
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 14 February 2025
Sec. Nephrology
Volume 12 - 2025 | https://doi.org/10.3389/fmed.2025.1526090
 Vincent Boima1
Vincent Boima1 Alex Baafi Agyekum2Khushali Ganatra3
Alex Baafi Agyekum2Khushali Ganatra3 Francis Agyekum1*Edward Kwakyi1Jalil Inusah3
Francis Agyekum1*Edward Kwakyi1Jalil Inusah3 Elmer Nayra Ametefe4
Elmer Nayra Ametefe4 Dwomoa Adu1
Dwomoa Adu1Chronic kidney disease (CKD) is a global public health issue characterized by progressive loss of kidney function, of which end-stage kidney disease (ESKD) is the last stage. The global increase in the prevalence of CKD is linked to the increasing prevalence of traditional risk factors, including obesity, hypertension, and diabetes mellitus, as well as metabolic factors, particularly insulin resistance, dyslipidemia, and hyperuricemia. Mortality and comorbidities, such as cardiovascular complications, rise steadily as kidney function deteriorates. Patients who progress to ESKD require long-term kidney replacement therapy, such as transplantation or hemodialysis/peritoneal dialysis. It is currently understood that a crucial aspect of CKD involves persistent, low-grade inflammation. In addition, increased oxidative and metabolic stress, endothelial dysfunction, vascular calcification from poor calcium and phosphate metabolism, and difficulties with coagulation are some of the complex molecular pathways underlying CKD-related and ESKD-related issues. Novel mechanisms, such as microbiome dysbiosis and apolipoprotein L1 gene mutation, have improved our understanding of kidney disease mechanisms. High kidney disease risk of Africa has been linked to APOL1 high-risk alleles. The 3-fold increased risk of ESKD in African Americans compared to European Americans is currently mainly attributed to variants in the APOL1 gene in the chromosome 22q12 locus. Additionally, the role of new therapies such as SGLT2 inhibitors, mineralocorticoid receptor antagonists, and APOL1 channel function inhibitors offers new therapeutic targets in slowing down the progression of chronic kidney disease. This review describes recent molecular mechanisms underlying CKD and emerging therapeutic targets.
Chronic kidney disease (CKD) is characterized by progressive loss of kidney function, ultimately leading to end-stage kidney disease (ESKD), necessitating long-term kidney replacement therapy such as transplantation or hemodialysis/peritoneal dialysis. As kidney function declines, mortality and comorbidities, particularly cardiovascular complications, rise steadily (1).
CKD is a significant global public health challenge, particularly affecting the elderly population, with nearly half of CKD patients aged over 70 years. However, while younger patients with CKD typically experience progressive loss of kidney function, 30% of patients over 65 years of age with CKD have stable disease (2–7). Currently, CKD affects 10–15% of the global population, significantly impacting overall health. The surge in CKD prevalence worldwide is primarily attributed to the escalating prevalence of traditional risk factors, such as obesity, hypertension, and diabetes mellitus (2). Additionally, metabolic factors, including insulin resistance, dyslipidemia, and hyperuricemia, have been associated with CKD development and progression. Some studies indicate a higher prevalence of CKD among men, with African Americans exhibiting a higher predisposition to kidney damage than Caucasians (8).
International guidelines currently describe CKD as a serious condition that typically progresses asymptomatically. It is characterized by reduced kidney function, indicated by a glomerular filtration rate (GFR) of less than 60 mL/min per 1.73 m2, or markers of kidney damage, such as albuminuria (albumin: creatinine ratio ≥ 30 mg/g), or both, that persist for a minimum of 3 months, irrespective of the underlying cause (2, 4, 7, 9).
The primary challenges associated with CKD include progression to kidney failure and the development of cardiovascular and metabolic diseases. Emerging evidence suggests that early detection and treatment can prevent or slow down some of these adverse outcomes. Blood pressure monitoring, urinalysis, and serum creatinine measurement with an estimation of GFR are some of the recommended screening measures for high-risk populations, which include people with hypertension, diabetes mellitus, and those older than 65 years (8).
This paper describes the recent advances in the molecular mechanisms underlying chronic kidney disease and new therapeutic targets that have emerged from these insights. These molecular mechanisms include oxidative stress, the role of the inflammatory cells, neutrophil gelatinase-associated lipocalin, matrix metalloproteinases, genetic mutations, and the gut–kidney axis (2, 8).
The kidneys play a crucial role in the ability of the human body to maintain homeostasis. To accomplish this, a wide range of cell types are arranged in the intricate three-dimensional structure of the nephron, the functional unit of the kidney, enabling it to react with various intracellular and intercellular signals, as well as hormonal, neurological, and inflammatory stimuli (10).
CKD and ESKD are characterized by a complex interplay of molecular pathways. Inflammation, increased oxidative and metabolic stress, endothelial dysfunction, vascular calcification resulting from poor calcium and phosphate metabolism, and difficulties with coagulation contribute significantly to the pathogenesis of CKD- and ESKD-related complications. Furthermore, the decline in GFR in advanced stages of CKD leads to the accumulation of drugs and chemical compounds that are typically metabolized or eliminated by the kidneys. This accumulation exacerbates renal dysfunction and contributes to disease progression [1]This article will discuss the exogenous and endogenous substances, cell injury, and genetic-related mechanisms relevant to CKD and ESKD. Each section will also delve into the available treatments.
Perfluoroalkyl chemicals, including perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS), are found in all populations worldwide, regardless of geographical location, due to their extensive use (11). The plasma concentrations of these substances exhibit geographical variation (12, 13). According to reports, the levels of per- and poly-fluoroalkyl substances (PFAS) in drinking water are consistent with PFAS exposure (14). Consequently, multiple regulatory entities have established the approved threshold for plasma concentration. For example, the National Food Agency in Sweden and the Environmental Protection Agency in the United States have suggested limits of 90 ng/L and a range of 13 to 1,000 ng/L, respectively (15, 16). PFOA has a long half-life that may last for several years (1.2 to 14.9 years) (17–21). The long half-life of PFOA is primarily due to its significant reabsorption in the renal tubules, which leads to sluggish urine excretion (22). PFAS substances build up in breast milk, liver, and kidneys upon absorption. Upon introduction into the body, perfluorooctanoic acid (PFOA) typically binds to proteins instead of lipids. As a result, it typically accumulates in tissues and organs that have a high protein content (21, 23, 24). Animal studies have demonstrated that PFAS are most highly concentrated in the kidney, liver, and lungs. The human body does not metabolize PFAS, leading to its excretion without any metabolic transformations (13, 22). The reabsorption of PFAS in the kidneys may result in progressive kidney damage over time. Epidemiological studies have demonstrated a correlation between exposure to perfluorooctanoic acid (PFOA) and various forms of kidney disease (25). PFOA exposure causes renal hypertrophy, tissue proliferation, and microvascular dysfunction (26).
Exposure to perfluorooctanoic acid (PFOA) alters many signaling pathways. These pathways include the inflammatory pathway, the oxidative stress pathway, the peroxisome proliferator-activated receptor pathway, DNA methylation, and the autophagy pathway (26). Animal experiments have found that PFOA causes oxidative stress in the kidney and liver. Reactive oxygen species cause oxidation that exceeds the capacity of antioxidant defense system, leading to oxidative stress (26). This leads to detrimental effects on the peroxide of membrane phospholipids, DNA damage and mutation, oxidation and deactivation of proteins and enzymes, and the commencement of the apoptosis process (27). In order to demonstrate the causal relationship between PFAO and oxidative stress, scientists conducted an experiment where they administered an antioxidant known as N-acetylcysteine (NAC). The purpose of this experiment was to observe whether NAC might mitigate or reduce the biomarkers associated with liver and kidney damage caused by PFOA. The authors demonstrated that NAC decreased the biomarkers of PFOA-induced kidney and liver toxicity (28). PFOA has also been shown to activate the nuclear receptor peroxisome proliferator α (PPAR α), which changes how the kidneys work. However, the precise workings of this pathway are still poorly understood (29, 30). PPAR receptors have subtypes, namely PPARα, PPARβ, and PPARγ, which share the same core structure (31). Previous research suggests that the levels of PPARα are elevated in the kidney and adrenal glands. Additional studies have demonstrated that PFOA has the ability to stimulate the activation of mouse and human PPARα and PPARδ/γ in mouse models (32). The immune-damaging effects that PFAO caused in zebrafish kidneys showed that it changed the activity of NF-κB transcription factors, which, in turn, changed the transcription of cytokines. PFOA initially modulates the Toll-like receptors (TLR), hence regulating the MyD88 and NF-κB pathways to govern cytokine transcription and stimulate the immune system in zebrafish (33).
Aluminum (Al) has favorable physical and chemical properties, making it the most widely used element in medicine, industry, and everyday life. The kidney primarily excretes Al, making it the primary location for Al accumulation and, consequently, a major site of Al-induced organ damage. A previous study found that chronic exposure to Al leads to kidney accumulation and, consequently, impairment in kidney function. On the other hand, our understanding of cause of kidney damage of Al remains incomplete. However, it may be because extracellular matrix (ECM) accumulation and apoptosis work together in several different pathogenic mechanisms to cause the injury and progression of kidney disease (34). In a related study, exposure to Al was found to upregulate TGF-β, thus inducing oxidative stress with an attendant increase in apoptosis-related protein expression and subsequent kidney cell apoptosis (35). Another study also showed increased ECM protein expression in animals exposed to Al (36). A recent study on animals showed that Al treatment increased apoptosis and increased TGF-β1 and its downstream Smad2 mediators (34). This suggests that an abnormality in the TGF-1/Smad2 signaling pathway likely causes Al-induced kidney damage. One of the main ways that progressive tubular and interstitial fibrosis occurs is through apoptotic death and the buildup of ECM (37, 38). Arsenic (As) is a noxious metallic element that is abundantly present on our planet and typically forms chemical bonds with oxygen, chlorine, and sulfur. Therefore, it is referred to as an inorganic arsenic (39). Humans are exposed to arsenic through dietary sources, the environment, and contaminated drinking water. Common dietary sources of arsenic, such as fish and other shellfish, may have elevated quantities of this element. In addition, youngsters may come into contact with As (arsenic) as a result of their regular interaction with sand (40–42). Following ingestion, the kidneys play a crucial role in eliminating it, making them a key site for absorption and buildup. Prior animal investigations indicate that glucose transporters GLUT1 (SLC2A1) and GLUT5 (SLC2A5) are likely to have important functions in the uptake of As at the basolateral membrane of the proximal tubular cells as well as at the peritubular capillaries into the proximal tubular cells (43). Additional animal research indicates that aquaporin 3 (AQP3) channels may also take in arsenic (As). Additional transporters potentially essential for arsenic uptake include inorganic anion-transporting peptides, such as OATP2B1 (SLCO2B1). The mechanism by which As exits the kidney is still unclear. However, it is possible that GLUT1 and GLUT5, which have the ability to transport substances in both directions, may play a role in transporting As out of the renal tubular cells (44). In vitro studies demonstrate that arsenic (As) export relies on the interaction between glutathione (GSH) and As, forming a complex. The As-GSH complex is subsequently removed in transportable forms such as As(GS)3 and MAs(GS)2 (45). The metal and toxicant extrusion protein (MATE; SLC47A1) is another transporter found on the apical membrane of the proximal tubular cells that export As out of the renal tubular cells (46).
Acute poisoning can cause damage to the tubules and interstitium of the kidneys, leading to hypercalciuria, albuminuria, nephrocalcinosis, and renal papillary necrosis. Internalization of As can lead to alterations in intracellular signaling pathways (47, 48). Exposure to arsenic (As) leads to an increase in the generation of ROS and raises the levels of heme oxygenase (HMOX1), a crucial modulator of heme oxidation and reaction to stress in kidney epithelial cells (49). It also increases the likelihood of developing hypertension, kidney damage, albuminuria, and chronic kidney disease (CKD), ascribed to the death of nephrons and subsequent hyperfiltration in the surviving nephrons (50, 51).
Mercury, a poisonous metal, can be found in several industrial and ecological contexts. It can be found in several organic and inorganic forms. Methylmercury (CH3Hg+) is the predominant form of organic mercury that humans are typically exposed to in the environment (52, 53). Mercury exposure can also happen when people come into contact with polluted water, consume contaminated food, engage in certain occupations, or interact with contaminated soil (54). After 2 weeks of being consumed, the body transforms CH3Hg + into Hg2+ (55). The kidneys are the primary location of mercury accumulation and toxicity, as they are responsible for eliminating both organic and inorganic forms of mercury from the body (47, 56). Exposure to any form of mercury can lead to renal diseases. However, kidney problems caused by Hg2+ conjugates are particularly severe. The first segment of the proximal tubule that is impacted upon exposure is the pars recta, which is particularly vulnerable to the toxic effects of mercury. Minimal amounts of mercury have no impact on the distal nephron segment and the pars convoluta. However, elevated levels can lead to damage and necrosis in these areas (57–59). Observable proof of kidney damage caused by mercury exposure includes alterations in mitochondrial morphology and the presence of pyknotic nuclei (60). After being exposed for a few hours, the cells experience a loss of microvilli, swelling of the mitochondria, and dilation of the endoplasmic reticulum. During the final phases following exposure, the plasma membrane ruptures, resulting in reduced interaction with the basement membrane (60). Prolonged exposure to mercury might also impact the glomeruli leading to glomerular fibrosis and membranous nephropathy (60).
Research increasingly suggests a bidirectional relationship between the gut microbiome and kidney health. Chronic kidney disease can alter the gut environment, which further promotes dysbiosis, which increases the risk of CKD progression and other CKD-related comorbidities, such as cardiovascular disease. These CKD-related events happen through many different mechanisms, such as microbiome metabolites, weakened intestinal barriers, and changes in the neuroendocrine immune system (61, 62). Three naturally occurring microbiome-derived toxins—indoxyl sulfate (IS), p-cresyl sulfate (pCS), and trimethylamine N-oxide (TMAO)—are linked to the development of cardiovascular disease, the worsening of kidney disease, and death from these conditions (61, 62).
IS is a uremic toxin that forms complexes with proteins. It is produced when bacteria digest tryptophan in meals and is eliminated from the body through urine (61, 62). The liver metabolizes IS into indole, which raises the likelihood of peripheral vascular disease and vascular access thrombosis (61–63). As renal function deteriorates, the level of IS in the plasma rises, confirming previous findings that the baseline concentration of IS can serve as an indicator of renal insufficiency (63). A scientific study has demonstrated that IS controls the expression of genes in the kidneys that are linked to tubulointerstitial fibrosis, such as transforming growth factor β1 and a tissue inhibitor of metalloproteinases (62, 64). Another study demonstrated that mouse podocytes, when exposed to IS for a prolonged duration, exhibited indications of a pro-inflammatory phenotype, a disrupted actin cytoskeleton, decreased expression of podocyte-specific genes, and diminished cell survival (65).
During the later stages of erythropoiesis, it has been observed that human primary CD34+ cells experience the apoptotic impact of IS on erythropoiesis. Furthermore, both human primary CD34+ cells treated with IS and a mouse model with 5/6 Nx exhibited a blockage at the BFU-E stage of erythropoiesis. Ultimately, IS eliminates regulatory mechanisms on several genes associated with erythropoiesis. The proteins involved are GATA-1, EPO-R, and β-globin. IS may impair the viability and differentiation of erythroid progenitor cells. This could hinder the process of erythropoiesis and contribute to the development of anemia in individuals with CKD (66).
Tyrosine and phenylalanine undergo anerobic bacterial fermentation in the colon, resulting in the production of pCS. Following absorption, the liver undergoes conjugation of pCS with other molecules through the addition of sulfate (67–69). In animal models of CKD, pCS increased the formation of reactive oxygen species (ROS), which triggered nicotinamide adenine dinucleotide phosphate oxidase and elevated caspase-3 activity, leading to accelerated apoptosis (70). In a prior investigation involving mice with partial nephrectomy, the activation of either IS or pCS stimulated the renin–angiotensin–aldosterone system (RAAS) in the kidneys, leading to interstitial fibrosis and glomerulosclerosis (71).
TMAO is produced by the consumption of dietary choline, phosphatidylcholine, and L-carnitine. Prior investigations discovered an inverse relationship between TMAO and glomerular filtration rate in individuals with CKD and established a connection between elevated levels of TMAO and tubulointerstitial fibrosis, suggesting an unfavorable prognosis for CKD patients (72, 73). Another study indicates that TMAO enhances the synthesis of SMAD3, a crucial regulator of fibrosis, and elevates the likelihood of atherosclerosis and thrombosis, hence increasing the risk of ischemic heart disease (74). Therefore, for individuals with chronic kidney disease TMAO was identified as an indicator of cardiovascular disease risk in the early stages.
Research has shown that epigenetic disruptions play a crucial role in the progression of CKD, and metabolic conditions such as uremia can trigger changes in epigenetic-regulated gene expression (75). The end result is the creation of uremic memory, which has the potential to initiate DNA methylation (76). This process creates an enduring epigenetic memory that can significantly alter the expression of genes (77). This involves a network of epigenetic regulators and transcription factors, specifically SIX2, HNF, and TCFAP, located within the methylation areas of DNA (78, 79). Researchers have demonstrated that DNA methylation alters the expression of genes involved in inflammation, fibrosis, kidney development, and renal function. Additionally, elevated levels of homocysteine, hypoxia, and inflammation have the potential to alter the epigenetic control of genes in CKD (76, 80, 81). As a result, it can initiate the progression of CKD.
Multiple studies have demonstrated that the gut microbiota affects the nutrition, metabolism, and immune system under physiological settings of the host. On the contrary, diseases such as obesity, diabetes, and cardiovascular diseases, have been linked to microbiome disturbances in the gut. The capacity of the gut microbiota to adapt is crucial for maintaining gut homeostasis, although drastic alterations caused by antibiotics or food might be harmful (2, 82–84). The gut microbiota, as an ecosystem, primarily plays trophic and defensive roles, but it also has several impacts on human physiology. One of these is the ability of commensal bacteria to enhance the intestinal epithelial barrier (8, 85).
