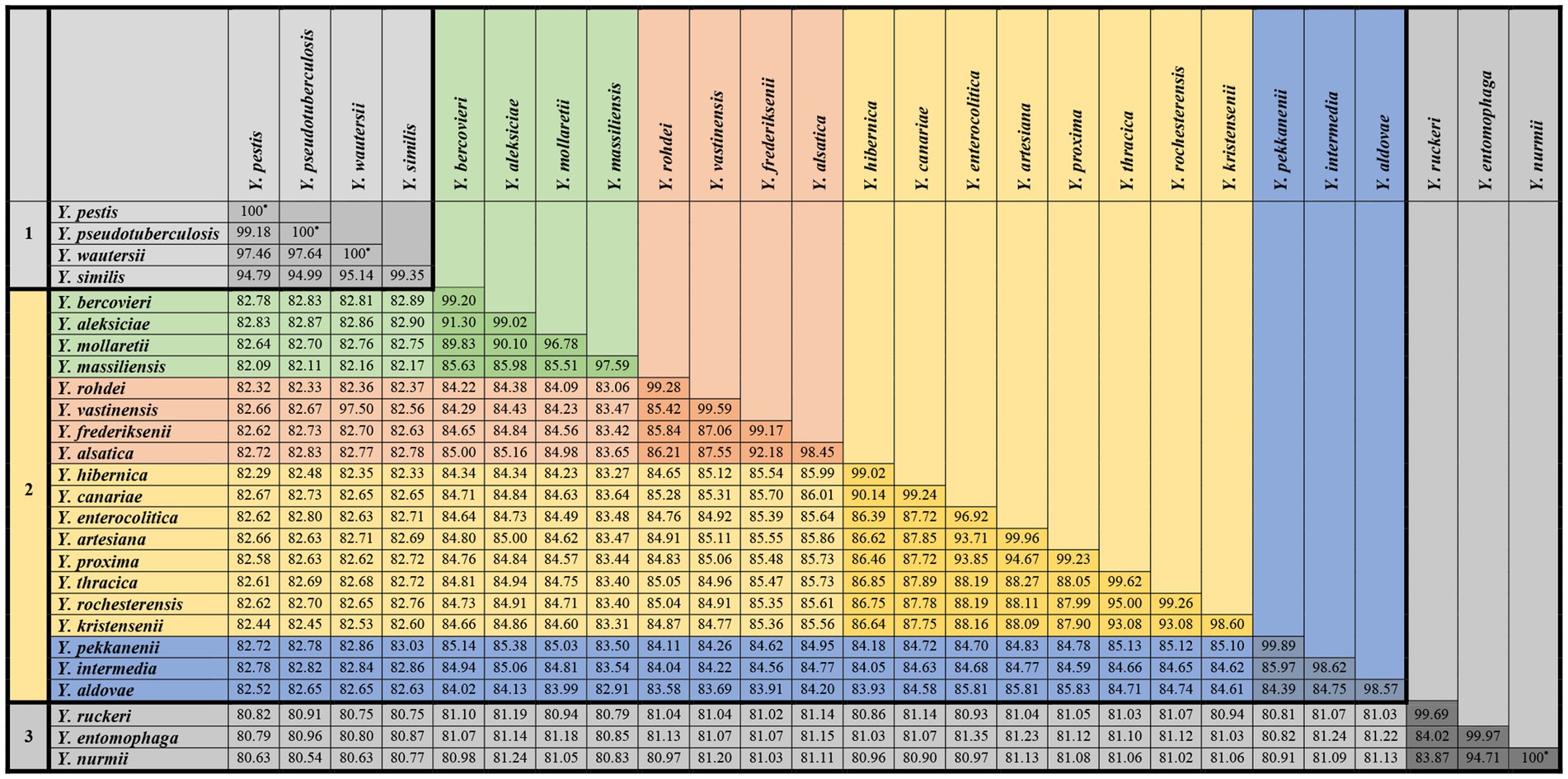
Figure 3. Average ANI values for all genomes of each species Yersinia were determined in relation to those strains belonging to other species.
The second group, consisting of four species, Y. pseudotuberculosis, Y. pestis, Y. wautersii, and Y. similis, was distinguished from other Yersinia species. The ANI value between the genomes of Y. pseudotuberculosis IP 32953, Y. pestis CO92, and Y. wautersii WP-931201 is more 97%, which is above the species threshold. These species are sometimes integrated into the Y. pseudotuberculosis complex, and a high ANI value is a significant reason for combining these species into one complex. The ANI of Y. similis ranges from 94.71 to 95.20% relatively to Y. pseudotuberculosis complex, demonstrating a close relationship. The ANI values between this group and other Yersinia species fluctuated from 81.94 to 83.26%.
