 Zhu-Ming Jiang1,2†
Zhu-Ming Jiang1,2† Yu-Qin Zhang
Yu-Qin Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 01 December 2023
Sec. Extreme Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1267771
This article is part of the Research Topic13th International Congress on Extremophiles: From Extremophilic Biomolecules and Microorganisms to Biotechnological and Sustainable ApplicationsView all 11 articles
Solirubrobacter spp. were abundant in soil samples collected from deserts and other areas with high UV radiation. In addition, a novel Solirubrobacter species, with strain CPCC 204708T as the type, was isolated and identified from sandy soil sample collected from the Badain Jaran Desert of the Inner Mongolia autonomous region. Strain CPCC 204708T was Gram-stain positive, rod-shaped, non-motile, non-spore-forming, and grew optimally at 28–30°C, pH 7.0–8.0, and in the absence of NaCl. Analysis of the 16S rRNA gene sequence of strain CPCC 204708T showed its identity within the genus Solirubrobacter, with highest nucleotide similarities (97.4–98.2%) to other named Solirubrobacter species. Phylogenetic and genomic analyses indicated that the strain was most closely related to Solirubrobacter phytolaccae KCTC 29190T, while represented a distinct species, as confirmed from physiological properties and comparison. The name Solirubrobacter deserti sp. nov. was consequently proposed, with CPCC 204708T (= DSM 105495T = NBRC 112942T) as the type strain. Genomic analyses of the Solirubrobacter spp. also suggested that Solirubrobacter sp. URHD0082 represents a novel species, for which the name Candidatus “Solirubrobacter pratensis” sp. nov. was proposed. Genomic analysis of CPCC 204708T revealed the presence of genes related to its adaptation to the harsh environments of deserts and may also harbor genes functional in plant-microbe interactions. Pan-genomic analysis of available Solirubrobacter spp. confirmed the presence of many of the above genes as core components of Solirubrobacter genomes and suggests they may possess beneficial potential for their associate plant and may be important resources for bioactive compounds.
The genus Solirubrobacter, belonging to the family Solirubrobacteraceae, the order Solirubrobacterales, the phylum Actinomycetota, was first identified by Singleton et al. (2003) with Solirubrobacter pauli as the type species. Two other species, Solirubrobacter soli and Solirubrobacter ginsenosidimutans, were subsequently reported to be identified from soil samples in ginseng fields in 2007 (Kim et al., 2007). And in 2014, two additional species (S. phytolaccae and S. taibaiensis) were identified after isolation from the roots and stems of Phytolacca acinosa Roxb., respectively (Wei et al., 2014; Zhang L. et al., 2014). These five currently known species generally inhabit soil and plant-based ecosystems.1
The boom in cultivation-independent techniques and multi-omics technologies enable improved studies on the diversity of yet-uncultivated microorganisms, also termed Microbial Dark Matter (MDM) (Jiao et al., 2020). It was found that the genus Solirubrobacter abundantly inhabits an Indian desert, just below members of the genera Gaiella and Streptomyces (Sivakala et al., 2018). In five different types of natural soil ecosystems in Northwest China, Solirubrobacter was a dominant genus (Zhang et al., 2019). It was also noted that members of the Actinomycetales and Solirubrobacterales are more abundant in soils with lower organic carbon available in varied agricultural landscapes (Shange et al., 2012).
Besides Solirubrobacter show potential for applications such as plant growth promotion and discovery of unique bioactive compounds and activities (Franke-Whittle et al., 2015). S. ginsenosidimutans DSM 22325T, for instance, displayed ginsenoside conversion activity, transforming ginsenoside Rb1 into ginsenoside F2, a phytochemical with numerous pharmacological properties (Kim et al., 2021; Zhou et al., 2021).
Desert ecosystems worldwide are abundant and diverse sources of microbiota. Harsh conditions such as extreme ultraviolet radiation, carbon and nitrogen scarcity, limited energy, hyper-aridity, and extreme temperatures, characterize these ecosystems. However, these stressful ecosystems host remarkably diverse microbial communities. Therefore, deserts can be viewed as microbial resource hotspots (Bull et al., 2016; Leung et al., 2020). One primary stress factor in deserts is high ultraviolet radiation (UVR), which all desert inhabitants, including plants, have to mitigate. Interestingly, S. pauli JCM 13025T, was reported to be a radiation-resistant strain, which has drawn considerable research attention (Singleton et al., 2003).
So far, the genus Solirubrobacter has been detected in soils (Sánchez-Marañón et al., 2017), biocrust (Miralles et al., 2020), rhizosphere habitats of various crops (Aguiar et al., 2020; Feng et al., 2021; Lee et al., 2021) and medicinal plants (Dong et al., 2018). It is also proposed that Solirubrobacter spp. may be pioneering organisms that enable microbiome develop in plant rhizospheres, thereby playing important roles in maintaining host plant health in ecologically stressful environments (Sivakala et al., 2018; Zhang et al., 2019). Its potential as plant-promoting, UV-resistant microorganisms is regarded as beneficial for desert ecosystem health, especially in the context of changing climate patterns.
In the current study, we investigated the ecological distribution of Solirubrobacter across different environmental samples, including desert soils. The research also resulted in isolation of Solirubrobacter strains leading to the discovery of a new species. Furthermore, we also conducted a global pan-genomic analysis of the genus Solirubrobacter to evaluate the functional and biological resource potential.
Twelve rhizosphere soil samples from herbs (IMB15101S–IMB15112S) were collected from high-altitude barren hills in Xinjiang. And another twelve rhizosphere soil samples from Yunnan Ethno-Medicinal plants (IMB21101S–IMB21112S) were collected from Ailao mountain in Yunnan. While desert soil samples (IMB19101D–IMB19112D, IMB15201D–IMB15212D, IMB16101D–IMB16112D) were collected from the Gurbantunggut Desert, the Tengger Desert and the Badain Jaran Desert, respectively. Cow feces samples (IMB20301F–IMB20312F) were collected from Fangshan in Beijing, while crow feces samples (IMB20101F–IMB20112F) were collected from Nanhaizi Wetland Park, Beijing. Soil samples were collected from the area surrounding Plateau Lakes (IMB20201S–IMB20212S) in the Guizhou province and sediment soil samples (IMB22101S–IMB22112S) were collected from fresh water reservoirs in the Sichuan province. Finally, water samples (IMB19301W–IMB19312W) were collected from Erhai Lake and phycosphere samples (IMB14101E–IMB14112E) were collected from the phycosphere of agar cultures maintained in our laboratory. Detailed information for samples is provided in Supplementary Table 1. Soil and sand samples were collected into sterile envelopes. Water samples were filtered through 3.0 μm pore-size filters to exclude most cyanobacterial colonies and other small particles, followed by filtering water to concentrate biomass on 0.22 μm pore-size filters. Samples were returned to the laboratory within 3 days of collection, with microbial isolation and DNA extraction carrying out immediately.
Samples from the same biotopes were pooled into a single composite sample and subjected to DNA extraction for community compositional analyses. Total genomic DNA from each soil composite sample was extracted with a PowerSoil DNA isolation kit (MoBio, USA). The DNA from each composite water sample was extracted with a PowerWater DNA isolation kit (MoBio, USA) according to the manufacturer’s protocols. Total DNA was then used as template for PCR amplification of the V3 to V4 hypervariable regions of 16S rRNA genes using the universal bacterial primers 5′-ACTCCTACGGGAGGCAGCAG-3′ (338F) and 5′-GGACTACHVGGGTWTCTAAT-3′ (806R). PCR amplifications were performed using high fidelity TransStart Fastpfu DNA Polymerase (Transgen, China) in 20 μL reaction mixtures containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mmol/L dNTPs, 0.8 μL of each primer (5 μmol/L), 0.4 μL of FastPfu Polymerase (final concentration was 1 unit), and 10 ng of template DNA. PCR conditions comprised 5 min of an initial denaturation at 94°C followed by 35 cycles of denaturation at 94°C for 30 s, 45 s of primer annealing at 55°C, 40 s of elongation at 72°C, and then a final 10 min elongation step at 72°C.
Purified PCR amplicons were pooled in equimolar amounts and paired-end sequenced on the Illumina MiSeq PE300 platform (Illumina, San Diego, USA) using standard protocols at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). Raw FASTQ files were de-multiplexed using an in-house perl script and then quality-filtered and merged using the following criteria: (i) 300 bp reads were truncated at any site with an average quality score of < 20 over a 50 bp sliding window, and truncated reads < 50 bp were discarded, in addition to reads containing ambiguous characters; (ii) only overlapping sequences > 10 bp were merged. The maximum mismatch ratio of the overlapping region was set to 0.2 and reads that could not be assembled were discarded. (iii) Samples were distinguished based on barcoded primers, sequence direction was adjusted, and exact barcode matching was specified, in addition to setting 2 nucleotide mismatches as the maximum for primer matching. The quality-filtered sequences were clustered into operational taxonomic units (OTUs) at the 97% nucleotide sequence similarity level. The taxonomic classification of each OTU representative sequence was analyzed using the RDP Classifier (version 2.2), with comparison against the Silva 16S rRNA gene database (version 138) using a classification confidence threshold of 0.7. We carried out the high throughput sequencing analysis using Majorbio online analysis platform.2 The alpha diversity traits of each composite sample estimated by the Chao1 estimator, and the Shannon diversity index, in addition to the coverage estimated by Good’s coverage. Rarefaction analyses was used to show whether the amount of sequencing data in these composite samples is reasonable. Relative abundance referred to the proportion of reads of a particular taxon (OTU) in all reads of a composite sample.
The dilution plating method was used to isolate strains from each sample using previously described procedures (Deng et al., 2022). Strain CPCC 204708T was obtained from soils of the Badain Jaran Desert (39°21′ N, 102°19′ E, 1,550 m), using media containing (L–1): 2.0 g sodium malonate, 0.1 g NH4NO3, 0.1 g KCl, 0.05 g MgSO4⋅7H2O, 0.05 g FeSO4⋅7H2O, 0.38 g marine trace salt mixture, 15 g agar, and pH adjustment to 7.0–7.2. Aztreonam and potassium dichromate were added to the isolation medium to final concentrations of 25 and 50 mg L–1, respectively, to prevent fungal and Gram-negative bacterial growth. Distinct colonies were chosen for subsequent streaking on R2A agar (Difco) to identify isolated and uniform colonies. Pure cultures were cultivated and maintained on R2A medium at 4°C and stored in aqueous glycerol suspensions (20%, v/v) at −80°C.
The reference strains S. phytolaccae KCTC 29190T and S. taibaiensis KCTC 29222T were obtained from the Korean Collection for Type Cultures (KCTC), while S. pauli JCM 13025T was obtained from the RIKEN BioResource Research Center (JCM). S. ginsenosidimutans DSM 21036T and S. soli DSM 22325T were obtained from the DSMZ. The reference strains were included in subsequent assays in parallel with newly identified strains.
The whole genome of Solirubrobacter sp. URHD0082, which was isolated from Mediterranean grassland soil, was retrieved from the NCBI database (accession: AUEK000000003), and was included in the pan-genomic analysis of the genus Solirubrobacter. The 16S rRNA gene sequence extracted from the genome was used to identify Solirubrobacter sp. URHD0082 based on sequence comparisons and phylogenetic analyses. In addition, genomic comparisons were used to identify the taxon to which strain URHD0082 belonged.
The genomic DNA of new isolates was extracted and their 16S rRNA genes were PCR amplified, as previously described (Li et al., 2007). The sequences were then compared against those in GenBank using the BLAST program and the EzBioCloud4 platform (Yoon et al., 2017) to determine approximate phylogenetic affiliations. Multiple sequence alignments of isolate and closely related 16S rRNA genes were conducted using the molecular evolutionary genetics analysis (MEGA) software package (version 7.0) (Kumar et al., 2016). Phylogenetic trees were inferred using Neighbor-Joining (Saitou and Nei, 1987), Maximum Parsimony (Kluge and Farris, 1969), and Maximum-Likelihood (Felsenstein, 1981) methods. Phylogenetic reconstruction topologies were evaluated with the bootstrap resampling method (Felsenstein, 1985) and 1,000 replicates. A phylogenetic tree was also conducted based on the concatenation of 120 ubiquitous single-copy maker genes (bac120 maker set) (Parks et al., 2018) by a pipeline called EasyCGTree (Xue et al., 2021). Average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) values among the strains CPCC 204708T, URHD0082 and other validly described Solirubrobacter species were calculated using the ezbiocloud platform (Yoon et al., 2017) and the Genome-to-Genome Distance Calculator (GGDC, version 3.0)5 (Auch et al., 2010), respectively.
Physiological characteristics of strain CPCC 204708T were examined by observing growth at 28°C for 1–4 weeks on peptone yeast glucose (PYG; 3% trypticase soy broth, 0.3% yeast extract, 1.5% agar; pH 7.0–7.2), tryptone soy agar (TSA, Difco), ISP 2, yeast extract sucrose (YM, Difco), Luria-Bertani, R2A (Difco), ISP 4, and nutrient (Shirling and Gottlieb, 1966) media formulations. Growth temperature ranges were evaluated by incubation on R2A agar medium at 0, 4, 10, 20, 25, 28, 30, 32, 37, and 40°C for 14 d (Sun et al., 2017). The growth pH range was measured using a previously described buffer system (Xu et al., 2005), R2A as the basal medium, and evaluation over a pH range of 4.0–11.0 (at intervals of 1 pH unit). Salt (sodium chloride) tolerance was evaluated on R2A agar medium supplemented with NaCl concentrations of 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and 15% (w/v). UV radiation tolerance was evaluated using a UV-C radiation wavelength of 254 nm, following previously described procedures (Zhang et al., 2007). Growth after 10 days was determined to be either positive or negative for UV resistance compared to unirradiated controls. The strain Deinococcus radiodurans ATCC 13939T was also used as a positive control. The strain Escherichia coli ATCC 25922 was used as a negative control.