In addition to their trophic and defensive activities, the gut microbiota acts as an ecosystem that has several impacts on human physiology. Commensal bacteria perform a variety of functions, one of which is enhancing the intestinal epithelial barrier (8, 86). Protein fermentation by the bacteria is the principal pathomechanism. The process is responsible for the formation of urea solutes such as indoxyl sulfate, p-cresyl sulfate, phenyl sulfate, cholate, hippurate, dimethylglycine, Î3-guanidino-butyrate, glutarate, 2-hydroxy-pentanoate, trimethylamine N-oxide, and phenaceturate. Reduced function of the epithelial barrier has the potential to cause oxidative stress damage to the kidneys by increasing the transfer of uremic toxins made by bacteria. Endotoxemia is common in uremic patients, even when a clinical infection is not present (8, 87, 88).
.The “gut–kidney crosstalk” is about how CKD, the digestive system, and changes in the permeability of the intestinal epithelial barrier affect each other” (2). Gut dysbiosis and subsequent bacterial translocation can lead to chronic systemic inflammation in persons with CKD. It is also known that microbiome dysbiosis can result in cardiovascular disease, insulin resistance, and diabetes mellitus, increasing the risk of CKD. Gut dysbiosis is characterized by the excessive growth of harmful bacteria, leading to the increased release of substances such as LPS, peptidoglycans, bacterial DNA, and outer membrane proteins into the bloodstream of the host. This, in turn, causes prolonged activation of the immune system. The aforementioned harmful substances alter the ability of the intestines to allow substances to pass through and activate the immune system of the intestinal lining. This leads to the creation of substances that cause inflammation, such as interferon-γ (IFN-γ), TNF-α, and IL-6 (89, 90).
Additionally, the uremic milieu that results in elevated expression of TLR2 and TLR4 may be the reason why neutrophils and monocytes from CKD patients exhibit an excessive response to stimulation with lipopolysaccharides (LPS) (2, 87, 90–95).
In order to maintain the balance of the gastrointestinal system, it is crucial to stimulate the development of mucosal immune responses. This is achieved through the activation of pathogen sensors, such as TLRs, NLRs, and RIG-I-like receptors, which are distributed throughout the intestinal lining. These sensors are capable of identifying PAMPs and initiating a series of signaling pathways and molecular processes. As a result, the production of anti-infective cytokines and other defensive molecules in the intestinal mucosa is generated (96).
Dendritic cells, which are adept at presenting antigens, are part of the gut immune system together with intestinal intraepithelial lymphocytes and T lymphocytes in the lamina propria. Numerous immunological and epithelial cells exhibit the important inflammasome family member NLRP3. Reactive oxygen species and toxins from gut bacteria cause the NLRP3 inflammasome to make more IL-1 and IL-18, which are usually activated by caspase-1 downstream effector proteins (2, 96, 97).
Both CKD and the gut microbiome are influenced by one another. The gut microbial composition is greatly affected by chronic kidney disease (CKD) and is highly sensitive to the number of UTs, just as gut dysbiosis can affect kidney function (2, 98). Urease-positive microbes hydrolyze elevated quantities of urea to ammonium hydroxide in the intestinal lumen. The disruption of tight junctions accelerates the subsequent progression of kidney dysfunction. Furthermore, it impairs the IEB and makes it more permeable, which opens the door for bacterial toxins to enter the bloodstream (2, 99–104).
The inflammatory mediators produced by microbiome dysbiosis (IFN-γ, TNF-α, and IL-6) may increase the expression of apolipoprotein A-1 (APOL1). IFN-γ and TNF-α increase the expression of APOL1 in endothelial cells and podocytes (105, 106). The increased expression of G1 and G2 risk variants of APOL1 has been linked to a decline in kidney function and albuminuria (107).
Cellular metabolism has the capacity to generate acrolein, a compound that is commonly found in both the environment and food that we consume. It is an α,β-unsaturated aldehyde that is released during the breakdown of petroleum fuels, biofuel, plastic, paper, and wood (108). Direct contact with this primary constituent of tobacco smoke results in immediate damage to the skin and lungs. Individuals who are at high risk of exposure include cigarette users, firefighters, workers in the acrolein industry, and residents of densely polluted cities (108–110). Cooked, fried, and charred food and beer, wine, rum, and bread are noteworthy sources of acrolein, which is produced when vegetable and animal fats are overheated (111). Cells synthesize acrolein through various metabolic pathways, including the metabolism of polyamines such as spermine and spermidine by amine oxidase, the breakdown of threonine by neutrophil-derived myeloperoxidase, the breakdown of cancer drugs such as cyclophosphamide, and the lipid peroxidation of polyunsaturated fatty acids (PUFAs) (110, 112). Research conducted in vivo and in vitro has shown that acrolein causes oxidative stress, resulting in the rupture of cell membranes, DNA and mitochondria damage, endoplasmic reticulum (ER) stress, and the potential to initiate apoptosis (112, 113). Furthermore, both high and low doses of acrolein, as well as prolonged exposure to acrolein, result in cellular damage through immunological and inflammatory mechanisms. These processes involve (1) enhancing inflammatory responses by activating NF-κB, IL-8, COX-2, IL-1β, IL-6, TNF, IFN-γ, KC, MCP-1, 5-lipoxygenase, LTB4, and MMP, resulting in tissue damage and inflammation (114, 115); and (2) suppressing immune responses by activating NF-κB, TNFα, IL-10, IFN-γ, and GM-CSF, thereby increasing the susceptibility to infections (116, 117).
Acrolein can induce ischemia and reperfusion damage via inflammatory mechanisms (118). Acrolein contributes to the development of diabetic nephropathy by promoting the accumulation of extracellular matrix, increasing the production of angiotensin II (Ang II), activating MAPK signaling pathways that phosphorylate JNK, ERK, or p38, increasing the expression of inflammatory cytokines such as IL-6, IL-1beta, IL-18, and TNF-alpha, and cleaving caspase 9, caspase 3, and PARP (119). Cyclophosphamide and ifosfamide, both used in cancer treatment, undergo metabolism to produce acrolein, which is a significant concern because it induces oxidative stress and can lead to hemorrhagic cystitis (120).
AST-120 is commonly used in CKD patients as an oral charcoal adsorbent to absorb uremic toxins and their derivatives, including IS. Previous studies revealed that AST-120 contributes to changes in the gut microbiome composition, reduces ROS production from endothelial cells, and thus blocks the resultant oxidative stress and inflammation (121, 122). Again, other research reports indicate that AST-120 reduced proteinuria, signs of uremia, and prolonged time to dialysis (121, 123). Nevertheless, recent studies revealed that AST-120 reduced uremic symptoms but did not have much impact on renal function or all-cause mortality (124). Some studies have proven that phosphate binders such as sevelamer can bind uremic toxins, but their effectiveness in removing uremic toxins such as IS and pCS has been inconsistent in other studies (125).
Although some clinical trials have produced encouraging findings, there are currently few studies evaluating the impact of therapies meant to alter the microbiome in individuals with chronic kidney disease. A meta-analysis of studies investigating the effect of prebiotics on renal function revealed that supplementing with fiber markedly reduced serum urea levels. In pilot research, probiotics such as Lactobacillus acidophilus, Bifidobacterium longum, and Streptococcus thermophilus were administered, with favorable results showing significantly lowered blood urea levels. However, subsequent clinical trials failed to validate these findings.
The SYNERGY randomized trial aimed to assess whether symbiotic therapy, involving the combined use of pre- and probiotics, modifies the gut microbiota and lowers blood levels of uremic toxins produced by the microbiome in CKD patients. The findings of the study showed that while blood levels of P-Cresol sulfate (PCS) considerably decreased along with a shift in the microbiota of the stool toward a healthier one, levels of indoxyl sulfate (IS) did not change as a result of the intervention (126).
Scientists recently used gene sequencing to manufacture disease-specific probiotics. For example, the next generation of probiotics (NGP) has shown potential as disease-specific therapeutics and will help us understand the effectiveness and safety of probiotic microorganisms (127, 128). As a result, investigations have shown that nanoprobiotics and nanoprebiotics are effective therapies for dysbiosis (129). Researchers have demonstrated the potential benefits of synbiotic foods, a combination of prebiotic and probiotic foods, for both host organisms and human health (130). The benefits of synbiotic food will have to be examined in people with CKD.
High-amylose maize-resistant protein (HAMRS) is a type 2 starch found in potato, banana, and maize. It is resistant to digestion and reaches the large intestine, where it serves as an energy source for beneficial bacteria such as Bifidobacterium and Lactobacillus (131, 132). Prior studies have demonstrated that HAMRS may slow the advancement of CKD and enhance microbial diversity (131–133). Additionally, animals administered HAMRS exhibited a significant rise in the ratio of Bacteroidetes to Firmicutes. Another study exhibited a reduction in oxidative stress and inflammation (133). Moreover, diets high in fiber were advantageous for decreasing inflammation and overall mortality (134).
Inflammation and CKD are closely associated with elevated levels of oxidative stress. Immunological function is impaired when oxidative stress stimulates several inflammatory signaling pathways. Metabolic syndrome, insulin resistance, hyperuricemia, CKD, high blood pressure, and other health problems are all linked to chronic inflammation-induced pro-oxidative stress (2, 135–137). Overproduction of reactive oxygen and nitrogen species (ROS and RNS, respectively) is the principal cause of oxidative stress, characterized by an imbalance between antioxidants and pro-oxidants. Covalent crosslinks, single- and double-strand breaks, and disturbances in redox signaling can emerge from the oxidation and molecular damage that this causes to biological components such as lipids, proteins, and DNA (8, 138).
.The kidneys are especially vulnerable to redox imbalances and oxidative stress because ROS has a substantial impact on the physiological regulation of renal function. An overabundance of reactive oxygen species (ROS) intensifies the inflammatory response in kidney diseases by setting in motion pathways that promote inflammation. Normally, cells produce small amounts of pro-oxidative agents, which serve important defensive roles, but antioxidant enzyme systems like glutathione and others inactivate them due to their ability to neutralize free radicals. The main sources of ROS are enzymes like NADPH oxidase (NOX1, NOX2, NOX4, and NOX5) and the mitochondrial respiratory chain reaction (138–140).
Several uremic toxins have been associated with a decline in kidney function and an increase in oxidative stress in CKD. Indoxyl sulfate builds up in the blood of chronic kidney disease patients and triggers the production of superoxide by cells by activating nicotinamide adenine dinucleotide phosphate oxidases (NOX4). In addition, indoxyl sulfate raises levels of proalpha1(I) collagen, tissue inhibitor of metalloproteinase-1, transforming growth factor-beta1, and free radicals in vascular endothelium and smooth muscle cells. While renal dysfunction progresses, the most important indicators of oxidative stress are plasma F2-isoprostanes, 8-oxo-7,8-dihydro-2-deoxyguanosine, malondialdehyde (MDA), carbamylated proteins, advanced oxidation protein products, asymmetric dimethylarginine, and oxidized lipoprotein particles (138, 141, 142).
In the early stages of CKD, there is evidence of elevated oxidative stress, which is linked to the progression to end-stage renal disease. Plasma total F2-isoprostanes is the most reliable indicator of oxidative stress damage, which occurs as a result of lipid peroxidation. Furthermore, protein carbonylation may be a secondary occurrence rather than a direct contributor to pathology, even though protein carbonyl concentrations are often higher than other biomarkers. Protein carbonylation is a useful indicator of oxidative stress associated with chronic kidney disease (8, 143, 144). Inflammation in the kidneys not only triggers endothelial dysfunction and activates glomerular and tubular epithelial cells but also releases inflammatory substances. These substances draw additional immune cells to the damaged kidneys.
In chronic kidney disease, recent research has linked changes in the lipid metabolic profile to endothelial dysfunction. Obesity and diabetes mellitus, two metabolic diseases, frequently coexist and cause endothelial damage. Patients with higher proteinuria and lower eGFR have more pronounced dyslipidemia from the onset of the disease, which is associated with quantitative and qualitative changes in lipoproteins, lipolytic enzymes, and lipoprotein receptors. These alterations may have contributed to the progression of the disease. Increased inflammation leads to worsening renal function, which, in turn, causes higher triglyceride levels, lower HDL-C, and variable amounts of oxidized LDL-C. CKD leads to changes in not only lipid and lipoprotein concentrations but also structural changes that alter the function of HDL and LDL that trigger pro-inflammatory and pro-atherogenic processes and oxidative stress. Serum fatty acid levels are also altered, leading to changes in fatty acid metabolism, causing mitochondrial dysfunction and cellular damage. Combining other metabolic conditions, such as diabetes and obesity, with an imbalanced fat metabolism—which is a pro-atherosclerotic factor—may significantly increase the risk of cardiovascular disease (CVD), especially in people with CKD (145).
.Excess extracellular matrix deposition, a hallmark of kidney fibrosis, is a significant contributor to CKD. In CKD patients, the degree of tubulointerstitial fibrosis is the best predictor of future renal function decline. Renal fibrosis development is influenced by elements such as oxidative stress, cytokines, and cell growth factors, particularly transforming growth factor-β1 (TGF-β1). TGF-β1 is a crucial protein that impacts fibroblast transition into myofibroblasts. ROS, a byproduct of NAD(P)H oxidase, supports the conversion of fibroblasts to myofibroblasts, making it similar to TGF-β1. TGF-β1 enhances NOX2 and NOX4 expression, as well as NADPH oxidase activity. P-cresyl sulfate, another uremic toxin linked to CKD progression, helps renal tubular cells make more NOX4, p22phox-NADPH, and ROS. Inflammatory and profibrotic cytokines cause reduced cell viability. Oxidative stress and TGF-1 produce chronic kidney damage, leading to kidney fibrosis (8, 146–148).
.Nitric oxide synthase in endothelial cells transforms arginine into nitric oxide. It reduces oxidative stress by inhibiting cytochrome C oxidase, the final enzyme in the electron transport chain connected to membrane potential of mitochondria. Asymmetric dimethylarginine (ADMA) accumulates in the plasma of CKD patients, potentially decreasing endothelial NO production. ADMA causes ROS generation to rise when NO levels fall. There is an inverse connection between GFR and ADMA concentrations (8, 149).
Inflammation is a characteristic aspect of deteriorating renal function (2, 150–152). CKD leads to the development of an environment that promotes inflammation, which can be caused by tissue ischemia, the presence of uremic substances, or infection. Sterile inflammation frequently occurs as a consequence of several clinical diseases associated with kidney disorders and nephropathies caused by toxic substances, ischemia, hypertension, or diabetes.
An increase in pro-inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), which are negatively associated with a decrease in GFR, is the characteristic feature of CKD. The worldwide STABILITY trial found that lower glomerular filtration rate (GFR) and higher interleukin-6 (IL-6) levels were signs of acute myocardial infarction (AMI), stroke, and death from any cause. It is believed that IL-6 is the most powerful inflammatory biomarker for chronic kidney disease. The risk of cardiovascular events and all-cause mortality is increased in patients with CKD when there are elevated levels of cytokines such as TNF-α and IL-6 and when interleukin-1α (IL-1α) is expressed on the surface of circulating monocytes. Thus, inflammation is a “non-traditional” risk factor for cardiovascular disease in CKD (2, 153–155). Research has shown that interleukin-6 (IL-6) has a role in renal damage and contributes to the pathogenesis of CKD. The levels of IL-6 in the blood rise as chronic kidney disease progresses. In addition, individuals in the latter stages of CKD are more likely to have adverse outcomes and death if their circulating IL-6 levels are higher.
By stimulating the innate immune system and encouraging the infiltration of inflammatory cells, interleukin-1 (IL-1) is an essential mediator of inflammation, host defense, and acute-phase responses. A study in animal models of CKD found that the degree of IL-1 expression affects anemia and kidney damage. In this study, elevated levels of IL-1, which impair kidney function, were more strongly associated with anemia and kidney failure. Researchers found that interleukin-1 regulates the accumulation of macrophages and neutrophils in tissues, which, in turn, regulates inflammatory damage in cardiorenal disorders. IL-1 also promotes renal tissue fibrosis, which is the ultimate pathological process for kidney damage (8, 156–159).
.A new study shows that IL-20, another interleukin, can affect the development of chronic kidney disease. Serum IL-20 levels were much higher in people with advanced-stage chronic kidney disease. A study on animals with CKD supports this claim. The study found that immune cells in the interstitium, mesangial cells in the glomeruli, and tubule epithelial cells all increased the amount of IL-20. IL-20 also caused tubular epithelial cells to die and increased mesangial cells’ production of pro-inflammatory molecules. IL-20 also causes kidney fibrosis by producing more TGF-1 and other growth factors that cause chronic inflammation. Animal studies show that IL-20 may cause kidney fibrosis, damage, and renal insufficiency by activating interstitial fibroblasts in the kidneys. More research is necessary to confirm the potential link between changes in IL-20 levels and CKD (83, 160–162).
.In the development of CKD, macrophages and nod-like receptor protein 3 (NLRP3) play an essential role. The mononuclear phagocyte system includes macrophages and monocytes, both of which are innate immune cells. Normally, monocytes are present in the blood, bone marrow, and spleen; however, when inflammation is present, they rapidly recruit to inflamed tissues and undergo a process of differentiation into macrophages. It is possible for macrophages to release a range of substances, including fibrotic factors such as TGF-Î2, anti-inflammatory cytokines such as IL-10, and mediators of inflammation such as IL-1, IL-6, and TNF. Key to immune system function are two phenotypic types of macrophages, M1 and M2 (8, 163, 164). M1 macrophages kill pathogens, whereas M2 macrophages reduce inflammation and aid in tissue healing. It is common for macrophages to enter the kidneys in CKD. Because of this, all kidney diseases are marked by an excess of macrophages in renal tissue, which includes the glomerulus, renal cortex, and interstitium of the medulla. Inflammation begins with M1 macrophages, whereas M2 macrophages facilitate fibrosis and healing. According to research conducted in rats, the start of CKD may be influenced by the ratio of monocytes to macrophages (M1/M2) (8, 163–168).
The protein complex called nod-like receptor protein 3 (NLRP3) is another crucial component of the immune system. An extremely inflammatory type of programmed cell death in reaction to infectious stimuli, pyroptosis, is triggered by NLRP3-induced caspase-1 activation, which in turn triggers the production of pro-inflammatory cytokines. CKD is one of the prevalent human disorders associated with NLRP3 dysregulation, which, in turn, compromises the ability of host immune system to fight infections. CKD and AKI both have ischemia–reperfusion injury (IRI) as a contributing component. Literature reviews have shown that NLRP3 plays a role in IRI (154). The study by Zheng et al. established a link between NLRP3 and insufficient recovery after AKI (166). Overexpression of tubular NLRP3 has been linked to inflammation, fibrosis, and poor tubular repair in mouse models of mild or severe acute kidney injury. Consequently, research demonstrated a persistent overexpression of NLRP3 in post-AKI kidneys. The NLRP3 inflammasome is likely a target for treatment in chronic kidney disease; thus, it would be good to understand its full mechanism in kidney illness. This would help us comprehend the pathophysiology of renal disease (8, 166, 169, 170).
