 Yang Zhao
Yang Zhao Wen-Jing Ding1
Wen-Jing Ding1 Ji-Quan Sun
Ji-Quan Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 16 May 2024
Sec. Microbe and Virus Interactions with Plants
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1395477
Stenotrophomonas strains, which are often described as plant growth promoting (PGP) bacteria, are ubiquitous in many environments. A total of 213 genomes of strains of Stenotrophomonas were analyzed using comparative genomics to better understand the ecological roles of these bacteria in the environment. The pan-genome of the 213 strains of Stenotrophomonas consists of 27,186 gene families, including 710 core gene families, 11,039 unique genes and 15,437 accessory genes. Nearly all strains of Stenotrophomonas harbor the genes for GH3-family cellulose degradation and GH2- and GH31-family hemicellulose hydrolase, as well as intact glycolysis and tricarboxylic acid cycle pathways. These abilities suggest that the strains of this genus can easily obtain carbon and energy from the environment. The Stenotrophomonas strains can respond to oxidative stress by synthesizing catalase, superoxide dismutase, methionine sulfoxide reductase, and disulfide isomerase, as well as managing their osmotic balance by accumulating potassium and synthesizing compatible solutes, such as betaine, trehalose, glutamate, and proline. Each Stenotrophomonas strain also contains many genes for resistance to antibiotics and heavy metals. These genes that mediate stress tolerance increase the ability of Stenotrophomonas strains to survive in extreme environments. In addition, many functional genes related to attachment and plant colonization, growth promotion and biocontrol were identified. In detail, the genes associated with flagellar assembly, motility, chemotaxis and biofilm formation enable the strains of Stenotrophomonas to effectively colonize host plants. The presence of genes for phosphate-solubilization and siderophore production and the polyamine, indole-3-acetic acid, and cytokinin biosynthetic pathways confer the ability to promote plant growth. These strains can produce antimicrobial compounds, chitinases, lipases and proteases. Each Stenotrophomonas genome contained 1–9 prophages and 17–60 genomic islands, and the genes related to antibiotic and heavy metal resistance and the biosynthesis of polyamines, indole-3-acetic acid, and cytokinin may be acquired by horizontal gene transfer. This study demonstrates that strains of Stenotrophomonas are highly adaptable for different environments and have strong potential for use as plant growth-promoting bacteria.
Stenotrophomonas is a genus with versatile metabolic capabilities, which belongs to the family Lysobacteraceae in the order Lysobacterales and is a common genus in various environments (Ryan et al., 2009). Many strains of Stenotrophomonas have been described as plant growth-promoting (PGP) bacteria owing to their various mechanisms (Singh and Jha, 2017; An and Berg, 2018; Alexander et al., 2019). For example, S. maltophilia SBP-9, a strain that simultaneously solubilizes inorganic phosphate and produces indole-3-acetic acid (IAA), gibberellic acid, ACC deaminase, and siderophores, significantly increased the biomass and chlorophyll content of wheat (Triticum aestivum) (Singh and Jha, 2017). Stenotrophomonas sp. CV83, which produces IAA, ACC deaminase and siderophores, can significantly enhance the growth of chickpea (Cicer arietinum) under drought stress conditions (Sharma et al., 2023). S. maltophilia SR1, a bacterium that can degrade aromatic hydrocarbons, exhibited PGP properties (Bashandy et al., 2020). Many strains of Stenotrophomonas have also been demonstrated to protect plants from phytopathogens via multiple ways. For example, S. maltophilia UN1512 can inhibit Colletotrichum nymphaeae, a fungal fruit and leaf pathogen, by secreting volatile and non-volatile organic compounds (Alijani et al., 2020). S. maltophilia C3 can decrease the incidence of Bipolaris leaf spot caused by the fungal pathogen Bipolaris sorokiniana by producing chitinases (Zhang et al., 2001). S. maltophilia MB9 can protect plants from multiple pathogens, including Curvularia sp., Aspergillus niger, Fusarium oxysporum, Diploidia sp., and Rhizoctonia solani, by producing the broad-spectrum antifungal compound dodecanoic acid (John and Thangavel, 2017).
Currently, a substantial amount of comparative genomic research on the species of Stenotrophomonas provides new insight on the formation of biofilm (Flores-Treviño et al., 2019), quorum sensing (QS) signaling and quenching (Huedo et al., 2018), genome evolution (Xu et al., 2023), PGP and antibiotic resistance mechanisms (Huang et al., 2018; Ulrich et al., 2021), and bioremediation (Mukherjee and Roy, 2016). For example, a comparison of the genome of the plant-associated strain S. rhizophila DSM 14405T with S. maltophilia K29a and R551-3 revealed that S. rhizophila DSM 14405T possesses unique genes for the biosynthesis and transportation of plant-protective spermidine, plant cell wall-degrading enzymes, and high salt tolerance, but it lacks several critical virulence factors and heat shock genes (Alavi et al., 2014). A comparative genomic analysis of 30 S. maltophilia and seven S. rhizophila strains revealed that 96 genes, including chitin-binding proteins and mechanosensitive channels protein genes, are unique to S. maltophilia, and 59 genes are unique to S. rhizophila. The strains within both species have a high potential for biocontrol owing to their production of proteases, chitinases and keratinases, as well as similar PGP properties provided by the biosynthesis of siderophores and spermidine (Pinski et al., 2020). A comparative genomic analysis based on the 14 genomes of S. maltophilia revealed that the antibiotic resistance genes were not significantly different between the clinical and environmental strains (Youenou et al., 2015).
Although a substantial amount of comparative genomic research related to Stenotrophomonas strains has been conducted, a systematic and comprehensive comparative genome of this genus, particularly comparative information on the PGP properties is still missing. To better understand the PGP mechanisms of the Stenotrophomonas strains and their ecological roles in the rhizosphere, a comprehensive comparative genomic analysis based on the genomes of 213 strains of Stenotrophomonas was conducted in this study.
By September 2023, there were 1,206 genome sequences of Stenotrophomonas in GenBank. A total of 727 unique genomes were downloaded. The downloaded genome sequences were then evaluated using CheckM (Parks et al., 2015). Only 160 genomes of S. maltophilia (>99.5% completeness and < 0.5% contamination) and 53 genomes of other species of Stenotrophomonas (>90.0% completeness and contamination <6.0%) were selected for subsequent comparative genomic analyses. More details about the 213 Stenotrophomonas genomes are listed in Supplementary Table S1. The global distribution and habitat preference of the genus Stenotrophomonas was evaluated using the analytical pipeline Microbe Atlas Project (MAP)1 (Matias Rodrigues et al., 2017).
All the downloaded genome sequences were annotated using Prodigal (Hyatt et al., 2010). The faa files that were produced were used for pan- and core-genome analyses using the Bacterial Pan Genome Analysis tool (BPGA) pipeline v. 1.3 (Chaudhari et al., 2016). In a pan-genome analysis, the number of accumulated genes that are related to the number of genomes can be predicted by Heaps’ law as (Tettelin et al., 2008). In the equation, is the number of genomes, while and are fitting parameters. When 0 < b < 1 indicates that the pan-genome is open, and only b < 0 indicates that the pan-genome is closed.
The core genome of Stenotrophomonas was constructed by aligning 213 genomes using USEARCH with a cut-off value 50% sequence identity. After the resulting core genes were concatenated and aligned using MUSCLE, a phylogenetic tree was constructed based on the concatenated core genes using the neighbor-joining (NJ) method (Chaudhari et al., 2016).
The amino acid sequences of the polyamines, IAA and cytokinin synthases were aligned using ClustalW (Hall, 2013). A phylogenetic tree based on these single coding sequences (CDS) was constructed using MEGA software v. 6.0 (Tamura et al., 2013) with the NJ algorithm (Saitou and Nei, 1987). The tree topology was evaluated using the bootstrap analysis based on 1,000 resampling replicates. The iTOL online server2 was used to display the phylogenetic tree (Letunic and Bork, 2021). The ANI values between these strains were calculated using fastANI v. 1.33 (Jain et al., 2018). The dDDH values were calculated using the Genome to Genome Distance Calculator (GGDC 2.5)3 (Auch et al., 2010).
A COG analysis of the sequences of proteins from the core, accessory and unique gene families was used to categorize them according to the COG database using BPGA (v. 1.3). To ensure the accuracy and completeness of the results, only 33 complete Stenotrophomonas genomes were selected for analysis of their primary metabolic profile and the prediction of secondary metabolite biosynthetic genes using KAAS (KEGG Automatic Annotation Server) and the antiSMASH version 6.0 online4 (Blin et al., 2021), respectively. The carbohydrate active enzymes (CAZymes) were identified using the HMMER method of the dbCAN online server5 (Zheng et al., 2023) with the e-value thresholds ≤1e−15 and coverage >0.35.
The genes related to PGP, oxidative and osmotic stresses, as well as resistance to heavy metals, were firstly searched in 33 complete genomes, and the sequences obtained were compared with the reference sequences in GenBank to determine the correctness of the sequences. Subsequently, the sequences that were obtained were used as the reference sequences to search their counterparts in the remaining genomes using BLAST.6 Only sequences that had >40% identity with the reference sequences indicated that the genome had the gene (Zhao et al., 2023).
The antibiotic resistance genes were identified using resistance gene identifier (RGI) of the CARD7 (Alcock et al., 2019), and the sequences with Perfect, Strict and Loose hits identities >70% were selected for further analysis.
Integrated prophages were identified using the PHASTER online server8 (Arndt et al., 2016). The predicted prophages with completeness score > 90 were thought to be intact; those with a completeness score of 60–90 were questionable prophages, and those with a completeness score < 60 were considered to be incomplete prophages. The genomic islands (GIs) of the genomes were predicted using the IslandViewer 4 online server9 with the IslandPick (Langille et al., 2008), SIGI-HMM (Waack et al., 2006), IslandPath-DIMOB (Hsiao et al., 2003), and Islander (Hudson et al., 2014) methods (Bertelli et al., 2017).
To learn the original source of the biosynthetic genes for polyamines, IAA, and cytokinin, the topologic differences of the phylogenetic tree between the single genes and the core genome were compared. The DNA genomic G + C content of the single genes and the whole genomes were compared (Syvanen, 1994; Garcia-Vallve, 2000).
Among the 213 strains, 78 were isolated from environmental habitats, such as soil, plants, sludge, water, and biofilm reactors, and 135 were from materials from clinical settings, such as urine, blood, sputum, wounds, lungs, and animals among others. The species S. maltophilia, S. rhizophila, S. indicatrix, S. pavanii and S. geniculata can be found in both clinical and environmental habitats, while S. acidaminiphila, S. lactitubi, S. nitritireducens, and S. bentonitica were only found in environmental habitats.
The genome size of the 213 strains of Stenotrophomonas ranged from 3.50 to 5.12 Mb with a G + C content of 63.8–69.3%. The number of CDS ranged from 3,099 to 5,180. The strains with the largest genome and CDS numbers were S. geniculata NWUBe21, which was isolated from seeds, and S. geniculate E119, which was isolated from feces, while the smallest was S. pictorum JCM 9942T, which isolated from soil. S. acidaminiphila Au-Ste40, which was isolated from soil, contained the highest content of genomic DNA G + C, while S. nitritireducens 2001 that was isolated from lake sediments had the lowest.
The phylogenetic tree based on core genes showed that the 213 strains of Stenotrophomonas can be subdivided into 10 clusters (Figure 1A). Among them, Clusters I, II, III, IV, V and IX are composed of two (S. indicatrix and S. lactitubi), two (S. chelatiphaga and S. tumulicola), four (S. terrae, S. nitritireducens, S. pictorum and S. humi), two (S. acidaminiphila and S. nitritireducens), two (S. bentonitica and S. rhizophila) and two (S. maltophilia and S. geniculate) species, respectively. Clusters VI, VIII, and X all consist of S. maltophilia, while Cluster VII only comprises strains of S. pavanii. The ANI values between all the strains tested were higher than 81%, which confirmed that the 213 strains of Stenotrophomonas belonged to a single genus (Supplementary Table S2). The topology of the tree based on ANI values was similar to that based on the core genome with only minor differences (Figure 1B). Cluster VII formed a clade with Cluster VI in the ANI phylogenetic tree.
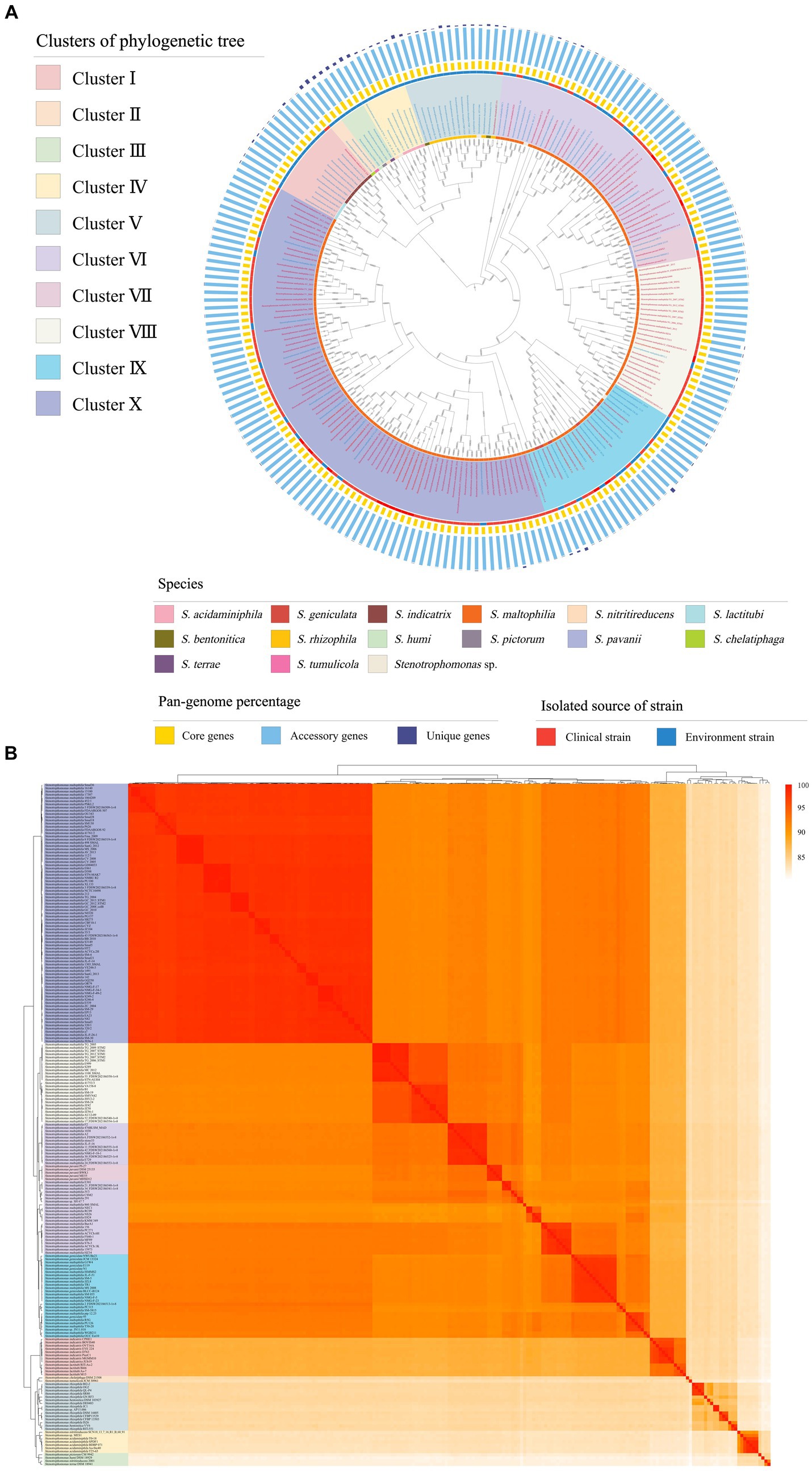
Figure 1. (A) Phylogenetic tree constructed based on core genomes. Clinical strains are marked with red font and environmental strains with blue font. The three colors in the bar graph represent the percentage of core, accessory, and unique genes in the pan-genome. (B) Pairwise average nucleotide identity comparison between the genomes of 213 strains of Stenotrophomonas. The ANI values were used to construct a dendrogram and a heatmap with the average linkage method and Euclidean distance used for clustering and correlation analyses, respectively.
