 Hao Wang1†
Hao Wang1† Mingxuan Teng1†
Mingxuan Teng1† Pingping Liu1,2Mingyang Zhao2
Pingping Liu1,2Mingyang Zhao2 Shi Wang1,2,3Jingjie Hu1,2
Shi Wang1,2,3Jingjie Hu1,2 Zhenmin Bao1,2,4
Zhenmin Bao1,2,4 Qifan Zeng1,2,4*
Qifan Zeng1,2,4*
- 1MOE Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 2Key Laboratory of Tropical Aquatic Germplasm of Hainan Province, Sanya Ocean Institute, Ocean University of China, Sanya, China
- 3Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 4Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
The Pacific white shrimp Litopenaeus vannamei is among the top aquatic species of commercial importance around the world. Over the last four decades, the breeding works of L. vannamei have been carried out intensively and have generated multiple strains with improved production and performance traits. However, signatures of domestication and artificial selection across the L. vannamei genome remain largely unexplored. In the present study, we conducted whole genomic resequencing of 180 Pacific white shrimps from two artificially selective breeds and four market-leading companies. A total of 37 million single nucleotide polymorphisms (SNPs) were identified with an average density of 22.5 SNPs/Kb across the genome. Ancestry estimation, principal component analysis, and phylogenetic inference have all revealed the obvious stratifications among the six breeds. We evaluated the linkage disequilibrium (LD) decay in each breed and identified the genetic variations driven by selection. Pairwise comparison of the fixation index (Fst) and nucleotide diversity (θπ) has allowed for mining the genomic regions under selective sweep in each breed. The functional enrichment analysis revealed that genes within these regions are mainly involved in the cellular macromolecule metabolic process, proteolysis, structural molecule activity, structure of the constituent ribosome, and responses to stimulus. The genome-wide SNP datasets provide valuable information for germplasm resources assessment and genome-assisted breeding of Pacific white shrimps, and also shed light on the genetic effects and genomic signatures of selective breeding.
Introduction
Aquaculture produces almost half of the seafood consumed by humans and is becoming one of the fastest-growing food production sectors in the world (FAO, 2020). To match the ever-increasing food demands of the growing population, aquaculture production should increase fivefold in the next three decades (Costello et al., 2020). The recent annual global production of farmed shrimps reached more than 7.7 million tons, representing a value of over 33 billion US dollars (FAO, 2020). Benefitting from technological innovation and policy reforms, shrimp farming has developed steeply from traditional and small-scale activities into a global industry, holding a great promise for enhancing the contribution of aquaculture production to food supply (Lotz, 1992; Briggs et al., 2005).
Pacific white shrimp (Litopenaeus vannamei) is the top shrimp species of commercial importance. The annual global yield of L. vannamei reached 4.4 million tons with a production value of about 26.7 billion USD, accounting for 80% of the total cultured shrimp production (FAO, 2020). The success of Pacific white shrimp aquaculture is largely attributed to a series of breeding programs since the 1970s (Lotz, 1992; FAO, 2011). Genetic improvements in performance traits and disease resistance have achieved remarkable progress over the last decade (Argue et al., 2002; Campos-Montes et al., 2012; Montaldo et al., 2013; Lillehammer et al., 2020). Several specific pathogen-free (SPF) L. vannamei breeds, with superior health and efficiency adapted to distinct farming conditions, have been cultivated and shipped worldwide (Briggs et al., 2005; Fletcher, 2020; Ren, 2020). Benefitting from the rapid development of high-throughput sequencing and genotyping technologies, several causative genes responsible for phenotypic variations of L. vannamei have been reported (Wang et al., 2019; Zhang X. et al., 2019; Lyu et al., 2021). For instance, genes encoding class C scavenger receptor (SRC), deoxycytidylate deaminase (dCMPD), and non-receptor protein tyrosine kinase (NPTK) were identified as potential genes related to the growth rate (Wang et al., 2019; Lyu et al., 2021). Whereas, genomic signatures underlying artificial selection remain largely unexplored. Dissection of the high-resolution genomic variation map is essential for a better understanding of genetic diversity, population structure, and genomic features during generations of selection.
In the present study, to profile the genome signatures of selection in L. vannamei, we performed a whole genomic resequencing analysis of shrimps from two artificially selective breeds (Renhai No. 1 and Kehai No. 1) and broodstocks of four market-leading companies (Benchmark Genetics, Charoen Pokphand, Shrimp Improvement Systems, and Top Aquaculture Technology). The genome-wide single nucleotide polymorphisms (SNP) datasets revealed the population structure and genetic effects during the breeding process, providing valuable information for germplasm resource assessment and genome-assisted breeding of Pacific white shrimps.
Materials and Methods
Sampling and DNA Extraction
A total of 180 samples from the L. vannamei broodstock of Renhai No. 1 (RH), Kehai No. 1 (KH), Benchmark Genetics (BMK), Charoen Pokphand (CP), Shrimp Improvement Systems (SIS), and Top Aquaculture Technology (TA) were collected from Hairen Aquatic Seed Industry Technology Co., Ltd (Hebei, China) (Supplementary Table 1). The muscle samples of each individual were collected for DNA extraction and genome resequencing. Genomic DNA was extracted using the TIANamp Marine Animal DNA Kits (TIANGEN Biotech Co., Ltd. Beijing, China). Paired-end sequencing libraries, with an insert size of 250 to 350 bp, were constructed by the VAHTS Universal Plus DNA Library Prep Kit for MGI (Vazyme Biotech Co., Ltd., Nanjing, China) in their lab, and sequenced on an MGI DNBSEQ-T7 system (BGI Genomics Co., Ltd., Shenzhen, China). All raw sequencing data were subject to quality control1. Low-quality bases and reads were trimmed by Trimmomatic with parameters “LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36” (Bolger et al., 2014).
Sequencing and Genotyping
The reference genome was downloaded from the National Center for Biotechnology Information (NCBI) with the accession of GCA_003789085.1 (Zhang X. et al., 2019). We anchored the contigs into scaffolds by the guidance of a genetic linkage maps (Yu et al., 2015; Jones et al., 2017), and all trimmed reads were aligned to this genomic assembly using the BWA (version 2.3.4.1), with the default parameters (Li and Durbin, 2009). The Binary Alignment Map (BAM) files were imported to the samtools (v0.1.19) for reference index building, format conversion, and reads sorting (Li et al., 2009). The Picard2 (v1.92) and the sambamba (v0.8.0) were used to assign read group information (Tarasov et al., 2015). PCR and optical duplicates were marked and filtered using GATK (v4.1). Variants and haplotypes were identified using the HaplotypeCaller algorithm in Genomic Varient Call Format (GVCF) mode (Mckenna et al., 2010). The GenotypeGVCFs were used subsequently for joint genotyping.
