 Liqing Hu1†
Liqing Hu1† Shan Lin2,3,4†Meng Zhang3,4Mengting Cai3,4Yuhang Shen3,4,5Peng Zeng3,6Xiaojun Song7
Shan Lin2,3,4†Meng Zhang3,4Mengting Cai3,4Yuhang Shen3,4,5Peng Zeng3,6Xiaojun Song7 Qiao Bian8
Qiao Bian8 Jina Gu9
Jina Gu9 Yun Luo10
Yun Luo10 Yu Chen3,4*
Yu Chen3,4* Dazhi Jin3,4,7*
Dazhi Jin3,4,7*- 1Department of Laboratory Medicine, The First Affiliated Hospital of Ningbo University, Ningbo, China
- 2TEDA Institute of Biological Sciences and Biotechnology, Nankai University, Tianjin, China
- 3School of Laboratory Medicine, Hangzhou Medical College, Hangzhou, China
- 4Key Laboratory of Biomarkers and In Vitro Diagnosis Translation of Zhejiang Province, Hangzhou, China
- 5Institute of Ageing Research, School of Basic Medical Sciences, Hangzhou Normal University, Hangzhou, China
- 6Department of Clinical Laboratory, Yuedong Hospital, The Third Affiliated Hospital of Sun Yat-sen University, Meizhou, China
- 7Laboratory Medicine Center, Department of Clinical Laboratory, Zhejiang Provincial People’s Hospital, Hangzhou Medical College, Hangzhou, China
- 8Zhejiang Provincial Center for Disease Control and Prevention, Hangzhou, China
- 9Department of Infectious Diseases, Ningbo No.2 Hospital, Ningbo, China
- 10School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW, Australia
Background: Nosocomial transmission of Clostridioides difficile infection (CDI) has been documented in Ningbo, China. However, data on molecular characteristics, clonal transmission, and risk factors of CDI in this region remain limited.
Methods: A cross-sectional study enrolled hospitalized patients with diarrhea during September to November 2021. Stool samples from all patients were tested for C. difficile, and isolated strains underwent toxin gene identification, genotyping, and antibiotic susceptibility testing. Whole-genome sequencing and epidemiological variables were analyzed in patients infected with C. difficile strains of the same sequence types (STs) to identify nosocomial transmission and risk factors for CDI.
Results: Of the 907 investigated patients, 115 (12.7%) had microbiologically proven CDI, as their diarrhea was associated with toxigenic C. difficile strains, which comprised 106 A+B+CDT−, 3 A−B+CDT−, and 6 A+B+CDT+. Predominant genotypes (ST2, ST3, ST35, and ST54) exhibited distinct antibiotic resistance patterns. ST54 strains showed higher resistance to erythromycin (100%) but lower resistance to moxifloxacin (18.2%) and gatifloxacin (18.2%) (χ2 = 10.24–16.65, p < 0.05). ST35 strains exhibited higher resistance to ciprofloxacin (66.7%) and tetracycline (33.3%) than other STs (χ2 = 13.30–20.19, p < 0.05). Genomic and epidemiological analysis revealed two nosocomial clonal transmission events caused by 5 ST35 strains (with ≤2 single nucleotide polymorphism differences), elucidating clonal transmission among different floors and buildings within the hospital. Prolonged hospitalization (> 10 days) (odds ratio [95% confidence interval], 1.76 [1.05–2.93]) and penicillin-class antibiotics (1.69 [1.11–2.58]) were risk factors for CDI, with the latter being an independent risk factor (1.57 [1.02–2.42]). For C. difficile ST35 infection, intensive care unit (12.00 [2.77–52.05]) and neurology departments (8.08 [1.46–44.65]) admissions were risk factors, with the latter as an independent risk factor (1.56 [1.01–2.40]).
Conclusion: Multiple C. difficile genotypes with varied antibiotic resistance patterns circulated in Ningbo, with ST35 causing nosocomial clonal transmission among different floors and buildings within the hospital. These findings and the identified risk factors necessitate enhanced surveillance and infection control in the region.
1 Introduction
Clostridioides difficile, a major nosocomial pathogen causing antibiotic-associated diarrhea, produces highly resistant spores that persist in hospital environments (Bi et al., 2023). C. difficile infection (CDI) manifests clinically from mild diarrhea to pseudomembranous colitis and even death (Shu et al., 2023). Posing an urgent threat to the U.S. healthcare system and public health, CDI resulted in approximately 223,900 cases, 12,800 deaths, and US$ 1 billion in medical costs in 2019 (Wu et al., 2022). Therefore, continuous surveillance of CDI and monitoring of its molecular epidemiology is crucial to detection of outbreaks and intuition of mitigation efforts toward its associated public health burden.
CDI has been an escalating concern in China, with nosocomial infection rates exceeding 10% (Wen et al., 2023). Despite variations in genotypes and antibiotic resistance among Asian countries (Luo et al., 2019), data on molecular characteristics of CDI across China still remain limited. Whole-genome sequencing (WGS) has revealed several nosocomial transmissions of specific STs in China. Previous studies have identified nosocomial transmission of ST1 in Beijing (Jia et al., 2016), ST81 in Shanghai (Qin et al., 2017), ST35 and ST37 in Zhejiang (Bi et al., 2023; Luo et al., 2024), suggesting both localized distribution and widespread dissemination of CDI. Common risk factors for CDI include advanced age, inappropriate antibiotic use, and proton pump inhibitor (PPI) administration (McDonald et al., 2018). In China, additional unique risk factors have been identified, such as lower age thresholds (Jin et al., 2017) and chronic diseases (Dai et al., 2020), and comorbidities (Saldanha et al., 2020). However, the risk factors of patients infected with nosocomial transmission-associated genotypes remain to be elucidated.
Ningbo, a major southeastern port city, has reported nosocomial transmission of multiple C. difficile genotypes, including ST3 and ST54 (Shu et al., 2023). To comprehensively understand the regional epidemiology of CDI, a cross-sectional study was conducted in a tertiary hospital. CDI prevalence, molecular characteristics, and clonal transmission were studied through genomic and epidemiological data. Furthermore, risk factors for CDI and for genotypes associated with nosocomial transmission were investigated to inform possible strategies for preventing and controlling CDI outbreaks.
2 Materials and methods
2.1 Study design and collection of stool samples
From September to November 2021, hospitalized patients with diarrhea were enrolled in this cross-sectional study at the Ningbo First Hospital, an academic tertiary care medical center. Diarrhea was defined as more than 3 loose, watery, or unformed stool passages within 24 h (McDonald et al., 2018). Stool samples were collected from hospitalized patients presenting with diarrhea. Only the first stool sample from each patient was included, exclusively for the purpose of research testing.
A case of CDI was defined as the presence of diarrhea concomitant with either a positive stool assay for toxigenic C. difficile or endoscopic/histopathological evidence of pseudomembranous colitis (McDonald et al., 2018). Parallel to stool collection, a standardized questionnaire was completed by clinicians for each patient with CDI to record clinical data (e.g., age, gender, previous antibiotic treatment within 8 weeks, hospital stay before sampling, and past medical history) as previously described (Jin et al., 2017). Ethical approval for this study was obtained from the Ethics Committee of Ningbo First Hospital (2021-R177).
2.2 C. difficile culture
Stool samples were treated with 95% alcohol, and the mixture was inoculated on cycloserine-cefoxitin-fructose agar with a selective supplement (Oxoid Inc., Basingstoke, UK), as previously described (Jin et al., 2017). After incubation for 48 h at 37°C in an anaerobic chamber (DW Microbiology Co., Ltd., Hangzhou, China), C. difficile strains were identified as described previously and stored at −80°C in brain-heart infusion broth with 10% glycerol until subsequent analysis (McDonald et al., 2005).
