 Gerald W. Tannock
Gerald W. Tannock- Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand
Human gut microbiomes (microbiotas) are highly individualistic in taxonomic composition but nevertheless are functionally similar. Thus, collectively, they comprise a “metacommunity.” In ecological terminology, the assembly of human gut microbiomes is influenced by four processes: selection, speciation, drift, and dispersal. As a result of fortuitous events associated with these processes, individual microbiomes are taxonomically “tailor-made” for each host. However, functionally they are “off-the-shelf” because of similar functional outputs resulting from metabolic redundancy developed in host-microbe symbiosis. Because of this, future microbiological and molecular studies of microbiomes should emphasize the metabolic interplay that drives the human gut metacommunity and that results in these similar functional outputs. This knowledge will support the development of remedies for specific functional dysbioses and hence provide practical examples of precision medicine.
Introduction
The colon of humans is colonized by a microbial community, commonly known as the gut microbiome (microbiota), that is composed mostly of bacterial species (Franzosa et al., 2015; Parizadeh and Arrieta, 2023). The community has been studied in detail from the 1970s using feces as a proxy for colon samples, with particular emphasis on taxonomic composition (“who is there?”) for the last 25 years. This became possible because of the availability and development of high throughput sequencing of bulk DNA extracted from feces, and subsequent sequence analysis (Franzosa et al., 2015; Parks et al., 2017; Asnicar et al., 2024; Parizadeh and Arrieta, 2023). In general, the community contains about twenty trillion bacterial cells in which members of the Bacillota (Firmicutes) and Bacteroidota (Bacteroidetes) form about 85% of the microbiome. Three bacterial families, the Lachnospiraceae, Ruminococcaceae, and Bacteroidaceae are well represented (White et al., 2014; Sender et al., 2016; Rampelli et al., 2020). Much of the research interest in the gut microbiome has focussed on defining the taxonomic composition of the “normal” or “healthy” microbiome but this is an impossible goal due to the huge variance in microbiome compositional diversity between individual humans at genus, species, and strain levels (Turnbaugh et al., 2009; Ursell et al., 2012; Shanahan et al., 2021). This article proposes that viewing the gut microbiome as a metacommunity which has functional consistency will aid the development of precision (personalized; individual) medicinal interventions to restore health.
What is a metacommunity?
Communities are interactive assemblages of species that are characteristic of a specific habitat or ecosystem. The formation of communities is influenced by four processes: environmental selection (deterministic fitness differences among species), ecological drift (stochastic changes in species abundance), local diversification (creation of new species), and dispersal (spatial movement of species) (Vellend, 2010). These processes have been studied in relation to the human gut microbiome and observations that are pertinent to this article follow.
1. Selection of bacterial species that have biochemical fitness determinants appropriate for catabolism of dietary components and host secretions is apparent among the members of the gut microbiome. Key indicators of selection are genetic features such as Polysaccharide Utilization Loci (PULs) that encode binding proteins, hydrolytic enzymes (carbohydrate-active enzymes), and transport proteins associated with the bacterial cell surface. Products of PULs sequester and degrade plant-derived glycans that are common in human food (for example, resistant starch, hemicelluloses such as complex xylans, and pectins) (El Kaoutari et al., 2013; Grondin et al., 2017). At least some of these specialized bacteria are keystone species that initiate the catabolism of complex carbohydrates that subsequently fuel the metabolism of consortia (guilds) (Ze et al., 2012).
2. Temporal drift in climax communities of individuals has been studied and, in general, the abundances of species is relatively constant over time (Faith et al., 2013). However, the influences of allochthonous factors such as dietary fiber, medications and environment are apparent (David et al., 2014; Tannock, 2021a; Nagata et al., 2022; Gacesa et al., 2022). The microbiomes of infants and children follow characteristic colonization patterns that are mostly influenced by trophic factors and are linked to dispersal of species (see below). The direct and indirect impact of endogenous factors such as predation by bacteriophages on species abundance has been measured in gnotobiotic mouse experiments and may have a modulating effect on some bacterial populations (Hsu et al., 2019). The ecological impact of bacteriocins and other antimicrobial substances encoded by biosynthetic gene clusters (BGC) is unknown (Tannock, 2022).
3. Speciation can best be understood by considering the widespread presence of bacterial strains (subsets of species) in the gut. Mutation of genes occurs commonly; de novo mutations are estimated at 2 × 109 to 6 × 1012 single nucleotide polymorphisms per microbiota per day. Gene loss and gene gain (by horizontal gene transfer) in bacterial species reveals a genetically mutable community in real time (Nielsen et al., 2014; Zhao et al., 2019). Consequently, strains of the same species differ in genetic characteristics where “core” genes common to all strains plus “dispensable” genes (variable presence) together comprising the pangenome, can be recognized (Medini et al., 2005; Truong et al., 2017).
4. Dispersal refers to the movement (transmission) of species that establish or augment communities in other sites. In terms of the human gut microbiome, the gut of each new-born infant offers a pristine environment for colonization by bacterial species. The maternal fecal microbiome is a major source of bacterial strains during the first few months of life on the assemblage of the “new” community. Although vertical transmission is very important in this dispersal, horizontal transmission from paternal, family and environmental sources also occurs (Song et al., 2013; Gaulke and Sharpton, 2018; Yassour et al., 2018; Tannock, 2021b; Gacesa et al., 2022; Valles-Colomer et al., 2023; Dubois et al., 2024). A community of low diversity emerges to begin with that, while infants are suckled at the breast, is dominated by species that can catabolize human milk components including human milk oligosaccharides (Mills et al., 2023). After weaning, this markedly trophic colonization is replaced by a more neutral (stochastic) model that is nevertheless driven by niche differentiation whereby bacterial species that catabolize dietary fiber and host secretions form the nucleus of a community that, especially in terms of emergent properties, eventually resembles that of adults in general (Leong et al., 2018; Lawley et al., 2019).
As a result of these processes, each person’s gut microbiome is like that of an island within an archipelago with a microbial assemblage that is individualistic with regards to taxonomic composition (Costello et al., 2012). These personal climax communities can be viewed as subsets of a “metacommunity” that are linked by the potential or actual dispersal of interacting species (Figure 1). Indeed, the dispersal of members of human microbiomes is not limited to early life. In work targeting 7,646 fecal samples from multinational sources, Valles-Colomer et al. (2023) provided strain-level metagenomic evidence of microbiome dispersal between adults who shared environments (strain sharing rate about 12%). Greater proportions of shared strains were detected in oral microbiomes than in the case of the gut, but the data nevertheless support the view that the human gut microbiome is a metacommunity whose subsets are linked by potential dispersal and acquisition throughout life.