The third group included the remaining species with ANI values of 81.93–95.16% between them. In this group, three pairs of species had high ANI values, indicating closer genetic relationships. These pairs of species were Y. aleksiciae and Y. bercovieri; Y. alsatica and Y. frederiksenii; Y. canariae and Y. hibernica. The two trios of species Y. enterocolitica, Y. artesiana, and Y. proxima; Y. kristensenii, Y. rochesterensis, and Y. thracica have higher 90% ANI values, indicating a close relationship.
The minimum and maximum ANI values within each Yersinia species were evaluated (Figure 4). The minimum ANI values relevant to the bacterial species threshold were found between the genomes of Y. mollaretii and Y. massiliensis at 94.95 and 95.42%, respectively.

Figure 4. Minimum and maximum ANI values within Yersinia non-pathogenic species.
A study of non-pathogenic Yersinia genomes using the core SNPs showed the existence of separate lineages (probable subspecies) within the species Y. mollaretii, Y. massiliensis, Y. intermedia, and Y. kristensenii. The separate phylogenetic trees (SplitsTree4 software, NJ method) (SplitsTree4, 1996) were constructed for these species, and ANI values were determined (Figure 5).
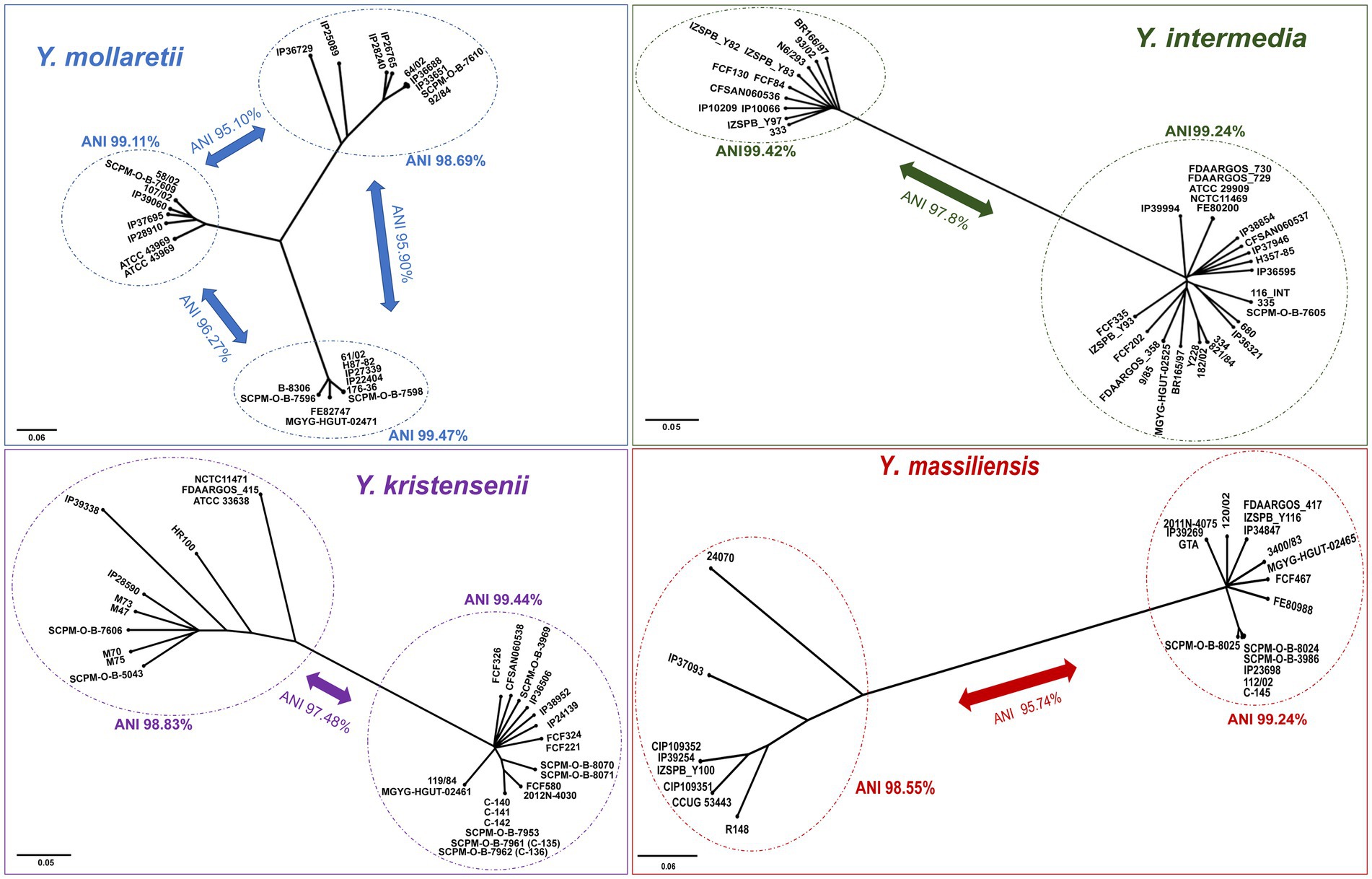
Figure 5. Phylogenetic trees of Y. mollaretii, Y. massiliensis, Y. intermedia, and Y. kristensenii showing the existence of the lineages into the species. A phylogenetic tree was conducted using the neighbor-joining (NJ) algorithm based on the core SNPs generated by the Snippy 4.6.0 software (Y. mollaretii – 259,428 SNPs, Y. massiliensis – 199,117 SNPs, Y. intermedia – 111,428 SNPs, and Y. kristensenii – 148,020 SNPs). The scale bar shows the expected number of substitutions per site. Bar, 0.05 or 0.06 substitutions per nucleotide position. The ANI values of the lineages are indicated.
The comparison of the core SNPs showed that the genomes of Y. mollaretii and Y. massiliensis are divided into three and two lineages, respectively. The ANI values between the three Y. mollaretii lineages ranged from 95.10 to 96.27%, whereas the values within each lineage were higher (98.68%). The ANI value between two lineages of the Y. massiliensis genome was 95.74%, whereas the values within each lineage were 99.24 and 98.55%.