2.3 Detection of C. difficile toxin genes
Genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen Inc., CA, USA). The housekeeping gene tpi, toxin genes tcdA and tcdB, and binary toxin genes (CDT) cdtA and cdtB were detected by PCR using previously described primer sequences (Persson et al., 2008). For the tcdA primers, a 369-bp amplicon was obtained for the tcdA-positive/tcdB-positive (A+B+) strains, and a 100-bp amplicon was obtained for the tcdA-negative/tcdB-positive (A−B+) strains. Standard C. difficile strains (ATCC 43255, ATCC 700057, BAA-1801, BAA-1803, and BAA-1870) were used as positive and negative controls (Jin et al., 2017). Blank, positive, and negative controls were examined in parallel for each test.
2.4 Multi-locus sequence typing (MLST)
Seven housekeeping genes (adk, atpA, dxr, glyA, recA, sodA, and tpi) were amplified by PCR and MLST was performed as previously reported (Griffiths et al., 2010). The sequencing results were submitted to the MLST database1 to obtain the C. difficile alleles and STs. Based on the STs, the minimum spanning tree was constructed using the MSTree V2 algorithm with the GrapeTree software (Fernandes et al., 2018).
2.5 WGS and assembly
Genomic DNA was extracted from strains with identical STs collected from the same ward, using the method described previously. WGS libraries were prepared using the TruePrep™ DNA library prep kit V2 (Illumina, Santiago, CA, USA), and WGS was performed by the Illumina Hiseq X Ten platform with 150-base paired end reads. Sequence data were processed and quality controlled according to a standard pipeline as previously described (Preston et al., 2014). Raw sequence data were processed and quality controlled, as previously described (Bolger et al., 2014). Briefly, FASTQ-formatted Illumina sequence reads were quality controlled with a minimum quality Phred score of 30 (as a rolling average over four bases), and adapters and low-quality sequences were removed using Trimmomatic v0.36, as previously described (Bolger et al., 2014). Totally, 96 Gb of clean bases were ultimately generated (1.32 Gb/per isolate, Q20 ≥ 95%). Genomic sequence data were de novo assembled using Velvet 1.2.10. Raw data were deposited in the NCBI database under the BioProject accession number PRJNA902108.
2.6 SNP calling and identification of clonal transmission
The genome sequences of all sequenced strains were aligned to the C. difficile publicly available completed genomes as described in the “Results” section. Single nucleotide polymorphism (SNP) identification was performed using a section of the recombination event tree (SaRTree) pipeline at a proportion threshold of 100 (Hu et al., 2020). In accordance with the guiding principles, pairs of strains that differed by 0–2 SNPs and were separated by less than 124 days were considered the result of direct clonal transmission, whereas pairs of strains that differed by 3–10 SNPs were considered potential clonal transmission (Eyre et al., 2013).
2.7 Antibiotic susceptibility testing
Antibiotic susceptibility testing was performed using the agar dilution assay according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (CLSI, 2020). The 12 tested antibiotics were clindamycin, ciprofloxacin, erythromycin, fusidic acid, gatifloxacin, levofloxacin, metronidazole, moxifloxacin, piperacillin, rifampicin, tetracycline, and vancomycin, with C. difficile ATCC 700057 and Bacteroides fragilis ATCC 25285 included as control strains. Minimal inhibitory concentration (MIC) results were interpreted using the CLSI M100 recommendations for clindamycin, metronidazole, moxifloxacin, piperacillin, and tetracycline (CLSI, 2020); EUCAST guidelines2 for fusidic acid, rifampicin, and vancomycin; and CLSI recommendations for moxifloxacin in anaerobes applied to ciprofloxacin, gatifloxacin, and levofloxacin. The erythromycin breakpoint was set at 8 μg/mL as previously reported (Mutlu et al., 2007). Multidrug resistance (MDR) was defined as resistance to at least three antibiotic classes (Magiorakos et al., 2012).
2.8 Data analysis
Statistical analysis was conducted using SPSS Statistics 26.0 (IBM Corp., NY, USA). The χ2 test or Fisher’s exact test was employed to assess correlations between STs and antibiotic susceptibility patterns. Bivariate analysis used the χ2 test or Fisher’s exact test to evaluate differences between CDI and non-CDI cases, and Fisher’s exact test to evaluate differences between nosocomial transmission STs and non-nosocomial transmission STs. For these comparisons, odds ratios (ORs), 95% confidence intervals (CIs), and p-values were calculated. Variables showing significance in bivariate analysis were further examined using bivariate logistic regression to identify independent risk factors for CDI. A p-value <0.05 was considered statistically significant.
3 Results
3.1 Molecular characteristic
3.1.1 Collection and genotyping of C. difficile strains
A total of 907 investigated patients with diarrhea from the Ningbo First Hospital were enrolled in this cross-sectional study. The flow diagram and clinical information were shown in Figure 1 and Supplementary Table S1, respectively. Among them, 115 C. difficile strains were recovered with a prevalence of 12.7%. Toxin gene analysis revealed that 106 (92.2%) strains were A+B+CDT−, 3 (2.6%) were A−B+CDT−, and 6 (5.2%) were A+B+CDT+, and non-toxigenic strains were not found. MLST analysis identified 25 different STs, with ST3 (n = 16, 13.9%), ST35 (n = 15, 13.0%), and ST2 (n = 12, 10.4%) being the predominant genotypes.