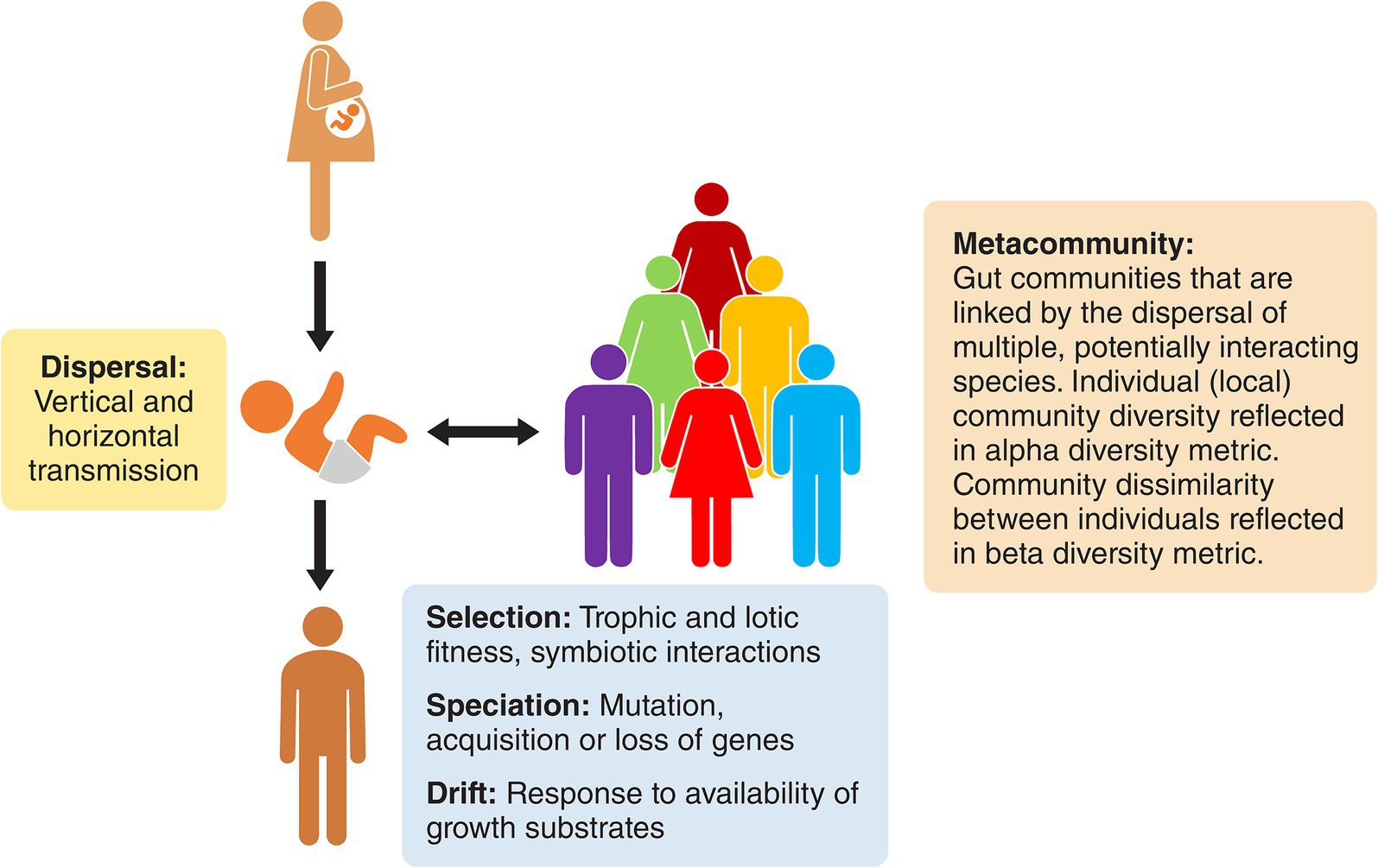
Figure 1. The gut microbiome of humans represented as a metacommunity. Each human is inhabited by an ecological community that is different from that of other humans, but the communities are nevertheless linked by dispersal and modified by drift, speciation, and selection. They have similar emergent properties due to metabolic redundancy among the otherwise disparate bacterial constituents.
Despite variation in taxonomic composition, metacommunity islands have common functional attributes derived from symbiotic relationships between microbiome and host that have developed during co-evolution (Tannock, 2024a). This is particularly apparent through study of the interaction of species in food webs. Community food webs in the gut are interlocking and interdependent food chains that originate in the catabolism of complex dietary components (plant glycans) and host secretions (bile acids, mucins) and develop through cross feeding of simple carbohydrates, amino acids, vitamins, and organic acids (e.g., formate, fumarate, succinate, lactate, succinate) between species (Lindstad et al., 2021; Culp and Goodman, 2023). These trophic interactions are consistent with the concept of the provision of “public good services” by some species for collective benefit of the community (Morris et al., 2012).
Food webs, like pathways in cellular processes, are nonlinear networks that are controlled by feedback loops. “Interspecies hydrogen transfer” is a good example of a feedback loop in the gut. Hydrogen is produced during fermentations, but the environmental concentration is kept low in the gut ecosystem by hydrogen consumers using acetogenesis, dissimilatory sulfate reduction, hydrogenotrophic respiration, or methanogenesis. Fermentative bacteria can live in the colon at the limits of what is thermodynamically possible because NADH oxidation can be coupled to proton reduction under these low hydrogen concentrations (Stams and Plugge, 2009).
Some metabolic functions within networks can be achieved by different biochemical pathways, hence different species may carry out the same function (metabolic redundancy, such as in butyrate production) (Pryde et al., 2002; Tian et al., 2020). Unrelated humans share 82% of bacterial metabolic pathways detected in fecal DNA but only 43% of bacterial species (Visconti et al., 2019). This helps to explain the taxonomic individualism, but similar metabolic outputs, of human gut microbiomes.
Measuring the “health” of the metacommunity
Most members of human gut microbiomes are obligate anaerobes that die within a short time under aerobic conditions, and many are autotrophs that require cross-feeding of nutrients from other community members (Tramontano et al., 2018; Nayfach et al., 2019). The logistical and technical difficulties of conducting large scale, culture-based analysis under these circumstances led to the adoption of metagenomic, phylogenetic comparisons of fecal microbiomes. Some studies using this technology indicated that the relative abundances of certain bacterial groups in fecal microbiomes indicated health or disease (Manor et al., 2020). Whether these differences were causative of disease or collateral damage due to disease could not be established by these observational studies. Relatively few human participants were sampled and there are numerous confounding factors that render it difficult to reproduce the results from one study to another (Sze and Schloss, 2016). More recent studies, using machine learning procedures, use data from larger groups of people and focus on bacterial strain differences (Manor et al., 2020). However, repeatability of these studies is also untested, and they sometimes seem to be inventories of microbial diversity rather than critical testing of hypotheses (Prosser, 2022).
