 Xin-yu Zhang1
Xin-yu Zhang1 Yu-ying Li1
Yu-ying Li1 Hai-xin Huang1,2
Hai-xin Huang1,2 Chen-chen Zhao1
Chen-chen Zhao1 Xiao-xiao Lei1Bao-peng Zhao1Jing-yi Lu1Tian Lan1*
Xiao-xiao Lei1Bao-peng Zhao1Jing-yi Lu1Tian Lan1* Wen-chao Sun1*
Wen-chao Sun1*- 1Institute of Virology, Wenzhou University, Wenzhou, China
- 2College of Veterinary Medicine, Northwest A&F University, Xianyang, China
Seneca Valley virus (SVV), a member of the Picornaviridae family, may cause serious water blister diseases in pregnant sows and acute death in newborn piglets, which have resulted in economic losses in pig production. The 3C protease is a vital enzyme for SVV maturation and is capable of regulating protein cleavage and RNA replication of the virus. Additionally, this protease can impede the host’s innate immune response by targeting the interferon pathway’s principal factor and enhance virus replication by modulating the host’s RNA metabolism while simultaneously triggering programmed cell death. This article reviews recent studies on SVV 3C functions, which include viral replication promotion, cell apoptosis modulation and host immune response evasion, and provides a theoretical basis for research on preventing and controlling SVV infection.
1. Introduction
In 2002, laboratory personnel at Gaithersburg, Maryland, United States, serendipitously discovered the Seneca Valley virus (SVV) when culturing a vector derived from adenovirus-5 (Ad5) in PER.C6 cells (Hales et al., 2008). Research has revealed that SVV exerts an oncolytic effect that holds promising prospects for the treatment of neuroendocrine tumours in humans (Reddy et al., 2007). SVV infection can result in vesicular disease in sows and sudden neonatal piglet mortality. The initial stages of SVV may be characterized by anorexia, lethargy, and fever, and these signs are followed by vesicular lesions presenting on the skin and the mucous membranes of the hoof, mouth, and nose, which may cause mouth and nose ulceration and even hoof horn loss and thus lead to lameness in severe cases. The symptoms of SVV are similar to those of foot-and-mouth disease virus (FMDV) infection and the two viral infections can only be distinguished by laboratory diagnosis (Reddy et al., 2007; Joshi et al., 2016; Maggioli et al., 2018; Wu et al., 2022). SVV has been spreading in pig herds in the United States since 1988, but water blister disease caused by SVV was not found until 2007 (Knowles et al., 2006; Pasma et al., 2008). Sporadic cases of SVV infection have been reported for more than 10 years since the discovery of this virus; however, a sudden outbreak was reported in Brazil at the end of 2014 and the beginning of 2015 (Vannucci et al., 2015; Leme et al., 2016) and a rapid and large-scale spread of SVV to countries such as the United States (Hause et al., 2016), Canada (Xu et al., 2017), Colombia (Sun et al., 2017), China (Wu et al., 2017), Thailand (Saeng-Chuto et al., 2018), and Vietnam (Arzt et al., 2019) then occurred. Notably, SVV frequently causes water blister disease outbreaks in the pig herds of the world’s leading pig-producing countries.
SVV, which belongs to the Picornaviridae family and the Senecavirus genus, is an unenveloped single-stranded positive-sense RNA virus. The viral genome consists of an ORF, a 5′ untranslated region (5′ UTR), and a 3′ untranslated region (3′ UTR). The viral genome initially encodes a polyprotein that is then cleaved into structural and nonstructural proteins during cotranslational and posttranslational processes. Structural proteins comprise VP1, VP2, VP3, and VP4, whereas nonstructural proteins comprise 2A, 2B, 2C, 3A, 3B, 3C, and 3D as well as the leader protein L (Hales et al., 2008; Meng et al., 2022). The nonstructural protein 3C is a cysteine protease encoded by the virus that has a conserved catalytic triad of His, Asp., and Cys (Meng et al., 2022). This protease has been shown to be involved in various pathological processes of SVV, and SVV 3Cpro employs complex strategies to modulate host antiviral immunity, which is essential for effective virus replication.
2. Structure of the 3C protease and virus amplification
Similar to the 3Cpro of other Picornaviridae viruses, SVV 3Cpro adopts the typical trypsin fold, but it lacks the characteristic KFRDI motif. H48 is one of the catalytic residues, and G158 and G161 are typical cysteine protease motifs with the sequence G-X-C/S-G (Ng et al., 2021; Meng et al., 2022). Furthermore, the catalytic cysteine residue typically participates in substrate recognition with the downstream GΦH motif (SVVGLH 3C176–178), which consists of 10–20 amino acids (Hales et al., 2008).
The SVV genome has an open reading frame encoding a polyprotein that consists of a leader protein (L), a structural protein region (P1) and nonstructural protein regions (P2 and P3) (Hales et al., 2008; Meng et al., 2022). The virus-encoded 3Cpro cleaves the polyprotein into its active structural and nonstructural proteins, and RNA replication is a crucial stage in virus maturation (Pathak et al., 2007). The structural protein region P1 undergoes cleavage, which produces VP0, VP3, and VP1. The precursor VP0 undergoes subsequent cleavage to yield VP4 and VP2, and the P2-P3 region undergoes cleavage, leading to the formation of 7 nonstructural proteins: 2A, 2B, 2C, 3A, 3B, 3C, and 3D. Virus-encoded 3Cpro cleaves specific sites, such as VP2-VP3, VP3-VP1, 2B-2C, 2C-3A, 3A-3B, 3B-3C, and 3C-3D. However, the cleavage mechanisms of L-VP4, VP1-2A, and 2A-2B differ among distinct Picornaviridae viruses. SVV 3Cpro cleaves both L-VP4 and VP1-2A, whereas 2A-2B cleavage relies on 2Apro. Unfortunately, the cleavage mechanism of VP4-VP2 remains elusive (Hales et al., 2008; Meng et al., 2022).
Picornaviruses contain a VPg protein that is covalently linked to the 5′ end of their genome. During replication of the genome of picornaviruses, VPg undergoes uridylylation through oriI to form VPg-pUpU, which is believed to enhance the replication of viral RNA. Either the precursor protein 3CD or its derivative 3C triggers this reaction. Both 3CD and 3C contain RNA binding and proteinase activities. Research has shown that the 3C domain exhibits specificity for OriI, whereas the 3D domain enhances its overall affinity. OriI binds to 3C(D) and recruits a polymerase to this site (Pathak et al., 2007). 3Cpro has a crucial function in the Picornaviridae virus maturation process.
3. SVV 3C functions in host antiviral immunity
3.1. Role of 3C proteases in regulating mRNA
The DEAD-box protein family is a conserved family of RNA helicases containing a DEAD-box domain, named after the conserved Asp-Glu-Ala-Asp (DEAD) sequence (Pathak et al., 2007). DDX21 mediates innate immunity and regulates the viral replication of viruses such as foot-and-mouth disease virus (FMDV) (Abdullah et al., 2021), dengue virus (DENV) (Dong et al., 2016) and Borna disease virus (BDV) (Watanabe et al., 2009). Furthermore, DDX21 has been shown to inhibit SVV replication, but its effects can be weakened by SVV 3Cpro, which induces caspase-dependent degradation of DDX21 and suppresses host antiviral immunity, limiting the cell antiviral response (Zhao et al., 2022). DHX30, a DEAD family member, contributes to the biosynthesis of mitochondrial ribosomes. Zinc-finger antiviral protein (ZAP) recruits DHX30 to unfold and degrade viral RNA (Ye et al., 2010). Although DHX30 inhibits SVV replication through its helicase activity, SVV 3Cpro can mediate the cleavage of DHX30 helicase at the Q220 site, leading to loss of its ability to inhibit SVV replication (Wen et al., 2022).
hnRNPs are a widely functional family of RNA-binding proteins mainly located in the cell nucleus (Jean-Philippe et al., 2013), hnRNP A1 can bind to viral proteins and modulate the replication of viruses such as the nucleocapsid proteins of porcine epidemic diarrhoea virus (PEDV) (Li et al., 2018). In addition, hnRNP A1 can interact directly with viral RNA sequences, including the coding region of the human papillomavirus 16 (HPV16) E7 sequence (Zheng et al., 2020). Research has shown that the protease activity of SVV 3Cpro mediates the degradation and translocation of hnRNP A1, which enhances the replication of SVV (Song et al., 2021). Similarly, hnRNP K participates in virus replication through interaction with the 5′ UTR of enterovirus 71 (EV71) (Lin et al., 2008) and FMDV (Liu et al., 2020). Knockdown of hnRNP K significantly inhibits SVV replication, whereas hnRNP K overexpression promotes virus proliferation. These studies suggest that intracellular hnRNP K contributes to SVV replication. SVV infection leads to the cleavage, degradation, and cytoplasmic redistribution of hnRNP K due to the activity of 3Cpro. 3Cpro induces the degradation of hnRNP K through the caspase pathway and cleaves hnRNP K at Q364. The cleaved fragment hnRNP K (365–464) promotes virus replication, whereas full-length hnRNP K exerts a similar effect. In contrast, the noncleaved fragment hnRNP K (1–364) tends to inhibit virus replication (Song et al., 2022b).