The genomes of species Y. kristensenii and Y. intermedia were divided into two lineages by the results of core SNPs. The ANI value between lineages of Y. kristensenii was 97.40%. The mean ANI value within the genomes of the first lineage was 99.44%, the second lineage is more heterogeneous, the mean ANI value was 98.83%. The genomes of Y. intermedia can also be divided into two lineages with mean ANI value of 97.80%. The ANI values within the first and second lineages are 99.24 and 99.42%, respectively.
Comparison of identification based on the 16S rRNA gene, core SNPs, and ANI
According to the clustering results, some groups included strains belonging to different species, as indicated in the NCBI database. Thus, group 2a included all 16S rRNA sequences of Y. massiliensis strains, as well as 11 genes of Y. frederiksenii (7 gene copies from the complete genome of strain FDAARGOS_417, as well as one copy each from the draft genomes of strains - FE80988, FCF467, 120/02, SCPM-O-B-3986) and one gene from the draft genome of Y. intermedia R148. According to the core SNPs and ANI analyses, these strains belong to the Y. massiliensis.
Some genes of Y. frederiksenii strains CFSAN060534, 28/85, 22,714/85, IP23047, SCPM-O-B-7604, and SCPM-O-B-8031 were grouped with the genes of Y. alsatica. Genes of Y. frederiksenii strains 3,430, FCF208, CFSAN060535, and FCF224 were clustered with Y. vastinensis as well. Based on core SNPs and ANI analyses, these Y. frederiksenii strains belong to confirmed species. In the same way the strains Y. kristensenii CFSAN060539 (Y. hibernica), Y. intermedia R148 (Y. massiliensis) Y. intermedia 58735 (Y. proxima) were misidentified.
However, identical 16S rRNA gene sequences were identified for Y. intermedia and Y. rochesterensis in the two groups of strain. The first group included Y. intermedia (IP39994, IZSPB_Y97, 93/02, FDAARGOS_729, FDAARGOS_730, NCTC11469, FDAARGOS_358, SCPM-O-B-10209, N6/293), Y. rochesterensis (IP37484, IP35638, IP38810, SCPM-O-B-9106 (C-191), Y231, ATCC BAA-2637), and strains Y. frederiksenii Y225, Y. kristensenii (IP28581, MGYG-HGUT-02462, OK6311, FE80982). However, the strains Y. frederiksenii and Y. kristensenii belong to Y. rochesterensis, according to genome analysis. The second group of identical 16S rRNA gene sequences consisted of Y. intermedia FCF130, Y. intermedia FCF84, Y. rochesterensis IP38921, Y. rochesterensis ATCC BAA-2637, Y. rochesterensis Y231, and Y. rochesterensis ATCC 33639.
