 Shubhra Chaturvedi
Shubhra Chaturvedi Anil K. Mishra
Anil K. Mishra- Division of Cyclotron and Radiopharmaceutical Sciences, Institute of Nuclear Medicine and Allied Sciences, Defence Research and Development Organisation, Delhi, India
Radiopharmaceuticals are an integral component of nuclear medicine and are widely applied in diagnostics and therapy. Though widely applied, the development of an “ideal” radiopharmaceutical can be challenging. Issues such as specificity, selectivity, sensitivity, and feasible chemistry challenge the design and synthesis of radiopharmaceuticals. Over time, strategies to address the issues have evolved by making use of new technological advances in the fields of biology and chemistry. This review presents the application of few advances in design and synthesis of radiopharmaceuticals. The topics covered are bivalent ligand approach and lipidization as part of design modifications for enhanced selectivity and sensitivity and novel synthetic strategies for optimized chemistry and radiolabeling of radiopharmaceuticals.
Introduction
Radiopharmaceuticals are being used in diagnostics and therapeutics for more than half a century. They are widely used in the delineation of neurodegenerative diseases, myocardial imaging and diagnosis, and treatment of cancer. Due to their wide application, the development of an “ideal” radiopharmaceutical continues to be the foremost challenge of the research frontier in nuclear medicine. The key issues confronting the research community in radiopharmaceutical chemistry is to develop highly specific and selective ligands with high specific activity capable of targeting and overcoming biological barriers.
The challenges emanate at the different stages of developing radiopharmaceutical, viz., design, modification, and radiolabeling. Selection of the type of molecule (antibody and their fragments, peptides, nucleosides, aptamers, small molecules), surface modifications, multivalency, and labeling reactions optimization are few variations that have been used to address the challenges. Based on these variations, the review presents three emerging approaches that address the challenges: high selectivity and sensitivity through design optimization using bivalent ligands (BLs), targeting against natural barriers through modification using lipidization, and high specific activity while radiolabeling using sophisticated chemistries, viz., bioorthogonal and cross-coupling reactions. These approaches have the potential to be integrated into radiopharmaceutical development. We describe each of these approaches seriatim in along with avenues for future research in Sections 1–3.
1. High Selectivity through Bivalent Ligand Approach
Bivalent Ligand Approach
In simplest terms, a BL consists of two pharmacophores linked through a spacer. The two pharmacophores can be identical resulting in a homobivalent ligand or different resulting in a heterobivalent ligand. The BL benefits from the collaborative binding of the two pharmacophores, resulting in favorable thermodynamics as compared to that of a monovalent ligand (1). Figure 1 presents binding modes a BL can exhibit.
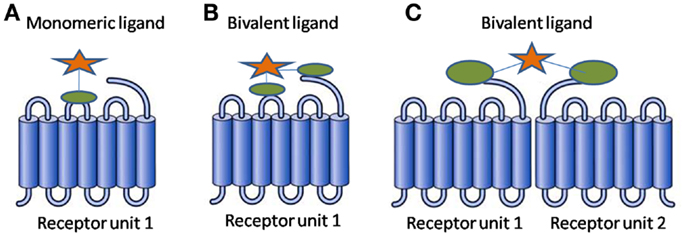
Figure 1. Binding modes for a ligand (A) monovalent ligand with one receptor unit (B) bivalent ligand with one receptor unit (C) bivalent ligand with receptor dimer.
Selectivity through BLA
Bivalent ligands are examples of multimeric interactions. Multimeric interactions are known to enhance the binding affinity of the ligands through multiple mechanisms, e.g., receptor clustering, chelating effect on receptors, ligand–receptor steric stabilization, and ligand accumulation near the receptor (2). Overall, the effect is enhanced selectivity and enhanced binding affinity (1). The multivalent concept has been extensively validated for peptides. Successful reports for multimeric peptides as diagnostics agent are included in Table 1.

Table 1. Few reports on successful targeting using multimeric peptides.
Reviews regarding the development of homo-multimeric and hetero-multimeric peptidic ligands are many, and hence, for peptidic multimeric ligands readers may refer reviews (4, 9). The multimeric concept is now being extended to small molecules as well. Small molecule-based BLs are capable of multimeric interactions, thereby having higher sensitivity and selectivity.
Applications of BLA
A BL functions best when multiple binding pockets are present in the target. Depending on the pharmacophores, a BL can target one or multiple biomarkers. Tumor targeting can benefit from the high binding avidity and selectivity of BL. Furthermore, hetero-BL can result in more specificity as it targets different receptors simultaneously.
Receptor-based imaging, especially for neuroreceptors, can also benefit from the bivalent approach. Many receptors/neuroreceptors belong to G-protein coupled receptor (GPCR) family (10). After the reports about the existence of GPCRs as oligomers and higher-orders started pouring (11), BLs were successfully developed and validated against them. The approach has been of high relevance in the design and development of second generation antipsychotics (12, 13). A BL can target both homo- and hetero-dimeric receptor systems depending on the pharmacophores.
Another target for BLs is β-amyloid plaques because of the presence of multiple binding sites (14).
Development Considerations for BLA
The key factors for BL design are (a) selection of pharmacophores, (b) optimization of linker length and its biocompatibility, and (c) spatial parameters of the final compound (2). As a radiopharmaceutical, a BL has to be evaluated for its in vitro and in vivo properties.
A series of small molecule-based dimeric and multimeric ligands have been developed and reported in recent past for targeted imaging of tumors, receptors, and β-amyloid plaques. Figure 2 summarizes radiolabeled small molecule-based BLs.
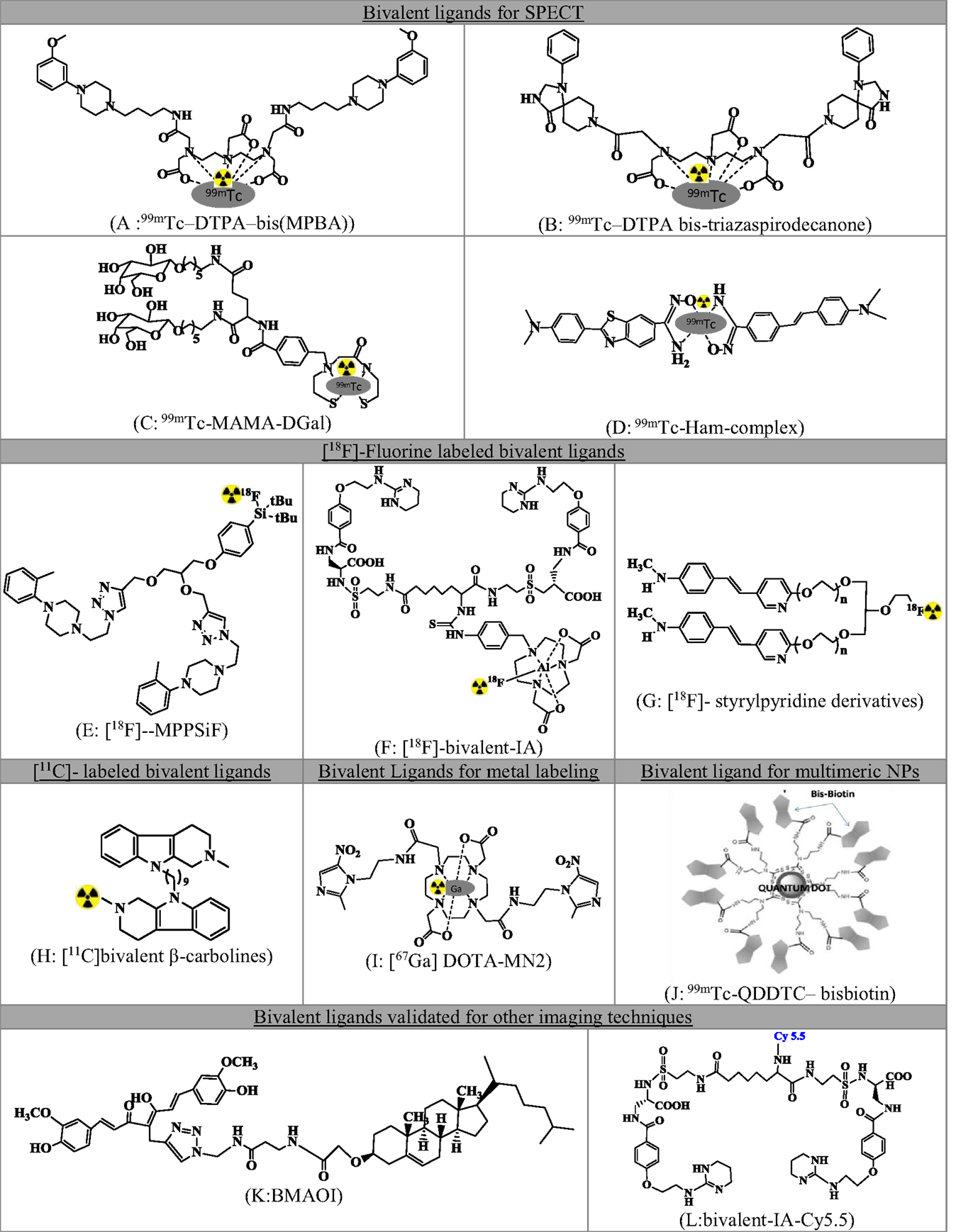
Figure 2. Comprehensive list of small molecule-based bivalent ligands for diagnostics. (A)99mTc-DTPA-bis(MPBA), (B)99mTc-DTPA bis-triazaspirodecanone, (C)99mTc-MAMA-DGal, (D)99mTc-Ham, (E) [18F]-MPPSiF, (F) [18F]-bivalent-IA, (G) [18F]-styrylpyridine derivatives, (H) [11C]bivalent β-carbolines, (I) [67Ga]DOTA-MN2, (J)99mTc-QDDTC-bisbiotin, (K) BMAOI, and (L) bivalent-IA-Cy5.5.
Bivalent Ligands Demonstrated for SPECT
Receptor Imaging
Singh et al. (15) demonstrated the proof-of-concept for 5HT1A receptors using homodimeric ligand and validated the ligand as a SPECT imaging agent. Two identical pharmacophores based on 1-(2-methoxyphenyl)piperazine (MPP) were linked using an aliphatic linker of four carbon atoms to the acyclic chelating agent DTPA and validated as SPECT agent after technetium labeling [99mTc-DTPA-bis(MPBA) Figure 2A]. The authors were able to demonstrate (a) 1000 times high selectivity toward 5HT1A receptors than 5HT2A receptors, (b) involvement of both the pharmacophores for bivalent binding using hill slope analysis, and (c) high labeling efficiency.
On similar lines, using DTPA as an acyclic chelator for technetium (16), reported the synthesis of bis-triazaspirodecanone (Figure 2B). The ligand showed enhanced binding affinity theoretically using docking and MM-GBSA calculations. Furthermore, the compound showed selective striatum uptake in the brain and selective dopamine D2 targeting.
Similarly, the divalent ligand with two units of galactose derivatives (99mTc-MAMA-DGal, Figure 2C) showed higher specific binding to asialoglycoprotein receptors (ASGPR) in dynamic microSPECT imaging and biodistribution studies of liver fibrosis (17). The monovalent ligand 99mTc-MAMA-MGal was also validated for comparison. The divalent ligand showed better binding affinity in vitro and fast pharmacokinetics.
β-Amyloid Imaging
To assess the amyloid aggregation (18), synthesized bivalent amyloid ligand and labeled with 99mTc leading to the formation of 99mTc-Ham (Figure 2D). Stilbene (SB) and benzothiazole (BT) derivatives were selected as amyloid binding units. These were conjugated to a hydroxamamide (Ham) and labeled for SPECT imaging using 99mTc. Five analogs were synthesized and evaluated for binding affinity and brain uptake.
[18F]-Fluorine-Labeled Bivalent Ligands
Receptor Imaging
In another study of Hazari et al. (19), bis-MPP (Figure 2E) derivative has been synthesized to image serotonin receptors. The duplication of the pharmacophores leads to a supra-additive increase in binding and potency as compared to monovalent analog. Thus, the bis-compound had sub-nanomolar affinity for the receptor, 1000 times more selectivity for 5HT1A as compared to D2, 5-HTT, or 5HT2A. The compound was validated as PET imaging agent.