Nucleolin (NCL) is involved in several cellular processes, such as RNA transcription, ribosome formation, nuclear-cytoplasmic transport, and posttranscriptional regulation of mRNA (Becherel et al., 2006; Durut and Sáez-Vásquez, 2015). Moreover, NCL is linked with viral proliferation and plays a crucial role in virus replication through nucleocytoplasmic redistribution (Greco et al., 2012; Han et al., 2021). An increase in the NCL expression levels and its cleavage are induced by SVV, which drives NCL redistribution outside the cell nucleus. The 3Cpro protein of SVV relocates NCL to the cytoplasm and cleaves it at Q545. Cleaved NCL facilitates virus replication, and the proteolytic activity of the 3Cpro protein regulates the cleavage and relocalization of NCL (Song et al., 2022c).
Cytoplasmic poly (A)-binding protein 1 (PABPC1) interacts with eukaryotic translation initiation factor 4G (eIF4G) and promotes the binding of the 60S ribosomal subunit to the 48S preinitiation complex in the final step of initiation, facilitating translation initiation (Tahiri-Alaoui et al., 2014; Smith et al., 2017). SVV replication is inhibited by PABPC1; however, SVV 3Cpro cleaves PABPC1 at Q437, which disrupts protein synthesis and interferes with host immunity, thereby promoting virus replication (Xue et al., 2020).
SVV 3Cpro inhibits RNA metabolism regulation, gene expression, and the immune response mediated by the aforementioned factors, thereby evading host defence mechanisms and providing a conducive environment for its replication.
3.2. Role of 3C protease in cell intrinsic innate immunity signalling
The innate immune response represents the crucial initial defence against pathogenic invasion in the host organism. RIG-I-like receptors (RLRs), including retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5), detect viral RNA and trigger the activation of mitochondrial antiviral signalling protein (Yoneyama et al., 2005; Paludan and Bowie, 2013). Upon activation, MAVS undergoes dimerization on the mitochondrial membrane, where it subsequently associates with TNF receptor-associated factor 3 (TRAF3) (Yoneyama et al., 2005; Tang and Wang, 2009). TRAF3 then recruits TRAF-related NF-κB-activating kinase (TANK), and TANK transduces upstream signals to TANK-binding kinase 1 (TBK1). TBK1 induces the phosphorylation of interferon regulatory factor 3/7 (IRF-3/7) and nuclear transcription factor kappa B (NF-κB), resulting in their dimerization and nuclear translocation (Pomerantz and Baltimore, 1999; Taniguchi et al., 2001; Shu et al., 2013). Ubiquitination is a crucial regulatory element in this process. For RIG-I to bind to RNA and become active, TRIM25 must undergo K63 ubiquitination. TRAF3 is K63-ubiquitinated, leading to the stimulation of IRF-3 phosphorylation (Castanier et al., 2012; Zhu et al., 2020). TIR domain containing adaptor molecule 1 (TRIF) responds to dsRNA and LPS, resulting in the activation of NF-κB (Schwandner et al., 1999; Alexopoulou et al., 2001).
The 3Cpro of SVV extensively inhibits the type I interferon (IFN) pathway by impairing vital factors. SVV 3Cpro inhibits MAVS-mediated downstream signal transduction by degrading RIG-I, which is responsible for recognizing viral RNA. Second, by cleaving MAVS (Qian et al., 2017) at Q148, SVV 3Cpro interrupts its interaction with RIG-I (Wen et al., 2019), leading to reduced activation of downstream pathways. Recent studies have shown that SVV infection suppresses the interaction between MAVS and RIG-I by promoting lactate production through glycolysis, thereby facilitating virus replication (Li et al., 2023). SVV 3Cpro also possesses deubiquitinase activity, which inhibits the ubiquitination of RIG-I, TBK1, and TRAF3 (Xue et al., 2018b). In addition, SVV 3Cpro cleaves TRIF at Q159 (Qian et al., 2017). The N-and C-terminal domains of TRIF play different roles in the regulation of innate immunity and cell apoptosis. The N-terminal domain of TRIF is essential for the initiation of IFN promoter activity, whereas both the N-and C-terminal domains are involved in NF-κB activation. Despite the N-terminal domain separating from TRIF, the C-terminal domain still plays a role in initiating interferon signals, activating NF-κB, and inducing cell apoptosis. SVV 3Cpro interacts with IRF3 and IRF7, which inhibits their phosphorylation and leads to their degradation, ultimately affecting the regulation of IFN and other inflammatory genes (Xue et al., 2018a). In addition, SVV 3Cpro cleaves TANK at the E272 and Q291 sites (Qian et al., 2017), affecting its regulation of TBK1/IKKε and IRF3/7, and resulting in the inhibition of IFN production. Moreover, SVV 3Cpro mediates the cleavage of NF-κB by caspase 3 (Fernandes et al., 2019), suppressing its regulation of cytokine expression. SVV 3Cpro utilizes multiple mechanisms to deregulate crucial factors in the IFN pathway and thus widely inhibits the production and transfer of IFN, which enables it to evade the host’s immune response (Figure 1).
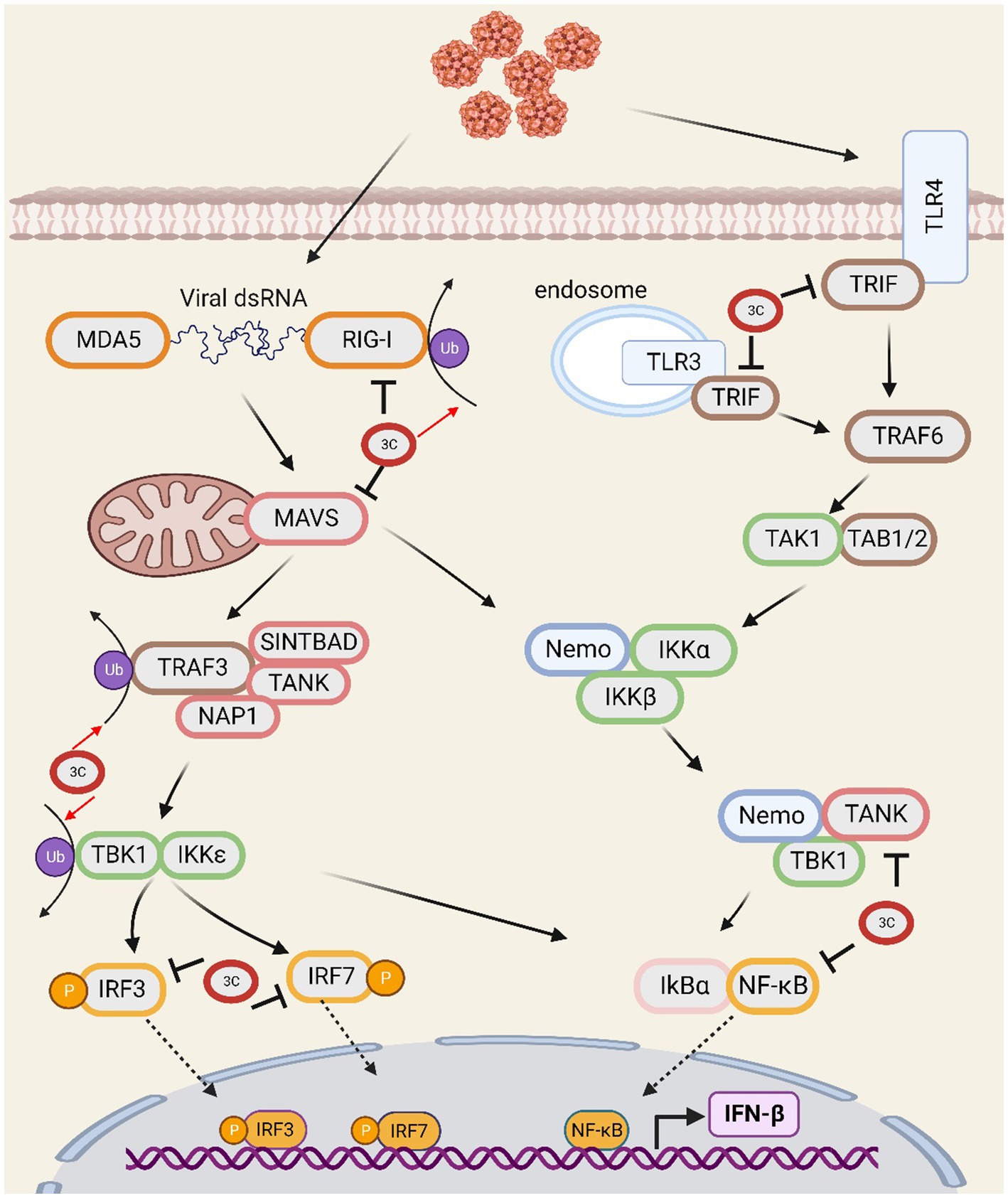
Figure 1. The SVV 3C protease is involved in the interferon signal transduction pathway and affects antiviral responses.