Gram stains were conducted as previously described (Magee et al., 1975). Colony appearance and pigment production were evaluated after incubation at 28°C on R2A medium. Cellular morphological features were observed with 5–7-day-old cultures using light microscopy (Zeiss Axio Scope. A1 Vario) and transmission electron microscopy (JEOL JEM-1010). Cell motility was evaluated with inverted microscopic observations of cells suspended in a 0.85% NaCl solution. Oxidase activity was evaluated using an analytical profile index (API) oxidase reagent (bioMeriéux) according to the manufacturer’s instructions, while catalase activity was determined by the presence of bubble production after application of 3% (v/v) H2O2. Metabolic characteristics were determined using Biolog GEN III (MicroPlate), API 50CH, and API ZYM (bioMerieux) test kits according to the manufacturer’s instructions. The abilities of the strain to hydrolyze gelatin, cellulose, and starch, in addition to producing H2S and indole were evaluated, as previously described (Gonzalez et al., 1978). The type strain S. phytolaccae KCTC 29190T was included in the physiological and biochemical tests for comparison.
The ability of the strains to produce indole-3-acetic acid (IAA) was evaluated with colorimetric methods (Bric et al., 1991). Briefly, log-phase cells were inoculated into 1% tryptone aqueous solutions containing 3 mmol/L L-tryptophan and then cultured at 28°C for 4 days. Absorbance at 530 nm was then plotted against IAA standard solution concentration solutions (0, 0.625, 1.0, 1.25, 2.0, 2.5, 4.0, and 5.0 mg/L), followed by linear regression to obtain a standard curve for IAA (Supplementary Figure 1). IAA quantification was based on a linear regression equation (y = 0.02x-0.001) for the colorimetric IAA content assay that exhibited a good fit (R2 = 0.9994), enabling quantification.
Chemotaxonomic and molecular systematic studies of strain CPCC 204708T were conducted with cells after cultivation in TSB medium at 28°C for 7 days in shake flasks on a rotary shaker (150 r/min) until cells reached the logarithmic growth phase. Amino acids and peptides in whole cell hydrolysates were analyzed by two-dimensional ascending thin-layer chromatography (TLC) on cellulose plates using solvent systems described by Schleifer and Kandler (1973). Sugar profiles were evaluated with TLC, as previously described (Komagata and Suzuki, 1988). Polar lipids were extracted, as previously described and identified by two-dimensional TLC (Minnikin et al., 1984). Menaquinones were extracted as previously described (Collins et al., 1977) and analyzed by HPLC (Groth et al., 1997). Cellular fatty acids analysis was performed using the Microbial Identification System (Sherlock Version 6.0; MIDI database: ACTIN1) (Kroppenstedt, 1985; Sasser, 1990).
Whole-genome sequencing of the new isolate CPCC 204708T and the reference strains S. phytolaccae KCTC 29190T, S. taibaiensis KCTC 29222T, S. ginsenosidimutans DSM 21036T were conducted on the Illumina HiSeq 4000 platform (Illumina, San Diego, CA, USA) at the Beijing Genomics Institute (Beijing, China) in this study. Genomic DNA was randomly sheared to construct three read libraries of length 300 bp using a Bioruptor ultrasonicator (Diagenode, Denville, NJ, USA) and physico-chemical methods. Paired-end fragment libraries were then sequenced using manufacturer protocols. Low quality reads (those with consecutive bases covered by fewer than five reads) were discarded and the remaining reads were assembled with the SOAPdenovo v1.05 software program (Xie Y. et al., 2014). The assembled genomes of strains S. pauli JCM 13025T and S. soli DSM 22325T were downloaded from NCBI database. The quality (index: completeness and contamination) of the draft genomes of the genus Solirubrobacter were accurately assessed by the CheckM pipeline (Parks et al., 2015).
The assembled genomes of strains CPCC 204708T, S. phytolaccae KCTC 29190T, S. pauli JCM 13025T, S. taibaiensis KCTC 29222T, S. ginsenosidimutans DSM 21036T, S. soli DSM 22325T, and URHD0082 were subjected to gene prediction using Hidden Markov models in the glimmer3 software program6 (Delcher et al., 2007), followed by functional annotation through comparison against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database7 (Moriya et al., 2007). Functional genes related to stress response were identified in these genomes by comparison to the Uniprot8 (UniProt Consortium, 2023) and Interpro9 databases (Paysan-Lafosse et al., 2023). Further, biosynthetic gene clusters (BGCs) were detected and characterized using the antibiotics and secondary metabolite analysis shell platform (antiSMASH; version 6.0)10 (Blin et al., 2021).
The bacterial pan-genome analysis (BPGA) pipeline (version 1.3) was used to analyze Solirubrobacter genomic diversity and characteristics. Protein sequences used for pan-genomic analysis were annotated with the Rapid Annotation using Subsystem Technology (RAST) server (version 2.0).11 BPGA was conducted with default settings, as previously described (Chaudhari et al., 2016). Proteins encoded by the seven genomes were used to generate orthologous gene/protein clusters (homologous families) using the USEARCH clustering tool and then to construct phylogenetic trees using concatenations of core genes to generate a pan-matrix in BPGA. Each homolog family was assigned a homologous gene family conservation value (HGFCV) based on its frequency in the three genomes. Different conservation values (CVs) reflect the distribution frequency of the gene homologs among the 7 strains, wherein higher CVs indicate a more widely conserved gene in the 7 Solirubrobacter strains. Gene families with HGFCVs of 7 were considered components of the core genome, while those with values of 2–6 or 1 were considered accessory or unique genes, respectively. Core, accessory, unique, and exclusively absent genes were retrieved from the genomes using the USEARCH clustering tool. BPGA was then used for evolutionary analysis based on concatenated core gene alignments and the binary pan-matrix. The gene matrix was calculated using shared gene value presence or absence within the orthologous gene clusters. The core genome phylogenetic tree was constructed in BPGA by first extracting the protein sequences (excluding paralogs) from 20 random orthologous gene clusters. MUSCLE was then used to generate multiple sequence alignments for each gene cluster. The alignments were concatenated and a Neighbor-Joining phylogenetic tree was constructed from the concatenated matrix.
Solirubrobacter spp. were detected in ecosystems based on high-throughput sequencing of 16S rRNA genes recovered from various environments. The alpha diversity of these composite samples from the different ecosystems exhibited different Chao1 and Shannon index values, suggesting significant difference in the richness or diversity of bacteria among these biotopes. Rarefaction analyses using the Shannon index as a diversity metric indicated that our sequencing efforts covered nearly all of the diversity that would be expected to be found in these composite samples. The highest abundances of Solirubrobacter spp. were observed in rhizosphere soils attached to medicinal plants in high-altitude areas of Xinjiang and Yunnan. They were also frequently observed in desert sandy soil samples. Low Solirubrobacter richness was observed in other ecosystems, including in crow feces and aquatic habitats (Figure 1). Further, Solirubrobacter spp. were not detected at all in the phycosphere of laboratory culture-systems, nor from cow feces collected from a Beijing farm. These results suggest that Solirubrobacter spp. may be adapted to life within extreme ecosystems like the high altitude barren hills of Xinjiang and Yunnan that exhibit strong solar radiation, in addition to arid deserts.
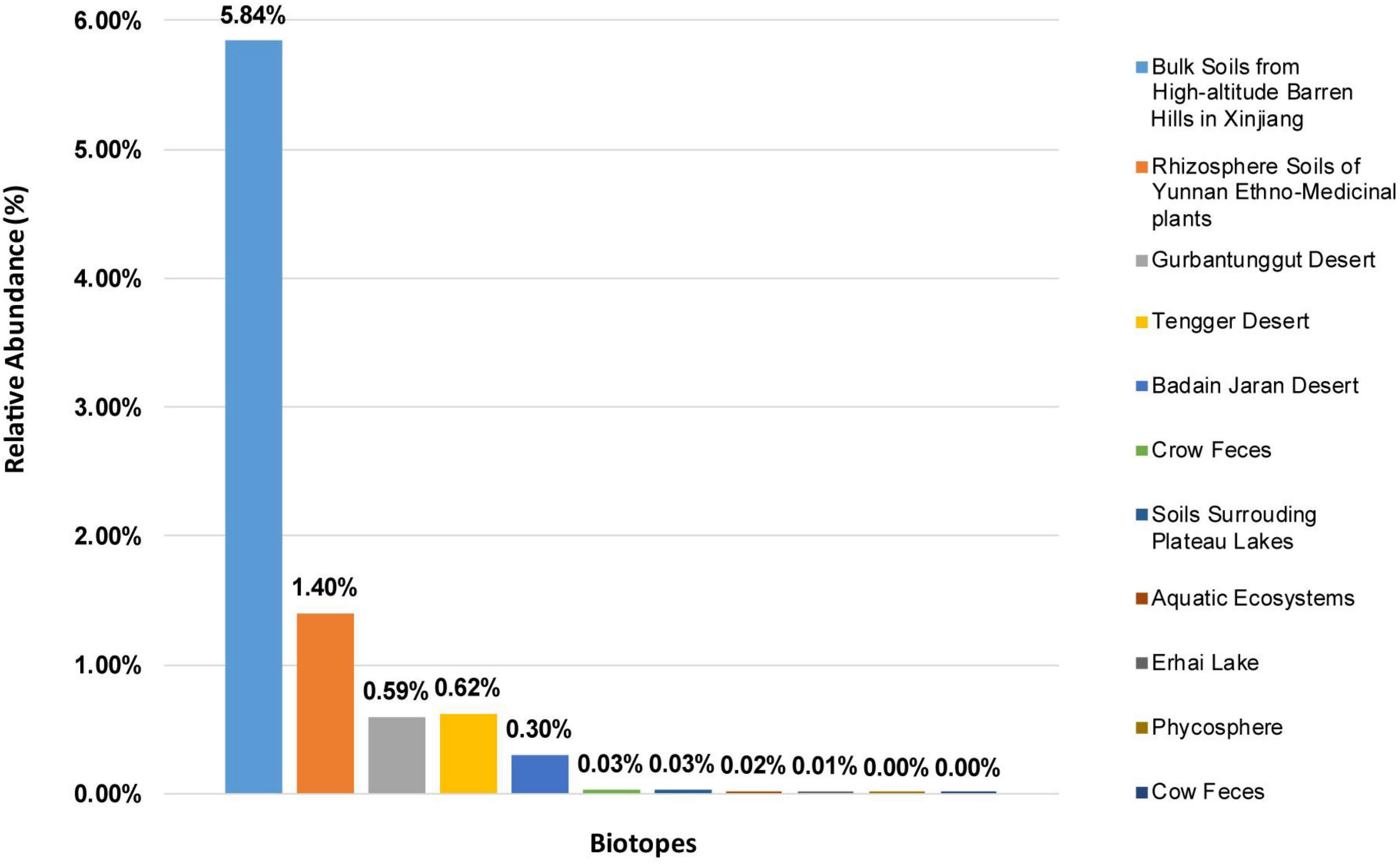
Figure 1. The relative abundances of Solirubrobacter among communities from eleven different biotopes, based on 16S rRNA gene analyses. The relative abundances are shown for composite samples from various environments, as indicated by the legend on the right.
Strain CPCC 204708T was isolated from a desert sandy soil sample (IMB16109D) collected from the Badain Jaran Desert (39°56′12″ N, 102°05′39″ E; 1,157 mH) from within the Inner Mongolia autonomous region. An almost complete 16S rRNA gene sequence (1,500 bp) of strain CPCC 204708T was obtained, and comparison against available sequences in GenBank revealed highest 16S rRNA gene sequence similarity to that of Solirubrobacter phytolaccae KCTC 29190T (98.3% nucleotide identity). The 16S rRNA gene sequence (1,427 bp) from the genome of Solirubrobacter sp. URHD0082 (AUEK00000000) was also extracted and compared against the Genbank database, revealing highest 16S rRNA gene sequence to that of S. ginsenosidimutans DSM 21036T (97.4% nucleotide similarity).
The draft genome sequences of strains CPCC 204708T, S. phytolaccae KCTC 29190T, S. taibaiensis KCTC 29222T and S. ginsenosidimutans DSM 21036T, were deposited in NCBI under the accessions JAPCID000000000, JAPDDP000000000, JAPDDQ000000000, and JAPDOD000000000, respectively (Table 1). The whole genome shotgun project accession number for strain URHD0082 is AUEK00000000. The GenBank accession numbers for the 16S rRNA gene sequences of strains CPCC 204708T and URHD0082 are MH509728 and OQ674416, respectively.
