 Mengdi Qu
Mengdi Qu Hao Zhang*†
Hao Zhang*† Zhaoyuan Chen
Zhaoyuan Chen Shuainan Zhu
Shuainan Zhu Wankun Chen
Wankun Chen Changhong Miao
Changhong Miao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 07 May 2021
Sec. Pulmonary Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.651552
This article is part of the Research Topic Early Origins of Lung Disease - An Interdisciplinary Approach View all 15 articles
Ferroptosis is a newly discovered type of regulated cell death that is different from apoptosis, necrosis and autophagy. Ferroptosis is characterized by iron-dependent lipid peroxidation, which induces cell death. Iron, lipid and amino acid metabolism is associated with ferroptosis. Ferroptosis is involved in the pathological development of various diseases, such as neurological diseases and cancer. Recent studies have shown that ferroptosis is also closely related to acute lung injury (ALI)/ acute respiratory distress syndrome (ARDS), suggesting that it can be a novel therapeutic target. This article mainly introduces the metabolic mechanism related to ferroptosis and discusses its role in ALI/ARDS to provide new ideas for the treatment of these diseases.
Ferroptosis, a new form of regulated cell death that can be triggered by erastin or RSL3 [(1S,3R)-RSL3], was first reported in 2012 by Dixon (1). Ferroptosis is characterized by the iron-dependent accumulation of lethal levels of lipid peroxides, while the morphology, biology and genetics are obviously different from those of apoptosis, necrosis, autophagy, and other forms of cell death (1). Amino acid, iron and lipid peroxide metabolism and other metabolic processes are closely related to ferroptosis (2). Studies have shown that ferroptosis, as the main cause of organ damage-related cell death, is involved in many pathological processes, such as neurodegenerative diseases, cancer and ischemia-reperfusion injury (2, 3).
Acute lung injury (ALI), resulting from both direct (e.g., pneumonia) and indirect (e.g., sepsis) pulmonary injuries, refers to pulmonary edema and atelectasis caused by diffuse alveolar-capillary injury and is characterized by refractory hypoxemia and pulmonary infiltration (4). Acute respiratory distress syndrome (ARDS) is a serious form of ALI and is described by the 2012 Berlin unified definition (5). The prevalence of ARDS in intensive care units is 10.4%, and there is a high mortality rate (35–46%) (6) but a lack of effective treatments (7).
In recent years, the role of ferroptosis in ARDS has been gradually revealed, and increasing attention has been given to the importance of regulating ferroptosis in the treatment of ARDS.
Ferroptosis is a form of cell death that is regulated by multiple genes and involves multiple metabolic processes, such as iron homeostasis, amino acid metabolism and lipid peroxidation. The mechanism is very complex as is shown in Figure 1, and it will be better explained in the following aspects.
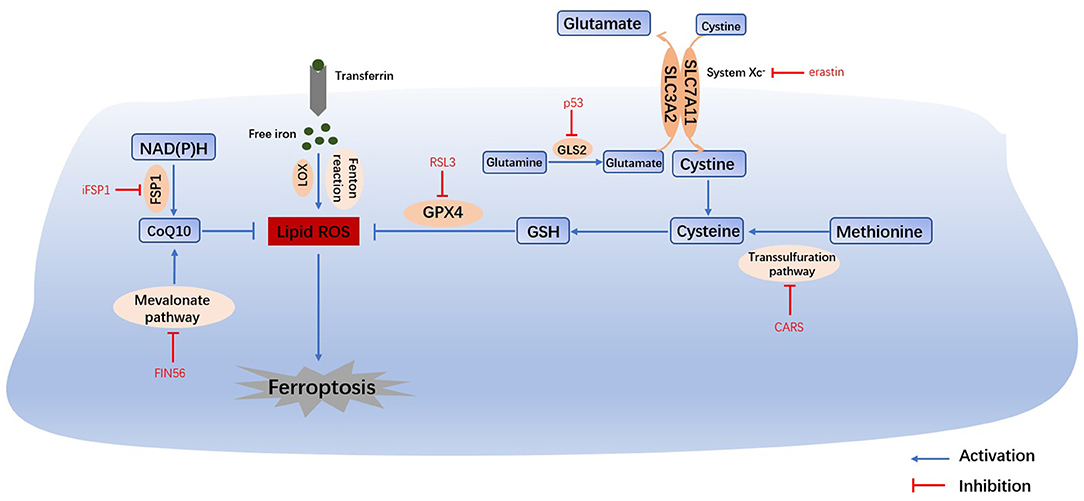
Figure 1. Main mechanisms of ferroptosis. The Fenton reaction, LOX and PUFAs facilitate the generation of lipid ROS. Cysteine can be generated from the uptake of cystine via system Xc- or the transsulfuration pathway. Amino acid metabolism and NAD(P)H, suppresses the synthesis of GSH and CoQ10, thus inhibiting the reduction in lipid ROS. The accumulation of lipid ROS leads to ferroptosis. Therefore, iron homeostasis, lipid peroxidation and amino acid metabolism are the main regulators of ferroptosis. System Xc-, cystine/glutamate transporter; GLS, glutaminase; GSH, glutathione; GPX4, glutathione peroxidase 4; LOX, lipoxygenase; FSP1, ferroptosis suppressor protein 1; CoQ10, coenzyme Q10.