3.3. Role of 3C protease in stress granules
Stress granules (SGs) are cytoplasmic structures composed of RNA and RNA-binding proteins that form in response to various stressors, including hypoxia, environmental toxins, and viral infections (Wen et al., 2020). The formation of SGs induced by viral infection inhibits the synthesis of viral proteins, exerting antiviral effects prior to the upregulation of host antiviral protein transcription. During infection, the double-stranded RNA (dsRNA) formed by the virus activates PKR, leading to eIF2α phosphorylation and subsequent promotion of SGs formation (McEwen et al., 2005; Yamasaki and Anderson, 2008). The RNA-binding protein GTPase-activating protein (SH3 domain)-binding protein 1 (G3BP1) binds to mRNA and aggregates, forming the core of SGs (Yamasaki and Anderson, 2008). Eukaryotic translation initiation factor 4G 1 (eIF4G1) is a key regulatory protein involved in translation initiation. G3BP1 can bind to and sequester eIF4G1, promoting stress-induced SGs formation and playing a role in mRNA transport and stability (Kedersha et al., 2016). Picornavirus 2A or L protein inhibits SGs formation by interfering with the eIF4GI-G3BP interaction (Yang et al., 2019). SVV infection induces the formation of stress granules (SGs) during the early stages of infection, but these SGs dissipate as the infection progresses to the late stages. Additionally, SVV infection transiently induces PKR and eIF2α phosphorylation-dependent SGs formation. SVV replication plays a crucial role in SGs formation. The cleavage of eIF4G1 by SVV. 3Cpro disrupts the eIF4GI-G3BP1 interaction, thus impairing SGs formation and contributing to enhanced viral replication (Wen et al., 2020).
3.4. The role of 3C protease in pyroptosis
The NLRP3 inflammasome, which is a large multiprotein complex that senses danger signals inside and outside the cell and initiates an inflammatory response, mainly consists of three components: NLRP3 (NOD-like receptor protein 3), ASC (apoptotic speck-like protein containing a caspase recruitment domain), and caspase-1. Upon the appearance of danger signals, such as pathogen infection, inflammatory stimulation or cell damage, the NLRP3 inflammasome can be activated, and once activated, the inflammasome can activate inflammatory cytokines such as IL-1β, IL-18 and gasdermin D (GSDMD) by stimulating caspase-1 activation. When activated, GSDMD forms pores and triggers pyroptosis, which is characterized by the rupture of the cell membrane and the release of intracellular contents. GSDMD belongs to the gasdermin family, which is a group of proteins with functions in cell membrane perforation and induction of inflammation. GSDMD has a self-inhibitory structure located between the N-and C-termini. When cleaved by proteases such as caspase-1, the N-terminus of GSDMD (GSDMD-N) is released and inserted into the cell membrane, forming pores. This process consequently results in the exchange of intracellular and extracellular substances, which ultimately leads to pyroptosis (Shi et al., 2015; Choudhury et al., 2022).
SVV 3Cpro cleaves NLRP3 at Q305, resulting in the inhibition of NLRP3 inflammasome formation. Concurrently, the protease directly cleaves pig GSDMD at Q193 and Q277, leading to release of the N-terminus of GSDMD. The cleavage of GSDMD 1–275 by caspase-1 triggers pyroptosis. Additionally, because GSDMD Q277 is proximal to the caspase-1-mediated cleavage site, GSDMD 1–277 may induce pyroptosis (Wen et al., 2021a). Research has shown that after infection with SVV markedly increases pig GSDMD cleavage and cell pyroptosis, confirming the inducible effect of SVV 3Cpro on GSDMD cleavage and pyroptosis. This finding suggests that SVV 3Cpro can independently trigger the inflammatory response of the NLRP3 inflammasome (Figure 2).
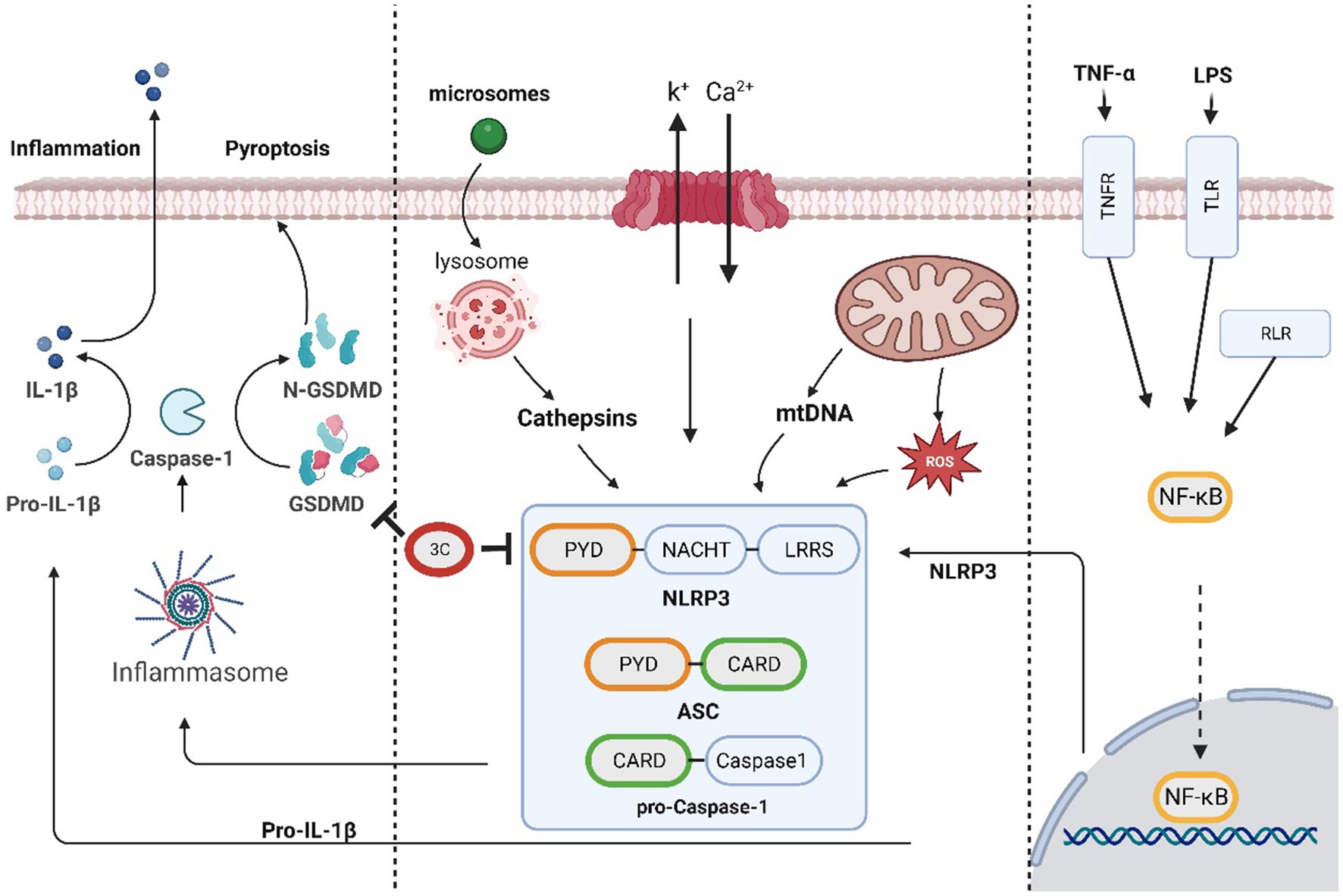
Figure 2. The SVV 3C protease is involved in the pyroptosis pathway and affects antiviral responses.
3.5. Role of 3C protease in apoptosis
Apoptosis is an essential programmed cell death mechanism involved in physiological and pathological processes, such as development, growth, immunity, and disease. Apoptosis is a widely recognized defence mechanism of the host and serves to ensure the death of cells that have been infected by viruses. There are two types of pathways that facilitate cell apoptosis, extrinsic and intrinsic, and these pathways are executed by the caspase family of cysteine proteases (Cohen, 1997; Elmore, 2007). The extrinsic pathway is initiated by external triggers and is regulated by membrane death receptors such as CD95 (Fas/APO-1) and tumour necrosis factor receptor 1 (TNFR1). When apoptosis is induced, caspase-8 is activated and triggers downstream effector molecules, including caspase-1, caspase-3, caspase-6, and caspase-7 (Muzio et al., 1996). The intrinsic pathway involves the activation of mitochondrial-related proteins. When host cells are subjected to factors such as infection and injury, the mitochondrial apoptosis pathway is stimulated, resulting in changes in mitochondrial membrane permeability and the release of various apoptotic factors, including cytochrome C (Galluzzi et al., 2012). This process leads to the activation of caspase-9 and downstream caspase-3, ultimately leading to cell apoptosis.