An alternative school of thought in relation to defining a healthy microbiome has recently been published together with an overall framework for future developmental research (Tannock, 2024b). In brief, the proposal states that bioassays should be developed to measure the functioning of microbiomes. This would be analogous to the use of “lab tests” of peripheral blood, widely used in medical diagnostics. A range of metacommunity “normal values” would first be established to which values from individual microbiomes would be compared. Potential bioassays to assess the health of the symbiosis between gut microbiome and human host include measuring undegraded plant glycans in fecal samples, fecal SCFA profiles, fecal bile acid profiles (for example, proportion of secondary bile acids), fecal mucin profiles, fecal agonists of G protein-coupled receptors (for example, short chain fatty acids and N-acyl amides), plasma/serum metabolomes, qPCR quantitation of bacterial genetic loci known to underpin catabolic features of symbiosis (for example, genes encoding carbohydrate-active enzymes [CAZymes]), and immune factors present in feces (for example, amounts of secretory IgA and calprotectin). It is proposed that these kinds of assays, informed by knowledge of community function, are likely to be more useful than taxonomic comparisons in differentiating healthy microbiotas from unhealthy because functional outputs of microbiomes are similar across the metacommunity of healthy humans.
Current research gaps in understanding the gut metacommunity
As mentioned previously, changes to gut community ecology may contribute to the increased prevalence of metabolic conditions (for example, obesity, cardiovascular disease, type 2 diabetes) in humans living in, or adopting, industrialized lifestyles. Altered functioning of the microbiome and hence altered host-microbe equilibrium (“dysbiosis”) might be involved. If that is correct, remedial action might be possible.
Restoration in westerners of “missing microbes” cultured from people following non-industrialized lifestyles has been suggested but would probably not succeed because the diet of recipients is unlike that of the donors, so niches for them in the gut are lacking (De Filippo et al., 2010). It would take some strong arguments to persuade people in industrialized countries to make transitions to ancestral diets and lifestyle (Burger et al., 2012). Modulation of gut microbiome function of humans by less dramatic dietary intervention is realistic (Tannock, 2021a), but there needs to be much better evaluation of habitual diets of humans participating in microbiome trials (Renall et al., 2023). Ethnicity of human participants also needs to be recorded because dietary preferences and lifestyles may be influenced (Gaulke and Sharpton, 2018; Xu et al., 2020). Control human cohorts in gut microbiota studies are frequently described as “healthy” but anthropometric tests that show this to be valid, rather than relying on personal perceptions, should be used to confirm health status (Renall et al., 2023).
Gut transit time should be measured in every study of the gut microbiome because it is variable between humans and is influenced by the amount of dietary fiber that is consumed. Community function is influenced by transit time because slower passage of digesta through the colon allows time for catabolism of most dietary carbohydrates in the proximal colon. Bacterial metabolism in the distal colon then turns to the use of amino acids as substrates with the generation of branched SCFAs (isobutyrate, isovalerate). Thus, the metabolic profile of the community can vary according to temporal loading of nutrients in colonic regions (Roager et al., 2016; Vandeputte et al., 2016; Asnicar et al., 2021).
The gut metacommunity of humans continues to evolve. This is revealed by comparison of fecal microbiomes in samples collected from people who are nomadic hunter-gatherers in Africa, agrarian populations living in countries where industrialization is minimal, in developed countries that are highly industrialized, and people who are in transition (migrants) between non-industrialized and industrialized regions or countries (De Filippo et al., 2010; Vangay et al., 2018; Schnorr et al., 2014; Shanahan et al., 2022; Tamburini et al., 2022; Blanco-Míguez et al., 2023; Carter et al., 2023). In general, industrialization selects for gut communities of lower diversity due to consumption of diets containing more refined grains, and less coarse dietary fiber (De Filippo et al., 2010). More studies on the microbiomes of non-western societies are required because they may reveal further molecular specializations of gut bacteria that are critical to catabolism of plant glycans. These studies will also define the “normal values” of ecosystem function for Asian and other societies.
Curing dysbiosis means that restoration of the gut ecosystem must occur. To do this, we need to know what the ecosystem was like, especially how it functioned, when it was healthy (Wainwright et al., 2018). For example, we might gain an appreciation of ecosystem function by focussing on nutritional features, such as cross-feeding, that are essential for promoting community diversity (Germerodt et al., 2016). There is a need to continue to investigate the ecological importance of spatial heterogeneity in the colon, especially the possibility that there are multiple habitats associated with complex plant glycan molecules (Pereira and Berry, 2017; Tannock and Taylor, 2017).
As summarized in Figure 2, the use of culture-based experiments with “synthetic” microbial communities may be advantageous in future research because they could model specific functions occurring in the gut ecosystem that can in turn be modulated by interventions (Widder et al., 2016; Brüls et al., 2021). These model, in vitro communities could have simplified bacterial diversity yet perform the complex functions observed in vivo and could be investigated by gene transcription and biochemistry to generate regulatory information (“how does it work?”) especially in relation to temporal nutrient switching, which may be an important feature of bacterial residency in the colon (Centanni et al., 2020a). Enrichment cultures, from which models could be derived using medium containing a specific plant glycan and a fecal inoculum, could be useful because metabolically cohesive, bacterial consortia, about which we know little, would be enriched and the rules that drive their formation would be revealed (Pascual-García et al., 2020). Work with co-cultures, preferably performed under lotic conditions such as in chemostats (steady-state conditions), provide opportunities for replication of experiments and hence improve reproducibility and statistical confidence, and potential for mathematical modeling (Kettle et al., 2015; Zomorrodi and Segrè, 2016; Centanni et al., 2019; Centanni et al., 2020b; Liu et al., 2020; Mabwi et al., 2020; Sims and Tannock, 2020; Shetty et al., 2022; Gianetto-Hill et al., 2023). Mutation of specific genes can test ecological fitness of bacteria showing the importance of specific bacterial attributes in underpinning symbiont life in the gut (Sims et al., 2011; Wilson et al., 2012; Tannock et al., 2012; Wilson et al., 2014). Overall, studying molecular details of the metacommunity should reveal intriguing details about how it came to be the way it is today, how its “health” can be better measured, and how it might be remediated for medical purposes.
