 Ruijuan Hao1Xiaowen Zhu2,3Changxu Tian2,3,4Mouyan Jiang2,3,4Yang Huang1,2Guangli Li2,3,4Chunhua Zhu1,2*
Ruijuan Hao1Xiaowen Zhu2,3Changxu Tian2,3,4Mouyan Jiang2,3,4Yang Huang1,2Guangli Li2,3,4Chunhua Zhu1,2*- 1Development and Research Center for Biological Marine Resources, Southern Marine Science and Engineering Guangdong Laboratory (Zhanjiang), Zhanjiang, China
- 2Fisheries College, Guangdong Ocean University, Zhanjiang, China
- 3Guangdong Provincial Engineering Laboratory for Mariculture Organism Breeding, Guangdong Ocean University, Zhanjiang, China
- 4Guangdong Research Center on Reproductive Control and Breeding Technology of Indigenous Valuable Fish Species, Guangdong Ocean University, Zhanjiang, China
Long non-coding RNAs (lncRNAs) play a multifaceted role in transcriptional regulation, and the potential molecular regulatory mechanisms of lncRNAs and lncRNA–miRNA–mRNA networks in body color formation are of great significance for its selective breeding. Therefore, lncRNAs and lncRNA-miRNA-mRNA ceRNA network of red- and black-colored Plectropomus leopardus were identified and analyzed. Sequencing analyses identified 167 differentially expressed lncRNAs (DELs) between red- and black-colored P. leopardus, including 89 upregulated and 78 downregulated DELs in the red-colored group (false discovery rate (FDR) < 0.05 and |log2FC| > 1). Differentially expressed miRNA (DEM), genes (DEG), and DEL analyses found 605 and 125 negatively co-expressed miRNA–mRNA pairs and lncRNA–miRNA pairs, respectively. Further correlation analysis with Spearman’s correlation coefficient >0.9 as the threshold identified 3,721 lncRNA–mRNA pairs. Then, a competitive endogenous RNA (ceRNA) network of 325 pairs (p < 0.05) was obtained. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of network DEGs showed that melanin metabolic process, lipid metabolism, and immune-related pathway were enriched. The ceRNA network provided interactions among lncRNAs, miRNAs, and mRNAs and extended the molecular foundation of body color formation.
1 Introduction
Body color formation is critical in many aquatic animals’ biological processes, including camouflage, thermoregulation, predator avoidance, selective mating, species identification, conspecific communication, and photoreception (Leclercq et al., 2010). Bright or red-colored skin is considered one of the important economic characteristics affecting human consumption and preference (Vissio et al., 2021). The pigmentation process is affected by multiple factors, including genes, such as forkhead box D3, tyrosinase (TYR), and tyrosinase-related protein 1 (TYRP1); miRNAs, such as miR-2188, miR-122, novel-m0118, miR-194, and miR-215; and metabolites, such as tyrosine, PGG2, PGH2, and traumatic acid (Braasch et al., 2009; Curran et al., 2009; Zhu et al., 2021; Hao et al., 2022a; Hao et al., 2022b). However, the underlying mechanisms regulating body color formation are still incompletely understood.
LncRNA is a group of non-coding RNA molecules widely transcribed in most genomes of Homo sapiens, Mus musculus, and Danio rerio (Ulitsky et al., 2011; Iyer et al., 2015; Chou et al., 2016). As a major class of ncRNAs, lncRNAs function as important gene expression regulators in energy balance, cell proliferation, skeletal muscle differentiation and growth, immune responses, and fillet quality traits (Paneru et al., 2016; Ali et al., 2018; Leiva et al., 2020). For example, lncRNA SPRIGHTLY regulates primary human melanocyte proliferation (Zhao et al., 2016). A decrease in lncRNA SPRY4-IT1 expression inhibits invasion and proliferation but increases apoptosis of melanoma cells (Mazar et al., 2014). Fish lncRNAs play critical roles in pathogen infestation, muscle firmness, sex determination, and gonadal development (Gan et al., 2020; Song et al., 2022). In Larimichthys crocea, a stringent set of 210 lncRNAs were detected as being specifically expressed in the spleen and potentially indicated in immune response (Jiang et al., 2016). In peripheral blood leukocytes after chitosan oligosaccharide treatment, as well as in peripheral blood and spleen after Vibrio anguillarum stimulation, lncRNAs and mRNAs showed significant differential expression in half-smooth tongue sole (Cynoglossus semilaevis), which indicated that they may be related to the immune response (Wei et al., 2021). LncRNAs showed a similar expression pattern with coding RNAs and participated in the development of the Choromytilus chorus (Nunez-Acuna et al., 2022). However, the mechanisms of lncRNAs regulating body color formation remain elusive.
Multiple reports presented that lncRNAs regulate gene expression through a competitive endogenous RNA (ceRNA) mechanism as miRNA sponge, mRNA interactor, and RNA binding protein sponge/transporter (Salmena et al., 2011; Wang and Chang, 2011). Researchers have shown that molecular sponging or miRNA sequestration is an important function mode of lncRNAs (Ali et al., 2018). Competition for miRNAs targeting between lncRNAs and mRNAs inhibits miRNAs and leads to the formation of a lncRNA–miRNA–mRNA axis, thus preventing miRNA-induced mRNA destabilization (Jalali et al., 2013; Bartel, 2018). Therefore, in the present study, lncRNAs were detected and analyzed with the data of miRNA (Hao et al., 2022b) and mRNA. Studies on lncRNA–miRNA–mRNA networks could accelerate our understanding of lncRNA–miRNA regulation in mRNA (Salmena et al., 2011; Jeggari et al., 2012).
Plectropomus leopardus, as an important member of the genus Plectropomus, is characterized by different body colors, which determine its economic and ornamental values (Maoka et al., 2017; Zhou et al., 2020). Recent studies on aquatic organisms’ body color formation mainly focus on miRNAs or mRNAs, and many related genes and miRNAs associated with pigmentation or color variation were characterized in P. leopardus (Wang et al., 2015; Yang et al., 2020; Zhu et al., 2021; Hao et al., 2022a; Hao et al., 2022b). However, the regulatory mechanisms of ceRNA in coral grouper P. leopardus remain unclear. Therefore, we sequenced lncRNAs of them with the total RNA library construction method, which detected more RNAs than poly(A) RNA library construction (Guo et al., 2015; Keel et al., 2020) and explored the regulatory mechanisms of lncRNAs and lncRNA–miRNA–mRNA ceRNA network to provide a foundation for resolving body color formation mystery in coral grouper.