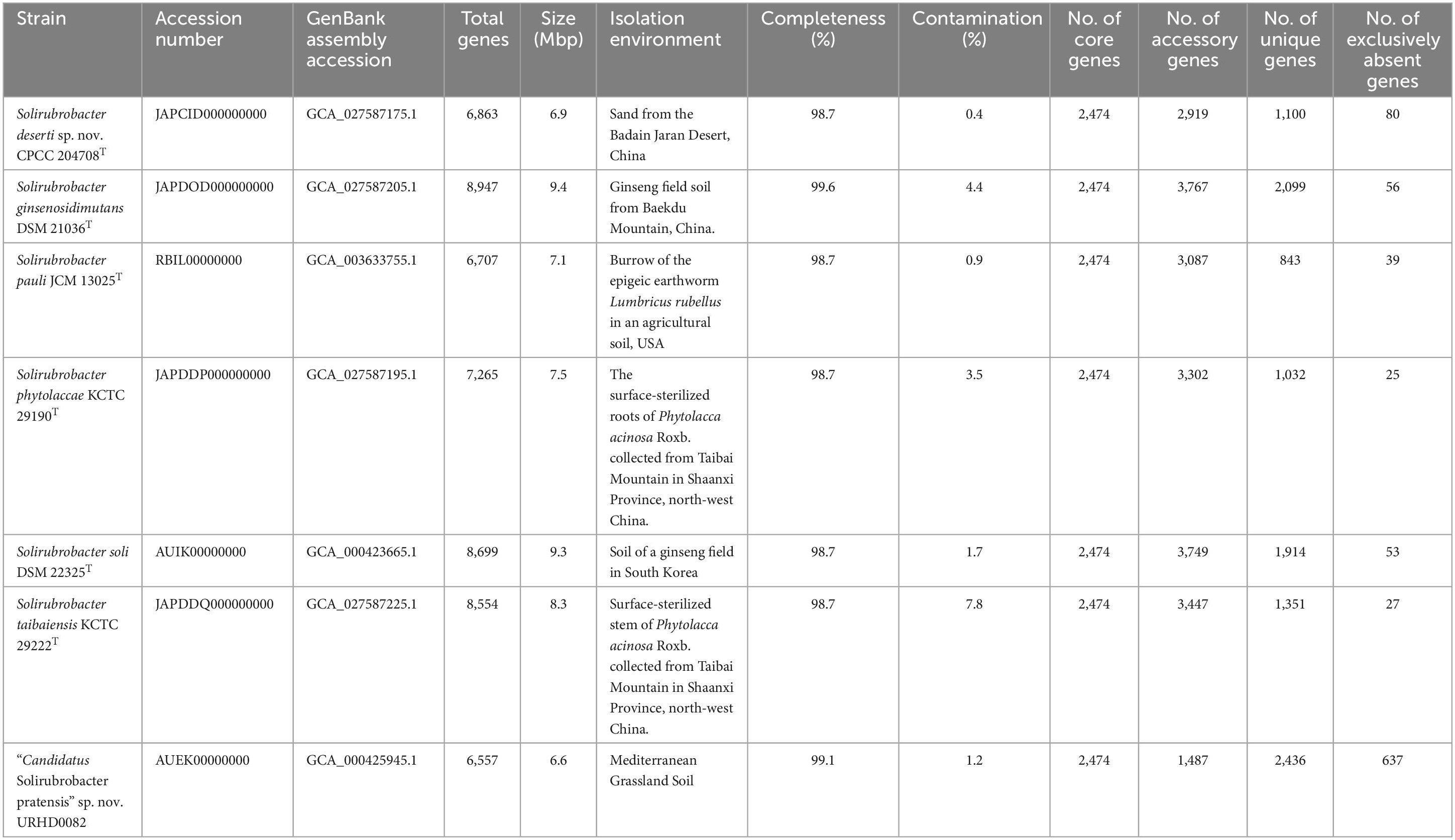
Table 1. Characteristics and distribution of core, accessory, unique, and exclusively absent genes of seven genomes included in the pan-genome of the genus Solirubrobacter.
Phylogenetic analysis of 16S rRNA gene sequences revealed that strains CPCC 204708T and URHD0082 formed a distinct group with the five Solirubrobacter species, regardless of phylogenetic reconstruction method (Supplementary Figure 2), as confirmed by the pan-matrix constructed from BPGA (Supplementary Figure 3). Thus, strains CPCC 204708T and URHD0082 were both phylogenetically affiliated to Solirubrobacter. In the core gene tree based on 120 ubiquitous single-copy maker genes (bac120 maker set) from whole genome sequences, strains CPCC 204708T and URHD0082 occupied distinct species positions in the genus Solirubrobacter (Figure 2), which was supported by the phylogenetic tree based on the 16S rRNA gene sequences (Supplementary Figure 2). The ANI values between strain CPCC 204708T, URHD0082, and the other validly described Solirubrobacter species were in the range of 77.3–84.4%, all being far lower than the threshold used for bacterial species delineation (ANI < 95%) (Kim et al., 2014). Further, the corresponding dDDH values ranged from 20.5 to 27.6% (Supplementary Table 2), which were also far below the threshold value (70%) used to identify bacterial strains of the same species (Auch et al., 2010). Based on these analyses, we proposed that the two strains identified here represent novel Solirubrobacter species, for which the epithets Solirubrobacter deserti sp. nov. and Candidatus “Solirubrobacter pratensis” sp. nov. are suggested, with strains CPCC 204708T and URHD0082T as the types, respectively.
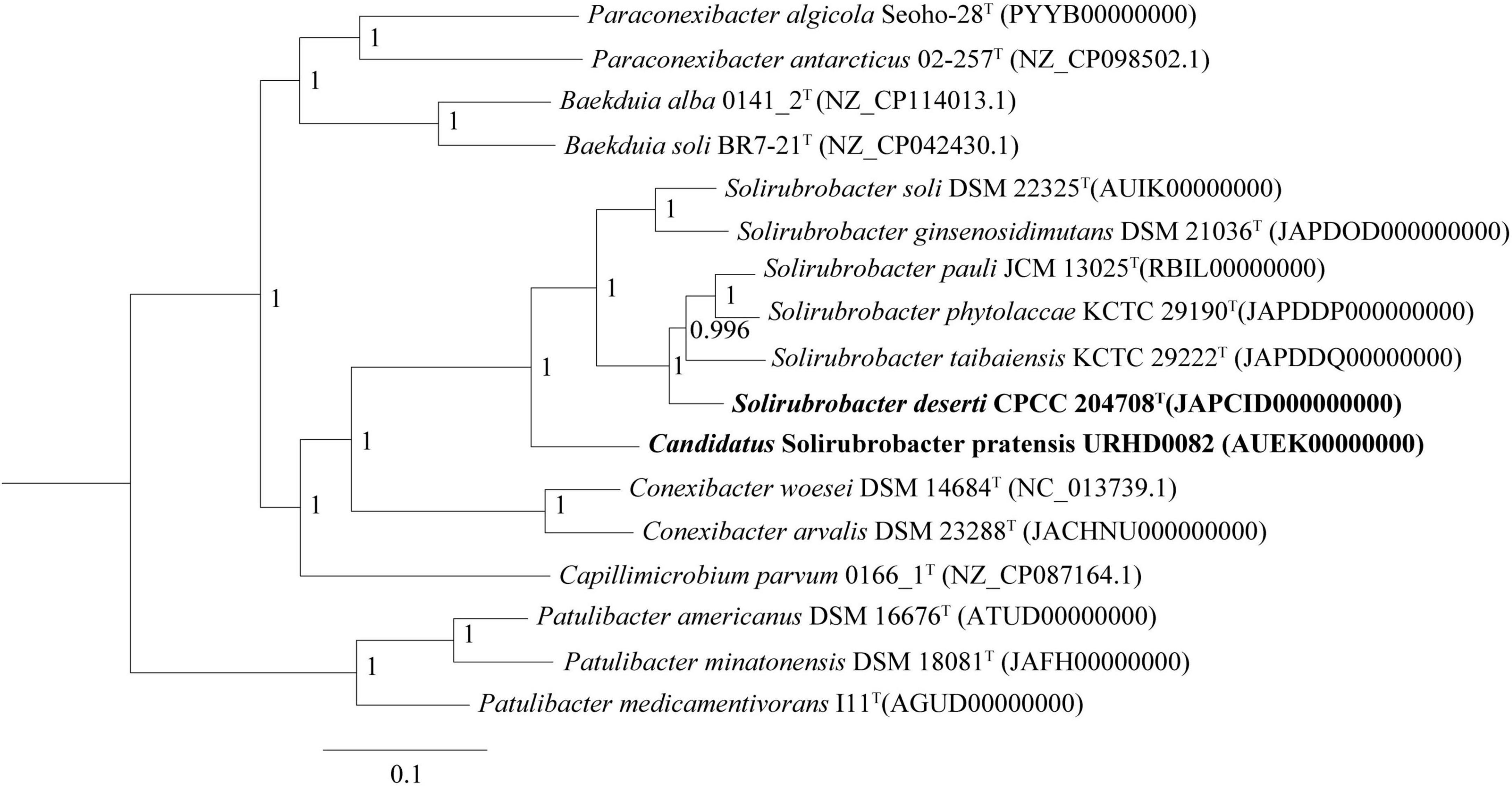
Figure 2. Core gene tree based on 120 ubiquitous single-copy maker genes (bac120 maker set) from whole genome sequences showing the relationship of strain CPCC 204708T and URHD0082 with other species in the genus Solirubrobacter and other related species in the order Solirubrobacterales. Bootstrap values (those above 50%) are shown as percentages of 1,000 replicates. Micrococcus luteus ATCC 4698T (GenBank accession no. QVMY00000000) was used as an outgroup (not shown). Bar, 0.1 nt substitution per nt.
Strain CPCC 204708T grew well on R2A, TSA and YM media, with moderate growth on ISP4 and PYG media, while no growth was observed on ISP 2, Luria-Bertani, and nutrient media. Strain CPCC 204708T growth was observed at 20–37°C, pH 5.0–8.0, and in presence of 0–10% (w/v) NaCl. Optimum growth occurred at 28–30°C, pH 7.0–8.0, and without NaCl addition. Strain CPCC 204708T colonies on R2A medium were circular, convex, and smooth, with pale pink color and approximately 1.9 mm in diameter. Diffusible pigments were not produced in the media. When grown on R2A medium, cells were aerobic, Gram-stain positive, non-motile, non-spore-forming, and rod-shaped, with sizes of 0.6–0.8 × 1.2–1.9 μm (Supplementary Figure 4). The cells were positive for catalase activity, but negative for oxidase activity. Detailed physiological and biochemical characteristics of strain CPCC 204708T are shown in Table 2 and in the species description.
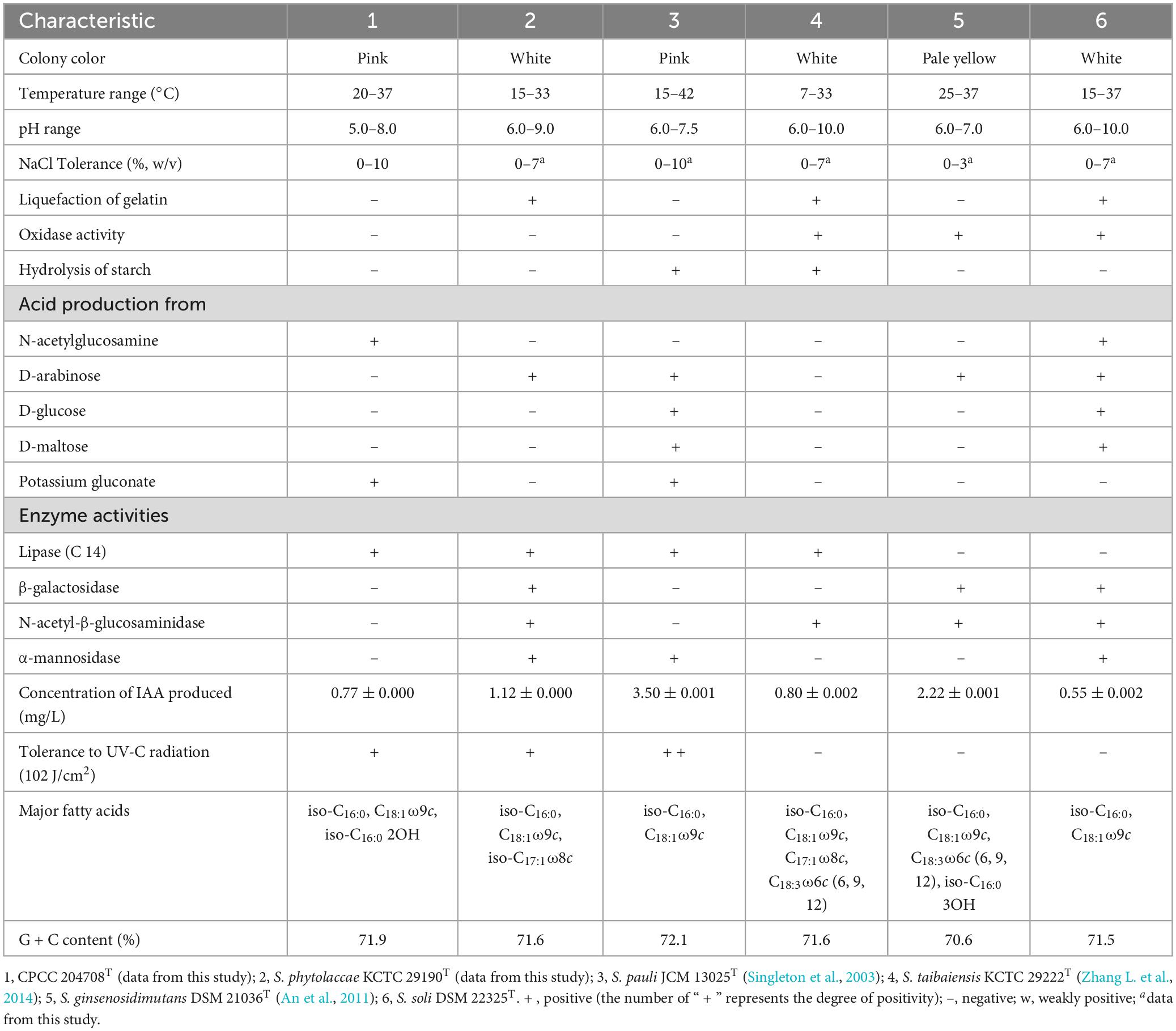
Table 2. Differentiating characteristics between strain CPCC 204708T and the type strains of other Solirubrobacter species.