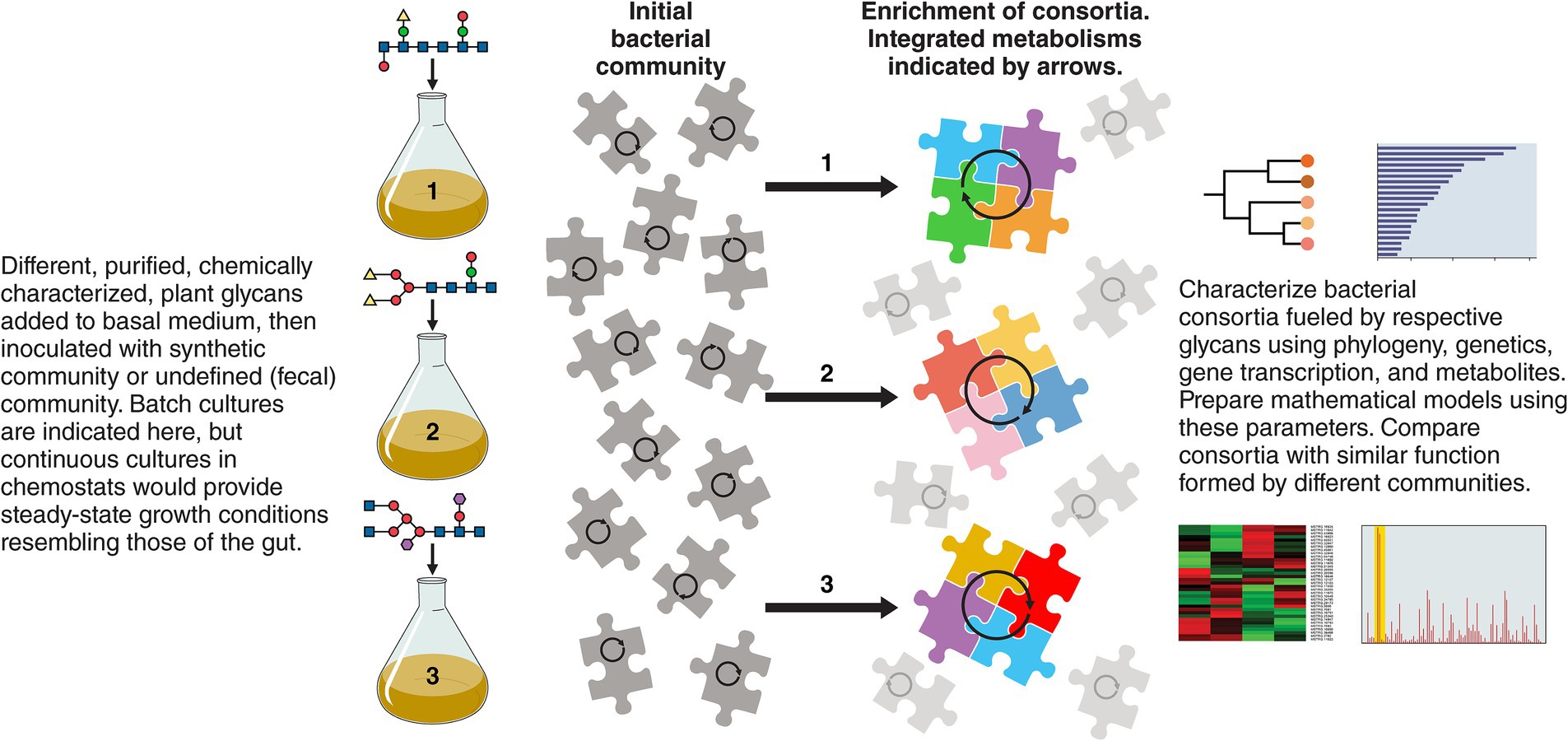
Figure 2. Research with cultured, defined or non-defined bacterial communities fed plant glycans and characterized phylogenetically, genetically, transcriptionally, and metabolically are suggested as starting points to reveal the consortia that drive the energetics of the community in relation to specific growth substrates. These experiments could identify keystone species and explain the individualism of gut microbiomes which are nevertheless similar in emergent properties because of metabolic redundancy. Potentially useful information about bacterial growth substrates for use in ecosystem restoration could be gained.
Potential future developments to restore microbiome function using precision medicine
Personal gut microbiomes, collectively, form a conceptual metacommunity that has functional criteria consistent with “health” of the microbial community. The taxonomic composition of personal microbiomes is “tailor-made” by fortuitous events associated with selection, drift, speciation, diversification, and dispersal. However, ecological functions are similar across the metacommunity due to selection of metabolic redundancies, so functionality is “off-the-shelf” regardless of taxonomic composition. This means that restorative measures to achieve health can avoid the difficulties associated with altering the taxonomic composition of the microbiome (which is vastly different between individual humans) and can focus instead on correction of specific functional differences. However, an understanding of how the metacommunity “works” is required to develop practical solutions.
The idea that health care can be tailored according to the patient’s genotype, environment and lifestyle rather than the expected responses of an “average patient” underpins the concept of “precision” (“personalised,” “individualised”) medicine (Kuntz and Gilbert, 2017; Petrosino, 2018; McCormick and Chang, 2021). Applied to the gut microbiome, the aim would be to remediate dysbiosis revealed by bioassays of microbiome functions using personalized treatments, rather than traditional probiotic and/or prebiotic approaches based on merchandising “wellness.” Precision medical approaches are feasible because responses to dietary supplementation with plant glycans (for example, response to doses of arabinoxylan or resistant starch) are variable among humans (Martínez et al., 2010; Nguyen et al., 2020; Holmes et al., 2022; Lancaster et al., 2022) thus providing possibilities of personalized nutrition. The scope of this kind of work has expanded recently because of better analytical methods to determine and modify glycan chemistry (Amicucci et al., 2019; Michalak et al., 2020; Tuncil et al., 2020; Deehan et al., 2020; Cantu-Jungles et al., 2021; Castillo et al., 2022; Couture et al., 2024), coupled with ever expanding knowledge of the biochemical diversity of plant cultivars, as well as an interest in using processed plant wastes to prepare novel foods for human consumption (Delannoy-Bruno et al., 2021).
Admittedly, a dysbiotic gut community might lack the microbial mechanisms that drive normal functions (for example, catabolism of resistant starch) (Walker et al., 2011). There could, therefore, be a need to prepare commercial, multi-component bacterial consortia with prescribed functional attributes and to transfer the artificial community to specific humans. This might be accomplished by a dose of a “defined function consortium” administered orally or by enema. This is analogous to restoring a disturbed colon community by means of “faecal microbiota transplant” (FMT) which is useful in treating some cases of Clostridioides difficile infection and may also be useful in ameliorating disrupted transfer of gut bacteria to children that have been delivered by cesarean section. However, the biology of FMT is ill-defined and not free of medical risk so inoculation with laboratory assembled consortia would be a sensible development (Gilbert and Lynch, 2019; Burke and Lamont, 2013; Smillie et al., 2018). Much work is required to not only develop pertinent consortia to achieve this goal, but also to prepare preparations that retain viability for use as inoculants, as well as details of dose and dosing schedule. Although the microbiome may be more malleable temporally than previously thought (Valles-Colomer et al., 2023), colonization resistance may be a limiting factor.
Considerations of the restoration of dysbiotic microbiomes based on knowledge of the human gut metacommunity has implications in another area of precision medicine, that of responses to cancer therapy. Receptor-ligand interactions (for example, PD-1, PD-L1) are associated with the ability of T-cells to differentiate between healthy human cells and potential pathogens. Unfortunately, cancer cells can co-opt this “immune checkpoint” system to avoid the destructive attentions of T-cells. Immunoglobulin administration is used to enhance therapy of several types of cancer by interfering with this immune checkpoint blockade (ICB). Destruction of cancer cells by T-cells is enhanced, although healthy cells are also affected. Curiously, not all patients respond equally to ICB treatment. Prior treatment with antibiotics reduces efficacy, indicating an influence of the gut microbiome. Research results using germfree and gnotobiotic mice suggest that some members of the microbiome promote response to ICB but this work is difficult to reconcile with human patients because the murine gut microbiome is dominated by different taxa and is differently distributed in the gut compared to humans (Cammarota et al., 2020; Fernandes et al., 2022; Park et al., 2023). Nevertheless, precision modulation of the human gut microbiome of cancer patients to enhance ICB could be useful spin-off technology from development of functional adjustments to restore healthy microbiomes.