2 Materials and methods
2.1 Samples used in the present experiment
Black- and red-colored 4-month-old leopard coral grouper (P. leopardus) samples were reared with the Dongwan grouper diet (Guangdong Yuequn Biotechnology Co., Ltd.). Fish were anesthetized with eugenol and sacrificed. Six skin tissues of similar parts of fishes from each group (red- and black-colored groups) were utilized, and every two samples were mixed to make one sample for the lncRNA analysis.
2.2 LncRNA sequencing and analysis
Total RNAs from skin tissues of leopard coral groupers with different body colors were extracted and qualified with TRIzol reagent and Agilent 2100 Bioanalyzer, respectively. After total RNA was extracted, rRNAs were removed to retain mRNAs and ncRNAs. The enriched mRNAs and ncRNAs were fragmented into short fragments by using a fragmentation buffer and were reverse-transcribed into cDNA with random primers. Second-strand cDNA was synthesized by DNA polymerase I, RNase H, dNTP (dUTP instead of dTTP), and buffer. Next, the cDNA fragments were purified with a QiaQuick PCR extraction kit (Qiagen, Venlo, The Netherlands), end-repaired, poly(A) added, and ligated to Illumina sequencing adapters. Then, UNG (Uracil-N-Glycosylase) was used to digest the second-strand cDNA. The digested products were size selected by agarose gel electrophoresis, PCR-amplified, and sequenced using Illumina HiSeq™ 4000. The sequenced data were filtered with fastp (version 0.18.0) (Chen et al., 2018). Then, HISAT2 (version 2.1.0) was utilized to map the clean reads to the reference genome (Kim et al., 2015; Zhou et al., 2020); Stringtie (version 1.3.4) (Pertea et al., 2015) and HISAT2 were used to reconstruct novel reliable genes. The coding potential of the novel transcripts was evaluated with CNCI (version 2) (Sun et al., 2013) and CPC (version 0.9-r2) (Kong et al., 2007). The intersection of two potential non-coding transcripts was chosen as lncRNAs. LncRNA expression was calculated with StringTie, and differentially expressed lncRNAs (DELs) were analyzed by DESeq2 software (Love et al., 2014) (false discovery rate (FDR) < 0.05 and |log2FC| > 1).
2.3 Integrated analysis of DEMs with DEGs and DELs
Differentially expressed miRNAs (DEMs) (Hao et al., 2022b), differentially expressed genes (DEGs) (FDR < 0.05 and |log2FC| > 1), and DELs were analyzed to identify the correlation between DEMs and DEGs, and DEMs and DELs. MiRNA targets were predicted using miRanda (version 3.3a), TargetScan (version 7.0), and RNAhybrid (version 2.1.2) + svm_light (version 6.01). LncRNA–miRNA pairs and miRNA–mRNA pairs with a Spearman’s correlation coefficient ≤−0.7 were considered negatively co-expressed.
2.4 Competing endogenous RNA network analysis
Correlations between lncRNAs and mRNAs were assessed using Pearson’s correlation coefficient (PCC). CeRNAs were defined as those with a PCC > 0.9. The significance of ceRNAs with common miRNA sponges was determined using the hypergeometric cumulative distribution function test (p < 0.05). qRT-PCR was utilized to analyze the expression of lncRNAs. Seven lncRNAs were selected with primers in Supplementary Table 1. qRT-PCR was performed using 2× SG Fast qPCR Master Mix (B639271, BBI, Roche, Basel, Switzerland) on Roche LightCycler 480. β-Actin was used as an endogenous control for lncRNA. The expression level of lncRNAs was calculated with 2−(ΔΔCt). The expression levels of four mRNAs and five miRNAs in the ceRNA network were also analyzed and calculated with 2−(ΔΔCt) with a similar method. Cytoscape (version 3.6.0) was utilized to visualize the lncRNA–miRNA–mRNA network. Then, DEGs of the network were analyzed by Gene Ontology (GO) (p < 0.05) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment.
3 Results
3.1 LncRNA sequencing analysis
LncRNA libraries were sequenced, and all reads were aligned to the reference genome according to their distribution information and divided into the intron, exon, and intergenic regions. The raw data presented in the study are deposited in the Sequence Read Archive of the NCBI (SRR22522288, SRR22522289, SRR22522290, SRR22522291, SRR22522292, SRR22522293). The accession number is PRJNA908242. The result showed that most reads were mapped to the exon regions (Figure 1A). The combined prediction results of CPC and CNCI identified 3,653 lncRNAs, which were all novel lncRNAs (Figure 1B). Among them, there are 388 senses, 54 antisenses, 2 intronics, 15 bidirectionals, and 3,069 intergenics (Figure 1C). The expression analysis of these lncRNAs indicated that they could be used for further analysis (Figure 1D).