At the early stages of SVA infection, the apoptotic pathway is not activated, which may allow the virus to complete its replication cycle before cell death/lysis and at the later stages of infection, the induction of cell apoptosis by SVV may function as a mechanism for facilitating the release and/or dissemination of the virus from infected cells (Kaminskyy and Zhivotovsky, 2010; Sun et al., 2016; Fernandes et al., 2019). The ability of 3Cpro to induce apoptosis in cells seems to remain consistent in small RNA viruses, including enterovirus 71 and poliovirus. Studies suggest that the SVV 3Cpro protein stimulates caspase-3, caspase-8, and caspase-9 and indicate that this protein invokes cell apoptosis through both mitochondrial and exogenous death receptor pathways (Liu et al., 2019). Although SVV 3Cpro induces cell apoptosis through its protease activity, it does not directly cleave PARP 1, which is a hallmark of cell apoptosis (Oliver et al., 1998; Liu et al., 2019). Emerging studies have shown that NF-κB likely plays a vital role in safeguarding host cells from small nuclear RNA virus-induced apoptosis (Oliver et al., 1998; Schwarz et al., 1998). A few studies have postulated that caspase 3-mediated cleavage of NF-κB encourages cell apoptosis (Kang et al., 2001; Kim et al., 2005), whereas SVV 3Cpro might impede innate immunity while promoting cell apoptosis by mediating the cleavage of caspase 3 through NF-κB (Fernandes et al., 2019; Figure 3). Notably, infection with human immunodeficiency virus (HIV) and African swine fever virus (ASFV) induces caspase-mediated NF-κB-p65 cleavage, thereby enhancing viral replication or inducing cell apoptosis after completion of the viral replication cycle (Vallée et al., 2001; Coiras et al., 2008). 3Cpro has a unique structural domain that binds to native phospholipid molecules, such as cardiolipin (CL) and phosphatidylinositol-4-phosphate (PI4P). CL can activate SVA 3Cpro activity in a homologous manner, leading to the cleavage of viral polyproteins and host proteins (such as NLRP3 and MAVS), disrupting host responses and ensuring viral replication. This binding serves as a positive regulatory mechanism for 3Cpro activity and promotes viral replication (Zhao et al., 2023). The replication of most positive-sense RNA viruses necessitates remodelling of cell membranes, converting them into virus replication organelles (ROs). The lipid microenvironment within ROs, which are rich in PI4P, is crucial for the replication of enterovirus RNA (Belov and van Kuppeveld, 2012). PV 3Cpro exhibits a wide and specific PIP-binding affinity for phospholipids, including PI4P. The binding of SVA 3Cpro to PI4P is hypothesized to induce membrane transformation and facilitate viral genome replication (Belov and van Kuppeveld, 2012; Zhao et al., 2023).
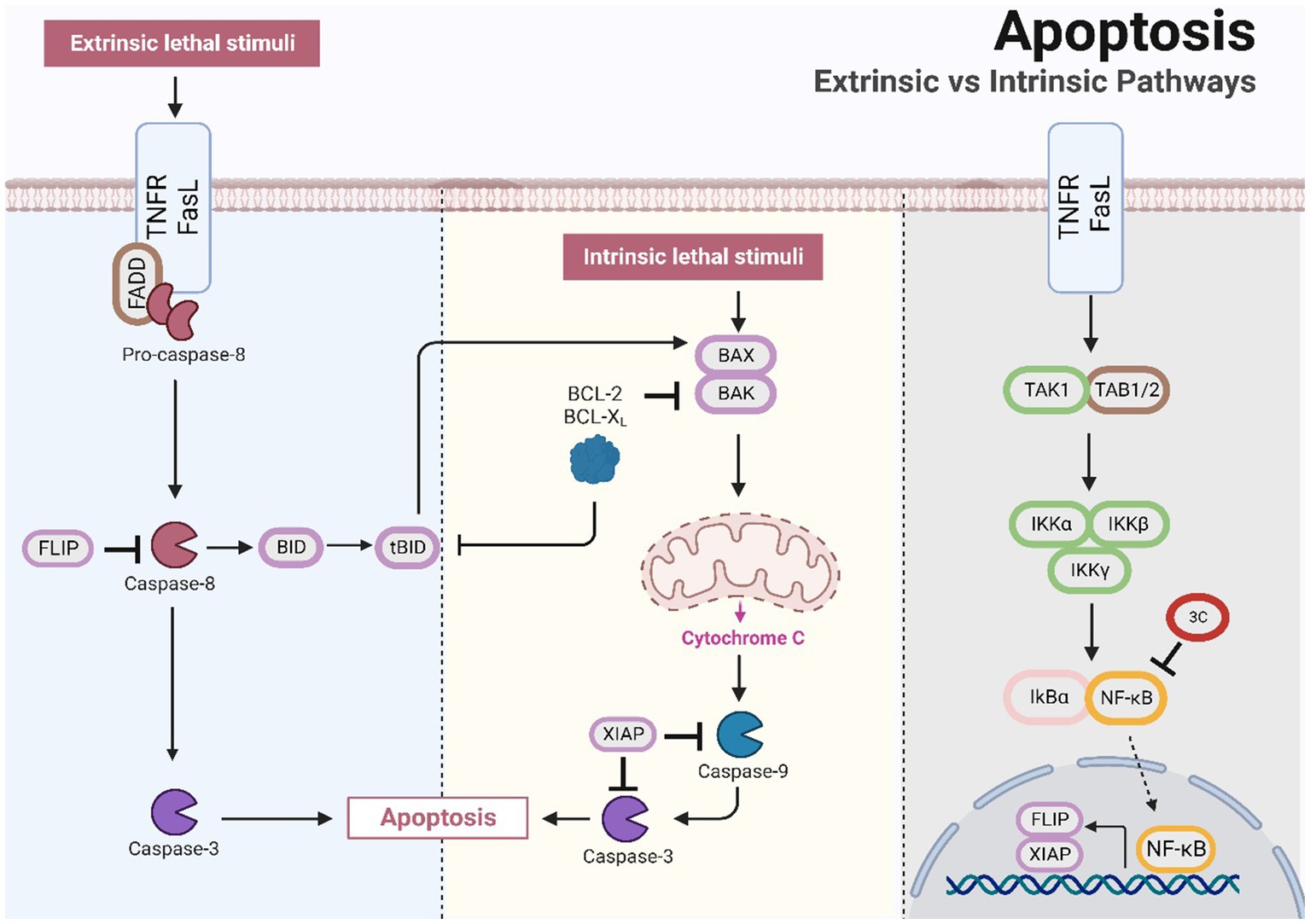
Figure 3. The SVV 3C protease is involved in the apoptotic pathway and affects antiviral responses.
3.6. Role of 3C protease in autophagy
Autophagy is a cellular degradation process that breaks down proteins, organelles, and other substances into small molecules for cellular metabolism. During autophagy, the cell forms and releases autophagosomes containing degraded substances (Klionsky and Codogno, 2013). Modulating the mTOR pathway may affect the virus-induced autophagy response (Zhu et al., 2012). Autophagy enhancement can result from inhibiting AKT activation, whereas AKT activation can decrease autophagy (Hay and Sonenberg, 2004). AMP-activated protein kinase (AMPK) modulates autophagy by inhibiting the mTOR pathway. Phosphorylation of the mTOR binding partner Raptor, which is controlled by AMPK, is necessary to inhibit the mTOR pathway (Yang and Klionsky, 2010). The MAPK signalling pathway, which is essential in the signal transduction network of eukaryotes, plays a vital role in vital processes such as cell proliferation, differentiation, autophagy, apoptosis and the stress response (Gwinn et al., 2008).
A previous study revealed that SVV infection induces autophagy via the PKR-like ER protein kinase (PERK) and the activating transcription factor 6 (ATF6) signalling pathways associated with endoplasmic reticulum stress. Autophagy promotes SVV infection in pig cells, and the research results further demonstrate the involvement of the PERK and ATF6 pathways in autophagy induction. Decreasing the expression of PERK or ATF6 can inhibit SVV replication (Hou et al., 2019). However, in cultured human cells, SVV triggers an autophagic response that inhibits viral replication (Wen et al., 2021b). Research has demonstrated that VP1, VP3, and 3Cpro can cooperatively activate the AKT-AMPK-MAPK-mTOR signalling pathway during SVV infection in host cells, leading to the induction of cellular autophagy. The expression of 3Cpro upregulates the levels of p-ERK1/2 MAPK, p-p38 MAPK, and p-AKT and induces no significant change in the p-AMPK levels. The synergistic effect of p-ERK1/2 MAPK, p-p38 MAPK, and p-AKT causes a reduction in the p-MTOR levels. The transfection of cells with 3Cpro alone has no significant effect on the LC3-II level. However, transfection with SVV VP1 or 3D alone results in a significant alteration in the LC3-II level. VP1 expression promotes the phosphorylation of AKT and AMPK, leading to a reduction in the p-MTOR levels. Moreover, VP3 can activate the ERK1/2 MAPK-MTOR and p38 MAPK-MTOR pathways to facilitate autophagy. Therefore, the findings suggest that viral proteins can work synergistically to initiate autophagy, which in turn enhances viral replication (Song et al., 2022a). Currently, no studies have been investigated the impact of 3Cpro on the PERK and ATF6 UPR signalling pathways.
