 Fangrui Lou
Fangrui Lou Li Wang2,3
Li Wang2,3 Linlin Zhao
Linlin Zhao Zhichuang Lu
Zhichuang Lu- 1School of Ocean, Yantai University, Yantai, China
- 2School of Municipal and Environmental Engineering, Shenyang Jianzhu University, Shenyang, China
- 3First Institute of Oceanography, Ministry of Natural Resources, Qingdao, China
- 4Qingdao Polar Haichang Ocean Park, Qingdao, China
- 5Liaoning Ocean and Fisheries Science Research Institute, Dalian, China
Rhincodon typus is a keystone and indicator species in marine ecosystems. Meanwhile, R. typus has been listed on the IUCN red list of vulnerable species. Here we used ONT platform to determine the full-length (FL) transcriptome of R. typus and obtained 14,930 FL transcripts. Among all FL transcripts, 14,915 transcripts were covered 11,892 genetic loci and 1,642 novel genetic loci were further found. Meanwhile, we identified 714 novel transcripts by compared FL transcripts with the R. typus genome. Based on FL transcripts, we also predicted the distribution patterns of ASs, LncRNAs, polyAs, CDSs and methylation sites on FL transcriptome of R. typus. Furthermore, a total of 31,021 (97.86%) CDSs can obtained annotation information. Overall, our work firstly provided the FL transcriptome and these sequences complete the annotated R. typus genome information. Furthermore, these information are a potential resource to study biological processes of R. typus.
Introduction
The whale shark, Rhincodon typus is the exclusive species of the genus Rhincodon and it belonging to the family Rhincodontidae under the order Orectolobiformes (Smith, 1829). Rhincodon typus is widely distributed in the tropical and temperate seas from latitude 30°N to 35°S and has rarely been found in waters with surface temperature below 21°C (Compagno, 2001; Colman, 2005; Rowat and Brooks, 2012; Sequeira et al., 2014). As the largest extant fish, the maximum length and weight of R. typus can reach 20 meters and 42 tons, respectively (Hsu et al., 2014). The head of R. typus is flat and wide, and there are many checkerboard shaped white spots and horizontal stripes on the dorsal part (Compagno, 2001). Previous study has demonstrated that the feeding habits of bait at the food chain bottom and the unique branchial cleft structure enables the R. typus to have a filter-feeding lifestyle (Stevens, 2007; Nozu et al., 2015). In conclusion, R. typus plays an important role in marine ecosystem due to its special biological characteristics and lifestyle, which can be used as an indicator of marine ecosystem stability.
The life-history strategy of R. typus has been researched extensively over the years (Weber et al., 2020). Earlier studies have found that the R. typus grow slowly and reach maturity at approximately 30 years old, and its maximum lifespan estimated at 70 to 80 years (Hsu et al., 2014). The R. typus is ovoviviparous and the pregnancy interval is also longer. Together, the life-history strategy of R. typus can be considered as K-selection (Cavanagh et al., 2003). Longer lifespan of R. typus have been suspected to be benefit from metabolic rate regulation caused by K-selection life-history strategy (including decreased growth rate, longer generational time, and increased body size) (Weber et al., 2020). However, K-selection life-history strategy may also has propelled the R. typus into the IUCN red list of vulnerable species (Cavanagh et al., 2003). This is the case because R. typus has difficulty responding quickly to habitat destruction caused by human activity and climate change, and ultimately leads to an inability to recover quickly from population declines (Norman, 2004). In conclusion, it would be interesting to accurately elucidate the effects of the K-selection life-history strategy on longevity and population size of R. typus.
The perfect genetic information for exploring the question mentioned above, thus it is essential to have access to high-quality genome resource of R. typus. Recently developed next-generation sequencing technology has provided a convenient and highly effective solution for deciphering the whole-genome information of R. typus. Weber et al. (2020) first assembled the R. typus genome using a combination of Illumina short-insert, mate-pair, and TruSeq Synthetic Long Read (TSLR) libraries and the estimated genome size was3.2-Gb. Meanwhile, the researcher also has uncovered several genetic traits associated with body size, metabolic rate, and lifespan of R. typus based on comparative genomics. Although the published genome has enriched the genetic information database of R. typus, the researchers predicted the incomplete protein-coding genes of R. typus only based on two published candidate gene sets (Stanke and Morgenstern, 2005; Haas et al., 2008). Meanwhile, the low completeness of R. typus genome annotation information also due to the lack of transcript data (Lou et al., 2020). Short transcripts were usually applied to the early genome annotation (Ozsolak and Milos, 2011; Duitama et al., 2012), but it is undeniable that the short transcripts have deficient in the recognition of alternative splicing, alternative transcription initiation or alternative transcription termination sites (Steijger et al., 2013; Tilgner et al., 2013). Meanwhile, the highly similar transcripts resulting from genome-wide replication events also limit the genome annotation ability of short transcripts (Postlethwait et al., 1998). The third generation sequencing technology has revolutionized the genome annotation due to full-length (FL) transcripts can be obtained to remedy the deficiency of short transcripts (Li et al., 2018). To date, FL transcripts has been successfully used to perfect the genome annotation information of many animals, such as Tachypleus tridentatus (Lou et al., 2020), Sus scrofa (Li et al., 2018), Danio rerio (Nudelman et al., 2018). Therefore, we have every reason to believe that FL transcripts will contribute to high-resolution R. typus genome annotation and provide an impeccable information for further elaboration of R. typus biology. Since 2012, the third-generation sequencing technology been gradually applied to long-read sequencing due to this technology showing high reads yield and having clusters representing individual molecules rather than amplifications (Eid et al., 2009; Feng et al., 2015). Different third-generation sequencing platforms have superior specificity. As a representative sequencing platform, Oxford Nanopore (ONT) platform has 1-Mb of sequencing reads (Wyman et al., 2019).
Oxford Nanopore (ONT) platform was applied in the present study to achieve more efficient R. typus FL transcripts. These results not only constitute a rich dataset of FL transcripts that extends our knowledge of the R. typus transcriptome, but also help to discover novel genes/transcripts to improve the genome annotation efficiency of R. typus. The ultimate goal of the present study is provide perfect genome annotation information for the exploration of R. typus biology.
Materials and Methods
Ethics Approval and Participation Consent
We have read the policies relating to animal experiments and confirmed the present study complied. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in the present study were approved by the Institutional Animal Care and Use Committee of Yantai University. All experimental methods were performed according to relevant guidelines and regulations established by the Institutional Animal Care and Use Committee of Yantai University. In accordance to the directive of Reporting of In vivo Experiments (ARRIVE) guidelines 2.0, we have designed this experiment. Meanwhile, our study did not require specific authorization.
Blood Collection, RNA Extraction, Library Construction and Oxford Nanopore Sequencing
The blood was extracted from a male R. typus (approximately 15 ∼ 20 years old, seven meters) from the Haichang aquarium (Yantai, China). The blood sample was then snap-frozen in liquid nitrogen and stored at −80°C for subsequent RNA extraction. Total RNA of R. typus blood was extracted using the standard TRIzol Reagent Kit (Invitrogen, CA, United States) and following the manufacturer’s protocol. The RNA concentration was measured using the NanoDrop 2000 (Thermo Fisher Scientific, MA, United States) and the RNA integrity was assessed using the RNA Nano 6000 Assay Kit and Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, United States). Approximately 900 ng of poly(A)-tailed mRNAs were purified using the standard Dynabeads mRNA Purification kit (Invitrogen, CA, United States), and the sequencing adapters and motor proteins were then immediately added to construct the sequencing library. The constructed library was added to a flow cell and transferred to Oxford Nanopore PromethION sequencing platform for direct RNA sequencing.
Oxford Nanopore Sequencing Data Processing
All raw reads in ‘‘fast5’’ format generated by the Oxford Nanopore PromethION sequencing platform were converted to ‘‘fastq’’ format using GUPPY software (version 3.2.61). The NanoFilt software (version 2.6.02) was further applied to filter out adapter sequences and low-quality raw reads (raw reads with quality score less than 7 or length less than 50bp) to obtain the clean reads for subsequent analyses, and the parameters as follows: -q 7 -l 50. These highly similar clean reads derived from the similar isoform were clustered into consensus sequences using Genomic Mapping and Alignment Program (GMAP; Wu and Watanabe, 2005) with the following parameters: –cross-species and –allow-close-indels 0. The consensus sequences were then mapped to the reference R. typus genome (Weber et al., 2020) using the GMAP (Wu and Watanabe, 2005), and those consensus sequences with different 5′-end exons were identified as redundant sequences. The stringTie software (version 2.1.23) was used to merge redundant consensus sequences to obtain the FL transcripts of R. typus, and the parameters as follows: –conservative -L -R.
The Distribution Analyses of Full-Length Transcripts on the Reference Genome
The minimap2 software (version 2.17-r9414) was used to compare the FL transcripts with the reference genome, and the parameters as follows: -ax splice -uf -k14. The comparing results of all FL transcript can be divided into “unmapped,” “mapped to ‘+’,” and “mapped to ‘−’,” which represent FL transcripts that are not mapped to the genome, mapped to positive and negative strands of the genome, respectively. Meanwhile, the distribution of FL transcripts in the reference genome was investigated based on the consensus sequence density successfully mapped to each chromosome, and the density distribution of the longest 15 consensus sequences were visualized.
Gffcompare software (version 0.11.25) was applied to annotate all FL consensus sequences and then to identify the known genes/transcripts and novel genes/transcripts. According to the mapping information of FL consensus sequences in the R. typus genome, those FL consensus sequences mapped to the unannotated genome region were defined as novel genes. Meanwhile, the FL consensus sequences with structures different from transcripts in genome “gtf” files were defined as novel transcripts.
Full-Length Transcriptome Structure Analyses
Optimization of Transcript Structure
Optimize the original annotated transcript structure is necessary to improve the annotation accuracy of the R. typus genome. In the present study, FL transcripts were compared with known transcripts of the reference genome based on Gffcompare software [version 0.11.2 (see text footnote 5)], and the parameters as follows: -R -C -K –M. The aim was to discover the new transcripts and eventually supplement the annotated information of existing transcripts. When the regions outside the original gene boundary were supported by the FL transcripts, the untranslated regions (UTRs) of the transcript were extended upward and downstream to complete the modification of the transcript boundary. In general, the novel transcripts consists of five types: (1) other parts of the same chain that overlap with the reference exon (O); (2) at least one matching multiple exons (J); (3) the exons on the anti-chain overlap (X); (4) completely contained in introns of the reference genes (I); (5) unknown novel transcripts (U) (Figure 1).