Neutrophil–gelatinase-associated lipocalin (NGAL) levels are typically low in healthy tubules; however, NGAL production rises in response to renal tubular epithelial cell injury. Because tubular cells express NGAL due to kidney injury, elevated gene transcription in this chronic disease could suggest ongoing kidney damage. NGAL is an emerging biomarker for kidney injury, including AKI and CKD. Furthermore, there is a direct link between high NGAL levels and albuminuria in people with chronic kidney disease, and NGAL as a biomarker for AKI has been extensively studied. Research into the role of NGAL in kidney injury could lead to novel approaches to treating chronic kidney disease (8, 171–181). Additionally, NGAL is a promising biomarker for CKD and its progression (182).
Several physiological processes rely on matrix metalloproteinases (MMPs), a class of proteolytic enzymes. These include cell differentiation, angiogenesis, inflammation, proliferation, vascular damage, and apoptosis. Collagenases, gelatinases, stromelysins, matrilysins, and other matrix metalloproteinases (MMPs) are among the about 20 varieties of mammalian MMPs. MMPs affect some clinicopathological conditions, including kidney diseases. Inflammation, matrix deposition, fibrosis, and fibroblast/myofibroblast activation, are all stages of CKD that involve multiple matrix metalloproteinases (MMPs) (8, 183, 184) (Table 1).
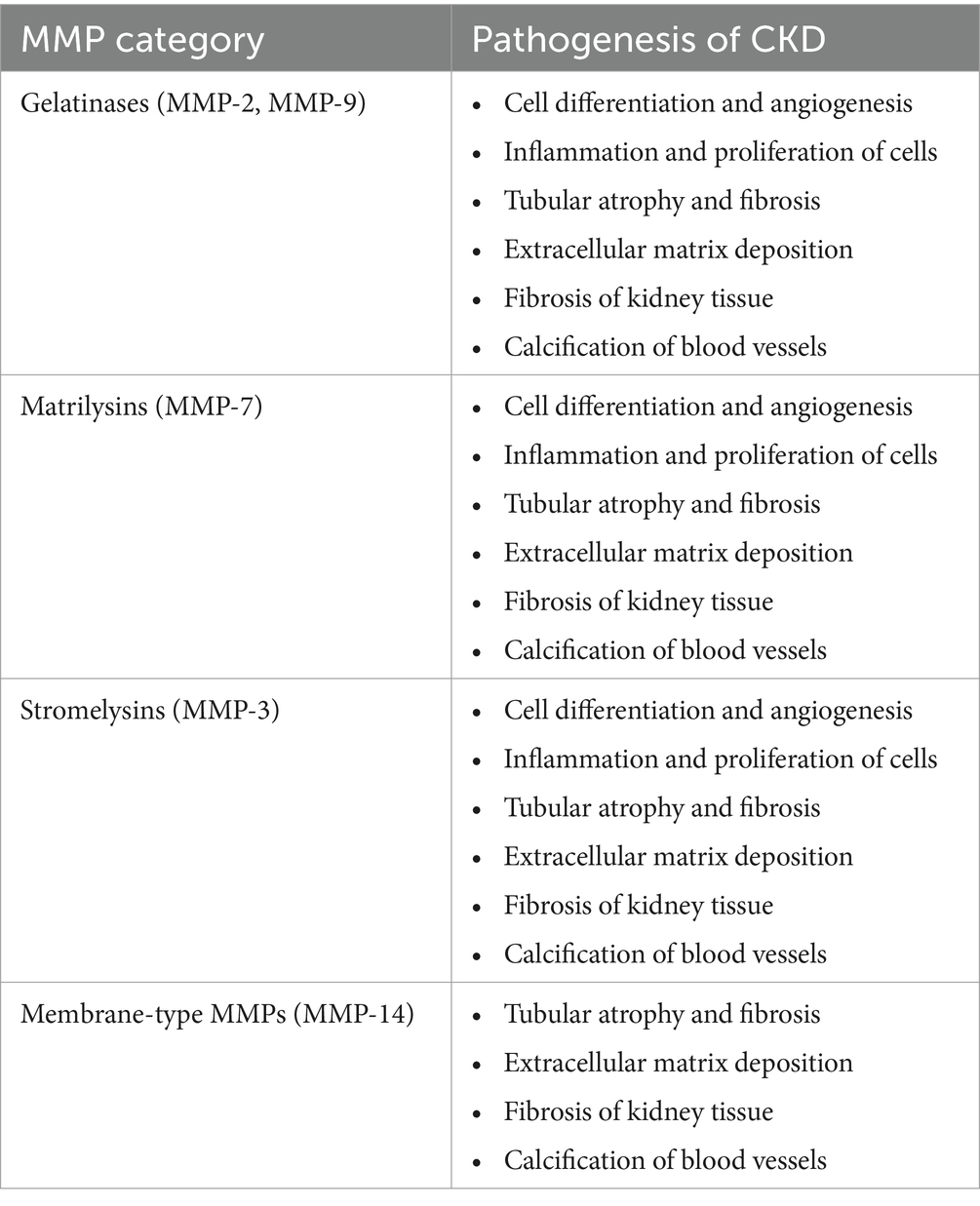
Table 1. Pathophysiological mechanisms in CKD due to different MMPs are divided into groups (8).
Many scientists have hypothesized that MMP-7-induced alterations to the extracellular matrix contribute to the onset of chronic kidney disease (CKD). It is possible that MMP-7 has a more important function than other MMPs in the development of kidney diseases (8, 184, 185).
.According to Tan et al., normal mice injected with an MMP-7 expression vector experienced proteinuria. Additionally, removing MMP-7 shielded the mice against glomerular damage and proteinuria (186). As stated earlier, new biomarkers must be created to identify kidney diseases early and determine their prognosis (170). They highlighted the possibility of MMP-7 levels in urine as a non-invasive marker of renal impairment. Furthermore, MMP-7 in urine may be a useful indicator of acute kidney injury, according to some studies (8, 187). Renal fibrosis non-invasively can be detected by measuring urinary MMP-7 levels because urine MMP-7 levels were shown to be positively associated with renal fibrosis scores and inversely associated with renal function, according to research by Zhou et al. (188). Gelatinases MMP-2 and MMP-9 are produced by tubular and glomerular cells, respectively. Research has shown that the activation of matrix metalloproteinases 2 and 9 (MMP-2 and MMP-9) sets in motion events that cause inflammation, abnormalities in the extracellular matrix, tubular atrophy, and fibrosis (84, 189–191).
Mincle cell receptor is an innate immune protein expressed by macrophages such as monocytes, neutrophils, and dendritic cells, and some types of B cells also upregulate it. It is an innate immunity protein in the cell membrane, and a number of factors trigger its expression (192). The mincle receptor identifies necrotic cells by binding to Sap-130. For instance, mincle, in conjunction with splenic tyrosine kinase (Syk) and caspase recruitment domain proteins, induces an inflammatory response to infections by mycobacterium and fungi is controlled (193).
A study demonstrated that during the early phases of cisplatin-associated acute kidney injury (AKI), mincle cells were produced mainly in the macrophages of the kidney. Using Immunofluorescence techniques, the authors were able to show that macrophages that expressed mincle entered the damaged kidney on the third day after cisplatin was introduced. They noted a rise in serum creatinine on the third day of cisplatin intake. Additionally, an elevation of mincle protein was found on day 1 of the kidney injury. The authors used flow cytometry and immunohistochemistry to demonstrate that macrophages that entered the kidneys (F4/80+ or CD68+) largely created mincle. They discovered that M1 macrophages were responsible for mincle production. Furthermore, a study found an association between AKI and macrophages that express mincle (192), while another research also showed that macrophages deprived of mincle could protect against kidney damage caused by cisplatin. In addition, the researchers showed that adoptive transfer with macrophages lacking mincle greatly decreased AKI (192). Again, through both gain-of-function and loss-of-function reactions, it was found that regulating the expression of mincle in macrophages can have an effect on the degree of AKI. Largely, M1 macrophage mincle expression is a critical trigger for acute kidney injury (AKI). This could potentially slow down the progression of AKI to CKD. A recent study (2024) found that macrophages and neutrophils expressed mincle throughout the transition from AKI to CKD, revealing its impact on the process. The authors demonstrated a substantial elevation of mincle level on day 1 of AKI and another elevation on day 14. These mincle-laden neutrophils and macrophages promoted kidney tissue inflammation by secreting tumor necrosis factor (TNF). They also discovered that mincle-deficient mice had no significant renal injury or fibrosis (194). Thus, mincle may become a future therapeutic target for the prevention of AKI transitioning to CKD.
Antioxidants such as edaravone, which lowers ROS levels, and ebselen, a glutathione peroxidase mimetic, have shown promising results in studies involving kidney disease models, improving renal function, lowering lipid peroxidation, and increasing endothelial and epithelial cell survival (126).
Various lipid-soluble tocopherols in vitamin E stop lipid peroxidation chain reactions and remove oxygen-free radicals. They do this by entering the plasma membrane (195). Vitamin E-rich foods contain antioxidant-rich such as α-tocotrienols (196). CKD patients do not have enough α-tocotrienol (197). Extra α-tocotrienol supplementation for end-stage kidney disease or dialysis patients reduces heart disease risk and oxidative stress and boosts antioxidants such as SOD, Gpx, and CAT (198). Some studies found no mortality benefits from high-dose vitamin E, whereas others found an increased prostate cancer risk. Trolox (± − 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid), a chemical comparable to α-tocopherol, helps eliminate free radicals more effectively. Due to its significant water solubility, studies have shown it can treat acute renal damage resulting from ischemia reperfusion (199). Combining α-tocopherol and Trolox may be more effective due to fast-acting qualities of Trolox and sustained scavenging abilities of α-tocopherol (200).
Omega-3 polyunsaturated fatty acids such as docosahexaenoic and eicosapentaenoic acids are anti-inflammatory and antioxidant (201). These compounds boost glutamyl-cysteinyl ligase and glutathione reductase (202). Eicosapentaenoic and docosahexaenoic acidtherapy decreases inflammation and oxidative stress, increasing kidney function and reducing renal fibrosis risk (201).
N-acetyl cysteine (NAC) decreases oxidative stress and boosts cell glutathione (203). Research on NAC therapy for CKD is unclear. Uremic toxins cause endothelial damage, whereas NAC therapy reduces NF-κB upregulation, which requires reactive oxygen species (203). In end-stage kidney disease and dialysis patients, NAC lowered serum 8-isoprostane and IL-6 (204, 205). Studies have shown that allopurinol protects against diseases where oxidative stress plays a role in their pathogenesis (206). Treatment of diabetic patients with allopurinol reduced high uric acid levels, albuminuria, and tubulointerstitial damage (207).
The kidneys have elevated concentrations of CoQ9 and CoQ10 due to their strong dependence on aerobic metabolism and high mitochondrial density (208). CoQ10 has two primary antioxidant roles: directly preventing lipid peroxidation and indirectly interacting with α-tocopherol to prevent lipid peroxidation (209). In a study conducted by Ishikawa et al. (210), it was discovered that CoQ10 supplementation had a positive impact on renal function and reduced kidney O2 levels in rats that had undergone hemi-nephrectomy, although the effects were not always consistent.
An expanding understanding of molecular mechanisms of chronic kidney disease has unveiled new therapeutic options. However, an incomplete comprehension of the pathophysiology impedes the quest for treatment targets for inflammation in the kidney (8). Two drugs, sirukumab and siltuximab, that directly target IL-6 ligands and block classical signaling and trans-signaling can be distinguished based on the inflammatory mechanism of CKD development. Moreover, antibodies such as tocilizumab and sarilumab block all three forms of IL-6 signaling (8, 158). Hsu et al. described another antibody, finding that anti-IL-20 (7E) therapy reduced glomerular area and blood glucose levels in mice with diabetic nephropathy, alongside improvements in kidney function (8, 161). The non-inflammatory mechanisms of CKD are linked to initially marked increases in glomerular permeability, which subsequently cause proteinuria or proliferation. It is well understood that podocyte depletion leads to proteinuria. When more than 40% of podocytes are damaged, it results in numerous dangerous side effects, including mesangial growth, adhesions, focal segmental glomerulosclerosis, or global sclerosis (211). Given that nephrotic non-inflammatory glomerulonephritis is a hallmark of many glomerular disorders, treatment approaches aimed at modifying podocyte activity are likely to be beneficial (8).
A complex network of cytokines/chemokines, growth factors, adhesion molecules, and signaling pathways is involved in kidney fibrosis (8, 212). Studies by Moon et al. demonstrate the potential to modulate TGF-β signaling in progressive fibrosis in the kidney. Their findings suggest that kidney damage from unilateral ureteral blockage can be significantly reduced by molecularly targeting the transforming growth factor-beta1 signaling pathway. An effective treatment option to prevent or mitigate the progression of renal fibrosis is IN-1130, an ALK5 inhibitor (8, 213).
In a diabetic nephropathy (DN) model, rats treated with coenzyme Q10 (CoQ10) or similar drugs, such as mitoquinone mesylate (MitoQ), exhibited improvements in renal function and tubular damage. Another mitochondria-targeting drug, dithiol a-lipoic acid, demonstrated renoprotective benefits in an animal model of hypertension and renal illness. Additionally, in mice with experimental tubulointerstitial nephritis, renoprotection was observed in conjunction with a reduction in the expression of inflammatory molecules when allopurinol or the blockade of genes linked to the NLRP3 inflammasome response (apoptosis-associated speck-like protein containing C-terminal caspase recruitment domain [CARD] (ASC) and caspase 1) was administered (126).
High kidney disease risk of Africa has been linked to APOL1 high-risk alleles. Recent studies have shown that Africans with two apolipoproteins L1 (APOL1) variations (G1 or G2) have a higher risk of CKD with proteinuria and ESKD than those with low-risk alleles. Substitution of two amino acids (S342G and 1,384 M) in the C terminus of the APOL1 gene causes G1 risk variants (214, 215). Like G1, the G2 risk variant has two amino acids (N388del and Y389del) deleted at the same APOL1 position (214). The G0 APOL1 allele is non-risk despite having several functional sequences. One possesses zero, one, or two APOL1 risk alleles because each parent transmits the gene. Two high-risk APOL1 alleles (G1G1, G2G2, or G1G2) raise kidney disease risk significantly while inheriting one low-risk allele (G0G1 and G0G2) does not increase the risk of CKD. However, these high-risk polymorphisms improve APOL1 channel function, which promotes podocyte injury (216–218) and progressive glomerular dysfunction and proteinuria. It is associated with various histological patterns such as FSGS, hypertension-associated CKD, HIV-associated nephropathy, COVID-19-associated nephropathy, and end-stage kidney disease risk (219–222).
Research has demonstrated that human embryonic kidney cells are capable of expressing APOL1 in the G0, G1, or G2 alleles. Additionally, APOL1 is responsible for the formation of cation channels in mammalian cells’ plasma membranes. The specific mechanism by which high-risk variants of G1 and G2 cause kidney disease is, for the most part, not well understood. It has been proven through the utilization of cell-based and transgenic animal models that high-risk variants are responsible for cellular damage and mortality, whereas the reference APOL1 G0 is relatively non-toxic (105, 107, 216). It is thought that high-risk variants cause APOL1-mediated kidney disease in a way that is very similar to how it causes cytotoxicity in laboratory animals. Researchers attribute the trypanolytic potential of the APOL1 risk variants to the passage of cations through these pores (223–225). The results of previous studies show that APOL1 risk variants can create a pore in a lipid layer that only allows the passage of Na+ and K+ ions (223, 224). The only two cells that are capable of causing an anomalous outflow of sodium and potassium ions are G1 and G2. This process ultimately leads to cell death, activation of c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), and swelling of the cell (216, 226). These studies directly attribute the cytotoxicity of high-risk variants to their cation pore function. However, these studies have not established whether or not this is the primary mechanism by which these alleles cause cytotoxicity (227, 228). Other investigations have shown that high-risk variations induce K+ to efflux mammalian cells and Na+ influx. Nevertheless, a recent study found that the APOL1 alleles (G0, G1, and G2) can facilitate the passage of Ca2+ through a lipid bilayer (218). Furthermore, the researchers found that the expression of G1 and G2 resulted in a consistent increase in the quantity of cytoplasmic Ca2+ in cell-based models. They concluded that the main cause of cell death is the import of Ca2+ and Na+ through APOL1 channels (218).
Not all people with high-risk APOL1 variants develop kidney disease. Thus, the development of APOL1-associated nephropathy requires the presence of a second factor (229). These second-hit factors may be viral or non-viral. Some of the non-viral factors may include the toxins mentioned in this manuscript, inflammatory mediators, and oxidative stress. Additionally, the APOL1 gene may interact with other genes to cause kidney disease.
In individuals with CKD, anemia is a frequent consequence that increases morbidity and mortality rates. The glycoprotein hormone erythropoietin (EPO) primarily regulates erythropoiesis. The plasma EPO level is disproportionate to the level of anemia. The liver is the primary site for the production of EPO in a fetus; however, the kidney takes over this role after birth. The kidney regulates EPO production at the mRNA level. Experiments performed in mice revealed that phlebotomy and anemia were associated with increased expression of EPO mRNA. Several recognized factors contribute to anemia in CKD; however, EPO insufficiency is the most important (230–232). The erythropoietin-producing cells within the kidneys diminish as kidney disease progresses (233). Reduced oxygen sensing and, consequently, reduced erythropoietin-producing cell (REPC) production in the kidney have been associated with low levels of EPO. Regardless of the original underlying condition that causes kidney injury, interstitial fibrosis is present in all cases of chronic kidney disease (CKD). This leads to the irreversible loss of normal kidney tissue and function (230).
Renal hypoxia is the primary trigger for erythropoietin synthesis. Hypoxia stops the degradation of HIF-1α, which lets HIF-1α attach to hypoxia-responsive parts of oxygen-regulated genes when hypoxia is present. The erythropoietin gene in the kidneys is controlled by these response elements, and when HIF-1α binds, it makes more erythropoietin (233–237). The bone marrow is where erythropoietin works best. It boosts erythropoiesis by attaching to its receptor, which is found on the surface of erythroid progenitor cells. Colony-forming unit-erythroid (CFU-E) cells are the most responsive to erythropoietin because they have the highest concentration of erythropoietin receptors (233, 238).