The receptor protein SQSTM1/p62 plays a significant role in the process of selective autophagy by recruiting ubiquitinated proteins or pathogens to autophagosomes for degradation (Bjørkøy et al., 2005; Lamark et al., 2017). Previous research reports have shown that SQSTM1 targets ubiquitin-dependent and ubiquitin-independent viral capsids for autophagic clearance, including FMDV, chikungunya virus (CHIKV) (Berryman et al., 2012; Judith et al., 2013). By targeting the SVV VP1 and VP3 proteins to autophagosomes, SQSTM1 suppresses viral replication. Research has revealed that SVV 3Cpro proactively cleaves SQSTM1/p62 at E355, Q392, and Q395, thereby producing an SQSTM1 cleavage product that cannot effectively induce selective autophagy or inhibit SVV replication (Wen et al., 2021b; Figure 4).
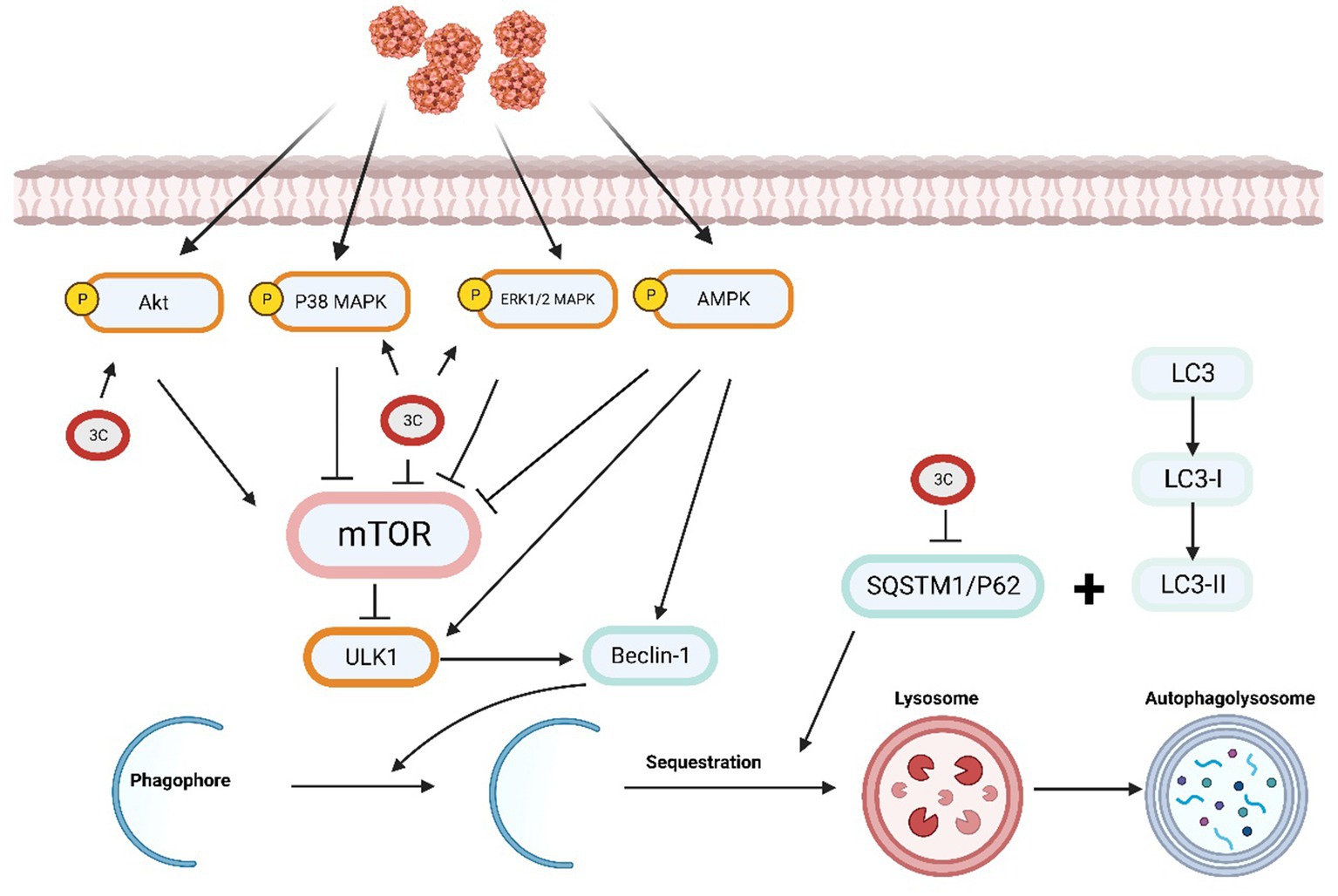
Figure 4. The SVV 3C protease is involved in the autophagy pathway and affects antiviral responses.
4. Conclusion
The 3Cpro of SVV is considered to play a significant role in SVV pathogenicity. This protease can affect host cell function through multiple mechanisms, inhibiting the immune response and promoting virus replication. Specifically, the protease can broadly inhibit the type I IFN pathway, enabling the virus to effectively evade host immune system attacks (Qian et al., 2017; Xue et al., 2018a,b; Wen et al., 2019). Additionally, SVV 3Cpro affects various processes, such as cell mRNA translation (Xue et al., 2020; Song et al., 2021, 2022b; Wen et al., 2022; Zhao et al., 2022), Pyroptosis (Wen et al., 2021a), apoptosis (Liu et al., 2019; Zhao et al., 2023), and autophagy (Wen et al., 2021b; Zhao et al., 2023), which may facilitate virus replication. Notably, this protease interacts with many crucial host proteins, cleaving them to exploit host cell resources while avoiding or mitigating immune responses (Table 1).
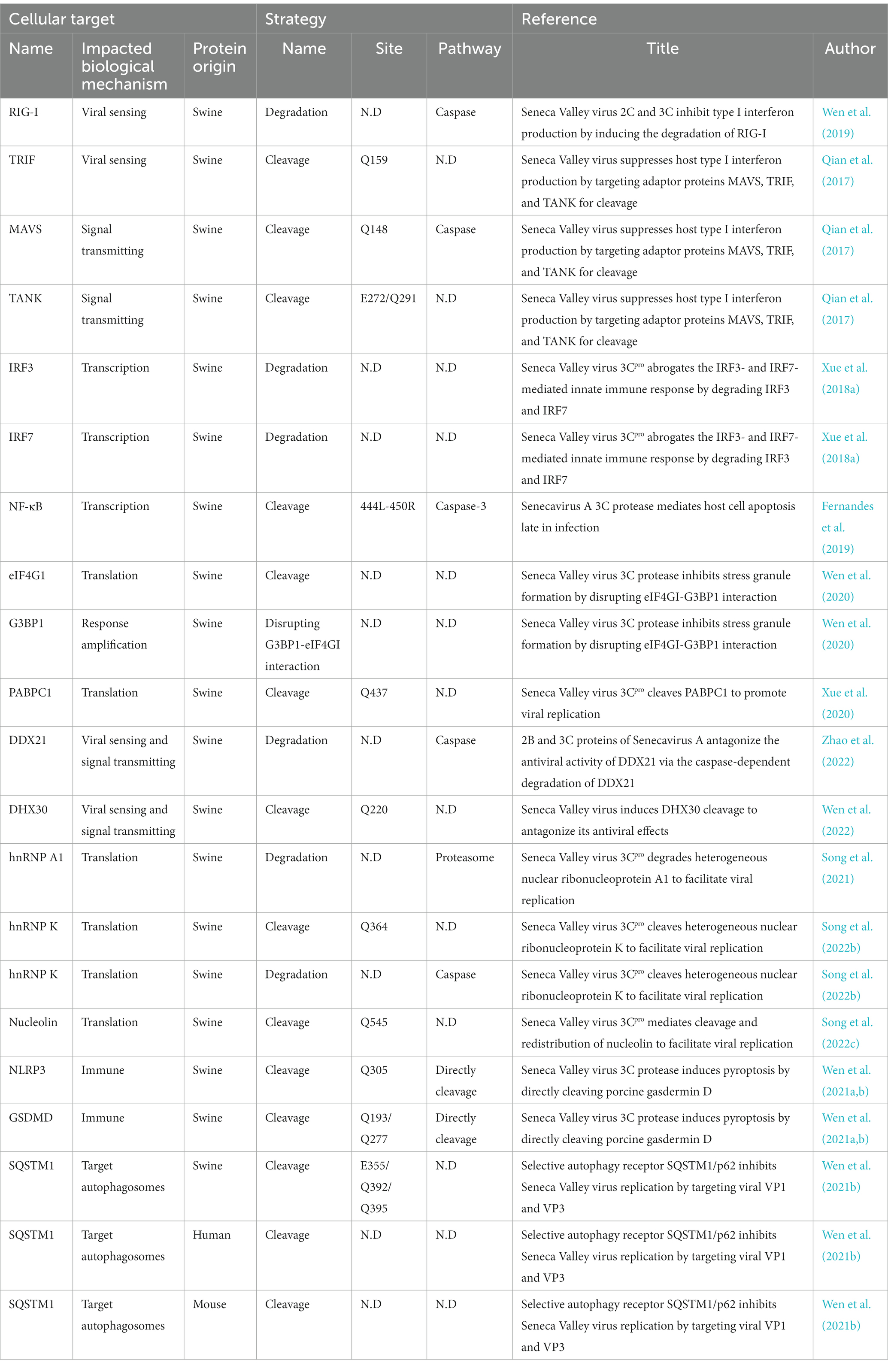
Table 1. Cleavage of cellular transcription factors by 3Cpro.