For the imaging of αvβ3, a non-peptidic BL was reported by Wang et al. (20) (Figure 2F). This molecule consisted of two units of antagonist 4-[2-(3,4,5,6-tetrahydropyrimidine-2-lamino)-ethyloxy]benzoyl-2-(S)-aminoethylsulfonyl-amino-h-alanine (IA) and radiolabeled using 18F-AlF/NODA chelation reaction.
β-Amyloid Imaging
A series of bivalent (Figure 2G) and trivalent 18F-styrylpyridine derivatives were developed for imaging β-amyloid plaques in the brain. The BL displayed high binding affinity. The study demonstrated the effect of linkers and the geometry of the molecule on the binding affinity. An ether linkage was found to have higher binding affinity vis-à-vis an amide linkage. The trivalent molecule had a reduced binding affinity as compared to the BL (14).
[11C]-Labeled Bivalent Ligands
Enzyme Imaging
β-Carboline bivalent derivatives that are known inhibitors for acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) were developed for imaging of cholinesterase in Alzheimer’s disease. The derivatives were radiolabeled at the nitrogen position of the amine precursor through N-[11C]methylation using [11C]CH3I (Figure 2H). Radiolabeling parameters of three derivatives of variable linker length were reported (21).
Bivalent Ligands for Metal Labeling
Tumor Imaging
Bivalent ligand concept has also been validated for metal-based radiopharmaceuticals. Metronidazole was conjugated to DOTA (DOTA-MN2, Figure 2I) and developed as radiogallium–DOTA complex without reducing the radiogallium complex stability for the imaging of hypoxic lesions using PET/SPECT (22). The complex showed significant tumor uptake and low non-target accumulation.
Bivalent Ligand for Multimeric Nanoparticles
Tumor Imaging
The concept of enhanced binding via multivalency using small molecules and nanoparticles (NPs) has also been reported. Nanoparticles (Quantum dots), as reported in the work of Bag et al. (23), were conjugated with multiple biotin units (bisbiotin) to have enhanced selectivity.99mTc-QDDTC-bisbiotin showed significantly higher tumor uptake, better tumor retention, and enhanced pharmacokinetics as compared to DTC–bisbiotin ligand. The work illustrates the bivalent effect of bisbiotin ligand for high tumor uptake. Other effects, viz., better tumor retention and enhanced pharmacokinetics were the results of the enhanced permeable and retention (EPR) effect due to the QD (Figure 2J).
Potential Bivalent Ligands Validated for Other Imaging Techniques
β-Amyloid Imaging
Though not as a radiopharmaceutical, amyloid-β plaque imaging was accomplished using curcumin and cholesterol BL (BMAOI, Figure 2K), which could bind to various Aβ42 species with micromolar binding affinity and has appropriate fluorescence properties for labeling and imaging Aβ plaques in situ (24).
Receptor Imaging
NIR imaging probe for αvβ3 (25): Figure 2L was reported for cancer imaging. The non-peptidic small molecule bivalent antagonist demonstrated improved binding avidity relative to the monovalent ligand.
Bivalent Ligands for Radiotherapy
As above-mentioned BLs alone are being used for the development of atypical antipsychotics. Bivalent peptide-based ligands are reported for radiotherapy applications (9). However, to the best of our knowledge, examples of small molecule-based BLs for radiotherapy have not been reported.
Future Directions
The advantages of high sensitivity, selectivity, and favorable pharmacodynamics make radiolabeled BLs promising candidates for diagnostics and possibly therapy. However, knowledge gaps in receptor expression patterns, receptor’s higher order structures, and binding pattern on receptors need to be filled for full utilization of the approach. In terms of ligands itself, an exact mechanistic aspect of the binding of ligand need to be understood. The structural features, pharmacophore, the cooperative effect on the binding of pharmacophore, linker length, and geometry effect all have to be considered in the design of the ligand. Such studies can take lead from theoretical screening-like docking and high-throughput screening or through control experiments, which include comparative studies with a monovalent ligand. The approach still needs to be extended to radiotherapy.
The radiolabeled BLs are promising candidates in diagnostics and can enhance the binding affinity and enable multi-targeting. However, the penetration ability across the cellular membrane and the circulation time that determines the serum availability of the radiopharmaceutical are also important for the efficacy. The following section discusses the efforts in delivering the radiopharmaceutical to the target site through lipidization and surface modification.
2. Enhanced Targeting through Lipidization and Surface Modification
Lipidization
Lipidization is a chemical approach to alter the solubility and pharmacokinetic behavior of a molecule. It involves attachment of lipid at the polar end of a molecule, thereby conferring lipophilic nature to the molecule.
Enhanced Targeting through Lipidization
Lipidization of drugs in the form of (a) Prodrug Strategy and (b) lipid-based carriers’ viz. Liposomes and lipidized NPs can enhance the drug targeting. This is because of (a) enhanced permeability across biological barriers, namely, the membranes, (b) improved pharmacokinetics that includes enhanced circulation time, (c) slow release, thereby prolonging drug action, and (d) enhanced bioactivity through passive targeting. This approach has been used for developing anticancer drugs, drugs for liver diseases, and the lymphatic system. The strategy can also provide a solution for CNS targeting due to BBB penetration (26).
Lipidic Prodrugs for Imaging/Radiopharmaceuticals
The lipidic modification can lead to enhanced permeability in the brain, and hence, has potential for brain imaging. However, in literature, examples highlighting the utility of lipidic prodrug for imaging are rare. In 2002, Kao et al. (27) demonstrated that an additional lipophilic character by benzoylation at 3′ and 5′ of FBAU enhanced the uptake in brain having normal blood–brain barrier. The prodrug FBAU 3′,5′-dibenzoate was radiolabeled with 76Br. Biodistribution studies indicated a higher brain accumulation of radioactivity (up to two times) at all time points in rats injected with [76Br]FBAU 3′,5′-dibenzoate (Figure 3A) than with [76Br]FBAU.
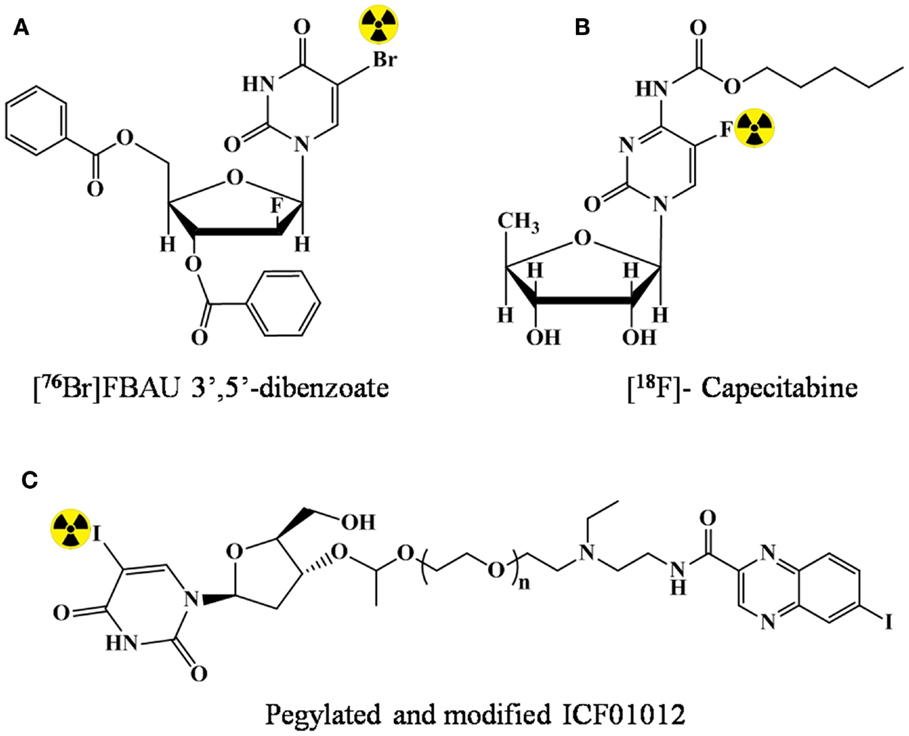
Figure 3. Lipidic modification of the nucleosides for prodrug strategy. (A) [76Br]FBAU 3′,5′-dibenzoate, (B) [18F]-Capecitabine, and (C) pegylated and modified ICF01012.
In 2005 (28), in order to reduce the toxicity and enhance the tumor penetration capability of 5-FU, prodrug strategy was validated. Capecitabine (N4-n-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine), which happened to be the first and the only orally administered fluoropyrimidine approved for the use as a second-line cancer therapy was labeled with 18F (Figure 3B). However, the study only included radiolabeling optimization, and no data for the capability of enhanced penetration/reduced was presented. In a present study of André et al. (29), N,N-diethyl N,N-diethylaminoethyleneheteroarylamide derivatives (e.g., ICF01012) was pegylated and conjugated with anti-metabolite 5-iodo-2′-deoxyuridine (IUdR). Enhanced and prolonged tumor uptake (melanoma) was observed after radiolabeling with 125I (Figure 3C).
Lipidic Nanoparticles for Imaging/Radiopharmaceuticals with Surface Modification
Lipidization can lead to enhanced efficiency of drug delivery systems. Lipid-based NPs consist of two types (a) liposomes and (b) solid lipid NPs (30). Encapsulation of drugs in these NPs protect from hydrolysis and aid in sustained controlled release at the site of interest. Reviews regarding lipid-based NPs are listed in Table 2. When radiolabeled the liposomes and NPs can prove to be effective theranostic agents.

Table 2. Reviews regarding lipid-based nanoparticles.
Liposomes are further surface modified for both enhanced pharmacokinetics and enhanced penetration. The modifications can include pegylation, squalenolation, and peptidization.
Pegylation
For surface modification of NPs (liposomes), pegylation is one of the most successful strategies. Pegylation is known to enhance the circulation time for NPs. Few examples of pegylation, especially in context with radio imaging are being discussed covering the following aspects:
(a) Pegylated liposomes with enhanced pharmacokinetics for imaging
(b) Pegylated liposomes with enhanced BBB permeation and with enhanced pharmacokinetics for imaging.
Pegylated Liposomes with Enhanced Pharmacokinetics for Imaging
Pegylated Nucleolipids for Imaging with Improved Pharmacokinetics
Nucleolipids are an emerging class of drug delivery systems. Recently, liposomes using the hybrid nucleoside lipids (NLs) were developed in which nucleosides were pegylated and targeted against folic acid. These liposomes (Figure 4A) were developed as the theranostic agent by encapsulating cisplatin as the therapeutic agent and 99mTc radiolabeled using the uridine rings at the outer surface of the liposomes. Enhanced uptake at the tumor site was observed along with the favorable pharmacokinetics, which included enhanced circulation time (35).