HIF is a basic helix–loop–helix heterodimer protein that belongs to the PER-ARNT-SIM (PAS) family. In addition to erythropoiesis, HIF binding across the genome turns on many target genes transcriptionally and helps with many physiological and developmental processes, such as blood vessel growth, energy metabolism, iron homeostasis, cell proliferation, differentiation, and iron homeostasis. There are three isoforms of HIF: HIF-1, HIF-2, and HIF-3. Each isoform possesses a common β-subunit and a unique α-subunit. The isoform that regulates the production of EPO is HIF-2. HIF-2 also helps the duodenum absorb iron by increasing the transcription of genes that make proteins that help move iron around, such as duodenal cytochrome B and divalent metal transporter 1. HIF-1 promotes the transcription of other genes, such as transferrin and ceruloplasmin, that code for iron-mobilizing proteins (239).
One of the most important hormones for preserving bodily homeostasis is vasopressin, sometimes referred to as antidiuretic hormone or arginine vasopressin. Experimental investigations have demonstrated that vasopressin directly influences cyst formation, and researchers have linked elevated vasopressin concentrations to both disease severity and illness progression in polycystic kidney disease (240).
Vasopressin has three distinct known receptors, all of which belong to the G-protein-coupled receptor subgroup of rhodopsin. Nonetheless, the V2 receptor is the receptor that matters in this conversation. The thick ascending limbs of the loops of Henle and the collecting ducts are home to the majority of the V2 receptor (240, 241). Recent studies on PKD have focused on two possible treatments: lowering the amount of vasopressin in the blood and stopping vasopressin from working on the kidneys through the vasopressin V2 receptor (240).
Inaxaplin (VX-147) was recently, shown to be a small-molecule blocker of APOL1 channel function (242). This APOL1 channel inhibitor prevents cell swelling and preserves cell viability and thus blocks cytotoxicity caused by G1 and G2 variants (243). A recent study by Egbuna et al. among participants with focal segmental glomerulosclerosis showed a significant reduction in proteinuria (244). Pharmacological strategies for slowing down the progression of kidney disease involve the use of drugs such as angiotensin-converting enzyme inhibitors (ACEi), angiotensin receptor blockers (ARB), and sodium-glucose transporter-2 inhibitors (SGLT-2i) to reduce proteinuria and retard the progression of kidney disease. Thus, the APOL1 pore function inhibitor can potentially complement the effects of these drugs in the treatment of kidney disease patients, especially among Africans with a high burden of kidney disease and where the frequency of APOL1 high-risk alleles is high.
Iron supplements and erythropoiesis-stimulating agents (ESAs) are the two main therapies currently available for renal anemia. For example, there are concerns about the use of exogenous ESAs, which can lead to more death and heart problems in patients who do not respond well or who have cancer. This means that a new treatment method for renal anemia is needed. For the treatment of renal anemia, prolyl hydroxylase (PHD) inhibitors offer a novel therapeutic option. PHD inhibitors boost the transcription of EPO mRNA in REPCs by blocking the proteasomal degradation of HIFα, which activates the HIF pathway (230). Two of the most used ESAs for treating anemia in CKD patients are recombinant human erythropoietin and darbepoetin alfa. Except for longer half-life of darbepoetin alfa, which permits less frequent dosage, they are substantially comparable in terms of efficacy and adverse effect profile (245).
The half-life of human erythropoietin is approximately 6–10 h. It is a 30,400-Dalton glycosylated protein with a backbone consisting of 165 amino acid residues. The amino acid sequence of natural hormone is identical to that of recombinant human erythropoietin (rHuEPO) products. Darbepoetin alfa (Aranesp., Amgen), epoetin beta (Neo-Recormon, Roche), epogen (Amgen; Procrit, Centocor Ortho Biotech Products; Eprex, Janssen), and continuous erythropoietin receptor activators (Mircera, Roche) are some of the recombinant erythropoietin products that are on the market (233, 246, 247). There are specific negative effects associated with epoetin. Some of these negative effects are similar to both IV and SC; however, they vary in frequency and degree. Both routes share the following common outcomes: pain at the injection site, hypertension development, arteriovenous fistulae thrombosis, hyperkalemia, iron store depletion, flu-like symptoms, prolonged dialysis, and, infrequently, pure red cell aplasia (PRCA) and seizures. There is also an increased risk of thrombotic, cardiovascular, and cerebrovascular events overall (248).
To trigger the transcription of HIF-responsive element genes, the HIF-α and HIF-β subunits travel together in a heterodimer to the cell nucleus. Oxygen causes the prolyl hydroxylase (PH) enzyme to become active, which leads to the hydroxylation of two proline residues on HIF-α, making it susceptible to degradation. HIF-α survives degradation in the absence of oxygen and can dimerize with the always-available HIF-β. For this PH, 2-oxoglutarate is a necessary cofactor. It has been demonstrated that small-molecule oral 2-oxoglutarate analogs inactivate HIF-PH in the presence of oxygen, serving as HIF stabilizers. These substances are referred to as HIF-PH inhibitors (HIF-PHIs) based on their mode of action (239, 249, 250). By imitating hypoxia through HIF prolyl hydroxylase domain enzyme (HIF-PHD) inhibition, HIF stabilizer increases endogenous erythropoietin (EPO). HIF stabilizers have been demonstrated in phase 2 and phase 3 clinical trials to be equally effective as ESA in treating renal anemia (251). Numerous HIF stabilizers have been studied in a number of clinical trials, including enarodustat, molidustat, desidustat, vadadustat, roxadustat, and daprodustat (252).
One clinically proven mechanism of action for the therapy of autosomal dominant polycystic kidney disease is vasopressin V2 receptor inhibition (253). Tolvaptan, which is a V2-receptor antagonist, has been demonstrated in experimental studies and a large randomized controlled trial involving 1,445 patients with autosomal dominant PKD to limit the progression of the disease. There was also a considerable drop in the size of the kidneys from 5.5 to 2.8% as well as the reciprocal slope of the serum creatinine level from −3.81 to −2.61 mg per mL-1/year (240, 254).
Recent research has focused on the therapeutic effects of glucose-lowering therapy in kidney injury using sodium-glucose cotransporter-2 (SGLT2) inhibitors. Research on diabetic rats administered with streptozotocin (STZ) demonstrated that phlorizin and empagliflozin, which block SGLT2, reduce glomerular hyperfiltration and hypertrophy, oxidative stress, inflammation, and fibrosis. Empagliflozin therapy reduced albuminuria and mesangial matrix growth in hypertensive BTBR ob/ob mice. The benefits of SGLT2 inhibitors occur through various mechanisms (255).
Autophagy is the cellular process of breaking down and recycling cytosol components, which are used as building blocks for the regeneration of tissue (256). This process involves lysosomes. Defectiveness or absence of autophagy leads to kidney damage. Recent studies have demonstrated that SGLT2 inhibitors trigger autophagy via the mammalian target of rapamycin (mTOR), 5′adenosine monophosphate-activated protein kinase (AMPK), sirtuin 1 (SIRT1), and hypoxia-inducible factor (HIF) signaling pathways (257–259).
The mTOR protein complex 1 (mTORC1) is a protein complex that acts as a serine–threonine kinase and plays a crucial role in integrating signals related to nutrients such as glucose and amino acids. In a state of calorie excess, mTORC1 promotes anabolism (the synthesis of complex molecules) and inhibits autophagy (260). The role of proximal tubular mTORC1 in DKD is considered significant (261). SGLT2 inhibitors decrease mTORC1 function in proximal tubule cells, preventing tubulointerstitial fibrosis (261). AMPK, in contrast to mTOR, functions as a detector of insufficient cellular energy (low ATP to 5′adenosine monophosphate ratio). It promotes the breakdown of molecules and triggers autophagy by suppressing mTORC1 when the body is in a low-calorie state (262). AMPK activates the catabolic process, which, in turn, provides ATP to cells that need sufficient energy. Canagliflozin stimulates AMPK-mediated autophagic stimulation, presumably via increasing calorie loss, without relying on insulin or glucagon signaling (263). Various studies have shown that SGLT2 inhibitors promote autophagy by using mTOR-AMPK-mediated pathways, which, in turn, help protect the kidneys against various types of renal damage (264–266).
SIRT1 relies on nicotinamide adenine dinucleotide to deacetylate and serve as a nutrient deficiency sensor (267). It removes acetyl groups from tumor protein 53, which augments the autophagy signaling pathway (268). In glucose deficiency states, SIRT1 and AMPK activate each other to increase autophagy and biosynthesis through mitochondria (269). SIRT1 can inactivate mTORC1 in the absence of AMPK (270). Activation of SIRT1 reduces kidney injury (271). Animal studies have shown that SGLT2 inhibitors increase the expression of SIRT1 (272, 273).
The transcription factors that react to low oxygen levels in cells are from the HIF family. HIF-1α and HIF-2α are isoforms that are activated by low oxygen levels and start processes that improve oxygen delivery and reduce oxygen usage (274). Activation of HIF-1 upregulates inflammation, fibrosis, angiogenesis, and mitochondrial clearance through autophagy, whereas activation of HIF-2 reduces inflammation and fibrosis and increases erythropoietin synthesis clearance of the peroxisome by autophagy (274). The inhibition of SGLT2 decreases the expression of HIF-1α in human proximal tubules under hypoxic conditions, leading to a reduction in tubular damage and interstitial fibrosis (274, 275). Furthermore, SGLT2 inhibitors can enhance HIF-2α activity via a mechanism that relies on SIRT1 (276). Therefore, SGLT2 inhibitors could offer kidney protection by re-establishing the equilibrium of HIF-1α and HIF-2α activities in renal cells.
SGLT inhibitors may protect kidneys by remodeling F-actin and α-actinin-4 filaments, reducing β1-integrin loss on podocyte surfaces, blocking macrophage infiltration, activating M1-M2 polarization, and preventing profibrotic M2 macrophages. They also maintain cellular redox homeostasis and reduce oxidative stress by activating Kelch-L. (276–279).
SGLT2 inhibitors have revolutionized CKD management. Regardless of their impact on glucose regulation, these medications prevent the deterioration of kidney function by lowering glomerular hypertension, which is mediated by tubuloglomerular feedback (280). Research has shown that SGLT2 inhibitors benefit patients with and without type 2 diabetes mellitus (T2DM) by reducing proteinuria and slowing the progression of CKD (239, 281). In a 12-week study, researchers randomly assigned 52 T2DM patients 1:1 to either dapagliflozin or a placebo; during this period, dapagliflozin significantly raised transferrin levels and reduced hepcidin and ferritin levels. The rise in the hemoglobin levels versus placebo was 0.5 g/dL (p = 0.02) (239, 282).
In a systematic review and meta-analysis, Mavrakanas et al. found that SGLT2 inhibitors were associated with a decreased incidence of CKD progression among patients with preexisting CKD (RR: 0.77; 95% CI: 0.68–0.88), compared with placebo. SGLT2 inhibitors were also linked to a lower risk of AKI (RR: 0.82; 95% CI: 0.72–0.93) and stopping treatment in CKD patients than a placebo. For patients with CKD, SGLT2 inhibitors provide significant protection against cardiovascular and renal outcomes. These findings provide compelling evidence in support of its use in patients with CKD and its continued use as renal function diminishes (283). Examples of SGLT2 inhibitors, or gliflozins, are canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin. A study called DAPA-CKD (dapagliflozin in patients with CKD) and EMPA-KIDNEY (empagliflozin in patients with CKD) found that dapagliflozin and empagliflozin help patients with and without T2D by protecting the kidneys (284, 285) (Figure 1).
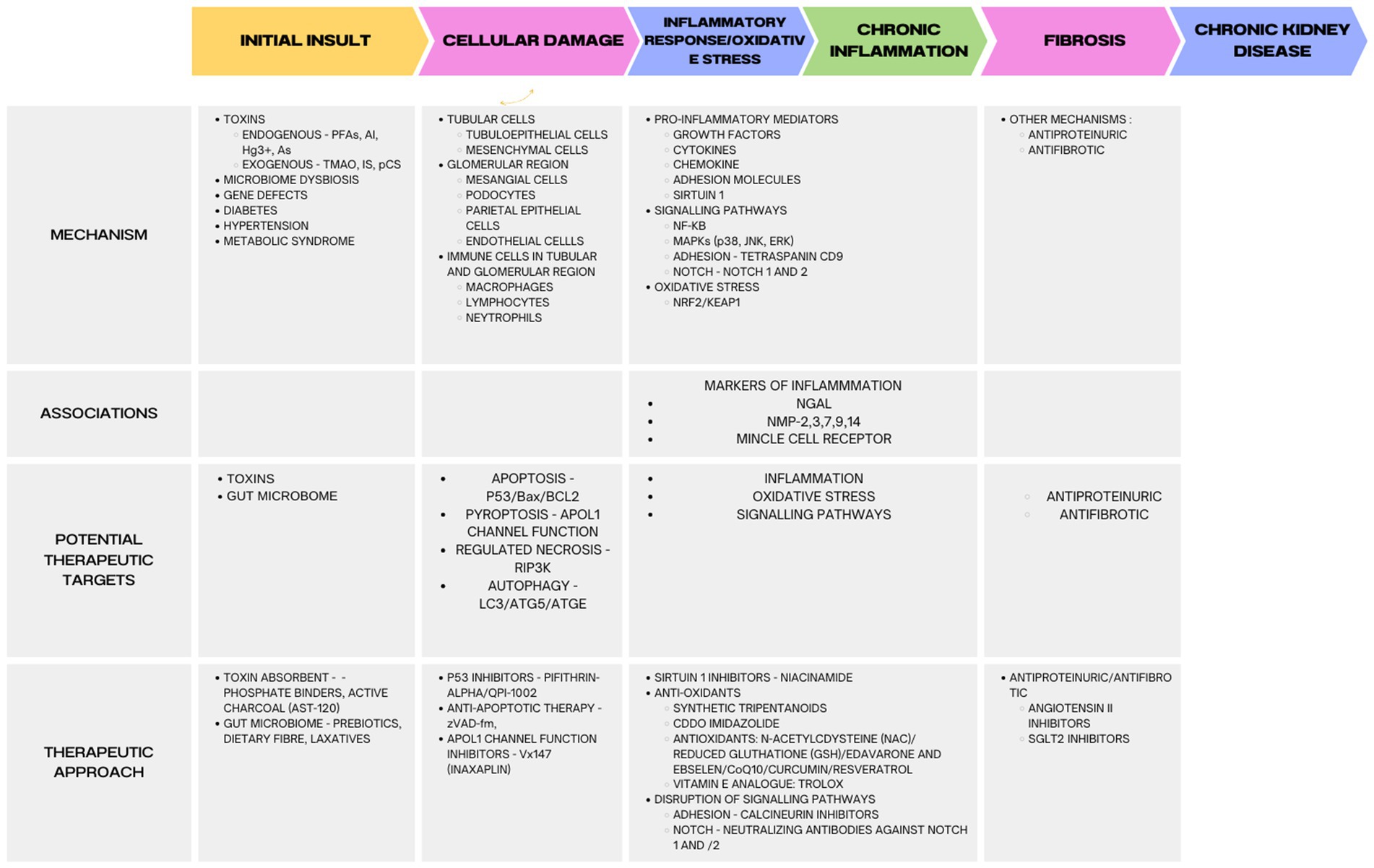
Figure 1. Molecular mechanisms and targets for treatment of kidney diseases. Indoxyl sulfate (IS), p-cresyl sulfate (pCS), trimethylamine N-oxide (TMAO), arsenic (As), mercury (Hg3+), per- and polyfluoroalkyl substances (PFAS), aluminum (Al), sodium-glucose cotransporter 2 (SGLT2).
CKD is a debilitating illness that increases the risk of cardiovascular problems and is typified by persistent inflammation. It is currently understood that a crucial aspect of CKD involves persistent, low-grade inflammation. Inflammation contributes to cardiovascular and all-cause mortality in CKD and has a distinct function in its pathophysiology. Chronic and recurring infections, altered adipose tissue metabolism, intestinal dysbiosis, increased synthesis and impaired clearance of pro-inflammatory cytokines, oxidative stress, and acidosis are some of the variables that lead to chronic inflammatory status in CKD. There is evidence of a reciprocal relationship between gut dysbiosis and CKD, which could influence the development and course of CKD by producing uremic toxins and/or mediating elevated inflammation. Additionally, APOL1 genetic polymorphism with its attendant cytotoxicity has been linked to the excess risk of CKD among people of recent African descent. Furthermore, not everyone with high-risk APOL1 variants develops kidney disease. Therefore, the development of kidney disease in people with high-risk alleles may require additional factors, known as secondary hits, such as infections, environmental factors (heavy metals), infections, and the microbiome, among others.
To better understand the course of CKD and identify new therapy targets, it is crucial to unravel the molecular interplay between inflammation, oxidative stress, MMPs, and other contributing factors (2, 8).
VB: Conceptualization, Project administration, Writing – original draft, Writing – review & editing. AA: Data curation, Writing – review & editing. KG: Writing – original draft, Writing – review & editing. FA: Writing – review & editing. EK: Writing – review & editing. JI: Data curation, Writing – review & editing. EA: Writing – review & editing. DA: Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Glorieux, G, Mullen, W, Duranton, F, Filip, S, Gayrard, N, Husi, H, et al. New insights in molecular mechanisms involved in chronic kidney disease using high-resolution plasma proteome analysis. Nephrol Dial Transplant. (2015) 30:1842–52. doi: 10.1093/ndt/gfv254
2. Altamura, S, Pietropaoli, D, Lombardi, F, Del Pinto, R, and Ferri, C. An overview of chronic kidney disease pathophysiology: the impact of Gut Dysbiosis and Oral disease. Biomedicines. (2023) 11:3033. doi: 10.3390/biomedicines11113033
3. Arora, P. Chronic kidney disease (CKD): Practice essentials, pathophysiology, etiology. Medscape. (2021). Available at: https://emedicine.medscape.com/article/238798-overview.