To keep the most reliable SNPs for subsequent analysis, variant sites were marked and filtered by the vcffilter with the following criteria: low quality score (GQ < 20); low quality by depth score (QD < 10); high Fisher strand score (FS > 10); low mapping quality (MQ < 40); low read position rank sum score (ReadPosRankSum < 8); high strand odds ratio (SOR > 4); and low mapping quality rank-sum score (MQRankSum < 12.5). The SNP loci with multiple alleles, low minor allele frequency (MAF < 0.05), and missing genotypes (max-missing < 1) were also removed using the VCFtools (v0.1.16) (Danecek et al., 2011). The potential effect of each SNP was annotated using the SNPEff (Cingolani et al., 2012).
Population Structure Analysis
Principal component analysis (PCA) was performed using the PLINK (Purcell et al., 2007). Ancestry estimation and population structure were analyzed using the ADMIXTURE (Alexander et al., 2009). Pairwise fixation index (Fst) was calculated by VCFtools with parameters –fst-window-size 50,000 and –fst-window-step 10,000 (Danecek et al., 2011). The genomic observed heterozygosity, expected heterozygosity, and inbreeding coefficient were evaluated by the VCFtools with parameter –het (Danecek et al., 2011). For the phylogenetic analysis, representative SNP markers were extracted using the VCFtools with parameter –thin 10,000 to mitigate the effects of LD, homozygous sites were removed from the alignment file. Maximum likelihood phylogenetic inference was carried out by IQ-TREE (Nguyen et al., 2015).
The SNPs with specific alleles in one population were defined as population-specific SNPs (ps-SNPs). If the frequency of the population-specific allele was greater than 50% in a population, the SNPs were defined as common population-specific SNPs (cps-SNPs). The LD decay was estimated for each population using the PopLDdecay (Zhang C. et al., 2019). Genomic inbreeding coefficients FROH were calculated according to the following equation, with a minimum window of 100 kb (McQuillan et al., 2008):
Selective Sweep Analysis
The pairwise Fst and the ratio of genetic polymorphisms (θπ) were calculated using a 50 kb window and a 10 kb step between each breed. Genomic regions with signals of differences in the spectrum of genetic polymorphisms were identified as regions that are potentially under selection. Empirical cut-offs for Fst and θπ ratio were set as the top 2% largest and the top 5% largest or smallest, respectively (Li L. et al., 2018; Wang et al., 2021). Candidate genes under selection were defined as within or overlapping with the regions showing signals of selection. Candidate genes were characterized with Gene Ontology (GO) enrichment analysis implemented in the EnrichPipeline based on Fisher’s exact test (Huang et al., 2009). The GO terms with a P-value < 0.05 were considered significantly enriched.
Results
SNP Identification and Annotation
An average of 164 million 300 bp paired-end reads were generated for each library, representing ∼18.7 × coverage. After quality control, approximately 80% of reads could be aligned to the reference genome (∼12.34 × mean unique coverage) and used for variant calling. A total of 37,308,098 SNPs were identified in the six populations. The number of SNPs in the chromosomes ranged from 182,599 to 1,274,977, and the density was between 20.60 SNPs/Kb and 24.58 SNPs/kb. Of these, 4,509,526 reliable SNPs were kept after the removal of low-quality samples and those within repetitive regions of missing genotypes. The transitions/transversions ratio in this biallelic SNPs set was found to be 1.63, with 1,117,185,030 transitions and 72,199,828 transversions, respectively. The SNPs were unevenly distributed across the genome, with over 10.56% (1,460,366) located in the intergenic regions, about 55.43% (7,662,564) located in the introns, and only 2.283% (238,482) located in the exons (Table 1). More than 73.89% of the SNPs (221,021) were silent mutations, the remaining 25.82 (77,219) and.30% of SNPs (886) could result in missense and non-sense mutations, respectively (Table 1).
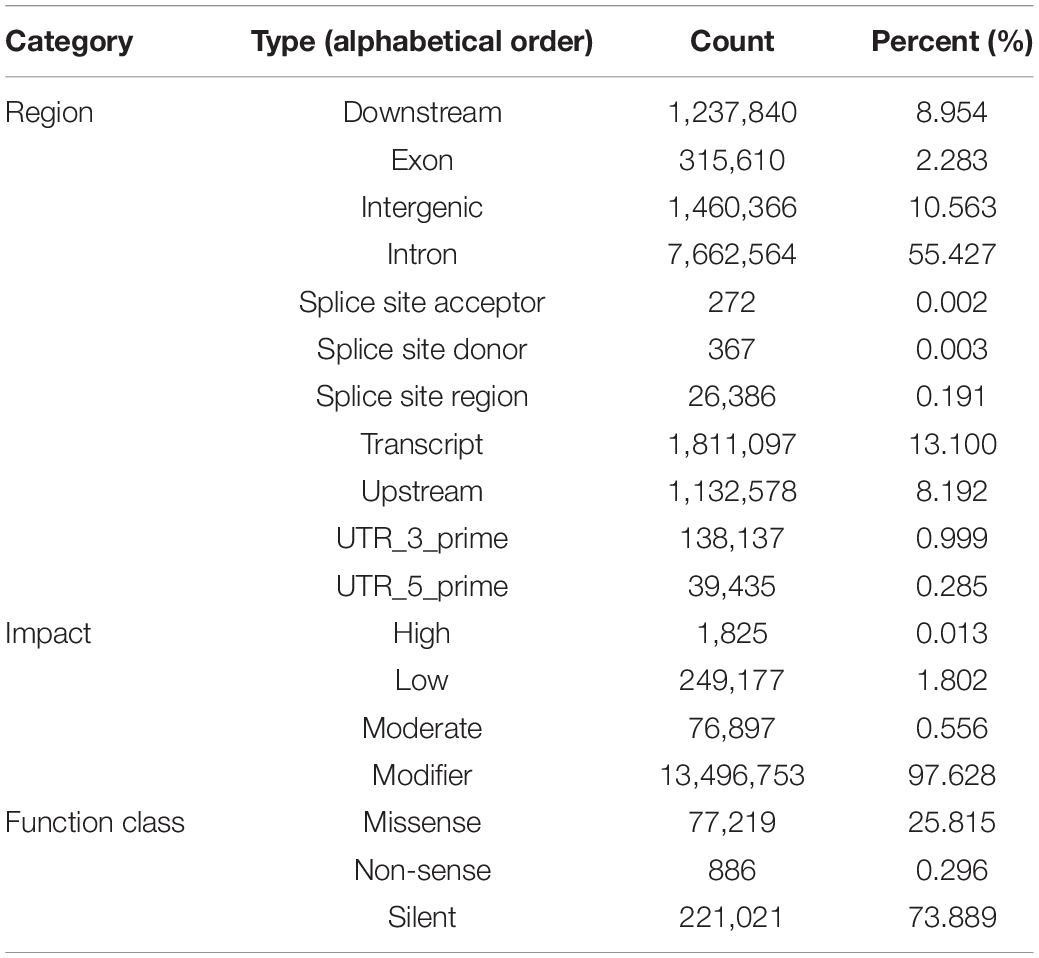
Table 1. SNP annotation by genomic region, impact and function class.