Analysis of 16S rRNA gene variants in non-pathogenic Yersinia species
The sequences of 16S rRNA genes within the species were also analyzed according to species identification using core SNPs and ANI values. Table 5 shows the minimum and average 16S rRNA homology values and, the number of sequence variants within the Yersinia species. For each species, the most common 16S rRNA sequence was determined.

Table 5. Variability of 16S rRNA genes within non-pathogenic Yersinia species.
The average 16S rRNA homology inside non-pathogenic Yersinia species exceeded 99%. The number of 16S rRNA variants in the species is likely depends on the total number of gene sequences. The largest number of 16S rRNA variants was 18 in Y. ruckeri (total 203 sequences). However, in Y. aldovae, 12 gene variants were identified among 14 sequences, and this species also has the smallest gene homology of 98.43%. 15 gene variants were found in Y. intermedia, but three of them are predominant, one of them observed in 14 cases, the second in 12, and the third 11. Two predominant sequences of Y. mollaretii were determined. They were found in 12 and 11 cases, respectively. Two dominant sequences were also found in Y. bercovieri, and both were observed in seven cases.
Discussion
Since the 1980s, the 16S rRNA gene has been used in phylogenetic studies of bacteria (Clarridge, 2004). This gene has been considered as the best target for identification because it exists in all known prokaryotic genomes and has conserved and variable regions (Srinivasan et al., 2015). These properties made the 16S rRNA gene suitable for taxonomy studies. However, the presence of multiple copies of rRNA operons and intragenomic heterogenicity of 16S rRNA genes are the limiting factors for species identification (Muhamad Rizal et al., 2020; Watts et al., 2017). In the study were analyzed 2013 complete genomes of bacteria and archaea, and intragenomic heterogenicity was found in 952 genomes (585 species), but the divergence was less than 1% in 87.5% of the genomes (Sun et al., 2013).
Although the genus Yersinia contains three known human pathogens (Y. pestis, Y. enterocolitica and Y. pseudotuberculosis), the remaining species have not been studied sufficiently. Recently, with the expansion of genome studies, the taxonomy of the Yersinia genus has been continuously refined. In previous years, a few new species of Yersinia have been described. The genetic homology of 16S rRNA genes in the genus Yersinia is high, and even identical 16S rRNA gene sequences can be found between distinct species (Clarridge, 2004; Rodriguez-R et al., 2018). However, studies of 16S rRNA genes sequences, and using this gene for the identification of non-pathogenic Yersinia with verification by methods based on whole genome analysis are lacking. Hao et al. studied the identification of Yersinia spp. using copy diversity in the chromosomal 16S rRNA gene sequence. In this study, we used complete and draft genomes deposed from the NCBI Genome database, which is used by researchers worldwide. We analyzed the 16S rRNA genes and used ANI and core SNPs to identify the species of Yersinia. First, we compared the sequences of 16S rRNA gene copies in the complete genomes of non-human-pathogenic Yersinia from NCBI. The homology of 16S rRNA copies in each genome exceeded 99.5% for all strains except for one. It has been shown that many species have gene copies in their genomes that differ by 1–1.3% (Sun et al., 2013). The intragenomic heterogenicity of the 16S rRNA gene in complete Yersinia genomes is above the threshold for species determination and should not affect species identification. The intragenomic heterogenicity of the species Y. wautersii, Y. vastinensis, Y. artesiana, Y. proxima, Y. pekkanenii, Y. thracica, and Y. nurmii is unknown due to the absence of complete genomes available from the NCBI database. Only one complete genome of Y. aldovae, Y. aleksiciae, Y. alsatica, Y. entomophaga, Y. kristensenii, Y. mollaretii, Y. rohdei, and Y. similis was available; other non-pathogenic species were presented with only a few complete genomes. However, in a small cohort of Yersinia, above 50% of genomes have four or more variants of the 16S rRNA gene. It was revealed that among 327 investigated draft genomes of non-pathogenic Yersinia, 11% did not have a full-length 16S rRNA gene. Generally, the gene consists of two separate parts or is not assembled at 100% length. One copy of 16S rRNA contains 287 (87.8%) draft genomes. Typically generated by WGS short reads (100–600 bp) related to repeat regions of the genome are assembled into one variant. Generally, the algorithm used for assembly software eliminates variable nucleotides of lower frequency. In this case, it was impossible to evaluate the intragenomic heterogenicity of the 16S rRNA gene because it was compiled from single reads from seven probable different copies.