Ultraviolet radiation tolerance experiments showed that strains CPCC 204708T, S. phytolaccae KCTC 29190T and S. pauli JCM 13025T could all survive when exposed to the dose of ultraviolet radiation (102 J/cm2), with survival rates following the order of S. pauli JCM 13025T, CPCC 204708T and S. phytolaccae KCTC 29190T. JCM 13025T and CPCC 204708T colony colors were both pink when grown on R2A media. Some radiation-resistant bacteria isolated from irradiated soils contain multiple pigments and are also more resistant to radiation than non-pigmented bacteria (An et al., 2011; Pulschen et al., 2015). For instance, the UV and cold tolerance of a purple violet pigment (PVP)-producing Antarctic bacterium Janthinobacterium sp. Ant5-2 was reported previously. Compared with the wild type Janthinobacterium sp. Ant5-2 PVP(+), the survival rate of mutant strain [PVP(-)] after ultraviolet irradiation (UV-B and UV-C) was significantly reduced (Mojib et al., 2013). Saxena et al. (2002) found that survival of Bt-m (an UV-resistant mutant of Bacillus thuringiensis subsp. kurstaki, producing a dark brown pigment, identified as melanin) spores and their insecticidal activity to irradiation at 254 nm and 366 nm were higher than those of the parent. Reis-Mansur et al. (2019) found that the increased survival of DNA repair-proficient E. coli grown overnight with added carotenoids (pigment extract) produced by Microbacterium sp. LEMMJ01 (isolated from Antarctic soil) revealed that part of the resistance of Microbacterium sp. LEMMJ01 against UV-B radiation seems to be connected with photoprotection by its pigments (carotenoids). Consequently, the pigment of strains JCM 13025T and CPCC 204708T may contribute greatly to their higher survival under UV radiation.
Indole-3-acetic acid is an important phytohormone that benefits plant growth and development. Many bacteria produce IAA, which when provided in an optimal concentration range, can stimulate plant root hair formation and increase the numbers and lengths of lateral roots and taproots (Davies, 1995). A positive correlation between the genus Solirubrobacter and plant growth was also observed (Franke-Whittle et al., 2015). Subsequently, the genus Solirubrobacter was detected as a dominant group in soils (Sánchez-Marañón et al., 2017), rhizosphere habitats of various crops and medicinal plants (Aguiar et al., 2020; Barajas et al., 2020; Feng et al., 2021; Lee et al., 2021), and was recognized as a kind of plant probiotic (Li et al., 2022). Several multifunctional rhizosphere soil microorganisms including Bacillus, Solirubrobacter, and Lysobacter with higher abundance in commercial organic fertilizer plus bioorganic fertilizer (CBF) were shown to promote plant growth (Franke-Whittle et al., 2015) by producing hormones such as IAA (indole-3-acetic acid), gibberellin, and cytokinin (Fabra et al., 2010; Mhatre et al., 2018). Our phenotypic experiments demonstrated that six strains of the genus Solirubrobacter could produce IAA. IAA was detected in the fermentation broths of strains CPCC 204708T, S. phytolaccae KCTC 29190T, S. taibaiensis KCTC 29222T, S. pauli JCM 13025T, S. ginsenosidimutans DSM 21036T, and S. soli DSM 22325T at concentrations of 0.77 ± 0.000, 1.12 ± 0.001, 0.80 ± 0.002, 3.50 ± 0.001, 2.22 ± 0.001, and 0.55 ± 0.002 mg/L, respectively (Supplementary Figure 1). Production of phytohormones (such as IAA) of plant endophytes (Crozier et al., 1988) stimulate growth and/or ameliorate the plant under harsh stressful conditions (Cohen et al., 2009; Piccoli et al., 2011). Given that all reported strains of the genus Solirubrobacter so far have been isolated from soils or the ecosystems that were vegetated or associated with crops, especially medicinal plants, we infer that the production of small amounts of IAA could represent an important mechanism of plant-microbe interaction for this genus (Duca et al., 2014). In addition, at the genomic level, we found that the IAA-production pathway was encoded by the seven Solirubrobacter strains. Specifically, the genes for aldehyde dehydrogenases (EC 1.2.1.3; aldH) and amidases (EC 3.5.1.4; amiE) were both present within the core genome of Solirubrobacter strains. Combining phenotypic and genotypic characteristics, the complete IAA production pathway (Figure 3) within the broader tryptophan metabolism pathways (Supplementary Figure 5) were identified for the strains based on annotation against the KEGG database.
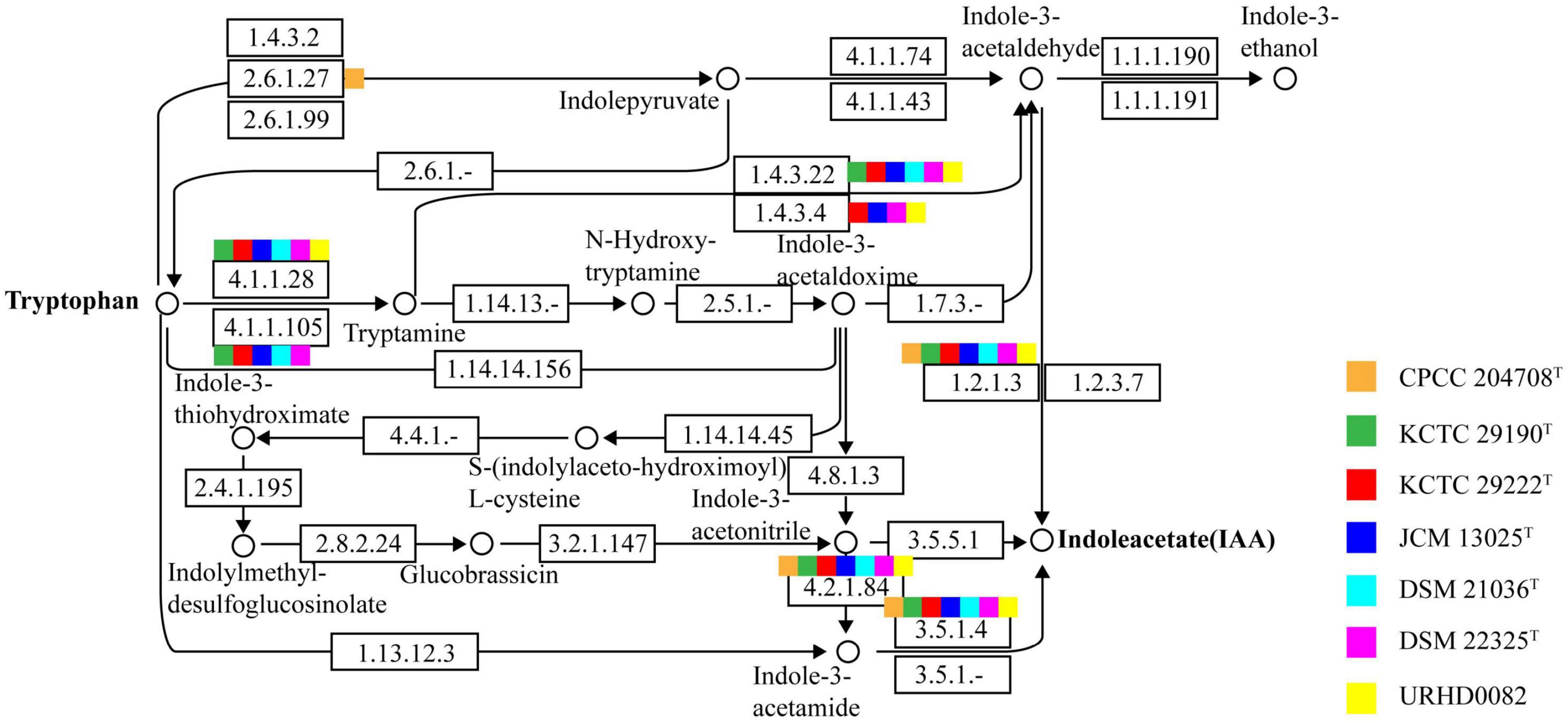
Figure 3. The indole-3-acetic acid (IAA) production pathway of Solirubrobacter species. Squares in orange, green, red, blue, light blue, pink, and yellow correspond to strains CPCC 204708T, KCTC 29190T, KCTC 29222T, JCM 13025T, DSM 21036T, DSM 22325T, and URHD0082, respectively.
Strain CPCC 204708T showed chemotaxonomic features consistent with the genus Solirubrobacter. In the whole cell hydrolysates of strain CPCC 204708T, meso-diaminopimelic acid was detected as the signature amino acid, and galactose, xylose, rhamnose, and ribose were the components of the sugar profile. Diphosphatidylglycerol (DPG), phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylinositol mannosides (PIM), an unidentified aminophospholipid (APL), and an unidentified phospholipid (PL) were the primary components of the polar lipid profile (Supplementary Figure 6). The predominant menaquinone of cells was MK-7(H4), which is consistent with other species of the genus Solirubrobacter (Kim et al., 2007; An et al., 2011; Wei et al., 2014; Zhang L. et al., 2014). The major cellular fatty acids (>10%) were iso-C16:0, C18:1ω9c, and iso-C16:0 2-OH, and the detailed composition is provided in the species description and in Supplementary Table 3. It was obvious that strain CPCC 204708T shared the major fatty acids of iso-C16:0 and C18:1ω9c with other validly described Solirubrobacter species, while the detailed fatty acids profiles could differentiate them from each other (Table 2; Supplementary Table 3). Overall, the chemotaxonomic analyses supported the classification of strain CPCC 204708T as a new member of the genus Solirubrobacter, consistent with the 16S rRNA gene sequence and phylogenetic analyses.
The genomic DNA G + C content of strain CPCC 204708T was 71.9% based on its draft genome sequence. Further detailed genomic characteristics for the seven strains were summarized in Table 1 and Supplementary Appendix 1.
Putative genes encoding catalase (katG) and superoxide dismutase (sodA) were identified in all genomes that likely help mitigate oxidative stress. In addition, the UvrABC repair system (Truglio et al., 2006) and other DNA recombination and repair-related genes were identified. As well, genes were identified that were associated with polyamine transport, osmoprotectant capacity, carotenoid biosynthesis, IAA production, iron-siderophore transport system, carbon monoxide dehydrogenase, carbon storage regulation, nitrogen assimilation, and phosphate-transport and solubilization (Figure 4).
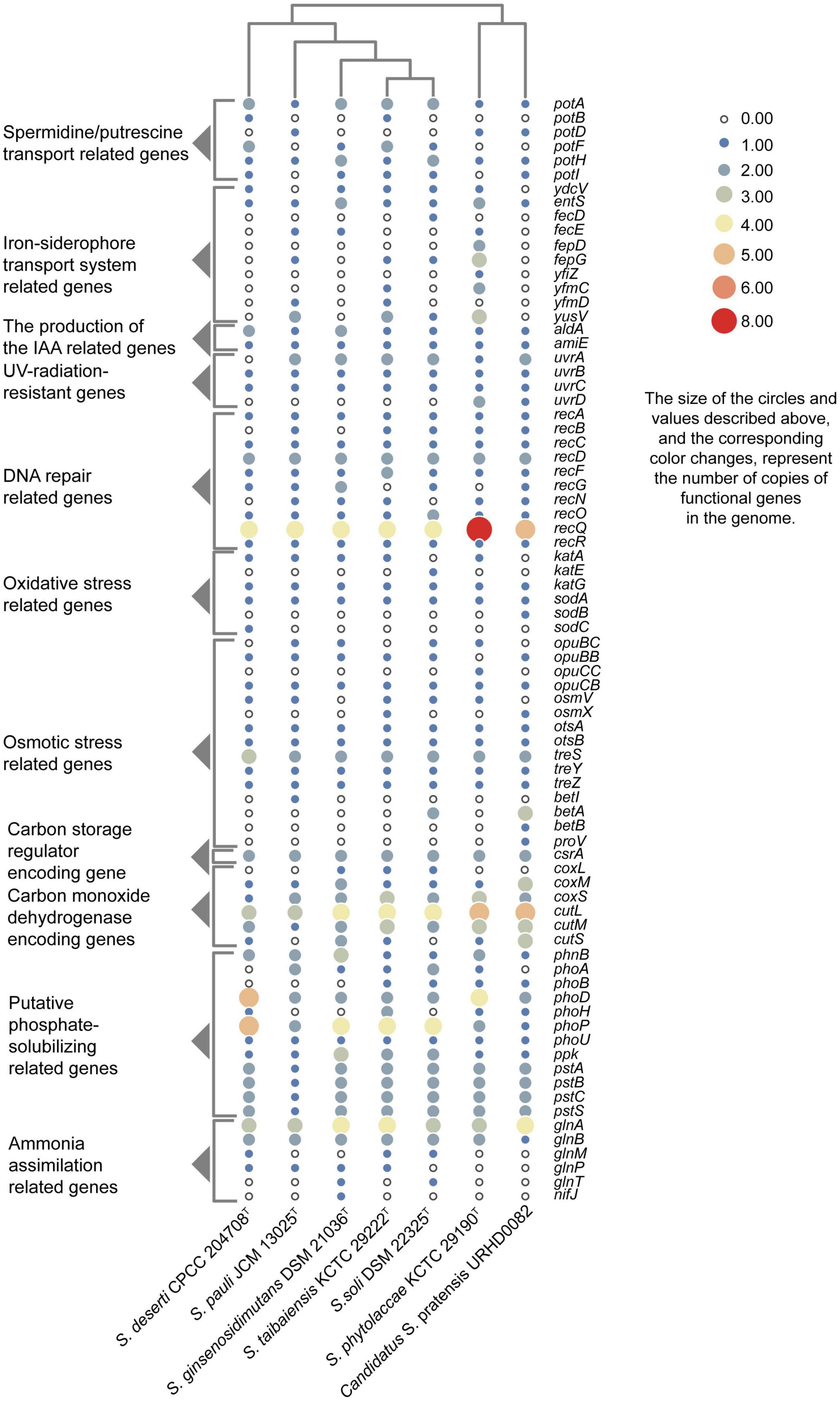
Figure 4. Heatmap showing the copy numbers of functional genes predicted in the genomes of seven Solirubrobacter species. Functional genes were identified based on comparison to the KEGG database.