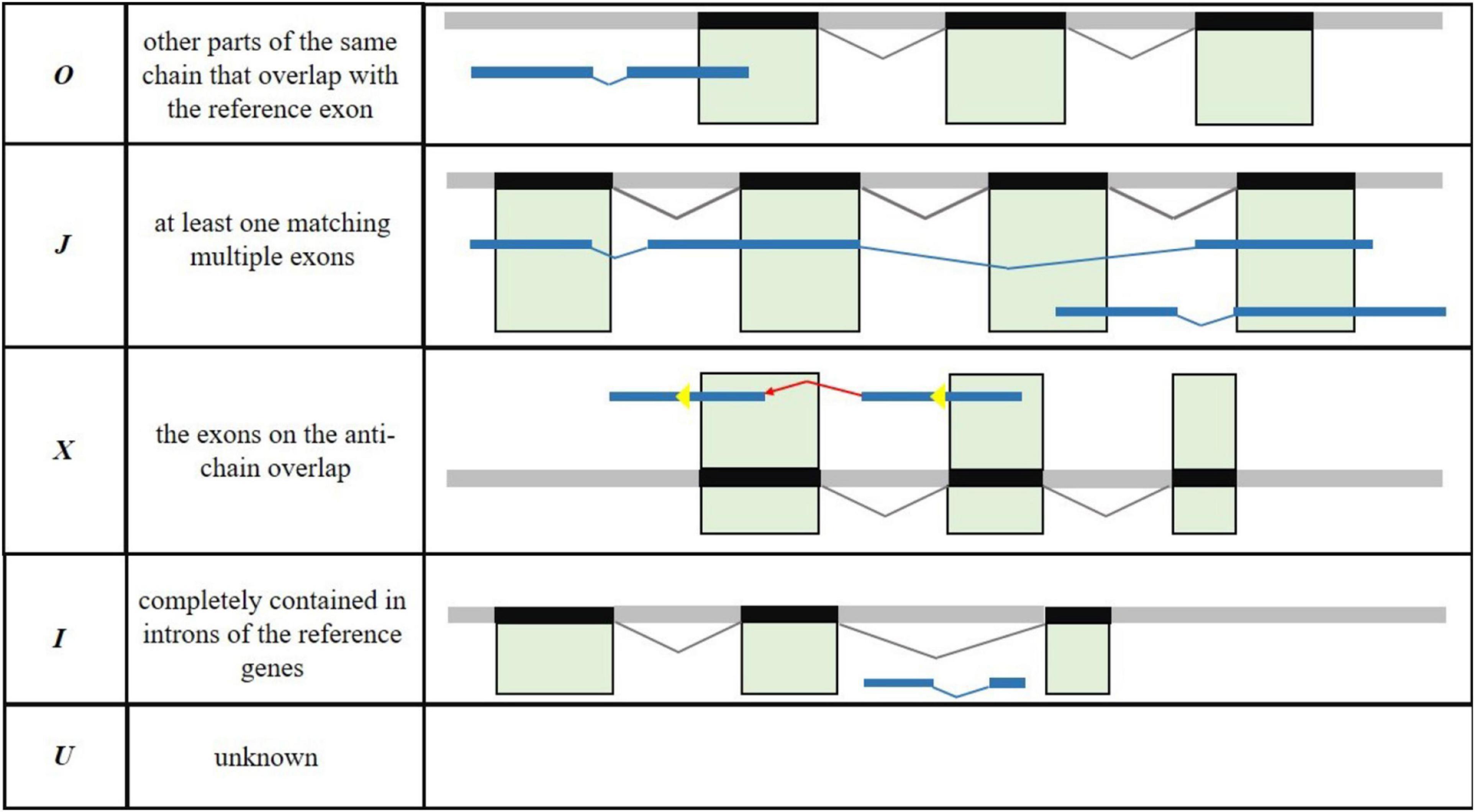
Figure 1. Five types of novel transcripts.
Alternative Splicing Analyses
Alternative splicing refers to multiple splicing types of precursor mRNA (pre-mRNA) can produce different mature mRNAs, which can be further translated into different proteins, and ultimately leading to diversity of biological traits. In the present study, suppa26 software was applied to predict the potentially alternative splicing in R. typus FL transcriptome, and the parameters as follows: -f ioe -e SE SS MX RI FL.
Fusion Transcripts Analyses
Fusion transcripts are always formed by having the coding regions of two or more transcripts joined end to end. It is worth noting that the fusion transcripts are uniformly regulated. Tofu software (version 13.0.0; Wang et al., 2016) was used to find fusion transcripts and the identification criteria as follows: (1) a fusion transcript must be mapped to two or more gene locus in the R. typus genome; (2) each gene loci must be aligned to 10% region of a fusion transcript; (3) the fusion transcript must be more than 99% of coverage on the R. typus genome; (4) distance between two mapped locus must be at least 100 kb.
PolyA Length Analyses
The PloyA tail is located in the downstream of the 3′ untranslated region of mRNA, and it contributes to mRNA stabilization and cytoplasmic transport. The length of PolyA can affects the translation efficiency of mRNA and therefore it is necessary to analyze the length of PolyA. In the present study, Nanopolish software (Version: 0.12.57) was used to calculate the length of PolyA and the parameter as follows: polya. Furthermore, we also analyzed the association between median length and expression level of PolyA.
Long Non-coding RNA Prediction
Long Non-coding RNA (LncRNA) is a generic term for RNAs that are more than 200nt long and cannot encode proteins. In the present study, LncRNAs were screened using CNCI software8, CPC2 software9, and Pfam database [version 33.1; (25)].
Coding Sequence Prediction
In the present study, TransDecoder software10 was used to identify the potential Coding Sequences (CDSs) of the R. typus FL transcripts based on the open reading frame (ORF) length, log-likelihood score, and the comparing information between amino acid sequence and protein domain sequence of Pfam database. Annotate the potential functions of CDSs and the metabolic pathways that may be involved based on existing protein databases. In the present study, we annotated the CDSs using the BLAST (Altschul et al., 1990) and based on the reference genome (Weber et al., 2020), Pfam (Protein Families; Finn et al., 2013), Nr (Non-Redundant Protein Sequences; Deng et al., 2006), KEGG (Kyoto Encyclopedia of Genes and Genomes; Kanehisa et al., 2004), GO (Gene Ontology; The Gene Ontology Consortium et al., 2000), and COG (Cluster of Orthologous Groups of Proteins; Tatusov et al., 2000) databases, and the parameter as follows: E-value < 0.00001.
Methylation Analyses
Direct RNA sequencing not only can sequenced the transcriptome information, but also can obtain the methylation modification information. In the present study, the m5C methylation site was predicted based on the alternative model in Tombo software (version 1.511), and the top 1000 sites with the highest alternate value were regarded as the credible sites. Meanwhile, the m6A methylation site was predicted based on the MINES pipeline,12 and those sites with alternate values greater than 0.7 were identified as credible sites. Meanwhile, we also mapped the predicted m5C and m6A methylation sites to the reference genome and then visualized the distribution of these sites on the genome. Additionally, two bases were extended upstream and downstream of the m5C and m6A methylated sites to obtain motifs that consisting of five bases. The meme software13 was applied to calculate the sequence characteristics of the motifs.
Results
Rhincodon typus Full-Length Transcriptome Sequencing
High-quality RNA was extracted from R. typus blood and then sequenced on the Nanopore PromethION platform. A total of 10,778,080 raw reads were generated from the platform, corresponding with 7,946,301,774 bp. The max length, average length, N50, N90 and mean quality value of all raw reads was 78,083 bp, 737.26 bp, 808 bp, 443 bp and 9.9, respectively (Figure 2). All raw reads were released in the NCBI Sequence Read Archive under BioProject number PRJNA765716, with accession number of SRR16036159. After filtering out the low-quality raw reads, a total of 9,579,943 clean reads were obtained, corresponding with 7,537,272,963 bp. The max length, average length, N50, N90 and mean quality value of all clean reads was 14,238 bp, 786.77 bp, 822 bp, 466 bp and 10.5, respectively. The length distribution of sequencing reads was showed in Figure 2. Highly similar clean reads were then clustered into 22,868 consensus sequences, with a max length, mean length and N50 of 14,089 bp, 2252.4 bp and 3,076 bp, respectively (Figure 3). All consensus sequences were further compared to the reference R. typus genome for remove redundant, and 14,930 FL transcripts were ultimately obtained.