There were 14 different Stenotrophomonas species found in 10 clusters. Among them, S. lactitubi, S. indicatrix, S. acidaminiphila, S. nitritireducens, S. bentonitica, S. rhizophila, S. maltophilia, S. pavanii, and S. geniculata contain multiple strains. The ANI values of strains within the same species of S. lactitubi (95.1–96.9%), S. indicatrix (96.0–98.8%), S. acidaminiphila (97.1–98.9%), and S. pavanii (98.6–98.7%) were > 95.0% (Supplementary Table S2), which indicated that the non-type strains of these species were accurately classified. In contrast, the values between some non-type strains within the other species of Stenotrophomonas compared to their type strain were lower than the threshold value for the delineation of a species (ANI value <95% and dDDH value <70%), which indicated that these strains had been misidentified. For example, strains of S. nitritireducens are present in both Clusters III and IV, and some S. maltophilia strains tightly clustered with S. geniculata in Cluster IX. Strains 2001 and SCN18_13_7_16_R1_B_68_91, which are currently both identified as S. nitritireducens, are scattered in Clusters III and IV, respectively. The ANI and dDDH values of strain 2001 to the type strain of S. nitritireducens were only 84.9 and 27.0%, respectively, indicating that it had been misidentified. Nine of 12 S. rhizophila strains had <90% ANI and < 35% dDDH values to the type strain of S. rhizophila (DSM 14405T), which suggested that these nine strains should not be identified as S. rhizophila. Strains of S. maltophilia were widely scattered in Clusters VI, VIII, IX, and X. Only those in Cluster X shared high ANI (>95%) and dDDH (>70%) values with the type strain, while the two values of the other three cluster strains with S. maltophilia NBRC 14161T ranged from 91.2–94.0% and 41.0–52.0%, respectively. The low values indicate that these strains were misidentified as S. maltophilia (Supplementary Table S2). Among them, the strains in Cluster VIII share high values of ANI and dDDH with each other and low values with the other strains, indicating that they belong to an unidentified novel species, while the ANI and the dDDH values between the strains in Cluster VI were 90.7–99.9% and 39.5–99.5%, respectively, indicating that the strains in this cluster can be further divided into two or more different species, and the classification of the Cluster IX strains was similar to that of Cluster VI.
The pan-genome of the 213 Stenotrophomonas strains contained 27,186 gene families. There were 710, 15,437, and 11,039 core, accessory, and unique genes, respectively. Each of the 213 Stenotrophomonas genomes were composed of 15.1–24.3% core genes and 69.0–83.5% accessory genes, while the unique genes only accounted for 0–11.1% of the genomes. According to Heaps’ law, the pan-genome of Stenotrophomonas was open and increasing (b = 0.48) (Figure 2A).
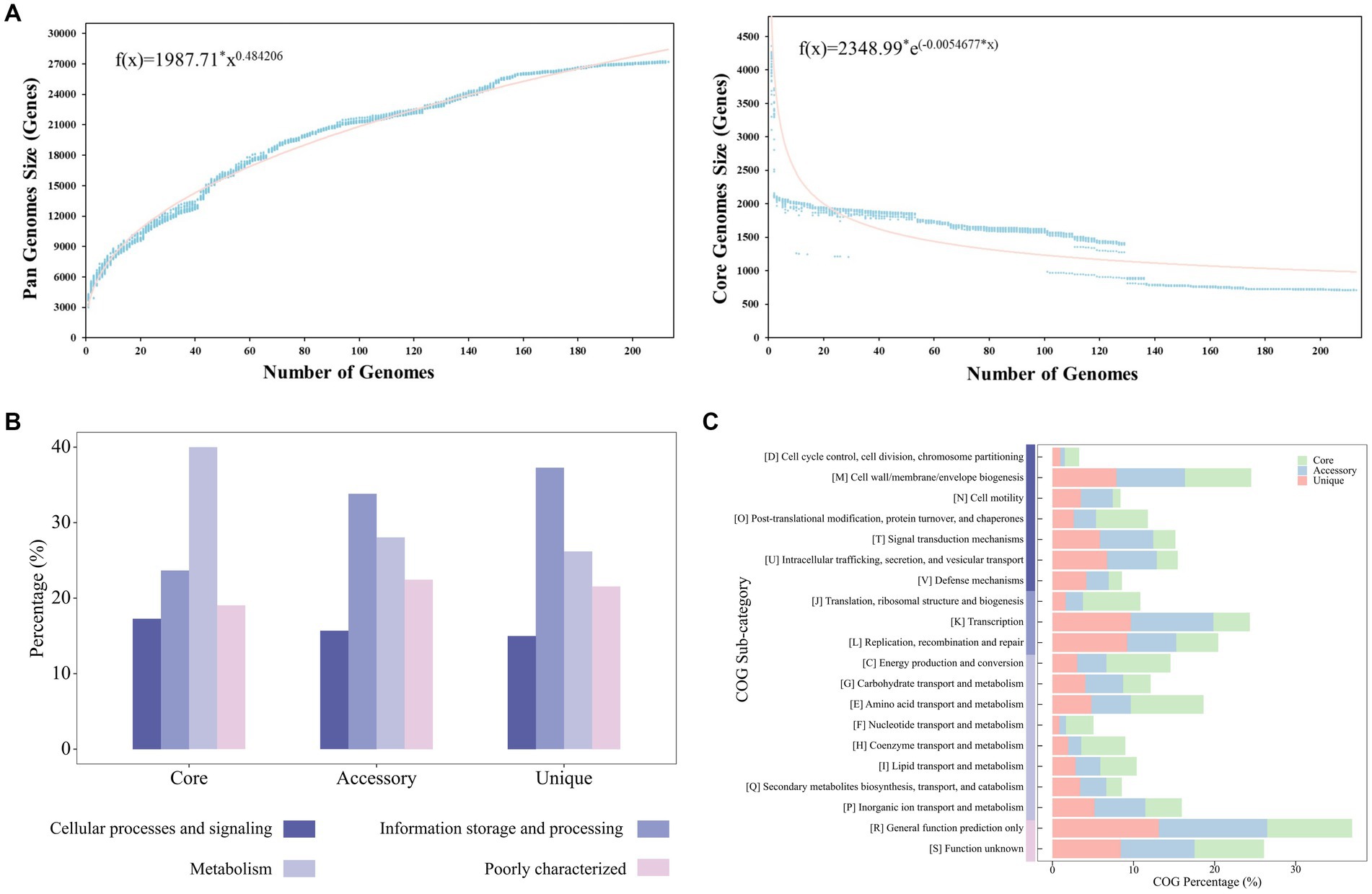
Figure 2. (A) Pan-genome and core genome plots of the 213 strains of Stenotrophomonas. The plot shows the equations fitting the total and core gene families, as well as how the number of gene families increase and decline in the pan and core genome with each consecutive addition of a Stenotrophomonas genome. The functional proportions of core, accessory and unique genes in COG categories (B) and COG sub-categories (C).
The most common categories of COGs in the core genome of Stenotrophomonas (40.0%) were related to metabolism, while those of the accessory genomes (33.8%) and unique genomes (37.3%) were both related to information storage and processing (Figure 2B). The most abundant components of the core genome were related to the maintenance of primary cellular processes, such as class E (9.0%; amino acid transport and metabolism), class C (7.9%; energy production and conversion) and class J (7.1%; translation, ribosomal structure and biogenesis) (Figure 2C). The percentages of classes E, C, and J in the accessory and unique genomes were 4.9 and 4.8%, 3.6 and 3.1%, and 2.1 and 1.6%, respectively. In contrast to the core genome, the accessory and unique genomes were primarily related to the functions of adapting to environmental niches. They were the most enriched in class K (transcription), class U (intracellular trafficking, secretion, and vesicular transport), class T (signal transduction mechanisms), class Q (secondary metabolites biosynthesis, transport, and catabolism) and class V (defense mechanisms), which accounted for 10.2 and 9.7% (4.5%), 6.1 and 6.7% (2.6%), 6.6 and 5.9% (2.7%), 3.2 and 3.4% (1.9%), as well as 2.8 and 4.2% (1.6%), respectively (Figure 2C).
All 33 strains with complete genome sequences harbored the intact pentose phosphate pathway, fructose metabolism pathway, tricarboxylic acid cycle (TCA cycle), fatty acid, lipopolysaccharide, and peptidoglycan biosynthetic pathways. Only S. geniculata E119 lacked an intact glycolysis pathway owing to a deficiency in the glyceraldehyde 3-phosphate dehydrogenase (gapA) gene that converts glyceraldehyde-3P to glycerate-1,3P2 (Figure 3; Supplementary Table S3). In terms of nitrogen metabolism, all 33 strains harbored the Amt family ammonia transporter gene, which is responsible for the uptake of ammonia. In particular, S. indicatrix MGMM10, S. nitritireducens 2001, S. maltophilia strains PSKL2, FDAARGOS_92, and FDAARGOS_507, and S. rhizophila strains DSM 14405T, Bl2-2, JC1, PI-27, and QL-P4 lacked both nitrate reductase and nitrite reductase, while the other 23 strains only lacked nitrite reductase. This indicated that all these strains cannot utilize nitrate as a nitrogen source. The metabolism of sulfur also varies between strains, and only S. acidaminiphila strains T0-18, SPDF1, and T25-65, and S. nitritireducens 2001 had an intact sulfate reduction pathway, while the remaining 29 strains lacked the genes involved in the conversion of sulfate to sulfite. However, they harbored the genes to convert sulfite to sulfide. In terms of the biosynthesis of amino acids, all 33 strains of Stenotrophomonas can synthesize 20 types of amino acids. There was only one exception. S. geniculata E119 cannot synthesize histidine because it lacks histidinol-phosphatase. In addition, the phosphate-specific ABC transporter complex PstSABC and the two-component system PhoRB were identified in the core genome. This indicates that the strains of Stenotrophomonas can take up inorganic phosphate and regulate phosphate homeostasis.
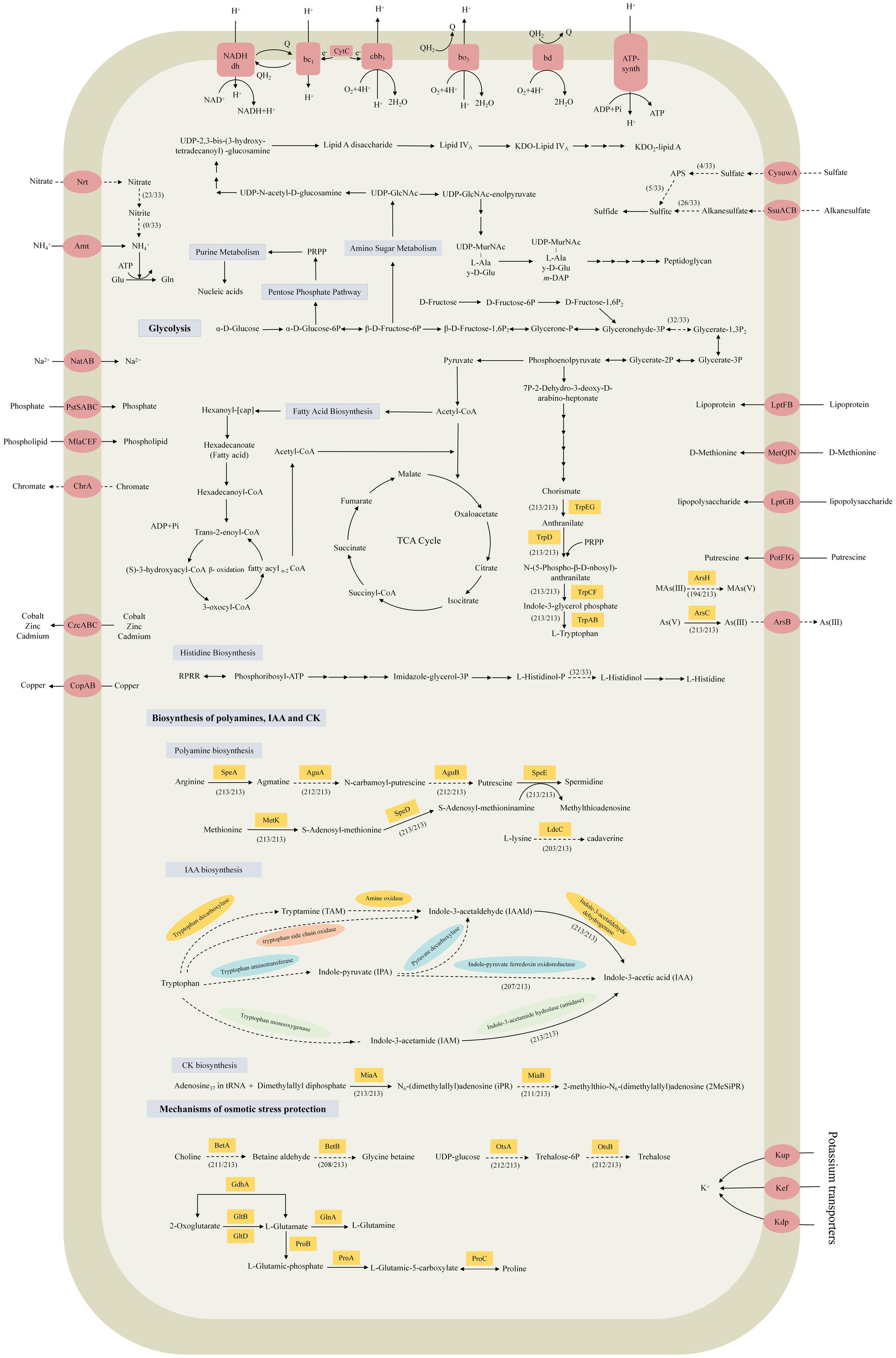
Figure 3. Prediction of the central metabolic potential of 33 complete genomes of Stenotrophomonas strains, as well as the possible biosynthetic pathway for phytohormones and the mechanism of hyperosmolar adaptation of the 213 strains. Solid lines, pathways common to all bacteria; dashed lines, partial presence.
Bacterial chemotaxis is an important prelude to metabolism, competition, symbiosis, infection and other ecological interactions in bacterial communities (Kato et al., 2008). The genomic analysis revealed that all 33 strains of Stenotrophomonas had the genes for flagellar proteins and the chemotaxis signaling system. In particular, all 33 strains of Stenotrophomonas harbored the flg operon (flagellar motif), the fli cluster, and flhAB that encodes the structural proteins for the flagella, as well as the motAB and filA genes that encode the quorum unit MotAB and the sigma factor FliA, respectively. The chemotaxis genes had the cheABYWR and mcp genes that encode the chemotaxis and methyl-accepting chemotaxis proteins, respectively. The two-component system DesK-DesR genes, which regulate the biosynthesis of fatty acid desaturase (Des), was found in all 33 strains (Beranová et al., 2010). In addition, the genes for DNA mismatch repair, glutathione and folate biosynthesis, peroxidase, and resistance to cationic antimicrobial peptides (CAMP) and β-lactams were also found in the core genome, which enables the strains of Stenotrophomonas to adapt to complex and variable environments more efficiently.