Clearly, the starting point for future research is the development of bioassays (such as those outlined above) and others (such as catabolism of resistant starch and hemicelluloses) (Walker et al., 2011), by which health standards can be set in relation to the human gut metacommunity. Functional dysbiosis will then be recognizable in personal microbiomes of patients, and restorative procedures could be developed. “Precision functional restoration” will be the goal of this exciting research focussing on the human-microbiome symbiosis.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
GT: Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
Gerald W. Tannock is Professor Emeritus of the University of Otago and is hosted by the Department of Microbiology and Immunology.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amicucci, M. J., Nandita, E., and Lebrilla, C. B. (2019). Function without structures: the need for in-depth analysis of dietary carbohydrates. J. Agric. Food Chem. 67, 4418–4424. doi: 10.1021/acs.jafc.9b00720
Asnicar, F., Leeming, E. R., Dimidi, E., Mazidi, M., Franks, P. W., Al Khatib, H., et al. (2021). Blue poo: impact of gut transit time on the gut microbiome using a novel marker. Gut 70, 1665–1674. doi: 10.1136/gutjnl-2020-323877
Asnicar, F., Thomas, A. M., Passerini, A., Waldron, L., and Segata, N. (2024). Machine learning for microbiologists. Nat. Rev. Microbiol. 22, 191–205. doi: 10.1038/s41579-023-00984-1
Blanco-Míguez, A., Gálvez, E. J. C., Pasolli, E., De Filippis, F., Amend, L., Huang, K. D., et al. (2023). Extension of the Segatella copri complex to 13 species with distinct large extrachromosomal elements and associations with host conditions. Cell Host Microbe 31, 1804–1819.e9. doi: 10.1016/j.chom.2023.09.013
Brüls, T., Baumdicker, F., and Smidt, H. (2021). Editorial: synthetic microbial ecology. Front. Microbiol. 12:757848. doi: 10.3389/fmicb.2021.757848
Burger, O., Baudisch, A., and Vaupel, J. W. (2012). Human mortality improvement in evolutionary context. Proc. Natl. Acad. Sci. USA 109, 18210–18214. doi: 10.1073/pnas.1215627109
Burke, K. E., and Lamont, J. T. (2013). Fecal transplantation for recurrent Clostridium difficile infection in older adults: a review. J. Am. Geriatr. Soc. 61, 1394–1398. doi: 10.1111/jgs.12378
Cammarota, G., Ianiro, G., Ahern, A., Carbone, C., Temko, A., Claesson, M. J., et al. (2020). Gut microbiome, big data and machine learning to promote precision medicine for cancer. Nat. Rev. Gastroenterol. Hepatol. 17, 635–648. doi: 10.1038/s41575-020-0327-3
Cantu-Jungles, T. M., Bulut, N., Chambry, E., Ruthes, A., Iacomini, M., Keshavarzian, A., et al. (2021). Dietary Fiber hierarchical specificity: the missing link for predictable and strong shifts in gut bacterial communities. mBio 12:e0102821. doi: 10.1128/mBio.01028-21
Carter, M. M., Olm, M. R., Merrill, B. D., Dahan, D., Tripathi, S., Spencer, S. P., et al. (2023). Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Cell 186, 3111–3124.e13. doi: 10.1016/j.cell.2023.05.046
Castillo, J. J., Couture, G., Bacalzo, N. P. Jr., Chen, Y., Chin, E. L., Blecksmith, S. E., et al. (2022). The development of the Davis food Glycopedia-A glycan encyclopedia of food. Nutrients 14:1639. doi: 10.3390/nu14081639
Centanni, M., Bell, T. J., Sims, I. M., and Tannock, G. W. (2020a). Preferential use of plant glycans for growth by Bacteroides ovatus. Anaerobe 66:102276. doi: 10.1016/j.anaerobe.2020.102276
Centanni, M., Ferguson, S. A., Sims, I. M., Biswas, A., and Tannock, G. W. (2019). Bifidobacterium bifidum ATCC 15696 and Bifidobacterium breve 24b metabolic interaction based on 2'-O-Fucosyl-lactose studied in steady-state cultures in a Freter-style Chemostat. Appl. Environ. Microbiol. 85, e02783–e02718. doi: 10.1128/AEM.02783-18
Centanni, M., Sims, I. M., Bell, T. J., Biswas, A., and Tannock, G. W. (2020b). Sharing a β-glucan meal: transcriptomic eavesdropping on a Bacteroides ovatus-Subdoligranulum variabile-Hungatella hathewayi consortium. Appl. Environ. Microbiol. 86, e01651–e01620. doi: 10.1128/AEM.01651-20
Costello, E. K., Stagaman, K., Dethlefsen, L., Bohannan, B. J., and Relman, D. A. (2012). The application of ecological theory toward an understanding of the human microbiome. Science 336, 1255–1262. doi: 10.1126/science.1224203
Couture, G., Cheang, S. E., Suarez, C., Chen, Y., Bacalzo, N. P. Jr., Jiang, J., et al. (2024). A multi-glycomic platform for the analysis of food carbohydrates. Nat. Protoc. doi: 10.1038/s41596-024-01017-8
Culp, E. J., and Goodman, A. L. (2023). Cross-feeding in the gut microbiome: ecology and mechanisms. Cell Host Microbe 31, 485–499. doi: 10.1016/j.chom.2023.03.016
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 107, 14691–14696. doi: 10.1073/pnas.1005963107
Deehan, E. C., Yang, C., Perez-Muñoz, M. E., Nguyen, N. K., Cheng, C. C., Triador, L., et al. (2020). Precision microbiome modulation with discrete dietary Fiber structures directs short-chain fatty acid production. Cell Host Microbe 27, 389–404.e6. doi: 10.1016/j.chom.2020.01.006
Delannoy-Bruno, O., Desai, C., Raman, A. S., Chen, R. Y., Hibberd, M. C., Cheng, J., et al. (2021). Evaluating microbiome-directed fibre snacks in gnotobiotic mice and humans. Nature 595, 91–95. doi: 10.1038/s41586-021-03671-4
Dubois, L., Valles-Colomer, M., Ponsero, A., Helve, O., Andersson, S., Kolho, K. L., et al. (2024). Paternal and induced gut microbiota seeding complement mother-to-infant transmission. Cell Host Microbe 32, 1011–1024.e4. doi: 10.1016/j.chom.2024.05.004
El Kaoutari, A., Armougom, F., Gordon, J. I., Raoult, D., and Henrissat, B. (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 11, 497–504. doi: 10.1038/nrmicro3050
Faith, J. J., Guruge, J. L., Charbonneau, M., Subramanian, S., Seedorf, H., Goodman, A. L., et al. (2013). The long-term stability of the human gut microbiota. Science 341:1237439. doi: 10.1126/science.1237439