The investigations of 16S rRNA revealed that homology of genes belonging to different bacterial species is very high (Hao et al., 2016). In our study, the average degree of gene homology in non-pathogenic Yersinia was 98.76%, and the maximum variability was 2.85%. This high homology could limit the use of 16S rRNA for species identification of these bacteria. The low degree of genetic heterogenicity of the gene (0.36%) was determined in group Y. pekkanenii/Y. proxima/Y. aldovae/Y. intermedia/Y. kristensenii/Y. rochesterensis. Previously, it was reported that some bacterial species have identical 16S rRNA sequences (Rodriguez-R et al., 2018; Hao et al., 2016). In our study, identical gene sequences were found in the genomes of the Y. intermedia and Y. rochesterensis strains identified using ANI and core SNPs analyses.
16S rRNA genes are considered as species specific markers for prokaryotes phylogenetic studies. The studies have confirmed the existence of 16S rRNA horizontal transfer between different species, as well as intergenomic and intragenomic recombination of 16S rRNA gene regions (Kitahara and Miyazaki, 2013; Sun et al., 2013). All these facts indicate that using only the 16S rRNA gene for identification or phylogenetic studies is not rational.
In our study, the phylogenetic tree based on 16S rRNA genes differed from the tree based on core SNPs of the genomes. The phylogenetic analysis based on core SNPs showed that three species, Y. ruckeri, Y. entomophaga, and Y. nurmii, form a separate clade of the genus Yersinia, which is consistent with other studies. Y. ruckeri clusters with part of the genomes of Y. kristensenii, whereas Y. entomophaga and Y. nurmii are grouped with Y. vastinensis on the 16S rRNA phylogenetic tree. The species Y. vastinensis was recently described and genetically related to Y. frederiksenii and Y. alsatica. However, these two species are in another group, with Y. pseudotuberculosis, Y. pestis, Y. similis, and Y. wautersii, according to the 16S rRNA-based phylogenetic tree. Based on the results of core SNPs and ANI analyses, the Y. pseudotuberculosis complex and Y. similis form a separate clade of the genus Yersinia, which is not related to other species.
The species Y. aldovae, Y. intermedia, Y. kristensenii, Y. rochesterensis, Y. pekkanenii, and Y. proxima have 99.64% homology with 16S rRNA genes and are grouped together in the tree. On the phylogenetic tree based on core SNPs, the species Y. aldovae, Y. intermedia, and Y. pekkanenii form a clade consisting of separated branches corresponding to each species. The closely related species Y. kristensenii, Y. thracica, and Y. rochesterensis were grouped together; Y. proxima was located with Y. enterocolitica and its related species, Y. artesiana and Y. canariae.
The 16S rRNA gene phylogenetic tree, shows that the Y. kristensenii strains are separated into two groups. The first group forms a separate branch within group 1a, and the second group is included in the third group of 1b, consisting of high homologous 16S rRNA sequences of Y. pekkanenii, Y. proxima, Y. aldovae, Y. intermedia, Y. kristensenii, and Y. rochesterensis. The 16S rRNA gene sequences of strains Y. rohdei 68/02 and Y. rohdei 3,343 are clustered separately from other gene sequences of Y. rohdei strains that are grouped with Y. thracica.
In addition, in the phylogenetic tree based on the 16S rRNA sequences, some gene copies from complete genomes are located very far from each other because of high variability. Six gene copies of Y. frederiksenii Y225 were clustered into group 1b, and the seventh into group 3a. Six gene copies of Y. aldovae IP07632 were clustered into group 1b, and one into group 3b.
In our study, species identification of non-pathogenic Yersinia using core SNPs was correlated with ANI results. These methods are based on whole genome comparisons and allow to determine species identification and the relationship between strains. For species identification of poorly studied bacteria or those with high 16S rRNA gene homology, such as non-pathogenic Yersinia, it is better to use a set of methods based on whole genome analysis.
WGS provides information about the whole genome, and in addition to species identification, it can be used to study phylogeny and, identify resistance genes and, virulence factors, as well as plasmids, prophages, and other significant genetic traits.
Although the 16S rRNA gene is not well suited for studying phylogenetic relationships, this method compared with WGS is not expensive, performs easier, and does not require special technical staff. Nevertheless, extensive experience has been accumulated in using 16S rRNA genes to determine species identity, and convenient databases exist for interpreting the data. Because of the presence of this gene in all known microorganisms and the existence of universal primers, the 16S rRNA gene is a suitable target for studying metagenomic communities.