Secondary metabolites can enhance the environmental adaptability of the strains and provide them with an evolutionary advantage (O’Connor, 2015). A total of 149 smBGCs of 14 major types were predicted in the 33 Stenotrophomonas genomes, and each strain contained three to seven smBGCs (Supplementary Table S4). Among them, S. maltophilia strains JZL8 and ISMMS2 had the highest number of smBGCs with seven, while each strain of S. nitritireducens 2001, and S. rhizophila strains Bl2-2, JC1, and QL-P4 had only three smBGCs. In detail, 55 RiPP-like smBGCs were found in 32 strains of Stenotrophomonas (Supplementary Figure S1), and 22 of the clusters were similar to the entolysin BGCs, which encode a cyclic lipopeptide (CLP) that can be fatal to bacteria, fungi, and viruses by disrupting their membranes (Oni et al., 2019). The clusters responsible for the biosynthesis of aryl polyenes (APEs), which help bacteria to evade the host immune system (Lee et al., 2021a,b), were found in 31 strains except for S. maltophilia CYZ and S. rhizophila JC1. The NRP-metallophore/NRPS heterozygous clusters were found in 29 Stenotrophomonas strains, and all of them were similar to the 2,3-dihydroxybenzoylserine (DHBS) BGCs, which is a cluster that is responsible for the biosynthesis of catecholate siderophore enterobactin (Ahire et al., 2011). In addition, some smBGCs are strain-specific. For example, the cluster responsible for ranthipeptide, lanthipeptide-class-i and lanthipeptide-class-iii biosynthesis was only detected in S. maltophilia strains CSM2, ISMMS2, and JV3, respectively.
Carbohydrates are the primary source of carbon for most microbes. Carbohydrate active enzymes (CAZymes), a class of enzymes that is involved in the metabolism of carbohydrates, is not only involved in the biosynthesis and degradation of biopolymers but also in the formation of bacterial biofilms and the glycosylation of proteins and lipids (Benini, 2020). A total of 118 different CAZyme genes family were identified in the 213 Stenotrophomonas genomes, including 63 glycoside hydrolases (GHs), 21 glycosyltransferases (GTs), 11 carbohydrate esterases (CEs), 9 polysaccharide lyases (PLs), 8 auxiliary activities (AAs), and 6 carbohydrate-binding modules (CBMs) (Supplementary Figure S2). S. rhizophila CFBP 13503 was identified as harboring the most CAZyme genes at 124, while S. pictorum JCM 9942T had the fewest (68). The CAZyme number of the strains from clinical sites (69 to118, average 106) was similar to that of the strains from the environmental strains (68 to 124, average 104).
In particular, this study explored the CAZymes involved in the degradation of complex polysaccharides. Lignocellulolytic enzymes can be subdivided into cellulases, hemicelluloses, and lignin-modifying enzymes. Most cellulases and hemicelluloses are members of the GHs family, while the lignin-modifying enzymes are generally in the AAs protein family (López-Mondéjar et al., 2016; Zhou et al., 2018). Among them, all 213 strains produced GH3-family enzymes that degraded cellulose, while only some of the 213 strains harbored GH5- (45 strains), GH8- (44 strains), GH1- (19 strains), GH9- (4 strains) and GH74-family (5 strains) enzymes involved in the degradation of cellulose. The hemicellulose hydrolases produced by these species primarily include GH2 (211 strains), GH31 (209 strains), GH10 (162 strains), GH43 (100 strains), GH16 (13 strains) and GH39 (2 strains) families. Lignin-modifying enzymes were primarily represented by the families for laccase-like multi-copper (AA1) and lignin-modifying peroxidases (AA2) (Ma et al., 2021). Genes for the AA1 family were found in all 213 genomes, while AA2 was not found in any of them. In addition, the GH18, GH19, and GH20 families, which are responsible for the degradation of chitin (Swiontek Brzezinska et al., 2013), were detected in 186, 61, and 210 strains, respectively. The analysis revealed that the strains of Stenotrophomonas produced a large number of CAZymes involved in the degradation of polysaccharides, which indicates that these strains can easily obtain carbon and energy from their environment.
To survive in environments, particularly those that are extreme, microbes have evolved the ability to surmount oxidative stress and osmotic pressure (Zhao et al., 2023). Among these, oxidative stress is the result of an imbalance between the production of reactive oxygen species (ROS) and the ability of biological systems to detoxify them (Chautrand et al., 2022). Bacteria use two primary mechanisms to detoxify the ROS. One is to scavenge the ROS using superoxide dismutase (SOD) and catalase (CAT) (Arts et al., 2015). The sodB and sodC genes, which are responsible for catalyzing the decomposition of superoxide anion (O2−) to O2 and hydrogen peroxide (H2O2), were found in all 213 strains of Stenotrophomonas (Figure 4). The katE, ahpC and bcp genes, which are responsible for the conversion of H2O2, were also found in all the strains. The gpx gene encodes glutathione peroxidase (GPX), which catalyzes the reduction of many oxidants, including H2O2 and peroxynitrite (ONOO−) (Panday et al., 2020), was found in all the strains tested except for S. maltophilia HZ34. Another is the timely repair of the cysteine and methionine residues that have been damaged by ROS oxidation. The disulfide isomerase (DsbC) and methionine sulfoxide reductase (Msr) systems, which can repair oxidized cysteine and methionine residues, respectively (Darby et al., 1998; Grimaud et al., 2001). The dsbC, msrA and msrB genes were found in all 213 strains.
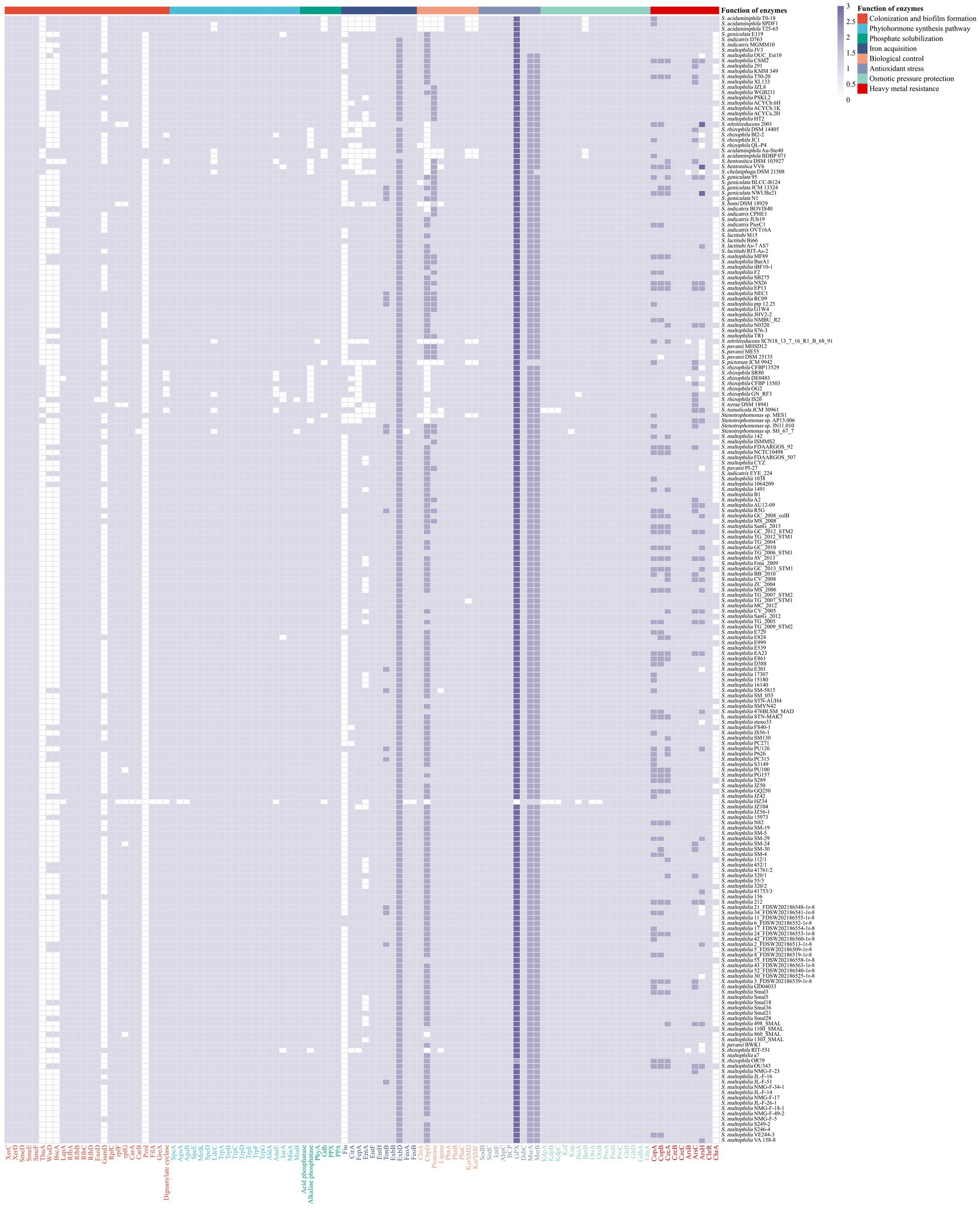
Figure 4. Distribution of enzymes with functions related to plant growth promotion and environmental adaptation in 213 strains of Stenotrophomonas. Enzymes associated with colonization and biofilm formation, XerC, XerD, SmeDEF, ThuA, WssD, BscA, LapA, RfbA, RfbB, RfbC, RfbD, ExoD, GumD, RpfC, RpfF, RpfG, CarA, CarB, PcoI, GreA, FilA, and diguanylate cyclase; polyamine synthases, SpeA, AguA, AguB, SpeE, MetK, SpeD, LdcC; IAA synthases, TrpAB, TrpC, TrpD, TrpEG, TrpF, IorA, AmiE, and AldA; CK synthases, MiaA and MiaB; phosphate-solubilizing enzymes, acid phosphatase, alkaline phosphatase, PhyA, GDH, exopolyphosphatase, and inorganic pyrophosphatase; iron acquisition proteins, Fiu, CirA, FepA, EntA, EntF, EntH, TonB, ExbB, ExbD, FeoA, and FeoB; biological control enzymes, ChiA, CbpD, protease, lipase, PhaA, PhaB, PhaC, KerSMD, and KerSMF; antioxidant stress kinases, SodB, SodC, KatE, AhpC, BpC, GPX, DsbC, MrsA, and MrsB; potassium ion transporters, KdpA, KdpB, KdpC, Kef, Kup; compatible solute synthases, BetA, BetB, OtsA, OtsB, ProA, ProB, ProC, GltB, GltD, GdhA, and GlnA; heavy metal resistance enzymes, CopA, CopB, CzcA, CzcB, CzcC, ArsB, ArsC, ArsH, ChrA, and ChrR.
Bacteria usually adopt two main strategies to maintain their osmotic balance (Gunde-Cimerman et al., 2018). One is to take up and accumulate a large amount of potassium (Vyrides and Stuckey, 2017). All 213 strains of Stenotrophomonas were found to have K+ uptake transporter proteins genes, including the Kdp-ATPase system (kdp), the potassium efflux system (kef), and the potassium transport system gene (kup) (Sleator and Hill, 2002). Another strategy relies on the biosynthesis and accumulation of compatible solutes (Figure 3), including sugars, such as trehalose; amino acids, such as proline and glutamate; and their derivatives, such as betaine (Zhao et al., 2023). Glycine betaine, a common compatible solute, can be synthesized by choline dehydrogenase (BetA) and betaine-aldehyde dehydrogenase (BetB) (Lamark et al., 1991). The betA gene was detected in 211 strains and was only absent in S. rhizophila GN_RF3 and S. maltophilia HZ34. In contrast, the betB gene was identified in 208 strains and only absent in five strains of S. acidaminiphila. Trehalose can be produced from UDP-glucose via the OstA-OstB pathway (Paul et al., 2008). The ostA and ostB genes were only absent in S. maltophilia HZ34. In addition, the glutamate (gdhA, gltBD and glnA) and proline (proABC) biosynthetic genes were detected in the genomes of all 213 strains of Stenotrophomonas. These results indicate that all the strains of Stenotrophomonas can eliminate ROS and tolerate osmotic stress, which endows them with the ability to survive more effectively in extreme environments.
Antibiotics are used extensively to treat bacterial infections in agriculture and animal husbandry. Bacteria usually harbor many antibiotic resistance genes (ARGs) to survive (Zhuang et al., 2021). The number of ARGs per strain of Stenotrophomonas ranged from 3 to 23. Among them, Stenotrophomonas sp. strain SH_67_7 harbored the most ARGs, and S. nitrodeiducens CN18_13_7_16_R1_B_68_91 harbored the least (Supplementary Table S5). There were somewhat more ARGs in the clinical strains (average 14 per strain) than in the environmental strains (average 11 per strain), and S. maltophilia (8–23), S. geniculata (9–20), and S. pavanii (15–16) species had dramatically higher number of ARGs than the other species (4–13). The antibiotic efflux pumps genes, including the resistance-nodulation-cell division (RND) type and the major facilitator superfamily (MFS) type, were identified in all the strains of Stenotrophomonas. rsmA, an RND efflux pump gene involved in the resistance to fluoroquinolones, diaminopyrimidines and phenol antibiotics, was found in all 213 strains except for S. geniculata NWUBe21 and S. rhizophila DE0483. smeDEF, an RND efflux pump gene cluster involved in the resistance to macrolide, fluoroquinolone, tetracycline, and phenol antibiotics (Alonso and Martínez, 2000), was identified in 201 strains of Stenotrophomonas. Another RND efflux pump gene cluster smeABC, which is involved in resistance to aminoglycoside, β-lactam, and fluoroquinolone antibiotics (Li et al., 2002), was found in most of the clinical strains (124) and a minority of the environmental strains (28).
The resistance of bacteria to aminoglycosides (AGs) is primarily caused by AG-modifying enzymes (AMEs), which include O-phosphotransferases (APHs), N-acetyltransferases (AACs) and O-adenyltransferases (ANTs) (Wright, 1999). The aph gene was identified in 134 clinical strains and 53 environmental strains, while the aac gene was found in 59 clinical and 17 environmental strains. In particular, the aph(9)-Ic, aph(3′)-IIc, aph(3″)-Ib, aph(6)-Id, aph(3′)-IIa, aph(3′)-VIa, and aph(6)-Ic genes that encode variants of APH were identified in 185, 183, 5, 4, 1, 1 and 1 strains, respectively, while the aac(6′)-Iz, aac(6′)-Ib8, aac(6′)-Iak, aac(6′)-Iap and aac(6′)-31 genes that encode variants of AAC were identified in 62, 12, 8, 3 and 1 strains, respectively. The ant(2″)-Ia gene was only found in S. maltophilia CV_2005, S. maltophilia 476BLSM_MAD, S. maltophilia 212 and S. maltophilia Smal28. In the Stenotrophomonas resistome, the highest numbers of types of genes for resistance were associated with aminocoumarin, phenicol, and tetracycline antibiotics, with 25, 14, and 12, respectively, while sulfonamide, monobactam, and mupirocin-like had the fewest types of resistance genes. The results indicated that the strains of Stenotrophomonas have multidrug resistance that can be adapted to respond to antibiotic pressure in the external environment.