Fernandes, M. R., Aggarwal, P., Costa, R. G. F., Cole, A. M., and Trinchieri, G. (2022). Targeting the gut microbiota for cancer therapy. Nat. Rev. Cancer 22, 703–722. doi: 10.1038/s41568-022-00513-x
Franzosa, E. A., Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X. C., et al. (2015). Sequencing and beyond: integrating molecular 'omics' for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372. doi: 10.1038/nrmicro3451
Gacesa, R., Kurilshikov, A., Vich Vila, A., Sinha, T., Klaassen, M. A. Y., Bolte, L. A., et al. (2022). Environmental factors shaping the gut microbiome in a Dutch population. Nature 604, 732–739. doi: 10.1038/s41586-022-04567-7
Gaulke, C. A., and Sharpton, T. J. (2018). The influence of ethnicity and geography on human gut microbiome composition. Nat. Med. 24, 1495–1496. doi: 10.1038/s41591-018-0210-8
Germerodt, S., Bohl, K., Lück, A., Pande, S., Schröter, A., Kaleta, C., et al. (2016). Pervasive selection for cooperative cross-feeding in bacterial communities. PLoS Comput. Biol. 12:e1004986. doi: 10.1371/journal.pcbi.1004986
Gianetto-Hill, C. M., Vancuren, S. J., Daisley, B., Renwick, S., Wilde, J., Schroeter, K., et al. (2023). The Robogut: A bioreactor model of the human Colon for evaluation of gut microbial community ecology and function. Curr. Protocols 3:e737. doi: 10.1002/cpz1.737
Gilbert, J. A., and Lynch, S. V. (2019). Community ecology as a framework for human microbiome research. Nat. Med. 25, 884–889. doi: 10.1038/s41591-019-0464-9
Grondin, J. M., Tamura, K., Déjean, G., Abbott, D. W., and Brumer, H. (2017). Polysaccharide utilization loci: fueling microbial communities. J. Bacteriol. 199, e00860–e00816. doi: 10.1128/JB.00860-16
Holmes, Z. C., Villa, M. M., Durand, H. K., Jiang, S., Dallow, E. P., Petrone, B. L., et al. (2022). Microbiota responses to different prebiotics are conserved within individuals and associated with habitual fiber intake. Microbiome 10:114. doi: 10.1186/s40168-022-01307-x
Hsu, B. B., Gibson, T. E., Yeliseyev, V., Liu, Q., Lyon, L., Bry, L., et al. (2019). Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe 25, 803–814.e5. doi: 10.1016/j.chom.2019.05.001
Kettle, H., Louis, P., Holtrop, G., Duncan, S. H., and Flint, H. J. (2015). Modelling the emergent dynamics and major metabolites of the human colonic microbiota. Environ. Microbiol. 17, 1615–1630. doi: 10.1111/1462-2920.12599
Kuntz, T. M., and Gilbert, J. A. (2017). Introducing the microbiome into precision medicine. Trends Pharmacol. Sci. 38, 81–91. doi: 10.1016/j.tips.2016.10.001
Lancaster, S. M., Lee-McMullen, B., Abbott, C. W., Quijada, J. V., Hornburg, D., Park, H., et al. (2022). Global, distinctive, and personal changes in molecular and microbial profiles by specific fibers in humans. Cell Host Microbe 30, 848–862.e7. doi: 10.1016/j.chom.2022.03.036
Lawley, B., Otal, A., Moloney-Geany, K., Diana, A., Houghton, L., Heath, A. M., et al. (2019). Fecal microbiotas of Indonesian and New Zealand children differ in complexity and Bifidobacterial taxa during the first year of life. Appl. Environ. Microbiol. 85, e01105–e01119. doi: 10.1128/AEM.01105-19
Leong, C., Haszard, J. J., Lawley, B., Otal, A., Taylor, R. W., Szymlek-Gay, E. A., et al. (2018). Mediation analysis as a means of identifying dietary components that differentially affect the fecal microbiota of infants weaned by modified baby-led and traditional approaches. Appl. Environ. Microbiol. 84, e00914–e00918. doi: 10.1128/AEM.00914-18
Lindstad, L. J., Lo, G., Leivers, S., Lu, Z., Michalak, L., Pereira, G. V., et al. (2021). Human gut Faecalibacterium prausnitzii deploys a highly efficient conserved system to cross-feed on β-Mannan-derived oligosaccharides. mBio 12:e0362820. doi: 10.1128/mBio.03628-20
Liu, Y., Heath, A. L., Galland, B., Rehrer, N., Drummond, L., Wu, X. Y., et al. (2020). Substrate use prioritization by a Coculture of five species of gut Bacteria fed mixtures of Arabinoxylan, xyloglucan, β-glucan, and pectin. Appl. Environ. Microbiol. 86, e01905–e01919. doi: 10.1128/AEM.01905-19
Mabwi, H. A., Kim, E., Song, D. G., Yoon, H. S., Pan, C. H., Komba, E. V. G., et al. (2020). Synthetic gut microbiome: advances and challenges. Comput. Struct. Biotechnol. J. 19, 363–371. doi: 10.1016/j.csbj.2020.12.029
Manor, O., Dai, C. L., Kornilov, S. A., Smith, B., Price, N. D., Lovejoy, J. C., et al. (2020). Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 11:5206. doi: 10.1038/s41467-020-18871-1
Martínez, I., Kim, J., Duffy, P. R., Schlegel, V. L., and Walter, J. (2010). Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS One 5:e15046. doi: 10.1371/journal.pone.0015046
McCormick, B. A., and Chang, E. B. (2021). The gut microbiome: reaching the promise through discovery- advancing knowledge and discovery of the gut microbiome in the age of precision medicine. Gastroenterology 160, 479–482. doi: 10.1053/j.gastro.2020.12.035
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Michalak, L., Gaby, J. C., Lagos, L., La Rosa, S. L., Hvidsten, T. R., Tétard-Jones, C., et al. (2020). Microbiota-directed fibre activates both targeted and secondary metabolic shifts in the distal gut. Nat. Commun. 11:5773. doi: 10.1038/s41467-020-19585-0
Mills, D. A., German, J. B., Lebrilla, C. B., and Underwood, M. A. (2023). Translating neonatal microbiome science into commercial innovation: metabolism of human milk oligosaccharides as a basis for probiotic efficacy in breast-fed infants. Gut Microbes 15:2192458. doi: 10.1080/19490976.2023.2192458
Morris, J. J., Lenski, R. E., and Zinser, E. R. (2012). The black queen hypothesis: evolution of dependencies through adaptive gene loss. mBio 3, e00036–e00012. doi: 10.1128/mBio.00036-12