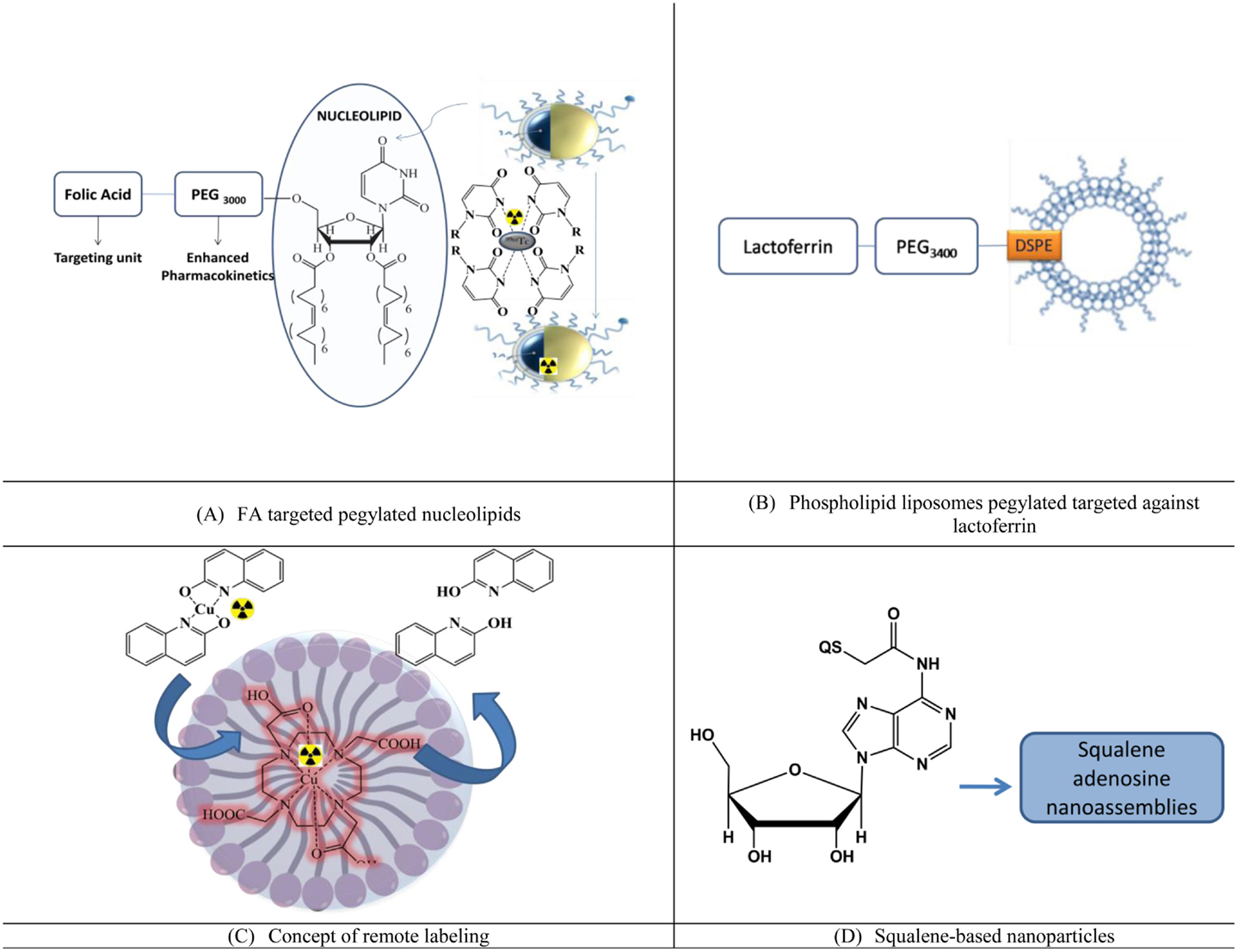
Figure 4. Lipidic nanoparticles for imaging and enhanced pharmacokinetics. (A) FA targeted pegylated nucleolipids, (B) phospholipid liposomes pegylated targeted against lactoferrin, (C) concept of remote labeling, and (D) squalene-based nanoparticles.
Pegylated Liposomes with Enhanced BBB Permeation with Enhanced Pharmacokinetics
Pegylated Phospholipidic
Lactoferrin targeted pegylated phospholipdic liposomes (Lf-PL-99mTc) based on distearoylphosphatidylcholine (DSPC), cholesterol, and distearoylphosphatidylethanolamine were radiolabeled and evaluated for BBB penetration and effect on pharmacokinetics [(36), Figure 4B].
1. BBB penetration: bEnd.3 cells, which is an immortalized mouse brain endothelial cell line was used as a mimic for BBB. The cellular uptake was significantly higher for the targeted liposome. Biodistribution studies indicated an enhanced uptake of the lipidic liposomes, which were targeted with lactoferrin, and approximately 1.47 times more uptake was reported than the non-targeted pegylated liposomes. However, the study did not comment on the penetration ability due to pegylation.
2. Pharmacokinetics: the area under the curve (AUC0 → 24 h) and the clearance rate (Cl) from Lf-PL-99mTc was found to be similar to PL-99mTc with p-values of 0.89 and 0.31, respectively. Thus, the Lf-conjugated liposomes could provide the similar long-circulation property in vivo. For designing a better Lf-PL-99mTc, the number of Lf ligand on the liposomes should have a suitable level.
Several other targeted pegylated liposomal preparations have also been reported (34).
Pegylated liposomes consisting of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol (Chol), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000) were used for remote loading of radionuclide. In the work of Petersen et al. (37) (Figure 4C), [64]Cu was crossed across the membrane of preformed liposomes into the aqueous cavity using a new ionophore, 2-hydroxyquinoline, in order to achieve high and stable loading of radionuclides.
Squalenolation
Though not with liposomes, squalene adenosine nano-assemblies (SqAdNA) were studied for their interaction with endothelial cells of the human brain to assess the mechanism of penetration (38). The internalization was mainly mediated by the LDL receptors-mediated endocytosis, after which the NA disassembled inside the cells and exocytosed as single molecules. Such assemblies were also prepared with an array of nucleosides (deoxycytidine-Sq, thymidine-Sq, gemcitabine MP-Sq, ddI-Sq, and deoxycytidine-5′-Sq) and studied to assess the influence of the nucleoside nature and position with respect to squalene on the structure of the NAs (39), Figure 4D. However, the utilization of the assemblies for imaging and brain penetration in vivo is yet to be validated.
Peptidization
Liposomal vector was modified as a novel bi-ligand having transferrin for targeting and poly-l-arginine for enhanced uptake in the brain (40). The bi-ligand liposomes accumulated in the rat brain at significantly (p < 0.05) higher concentrations as compared to the single-ligand (transferrin) or plain liposomes.
Future Directions
The prodrug approach needs to be exploited for design of lipidic-based radiopharmaceutical. From design perspective choice of lipids can be important for effectiveness. Literature available till date does highlight features of fatty acids (FA) required for effectiveness. For example, the importance of carboxylate in cellular internalization, an effect of chain length (longer chain fatty acid are more stable than shorter chain FA and better suited for lymphatic targeting) and pros and cons when exploiting carboxylate or ω-position for drug conjugation. Further studies on the effect of chain length of FA on targeting the type of membrane will be helpful. Prodrugs whether radiolabeled or not, also suffer from one challenge, guarantee of conversion from inactive to active form in the living system.
The drug delivery systems, liposomes and solid lipid NPs are expensive options with limited shelf life. Second, their toxicity, especially for cationic liposomes, and cellular interaction need to be addressed. Future work needs to address the issues for successful utilization of liposomes and solid lipid NPs as drug delivery systems and radiopharmaceuticals.
The concluding step in the synthesis of any radiopharmaceutical is the radiolabeling. A molecule with good selectivity and sensitivity and also with good penetration ability may not prove to be an ideal radiopharmaceutical because of the poor specific activity after radiolabeling. Hence, novel and optimized radiolabeling conditions play an important role in the development of a radiopharmaceutical. A lot of work has been done in this regard. The following section gives an overview of the development in radiolabeling chemistry.
3. Synthesis and Radiolabeling Optimization
Radiolabeling
Radiolabeling is the incorporation of the radioactive moiety in a compound in order to track the compound. With the growing utilization of diverse molecules as radiotracers, there is a growing need for new or modified radiolabeling methods that require low quantities of bioactive compounds, employ mild conditions to avoid loss of bioactivity, have short reaction times for short-lived radionuclides, and result in high specific activity. At the same time, for human application, the new or modified radiolabeling methods need to focus on toxic free reagents or supplemented with better purification procedures. Bioorthogonal and cross-coupling are upcoming approaches in order to meet the above requirements.
Radiolabeling can proceed in two ways: (a) using radiolabeled prosthetic groups that are coupled to bioactive molecules using bioorthogonal reactions and (b) direct labeling of bioactive molecules using cross-coupling reactions.
Bioorthogonal Approaches
Bioorthogonal reactions can proceed in the living systems without influencing or getting influenced by the biological processes, the efficacy of the ligands is retained and can demonstrate fast kinetics especially when used for monitoring.
It may be noted that a large number of reviews have already been published, which cover the detailed aspects (41, 42). Hence, here a summarization along with few additions is being given for different types of bioorthogonal approaches.
Copper-Based Click Ligation
Click chemistry as described by K. Barry Sharpless is “a set of powerful, virtually 100% reliable, selective reactions for the rapid synthesis of new compounds” (29, 43). Click chemistry reports in radiopharmaceutical sciences were first published in 2006 (44). It has been extensively studied and published. Many comprehensive reviews are available. Some examples are (a) click chemistry mechanism (45), (b) application in radiopharmaceuticals (43, 44), (c) application with specific precursors-glycobiology (46), (d) click chemistry in chelate development (44), and (e) patent analysis (47).
An overview of click chemistry for radiopharmaceuticals is as follows:
(a) Due to its bioorthogonal nature, click chemistry has been widely applied with different types of precursors.
(b) Its application extends from
i. Linking two biomolecules without compromising the bio-efficacy
ii. Developing prosthetic groups that serve as radiolabeling precursors for fluorine-18 and carbon-11. Choice of a prosthetic group can influence (a) metabolic profile (b) in vivo behavior (41).
iii. Novel chelate development wherein the triazole moiety acts as an electron donor to the metal.
Few representative structures developed using click chemistry are shown in Figure 5 [structures referenced in Kettenbach et al. (41) and Pretze et al. (42)] covering the aspects b (ii) and b (iii). Though most popular as copper (I) catalyzed click chemistry leading to selective formation of 1,4 regioisomer, another variation using ruthenium complexes which leads to selective 1,5 regioisomer has also been explored.

Strain-Promoted Click Chemistry/Strain-Promoted Azide Alkyne Cycloaddition
Largely driven by the requirement of copper-free click chemistry due to copper linked toxicity (cytotoxicity, non-compatibility with oligonucleotides, hepatitis, and implications in Alzheimer’s disease and neurological diseases), strain-promoted, and copper-free variants of click chemistry are being validated in radiopharmacy (42). Apart from being copper-free, the reaction proceeds at a faster rate and can be used for short-lived radioisotopes like 64Cu (67); it is efficient, has high specificity, and requires mild reaction conditions (68). These were first reported in 2011 (69). Since then, the reaction has been used for radiolabeling of peptides [BBN (70), RGD (67, 71), c-Met-binding peptide (71), apoptosis-targeting peptide (ApoPep) (72), somatostatin analogs (72), DOTA-biotin conjugate (73), and NPs (68, 74)]. However, the concern for strain-promoted azide alkyne cycloaddition (SPAAC) include (a) effect of bulky moieties such as DBCO and ADIBO on lipophilicity, binding affinity with the target and the variation on pharmacokinetic behavior, and (b) non-regioselective product formation consisting both 1,4 and 1,5 regioisomers (72). Figure 6 presents precursors for fluorine labeling and radiopharmaceuticals developed using SPAAC.
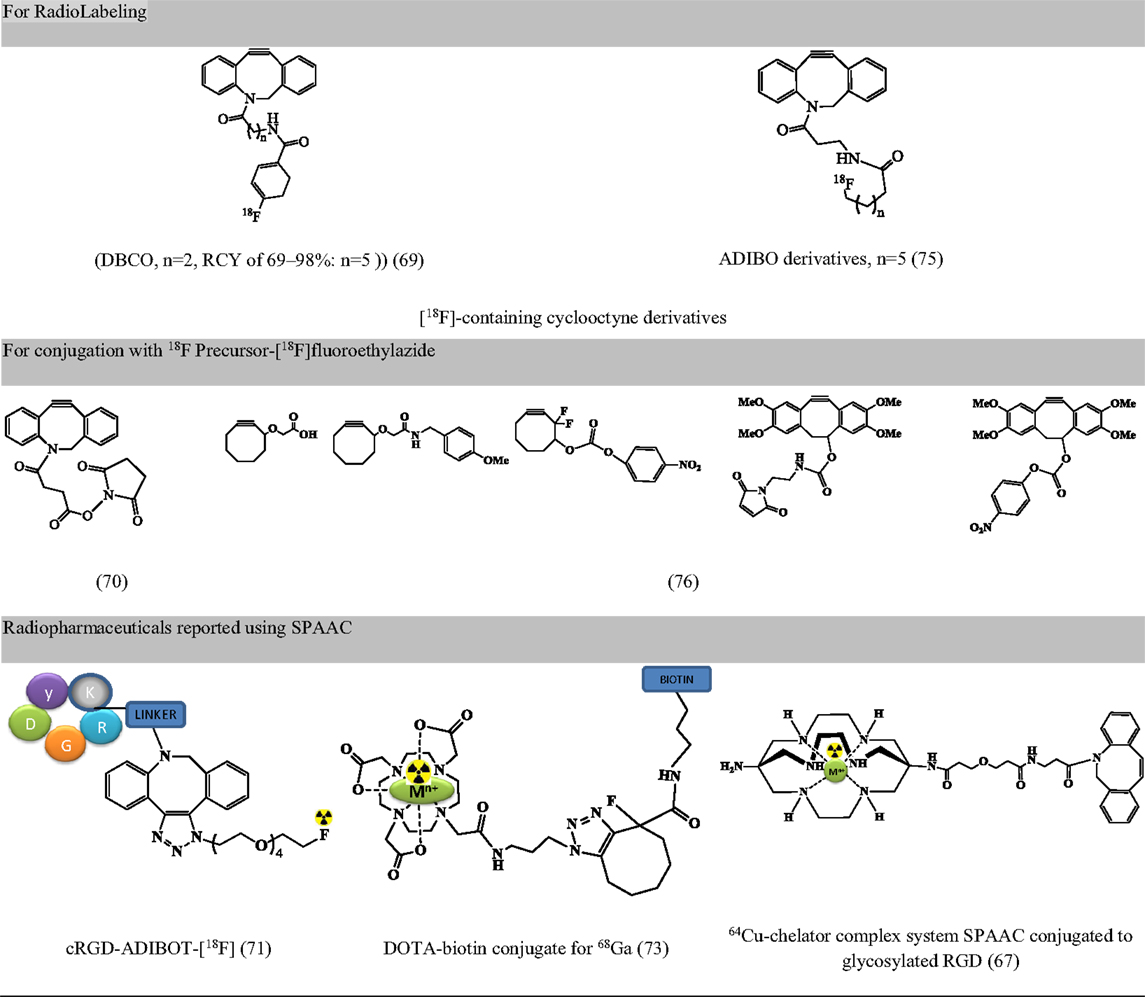
Figure 6. Representative examples for radiopharmaceuticals using SPAAC. Structures referenced in (42, 67, 69–71, 73, 75, 76).