Most heavy metals are lethal in the environment (Gadd, 2010). The CopAB proteins are responsible for resistance to copper. In particular, CopA can bind copper ions in the periplasm to sequester the copper, while CopB is a transmembrane protein that can serve as an exporter of copper (Ge et al., 2021). The copA gene was identified in all 213 strains of Stenotrophomonas, and the copB gene was found in 212 strains (Figure 4). The CzcABC-type efflux pump proteins are involved in exporting cobalt, zinc and cadmium from the cells (Hussain et al., 2022). All 213 strains of Stenotrophomonas also harbored czcA, czcB and czcC genes. The ars operon ensures that various bacteria are resistant to arsenic. Some bacteria can reduce As(V) (arsenate) to As(III) (arsenite), which is catalyzed by arsenate reductase (ArsC). The As(III) that is produced can be directly pumped out of the cell via ArsB. In addition, MAs(III) (methylarsenite) can be oxidized to the less toxic MAs(V) (methylarsenate) via ArsH (Hao et al., 2021). The arsC, arsB and arsH genes were identified in 213, 209 and 194 genomes of Stenotrophomonas, respectively. The chrR gene encodes chromate reductase, which catalyzes the conversion of soluble and toxic Cr(VI) to insoluble and less toxic Cr(III), respectively (Baldiris et al., 2018), was found in all strains. In contrast, the chrA gene that encodes the chromate efflux transport protein, which pumps chromate out of the cytoplasm (Díaz-Pérez et al., 2007), was found in 55 strains. These genes in strains of Stenotrophomonas endow the bacteria with tolerance to heavy metals in the environment.
MGEs are the direct evidence of the evolution and adaptation of microbial populations through the horizontal transfer of genes (Sobecky and Hazen, 2009). The MGEs in microbes usually include prophages, GIs, and insertion sequence (IS). A total of 637 prophage regions, including 79 questionable, 354 incomplete, and 204 intact prophages, were detected in these Stenotrophomonas genomes, thus, indicating that inducible or transferable functional prophages are widely distributed in strains of Stenotrophomonas. The number of prophages in each strain ranged from one to nine. Among them, 204 intact prophages were found in 60.6% (129/213 strains) of the strains of Stenotrophomonas, and they ranged from one to five intact prophages per strain (Supplementary Table S6). In particular, the largest proportion was composed of Verrucomicrobia phage P862 (NC_029047) followed by Burkholderia phage phi1026b (NC_005284), Vibrio phages VHML (NC_004456) and vB_VpaM_MAR (NC_019722). The major proteins of the prophages that were detected were the structural proteins of tail, capsid, portal, and head, and the functional enzymes terminase, integrase and lysin.
A total of 6,663 GIs were predicted in the 213 Stenotrophomonas strains. The number of GIs per strain ranged from 17 to 60 (Supplementary Table S1). S. rhizophila JC1 contained the highest number of GIs, while S. chelatiphaga DSM 21508T contained the fewest (17). There are many vital functional protein genes in these GIs, including kinases (201 strains), MFS transporters (192 strains), GNAT family N-acetyltransferase (171 strains), ATP-binding proteins (166 strains), response regulator transcription factor (156 strains), efflux RND transporter (118 strains), siderophore receptor (112 strains), RNA polymerase (111 strains), nucleotidyl transferase (101 strains), DNA polymerase III subunit beta (70 strains), DNA replication/repair protein RecF (64 strains), lysozyme (54 strains), type II toxin-antitoxin system (52 strains), CusA/CzcA family heavy metal efflux RND transporter (68 strains), copper resistance protein (62 strains), arsenical resistance protein (40 strains), mercury reductase (31 strains) and chromate efflux transporter (25 strains). These genes are involved in the physiology at the transcriptional and translational levels, as well as confer pathogenicity, drug and heavy metal resistance to the strain (Supplementary Table S7). In addition, some Gls include the genes that encode integrases (205 strains), transposases (185 strains) and recombinases (163 strains), which may mediate the movement of GIs among the host bacteria. Thus, the prophages and GIs enhanced the genetic diversity of the Stenotrophomonas strains and enabled them to rapidly adapt to diverse ecological niches.
Colonization of the rhizosphere by bacterial strains is the first and the foremost step. Genes involved in motility, chemotaxis, adhesion and biofilm formation are thought to contribute to this colonization (Kandel et al., 2017). The two site-specific tyrosine recombinases XerC and XerD are associated with competitive colonization on the root surface (Martínez-Granero et al., 2005). The two genes xerC and xerD were both found in the genomes of all 213 strains of Stenotrophomonas (Figure 4). The SmeDEF efflux pump, which is responsible for the microbial resistance to quinolones, has been found to be involved in the endophytic colonization of plant roots (García-León et al., 2014). The smeDEF gene was identified in all 213 strains. In addition, the thuA gene, which is involved in the utilization of trehalose, enhances the ability of the strain to colonize plants at the early stages (Pinski et al., 2020). The thuA gene was detected in 15 strains, including 10 S. rhizophila strains, S. bentonitica DSM 103927T, S. bentonitica VV6, S. chelatiphaga DSM 21508T, S. tumulicola JCM 30961T and Stenotrophomonas sp. AP15.006.
As a physical barrier, biofilm can protect the embedded bacteria. Thus, the ability of bacteria to form biofilms determines their ability to colonize the root surface. Cellulose is an important component of biofilms in some species. The bcsA and wssD genes are two genes that are responsible for the biosynthesis of cellulose. Their inactivation significantly reduced the formation of biofilm and decreased the ability of strains to colonize the plant host (Barak et al., 2007; Monteiro et al., 2012). However, these two genes were only found in 43 strains of Stenotrophomonas (21 environmental and 22 clinical strains). In addition, lipopolysaccharides (LPS) and exopolysaccharides (EPS) facilitate the attachment of bacterial cells to the root surface (Kandel et al., 2017). The lapA gene, a key gene responsible for the assembly of LPS, was found in 210 strains. Rhamnose is an important subcomponent of LPS. The rfbABCD gene, a cluster that is responsible for the biosynthesis of rhamnose (Balsanelli et al., 2010), was found in all the strains of Stenotrophomonas. The exoD gene that encodes the protein for the biosynthesis of EPS (Reed and Walker, 1991) was found in 212 strains. The gumD gene, which is also responsible for the catalysis of the biosynthesis of EPS, (Meneses et al., 2011) was found in the genomes of 50 strains of Stenotrophomonas (33 environmental and 17 clinical strains). The results of genomic analyses showed that all the strains of Stenotrophomonas have a range of biofilm formation genes that can promote their ability to colonize plants. In addition, the ability to form biofilms has also been shown to be one of the main pathogenesis-related virulence factors (Wall et al., 2019). Inhibiting the expression of virulence-associated genes is reportedly correlated with the attenuation of the biofilm formation ability of S. maltophilia (Kim et al., 2018).
The formation of biofilm and cell adhesion are usually regulated by QS at the bacterial community level. The diffusible signal factor (DSF) is a QS molecule that can regulate the genes related to plant colonization, such those involved in chemotaxis, motility, the formation of biofilm and production of the multidrug efflux pump (Alavi et al., 2013a). The rpf gene cluster, which encodes all the components of the DSF system, was detected in 213 strains of Stenotrophomonas (Figure 4). Carbamoyl phosphate synthase, which is encoded by the carAB genes, can be used to degrade DSF (Newman et al., 2008). The carA gene was found in 212 strains (S. maltophilia HZ34 lacked), while the carB gene was found in 211. S. maltophilia HZ34 and Stenotrophomonas sp. SH_67_7 lack this gene. pcoI is a key gene related to the population sensing signals. Its deletion led to significant deficiencies in the formation of biofilm at the wheat rhizosphere (Wei and Zhang, 2006). The pcoI gene was only found in seven environmental strains, including five strains of S. acidaminiphila, and S. nitritireducens SCN18_13_7_16_R1_B_68_91 and Stenotrophomonas sp. MES1.
Some prevalent bacterial transcriptional regulators are also associated with plant-rhizobacterial interactions. The sigma-28 factor encoded by the filA gene is involved in the regulation of the expression of flagellin, chemotaxis, and motility-related genes (Yi et al., 2017). The filA gene was identified in the genomes of all the strains of Stenotrophomonas. The greA gene, another important transcription factor, determines the ability of host to adapt to hyperosmolarity and salt stress. The strains that lack this gene cannot establish an effective symbiotic relationship with plants (Nogales et al., 2002). The greA gene was found in 212 strains and was only absent from S. maltophilia HZ34. Cyclic diguanylate (c-di-GMP), a universal secondary messenger in bacteria, is involved in the regulation of a variety of physiological functions, including cell differentiation, biofilm formation, motility, adhesion, colonization of host tissues and the generation of pathogenic factors (Pinski et al., 2020). The biosynthesis of c-di-GMP is catalyzed by the gene that encodes diguanylate cyclase, which was found in 205 strains of Stenotrophomonas. The results indicate that strains of Stenotrophomonas have a range of transcriptional regulators that are related to the colonization of plants.
Polyamines, including putrescine, spermidine, spermine and cadaverine, may be involved in the promotion and protection of plant growth (Couée et al., 2004; Xie et al., 2014). Bacteria could synthesize putrescine from arginine step by step catalyzing by arginine decarboxylase (SpeA), agmatine deiminase (AguA), and N-carbamoyl-putrescine amidase (AguB) (Burrell et al., 2010; Michael, 2016) (Figure 3). The speA, aguA, and aguB genes were present in 213, 212, and 212 strains, respectively. Only S. maltophilia HZ34 lacked both the aguA and aguB genes (Figure 4). The speE gene, which is responsible for the conversion of putrescine to spermidine (Lee et al., 2012), was found in all the strains of Stenotrophomonas. The MetK and SpeD enzymes are responsible for the conversion of methionine to S-adenosyl-methioninamine (dcSAM), which is required for the biosynthesis of spermine (Xie et al., 2014). The metK and speD genes are found in all 213 strains. In addition, lysine decarboxylase (LdcC) catalyzes the conversion of L-lysine to cadaverine (Liu et al., 2022). The ldcC gene was found in the genomes of 203 strains of Stenotrophomonas. The presence of these genes suggests that strains of Stenotrophomonas can produce multiple types of polyamines.
IAA and cytokinin (CK) are the two most common phytohormones that influence various traits of plant growth and development, including cell division and elongation, seed germination, fruit development and the delay of senescence (Grossmann, 2009; McSteen, 2010; Akhtar et al., 2020). There are five different pathways for the biosynthesis of IAA from tryptophan. Among them, the indole-3-acetamide pathway (IAM), the indole-3-pyruvic acid pathway (IPA/IPyA) and the tryptamine pathway (TAM) are the most common pathways in bacteria (Zhang et al., 2019; Tang et al., 2023). The vital genes for the biosynthesis of tryptophan, including trpAB, trpC, trpD, trpEG, and trpF, were present in all 213 genomes of Stenotrophomonas. For the IPA pathway, the gene that is responsible for the conversion of tryptophan to IAAld was not detected in all 213 genomes, but the gene for indole-3-acetaldehyde dehydrogenase (AldA) that converts IAAld to IAA was found in all these genomes (Figure 3). In addition, the iroA gene that encodes indole-pyruvate ferredoxin oxidoreductase (IorA), which can directly convert IPA to IAA (Imada et al., 2017), was identified in 207 strains. As the IAM pathway, all 213 strains lacked the tryptophan monooxygenase that catalyzes the initial conversion of tryptophan to IAM, but the amidase gene responsible for the conversion of IAM to IAA was detected in all 213 genomes. For the TAM and TSO (tryptophan side-chain oxidase) pathway, only the aldA gene of the final step was detected, but the gene responsible for the conversion of tryptophan to IAAld was not detected.
The miaA gene encodes a tRNA dimethylallyltransferase that is responsible for the conversion of dimethylallyl diphosphate to N6-(dimethylallyl) adenosine (iPR). The miaB gene, which encodes tRNA-2-methylthio-N(6)-dimethylallyladenosine synthase, converts iPR to 2-methylthio-N6-(dimethylallyl)adenosine (2MeSiPR). Both genes are involved in the biosynthesis and conversion of CK (Nascimento et al., 2020) and were identified in all the Stenotrophomonas genomes except for S. rhizophila IS26 and Stenotrophomonas SH_67_7, which both lacked miaB.
Phosphorus (P) is an essential macronutrient for plant growth and development. Although there are adequate amounts of P in the soil, most of the organic and inorganic phosphate that is found in the soil is not directly available to plants. Many bacteria that are associated with plants can solubilize P into a form that can be utilized by plants (Sharma et al., 2013). Organic phosphates are primarily degraded by the enzyme acid phosphatases, alkaline phosphatases and phytase that are produced by bacteria (Sharma et al., 2013), while inorganic phosphates can be solubilized by the production of organic acids, such as gluconic acid, citric acid, malic acid, and succinic acid (Vassilev et al., 2006). There are two categories of phosphatases that are based on their optimal pH, including acid phosphatase and alkaline phosphatase, which both dephosphorylate phosphate ester or the phosphoric anhydride bonds in organic compounds (Jorquera et al., 2008). The acid phosphatase and alkaline phosphatase genes were found in 213 and 197 strains, respectively (Figure 4). The phytase encoded by phyA is responsible for the catalysis of the degradation of phytate to release P, and this gene was identified in 212 strains. S. nitritireducens SCN18_13_7_16_R1_B_68_91 does not produce phyA. In addition, the gdh gene encodes a phosphate starvation-inducible glucose dehydrogenase (GDH), which may be involved in the solubilization of mineral phosphate (Sharma et al., 2005). This gene was identified in 205 strains of Stenotrophomonas. Among them, five S. acidaminiphila strains, S. pictorum JCM 9942T, Stenotrophomonas sp. MES1 and S. maltophilia HZ34 lacked gdh gene. An exopolyphosphatase and an inorganic pyrophosphatase are encoded by the ppx and ppa genes, respectively. They catalyze the hydrolysis of inorganic pyrophosphate. Both genes were found in all 213 genomes of Stenotrophomonas. These genes enable these strains of Stenotrophomonas to solubilize soil phosphate, which provides adequate amounts of P for their own metabolism and that of their host plant.
Similar to P, iron is an essential nutrient for plant and microbial growth, but its bioavailability is limited owing to the low solubility of ferric oxide (Fe3+) ions (Behnke and LaRoche, 2020). Siderophores, iron chelators that are produced by microorganisms, can convert the iron into soluble complexes that can be utilized by plants (Ahmed and Holmström, 2014). The comparative analysis revealed that most strains of Stenotrophomonas possessed the genes to biosynthesize catecholate siderophores. In particular, 26 and 189 strains possessed the catecholate siderophore biosynthetic genes fiu and cirA, respectively, and 171 and 187 strains had the biosynthetic genes entAFH and fepA, respectively, for the catecholate siderophore enterobactin (Patzer et al., 2003; Pakarian and Pawelek, 2016). In Gram-negative bacteria, the uptake and transfer of iron ions (Fe3+) by siderophores is powered by the Ton complex (TonB-ExbB-ExbD) (Faraldo-Gómez and Sansom, 2003). The tonB, exbB and exbD genes were present in all 213 genomes (Figure 4). In addition, many bacteria can take up ferrous iron (Fe2+) using the Feo system under anaerobic and acidic environments (Lau et al., 2016). The Feo system is primarily composed of FeoA and FeoB, (Kalidasan et al., 2018). The feoB gene was detected in all 213 strains of Stenotrophomonas except for S. maltophilia HZ34, while the feoA gene was not found in Stenotrophomonas SH_67_7 and S. maltophilia HZ34. The results suggest that the strains of Stenotrophomonas can enhance the availability of Fe3+ in an environment deficient in iron, which benefits both their survival and the growth of plant.