However, this review have several limitations. First, the referenced studies have heavily relied on in vitro cell experiments, and the results have not yet been validated in animal models. Second, investigating the existence of a synergistic effect between 3Cpro and other proteins has certain limitations. Furthermore, the described results from studies on the structural and functional changes of specific target proteins are not adequately comprehensive. Moreover, clarifying the differences in reactions among different hosts and cells is crucial. Additionally, due to limited research on the effects of SVV 3C protein on SGs, autophagy, and apoptosis, a comprehensive and in-depth understanding of its impact on these processes is still lacking.
It is crucial to examine how the virus employs 3Cpro to impair the host’s immune response. The related studies not only enhance our comprehension of the virus-and-host-cell interaction but also generate novel drug targets for antiviral agents. 3Cpro inhibitors restore the immune function of host cells and inhibit the replication of SVV. Several 3CLpro inhibitors have been shown to inhibit coronavirus replication in vitro and enhance the survival rate of mice infected with Middle East respiratory syndrome coronavirus (MERS-CoV). Additionally, the capsid binder vapendavir and the newly developed 3Cpro inhibitor SG85 potently inhibit enterovirus 71 replication (Tijsma et al., 2014; Rathnayake et al., 2020). Similarly, understanding the impact of SVV 3Cpro on processes such as autophagy and apoptosis would expose additional details about virus-to-cell interactions., and this research outcome is vital for the development of new antiviral treatments. As an oncolytic virus, studying the ability of SVV to infect human cells and replicate, as well as the impact of 3Cpro on cellular immunity and programmed cell death, can provide a theoretical basis for tumour-related research in therapy (Reddy et al., 2007). The effect of SVV 3Cpro on the host is complex and interconnected and disrupts signal transduction, nuclear transport, transcription, translation, and protein stability. These effects are presumed to synergize with one another, but the underlying mechanisms require additional exploration.
Author contributions
X-yZ and Y-yL jointly drafted the manuscript. H-xH, C-cZ, X-xL, B-pZ, and J-yL provided ideas that contributed to the conceptualization of this article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Wenzhou Basic Medical Health Science and Technology Project (grant number Y20220136).
Acknowledgments
All pathway maps are created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdullah, S. W., Wu, J., Zhang, Y., Bai, M., Guan, J., Liu, X., et al. (2021). DDX21, a host restriction factor of FMDV IRES-dependent translation and replication. Viruses 13:1765. doi: 10.3390/v13091765
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nature 413, 732–738. doi: 10.1038/35099560
Arzt, J., Bertram, M. R., Vu, L. T., Pauszek, S. J., Hartwig, E. J., Smoliga, G. R., et al. (2019). First detection and genome sequence of Senecavirus A in Vietnam. Microbiol. Resour. Announc. 8, e01247–e01218. doi: 10.1128/MRA.01247-18
Becherel, O. J., Gueven, N., Birrell, G. W., Schreiber, V., Suraweera, A., Jakob, B., et al. (2006). Nucleolar localization of aprataxin is dependent on interaction with nucleolin and on active ribosomal DNA transcription. Hum. Mol. Genet. 15, 2239–2249. doi: 10.1093/hmg/ddl149
Belov, G. A., and van Kuppeveld, F. J. (2012). (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2, 740–747. doi: 10.1016/j.coviro.2012.09.006
Berryman, S., Brooks, E., Burman, A., Hawes, P., Roberts, R., Netherton, C., et al. (2012). Foot-and-mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3-kinase-independent pathway. J. Virol. 86, 12940–12953. doi: 10.1128/JVI.00846-12
Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., et al. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614. doi: 10.1083/jcb.200507002
Castanier, C., Zemirli, N., Portier, A., Garcin, D., Bidère, N., Vazquez, A., et al. (2012). MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 10:44. doi: 10.1186/1741-7007-10-44
Choudhury, S. M., Ma, X., Zeng, Z., Luo, Z., Li, Y., Nian, X., et al. (2022). Senecavirus A 3D interacts with NLRP3 to induce IL-1β production by activating NF-κB and ion channel signals. Microbiol. Spectr. 10:e0209721. doi: 10.1128/spectrum.02097-21
Cohen, G. M. (1997). Caspases: the executioners of apoptosis. Biochem. J. 326, 1–16. doi: 10.1042/bj3260001
Coiras, M., López-Huertas, M. R., Mateos, E., and Alcamí, J. (2008). Caspase-3-mediated cleavage of p65/RelA results in a carboxy-terminal fragment that inhibits IκBα and enhances HIV-1 replication in human T lymphocytes. Retrovirology 5:109. doi: 10.1186/1742-4690-5-109
Dong, Y., Ye, W., Yang, J., Han, P., Wang, Y., Ye, C., et al. (2016). DDX21 translocates from nucleus to cytoplasm and stimulates the innate immune response due to dengue virus infection. Biochem. Biophys. Res. Commun. 473, 648–653. doi: 10.1016/j.bbrc.2016.03.120
Durut, N., and Sáez-Vásquez, J. (2015). Nucleolin: dual roles in rDNA chromatin transcription. Gene 556, 7–12. doi: 10.1016/j.gene.2014.09.023
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Fernandes, M. H. V., Maggioli, M. F., Otta, J., Joshi, L. R., Lawson, S., and Diel, D. G. (2019). Senecavirus A 3C protease mediates host cell apoptosis late in infection. Front. Immunol. 10:363. doi: 10.3389/fimmu.2019.00363
Galluzzi, L., Kepp, O., Trojel-Hansen, C., and Kroemer, G. (2012). Mitochondrial control of cellular life, stress, and death. Circ. Res. 111, 1198–1207. doi: 10.1161/CIRCRESAHA.112.268946
Greco, A., Arata, L., Soler, E., Gaume, X., Couté, Y., Hacot, S., et al. (2012). Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J. Virol. 86, 1449–1457. doi: 10.1128/JVI.06194-11
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hales, L. M., Knowles, N. J., Reddy, P. S., Xu, L., Hay, C., and Hallenbeck, P. L. (2008). Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J. Gen. Virol. 89, 1265–1275. doi: 10.1099/vir.0.83570-0
Han, S., Wang, X., Guan, J., Wu, J., Zhang, Y., Li, P., et al. (2021). Nucleolin promotes IRES-driven translation of foot-and-mouth disease virus by supporting the assembly of translation initiation complexes. J. Virol. 95:e0023821. doi: 10.1128/JVI.00238-21
Hause, B. M., Myers, O., Duff, J., and Hesse, R. A. (2016). Senecavirus A in pigs, United States, 2015. Emerg. Infect. Dis. 22, 1323–1325. doi: 10.3201/eid2207.151591
Hay, N., and Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704
Hou, L., Dong, J., Zhu, S., Yuan, F., Wei, L., Wang, J., et al. (2019). Seneca Valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology 537, 254–263. doi: 10.1016/j.virol.2019.08.029
Jean-Philippe, J., Paz, S., and Caputi, M. (2013). hnRNP A1: the Swiss army knife of gene expression. Int. J. Mol. Sci. 14, 18999–19024. doi: 10.3390/ijms140918999
Joshi, L. R., Fernandes, M. H. V., Clement, T., Lawson, S., Pillatzki, A., Resende, T. P., et al. (2016). Pathogenesis of Senecavirus A infection in finishing pigs. J. Gen. Virol. 97, 3267–3279. doi: 10.1099/jgv.0.000631
Judith, D., Mostowy, S., Bourai, M., Gangneux, N., Lelek, M., Lucas-Hourani, M., et al. (2013). Species-specific impact of the autophagy machinery on chikungunya virus infection. EMBO Rep. 14, 534–544. doi: 10.1038/embor.2013.51
Kaminskyy, V., and Zhivotovsky, B. (2010). To kill or be killed: how viruses interact with the cell death machinery. J. Intern. Med. 267, 473–482. doi: 10.1111/j.1365-2796.2010.02222.x
Kang, K. H., Lee, K. H., Kim, M. Y., and Choi, K. H. (2001). Caspase-3-mediated cleavage of the NF-κB subunit p65 at the NH2 terminus potentiates naphthoquinone analog-induced apoptosis. J. Biol. Chem. 276, 24638–24644. doi: 10.1074/jbc.M101291200
Kedersha, N., Panas, M. D., Achorn, C. A., Lyons, S., Tisdale, S., Hickman, T., et al. (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 212, 845–860. doi: 10.1083/jcb.201508028
Kim, H. S., Chang, I., Kim, J. Y., Choi, K. H., and Lee, M. S. (2005). Caspase-mediated p65 cleavage promotes TRAIL-induced apoptosis. Cancer Res. 65, 6111–6119. doi: 10.1158/0008-5472.CAN-05-0472
Klionsky, D. J., and Codogno, P. (2013). The mechanism and physiological function of macroautophagy. J. Innate Immun. 5, 427–433. doi: 10.1159/000351979
Knowles, N. J., Hales, L. M., Jones, B. H., Landgraf, J. G., House, J. A., Skele, K. L., et al. (2006). Epidemiology of Seneca Valley virus: identification and characterization of isolates from pigs in the United States. Northern Lights EUROPIC.