Population Structure
The population structure was inferred based on the genome-wide biallelic SNPs. As shown by the PCA, the first three components cumulatively explained 24.56% of the variance (Figure 1A and Supplementary Figures 1, 2). Samples from the six populations were grouped into three major clusters, which largely restored the sampling sources. Obvious stratifications were observed among BMK, SIS, and the Asian breeds (RH, KH, TA, and CP). Although RH, KH, TA, and CP were distributed closely on the first two components, they possessed distinct population structures (Figure 1 and Supplementary Figure 2). The pairwise comparison revealed that the Fst values among the six populations ranged from 0.0005 to 0.13415, and that BMK had the largest genetic distances from the others. The KH and TA share a relatively higher similarity in genetic structure (Figure 1B). Populations defined by estimated genetic ancestries also confirmed the distribution of samples, where six ancestral populations were inferred to be the optimal scenario by cross-validation (Figure 1C and Supplementary Figure 3). The phylogenetic relationships were reconstructed via the maximum likelihood inference. Except for two samples of TA clustered together with CP, samples from each population formed monophyletic clades, which is consistent with the population structure inferred by PCA and ancestral estimation (Figure 1D).
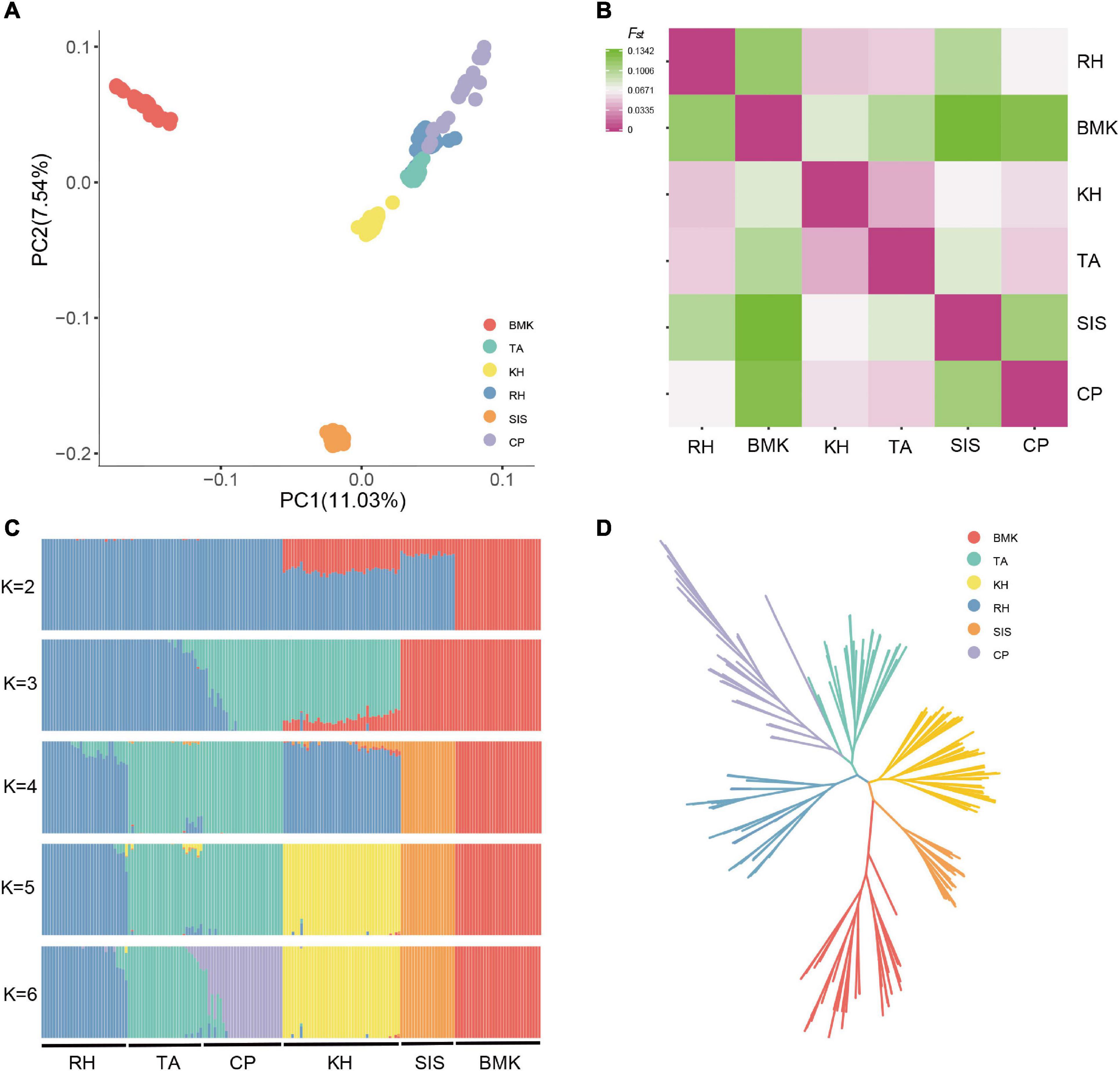
Figure 1. Population structure analysis of the six populations by principal component analysis (PCA) (A), Pairwise Fst distance (B), ancestral estimation (C), and phylogenetic inference (D).
Genome Selection Signatures
As shown in Figure 2A, population-specific SNPs were identified in each breed. The BMK and SIS possessed 83,273 and 15,980 cps-SNPs, respectively, which are much larger than that of the other four breeds. The RH, KH, TA, and CP only had 3,153, 1,889, 485, and 836 cps-SNPs, respectively (Table 2). We evaluated the genetic diversity by the observed heterozygosity, expected heterozygosity, and inbreeding coefficient (Supplementary Data Sheet 1). These six populations have an average genomic observed heterozygosity of 0.122, 0.137, 0.144, 0.147, 0.149; and 0.156 in BMK, CP, TA, RH, KH, and SIS. The extent of genome-wide LD decay was measured against the physical distance in each population (Figure 2B). Asian breeds (RH, KH, TA, and CP) typically exhibited an overall lower level of LD than the others. Their LD levels (r2) fell below 0.1 at less than 15Kb, whereas, the physical distances reached over 70 kb in BMK and SIS. Analysis of runs of homozygosity (ROH) revealed that BMK and CP reserved higher levels of inbreeding than the others (Figure 2C and Supplementary Figure 4). Homozygous segments larger than 1Mb were mostly identified in these two populations. Despite that the genome-wide distribution of ROH is distinct in each population, peaks in specific regions of chromosomes 10, 15, and 42 were identified in each population (Figure 2D).