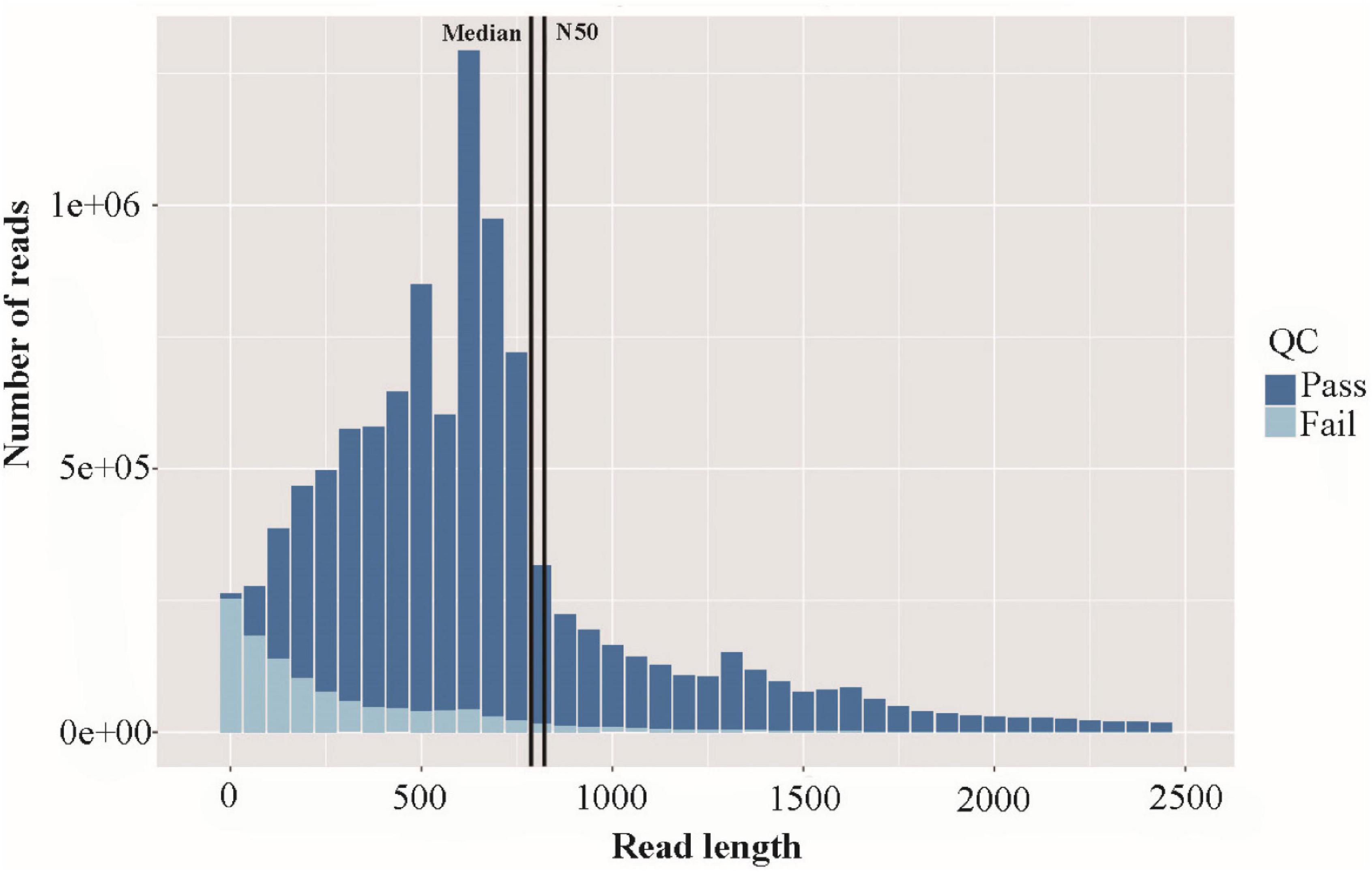
Figure 2. Histogram showing the length distribution of sequencing reads across quality passing sequences.
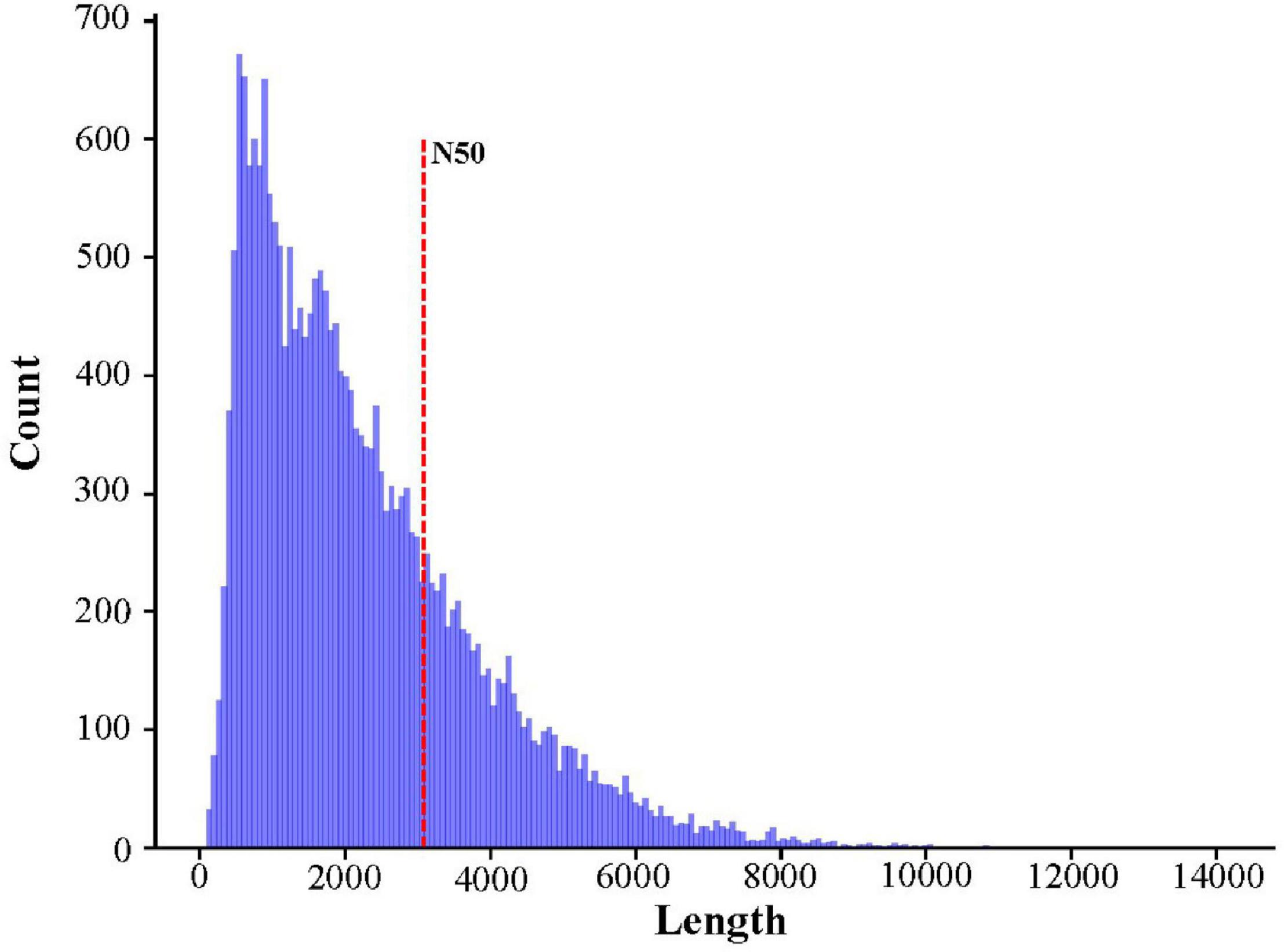
Figure 3. Histogram showing the length distribution of consensus sequences.
Comparison of Transcriptome With Reference Genome
The newly constructed 14,930 FL transcripts were mapped to the reference genome and the aligned results showed that 14,915 sequences were covered 11,892 genetic loci of R. typus genome, and the successfully comparison number of “unmapped,” “mapped to ‘ + ‘,” and “mapped to ‘−”’ was 15, 7,566 and 6,970, respectively. Meanwhile, 1,642 novel genetic loci were found based on the FL transcripts. Finally, we have counted the FL transcripts distribution on the reference genome and drew the density distribution of known and novel transcripts on the 15 longest FL transcripts (Figure 4).
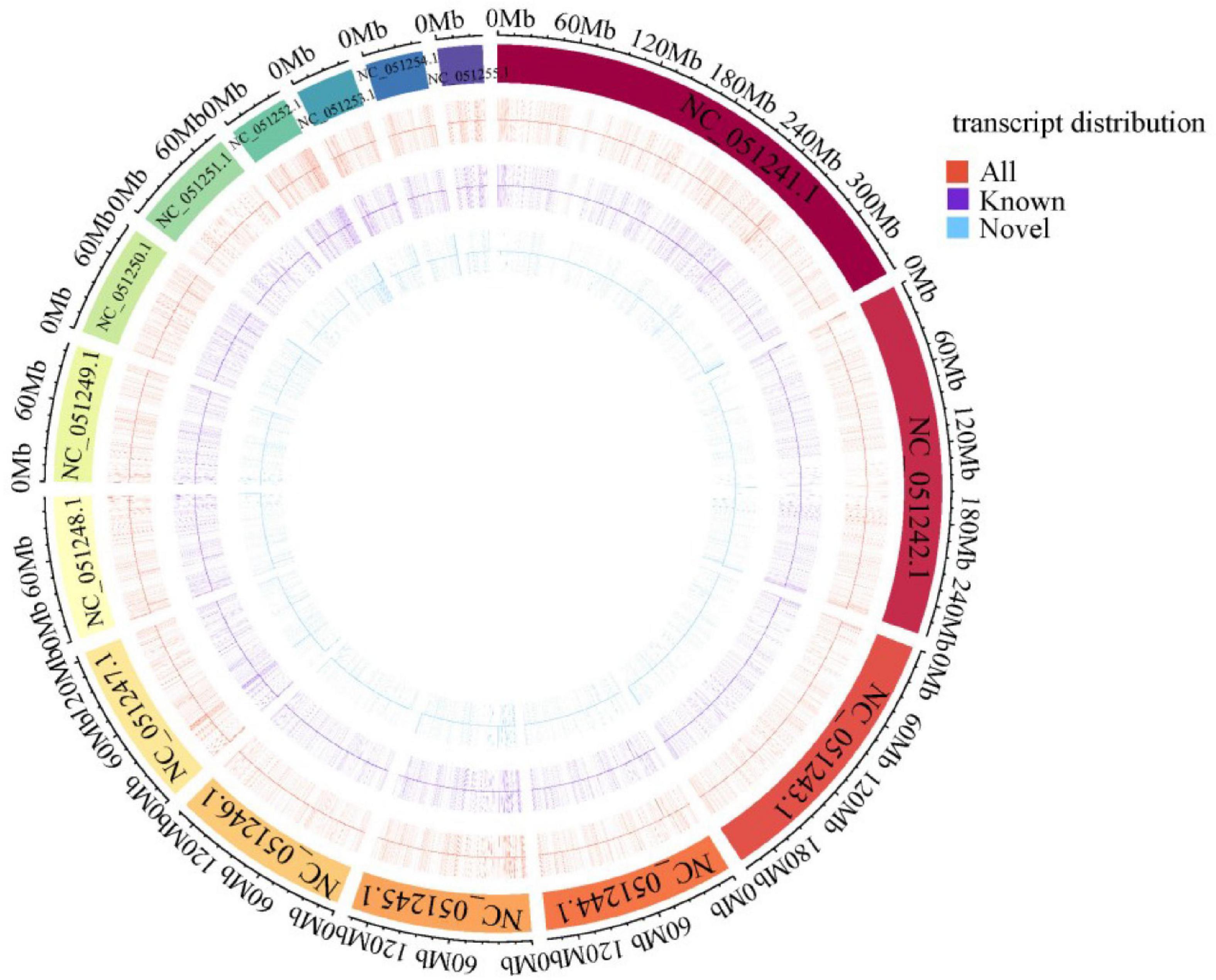
Figure 4. The density distribution of known and novel transcripts on the longest FL transcripts.
