 Beining Zhang
Beining Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 April 2025
Sec. Comparative Immunology
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1587414
Heat shock protein 70 (HSP70) is a highly conserved molecular chaperone that plays a core role in assisting protein folding and maintaining cellular homeostasis. In recent years, studies have revealed that HSP70 has dual functions in immune regulation: on the one hand, it enhances immune responses by activating non-specific immunity (such as Toll-like receptor 2/4 (TLR2/4) signaling pathways) and specific immunity (such as cross-presentation of antigens, T helper 1 (Th1)/T helper 17 (Th17) differentiation); on the other hand, it inhibits excessive immune reactions by inducing the differentiation of regulatory T cells (Treg) and promoting the secretion of anti-inflammatory factors [such as interleukin-10 (IL-10)]. In cancer, the duality of HSP70 is also very prominent: it can drive tumor progression through pathways such as inhibiting apoptosis, promoting angiogenesis, and tumor metastasis, and it can also inhibit tumor growth by activating immunogenic cell death (ICD), enhancing antigen presentation, and natural killer (NK) cell activity. This review aims to systematically analyze the immune regulatory functions of HSP70, focusing on its dual regulatory mechanisms and the “double-edged sword” nature of HSP70 in tumor immunotherapy and the innovative nature of targeted strategies, as well as providing a theoretical basis and research directions for precision medicine in the treatment strategies of related diseases.
The heat shock protein family (HSPs), as a group of highly conserved molecular chaperones, exhibits remarkable conservation throughout biological evolution (1–4). Studies have shown that HSPs are expressed in a wide range of organisms, from prokaryotes to eukaryotes, including bacteria and humans (1, 3, 5). HSPs play a crucial role in maintaining cellular protein homeostasis under both physiological and stress conditions. Their functions are mainly reflected in two aspects: one is to assist the correct folding of newly synthesized proteins, and the other is to prevent the aggregation of misfolded proteins (6–8), thus effectively avoiding cellular damage (5).
Based on the differences in molecular weight, HSPs can be divided into several subfamilies, including HSP100, HSP90, HSP70, HSP60, and small HSPs. Among them, HSP70 (70 kDa heat shock protein) has attracted much attention due to its multiple functions in protein folding, degradation, and stress adaptation (9). This protein can be induced by a variety of stress factors, including heat shock, oxidative stress, inflammation, and hypoxia (10). Inside the cell, HSP70 mainly functions as a molecular chaperone (11–13). Its main mode of action is to bind to the exposed hydrophobic regions of nascent or misfolded polypeptides, using ATP-dependent conformational changes to promote the correct folding of proteins (14)and prevent the aggregation of misfolded proteins (15, 16). The roles of HSP70 in non-tumor diseases and physiological processes are also remarkable. For instance, in neurodegenerative diseases like Alzheimer’s and Parkinson’s, HSP70 promotes the clearance of misfolded proteins (17–19). In cardiovascular diseases, HSP70 also mitigates myocardial ischemia-reperfusion injury by stabilizing mitochondrial integrity (20).
It is worth noting that in addition to its function in molecular chaperone, non-tumor diseases and physiological processes, studies have also found that under specific conditions, HSP70 can participate in the immune regulation process.
The immune system, as the body’s defense mechanism, consists of two main parts: non-specific immunity and specific immunity. Non-specific immunity rapidly recognizes pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through pattern recognition receptors (PRRs), thereby activating inflammatory responses and initiating adaptive immune responses (21–23). Specific immunity, on the other hand, establishes long-term immune memory through antigen-specific responses of T lymphocytes and B lymphocytes. The synergistic effect of these two immune mechanisms can not only effectively resist pathogen infections but also eliminate abnormal cells (such as tumor cells) and avoid self-tissue damage through immune tolerance mechanisms. However, dysfunction of the immune system can lead to a variety of diseases. For example, excessive activation may trigger autoimmune diseases, while immune suppression may promote tumor immune escape (24, 25).
Research has found that HSP70 exhibits dual roles in immune regulation: on the one hand, it can enhance antigen presentation efficiency and activate non-specific immune responses (25–27); on the other hand, it can also exert immune suppressive effects by promoting the differentiation of regulatory T cells (Treg) (28, 29) and inhibiting pro-inflammatory cytokines (30, 31). This dual regulatory characteristic is particularly prominent in tumor immunity: HSP70 can both promote anti-tumor immune responses (through cross-presentation of tumor antigens) and support tumor progression by facilitating immune escape and angiogenesis.
This review aims to systematically analyze the immune regulatory functions of HSP70, focusing on its dual regulatory mechanisms and the “double-edged sword” nature of HSP70 in tumor immunotherapy and the innovative nature of targeted strategies, as well as providing a theoretical basis and research directions for precision medicine in the treatment strategies of related diseases.
HSP70 comprises two key functional domains: the N-terminal ATPase domain (NBD, approximately 45 kDa) and the C-terminal substrate-binding domain (SBD, approximately 25 kDa) (32). The N-terminal domain possesses ATP binding and hydrolysis activities, providing the essential energy source for protein conformational changes (33, 34). The C-terminal domain consists of a β-sheet subdomain and an α-helical “lid” region. The former is responsible for recognizing and binding to unfolded or misfolded substrate proteins, while the latter regulates the binding and release of substrates (35). These two domains are connected by a linker (13 amino acids) (36) and function independently in both free and bound states.
The molecular function of HSP70 relies on the allosteric regulation mechanism between the N-terminal and C-terminal domains, which is driven by the conversion of ATP to ADP (37). Through this precise allosteric regulation, HSP70 can effectively facilitate the binding and release of substrates, providing misfolded proteins with the opportunity to correctly refold, thereby fulfilling its molecular chaperone function (38, 39).
As an essential molecular chaperone within cells, HSP70 is widely distributed in the cytoplasm, endoplasmic reticulum, and mitochondria. It assists in the correct folding of nascent polypeptide chains after their release from the ribosome, promoting the folding and formation of the three-dimensional structure of newly synthesized polypeptide chains (11, 12, 38). Under adverse conditions such as heat shock, hypoxia, or oxidative stress, the expression level of HSP70 is significantly upregulated. It maintains cellular homeostasis by repairing misfolded proteins or directing them to the proteasome for degradation.
HSP70 exerts anti-apoptotic and pro-survival functions through multiple pathways: it can directly bind to apoptotic protease activating factor-1 (Apaf-1), inhibiting the activation of caspase-9 and thus blocking the transmission of apoptotic signals. HSP70 also stabilizes the mitochondrial membrane by preventing the oligomerization of Bax/Bak proteins, effectively inhibiting the release of cytochrome c. Additionally, HSP70 is involved in regulating the balance between autophagy and apoptosis, with mechanisms that may involve inhibiting JNK phosphorylation or interacting with Beclin-1. These multiple protective mechanisms together form the important molecular basis for HSP70 to maintain cell survival.
HSP70 not only possesses the canonical functions of a molecular chaperone, such as participating in the regulation of protein homeostasis and stress responses, but also plays a complex bidirectional regulatory role in the immune system (Figure 1). The dual nature of its immune regulatory function is reflected in two aspects: On the one hand, it enhances immune responses by activating pattern recognition receptors and the antigen presentation system. On the other hand, it maintains immune tolerance by inducing the differentiation of immunosuppressive cells. This dynamic regulatory mechanism makes HSP70 a key molecule linking innate and adaptive immunity.