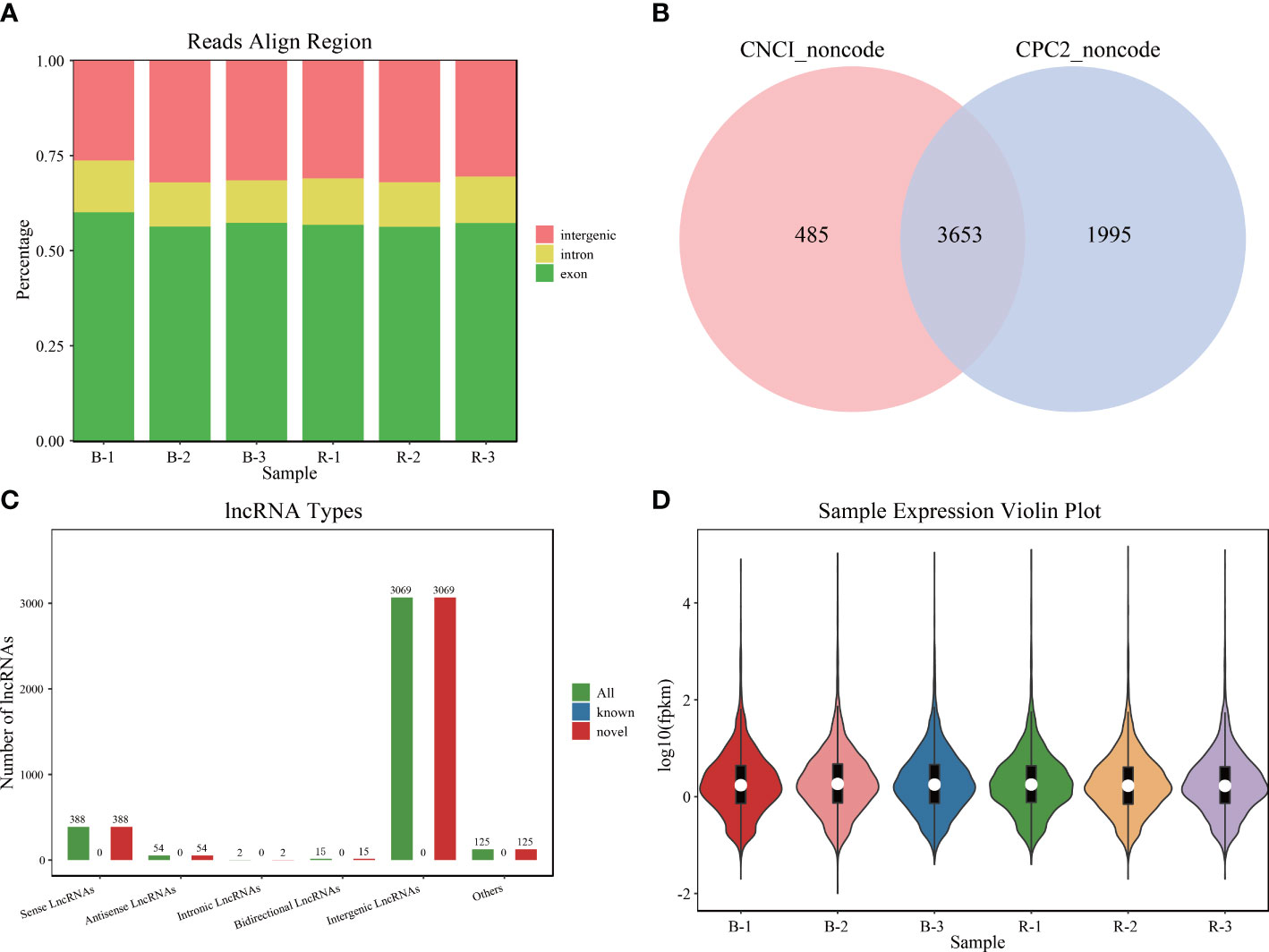
Figure 1 Sequencing data of Plectropomus leopardus lncRNA analysis. (A) The distribution position of all reads in the reference genome. (B) LncRNA number analyzed by software CPC and CNCI. (C) Different types of lncRNA number distribution. (D) LncRNA expression character. B and R represent black-colored and red-colored groups, respectively. lncRNA, long non-coding RNA.
3.2 LncRNA expression analysis
The expression pattern of lncRNAs in black- and red-colored P. leopardus was analyzed. A total of 167 lncRNAs were identified as DELs (FDR < 0.05 and |log2FC| > 1) (Supplementary Table 2; Figure 2A). Compared with the black-colored P. leopardus, the red-colored fish have 89 upregulated and 78 downregulated DELs, respectively (Figure 2B). Seven lncRNAs were validated and showed the same expression trend with the sequencing data, which indicated that the lncRNA sequencing data reliably presented the differences in lncRNA expression (Supplementary Figure 1A).
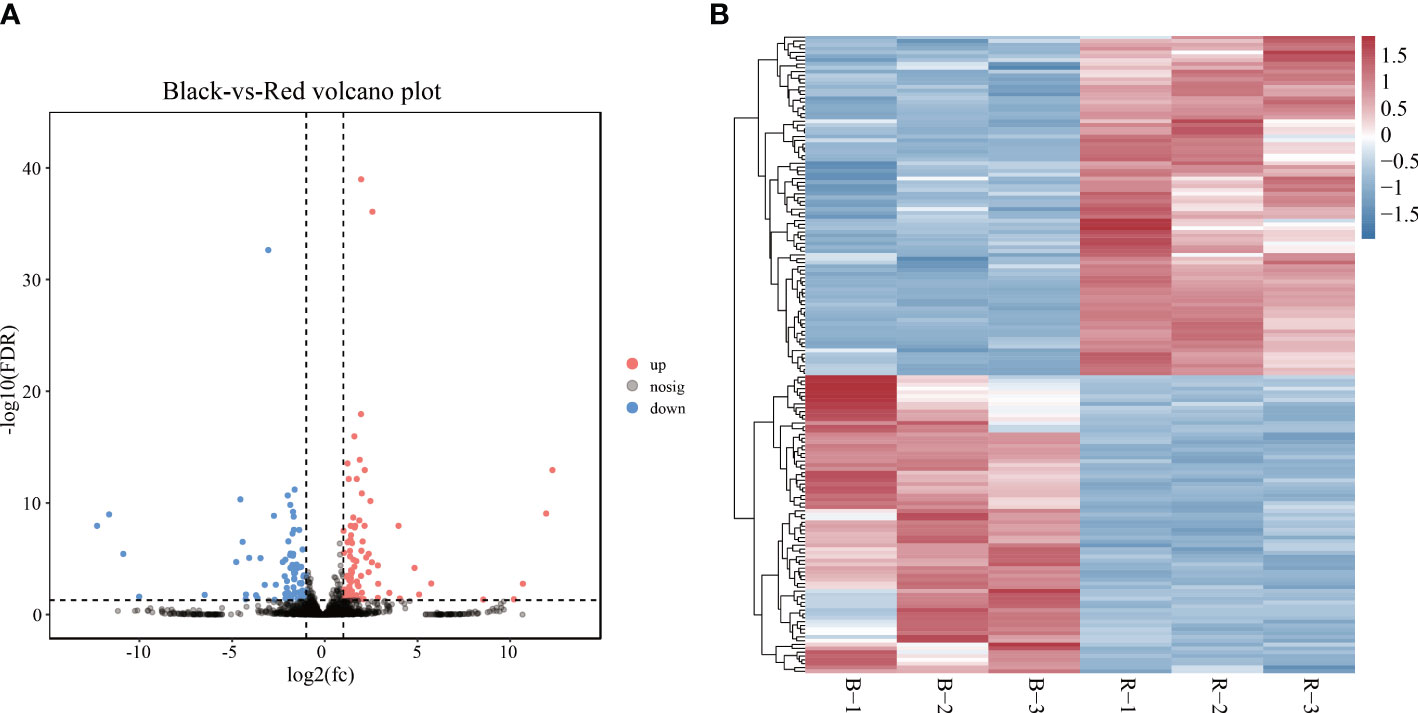
Figure 2 LncRNA expression analysis. (A) The volcano plot analysis of lncRNAs. (B) Heatmaps of the expression of lncRNAs, where red and blue indicate relatively high and low expression among samples, respectively. B and R represent black-colored and red-colored groups, respectively. lncRNA, long non-coding RNA.