Besides lack of precipitation in such harsh ecosystems, heterotrophic microorganisms have to challenge extreme starvation for carbon and other energy substrates. Diverseand viable microbial communities are present in the sandy soils of most deserts. To explore the potential of the desert-derived strains of this study to assimilate carbon and other energy substrates, genes associated with these capacities were annotated and identified. Carbon monoxide dehydrogenase encoding genes (coxL, coxM, coxS, cutL, cutM, and cutS) and a carbon storage regulation coding gene (csrA) were retrieved from the genomes (Figure 4). In addition, several genes related to acquisition and assimilation of phosphorus were identified, including phnB, phoA, phoB, phoD, phoH, phoP, phoU, ppk, pstA, pstB, pstC, and pstS (Figure 4). Consistently, phenotypic experiments indicated that strain CPCC 204708T encoded acid phosphatase and alkaline phosphatase. Nitrogen assimilation genes were also identified in the genomes of the seven strains (Figure 4). Specifically, genes encoding glutamine synthetase (glnA), the nitrogen regulatory protein P-II 1 (glnB), a probable glutamine ABC transporter permease protein (glnM and glnP), and the probable sodium/glutamine symporter GlnT (glnT) were identified. Glutamine synthetase encoded by glnA is a key multitasking protein involved in ammonium assimilation and in the regulation of genes involved in nitrogen metabolism (Schumacher et al., 2015). GlnA may bethat glnA is involved in ammonia assimilation under ammonia-starvation conditions, while P-II indirectly controls the transcription of glnA. The proteins encoded by glnM and glnP form components of the ABC transporter complex GlnHMPQ involved in glutamine transport (Yoshida et al., 2003). Moreover, plants have evolved sophisticated mechanisms to mitigate stress from fluctuating nitrate levels and can recruit microorganisms to improve nitrogen uptake (Chai et al., 2022). The bacteria associated with nitrogen transformation, such as Solirubrobacter spp., etc., were highly abundant; these bacteria may possess the ability to increase nitrogen availability in the crude oil-contaminated soil (Gao et al., 2022). Nitrogen fixation related gene nifJ was only retrieved from the genome of the strain S. ginsenosidimutans DSM 21036T. Thus, these Solirubrobacter strains may potentially promote nitrogen absorption by their symbiotics in the desert environments, thereby potentially improving their growth in these niches. Consequently, these genomic observations highlight the potential important contributions of the proteins toward adaptation to carbon, nitrogen, and energy starvation in Solirubrobacter.
ATP-binding cassette (ABC) transporters are one of the largest known protein families, and are ubiquitous among bacteria. The transporters couple ATP hydrolysis to active transport of diverse substrates including ions, sugars, lipids, sterols, peptides, proteins, and drugs. To explore the transport potential of the seven strains, we analyzed the related genome sequences with KEGG database and blasted with TCDB12 (Supplementary Appendix 2), and genes encoding ABC transporters were identified (Supplementary Figure 7). Annotation against the KEGG database revealed the presence of spermidine/putrescine transport related genes (potA, potB, potC, and potD) in the seven Solirubrobacter genomes (Figure 4). Higher polyamine levels in plants can minimize harmful effects resultant from biotic and abiotic stresses (salt, drought, UV, temperature, heavy metals, etc.) (Takahashi and Kakehi, 2010; Hussain et al., 2011; Shi and Chan, 2014). Inoculation with beneficial rhizobacterium Pseudomonas putida GAP-P45 led to increased levels of the expression of most polyamine biosynthetic genes and cellular polyamine levels in Arabidopsis thaliana, resulting in resistance to water-stressed conditions (Sen et al., 2018). Rhizobacteria also could modulate the redox state of salinity-affected plants by enhancing polyamines and antioxidants, which leads to increased photosynthetic efficiency (Radhakrishnan and Baek, 2017). Given that all reported strains of the genus Solirubrobacter so far were isolated from soils or the ecosystems that were vegetated or associated with crops, especially medicinal plants, and that polyamine transport-related genes were retrieved from the genomes of all seven strains of the genus, we concluded that Solirubrobacter spp. might have the potential or relate to the growth promotion of plants through the transport of polyamines. Other complete transport-related genes were identified in the seven genomes of the genus Solirubrobacter, including those related to transport of osmoprotectants, raffinose/stachyose/melibiose (msmE, msmF, msmG and msmK), nucleosides (bmpA, nupB, nupC, and nupA), D-xylose (xylF, xylH, and xylG), erythritol (eryG, eryF, and eryE), phosphate, branched-chain amino acids (livK, livH, livG, livM, and livF), D-methionine (metQ, metI, and metN), iron-siderophores, and lipo-oligosaccharides (nodJ and nodI).
Salt tolerance assays revealed that strains CPCC 204708T, S. phytolaccae KCTC 29190T, S. taibaiensis KCTC 29222T, S. pauli JCM 13025T, S. ginsenosidimutans DSM 21036T, and S. soli DSM 22325T grew in the presence of 0–10, 0–7, 0–7, 0–10, 0–3, and 0–7% NaCl (w/v), respectively. Thus, most Solirubrobacter species were able to tolerate certain levels of osmotic pressure. Genomic analysis revealed the presence of several genes related to osmoprotectant capacity, including genes encoding the choline transport system permease protein OpuBB (opuBB), choline ABC transporter substrate-binding lipoprotein OpuBC (opuBC), glycine betaine/carnitine/choline transport system permease protein OpuCB (opuCB), glycine betaine/carnitine/choline-binding protein OpuCC (opuCC), osmoprotectant import ATP-binding protein OsmV (osmV), osmoprotectant-binding protein OsmX (osmX), trehalose-6-phosphate synthase (otsA), trehalose-phosphate phosphatase (otsB), malto-oligosyltrehalose trehalohydrolase (treZ), trehalose synthase/amylase TreS (treS), maltooligosyl trehalose synthase (treY), HTH-type transcriptional regulator BetI (betI), oxygen-dependent choline dehydrogenase (betA), betaine aldehyde dehydrogenase (betB), and glycine betaine/proline betaine transport system ATP-binding protein ProV (proV) (Figure 4). Notably, many of these proteins are involved in trehalose uptake and associated biosynthesis pathways. Trehalose is a major compatible solute involved in osmotic stress responses of cells, cellular adaptation, and survival under heat and desiccation stress (Reina-Bueno et al., 2012). Lee et al. (2021) found that in high-salinity rhizosphere soil habitats planted with tomato, the abundance of some actinobacteria (such as Solirubrobacter) increased and the community structure tended to be stable, indicating that strains of these groups could tolerate high osmotic pressure and potentially help plants tolerate high salt environment through complex plant-microbial interaction.
A total of 50,768 protein-coding genes (Table 1) were identified among the seven Solirubrobacter genomes that comprised 19,800 homologous families based on cluster analysis. Homologous gene family conservation values (HGFCVs) were evaluated among the homolog clusters (Supplementary Figure 8A). A total of 2,474 core genes were shared by the seven strains (HGFCV = 7), accounting for ∼12.5% of the total homologous gene families, while accessory genes (HGFCVs = 2–6) accounted for ∼33.1% of the gene families (6,551 genes) in the genus Solirubrobacter. In addition, unique genes (HGFCV = 1) comprised ∼54.4% of the total (10,775 genes).
The functional relationship between pan-genome size (fpan) and the number of genomes (n) was obtained by evaluating the following equation:
In addition, the functional relationship between the number of core genes (fcore) and the number of genomes (n) was obtained by evaluating the following equation:
Please refer to the reference (Chaudhari et al., 2016) for detailed derivation. With increasing numbers of sequenced genomes, the pan-genome size increased, rather than plateauing (Supplementary Figure 8B), suggesting that pan-genome size may continue to enlarge if the number of genomes of this genus continue to increase. Thus, the pan-genome of Solirubrobacter may be of an open type. Open pan-genomes are associated with species of the genus inhabiting multiple environments and having multiple ways of exchanging genetic material (Medini et al., 2005). Therefore, it is reasonable to infer that many unknown species of the genus Solirubrobacter inhabited in other biotopes have yet to be discovered.
Of the 19,800 genes (clusters), BPGA mapped 6,216 (31.4%) to KEGG database pathways, including for 2,087 core genes (33.6%), 2,017 accessory genes (32.4%), and 2,112 unique genes (34.0%). KEGG pathway results related to eukaryotes (e.g., Human Diseases and Organismal Systems) were removed in order to construct a metabolic reconstruction of the Solirubrobacter pan-genome. Many core genes (1,997) were involved in carbohydrate metabolism (17.0%), other carbon metabolism pathways (biosynthesis of amino acids, 5.8%; carbon metabolism, 4.9%; 2-oxocarboxylic acid metabolism, 1.6%; fatty acid metabolism, 1.3%; and degradation of aromatic compounds, 0.4%) (14.0% total), amino acid metabolism (13.7%), energy metabolism (7.6%), metabolism of cofactors and vitamins (5.8%), nucleotide metabolism (4.9%), lipid metabolism (4.4%), replication and repair (4.3%), signal transduction (4.1%), xenobiotic biodegradation and metabolism (4.0%), translation (3.8%), membrane transport (2.8%), metabolism of other amino acids (2.7%), metabolism of terpenoids and polyketides (2.5%), biosynthesis of other secondary metabolites (1.9%), cell motility (1.9%), folding, sorting, and degradation (1.8%) and glycan biosynthesis and metabolism (1.3%). Accessory and unique genes were enriched in pathways related to carbohydrate metabolism, amino acid metabolism, and the metabolism of other substrates.
Among the accessory genes (1,886), most encoded proteins related to the pathways of carbohydrate metabolism (15.0%), amino acid metabolism (14.3%), other specific carbon metabolism pathways (carbon metabolism, 3.8%; biosynthesis of amino acids, 2.6%; fatty acid metabolism, 1.7%; degradation of aromatic compounds, 0.9% and 2-oxocarboxylic acid metabolism, 0.6%) (9.8%), signal transduction (8.8%), membrane transport (7.4%), xenobiotic biodegradation and metabolism (7.3%), lipid metabolism (6.3%), energy metabolism (6.0%), metabolism of cofactors and vitamins (5.6%), metabolism of other amino acids (3.2%), nucleotide metabolism (2.9%), biosynthesis of other secondary metabolites (2.6%), metabolism of terpenoids and polyketides (2.3%), cell motility (2.2%), translation (1.3%), transport and catabolism (1.1%), and glycan biosynthesis and metabolism (1.0%).
Among the 1,876 unique genes, their encoded proteins were related to pathways associated with carbohydrate metabolism (17.0%), amino acid metabolism (11.8%), other specific carbon compound metabolism (carbon metabolism, 4.3%; biosynthesis of amino acids, 2.8%; fatty acid metabolism, 2.3%; 2-oxocarboxylic acid metabolism, 0.7% and degradation of aromatic compounds, 0.6%) (10.7%), signal transduction (8.8%), membrane transport (7.1%), lipid metabolism (6.4%), energy metabolism (5.6%), xenobiotic biodegradation and metabolism (5.6%), metabolism of cofactors and vitamins (4.5%), biosynthesis of other secondary metabolites (2.9%), nucleotide metabolism (2.6%), metabolism of terpenoids and polyketides (2.6%), metabolism of other amino acids (2.1%), folding, sorting, and degradation (1.9%), glycan biosynthesis and metabolism (1.8%), transport and catabolism (1.5%), signaling molecules and interactions (1.4%), cellular community (1.3%), cellular motility (1.2%), translation (1.2%) and replication and repair (1.1%) (Figure 5).