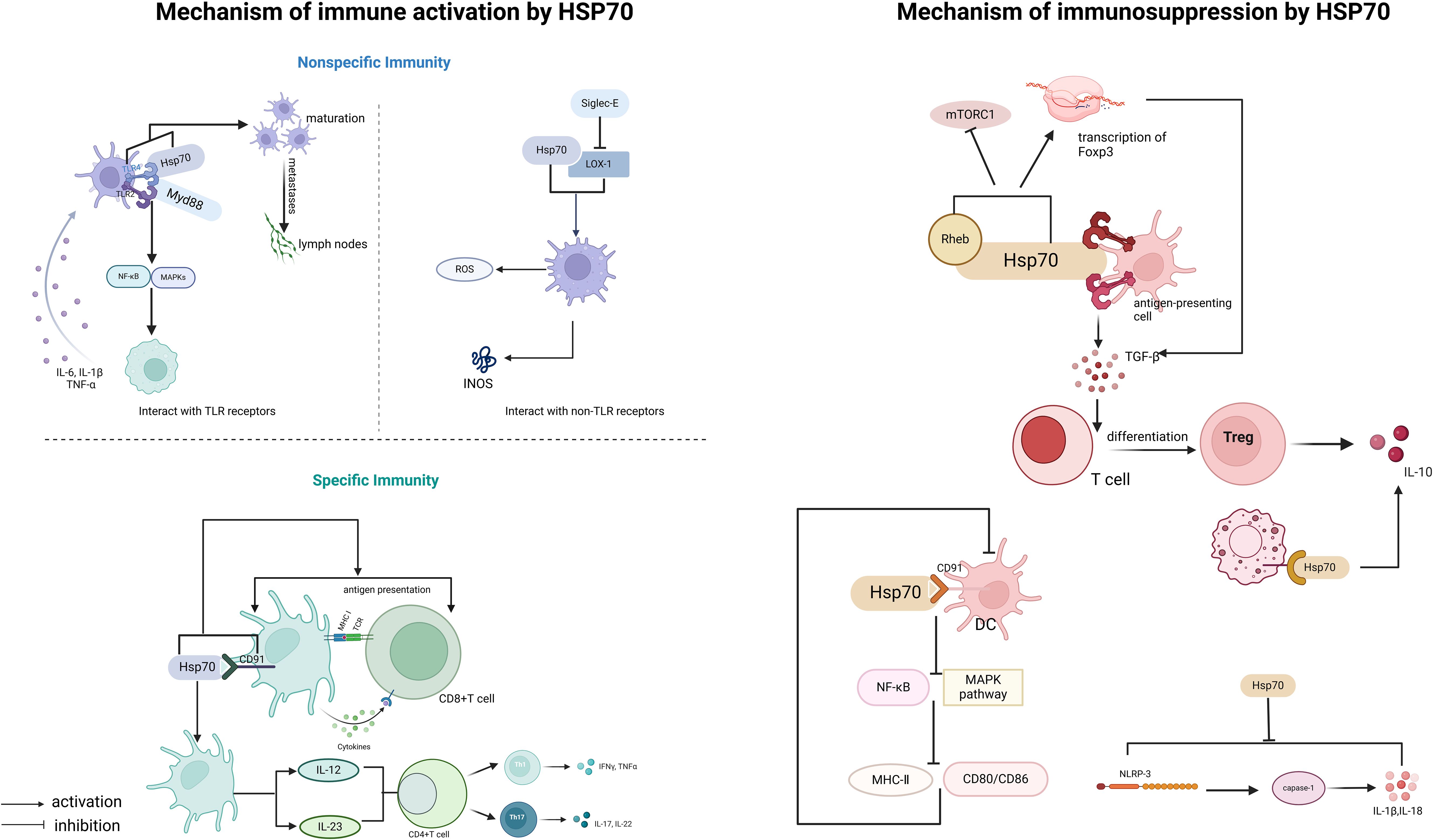
Figure 1. Immunoregulatory functions of HSP70.
The immune-activating function of HSP70 is mainly reflected in two aspects: non-specific immunity and specific immunity (40). Its function is characterized by multidimensionality: In terms of space, it can initiate innate immune responses through cell membrane surface receptors [such as Toll-like receptor 2/4 (TLR2/4)] and also activate specific immunity through the intracellular antigen processing system. In terms of time, it is involved in triggering early inflammatory responses and also affects the formation of late immune memory. The specific mechanisms of action include:
Non-specific immunity is the body’s innate defense system, characterized by broad-spectrum, rapid response, and lack of memory. Its defense mechanisms include physical barriers (such as skin and mucous membranes), chemical barriers (such as lysozyme and stomach acid), immune cells (such as macrophages and natural killer cells), and cytokines.
Damage-associated molecular patterns (DAMPs) are endogenous molecules released by damaged or dying cells, which can be recognized by the immune system and trigger immune responses (21, 41). DAMPs are mainly recognized by the immune system through two mechanisms: one is through pattern recognition receptors (PRRs) (41), and the other is through inducing the activation of pro-inflammatory cytokines such as IL-18 (42, 43). Studies have found that extracellular HSP70 (eHSP70), as a DAMP, can bind to antigen-presenting cells (such as dendritic cells (DCs) and macrophages) through TLR2 and TLR4 and be recognized by the immune system (44), thereby activating signaling pathways.
This binding can trigger a MyD88-dependent signaling cascade, leading to the activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs) (30). The activation of NF-κB further promotes the expression and secretion of pro-inflammatory cytokines (such as tumor necrosis factor-alpha(TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β)), amplifying the inflammatory response and enhancing the activation of antigen-presenting cells (APCs) (45). Thus, eHSP70 regulates the inflammatory response through the TLR/MyD88 signaling pathway. Studies have also found that in airway epithelial cells, eHSP70 inhibits the pro-inflammatory ERK1/2-CREB signaling pathway activated by TGF-β1 through the TLR4-GR-DUSP1 signaling pathway (31), reducing the secretion of inflammatory factors. Moreover, the HSP70-TLR4 signaling pathway promotes DC maturation, upregulates the expression of co-stimulatory molecules (such as CD80/86), and promotes the migration of DCs to the lymph nodes (46, 47), thereby bridging non-specific immunity and specific immunity.
HSP70 can also regulate non-specific immune responses through interactions with Siglec-E and oxidized low-density lipoprotein receptor (LOX-1). The binding of HSP70 to LOX-1 can activate macrophages (48), which produce reactive oxygen species (ROS) during the inflammatory response; meanwhile, macrophages, especially the M1 type, produce a large amount of NO through the expression of inducible nitric oxide synthase (iNOS), enhancing their antimicrobial defense capabilities (49). In contrast, Siglec-E acts as a negative regulator of LOX-1-mediated non-specific immune responses when bound to HSP70 or oxidized LDL. This mechanism helps to activate the immune response, regulate the inflammatory response, and avoid excessive inflammation (50).
Specific immunity is acquired and mainly mediated by T cells and B cells, characterized by high specificity and memory. According to the cell types mediating the immune response, specific immunity is divided into humoral immunity (mediated by B cells) and cellular immunity (mediated by T cells).
Cross-presentation of antigens refers to the process by which antigen-presenting cells (mainly DCs) process exogenous antigens (such as viral or tumor antigens) and present them to CD8+ T cells, thereby activating adaptive immunity. As a molecular chaperone, HSP70 can assist in the folding and stabilization of antigens, promoting their intracellular transport and processing.
CD91 is a multifunctional receptor on the surface of dendritic cells, involved in endocytosis and intercellular signaling (51, 52). Studies have found that the binding of HSP70 to CD91 can promote the internalization of HSP70-antigen complexes by DCs, enhancing antigen processing and presentation efficiency (53). Sec61 is a transmembrane protein complex in the endoplasmic reticulum, mainly responsible for protein translocation and involved in the process of antigens being released from the cytosol into the cytoplasm. HSP70 can interact with Sec61 to promote antigen transport (54), playing a key role in the rocess of cross-antigen presentation.
T helper 1 (Th1) and T helper 17 (Th17) cells both originate from naive CD4+ T cells and differentiate into different effector T cell subsets with distinct functions under specific cytokine environments (55, 56). Th1 cells are mainly responsible for mediating cellular immune responses and clearing intracellular pathogens, while Th17 cells mediate pro-inflammatory responses by promoting the mobilization and activation of neutrophils (56).
HSP70 promotes T cell differentiation by regulating the cytokine environment. HSP70-activated DCs secrete IL-12 and IL-23 (57, 58), which induce the differentiation of Th1 and Th17 cells, respectively (59). Th1 cells secrete interferon-gamma (IFN-γ) to enhance the activity of cytotoxic T cells (CTLs), while Th17 cells recruit neutrophils through IL-17, working together to clear pathogens.
HSP70 not only plays a role in immune activation but also acts as an important regulator in immune suppression. Its immune-suppressive effects are mainly reflected in the induction of regulatory immune cells and the regulation of anti-inflammatory signaling pathways.
Regulatory T cells (Tregs) play a key role in immune tolerance and the suppression of excessive immune responses. Studies have found that HSP70 can bind to TLR2/TLR4 on the surface of antigen-presenting cells (APCs) (44, 60), inducing APCs to secrete TGF-β, which in turn promotes the differentiation of naive T cells into Tregs (61). Rheb is an upstream activator of mTORC1, and HSP70 can bind to Rheb and induce its degradation, thereby inhibiting the activity of mTORC1 (62) and enhancing Foxp3 transcription (63). Foxp3 can inhibit the production of pro-inflammatory cytokines while promoting the secretion of immune-suppressive cytokines (such as interleukin-10 (IL-10) and TGF-β), maintaining the immune-suppressive function of Treg cells and thus sustaining an immune-suppressive microenvironment. Additionally, extracellular HSP70 (eHSP70) binds to the CD91 receptor on the surface of DCs (53), inhibiting the NF-κB and MAPK pathways, downregulating the expression of co-stimulatory molecules (CD80/86) and major histocompatibility complex class II (MHC-II), leading to DC dysfunction and weakened antigen-presenting capabilities (64).
IL-10 is an important anti-inflammatory factor that can inhibit the production of pro-inflammatory cytokines and suppress the function of antigen-presenting cells. Studies have found that HSP70 in MSCs-Exo can reduce the inflammatory response after liver transplantation and improve liver function by activating IL-10 secretion (65). HSP70 can bind to receptors on the surface of macrophages, activate downstream signaling pathways, and promote IL-10 secretion (66), helping to maintain the balance of the immune system and prevent excessive immune responses from causing damage to the body.
The NLRP3 inflammasome is an important component of the non-specific immune system, capable of activating caspase-1 (interleukin-1β-converting enzyme), promoting the maturation and release of IL-1β and IL-18 (67), and triggering a strong inflammatory response. HSP70 can inhibit the NLRP3 inflammasome through several mechanisms (68–70). From the study, it is evident that the absence of HSP70 exacerbates NLRP3-dependent peritonitis and enhances caspase-1 activation and IL-1β production in bone marrow-derived macrophages (BMDMs) of mice, while also increasing the number and size of ASC/NLRP3 specks (68). Conversely, overexpression of HSP70 inhibits these processes. HSP70 can also directly interact with NLRP3, and this interaction disappears upon NLRP3 inflammasome activation (20, 68), suggesting that HSP70 may inhibit NLRP3 inflammasome formation by binding to NLRP3 and altering its conformation. Additionally, HSP70 significantly reduces the LPS (lipopolysaccharide)-induced elevation of NLRP3 protein levels, thereby suppressing the formation and activation of the NLRP3 inflammasome (20).