Lamark, T., Svenning, S., and Johansen, T. (2017). Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 61, 609–624. doi: 10.1042/EBC20170035
Leme, R. A., Oliveira, T. E., Alcântara, B. K., Headley, S. A., Alfieri, A. F., Yang, M., et al. (2016). Clinical manifestations of Senecavirus A infection in neonatal pigs, Brazil, 2015. Emerg. Infect. Dis. 22, 1238–1241. doi: 10.3201/eid2207.151583
Li, H., Lin, C., Qi, W., Sun, Z., Xie, Z., Jia, W., et al. (2023). Senecavirus A-induced glycolysis facilitates virus replication by promoting lactate production that attenuates the interaction between MAVS and RIG-I. PLoS Pathog. 19:e1011371. doi: 10.1371/journal.ppat.1011371
Li, Z., Zeng, W., Ye, S., Lv, J., Nie, A., Zhang, B., et al. (2018). Cellular hnRNP A1 interacts with nucleocapsid protein of porcine epidemic Diarrhea virus and impairs viral replication. Viruses 10:127. doi: 10.3390/v10030127
Lin, J. Y., Li, M. L., Huang, P. N., Chien, K. Y., Horng, J. T., and Shih, S. R. (2008). Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5′ untranslated region and participates in virus replication. J. Gen. Virol. 89, 2540–2549. doi: 10.1099/vir.0.2008/003673-0
Liu, T., Li, X., Wu, M., Qin, L., Chen, H., and Qian, P. (2019). Seneca Valley virus 2C and 3Cpro induce apoptosis via mitochondrion-mediated intrinsic pathway. Front. Microbiol. 10:1202. doi: 10.3389/fmicb.2019.01202
Liu, W., Yang, D., Sun, C., Wang, H., Zhao, B., Zhou, G., et al. (2020). hnRNP K is a novel internal ribosomal entry site-transacting factor that negatively regulates foot-and-mouth disease virus translation and replication and is antagonized by viral 3C protease. J. Virol. 94, e00803–e00820. doi: 10.1128/JVI.00803-20
Maggioli, M. F., Lawson, S., de Lima, M., Joshi, L. R., Faccin, T. C., Bauermann, F. V., et al. (2018). Adaptive immune responses following Senecavirus A infection in pigs. J. Virol. 92:e01717. doi: 10.1128/JVI.01717-17
McEwen, E., Kedersha, N., Song, B., Scheuner, D., Gilks, N., Han, A., et al. (2005). Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280, 16925–16933. doi: 10.1074/jbc.M412882200
Meng, K., Zhang, L., Xue, X., Xue, Q., Sun, M., and Meng, G. (2022). Structure of Senecavirus A 3C protease revealed the cleavage pattern of 3C protease in picornaviruses. J. Virol. 96:e0073622. doi: 10.1128/jvi.00736-22
Muzio, M., Chinnaiyan, A. M., Kischkel, F. C., O’Rourke, K., Shevchenko, A., Ni, J., et al. (1996). FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death—inducing signaling complex. Cells 85, 817–827. doi: 10.1016/S0092-8674(00)81266-0
Ng, C. S., Stobart, C. C., and Luo, H. (2021). Innate immune evasion mediated by picornaviral 3C protease: possible lessons for coronaviral 3C-like protease? Rev. Med. Virol. 31, 1–22. doi: 10.1002/rmv.2206
Oliver, F. J., de la Rubia, G., Rolli, V., Ruiz-Ruiz, M. C., de Murcia, G., and Murcia, J. M. (1998). Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J. Biol. Chem. 273, 33533–33539. doi: 10.1074/jbc.273.50.33533
Paludan, S. R., and Bowie, A. G. (2013). Immune sensing of DNA. Immunity 38, 870–880. doi: 10.1016/j.immuni.2013.05.004
Pasma, T., Davidson, S., and Shaw, S. L. (2008). Idiopathic vesicular disease in swine in Manitoba. Can. Vet. J. 49, 84–85.
Pathak, H. B., Arnold, J. J., Wiegand, P. N., Hargittai, M. R., and Cameron, C. E. (2007). Picornavirus genome replication: assembly and organization of the VPg uridylylation ribonucleoprotein (initiation) complex. J. Biol. Chem. 282, 16202–16213. doi: 10.1074/jbc.M610608200
Pomerantz, J. L., and Baltimore, D. (1999). NF-κB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18, 6694–6704. doi: 10.1093/emboj/18.23.6694
Qian, S., Fan, W., Liu, T., Wu, M., Zhang, H., Cui, X., et al. (2017). Seneca Valley virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J. Virol. 91, e00823–e00817. doi: 10.1128/JVI.00823-17
Rathnayake, A. D., Zheng, J., Kim, Y., Perera, K. D., Mackin, S., Meyerholz, D. K., et al. (2020). 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med. 12:eabc5332. doi: 10.1126/scitranslmed.abc5332
Reddy, P. S., Burroughs, K. D., Hales, L. M., Ganesh, S., Jones, B. H., Idamakanti, N., et al. (2007). Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J. Natl. Cancer Inst. 99, 1623–1633. doi: 10.1093/jnci/djm198
Saeng-Chuto, K., Rodtian, P., Temeeyasen, G., Wegner, M., and Nilubol, D. (2018). The first detection of Senecavirus A in pigs in Thailand, 2016. Transbound. Emerg. Dis. 65, 285–288. doi: 10.1111/tbed.12654
Schwandner, R., Dziarski, R., Wesche, H., Rothe, M., and Kirschning, C. J. (1999). Peptidoglycan-and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274, 17406–17409. doi: 10.1074/jbc.274.25.17406
Schwarz, E. M., Badorff, C., Hiura, T. S., Wessely, R., Badorff, A., Verma, I. M., et al. (1998). NF-κB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J. Virol. 72, 5654–5660. doi: 10.1128/JVI.72.7.5654-5660.1998
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shu, C., Sankaran, B., Chaton, C. T., Herr, A. B., Mishra, A., Peng, J., et al. (2013). Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure 21, 1137–1148. doi: 10.1016/j.str.2013.04.025
Smith, R. W. P., Anderson, R. C., Larralde, O., Smith, J. W. S., Gorgoni, B., Richardson, W. A., et al. (2017). Viral and cellular mRNA-specific activators harness PABP and eIF4G to promote translation initiation downstream of cap binding. Proc. Natl. Acad. Sci. U. S. A. 114, 6310–6315. doi: 10.1073/pnas.1610417114
Song, J., Hou, L., Quan, R., Wang, D., Jiang, H., and Liu, J. (2022a). Synergetic contributions of viral VP1, VP3, and 3C to activation of the AKT-AMPK-MAPK-MTOR signaling pathway for Seneca Valley virus-induced autophagy. J. Virol. 96:e0155021. doi: 10.1128/JVI.01550-21
Song, J., Quan, R., Wang, D., and Liu, J. (2022b). Seneca Valley virus 3C pro cleaves heterogeneous nuclear ribonucleoprotein K to facilitate viral replication. Front. Microbiol. 13:945443. doi: 10.3389/fmicb.2022.1038034
Song, J., Quan, R., Wang, D., and Liu, J. (2022c). Seneca Valley virus 3Cpro mediates cleavage and redistribution of nucleolin to facilitate viral replication. Microbiol. Spectr. 10:e0030422. doi: 10.1128/spectrum.00304-22
Song, J., Wang, D., Quan, R., and Liu, J. (2021). Seneca Valley virus 3Cpro degrades heterogeneous nuclear ribonucleoprotein A1 to facilitate viral replication. Virulence 12, 3125–3136. doi: 10.1080/21505594.2021.2014681
Sun, D., Chen, S., Cheng, A., and Wang, M. (2016). Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses 8:82. doi: 10.3390/v8030082
Sun, D., Vannucci, F., Knutson, T. P., Corzo, C., and Marthaler, D. G. (2017). Emergence and whole-genome sequence of Senecavirus A in Colombia. Transbound. Emerg. Dis. 64, 1346–1349. doi: 10.1111/tbed.12669