3.3 Integrated analysis of DEMs with DEGs and DELs
Integrated analysis of DEMs with DELs and DEGs was performed based on datasets of DEGs, DEMs, and DELs from the red- and black-colored P. leopardus. A total of 489 DEGs (FDR < 0.05 and |log2FC| > 1, Supplementary Table 3), 60 DEMs (p < 0.05 and |log2FC| > 1, Hao et al., 2022b), and 167 DELs (FDR < 0.05 and |log2FC| > 1, Supplementary Table 2) were utilized. The results showed that red- and black-colored P. leopardus possessed 2,173 mRNA–miRNA pairs and 531 lncRNA–miRNA pairs with target prediction relationships. The former included 379 DEGs and 59 DEMs, and the latter included 113 DELs and 59 DEMs. Among them, 605 miRNA–mRNA pairs and 125 miRNA–lncRNA pairs had a Spearman’s correlation coefficient ≤−0.7 and were subjected to network analysis (Supplementary Tables 4, 5).
3.4 Regulatory ceRNA network (DELs–DEMs–DEGs) analysis
The network analysis found 3,721 DEL–DEG pairs, including 69 lncRNAs and 272 mRNAs, which had a Spearman’s correlation coefficient >0.9. Further hypergeometric cumulative distribution function test with p < 0.05 as the threshold parameter identified a final ceRNA network with 325 pairs (Supplementary Table 6), involving 57 lncRNAs, 32 miRNAs, and 146 mRNAs (Supplementary Figure 2). The hub genes in the network held important roles and were subjected to connectivity analysis. As shown in Figure 3, in the ceRNA network, tyrosine family member gene TYRP1 (Dxb_GLEAN_10020133; Dxb_GLEAN_10003424) was regulated by five lncRNAs (MSTRG.12272.1, MSTRG.13340.1, MSTRG.16640.2, MSTRG.24705.2, and MSTRG.3042.1) and three miRNAs (miR-466-x, novel-m0095-3p, and miR-194-x). The expression levels of seven lncRNAs, five miRNAs, and four mRNAs from the ceRNA network were analyzed and showed the same expression correlation among them compared with the sequencing data (Supplementary Figures 1B, C). These results verified 10 lncRNA–miRNA pairs and five mRNA–miRNA pairs in the ceRNA network.
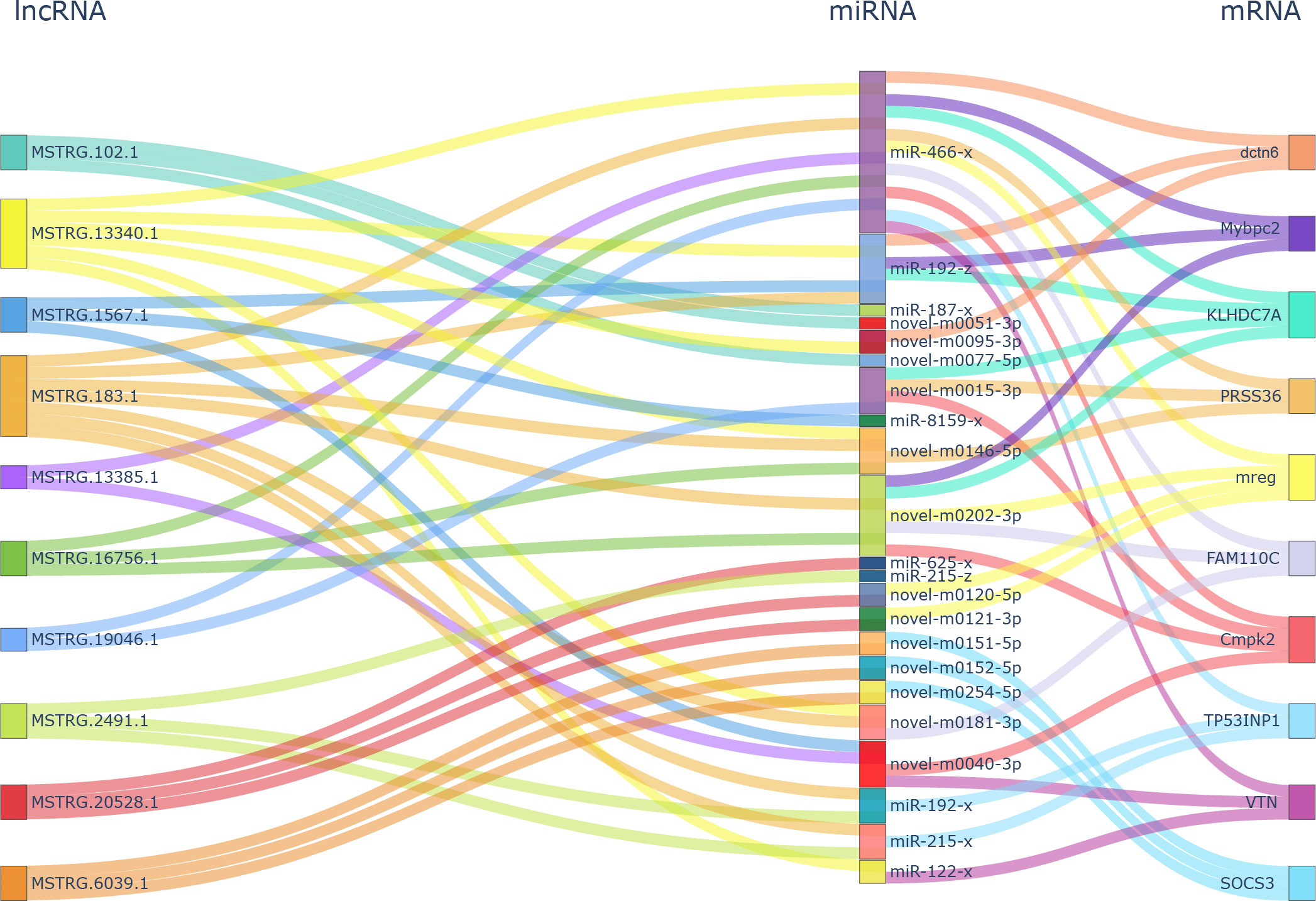
Figure 3 Hub gene analysis in ceRNA network analysis of DELs–DEMs–DEGs. ceRNA, competitive endogenous RNA; DELs, differentially expressed lncRNAs; DEMs, differentially expressed miRNAs; DEGs, differentially expressed genes.