4. Matovinović, MS. 1. Pathophysiology and classification of kidney diseases. EJIFCC. (2009) 20:2–11.
5. Shabaka, A, Cases-Corona, C, and Fernandez-Juarez, G. Therapeutic insights in chronic kidney disease progression. Front Med (Lausanne). (2021) 8:645187. doi: 10.3389/fmed.2021.645187
6. Tejeswini Sen, T, Kale, A, Lech, M, Anders, HJ, and Gaikwad, AB. Promising novel therapeutic targets for kidney disease: emphasis on kidney-specific proteins. Drug Discov Today. (2023) 28:103466. doi: 10.1016/j.drudis.2022.103466
7. Vaidya, SR, and Aeddula, NR. Chronic kidney disease In: SR Vaidya, editor. StatPearls. Treasure Island, FL: StatPearls Publishing LLC (2024)
8. Frąk, W, Kućmierz, J, Szlagor, M, Młynarska, E, Rysz, J, and Franczyk, B. New insights into molecular mechanisms of chronic kidney disease. Biomedicines. (2022) 10:846. doi: 10.3390/biomedicines10112846
9. Tuttle, KR, Agarwal, R, Alpers, CE, Bakris, GL, Brosius, FC, Kolkhof, P, et al. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. (2022) 102:248–60. doi: 10.1016/j.kint.2022.05.012
10. Schaub, JA, Hamidi, H, Subramanian, L, and Kretzler, M. Systems biology and kidney disease. Clin J Am Soc Nephrol. (2020) 15:695–703. doi: 10.2215/CJN.09990819
11. Giesy, JP, and Kannan, K. Peer reviewed: perfluorochemical surfactants in the environment. Environ Sci Technol. (2002) 36:146A–52A. doi: 10.1021/es022253t
12. Fromme, H, Midasch, O, Twardella, D, Angerer, J, Boehmer, S, and Liebl, B. Occurrence of perfluorinated substances in an adult German population in southern Bavaria. Int Arch Occup Environ Health. (2007) 80:313–9. doi: 10.1007/s00420-006-0136-1
13. Lau, C, Anitole, K, Hodes, C, Lai, D, Pfahles-Hutchens, A, and Seed, J. Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol Sci. (2007) 99:366–94. doi: 10.1093/toxsci/kfm128
14. Garnick, L, Massarsky, A, Mushnick, A, Hamaji, C, Scott, P, and Monnot, A. An evaluation of health-based federal and state PFOA drinking water guidelines in the United States. Sci Total Environ. (2021) 761:144107. doi: 10.1016/j.scitotenv.2020.144107
15. Banzhaf, S, Filipovic, M, Lewis, J, Sparrenbom, CJ, and Barthel, R. A review of contamination of surface-, ground-, and drinking water in Sweden by perfluoroalkyl and polyfluoroalkyl substances (PFASs). Ambio. (2017) 46:335–46. doi: 10.1007/s13280-016-0848-8
16. Cordner, A, De La Rosa, VY, Schaider, LA, Rudel, RA, Richter, L, and Brown, P. Guideline levels for PFOA and PFOS in drinking water: the role of scientific uncertainty, risk assessment decisions, and social factors. J Expo Sci Environ Epidemiol. (2019) 29:157–71. doi: 10.1038/s41370-018-0099-9
17. Worley, RR, Moore, SM, Tierney, BC, Ye, X, Calafat, AM, Campbell, S, et al. Per- and polyfluoroalkyl substances in human serum and urine samples from a residentially exposed community. Environ Int. (2017) 106:135–43. doi: 10.1016/j.envint.2017.06.007
18. Xu, Y, Fletcher, T, Pineda, D, Lindh, CH, Nilsson, C, Glynn, A, et al. Serum half-lives for short- and Long-chain Perfluoroalkyl acids after ceasing exposure from drinking water contaminated by firefighting foam. Environ Health Perspect. (2020) 128:77004. doi: 10.1289/EHP6785
19. Xu, Y, Nielsen, C, Li, Y, Hammarstrand, S, Andersson, EM, Li, H, et al. Serum perfluoroalkyl substances in residents following long-term drinking water contamination from firefighting foam in Ronneby, Sweden. Environ Int. (2021) 147:106333. doi: 10.1016/j.envint.2020.106333
20. Li, Y, Andersson, A, Xu, Y, Pineda, D, Nilsson, CA, Lindh, CH, et al. Determinants of serum half-lives for linear and branched perfluoroalkyl substances after long-term high exposure-a study in Ronneby, Sweden. Environ Int. (2022) 163:107198. doi: 10.1016/j.envint.2022.107198
21. Weiss, JM, Andersson, PL, Lamoree, MH, Leonards, PE, van Leeuwen, SP, and Hamers, T. Competitive binding of poly- and perfluorinated compounds to the thyroid hormone transport protein transthyretin. Toxicol Sci. (2009) 109:206–16. doi: 10.1093/toxsci/kfp055
22. Loccisano, AE, Campbell, JL Jr, Andersen, ME, and Clewell, HJ 3rd. Evaluation and prediction of pharmacokinetics of PFOA and PFOS in the monkey and human using a PBPK model. Regul Toxicol Pharmacol. (2011) 59:157–75. doi: 10.1016/j.yrtph.2010.12.004
23. Jones, PD, Hu, W, De Coen, W, Newsted, JL, and Giesy, JP. Binding of perfluorinated fatty acids to serum proteins. Environ Toxicol Chem. (2003) 22:2639–49. doi: 10.1897/02-553
24. Vanden Heuvel, JP, Kuslikis, BI, and Peterson, RE. Covalent binding of perfluorinated fatty acids to proteins in the plasma, liver and testes of rats. Chem Biol Interact. (1992) 82:317–28. doi: 10.1016/0009-2797(92)90003-4
25. Gong, X, Yang, C, Hong, Y, Chung, ACK, and Cai, Z. PFOA and PFOS promote diabetic renal injury in vitro by impairing the metabolisms of amino acids and purines. Sci Total Environ. (2019) 676:72–86. doi: 10.1016/j.scitotenv.2019.04.208
26. Cui, L, Zhou, QF, Liao, CY, Fu, JJ, and Jiang, GB. Studies on the toxicological effects of PFOA and PFOS on rats using histological observation and chemical analysis. Arch Environ Contam Toxicol. (2009) 56:338–49. doi: 10.1007/s00244-008-9194-6
27. Bonato, M, Corrà, F, Bellio, M, Guidolin, L, Tallandini, L, Irato, P, et al. PFAS environmental pollution and antioxidant responses: an overview of the impact on human field. Int J Environ Res Public Health. (2020) 17:8020. doi: 10.3390/ijerph17218020
28. Owumi, S, Bello, T, and Oyelere, AK. N-acetyl cysteine abates hepatorenal toxicities induced by perfluorooctanoic acid exposure in male rats. Environ Toxicol Pharmacol. (2021) 86:103667. doi: 10.1016/j.etap.2021.103667
29. Klaunig, JE, Babich, MA, Baetcke, KP, Cook, JC, Corton, JC, David, RM, et al. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. (2003) 33:655–780. doi: 10.1080/713608372
30. Andersen, ME, Butenhoff, JL, Chang, SC, Farrar, DG, Kennedy, GL Jr, Lau, C, et al. Perfluoroalkyl acids and related chemistries—Toxicokinetics and modes of action. Toxicol Sci. (2008) 102:3–14. doi: 10.1093/toxsci/kfm270
31. Abbott, BD, Wood, CR, Watkins, AM, Tatum-Gibbs, K, Das, KP, and Lau, C. Effects of perfluorooctanoic acid (PFOA) on expression of peroxisome proliferator-activated receptors (PPAR) and nuclear receptor-regulated genes in fetal and postnatal CD-1 mouse tissues. Reprod Toxicol. (2012) 33:491–505. doi: 10.1016/j.reprotox.2011.11.005
32. Takacs, ML, and Abbott, BD. Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci. (2007) 95:108–17. doi: 10.1093/toxsci/kfl135
33. Zhang, H, Shen, L, Fang, W, Zhang, X, and Zhong, Y. Perfluorooctanoic acid-induced immunotoxicity via NF-kappa B pathway in zebrafish (Danio rerio) kidney. Fish Shellfish Immunol. (2021) 113:9–19. doi: 10.1016/j.fsi.2021.03.004
34. Wei, H, Li, D, Luo, Y, Wang, Y, Lin, E, and Wei, X. Aluminum exposure induces nephrotoxicity via fibrosis and apoptosis through the TGF-β1/Smads pathway in vivo and in vitro. Ecotoxicol Environ Saf. (2023) 249:114422. doi: 10.1016/j.ecoenv.2022.114422
35. Ueshima, E, Fujimori, M, Kodama, H, Felsen, D, Chen, J, Durack, JC, et al. Macrophage-secreted TGF-β1 contributes to fibroblast activation and ureteral stricture after ablation injury. Am J Physiol. (2019) 317:F52–64. doi: 10.1152/ajprenal.00260.2018
36. Nasu, K, Kawakami, T, Shinohara, A, Sakamoto, T, and Nangaku, M. Munc18-1-interacting protein 3 mitigates renal fibrosis through protection of tubular epithelial cells from apoptosis. Nephrol Dial Transplant. (2019) 35:576–86. doi: 10.1093/ndt/gfz177
37. Yang, S-R, Hung, S-C, Chu, LJ, Hua, K-F, Wei, C-W, Tsai, I-L, et al. NSC828779 alleviates renal Tubulointerstitial lesions involving Interleukin-36 signaling in mice. Cells. (2021) 10:3060. doi: 10.3390/cells10113060
38. Chen, Y-T, Jhao, P-Y, Hung, C-T, Wu, Y-F, Lin, S-J, Chiang, W-C, et al. Endoplasmic reticulum protein TXNDC5 promotes renal fibrosis by enforcing TGF-β signaling in kidney fibroblasts. J Clin Invest. (2021) 131:645. doi: 10.1172/JCI143645
39. Briffa, J, Sinagra, E, and Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon. (2020) 6:e04691. doi: 10.1016/j.heliyon.2020.e04691
40. Sarkar, M, and Chandra Pal, S. Human health hazard assessment for high groundwater arsenic and fluoride intact in Malda district, Eastern India. Groundw Sustain Dev. (2021) 13:100565. doi: 10.1016/j.gsd.2021.100565
41. Bencko, V, and Yan Li Foong, F. The history of arsenical pesticides and health risks related to the use of agent blue. Ann Agric Environ Med. (2017) 24:312–6. doi: 10.26444/aaem/74715
42. Ahmed, MK, Shaheen, N, Islam, MS, Habibullah-Al-Mamun, M, Islam, S, Islam, MM, et al. A comprehensive assessment of arsenic in commonly consumed foodstuffs to evaluate the potential health risk in Bangladesh. Sci Total Environ. (2016) 544:125–33. doi: 10.1016/j.scitotenv.2015.11.133
43. Koepsell, H. Glucose transporters in the small intestine in health and disease. Pflugers Arch. (2020) 472:1207–48. doi: 10.1007/s00424-020-02439-5
44. Orr, SE, and Bridges, CC. Chronic kidney disease and exposure to nephrotoxic metals. Int J Mol Sci. (2017) 18:1039. doi: 10.3390/ijms18051039
45. Raab, A, Meharg, AA, Jaspars, M, Genney, DR, and Feldmann, J. Arsenic–glutathione complexes—their stability in solution and during separation by different HPLC modes. J Anal At Spectrom. (2004) 19:183–90. doi: 10.1039/B307945G
46. Staud, F, Cerveny, L, Ahmadimoghaddam, D, and Ceckova, M. Multidrug and toxin extrusion proteins (MATE/SLC47); role in pharmacokinetics. Int J Biochem Cell Biol. (2013) 45:2007–11. doi: 10.1016/j.biocel.2013.06.022
47. Zalups, RK. Autometallographic localization of inorganic mercury in the kidneys of rats: effect of unilateral nephrectomy and compensatory renal growth. Exp Mol Pathol. (1991) 54:10–21. doi: 10.1016/0014-4800(91)90039-Z
48. Roggenbeck, BA, Banerjee, M, and Leslie, EM. Cellular arsenic transport pathways in mammals. J Environ Sci (China). (2016) 49:38–58. doi: 10.1016/j.jes.2016.10.001
49. Leslie, EM, Deeley, RG, and Cole, SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. (2005) 204:216–37. doi: 10.1016/j.taap.2004.10.012
50. Jha, V, Garcia-Garcia, G, Iseki, K, Li, Z, Naicker, S, Plattner, B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. (2013) 382:260–72. doi: 10.1016/S0140-6736(13)60687-X
51. Zalups, RK. Enhanced renal outer medullary uptake of mercury associated with uninephrectomy: implication of a luminal mechanism. J Toxicol Environ Health. (1997) 50:173–94. doi: 10.1080/009841097160564
52. Liu, S, Wang, X, Guo, G, and Yan, Z. Status and environmental management of soil mercury pollution in China: a review. J Environ Manag. (2021) 277:111442. doi: 10.1016/j.jenvman.2020.111442
53. Long, C, Li, X, Jiang, Z, Zhang, P, Qing, Z, Qing, T, et al. Adsorption-improved MoSe(2) nanosheet by heteroatom doping and its application for simultaneous detection and removal of mercury (II). J Hazard Mater. (2021) 413:125470. doi: 10.1016/j.jhazmat.2021.125470
54. Fattima Al-Zahra Gabar, G. Mercury pollution: dangers and treatment In: M Monique, HHA Mohamed, B Teresa, and AA Ahmed, editors. Marine pollution. Rijeka: IntechOpen (2023). 10.
55. Taylor, VF, Bugge, D, Jackson, BP, and Chen, CY. Pathways of CH3Hg and hg ingestion in benthic organisms: an enriched isotope approach. Environ Sci Technol. (2014) 48:5058–65. doi: 10.1021/es404159k
56. Zalups, RK. Early aspects of the intrarenal distribution of mercury after the intravenous administration of mercuric chloride. Toxicology. (1993) 79:215–28. doi: 10.1016/0300-483X(93)90213-C
57. Danscher, G, Hørsted-Bindslev, P, and Rungby, J. Traces of mercury in organs from primates with amalgam fillings. Exp Mol Pathol. (1990) 52:291–9. doi: 10.1016/0014-4800(90)90070-T
58. Aslamkhan, AG, Han, YH, Yang, XP, Zalups, RK, and Pritchard, JB. Human renal organic anion transporter 1-dependent uptake and toxicity of mercuric-thiol conjugates in Madin-Darby canine kidney cells. Mol Pharmacol. (2003) 63:590–6. doi: 10.1124/mol.63.3.590
59. Zalups, RK, and Ahmad, S. Handling of cysteine S-conjugates of methylmercury in MDCK cells expressing human OAT1. Kidney Int. (2005) 68:1684–99. doi: 10.1111/j.1523-1755.2005.00585.x
60. Carranza-Rosales, P, Said-Fernández, S, Sepúlveda-Saavedra, J, Cruz-Vega, DE, and Gandolfi, AJ. Morphologic and functional alterations induced by low doses of mercuric chloride in the kidney OK cell line: ultrastructural evidence for an apoptotic mechanism of damage. Toxicology. (2005) 210:111–21. doi: 10.1016/j.tox.2005.01.006
61. Koppe, L, Fouque, D, and Soulage, CO. The role of Gut microbiota and diet on uremic retention solutes production in the context of chronic kidney disease. Toxins (Basel). (2018) 10:155. doi: 10.3390/toxins10040155
62. Rukavina Mikusic, NL, Kouyoumdzian, NM, and Choi, MR. Gut microbiota and chronic kidney disease: evidences and mechanisms that mediate a new communication in the gastrointestinal-renal axis. Pflugers Arch. (2020) 472:303–20. doi: 10.1007/s00424-020-02352-x
63. Wu, IW, Hsu, KH, Lee, CC, Sun, CY, Hsu, HJ, Tsai, CJ, et al. P-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant. (2011) 26:938–47. doi: 10.1093/ndt/gfq580
64. Miyazaki, T, Ise, M, Seo, H, and Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-beta 1, TIMP-1 and pro-alpha 1(I) collagen in uremic rat kidneys. Kidney Int Suppl. (1997) 62:S15–22.