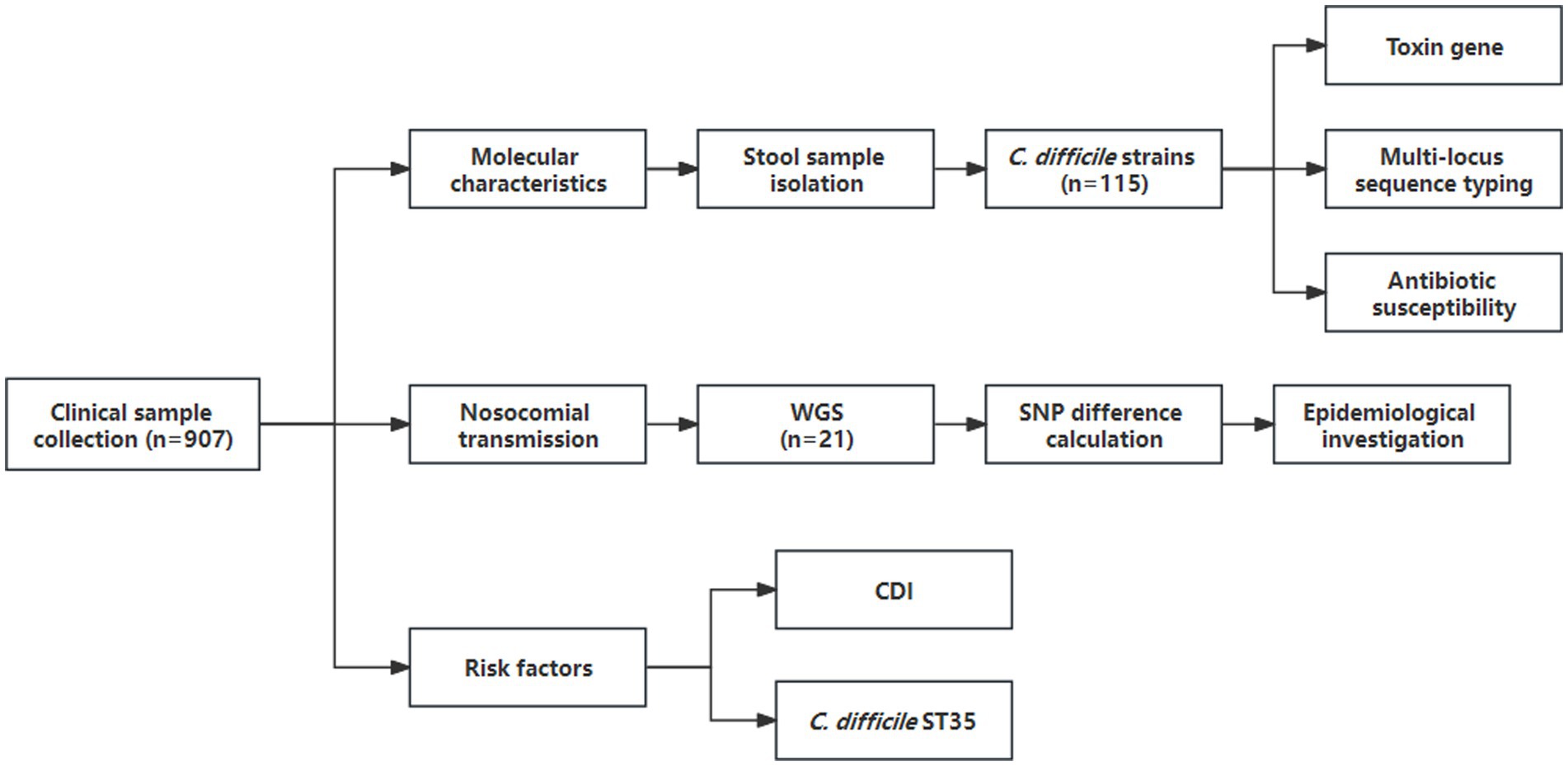
Figure 1. Flow diagram of data collected during the cross-sectional study (1 September 2021 to 30 November 2021). WGS, whole-genome sequencing; SNP, single nucleotide polymorphism.
3.1.2 Antibiotic susceptibility testing
The summary MIC distributions of the 115 C. difficile strains for the 12 antibiotics were presented in Figure 2, and the values of the MIC50, MIC90, MIC mode and geometric mean MIC (GM) were shown in Table 1. ST35 had higher GM values for ciprofloxacin (6.35 μg/mL) and tetracycline (6.33 μg/mL) compared to all the other STs (2.84 μg/mL, 0.43 μg/mL, respectively). The ST54 had higher GM values for clindamycin (56.42 μg/mL) and erythromycin (105.95 μg/mL) compared to all the other STs (18.61 μg/mL, 12.54 μg/mL, respectively). The GM values of ST1 against metronidazole, vancomycin, rifampicin, piperacillin, and three quinolones (levofloxacin, moxifloxacin, and gatifloxacin) were higher than those of other STs.
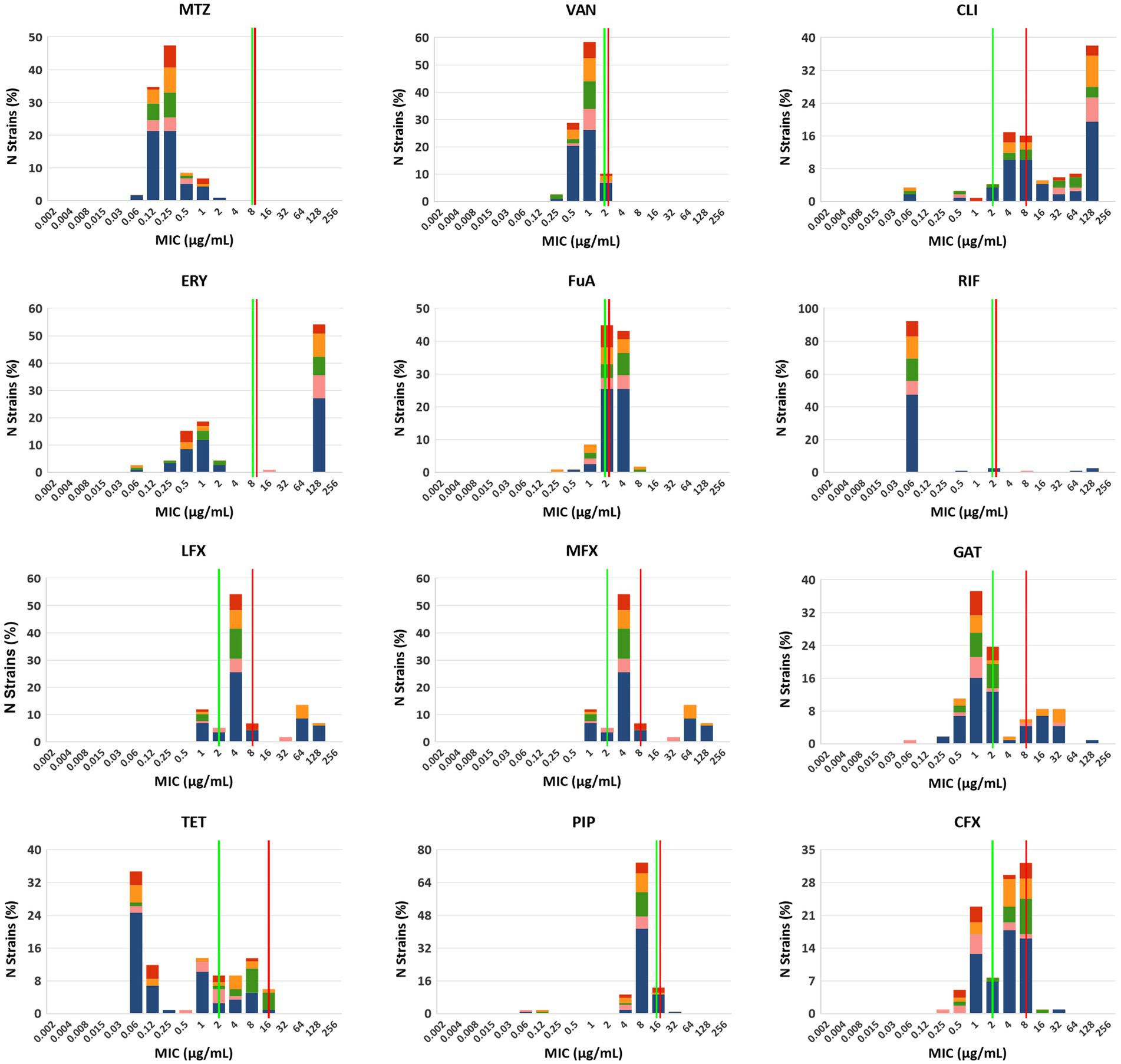
Figure 2. MIC distributions for 12 antibiotics among different STs, red: ST2, orange: ST3, green: ST35, pink: ST54 and blue: other STs. MTZ, metronidazole; VAN, vancomycin; CLI, clindamycin; ERY, erythromycin; FuA, fusidic acid; RIF, rifampicin; LFX, levofloxacin; MFX, moxifloxacin; GAT, gatifloxacin; TET, tetracycline; PIP, piperacillin; CFX, ciprofloxacin. Established susceptible and resistant breakpoints were indicated by vertical green and red lines, respectively, where available.