Full-Length Transcriptome Structure
Novel Transcripts
We have provided a preliminary assessment of the novel transcripts based on the aligned results of FL transcripts on the reference genome and 714 novel transcripts were identified. Among the five transcript types, the novel transcript number of I, J, O, U, X type was 3, 630, 12, 55 and 14, respectively.
Alternative Splicing Events
Based on suppa2 software, 1,941 AS events were discovered in the R. typus FL transcriptome, which were divided into 7 types: (i) skipping exon (SE), (ii) mutually exclusive exons (MX), (iii) alternative 5′ splice site (A5), (iv) alternative 3′ splice site (A3), (v) retained intron (RI), (vi) alternative first exon (AF), and (vii) alternative last exon (AL). Among all AS types, A3 type predominated and accounting for 25.50% of all AS events, while MX type was the least frequent (1.55%) (Figure 5).
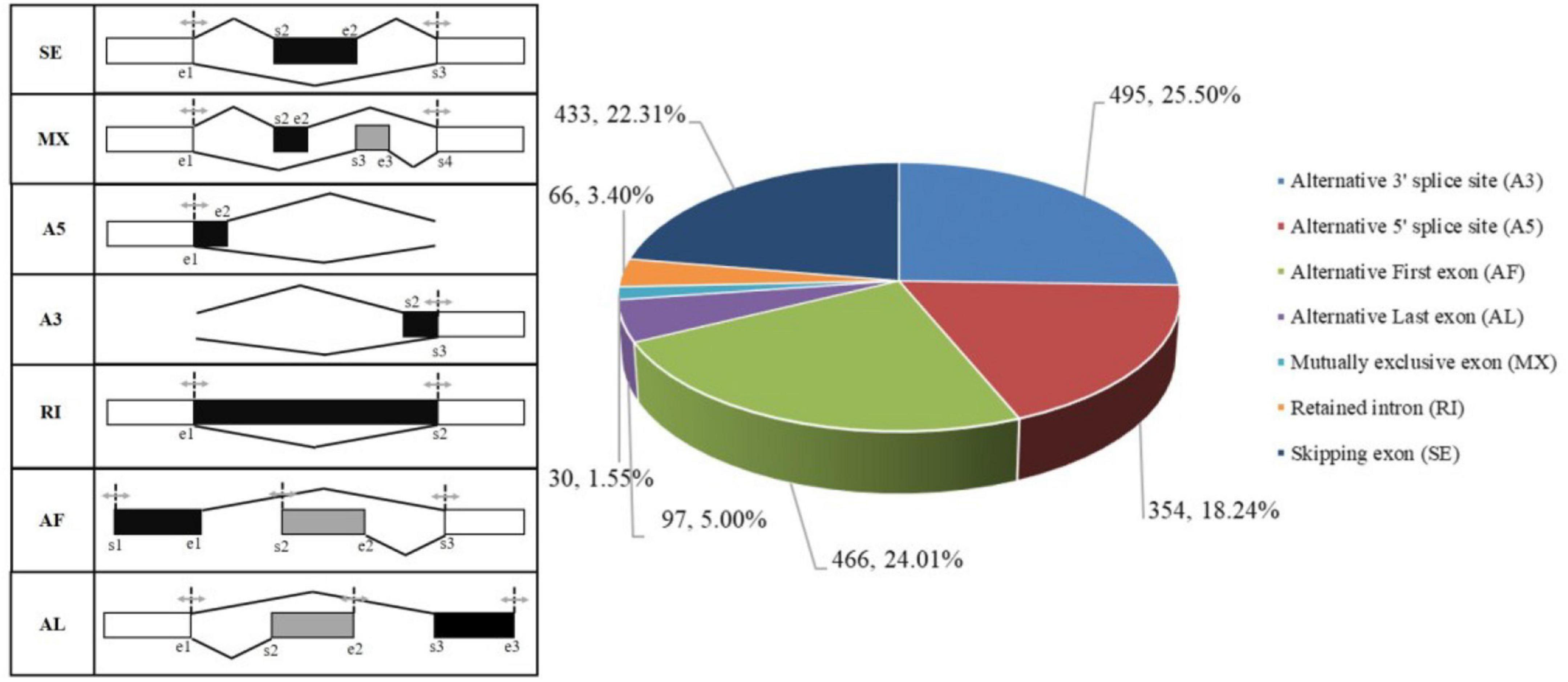
Figure 5. The types and number of AS events in the R. typus FL transcriptome.
Fusion Transcripts
Based on the identification criteria, we identified 175 fusion transcripts in the R. typus FL transcriptome, all of which consisted of two transcripts.
PolyAs
By comparing the FL transcripts with the R. typus reference genome, a total of 100 ployAs were obtained in the present study, with the mean length and N50 of 89.16 bp and 77.93 bp. The length distribution of polyAs was showed in the Figure 6A. Meanwhile, we conducted a correlation analysis between polyA N50 and expression level of FL transcripts, and the correlation was showed in the Figure 6B.
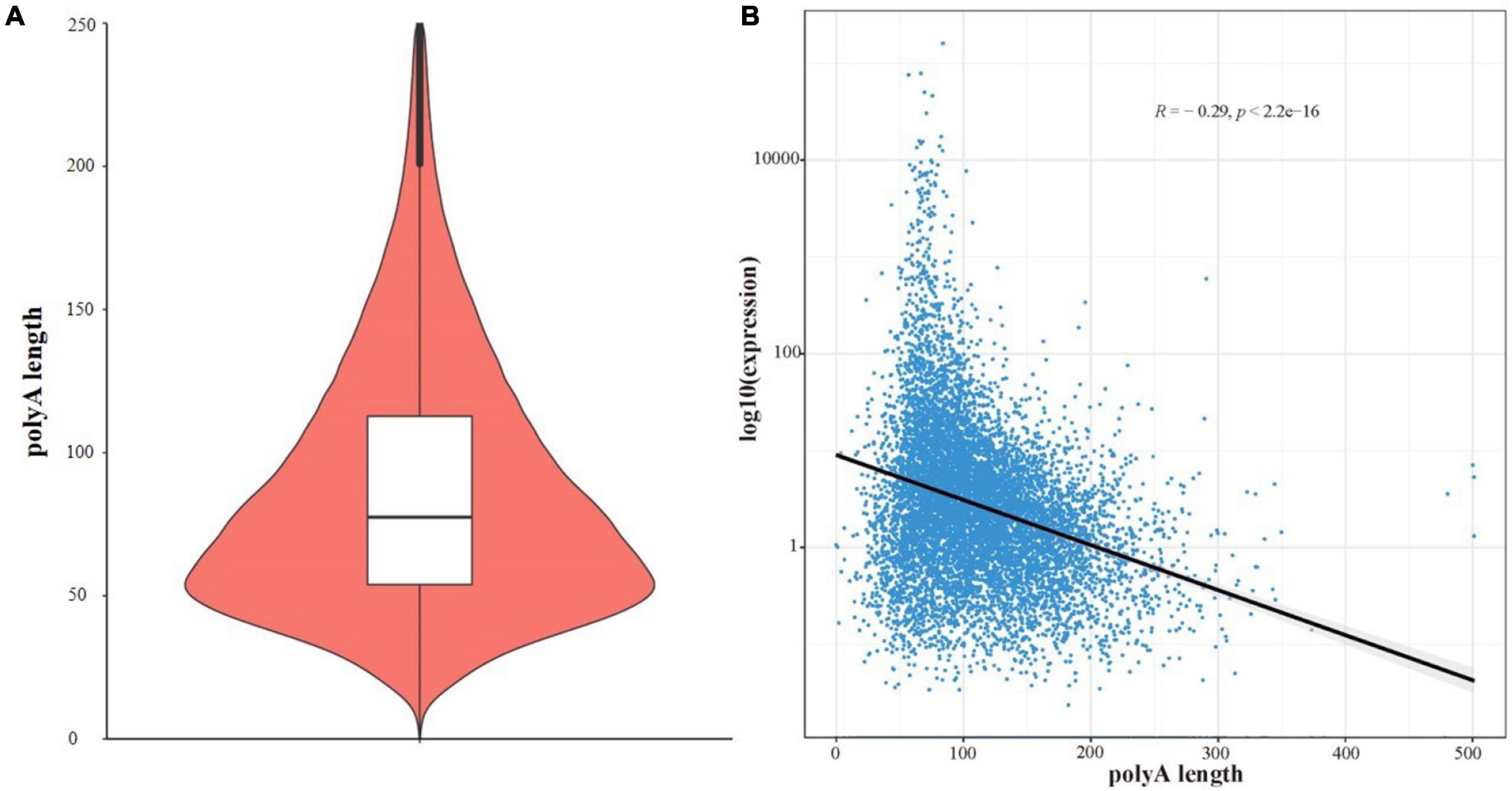
Figure 6. (A) The length distribution of polyAs. (B) The correlation between polyA N50 and expression level of FL transcripts.
LncRNAs
Three prediction approaches were applied to identify the LncRNA and a total of 308 were predicted in the FL transcripts. Specifically, 162, 210, and 212 LncRNAs with length greater than 200 bp were predicted in the CNCI software, CPC2 software and Pfam database, respectively (Figure 7). Furthermore, veen chart indicated that only 89 LncRNAs were shared among the three prediction approaches.