In summary, the 16S rRNA gene is not the most appropriate candidate gene for the accurate identification of Yersinia species. This is due to several reasons. The first limitation is the poorly studied non-pathogenic Yersinia and not enough numbers of genomes or sequences of 16S rRNA genes in the available databases. Second, in some cases, the results are difficult to interpret due to small differences between the 16S rRNA genes of the Yersinia genus (Clarridge, 2004). In our study, the average degree of gene homology was 98.76%. In some cases, identical 16S rRNA gene sequences correspond to different species (Rodriguez-R et al., 2018); in others, the distinction could be only one or a few nucleotides located outside the examined part of the gene. In addition, existing slightly different 16S rRNA gene variants within the species could be confused the researchers; especially if the species have limited available sequences or genomes in databases. The intragenomic heterogenicity of the 16S rRNA gene in complete Yersinia genomes exceeded the threshold for species determination and should not affect species identification. However, due to the small number of complete genomes in NCBI, this could not be clarified properly.
However, in a small cohort of Yersinia complete genomes, above 50% of them have four or more variants of the 16S rRNA gene in each. In most of the investigated draft genomes (87.8%), only one copy of 16S rRNA gene was present. The most likely this sequence was compiled from different copies of the gene. Besides this the full-length assembled 16S rRNA gene was absent in 36 drafts (9%). In additional, the other identification mismatches could be related to the development of the genus Yersinia taxonomy when there is no time to change old species names or revise data.
In our case, we determined that it was better to use core SNPs or ANI for accurate species identification of Yersinia strains than to sequence the 16S rRNA gene. Using for this purpose the methods based on whole genome comparison let to avoid misidentification. Of course, performing of whole genome sequencing and bioinformatics analysis requires expensive equipment and professionals, but in ambiguous and controversial cases, this is the best method for species identification and determining the phylogenetic relationship among strains.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
AK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft. AS: Data curation, Formal analysis, Investigation, Software, Validation, Writing – original draft. YS: Data curation, Formal analysis, Investigation, Validation, Writing – original draft. SD: Methodology, Project administration, Supervision, Writing – review & editing. AA: Funding acquisition, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Ministry of Science and Higher Education of the Russian Federation, agreement number 075-15-2019-1671.
Acknowledgments
We thank Lidia A. Shishkina and Viktor I. Solomentsev for assistance in genome sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1519733/full#supplementary-material
SUPPLEMENTARY TABLE S1 | The complete and draft genomes of Yersinia used in the study.
SUPPLEMENTARY TABLE S2 | Sequencing technologies, assembly methods and NCBI Genome accession numbers of Yersinia complete genomes used in the study.
SUPPLEMENTARY TABLE S3 | Pairwise comparisons of the 16S rRNA genes of the Yersinia genomes used in the study.
SUPPLEMENTARY TABLE S4 | ANI pairwise for the Yersinia genomes used in the study.
SUPPLEMENTARY FIGURE S1 | Neighbor-joining phylogenetic tree based on 644 aligned genes of the 16S rRNA genus Yersinia.
SUPPLEMENTARY FIGURE S2 | Neighbor-joining phylogenetic tree based on 644 aligned genes of the 16S rRNA genus Yersinia. The branches composed of identical genes or genes with some nucleotide differences were compressed.
SUPPLEMENTARY FIGURE S3 | Neighbor joining tree based on 9494 core SNPs in Yersinia genomes. This tree was rooted using midpoint option. The groups of genomes observed in the tree were identified according to the species names of the type strains.