Other Ligations: Staudinger Ligation, Tetrazines (Tetrazine-Trans-Cyclooctene Ligation), and Radio-Kinugasa Reaction
Staudinger Ligation
Staudinger ligation is another example of the metal-free conjugation reaction (42). Two variants exist: the non-traceless with the inclusion of phosphine oxide and the traceless version without the inclusion of the phosphine oxide in the final product. Both lead to the formation of the amide bond. The non-traceless version has not been as widely applied as the traceless version. Furthermore, the reaction can be accomplished either through direct approach (azide of biomolecule reacted with 18F-phosphanes) or indirect approach (phosphane derivatized biomolecule reacted with 18F-azide). The range of radiopharmaceuticals developed using the Staudinger Ligation is covered in the review (42).
Tetrazines (Tetrazine-Trans-Cyclooctene Ligation) (41, 42)
Tetrazine-trans-cyclooctene ligation (TTCO ligation), introduced in 2010, is the inverse electron demand of the Diels–Alder (IEDDA) cycloaddition between a cyclooctene and a 1,2,4,5-tetrazine under the release of nitrogen (77). Here, the tetrazine functionalized biomolecule is reacted with an 18F-labeled cyclooctene (more preferred for radiolabeling). The approach has the advantages of fast reaction rates even without catalyst making it suitable for 11C-labeling reaction (78), non-reversibility because of nitrogen release, broad tolerance range, both aqueous and organic based high yields. Its mechanism and the application are covered in the review (42). In short, the reaction has been applied for labeling peptides (RGD, GLP-1, exendin), small molecules PARP1-targeting small molecule and DOTA derivatives [refer review (79)].
Radio-Kinugasa Reaction
A recent addition to the radio fluorination is the Kinugasa reaction validated in 2014 (80). Advantage includes fast kinetics and a broad spectrum of biological activities and low toxicity of β-lactams. Radiochemical yields of the Kinugasa reaction products could be significantly increased by the use of different Cu(I) ligands (81).
Novel Cross-Coupling Approaches
The transition metal-mediated cross-coupling reactions have been used as part of organic synthesis for the precursors for radiolabeling. The cross-coupling reactions came into the picture in 1995 with the work of Langstorm using Stille and Suzuki reactions for PET radiopharmaceuticals. Largely driven by mild conditions as opposed to the harsh conditions of conventional fluorine labeling, high radiochemical yields and fast kinetics, the metal-mediated cross-coupling reactions are being increasingly validated. The review presented by Doi (80) and Pretze et al. (82), cover the historical and development details for Stille, Suzuki coupling, Negishi coupling, and Sonogashira and Heck coupling. Among the reactions, Stille reaction has been widely applied in the synthesis of radiopharmaceuticals.
Figures 7–9 summarizes the major contribution of the cross-coupling reactions in the development of radiopharmaceuticals.
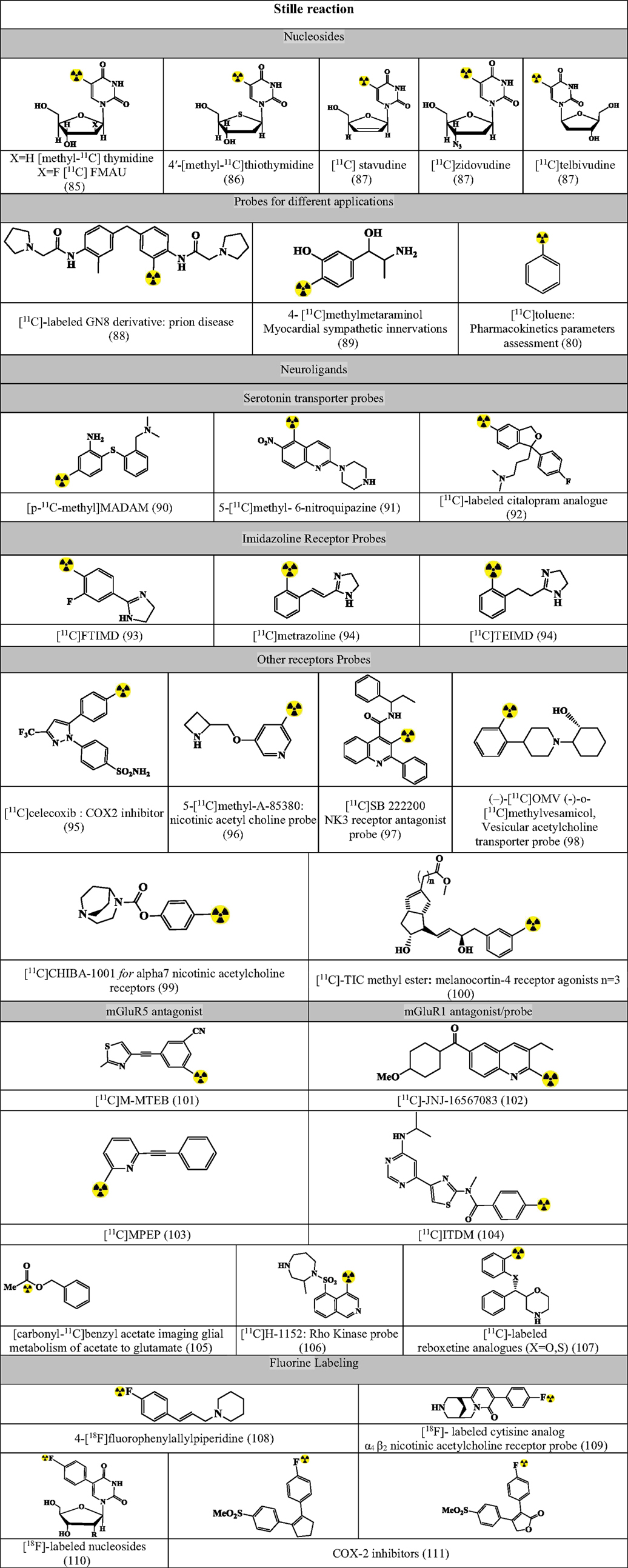
Figure 7. Selected structures developed using Stille reaction. Structures referenced in (80, 82, 85–111).
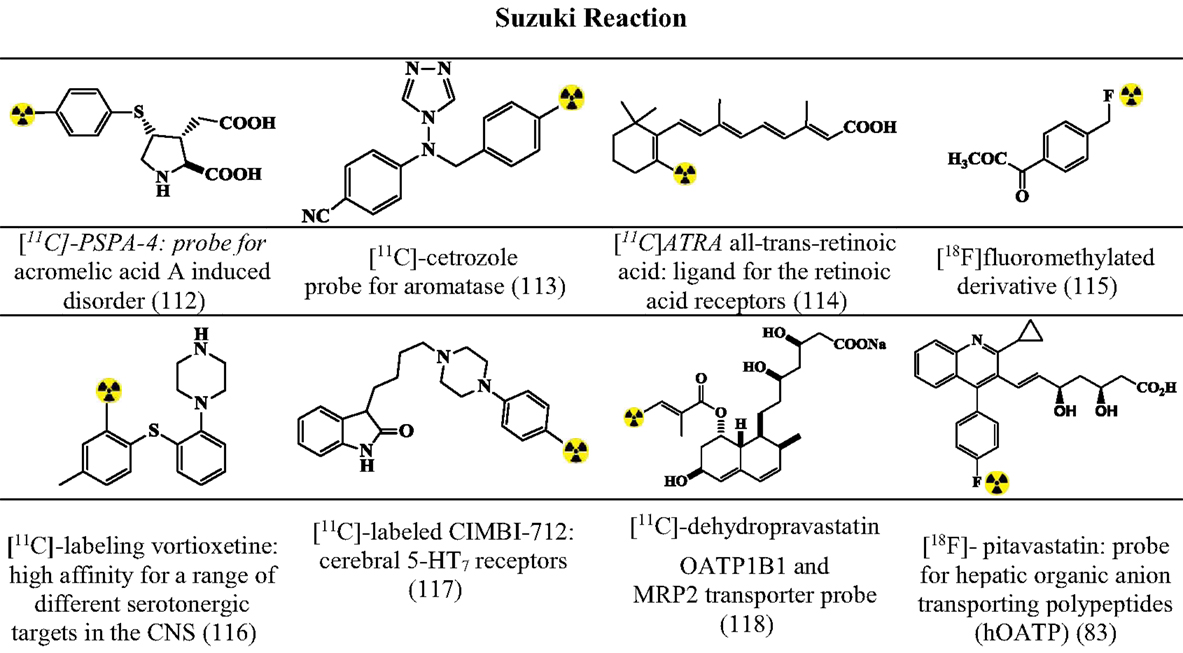
Figure 8. Selected structures developed using Suzuki reactions. Structures referenced in (80, 82, 83, 112–118).
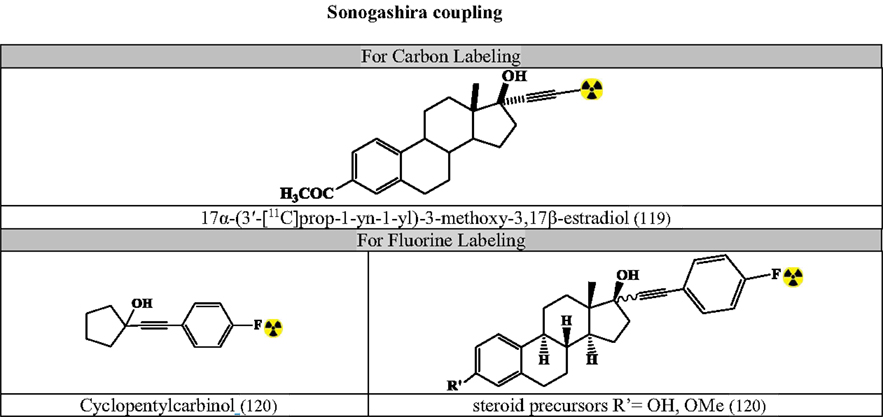
Figure 9. Selected radiopharmaceuticals developed using Sonogashira coupling. Structures referenced in (80, 82, 119–120).
Stille Reaction
Stille reaction involves coupling between an organotin compound with alkyl or aryl halogenide using Pd-catalyst and a phosphane-based coligand for the formation of both C–C bond and C–X bond (80, 82). The reaction has been tested with the following reaction conditions and validated for the synthesis of precursors as in Figure 7 (80, 82, 85–111).