Nagata, N., Nishijima, S., Miyoshi-Akiyama, T., Kojima, Y., Kimura, M., Aoki, R., et al. (2022). Population-level metagenomics uncovers distinct effects of multiple medications on the human gut microbiome. Gastroenterology 163, 1038–1052. doi: 10.1053/j.gastro.2022.06.070
Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S., and Kyrpides, N. C. (2019). New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510. doi: 10.1038/s41586-019-1058-x
Nguyen, N. K., Deehan, E. C., Zhang, Z., Jin, M., Baskota, N., Perez-Muñoz, M. E., et al. (2020). Gut microbiota modulation with long-chain corn bran arabinoxylan in adults with overweight and obesity is linked to an individualized temporal increase in fecal propionate. Microbiome 8:118. doi: 10.1186/s40168-020-00887-w
Nielsen, H. B., Almeida, M., Juncker, A. S., Rasmussen, S., Li, J., Sunagawa, S., et al. (2014). Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828. doi: 10.1038/nbt.2939
Parizadeh, M., and Arrieta, M. C. (2023). The global human gut microbiome: genes, lifestyles, and diet. Trends Mol. Med. 29, 789–801. doi: 10.1016/j.molmed.2023.07.002
Park, J. S., Gazzaniga, F. S., Wu, M., Luthens, A. K., Gillis, J., Zheng, W., et al. (2023). Targeting PD-L2-RGMb overcomes microbiome-related immunotherapy resistance. Nature 617, 377–385. doi: 10.1038/s41586-023-06026-3
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-7
Pascual-García, A., Bonhoeffer, S., and Bell, T. (2020). Metabolically cohesive microbial consortia and ecosystem functioning. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 375:20190245. doi: 10.1098/rstb.2019.0245
Pereira, F. C., and Berry, D. (2017). Microbial nutrient niches in the gut. Environ. Microbiol. 19, 1366–1378. doi: 10.1111/1462-2920.13659
Petrosino, J. F. (2018). The microbiome in precision medicine: the way forward. Genome Med. 10:12. doi: 10.1186/s13073-018-0525-6
Prosser, J. I. (2022). How and why in microbial ecology: an appeal for scientific aims, questions, hypotheses and theories. Environ. Microbiol. 24, 4973–4980. doi: 10.1111/1462-2920.16221
Pryde, S. E., Duncan, S. H., Hold, G. L., Stewart, C. S., and Flint, H. J. (2002). The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 217, 133–139. doi: 10.1111/j.1574-6968.2002.tb11467.x
Rampelli, S., Soverini, M., D'Amico, F., Barone, M., Tavella, T., Monti, D., et al. (2020). Shotgun metagenomics of gut microbiota in humans with up to extreme longevity and the increasing role of xenobiotic degradation. mSystems 5, e00124–e00120. doi: 10.1128/mSystems.00124-20
Renall, N., Lawley, B., Vatanen, T., Merz, B., Douwes, J., Corbin, M., et al. (2023). The fecal microbiotas of women of Pacific and New Zealand European ethnicities are characterized by distinctive enterotypes that reflect dietary intakes and fecal water content. Gut Microbes 15:2178801. doi: 10.1080/19490976.2023.2178801
Roager, H. M., Hansen, L. B., Bahl, M. I., Frandsen, H. L., Carvalho, V., Gøbel, R. J., et al. (2016). Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat. Microbiol. 1:16093. doi: 10.1038/nmicrobiol.2016.93
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5:3654. doi: 10.1038/ncomms4654
Sender, R., Fuchs, S., and Milo, R. (2016). Revised estimates for the number of human and Bacteria cells in the body. PLoS Biol. 14:e1002533. doi: 10.1371/journal.pbio.1002533
Shanahan, F., Ghosh, T. S., Molloy, M. G., and O'Toole, P. W. (2022). The nonindustrialised microbiome in a modern world. Clin. Sci. (Lond.) 136, 1683–1690. doi: 10.1042/CS20220203
Shanahan, F., Ghosh, T. S., and O'Toole, P. W. (2021). The healthy microbiome-what is the definition of a healthy gut microbiome? Gastroenterology 160, 483–494. doi: 10.1053/j.gastro.2020.09.057
Shetty, S. A., Kostopoulos, I., Geerlings, S. Y., Smidt, H., de Vos, W. M., and Belzer, C. (2022). Dynamic metabolic interactions and trophic roles of human gut microbes identified using a minimal microbiome exhibiting ecological properties. ISME J. 16, 2144–2159. doi: 10.1038/s41396-022-01255-2
Sims, I. M., Frese, S. A., Walter, J., Loach, D., Wilson, M., Appleyard, K., et al. (2011). Structure and functions of exopolysaccharide produced by gut commensal Lactobacillus reuteri 100-23. ISME J. 5, 1115–1124. doi: 10.1038/ismej.2010.201
Sims, I. M., and Tannock, G. W. (2020). Galacto- and Fructo-oligosaccharides utilized for growth by Cocultures of Bifidobacterial species characteristic of the infant gut. Appl. Environ. Microbiol. 86, e00214–e00220. doi: 10.1128/AEM.00214-20
Smillie, C. S., Sauk, J., Gevers, D., Friedman, J., Sung, J., Youngster, I., et al. (2018). Strain tracking reveals the determinants of bacterial engraftment in the human gut following fecal microbiota transplantation. Cell Host Microbe 23, 229–240.e5. doi: 10.1016/j.chom.2018.01.003
Song, S. J., Lauber, C., Costello, E. K., Lozupone, C. A., Humphrey, G., Berg-Lyons, D., et al. (2013). Cohabiting family members share microbiota with one another and with their dogs. eLife 2:e00458. doi: 10.7554/eLife.00458
Stams, A. J., and Plugge, C. M. (2009). Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat. Rev. Microbiol. 7, 568–577. doi: 10.1038/nrmicro2166
Sze, M. A., and Schloss, P. D. (2016). Looking for a signal in the noise: Revisiting Obesity and the Microbiome. mBio 7, e01018–e01016. doi: 10.1128/mBio.01018-16
Tamburini, F. B., Maghini, D., Oduaran, O. H., Brewster, R., Hulley, M. R., Sahibdeen, V., et al. (2022). Short- and long-read metagenomics of urban and rural south African gut microbiomes reveal a transitional composition and undescribed taxa. Nat. Commun. 13:926. doi: 10.1038/s41467-021-27917-x
Tannock, G. W. (2021a). Modulating the gut microbiota of humans by dietary intervention with plant Glycans. Appl. Environ. Microbiol. 87, e02757–e02720. doi: 10.1128/AEM.02757-20
Tannock, G. W. (2021b). Building robust assemblages of Bacteria in the human gut in early life. Appl. Environ. Microbiol. 87:e0144921. doi: 10.1128/AEM.01449-21