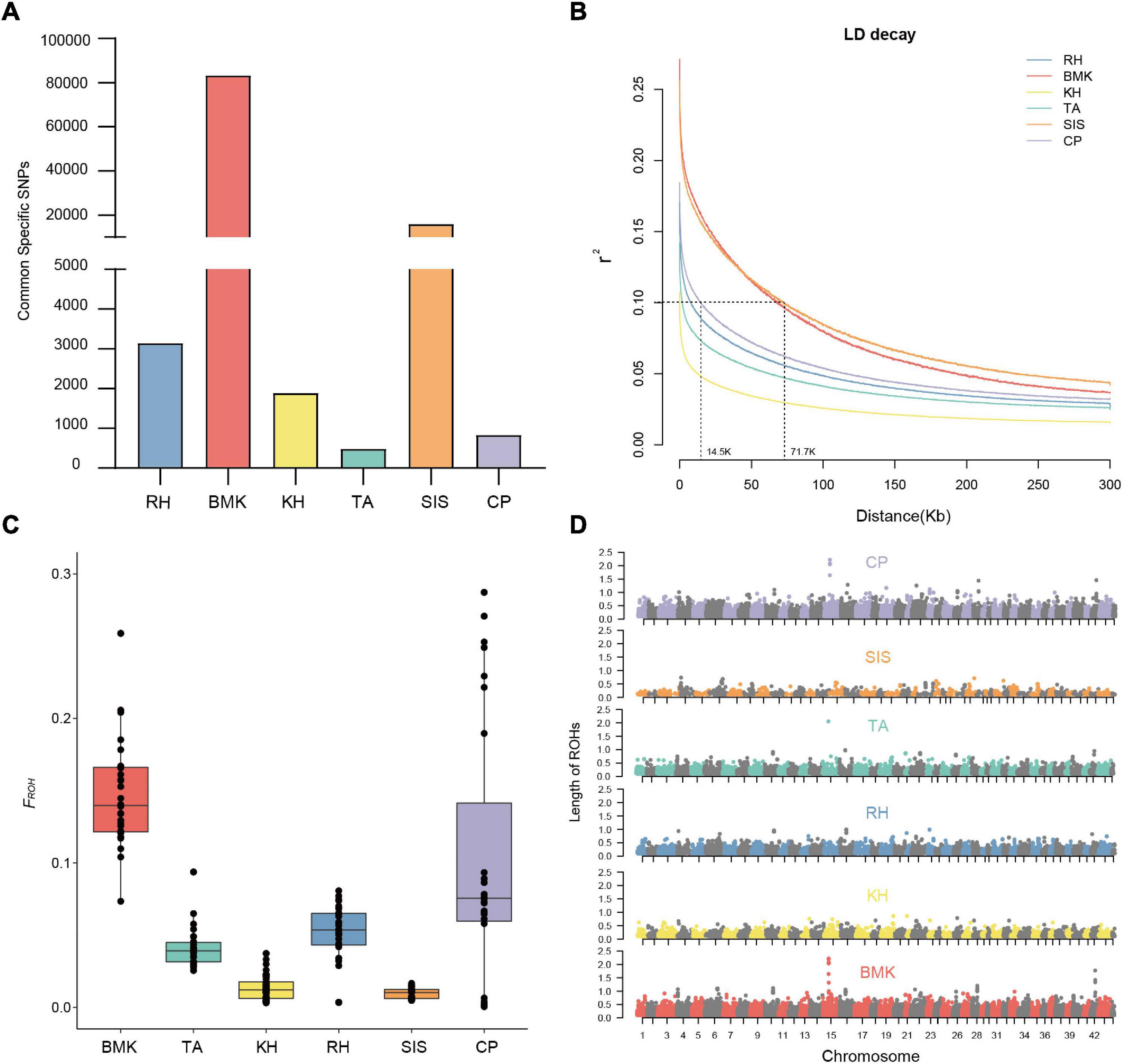
Figure 2. (A) cps-SNPs among the six populations. (B) linkage disequilibrium (LD) decay at a distance of 0–300Kb. (C) Box plot of FROH statistic. (D) Genome-wide distribution of ROHs in the six populations.
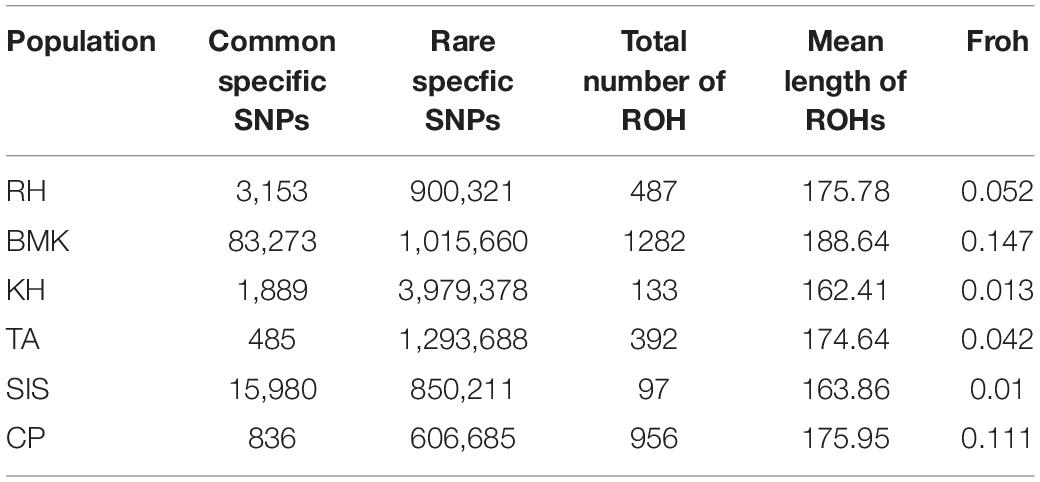
Table 2. Genetic statistical summary of the six population.