Optimized conditions for catalyst and solvents include: (80, 82)
(Temperature and time dependent on reactants)
[11C]-Labeling
[11C]-labeled methyl iodide
i. aromatictrimethylstannyl compounds in DMF or DMSO, Pd2(dba)3 with P(o-Tol)3 as coligand and CuCl/K2CO3 as additive in DMF
ii. aromatictrimethylstannyl compounds with Pd2(dba)3/P(o-Tol)3, DMF
carbonylative[11C]-CO coupling
i. organic iodides with organostannanes in DMSO with an excess of P(o-Tol)3 relative to Pd-catalyst
[11C]-acetyl chloride
i. organostannate with Pd2(dba)3 and coligand 2,8,9-trimethyl-2,5,8,9-tetraaza-1-phosphabicyclo[3.3.3]undecane hydrochloride in the ratio 1:0.5, respectively.
[18F]-Fluorination
1-[18F]fluoro-4-iodobenzene
i. hexamethylphosphoramide is used as solvent and Pd(PPh3)4 as catalyst
ii. Pd(PPh3)2Cl2/CuI as catalyst in dioxane
iii. DMF/dioxane (1:1) or THF/dioxane (1:1) mixture using Pd2(dba)3/CuI/AsPh3
iv. Pd2(dba)3/P(o-Tol)3/CuI, DMF/toluene (1:1)
1-[18F]fluoro-4-bromobenzene:
i. Pd2dba3/AsPh3 as mediator in a DMF:dioxane mixture (1:1)
ii. DMF/dioxane mixture and Pd(PPh3)4
iii. dioxane with PdCl(PPh3)2
Common conditions: DMF:dioxane mixture (1:1) as solvent and BnClPd(PPh3)2:CuI (ratio 1:1) as catalyst.
Advantages are (a) mild conditions, (b) wide tolerance of functional groups such as amino, hydroxyl, thiol, or carboxylate, and (c) stability of the organotin compounds.
Disadvantages include (a) metal linked toxicity and (b) kinetic and thermodynamic feasibility of the reaction.
Challenges are possible side reactions with different functional groups, difficult preparation and purification of stannyl compounds, reproducibility can be sensitive to the purification level of 11C-methyl iodide.
Suzuki Reactions
The Suzuki coupling is based on the conjugation of boron substrates (alkylborane/benzylborane/alkenylboranes) with alkyl halide leading to C–C bond formation or C–X bond formation (80, 82). General Optimized conditions (80, 82) for the synthesis of various precursors (Figure 8) include:
(Temperature and time dependent on reactants)
[11C]-Carbon Labeling
[11C]-methyl iodide
(i) aryl iodide or aryl boranes (reactant) with Pd(PPh3)4 as catalyst with THF as solvent under basic conditions.
(ii) aryl boranes (especially consisting acidic protons) in the presence of [Pd(dppf)Cl2] and K3PO4 in DMF under microwave heating
(iii) aryl boranes using Pd0-mediated conventional thermal heating method
(iv) pinacolphenylboronate/alkenylboranes/aryl boranewith Pd2(dba)3/P(o-tolyl)3/K2CO3 (1:4:4) in DMF or DMF/H2O (9:1)
[11C]-CO
(i) aryl iodides and phenylboronic acid (reactant) with Pd(PPh3)2Cl2(catalyst), K2CO3(base) and DMSO (solvent)
(ii) aryltriflate + alkyl boronic acid (reactant) with bases such as tetra-butylammonium fluoride or aryltriflate + aromatic boronic acid (reactant) with bases such as potassium tert-butoxide. Lithium bromide (promoter) may also be added.
Fluorine Labeling
[18F] fluoromethyl iodide ([18F]-FCH2I)
(i) pinacolphenylboronate with 1:3 ratio of Pd/P(o-tolyl)3
1-[18F]fluoro-4-iodobenzene
(i) organoboranes with Pd2(dba)3 as mediator, Cs2CO3 as base and acetonitrile as solvent.
Advantages are (a) borane derivatives that are less toxic than the stannous substrates, (b) organoborane has relatively high reactivity, especially in the presence of a base or a fluoride anion, (c) compatible with a wide variety of functionalities, and (d) water tolerant.
Sonogashira Coupling
Based on organocopper species that interacts with the Pd-catalyst in transmetalation step for conjugation of terminal alkynes with vinylic or aryl halides (Figure 9) (80, 82, 119–120).
Optimized conditions: (80, 82)
(Temperature and time dependent on reactants)
Carbon Labeling
[11C]-methyl iodide
i. terminal alkyne with Pd2(dba)3, AsPh3 and tetra-n-butyl-ammonium fluoride in THF (for Sonogashira-like coupling)
Fluorine Labeling
i. 4-[18F]fluoro-1-iodobenzene: THF as solvent and Et3N as base
Heck Reaction
Based on palladium-catalyzed C–C bond formation between olefins and aryl/vinyl halides (Figure 10) (82).
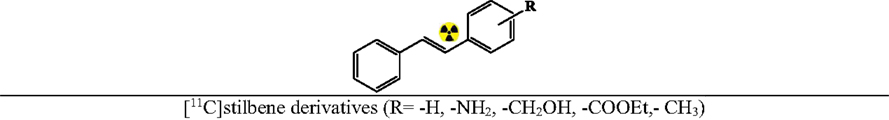
Negishi Reaction and Misc Reactions
Negishi coupling can be a coupling of choice when other couplings fail (80, 82). It is based on organozincs as nucleophiles and is palladium-catalyzed reaction. Disadvantages include (a) incompatible with common functional groups, such as hydroxyl, sulfhydryl, aldehyde, and carboxylic acid, hence limited scope and (b) sensitive to environmental conditions.
Carbon Labeling: (Temperature and Time Dependent on Reactants)
Arylzinc iodide and 11C labeled methyl iodide with Pd(PPh3)Cl2 in dimethylacetamide at room temperature or elevated temperature.
Apart from the above-mentioned cross-coupling reactions, there exist many miscellaneous reactions that can make an important contribution in near future. Figure 11 summarizes some contributions (80, 82, 84, 122–129).
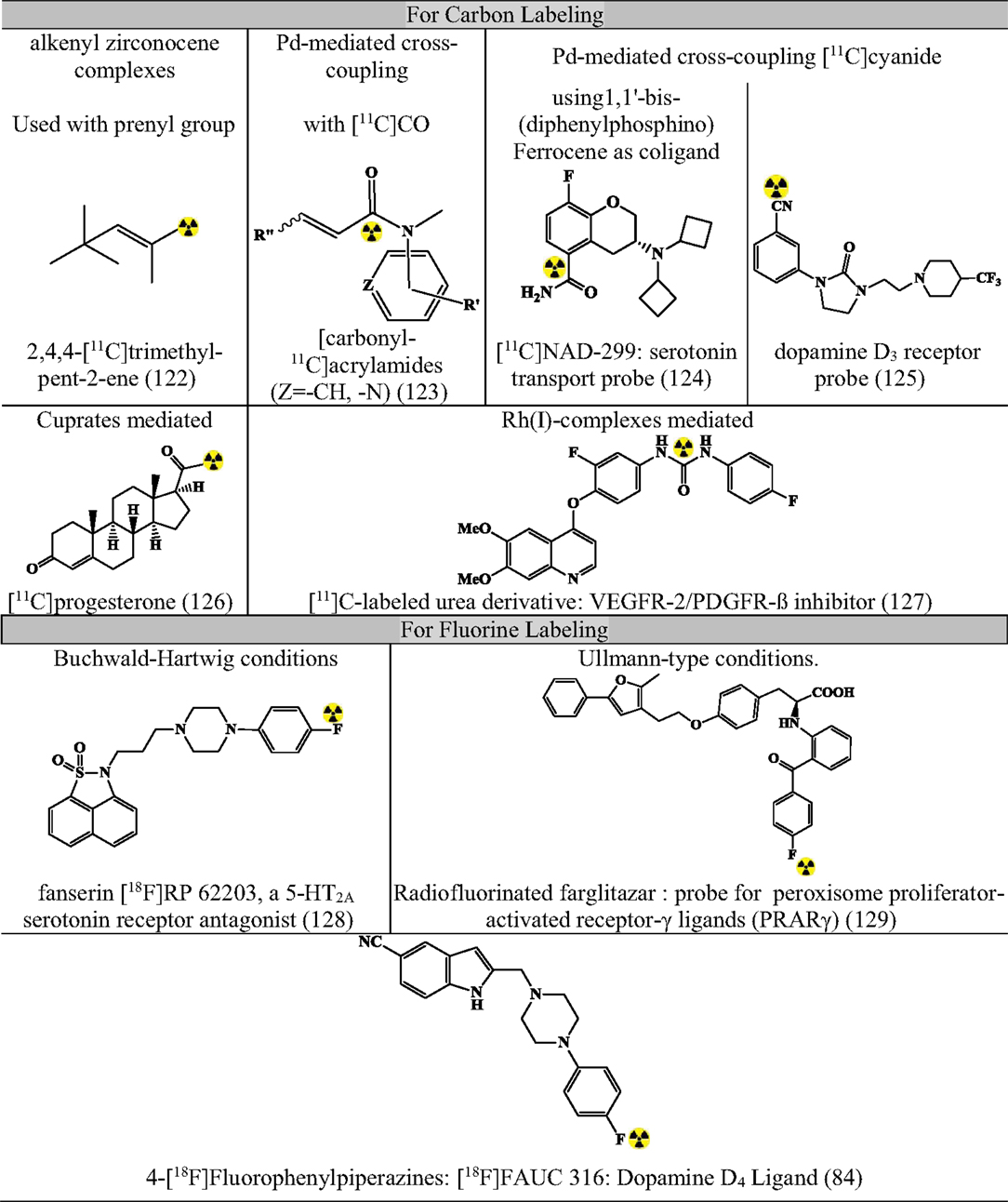
Figure 11. Representative radiopharmaceuticals developed using Misc reactions. Reference: (80, 82, 84, 122–129).
Future Directions
The future directions for successfully utilizing the novel chemistries include (a) easy synthesis of precursors for example cyclooctynes and tetrazines (b) better purification techniques to remove metal linked toxic species, especially using cartridges or scavenger resins that allow faster and easy purification (c) standardization of novel approaches toward automated synthesis (d) regioisomer selectivity, and (e) studies to understand the effect of bulky precursors on pharmacokinetics and biological efficacy of radiopharmaceuticals.
Conclusion
This review has summarized the applications and scope of the three approaches for the development of radiopharmaceuticals (a) bivalent ligand approach (BLA) for the novel design of the radiopharmaceuticals, (b) lipidization and surface modification, and (c) novel chemistries for radiolabeling. Despite the rise to prominence only 5–10 years ago all the above approaches have made a significant impact in radiolabeling and development of radiopharmaceuticals. The reactions have been tested with a wide variety of biomolecules-small molecules, steroids, nucleosides, glucose derivatives, peptides, and also with NPs. Selectivity, orthogonality, and fast kinetics are the key requirements for being a method of choice of novel chemistries.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Director, INMAS for providing the necessary facilities.
References
1. Leopoldo M, Lacivita E, Colabufo NA, Niso M, Berardi F, Perrone R. Bivalent ligand approach on 4-[2-(3-methoxyphenyl)ethyl]-1-(2-methoxyphenyl)piperazine: synthesis and binding affinities for 5-HT7 and 5- HT1A receptors. Bioorg Med Chem (2007) 15:5316–21. doi:10.1016/j.bmc.2007.05.010
2. Paolino M, Mennuni L, Giuliani G, Anzini M, Lanza M, Caselli G, et al. Dendrimeric tetravalent ligands for the serotonin-gated ion channel. Chem Commun (2014) 50(62):8582–5. doi:10.1039/c4cc02502d
3. Janssen M, Oyen WJ, Massuger LF, Frielink C, Dijkgraaf I, Edwards DS, et al. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm (2004) 17(6):641–6. doi:10.1089/108497802320970244
4. Liu S. Radiolabeled multimeric cyclic RGD peptides as integrin αvβ3 targeted radiotracers for tumor imaging. Mol Pharm (2006) 3(5):472–87. doi:10.1021/mp060049x
5. Li ZB, Cai W, Cao Q, Chen K, Wu Z, He L, et al. 64Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor αvβ3 integrin expression. J Nucl Med (2007) 48(7):1162–71. doi:10.2967/JNUMED.107.039859
6. Zhang Y, Xiao L, Chordia MD, Locke LW, Williams MB, Berr SS, et al. Neutrophil targeting heterobivalent SPECT imaging probe: cFLFLF-PEG-TKPPR-99mTc. Bioconjug Chem (2010) 21(10):1788–93. doi:10.1021/BC100063A
7. Brabez N, Lynch RM, Xu L, Gillies RJ, Chassaing G, Lavielle S, et al. Design, synthesis, and biological studies of efficient multivalent melanotropin ligands: tools toward melanoma diagnosis and treatment. J Med Chem (2011) 54(20):7375–84. doi:10.1021/jm2009937
8. Abiraj K, Jaccard H, Kretzschmar M, Helm L, Maecke HR. Novel DOTA-based prochelator for divalent peptide vectorization: synthesis of dimericbombesin analogues for multimodality tumor imaging and therapy. Chem Commun (2008) (28):3248–50. doi:10.1039/B805281F
9. Fischer G, Schirrmacher R, Wängler B, Wängler C. Radiolabeled heterobivalent peptidic ligands: an approach with high future potential for in vivo imaging and therapy of malignant diseases. ChemMedChem (2013) 8:883–90. doi:10.1002/cmdc.201300081
10. Iwama H, Gojobori T. Identification of neurotransmitter receptor genes under significantly relaxed selective constraint by orthologous gene comparisons between humans and rodents. Mol Biol Evol (2002) 19(11):1891–901.