3.5 GO analysis of ceRNA network DEGs
GO analysis indicated that DEGs in the ceRNA network were significantly enriched in 212 GO terms, including cellular components, molecular functions, and biological processes (p < 0.05). These 212 GO terms included pigment-related terms (melanin metabolic process, pigment cell differentiation, developmental pigmentation, and tyrosine metabolic process), immune-related terms (leukocyte differentiation, response to bacterium, and response to oxidative stress), and lipid-related terms (long-chain fatty acid metabolic process, short-chain fatty acid metabolic process, and fatty acid metabolic process) (Figure 4A; Supplementary Table 7).
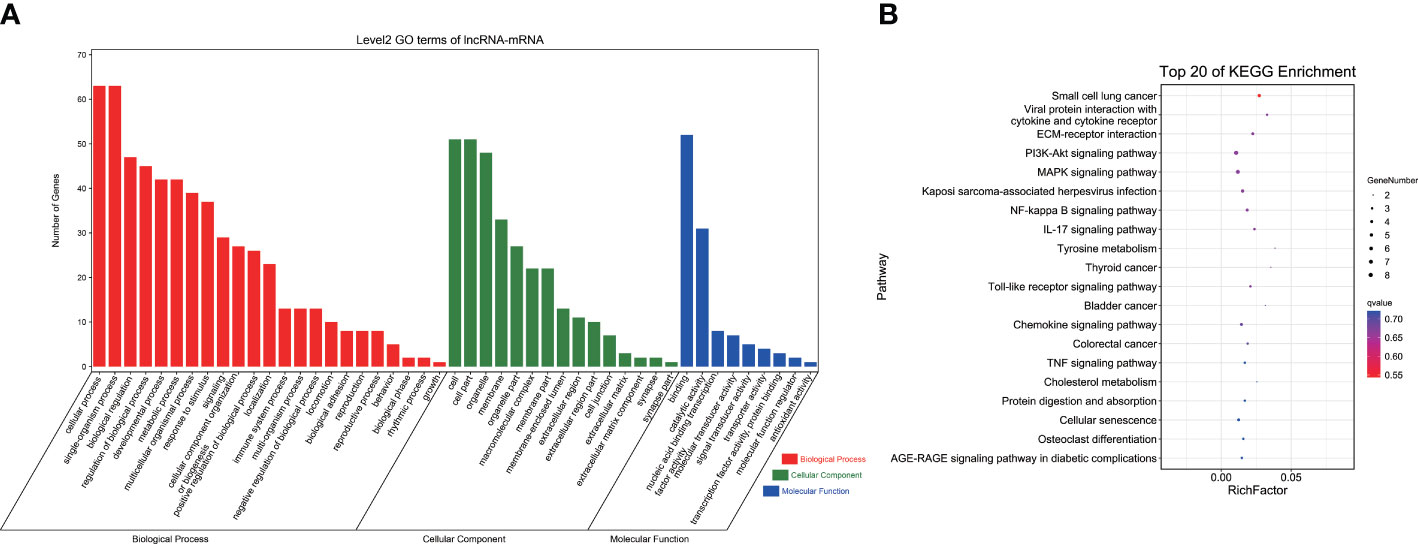
Figure 4 GO and KEGG analyses of DEGs in ceRNA network. (A) GO term analysis. (B) KEGG analysis of DEGs. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, differentially expressed genes; ceRNA, competitive endogenous RNA.
3.6 KEGG pathway analysis of DEGs in the ceRNA network
KEGG pathway analysis revealed a total of 174 enriched pathways, containing pigmentation-related pathways (tyrosine metabolism and melanogenesis), immune-related pathways (C-type lectin receptor signaling pathway, toll-like receptor signaling pathway, IL-17 signaling pathway, and NOD-like receptor signaling pathway), and lipid metabolism pathways (biosynthesis of unsaturated fatty acids, fatty acid elongation, and arachidonic acid metabolism) (Figure 4B; Supplementary Table 8). These findings indicated that lncRNAs and miRNAs might regulate related gene expression to participate in body color formation.
4 Discussion
The underlying molecular mechanism of body coloration has been widely explored in multiple species, including coral leopard grouper, medaka, carp, and zebrafish (Nagao et al., 2014; Wang et al., 2015; Yang et al., 2020). Multiple miRNAs and lncRNAs are involved in the bioprocesses by regulating gene expressions (Qin et al., 2019). Target prediction and functional analysis of color-related miRNAs such as miR-200b, miR-206, and miR-196a highlighted putative target genes in Koi carp (Cyprinus carpio L.), including Mitf, Mc1r, Foxd3, and Sox10, which are potentially related to pigmentation (Luo et al., 2018). MiR-206 plays a regulatory role in skin color pigmentation by targeting the Mc1r gene in Koi Carp (Cyprinus carpio L.) (Dong et al., 2020). Luo et al. (2019) conducted lncRNA sequencing of black-, white-, and red-skin koi carp (C. carpio L.) and showed that membrane, pigment cell development, cAMP signaling, melanogenesis, and tyrosine metabolism appear to affect skin pigmentation. However, the specific regulatory mechanism of lncRNAs and lncRNA–miRNA–mRNA networks in marine animals, especially those with diverse body colors, remains limited. Therefore, we identified lncRNAs involved in P. leopardus and analyzed their regulatory roles in body color formation via regulating gene expression. Our functional enrichment analyses revealed the potential functions of lncRNAs and lncRNA–miRNA–mRNA networks in body color formation, which provided a base for research on the regulatory mechanism of ceRNA in marine animals’ body color formation.
MiRNAs and lncRNAs have been reported to contribute to epigenetic modifications and transcriptional and translational regulations (Birney et al., 2007; Peschansky and Wahlestedt, 2014; Schmitz et al., 2016). LncRNAs have gained extensive attention from researchers as an important regulator of gene expression levels (Kopp and Mendell, 2018). LncRNAs act as sponges, binding to related miRNAs to affect their expression and downstream target genes (Zhu et al., 2020). Analysis of lncRNAs in coral leopard grouper P. leopardus with different body colors identified 3,635 lncRNAs. We found 89 upregulated and 78 downregulated lncRNAs between P. leopardus with two different colors, suggesting that they are important in regulating body color formation. Furthermore, body color formation-related genes were differentially expressed, further proving that our sequencing data were reliable.
NcRNAs, including miRNAs and lncRNAs, play important roles in regulating gene expression (Carrington and Ambros, 2003). LncRNAs and mRNAs can act as sponge molecules with common miRNA binding sites, usually leading to the inhibition of target gene expression (Yao et al., 2012; Paneru et al., 2018). Whole-transcriptome analysis of grass carp with different growth rates obtained a lncRNA–miRNA–mRNA network associated with nutrient metabolism and brain development (Ye et al., 2021). A complete endogenous lncRNA–miRNA–mRNA network of the female ovary and male testis of Pelodiscus sinensis identified several genes involved in gonadal development (Ma et al., 2020). The comparison of red- and black-colored P. leopardus revealed significantly different quantities and expression levels of DELs, indicating that P. leopardus with different body colors exhibited different numbers and expression levels of various RNAs, including some important genes and non-coding RNAs in the ceRNA network.