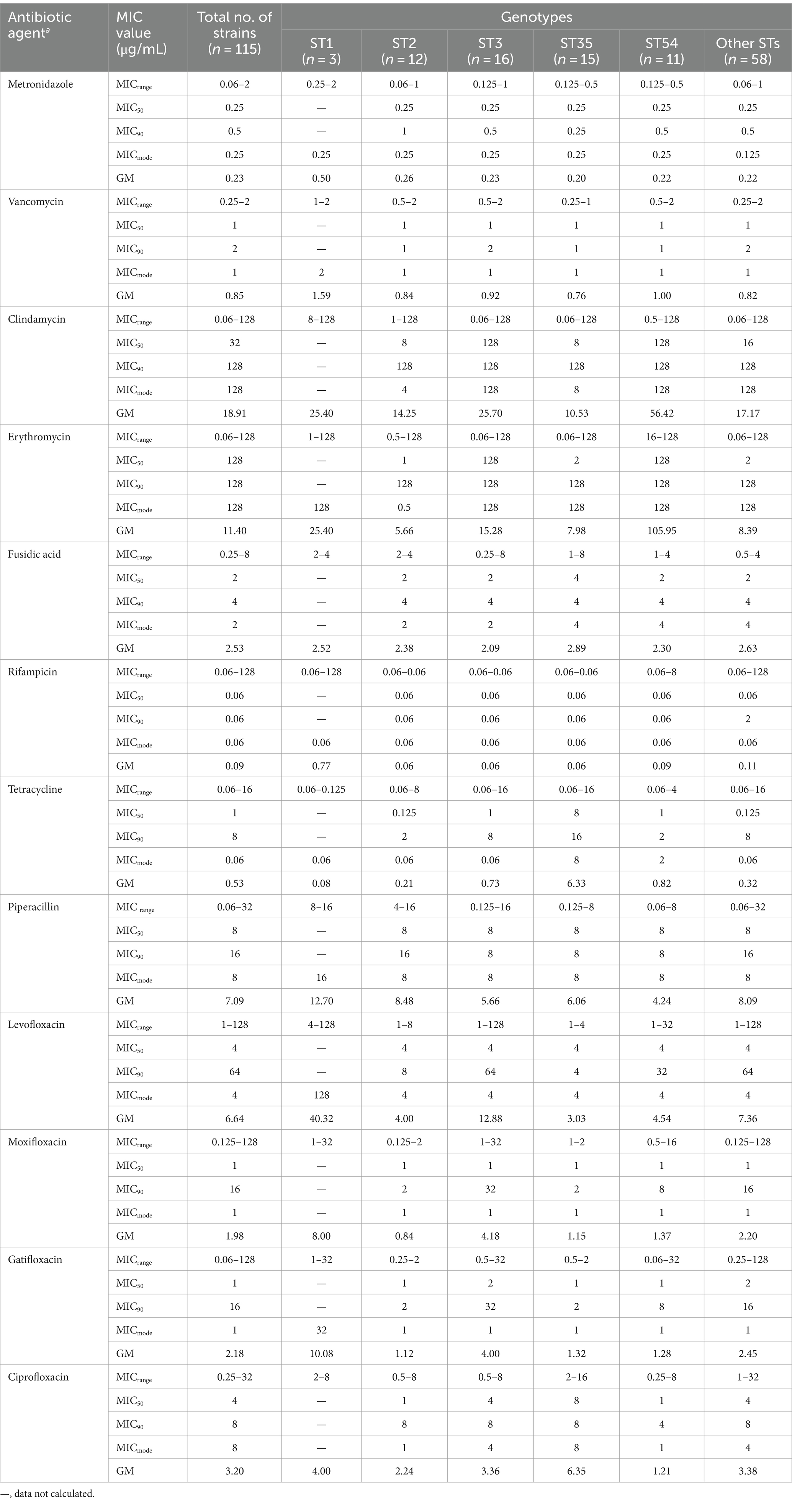
Table 1. MIC parameters of genotypes against 12 antibiotic agents.
Correlations between antibiotic resistance and predominant STs were also determined (Supplementary Table S2). For ST54, the rate of erythromycin resistance (100%) was significantly higher than those in other STs (χ2 = 10.24, p = 0.02). However, the rates of resistance to moxifloxacin (18.2%) and gatifloxacin (18.2%) were significantly lower than those in other STs (χ2 = 16.65 and 14.91, p = 0.001 and 0.002, respectively). For ST35, the rates of ciprofloxacin resistance (66.7%) and tetracycline resistance (33.3%) were distinctly higher than those in other STs (χ2 = 13.30 and 20.19, p = 0.004 and < 0.001, respectively). Notably, of 7 tetracycline-resistant strains, 5 belonged to ST35. Moreover, 80 (69.6%) of strains were MDR with significant differences in the distribution of STs (χ2 = 9.88, p = 0.02).
3.2 Clonal transmission
3.2.1 Genetic relatedness of STs across different departments
The minimum spanning tree revealed the same STs distributed across multiple departments (Figure 3). Among these, ST48 detected in the neurology department (NEU), was identified as a putative ancestor of all STs. Further analysis showed that ST48 exhibited one allelic difference from both ST42 and ST3. Specifically, ST42 was detected in the infectious disease department (INF) and NEU, while ST3 was detected in INF, cardiothoracic surgery (CTS), hepatobiliary surgery (HBS), intensive care unit (ICU), hematology (HEM), gastroenterology (GAS), and other departments. The distribution of the main hospitalized departments was illustrated in Supplementary Figure S1. Building A mainly contained ICU, HBS, GAS, and NEU, while Building B mainly contained ICU, CTS, and HEM. A connecting platform linked Building A and Building B. The two INF were situated in Buildings C and D. The other departments were distributed across these four buildings.
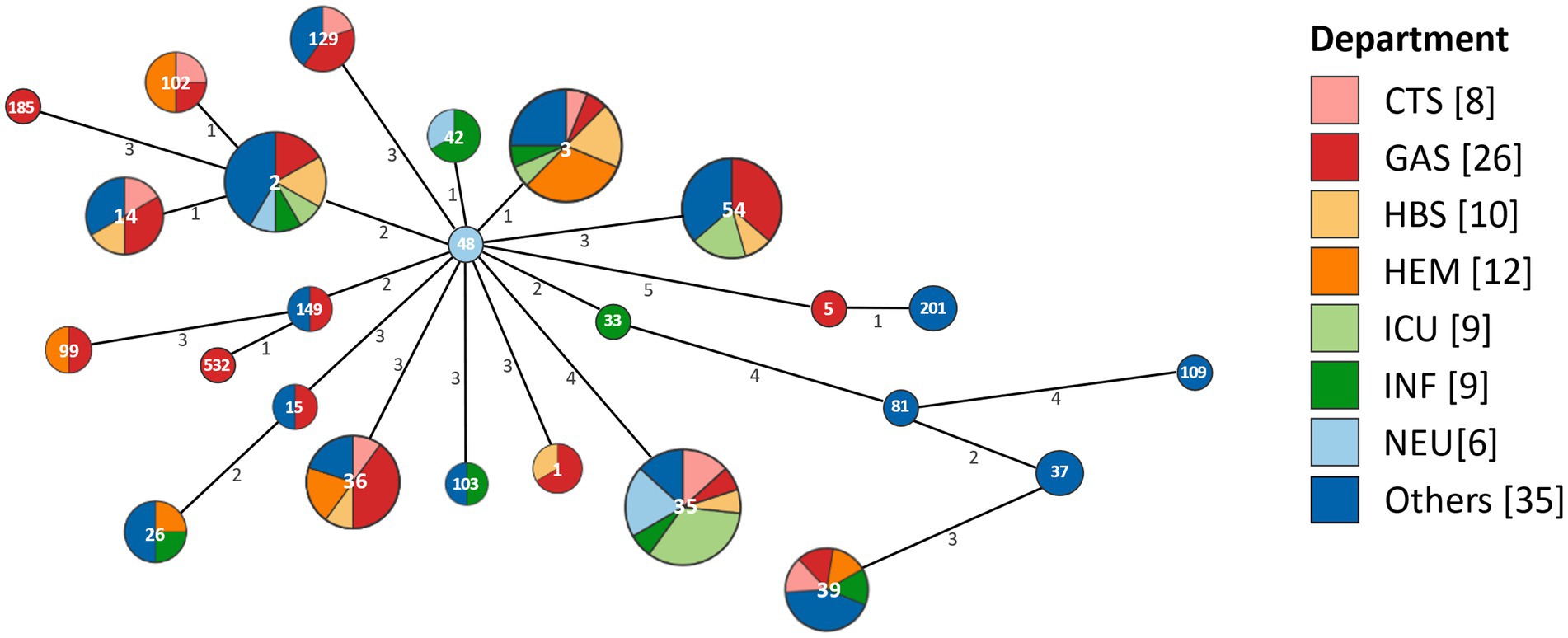
Figure 3. The minimum spanning tree based on the distribution of STs across different departments. Each circle represents an ST, with the size of the circle and the number of its segments indicating the quantity of corresponding isolates. The numerical values on the lines connecting the circles represent the number of allelic differences between adjacent STs. CTS, cardiothoracic surgery department; GAS, gastroenterology department; HBS, hepatobiliary surgery department; HEM, hematology department; ICU, intensive care unit; INF, infectious diseases department; NEU, neurology department.