Upon comprehensively analyzing the roles of HSP70 in immune activation and immune suppression, we found that HSP70 binding to the same type of receptor on the same kind of cell may induce different cytokines and even exert completely opposite effects on immune regulation. For example, HSP70 binding to TLR2/TLR4 on antigen-presenting cells (APCs) can trigger the MyD88 cascade to activate the immune response (71), as well as induce APCs to secrete TGF-β (61), which promotes the differentiation of naive T cells into regulatory T cells (Tregs) to suppress the immune response. Similarly, HSP70 binding to the CD91 receptor can enhance antigen presentation capacity, but it can also weaken this capacity by inhibiting the NF-κB and MAPK pathways. Its effects are highly dependent on factors such as co-stimulatory signals in the microenvironment (e.g., cytokine concentrations, oxidative stress levels).
HSP70 can not only promote tumor progression by facilitating cancer cell proliferation, metastasis, and drug resistance but also enhance the body’s anti-tumor immune response by activating immune cells and mediating immune responses (72, 73).
Studies have shown that HSP70 is highly expressed in various tumor cells and exerts pro-cancer effects by inhibiting apoptosis (74). HSP70 exerts its anti-apoptotic effects by regulating key effector molecules in apoptotic signaling pathways.
In the intrinsic apoptosis pathway, the HSP70-CHIP complex can promote the proteasomal degradation of apoptosis signal-regulating kinase 1 (ASK1), thereby inhibiting the activation of c-Jun N-terminal kinase (JNK) and p38 (75). Notably, the activation of JNK is crucial for the release of cytochrome C from mitochondria and the initiation of the endogenous apoptotic pathway (76). HSP70 can also inhibit apoptosis through the following mechanisms: ① inhibiting the translocation of pro-apoptotic protein Bax to the mitochondrial outer membrane and preventing Bax oligomerization to form pores, thereby inhibiting the release of cytochrome C and other apoptotic factors from mitochondria to the cytoplasm (77); ② inhibiting Bcl-2 transcription and blocking Bax translocation to mitochondria, thus preventing an increase in mitochondrial outer membrane permeability (78, 79); ③ binding to the CARD domain of Apaf-1, preventing the recruitment of caspase-9 to the apoptosome (73).
In the extrinsic apoptotic pathway, HSP70 exerts anti-apoptotic effects through the following mechanisms: ① interacting with TNF-related apoptosis-inducing ligand receptors 1 (TRAIL-R1) and TRAIL-R2, blocking the formation of the death-inducing signaling complex (DISC); ② forming a complex with FANCC protein, inhibiting the activation of the extrinsic apoptotic pathway (80, 81); ③ in BCR-ABL expressing cells, binding to death receptors DR4 and DR5, inhibiting the formation of apoptotic signaling complexes (82). Additionally, HSP70 can also inhibit the activation of JNK and p38 induced by DNA damage (83), which play key roles in apoptotic signaling (Figure 2).

Figure 2. Key effector molecules regulated by HSP70 in intrinsic and extrinsic apoptosis signaling pathways.
The epithelial-mesenchymal transition (EMT) is an important process for tumor cells to acquire invasive and metastatic capabilities, characterized by changes in cell morphology, reduced cell adhesion, and enhanced migration and invasion abilities. It has been found that HSP70 overexpression can promote the EMT process and tumor metastasis by activating signaling pathways such as NF-κB and upregulating the expression of mesenchymal markers like N-cadherin (84).
The interaction between HSP70 and vascular endothelial growth factor (VEGF) is also noteworthy. VEGF, a member of the platelet-derived growth factor family, plays a crucial role in tumor angiogenesis and vascular permeability regulation. VEGF is highly expressed in the tumor microenvironment, promoting tumor angiogenesis to provide nutrients and oxygen to tumors, thereby facilitating tumor growth and metastasis.
Clinical studies have found that the levels of HSP70 and VEGF in the serum of pancreatic cancer patients are significantly increased (85), indicating that HSP70 may promote VEGF expression through specific mechanisms.In hepatocellular carcinoma cells HepG2, extracellular HSP70/HSP70-PCs can upregulate VEGF expression through the HIF-1α (hypoxia-inducible factor-1α) signaling pathway. However, in some cases, HSP70 may have the opposite effect. For example, in osteoblasts, HSP70 negatively regulates VEGF synthesis by inhibiting p38 MAPK phosphorylation (86). From a cellular functional perspective, tumor cells typically reside in a hypoxic and nutrient-deprived tumor microenvironment, which drives HSP70 to promote angiogenesis to support tumor cell proliferation, invasion, and metastasis. Specifically, HSP70 stabilizes hypoxia-inducible factor-1α (HIF-1α), preventing its degradation via the ubiquitin-proteasome pathway, thereby facilitating the direct binding of HIF-1α to the VEGF promoter, which drives VEGF transcription and expression, ultimately enhancing angiogenesis. In contrast, in osteoblasts, due to the demands of mechanical stress and bone homeostasis maintenance, HSP70 tends to suppress angiogenesis to prioritize bone mineralization and repair processes. In osteoblasts, HSP70 negatively regulates VEGF synthesis by inhibiting the activation of p38 MAP kinase. Based on these findings, we hypothesize that the differential regulatory effects of HSP70 on VEGF may stem from the distinct microenvironments, functional requirements, and signaling pathways mediated by different cell types.
Therefore, the regulatory role of HSP70 in VEGF expression exhibits significant microenvironment dependency, and its specific mechanisms require in-depth analysis based on specific experimental conditions, disease contexts, and research objectives. However, current research on HSP70-mediated VEGF regulation in non-tumor cells remains limited. Future studies are needed to further explore the diverse functions of HSP70 across different cell types and physiological or pathological conditions, to comprehensively elucidate its regulatory networks, and to provide new theoretical foundations and potential therapeutic targets for precision medicine in related diseases.
Tumor immune escape is a key process in the development of malignant tumors, essentially involving tumor cells evading recognition and elimination by the host immune system through various molecular mechanisms, thereby allowing continuous growth and spread within the host. This complex process involves multiple immune checkpoints, where HSP70 plays a significant role.
The tumor microenvironment (TME) is a complex ecosystem, including the extracellular matrix, immune cells, fibroblasts, vascular endothelial cells, and various cytokines and metabolic products (87, 88). In this unique environment, HSP70 can promote immune escape through multiple mechanisms, such as inhibiting the activity and function of T cells and natural killer (NK) cells; HSP70 can also affect the maturation of dendritic cells (DCs), interfering with their antigen-presenting functions, thereby reducing the activation efficiency of T cells (73).
In the tumor microenvironment, myeloid-derived suppressor cells (MDSCs) are a group of myeloid cells with immune-suppressive functions. HSP70 can promote immune escape by interacting with MDSCs. Studies have found that 5-fluorouracil (5-FU) can selectively eliminate MDSCs while activating the NLRP3 inflammasome (89). This process is accompanied by the activation of caspase-1 and the production of IL-1β, ultimately exerting anti-tumor effects (89). Research has shown that HSP70 on the surface of exosomes can interact with TLR2 on MDSCs, thereby activating the immune-suppressive functions of MDSCs (90). The mechanisms are as follows. After tumor - derived exosomal HSP70 binds to TLR2 on the surface of MDSCs, it activates STAT3 phosphorylation, induces the expression of arginase 1 (Arg1) and inducible nitric oxide synthase (iNOS), and enhances the immunosuppressive activity of MDSCs (90). Meanwhile, the HSP70 - TLR2 signal can also promote the secretion of IL - 10 through the ROS - dependent ERK1/2 pathway, further inhibiting the function of CD8+ T cells. The tumor microenvironment is often in a hypoxic and acidic state. Based on the activation of the STAT3 pathway by HSP70 - exosomes through the TLR2 - MDSCs axis (91), it may further amplify the immunosuppressive effect mediated by HSP70 by regulating the membrane localization or signal transduction efficiency of TLR2.
The programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) signaling pathway, as a key immune checkpoint, plays a core role in maintaining immune homeostasis and tumor immune escape. HSP70 participates in the regulation of this pathway through various molecular mechanisms:
1. Epigenetic regulation: HSP70 can enhance histone acetyltransferase (HAT) activity, promoting the histone H3K27ac modification in the PD-L1 promoter region, thereby upregulating PD-L1 transcription.
2. Signal transduction pathway: HSP70 binding to TLR4 activates the NF-κB pathway (45), inducing the co-expression of PD-L1 and IL-10, forming an immune-suppressive microenvironment (92).
3. Protein stability regulation: As a molecular chaperone, HSP70 can stabilize PD-L1 protein, preventing its degradation, thereby increasing PD-L1 expression on the cell surface (73).
However, Hsc70, a member of the HSP70 family, plays an opposing regulatory role. It can promote the lysosomal degradation of PD-L1 by competitively binding to it, thereby reducing the expression of PD-L1 on the cell membrane (93). Specifically, Hsc70 targets PD-L1 for lysosomal degradation through the endosomal microautophagy (eMI) pathway and competitively inhibits CMTM6-mediated recycling of PD-L1 (93).