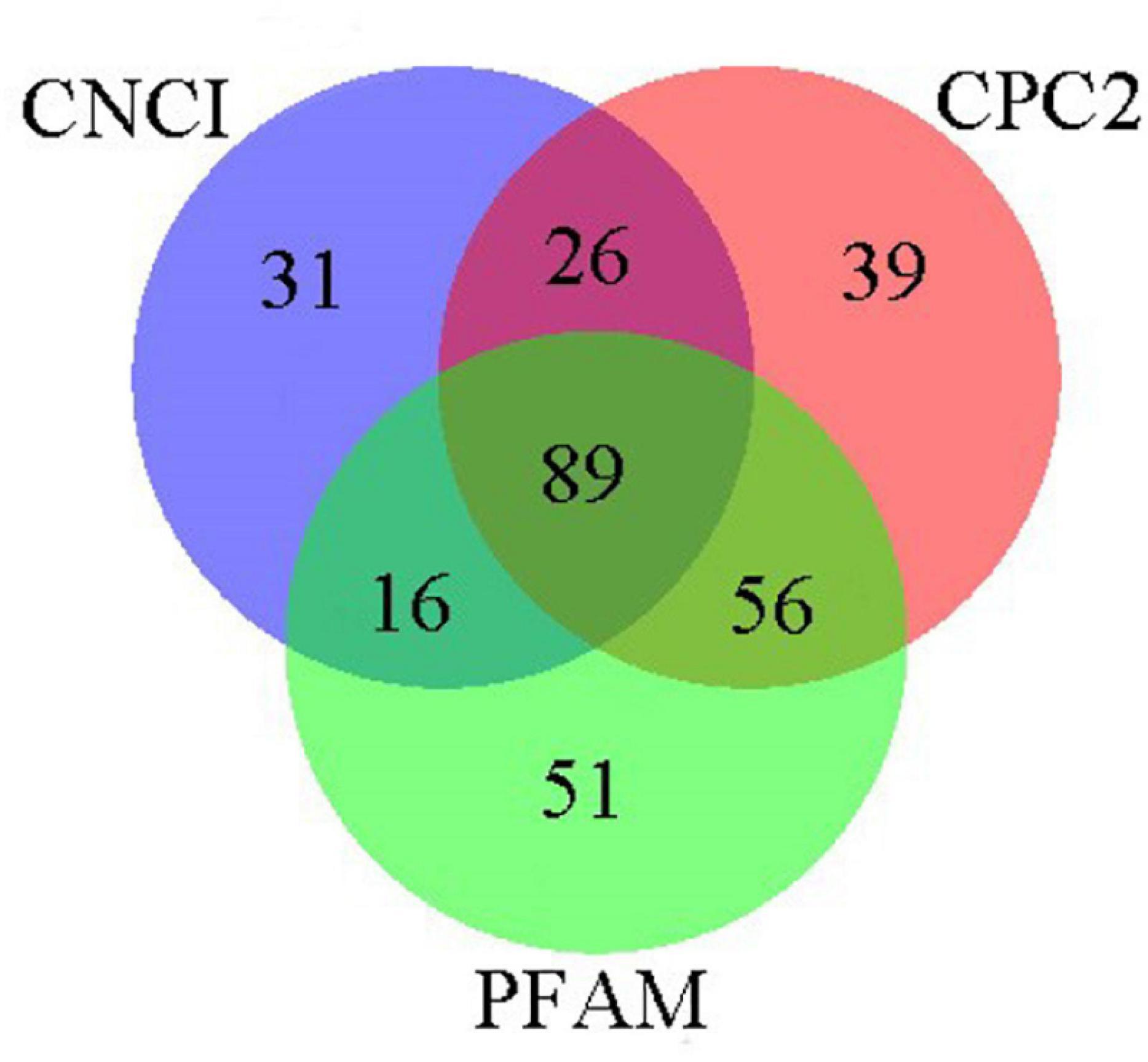
Figure 7. Veen chart showing the predicted LncRNAs shared among the CNCI software, CPC2 software and Pfam database.
Coding Sequences
We have predicted 31,698 potential CDSs from the R. typus FL transcripts based on the TransDecoder software and Pfam database, corresponding with a total length, mean length, and N50 of 41,659,911 bp, 1314.28 bp and 6,459 bp. Among all CDSs, a large proportion (26,138, 82.46%) of CDSs are less than 2,000 bp in length (Figure 8).
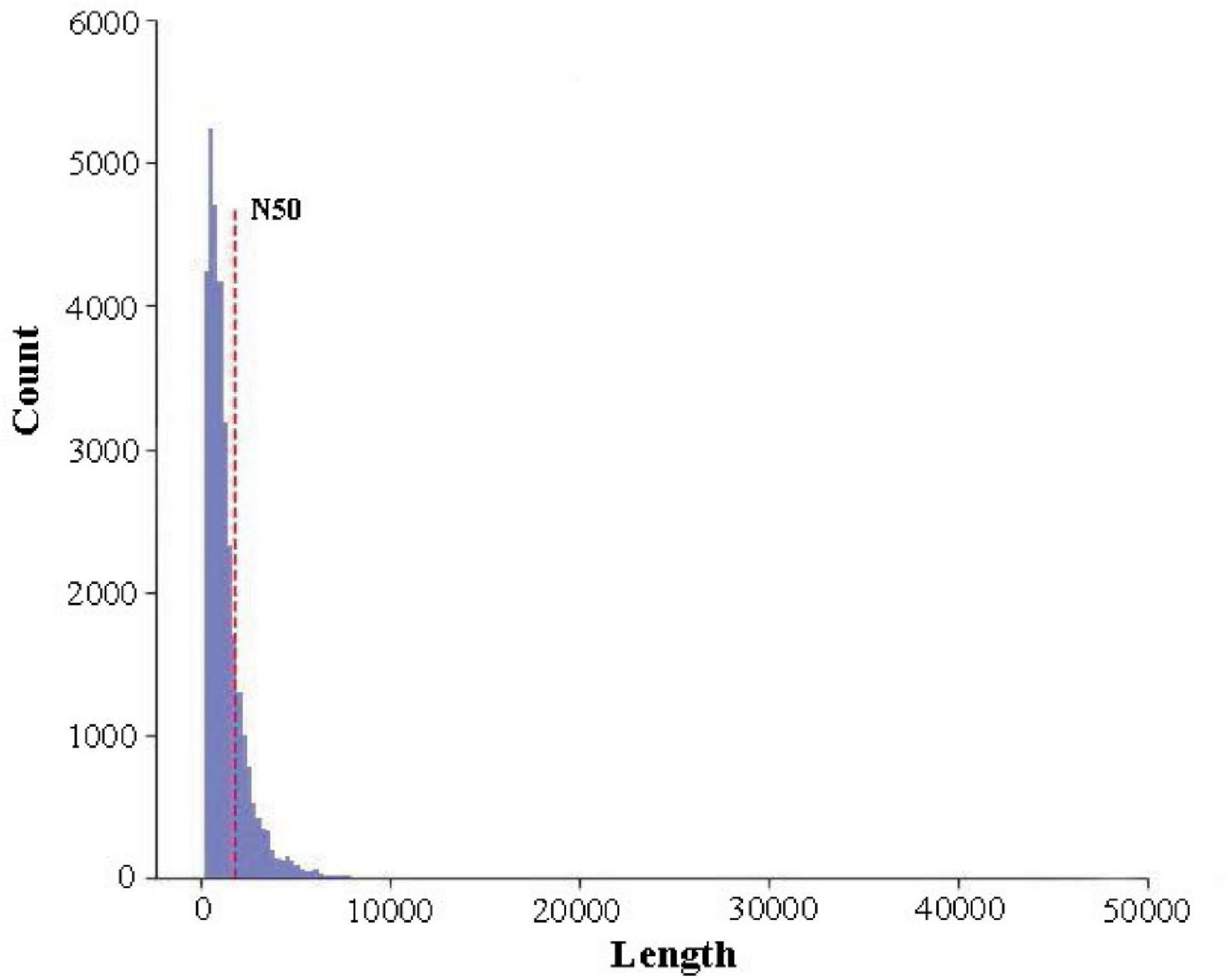
Figure 8. Histogram showing the length distribution of CDSs.
Coding Sequence Annotation
The lack of previous transcripts may have limited the completeness of the R. typus genome annotation information, it is necessary to annotate the CDSs. All the 31,698 potential CDSs were further compared to the afore-mentioned databases and a total of 31,021 (97.86%) CDSs can obtained annotation information. Specifically, we have evaluated the genetic sequence similarity of R. typus and other species by comparing all FL transcripts against the Nr database. Result showed that 30,189 CDSs were found in Nr database and 94.98% CDSs have a strong similarity with the existing sequences from R. typus (Figure 9A). GO classification can exactly define the gene characteristic and can help us understand which genes might be associated with biological processes. GO classification result showed that the terms of “positive regulation of transcription DNA-templated,” “nucleus,” and “metal ion binding” were dominant in “biological process,” “cellular component,” and “molecular function,” respectively (Figure 9B). The COG database have divided the homologous genes of different species into different ortholog transcripts based on evolutionary relationship. In the present study, 6,561 CDSs were categorized into 23 COG categories (Figure 9C). Among these categories, the first three largest groups were R category, T category and J category, representing “General function prediction only,” “Signal transduction mechanisms,” and “Translation, ribosomal structure and biogenesis,” respectively. KEGG analysis was used to analyze the functions and their metabolic pathways of gene products in cells. In this study, a total of 27,233 CDSs were assigned to 5 terms according to metabolic pathways: cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems (Figure 9D). Meanwhile, the most significant pathways in 5 terms were “cellular community-eukaryotes,” “signal transduction,” “folding, sorting and degradation,” “carbohydrate metabolism” and “endocrine system,” respectively.