11. George SR, O’Dowd BF, Lee SP. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov (2002) 1:808–20. doi:10.1038/NRD913
12. Kühhorn J, Hübner H, Gmeiner P. Bivalent dopamine D2 receptor ligands: synthesis and binding properties. J Med Chem (2011) 54(13):4896–903. doi:10.1021/jm2004859
13. Hiller C, Kühhorn J, Gmeiner P. Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J Med Chem (2013) 56(17):6542–59. doi:10.1021/jm4004335
14. Zha Z, Choi SR, Ploessl K, Lieberman BP, Qu W, Hefti F, et al. (18)F-polypegylated styrylpyridines as imaging agents for Aβ plaques in cerebral amyloid angiopathy (CAA). J Med Chem (2011) 54(23):8085–98. doi:10.1021/jm2009106
15. Singh N, Hazari PP, Prakash S, Chuttani K, Khurana H, Chandra H, et al. A homodimeric bivalent radioligand derived from 1-(2-methoxyphenyl) piperazine with high affinity for in vivo 5-HT1A receptor imaging. Med Chem Commun (2012) 3(7):814–23. doi:10.1039/C2MD20062G
16. Sethi SK, Varshney R, Rangaswamy S, Chadha N, Hazari PP, Kaul A, et al. Design, synthesis and preliminary evaluation of a novel SPECT DTPA-bis-triazaspirodecanone conjugate for D2 receptor imaging. RSC Adv (2014) 4(91):50153–62. doi:10.1039/C4RA07004F
17. Chang WY, Kao HW, Wang HE, Chen JT, Lin WJ, Wang SJ, et al. Synthesis and biological evaluation of technetium-99m labeled galactose derivatives as potential asialoglycoprotein receptor probes in a hepatic fibrosis mouse model. Bioorg Med Chem Lett (2013) 23(23):6486–91. doi:10.1016/j.bmcl.2013.09.012
18. Iikuni S, Ono M, Watanabe H, Matsumura K, Yoshimura M, Harada N, et al. Enhancement of binding affinity for amyloid aggregates by multivalent interactions of 99mTc-hydroxamamide complexes. Mol Pharm (2014) 11(4):1132–9. doi:10.1021/mp400499y
19. Hazari PP, Schulz J, Vimont D, Chadha N, Allard M, Szlosek-Pinaud M, et al. A new SiF-dipropargyl glycerol scaffold as a versatile prosthetic group to design dimeric radioligands: synthesis of the [18F] BMPPSiF tracer to image serotonin receptors. ChemMedChem (2014) 9(2):337–49. doi:10.1002/cmdc.201300458
20. Wang W, Liu Z, Li Z. One-step (18)F labeling of non-peptidic bivalent integrin αvβ3 antagonist for cancer imaging. Bioconjug Chem (2015) 26(1):24–8. doi:10.1021/bc500590f
21. Wang M, Zheng DX, Gao M, Hutchins GD, Zheng QH. Synthesis of carbon-11-labeled bivalent β-carbolines as new PET agents for imaging of cholinesterase in Alzheimer’s disease. Appl Radiat Isot (2011) 69(4):678–85. doi:10.1016/j.apradiso.2011.01.004
22. Mukai T, Suwada J, Sano K, Okada M, Yamamoto F, Maeda M. Design of Ga-DOTA-based bifunctional radiopharmaceuticals: two functional moieties can be conjugated to radiogallium-DOTA without reducing the complex stability. Bioorg Med Chem (2009) 17(13):4285–9. doi:10.1016/j.bmc.2009.05.041
23. Bag N, Mathur R, Singh S, Hussain F, Chauhan RP, Chuttani K, et al. Design, synthesis and evaluation of the QD-DTC-bisbiotinnanobioconjugate as a potential optical-SPECT imaging agent. Med Chem Commun (2015) 6(2):363–71. doi:10.1039/C4MD00294F
24. Liu K, Guo TL, Chojnacki J, Lee HG, Wang X, Siedlak SL, et al. Bivalent ligand containing curcumin and cholesterol as fluorescence probe for Aβ plaques in Alzheimer’s disease. ACS Chem Neurosci (2012) 3(2):141–6. doi:10.1021/cn200122j
25. Li F, Liu J, Jas GS, Zhang J, Qin G, Xing J, et al. Synthesis and evaluation of a near-infrared fluorescent non-peptidic bivalent integrin alpha(v)beta(3) antagonist for cancer imaging. Bioconjug Chem (2010) 21(2):270–8. doi:10.1021/bc900313d
26. Zaro JL. Lipid-based drug carriers for prodrugs to enhance drug delivery. AAPS J (2015) 17(1):83–92. doi:10.1208/s12248-014-9670-z
27. Kao CH, Waki A, Sassaman MB, Jagoda EM, Szajek LP, Ravasi L, et al. Evaluation of [76Br]FBAU 3’,5’-dibenzoate as a lipophilic prodrug for brain imaging. Nucl Med Biol (2002) 29(5):527–35. doi:10.1016/S0969-8051(02)00324-4
28. Moon BS, Shim AY, Lee KC, Lee HJ, Lee BS, An GI, et al. Synthesis of F-18 labeled capecitabine using [18F] F2 gas as a tumor imaging agent. Bull Korean Chem Soc (2005) 26(11):1865–8. doi:10.1002/chin.200615200
29. André M, Besse S, Chezal JM, Mounetou E. PEGylation enhances the tumor selectivity of melanoma-targeted conjugates. Org Biomol Chem (2015) 13(2):388–97. doi:10.1039/c4ob01751j
30. de Barros AB, Tsourkas A, Saboury B, Cardoso VN, Alavi A. Emerging role of radiolabeled nanoparticles as an effective diagnostic technique. EJNMMI Res (2012) 2(1):39. doi:10.1186/2191-219X-2-39
31. Masserini M. Nanoparticles for brain drug delivery. ISRN Biochem (2013) 2013:238428. doi:10.1155/2013/238428
32. Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev (2015) 115(19):10938–66. doi:10.1021/acs.chemrev.5b00046
33. Luk BT, Fang RH, Zhang L. Lipid- and polymer-based nanostructures for cancer theranostics. Theranostics (2012) 2(12):1117–26. doi:10.7150/thno.4381
34. Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond) (2011) 6(4):715–28. doi:10.2217/nnm.11.19
35. Oumzil K, Khiati S, Camplo M, Koquely M, Chuttani K, Chaturvedi S, et al. Nucleolipids as building blocks for the synthesis of 99mTc-labeled nanoparticles functionalized with folic acid. New J Chem (2014) 38(11):5240–6. doi:10.1039/C4NJ00559G
36. Huang F-YJ, Chen W-J, Lee W-Y, Lo S-T, Lee T-W, Lo J-M. In vitro and in vivo evaluation of lactoferrin-conjugated liposomes as a novel carrier to improve the brain delivery. Int J Mol Sci (2013) 14(2):2862–74. doi:10.3390/ijms14022862
37. Petersen AL, Binderup T, Rasmussen P, Henriksen JR, Elema DR, Kjær A, et al. 64Cu loaded liposomes as positron emission tomography imaging agents. Biomaterials (2011) 32(9):2334–41. doi:10.1016/j.biomaterials.2010.11.059
38. Gaudin A, Tagit O, Sobot D, Lepetre-Mouelhi S, Mougin J, Martens TF, et al. Transport mechanisms of squalenoyl-adenosine nanoparticles across the blood-brain barrier. Chem Mater (2015) 27(10):3636–47. doi:10.1021/acs.chemmater.5b00267
39. Lepeltier E, Bourgaux C, Rosilio V, Poupaert JH, Meneau F, Zouhiri F, et al. Self-assembly of squalene-based nucleolipids: relating the chemical structure of the bioconjugates to the architecture of the nanoparticles. Langmuir (2013) 29(48):14795–803. doi:10.1021/la403338y
40. Sharma G, Modgil A, Layek B, Arora K, Sun C, Law B, et al. Cell penetrating peptide tethered bi-ligand liposomes for delivery to brain in vivo: biodistribution and transfection. J Control Release (2013) 167(1):1–10. doi:10.1016/j.jconrel.2013.01.016
41. Kettenbach K, Schieferstein H, Ross Tobias L. 18F-labeling using click cycloadditions. Biomed Res Int (2014) 2014:361329. doi:10.1155/2014/361329
42. Pretze M, Pietzsch D, Mamat C. Recent trends in bioorthogonal click-radiolabeling reactions using fluorine-18. Molecules (2013) 18(7):8618–65. doi:10.3390/molecules18078618
43. Zeng D, Zeglis BM, Lewis JS, Anderson CJ. Th e growing impact of bioorthogonalclick chemistry on the development of radiopharmaceuticals. J Nucl Med (2013) 54(6):829–32. doi:10.2967/jnumed.112.115550
44. Kluba CA, Mindt TL. Click-to-chelate: development of technetium and rhenium-tricarbonyl labeled radiopharmaceuticals. Molecules (2013) 18(3):3206–26. doi:10.3390/molecules18033206
45. Worrell BT, Malik JA, Fokin VV. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science (2013) 340(6131):457–60. doi:10.1126/science.1229506
46. Zhang X, Zhang Y. Applications of azide-based bioorthogonal click chemistry in glycobiology. Molecules (2013) 18(6):7145–9. doi:10.3390/molecules18067145
47. Xu H, Jones LH. Click chemistry patents and their impact on drug discovery and chemical biology. Pharm Pat Anal (2015) 4(2):109–19. doi:10.4155/ppa.14.59
48. Struthers H, Spingler B, Mindt TL, Schibli R. ‘Click-to-chelate’: design and incorporation of triazole-containing metal-chelating systems into biomolecules of diagnostic and therapeutic interest. Chemistry (2008) 14(20):6173–83. doi:10.1002/chem.200702024
49. Glaser M, Årstad E. Click labeling with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjug Chem (2007) 18(3):989–93. doi:10.1021/bc060301j
50. Sirion U, Kim HJ, Lee JH, Seo JW, Lee BS, Lee SJ, et al. An efficient F-18 labeling method for PET study: Huisgen 1,3-dipolar cycloaddition of bioactive substances and F-18-labeled compounds. Tetrahedron Lett (2007) 48(23):3953–7. doi:10.1016/j.tetlet.2007.04.048
51. Ramenda T, Bergmann R, Wuest F. Synthesis of 18F-labeled neurotensin(8-13) via copper-mediated 1,3-dipolar [3+2]cycloaddition reaction. Lett Drug Des Discov (2007) 4(4):279–85. doi:10.2174/157018007784619998
52. Vaidyanathan G, White BJ, Zalutsky MR. Propargyl 4-[18F] fluorobenzoate: a putatively more stable prosthetic group for the fluorine-18 labeling of biomolecules via click chemistry. Curr Radiopharm (2009) 2(1):63–74. doi:10.2174/1874471010902010063
53. Thonon D, Kech C, Paris J, Lemaire C, Luxen A. New strategy for the preparation of clickable peptides and labeling with 1-(azidomethyl)-4-[18F]-fluorobenzene for PET. Bioconjugate Chem (2009) 20(4):817–23.