Genes Under the Selective Sweep
The pairwise Fst and θπ ratio were calculated to scan the genomic regions with genetic signals of divergence. The top 5% of the windows, with high values of Fst and differentiation of polymorphism frequency spectrum, were identified as regions that are potentially under selection (Figure 3A and Supplementary Figure 5). Genome-wide selective sweeps in BMK were illustrated in Figure 3A. We detected 271, 440, 257, 269, 431, and 429 candidate genes under a strong selection from 166,258 genomic windows in RH, BMK, KH, TA, SIS, and CP, respectively (Supplementary Data Sheet 2). Interestingly, 206 genes were under selection in more than one population (Supplementary Data Sheet 2). Function enrichment analysis of these candidate genes provided 631 significant enriched categories (Supplementary Data Sheet 3). For example, genes under selection in BMK were enriched in response to stimulus, carbohydrate transport, and several biological processes (Figure 3B). Selective genes in RH were mainly enriched in trialkyl sulfonium hydrolase activity, protein modification by a small protein conjugation, and phospholipid binding. Selective genes in KH were mainly enriched in the structural constituent of ribosome, structure molecule activity, and cellular macromolecule metabolic process. Selective genes in TA were enriched in the pyruvate kinase activity, peptidase inhibitor activity, and ATP metabolic process. Selective genes in SIS were enriched in the ribonuclease H2 complex, GTP cyclohydrolase I activity, and cellular macromolecule metabolic process. Selective genes in CP were mainly enriched in the structural molecule activity, regulation of ATPase activity, and 5′-deoxynucleotidase activity (please check Supplementary Data Sheet 3 for detailed in section “Results”). Notably, B3GT5, CDC42, PXDN, and several genes that play important roles in cellular macromolecule metabolic process, proteolysis, and responses to stimulus were identified under selection in more than one population (Figure 4).
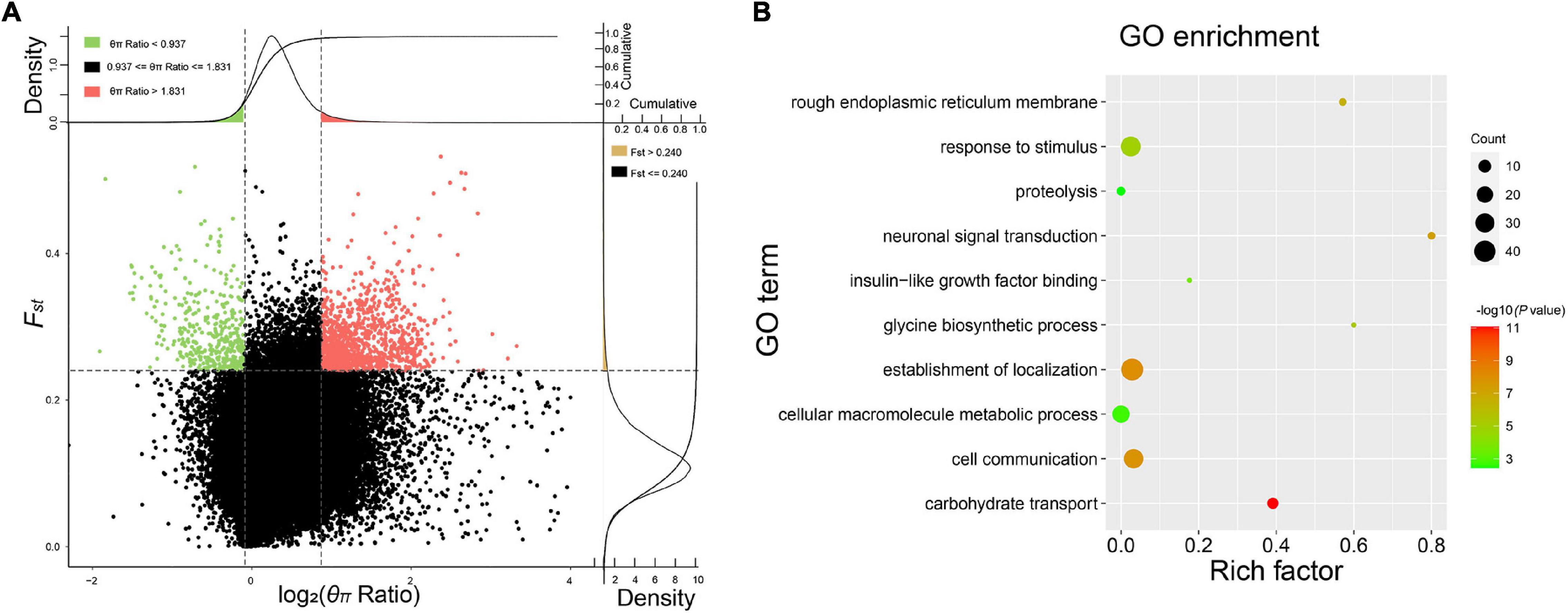
Figure 3. Selective sweep and Gene Ontology (GO) enrichment analysis of the BMK samples. (A) Distribution of θπ ratios (θπ reference population/θπ selective breed) and Fst values calculated in 50 kb window with a 10 kb step. Windows with the 5% largest or smallest of θπ ratio, and the top 2% largest Fst were highlighted as regions under selection. (B) Enriched GO terms of genes under selection.
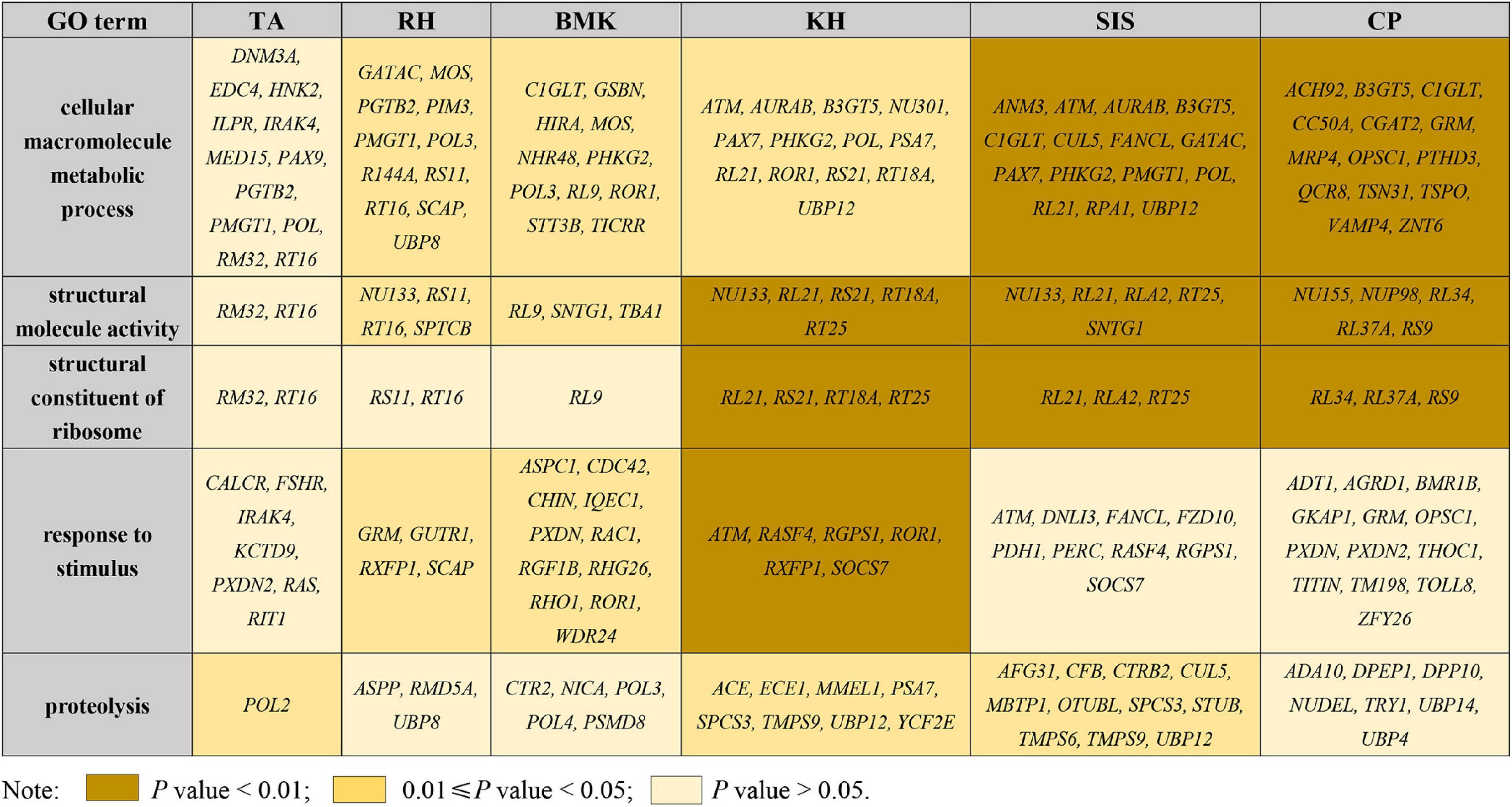
Figure 4. Shared enriched GO terms of selective genes among each breed.