65. Kanbay, M, Onal, EM, Afsar, B, Dagel, T, Yerlikaya, A, Covic, A, et al. The crosstalk of gut microbiota and chronic kidney disease: role of inflammation, proteinuria, hypertension, and diabetes mellitus. Int Urol Nephrol. (2018) 50:1453–66. doi: 10.1007/s11255-018-1873-2
66. Hamza, E, Vallejo-Mudarra, M, Ouled-Haddou, H, García-Caballero, C, Guerrero-Hue, M, Santier, L, et al. Indoxyl sulfate impairs erythropoiesis at BFU-E stage in chronic kidney disease. Cell Signal. (2023) 104:110583. doi: 10.1016/j.cellsig.2022.110583
67. Ichii, O, Otsuka-Kanazawa, S, Nakamura, T, Ueno, M, Kon, Y, Chen, W, et al. Podocyte injury caused by Indoxyl sulfate, a uremic toxin and aryl-hydrocarbon receptor ligand. PLoS One. (2014) 9:e108448. doi: 10.1371/journal.pone.0108448
68. Yang, T, Richards, EM, Pepine, CJ, and Raizada, MK. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol. (2018) 14:442–56. doi: 10.1038/s41581-018-0018-2
69. Plata, C, Cruz, C, Cervantes, LG, and Ramírez, V. The gut microbiota and its relationship with chronic kidney disease. Int Urol Nephrol. (2019) 51:2209–26. doi: 10.1007/s11255-019-02291-2
70. Han, H, Zhu, J, Zhu, Z, Ni, J, Du, R, Dai, Y, et al. P-Cresyl sulfate aggravates cardiac dysfunction associated with chronic kidney disease by enhancing apoptosis of cardiomyocytes. J Am Heart Assoc. (2015) 4:e001852. doi: 10.1161/JAHA.115.001852
71. Sun, CY, Chang, SC, and Wu, MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS One. (2012) 7:e34026. doi: 10.1371/journal.pone.0034026
72. Pelletier, CC, Croyal, M, Ene, L, Aguesse, A, Billon-Crossouard, S, Krempf, M, et al. Elevation of trimethylamine-N-oxide in chronic kidney disease: contribution of decreased glomerular filtration rate. Toxins (Basel). (2019) 11:635. doi: 10.3390/toxins11110635
73. Tang, WH, Wang, Z, Kennedy, DJ, Wu, Y, Buffa, JA, Agatisa-Boyle, B, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. (2015) 116:448–55. doi: 10.1161/CIRCRESAHA.116.305360
74. Stubbs, JR, House, JA, Ocque, AJ, Zhang, S, Johnson, C, Kimber, C, et al. Serum trimethylamine-N-oxide is elevated in CKD and correlates with coronary atherosclerosis burden. J Am Soc Nephrol. (2016) 27:305–13. doi: 10.1681/ASN.2014111063
75. Feehally, J, Farrall, M, Boland, A, Gale, DP, Gut, I, Heath, S, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. (2010) 21:1791–7. doi: 10.1681/ASN.2010010076
76. Chen, XJ, Zhang, H, Yang, F, Liu, Y, and Chen, G. DNA methylation sustains "inflamed" memory of peripheral immune cells aggravating kidney inflammatory response in chronic kidney disease. Front Physiol. (2021) 12:637480. doi: 10.3389/fphys.2021.637480
77. Wanner, N, and Bechtel-Walz, W. Epigenetics of kidney disease. Cell Tissue Res. (2017) 369:75–92. doi: 10.1007/s00441-017-2588-x
78. Schiffer, M, von Gersdorff, G, Bitzer, M, Susztak, K, and Böttinger, EP. Smad proteins and transforming growth factor-beta signaling. Kidney Int Suppl. (2000) 58:S45–52. doi: 10.1046/j.1523-1755.2000.07708.x
79. Inazaki, K, Kanamaru, Y, Kojima, Y, Sueyoshi, N, Okumura, K, Kaneko, K, et al. Smad3 deficiency attenuates renal fibrosis, inflammation,and apoptosis after unilateral ureteral obstruction. Kidney Int. (2004) 66:597–604. doi: 10.1111/j.1523-1755.2004.00779.x
80. Ding, N, Xie, L, Ma, F, Ma, S, Xiong, J, Lu, G, et al. miR-30a-5p promotes glomerular podocyte apoptosis via DNMT1-mediated hypermethylation under hyperhomocysteinemia. Acta Biochim Biophys Sin Shanghai. (2022) 54:126–36. doi: 10.3724/abbs.2021005
81. Tanaka, S, Tanaka, T, and Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am J Physiol Renal Physiol. (2014) 307:F1187–95. doi: 10.1152/ajprenal.00425.2014
82. Belkaid, Y, and Harrison, OJ. Homeostatic immunity and the microbiota. Immunity. (2017) 46:562–76. doi: 10.1016/j.immuni.2017.04.008
83. Hsu, C-N, and Tain, Y-L. Chronic kidney disease and Gut microbiota: what is their connection in early life? Int J Mol Sci. (2022) 23:3954. doi: 10.3390/ijms23073954
84. Hu, Y, and Ivashkiv, LB. Costimulation of chemokine receptor signaling by matrix Metalloproteinase-9 mediates enhanced migration of IFN-α dendritic cells. J Immunol. (2006) 176:6022–33. doi: 10.4049/jimmunol.176.10.6022
85. Vaziri, ND, Wong, J, Pahl, M, Piceno, YM, Yuan, J, Desantis, TZ, et al. Chronic kidney disease alters intestinal microbial flora. Kidney Int. (2013) 83:308–15. doi: 10.1038/ki.2012.345
86. Rysz, J, Franczyk, B, Ławiński, J, Olszewski, R, Ciałkowska-Rysz, A, and Gluba-Brzózka, A. The impact of CKD on uremic toxins and Gut microbiota. Toxins. (2021) 13:252. doi: 10.3390/toxins13040252
87. Gonçalves, S, Pecoits-Filho, R, Perreto, S, Barberato, SH, Stinghen, AEM, Lima, EGA, et al. Ssociations between renal function, volume status and endotoxaemia in chronic kidney disease patients. Nephrol Dial Transplant. (2006) 21:2788–94. doi: 10.1093/ndt/gfl273
88. Mishima, E, Fukuda, S, Mukawa, C, Yuri, A, Kanemitsu, Y, Matsumoto, Y, et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. (2017) 92:634–45. doi: 10.1016/j.kint.2017.02.011
89. Tanaka, S, Watanabe, H, Nakano, T, Imafuku, T, Kato, H, Tokumaru, K, et al. Indoxyl sulfate contributes to adipose tissue inflammation through the activation of NADPH oxidase. Toxins (Basel). (2020) 12:502. doi: 10.3390/toxins12080502
90. Hand, TW, Vujkovic-Cvijin, I, Ridaura, VK, and Belkaid, Y. Linking the microbiota, chronic disease, and the immune system. Trends Endocrinol Metab. (2016) 27:831–43. doi: 10.1016/j.tem.2016.08.003
91. Anders, HJ, Andersen, K, and Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. (2013) 83:1010–6. doi: 10.1038/ki.2012.440
92. Donadei, C, Angeletti, A, Pizzuti, V, Zappulo, F, Conte, D, Cappuccilli, M, et al. Impact of single hemodialysis treatment on immune cell subpopulations. J Clin Med. (2023) 12:3107. doi: 10.3390/jcm12093107
93. Grabulosa, CC, Manfredi, SR, Canziani, ME, Quinto, BMR, Barbosa, RB, Rebello, JF, et al. Chronic kidney disease induces inflammation by increasing toll-like receptor-4, cytokine and cathelicidin expression in neutrophils and monocytes. Exp Cell Res. (2018) 365:157–62. doi: 10.1016/j.yexcr.2018.02.022
94. Rossi, M, Campbell, KL, Johnson, DW, Stanton, T, Vesey, DA, Coombes, JS, et al. Protein-bound uremic toxins, inflammation and oxidative stress: a Cross-sectional study in stage 3–4 chronic kidney disease. Arch Med Res. (2014) 45:309–17. doi: 10.1016/j.arcmed.2014.04.002
95. Sabatino, A, Regolisti, G, Brusasco, I, Cabassi, A, Morabito, S, and Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol Dial Transplant. (2015) 30:924–33. doi: 10.1093/ndt/gfu287
96. Kurashima, Y, and Kiyono, H. Mucosal ecological network of epithelium and immune cells for Gut homeostasis and tissue healing. Annu Rev Immunol. (2017) 35:119–47. doi: 10.1146/annurev-immunol-051116-052424
97. Lin, T, Chen, L, Yang, X, Fu, C, Yuk, H-G, Fan, R, et al. NLRP3 and Gut microbiota homeostasis: Progress in research. Cells. (2022) 11:3758. doi: 10.3390/cells11233758
98. Melekoglu, E, and Samur, FG. Dietary strategies for gut-derived protein-bound uremic toxins and cardio-metabolic risk factors in chronic kidney disease: a focus on dietary fibers. Crit Rev Food Sci Nutr. (2023) 63:3994–4008. doi: 10.1080/10408398.2021.1996331
99. Burnier, M, and Damianaki, A. Hypertension as cardiovascular risk factor in chronic kidney disease. Circ Res. (2023) 132:1050–63. doi: 10.1161/CIRCRESAHA.122.321762
100. Lau, WL, and Vaziri, ND. The leaky Gut and altered microbiome in chronic kidney disease. J Ren Nutr. (2017) 27:458–61. doi: 10.1053/j.jrn.2017.02.010
101. Spoto, B, Pisano, A, and Zoccali, C. Insulin resistance in chronic kidney disease: a systematic review. Am J Physiol Ren Physiol. (2016) 311:F1087–108. doi: 10.1152/ajprenal.00340.2016
102. Vaziri, ND, Yuan, J, and Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol. (2013) 37:1–6. doi: 10.1159/000345969
103. Yan, MT, and Chao, CT. Lin SH: chronic kidney disease: strategies to retard progression. Int J Mol Sci. (2021) 22:84. doi: 10.3390/ijms221810084
104. Yang, J, Lim, SY, Ko, YS, Lee, HY, Oh, SW, Kim, MG, et al. Intestinal barrier disruption and dysregulated mucosal immunity contribute to kidney fibrosis in chronic kidney disease. Nephrol Dial Transplantat. (2019) 34:419–28. doi: 10.1093/ndt/gfy172
105. Nichols, B, Jog, P, Lee, JH, Blackler, D, Wilmot, M, D'agati, V, et al. Innate immunity pathways regulate the nephropathy gene apolipoprotein L1. Kidney Int. (2015) 87:332–42. doi: 10.1038/ki.2014.270
106. Zhaorigetu, S, Wan, G, Kaini, R, Wan, G, and Jiang, Z. Hu C-aA: ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. (2008) 4:1079–82. doi: 10.4161/auto.7066
107. Beckerman, P, Bi-Karchin, J, Park, ASD, Qiu, C, Dummer, PD, Soomro, I, et al. Hu C-AA: transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med. (2017) 23:429–38. doi: 10.1038/nm.4287
108. Kassem, NO, Daffa, RM, Liles, S, Jackson, SR, Kassem, NO, Younis, MA, et al. Children's exposure to secondhand and thirdhand smoke carcinogens and toxicants in homes of hookah smokers. Nicotine Tob Res. (2014) 16:961–75. doi: 10.1093/ntr/ntu016
109. Cahill, TM. Ambient acrolein concentrations in coastal, remote, and urban regions in California. Environ Sci Technol. (2014) 48:8507–13. doi: 10.1021/es5014533
110. Stevens, JF, and Maier, CS. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol Nutr Food Res. (2008) 52:7–25. doi: 10.1002/mnfr.200700412
111. Abraham, K, Andres, S, Palavinskas, R, Berg, K, Appel, KE, and Lampen, A. Toxicology and risk assessment of acrolein in food. Mol Nutr Food Res. (2011) 55:1277–90. doi: 10.1002/mnfr.201100481
112. Uchida, K, Kanematsu, M, Morimitsu, Y, Osawa, T, Noguchi, N, and Niki, E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. (1998) 273:16058–66. doi: 10.1074/jbc.273.26.16058
113. Adams, JD, and Klaidman, LK. Acrolein-induced oxygen radical formation. Free Radic Biol Med. (1993) 15:187–93. doi: 10.1016/0891-5849(93)90058-3
114. Ong, FHC, Henry, P, and Burcham, P. / prior exposure to acrolein accelerates pulmonary inflammation in influenza A-infected mice. Toxicol Lett. (2012) 212:241–51. doi: 10.1016/j.toxlet.2012.06.003
115. Sarkar, P, and Hayes, BE. Induction of COX-2 by acrolein in rat lung epithelial cells. Mol Cell Biochem. (2007) 301:191–9. doi: 10.1007/s11010-007-9411-z
116. Lambert, C, Li, J, Jonscher, K, Yang, TC, Reigan, P, Quintana, M, et al. Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-kappaB1 DNA binding domain. J Biol Chem. (2007) 282:19666–75. doi: 10.1074/jbc.M611527200
117. Valacchi, G, Pagnin, E, Phung, A, Nardini, M, Schock, BC, Cross, CE, et al. Inhibition of NFkappaB activation and IL-8 expression in human bronchial epithelial cells by acrolein. Antioxid Redox Signal. (2005) 7:25–31. doi: 10.1089/ars.2005.7.25
118. Aihara, S, Torisu, K, Hirashima, Y, Kitazono, T, and Nakano, T. Acrolein produced during acute kidney injury promotes tubular cell death. Biochem Biophys Res Commun. (2023) 666:137–45. doi: 10.1016/j.bbrc.2023.05.029
119. Tong, ZJ, Kuo, CW, Yen, PC, Lin, CC, Tsai, MT, Lu, SH, et al. Acrolein plays a culprit role in the pathogenesis of diabetic nephropathy in vitro and in vivo. Eur J Endocrinol. (2022) 187:579–92. doi: 10.1530/EJE-22-0493
120. Korkmaz, A, Topal, T, and Oter, S. Pathophysiological aspects of cyclophosphamide and ifosfamide induced hemorrhagic cystitis; implication of reactive oxygen and nitrogen species as well as PARP activation. Cell Biol Toxicol. (2007) 23:303–12. doi: 10.1007/s10565-006-0078-0
121. Liu, WC, Tomino, Y, and Lu, KC. Impacts of Indoxyl sulfate and p-cresol sulfate on chronic kidney disease and mitigating effects of AST-120. Toxins (Basel). (2018) 10:367. doi: 10.3390/toxins10090367
122. Sato, E, Hosomi, K, Sekimoto, A, Mishima, E, Oe, Y, Saigusa, D, et al. Effects of the oral adsorbent AST-120 on fecal p-cresol and indole levels and on the gut microbiota composition. Biochem Biophys Res Commun. (2020) 525:773–9. doi: 10.1016/j.bbrc.2020.02.141
123. Cha, RH, Kang, SW, Park, CW, Cha, DR, Na, KY, Kim, SG, et al. Sustained uremic toxin control improves renal and cardiovascular outcomes in patients with advanced renal dysfunction: post-hoc analysis of the Kremezin study against renal disease progression in Korea. Kidney Res Clin Pract. (2017) 36:68–78. doi: 10.23876/j.krcp.2017.36.1.68
124. Chen, YC, Wu, MY, Hu, PJ, Chen, TT, Shen, WC, Chang, WC, et al. Effects and safety of an Oral adsorbent on chronic kidney disease progression: a systematic review and Meta-analysis. J Clin Med. (2019) 8:718. doi: 10.3390/jcm8101718
125. Rodríguez-Osorio, L, Zambrano, DP, Gracia-Iguacel, C, Rojas-Rivera, J, Ortiz, A, Egido, J, et al. Use of sevelamer in chronic kidney disease: beyond phosphorus control. Nefrologia. (2015) 35:207–17. doi: 10.1016/j.nefro.2015.05.022
126. Rayego-Mateos, S, and Valdivielso, JM. New therapeutic targets in chronic kidney disease progression and renal fibrosis. Expert Opin Ther Targets. (2020) 24:655–70. doi: 10.1080/14728222.2020.1762173
127. Mandarino, FV, Sinagra, E, Raimondo, D, and Danese, S. The role of microbiota in upper and lower gastrointestinal functional disorders. Microorganisms. (2023) 11:980. doi: 10.3390/microorganisms11040980
128. Chang, CJ, Lin, TL, Tsai, YL, Wu, TR, Lai, WF, Lu, CC, et al. Next generation probiotics in disease amelioration. J Food Drug Anal. (2019) 27:615–22. doi: 10.1016/j.jfda.2018.12.011
129. Durazzo, A, Nazhand, A, Lucarini, M, Atanasov, AG, Souto, EB, Novellino, E, et al. An updated overview on Nanonutraceuticals: focus on Nanoprebiotics and Nanoprobiotics. Int J Mol Sci. (2020) 21:285. doi: 10.3390/ijms21072285
130. Gomez Quintero, DF, Kok, CR, and Hutkins, R. The future of Synbiotics: rational formulation and design. Front Microbiol. (2022) 13:919725. doi: 10.3389/fmicb.2022.919725
131. Kalmokoff, M, Zwicker, B, O'Hara, M, Matias, F, Green, J, Shastri, P, et al. Temporal change in the gut community of rats fed high amylose cornstarch is driven by endogenous urea rather than strictly on carbohydrate availability. J Appl Microbiol. (2013) 114:1516–28. doi: 10.1111/jam.12157
132. Kieffer, DA, Piccolo, BD, Vaziri, ND, Liu, S, Lau, WL, Khazaeli, M, et al. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am J Physiol Renal Physiol. (2016) 310:F857–71. doi: 10.1152/ajprenal.00513.2015
133. Vaziri, ND, Liu, S-M, Lau, WL, Khazaeli, M, Nazertehrani, S, Farzaneh, SH, et al. High amylose resistant starch diet ameliorates oxidative stress, inflammation, and progression of chronic kidney disease. PLoS One. (2014) 9:e114881. doi: 10.1371/journal.pone.0114881
134. Krishnamurthy, VM, Wei, G, Baird, BC, Murtaugh, M, Chonchol, MB, Raphael, KL, et al. High dietary fiber intake is associated with decreased inflammation and all-cause mortality in patients with chronic kidney disease. Kidney Int. (2012) 81:300–6. doi: 10.1038/ki.2011.355
135. Carlstrom, M, and Montenegro, MF. Therapeutic value of stimulating the nitrate-nitrite-nitric oxide pathway to attenuate oxidative stress and restore nitric oxide bioavailability in cardiorenal disease. J Intern Med. (2019) 285:2–18. doi: 10.1111/joim.12818
136. Gherghina, ME, Peride, I, Tiglis, M, Neagu, TP, Niculae, A, and Checherita, IA. Uric acid and oxidative stress-relationship with cardiovascular, metabolic, and renal impairment. Int J Mol Sci. (2022) 23:188. doi: 10.3390/ijms23063188
137. Jung, SW, Kim, SM, Kim, YG, Lee, SH, and Moon, JY. Uric acid and inflammation in kidney disease. Am J Physiol Renal Physiol. (2020) 318:F1327–f1340. doi: 10.1152/ajprenal.00272.2019
138. Rapa, SF, Di Iorio, BR, Campiglia, P, Heidland, A, and Marzocco, S. Inflammation and oxidative stress in chronic kidney disease-potential therapeutic role of minerals, vitamins and plant-derived metabolites. Int J Mol Sci. (2019) 21:263. doi: 10.3390/ijms21010263
139. Gorin, Y, and Wauquier, F. Upstream regulators and downstream effectors of NADPH oxidases as novel therapeutic targets for diabetic kidney disease. Mol Cells. (2015) 38:285–96. doi: 10.14348/molcells.2015.0010