3.2.2 Analysis of genetic relationship and identification of clonal transmission of CDI
WGS and SNP analysis was performed on 21 strains with identical STs from the same ward, including 7 strains of ST35, 6 of ST36, 4 of ST3, 2 of ST14, and 2 of ST54. These strains were collected within a 124-day period. The genome sequences of the seven ST35 strains were aligned to the C. difficile ST35 complete genome A9 (SRR18235872). Among these, three strains (NB184, NB191, and NB197) showed ≤2 SNP differences, while no SNP difference was found between NB375 and NB437 (Figure 4A). By integrating SNP results with patient hospitalization timelines (Figure 4B), two nosocomial clonal transmission events were identified (Figure 4C). The first event occurred between September 9 and 17, involving the spread of C. difficile ST35 between patient NB197 and NB191 in the NEU, and subsequently to NB184 in the CTS. The second event, occurring between October 4 and 11, involved the clonal transmission of C. difficile ST35 between patient NB437 in the CTS and NB375 in the ICU. In addition, two ST36 strains (NB454 and NB647), which were isolated less than 124 days apart, had 8 SNP differences. The genome sequences of the other non-ST35 strains were aligned to the C. difficile 630 genome (AM180355.1) (Supplementary Figure S2).
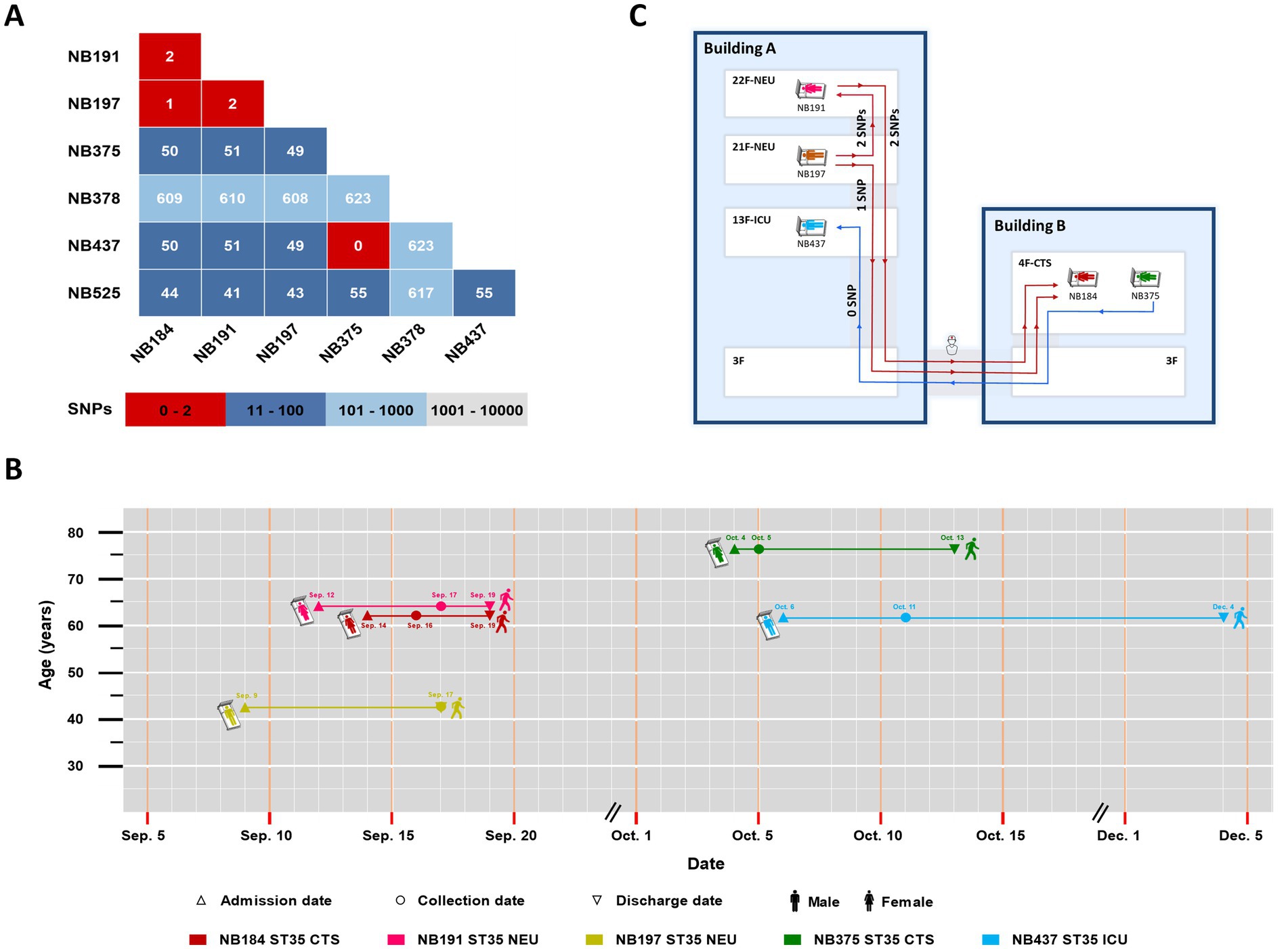
Figure 4. (A) The number of SNP differences in pairs was determined in the genomes of seven ST35 strains; (B) The timelines of five hospitalized patients with genetically related strains. Patient admission dates are marked by equilateral triangles, collection dates by circles and discharge dates by inverted triangles; (C) The transmission map of two nosocomial clonal transmission events among different floors and buildings. CTS, cardiothoracic surgery department; NEU, neurology department; ICU, intensive care unit.