Iron overload is one of the key events in ferroptosis. Iron is necessary for the accumulation of lipid peroxides, and iron ingestion, storage and transport all affect ferroptosis (2). Iron homeostasis is regulated by a series of iron regulatory proteins (IRPs). Extracellular iron enters the cell through transferrin (TF) and its receptors, and then Fe2+ can produce lipid peroxides via the Fenton reaction or the iron-containing enzyme lipoxygenase (LOX) (8). Most intracellular Fe2+ is stored in ferritin (FT), and so there is very little free Fe2+ (9). The degradation of FT increases the level of intracellular Fe2+, enhances lipid peroxidation, and induces ferroptosis. This process is related to autophagy and is regulated by nuclear receptor coactivator 4 (NCOA4) (8). Iron response element binding protein 2 (IREB2) (1) and other proteins related to iron metabolism (2) (HSPB1, CISD1, etc.) can also increase the sensitivity of cells to ferroptosis.
Glutathione (GSH) depletion is another key event in ferroptosis. The cystine/glutamate antiporter System Xc-, which is mainly composed of SLC3A2 (solute carrier family 3 member 2) and SLC7A11 (solute carrier family 7 member 11) (10), is located on the cell membrane and transports extracellular cystine and intracellular glutamate at a ratio of 1:1. Extracellular cystine and intracellular cysteine are essential for the biosynthesis of GSH. Cystine ultimately generates GSH through a series of enzymatic reactions, and GSH is the essential substrate for glutathione peroxide enzyme 4 (GPX4) to degrade phospholipid hydroperoxide (PLOOH) (11). GPX4 is at the intersection of GSH metabolism and lipid peroxidation, both of which are related to ferroptosis. Downregulation of SLC7A11 can also lead to ferroptosis through a decrease in GPX4 activity (12). In addition, methionine can transfer sulfur atoms to serine to generate cysteine through the transsulfuration pathway, which can be upregulated by the knockout of cysteinyl-tRNA synthetase (CARS), thus making cells resistant to ferroptosis (13).
Under physiological conditions, a high level of extracellular glutamate can inhibit the activity of System Xc- and prevent the uptake of cysteine (14). Therefore, glutamate is a natural trigger for ferroptosis and has the same effect as erastin and other System Xc- inhibitors (8). In addition, glutamine is abundant in tissue and plasma and can be converted into glutamate by glutaminase (GLS1 and GLS2) catalysis. Glutaminolysis is necessary for the tricarboxylic acid cycle and lipid biosynthesis, and α-ketoglutarate, as the product of decomposition, is involved in ferroptosis (15). Therefore, when glutamine is deficient or its decomposition is inhibited, reactive oxygen species (ROS) accumulation, lipid peroxidation and ferroptosis are also inhibited. Furthermore, GLS2, as a target gene of the tumor suppressor p53, is closely related to ferroptosis (16). In summary, the metabolism of amino acids (especially glutamate and cystine) plays an important role in the pathological process of ferroptosis.
The most prominent feature of ferroptosis is plasma membrane damage caused by the production of iron-dependent lipid peroxides (lipid ROS) (8). ROS include products of oxygen reduction, such as O2−, H2O2 and ·OH. Oxygen homeostasis is crucial to normal cellular functions, and the abnormal accumulation of ROS is harmful to the body (3). During ferroptosis, the reduction reaction mediated by GPX4 and ferroptosis suppressor protein 1 (FSP1, formerly known as mitochondrial apoptosis inducing factor 2, AIFM2) is inhibited, and the oxidation reaction catalyzed by Fe2+ and a series of iron-dependent enzymes (mainly LOX) is enhanced, inducing the accumulation of polyunsaturated fatty acids (PUFAs) (8). Then, lipid peroxidation drove by PUFAs increases the permeability of the cell membrane and makes the cell more sensitive to oxidation, which eventually leads to ferroptosis (17, 18). The inhibition of lipid peroxidation and the consumption of PUFAs can inhibit ferroptosis (2).
Bersuker and Doll found that FSP1 and GPX4 had a strong synergistic effect (19, 20). In the FSP1-NAD(P)H pathway, coenzyme Q10 (CoQ10) can reduce lipid peroxidation by inhibiting the accumulation of free radicals, and FSP1 catalyzes the production of CoQ10 through NAD(P)H. iFSP1, an inhibitor of FSP1, can induce ferroptosis in cells that overexpress FSP1 (20). In conclusion, the FSP1-CoQ10-NAD(P)H pathway cooperates with GPX4 and GSH to inhibit lipid peroxidation and ferroptosis. Moreover, CoQ10 can also be generated by the mevalonate (MVA) pathway. FIN56 can not only accelerate the degradation of GPX4 but also consume CoQ10 by affecting the MVA pathway, ultimately leading to excessive lipid peroxide accumulation and ferroptosis (21).