54. Maschauer S, Prante O. A series of 2-O-trifluoromethylsulfonyl-d-mannopyranosides as precursors for concomitant 18F-labeling and glycosylation by click chemistry. Carbohydr Res (2009) 344(6):753–61. doi:10.1016/j.carres.2009.02.001
55. Li Y, Guo J, Tang S, Lang L, Chen X, Perrin DM. One-step and one-pot-two-step radiosynthesis of cyclo-RGD-18F-aryltrifluoroborate conjugates for functional imaging. Am J Nucl Med Mol Imaging (2013) 3(1):44–56.
56. Schieferstein H, Ross TL. A polar 18F-labeled amino acid derivative for click labeling of biomolecules. Eur J Org Chem (2014) 17:3546–50. doi:10.1002/ejoc.201400071
57. Pretze M, Mamat C. Automated preparation of [18F]AFP and [18F]BFP: two novel bifunctional18F-labeling building blocks for Huisgen-click. J Fluor Chem (2013) 150:25–35. doi:10.1016/j.jfluchem.2013.02.028
58. Liu Z, Amouroux G, Zhang Z, Pan J, Hundal-Jabal N, Colpo N, et al. (18)F-trifluoroborate derivatives of [des-arg(10)]kallidin for imaging bradykinin b1 receptor expression with positron emission tomography. Mol Pharm (2015) 12(3):974–82. doi:10.1021/acs.molpharmaceut.5b00003
59. Pourghiasian M, Liu Z, Pan J, Zhang Z, Colpo N, Lin KS, et al. 18F-AmBF3-MJ9: a novel radiofluorinatedbombesin derivative for prostate cancer imaging. Bioorg Med Chem (2015) 23(7):1500–6. doi:10.1016/j.bmc.2015.02.009
60. Inkster JAH, Guérin B, Ruth TJ, Adam MJ. Radiosynthesis and bioconjugation of [18F]FPy5yne, a prosthetic group for the 18F labeling of bioactive peptides. J Label Compd Radiopharm (2008) 51(14):444–52. doi:10.1002/jlcr.1561
61. Valdivia AC, Estrada M, Hadizad T, Stewart DJ, Beanlands RS, DaSilva JN. A fast, simple, and reproducible automated synthesis of [18F]FPyKYNE-c(RGDyK) for αvβ3 receptor positron emission tomography imaging. J Label Compd Radiopharm (2012) 55:57–60. doi:10.1002/jlcr.1948
62. Daumar P, Wanger-Baumann CA, Pillarsetty N, Fabrizio L, Carlin SD, Andreev OA, et al. Efficient 18F-labeling of large 37-amino-acid pHLIP peptide analogues and their biological evaluation. Bioconjug Chem (2012) 23:1557–66. doi:10.1021/bc3000222
63. Schirrmacher R, Lakhrissi Y, Jolly D, Goodstein J, Lucas P, Schirrmacher E. Rapid in situ synthesis of [11C]methylazide and its application in 11C click-chemistry. Tetrahedron Lett (2008) 49(33):4824–7. doi:10.1016/j.tetlet.2008.06.020
64. Bordenave T, Hazari PP, James D, Mishra AK, Szlosek-Pinaud M, Fouquet E. 11C click chemistry using [11C] methyl azide: simplified, versatile, and practical alternative access to [11C] nucleosides and [11C] oligonucleotides for PET imaging. Eur J Org Chem (2013) 2013(7):1214–7. doi:10.1002/ejoc.201201379
65. Mindt TL, Struthers H, Brans L, Anguelov T, Schweinsberg C, Maes V, et al. “Click to chelate”: synthesis and installation of metal chelates into biomolecules in a single step. J Am Chem Soc (2006) 128(47):15096–7. doi:10.1021/ja066779f
66. Cai Z, Li BT, Wong EH, Weisman GR, Anderson CJ. Cu(I)-assisted click chemistry strategy for conjugation of non-protected cross-bridged macrocyclicchelators to tumour-targeting peptides. Dalton Trans (2015) 44(9):3945–8. doi:10.1039/C4DT03897E
67. Chen K, Wang X, Lin WY, Shen CKF, Yap LP, Hughes LD, et al. Strain-promoted catalyst-free click chemistry for rapid construction of 64Cu-labeled PET imaging probes. ACS Med Chem Lett (2012) 3(12):1019–23. doi:10.1021/ml300236m
68. Lee DE, Na JH, Lee S, Kang CM, Kim HN, Han SJ, et al. Facile method to radiolabel glycol chitosan nanoparticles with 64Cu via copper-free click chemistry for MicroPET imaging. Mol Pharm (2013) 10(6):2190–8. doi:10.1021/mp300601r
69. Bouvet V, Wuest M, Wuest F. Copper-free click chemistry with the short-lived positron emitter fluorine-18. Org Biomol Chem (2011) 9:7393–9. doi:10.1039/C1OB06034A
70. Campbell-Verduyn LS, Mirfeizi L, Schoonen AK, Dierckx RA, Elsinga PH, Feringa BL. Strain-promoted copper-free “click” chemistry for 18F radiolabeling of bombesin. Angew Chem Int Ed (2011) 50(47):11117–20. doi:10.1002/anie.201105547
71. Sachin K, Jadhav VH, Kim EM, Kim HL, Lee SB, Jeong HJ, et al. F-18 labeling protocol of peptides based on chemically orthogonal strain-promoted cycloaddition under physiologically friendly reaction conditions. Bioconjug Chem (2012) 23(8):1680–6. doi:10.1021/bc3002425
72. Cai Z, Ouyang Q, Zeng D, Nguyen KN, Modi J, Wang L, et al. 64Cu-labeled somatostatin analogues conjugated with cross-bridged phosphonate-based chelators via strain-promoted click chemistry for PET imaging: in silico through in vivo studies. J Med Chem (2014) 57(14):6019–29. doi:10.1021/jm500416f
73. Schultz MK, Parameswarappa SG, Pigge FC. Synthesis of a DOTA-biotin conjugate for radionuclide chelation via Cu-free click chemistry. Org Lett (2010) 12(10):2398–401. doi:10.1021/ol100774p
74. Zeng D, Lee NS, Liu Y, Zhou D, Dence CS, Wooley KL, et al. 64Cu core-labeled nanoparticles with high specific activity via metal-free click chemistry. ACS Nano (2012) 6(6):5209–19. doi:10.1021/nn300974s
75. Arumugam S, Chin J, Schirrmacher R, Popik VV, Kostikov AP. [18F] Azadibenzocyclooctyne ([18F] ADIBO): A biocompatible radioactive labeling synthon for peptides using catalyst free [3+ 2] cycloaddition. Bioorg Med Chem Lett (2011) 21(23):6987–91.
76. Evans HL, Slade RL, Carroll L, Smith G, Nguyen Q-D, Iddon L, et al. Copper-free click – a promising tool for pre-targeted PET imaging. Chem Commun (2012) 48:991–3. doi:10.1039/C1CC16220A
77. Li Z, Cai H, Hassink M, Blackman ML, Brown RC, Conti PS, et al. Tetrazine-trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chem Commun (Camb) (2010) 46:8043–5. doi:10.1039/C0CC03078C
78. Herth MM, Andersen VL, Lehel S, Madsen J, Knudsen GM, Kristensen JL. Development of a11C-labeled tetrazine for rapid tetrazine-trans-cyclooctene ligation. Chem Commun (Camb) (2013) 49(36):3805–7. doi:10.1039/C3CC41027G
79. Reiner T, Zeglis BM. The inverse electron demand Diels-Alder click reaction in radiochemistry. J Labelled Comp Radiopharm (2014) 57(4):285–90. doi:10.1002/jlcr.3149
80. Doi H. Pd-mediated rapid cross-couplings using [11C]methyl iodide: groundbreaking labeling methods in 11C radiochemistry. J Labelled Comp Radiopharm (2015) 58(3):73–85. doi:10.1002/jlcr.3253
81. Zlatopolskiy BD, Krapf P, Richarz R, Frauendorf H, Mottaghy FM, Neumaier B. Synthesis of 18F-labelled β-lactams by using the Kinugasa reaction. Chemistry (2014) 20(16):4697–703. doi:10.1002/chem.201304056
82. Pretze M, Grosse-Gehling P, Mamat C. Cross-coupling reactions as valuable tool for the preparation of PET radiotracers. Molecules (2011) 16(2):1129–65. doi:10.3390/molecules16021129
83. Yagi Y, Kimura H, Arimitsu K, Ono M, Maeda K, Kusuhara H, et al. The synthesis of [18F]pitavastatin as a tracer for hOATP using the Suzuki coupling. Org Biomol Chem (2015) 13:1113–21. doi:10.1039/C4OB01953A
84. Kügler F, Ermert J, Kaufholz P, Coenen HH. 4-[18F]Fluorophenylpiperazines by improved Hartwig-Buchwald N-arylation of 4-[18F]fluoroiodobenzene, formed via hypervalent λ3-iodane precursors: application to build-up of the dopamine D4 ligand [18F]FAUC 316. Molecules (2014) 20(1):470–86. doi:10.3390/molecules20010470
85. Samuelsson L, Långström B. Synthesis of 1-(2’-deoxy-2’-fluoro-ß-d-arabinofuranosyl)-[Methyl-11C]thymine ([11C]FMAU) via a Stille cross-coupling reaction with [11C]methyl iodide. J Labelled Comp Radiopharm (2003) 46(3):263–72. doi:10.1002/jlcr.668
86. Toyohara J, Okada M, Toramatsu C, Suzuki K, Irie T. Feasibility studies of 4’-[methyl-11C] thiothymidine as a tumor proliferation imaging agent in mice. Nucl Med Biol (2008) 35(1):67–74. doi:10.1016/j.nucmedbio.2007.10.001
87. Zhang Z, Koyama H, Watanabe Y, Suzuki M. Efficient syntheses of [11C] zidovudine and its analogs by convenient one-pot palladium (0)–copper (I) co-mediated rapid C-[11C] methylation. J Labelled Comp Radiopharm (2014) 57(8):540–9. doi:10.1002/jlcr.3213
88. Kimura T, Sako T, Hosokawa-Muto J, Cui YL, Wada Y, Kataoka Y, et al. Synthesis of an 11C-labeled antiprion GN8 derivative and evaluation of its brain uptake by positron emission tomography. ChemMedChem (2013) 8(7):1035–9. doi:10.1002/cmdc.201300167
89. Langer O, Forngren T, Sandell J, Dollé F, Långström B, Någren K, et al. Preparation of 4-[11C] methylmetaraminol, a potential PET tracer for assessment of myocardial sympathetic innervation. J Labelled Comp Radiopharm (2003) 46(1):55–65. doi:10.1002/jlcr.642
90. Tarkiainen J, Vercouillie J, Emond P, Sandell J, Frangin Y, Guilloteau D, et al. Carbon-11 labelling of madam in two different positions: a highly selective pet radioligand for the serotonin transporter. J Labelled Comp Radiopharm (2001) 44(14):1013–23. doi:10.1002/jlcr.523
91. Sandell J, Yu M, Emond P, Garreau L, Chalon S, Någren K, et al. Synthesis, radiolabeling and preliminary biological evaluation of radiolabeled 5-methyl-6-nitroquipazine, a potential radioligand for the serotonin transporter. Bioorg Med Chem Lett (2002) 12(24):3611–3. doi:10.1016/S0960-894X(02)00787-4
92. Madsen J, Merachtsaki P, Davoodpour P, Bergström M, Långström B, Andersen K, et al. Synthesis and biological evaluation of novel carbon-11-labelled analogues of citalopram as potential radioligands for the serotonin transporter. Bioorg Med Chem (2003) 11(16):3447–56. doi:10.1016/S0968-0896(03)00307-9
93. Kawamura K, Naganawa M, Konno F, Yui J, Wakizaka H, Yamasaki T, et al. Imaging of I 2-imidazoline receptors by small-animal PET using 2-(3-fluoro-[4-11C] tolyl)-4, 5-dihydro-1H-imidazole ([11C] FTIMD). Nucl Med Biol (2010) 37(5):625–35. doi:10.1016/j.nucmedbio.2010.02.013
94. Kawamura K, Yui J, Konno F, Yamasaki T, Hatori A, Wakizaka H, et al. Synthesis and evaluation of PET probes for the imaging of I 2 imidazoline receptors in peripheral tissues. Nucl Med Biol (2012) 39(1):89–99. doi:10.1016/j.nucmedbio.2011.06.001