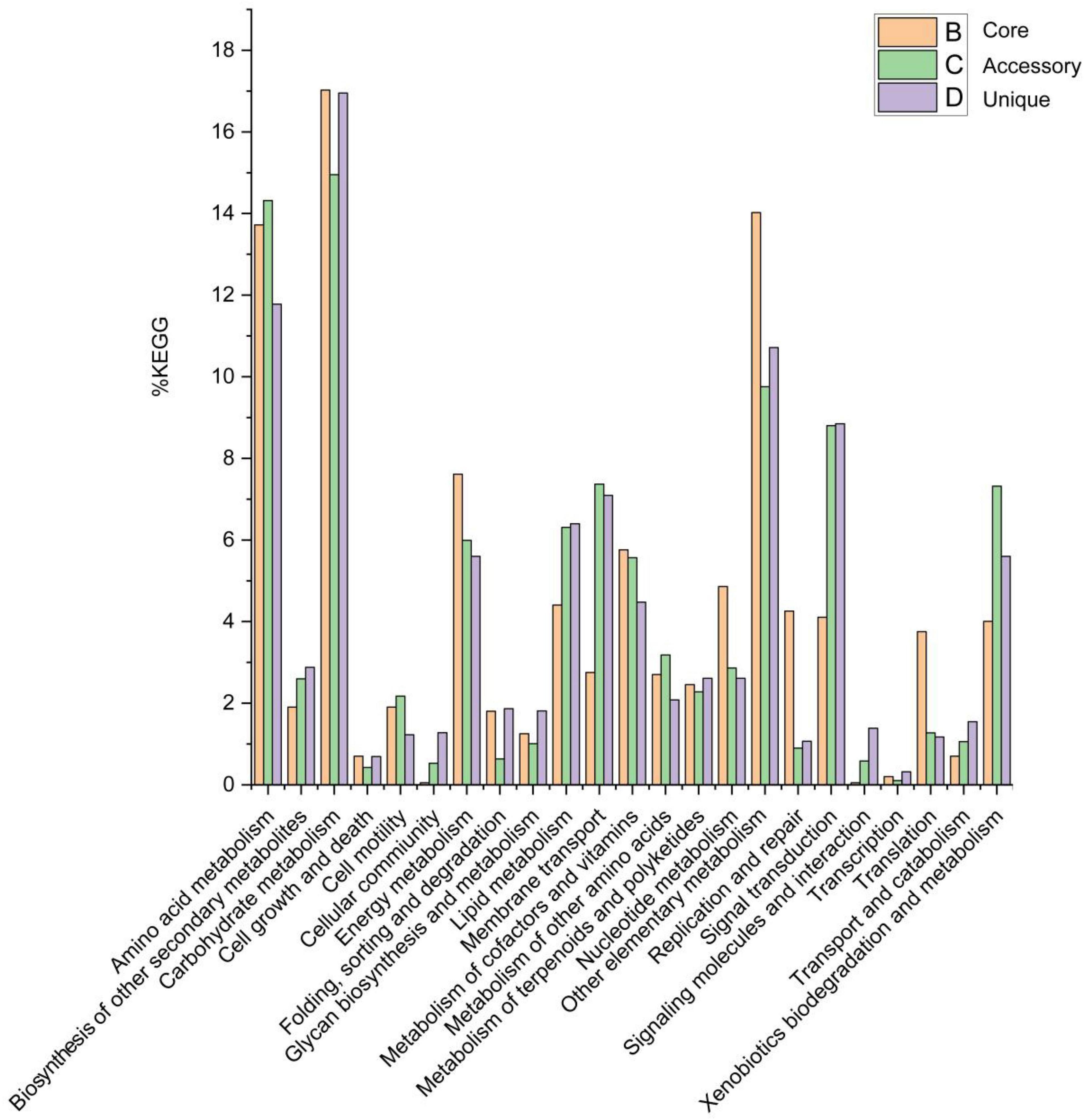
Figure 5. Abundances of metabolic pathways associated with core, accessory, and unique genes within the Solirubrobacter pan-genomic analyses. Metabolic pathways were identified from the KEGG database.
The pan-genome of the genus Solirubrobacter is characterized by a high proportion of carbohydrate metabolism, amino acid metabolism, and energy metabolism, these genomic features suggest that these strains have the potential to assimilate more sources of carbon and nitrogen to cope with extreme starvation of carbon and other energy substrates. These genomic-level characteristics also suggest that members of the genus Solirubrobacter might play an important role in soil organic matter assimilation and biogeochemical cycling.
Compared with the core genomes of the species Modestobacter deserti (Jiang et al., 2021) and the genus Geminicoccus (Jiang et al., 2022) by BPGA in our previous studies, we found that spermidine/putrescine transport related genes (potA, potB, potC and potD) were retrieved only in the core genome of seven strains of the genus Solirubrobacter, with no spermidine/putrescine transport related genes in the genomes of Modestobacter deserti and the genus Geminicoccus. While spermidine/putrescine transport related genes were present in the genomes of Conexibacter spp., a group of actinobacteria mostly isolated from the vegetated biotopes, a small number of Conexibacter spp. isolated from desert and aquatic habitats had only potA genes, or were missing any pot genes (data unpublished). Integrating the information from all validly described strains of the genus Solirubrobacter derived from the vegetated biotopes or the ecosystems associated with crops, we supposed that spermidine/putrescine transporter encoding genes might correlate to vegetation (Leontidou et al., 2020). Leontidou et al. (2020) experimentally verified that most selected Plant Growth Promoting Rhizobacteria (PGPR) harbored the genes responsible for polyamine biosynthesis. Accordingly, we supposed that the polyamine related pathway probably act as an additional PGPR-related mechanism involved in plant growth promotion, which need to be further explored. Polyamine (e.g., spermidine) is essential for eukaryotic cell viability and is correlated with lateral root development, pathogen resistance and alleviation of oxidative, osmotic and acidic stresses (Xie S. S. et al., 2014).
Genomes are subject to damage by chemical and physical agents in environments (e.g., UV and ionizing radiation, fungal or bacterial toxins, and chemical mutagens) and by free radicals endogenously generated during cellular metabolism (Tuteja et al., 2001). A variety of different DNA repair pathways help mitigate DNA damage and enable cells to withstand the high solar radiation encountered in desert habitats. UV radiation analysis revealed that strain CPCC 204708T, S. phytolaccae KCTC 29190T, and S. pauli JCM 13025T survived exposure to 254 nm ultraviolet radiation. Within the core genome of the seven strains analyzed here, genes were identified associated with the UvrABC repair system (Truglio et al., 2006; Supplementary Figure 9) (uvrA, uvrB, uvrC, and uvrD) and other DNA recombination and repair pathways (recA, recB, recC, recD, recF, recG, recO, and recR) (Taylor and Smith, 1999; Supplementary Figure 10). The DNA repair pathways related genes uvr and rec were also retrieved in the core-genomes of the genus Geminicoccus, the genus Herbiconiux, the genus Conexibacter and the species Modestobacter deserti.
β-Glucosidase activity was also observed for strain DSM 21036T and this activity was demonstrated as responsible for the gradual conversion of ginsenoside Rb1 to the compound F2 (An et al., 2011). β-glucosidase activity was also observed for strain CPCC 204708T and the five other Solirubrobacter strains. Consistently, beta-glucosidase encoding genes (bgl) were identified in the core genome of the Solirubrobacter strains. The bgl gene (encoding β-glucosidase) was retrieved in the core-genomes of the genus Geminicoccus, the genus Herbiconiux, and the species Modestobacter deserti, but not in the core-genome of the genus Conexibacter.
The secondary metabolism of actinobacteria, especially actinobacteria from extreme habitats, is a rich source of novel bioactive compounds with potential medicinal value. In order to identify new drug candidates, microbiologists are increasingly combining multi-omics techniques to predict the potential for secondary metabolite synthesis by sequencing the genomes of various microorganisms. Here, we identified the secondary metabolite biosynthetic gene clusters (BGCs) of genus Solirubrobacter by antiSMASH.
The annotation of secondary metabolite biosynthesis gene clusters in the seven Solirubrobacter genomes revealed the presence within each genome of eight to thirteen secondary metabolite gene clusters, which exhibited low similarities to previously described secondary metabolite biosynthetic gene clusters. Specifically, the gene clusters exhibited 5–40% nucleotide similarities to known secondary metabolite biosynthetic gene clusters including those for accramycin A, microansamycin, calcium-dependent lipopeptide, lomofungin, tiancimycin, schizokinen, lankacidin C, linfuranone B/C, kitasetaline, fulvuthiacene A/B, macrotermycins. In addition, other unidentified secondary metabolite clusters were identified that were attributable to those encoding NAPS-independent-siderophores, lassopeptides, LAP thiopeptides, terpenes, NAPAAs, redox-cofactors, RiPP-like compounds, RRE-containing compounds, ranthipeptides, indole, and lanthipeptide-class-iv types (Supplementary Table 4). The analysis with antiSMASH also revealed the presence of a microansamycin gene cluster in strain CPCC 204708T. Pentaketide ansamycins have rarely been reported and include compounds like the antioxidant Q-1047, lipoxygenase inhibitor tetrapetalones (Komoda et al., 2004), radical scavenger ansaetherones (Komoda et al., 2008), cebulactams (Pimentel-Elardo et al., 2009), and the macrodilactam juanlimycins (Zhang J. et al., 2014). The novel pentaketide ansamycin, Microansamycin D, was recently identified and its antioxidant activity was confirmed (Wang et al., 2018). Accramycin A is a new naphthacene-type aromatic natural product and was identified in the antiSMASH analysis of strain CPCC 204708T. Accramycin A was first discovered from the metabolites of Streptomyces sp. MA37 (Maglangit et al., 2019). The antibacterial activities of accramycin A were also preliminarily evaluated against Group B Streptococcus, revealing a minimum inhibitory concentration (MIC) of 27 μg/mL, providing the first evidence of naphthacene-type aromatic polyketide bioactivity. The Lomofungin gene cluster was also identified in the antiSMASH analysis of strain S. phytolaccae KCTC 29190T. This antibiotic exhibits antibacterial activity against fungi, yeast, and bacteria (Klo et al., 1973). Further, macrotermycins A and C have exhibited antimicrobial activity against human pathogenic Staphylococcus aureus in addition to selective antifungal activity against a fungal parasite from termite fungal gardens (Beemelmanns et al., 2017).
The pangenomes of the genera Geminicoccus, Herbiconiux and Conexibacter, as well as the species Modestobacter deserti were used as the control genomes of the genus Solirubrobacter. Upon comparison, it was discovered that secondary metabolism cluster profiles in the pangenome of the genus indeed showed specific. The secondary metabolite gene clusters responsible for accramycin A, microansamycin, calcium-dependent lipopeptide, lomofungin, tiancimycin, schizokinen, lankacidin C, linfuranone B/C, kitasetaline, fulvuthiacene A/B, and macrotermycins, were only retrieved from the genomes of genus Solirubrobacter, but not from those of the genus Herbiconiux, Geminicoccus, Conexibacter (data unpublished), and the species Modestobacter deserti.
In this study, the distribution of the genus Solirubrobacter was evaluated across numerous environments, revealing their enrichment in soils of areas with high UV radiation (e.g., desert soils). In addition, the novel strain Solirubrobacter deserti sp. nov. CPCC 204708T was isolated, identified, and characterized, in addition to subsequent characterization of the genetic basis of Solirubrobacter adaptations to harsh environments and their potential mediation of plant-microbe interactions. Further, strain URHD0082 was identified as Candidatus “Solirubrobacter pratensis” based on genome information. Genome-scale analysis of strain CPCC 204708T revealed the molecular basis for their adaptations to desert environments via mitigation of stress from UV radiation, carbon starvation, desiccation, and osmotic stress. In the absence of macrophytic phototrophs, such as in desert soils, such microorganism could potentially serve as significant contributors to both primary productivity and biogeochemical activities, thereby assuming the role of pioneering organisms. Global analysis of Solirubrobacter genomes and their environmental distributions suggest they are abundant in ecosystems associated with plants, where they may promote plant health.
Solirubrobacter deserti (de.ser’ti. L. gen. n. deserti of a desert).
Cells are aerobic, Gram-stain positive, non-motile, non-spore-forming, and rod-shaped, with sizes of 0.6–0.8 × 1.2–1.9 μm. Diffusible pigments are not produced on any media. Colonies on R2A medium are circular, convex, smooth, and entire, with pale pink coloration and diameters of approximately 1.9 mm. Growth occurs at 20–37°C (optimum: 28–30°C) and at pH 5.0–8.0 (optimum: 7.0–8.0), and up to 10% (w/v) NaCl. Cells are catalase-positive and oxidase-negative, but negative for H2S and indole production. Cells cannot hydrolyze cellulose, starch, Tween 40, and gelatin. API ZYM strip analysis indicated that cells are positive for enzymatic activities including acid phosphatase, alkaline phosphatase, cystine arylamidase, esterase (C4), esterase lipase (C8), N-acetyl-β-glucosaminidase, β-glucosidase, leucine arylamidase, lipase (C 14), trypsin, and valine arylamidase. Acetoacetic acid, D-cellobiose, dextrin, D-fructose, D-galactose, D-galacturonic acid, D-galacturonic acid, D-gluconic acid, D-glucose-6-PO4, D-glucuronic acid, D-maltose, D-mannitol, D-mannose, D-melibiose, D-saccharic acid, D-trehalose, D-turanose, gelatin, glucuronamide, glycerol, L-aspartic acid, L-fucose, L-histidine, L-rhamnose, mucic acid, N-Acetyl-D-glucosamine, pectin, quinic acid, sucrose, α-D-glucose, and α-keto-glutaric acid can be used as sole carbon sources. Alanine, glycine, glutamate, asparagine, and meso-diaminopimelic acid were identified in the whole cell hydrolysates. The whole cell sugar profiles contained galactose, xylose, rhamnose and ribose. Polar lipids comprise diphosphatidylglycerol, phosphatidylglycerol, an unidentified aminophospholipid, and an unidentified phospholipid. The predominant menaquinone is MK-7(H4). The cellular fatty acids profile contains iso-C16:0, C18:1ω9c, and iso-C16:0 2-OH as the major (>10%) components, with moderate (3–9%) amounts of iso-C16:1 H, C17:1 ω8c and 10-methyl C17:0. The G + C content of the genomic DNA is 71.9%. The type strain CPCC 204708T (= DSM 105495 T = NBRC 112942T) was isolated from sandy soil collected from the Badain Jaran Desert in the Inner Mongolia autonomous region.