The most common cause of ARDS is bacterial or viral pneumonia, while sepsis, severe trauma and gastric reflux and aspiration are also common factors (22). The inflammatory response is activated by infection, trauma, or damage to the lung. Moderate inflammation is conducive to the clearance of pathogens, but excessive inflammation may lead to alveolar damage and increased permeability of the pulmonary capillary endothelium and alveolar epithelium, after which protein-rich fluid exudes from the alveolar cavity, leading to pulmonary edema (22). Therefore, ARDS is the pulmonary manifestation of systemic inflammatory response syndrome (SIRS) (23), which involves various inflammatory cells (macrophages, neutrophils, vascular endothelial cells, and platelets), and the inflammatory mediators and cytokines released by these cells indirectly mediate inflammation in the lung.
The levels of proinflammatory cytokines (IL-1β, IL-8, TNFα, TGFβ1, etc.) are very high in the pulmonary edema fluid in ARDS patients, and cytokines can activate the innate immune system. Activated neutrophils can produce toxic substances such as ROS and proteases, leading to pulmonary endothelial and alveolar epithelial damage and even necrosiss (24). Necrosis and the accumulation of edema fluid, in turn, trigger more severe inflammation and immune responses. Many clinical trials have evaluated the potential effect of anti-inflammatory therapy to treat ARDS (25–27). In summary, excessive inflammation and increased permeability of the pulmonary capillary endothelium and alveolar epithelium lead to alveolar damage, which is the main pathological mechanism of ARDS.
Various cell types in the lung, including epithelial cells and macrophages, can produce iron metabolism-related proteins to regulate iron homeostasis and protect lung tissue from oxidative stress (28). Iron metabolism disorders are closely related to lung tissue damage in ARDS patients (29, 30); that is, too much iron can generate ROS and cytotoxicity through the Fenton reaction. Many clinical studies have shown that the severity of ARDS is associated with the levels of iron and iron-related proteins (31). One study indicated that iron in blood products leads to an increase in iron in blood recipients, which promotes the occurrence of blood transfusion-related ALI (32). Elevated levels of Fe2+ and iron regulators, such as TF and FT, can be detected in the bronchoalveolar lavage fluid (BALF) of ARDS patients (28, 29, 33–35). In an oleic acid-induced ALI model in mice, iron overload was detected in the lung tissue (36). Moreover, supplementing mice with iron in advance exacerbates damage to the lung (12). A recent study also showed that increased apoptosis in mice with iron overload exacerbated ALI. However, this effect was quite transient and did not affect the degree of inflammation or speed of recovery in ALI (37). Ferroptosis is an iron-dependent process, and iron overload is the driving factor of it. In ARDS, iron overload leads to ferroptosis, which aggravates lung injury. In other words, the cells appear to be overloaded with iron due to ferroptosis and the disease becomes increasingly worse. Therefore, we have enough reason to believe that ferroptosis plays a crucial role in ARDS. And whether iron overload actually causes lung injury or is just a byproduct of ferroptosis remains to be confirmed.
Exhaled breath analysis is expected to be clinically used for the early diagnosis and prediction of ARDS, and most candidate markers are related to oxidative stress (38). Oxidative stress causes damage to the barriers of the pulmonary epithelium and endothelium, and neutrophils accumulate in large quantities in the alveolar fluid, producing proinflammatory cytokines and ROS. Moreover, ROS can further increase the level of cytokines, exacerbating tissue damage and edema. Therefore, oxidative stress plays an important role in the pathogenesis of ARDS (39, 40). ROS are known as important mediators of ARDS (41–44), and enzymes related to the production of ROS (xanthine oxidase (XOR) (45), endothelial nitric oxide synthase (eNOS) (46), cytochrome P450 (CYP) (7), and NADPH oxidase (NOX) (47)) have been reported to be involved in ARDS. The level of malondialdehyde (MDA), a product of lipid peroxidation, is increased in the ALI mouse model (36, 48). In fact, MDA is commonly regarded as a marker of ferroptosis. Increases in both neutrophils and ROS can be detected in ARDS patients (49).
GSH is the most important antioxidant in the airway epithelium and exerts antioxidant effects through the removal of ROS (50) and the repair of cellular damage (51), thus helping to alleviate inflammation (52). Decreased GSH and increased oxidized GSH (GSSG) were observed in both ALI patients and animal models (12, 36, 42). A lack of GSH in alveolar fluid made ARDS patients more susceptible to lung injury (49). Moreover, ROS and GSH metabolism is the main feature of ferroptosis. Whether these metabolic processes are also involved in the pathogenesis of ARDS by regulating ferroptosis needs to be investigated. Inhibition of ferroptosis can inhibit the production of these peroxides, thus reducing the severity of ARDS, which is a potential therapeutic strategy.
Indicators related to ferroptosis were detected in ALI animal models, including increased Fe2+, ROS, MDA and decreased GSH. And inhibitors of ferroptosis have the potential to alleviate lung damage. These results show that ferroptosis is indeed associated with ARDS. However, the specific mechanism by which ferroptosis affects the onset of ARDS is still unclear.