140. Wang, D, Li, J, Luo, G, Zhou, J, Wang, N, Wang, S, et al. Nox4 as a novel therapeutic target for diabetic vascular complications. Redox Biol. (2023) 64:102781. doi: 10.1016/j.redox.2023.102781
141. Dounousi, E, Papavasiliou, E, Makedou, A, Ioannou, K, Katopodis, KP, Tselepis, A, et al. Oxidative stress is progressively enhanced with advancing stages of CKD. Am J Kidney Dis. (2006) 48:752–60. doi: 10.1053/j.ajkd.2006.08.015
142. Locatelli, F, Canaud, B, Eckardt, KU, Stenvinkel, P, Wanner, C, and Zoccali, C. Oxidative stress in end-stage renal disease: an emerging threat to patient outcome. Nephrol Dial Transplant. (2003) 18:1272–80. doi: 10.1093/ndt/gfg074
143. Roberts, LJ, and Morrow, JD. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med. (2000) 28:505–13. doi: 10.1016/S0891-5849(99)00264-6
144. Schei, J, Fuskevåg, OM, Stefansson, VTN, Solbu, MD, Jenssen, TG, Eriksen, BO, et al. Urinary markers of oxidative stress are associated with albuminuria but not GFR decline. Kidney Int Rep. (2018) 3:573–82. doi: 10.1016/j.ekir.2017.11.020
146. Bondi, CD, Manickam, N, Lee, DY, Block, K, Gorin, Y, Abboud, HE, et al. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. (2010) 21:93–102. doi: 10.1681/ASN.2009020146
147. Mack, M, and Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. (2015) 87:297–307. doi: 10.1038/ki.2014.287
148. Wang, H, Chen, X, Su, Y, Paueksakon, P, Hu, W, Zhang, MZ, et al. p47(phox) contributes to albuminuria and kidney fibrosis in mice. Kidney Int. (2015) 87:948–62. doi: 10.1038/ki.2014.386
149. Modlinger, PS, Wilcox, CS, and Aslam, S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin Nephrol. (2004) 24:354–65. doi: 10.1016/j.semnephrol.2004.04.007
150. Amdur, RL, Feldman, HI, Gupta, J, Yang, W, Kanetsky, P, Shlipak, M, et al. Inflammation and progression of CKD: the CRIC study. Clin J Am Soc Nephrol. (2016) 11:1546–56. doi: 10.2215/CJN.13121215
151. Graterol Torres, F, Molina, M, Soler-Majoral, J, Romero-González, G, Rodríguez Chitiva, N, Troya-Saborido, M, et al. Evolving concepts on inflammatory biomarkers and malnutrition in chronic kidney disease. Nutrients. (2022) 14:297. doi: 10.3390/nu14204297
152. Kadatane, SP, Satariano, M, Massey, M, Mongan, K, and Raina, R. The role of inflammation in CKD. Cells. (2023) 12:581. doi: 10.3390/cells12121581
153. Batra, G, Lakic, TG, Lindbäck, J, Held, C, White, HD, Stewart, RAH, et al. Interleukin 6 and cardiovascular outcomes in patients with chronic kidney disease and chronic coronary syndrome. JAMA Cardiol. (2021) 6:1440–5. doi: 10.1001/JAMACARDIO.2021.3079
154. Abais, JM, Xia, M, Li, G, Chen, Y, Conley, SM, Gehr, TWB, et al. Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J Biol Chem. (2014) 289:27159–68. doi: 10.1074/jbc.M114.567537
155. Sun, J, Axelsson, J, Machowska, A, Heimbürger, O, Bárány, P, Lindholm, B, et al. Biomarkers of cardiovascular disease and mortality risk in patients with advanced CKD. Clin J Am Soc Nephrol. (2016) 11:1163–72. doi: 10.2215/CJN.10441015
156. Barreto, DV, Barreto, FC, Liabeuf, S, Temmar, M, Lemke, HD, Tribouilloy, C, et al. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. (2010) 77:550–6. doi: 10.1038/ki.2009.503
157. Jones, SA, Fraser, DJ, Fielding, CA, and Jones, GW. Interleukin-6 in renal disease and therapy. Nephrol Dial Transplant. (2014) 30:564–74. doi: 10.1093/ndt/gfu233
158. Kreiner, FF, Kraaijenhof, JM, von Herrath, M, Hovingh, GKK, and von Scholten, BJ. Interleukin 6 in diabetes, chronic kidney disease, and cardiovascular disease: mechanisms and therapeutic perspectives. Expert Rev Clin Immunol. (2022) 18:377–89. doi: 10.1080/1744666X.2022.2045952
159. Mima, T, and Nishimoto, N. Clinical value of blocking IL-6 receptor. Curr Opin Rheumatol. (2009) 21:224–30. doi: 10.1097/BOR.0b013e3283295fec
160. Chang, MS, and Hsu, YH. The role of IL-20 in chronic kidney disease and diabetic nephropathy: pathogenic and therapeutic implications. J Leukoc Biol. (2018) 104:919–23. doi: 10.1002/JLB.MR1217-489R
161. Hsu, YH, Wu, CH, Chiu, CJ, Chen, WT, Chang, YC, Wabitsch, M, et al. IL-20 is involved in obesity by modulation of adipogenesis and macrophage dysregulation. Immunology. (2021) 164:817–33. doi: 10.1111/imm.13403
162. Romanova, Y, Laikov, A, Markelova, M, Khadiullina, R, Makseev, A, Hasanova, M, et al. Salafutdinov I: proteomic analysis of human serum from patients with chronic kidney disease. Biomol Ther. (2020) 10:257. doi: 10.3390/biom10020257
163. Chiu, S, and Bharat, A. Role of monocytes and macrophages in regulating immune response following lung transplantation. Curr Opin Organ Transplant. (2016) 21:239–45. doi: 10.1097/MOT.0000000000000313
164. Geissmann, F, Manz, MG, Jung, S, Sieweke, MH, Merad, M, and Ley, K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
165. Caillon, A, Paradis, P, and Schiffrin, EL. Role of immune cells in hypertension. Br J Pharmacol. (2019) 176:1818–28. doi: 10.1111/bph.14427
166. Kim, HJ, Lee, DW, Ravichandran, K, Keys, DO, Akcay, A, Nguyen, Q, et al. NLRP3 Inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther. (2013) 346:465–72. doi: 10.1124/jpet.113.205732
167. Kim, SY, and Nair, MG. Macrophages in wound healing: activation and plasticity. Immunol Cell Biol. (2019) 97:258–67. doi: 10.1111/imcb.12236
168. Stout, RD, and Suttles, J. Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunol Rev. (2005) 205:60–71. doi: 10.1111/j.0105-2896.2005.00260.x
169. Anders, HJ, Suarez-Alvarez, B, Grigorescu, M, Foresto-Neto, O, Steiger, S, Desai, J, et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1–mediated tissue injury. Kidney Int. (2018) 93:656–69. doi: 10.1016/j.kint.2017.09.022
170. Liu, D, Xu, M, Ding, LH, Lv, LL, Liu, H, Ma, KL, et al. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: a novel mechanism of albumin-induced tubulointerstitial inflammation. Int J Biochem Cell Biol. (2014) 57:7–19. doi: 10.1016/j.biocel.2014.09.018
171. Albert, C, Zapf, A, Haase, M, Röver, C, Pickering, JW, Albert, A, et al. Neutrophil gelatinase-associated Lipocalin measured on clinical laboratory platforms for the prediction of acute kidney injury and the associated need for Dialysis therapy: a systematic review and Meta-analysis. Am J Kidney Dis. (2020) 76:826–841.e1. doi: 10.1053/j.ajkd.2020.05.015
172. Anderson, AH, Xie, D, Wang, X, Baudier, RL, Orlandi, P, Appel, LJ, et al. Novel risk factors for progression of diabetic and nondiabetic CKD: findings from the chronic renal insufficiency cohort (CRIC) study. Am J Kidney Dis. (2021) 77:56–73.e1. doi: 10.1053/j.ajkd.2020.07.011
173. Banai, A, Rozenfeld, KL, Lewit, D, Merdler, I, Loewenstein, I, Banai, S, et al. Neutrophil gelatinase-associated lipocalin (NGAL) for the prediction of acute kidney injury in chronic kidney disease patients treated with primary percutaneous coronary intervention. IJC Heart Vasc. (2021) 32:100695. doi: 10.1016/j.ijcha.2020.100695
174. Glassford, NJ, Schneider, AG, Eastwood, G, Peck, L, Young, H, and Bellomo, R. Neutrophil gelatinase-associated lipocalin as a marker of tubular damage appears to be unrelated to fractional excretion of sodium as a marker of tubular function in septic patients, with or without AKI. Crit Care. (2011) 15:10. doi: 10.1186/cc10379
175. Marouf, R, Adekile, AD, El-Muzaini, H, Abdulla, R, and Mojiminiyi, OA. Neutrophil gelatinase-associated lipocalin as a biomarker of nephropathy in sickle cell disease. Ann Hematol. (2021) 100:1401–9. doi: 10.1007/s00277-021-04500-4
176. Schmidt-Ott, KM, Mori, K, Kalandadze, A, Li, JY, Paragas, N, Nicholas, T, et al. Neutrophil gelatinase-associated lipocalin-mediated iron traffic in kidney epithelia. Curr Opin Nephrol Hypertens. (2006) 15:442–9. doi: 10.1097/01.mnh.0000232886.81142.58
177. Sise, ME, Barasch, J, Devarajan, P, and Nickolas, TL. Elevated urine neutrophil gelatinase-associated lipocalin can diagnose acute kidney injury in patients with chronic kidney diseases. Kidney Int. (2009) 75:115–6. doi: 10.1038/ki.2008.529
178. Tecson, KM, Erhardtsen, E, Eriksen, PM, Gaber, AO, Germain, M, Golestaneh, L, et al. Optimal cut points of plasma and urine neutrophil gelatinase-associated lipocalin for the prediction of acute kidney injury among critically ill adults: retrospective determination and clinical validation of a prospective multicentre study. BMJ. (2017) 7:e016028. doi: 10.1136/bmjopen-2017-016028
179. Zeng, F, Singh, AB, and Harris, RC. The role of the EGF family of ligands and receptors in renal development, physiology and pathophysiology. Exp Cell Res. (2009) 315:602–10. doi: 10.1016/j.yexcr.2008.08.005
180. Zhang, A, Cai, Y, Wang, PF, Qu, JN, Luo, ZC, Chen, XD, et al. Diagnosis and prognosis of neutrophil gelatinase-associated lipocalin for acute kidney injury with sepsis: a systematic review and meta-analysis. Crit Care. (2016) 20:1212. doi: 10.1186/s13054-016-1212-x
181. Zhou, F, Luo, Q, Wang, L, and Han, L. Diagnostic value of neutrophil gelatinase-associated lipocalin for early diagnosis of cardiac surgery-associated acute kidney injury: a meta-analysis. Eur J Cardiothorac Surg. (2016) 49:746–55. doi: 10.1093/ejcts/ezv199
182. Bansal, A, Nigoskar, S, and Thalquotra, M. Comparison of BTP, NGAL, KIM-1, & ADMA biomarkers in CKD and non-CKD subjects. Int J Biochem Mol Biol. (2023) 14:32–9.
183. Provenzano, M, Andreucci, M, Garofalo, C, Faga, T, Michael, A, Ielapi, N, et al. The Association of Matrix Metalloproteinases with chronic kidney disease and peripheral vascular disease: a light at the end of the tunnel? Biomol Ther. (2020) 10:154. doi: 10.3390/biom10010154
184. Thrailkill, KM, Clay Bunn, R, and Fowlkes, JL. Matrix metalloproteinases: their potential role in the pathogenesis of diabetic nephropathy. Endocrine. (2009) 35:1–10. doi: 10.1007/s12020-008-9114-6
185. Zheng, CM, Lu, KC, Chen, YJ, Li, CY, Lee, YH, and Chiu, HW. Matrix metalloproteinase-7 promotes chronic kidney disease progression via the induction of inflammasomes and the suppression of autophagy. Biomed Pharmacother. (2022) 154:113565. doi: 10.1016/j.biopha.2022.113565
186. Tan, RJ, Li, Y, Rush, BM, Cerqueira, DM, Zhou, D, Fu, H, et al. Tubular injury triggers podocyte dysfunction by β-catenin–driven release of MMP-7. JCI. Insight. (2019) 4:399. doi: 10.1172/jci.insight.122399
187. Liu, Z, Tan, RJ, and Liu, Y. The many faces of matrix Metalloproteinase-7 in kidney diseases. Biomol Ther. (2020) 10:960. doi: 10.3390/biom10060960
188. Zhou, D, Tian, Y, Sun, L, Zhou, L, Xiao, L, Tan, RJ, et al. Matrix metalloproteinase-7 is a urinary biomarker and pathogenic mediator of kidney fibrosis. J Am Soc Nephrol. (2017) 28:598–611. doi: 10.1681/ASN.2016030354
189. Andreucci, M, Provenzano, M, Faga, T, Michael, A, Patella, G, Mastroroberto, P, et al. Aortic aneurysms, chronic kidney disease and metalloproteinases. Biomol Ther. (2021) 11:194. doi: 10.3390/biom11020194
190. Peiskerová, M, Kalousová, M, Kratochvílová, M, Dusilová-Sulková, S, Uhrová, J, Bandúr, S, et al. Fibroblast growth factor 23 and matrix-metalloproteinases in patients with chronic kidney disease: are they associated with cardiovascular disease? Kidney Blood Press Res. (2009) 32:276–83. doi: 10.1159/000243050
191. Sampieri, CL, and Orozco-Ortega, RA. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in chronic kidney disease and acute kidney injury: a systematic review of the literature. Hippokratia. (2018) 22:99–104.
192. Lv, LL, Tang, PM-K, Li, CJ, You, YK, Li, J, Huang, X-R, et al. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. (2017) 91:587–602. doi: 10.1016/j.kint.2016.10.020
193. Arumugam, TV, Manzanero, S, Furtado, M, Biggins, PJ, Hsieh, Y-H, Gelderblom, M, et al. An atypical role for the myeloid receptor Mincle in central nervous system injury. J Cereb Blood Flow Metab. (2017) 37:2098–111. doi: 10.1177/0271678X16661201
194. Wang, C, Zhang, Y, Shen, A, Tang, T, Li, N, Xu, C, et al. Mincle receptor in macrophage and neutrophil contributes to the unresolved inflammation during the transition from acute kidney injury to chronic kidney disease. Front Immunol. (2024) 15:1385696. doi: 10.3389/fimmu.2024.1385696
195. Serbinova, E, Kagan, V, Han, D, and Packer, L. Free radical recycling and intramembrane mobility in the antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Free Radic Biol Med. (1991) 10:263–75. doi: 10.1016/0891-5849(91)90033-Y
196. Machlin, L, and Gabriel, E. Kinetics of tissue alpha-tocopherol uptake and depletion following administration of high levels of vitamin E. Ann N Y Acad Sci. (1982) 393:48–60. doi: 10.1111/j.1749-6632.1982.tb31231.x
197. Karamouzis, I, Sarafidis, PA, Karamouzis, M, Iliadis, S, Haidich, A-B, Sioulis, A, et al. Increase in oxidative stress but not in antioxidant capacity with advancing stages of chronic kidney disease. Am J Nephrol. (2008) 28:397–404. doi: 10.1159/000112413
198. Giray, B, Kan, E, Bali, M, Hincal, F, and Basaran, N. The effect of vitamin E supplementation on antioxidant enzyme activities and lipid peroxidation levels in hemodialysis patients. Clin Chim Acta. (2003) 338:91–8. doi: 10.1016/j.cccn.2003.07.020
199. Wongmekiat, O, Thamprasert, K, and Lumlertgul, D. Renoprotective effect of trolox against ischaemia–reperfusion injury in rats. Clin Exp Pharmacol Physiol. (2007) 34:753–9. doi: 10.1111/j.1440-1681.2007.04651.x
200. Small, DM, Coombes, JS, Bennett, N, Johnson, DW, and Gobe, GC. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology. (2012) 17:311–21. doi: 10.1111/j.1440-1797.2012.01572.x
201. Peake, JM, Gobe, GC, Fassett, RG, and Coombes, JS. The effects of dietary fish oil on inflammation, fibrosis and oxidative stress associated with obstructive renal injury in rats. Mol Nutr Food Res. (2011) 55:400–10. doi: 10.1002/mnfr.201000195
202. Arab, K, Rossary, A, Flourié, F, Tourneur, Y, and Steghens, J-P. Docosahexaenoic acid enhances the antioxidant response of human fibroblasts by upregulating γ-glutamyl-cysteinyl ligase and glutathione reductase. Br J Nutr. (2006) 95:18–26. doi: 10.1079/BJN20051626
203. Tumur, Z, Shimizu, H, Enomoto, A, Miyazaki, H, and Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-ĸB activation. Am J Nephrol. (2010) 31:435–41. doi: 10.1159/000299798
204. Hsu, S-P, Chiang, C-K, Yang, S-Y, and Chien, C-T. N-acetylcysteine for the management of anemia and oxidative stress in hemodialysis patients. Nephron Clin Pract. (2010) 116:c207–16. doi: 10.1159/000317201
205. Nascimento, MM, Suliman, ME, Silva, M, Chinaglia, T, Marchioro, J, Hayashi, SY, et al. Effect of oral N-acetylcysteine treatment on plasma inflammatory and oxidative stress markers in peritoneal dialysis patients: a placebo-controlled study. Perit Dial Int. (2010) 30:336–42. doi: 10.3747/pdi.2009.00073
206. El-Sheikh, AA, Van Den Heuvel, JJ, Koenderink, JB, and Russel, FG. Effect of hypouricaemic and hyperuricaemic drugs on the renal urate efflux transporter, multidrug resistance protein 4. Br J Pharmacol. (2008) 155:1066–75. doi: 10.1038/bjp.2008.343
207. Kosugi, T, Nakayama, T, Heinig, M, Zhang, L, Yuzawa, Y, Sanchez-Lozada, LG, et al. Effect of lowering uric acid on renal disease in the type 2 diabetic db/db mice. Am J Physiol. (2009) 297:F481–8. doi: 10.1152/ajprenal.00092.2009
208. Lass, A, Forster, MJ, and Sohal, RS. Effects of coenzyme Q10 and α-tocopherol administration on their tissue levels in the mouse: elevation of mitochondrial α-tocopherol by coenzyme Q10. Free Radic Biol Med. (1999) 26:1375–82. doi: 10.1016/S0891-5849(98)00330-X
209. Frei, B, Kim, MC, and Ames, BN. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc Natl Acad Sci. (1990) 87:4879–83. doi: 10.1073/pnas.87.12.4879
210. Ishikawa, A, Kawarazaki, H, Ando, K, Fujita, M, Fujita, T, and Homma, Y. Renal preservation effect of ubiquinol, the reduced form of coenzyme Q10. Clin Exp Nephrol. (2011) 15:30–3. doi: 10.1007/s10157-010-0350-8