Black body color formation is significantly correlated with melanin synthesis in fish (Henning et al., 2013; Hao et al., 2022b). In the present study, DEGs are involved in the ceRNA network-enriched tyrosine metabolism, an essential pathway in melanin synthesis (Zhu et al., 2021). The hub gene analysis of the ceRNA network identified two TYRP1 genes that hold higher expression in the black-colored group. TYRP1, an important gene of the tyrosine gene family, participates in melanin synthesis (Kwon, 1993) and functions in color patterning and pigmentation, including eumelanin synthesis and tyrosinase activity regulation (Rad et al., 2004).
Furthermore, GO and KEGG pathway enrichment analyses showed that the DEGs of the ceRNAs network were enriched in lipid metabolism and immune response, consistent with previous findings (Zhu et al., 2021; Hao et al., 2022a). Xing et al. (2021) reported that lipid metabolism-related pathways, such as fatty acid biosynthesis and biosynthesis of unsaturated fatty acids, were identified in Apostichopus japonicus with different body wall color morphs. Variations in arachidonic acid concentration could influence fucoxanthin production, an important marine carotenoid (Zhang et al., 2017). Linoleic acid participates in the bioprocess of promoting tyrosinase depletion and inhibiting hyperpigmentation (Ando et al., 1999; 2004). A total of 202 immune-related DELs were acquired in the immune process after Aeromonas hydrophila infection of the yellow catfish Pelteobagrus fulvidraco (Zhong and Gao, 2022). LncRNAs regulated hosts’ responses in the infective process of the infectious salmon anemia virus (Boltana et al., 2016). Multiple immune corresponding pathways were enriched in the DEGs of the ceRNA network in the present study, showing the potential of ceRNAs in the fish immune regulation with a different color. In Atlantic salmon, the lncRNA–miRNA–mRNA axis modulates its immune response (Xia et al., 2020). Previous reports have shown that darker eumelanic individuals potentially possess a more excellent immune activity than lighter individuals (McGraw, 2005; Ducrest et al., 2008). However, whether these lncRNAs and the ceRNA network mediate or regulate P. leopardus body color formation and their underlying molecular regulatory mechanisms require further investigations using RNA interference, silencing, and other methods.
5 Conclusions
The regulatory potential function of ceRNAs in coral leopard P. leopardus with different body colors was analyzed. A total of 3,653 novel lncRNAs were detected, and among them, 89 were upregulated and 78 were downregulated in red-colored P. leopardus compared with black-colored P. leopardus. CeRNA network analysis of DEGs, DELs, and DEMs revealed the DEGs of the network enriched in pigmentation processes, lipid metabolism, and immune response, which improved the understanding of body color breeding selection.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was reviewed and approved by the respective Animal Care and Use Committee of Guangdong Ocean University, China.
Author contributions
CZ designed and supervised the research. RH conducted the research, analyzed the data, and wrote the manuscript. XZ, CT, MJ, YH, and GL contributed to the final writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Nature Science Foundation of China (Grant No. 32102766); Southern Marine Science and Engineering Guangdong Laboratory (Zhanjiang) (Grant No. ZJW-2019-06); Jieyang City Science and Technology Plan Project (Grant No. 2019066).
Acknowledgments
We would like to thank Robert Mkuye for the linguistic assistance of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1170762/full#supplementary-material
Supplementary Figure 1 | Expression analysis of DELs, DEMs and DEGs. (A), (B), and (C) showed the expression analysis of DELs, DEMs, and DEGs, respectively.
Supplementary Figure 2 | ceRNA analysis of DELs-DEMs-DEGs network.
References
Ali A., AI-Tobasei R., Kenney B., Leeds T. D., Salem M. (2018). Integrated analysis of lncRNA and mRNA expression in rainbow trout families showing variation in muscle growth and fillet quality traits. Sci. Rep. 8, 12111. doi: 10.1038/s41598-018-30655-8
Ando H., Funasaka Y., Oka M., Ohashi A., Furumura M., Matsunaga J., et al. (1999). Possible involvement of proteolytic degradation of tyrosinase in the regulatory effect of fatty acids on melanogenesis. J. Lipid Res. 40, 1312–1316. doi: 10.1016/S0022-2275(20)33493-3
Ando H., Watabe H., Valencia J. C., Yasumoto K., Furumura M., Funasaka Y., et al. (2004). Fatty acids regulate pigmentation via proteasomal degradation of tyrosinase: a new aspect of ubiquitin-proteasome function. J. Biol. Chem. 279, 15427–15433. doi: 10.1074/jbc.M313701200
Birney E., Stamatoyannopoulos J. A., Dutta A., Guigó R., Gingeras T. R., Margulies E. H., et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 (7146), 799–816. doi: 10.1038/nature05874
Boltana S., Valenzuela-Miranda S., Aguilar A., Mackenzie S., Gallardo-Escarate C. (2016). Long noncoding RNAs (lncRNAs) dy-namics evidence immunomodulation during ISAV-infected Atlantic salmon (Salmo salar). Sci. Rep. 6, 22698. doi: 10.1038/srep22698
Braasch I., Liedtke D., Volff J., Schartl M. (2009). Pigmentary function and evolution of tyrp1 gene duplicates in fish. Pigment Cell Melanoma Res. 22, 839–850. doi: 10.1111/j.1755-148X.2009.00614.x
Carrington J. C., Ambros V. (2003). Role of microRNAs in plant and animal development. Science 301, 336–338. doi: 10.1126/science.1085242
Chen S., Zhou Y., Chen Y., Gu J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinfomatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chou J., Wang B., Zheng T., Li X., Zheng L., Hu J., et al. (2016). MALAT1 induced migration and invasion of human breast cancer cells by competitively binding miR-1 with cdc42. Biochem. Biophys. Res. Commun. 472, 262–269. doi: 10.1016/j.bbrc.2016.02.102