Candidatus “Solirubrobacter pratensis” (pra.ten’sis. L. fem. adj. pratensis growing in a meadow, referring to the isolation of the strain from grassland).
URHD0082 is temporarily proposed as the type genome for the species. The strain URHD0082 was isolated from the Mediterranean grassland soil. The accession number of the genome of the isolate URHD0082 is available in the DDBJ/ENA/Genbank database under accession AUEK00000000 and the Genbank accession number for the 16S rRNA gene sequence (extracted from the genome) is OQ674416. The genome of URHD0082 has the following characteristics: a draft genome of 6,640,086 bp, assembled from 28 qualified scaffolds, with total 6,557 genes, including 6,470 protein-coding genes and 87 RNA genes (consisting of 3 rRNA genes, 76 tRNA genes and 8 other RNA genes). The G + C content in the genomic DNA of URHD0082 is 72.2%.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The manuscript presents research on animals that do not require ethical approval for their study.
Z-MJ: Conceptualization, Data curation, Formal analysis, Investigation, Validation, Visualization, Writing—original draft, Writing—review and editing. TM: Data curation, Investigation, Validation, Writing—original draft. YS: Formal analysis, Investigation, Validation, Writing—review and editing. JS: Resources, Validation, Investigation, Writing—review and editing. L-YY: Resources, Validation, Writing—review and editing. Y-QZ: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Validation, Writing—original draft, Writing—review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS, 2021-I2M-1-055), the National Natural Science Foundation of China (32170021; 31670010), the Beijing Natural Science Foundation (5212018), Key Project at central government level-the ability establishment of sustainable use for valuable Chinese Medicine Resources (2060302), and the National Infrastructure of Microbial Resources (NIMR-2022-3).
We sincerely thank Institute of Microbiology, Chinese Academy of Sciences, for the kind assistance in the cells morphology observation using transmission electron microscopy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1267771/full#supplementary-material
Aguiar, L. M., Souza, M. F., de Laia, M. L., de Oliveira Melo, J., da Costa, M. R., Gonçalves, J. F., et al. (2020). Metagenomic analysis reveals mechanisms of atrazine biodegradation promoted by tree species. Environ. Pollut. 267:115636. doi: 10.1016/j.envpol.2020.115636
An, D. S., Wang, L., Kim, M. S., Bae, H. M., Lee, S. T., and Im, W. T. (2011). Solirubrobacter ginsenosidimutans sp. nov., isolated from soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 61, 2606–2609. doi: 10.1099/ijs.0.028431-0
Auch, A. F., von Jan, M., Klenk, H. P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Barajas, H. R., Martínez-Sánchez, S., Romero, M. F., Álvarez, C. H., Servín-González, L., Peimbert, M., et al. (2020). Testing the two-step model of plant root microbiome acquisition under multiple plant species and soil sources. Front. Microbiol. 11:542742. doi: 10.3389/fmicb.2020.542742
Beemelmanns, C., Ramadhar, T. R., Kim, K. H., Klassen, J. L., Cao, S., Wyche, T. P., et al. (2017). Macrotermycins A-D, glycosylated macrolactams from a termite-associated Amycolatopsis sp. M39. Org. Lett. 19, 1000–1003. doi: 10.1021/acs.orglett.6b03831
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M. H., et al. (2021). Antismash 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/nar/gkab335
Bric, J. M., Bostock, R. M., and Silverstone, S. E. (1991). Rapid in situ assay for indoleacetic Acid production by bacteria immobilized on a nitrocellulose membrane. Appl. Environ. Microbiol. 57, 535–538. doi: 10.1128/aem.57.2.535-538.1991
Bull, A. T., Asenjo, J. A., Goodfellow, M., and Gomez-Silva, B. (2016). The atacama desert: Technical resources and the growing importance of novel microbial diversity. Annu. Rev. Microbiol. 70, 215–234. doi: 10.1146/annurev-micro-102215-095236
Chai, X., Wang, X., Pi, Y., Wu, T., Zhang, X., Xu, X., et al. (2022). Nitrate transporter MdNRT2.4 interacts with Rhizosphere bacteria to enhance nitrate uptake in apple rootstocks. J. Exp. Bot. 73, 6490–6504. doi: 10.1093/jxb/erac301
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Cohen, A. C., Travaglia, C. N., Bottini, R., and Piccoli, P. N. (2009). Participation of abscisic acid and gibberellins produced by endophytic Azospirillum in the alleviation of drought effects in maize. Bot. Bot. 87, 455–462. doi: 10.1139/B09-023
Collins, M. D., Pirouz, T., Goodfellow, M., and Minnikin, D. E. (1977). Distribution of menaquinones in actinomycetes and corynebacteria. J. Gen. Microbiol. 100, 221–230. doi: 10.1099/00221287-100-2-221
Crozier, A., Arruda, P., Jasmim, J. M., Monteiro, A. M., and Sandberg, G. (1988). Analysis of Indole-3-acetic acid and related indoles in culture medium from Azospirillum lipoferum and Azospirillum brasilense. Appl. Environ. Microbiol. 54, 2833–2837. doi: 10.1128/aem.54.11.2833-2837.1988
Davies, P. J. (1995). Plant Hormones: Physiology, Biochemistry and Molecular Biology. Boston, MA: Kluwer Academic.
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Deng, Y., Han, X. F., Jiang, Z. M., Yu, L. Y., Li, Y., and Zhang, Y. Q. (2022). Characterization of three Stenotrophomonas strains isolated from different ecosystems and proposal of Stenotrophomonas mori sp. nov. and Stenotrophomonas lacuserhaii sp. nov. Front. Microbiol. 13:1056762. doi: 10.3389/fmicb.2022.1056762
Dong, L., Cheng, R., Xiao, L., Wei, F., Wei, G., Xu, J., et al. (2018). Diversity and composition of bacterial endophytes among plant parts of Panax notoginseng. Chin. Med. 13:41. doi: 10.1186/s13020-018-0198-5
Duca, D., Lorv, J., Patten, C. L., Rose, D., and Glick, B. R. (2014). Indole-3-acetic acid in plant-microbe interactions. Antonie Van Leeuwenhoek 106, 85–125. doi: 10.1007/s10482-013-0095-y
Fabra, A., Castro, S., Taurian, T., Angelini, J., Ibañez, F., Dardanelli, M., et al. (2010). Interaction among Arachis hypogaea L. (peanut) and beneficial soil microorganisms: How much is it known? Crit. Rev. Microbiol. 36, 179–194. doi: 10.3109/10408410903584863
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Felsenstein, J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Feng, W. M., Liu, P., Yan, H., Zhang, S., Shang, E. X., Yu, G., et al. (2021). Impact of bacillus on phthalides accumulation in Angelica sinensis (Oliv.) by stoichiometry and microbial diversity analysis. Front. Microbiol. 11:611143. doi: 10.3389/fmicb.2020.611143
Franke-Whittle, I. H., Manici, L. M., Insam, H., and Stres, B. (2015). Rhizosphere bacteria and fungi associated with plant growth in soils of three replanted apple orchards. Plant Soil 395, 317–333. doi: 10.1007/s11104-015-2562-x
Gao, Y., Yuan, L., Du, J., Wang, H., Yang, X., Duan, L., et al. (2022). Bacterial community profile of the crude oil-contaminated saline soil in the Yellow River Delta Natural Reserve, China. Chemosphere 289:133207. doi: 10.1016/j.chemosphere.2021.133207
Gonzalez, C., Gutierrez, C., and Ramirez, C. (1978). Halobacterium vallismortis sp. nov. An amylolytic and carbohydrate-metabolizing, extremely halophilic bacterium. Can. J. Microbiol. 24, 710–715. doi: 10.1139/m78-119
Groth, I., Schumann, P., Rainey, F. A., Martin, K., Schuetze, B., and Augsten, K. (1997). Demetria terragena gen. nov., sp. nov., a new genus of actinomycetes isolated from compost soil. Int. J. Syst. Bacteriol. 47, 1129–1133. doi: 10.1099/00207713-47-4-1129
Hussain, S. S., Ali, M., Ahmad, M., and Siddique, K. H. M. (2011). Polyamines: Natural and en-gineered abiotic and biotic stress tolerance in plants. Biotechnol. Adv. 29, 300–311. doi: 10.1016/j.biotechadv.2011.01.003
Jiang, Z. M., Deng, Y., Han, X. F., Su, J., Wang, H., Yu, L. Y., et al. (2022). Geminicoccus flavidas sp. nov. and Geminicoccus harenae sp. nov., two IAA-producing novel rare bacterial species inhabiting desert biological soil crusts. Front. Microbiol. 13:1034816. doi: 10.3389/fmicb.2022.1034816
Jiang, Z. M., Zhang, B. H., Sun, H. M., Zhang, T., Yu, L. Y., and Zhang, Y. Q. (2021). Properties of Modestobacter deserti sp. nov., a Kind of novel phosphate-solubilizing actinobacteria inhabited in the desert biological soil crusts. Front. Microbiol. 12:742798. doi: 10.3389/fmicb.2021.742798
Jiao, J. Y., Liu, L., Hua, Z. S., Fang, B. Z., Zhou, E. M., Salam, N., et al. (2020). Microbial dark matter coming to light: Challenges and opportunities. Natl. Sci. Rev. 8:nwaa280. doi: 10.1093/nsr/nwaa280
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kim, M. K., Na, J. R., Lee, T. H., Im, W. T., Soung, N. K., and Yang, D. C. (2007). Solirubrobacter soli sp. nov., isolated from soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 57, 1453–1455. doi: 10.1099/ijs.0.64715-0
Kim, T. J., Kim, H. J., Kang, M., Cho, J. H., Kim, Y. G., Lee, S. M., et al. (2021). Ginsenoside F2 induces cellular toxicity to glioblastoma through the impairment of mitochondrial function. Phytomedicine 83:153483. doi: 10.1016/j.phymed.2021.153483
Klo, S. C., Cano, F. R., and Lampen, J. O. (1973). Lomofungin, an inhibitor of ribonucleic acid synthesis in yeast protoplasts: Its effect on enzyme formation. Antimicrob Agents Chemother. 3, 716–722. doi: 10.1128/aac.3.6.716
Kluge, A. G., and Farris, J. S. (1969). Quantitative phyletics and the evolution of anurans. Syst. Biol. 18, 1–32. doi: 10.1093/sysbio/18.1.1
Komagata, K., and Suzuki, K. I. (1988). 4 lipid and cell-wall analysis in bacterial systematics. Methods Microbiol. 19, 161–207. doi: 10.1016/S0580-9517(08)70410-0
Komoda, T., Akasaka, K., and Hirota, A. (2008). Ansaetherone, a new radical scavenger from Streptomyces sp. Biosci. Biotechnol. Biochem. 72, 2392–2397. doi: 10.1271/bbb.80282
Komoda, T., Yoshida, K., Abe, N., Sugiyama, Y., Imachi, M., Hirota, H., et al. (2004). Tetrapetalone A, a novel lipoxygenase inhibitor from Streptomyces sp. Biosci. Biotechnol. Biochem. 68, 104–111. doi: 10.1271/bbb.68.104
Kroppenstedt, R. M. (1985). Fatty acid and menaquinone analysis of actinomycetes and related organisms in society for applied bacteriology technical series: Chemical methods in bacterial systematics. Soc. Appl. Bacteriol. Tech. Ser. 20, 173–199.