3.3 Risk factors
Risk factors for CDI were assessed through a blinded review of each patient’s medical records. Bivariate analysis was performed between the 115 CDI cases and 792 non-CDI cases, as shown in Table 2. The following parameters were found to be statistically significant between CDI cases and non-CDI cases: hospitalization over 10 days before sampling (OR = 1.757, 95% CI = 1.053–2.931, p = 0.029), the use of penicillin-class antibiotics (OR = 1.690, 95% CI = 1.107–2.579, p = 0.019), and use of esomeprazole (OR = 1.942, 95% CI = 1.159–3.253, p = 0.014). Subsequent bivariate logistic analysis showed that the use of penicillin-class antibiotics remained a significant factor associated with CDI (OR = 1.573, 95% CI = 1.020–2.424, p = 0.040).
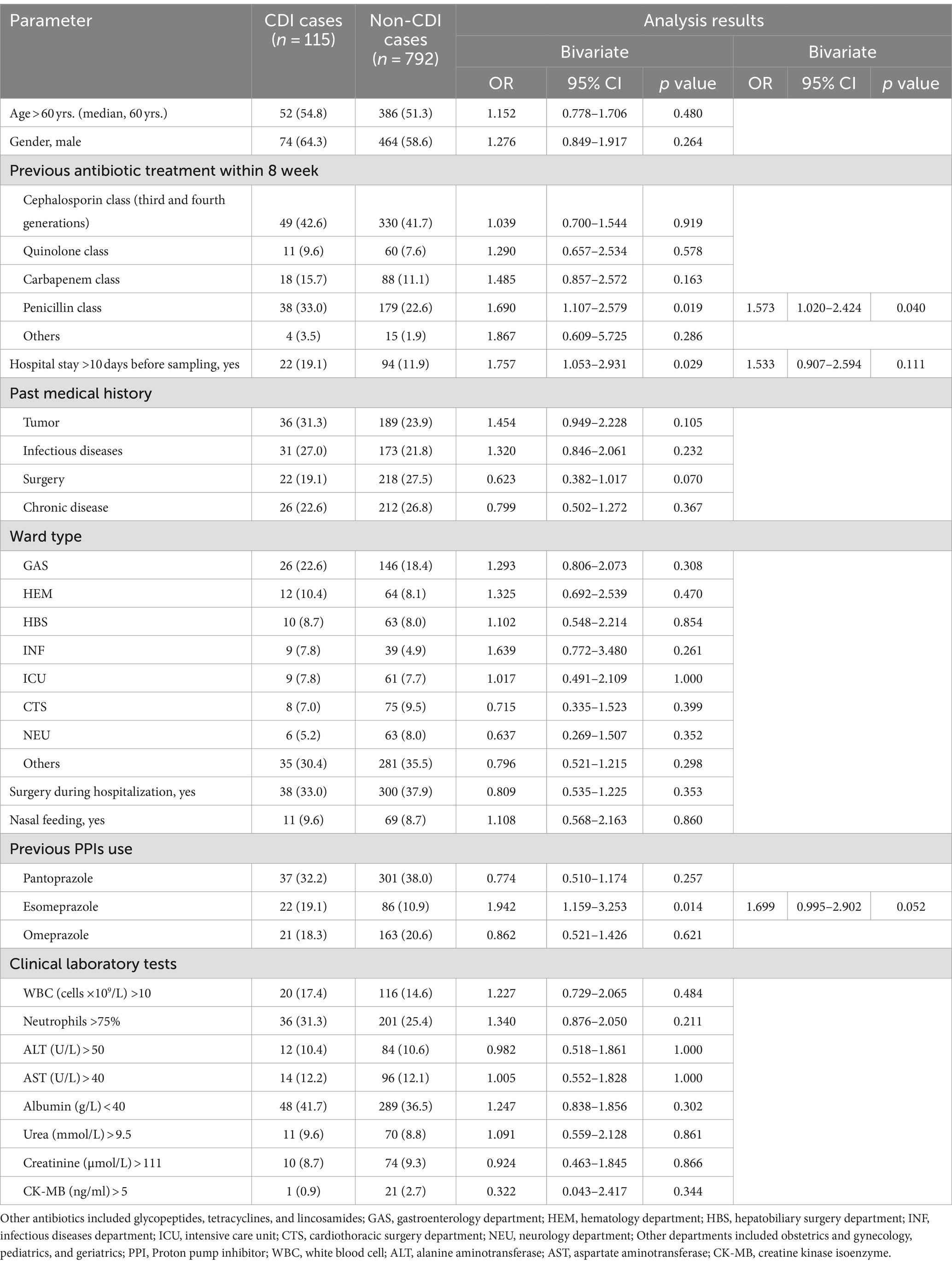
Table 2. Parameters and risk factors in 907 hospitalized patients with diarrhea.
Significant differences were revealed between 15 ST35 and 100 non-ST35 CDI cases in rates of admission to the ICU (OR = 12.000, 95% CI = 2.767–52.047, p = 0.002) and the NEU (OR = 8.083, 95% CI = 1.463–44.648, p = 0.037). Subsequent bivariate logistic analysis showed that admission to the NEU was a significant risk factor between ST35 and non-ST35 CDI cases (OR = 1.556, 95% CI = 1.011–2.396, p = 0.044).
4 Discussion
The previous study did not address CDI risk factors among hospitalized patients in Ningbo, China (Shu et al., 2023). To fill this knowledge gap, we conducted a further analysis of molecular characteristics, clonal transmission, and risk factors in a tertiary hospital. Our study provided the first genomic evidence of C. difficile ST35 among different floors and buildings within the hospital. Notably, NEU admission was identified as an independent risk factor for C. difficile ST35 infection, informing prevention strategies for large-scale outbreaks in this region.
This study revealed a CDI prevalence of 12.7%, comparable to rates reported among Chinese patients with diarrhea in a meta-analysis (11.4%) (Wen et al., 2023). Our previous cross-sectional study (2013–2015) identified C. difficile ST37 as a dominant genotype (16.5%) (Jin et al., 2017), contrasting with the current study, which found ST37 at a prevalence of 1.7%. C. difficile ST81, a single-allelic atpA variant of ST37, has been predominant in Shanghai for a decade (Yang et al., 2020), but our study found only 0.9% of strains were ST81. A recent meta-analysis observed significant molecular differences in C. difficile between northern and southern China (Wen et al., 2023). Regional variations might be attributable to differences on genotypes and antibiotic use patterns in C. difficile epidemiology (Luo et al., 2019). Therefore, further regional studies are crucial to fully elucidate CDI molecular characteristics in this region and understand the factors driving these regional variations in China.
Our study revealed significant changes in antibiotic resistance patterns of C. difficile strains, highlighting the dynamic nature of CDI epidemiology in this region. The antibiotic resistance pattern in this study showed that the rates of resistance to ciprofloxacin, levofloxacin, and tetracycline were significantly lower than data from our previous study (Jin et al., 2017). While the rates on clindamycin resistance were prevalent in Asia-Pacific countries (80.7%) and China (70.8–87.9%), the quinolone resistance rates (24.3–34.8%) in this study were lower than those reported in China (Luo et al., 2019; Wu et al., 2022). Similarly, the tetracycline resistance rate (6.1%) aligned with the coastal region data but differed from other domestic studies (6.0–46.9%) (Yan et al., 2017; Wu et al., 2022). These findings highlighted variations in CDI epidemiology and antibiotic resistance patterns in this region. Notably, we identified one ST1 isolate with a metronidazole MIC value of 2 μg/mL. The emergence of metronidazole-resistant C. difficile, predominantly C. difficile ST1, has been a growing concern worldwide (Abdrabou et al., 2021). Given that metronidazole is currently not recommended as a first-line drug according to the IDSA/SHEA guideline in 2017 (McDonald et al., 2018), dynamic changes in its MIC values should be continuously monitored.