Footnotes
References
Abellan-Schneyder, I., Matchado, M. S., Reitmeier, S., Sommer, A., Sewald, Z., Baumbach, J., et al. (2021). Primer, pipelines, parameters: issues in 16S rRNA gene sequencing. mSphere 6:e01202-20. doi: 10.1128/mSphere.01202-20
Achtman, M., Zurth, K., Morelli, G., Torrea, G., Guiyoule, A., and Carniel, E. (1999). Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 96, 14043–14048. doi: 10.1073/pnas.96.24.14043
Atkinson, S., and Williams, P. (2016). Yersinia virulence factors - a sophisticated arsenal for combating host defences. F1000Res 5:F1000 Faculty Rev-1370. doi: 10.12688/f1000research.8466.1
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Chen, P. E., Cook, C., Stewart, A. C., Nagarajan, N., Sommer, D. D., Pop, M., et al. (2010). Genomic characterization of the Yersinia genus. Genome Biol. 11:R1. doi: 10.1186/gb-2010-11-1-r1
Church, D. L., Cerutti, L., Gürtler, A., Griener, T., Zelazny, A., and Emler, S. (2020). Performance and application of 16S rRNA gene cycle sequencing for routine identification of bacteria in the clinical microbiology laboratory. Clin. Microbiol. Rev. 33, e00053–e00019. doi: 10.1128/CMR.00053-19
Clarridge, J. E. 3rd. (2004). Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 17, 840–862. doi: 10.1128/CMR.17.4.840-862.2004
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Cunningham, S. A., Jeraldo, P., and Patel, R. (2019). Yersinia kristensenii subsp. rochesterensis subsp. nov., isolated from human feces. Int. J. Syst. Evol. Microbiol. 69, 2292–2298. doi: 10.1099/ijsem.0.003464
Edgar, R. (2018). Taxonomy annotation and guide tree errors in 16S rRNA databases. PeerJ 6:e5030. doi: 10.7717/peerj.5030
FigTree 1.4.3 (2017). Available at: http://tree.bio.ed.ac.uk/software/figtree (Accessed March 12, 2024)
Gonzalez, E., Pitre, F. E., and Brereton, N. J. B. (2019). ANCHOR: a 16S rRNA gene amplicon pipeline for microbial analysis of multiple environmental samples. Environ. Microbiol. 21, 2440–2468. doi: 10.1111/1462-2920.14632
Hao, H., Liang, J., Duan, R., Chen, Y., Liu, C., Xiao, Y., et al. (2016). Yersinia spp. identification using copy diversity in the chromosomal 16S rRNA gene sequence. PLoS One 11:e0147639. doi: 10.1371/journal.pone.0147639
Hsieh, Y. P., Hung, Y. M., Tsai, M. H., Lai, L. C., and Chuang, E. Y. (2022). 16S-ITGDB: an integrated database for improving species classification of prokaryotic 16S ribosomal RNA sequences. Front. Bioinform. 2:905489. doi: 10.3389/fbinf.2022.905489
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10:5029. doi: 10.1038/s41467-019-13036-1
Kitahara, K., and Miyazaki, K. (2013). Revisiting bacterial phylogeny: natural and experimental evidence for horizontal gene transfer of 16S rRNA. Mob. Genet. Elements 3:e24210. doi: 10.4161/mge.24210
Le Guern, A. S., Savin, C., Angermeier, H., Brémont, S., Clermont, D., Mühle, E., et al. (2020). Yersinia artesiana sp. nov., Yersinia proxima sp. nov., Yersinia alsatica sp. nov., Yersina vastinensis sp. nov., Yersinia thracica sp. nov. and Yersinia occitanica sp. nov., isolated from humans and animals. Int. J. Syst. Evol. Microbiol. 70, 5363–5372. doi: 10.1099/ijsem.0.004417
Mares, C. A., Lugo, F. P., Albataineh, M., Goins, B. A., Newton, I. G., Isberg, R. R., et al. (2021). Heightened virulence of Yersinia is associated with decreased function of the YopJ protein. Infect. Immun. 89:e0043021. doi: 10.1128/IAI.00430-21
Mignard, S., and Flandrois, J. P. (2006). 16S rRNA sequencing in routine bacterial identification: a 30-month experiment. J. Microbiol. Methods 67, 574–581. doi: 10.1016/j.mimet.2006.05.009
Muhamad Rizal, N. S., Neoh, H. M., Ramli, R., A/L K Periyasamy, P. R., Hanafiah, A., Abdul Samat, M. N., et al. (2020). Advantages and limitations of 16S rRNA next-generation sequencing for pathogen identification in the diagnostic microbiology laboratory: perspectives from a middle-income country. Diagnostics 10:816. doi: 10.3390/diagnostics10100816
Nguyen, S. V., Greig, D. R., Hurley, D., Donoghue, O., Cao, Y., McCabe, E., et al. (2020a). Yersinia canariae sp. nov., isolated from a human yersiniosis case. Int. J. Syst. Evol. Microbiol. 70, 2382–2387. doi: 10.1099/ijsem.0.004047