Discussion
Genetic breeding of L. vannamei has achieved remarkable progress and cultivated multiple novel breeds with significant improvement in growth and adaptation to modern farming conditions after generations of artificial selection (Argue et al., 2002; Campos-Montes et al., 2012; Montaldo et al., 2013; Lillehammer et al., 2020). As exploring the full range of genetic diversity has practical implications for the management of germplasm resources and breeding programs, several studies have been carried out to decipher the spectrum of genetic polymorphism (Castillo-Juarez et al., 2015; Garcia et al., 2021). Whereas, these studies were mainly based on a limited number of markers by targeted or by reduced-representation genotyping (Li and Wu, 2003; Perez-Enriquez et al., 2009, 2018b; Garcia et al., 2021; Prithvisagar et al., 2021). The genome-wide landscape of variations shaped by artificial selection remains largely unexplored. In this study, we conducted whole genomic resequencing of L. vannamei and investigated the distribution of SNP sites among six populations. In total, 4.5 million high confidence SNPs were identified across the genome, reaching an average density of 22.5 SNPs/kb across the genome. Interestingly, population structures inferred by PCA, ancestry estimation, and phylogenetic analysis provided strong evidence for interpopulation stratifications. Considering that the founders of L. vannamei breeds were originally introduced from native populations in the Pacific coast from Mexico to Northern Peru in the late 1970s and 1980s (Wyban and Sweeney, 1991; Rosenberry, 2000), it was suggested that genetic drift and artificial selection could potentially contribute to the differentiation. The genomic heterozygosity of the six populations ranges from 0.122 to 0.156. The BMK and CP exhibit the lowest level of heterozygosity and high inbreeding coefficient, which is consistent with the ROH analysis. The observed heterozygosity levels are relatively lower than previous estimations (De Freitas and Galetti, 2005; Artiles et al., 2011; Rezaee et al., 2016; Perez-Enriquez et al., 2018a). The decay of LD provides important information about the historical recombination of population and is widely used to understand the evolutionary and demographic processes (Slatkin, 2008). Our results revealed that the overall LD levels of the four Asian populations are comparable to the breeding population of Ecuador (Garcia et al., 2021), while LD at long-range was stronger in BMK and SIS. Despite the biases in sampling size and the relatedness among individuals, LD extension could be attributed to demographic and biological factors, such as the admixture, mutation, and fluctuation of the effective population size (Qanbari, 2019). As the natural distribution of L. vannamei is restricted in America, we presumed that the observed long-range LD in American strains may be introduced by recurrent inclusion of wild strains, which was absent in the Asian populations (Moss et al., 2012). This phenomenon was also reported in salmonids, where admixture is the major factor contributing to long-range LD (Odegard et al., 2014; Barria et al., 2018; Vallejo et al., 2018).
The Fst values and θπ ratios have been broadly used to identify genetic differentiation and genome-wide selective sweeps (Barreiro et al., 2008; Li L. et al., 2018). In the present study, we revealed numerous breeding signatures reflecting the complex genomic architectures from six breeds with distinct genetic backgrounds. A series of positive selective genes, such as B1GT3, C1GLT, NU133, PXDN, SOCS7, and UBP8, etc., have been identified in more than one breed. Previous studies in arthropods have revealed that many of these genes are involved in important physiological and biochemical functions. For example, PXDN, SCAP (SREPF Chaperone), SOCS7, and TOLL8 are related to antiviral or antibacterial immunity in Pacific white shrimps and other Crustaceans (Du et al., 2013; Wang et al., 2016; Li H. et al., 2018; Aweya et al., 2020). The CDC42 plays an important role in Reactive oxygen species (ROS) production and apoptosis by Mitogen-activated protein kinases (MAPK) pathway in L. vannamei (Peng et al., 2015). As selective breeding of these populations has been carried out for generations, the identified selective genes seem to coincide with the improved performance traits. Previous studies revealed that target genotyping of trait-linked markers can improve the accuracy of breeding value estimation (Li et al., 2017). Thus, we expect that these loci could serve as important markers for genetic improvement via targeted genotyping.
Conclusion
We conducted a population genetic study on six L. vannamei breeds by whole genomic resequencing and identified over 37 million SNPs. Population structure revealed by PCA, ancestral estimation, and phylogenetic analysis confirmed the stratification among these populations. Selective sweep analysis detected that 206 genes were under selection in more than one population. Among them, several candidate genes act as key regulators in cellular macromolecule metabolic process, proteolysis, and responses to stimulus, which could be responsible for the improved economic traits and adaptation to modern aquaculture. This study not only provides valuable information for germplasm resources assessment and genome-assisted breeding of L. vannamei, but also sheds light on the genetic effects and genomic signatures of selective breeding.
Data Availability Statement
The data presented in the study are deposited in the repository of Aquaculture Molecular Breeding Platform (AMBP, http://mgb.qnlm.ac).