The biological control of microbes in the rhizosphere provides their hosts with natural protection from soilborne phytopathogens (Pandit et al., 2022). The production of cell wall-degrading enzymes by rhizobacteria is one of the primary mechanisms that they use to destroy pathogens. The chitinase encoded by the chiA gene can degrade the chitin in the cell walls of pathogenic fungi (Kharade and McBride, 2014). The chiA gene was detected in 204 genomes of Stenotrophomonas. In addition, the chitin-binding proteins (CBPs), encoded by the cbpD gene, can increase the efficiency of some specific chitinases (Manjeet et al., 2013). The cbpD gene is found in 183 strains of Stenotrophomonas (Figure 4). Proteases and lipases also play an important role in biocontrol (Friedrich et al., 2012). Among them, protease genes were identified in 205 strains of Stenotrophomonas, while lipase genes were identified in 200 strains. Six strains lacked the chitinase, protease, and lipase genes, including S. acidaminiphila T0-18, S. acidaminiphila T25-65 and S. nitritireducens SCN18_13_7_16_R1_B_68_91 isolated from bioreactors, S. pictorum JCM 9942T isolated from the soil and S. acidaminiphila SPDF1 isolated from cell culture media.
Polyhydroxybutyrate (PHB), the most abundant and well-characterized polymer of bacterial polyhydroxyalkanoate (PHA) (Jendrossek, 2009), has proven to have both antibacterial and antifungal activities (Ma et al., 2019). The biosynthesis of PHB is a three-step process that begins with acetyl-CoA acetyltransferase (PhaA), which catalyzes the condensation of two molecules of acetyl-CoA to acetoacetyl-CoA; this compound is then reduced to 3-hydroxybutyryl-CoA by acetoacetyl-CoA reductase (PhaB) and finally catalyzed by PhaC (poly(3-hydroxyalkanoate) polymerase) to produce PHB (Jendrossek and Pfeiffer, 2014). The three genes are found in the genomes of all strains of Stenotrophomonas. The keratinases KerSMD and KerSMF have broad substrate specificity and can be used as pesticides (Gupta and Ramnani, 2006; Yue et al., 2011). Both genes kerSMD and kerSMF were detected in 205 strains of Stenotrophomonas. The results suggest that Stenotrophomonas may be a useful tool for the integrated management of various plant pathogens and pests.
The genus Stenotrophomonas is widely distributed in various habitants on the globe, including plant, animal, soil and aquatic environments (Figure 5A). Among them, strains of Stenotrophomonas were detected in 1,712 plant samples (24.3%), while only 428 samples were detected in aquatic environments (6.09%); the rhizosphere is the primary habitat among plant environments that has been shown to harbor these bacteria (Figure 5B). This suggests that the strains of the genus are closely associated with plants. Previous studies have also shown that strains of Stenotrophomonas can promote plant growth, as well as manage pests and pathogens (Kumar et al., 2023). However, there have been no systematic analyses of the abilities of this genus to adapt to the environment and promote the growth of plants. To our knowledge, this is the first study that systematically and comprehensively elucidates the possible adaptive and plant-promoting mechanisms of the genus Stenotrophomonas using the comparative genomic method.
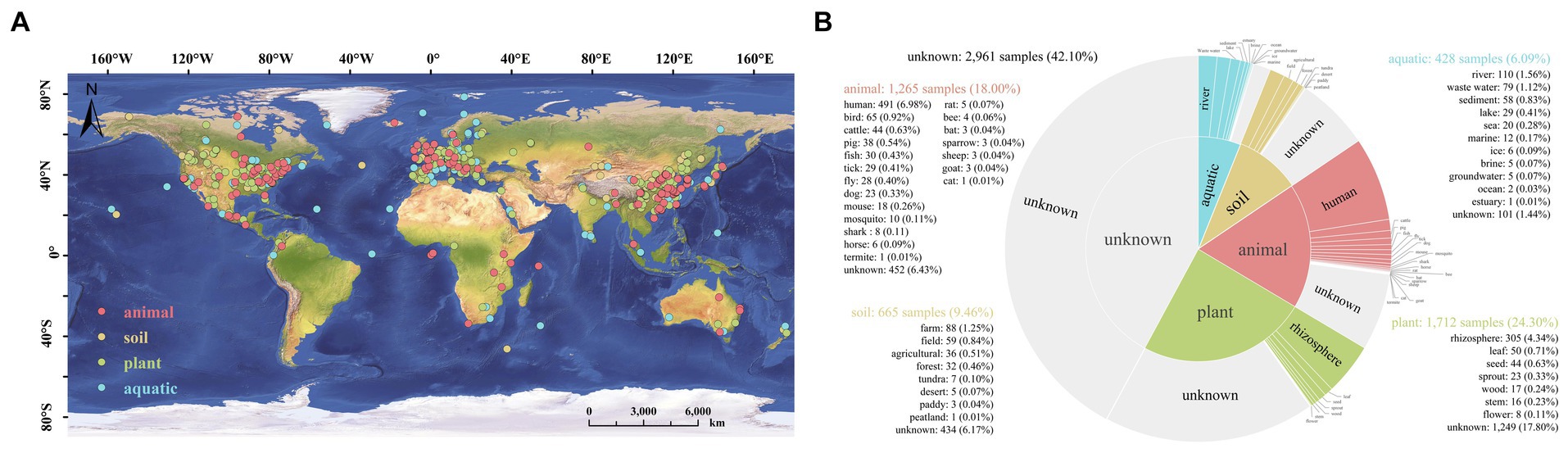
Figure 5. Biogeographic distribution analysis of the genus Stenotrophomonas based on the Microbe Atlas Project (MAP) database and pipeline. (A) The global distribution of Stenotrophomonas. (B) Number of samples that contain the representative OTU sequence per habitat and sub-habitat. OTU, operational taxonomic unit.
Stenotrophomonas was named for its tolerance to oligotrophic conditions. The genomic analysis showed that almost all of the strains that have been analyzed can synthesize all 20 amino acids, which suggests that most of them can grow well in simple nutrients. In addition, the comparative results suggest that the strains of this genus are extremely effective at utilizing carbon sources. In detail, nearly all of the Stenotrophomonas strains that have been analyzed harbor the GH3-family cellulose-degrading genes and the GH2- and GH31-family genes for hemicellulose hydrolase. Most of the strains also contained intact EMP and TCA pathways to utilize the monomer carbohydrates. This indicates that the strains of Stenotrophomonas can not only degrade two types of abundant carbohydrates in the biosphere, including both cellulose and hemicellulose, but also their monomers. The high ability of these strains to utilize carbon sources enables these them to easily survive in the soil. Alternatively, all the strains of Stenotrophomonas contain SOD and CAT genes to scavenge excess ROS, as well as the dsbC and msrAB genes to repair the cysteine and methionine residues that been damaged owing to oxidation by ROS. These genes ensure that the strains of Stenotrophomonas can remove the ROS produced during their rapid growth on eutrophic conditions, which suggests that the strains of this genus can also thrive in eutrophic media.
Extreme conditions, including osmotic pressure, antibiotics and heavy metals, are another important limiting factor for the survival of the strain in the environment. All the strains of Stenotrophomonas possess the mechanism to pump in potassium ions using a specific transport system (kdp, kup and kef) to maintain their osmotic balance. In addition, most of them also harbored genes to synthesize compatible solutes, such as betaine, trehalose, glutamate and proline, to manage osmotic stress. The numerous genes for antibiotic resistance that are present in the genomes increase the ability of Stenotrophomonas to survive in high-stress environments that contain antibiotics. Previous studies have shown that SmeABC and SmeDEF are the two most common efflux pumps in S. maltophilia (Wang et al., 2018). The results of this study confirmed these two efflux pumps are common not only in S. maltophilia but also in the entire genus of Stenotrophomonas. In addition, all the genomes of Stenotrophomonas harbored AG-modifying enzyme genes, including APHs and AACs, which enable their resistance to broad-spectrum aminoglycoside antibiotics. It is notable that fewer strains of Stenotrophomonas harbor the genes for resistance to monobactam, mupirocin and sulfonamide antibiotics, which suggests that they could be used to select clinical candidates and develop them to some extent. cop, czc, ars and chr operon units were identified in all the genomes of Stenotrophomonas, and they provide resistance to copper, cobalt, zinc, cadmium, arsenic and chromium. In summary, the genes related to carbon source utilization, antioxidative stress, osmotic protection, antibiotic and heavy metal resistance enhance the ability of the strains of Stenotrophomonas to survive in extreme environments, as well as to provide a foundation for its colonization of the rhizosphere and promotion of plant growth.
Chemotaxis and motility are vitally important for free-living bacteria because the levels of nutrients vary, and nutrients are often present as point sources over time (Blackburn et al., 1998). All the strains of Stenotrophomonas have chemotaxis and motile device flagella, which suggests that these strains can sense a variety of physicochemical cues and rapidly move to inhabit a niche that is more conducive to their survival. Furthermore, the active motility facilitated by the flagella and guided by chemotactic responses promotes the initial contact of strains of Stenotrophomonas with their host root surface, which increases the efficiency of colonization. Similarly, Stenotrophomonas can form biofilms that increase their adhesion to biotic and abiotic surfaces and result in their resistance to heat, antibiotics, UV and other environmental stresses, which leads to a promotion in the colonization and biocontrol efficacy of roots (Shaheen et al., 2010; Gao et al., 2019). A series of transcriptional regulators were identified in all the genomes of Stenotrophomonas that regulate and enhance the levels of expression of the genes related to motility, chemotaxis, adhesion, and biofilm formation to improve the survival of these strains and their efficiency of colonization. Altogether, the presence of genes related to motility, chemotaxis and biofilm formation contribute to the resistance of strains of Stenotrophomonas to environmental stresses and facilitate their efficient colonization of plant hosts.
Rhizobacteria can promote the growth of host plants through a variety of direct and indirect mechanisms, with direct mechanisms that include the biosynthesis of phytohormones and the facilitation of acquiring resources (Figure 6). The genomic analysis revealed that all the strains of Stenotrophomonas harbor the genes involved in the biosynthesis of L-tryptophan and the final step of IAA biosynthesis. This result was supported by a previous study that showed that Stenotrophomonas sp. 169 and S. maltophilia R551-3 can produce IAA, but the genes that catalyze the first two steps of IAA biosynthesis were not found in their genomes (Taghavi et al., 2009; Ulrich et al., 2021). This suggests that Stenotrophomonas may have unidentified enzymes involved in the initial conversion of tryptophan or the existence of novel pathways for the biosynthesis of IAA. The miaAB gene for the biosynthesis of cytokinin and several genes involved in the biosynthesis of polyamines, such as putrescine, spermidine and cadaverine, were also identified in all the genomes of Stenotrophomonas. Polyamines are not only associated with the establishment of biological interactions between the roots and rhizosphere microorganisms (Jang et al., 2002; Alavi et al., 2013a,b), but they also protect plants from various environmental stresses, including acidic, oxidative, cold and osmotic stresses (Kasukabe et al., 2004; Alavi et al., 2013a). In addition, each of the 213 Stenotrophomonas genomes analyzed harbored multiple genes that were responsible for phosphate-solubilization, which suggests that the genus has a strong ability to solubilize phosphate to meet the plant requirements for P. All the strains of Stenotrophomonas can also secrete a catecholate siderophore to chelate Fe3+ and utilize the Feo system to absorb Fe2+. These iron acquisition systems also play an important role in biological control in addition to providing iron to plants because they can reduce availability of iron to plant pathogens and limit their growth in the rhizosphere by promoting the uptake of iron (Aznar et al., 2015).
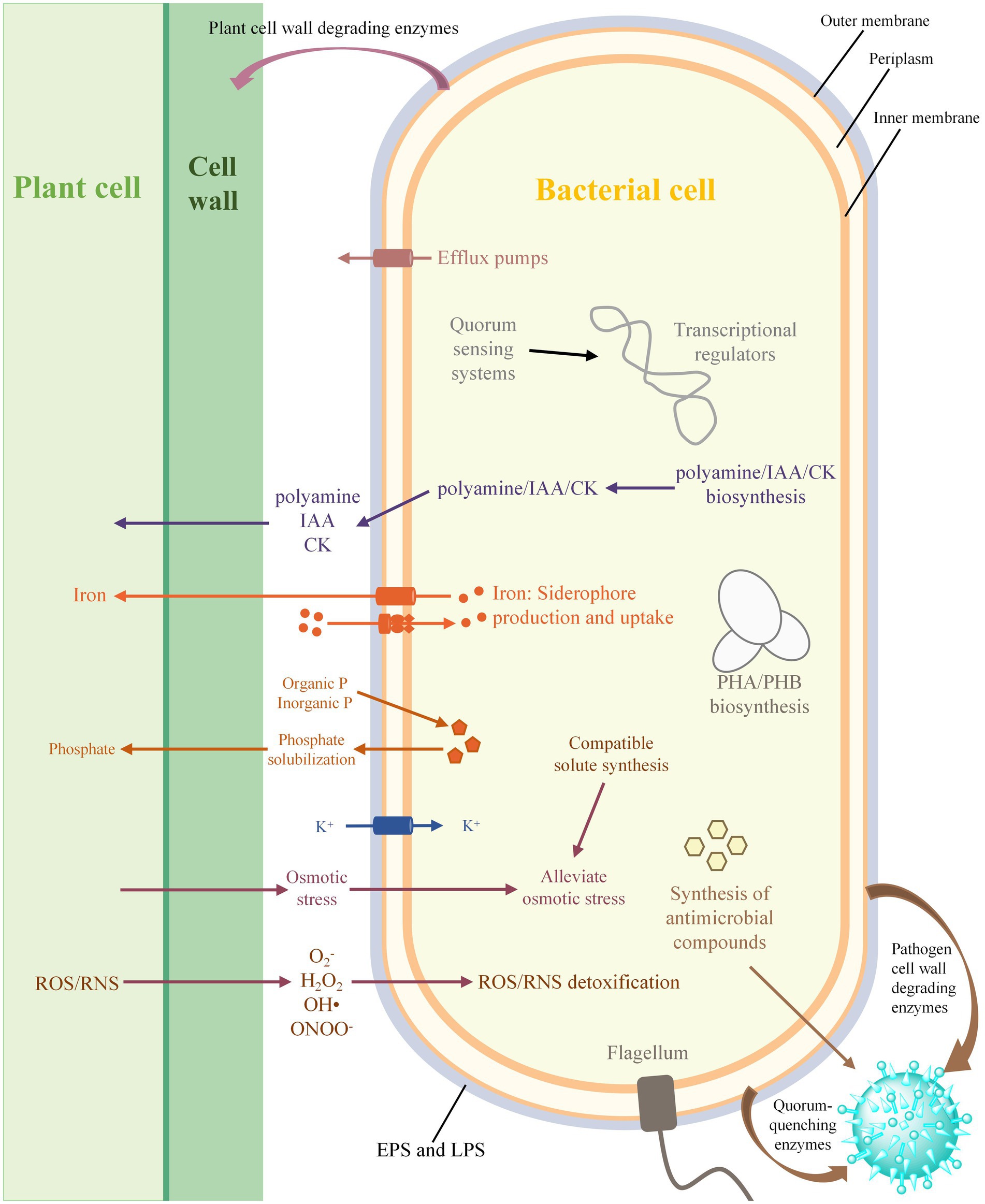
Figure 6. Schematic diagram of plant-growth promoting bacterial interactions. IAA, indole-3-acetic acid; CK, cytokinin; EPS, exopolysaccharide; LPS, lipopolysaccharide; PHA, polyhydroxyalkanoate; PHB, polyhydroxybutyrate; ROS, reactive oxygen species; RNS, reactive nitrogen species.