Although Hsc70 and Hsp70 exhibit only minor differences in amino acid sequences, they display significant distinctions in expression patterns, subcellular localization, functions, and regulatory mechanisms. Firstly, Hsc70 is constitutively expressed, whereas the expression of Hsp70 is typically induced under stress conditions. Hsc70 is primarily localized in the cytoplasm but can shuttle between the cytoplasm and the nucleus under specific conditions, participating in the regulation of nucleocytoplasmic transport. In contrast, Hsp70 predominantly functions as a molecular chaperone under stress conditions.
In terms of function, Hsc70 is not only involved in classical chaperone activities but also plays a critical role in protein degradation, particularly by facilitating the degradation of specific proteins through chaperone-mediated autophagy (CMA) and endosome-associated microautophagy (EmiA) (94, 95). Additionally, Hsc70 protects the integrity of lysosomal membranes (96). In comparison, Hsp70 is more inclined to stabilize proteins or regulate their functions to support cell survival and proliferation.
Moreover, Hsc70 exhibits unique characteristics in secretion and plasma membrane localization. Its secreted form can be induced by contact inhibition or serum deprivation, and this secreted form has been shown to inhibit cell proliferation and promote contact inhibition (97), a feature that Hsp70 does not possess. Regarding immune regulation, both Hsc70 and Hsp70 can be released into the extracellular space during viral infections or upon cytokine stimulation. These differences enable Hsc70 and Hsp70 to play both overlapping and distinct roles in maintaining cellular homeostasis and contributing to disease pathogenesis (97).
The MerTK signaling pathway, as an important cell signaling system, plays a key role in regulating apoptosis, phagocytosis, and immune regulation. In the tumor microenvironment, the expression level of MerTK is closely related to immune escape, mainly involving the functional regulation of tumor-associated macrophages (TAMs). Studies have shown that MerTK-positive TAMs typically exhibit significant immune-suppressive characteristics, inhibiting T cell function by expressing immune checkpoint molecules such as PD-L1 (98), thereby promoting tumor immune escape. This finding makes MerTK a potential target for cancer therapy.
Recent studies have found that HSP70 secreted by tumor cells can bind to TLR2 on the surface of macrophages, forming an HSP70-TLR2 complex, which upregulates MerTK expression and promotes the polarization of macrophages to the M2 type (66). This process further enhances the immune-suppressive characteristics of the tumor microenvironment. Therefore, intervention in the HSP70-TLR2-MerTK signaling pathway may provide new strategies for cancer treatment.
The anti-tumor effects of HSP70 in tumor immunity are mainly reflected in its ability to activate the immune system and enhance the body’s immune surveillance and killing capabilities against tumors.
Immunogenic cell death is a unique form of cell death that can trigger specific immune responses in hosts with normal immune functions. As an important member of damage-associated patterns, HSP70 plays a key role in the ICD process. When tumor cells undergo ICD, the released HSP70 can activate APCs and enhance their antigen-presenting capabilities. By promoting ICD, HSP70 can significantly increase the immunogenicity of tumor cells, thereby enhancing the anti-tumor immune response.
Tumor-derived exosomes are rich in HSP70, and these exosomes can significantly induce the body’s anti-tumor immune response (99). Studies have found that HSP70 can selectively activate NK cells by binding to receptors on the surface of NK cells. Notably, exosomes or tumor cell lysates lacking HSP70 do not possess this activation capability (100). Further research has indicated that TREM-1 (Triggering Receptor Expressed on Myeloid cells-1) receptors expressed on the surface of myeloid cells and NK cells may be involved in this process (101), but the specific molecular mechanisms still need to be further clarified.
HSP70 can form stable complexes with tumor antigen peptides. These complexes are internalized by DCs through the CD91 receptor on their surface and then transported to the cytoplasm via the endosomal escape mechanism. In the cytoplasm, these antigen peptides are processed by proteasomes and bind to MHC-I molecules, ultimately activating CD8+ T cells and mediating specific anti-tumor immune responses. This mechanism has been verified in melanoma and lung cancer models, confirming that HSP70-antigen complexes can elicit a strong anti-tumor immune response (102).
Current research findings indicate that HSP70 exhibits a complex bidirectional regulatory effect on immune-related proteins, such as its bidirectional regulation of VEGF and the PD-1/PD-L1 axis. This regulatory characteristic may be dynamically regulated by epigenetic modifications in the tumor microenvironment, suggesting that the pro-cancer or anti-cancer effects of HSP70 are environment-dependent. This discovery provides a new perspective for a deeper understanding of the role of HSP70 in tumor immunity and also presents challenges for the development of precision treatment strategies based on HSP70.
Based on the dual role of HSP70 in immune regulation, researchers have developed various potential therapeutic strategies, providing new ideas and directions for tumor treatment (Figure 3).
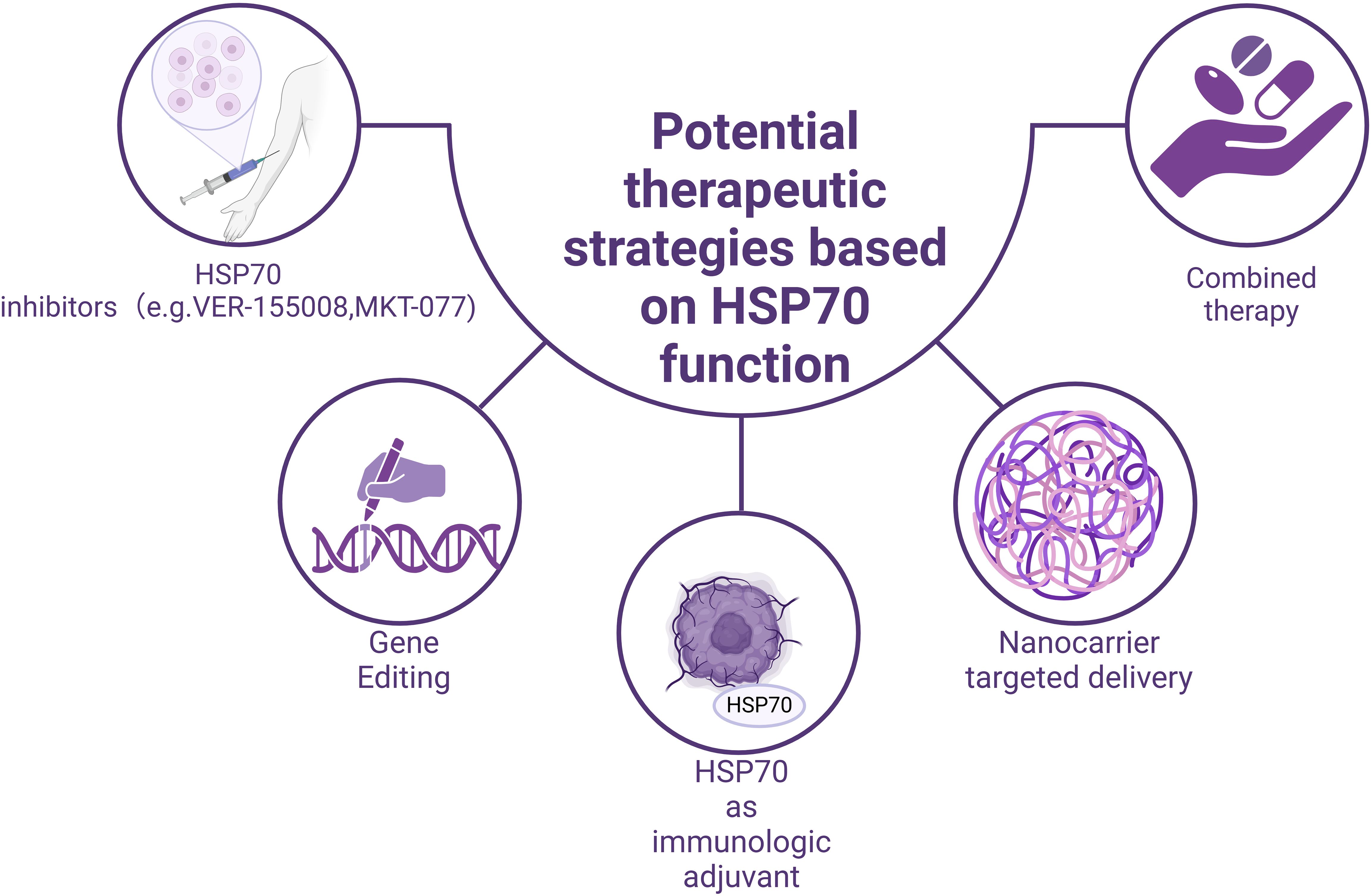
Figure 3. Potential therapeutic strategies based on the functions of HSP70.