Tannock, G. W. (2022). Exploring bacterial attributes that underpin symbiont life in the Monogastric gut. Appl. Environ. Microbiol. 88:e0112822. doi: 10.1128/aem.01128-22
Tannock, G. W. (2024a). Understanding the gut microbiota by considering human evolution: a story of fire, cereals, cooking, molecular ingenuity, and functional cooperation. Microbiol. Mol. Biol. Rev. 88:e0012722. doi: 10.1128/mmbr.00127-22
Tannock, G. W. (2024b). Scoring microbiota function: A proposal to use features of evolutionary, symbiotic innovation to recognize a “healthy” human gut microbiota. Gut Microbes Reports 1, 1–7. doi: 10.1080/29933935.2024.2376543
Tannock, G. W., and Taylor, M. W. (2017). Embracing the co-operative society to better understand assembly of the gut microbiota. Environ. Microbiol. 19, 2924–2925. doi: 10.1111/1462-2920.13752
Tannock, G. W., Wilson, C. M., Loach, D., Cook, G. M., Eason, J., O'Toole, P. W., et al. (2012). Resource partitioning in relation to cohabitation of Lactobacillus species in the mouse forestomach. ISME J. 6, 927–938. doi: 10.1038/ismej.2011.161
Tian, L., Wang, X. W., Wu, A. K., Fan, Y., Friedman, J., Dahlin, A., et al. (2020). Deciphering functional redundancy in the human microbiome. Nat. Commun. 11:6217. doi: 10.1038/s41467-020-19940-1
Tramontano, M., Andrejev, S., Pruteanu, M., Klünemann, M., Kuhn, M., Galardini, M., et al. (2018). Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat. Microbiol. 3, 514–522. doi: 10.1038/s41564-018-0123-9
Truong, D. T., Tett, A., Pasolli, E., Huttenhower, C., and Segata, N. (2017). Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 27, 626–638. doi: 10.1101/gr.216242.116
Tuncil, Y. E., Thakkar, R. D., Arioglu-Tuncil, S., Hamaker, B. R., and Lindemann, S. R. (2020). Subtle variations in dietary-Fiber fine structure differentially influence the composition and metabolic function of gut microbiota. mSphere 5, e00180–e00120. doi: 10.1128/mSphere.00180-20
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Ursell, L. K., Metcalf, J. L., Parfrey, L. W., and Knight, R. (2012). Defining the human microbiome. Nutr. Rev. 70 Suppl 1, S38–S44. doi: 10.1111/j.1753-4887.2012.00493.x
Valles-Colomer, M., Blanco-Míguez, A., Manghi, P., Asnicar, F., Dubois, L., Golzato, D., et al. (2023). The person-to-person transmission landscape of the gut and oral microbiomes. Nature 614, 125–135. doi: 10.1038/s41586-022-05620-1
Vandeputte, D., Falony, G., Vieira-Silva, S., Tito, R. Y., Joossens, M., and Raes, J. (2016). Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65, 57–62. doi: 10.1136/gutjnl-2015-309618
Vangay, P., Johnson, A. J., Ward, T. L., Al-Ghalith, G. A., Shields-Cutler, R. R., Hillmann, B. M., et al. (2018). US immigration westernizes the human gut microbiome. Cell 175, 962–972.e10. doi: 10.1016/j.cell.2018.10.029
Vellend, M. (2010). Conceptual synthesis in community ecology. Q. Rev. Biol. 85, 183–206. doi: 10.1086/652373
Visconti, A., Le Roy, C. I., Rosa, F., Rossi, N., Martin, T. C., Mohney, R. P., et al. (2019). Interplay between the human gut microbiome and host metabolism. Nat. Commun. 10:4505. doi: 10.1038/s41467-019-12476-z
Wainwright, C. E., Staples, T. L., Charles, L. S., Flanagan, T. C., Lai, H. R., Loy, X., et al. (2018). Links between community ecology theory and ecological restoration are on the rise. J. Appl. Ecol. 55, 570–581. doi: 10.1111/1365-2664.12975
Walker, A. W., Ince, J., Duncan, S. H., Webster, L. M., Holtrop, G., Ze, X., et al. (2011). Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 5, 220–230. doi: 10.1038/ismej.2010.118
White, B. A., Lamed, R., Bayer, E. A., and Flint, H. J. (2014). Biomass utilization by gut microbiomes. Ann. Rev. Microbiol. 68, 279–296. doi: 10.1146/annurev-micro-092412-155618
Widder, S., Allen, R. J., Pfeiffer, T., Curtis, T. P., Wiuf, C., Sloan, W. T., et al. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J. 10, 2557–2568. doi: 10.1038/ismej.2016.45
Wilson, C. M., Aggio, R. B., O'Toole, P. W., Villas-Boas, S., and Tannock, G. W. (2012). Transcriptional and metabolomic consequences of LuxS inactivation reveal a metabolic rather than quorum-sensing role for LuxS in Lactobacillus reuteri 100-23. J. Bacteriol. 194, 1743–1746. doi: 10.1128/JB.06318-11
Wilson, C. M., Loach, D., Lawley, B., Bell, T., Sims, I. M., O'Toole, P. W., et al. (2014). Lactobacillus reuteri 100-23 modulates urea hydrolysis in the murine stomach. Appl. Environ. Microbiol. 80, 6104–6113. doi: 10.1128/AEM.01876-14
Xu, J., Lawley, B., Wong, G., Otal, A., Chen, L., Ying, T. J., et al. (2020). Ethnic diversity in infant gut microbiota is apparent before the introduction of complementary diets. Gut Microbes 11, 1362–1373. doi: 10.1080/19490976.2020.1756150
Yassour, M., Jason, E., Hogstrom, L. J., Arthur, T. D., Tripathi, S., Siljander, H., et al. (2018). Strain-level analysis of mother-to-child bacterial transmission during the first few months of life. Cell Host Microbe 24, 146–154.e4. doi: 10.1016/j.chom.2018.06.007
Ze, X., Duncan, S. H., Louis, P., and Flint, H. J. (2012). Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 6, 1535–1543. doi: 10.1038/ismej.2012.4
Zhao, S., Lieberman, T. D., Poyet, M., Kauffman, K. M., Gibbons, S. M., Groussin, M., et al. (2019). Adaptive evolution within gut microbiomes of healthy people. Cell Host Microbe 25, 656–667.e8. doi: 10.1016/j.chom.2019.03.007
Keywords: metacommunity, microbiome, microbiota, precision medicine, symbiosis, dysbiosis
Citation: Tannock GW (2024) The human gut metacommunity as a conceptual aid in the development of precision medicine. Front. Microbiol. 15:1469543. doi: 10.3389/fmicb.2024.1469543
Edited by:
Franck Carbonero, Washington State University Health Sciences Spokane, United StatesReviewed by:
Silvia Turroni, University of Bologna, ItalyCopyright © 2024 Tannock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerald W. Tannock, Z2VyYWxkLnRhbm5vY2tAb3RhZ28uYWMubno=