Nguyen, S. V., Muthappa, D. M., Eshwar, A. K., Buckley, J. F., Murphy, B. P., Stephan, R., et al. (2020b). Comparative genomic insights into Yersinia hibernica - a commonly misidentified Yersinia enterocolitica-like organism. Microb. Genom. 6:mgen000411. doi: 10.1099/mgen.0.000411
Park, S. C., and Won, S. (2018). Evaluation of 16S rRNA databases for taxonomic assignments using mock community. Genomics Inform. 16:e24. doi: 10.5808/GI.2018.16.4.e24
Reuter, S., Connor, T. R., Barquist, L., Walker, D., Feltwell, T., Harris, S. R., et al. (2014). Parallel independent evolution of pathogenicity within the genus Yersinia. Proc. Natl. Acad. Sci. U. S. A. 111, 6768–6773. doi: 10.1073/pnas.1317161111
Riesco, R., and Trujillo, M. E. (2024). Update on the proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 74:006300. doi: 10.1099/ijsem.0.006300
Rodriguez-R, L. M., Castro, J. C., Kyrpides, N. C., Cole, J. R., Tiedje, J. M., and Konstantinidis, K. T. (2018). How much do rRNA gene surveys underestimate extant bacterial diversity? Appl. Environ. Microbiol. 84, e00014–e00018. doi: 10.1128/AEM.00014-18
Savin, C., Criscuolo, A., Guglielmini, J., Le Guern, A. S., Carniel, E., Pizarro-Cerdá, J., et al. (2019). Genus-wide Yersinia core-genome multilocus sequence typing for species identification and strain characterization. Microb. Genom. 5:e000301. doi: 10.1099/mgen.0.000301
Snippy (2015). Available at: https://github.com/tseemann/snippy (Accessed March 12, 2024).
SplitsTree4 (1996). Available at: https://github.com/husonlab/splitstree4 (Accessed March 12, 2024).
Srinivasan, R., Karaoz, U., Volegova, M., MacKichan, J., Kato-Maeda, M., Miller, S., et al. (2015). Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PLoS One 10:e0117617. doi: 10.1371/journal.pone.0117617
Sulakvelidze, A. (2000). Yersiniae other than Y. enterocolitica, Y. pseudotuberculosis, and Y. pestis: the ignored species. Microbes Infect. 2, 497–513. doi: 10.1016/s1286-4579(00)00311-7
Sun, D. L., Jiang, X., Wu, Q. L., and Zhou, N. Y. (2013). Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 79, 5962–5969. doi: 10.1128/AEM.01282-13
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tatusova, T., Ciufo, S., Federhen, S., Fedorov, B., McVeigh, R., O'Neill, K., et al. (2015). Update on RefSeq microbial genomes resources. Nucleic Acids Res. 43, D599–D605. doi: 10.1093/nar/gku1062
van Baarlen, P., van Belkum, A., Summerbell, R. C., Crous, P. W., and Thomma, B. P. (2007). Molecular mechanisms of pathogenicity: how do pathogenic microorganisms develop cross-kingdom host jumps? FEMS Microbiol. Rev. 31, 239–277. doi: 10.1111/j.1574-6976.2007.00065.x
Van Ert, M. N., Easterday, W. R., Huynh, L. Y., Okinaka, R. T., Hugh-Jones, M. E., Ravel, J., et al. (2007). Global genetic population structure of Bacillus anthracis. PLoS One 2:e461. doi: 10.1371/journal.pone.0000461
Watts, G. S., Youens-Clark, K., Slepian, M. J., Wolk, D. M., Oshiro, M. M., Metzger, G. S., et al. (2017). 16S rRNA gene sequencing on a benchtop sequencer: accuracy for identification of clinically important bacteria. J. Appl. Microbiol. 123, 1584–1596. doi: 10.1111/jam.13590
Keywords: Yersinia , genome, taxonomy, phylogeny, WGS, 16S rRNA, core SNPs, ANI
Citation: Kislichkina AA, Sizova AA, Skryabin YP, Dentovskaya SV and Anisimov AP (2025) Evaluation of 16S rRNA genes sequences and genome-based analysis for identification of non-pathogenic Yersinia. Front. Microbiol. 15:1519733. doi: 10.3389/fmicb.2024.1519733
Edited by:
Haruo Suzuki, Keio University Shonan Fujisawa Campus, JapanReviewed by:
Jing Yang, National Institute for Communicable Disease Control and Prevention (China CDC), ChinaScott Van Nguyen, American Type Culture Collection, United States
Copyright © 2025 Kislichkina, Sizova, Skryabin, Dentovskaya and Anisimov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrey P. Anisimov, YW5pc2ltb3ZAb2JvbGVuc2sub3Jn; Angelina A. Kislichkina, YW5nZWxpbmFraXNsaWNoa2luYUB5YW5kZXgucnU=