Given that HSP70 mediates tumor immune evasion in the tumor microenvironment through mechanisms such as promoting the differentiation of Treg cells, the development of specific HSP70 inhibitors may help restore the body’s anti-tumor capabilities. Several small-molecule inhibitors have shown promising application prospects (Table 1). For example,VER-155008, a highly selective HSP70 inhibitor, exerts its anti-tumor function by competitively binding to the ATP-binding site of HSP70 (114–117). In experiments related to pheochromocytoma, VER-155008 was found to significantly inhibit the proliferation of PC12 cells (a cell line derived from rat adrenal pheochromocytoma, PHEO) in vitro. After two weeks of treatment, the tumor volume was markedly reduced from 2584.7 ± 525.6 mm³ to 1253.9 ± 157.8 mm³ (P < 0.001) (103); Another representative inhibitor, MKT-077, a cationic rhodamine dye (118), regulates tumor cell proliferation by inhibiting the chaperone function of the HSP70 family (119, 120). The study found that the concentration of MKT-077 was positively correlated with its inhibitory effect on tumor cells. When the concentration of MKT-077 reached 2 µg/ml or higher, it exhibited significant cytotoxicity against multiple tumor types (121), for example, MKT-077 selectively accumulates in mitochondria within medullary thyroid carcinoma (MTC) cells, disrupts mitochondrial activity, induces oxidative stress and apoptosis, thereby inhibiting cell proliferation and survival. Experimental data show that after 48 hours of MKT-077 treatment, the survival rate of TT cells significantly decreased, with an IC50 value of 0.74 μm, indicating its potent cytotoxicity against MTC cells (104, 105). Besides, in estrogen receptor-positive breast cancer, S1g-2, acting as a specific inhibitor of the Hsp70-Bim protein-protein interaction, can overcome tamoxifen resistance. Studies using xenograft models have shown that disrupting the Hsp70-Bim interaction with S1g-2 can reduce tumor volume by approximately threefold (106). Furthermore, experiments have revealed that S1g-2 can reverse the specific protective effect of Hsp70-Bim protein-protein interaction (PPI) in chronic myeloid leukemia (CML). S1g-2 exhibits 5-10 times higher apoptosis-inducing activity in CML cells compared to other cancer cells, normal lymphocytes, and BaF3 cells (107, 108).
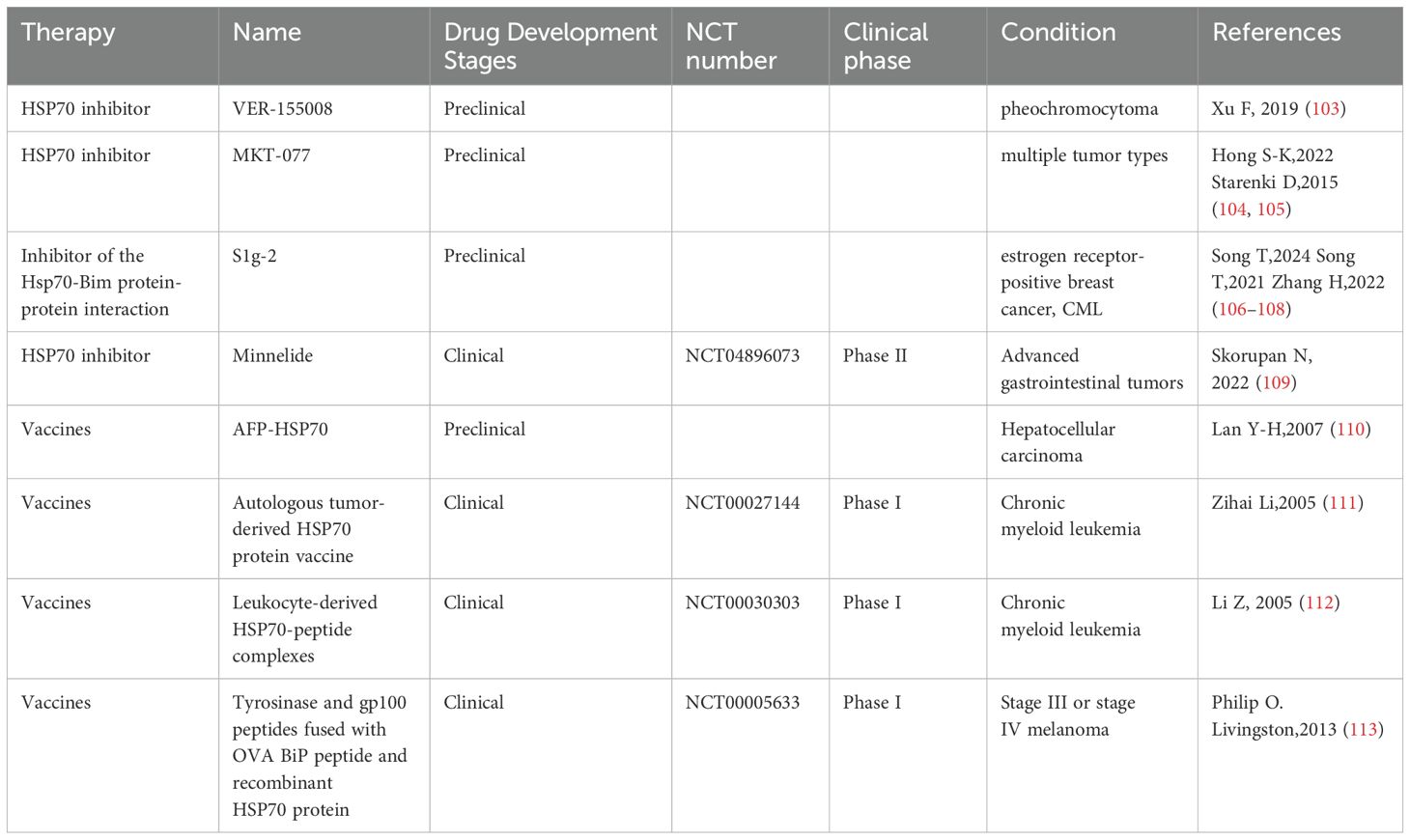
Table 1. Preclinical and clinical trials of HSP70-related therapies.
Moreover, VER-155008 does not inhibit HSP90, which is advantageous for specifically targeting HSP70 without affecting the normal functions of HSP90. This specificity allows the drug to precisely interfere with the survival mechanisms of tumor cells while minimizing interference with normal cells, thereby enhancing the precision and efficacy of treatment. Additionally, since HSP90 plays important physiological roles in normal cells, inhibiting it could lead to toxicity in these cells. The lack of effect of VER-155008 on HSP90 reduces toxicity to normal cells, improves treatment safety, and minimizes potential side effects.
The unique role of HSP70 in antigen presentation and immune activation makes it a promising candidate for an immunological adjuvant. Its mechanism primarily involves forming HSP70-antigen complexes, which promote cross-presentation of antigens and subsequently activate CD8+ T cells (122). In melanoma and lung cancer models, HSP70-antigen complexes have shown good anti-tumor effects. A previous study constructed a therapeutic peptide vaccine, HSP70-P/AFP-P, by linking the functional peptide of heat shock protein 70 (HSP70) with an epitope peptide of alpha-fetoprotein (AFP) to enhance the immune response against AFP-expressing tumors. In a BALB/c mouse model, the HSP70-P/AFP-P vaccine significantly increased the number of AFP-specific CD8+ T cells. The HSP70-P/AFP-P vaccine significantly prolonged survival, with vaccinated mice surviving for over 60 days, whereas all mice in the control group died within 35 days (123), Studies have also found that mice vaccinated with pcDNA3.1-AFP-HSP70 exhibited significantly higher cytotoxic T lymphocyte (CTL) activity compared to the control group. Moreover, CTL activity increased markedly with the rise in the effector-to-target cell ratio (E/T) (P < 0.001) (110).These findings provide a theoretical basis for developing novel cancer vaccines.
However, clinical research and trials on HSP70 inhibitors and vaccines are scarce at present. Current clinical trials have found that Minnelide, a water-soluble pro-drug derivative of Triptolide, exerts its anti-tumor effects by suppressing the expression of HSP70 (124). Minnelide has demonstrated significant anti-tumor efficacy in various cancers (125, 126). In preclinical studies, Minnelide markedly suppressed tumor growth and metastasis (127–129), and it has also shown potential therapeutic effects in clinical trials targeting advanced refractory adenosquamous carcinoma of the pancreas (109); Compared to HSP70 inhibitors, HSP70 vaccines have relatively more clinical trials (Table 1). For example, the autologous tumor-derived HSP70 protein vaccine and the leukocyte-derived HSP70-peptide complexes have demonstrated favorable tolerability and potential immunotherapeutic efficacy in clinical trials involving patients with chronic myeloid leukemia (CML) (112). Besides, Tyrosinase and gp100 peptides fused with OVA BiP peptide and recombinant HSP70 protein have also demonstrated promising therapeutic potential in melanoma. It is expected that more clinical researchers will explore its application value in clinical treatment in the future.
With the development of gene-editing technologies such as CRISPR-Cas9, therapeutic strategies targeting the HSP70 gene have shown great potential. Given the close association between the high expression of HSP70 in tumor cells and their anti-apoptotic characteristics, precise regulation of HSP70 expression through gene-editing techniques can significantly enhance the sensitivity of tumor cells to apoptosis. Moreover, combining gene-editing technologies with other treatments, such as magnetic hyperthermia, can further improve therapeutic outcomes while reducing damage to normal tissues, offering a new precision medicine approach for cancer treatment. Recently, a research team developed a magnetothermal-activated CRISPR-Cas9 gene-editing system to target the HSP70 and BCL2 genes in tumor cells. This system, based on a magnetothermal nanoparticle platform, utilizes the mild thermal effect (42°C) generated by an alternating magnetic field to activate the CRISPR-Cas9 system, thereby precisely targeting the HSP70 and BCL2 genes and significantly enhancing tumor cell apoptosis (130).