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lee, S. A., Kim, H. S., Sang, M. K., Song, J., and Weon, H. Y. (2021). Effect of Bacillus mesonae H20-5 treatment on rhizospheric bacterial community of tomato plants under salinity stress. Plant Pathol. J. 37, 662–672. doi: 10.5423/PPJ.FT.10.2021.0156
Leontidou, K., Genitsaris, S., Papadopoulou, A., Kamou, N., Bosmali, I., Matsi, T., et al. (2020). Plant growth promoting rhizobacteria isolated from halophytes and drought-tolerant plants: Genomic characterisation and exploration of phyto-beneficial traits. Sci. Rep. 10:14857. doi: 10.1038/s41598-020-71652-0
Leung, P. M., Bay, S. K., Meier, D. V., Chiri, E., Cowan, D. A., Gillor, O., et al. (2020). Energetic basis of microbial growth and persistence in desert ecosystems. mSystems 5:e00495-19. doi: 10.1128/mSystems.00495-19
Li, W. J., Xu, P., Schumann, P., Zhang, Y. Q., Pukall, R., Xu, L. H., et al. (2007). Georgenia ruanii sp. nov., a novel actinobacterium isolated from forest soil in Yunnan (China), and emended description of the genus Georgenia. Int. J. Syst. Evol. Microbiol. 57, 1424–1428. doi: 10.1099/ijs.0.64749-0
Li, X., Chu, C., Ding, S., Wei, H., Wu, S., and Xie, B. (2022). Insight into how fertilization strategies increase quality of grape (Kyoho) and shift microbial community. Environ. Sci. Pollut. Res. Int. 29, 27182–27194. doi: 10.1007/s11356-021-17759-x
Magee, C. M., Rodeheaver, G., Edgerton, M. T., and Edlich, R. F. (1975). A more reliable gram staining technic for diagnosis of surgical infections. Am. J. Surg. 130, 341–346. doi: 10.1016/0002-9610(75)90398-0
Maglangit, F., Fang, Q., Leman, V., Soldatou, S., Ebel, R., Kyeremeh, K., et al. (2019). Accramycin A, a new aromatic polyketide, from the soil bacterium, Streptomyces sp. MA37. Molecules 24:3384. doi: 10.3390/molecules24183384
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Mhatre, P. H., Karthik, C., Kadirvelu, K., Divya, K. L., Venkatasalam, E. P., Srinivasan, S., et al. (2018). Plant growth promoting rhizobacteria (PGPR): A potential alternative tool for nematodes bio-control. Biocatal. Agric. Biotechnol. 17, 119–128. doi: 10.1016/j.bcab.2018.11.009
Minnikin, D. E., O’Donnell, A. G., Goodfellow, M., Alderson, G., Athalye, M., Schaal, A., et al. (1984). An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 2, 233–241. doi: 10.1016/0167-7012(84)90018-6
Miralles, I., Lázaro, R., Sánchez-Marañón, M., Soriano, M., and Ortega, R. (2020). Biocrust cover and successional stages influence soil bacterial composition and diversity in semiarid ecosystems. Sci. Total Environ. 709:134654. doi: 10.1016/j.scitotenv.2019.134654v
Mojib, N., Farhoomand, A., Andersen, D. T., and Bej, A. K. (2013). UV and cold tolerance of a pigment-producing Antarctic Janthinobacterium sp. Ant5-2. Extremophiles 17, 367–378. doi: 10.1007/s00792-013-0525-9
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Paysan-Lafosse, T., Blum, M., Chuguransky, S., Grego, T., Pinto, B. L., Salazar, G. A., et al. (2023). InterPro in 2022. Nucleic Acids Res. 51, D418–D427. doi: 10.1093/nar/gkac993
Piccoli, P., Travaglia, C., Cohen, A., Sosa, L., Cornejo, P., Masuelli, R., et al. (2011). An endophytic bacterium isolated from roots of the halophyte Prosopis strombulifera produces ABA, IAA, gibberellins A 1 and A 3 and jasmonic acid in chemically-defined culture medium. Plant Growth Regul. 64, 207–210. doi: 10.1007/s10725-010-9536-z
Pimentel-Elardo, S. M., Gulder, T., Hentschel, U., and Bringmann, G. J. C. (2009). Cebulactams A1 and A2, new macrolactams isolated from Saccharopolyspora cebuensis, the first obligate marine strain of the genus Saccharopolyspora. Cheminform 49, 6889–6892. doi: 10.1016/j.tetlet.2008.09.094
Pulschen, A. A., Rodrigues, F., Duarte, R. T., Araujo, G. G., Santiago, I. F., Paulino-Lima, I. G., et al. (2015). UV-resistant yeasts isolated from a high-altitude volcanic area on the Atacama Desert as eukaryotic models for astrobiology. Microbiologyopen 4, 574–588. doi: 10.1002/mbo3.262
Radhakrishnan, R., and Baek, K. H. (2017). Physiological and biochemical perspectives of non-salt tolerant plants during bacterial interaction against soil salinity. Plant Physiol. Biochem. 116, 116–126. doi: 10.1016/j.plaphy.2017.05.009
Reina-Bueno, M., Argandona, M., Nieto, J. J., Hidalgo-Garcia, A., Iglesias-Guerra, F., Delgado, M. J., et al. (2012). Role of trehalose in heat and desiccation tolerance in the soil bacterium Rhizobium etli. BMC Microbiol. 12:207. doi: 10.1186/1471-2180-12-207
Reis-Mansur, M. C. P. P., Cardoso-Rurr, J. S., Silva, J. V. M. A., de Souza, G. R., Cardoso, V. D. S., Mansoldo, F. R. P., et al. (2019). Carotenoids from UV-resistant Antarctic Microbacterium sp, LEMMJ01. Sci. Rep. 9:9554. doi: 10.1038/s41598-019-45840-6
Saitou, N., and Nei, M. (1987). The Neighbor-Joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sánchez-Marañón, M., Miralles, I., Aguirre-Garrido, J. F., Anguita-Maeso, M., Millán, V., Ortega, R., et al. (2017). Changes in the soil bacterial community along a pedogenic gradient. Sci. Rep. 7:14593. doi: 10.1038/s41598-017-15133-x
Sasser, M. (1990). Identification of bacteria by gas chromatography of cellular fatty acids. Usfcc Newsl. 20, 1–6.
Saxena, D., Ben-Dov, E., Manasherob, R., Barak, Z., Boussiba, S., and Zaritsky, A. (2002). A UV tolerant mutant of Bacillus thuringiensis subsp. kurstaki producing melanin. Curr. Microbiol. 44, 25–30. doi: 10.1007/s00284-001-0069-6
Schleifer, K. H., and Kandler, O. (1973). Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 37:258. doi: 10.1128/br.37.2.258-258.1973
Schumacher, M. A., Chinnam, N. B., Cuthbert, B., Tonthat, N. K., and Whitfill, T. (2015). Structures of regulatory machinery reveal novel molecular mechanisms controlling B. subtilis nitrogen homeostasis. Genes Dev. 29, 451–464. doi: 10.1101/gad.254714.114
Sen, S., Ghosh, D., and Mohapatra, S. (2018). Modulation of polyamine biosynthesis in Arabidopsis thaliana by a drought mitigating Pseudomonas putida strain. Plant Physiol. Biochem. 129, 180–188. doi: 10.1016/j.plaphy.2018.05.034
Shange, R. S., Ankumah, R. O., Ibekwe, A. M., Zabawa, R., and Dowd, S. E. (2012). Distinct soil bacterial communities revealed under a diversely managed agroecosystem. PLoS One 7:e40338. doi: 10.1371/journal.pone.0040338
Shi, H., and Chan, Z. (2014). Improvement of plant abiotic stress tolerance through modulation of the polyamine pathway. J. Integr. Plant. Biol. 56, 114–121. doi: 10.1111/jipb.12128
Shirling, E. B., and Gottlieb, D. (1966). Methods for characterization of Streptomyces species. Int. J. Syst. Bacteriol. 16, 313–340. doi: 10.1099/00207713-16-3-313
Singleton, D. R., Furlong, M. A., Peacock, A. D., White, D. C., Coleman, D. C., and Whitman, W. B. (2003). Solirubrobacter pauli gen. nov., sp. nov., a mesophilic bacterium within the Rubrobacteridae related to common soil clones. Int. J. Syst. Evol. Microbiol. 53, 485–490. doi: 10.1099/ijs.0.02438-0
Sivakala, K. K., Jose, P. A., Anandham, R., Thinesh, T., Jebakumar, S. R. D., Samaddar, S., et al. (2018). Spatial physiochemical and metagenomic analysis of desert environment. J. Microbiol. Biotechnol. 28, 1517–1526. doi: 10.4014/jmb.1804.04005
Sun, Y., Chen, H. H., Sun, H. M., Ai, M. J., Su, J., Yu, L. Y., et al. (2017). Naumannella huperziae sp. nov., an endophytic actinobacterium isolated from Huperzia serrata (Thunb.). Int. J. Syst. Evol. Microbiol. 67, 1867–1872. doi: 10.1099/ijsem.0.001882
Takahashi, T., and Kakehi, J. (2010). Polyamines: Ubiquitous polycations with unique roles in growth and stress responses. Ann. Bot. 105, 1–6. doi: 10.1093/aob/mcp259
Taylor, A. F., and Smith, G. R. (1999). Regulation of homologous recombination: Chi inactivates RecBCD enzyme by disassembly of the three subunits. Genes Dev. 13, 890–900. doi: 10.1101/gad.13.7.890
Truglio, J. J., Croteau, D. L., Van Houten, B., and Kisker, C. (2006). Prokaryotic nucleotide excision repair: The UvrABC system. Chem. Rev. 106, 233–252. doi: 10.1021/cr040471u
Tuteja, N., Singh, M. B., Misra, M. K., Bhalla, P. L., and Tuteja, R. (2001). Molecular mechanisms of DNA damage and repair: Progress in plants. Crit. Rev. Biochem. Mol. Biol. 36, 337–397. doi: 10.1080/20014091074219
UniProt Consortium (2023). UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 251, D523–D531. doi: 10.1093/nar/gkac1052
Wang, J., Li, W., Wang, H., and Lu, C. (2018). Pentaketide ansamycin microansamycins A-I from Micromonospora sp. Reveal Diverse Post-PKS modifications. Org. Lett. 20, 1058–1061. doi: 10.1021/acs.orglett.7b04018
Wei, L., Ouyang, S., Wang, Y., Shen, X., and Zhang, L. (2014). Solirubrobacter phytolaccae sp. nov., an endophytic bacterium isolated from roots of Phytolacca acinosa Roxb. Int. J. Syst. Evol. Microbiol. 64, 858–862. doi: 10.1099/ijs.0.057554-0
Xie, S. S., Wu, H. J., Zang, H. Y., Wu, L. M., Zhu, Q. Q., and Gao, X. W. (2014). Plant growth promotion by spermidine-producing Bacillus subtilis OKB105. Mol. Plant Microbe Interact. 27, 655–663. doi: 10.1094/MPMI-01-14-0010-R
Xie, Y., Wu, G., Tang, J., Luo, R., Patterson, J., Liu, S., et al. (2014). SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666. doi: 10.1093/bioinformatics/btu077
Xu, P., Li, W. J., Tang, S. K., Zhang, Y. Q., Chen, G. Z., Chen, H. H., et al. (2005). Naxibacter alkalitolerans gen. nov., sp. nov., a novel member of the family ‘Oxalobacteraceae’ isolated from China. Int. J. Syst. Evol. Microbiol. 55, 1149–1153. doi: 10.1099/ijs.0.63407-0
Xue, H. P., Zhang, D. F., Xu, L., Wang, X. N., Zhang, A. H., Huang, J. K., et al. (2021). Actirhodobacter atriluteus gen. nov., sp. nov., isolated from the surface water of the Yellow Sea. Antonie Van Leeuwenhoek 114, 1059–1068. doi: 10.1007/s10482-021-01576-w
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yoshida, K., Yamaguchi, H., Kinehara, M., Ohki, Y. H., Nakaura, Y., and Fujita, Y. (2003). Identification of additional TnrA-regulated genes of Bacillus subtilis associated with a TnrA box. Mol. Microbiol. 49, 157–165. doi: 10.1046/j.1365-2958.2003.03567.x
Zhang, B. L., Wu, X. K., Tai, X. S., Sun, L. K., Wu, M. H., Zhang, W., et al. (2019). Variation in actinobacterial community composition and potential function in different soil ecosystems belonging to the arid heihe river basin of Northwest China. Front. Microbiol. 10:2209. doi: 10.3389/fmicb.2019.02209
Zhang, J., Qian, Z., Wu, X., Ding, Y., Li, J., Lu, C., et al. (2014). Juanlimycins A and B, ansamycin macrodilactams from Streptomyces sp. Org. Lett. 16, 2752–2755. doi: 10.1021/ol501072t
Zhang, L., Zhu, L., Si, M., Li, C., Zhao, L., Wei, Y., et al. (2014). Solirubrobacter taibaiensis sp. nov., isolated from a stem of Phytolacca acinosa Roxb. Antonie Van Leeuwenhoek 106, 279–285. doi: 10.1007/s10482-014-0194-4
Zhang, Y. Q., Sun, C. H., Li, W. J., Yu, L. Y., Zhou, J. Q., Zhang, Y. Q., et al. (2007). Deinococcus yunweiensis sp. nov., a gamma- and UV-radiation-resistant bacterium from China. Int. J. Syst. Evol. Microbiol. 57, 370–375. doi: 10.1099/ijs.0.64292-0
Keywords: Solirubrobacter, pangenome, UV resistance, microbial resources, distribution
Citation: Jiang Z-M, Mou T, Sun Y, Su J, Yu L-Y and Zhang Y-Q (2023) Environmental distribution and genomic characteristics of Solirubrobacter, with proposal of two novel species. Front. Microbiol. 14:1267771. doi: 10.3389/fmicb.2023.1267771
Received: 27 July 2023; Accepted: 03 November 2023;
Published: 01 December 2023.
Edited by:
Melina Kerou, University of Vienna, AustriaReviewed by:
Maher Gtari, Carthage University, TunisiaCopyright © 2023 Jiang, Mou, Sun, Su, Yu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Qin Zhang, eXpoYW5nQGltYi5wdW1jLmVkdS5jbg==
†These; authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.