Author Contributions
ZB, QZ, JH, and SW designed the experiments. MT, MZ, and PL collected the samples. HW, MT, and PL performed the experiments. HW, MT, and MZ analyzed the data. HW and QZ wrote the manuscript. All authors have read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank graduate students Jing Liu, Baojun Zhao, Hongyu Lv, and Yantong Cai for their assistance in sample processing. We acknowledge the grant support from the Project of Sanya Yazhouwan Science and Technology City Management Foundation (SKJC-KJ-2019KY01), National Key Research and Development Program of China (2021YFD1200805), Major Science and Technology Program of Hainan Province (ZDKJ2021017), and Agricultural Variety Improvement Project of Shandong Province (2019LZGC014).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.844597/full#supplementary-material
Footnotes
References
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Argue, B. J., Arce, S. M., Lotz, J. M., and Moss, S. M. (2002). Selective breeding of Pacific white shrimp (Litopenaeus vannamei) for growth and resistance to Taura Syndrome Virus. Aquaculture 204, 447–460.
Artiles, A., Rodríguez, I., Pérez, A., Pérez, L., and Espinosa, G. (2011). Low genetic variability in the fifth introduction of Litopenaeus vannamei in Cuba, as estimated with microsatellite markers. Biotecnología Aplicada 28, 147–150.
Aweya, J. J., Zheng, X., Zheng, Z., Wang, W., Fan, J., Yao, D., et al. (2020). The sterol regulatory element binding protein homolog of Penaeus vannamei modulates fatty acid metabolism and immune response. Biochimica et Biophysica Acta - Mol. Cell Biol. Lipids 1865:158757. doi: 10.1016/j.bbalip.2020.158757
Barreiro, L. B., Laval, G., Quach, H., Patin, E., and Quintana-Murci, L. (2008). Natural selection has driven population differentiation in modern humans. Nat. Genet. 40, 340–345. doi: 10.1038/ng.78
Barria, A., Lopez, M. E., Yoshida, G., Carvalheiro, R., Lhorente, J. P., and Yanez, J. M. (2018). Population genomic structure and genome-wide linkage disequilibrium in farmed atlantic salmon (Salmo salar L.) Using Dense SNP Genotypes. Front. Genet. 9:649. doi: 10.3389/fgene.2018.00649
Bolger, A., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Briggs, M., Funge-Smith, S., Subasinghe, R. P., and Phillips, M. (2005). “Introductions and movement of two penaeid shrimp species in Asia and the Pacific,” in FAO Fisheries Technical Paper. Rome: FAO.
Campos-Montes, G. R., Montaldo, H. H., Martínez-Ortega, A., Jiménez, A. M., and Castillo-Juárez, H. (2012). Genetic parameters for growth and survival traits in Pacific white shrimp Penaeus (Litopenaeus) vannamei from a nucleus population undergoing a two-stage selection program. Aquacult. Int. 21, 299–310.
Castillo-Juarez, H., Campos-Montes, G. R., Caballero-Zamora, A., and Montaldo, H. H. (2015). Genetic improvement of Pacific white shrimp [Penaeus (Litopenaeus) vannamei]: perspectives for genomic selection. Front. Genet. 6:93. doi: 10.3389/fgene.2015.00093
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Tung, N., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Costello, C., Cao, L., Gelcich, S., Cisneros-Mata, M. A., Free, C. M., Froehlich, H. E., et al. (2020). The future of food from the sea. Nature 588, 95–100.
Danecek, P., Auton, A., Abecasis, G. R., Albers, C. A., Banks, E., Depristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
De Freitas, P. D., and Galetti, P. M. (2005). Assessment of the genetic diversity in five generations of a commercial broodstock line of Litopenaeus vannamei shrimp. African J. Biotechnol. 4, 1362–1367.
Du, Z. Q., Ren, Q., Huang, A. M., Fang, W. H., Zhou, J. F., Gao, L. J., et al. (2013). A novel peroxinectin involved in antiviral and antibacterial immunity of mud crab, Scylla paramamosain. Mol. Biol. Rep. 40, 6873–6881. doi: 10.1007/s11033-013-2805-y
Fletcher, R. (2020). Trends in the Specific Pathogen-Free Shrimp Trade Revealed. The Fish Site. Available online at: https://thefishsite.com/articles/trends-in-the-specific-pathogen-free-shrimp-trade-revealed
Garcia, B. F., Bonaguro, A., Araya, C., Carvalheiro, R., and Yanez, J. M. (2021). Application of a novel 50K SNP genotyping array to assess the genetic diversity and linkage disequilibrium in a farmed Pacific white shrimp (Litopenaeus vannamei) population. Aquacult. Rep. 20:100691.
Huang, D., Sherman, B. T., and Lempicki, R. A. (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1093/nar/gkn923
Jones, D. B., Jerry, D. R., Khatkar, M. S., Raadsma, H. W., Steen, H. V., Prochaska, J., et al. (2017). A comparative integrated gene-based linkage and locus ordering by linkage disequilibrium map for the Pacific white shrimp, Litopenaeus vannamei. Sci. Rep. 7:10360. doi: 10.1038/s41598-017-10515-7
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, H., Su, G., Jiang, L., and Bao, Z. (2017). An efficient unified model for genome-wide association studies and genomic selection. Genet. Select. Evol. 49:64. doi: 10.1186/s12711-017-0338-x
Li, H., Yin, B., Wang, S., Fu, Q., Xiao, B., Lu, K., et al. (2018). RNAi screening identifies a new Toll from shrimp Litopenaeus vannamei that restricts WSSV infection through activating Dorsal to induce antimicrobial peptides. PLoS Pathog. 14:e1007109. doi: 10.1371/journal.ppat.1007109
Li, L., Li, A., Song, K., Meng, J., Guo, X., Li, S., et al. (2018). Divergence and plasticity shape adaptive potential of the Pacific oyster. Nat. Ecol. Evol. 2, 1751–1760. doi: 10.1038/s41559-018-0668-2
Li, Z. B., and Wu, Z. Q. (2003). Study on genetic diversity of cultivated populations in 4 species of shrimps. Acta Oceanol. Sinica 22, 97–101.
Lillehammer, M., Bangera, R., Salazar, M., Vela, S., Erazo, E. C., Suarez, A., et al. (2020). Genomic selection for white spot syndrome virus resistance in whiteleg shrimp boosts survival under an experimental challenge test. Sci. Rep. 10:20571. doi: 10.1038/s41598-020-77580-3
Lotz, J. (1992). “Developing specific-pathogen-free (SPF) animal populations for use in aquaculture: a case study for IHHN virus of penaeid shrimp,” in Diseases of Cultured Penaeid Shrimp in Asia and the United States, eds W. Fulks and K. L. Main 269–284. New York, NY: Springer.