Curran K., Raible D. W., Lister J. A. (2009). Foxd3 controls melanophore specification in the zebrafish neural crest by regulation of mitf. Dev. Biol. 332, 408–417. doi: 10.1016/j.ydbio.2009.06.010
Dong Z., Luo M., Wang L., Yin H., Zhu W., Fu J. (2020). MicroRNA-206 regulation of skin pigmentation in koi carp (Cyprinus carpio l.). Front. Genet. 11. doi: 10.3389/fgene.2020.00047
Ducrest A. L., Keller L., Roulin A. (2008). Pleiotropy in the melanocortin system, coloration and behavioural syndromes. Trends Ecol. Evol. 23, 502–510. doi: 10.1016/j.tree.2008.06.001
Gan L., Wang Y., Chen S., Lin Z., Sun J., He Y., et al. (2020). Identification and characterization of long non-coding RNAs in muscle sclerosis of grass carp, Ctenopharyngodon idellus fed with faba bean meal. Aquaculture 516, 734521. doi: 10.1016/j.aquaculture.2019.734521
Guo Y., Zhao S., Sheng Q., Guo M., Lehmann B., Pietenpol J., et al. (2015). RNAseq by total RNA library identifies additional RNAs compared to poly(A) RNA library. Biomed. Res. Int., 2015, 862130. doi: 10.1155/2015/862130
Hao R., Zhu X., Tian C., Jiang M., Huang Y., Zhu C. (2022b). Integrated analysis of the role of miRNA-mRNA in determining different body colors of leopard coral grouper (Plectropomus leopardus). Aquaculture 548, 737575. doi: 10.1016/j.aquaculture.2021.737575
Hao R., Zhu X., Tian C., Zhu C., Li G. (2022a). Analysis of body color formation of leopard coral grouper Plectropomus leopardus. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.964774
Henning F., Jones J. C., Franchini P., Meyer A. (2013). Transcriptomics of morphological color change in polychromatic Midas cichlids. BMC Genomics 14, 171. doi: 10.1186/1471-2164-14-171
Iyer M. K., Niknafs Y. S., Malik R., Singhal U., Sahu A., Hosono Y., et al. (2015). The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208. doi: 10.1038/ng.3192
Jalali S., Bhartiya D., Lalwani M. K., Sivasubbu S., Scaria V. (2013). Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PloS One 8, e53823. doi: 10.1371/journal.pone.0053823
Jeggari A., Marks D. S., Larsson E. (2012). miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 28, 2062–2063. doi: 10.1093/bioinformatics/bts344
Jiang L., Liu W., Zhu A., Zhang J., Zhou J., Wu C. (2016). Transcriptome analysis demonstrate widespread differential expression of long noncoding RNAs involve in Larimichthys crocea immune response. Fish Shellfish Immunol. 51, 1–8. doi: 10.1016/j.fsi.2016.02.001
Keel B. N., Oliver W. T., Keele J. W., Lindholm-Perry A. K. (2020). Evaluation of transcript assembly in multiple porcine tissues suggests optimal sequencing depth for RNA-seq using total RNA library. Anim. Gene 17-18, 200105. doi: 10.1016/j.angen.2020.200105
Kim D., Langmead B., Salzberg S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kong L., Zhang Y., Ye Z., Liu X., Zhao S., Wei L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, 345–349. doi: 10.1093/nar/gkm391
Kopp F., Mendell J. T. (2018). Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407. doi: 10.1016/j.cell.2018.01.011
Kwon B. S. (1993). Pigmentation genes: the tyrosinase gene family and the pmel 17 gene family. J. Invest. Dermatol. 100, 134S–140S. doi: 10.1111/1523-1747.ep12465022
Leclercq E., Taylor J. F., Migaud H. (2010). Morphological skin colour changes in teleosts. Fish Fish 11, 159–193. doi: 10.1111/j.1467-2979.2009.00346.x
Leiva F., Rojas-Herrera M., Reyes D., Bravo S., Garcia K. K., Moya J., et al. (2020). Identification and characterization of miRNAs and lncRNAs of coho salmon (Oncorhynchus kisutch) in normal immune organs. Genomics 112, 45–54. doi: 10.1016/j.ygeno.2019.07.015
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Luo M., Wang L., Yin H., Zhu W., Fu J., Dong Z. (2019). Integrated analysis of long non-coding RNA and mRNA expression in different colored skin of koi carp. BMC Genomics 20, 515. doi: 10.1186/s12864-019-5894-8
Luo M., Wang L., Zhu W., Fu J., Song F., Fang M., et al. (2018). Identification and characterization of skin color microRNAs in koi carp (Cyprinus carpio l.) by illumina sequencing. BMC Genomics 19, 779. doi: 10.1186/s12864-018-5189-5
Ma X., Cen S., Wang L., Zhang C., Wu L., Tian X., et al. (2020). Genome-wide identification and comparison of differentially expressed profiles of miRNAs and lncRNAs with associated ceRNA networks in the gonads of Chinese soft-shelled turtle, Pelodiscus sinensis. BMC Genomics 21, 443. doi: 10.1186/s12864-020-06826-1
Maoka T., Sato W., Nagai H., Takahashi T. (2017). Carotenoids of red, brown, and black specimens of Plectropomus leopardus, the coral trout (Suziara in Japanese). J. Oleo Sci. 66, 579–584. doi: 10.5650/jos.ess16179
Mazar J., Zhao W., Khalil A. M., Lee B., Shelley J., Govindarajan S. S., et al. (2014). The functional characterization of long noncoding RNA SPRY4-IT1 in human melanoma cells. Oncotarget 5, 8959–8969. doi: 10.18632/oncotarget.1863
McGraw K. J. (2005). The antioxidant function of many animal pigments: are there consistent health benefits of sexually selected colourants? Anim. Behav. 69, 757–764. doi: 10.1016/j.anbehav.2004.06.022
Nagao Y., Suzuki T., Shimizu A., Kimura T., Seki R., Adachi T., et al. (2014). Sox5 functions as a fate switch in medaka pigment cell development. PloS Genet. 10, 1004246. doi: 10.1371/journal.pgen.1004246
Nunez-Acuna G., Fernandez C., Sanhueza-Guevara S., Gallardo-Escarate C. (2022). Transcriptome profiling of the early developmental stages in the giant mussel choromytilus chorus exposed to delousing drugs. Mar. Genom. 65, 100970. doi: 10.1016/j.margen.2022.100970
Paneru B., Ali A., Al-Tobasei R., Kenney B., Salem M. (2018). Crosstalk among lncRNAs, microRNAs and mRNAs in the muscle 'degradome' of rainbow trout. Sci. Rep. 8, 8416. doi: 10.1038/s41598-018-26753-2