211. Headley, SA, Chapman, DJ, Germain, MJ, Evans, EE, Hutchinson, J, Madsen, KL, et al. The effects of 16-weeks of prebiotic supplementation and aerobic exercise training on inflammatory markers, oxidative stress, uremic toxins, and the microbiota in pre-dialysis kidney patients: a randomized controlled trial-protocol paper. BMC Nephrol. (2020) 21:1–9. doi: 10.1186/s12882-020-02177-x
212. Lee, SB, and Kalluri, R. Mechanistic connection between inflammation and fibrosis. Kidney Int. (2010) 78:S22–6. doi: 10.1038/ki.2010.418
213. Moon, JA, Kim, HT, Cho, IS, Sheen, YY, and Kim, DK. IN-1130, a novel transforming growth factor-β type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. (2006) 70:1234–43. doi: 10.1038/sj.ki.5001775
214. Genovese, G, Friedman, DJ, Ross, MD, Lecordier, L, Uzureau, P, Freedman, BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. (2010) 329:841–5. doi: 10.1126/science.1193032
215. Tzur, S, Rosset, S, Shemer, R, Yudkovsky, G, Selig, S, Tarekegn, A, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet. (2010) 128:345–50. doi: 10.1007/s00439-010-0861-0
216. Olabisi, OA, Zhang, J-Y, VerPlank, L, Zahler, N, DiBartolo, S III, Heneghan, JF, et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci. (2016) 113:830–7. doi: 10.1073/pnas.1522913113
217. Lan, X, Jhaveri, A, Cheng, K, Wen, H, Saleem, MA, Mathieson, PW, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol. (2014) 307:F326–36. doi: 10.1152/ajprenal.00647.2013
218. Giovinazzo, JA, Thomson, RP, Khalizova, N, Zager, PJ, Malani, N, Rodriguez-Boulan, E, et al. Apolipoprotein L-1 renal risk variants form active channels at the plasma membrane driving cytotoxicity. eLife. (2020) 9:e51185. doi: 10.7554/eLife.51185
219. Chen, TK, Tin, A, Peralta, CA, Appel, LJ, Choi, MJ, Lipkowitz, MS, et al. APOL1 risk variants, incident proteinuria, and subsequent eGFR decline in blacks with hypertension-attributed CKD. Clin J Am Soc Nephrol. (2017) 12:1771–7. doi: 10.2215/CJN.01180117
220. Freedman, BI, Kopp, JB, Langefeld, CD, Genovese, G, Friedman, DJ, Nelson, GW, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. (2010) 21:1422–6. doi: 10.1681/ASN.2010070730
221. Kopp, JB. Rethinking hypertensive kidney disease: arterionephrosclerosis as a genetic, metabolic, and inflammatory disorder. Curr Opin Nephrol Hypertens. (2013) 22:266–72. doi: 10.1097/MNH.0b013e3283600f8c
222. May, RM, Cassol, C, Hannoudi, A, Larsen, CP, Lerma, EV, Haun, RS, et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in coronavirus 2019 disease (COVID-19). Kidney Int. (2021) 100:1303–15. doi: 10.1016/j.kint.2021.07.015
223. Thomson, R, and Finkelstein, A. Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: relevance to trypanosome lysis. Proc Natl Acad Sci USA. (2015) 112:2894–9. doi: 10.1073/pnas.1421953112
224. Molina-Portela Mdel, P, Lugli, EB, Recio-Pinto, E, and Raper, J. Trypanosome lytic factor, a subclass of high-density lipoprotein, forms cation-selective pores in membranes. Mol Biochem Parasitol. (2005) 144:218–26. doi: 10.1016/j.molbiopara.2005.08.018
225. Rifkin, MR. Trypanosoma brucei: biochemical and morphological studies of cytotoxicity caused by normal human serum. Exp Parasitol. (1984) 58:81–93. doi: 10.1016/0014-4894(84)90023-7
226. Datta, S, Kataria, R, Zhang, JY, Moore, S, Petitpas, K, Mohamed, A, et al. Kidney disease-associated APOL1 variants have dose-dependent, dominant toxic gain-of-function. J Am Soc Nephrol. (2020) 31:2083–96. doi: 10.1681/ASN.2020010079
227. Ma, L, Chou, JW, Snipes, JA, Bharadwaj, MS, Craddock, AL, Cheng, D, et al. APOL1 renal-risk variants induce mitochondrial dysfunction. J Am Soc Nephrol. (2017) 28:1093–105. doi: 10.1681/ASN.2016050567
228. Granado, D, Müller, D, Krausel, V, Kruzel-Davila, E, Schuberth, C, Eschborn, M, et al. Intracellular APOL1 risk variants cause cytotoxicity accompanied by energy depletion. J Am Soc Nephrol. (2017) 28:3227–38. doi: 10.1681/ASN.2016111220
229. Kruzel-Davila, E, Wasser, WG, Aviram, S, and Skorecki, K. APOL1 nephropathy: from gene to mechanisms of kidney injury. Nephrol Dial Transplantat. (2015) 31:349–58. doi: 10.1093/ndt/gfu391
230. Shih, HM, Wu, CJ, and Lin, SL. Physiology and pathophysiology of renal erythropoietin-producing cells. J Formos Med Assoc. (2018) 117:955–63. doi: 10.1016/j.jfma.2018.03.017
231. Schuster, SJ, Badiavas, EV, Costa-Giomi, P, Weinmann, R, Erslev, AJ, and Caro, J. Stimulation of erythropoietin gene transcription during hypoxia and cobalt exposure. Blood. (1989) 73:13–6. doi: 10.1182/blood.V73.1.13.13
232. Bondurant, MC, and Koury, MJ. Anemia induces accumulation of erythropoietin mRNA in the kidney and liver. Mol Cell Biol. (1986) 6:2731–3. doi: 10.1128/MCB.6.7.2731
233. Chalhoub, S, Langston, C, and Eatroff, A. Anemia of renal disease: what it is, what to do and what's new. J Feline Med Surg. (2011) 13:629–40. doi: 10.1016/j.jfms.2011.07.016
234. Semenza, GL. Regulation of erythropoietin production. New insights into molecular mechanisms of oxygen homeostasis. Hematol Oncol Clin North Am. (1994) 8:863–84. doi: 10.1016/S0889-8588(18)30134-5
235. Semenza, GL, Agani, F, Booth, G, Forsythe, J, Iyer, N, Jiang, BH, et al. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. (1997) 51:553–5. doi: 10.1038/ki.1997.77
236. Zhu, H, and Bunn, HF. Oxygen sensing and signaling: impact on the regulation of physiologically important genes. Respir Physiol. (1999) 115:239–47. doi: 10.1016/S0034-5687(99)00024-9
237. Crugliano, G, Serra, R, Ielapi, N, Battaglia, Y, Coppolino, G, Bolignano, D, et al. Hypoxia-inducible factor stabilizers in end stage kidney disease: "can the promise be kept?". Int J Mol Sci. (2021) 22:590. doi: 10.3390/ijms222212590
238. Zhang, Y, Zhu, X, Huang, X, Wei, X, Zhao, D, Jiang, L, et al. Advances in understanding the effects of erythropoietin on renal fibrosis. Front Med (Lausanne). (2020) 7:47. doi: 10.3389/fmed.2020.00047
239. Wish, JB. Treatment of Anemia in kidney disease: beyond erythropoietin. Kidney Int Rep. (2021) 6:2540–53. doi: 10.1016/j.ekir.2021.05.028
240. van Gastel, MDA, and Torres, VE. Polycystic kidney disease and the vasopressin pathway. Ann Nutr Metab. (2017) 70:43–50. doi: 10.1159/000463063
241. Bichet, DG. Central vasopressin: dendritic and axonal secretion and renal actions. Clin Kidney J. (2014) 7:242–7. doi: 10.1093/ckj/sfu050
242. Brodney, M. EA, inventors;Vertex Pharmaceutical, assignee. Inhibitors of apol1 and methods of using same. U.S. Patent No. WO 2020/131807 A1. June 25, 2020. (2020).
243. Datta, S, Antonio, BM, Zahler, NH, Theile, JW, Krafte, D, Zhang, H, et al. APOL1-mediated monovalent cation transport contributes to APOL1-mediated podocytopathy in kidney disease. J Clin Invest. (2024) 134:262. doi: 10.1172/JCI172262
244. Egbuna, O, Zimmerman, B, Manos, G, Fortier, A, Chirieac, MC, Dakin, LA, et al. Inaxaplin for Proteinuric kidney disease in persons with two APOL1 variants. N Engl J Med. (2023) 388:969–79. doi: 10.1056/NEJMoa2202396
245. Shaikh, H, Hashmi, MF, and Aeddula, NR. Anemia of chronic renal disease In: H Shaikh, editor. StatPearls. Treasure Island, F): StatPearls Publishing Copyright (2024)
247. Jelkmann, W. Molecular biology of erythropoietin. Intern Med. (2004) 43:649–59. doi: 10.2169/internalmedicine.43.649
248. Shahab, MH, and Saifullah Khan, S. Erythropoietin Administration for Anemia due to chronic kidney disease - subcutaneous OR intravenous, what do we know so far? Cureus. (2020) 12:e10358. doi: 10.7759/cureus.10358
249. Haase, VH. Hypoxia-inducible factor-prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int Suppl. (2011) 11:8–25. doi: 10.1016/j.kisu.2020.12.002
250. Sanghani, NS, and Haase, VH. Hypoxia-inducible factor activators in renal Anemia: current clinical experience. Adv Chronic Kidney Dis. (2019) 26:253–66. doi: 10.1053/j.ackd.2019.04.004
251. Kuriyama, S, Maruyama, Y, and Honda, H. A new insight into the treatment of renal anemia with HIF stabilizer. Ren Replac Ther. (2020) 6:63. doi: 10.1186/s41100-020-00311-x
252. Takkavatakarn, K, Thammathiwat, T, Phannajit, J, Katavetin, P, Praditpornsilpa, K, Eiam-Ong, S, et al. The impacts of hypoxia-inducible factor stabilizers on laboratory parameters and clinical outcomes in chronic kidney disease patients with renal anemia: a systematic review and meta-analysis. Clin Kidney J. (2023) 16:845–58. doi: 10.1093/ckj/sfac271
253. Wang, X, Gattone, V 2nd, Harris, PC, and Torres, VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. (2005) 16:846–51. doi: 10.1681/ASN.2004121090
254. Furlano, M, Talavera, F, Aronoff, GR, Batuman, V, Mulloy, LL, and Torra, R. Polycystic kidney disease medication: Vasopressin antagonists, angiotensin-converting enzyme inhibitors, angiotensin II receptor antagonists, antibiotics, Carbapenems, phosphate binders. Medscape. (2023). Available at: https://emedicine.medscape.com/article/244907-medication.
255. Kawanami, D, Matoba, K, Takeda, Y, Nagai, Y, Akamine, T, Yokota, T, et al. SGLT2 inhibitors as a therapeutic option for diabetic nephropathy. Int J Mol Sci. (2017) 18:1083. doi: 10.3390/ijms18051083
256. Gonzalez, CD, Carro Negueruela, MP, Nicora Santamarina, C, Resnik, R, and Vaccaro, MI. Autophagy dysregulation in diabetic kidney disease: from pathophysiology to pharmacological interventions. Cells. (2021) 10:2497. doi: 10.3390/cells10092497
257. Tuttle, KR. Digging deep into cells to find mechanisms of kidney protection by SGLT2 inhibitors. J Clin Invest. (2023) 133:700. doi: 10.1172/JCI167700
258. Packer, M. Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: implications for understanding the effects of sodium-glucose cotransporter 2-inhibitors. J Am Soc Nephrol. (2020) 31:907–19. doi: 10.1681/ASN.2020010010
259. Packer, M. SGLT2 inhibitors produce cardiorenal benefits by promoting adaptive cellular reprogramming to induce a state of fasting mimicry: a paradigm shift in understanding their mechanism of action. Diabetes Care. (2020) 43:508–11. doi: 10.2337/dci19-0074
260. Kim, YC, and Guan, K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. (2015) 125:25–32. doi: 10.1172/JCI73939
261. Kogot-Levin, A, Hinden, L, Riahi, Y, Israeli, T, Tirosh, B, Cerasi, E, et al. Proximal tubule mTORC1 is a central player in the pathophysiology of diabetic nephropathy and its correction by SGLT2 inhibitors. Cell Rep. (2020) 32:107954. doi: 10.1016/j.celrep.2020.107954
262. Herzig, S, and Shaw, RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
263. Osataphan, S, Macchi, C, Singhal, G, Chimene-Weiss, J, Sales, V, Kozuka, C, et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and-independent mechanisms. JCI Insight. (2019) 4:130. doi: 10.1172/jci.insight.123130
264. Park, CH, Lee, B, Han, M, Rhee, WJ, Kwak, MS, Yoo, T-H, et al. Canagliflozin protects against cisplatin-induced acute kidney injury by AMPK-mediated autophagy in renal proximal tubular cells. Cell Death Discov. (2022) 8:12. doi: 10.1038/s41420-021-00801-9
265. Xu, J, Kitada, M, Ogura, Y, Liu, H, and Koya, D. Dapagliflozin restores impaired autophagy and suppresses inflammation in high glucose-treated HK-2 cells. Cells. (2021) 10:1457. doi: 10.3390/cells10061457
266. Liu, X, Xu, C, Xu, L, Li, X, Sun, H, Xue, M, et al. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism. (2020) 111:154334. doi: 10.1016/j.metabol.2020.154334
267. Nemoto, S, Fergusson, MM, and Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. (2004) 306:2105–8. doi: 10.1126/science.1101731
268. Liu, T, Ma, X, Ouyang, T, Chen, H, Lin, J, Liu, J, et al. SIRT1 reverses senescence via enhancing autophagy and attenuates oxidative stress-induced apoptosis through promoting p53 degradation. Int J Biol Macromol. (2018) 117:225–34. doi: 10.1016/j.ijbiomac.2018.05.174
269. Ruderman, NB, Julia, XX, Nelson, L, Cacicedo, JM, Saha, AK, Lan, F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. (2010) 298:E751–60. doi: 10.1152/ajpendo.00745.2009
270. Ghosh, HS, McBurney, M, and Robbins, PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. (2010) 5:e9199. doi: 10.1371/journal.pone.0009199
271. Zhang, T, Chi, Y, Kang, Y, Lu, H, Niu, H, Liu, W, et al. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1α mediated attenuation of mitochondrial oxidative stress. J Cell Physiol. (2019) 234:5033–43. doi: 10.1002/jcp.27306
272. Mohamed, HE, Asker, ME, Keshawy, MM, Hasan, RA, and Mahmoud, YK. Inhibition of tumor necrosis factor-α enhanced the antifibrotic effect of empagliflozin in an animal model with renal insulin resistance. Mol Cell Biochem. (2020) 466:45–54. doi: 10.1007/s11010-020-03686-x
273. Umino, H, Hasegawa, K, Minakuchi, H, Muraoka, H, Kawaguchi, T, Kanda, T, et al. High basolateral glucose increases sodium-glucose cotransporter 2 and reduces sirtuin-1 in renal tubules through glucose transporter-2 detection. Sci Rep. (2018) 8:6791. doi: 10.1038/s41598-018-25054-y
274. Packer, M. Mechanisms leading to differential hypoxia-inducible factor signaling in the diabetic kidney: modulation by SGLT2 inhibitors and hypoxia mimetics. Am J Kidney Dis. (2021) 77:280–6. doi: 10.1053/j.ajkd.2020.04.016
275. Bessho, R, Takiyama, Y, Takiyama, T, Kitsunai, H, Takeda, Y, Sakagami, H, et al. Hypoxia-inducible factor-1α is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci Rep. (2019) 9:14754. doi: 10.1038/s41598-019-51343-1
276. Chen, R, Xu, M, Hogg, RT, Li, J, Little, B, Gerard, RD, et al. The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. J Biol Chem. (2012) 287:30800–11. doi: 10.1074/jbc.M111.244780
277. Cassis, P, Locatelli, M, Cerullo, D, Corna, D, Buelli, S, Zanchi, C, et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight. (2018) 3:720. doi: 10.1172/jci.insight.98720
278. Chen, X, Hocher, C-F, Shen, L, Krämer, BK, and Hocher, B. Reno-and cardioprotective molecular mechanisms of SGLT2 inhibitors beyond glycemic control: from bedside to bench. Am J Phys Cell Phys. (2023) 325:C661–81. doi: 10.1152/ajpcell.00177.2023
279. Lopez-Ruiz, A, Chandrashekar, K, and Juncos, LA. Nrf2 activation in the glomeruli and podocytes: deciphering the renal mechanisms of Nrf2. Kidney360. (2023) 4:1350–2. doi: 10.34067/KID.0000000000000268
280. Yau, K, Dharia, A, Alrowiyti, I, and Cherney, DZI. Prescribing SGLT2 inhibitors in patients with CKD: expanding indications and practical considerations. Kidney Int Rep. (2022) 7:1463–76. doi: 10.1016/j.ekir.2022.04.094
281. Wheeler, DC, Stefánsson, BV, Jongs, N, Chertow, GM, Greene, T, Hou, FF, et al. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: a prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. (2021) 9:22–31. doi: 10.1016/S2213-8587(20)30369-7
282. Ghanim, H, Abuaysheh, S, Hejna, J, Green, K, Batra, M, Makdissi, A, et al. Dapagliflozin suppresses Hepcidin and increases erythropoiesis. J Clin Endocrinol Metab. (2020) 105:e1056–63. doi: 10.1210/clinem/dgaa057
283. Mavrakanas, TA, Tsoukas, MA, Brophy, JM, Sharma, A, and Gariani, K. SGLT-2 inhibitors improve cardiovascular and renal outcomes in patients with CKD: a systematic review and meta-analysis. Sci Rep. (2023) 13:15922. doi: 10.1038/s41598-023-42989-z
284. Empagliflozin in Patients with Chronic Kidney Disease. Empagliflozin in patients with chronic kidney disease. N Engl J Med. (2023) 388:117–27. doi: 10.1056/NEJMoa2204233
Keywords: advances, kidney disease, pathogenesis, therapy, APOL1 gene
Citation: Boima V, Agyekum AB, Ganatra K, Agyekum F, Kwakyi E, Inusah J, Ametefe EN and Adu D (2025) Advances in kidney disease: pathogenesis and therapeutic targets. Front. Med. 12:1526090. doi: 10.3389/fmed.2025.1526090
Edited by:
Anil Kumar Pasupulati, University of Hyderabad, IndiaReviewed by:
Nasar Alwahaibi, Sultan Qaboos University, OmanCopyright © 2025 Boima, Agyekum, Ganatra, Agyekum, Kwakyi, Inusah, Ametefe and Adu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francis Agyekum, ZnJhbmFneWVrdW1AZ21haWwuY29t; ZnJhZ3lla3VtQHVnLmVkdS5naA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.