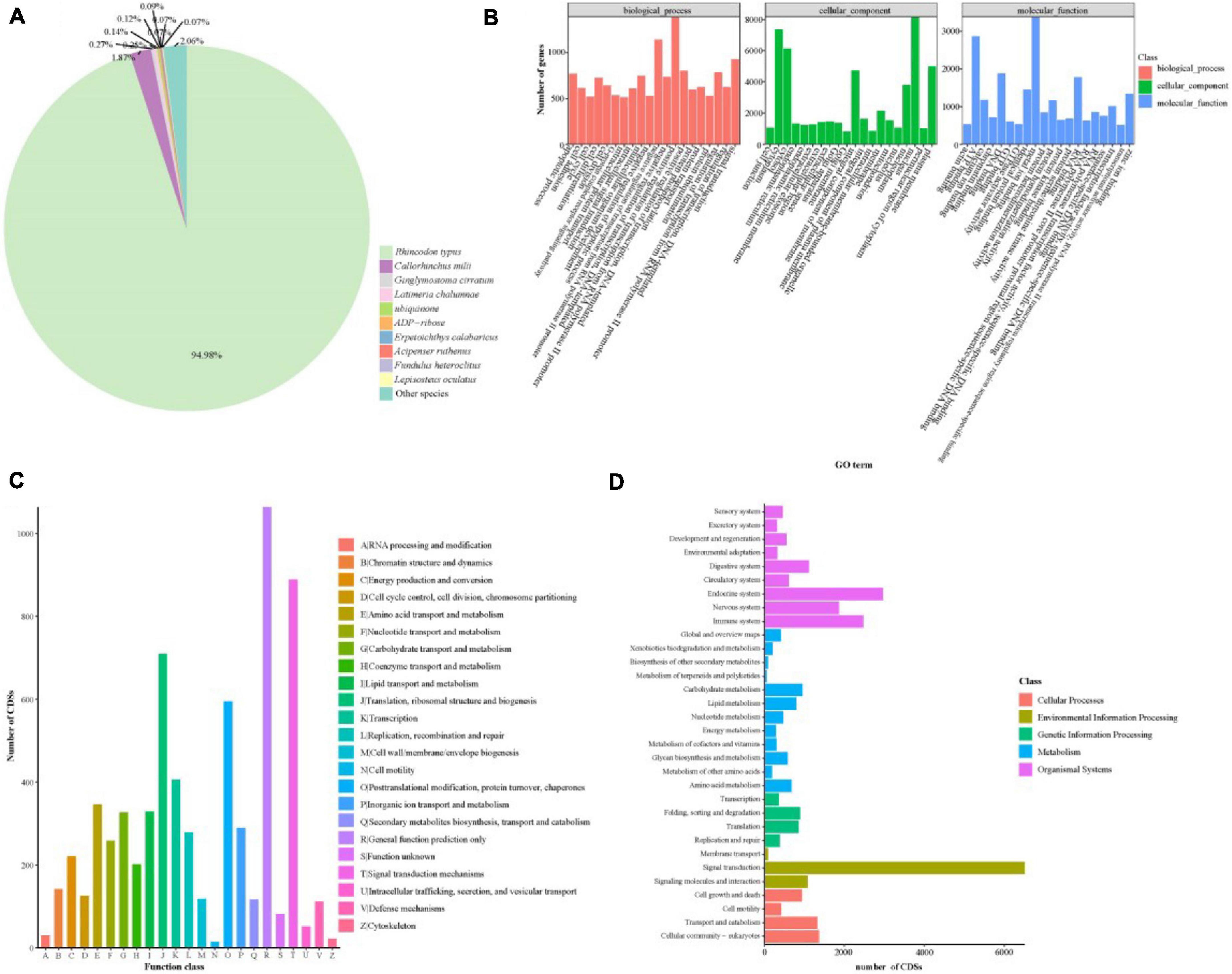
Figure 9. CDSs annotation information in Nr (A), GO (B), COG (C), and KEGG (D) database.
m5C and m6A Methylation Sites
In the present study, these m5C methylation sites with the top 1000 alternative value and 5,088 m6A methylation sites with alternative value greater than 0.7 were used to analyze the distribution of methylation sites. The methylation site distribution of 15 longest FL transcripts was showed in the Figure 10. Distribution results showed that m5C methylation sites were distributed in both the sense strand and antisense strand of the 15 longest FL transcripts, while m6A methylation sites were only predicted in the antisense strand of the 15 longest FL transcripts.
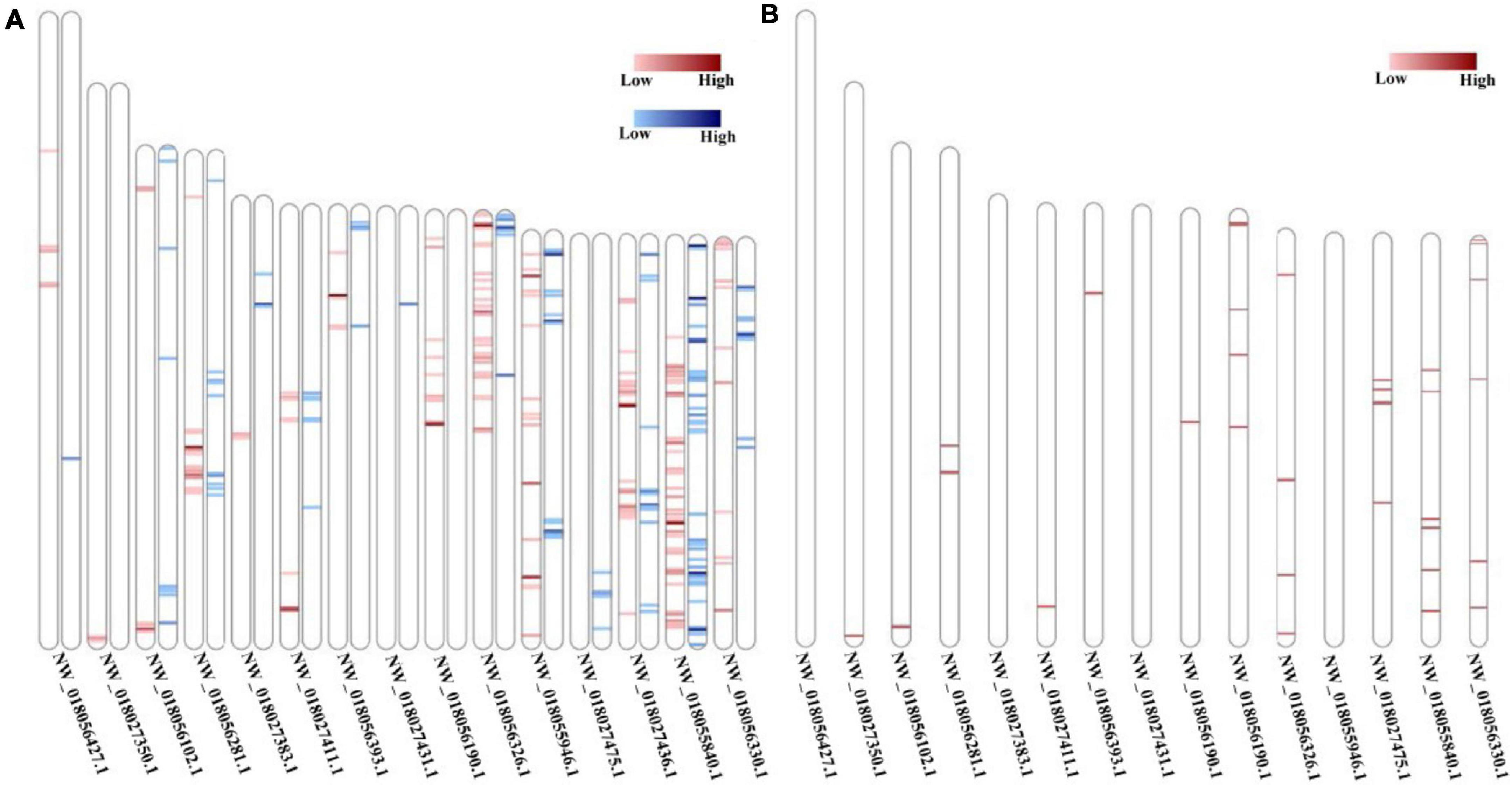
Figure 10. The m5C (A) and m6A (B) methylation sites distribution of the 15 longest FL transcripts.
Additionally, the motif of m5C and m6A methylation sites were identified by extending the upstream and downstream bases of the methylation sites. Results showed that the motifs of m5C methylated sites showed no regularity, while all motifs of m6A methylated sites were R(representing A or G)GACH(representing A or T or C) type (Figure 11).
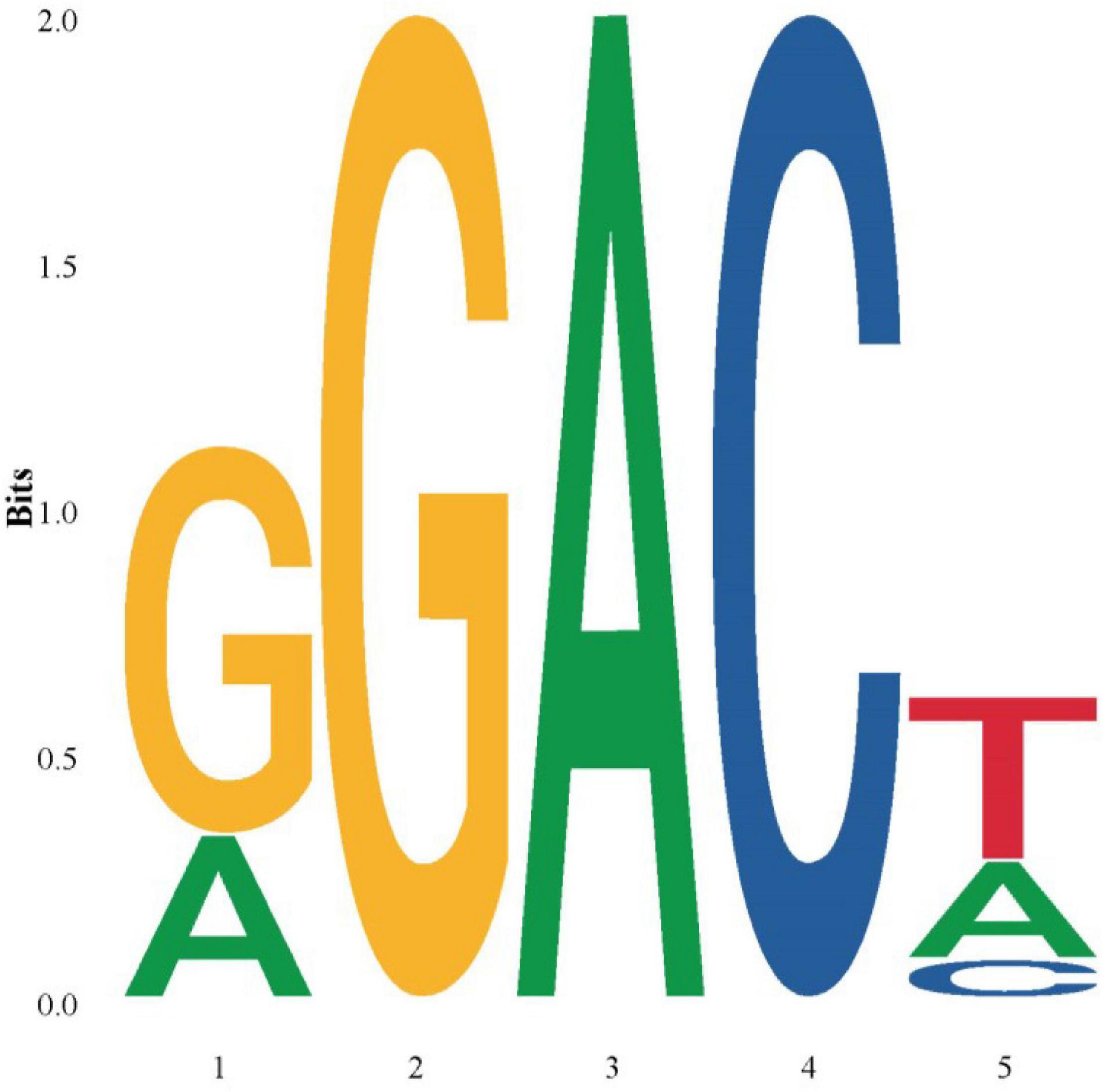
Figure 11. The consensus motifs of m6A methylation sites. The height of the base signal represents the relative base frequency of each site.