Stenotrophomonas can also indirectly promote the growth of plants by inhibiting phytopathogens, which is primarily accomplished through the production of antimicrobial compounds and cell wall-degrading enzymes and quenching the QS of pathogens (Figure 6). In particular, strains of Stenotrophomonas harbor many abundant BGCs with PiPP-like, arylpolyene, and NRP-metallophore/NRPS. The secondary metabolites produced by these BGCs may contribute to the prevention of plant damage by phytopathogens and enhance the competition for niche resources (Bulgarelli et al., 2013). All the strains of Stenotrophomonas can also synthesize PHB, which not only has antifungal and antibacterial properties, but also serves as carbon and energy storage compounds in bacteria that can improve their survival and tolerance to stress during starvation (Sadykov et al., 2017). In addition, the genes that encode various hydrolytic enzymes, such as keratinases, chitinases, proteases and lipases, were identified in most of the Stenotrophomonas genome. Previous studies have shown that keratinases can be used as effective biocontrol agents for plant-parasitic nematodes (Pinski et al., 2020), while chitinases, proteases, and lipases can protect plants against phytopathogens by destroying the cell walls of fungi (Zhang et al., 2001). The carAB gene required for the rapid degradation of DSF was also found in most strains of Stenotrophomonas, which controls these pathogens that produce DSF by quenching their QS abilities (Newman et al., 2008). Altogether, these findings suggest that Stenotrophomonas can have beneficial effects on plants through a variety of mechanisms.
MGEs may expand the species gene pool and enhance bacterial adaptation and competitiveness in specific habitats through a large number of mechanisms of horizontal gene transfer (HGT) (Dobrindt et al., 2004). Different numbers of horizontally transferred GIs and prophages were found in each Stenotrophomonas genome, and genes involved in the translocation and catabolism of various substrates, siderophore receptors, and resistance to antibiotics and heavy metals were identified in the GIs, which indicates that many strains of Stenotrophomonas have acquired the genes for resistance to antibiotics and heavy metals through HGT. In addition, the phylogenetic analysis revealed that the biosynthetic genes for polyamines (aguB, speE, and ldcC), IAA (iroA and aldA) and cytokinins (miaA and miaB) could have been acquired by HGT. In particular, the topology of the phylogenetic trees based on the sequences of AguB (Figure 7A), SpeE (Figure 7B), LdcC (Figure 7C), IroA (Figure 7E), AldA (Figure 7F), MiaA (Figure 7G), and MiaB (Figure 7H) differed significantly from that based on the core genome (Figure 1A). Strains that clustered together in the core genome tree were usually scattered in different branches in the trees that were constructed using single AguB, SpeE, LdcC, IroA, AldA, MiaA, and MiaB proteins. In contrast, the topology of the tree based on AmiE (Figure 7D) is similar to that of the core genome-based phylogenetic tree with only minor differences. The S. maltophilia strains RC09, NEC1, KMM_349, E824, NS26, and 291 located in Cluster VI in the core phylogenetic tree scattered separately in different clusters on the AimE tree. S. maltophilia PC315, which is a member of Cluster VI on the core genome tree, presents as Cluster IX in the AmiE tree. Although the phylogenetic tree based on the AmiE sequence differed slightly from the core genome phylogenetic tree in topology, the G + C content of the amiE gene (68.1–74.9%) is significantly higher than that of the host genome (63.8–69.3%) (Supplementary Figure S3D). This suggests that the amiE gene may also have been acquired through HGT. In addition, the open pan-genome and increasing number of novel genes suggest that the strains of Stenotrophomonas can introduce alien genes by the exchange of genetic material with other community members in their habitat and promote their broader genomic plasticity.
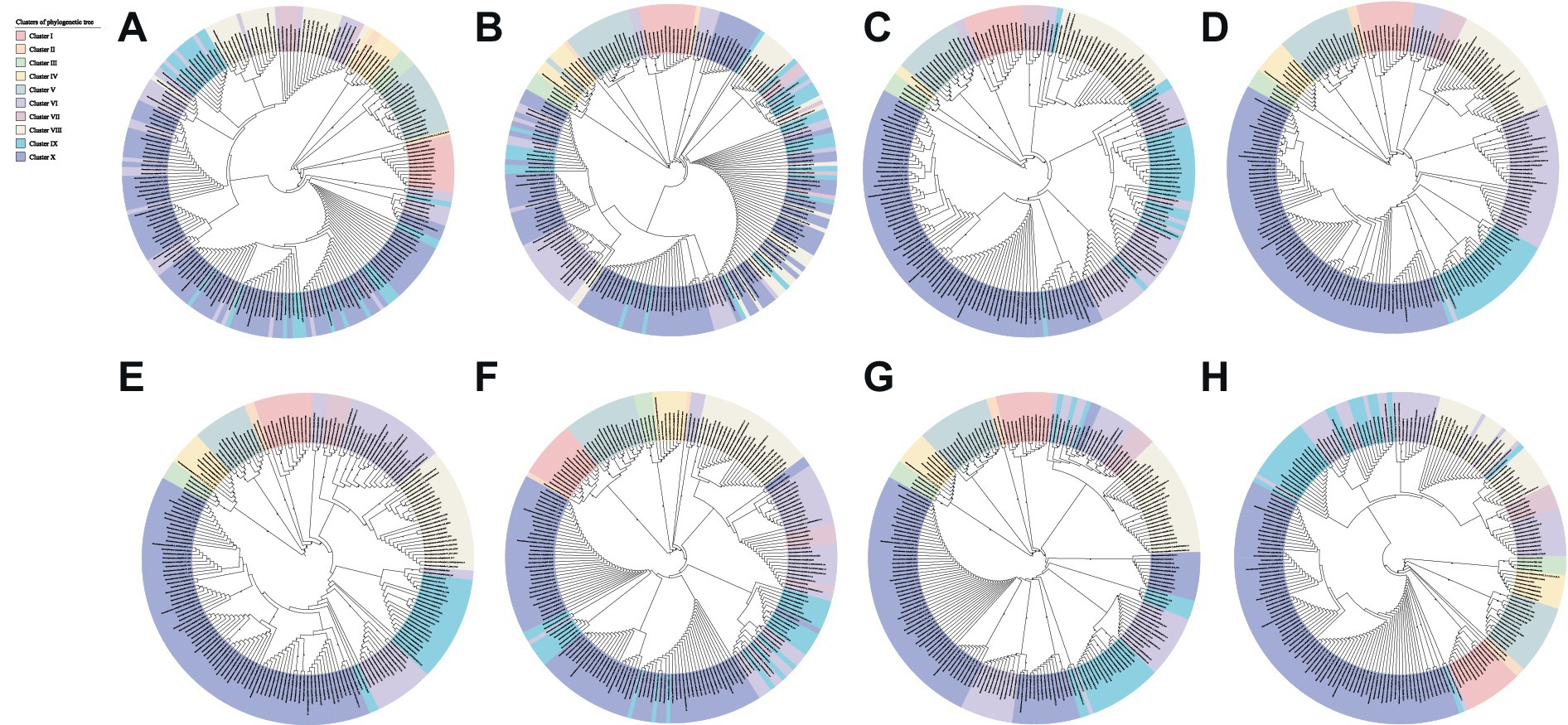
Figure 7. Phylogenetic tree of Stenotrophomonas constructed based on the amino acid sequences of AguB (A), SpeE (B), LdcC (C), AmiE (D), IroA (E), AldA (F), MiaA (G), MiaB (H).
In summary, the genus Stenotrophomonas has a high degree of environmental adaptability and may promote plant growth through one or more mechanisms, which indicate that this genus is a promising PGPB, and can be used as a viable alternative to synthetic pesticides and fertilizers in agriculture to some extent. Thus, this bacterium could provide an effective and environmentally friendly solution to address agricultural pollution and ensure food security. Moreover, the presence of large numbers of heavy metal resistance genes in Stenotrophomonas proves that it may have immense potential in assisting in the phytoremediation of heavy metal pollution. However, it is worth noting that the diversity of Stenotrophomonas strains makes it difficult to distinguish characteristics because of their advantageous interactions with plants and their facultatively harmful infections with humans (Berg and Martinez, 2015). Therefore, advanced molecular techniques should be developed to identify and discriminate between human pathogenic and non-pathogenic strains of Stenotrophomonas, so that non-pathogenic strains can be applied in future agricultural systems.
The comparative genomic analysis of 213 strains of Stenotrophomonas revealed that despite their geographical isolation and diverse hosts, genetic factors related to environmental adaptation and plant promotion were identified in all of them. In particular, nearly all the strains can utilize ubiquitous carbohydrates in the biosphere, such as cellulose and hemicellulose, as carbon and energy sources. The presence of functional genes associated with antioxidant stress, osmotic pressure protection, heavy metal and antibiotic resistance, motility, chemotaxis and biofilm formation enable them to colonize and survive in a variety of host environments. The genes involved in dissolving phosphates and producing siderophores, polyamines, phytohormones, antimicrobial compounds, and pathogen cell wall-degrading enzymes confer the Stenotrophomonas strain with plant growth-promoting and biocontrol properties. Overall, the results of this study suggest that Stenotrophomonas may have immense potential for use in sustainable agricultural practices and bioremediation technologies and can be used as a suitable candidate for a microbial biocontrol agent or an anti-stress agent for crops.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
YZ: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing, Visualization. W-JD: Investigation, Writing – review & editing. LX: Investigation, Writing – review & editing. J-QS: Conceptualization, Funding acquisition, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by National Natural Science Foundation of China (grant no. 31960020, and 32260022) and Inner Mongolia Science and Technology Plan (no. 2020GG0034).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1395477/full#supplementary-material
3. ^http://ggdc.dsmz.de/ggdc.php
4. ^https://antismash.secondarymetabolites.org/
5. ^https://bcb.unl.edu/dbCAN2
Ahire, J. J., Patil, K. P., Chaudhari, B. L., and Chincholkar, S. B. (2011). A potential probiotic culture ST2 produces siderophore 2,3-dihydroxybenzoylserine under intestinal conditions. Food Chem. 127, 387–393. doi: 10.1016/j.foodchem.2010.12.126
Ahmed, E., and Holmström, S. J. M. (2014). Siderophores in environmental research: roles and applications. Microb. Biotechnol. 7, 196–208. doi: 10.1111/1751-7915.12117
Akhtar, S. S., Mekureyaw, M. F., Pandey, C., and Roitsch, T. (2020). Role of cytokinins for interactions of plants with microbial pathogens and pest insects. Front. Plant Sci. 10:1777. doi: 10.3389/fpls.2019.01777
Alavi, P., Müller, H., Cardinale, M., Zachow, C., Sánchez, M. B., Martínez, J. L., et al. (2013a). The DSF quorum sensing system controls the positive influence of Stenotrophomonas maltophilia on plants. PLoS One 8:e67103. doi: 10.1371/journal.pone.0067103
Alavi, P., Starcher, M. R., Thallinger, G. G., Zachow, C., Müller, H., and Berg, G. (2014). Stenotrophomonas comparative genomics reveals genes and functions that differentiate beneficial and pathogenic bacteria. BMC Genomics 15:482. doi: 10.1186/1471-2164-15-482
Alavi, P., Starcher, M. R., Zachow, C., Müller, H., and Berg, G. (2013b). Root-microbe systems: the effect and mode of interaction of stress protecting agent (SPA) Stenotrophomonas rhizophila DSM14405T. Front. Plant Sci. 4:141. doi: 10.3389/fpls.2013.00141
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2019). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Alexander, A., Singh, V. K., Mishra, A., and Jha, B. (2019). Plant growth promoting rhizobacterium Stenotrophomonas maltophilia BJ01 augments endurance against N2 starvation by modulating physiology and biochemical activities of Arachis hypogea. PLoS One 14:e0222405. doi: 10.1371/journal.pone.0222405
Alijani, Z., Amini, J., Ashengroph, M., and Bahramnejad, B. (2020). Volatile compounds mediated effects of Stenotrophomonas maltophilia strain UN1512 in plant growth promotion and its potential for the biocontrol of Colletotrichum nymphaeae. Physiol. Mol. Plant Pathol. 112:101555. doi: 10.1016/j.pmpp.2020.101555
Alonso, A., and Martínez, J. L. (2000). Cloning and characterization of SmeDEF, a novel multidrug efflux pump from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 44, 3079–3086. doi: 10.1128/aac.44.11.3079-3086.2000
An, S., and Berg, G. (2018). Stenotrophomonas maltophilia. Trends Microbiol. 26, 637–638. doi: 10.1016/j.tim.2018.04.006
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Arts, I. S., Gennaris, A., and Collet, J.-F. (2015). Reducing systems protecting the bacterial cell envelope from oxidative damage. FEBS Lett. 589, 1559–1568. doi: 10.1016/j.febslet.2015.04.057
Auch, A. F., von Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Aznar, A., Chen, N. W. G., Thomine, S., and Dellagi, A. (2015). Immunity to plant pathogens and iron homeostasis. Plant Sci. 240, 90–97. doi: 10.1016/j.plantsci.2015.08.022
Baldiris, R., Acosta-Tapia, N., Montes, A., Hernández, J., and Vivas-Reyes, R. (2018). Reduction of hexavalent chromium and detection of chromate reductase (ChrR) in Stenotrophomonas maltophilia. Molecules 23:406. doi: 10.3390/molecules23020406
Balsanelli, E., Serrato, R. V., De Baura, V. A., Sassaki, G., Yates, M. G., Rigo, L. U., et al. (2010). Herbaspirillum seropedicae rfbB and rfbC genes are required for maize colonization. Environ. Microbiol. 12, 2233–2244. doi: 10.1111/j.1462-2920.2010.02187.x
Barak, J. D., Jahn, C. E., Gibson, D. L., and Charkowski, A. O. (2007). The role of cellulose and O-antigen capsule in the colonization of plants by Salmonella enterica. Mol. Plant-Microbe Interact. 20, 1083–1091. doi: 10.1094/mpmi-20-9-1083
Bashandy, S. R., Abd-Alla, M. H., and Dawood, M. F. A. (2020). Alleviation of the toxicity of oily wastewater to canola plants by the N2-fixing, aromatic hydrocarbon biodegrading bacterium Stenotrophomonas maltophilia-SR1. Appl. Soil Ecol. 154:103654. doi: 10.1016/j.apsoil.2020.103654
Behnke, J., and LaRoche, J. (2020). Iron uptake proteins in algae and the role of Iron starvation-induced proteins (ISIPs). Eur. J. Phycol. 55, 339–360. doi: 10.1080/09670262.2020.1744039
Benini, S. (2020). Carbohydrate-active enzymes: structure, activity, and reaction products. Int. J. Mol. Sci. 21:2727. doi: 10.3390/ijms21082727
Beranová, J., Mansilla, M. C., de Mendoza, D., Elhottová, D., and Konopásek, I. (2010). Differences in cold adaptation of Bacillus subtilis under anaerobic and aerobic conditions. J. Bacteriol. 192, 4164–4171. doi: 10.1128/jb.00384-10
Berg, G., and Martinez, J. L. (2015). Friends or foes: can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front. Microbiol. 6:241. doi: 10.3389/fmicb.2015.00241