As a clinically common respiratory disease, the main pathogenic mechanism of ARDS is the apoptosis of alveolar epithelial cells and pulmonary microvascular endothelial cells and the polarization of alveolar macrophages. Then, a large amount of ROS and inflammatory factors trigger an imbalance between the oxidation and antioxidant systems, and the “cytokine storm” leads to the disturbance in the local microenvironment of the lungs, resulting in a series of inflammatory reactions (53). Unlike apoptosis, ferroptosis is associated with a consistent release of damage-associated molecular patterns (DAMPs) and inflammatory cytokines, which promote a series of inflammatory responses. Therefore, ferroptosis is considered an immunogenic form of cell death (54). Inflammatory cytokines further promote ferroptosis and other forms of cell death, thus forming a self-amplifying loop that mutually promotes organ damage (55). Ferroptosis plays a key role in ALI in mice, and ferroptosis inducers can exacerbate pulmonary edema and alveolar inflammation, accompanied by high levels of cytokines (IL-1β, IL-6, and TNF-α), while these effects can be reversed by ferroptosis inhibitors (12, 41, 48). In the latest study (36), ARDS animal model was prepared by injecting oleic acid into the tail vein of mice. The results showed that the pulmonary cells of ARDS group showed mitochondrial shrinkage and rupture of the mitochondrial membrane. In addition, iron overload, GSH depletion and down-regulated expression of ferritin appeared in lung tissues. Similar results were observed in the model of lung ischemia-reperfusion injury (56). The above results suggest that ferroptosis is involved in the pathogenesis of lung injury, which will provide a new theoretical basis for the clinical treatment of ARDS. However, no clinical studies have examined the association of these ferroptosis indicators with severity and prognosis of ARDS.
Ferroptosis is one of the critical mechanisms contributing to sepsis-induced injuries in mice models, including heart, liver, intestine, and the inhibition of ferroptosis via enhancing GPX4 or nuclear factor erythroid 2-related factor 2 (Nrf2) alleviates these injuries (57–60). It contradicts that erastin attenuates the inflammatory response, resulting in inhibition of sepsis development (61). So what is the real role of ferroptosis? We know that sepsis is also an important inducer of ARDS, so it is natural to consider the role of ferroptosis in sepsis-induced lung injury. Furthermore, the lipid peroxidation in ferroptosis drives pyroptosis, indicating a crosstalk between ferroptosis and other forms of cell death in sepsis, and such interactions may also exist in ARDS (62).
Ultimately, ferroptosis causes cellular injury primarily through inflammation and oxidative stress, and the NOD-like receptor protein 3 (NLRP3) inflammasome and Nrf2 are key molecules in these processes (12, 41, 42, 63, 64). Both of them are important regulatory molecule in ARDS and can be used as targets for the treatment of ARDS (65–71). Of course, they could be recognized as mediators of ferroptosis in ARDS, as shown in Figure 2. Ferroptosis may promote inflammation and swelling of alveolar epithelial cells via the NLRP3 inflammasome, bringing about ARDS. NLRP3 is a key mediator in the process of pyroptosis, so crosstalk between ferroptosis and pyroptosis may occur in the pathogenesis of ARDS, aggravating lung injury. Further animal experiments and clinical studies are needed to verify these points. The regulation of the NLRP3 inflammasome by inhibiting ferroptosis to thereby alleviate ARDS may also be a new therapeutic strategy.

Figure 2. Possible relationship between ferroptosis and ARDS. Here is an ARDS lung. DAMPs, ROS, and iron overload contribute to ARDS, and they can activate the NLRP3 inflammasome, which promotes the maturation and secretion of inflammatory factors, forming a self-amplifying loop with ferroptosis. Nrf2 inhibits the generation of ROS and iron to negatively regulate ferroptosis. However, the direct link between ferroptosis and ARDS is unclear. DAMPs, damage-associated molecular patterns; ROS, reactive oxygen species; NLRP3, NOD-like receptor protein 3; Nrf2, nuclear factor erythroid 2-related factor 2.
At the same time, recent studies have shown that Nrf2 inhibits ferroptosis by regulating the expression of SLC7A11 and heme oxygenase-1 (HO-1), thus alleviating lung injury (12, 64). Nrf2 activators can cause a reduction in ROS and prevent GSH depletion and lipid peroxide accumulation. As a result, ferroptosis is inhibited, thereby alleviating ALI and producing the same effect as that of Fer-1 (42). In addition, inhibitor of apoptosis-stimulating protein of p53 (iASPP) could inhibit ferroptosis and ALI by upregulating Nrf2. Furthermore, the levels of a variety of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) were also decreased (41).
In balance, most studies have not directly focused on the relationship between ferroptosis and ARDS. Therefore, the direct relationship between them needs to be explored, together with more treatments targeting ferroptosis.
Disorders of iron homeostasis, the depletion of GSH, and oxidative stress are the key points leading to ferroptosis and could be used as targets for the treatment of ARDS (Figure 3).
Figure 3. Treatments for ARDS. Therapeutic targets for ARDS associated with ferroptosis, such as iron chelators, antioxidants and anti-inflammatory treatments.
Iron chelators (deferoxamine, deferiprone, and deferasirox), especially deferoxamine (DFO), have been approved by the FDA for the treatment of iron overload (72). In various animal models of infection, DFO has immunomodulatory effects to resist pathogens such as bacteria, viruses, and fungi, in addition to chelating iron (73). DFO can reduce the levels of inflammatory cytokines and ROS in vitro, exerting anti-inflammatory effects (74). DFO inhalation was shown to improve pulmonary fibrosis and prevent a decline in pulmonary function in mice (75). Simultaneous perfusion of DFO and FT could attenuate leakage syndrome in isolated mouse lungs (76). In summary, iron chelators may also be effective in treating ARDS, and the mechanism may be related to the suppression of ferroptosis.