The rapid development of nanotechnology provides new solutions for the precise delivery of HSP70 inhibitors. By designing nanotargeted delivery systems, the specific enrichment of HSP70 inhibitors at tumor sites can be achieved, thereby improving drug therapeutic effects and reducing systemic toxicity. These delivery systems can not only enhance drug bioavailability but also achieve further improvements in targeting through surface modifications. Additionally, nanotechnology can be used for the delivery of HSP70-antigen complexes, enhancing antigen presentation efficiency and immune activation effects.
The combined application of HSP70 inhibitors with other treatment methods shows significant synergistic effects. For example, when VER - 155008 and anti - PD - 1 antibody are used separately, drug resistance or side effect may occur (131), leading to a decline in efficacy. However, if the two are used in combination, the anti - tumor immune response may be significantly enhanced (131), and this combination may even be superior to single - agent therapies in terms of controlling tumor volume and weight. Moreover, the combination of HSP70 inhibitors with chemotherapeutic drugs also shows promising prospects, improving chemotherapy effects by inhibiting tumor cell drug resistance and promoting apoptosis. Notably, the synergistic effect of dual inhibition of HSP70 and autophagy with cisplatin treatment can significantly reduce tumor cell metabolic activity and growth (132). These combined treatment strategies not only improve therapeutic effects but also effectively reduce the side effects of monotherapy, providing new ideas for comprehensive cancer treatment.
Although therapies targeting HSP70 hold great promise, they face significant challenges in clinical translation, which may be mainly attributed to the inefficient drug delivery system, complex interactions within the tumor microenvironment, and the emergence of drug resistance mechanisms.
The abnormal vasculature and dense extracellular matrix in the tumor microenvironment severely impede the uniform distribution of HSP70 - targeted drugs, potentially resulting in suboptimal drug concentrations in the tumor area. Although current nanotechnology - based delivery systems (Section 5.4) have shown promising potential, further optimization is required to enhance tumor penetration and minimize off - target effects. Additionally, although HSP70 is overexpressed in many cancers, its basal expression in normal cells (especially under stress conditions) has a great impact on treatment specificity. For example, cardiomyocytes and neurons rely on HSP70 for stress adaptation, and its inhibition may exacerbate tissue damage. In this context, methods such as utilizing tumor - specific post - translational modifications or altering microenvironmental cues (such as the hypoxic environment) can be considered to improve selectivity.
Furthermore, the problem of drug resistance to HSP70 - targeted therapy also affects the treatment effect. This drug resistance may be attributed to multiple complex mechanisms: First, when HSP70 is inhibited, tumor cells may compensatorily upregulate the expression of other molecular chaperones (such as HSP90, HSP60, etc.) to maintain protein homeostasis and cell survival. This compensatory mechanism enables tumor cells to continue to survive and proliferate under the action of HSP70 - targeted drugs. Second, the intracellular localization of HSP70 has an important impact on its function. HSP70 - targeted therapy may change its subcellular localization, causing it to transfer to other cell regions under the action of drugs, thus evading drug inhibition. Tumor heterogeneity is also an important source of drug resistance to HSP70 - targeted therapy. Tumor cells show significant heterogeneity at the genetic and epigenetic levels, resulting in different sensitivities of different cells to HSP70 - targeted drugs. During the treatment process, tumor cell subsets with stronger drug resistance may gradually accumulate, forming drug tolerance. These drug resistance mechanisms make HSP70 - targeted drugs may show certain efficacy in the initial treatment, but over time, tumor cells may gradually adapt and develop drug resistance, leading to treatment failure. Consider combining HSP70 inhibitors with inhibitors of other molecular chaperones to reduce the possibility of tumor cells developing drug resistance through compensatory mechanisms. We can also deeply analyze the genetic and epigenetic characteristics of each patient’s tumor, understand its HSP70 expression level, the activation status of related signaling pathways, and the characteristics of the tumor microenvironment, and tailor the most effective treatment plan for the patient. This personalized treatment can not only improve the success rate of treatment but also reduce unnecessary drug side effects.
Despite the promising therapeutic potential of HSP70 in cancer treatment, several significant challenges remain to be addressed to fully exploit its role. First, the precision of targeted therapy is critical. It is essential to ensure that drugs specifically inhibit the function of HSP70 in tumor cells without affecting its normal physiological roles in healthy cells, thereby minimizing toxicity and side effects.
Second, the complexity of the tumor microenvironment poses another challenge. The heterogeneous expression of HSP70 in different tumor cells and the presence of various immune cells within the tumor microenvironment can influence treatment efficacy. Therefore, a deeper understanding of the microenvironmental factors that regulate HSP70 expression and function is necessary to design more effective therapeutic strategies.
Moreover, combination therapies based on HSP70, such as those integrating immunotherapy or chemotherapy, require careful consideration and thorough preclinical evaluation to optimize treatment protocols and elucidate their underlying mechanisms. Clinical trials are also needed to validate the safety and efficacy of these combination therapies in patients.
The immunomodulatory functions of HSP70 extend beyond the realm of oncology, and its potential roles in non-neoplastic diseases warrant further exploration. In autoimmune diseases, HSP70 may regulate excessive immune responses through mechanisms such as inducing regulatory T cell (Treg) differentiation or suppressing NLRP3 inflammasome activation. In chronic inflammatory diseases, its anti-inflammatory properties and protein homeostasis maintenance functions could serve as potential therapeutic targets. Future research should systematically elucidate the immunoregulatory networks of HSP70 across diverse pathological contexts, thereby expanding its therapeutic potential in immune-related disorders.
Furthermore, HSP70 holds significant promise as a predictive biomarker for immunotherapy responses. Future studies should employ multi-omics analyses and prospective cohort investigations to establish the correlation between dynamic HSP70 expression patterns and patient outcomes, while exploring its feasibility as a treatment response biomarker. For instance, monitoring changes in serum or exosomal HSP70 levels before and after treatment may provide valuable insights for optimizing individualized therapeutic strategies.
In summary, HSP70 functions as a “double-edged sword” in immunomodulation, with its functional complexity presenting both challenges and opportunities. Through interdisciplinary collaboration, technological innovation, and the deepening of translational research, we may unlock the multidimensional value of HSP70 in precision medicine, thereby opening new avenues for the treatment of cancer and other immune-related diseases.
BZ: Writing – original draft, Writing – review & editing, Conceptualization. RQ: Supervision, Writing – review & editing, Conceptualization.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The figures are created with Biorender.com. Figure1.Created in BioRender. zhang, b. (2025) https://BioRender.com/eapp2e8. Figure2.Created in BioRender. zhang, b. (2025) https://BioRender.com/i9vswo3. Figure3.Created in BioRender. zhang, b. (2025) https://BioRender.com/z47otn3.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Yu E-M, Yoshinaga T, Jalufka FL, Ehsan H, Mark Welch DB, Kaneko G. The complex evolution of the metazoan HSP70 gene family. Sci Rep. (2021) 11:17794. doi: 10.1038/s41598-021-97192-9