Tahiri-Alaoui, A., Zhao, Y., Sadigh, Y., Popplestone, J., Kgosana, L., Smith, L. P., et al. (2014). Poly(A) binding protein 1 enhances cap-independent translation initiation of neurovirulence factor from avian herpesvirus. PLoS One 9:e114466. doi: 10.1371/journal.pone.0114466
Tang, E. D., and Wang, C. Y. (2009). MAVS self-association mediates antiviral innate immune signaling. J. Virol. 83, 3420–3428. doi: 10.1128/JVI.02623-08
Taniguchi, T., Ogasawara, K., Takaoka, A., and Tanaka, N. (2001). IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19, 623–655. doi: 10.1146/annurev.immunol.19.1.623
Tijsma, A., Franco, D., Tucker, S., Hilgenfeld, R., Froeyen, M., Leyssen, P., et al. (2014). The capsid binder Vapendavir and the novel protease inhibitor SG85 inhibit enterovirus 71 replication. Antimicrob. Agents Chemother. 58, 6990–6992. doi: 10.1128/AAC.03328-14
Vallée, I., Tait, S. W., and Powell, P. P. (2001). African swine fever virus infection of porcine aortic endothelial cells leads to inhibition of inflammatory responses, activation of the thrombotic state, and apoptosis. J. Virol. 75, 10372–10382. doi: 10.1128/JVI.75.21.10372-10382.2001
Vannucci, F. A., Linhares, D. C., Barcellos, D. E., Lam, H. C., Collins, J., and Marthaler, D. (2015). Identification and complete genome of Seneca Valley virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound. Emerg. Dis. 62, 589–593. doi: 10.1111/tbed.12410
Watanabe, Y., Ohtaki, N., Hayashi, Y., Ikuta, K., and Tomonaga, K. (2009). Autogenous translational regulation of the Borna disease virus negative control factor X from polycistronic mRNA using host RNA helicases. PLoS Pathog. 5:e1000654. doi: 10.1371/journal.ppat.1000654
Wen, W., Li, X., Wang, H., Zhao, Q., Yin, M., Liu, W., et al. (2021a). Seneca Valley virus 3C protease induces pyroptosis by directly cleaving porcine gasdermin D. J. Immunol. 207, 189–199. doi: 10.4049/jimmunol.2001030
Wen, W., Li, X., Yin, M., Wang, H., Qin, L., Li, H., et al. (2021b). Selective autophagy receptor SQSTM1/p62 inhibits Seneca Valley virus replication by targeting viral VP1 and VP3. Autophagy 17, 3763–3775. doi: 10.1080/15548627.2021.1897223
Wen, W., Yin, M., Zhang, H., Liu, T., Chen, H., Qian, P., et al. (2019). Seneca Valley virus 2C and 3C inhibit type I interferon production by inducing the degradation of RIG-I. Virology 535, 122–129. doi: 10.1016/j.virol.2019.06.017
Wen, W., Zhao, Q., Yin, M., Qin, L., Hu, J., Chen, H., et al. (2020). Seneca Valley virus 3C protease inhibits stress granule formation by disrupting eIF4GI-G3BP1 interaction. Front. Immunol. 11:577838. doi: 10.3389/fimmu.2020.577838
Wen, W., Zheng, Z., Wang, H., Zhao, Q., Yin, M., Chen, H., et al. (2022). Seneca Valley virus induces DHX30 cleavage to antagonize its antiviral effects. J. Virol. 96:e0112122. doi: 10.1128/jvi.01121-22
Wu, H., Li, C., Ji, Y., Mou, C., Chen, Z., and Zhao, J. (2022). The evolution and global spatiotemporal dynamics of Senecavirus A. Microbiol. Spectr. 10:e0209022. doi: 10.1128/spectrum.02673-22
Wu, Q., Zhao, X., Bai, Y., Sun, B., Xie, Q., and Ma, J. (2017). The first identification and complete genome of Senecavirus A affecting pig with idiopathic vesicular disease in China. Transbound. Emerg. Dis. 64, 1633–1640. doi: 10.1111/tbed.12557
Xu, W., Hole, K., Goolia, M., Pickering, B., Salo, T., Lung, O., et al. (2017). Genome wide analysis of the evolution of Senecavirus A from swine clinical material and assembly yard environmental samples. PLoS One 12:e0176964. doi: 10.1371/journal.pone.0176964
Xue, Q., Liu, H., Zhu, Z., Xue, Z., Liu, X., and Zheng, H. (2020). Seneca Valley virus 3Cpro cleaves PABPC1 to promote viral replication. Pathogens 9:443. doi: 10.3390/pathogens9060443
Xue, Q., Liu, H., Zhu, Z., Yang, F., Ma, L., Cai, X., et al. (2018a). Seneca Valley virus 3Cpro abrogates the IRF3-and IRF7-mediated innate immune response by degrading IRF3 and IRF7. Virology 518, 1–7. doi: 10.1016/j.virol.2018.01.028
Xue, Q., Liu, H., Zhu, Z., Yang, F., Xue, Q., Cai, X., et al. (2018b). Seneca Valley virus 3C protease negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. Antivir. Res. 160, 183–189. doi: 10.1016/j.antiviral.2018.10.028
Yamasaki, S., and Anderson, P. (2008). Reprogramming mRNA translation during stress. Curr. Opin. Cell Biol. 20, 222–226. doi: 10.1016/j.ceb.2008.01.013
Yang, X., Hu, Z., Zhang, Q., Fan, S., Zhong, Y., Guo, D., et al. (2019). SG formation relies on eIF4GI-G3BP interaction which is targeted by picornavirus stress antagonists. Cell Discov. 5:1. doi: 10.1038/s41421-018-0068-4
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi: 10.1016/j.ceb.2009.11.014
Ye, P., Liu, S., Zhu, Y., Chen, G., and Gao, G. (2010). DEXH-box protein DHX30 is required for optimal function of the zinc-finger antiviral protein. Protein Cell 1, 956–964. doi: 10.1007/s13238-010-0117-8
Yoneyama, M., Kikuchi, M., Matsumoto, K., Imaizumi, T., Miyagishi, M., Taira, K., et al. (2005). Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858. doi: 10.4049/jimmunol.175.5.2851
Zhao, K., Guo, X. R., Liu, S. F., Liu, X. N., Han, Y., Wang, L. L., et al. (2022). 2B and 3C proteins of Senecavirus A antagonize the antiviral activity of DDX21 via the caspase-dependent degradation of DDX21. Front. Immunol. 13:951984. doi: 10.3389/fimmu.2022.1089200
Zhao, H. F., Meng, L., Geng, Z., Gao, Z. Q., Dong, Y. H., Wang, H. W., et al. (2023). Allosteric regulation of Senecavirus A 3Cpro proteolytic activity by an endogenous phospholipid. PLoS Pathog. 19:e1011411. doi: 10.1371/journal.ppat.1011411
Zheng, Y., Jönsson, J., Hao, C., Shoja Chaghervand, S., Cui, X., Kajitani, N., et al. (2020). Heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) and hnRNP A2 inhibit splicing to human papillomavirus 16 splice site SA409 through a UAG-containing sequence in the E7 coding region. J. Virol. 94, e01509–e01520. doi: 10.1128/JVI.01509-20
Zhu, Q., Yu, T., Gan, S., Wang, Y., Pei, Y., Zhao, Q., et al. (2020). TRIM24 facilitates antiviral immunity through mediating K63-linked TRAF3 ubiquitination. J. Exp. Med. 217:e20192083. doi: 10.1084/jem.20192083
Keywords: 3C protease, virus replication, interferon pathway, host RNA metabolism, programmed cell death
Citation: Zhang X-y, Li Y-y, Huang H-x, Zhao C-c, Lei X-x, Zhao B-p, Lu J-y, Lan T and Sun W-c (2023) Seneca Valley virus 3Cpro antagonizes host innate immune responses and programmed cell death. Front. Microbiol. 14:1235620. doi: 10.3389/fmicb.2023.1235620
Edited by:
Jose M. Jimenez-Guardeño, King’s College London, United KingdomReviewed by:
Jia Xue, China Agricultural University, ChinaTong-Yun Wang, Harbin Veterinary Research Institute (CAAS), China
Copyright © 2023 Zhang, Li, Huang, Zhao, Lei, Zhao, Lu, Lan and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tian Lan, ODI3MDAyMTUxQHFxLmNvbQ==; Wen-chao Sun, c3Vud2VuY2hhbzEzMUAxNjMuY29t