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Blackburn, N., Fenchel, T., and Mitchell, J. (1998). Microscale nutrient patches in planktonic habitats shown by chemotactic bacteria. Science 282, 2254–2256. doi: 10.1126/science.282.5397.2254
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., Van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/nar/gkab335
Bulgarelli, D., Schlaeppi, K., Spaepen, S., Van Themaat, E. V. L., and Schulze-Lefert, P. (2013). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
Burrell, M., Hanfrey, C. C., Murray, E. J., Stanley-Wall, N. R., and Michael, A. J. (2010). Evolution and multiplicity of arginine decarboxylases in polyamine biosynthesis and essential role in Bacillus subtilis biofilm formation. J. Biol. Chem. 285, 39224–39238. doi: 10.1074/jbc.m110.163154
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chautrand, T., Souak, D., Chevalier, S., and Duclairoir-Poc, C. (2022). Gram-negative bacterial envelope homeostasis under oxidative and nitrosative stress. Microorganisms 10:924. doi: 10.3390/microorganisms10050924
Couée, I., Hummel, I., Sulmon, C., Gouesbet, G., and El Amrani, A. (2004). Involvement of polyamines in root development. Plant Cell Tissue Organ Cult. 76, 1–10. doi: 10.1023/a:1025895731017
Darby, N. J., Raina, S., and Creighton, T. E. (1998). Contributions of substrate binding to the catalytic activity of DsbC. Biochemistry 37, 783–791. doi: 10.1021/bi971888f
Díaz-Pérez, C., Cervantes, C., Campos-García, J., Julián-Sánchez, A., and Riveros-Rosas, H. (2007). Phylogenetic analysis of the chromate ion transporter (CHR) superfamily. FEBS J. 274, 6215–6227. doi: 10.1111/j.1742-4658.2007.06141.x
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Faraldo-Gómez, J. D., and Sansom, M. S. P. (2003). Acquisition of siderophores in gram-negative bacteria. Nat. Rev. Mol. Cell Biol. 4, 105–116. doi: 10.1038/nrm1015
Flores-Treviño, S., Bocanegra-Ibarias, P., Camacho-Ortiz, A., Morfín-Otero, R., Salazar-Sesatty, H. A., and Garza-González, E. (2019). Stenotrophomonas maltophilia biofilm: its role in infectious diseases. Expert Rev. Anti-Infect. Ther. 17, 877–893. doi: 10.1080/14787210.2019.1685875
Friedrich, N., Hagedorn, M., Soldati-Favre, D., and Soldati, T. (2012). Prison break: pathogens’ strategies to egress from host cells. Microbiol. Mol. Biol. Rev. 76, 707–720. doi: 10.1128/mmbr.00024-12
Gadd, G. M. (2010). Metals, minerals and microbes: geomicrobiology and bioremediation. Microbiology 156, 609–643. doi: 10.1099/mic.0.037143-0
Gao, T., Ding, M., Yang, C.-H., Fan, H., Chai, Y., and Li, Y. (2019). The phosphotransferase system gene ptsH plays an important role in MnSOD production, biofilm formation, swarming motility, and root colonization in Bacillus cereus 905. Res. Microbiol. 170, 86–96. doi: 10.1016/j.resmic.2018.10.002
García-León, G., Hernández, A., Hernando-Amado, S., Alavi, P., Berg, G., and Martínez, J. L. (2014). A function of SmeDEF, the major quinolone resistance determinant of Stenotrophomonas maltophilia, is the colonization of plant roots. Appl. Environ. Microbiol. 80, 4559–4565. doi: 10.1128/aem.01058-14
Garcia-Vallve, S. (2000). Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 10, 1719–1725. doi: 10.1101/gr.130000
Ge, Q., Cobine, P. A., and De La Fuente, L. (2021). The influence of copper homeostasis genes copA and copB on Xylella fastidiosa virulence is affected by sap copper concentration. Phytopathology 111, 1520–1529. doi: 10.1094/phyto-12-20-0531-r
Grimaud, R., Ezraty, B., Mitchell, J. K., Lafitte, D., Briand, C., Derrick, P. J., et al. (2001). Repair of oxidized proteins. J. Biol. Chem. 276, 48915–48920. doi: 10.1074/jbc.m105509200
Grossmann, K. (2009). Auxin herbicides: current status of mechanism and mode of action. Pest Manag. Sci. 66, 113–120. doi: 10.1002/ps.1860
Gunde-Cimerman, N., Plemenitaš, A., and Oren, A. (2018). Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 42, 353–375. doi: 10.1093/femsre/fuy009
Gupta, R., and Ramnani, P. (2006). Microbial keratinases and their prospective applications: an overview. Appl. Microbiol. Biotechnol. 70, 21–33. doi: 10.1007/s00253-005-0239-8
Hall, B. G. (2013). Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 30, 1229–1235. doi: 10.1093/molbev/mst012
Hao, X., Zhu, J., Rensing, C., Liu, Y., Gao, S., Chen, W., et al. (2021). Recent advances in exploring the heavy metal(loid) resistant microbiome. Comput. Struct. Biotechnol. J. 19, 94–109. doi: 10.1016/j.csbj.2020.12.006
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. L. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Huang, Y.-T., Chen, J.-M., Ho, B.-C., Wu, Z.-Y., Kuo, R. C., and Liu, P.-Y. (2018). Genome sequencing and comparative analysis of Stenotrophomonas acidaminiphila reveal evolutionary insights into sulfamethoxazole resistance. Front. Microbiol. 9:1013. doi: 10.3389/fmicb.2018.01013
Hudson, C. M., Lau, B. Y., and Williams, K. P. (2014). Islander: a database of precisely mapped genomic islands in tRNA and tmRNA genes. Nucleic Acids Res. 43, D48–D53. doi: 10.1093/nar/gku1072
Huedo, P., Coves, X., Daura, X., Gibert, I., and Yero, D. (2018). Quorum sensing signaling and quenching in the multidrug-resistant pathogen Stenotrophomonas maltophilia. Front. Cell. Infect. Microbiol. 8:122. doi: 10.3389/fcimb.2018.00122
Hussain, S., Khan, M., Sheikh, T. M. M., Mumtaz, M. Z., Chohan, T. A., Shamim, S., et al. (2022). Zinc essentiality, toxicity, and its bacterial bioremediation: a comprehensive insight. Front. Microbiol. 13:1133733. doi: 10.3389/fmicb.2022.900740
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11:119. doi: 10.1186/1471-2105-11-119
Imada, E. L., Rolla dos Santos, A. A., Oliveira, A. L. M., Hungria, M., and Rodrigues, E. P. (2017). Indole-3-acetic acid production via the indole-3-pyruvate pathway by plant growth promoter Rhizobium tropici CIAT 899 is strongly inhibited by ammonium. Res. Microbiol. 168, 283–292. doi: 10.1016/j.resmic.2016.10.010
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jang, S. J., Choi, Y. J., and Park, K. Y. (2002). Effects of polyamines on shoot and root development in arabidopsis seedlings and carnation cultures. J. Plant Biol. 45, 230–236. doi: 10.1007/bf03030365
Jendrossek, D. (2009). Polyhydroxyalkanoate granules are complex subcellular organelles (carbonosomes). J. Bacteriol. 191, 3195–3202. doi: 10.1128/jb.01723-08
Jendrossek, D., and Pfeiffer, D. (2014). New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly(3-hydroxybutyrate). Environ. Microbiol. 16, 2357–2373. doi: 10.1111/1462-2920.12356
John, N., and Thangavel, M. (2017). Stenotrophomonas maltophilia: a novel plant growth promoter and biocontrol agent from marine environment. Int. J. Adv. Res. 5, 207–214. doi: 10.21474/ijar01/3797
Jorquera, M. A., Hernández, M. T., Rengel, Z., Marschner, P., and de la Luz Mora, M. (2008). Isolation of culturable phosphobacteria with both phytate-mineralization and phosphate-solubilization activity from the rhizosphere of plants grown in a volcanic soil. Biol. Fertil. Soils 44, 1025–1034. doi: 10.1007/s00374-008-0288-0
Kalidasan, V., Joseph, N., Kumar, S., Awang Hamat, R., and Neela, V. K. (2018). Iron and virulence in Stenotrophomonas Maltophilia: all we know so far. Front. Cell. Infect. Microbiol. 8:401. doi: 10.3389/fcimb.2018.00401
Kandel, S., Joubert, P., and Doty, S. (2017). Bacterial endophytecolonization and distribution within plants. Microorganisms 5:77. doi: 10.3390/microorganisms5040077
Kasukabe, Y., He, L., Nada, K., Misawa, S., Ihara, I., and Tachibana, S. (2004). Overexpression of spermidine synthase enhances tolerance to multiple environmental stresses and up-regulates the expression of various stress-regulated genes in transgenic Arabidopsis thaliana. Plant Cell Physiol. 45, 712–722. doi: 10.1093/pcp/pch083
Kato, J., Kim, H.-E., Takiguchi, N., Kuroda, A., and Ohtake, H. (2008). Pseudomonas aeruginosa as a model microorganism for investigation of chemotactic behaviors in ecosystem. J. Biosci. Bioeng. 106, 1–7. doi: 10.1263/jbb.106.1
Kharade, S. S., and McBride, M. J. (2014). Flavobacterium johnsoniae chitinase ChiA is required for chitin utilization and is secreted by the type IX secretion system. J. Bacteriol. 196, 961–970. doi: 10.1128/jb.01170-13
Kim, H.-R., Lee, D., and Eom, Y.-B. (2018). Anti-biofilm and anti-virulence efficacy of celastrol against Stenotrophomonas maltophilia. Int. J. Med. Sci. 15, 617–627. doi: 10.7150/ijms.23924
Kumar, A., Rithesh, L., Kumar, V., Raghuvanshi, N., Chaudhary, K., Abhineet, P., et al. (2023). Stenotrophomonas in diversified cropping systems: friend or foe? Front. Microbiol. 14:1214680. doi: 10.3389/fmicb.2023.1214680
Lamark, T., Kaasen, I., Eshoo, M. W., Falkenberg, P., McDougall, J., and Strøm, A. R. (1991). DNA sequence and analysis of the bet genes encoding the osmoregulatory choline-glycine betaine pathway of Escherichia coli. Mol. Microbiol. 5, 1049–1064. doi: 10.1111/j.1365-2958.1991.tb01877.x
Langille, M. G., Hsiao, W. W., and Brinkman, F. S. (2008). Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinf. 9:329. doi: 10.1186/1471-2105-9-329
Lau, C. K. Y., Krewulak, K. D., and Vogel, H. J. (2016). Bacterial ferrous iron transport: the Feo system. FEMS Microbiol. Rev. 40, 273–298. doi: 10.1093/femsre/fuv049
Lee, W. C., Choi, S., Jang, A., Son, K., and Kim, Y. (2021a). Structural comparison of Acinetobacter baumannii β-ketoacyl-acyl carrier protein reductases in fatty acid and aryl polyene biosynthesis. Sci. Rep. 11:7945. doi: 10.1038/s41598-021-86997-3
Lee, W. C., Choi, S., Jang, A., Yeon, J., Hwang, E., and Kim, Y. (2021b). Structural basis of the complementary activity of two ketosynthases in aryl polyene biosynthesis. Sci. Rep. 11:16340. doi: 10.1038/s41598-021-95890-y
Lee, M.-J., Yang, Y.-T., Lin, V., and Huang, H. (2012). Site-directed mutations of the gatekeeping loop region affect the activity of Escherichia coli spermidine synthase. Mol. Biotechnol. 54, 572–580. doi: 10.1007/s12033-012-9599-3
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, X.-Z., Zhang, L., and Poole, K. (2002). SmeC, an outer membrane multidrug efflux protein of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 46, 333–343. doi: 10.1128/aac.46.2.333-343.2002
Liu, S., Mi, J., Song, K., Qi, H., and Zhang, L. (2022). Engineering synthetic microbial consortium for cadaverine biosynthesis from glycerol. Biotechnol. Lett. 44, 1389–1400. doi: 10.1007/s10529-022-03306-2
López-Mondéjar, R., Zühlke, D., Becher, D., Riedel, K., and Baldrian, P. (2016). Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 6:25279. doi: 10.1038/srep25279
Ma, Y., Ling, T., Su, X., Jiang, B., Nian, B., Chen, L., et al. (2021). Integrated proteomics and metabolomics analysis of tea leaves fermented by Aspergillus niger, Aspergillus tamarii and Aspergillus fumigatus. Food Chem. 334:127560. doi: 10.1016/j.foodchem.2020.127560
Ma, L., Zhang, Z., Li, J., Yang, X., Fei, B., Leung, P. H. M., et al. (2019). A new antimicrobial agent: poly (3-hydroxybutyric acid) oligomer. Macromol. Biosci. 19:e1800432. doi: 10.1002/mabi.201800432
Manjeet, K., Purushotham, P., Neeraja, C., and Podile, A. R. (2013). Bacterial chitin binding proteins show differential substrate binding and synergy with chitinases. Microbiol. Res. 168, 461–468. doi: 10.1016/j.micres.2013.01.006
Martínez-Granero, F., Capdevila, S., Sánchez-Contreras, M., Martín, M., and Rivilla, R. (2005). Two site-specific recombinases are implicated in phenotypic variation and competitive rhizosphere colonization in Pseudomonas fluorescens. Microbiology 151, 975–983. doi: 10.1099/mic.0.27583-0
Matias Rodrigues, J. F., Schmidt, T. S. B., Tackmann, J., and von Mering, C. (2017). MAPseq: highly efficient k-mer search with confidence estimates, for rRNA sequence analysis. Bioinformatics 33, 3808–3810. doi: 10.1093/bioinformatics/btx517
McSteen, P. (2010). Auxin and monocot development. Cold Spring Harb. Perspect. Biol. 2:a001479. doi: 10.1101/cshperspect.a001479