Paneru B., Al-Tobasei R., Palti Y., Wiens G. D., Salem M. (2016). Differential expression of long non-coding RNAs in three genetic lines of rainbow trout in response to infection with Flavobacterium psychrophilum. Sci. Rep. 6, 36032. doi: 10.1038/srep36032
Pertea M., Perter G. M., Antonescu C. M., Chang T. C., Mendell J. T., Salzberg S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Peschansky V. J., Wahlestedt C. (2014). Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 9, 3–12. doi: 10.4161/epi.27473
Qin C., Xia X., Fan Y., Jiang Y., Chen Y., Zhang N., et al. (2019). A novel, noncoding-RNA-mediated, post-transcriptional mechanism of AMH regulation by the H19/let-7 axis. Biol. Reprod. 101, 257. doi: 10.1016/j.fertnstert.2017.07.091
Rad H. H., Yamashita T., Jin H. Y., Hirosaki K., Wakamatsu K., Ito S., et al. (2004). Tyrosinase-related proteins suppress tyrosinase-mediated cell death of melanocytes and melanoma cells. Exp. Cell Res. 298, 317–328. doi: 10.1016/j.yexcr.2004.04.045
Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P. P. (2011). A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell 146, 353–358. doi: 10.1016/j.cell.2011.07.014
Schmitz S. U., Grote P., Herrmann B. G. (2016). Mechanisms of long noncoding RNA function in development and disease. Cell Mol. Life Sci. 73, 2491–2509. doi: 10.1007/s00018-016-2174-5
Song F., Gu Y., Chen Y., Zhang K., Shi L., Sun J., et al. (2022). Transcriptome analysis provides insights into differentially expressed long noncoding RNAs between the testis and ovary in golden pompano (Trachinotus blochii). Aquacult. Rep. 22, 100971. doi: 10.1016/j.aqrep.2021.100971
Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41 (17), e166. doi: 10.1093/nar/gkt646
Ulitsky I., Shkumatava A., Jan C. H., Sive H., Bartel D. P. (2011). Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550. doi: 10.1016/j.cell.2011.11.055
Vissio P. G., Darias M. J., Di Yorio M. P., Perez Sirkin D. I., Delgadin T. H. (2021). Fish skin pigmentation in aquaculture: the influence of rearing conditions and its neuroendocrine regulation. Gen. Comp. Endocrinol. 301, 113662. doi: 10.1016/j.ygcen.2020.113662
Wang K. C., Chang H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. doi: 10.1016/j.molcel.2011.08.018
Wang L., Yu C., Guo L., Lin H., Meng Z. (2015). In silico comparative transcriptome analysis of two color morphs of the common coral trout (Plectropomus leopardus). PloS One 10, e145868. doi: 10.1371/journal.pone.0145868
Wei S., Chen Y., Huang L., Ma H., Qi L., Wang Q., et al. (2021). Analysis of lncRNA and mRNA expression profiles in peripheral blood leukocytes of the half-smooth tongue sole (Cynoglossus semilaevis) treated with chitosan oligosaccharide. Dev. Comp. Immunol. 120, 104043. doi: 10.1016/j.dci.2021.104043
Xia Y., Cheng J., Liu Y., Li C., Liu Y., Liu P. (2020). Genome-wide integrated analysis reveals functions of lncRNA-miRNA-mRNA interactions in Atlantic salmon challenged by Aeromonas salmonicida. Genomics 114, 328–339. doi: 10.1016/j.ygeno.2021.12.013
Xing L., Sun L., Liu S., Zhang L., Sun J., Yang H. (2021). Metabolomic analysis of white, green and purple morphs of sea cucumber Apostichopus japonicus during body color pigmentation process. Comp. Biochem. Physiol. Part D. Genomics Proteomics 39, 100827. doi: 10.1016/j.cbd.2021.100827
Yang Y., Wu L., Chen J., Wu X., Xia J., Meng Z., et al. (2020). Whole-genome sequencing of leopard coral grouper (Plectropomus leopardus) and exploration of regulation mechanism of skin color and adaptive evolution. Zool. Res. 41, 328–340. doi: 10.24272/j.issn.2095-8137.2020.038
Yao P., Potdar A., Arif A. (2012). Coding region polyadenylation generates a truncated tRNA synthetase that counters translation repression. Cell 149, 88–100. doi: 10.1016/j.cell.2012.02.018
Ye W., Duan Y., Zhang W., Cheng Y., Shi M., Xia X. (2021). Comprehensive analysis of hub mRNA, lncRNA and miRNA, and associated ceRNA networks implicated in grass carp (Ctenopharyngodon idella) growth traits. Genomics 113, 4004–4014. doi: 10.1016/j.ygeno.2021.10.001
Zhang Y., Liu J., Peng L., Ren L., Zhang H., Zou L., et al. (2017). Comparative transcriptome analysis of molecular mechanism underlying gray-to-red body color formation in red crucian carp (Carassius auratus, red var.). Fish Physiol. Biochem. 43, 1387–1398. doi: 10.1007/s10695-017-0379-7
Zhao W., Mazar J., Lee B., Sawada J., Li J., Shelley J., et al. (2016). The long non-coding RNA SPRIGHTLY regulates cell proliferation in primary human melanocytes. J. Invest. Dermatol. 136, 819–828. doi: 10.1016/j.jid.2016.01.018
Zhong A., Gao T. (2022). Characterization of immunomodulatory lncRNAs in the head kidney of yellow catfish (Pelteobagrus fulvidraco) infected by aeromonas hydrophila. Aquacult. Rep. 23, 101091. doi: 10.1016/j.aqrep.2022.101091
Zhou Q., Guo X., Huang Y., Gao H., Xu H., Liu S., et al. (2020). De novo sequencing and chromosomal-scale genome assembly of leopard coral grouper, Plectropomus leopardus. mol. Ecol. Resour. 20, 1403–1413. doi: 10.1111/1755-0998.13207
Zhu X., Hao R., Tian C., Zhang J., Zhu C., Li G. (2021). Integrated transcriptomics and metabolomics analysis of body color formation in the leopard coral grouper (Plectropomus leopardus). Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.726102
Keywords: Plectropomus leopardus, lncRNA, ceRNA network, body color, miRNA
Citation: Hao R, Zhu X, Tian C, Jiang M, Huang Y, Li G and Zhu C (2023) LncRNA–miRNA–mRNA ceRNA network of different body colors in Plectropomus leopardus. Front. Mar. Sci. 10:1170762. doi: 10.3389/fmars.2023.1170762
Received: 21 February 2023; Accepted: 26 April 2023;
Published: 12 May 2023.
Edited by:
Hongyu Ma, Shantou University, ChinaReviewed by:
Xianliang Meng, Yellow Sea Fisheries Research Institute (CAFS), ChinaSong Yang, Sichuan Agricultural University, China
Jie Mei, Huazhong Agricultural University, China
Copyright © 2023 Hao, Zhu, Tian, Jiang, Huang, Li and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunhua Zhu, chz416@163.com