Discussion
The acquisition of FL transcripts is essential to accurately investigate the gene function. Although the next-generation sequencing technology has improved the capture efficiency of transcripts, assembled transcripts were generally short and non-full-length, which may ultimately limit the accuracy and completeness of genome annotation information (Postlethwait et al., 1998; Steijger et al., 2013; Tilgner et al., 2013). The third-generation sequencing technology seems to offer an opportunity to solve above-mentioned difficulties, this is the case because it can obtain some transcripts large enough to cover the FL genes (Eid et al., 2009; Feng et al., 2015). Given the advantage of ONT platform in long-read sequencing (Wyman et al., 2019), thus it can be applied to sequence the FL transcriptome of R. typus. In the present study, a total of 14,930 FL transcripts were generated and these sequences helped us identify 714 novel transcripts and 1,642 novel genetic loci in the R. typus genome. Meanwhile, the longest transcript obtained in the present study is 14,089 bp, which is beyond the reach of next-generation sequencing technology.
Full-length (FL) transcripts provide the basis for the research of AS events. Gene can produce different mRNAs by different splicing and then increase the functional proteome variability in cells and tissues. This implies that AS events can alter the composition of transcribed genes without increasing the number of genes (Wang et al., 2008). Alternative splicing events have been proved to be involved in the regulation of gene expression in various biological processes such as growing development, sex differentiation and immune resistance (Modrek and Lee, 2002). However, short transcripts are difficult to identify the combinations of splice-site (Wang et al., 2008; Chacko and Ranganathan, 2009). Therefore, FL transcripts are necessary for the more complete and accurate identification of R. typus AS events. A total of 1,941 AS events were identified in this study and these results will greatly improve reference annotation. There is no denying that the proportion of AS events obtained by FL transcripts is low, which may be because the R. typus have slightly more single-exon isoform with lower coding ability, thus representing lncRNAs (Sharon et al., 2013). Meanwhile, considering that exons are more prone to methylation than introns, thus we suspected that alternatively splicing of a large proportion of multi-exon genes may increase the transcriptional diversity of R. typus, which has been demonstrated in many organisms (Pan et al., 2008; Gelfman and Ast, 2013).
Fusion transcripts are usually identified by RNA sequencing and play an important role in some physiological processes, whether protein-coding or non-coding RNAs (Wu et al., 2014). In the present study, 175 fusion transcripts were detected, which is relatively low. We suspected that the poor accuracy of ONT platform sequencing led to this finding (Wyman et al., 2019). Due to the lack of genomic data at the chromosomal level, we have not been able to determine whether these fusion transcripts are from intergenic splicing or chromosomal rearrangement (Xie et al., 2016). In fact, fusion transcripts have been identified in a variety of animals including humans and mouse (Babiceanu et al., 2016). Although the physiological function of fusion transcripts is still unknown, fusion transcripts with specific functions may be positively selected during the R. typus evolution. In fact, fusion transcripts can increase the diversity of the transcriptome (Ou et al., 2021). Future studies still need to verify the physiological function of the fusion transcripts to explore its regulatory mechanism for the life-history strategy of the R. typus.
The ability to estimate the relationship between polyA length and expression level is another advantage of the third-generation sequencing technology. Our study have confirmed that the N50 of polyA was negatively correlated with the expression level, meaning that highly expressed genes generally have shorter polyA length (Lima et al., 2017; Legnini et al., 2019). In fact, polyA are the recognized regulators of translation and transcriptional stability (Roach et al., 2020). A similar negative correlation was found in the larval stage of Caenorhabditis elegans, but not in the adult stage (Roach et al., 2020). The R. typus used in this study were 15-20 years old and belonged to the early development. Whether the correlation between polyA length and expression level of FL transcripts of R. typus would show age-specific is still worth studying. Additionally, the polyA length is species-specific and it is also of concern because mRNA with short polyA are easily enzymatic or translational dormancy (Elkon et al., 2013; Abdel-Ghany et al., 2016). In the present study, the mean polyA length of R. typus was only 89.16 bp and significantly lower than human (Mayr and Bartel, 2009). Previous study have confirmed that polyA length are closely related to the intron retention events involved in cell senescence regulation (Roach et al., 2020; Yao et al., 2020). Although the effect of intron retention on cell senescence is unclear, it does not prevent us to speculate that the short polyA may be related to the longevity of R. typus.
LncRNAs are these non-coding RNAs which structure is similar to mRNA and length is longer than 200 nucleotides (Wan et al., 2019). Although LncRNAs have no protein-coding ability, they still regulate gene expression at epigenetic, transcriptional and post-transcriptional levels, and therefore participate in many biological processes (Kapranov et al., 2007). To date, the LncRNAs of R. typus has never been reported. In our study, 89 LncRNAs were predicted by three methods. Fewer lncRNAs were identified may be due to fewer lncRNAs were annotated in the database. At present, lncRNAs are mostly found in humans and mice, but their regulatory mechanisms in other species have not been thoroughly studied (Zhang et al., 2017). A critical reason for this is that non-coding RNAs are not highly conserved among species (Liu et al., 2018). Therefore, LncRNAs obtained in this study will facilitate the function study of R. typus. However, it is undeniable that the real-time dynamic changes of lncRNAs in cells are difficult to be analyzed, which affects the annotation of the function and mechanism of R. typus lncRNAs.
RNA methylation refers to methylated modifications that occur at different sites on RNA, and it plays an important role in the regulation of RNA metabolism, such as precursor RNA splicing, RNA editing, RNA translation, and RNA stability (Liang et al., 2020). The two most common post-transcriptional modification of RNA, including m5C and m6A, are believed to play important roles in the regulation of adaptive functions of organisms (Cui et al., 2017; David et al., 2017). This study for the first time clarified the distribution patterns of m5C and m6A in R. typus FL transcripts, which can provide basic data for exploring the adaptive functions of R. typus. Meanwhile, the motifs of m5C methylated sites showed no regularity, although all motifs of m6A methylated site were R(representing A or G)GACH(representing A or T or C) type. The regular existence of m5C modification may imply that it is a specific modification and regulation mode of R. typus. Previous study has suggested that the distribution of m5C in mRNA is related to cis-acting elements and microRNA binding sites, and thus m5C may be involved in the metabolism process after mRNA transcription (Squires et al., 2012). As mentioned earlier, a low basal metabolic rate may contribute to the longevity of R. typus (Weber et al., 2020). Meanwhile, m6A has been confirmed not only to regulate transcriptome, but also to participate in DNA damage repair (Lee et al., 2015). Although the excavation of longevity-related m5C and m6A sites is necessary to unlock the longevity code of R. typus, the mechanisms involved are not yet clear. Future research needs to further discuss these uncertainties.
In conclusion, the present FL transcript information would be of great value to improve R. typus genome annotation and relevant biological research.
Conclusion
In the present study, the ONT platform was first applied to sequence the FL transcriptome of R. typus and these FL transcripts can compensate for short-read used in genomic annotation processes. Based on FL transcripts, we have identified the novel genes and novel transcripts of R. typus and further generated an updated reference genome annotation information. Meanwhile, our research revealed the R. typus-specificity distribution patterns of ASs, fusion transcripts, LncRNAs, and methylation sites, and these information provides a more comprehensive foundation to explain the transcriptome diversity of R. typus. In conclusion, our results not only significantly improve existing genome annotation information of R. typus, but also have revealed important transcriptome characteristics and generated novel resource and information with positive implications for biological research of R. typus.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA765716).
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Yantai University.
Author Contributions
FL: conceptualization, methodology, formal analysis, data curation, writing—original draft preparation, writing—review and editing, visualization, and project administration. LiW, ZW, LeiW, and LZ: software. ZL and YT: validation and supervision. QZ: resources. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ https://github.com/zhuyifei1999/guppy3
- ^ https://github.com/wdecoster/nanofilt
- ^ http://ccb.jhu.edu/software/stringtie/
- ^ https://github.com/lh3/minimap2
- ^ http://ccb.jhu.edu/software/stringtie/gffcompare.shtml
- ^ https://github.com/comprna/SUPPA
- ^ https://github.com/jts/nanopolish
- ^ https://github.com/www-bioinfo-org/CNCI
- ^ http://cpc2.cbi.pku.edu.cn/
- ^ http://transdecoder.github.io
- ^ https://github.com/nanoporetech/tombo
- ^ https://github.com/YeoLab/MINES
- ^ http://meme-suite.org/index.html
References
Abdel-Ghany, S. E., Michael, H., Jacobi, J. L., Ngam, P., Devitt, N., Schilkey, F., et al. (2016). A survey of the sorghum tranome using single-molecule long reads. Nat. Commun. 7:11706. doi: 10.1038/ncomms11706
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Babiceanu, M., Qin, F., Xie, Z., Jia, Y., Lopez, K., Janus, N., et al. (2016). Recurrent chimeric fusion RNAs in noncancer tissues and cells. Nucleic Acids Res. 44, 2859–2872. doi: 10.1093/nar/gkw032
Cavanagh, R. D., Kyne, P. M., Fowler, S. L., Musick, J. A., and Bennett, M. B. (2003). The Conservation Status of Australian Chondrichthyans. Report of the IUCN Shark Specialist Group Australian and Oceania Regional Red List Workshop. Brisbane, QLD: The University of Queensland, School of Biomedical Sciences.
Chacko, E., and Ranganathan, S. (2009). Genome-wide analysis of alternative splicing in cow: implications in bovine as a model for human diseases. BMC Genomics 10:S11. doi: 10.1186/1471-2164-10-S3-S11
Colman, J. G. (2005). A review of the biology and ecology of the whale shark. J. Fish Biol. 51, 1219–1234. doi: 10.1111/j.1095-8649.1997.tb01138.x
Compagno, L. J. V. (2001). Sharks of the World: An Annotated and Illustrated Catalogue of Shark Species Known to Date. Bullhead, Mackerel and Carpet Sharks (Heterodontiformes, Lamniformes and Orectolobiformes). FAO Species Catalogue for Fishery Purposes, Vol. 2. Rome: Food and Agriculture Organization of the United Nations.