2. Li Y, Yu Y, Li Y, Wang H, Li Q. Molecular evolution of the heat shock protein family and the role of HSP30 in immune response and wound healing in lampreys (Lethenteron reissneri). Fish Shellfish Immunol. (2024) 145:109323. doi: 10.1016/j.fsi.2023.109323
3. Robert J. Evolution of heat shock protein and immunity. Dev Comp Immunol. (2003) 27:449–64. doi: 10.1016/S0145-305X(02)00160-X
4. Bellini S, Barutta F, Mastrocola R, Imperatore L, Bruno G, Gruden G. Heat shock proteins in vascular diabetic complications: review and future perspective. Int J Mol Sci. (2017) 18:2709. doi: 10.3390/ijms18122709
5. Hu C, Yang J, Qi Z, Wu H, Wang B, Zou F, et al. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm. (2022) 3:e161. doi: 10.1002/mco2.161
6. T Z, L R, A S. Heat shock proteins as immunomodulants. Mol Basel Switz. (2018) 23(11):2846. doi: 10.3390/molecules23112846
7. Cw Y, Hj K, Jh L, Sh L. Heat shock proteins: agents of cancer development and therapeutic targets in anti-cancer therapy. Cells. (2019) 9(1):60. doi: 10.3390/cells9010060
8. Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. (2011) 475:324–32. doi: 10.1038/nature10317
9. Luengo TM, Mayer MP, Rüdiger SGD. The hsp70–hsp90 chaperone cascade in protein folding. Trends Cell Biol. (2019) 29:164–77. doi: 10.1016/j.tcb.2018.10.004
10. Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. ScientificWorldJournal. (2010) 10:1543–52. doi: 10.1100/tsw.2010.152
11. R R, Nb N, Mp M, B B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. (2019) 20(11):665–80. doi: 10.1038/s41580-019-0133-3
12. Clerico EM, Meng W, Pozhidaeva A, Bhasne K, Petridis C, Gierasch LM. Hsp70 molecular chaperones: multifunctional allosteric holding and unfolding machines. Biochem J. (2019) 476:1653–77. doi: 10.1042/BCJ20170380
13. Ambrose AJ, Chapman E. Function, therapeutic potential, and inhibition of hsp70 chaperones. J Med Chem. (2021) 64:7060–82. doi: 10.1021/acs.jmedchem.0c02091
14. Wang W, Liu Q, Liu Q, Hendrickson WA. Conformational equilibria in allosteric control of Hsp70 chaperones. Mol Cell. (2021) 81:3919–3933.e7. doi: 10.1016/j.molcel.2021.07.039
15. Larburu N, Adams CJ, Chen C-S, Nowak PR, Ali MMU. Mechanism of Hsp70 specialized interactions in protein translocation and the unfolded protein response. Open Biol. (2020) 10:200089. doi: 10.1098/rsob.200089
16. Gehrmann M, Marienhagen J, Eichholtz-Wirth H, Fritz E, Ellwart J, Jäättelä M, et al. Dual function of membrane-bound heat shock protein 70 (Hsp70), Bag-4, and Hsp40: protection against radiation-induced effects and target structure for natural killer cells. Cell Death Differ. (2005) 12:38–51. doi: 10.1038/sj.cdd.4401510
17. Sv S, Sn W. The unfolding story of the Escherichia coli Hsp70 DnaK: is DnaK a holdase or an unfoldase? Mol Microbiol. (2002) 45(5):1197–206. doi: 10.1046/j.1365-2958.2002.03093.x
18. F D, Wj N, K T, F L, Fu H, A T, et al. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U.S.A. (2003) 100(2):721–6. doi: 10.1073/pnas.242720499
19. Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. (2004) 279:4869–76. doi: 10.1074/jbc.M305838200
20. Song C, Zhang Y, Pei Q, Zheng L, Wang M, Shi Y, et al. HSP70 alleviates sepsis-induced cardiomyopathy by attenuating mitochondrial dysfunction-initiated NLRP3 inflammasome-mediated pyroptosis in cardiomyocytes. Burns Trauma. (2022) 10:tkac043. doi: 10.1093/burnst/tkac043
21. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
22. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
23. Remick BC, Gaidt MM, Vance RE. Effector-triggered immunity. Annu Rev Immunol. (2023) 41:453–81. doi: 10.1146/annurev-immunol-101721-031732
24. Sharma P, Zhang X, Ly K, Kim JH, Wan Q, Kim J, et al. Hyperglycosylation of prosaposin in tumor dendritic cells drives immune escape. Science. (2024) 383:190–200. doi: 10.1126/science.adg1955
25. Feng M, Jiang W, Kim BYS, Zhang CC, Fu Y-X, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. (2019) 19:568–86. doi: 10.1038/s41568-019-0183-z
26. Sharapova TN, Romanova EA, Ivanova OK, Yashin DV, Sashchenko LP. Hsp70 interacts with the TREM-1 receptor expressed on monocytes and thereby stimulates generation of cytotoxic lymphocytes active against MHC-negative tumor cells. Int J Mol Sci. (2021) 22:6889. doi: 10.3390/ijms22136889
27. Junprung W, Supungul P, Tassanakajon A. Structure, gene expression, and putative functions of crustacean heat shock proteins in innate immunity. Dev Comp Immunol. (2021) 115:103875. doi: 10.1016/j.dci.2020.103875
28. Z W, T L, Y Z, Q Y, H M, Y H, et al. Salidroside regulates tumor microenvironment of non-small cell lung cancer via Hsp70/Stub1/Foxp3 pathway in Tregs. BMC Cancer. (2023) 23(1):717. doi: 10.1186/s12885-023-11036-5
29. de Wolf C, van der Zee R, den Braber I, Glant T, Maillère B, Favry E, et al. An arthritis-suppressive and treg cell-inducing CD4+ T cell epitope is functional in the context of HLA-restricted T cell responses. Arthritis Rheumatol Hoboken NJ. (2016) 68:639–47. doi: 10.1002/art.39444
30. Sb Y, H H, Hj L, Jh C, Yp C, Sw S, et al. A novel IRAK4/PIM1 inhibitor ameliorates rheumatoid arthritis and lymphoid Malignancy by blocking the TLR/MYD88-mediated NF- κ B pathway. Acta Pharm Sin B. (2023) 13(3):1093–109. doi: 10.1016/j.apsb.2022.12.001
31. Zhou L, Fang L, Tamm M, Stolz D, Roth M. Extracellular heat shock protein 70 increases the glucocorticoid receptor and dual-specificity phosphatase 1 via toll-like receptor 4 and attenuates inflammation in airway epithelial cells. Int J Mol Sci. (2023) 24:11700. doi: 10.3390/ijms241411700
32. Osipiuk J, Walsh MA, Freeman BC, Morimoto RI, Joachimiak A. Structure of a new crystal form of human Hsp70 ATPase domain. Acta Crystallogr D Biol Crystallogr. (1999) 55:1105–7. doi: 10.1107/S0907444999002103
33. Arakawa A, Handa N, Shirouzu M, Yokoyama S. Biochemical and structural studies on the high affinity of Hsp70 for ADP. Protein Sci. (2011) 20:1367–79. doi: 10.1002/pro.663
34. Gao X-C, Zhou C-J, Zhou Z-R, Wu M, Cao C-Y, Hu H-Y. The C-terminal helices of heat shock protein 70 are essential for J-domain binding and ATPase activation. J Biol Chem. (2012) 287:6044–52. doi: 10.1074/jbc.M111.294728
35. Cheeseman MD, Westwood IM, Barbeau O, Rowlands M, Dobson S, Jones AM, et al. Exploiting protein conformational change to optimize adenosine-derived inhibitors of HSP70. J Med Chem. (2016) 59:4625–36. doi: 10.1021/acs.jmedchem.5b02001
36. Umehara K, Hoshikawa M, Tochio N, Tate S. Substrate binding switches the conformation at the lynchpin site in the substrate-binding domain of human hsp70 to enable allosteric interdomain communication. Molecules. (2018) 23:528. doi: 10.3390/molecules23030528
37. A A, N H, N O, M S, T K, F H, et al. The C-terminal BAG domain of BAG5 induces conformational changes of the Hsp70 nucleotide-binding domain for ADP-ATP exchange. Struct Lond Engl. (2010) 1993:18. doi: 10.1016/j.str.2010.01.004
38. Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. (2013) 14:630–42. doi: 10.1038/nrm3658
39. Mp M, Lm G. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J Biol Chem. (2019) 294(6):2085–97. doi: 10.1074/jbc.REV118.002810
40. Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. (2002) 2:185–94. doi: 10.1038/nri749
41. M M, W J, R Z. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity. (2024) 57(4):752–71. doi: 10.1016/j.immuni.2024.03.002
42. Shen J, Zhang Y, Tang W, Yang M, Cheng T, Chen Y, et al. Short IL-18 generated by caspase-3 cleavage mobilizes NK cells to suppress tumor growth. Nat Immunol. (2025) 26(3):416–28. doi: 10.1038/s41590-024-02074-7
43. Shi X, Sun Q, Hou Y, Zeng H, Cao Y, Dong M, et al. Recognition and maturation of IL-18 by caspase-4 noncanonical inflammasome. Nature. (2023) 624:442–50. doi: 10.1038/s41586-023-06742-w
44. Hulina A, Grdić Rajković M, Jakšić Despot D, Jelić D, Dojder A, Čepelak I, et al. Extracellular Hsp70 induces inflammation and modulates LPS/LTA-stimulated inflammatory response in THP-1 cells. Cell Stress Chaperones. (2018) 23:373–84. doi: 10.1007/s12192-017-0847-0
45. Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the toll/interleukin-1 receptor signal pathway. J Biol Chem. (2002) 277:15107–12. doi: 10.1074/jbc.M111204200
46. Aosai F, Rodriguez Pena MS, Mun H-S, Fang H, Mitsunaga T, Norose K, et al. Toxoplasma gondii-derived heat shock protein 70 stimulates maturation of murine bone marrow-derived dendritic cells via Toll-like receptor 4. Cell Stress Chaperones. (2006) 11:13–22. doi: 10.1379/csc-138r.1
47. Lee M-S, Park SM, Kim Y-J. Photothermal treatment-based heat stress regulates function of myeloid-derived suppressor cells. Sci Rep. (2024) 14:18847. doi: 10.1038/s41598-024-69074-3