Our study unveiled novel nosocomial clonal transmission among different floors and buildings within the hospital of C. difficile ST35, highlighting the complex dynamics of its spread in healthcare settings. C. difficile ST35 has caused severe outbreaks with high mortality in Sweden (Magnusson et al., 2022), and evidence suggested its widespread proliferation in Zhejiang through nosocomial and cross-species transmission (Luo et al., 2024). We identified two nosocomial clonal transmission events within a three-month period. This finding aligned with previous observations of C. difficile ST37 transmission among different floors in NEU departments in Zhejiang (Bi et al., 2023). Our epidemiological evidence suggested that cross-care provided by healthcare workers in the NEU likely served as the primary vector for this clonal transmission. Notably, we identified clonal transmission between patients NB191 and NB197 in the NEU and patient NB184 in the CTS, demonstrating bacterial spread among different floors and buildings within the hospital. Laboratory samples from both buildings were transported by the same staff, facilitating potential transmission among buildings. These observations indicated that healthcare worker mobility, environmental contamination, and indirect patient contact were key factors contributing to this clonal transmission.
The second clonal transmission event originated from patient NB437 from the CTS. Despite a one-day hospital stay, the patient, with three-year Crohn’s disease history, multiple prior admissions, and long-term use of antibiotics and immunosuppressants, might have been infected by C. difficile ST35 from other healthcare facilities prior to this admission. Subsequently, patient NB437 transmitted ST35 to susceptible ICU patient NB375 through transmission among buildings, linked to the same healthcare worker who transported the laboratory samples. Notably, C. difficile ST35 exhibited strong spore formation, a characteristic that significantly enhanced its environmental persistence and facilitates rapid spread in healthcare setting (Luo et al., 2024). These findings highlighted the need for strict infection control among healthcare workers moving among hospital areas, including enhanced hand hygiene and frequent disinfection of high-touch surfaces. Screening and monitoring ST35 cases on admission, especially those with inflammatory bowel disease, are also crucial.
For CDI risk, our findings corroborated previous reports identifying prolonged hospitalization (>10 days) as a risk factor for CDI (Yang et al., 2020). Bivariate logistic regression analysis revealed penicillin-class antibiotics as an independent risk factor for CDI, consistent with a study conducted in Hong Kong (Guo et al., 2021). Notably, all CDI patients in our study had a history of piperacillin-tazobactam usage, underscoring the necessity for stringent control and monitoring of this antibiotic to mitigate CDI risk. Further analysis of all C. difficile ST35 cases revealed novel risk factors. ICU admission was associated with increased ST35 infection risk, possibly due to the compromised immune status of ICU patients and frequent use of broad-spectrum antibiotics (Murphy et al., 2022). Notably, admission to the NEU emerged as an independent risk factor for ST35 infection. Recent research has shown that neurons play a crucial role in regulating CDI-induced inflammation (Manion et al., 2023), indicating neurological dysfunction may influence host susceptibility to CDI. However, the exact mechanisms warrant further investigation. These findings underscored the importance of judicious piperacillin-tazobactam use, routine CDI screening in high-risk patients, especially in the NEU, and prompt implementation of infection control measures.
This study has some limitations. First, clinical samples were collected over a three-month period, potentially missing seasonal variations and long-term molecular trends. Second, the absence of environmental sampling limited the understanding of direct nosocomial transmission pathways for ST35 strains. Additionally, the lack of clinical outcome data for ST35 cases limited the assessment of this genotype’s impact on disease severity.
In conclusion, this study elucidated multiple C. difficile genotypes were prevalent with varied antibiotic resistance patterns in Ningbo, China, and provided the genomic evidence of C. difficile ST35 clonal transmission among different floors and buildings within the hospital. Our findings also highlighted the importance of enhanced surveillance and targeted interventions for penicillin-class antibiotics use, and C. difficile ST35 cases in the NEU. Further studies should be conducted to investigate long-term molecular trends, environmental reservoirs, and clinical outcomes to comprehensively understand CDI dynamics and inform prevention strategies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving humans were approved by Ethics Committee of Ningbo First Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LH: Funding acquisition, Resources, Writing – original draft, Writing – review & editing. SL: Investigation, Methodology, Writing – original draft. MZ: Data curation, Methodology, Writing – original draft. MC: Data curation, Methodology, Writing – original draft. YS: Data curation, Methodology, Software, Validation, Writing – original draft. PZ: Data curation, Formal analysis, Software, Visualization, Writing – original draft. XS: Investigation, Methodology, Validation, Writing – original draft. QB: Conceptualization, Data curation, Writing – original draft. JG: Funding acquisition, Resources, Writing – original draft. YL: Data curation, Formal analysis, Visualization, Writing – review & editing. YC: Funding acquisition, Resources, Writing – review & editing, Data curation, Methodology, Validation, Writing – original draft. DJ: Funding acquisition, Methodology, Supervision, Visualization, Writing – review & editing, Conceptualization, Investigation, Validation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by the National Natural Science Foundation of China (82372299), the Key Project of Natural Science Foundation of Zhejiang Province (LXZ22H300001), and the Program for Major Science and Technology Medicine and Healthcare in Zhejiang (WKJ-ZJ-2107 and 2309), the Medical Scientific Research Foundation of Zhejiang Province, China (2019KY598 and 2024XY055).
Acknowledgments
We would like to thank Editage (www.editage.com) for manuscript editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1507128/full#supplementary-material
Footnotes
References
Abdrabou, A. M. M., Ul Habib Bajwa, Z., Halfmann, A., Mellmann, A., Nimmesgern, A., Margardt, L., et al. (2021). Molecular epidemiology and antimicrobial resistance of Clostridioides difficile in Germany, 2014-2019. Int. J. Med. Microbiol. 311:151507. doi: 10.1016/j.ijmm.2021.151507
Bi, X., Zheng, L., Yang, Z., Lv, T., Tong, X., and Chen, Y. (2023). Retrospective study of the epidemiology of Clostridioides difficile infection in the neurosurgery department of a tertiary hospital in China. Infect. Drug Resist. 16, 545–554. doi: 10.2147/idr.S397544
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
CLSI (2020). Performance standards for antimicrobial susceptibility testing. Wayne, PA: National Committee for Clinical and Laboratory Standards.