Cui, X., Liang, Z., Shen, L., Zhang, Q., Bao, S., Geng, Y., et al. (2017). 5-methylcytosine RNA methylation in Arabidopsis thaliana. Mol. Plant 10, 1387–1399. doi: 10.1016/j.molp.2017.09.013
David, R., Burgess, A., Parker, B., Li, J., Pulsford, K., Sibbritt, T., et al. (2017). Transcriptome-wide mapping of RNA 5-methylcytosine in Arabidopsis mRNAs and noncoding RNAs. Plant Cell 29, 445–460. doi: 10.1105/tpc.16.00751
Deng, Y. Y., Li, J. Q., Wu, S. F., Zhu, Y. P., and Fuchu, H. E. (2006). Integrated Nr database in protein annotation system and its localization. Comput. Eng. 32, 71–74. doi: 10.1109/INFOCOM.2006.241
Duitama, J., Srivastava, P. K., and Măndoiu, I. I. (2012). Towards accurate detection and genotyping of expressed variants from whole transcriptome sequencing data. BMC Genomics 13:S6. doi: 10.1186/1471-2164-13-s2-s6
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Elkon, R., Ugalde, A. P., and Agami, R. (2013). Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 14, 496–506. doi: 10.3233/JAD-2009-1076
Feng, Y., Zhang, Y., Ying, C., Wang, D., and Du, C. (2015). Nanopore-based fourth-generation DNA sequencing technology. Genomics Proteomics Bioinformatics 13, 4–16. doi: 10.1016/j.gpb.2015.01.009
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2013). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Gelfman, S., and Ast, G. (2013). When epigenetics meets alternative splicing: the roles of DNA methylation and GC architecture. Epigenomics 5, 351–353. doi: 10.2217/EPI.13.32
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Hsu, H. H., Joung, S. J., Hueter, R. E., and Liu, K. M. (2014). Age and growth of the whale shark (Rhincodon typus) in the north-western Pacific. Mar. Freshw. Res. 65, 1145–1154. doi: 10.1071/MF13330
Kanehisa, M., Susumu, G., Shuichi, K., Yasushi, O., and Masahiro, H. (2004). The KEGG resource fordeciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488. doi: 10.1126/science.1138341
Lee, A. S., Kranzusch, P. J., and Cate, J. H. (2015). eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114. doi: 10.1038/nature14267
Legnini, I., Alles, J., Karaiskos, N., Ayoub, S., and Rajewsky, N. (2019). FLAM-seq: FulllengthmRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods 16, 879–886. doi: 10.1038/s41592-019-0503-y
Li, Y., Fang, C. C., Fu, Y. H., Hu, A., Li, C. C., Zou, C., et al. (2018). A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Res. 25, 421–437. doi: 10.1093/dnares/dsy014
Liang, Z., Riaz, A., Chachar, S., Ding, Y., Du, H., and Gu, X. (2020). Epigenetic modifications of mRNA and DNA in plants. Mol. Plant 13, 14–30. doi: 10.1016/j.molp.2019.12.007
Lima, S. A., Chipman, L. B., Nicholson, A. L., Chen, Y. H., Yee, B. A., Yeo, G. W., et al. (2017). Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 24, 1057–1063. doi: 10.1038/nsmb.3499
Liu, Z. Y., Cao, A., Jiang, L. S., and Cao, S. Y. (2018). Biological function and regulatory mechanism of long non-coding RNA (LncRNA). J. Agric. Biotechnol. 26, 1419–1430. (In Chinese)
Lou, F. R., Song, N., Han, Z. Q., and Gao, T. X. (2020). Single-molecule real-time (SMRT) sequencing facilitates Tachypleus tridentatus genome annotation. Int. J. Biol. Macromol. 147, 89–97. doi: 10.1016/j.ijbiomac.2020.01.029
Mayr, C., and Bartel, D. P. (2009). Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenesin cancer cells. Cell 138, 673–684. doi: 10.1016/j.cell.2009.06.016
Modrek, B., and Lee, C. (2002). A genomic view of alternative splicing. Nat. Genet. 30, 13–19. doi: 10.1038/ng0102-13
Norman, B. M. (2004). Review of the current conservation concerns for the whale shark (Rhincodon typus): a regional perspective. Technical Report (NHT Coast & Clean Seas Project No. 2127), 74.
Nozu, R., Muakumo, K., Matsumoto, R., Nakamura, M., Ueda, K., and Sato, K. (2015). Gonadal morphology, histology, and endocrinological characteristics of immature female whale sharks, Rhincodon typus. Zool. Sci. 32, 455–458. doi: 10.2108/zs150040
Nudelman, G., Frasca, A., Kent, B., Sadler, K. C., Sealfon, S. C., Walsh, M. J., et al. (2018). High resolution annotation of zebrafish transcriptome using long-read sequencing. Genome Res. 28, 1415–1425. doi: 10.1101/gr.223586.117
Ou, M. Y., Xiao, Q., Ju, X. C., Zeng, P. M., Huang, J., Sheng, A. L., et al. (2021). The CTNNBIP1-CLSTN1 fusion transcript regulates human neocortical development. Cell Rep. 35:109290. doi: 10.1016/j.celrep.2021.109290
Ozsolak, F., and Milos, P. M. (2011). RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98. doi: 10.1038/nrg2934
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415. doi: 10.1038/ng.259
Postlethwait, J. H., Yan, Y. L., Gates, M. A., Horne, S., Amores, A., Brownlie, A., et al. (1998). Vertebrate genome evolution and the zebrafish gene map. Nat. Genet. 18, 345–349. doi: 10.1038/ng0498-345
Roach, N. P., Sadowski, N., Alessi, A. F., Timp, W., Taylor, J., and Kim, J. K. (2020). The full-length transcriptome of Caenorhabditis elegans using direct RNA sequencing. Genome Res. 30, 299–312. doi: 10.1101/gr.251314.119
Rowat, D., and Brooks, K. S. (2012). A review of the biology, fisheries and conservation of the whale shark Rhincodon typus. J. Fish Biol. 80, 1019–1056. doi: 10.1111/j.1095-8649.2012.03252.x
Sequeira, A. M., Mellin, C., Floch, L., Williams, P. G., and Bradshaw, C. J. (2014). Inter-ocean asynchrony in whale shark occurrence patterns. J. Exp. Mar. Biol. Ecol. 450, 21–29. doi: 10.1016/j.jembe.2013.10.019
Sharon, D., Tilgner, H., Grubert, F., and Snyder, M. (2013). A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 31, 1009–1014. doi: 10.1038/nbt.2705
Smith, A. (1829). Description of new, or imperfectly known objects of the animal kingdom, found in the south of Africa. Afr. Comm. Advertiser 3:2.
Squires, J. E., Patel, H. R., Marco, N., Humphreys, D. T., Parker, B. J., Suter, C. M., et al. (2012). Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40, 5023–5033.
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467. doi: 10.1093/nar/gki458
Steijger, T., Abril, J. F., Engström, P. G., Kokocinski, F., and Consortium, R. (2013). Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 10, 1177–1184. doi: 10.1038/nmeth.2714
Stevens, J. D. (2007). Whale shark (Rhincodon typus) biology and ecology: a review of the primary literature. Fish. Res. 84, 4–9. doi: 10.1016/j.fishres.2006.11.008
Tatusov, R. L., Galperin, M. Y., and Natale, D. A. (2000). The COG database: a tool for genomescale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
The Gene Ontology Consortium, Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., et al. (2000). Gene ontology: tool forthe unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Tilgner, H., Raha, D., Habegger, L., Mohiuddin, M., Gerstein, M., and Snyder, M. (2013). Accurate identification and analysis of human mRNA isoforms using deep long read sequencing. G3 3, 387–397. doi: 10.1534/g3.112.004812
Wan, H. F., Jia, X. W., Zou, P. E., Zhang, Z. P., and Wang, Y. L. (2019). The Single-molecule long-read sequencing of Scylla paramamosain. Sci. Rep. 9:12401. doi: 10.1038/s41598-019-48824-8
Wang, B., Tseng, E., Regulski, M., Clark, T. A., Hon, T., Jiao, Y. P., et al. (2016). Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 7:11708. doi: 10.1038/ncomms11708
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Weber, J. A., Park, S. G., Luria, V., Jeon, S., Kim, H. M., Jeon, Y., et al. (2020). The whale shark genome reveals how genomic and physiological properties scale with body size. Proc. Natl. Acad. Sci. U.S.A. 117, 20662–20671. doi: 10.1073/pnas.1922576117
Wu, C. S., Yu, C. Y., Chuang, C. Y., Hsiao, M., Kao, C. F., Kuo, H. C., et al. (2014). Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 24, 25–36. doi: 10.1101/gr.159483.113
Wu, T. D., and Watanabe, C. K. (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. doi: 10.1093/bioinformatics/bti310
Wyman, D., Balderrama-Gutierrez, G., Reese, F., Jiang, S., Rahmanian, S., Forner, S., et al. (2019). A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. bioRxiv [preprint] doi: 10.1101/672931
Xie, Z., Babiceanu, M., Kumar, S., Jia, Y., Qin, F., Barr, F. G., et al. (2016). Fusion transcriptome profiling provides insights into alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. U.S.A. 113, 13126–13131. doi: 10.1073/pnas.1612734113
Yao, J., Ding, D., Li, X. P., Shen, T., Fu, H. H., Zhong, H., et al. (2020). Prevalent intron retention fine-tunes gene expression and contributes to cellular senescence. Aging Cell 19:e13276. doi: 10.1111/acel.13276
Keywords: Rhincodon typus, full-length transcriptome, ONT sequencing, genome annotation, transcriptome structure
Citation: Lou F, Wang L, Wang Z, Wang L, Zhao L, Zhou Q, Lu Z and Tang Y (2022) Full-Length Transcriptome of the Whale Shark (Rhincodon typus) Facilitates the Genome Information. Front. Mar. Sci. 8:821253. doi: 10.3389/fmars.2021.821253
Received: 24 November 2021; Accepted: 23 December 2021;
Published: 07 February 2022.
Edited by:
Hui Zhang, Institute of Oceanology, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Chen Jiang, Dalian Ocean University, ChinaLingfeng Kong, Ocean University of China, China
Copyright © 2022 Lou, Wang, Wang, Wang, Zhao, Zhou, Lu and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongzheng Tang, MTM5MDYzODAwNjNAMTYzLmNvbQ==; Zhichuang Lu, bHV6aGljaHVhbmdAaG90bWFpbC5jb20=