Antioxidants can reduce the severity of ARDS (7). Several kinds of drugs decrease the levels of lipid peroxidation and ROS, attenuate inflammation and oxidative stress, and ultimately alleviate ARDS in mice and improve gas exchange (77–80). GSH supplementation could significantly alleviate mitochondrial dysfunction and oxidative damage in the LPS-induced ALI model (81). Animal experiments and clinical studies have shown that regulating the level of GSH (82) via N-acetylcysteine (NAC) could promote the production of GSH and alleviate ALI (7). NAC treatment resulted in increased pulmonary compliance and reduced pulmonary edema (83). In New York, two patients with ARDS caused by COVID-19 showed significant relief of dyspnea after oral and intravenous GSH supplementation, demonstrating that this is indeed a new treatment strategy for ARDS (52). Given the importance of GSH in ferroptosis, it is also worth investigating whether GSH plays a role in the treatment of ARDS by inhibiting ferroptosis.
Inhibiting inflammation is an important treatment strategy for ARDS. Combined inhibition of ferroptosis and inflammation has been reported to treat a variety of diseases, such as stroke, myocardial infarction, and pancreatitis (54, 55). In ALI mouse models, ferroptosis inhibitors reduced inflammatory cytokines and pulmonary edema to treat ALI (41, 48). The exact mechanism of ferroptosis and inflammation needs to be confirmed by additional experiments, and ways to modulate inflammation by controlling ferroptosis also need to be further explored. These studies will provide new strategies for the clinical treatment of ARDS.
Ferroptosis is a newly discovered form of cell death, and ferroptosis regulators provide new therapeutic directions for many refractory diseases (84). Ferroptosis is an abnormal metabolic process involving iron, lipids and amino acids, and the metabolism of these substances plays a key role in cell proliferation and differentiation. Ferroptosis is characterized by metabolic imbalances and disturbances in redox homeostasis, in which the metabolic process is not independent, but a part of a complex metabolic network. The results of animal experiments and clinical trials preliminarily show that a variety of diseases and pathological processes are closely related to ferroptosis, and intervention in ferroptosis can effectively delay the progression of the disease and improve clinical symptoms to a certain extent. However, research on ferroptosis is still in its infancy. Studies on ferroptosis and lung cancer have made some progress, and ferroptosis inducers as new adjuvants based on traditional treatments have shown their effectiveness. The development of new ferroptosis inducers and the application of multiple forms of combined treatment strategies may be expected to provide new ideas for the treatment of lung cancer.
Recent studies have shown that ferroptosis is closely related to ALI/ARDS, making it a potential target for the treatment of ALI/ARDS. The current studies are based on animal models, while there is a lack of clinical studies. In this context, it is worth noting that the precise role of ferroptosis in the development of ALI/ARDS, and the pharmacological inhibition of ferroptosis, but not necroptosis or apoptosis, protects lung tissues from injury, which remains to be fully elucidated. Considering that ferroptosis was proposed as a brand new concept, there are still large gaps that need to be filled. Clinically, whether the key molecules of ferroptosis can be used as biomarkers to predict the severity of ARDS are needed to investigate. Also, it is necessary to prove whether ferroptosis is the core mode of cell death in ARDS and their crosstalk mechanism.
MQ and HZ: conception and design. All authors: manuscript writing and Final approval of manuscript.
This research was supported by the National Key Research and Development Program of China (NO. 2020YFC2008400, 2020YFC2008403), National Natural Science Foundation of China (NO. 81873948, 81871591), Clinical Research Plan of SHDC (NO. SHDC2020CR4064, SHDC2020CR1005A, SHDC12018105), Key Technology and Development Program of Shanghai (NO.17411963400), Talent Program of Fudan University (JIF159607), Shanghai Sailing Program (21YF1406800).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
2. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
3. Gao M, Jiang X. To eat or not to eat-the metabolic flavor of ferroptosis. Curr Opin Cell Biol. (2018) 51:58–64. doi: 10.1016/j.ceb.2017.11.001
4. Sweeney RM, McAuley DF, Acute respiratory distress syndrome. Lancet. (2016) 388:2416–30. doi: 10.1016/S0140-6736(16)00578-X
5. Ferguson ND, Fan E, Camporota L, Antonelli M, Anzueto A, Beale R, et al. The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive Care Med. (2012) 38:1573–82. doi: 10.1007/s00134-012-2682-1
6. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. (2016) 315:788–800. doi: 10.1001/jama.2016.0291
7. Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, Black SM. ROS signaling in the pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv Exp Med Biol. (2017) 967:105–37. doi: 10.1007/978-3-319-63245-2_8
8. Ye Z, Liu W, Zhuo Q, Hu Q, Liu M, Sun Q, et al. Ferroptosis: Final destination for cancer? Cell Prolif . (2020) 53, e12761. doi: 10.1111/cpr.12761
9. Nakamura T, Naguro I, Ichijo H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj. (2019) 1863:1398–409. doi: 10.1016/j.bbagen.2019.06.010
10. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523.018
11. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium Utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. (2018) 172:409–22. doi: 10.1016/j.cell.2017.11.048
12. Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (Albany NY). (2020) 12:12943–59. doi: 10.18632/aging.103378
13. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. (2016) 23:270–8. doi: 10.1038/cdd.2015.93
14. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. (1989) 2:1547–58. doi: 10.1016/0896-6273(89)90043-3
15. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
16. Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. (2015) 30:918–30. doi: 10.1101/gad.275891.115
17. Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
18. Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. (2018) 8:5155. doi: 10.1038/s41598-018-23408-0
19. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575: 688–92. doi: 10.1038/s41586-019-1705-2
20. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
21. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. (2016) 12:497–503. doi: 10.1038/nchembio.2079
22. Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med. (2019) 40:31–9. doi: 10.1055/s-0039-1683996
23. Potera RM, Cao M, Jordan LF, Hogg RT, Hook JS, Moreland JG. Alveolar macrophage chemokine secretion mediates neutrophilic lung injury in Nox2-deficient mice. Inflammation. (2019) 42:185–98. doi: 10.1007/s10753-018-0883-7
24. Wohlrab P, Kraft F, Tretter V, Ullrich R, Markstaller K, Klein KU. Recent advances in understanding acute respiratory distress syndrome. F1000Res. (2018) 7:263. doi: 10.12688/f1000research.11148.1
25. Meduri GU, Headley AS, Golden E, Carson SJ, Umberger RA, Kelso T, et al. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. JAMA. (1998) 280:159–65. doi: 10.1001/jama.280.2.159
26. Steinberg KP, Hudson LD, Goodman RB, Hough CL, Lanken PN, Hyzy R, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. (2006) 354:1671–684. doi: 10.1056/NEJMoa051693
27. Mongardon N, Piagnerelli M, Grimaldi D, Perrot B, Lascarrou JB, Investigators Csg. Impact of late administration of corticosteroids in COVID-19 ARDS. Intensive Care Med. (2021) 47:110–2. doi: 10.1007/s00134-020-06311-z
28. Ali MK, Kim RY, Karim R, Mayall JR, Martin KL, Shahandeh A, et al. Role of iron in the pathogenesis of respiratory disease. Int J Biochem Cell Biol. (2017) 88:181–95. doi: 10.1016/j.biocel.2017.05.003
29. Quinlan GJ, Evans TW, Gutteridge JM. Iron and the redox status of the lungs. Free Radic Biol Med. (2002) 33:1306–13. doi: 10.1016/S0891-5849(02)00903-6
30. Khiroya H, Turner AM. The role of iron in pulmonary pathology. Multidiscip Respir Med. (2015) 10:34. doi: 10.1186/s40248-015-0031-2
31. Zhang V, Nemeth E, Kim A. Iron in lung pathology. Pharmaceuticals (Basel). (2019) 12:30. doi: 10.3390/ph12010030
32. Jenkins ZA, Hagar W, Bowlus CL, Johansson HE, Harmatz P, Vichinsky EP, et al. Iron homeostasis during transfusional iron overload in beta-thalassemia and sickle cell disease: changes in iron regulatory protein, hepcidin, and ferritin expression. Pediatr Hematol Oncol. (2007) 24:237–43. doi: 10.1080/08880010701360700
33. Ghio AJ, Carter JD, Richards JH, Richer LD, Grissom CK, Elstad MR. Iron and iron-related proteins in the lower respiratory tract of patients with acute respiratory distress syndrome. Crit Care Med. (2003) 31:395–400. doi: 10.1097/01.CCM.0000050284.35609.97
34. Connelly KG, Moss M, Parsons PE, Moore EE, Moore FA, Giclas PC, et al. Serum ferritin as a predictor of the acute respiratory distress syndrome. Am J Respir Crit Care Med. (1997) 155:21–5. doi: 10.1164/ajrccm.155.1.9001283
35. Sharkey RA, Donnelly SC, Connelly KG, Robertson CE, Haslett C, Repine JE. Initial serum ferritin levels in patients with multiple trauma and the subsequent development of acute respiratory distress syndrome. Am J Respir Crit Care Med. (1999) 159(5 Pt 1), 1506–9. doi: 10.1164/ajrccm.159.5.9809027
36. Zhou H, Li F, Niu JY, Zhong WY, Tang MY, Lin D, et al. Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng Li Xue Bao. (2019) 71:689–97. doi: 10.13294/j.aps.2019.0070