95. Prabhakaran J, Majo VJ, Simpson NR, Van Heertum RL, Mann JJ, Kumar JS. Synthesis of [11C] celecoxib: a potential PET probe for imaging COX-2 expression. J Labelled Comp Radiopharm (2005) 48(12):887–95. doi:10.1002/jlcr.1002
96. Karimi F, Långström B. Synthesis of 3-[(2S)-azetidin-2-ylmethoxy]-5-[11C]-methylpyridine, an analogue of A-85380, via a Stille coupling. J Labelled Comp Radiopharm (2002) 45(5):423–34. doi:10.1002/jlcr.569
97. Bennacef I, Perrio C, Lasne MC, Barré L. Functionalization through lithiation of (S)-N-(1-phenylpropyl)-2-phenylquinoline-4-carboxamide. Application to the labeling with carbon-11 of NK-3 receptor antagonist SB 222200. J Org Chem (2007) 72(6):2161–5. doi:10.1021/jo062285p
98. Kawamura K, Shiba K, Tsukada H, Nishiyama S, Mori H, Ishiwata K. Synthesis and evaluation of vesamicol analog (-)-O-[11C] methylvesamicol as a PET ligand for vesicular acetylcholine transporter. Ann Nucl Med (2006) 20(6):417–24. doi:10.1007/BF03027377
99. Hashimoto K, Nishiyama S, Ohba H, Matsuo M, Kobashi T, Takahagi M, et al. [11C] CHIBA-1001 as a novel PET ligand for α7 nicotinic receptors in the brain: a PET study in conscious monkeys. PLoS One (2008) 3(9):e3231. doi:10.1371/journal.pone.0003231
100. Björkman M, Andersson Y, Doi H, Sato K, Suzuki M, Noyori R, et al. Synthesis of 11C/13C-labelled prostacyclins. Acta Chem Scand (1998) 52(5):635–40. doi:10.3891/acta.chem.scand.52-0635
101. Hamill TG, Krause S, Ryan C, Bonnefous C, Govek S, Seiders TJ, et al. Synthesis, characterization, and first successful monkey imaging studies of metabotropic glutamate receptor subtype 5 (mGluR5) PET radiotracers. Synapse (2005) 56(4):205–16. doi:10.1002/syn.20147
102. Huang Y, Narendran R, Bischoff F, Guo N, Zhu Z, Bae SA, et al. A positron emission tomography radioligand for the in vivo labeling of metabotropic glutamate 1 receptor: (3-ethyl-2-[11C] methyl-6-quinolinyl)(cis-4-methoxycyclohexyl) methanone. J Med Chem (2005) 48(16):5096–9. doi:10.1021/jm050263+
103. Yu M, Tueckmantel W, Wang X, Zhu A, Kozikowski AP, Brownell AL. Methoxyphenylethynyl, methoxypyridylethynyl and phenylethynyl derivatives of pyridine: synthesis, radiolabeling and evaluation of new PET ligands for metabotropic glutamate subtype 5 receptors. Nucl Med Biol (2005) 32(6):631–40. doi:10.1016/j.nucmedbio.2005.05.004
104. Fujinaga M, Yamasaki T, Maeda J, Yui J, Xie L, Nagai Y, et al. Development of N-[4-[6-(isopropylamino) pyrimidin-4-yl]-1, 3-thiazol-2-yl]-N-methyl-4-[11C] methylbenzamide for positron emission tomography imaging of metabotropic glutamate 1 receptor in monkey brain. J Med Chem (2012) 55(24):11042–51. doi:10.1021/jm301597s
105. Lu S, Hong J, Itoh T, Fujita M, Inoue O, Innis RB, et al. [carbonyl-11C] benzyl acetate: automated radiosynthesis via Pd-mediated [11C] carbon monoxide chemistry and PET measurement of brain uptake in monkey. J Labelled Comp Radiopharm (2010) 53(8):548–51. doi:10.1002/jlcr.1779
106. Suzuki M, Takashima-Hirano M, Koyama H, Yamaoka T, Sumi K, Nagata H, et al. Efficient synthesis of [11C] H-1152, a PET probe specific for Rho-kinases, highly potential targets in diagnostic medicine and drug development. Tetrahedron (2012) 68(10):2336–41. doi:10.1016/j.tet.2012.01.033
107. Zeng F, Mun J, Jarkas N, Stehouwer JS, Voll RJ, Tamagnan GD, et al. Synthesis, radiosynthesis, and biological evaluation of carbon-11 and fluorine-18 labeled reboxetine analogues: potential positron emission tomography radioligands for in vivo imaging of the norepinephrine transporter. J Med Chem (2008) 52(1):62–73. doi:10.1021/jm800817h
108. Allain-Barbier L, Lasne MC, Perrio-Huard C, Moreau B, Barré L. Synthesis of [18F]fluorophenyl alkenes or arenes via palladium-catalyzed coupling of 4-[18F]fluoroiodobenzene with vinyl and aryl tin reagents. Acta Chem Scand (1998) 52(4):480–9. doi:10.3891/acta.chem.scand.52-0480
109. Marriere E, Rouden J, Tadino V, Lasne MC. Synthesis of analogues of (–)-cytisine for in vivo studies of nicotinic receptors using positron emission tomography. Org Lett (2000) 2(8):1121–4. doi:10.1021/ol005685m
110. Wüst FR, Kniess T. No-carrier added synthesis of 18F-labelled nucleosides using Stille cross-coupling reactions with 4-[18F] fluoroiodobenzene. J Labelled Comp Radiopharm (2004) 47(8):457–68. doi:10.1002/jlcr.834
111. Wüst FR, Höhne A, Metz P. Synthesis of 18F-labelled cyclooxygenase-2 (COX-2) inhibitors via Stille reaction with 4-[18F] fluoroiodobenzene as radiotracers for positron emission tomography (PET). Org Biomol Chem (2005) 3(3):503–7. doi:10.1039/B412871K
112. Kanazawa M, Furuta K, Doi H, Mori T, Minami T, Ito S, et al. Synthesis of an acromelic acid A analog-based 11C-labeled PET tracer for exploration of the site of action of acromelic acid A in allodynia induction. Bioorg Med Chem Lett (2011) 21(7):2017–20. doi:10.1016/j.bmcl.2011.02.018
113. Takahashi K, Hosoya T, Onoe K, Doi H, Nagata H, Hiramatsu T, et al. 11C-cetrozole: an improved C-11C-methylated PET probe for aromatase imaging in the brain. J Nucl Med (2014) 55(5):852–7. doi:10.2967/jnumed.113.131474
114. Suzuki M, Takashima-Hirano M, Ishii H, Watanabe C, Sumi K, Koyama H, et al. Synthesis of 11C-labeled retinoic acid,[11C] ATRA, via an alkenylboron precursor by Pd (0)-mediated rapid C-[11C] methylation. Bioorg Med Chem Lett (2014) 24(15):3622–5. doi:10.1016/j.bmcl.2014.05.041
115. Doi H, Goto M, Suzuki M. Pd 0-mediated rapid C-[18F] fluoromethylation by the cross-coupling reaction of a [18F] fluoromethyl halide with an arylboronic acid ester: novel method for the synthesis of a 18F-labeled molecular probe for positron emission tomography. Bull Chem Soc Jpn (2012) 85(11):1233–8. doi:10.1246/bcsj.20120151
116. Andersen VL, Hansen HD, Herth MM, Knudsen GM, Kristensen JL. 11C-labeling and preliminary evaluation of vortioxetine as a PET radioligand. Bioorg Med Chem Lett (2014) 24(11):2408–11. doi:10.1016/j.bmcl.2014.04.044
117. Andersen VL, Herth MM, Lehel S, Knudsen GM, Kristensen JL. Palladium-mediated conversion of para-aminoarylboronic esters into para-aminoaryl-11C-methanes. Tetrahedron Lett (2013) 54(3):213–6. doi:10.1016/j.tetlet.2012.11.001
118. Ijuin R, Takashima T, Watanabe Y, Sugiyama Y, Suzuki M. Synthesis of [11C] dehydropravastatin, a PET probe potentially useful for studying OATP1B1 and MRP2 transporters in the liver. Bioorg Med Chem (2012) 20(12):3703–9. doi:10.1016/j.bmc.2012.04.051
119. Wuest F, Zessin J, Johannsen B. A new approach for 11C–C bond formation: synthesis of 17α-(3’-[11C] prop-1-yn-1-yl)-3-methoxy-3, 17β-estradiol. J Labelled Comp Radiopharm (2003) 46(4):333–42. doi:10.1002/jlcr.674
120. Wüst FR, Kniess T. Synthesis of 4-[18F] fluoroiodobenzene and its application in sonogashira cross-coupling reactions. J Labelled Comp Radiopharm (2003) 46(8):699–713. doi:10.1002/jlcr.709
121. Björkman M, Långström B. Functionalisation of 11C-labelled olefins via a Heck coupling reaction. J Chem Soc Perkin Trans 1 (2000) 2000(18):3031–4. doi:10.1039/B003960H
122. Wuest FR, Berndt M. 11C–C bond formation by palladium-mediated cross-coupling of alkenylzirconocenes with [11C] methyl iodide. J Labelled Comp Radiopharm (2006) 49(2):91–100. doi:10.1002/jlcr.1044
123. Eriksson J, åberg O, Långström B. Synthesis of [11C]/[13C] acrylamides by palladium-mediated carbonylation. Eur J Org Chem (2007) 2007(3):455–61. doi:10.1002/ejoc.200600700
124. Sandell J, Halldin C, Hall H, Thorberg SO, Werner T, Sohn D, et al. Radiosynthesis and autoradiographic evaluation of [11C] NAD-299, a radioligand for visualization of the 5-HT 1A receptor. Nucl Med Biol (1999) 26(2):159–64. doi:10.1016/S0969-8051(98)00091-2
125. Bennacef I, Salinas CA, Bonasera TA, Gunn RN, Audrain H, Jakobsen S, et al. Dopamine D 3 receptor antagonists: the quest for a potentially selective PET ligand. Part 3: radiosynthesis and in vivo studies. Bioorg Med Chem Lett (2009) 19(17):5056–9. doi:10.1016/j.bmcl.2009.07.055
126. Lidström P, Neu H, Långström B. Syntheses of [21-11C] and (21-13C) progesterone. J Labelled Comp Radiopharm (1997) 39(8):695–704. doi:10.1002/(SICI)1099-1344(199708)39:8<695::AID-JLCR10>3.0.CO;2-4
127. Ilovich O, Åberg O, Långström B, Mishani E. Rhodium-mediated [11C] carbonylation: a library of N-phenyl-N’-{4-(4-quinolyloxy)-phenyl}-[11C]-urea derivatives as potential PET angiogenic probes. J Labelled Comp Radiopharm (2009) 52(5):151–7. doi:10.1002/jlcr.1582
128. Marrière E, Chazalviel L, Dhilly M, Toutain J, Perrio C, Dauphin F, et al. Synthesis of [18F] RP 62203, a potent and selective serotonin 5-HT 2 A receptor antagonist and biological evaluation with ex-vivo autoradiography. J Labelled Comp Radiopharm (1999) 42:S69–71.
129. Lee BC, Dence CS, Zhou H, Parent EE, Welch MJ, Katzenellenbogen JA. Fluorine-18 labeling and biodistribution studies on peroxisome proliferator-activated receptor-γ ligands: potential positron emission tomography imaging agents. Nucl Med Biol (2009) 36(2):147–53. doi:10.1016/j.nucmedbio.2008.11.002
Keywords: radiopharmaceuticals, multivalent ligands, bioorthogonal approaches, surface modification, cross-coupling reaction
Citation: Chaturvedi S and Mishra AK (2016) Small Molecule Radiopharmaceuticals – A Review of Current Approaches. Front. Med. 3:5. doi: 10.3389/fmed.2016.00005
Received: 12 August 2015; Accepted: 15 January 2016;
Published: 23 February 2016
Edited by:
Jean-Pierre Pouget, INSERM, FranceReviewed by:
William John McBride, Immunomedics Inc., USAEkaterina Dadachova, Albert Einstein College of Medicine, USA
Alain Faivre-Chauvet, University of Nantes and University Hospital of Nantes, France
Copyright: © 2016 Chaturvedi and Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anil K. Mishra, YWttaXNocmE2M0BnbWFpbC5jb20=, YWttaXNocmFAaW5tYXMuZHJkby5pbg==