Dai, W., Yang, T., Yan, L., Niu, S., Zhang, C., Sun, J., et al. (2020). Characteristics of Clostridium difficile isolates and the burden of hospital-acquired Clostridium difficile infection in a tertiary teaching hospital in Chongqing, Southwest China. BMC Infect. Dis. 20:277. doi: 10.1186/s12879-020-05014-6
Eyre, D. W., Cule, M. L., Wilson, D. J., Griffiths, D., Vaughan, A., O'connor, L., et al. (2013). Diverse sources of C. difficile infection identified on whole-genome sequencing. N. Engl. J. Med. 369, 1195–1205. doi: 10.1056/NEJMoa1216064
Fernandes, M. R., Sellera, F. P., Moura, Q., Gaspar, V. C., Cerdeira, L., and Lincopan, N. (2018). International high-risk clonal lineages of CTX-M-producing Escherichia coli F-ST648 in free-roaming cats, South America. Infect. Genet. Evol. 66, 48–51. doi: 10.1016/j.meegid.2018.09.009
Griffiths, D., Fawley, W., Kachrimanidou, M., Bowden, R., Crook, D. W., Fung, R., et al. (2010). Multilocus sequence typing of Clostridium difficile. J. Clin. Microbiol. 48, 770–778. doi: 10.1128/jcm.01796-09
Guo, C. L. T., Kwong, T. N. Y., Mak, J. W. Y., Zhang, L., Lui, G. C. Y., Wong, G. L. H., et al. (2021). Trends in incidence and clinical outcomes of Clostridioides difficile infection, Hong Kong. Emerg. Infect. Dis. 27, 3036–3044. doi: 10.3201/eid2712.203769
Hu, D., Liu, B., Wang, L., and Reeves, P. R. (2020). Living trees: high-quality reproducible and reusable construction of bacterial phylogenetic trees. Mol. Biol. Evol. 37, 563–575. doi: 10.1093/molbev/msz241
Jia, H., Du, P., Yang, H., Zhang, Y., Wang, J., Zhang, W., et al. (2016). Nosocomial transmission of Clostridium difficile ribotype 027 in a Chinese hospital, 2012-2014, traced by whole genome sequencing. BMC Genomics 17:405. doi: 10.1186/s12864-016-2708-0
Jin, D., Luo, Y., Huang, C., Cai, J., Ye, J., Zheng, Y., et al. (2017). Molecular epidemiology of Clostridium difficile infection in hospitalized patients in eastern China. J. Clin. Microbiol. 55, 801–810. doi: 10.1128/jcm.01898-16
Luo, Y., Chen, Y., Lin, S., Hu, H., Song, X., Bian, Q., et al. (2024). Genomic epidemiology of Clostridioides difficile sequence type 35 reveals intraspecies and interspecies clonal transmission. Emerg. Microbes Infect. 13:2408322. doi: 10.1080/22221751.2024.2408322
Luo, Y., Cheong, E., Bian, Q., Collins, D. A., Ye, J., Shin, J. H., et al. (2019). Different molecular characteristics and antimicrobial resistance profiles of Clostridium difficile in the Asia-Pacific region. Emerg. Microbes Infect. 8, 1553–1562. doi: 10.1080/22221751.2019.1682472
Magiorakos, A. P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Magnusson, C., Mernelius, S., Bengnér, M., Norén, T., Serrander, L., Forshell, S., et al. (2022). Characterization of a Clostridioides difficile outbreak caused by PCR ribotype 046, associated with increased mortality. Emerg. Microbes Infect. 11, 850–859. doi: 10.1080/22221751.2022.2049981
Manion, J., Musser, M. A., Kuziel, G. A., Liu, M., Shepherd, A., Wang, S., et al. (2023). C. difficile intoxicates neurons and pericytes to drive neurogenic inflammation. Nature 622, 611–618. doi: 10.1038/s41586-023-06607-2
Mcdonald, L. C., Gerding, D. N., Johnson, S., Bakken, J. S., Carroll, K. C., Coffin, S. E., et al. (2018). Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 66, e1–e48. doi: 10.1093/cid/cix1085
McDonald, L. C., Killgore, G. E., Thompson, A., Owens, R. C., Kazakova, S. V., Sambol, S. P., et al. (2005). An epidemic, toxin gene variant strain of Clostridium difficile. N. Engl. J. Med. 353, 2433–2441. doi: 10.1056/NEJMoa051590
Murphy, C. V., Reed, E. E., Herman, D. D., Magrum, B., Beatty, J. J., and Stevenson, K. B. (2022). Antimicrobial stewardship in the ICU. Semin. Respir. Crit. Care Med. 43, 131–140. doi: 10.1055/s-0041-1740977
Mutlu, E., Wroe, A. J., Sanchez-Hurtado, K., Brazier, J. S., and Poxton, I. R. (2007). Molecular characterization and antimicrobial susceptibility patterns of Clostridium difficile strains isolated from hospitals in south-east Scotland. J. Med. Microbiol. 56, 921–929. doi: 10.1099/jmm.0.47176-0
Persson, S., Torpdahl, M., and Olsen, K. E. (2008). New multiplex PCR method for the detection of Clostridium difficile toxin a (tcdA) and toxin B (tcdB) and the binary toxin (cdtA/cdtB) genes applied to a Danish strain collection. Clin. Microbiol. Infect. 14, 1057–1064. doi: 10.1111/j.1469-0691.2008.02092.x
Preston, M. D., Assefa, S. A., Ocholla, H., Sutherland, C. J., Borrmann, S., Nzila, A., et al. (2014). PlasmoView: a web-based resource to visualise global plasmodium falciparum genomic variation. J. Infect. Dis. 209, 1808–1815. doi: 10.1093/infdis/jit812
Qin, J., Dai, Y., Ma, X., Wang, Y., Gao, Q., Lu, H., et al. (2017). Nosocomial transmission of Clostridium difficile genotype ST81 in a general teaching hospital in China traced by whole genome sequencing. Sci. Rep. 7:9627. doi: 10.1038/s41598-017-09878-8
Saldanha, G. Z., Pires, R. N., Rauber, A. P., De Lima-Morales, D., Falci, D. R., Caierão, J., et al. (2020). Genetic relatedness, virulence factors and antimicrobial resistance of C. difficile strains from hospitalized patients in a multicentric study in Brazil. J. Glob. Antimicrob. Resist. 22, 117–121. doi: 10.1016/j.jgar.2020.01.007
Shu, C., Zhou, J., Yu, H., Fu, W., Shen, J., Liang, L., et al. (2023). Genomic epidemiology and antimicrobial resistance profiles of Clostridioides difficile from multi-hospitals in a city in eastern China. Infect. Drug Resist. 16, 3379–3388. doi: 10.2147/idr.S407497
Wen, B. J., Dong, N., Ouyang, Z. R., Qin, P., Yang, J., Wang, W. G., et al. (2023). Prevalence and molecular characterization of Clostridioides difficile infection in China over the past 5 years: a systematic review and meta-analysis. Int. J. Infect. Dis. 130, 86–93. doi: 10.1016/j.ijid.2023.03.009
Wu, Y., Wang, Y. Y., Bai, L. L., Zhang, W. Z., Li, G. W., and Lu, J. X. (2022). A narrative review of Clostridioides difficile infection in China. Anaerobe 74:102540. doi: 10.1016/j.anaerobe.2022.102540
Yan, J., Liang, J., Lv, T., Gu, S., Jiang, T., Huang, L., et al. (2017). Epidemiology of Clostridium difficile in a county level hospital in China. Jundishapur. J. Microbiol. 10:e14376. doi: 10.5812/jjm.14376
Keywords: Clostridioides difficile infection, molecular characteristics, whole-genome sequencing, clonal transmission, risk factors
Citation: Hu L, Lin S, Zhang M, Cai M, Shen Y, Zeng P, Song X, Bian Q, Gu J, Luo Y, Chen Y and Jin D (2024) Molecular characteristics, clonal transmission, and risk factors of Clostridioides difficile among hospitalized patients in a tertiary hospital in Ningbo, China. Front. Microbiol. 15:1507128. doi: 10.3389/fmicb.2024.1507128
Edited by:
Samuel Sunday Taiwo, Ladoke Akintola University of Technology, NigeriaReviewed by:
Samuel Adetona Fayemiwo, University of Ibadan, NigeriaAbayomi Fadeyi, University of Ilorin, Nigeria
Copyright © 2024 Hu, Lin, Zhang, Cai, Shen, Zeng, Song, Bian, Gu, Luo, Chen and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dazhi Jin, amluZEBobWMuZWR1LmNu; Yu Chen, eWNoZW5AaG1jLmVkdS5jbg==
†These authors have contributed equally to this work