37. Zhang V, Ganz T, Nemeth E, Kim A. Iron overload causes a mild and transient increase in acute lung injury. Physiol Rep. (2020) 8:e14470. doi: 10.14814/phy2.14470
38. Bos LDJ. Diagnosis of acute respiratory distress syndrome by exhaled breath analysis. Ann Transl Med. (2018) 6:33. doi: 10.21037/atm.2018.01.17
39. Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med. (1999) 27:1738–44. doi: 10.1097/00003246-199909000-00007
40. Carpenter CT, Price PV, Christman BW. Exhaled breath condensate isoprostanes are elevated in patients with acute lung injury or ARDS. Chest. (1998) 114:1653–9. doi: 10.1378/chest.114.6.1653
41. Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping F, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. (2020) 27:2635–50. doi: 10.1038/s41418-020-0528-x
42. Qiu YB, Wan BB, Liu G, Wu YX, Chen D, Lu MD, et al. Nrf2 protects against seawater drowning-induced acute lung injury via inhibiting ferroptosis. Respir Res. (2020) 21:232. doi: 10.1186/s12931-020-01500-2
43. Erol N, Saglam L, Saglam YS, Erol HS, Altun S, Aktas MS, et al. The protection potential of antioxidant vitamins against acute respiratory distress syndrome: a rat trial. Inflammation. (2019) 42:1585–94. doi: 10.1007/s10753-019-01020-2
44. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun. (2019) 519:240–5. doi: 10.1016/j.bbrc.2019.08.165
45. Abdulnour R-EE, Peng X, Finigan JH, Han EJ, Hasan EJ, Birukov KG, et al. Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am J Physiol Lung Cell Mol Physiol. (2006). 291:L345–53. doi: 10.1152/ajplung.00453.2005
46. Gross CM, Rafikov R, Kumar S, Aggarwal S, Ham PB 3rd, Meadows ML, et al. Endothelial nitric oxide synthase deficient mice are protected from lipopolysaccharide induced acute lung injury. PLoS ONE. (2015) 10:e0119918. doi: 10.1371/journal.pone.0119918
47. Sato K, Kadiiska MB, Ghio AJ, Corbett J, Fann YC, Holland SM, et al. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: a model for ARDS. FASEB J. (2002) 16:1713–20. doi: 10.1096/fj.02-0331com
48. Liu P, Feng Y, Li H, Chen X, Wang G, Xu S, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett. (2020) 25:10. doi: 10.1186/s11658-020-00205-0
49. Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. (1991) 100:1397–403. doi: 10.1378/chest.100.5.1397
50. Syrkina O, Jafari B, Hales CA, Quinn DA. Oxidant stress mediates inflammation and apoptosis in ventilator-induced lung injury. Respirology. (2008) 13:333–40. doi: 10.1111/j.1440-1843.2008.01279.x
51. Gill SS, Tuteja N. (2010). Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol Biochem. 48:909–30. doi: 10.1016/j.plaphy.2010.08.016
52. Horowitz RI, Freeman PR, Bruzzese J. Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: a report of 2 cases. Respir Med Case Rep. (2020) 30:101063. doi: 10.1016/j.rmcr.2020.101063
53. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-018-0102-y
54. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
55. Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. (2014) 14:759–67. doi: 10.1038/nri3743
56. Xu Y, Li X, Cheng Y, Yang M, Wang R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. (2020) 34:16262–75. doi: 10.1096/fj.202001758R
57. Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. (2020) 160:303–18. doi: 10.1016/j.freeradbiomed.2020.08.009
58. Li J, Lu K, Sun F, Tan S, Zhang X, Sheng W, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. (2021) 19:96. doi: 10.1186/s12967-021-02745-1
59. Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang CF, et al. Dexmedetomidine alleviated sepsisinduced myocardial ferroptosis and septic heart injury. Mol Med Rep. (2020). 22:175–84. doi: 10.3892/mmr.2020.11114
60. Wei S, Bi J, Yang L, Zhang J, Wan Y, Chen X, et al. Serum irisin levels are decreased in patients with sepsis, and exogenous irisin suppresses ferroptosis in the liver of septic mice. Clin Transl Med. (2020) 10:e173. doi: 10.1002/ctm2.173
61. Oh BM, Lee SJ, Park GL, Hwang YS, Lim J, Park ES, et al. Erastin inhibits septic shock and inflammatory gene expression via suppression of the NF-kappaB pathway. J Clin Med. (2019) 8:2210. doi: 10.3390/jcm8122210
62. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. (2018) 24:97–108. e104. doi: 10.1016/j.chom.2018.05.009
63. Lee S, Suh GY, Ryter SW, Choi AM. Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. Am J Respir Cell Mol Biol. (2016) 54:151–60. doi: 10.1165/rcmb.2015-0231TR
64. Qiang Z, Dong H, Xia Y, Chai D, Hu R, Jiang H. Nrf2 and STAT3 alleviates ferroptosis-mediated IIR-ALI by regulating SLC7A11. Oxid Med Cell Longev. (2020) 2020:5146982. doi: 10.1155/2020/5146982