Meneses, C. H. S. G., Rouws, L. F. M., Simões-Araújo, J. L., Vidal, M. S., and Baldani, J. I. (2011). Exopolysaccharide production is required for biofilm formation and plant colonization by the nitrogen-fixing endophyte Gluconacetobacter diazotrophicus. Mol. Plant-Microbe Interact. 24, 1448–1458. doi: 10.1094/mpmi-05-11-0127
Michael, A. J. (2016). Polyamines in eukaryotes, bacteria, and archaea. J. Biol. Chem. 291, 14896–14903. doi: 10.1074/jbc.r116.734780
Monteiro, R. A., Balsanelli, E., Tuleski, T., Faoro, H., Cruz, L. M., Wassem, R., et al. (2012). Genomic comparison of the endophyte Herbaspirillum seropedicae SmR1 and the phytopathogen Herbaspirillum rubrisubalbicans M1 by suppressive subtractive hybridization and partial genome sequencing. FEMS Microbiol. Ecol. 80, 441–451. doi: 10.1111/j.1574-6941.2012.01309.x
Mukherjee, P., and Roy, P. (2016). Genomic potential of Stenotrophomonas maltophilia in bioremediation with an assessment of its multifaceted role in our environment. Front. Microbiol. 7:907. doi: 10.3389/fmicb.2016.00967
Nascimento, F. X., Hernandez, A. G., Glick, B. R., and Rossi, M. J. (2020). The extreme plant-growth-promoting properties of Pantoea phytobeneficialis MSR2 revealed by functional and genomic analysis. Environ. Microbiol. 22, 1341–1355. doi: 10.1111/1462-2920.14946
Newman, K. L., Chatterjee, S., Ho, K. A., and Lindow, S. E. (2008). Virulence of plant pathogenic bacteria attenuated by degradation of fatty acid cell-to-cell signaling factors. Mol. Plant-Microbe Interact. 21, 326–334. doi: 10.1094/mpmi-21-3-0326
Nogales, J., Campos, R., BenAbdelkhalek, H., Olivares, J., Lluch, C., and Sanjuan, J. (2002). Rhizobium tropici genes involved in free-living salt tolerance are required for the establishment of efficient nitrogen-fixing symbiosis with Phaseolus vulgaris. Mol. Plant-Microbe Interact. 15, 225–232. doi: 10.1094/mpmi.2002.15.3.225
O’Connor, S. E. (2015). Engineering of secondary metabolism. Annu. Rev. Genet. 49, 71–94. doi: 10.1146/annurev-genet-120213-092053
Oni, F. E., Geudens, N., Omoboye, O. O., Bertier, L., Hua, H. G. K., Adiobo, A., et al. (2019). Fluorescent Pseudomonas and cyclic lipopeptide diversity in the rhizosphere of cocoyam (Xanthosoma sagittifolium). Environ. Microbiol. 21, 1019–1034. doi: 10.1111/1462-2920.14520
Pakarian, P., and Pawelek, P. D. (2016). Subunit orientation in the Escherichia coli enterobactin biosynthetic EntA-EntE complex revealed by a two-hybrid approach. Biochimie 127, 1–9. doi: 10.1016/j.biochi.2016.04.007
Panday, S., Talreja, R., and Kavdia, M. (2020). The role of glutathione and glutathione peroxidase in regulating cellular level of reactive oxygen and nitrogen species. Microvasc. Res. 131:104010. doi: 10.1016/j.mvr.2020.104010
Pandit, M. A., Kumar, J., Gulati, S., Bhandari, N., Mehta, P., Katyal, R., et al. (2022). Major biological control strategies for plant pathogens. Pathogens 11:273. doi: 10.3390/pathogens11020273
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patzer, S. I., Baquero, M. R., Bravo, D., Moreno, F., and Hantke, K. (2003). The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, Cir, Fiu and IroN. Microbiology 149, 2557–2570. doi: 10.1099/mic.0.26396-0
Paul, M. J., Primavesi, L. F., Jhurreea, D., and Zhang, Y. (2008). Trehalose metabolism and signaling. Annu. Rev. Plant Biol. 59, 417–441. doi: 10.1146/annurev.arplant.59.032607.092945
Pinski, A., Zur, J., Hasterok, R., and Hupert-Kocurek, K. (2020). Comparative genomics of Stenotrophomonas maltophilia and Stenotrophomonas rhizophila revealed characteristic features of both species. Int. J. Mol. Sci. 21:4922. doi: 10.3390/ijms21144922
Reed, J. W., and Walker, G. C. (1991). The exoD gene of Rhizobium meliloti encodes a novel function needed for alfalfa nodule invasion. J. Bacteriol. 173, 664–677. doi: 10.1128/jb.173.2.664-677.1991
Ryan, R. P., Monchy, S., Cardinale, M., Taghavi, S., Crossman, L., Avison, M. B., et al. (2009). The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525. doi: 10.1038/nrmicro2163
Sadykov, M. R., Ahn, J., Widhelm, T. J., Eckrich, V. M., Endres, J. L., Driks, A., et al. (2017). Poly(3-hydroxybutyrate) fuels the tricarboxylic acid cycle and de novo lipid biosynthesis during Bacillus anthracis sporulation. Mol. Microbiol. 104, 793–803. doi: 10.1111/mmi.13665
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Shaheen, R., Svensson, B., Andersson, M. A., Christiansson, A., and Salkinoja-Salonen, M. (2010). Persistence strategies of Bacillus cereus spores isolated from dairy silo tanks. Food Microbiol. 27, 347–355. doi: 10.1016/j.fm.2009.11.004
Sharma, V., Kumar, V., Archana, G., and Kumar, G. N. (2005). Substrate specificity of glucose dehydrogenase (GDH) of Enterobacter asburiae PSI3 and rock phosphate solubilization with GDH substrates as C sources. Can. J. Microbiol. 51, 477–482. doi: 10.1139/w05-032
Sharma, S. B., Sayyed, R. Z., Trivedi, M. H., and Gobi, T. A. (2013). Phosphate solubilizing microbes: sustainable approach for managing phosphorus deficiency in agricultural soils. Springerplus 2:587. doi: 10.1186/2193-1801-2-587
Sharma, A., Vaishnav, A., Jamali, H., Keswani, C., Srivastava, A. K., Kaushik, R., et al. (2023). Unraveling the plant growth-promoting mechanisms of Stenotrophomonas sp. CV83 for drought stress tolerance and growth enhancement in chickpea. J. Plant Growth Regul. 42, 6760–6775. doi: 10.1007/s00344-023-11010-2
Singh, R. P., and Jha, P. N. (2017). The PGPR Stenotrophomonas maltophilia SBP-9 augments resistance against biotic and abiotic stress in wheat plants. Front. Microbiol. 8:1945. doi: 10.3389/fmicb.2017.01945
Sleator, R. D., and Hill, C. (2002). Bacterial osmoadaptation: the role of osmolytes in bacterial stress and virulence. FEMS Microbiol. Rev. 26, 49–71. doi: 10.1111/j.1574-6976.2002.tb00598.x
Sobecky, P. A., and Hazen, T. H. (2009). “Horizontal gene transfer and mobile genetic elements in marine systems” in Horizontal Gene Transfer. eds. M. B. Gogarten, J. P. Gogarten, and L. Olendzenski (Totowa, NJ: Genomes in Flux), 435–453.
Swiontek Brzezinska, M., Jankiewicz, U., Burkowska, A., and Walczak, M. (2013). Chitinolytic microorganisms and their possible application in environmental protection. Curr. Microbiol. 68, 71–81. doi: 10.1007/s00284-013-0440-4
Syvanen, M. (1994). Horizontal gene transfer: evidence and possible consequences. Annu. Rev. Genet. 28, 237–261. doi: 10.1146/annurev.ge.28.120194.001321
Taghavi, S., Garafola, C., Monchy, S., Newman, L., Hoffman, A., Weyens, N., et al. (2009). Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl. Environ. Microbiol. 75, 748–757. doi: 10.1128/aem.02239-08
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tang, J., Li, Y., Zhang, L., Mu, J., Jiang, Y., Fu, H., et al. (2023). Biosynthetic pathways and functions of indole-3-acetic acid in microorganisms. Microorganisms 11:2077. doi: 10.3390/microorganisms11082077
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Ulrich, K., Kube, M., Becker, R., Schneck, V., and Ulrich, A. (2021). Genomic analysis of the endophytic Stenotrophomonas strain 169 reveals features related to plant-growth promotion and stress tolerance. Front. Microbiol. 12:687463. doi: 10.3389/fmicb.2021.687463
Vassilev, N., Vassileva, M., and Nikolaeva, I. (2006). Simultaneous P-solubilizing and biocontrol activity of microorganisms: potentials and future trends. Appl. Microbiol. Biotechnol. 71, 137–144. doi: 10.1007/s00253-006-0380-z
Vyrides, I., and Stuckey, D. C. (2017). Compatible solute addition to biological systems treating waste/wastewater to counteract osmotic and other environmental stresses: a review. Crit. Rev. Biotechnol. 37, 865–879. doi: 10.1080/07388551.2016.1266460
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinf. 7:142. doi: 10.1186/1471-2105-7-142
Wall, G., Montelongo-Jauregui, D., Bonifacio, B. V., Lopez-Ribot, J. L., and Uppuluri, P. (2019). Candida albicans biofilm growth and dispersal: contributions to pathogenesis. Curr. Opin. Microbiol. 52, 1–6. doi: 10.1016/j.mib.2019.04.001
Wang, Y., He, T., Shen, Z., and Wu, C. (2018). Antimicrobial resistance in Stenotrophomonas spp. Microbiol. Spectrum 6:ARBA-0005-2017. doi: 10.1128/microbiolspec.arba-0005-2017
Wei, H.-L., and Zhang, L.-Q. (2006). Quorum-sensing system influences root colonization and biological control ability in Pseudomonas fluorescens 2P24. Antonie Van Leeuwenhoek 89, 267–280. doi: 10.1007/s10482-005-9028-8
Wright, G. D. (1999). Aminoglycoside-modifying enzymes. Curr. Opin. Microbiol. 2, 499–503. doi: 10.1016/s1369-5274(99)00007-7
Xie, S.-S., Wu, H.-J., Zang, H.-Y., Wu, L.-M., Zhu, Q.-Q., and Gao, X.-W. (2014). Plant growth promotion by spermidine-producing Bacillus subtilis OKB105. Mol. Plant-Microbe Interact. 27, 655–663. doi: 10.1094/mpmi-01-14-0010-r
Xu, Y. Y., Cheng, T., Rao, Q. Y., Zhang, S. Q., and Ma, Y. Y. (2023). Comparative genomic analysis of Stenotrophomonas maltophilia unravels their genetic variations and versatility trait. J. Appl. Genet. 64, 351–360. doi: 10.1007/s13353-023-00752-0
Yi, Y., de Jong, A., Frenzel, E., and Kuipers, O. P. (2017). Comparative transcriptomics of Bacillus mycoides strains in response to potato-root exudates reveals different genetic adaptation of endophytic and soil isolates. Front. Microbiol. 8:1487. doi: 10.3389/fmicb.2017.01487
Youenou, B., Favre-Bonté, S., Bodilis, J., Brothier, E., Dubost, A., Muller, D., et al. (2015). Comparative genomics of environmental and clinical Stenotrophomonas maltophilia strains with different antibiotic resistance profiles. Genome Biol. Evol. 7, 2484–2505. doi: 10.1093/gbe/evv161
Yue, X. Y., Zhang, B., Jiang, D. D., Liu, Y. J., and Niu, T. G. (2011). Separation and purification of a keratinase as pesticide against root-knot nematodes. World J. Microbiol. Biotechnol. 27, 2147–2153. doi: 10.1007/s11274-011-0680-z
Zhang, P., Jin, T., Kumar Sahu, S., Xu, J., Shi, Q., Liu, H., et al. (2019). The distribution of tryptophan-dependent indole-3-acetic acid synthesis pathways in bacteria unraveled by large-scale genomic analysis. Molecules 24:1411. doi: 10.3390/molecules24071411
Zhang, Z., Yuen, G. Y., Sarath, G., and Penheiter, A. R. (2001). Chitinases from the plant disease biocontrol agent, Stenotrophomonas maltophilia C3. Phytopathology 91, 204–211. doi: 10.1094/phyto.2001.91.2.204
Zhao, Y., Wei, H.-M., Yuan, J.-L., Xu, L., and Sun, J.-Q. (2023). A comprehensive genomic analysis provides insights on the high environmental adaptability of Acinetobacter strains. Front. Microbiol. 14:1177951. doi: 10.3389/fmicb.2023.1177951
Zheng, J., Ge, Q., Yan, Y., Zhang, X., Huang, L., and Yin, Y. (2023). dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121. doi: 10.1093/nar/gkad328
Zhou, S., Zhang, J., Ma, F., Tang, C., Tang, Q., and Zhang, X. (2018). Investigation of lignocellulolytic enzymes during different growth phases of Ganoderma lucidum strain G0119 using genomic, transcriptomic and secretomic analyses. PLoS One 13:e0198404. doi: 10.1371/journal.pone.0198404
Zhuang, M., Achmon, Y., Cao, Y., Liang, X., Chen, L., Wang, H., et al. (2021). Distribution of antibiotic resistance genes in the environment. Environ. Pollut. 285:117402. doi: 10.1016/j.envpol.2021.117402
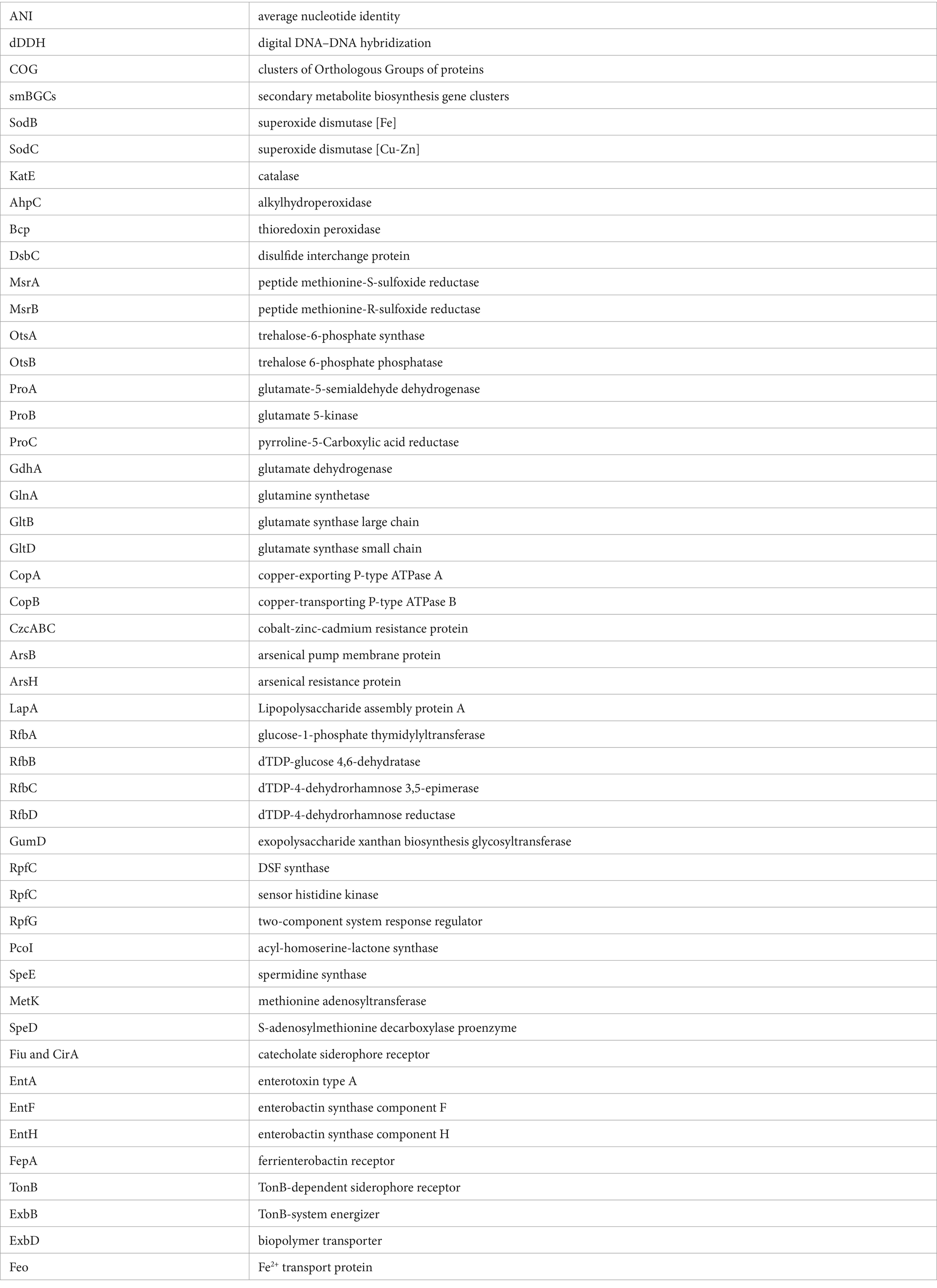
Keywords: Stenotrophomonas, comparative genomic analysis, environmental adaptability, plant growth promotion, horizontal gene transfer
Citation: Zhao Y, Ding W-J, Xu L and Sun J-Q (2024) A comprehensive comparative genomic analysis revealed that plant growth promoting traits are ubiquitous in strains of Stenotrophomonas. Front. Microbiol. 15:1395477. doi: 10.3389/fmicb.2024.1395477
Edited by:
Alessandra Alves De Souza, Secretariat of Agriculture and Food Supply of São Paulo State, BrazilReviewed by:
Helena Lima, Universidade Federal de Viçosa, BrazilCopyright © 2024 Zhao, Ding, Xu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Quan Sun, c3VuanE4MUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.