Lyu, D., Yu, Y., Wang, Q., Luo, Z., Zhang, Q., Zhang, X., et al. (2021). Identification of growth-associated genes by genome-wide association study and their potential application in the breeding of pacific white shrimp (Litopenaeus vannamei). Front. Genet. 12:611570. doi: 10.3389/fgene.2021.611570
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
McQuillan, R., Leutenegger, A. L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Human Genet. 83, 359–372. doi: 10.1016/j.ajhg.2008.08.007
Montaldo, H. H., Castillo-Juárez, H., Campos-Montes, G., and Pérez-Enciso, M. (2013). Effect of the data family structure, tank replication and the statistical model, on the estimation of genetic parameters for body weight at 28 days of age in the Pacific white shrimp (Penaeus (Litopenaeus) vannamei Boone, 1931). Aquacult. Res. 44, 1715–1723.
Moss, S. M., Moss, D. R., Arce, S. M., Lightner, D. V., and Lotz, J. M. (2012). The role of selective breeding and biosecurity in the prevention of disease in penaeid shrimp aquaculture. J. Inverteb. Pathol. 110, 247–250. doi: 10.1016/j.jip.2012.01.013
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Odegard, J., Moen, T., Santi, N., Korsvoll, S. A., Kjoglum, S., and Meuwissen, T. H. (2014). Genomic prediction in an admixed population of Atlantic salmon (Salmo salar). Front. Genet. 5:402. doi: 10.3389/fgene.2014.00402
Peng, T., Wang, W. N., Gu, M. M., Xie, C. Y., Xiao, Y. C., Liu, Y., et al. (2015). Essential roles of Cdc42 and MAPK in cadmium-induced apoptosis in Litopenaeus vannamei. Aquatic Toxicol. 163, 89–96. doi: 10.1016/j.aquatox.2015.03.023
Perez-Enriquez, R., Hernandez-Martinez, F., and Cruz, P. (2009). Genetic diversity status of White shrimp Penaeus (Litopenaeus) vannamei broodstock in Mexico. Aquaculture 297, 44–50.
Perez-Enriquez, R., Medina-Espinoza, J. A., Max-Aguilar, A., and Saucedo-Barron, C. J. (2018a). Genetic tracing of farmed shrimp (Decapoda: Penaeidae) in wild populations from a main aquaculture region in Mexico. Revista De Biologia Tropical 66, 381–393.
Perez-Enriquez, R., Robledo, D., Houston, R. D., and Llera-Herrera, R. (2018b). SNP markers for the genetic characterization of Mexican shrimp broodstocks. Genomics 110, 423–429. doi: 10.1016/j.ygeno.2018.10.001
Prithvisagar, K. S., Kumar, B. K., Kodama, T., Rai, P., Iida, T., Karunasagar, I., et al. (2021). Whole genome analysis unveils genetic diversity and potential virulence determinants in Vibrio parahaemolyticus associated with disease outbreak among cultured Litopenaeus vannamei (Pacific white shrimp) in India. Virulence 12, 1936–1949. doi: 10.1080/21505594.2021.1947448
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Human Genet. 81, 559–575. doi: 10.1086/519795
Qanbari, S. (2019). On the extent of linkage disequilibrium in the genome of farm animals. Front. Genet. 10:1304. doi: 10.3389/fgene.2019.01304
Ren, S. (2020). Genetic Improvement of the Pacific White Shrimp (Penaeus vannamei) in China. Doctor of Philosophy. Queensland: University of Technol.
Rezaee, S., Farahmand, H., and Nematollahi, M. A. (2016). Genetic diversity status of Pacific white shrimp (Litopenaeus vannamei) using SSR markers in Iran. Aquacult. Int. 24, 479–489.
Slatkin, M. (2008). Linkage disequilibrium–understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9, 477–485. doi: 10.1038/nrg2361
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J., and Prins, P. (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. doi: 10.1093/bioinformatics/btv098
Vallejo, R. L., Silva, R. M. O., Evenhuis, J. P., Gao, G., Liu, S., Parsons, J. E., et al. (2018). Accurate genomic predictions for BCWD resistance in rainbow trout are achieved using low-density SNP panels: Evidence that long-range LD is a major contributing factor. J. Animal Breed. Genet. 135, 263–274. doi: 10.1111/jbg.12335
Wang, H., Lv, J., Zeng, Q., Liu, Y., Xing, Q., Wang, S., et al. (2021). Genetic differentiation and selection signatures in two bay scallop (Argopecten irradians) breeds revealed by whole-genome resequencing analysis. Aquaculture 543:736944.
Wang, Q., Yu, Y., Zhang, Q., Zhang, X., Yuan, J., Huang, H., et al. (2019). A novel candidate gene associated with body weight in the pacific white shrimp litopenaeus vannamei. Front. Genet. 10:520. doi: 10.3389/fgene.2019.00520
Wang, S., Song, X., Zhang, Z., Li, H., Lu, K., Yin, B., et al. (2016). Shrimp with knockdown of LvSOCS2, a negative feedback loop regulator of JAK/STAT pathway in Litopenaeus vannamei, exhibit enhanced resistance against WSSV. Dev. Comp. Immunol. 65, 289–298. doi: 10.1016/j.dci.2016.07.021
Wyban, J. A., and Sweeney, J. N. (1991). Intensive shrimp production technology: the Oceanic Institute shrimp manual. Honolulu: Hawaii Institute.
Yu, Y., Zhang, X., Yuan, J., Li, F., Chen, X., Zhao, Y., et al. (2015). Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific white shrimp Litopenaeus vannamei. Sci. Rep. 5:15612. doi: 10.1038/srep15612
Zhang, C., Dong, S., Xu, J., He, W., and Yang, T. (2019). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. doi: 10.1093/bioinformatics/bty875
Keywords: Pacific white shrimp, SNP, population structure, linkage disequilibrium, selective sweep
Citation: Wang H, Teng M, Liu P, Zhao M, Wang S, Hu J, Bao Z and Zeng Q (2022) Selection Signatures of Pacific White Shrimp Litopenaeus vannamei Revealed by Whole-Genome Resequencing Analysis. Front. Mar. Sci. 9:844597. doi: 10.3389/fmars.2022.844597
Received: 28 December 2021; Accepted: 20 January 2022;
Published: 24 March 2022.
Edited by:
Anthony Lee Dellinger, Kepley BioSystems Inc., United StatesReviewed by:
Zhongkai Wang, Qingdao Agricultural University, ChinaQuanchao Wang, Yantai Institute of Coastal Zone Research (CAS), China
Chaozheng Li, Sun Yat-sen University, China
Copyright © 2022 Wang, Teng, Liu, Zhao, Wang, Hu, Bao and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qifan Zeng, emVuZ3FpZmFuQG91Yy5lZHUuY24=
†These authors have contributed equally to this work