65. Zhong WJ, Duan JX, Liu T, Yang HH, Guan XX, Zhang CY, et al. Activation of NLRP3 inflammasome up-regulates TREM-1 expression in murine macrophages via HMGB1 and IL-18. Int Immunopharmacol. (2020) 89(Pt A):107045. doi: 10.1016/j.intimp.2020.107045
66. Li D, Ren W, Jiang Z, Zhu L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol Med Rep. 18:4399–409. doi: 10.3892/mmr.2018.9427
67. He DK, Xu N, Shao YR, Shen J. NLRP3 gene silencing ameliorates phosgene-induced acute lung injury in rats by inhibiting NLRP3 inflammasome and proinflammatory factors, but not anti-inflammatory factors. J Toxicol Sci. (2020) 45:625–37. doi: 10.2131/jts.45.625
68. Yang H, Lv H, Li H, Ci X, Peng L. Oridonin protects LPS-induced acute lung injury by modulating Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-kappaB pathways. Cell Commun Signal. (2019) 17:62. doi: 10.1186/s12964-019-0366-y
69. Lv H, Liu Q, Wen Z, Feng H, Deng X, Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol. (2017) 12:311–24. doi: 10.1016/j.redox.2017.03.001
70. Lei J, Wei Y, Song P, Li Y, Zhang T, Feng Q, et al. Cordycepin inhibits LPS-induced acute lung injury by inhibiting inflammation and oxidative stress. Eur J Pharmacol. (2018) 818:110–4. doi: 10.1016/j.ejphar.2017.10.029
71. Rojo de la Vega M, Dodson M, Gross C, Mansour HM, Lantz RC, Chapman E, et al. Role of Nrf2 and autophagy in acute lung injury. Curr Pharmacol Rep. (2016) 2:91–101. doi: 10.1007/s40495-016-0053-2
72. Crisponi G, Nurchi VM, Lachowicz JI. Iron chelation for iron overload in Thalassemia. Met Ions Life Sci. (2019) 19:9. doi: 10.1515/9783110527872-009
73. Williams A, Meyer D. Desferrioxamine as immunomodulatory agent during microorganism infection. Curr Pharm Des. (2009) 15:1261–8. doi: 10.2174/138161209787846801
74. Ramezanpour M, Smith JLP, Ooi ML, Gouzos M, Psaltis AJ, Wormald PJ, et al. Deferiprone has anti-inflammatory properties and reduces fibroblast migration in vitro. Sci Rep. (2019) 9:2378. doi: 10.1038/s41598-019-38902-2
75. Ogger PP, Byrne AJ. Lung fibrosis enters the iron age(dagger). J Pathol. (2020) 252:1–3. doi: 10.1002/path.5489
76. Hybertson BM, Connelly KG, Buser RT, Repine JE. Ferritin and desferrioxamine attenuate xanthine oxidase-dependent leak in isolated perfused rat lungs. Inflammation. (2002) 26:153–9. doi: 10.1023/A:1016511611435
77. Ni YL, Shen HT, Su CH, Chen WY, Huang-Liu R, Chen CJ, et al. Nerolidol suppresses the inflammatory response during lipopolysaccharide-induced acute lung injury via the modulation of antioxidant enzymes and the AMPK/Nrf-2/HO-1 pathway. Oxid Med Cell Longev. (2019) 2019:9605980. doi: 10.1155/2019/9605980
78. Zhang Y, Yu W, Han D, Meng J, Wang H, Cao G. L-lysine ameliorates sepsis-induced acute lung injury in a lipopolysaccharide-induced mouse model. Biomed Pharmacother. (2019) 118:109307. doi: 10.1016/j.biopha.2019.109307
79. Vazquez-Medina JP, Tao JQ, Patel P, Bannitz-Fernandes R, Dodia C, Sorokina EM, et al. Genetic inactivation of the phospholipase A2 activity of peroxiredoxin 6 in mice protects against LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L656–68. doi: 10.1152/ajplung.00344.2018
80. Ma X, Liu X, Feng J, Zhang D, Huang L, Li D, et al. Fraxin alleviates LPS-induced ARDS by downregulating inflammatory responses and oxidative damages and reducing pulmonary vascular permeability. Inflammation. (2019) 42:1901–12. doi: 10.1007/s10753-019-01052-8
81. Aggarwal S, Dimitropoulou C, Lu Q, Black SM, Sharma S. Glutathione supplementation attenuates lipopolysaccharide-induced mitochondrial dysfunction and apoptosis in a mouse model of acute lung injury. Front Physiol. (2012) 3:161. doi: 10.3389/fphys.2012.00161
82. Qian M, Lou Y, Wang Y, Zhang M, Jiang Q, Mo Y, et al. PICK1 deficiency exacerbates sepsis-associated acute lung injury and impairs glutathione synthesis via reduction of xCT. Free Radic Biol Med. (2018) 118:23–34. doi: 10.1016/j.freeradbiomed.2018.02.028
83. Bernard GR. Potential of N-acetylcysteine as treatment for the adult respiratory distress syndrome. Eur Respir J Suppl. (1990) 11:496s−8.
Keywords: ferroptosis, acute respiratory distress syndrome, metabolism, inflammation, oxidative stress
Citation: Qu M, Zhang H, Chen Z, Sun X, Zhu S, Nan K, Chen W and Miao C (2021) The Role of Ferroptosis in Acute Respiratory Distress Syndrome. Front. Med. 8:651552. doi: 10.3389/fmed.2021.651552
Received: 10 January 2021; Accepted: 12 April 2021;
Published: 07 May 2021.
Edited by:
Mandy Laube, Leipzig University, GermanyReviewed by:
Brent Stockwell, Columbia University, United StatesCopyright © 2021 Qu, Zhang, Chen, Sun, Zhu, Nan, Chen and Miao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wankun Chen, Y2hlbndhbmtAMTYzLmNvbQ==; Changhong Miao, bWlhb2NoYW5naEAxNjMuY29t; Hao Zhang, ZnVzY2NfYW5lc3RoZXNpYUB5ZWFoLm5ldA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.