48. Murshid A, Borges TJ, Bonorino C, Lang BJ, Calderwood SK. Immunological outcomes mediated upon binding of heat shock proteins to scavenger receptors SCARF1 and LOX-1, and endocytosis by mononuclear phagocytes. Front Immunol. (2019) 10:3035. doi: 10.3389/fimmu.2019.03035
49. Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, et al. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity. (2002) 17:353–62. doi: 10.1016/s1074-7613(02)00388-6
50. Borges TJ, Lima K, Murshid A, Lape IT, Zhao Y, Rigo MM, et al. Innate extracellular Hsp70 inflammatory properties are mediated by the interaction of Siglec-E and LOX-1 receptors. bioRxiv. (2023), 569623. doi: 10.1101/2023.12.01.569623
51. Mantuano E, Azmoon P, Banki MA, Gunner CB, Gonias SL. The LRP1/CD91 ligands, tissue-type plasminogen activator, α2-macroglobulin, and soluble cellular prion protein have distinct co-receptor requirements for activation of cell-signaling. Sci Rep. (2022) 12:17594. doi: 10.1038/s41598-022-22498-1
52. Chang C, Tang X, Mosallaei D, Chen M, Woodley DT, Schönthal AH, et al. LRP-1 receptor combines EGFR signalling and eHsp90α autocrine to support constitutive breast cancer cell motility in absence of blood supply. Sci Rep. (2022) 12:12006. doi: 10.1038/s41598-022-16161-y
53. Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol. (2000) 1:151–5. doi: 10.1038/77835
54. Itskanov S, Wang L, Junne T, Sherriff R, Xiao L, Blanchard N, et al. A common mechanism of Sec61 translocon inhibition by small molecules. Nat Chem Biol. (2023) 19:1063–71. doi: 10.1038/s41589-023-01337-y
55. Jm D, Am H, Rr C. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. (2010) 1183:211–21. doi: 10.1111/j.1749-6632.2009.05133.x
56. Zhu J. T helper cell differentiation, heterogeneity, and plasticity. Cold Spring Harb Perspect Biol. (2018) 10:a030338. doi: 10.1101/cshperspect.a030338
57. Larosa M, Zen M, Gatto M, Jesus D, Zanatta E, Iaccarino L, et al. IL-12 and IL-23/Th17 axis in systemic lupus erythematosus. Exp Biol Med. (2019) 244:42. doi: 10.1177/1535370218824547
58. Revu S, Wu J, Henkel M, Rittenhouse N, Menk A, Delgoffe GM, et al. IL-23 and IL-1β Drive human th17 cell differentiation and metabolic reprogramming in absence of CD28 costimulation. Cell Rep. (2018) 22:2642. doi: 10.1016/j.celrep.2018.02.044
59. H C, M C-S, Kw S, Mp M. The Heat Shock Protein HSP70 Promotes Th17 Genes’ Expression via Specific Regulation of microRNA. Int J Mol Sci. (2020) 21(8):2823. doi: 10.3390/ijms21082823
60. Tukaj S. Heat shock protein 70 as a double agent acting inside and outside the cell: insights into autoimmunity. Int J Mol Sci. (2020) 21:5298. doi: 10.3390/ijms21155298
61. Feng H, Guo Z, Chen X, Liu K, Li H, Jia W, et al. Excessive HSP70/TLR2 activation leads to remodeling of the tumor immune microenvironment to resist chemotherapy sensitivity of mFOLFOX in colorectal cancer. Clin Immunol Orlando Fla. (2022) 245:109157. doi: 10.1016/j.clim.2022.109157
62. Ryu HH, Ha SH. HSP70 interacts with Rheb, inhibiting mTORC1 signaling. Biochem Biophys Res Commun. (2020) 533:1198–203. doi: 10.1016/j.bbrc.2020.07.053
63. Wachstein J, Tischer S, Figueiredo C, Limbourg A, Falk C, Immenschuh S, et al. HSP70 enhances immunosuppressive function of CD4+CD25+FoxP3+ T regulatory cells and cytotoxicity in CD4+CD25– T cells. PloS One. (2012) 7:e51747. doi: 10.1371/journal.pone.0051747
64. Ferat-Osorio E, Sánchez-Anaya A, Gutiérrez-Mendoza M, Boscó-Gárate I, Wong-Baeza I, Pastelin-Palacios R, et al. Heat shock protein 70 down-regulates the production of toll-like receptor-induced pro-inflammatory cytokines by a heat shock factor-1/constitutive heat shock element-binding factor-dependent mechanism. J Inflammation Lond Engl. (2014) 11:19. doi: 10.1186/1476-9255-11-19
65. Tian Y, Jin M, Ye N, Gao Z, Jiang Y, Yan S. Mesenchymal stem cells-derived exosomes attenuate mouse non-heart-beating liver transplantation through Mir-17-5p-regulated Kupffer cell pyroptosis. Stem Cell Res Ther. (2025) 16:57. doi: 10.1186/s13287-025-04169-w
66. Kaynak A, Vallabhapurapu SD, Davis HW, Smith EP, Muller P, Vojtesek B, et al. TLR2-bound cancer-secreted hsp70 induces merTK-mediated immunosuppression and tumorigenesis in solid tumors. Cancers. (2025) 17:450. doi: 10.3390/cancers17030450
67. Fu J, Wu H. Structural mechanisms of NLRP3 inflammasome assembly and activation. Annu Rev Immunol. (2023) 41:301–16. doi: 10.1146/annurev-immunol-081022-021207
68. Martine P, Chevriaux A, Derangère V, Apetoh L, Garrido C, Ghiringhelli F, et al. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. (2019) 10:256. doi: 10.1038/s41419-019-1491-7
69. Rodríguez-Iturbe B, Johnson RJ, Sánchez-Lozada LG. Relationship between hyperuricemia, HSP70 and NLRP3 inflammasome in arterial hypertension. Arch Cardiol Mex. (2023) 93:458–63. doi: 10.24875/ACM.22000174
70. Lu H, Lu X, Xie Q, Wan H, Sun Y. TTC4 inhibits NLRP3 inflammation in rheumatoid arthritis by HSP70. Int J Rheum Dis. (2023) 26:1751–9. doi: 10.1111/1756-185X.14818
71. Kim T-H, Shin SJ, Park Y-M, Jung ID, Ryu S-W, Kim D-J, et al. Critical role of TRIF and MyD88 in Mycobacterium tuberculosis Hsp70-mediated activation of dendritic cells. Cytokine. (2015) 71:139–44. doi: 10.1016/j.cyto.2014.09.010
72. M L, von EP S. The role of extracellular HSP70 in the function of tumor-associated immune cells. Cancers. (2021) 13(18):4721. doi: 10.3390/cancers13184721
73. Sha G, Jiang Z, Zhang W, Jiang C, Wang D, Tang D. The multifunction of HSP70 in cancer: Guardian or traitor to the survival of tumor cells and the next potential therapeutic target. Int Immunopharmacol. (2023) 122:110492. doi: 10.1016/j.intimp.2023.110492
74. Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B. Role of the Human Heat Shock Protein hsp70 in Protection against Stress-Induced Apoptosis. Mol Cell Biol. (1997) 17:5317–27. doi: 10.1128/MCB.17.9.5317
75. Y G, C H, H H, Y X, Y X, L L, et al. Heat shock protein 70 together with its co-chaperone CHIP inhibits TNF-alpha induced apoptosis by promoting proteasomal degradation of apoptosis signal-regulating kinase1. Apoptosis Int J Program Cell Death. (2010) 15(7):822–33. doi: 10.1007/s10495-010-0495-7
76. Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress- induced activation of the cytochrome c-mediated death pathway. Science. (2000) 288:870–4. doi: 10.1126/science.288.5467.870
77. Albakova Z, Armeev GA, Kanevskiy LM, Kovalenko EI, Sapozhnikov AM. HSP70 multi-functionality in cancer. Cells. (2020) 9:587. doi: 10.3390/cells9030587
78. Stankiewicz AR, Lachapelle G, Foo CPZ, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing bax translocation. J Biol Chem. (2005) 280:38729–39. doi: 10.1074/jbc.M509497200
79. Gotoh T, Terada K, Oyadomari S, Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. (2004) 11:390–402. doi: 10.1038/sj.cdd.4401369
80. Pang Q, Christianson TA, Keeble W, Koretsky T, Bagby GC. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. J Biol Chem. (2002) 277:49638–43. doi: 10.1074/jbc.M209386200
81. S K, J S, Up S, SG K, A A, U M, et al. Targeting Hsp70: A possible therapy for cancer. Cancer Lett. (2016) 374(1):156–66. doi: 10.1016/j.canlet.2016.01.056
82. F G, C S, P B, P G, W F, A S, et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood. (2005) 105(3):1246–55. doi: 10.1182/blood-2004-05-2041
83. Gabai VL, Mabuchi K, Mosser DD, Sherman MY. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol Cell Biol. (2002) 22:3415–24. doi: 10.1128/MCB.22.10.3415-3424.2002
84. Xiong L, Li D, Xiao G, Tan S, Xu L, Wang G. HSP70 promotes pancreatic cancer cell epithelial-mesenchymal transformation and growth via the NF-κB signaling pathway. Pancreas. (2024) 54(2):e89–96. doi: 10.1097/MPA.0000000000002398
85. Xiong L, Li D, Xiao G, Tan S, Wen J, Wang G. Serum HSP70 and VEGF levels are effective predictive factors of chemoradiosensitivity and prognosis of pancreatic cancer patients. Pancreas. (2024) 53:e713–22. doi: 10.1097/MPA.0000000000002358
86. Sakai G, Tokuda H, Fujita K, Kainuma S, Kawabata T, Matsushima-Nishiwaki R, et al. Heat Shock Protein 70 Negatively Regulates TGF-β-Stimulated VEGF Synthesis via p38 MAP Kinase in Osteoblasts. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. (2017) 44:1133–45. doi: 10.1159/000485418
87. Mz J, Wl J. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. (2020) 5(1):166. doi: 10.1038/s41392-020-00280-x
88. Peng C, Xu Y, Wu J, Wu D, Zhou L, Xia X. TME-related biomimetic strategies against cancer. Int J Nanomed. (2024) 19:109–35. doi: 10.2147/IJN.S441135
89. T P, A F, C T, A P, L D, E L, et al. Heat shock and HSP70 regulate 5-FU-mediated caspase-1 activation in myeloid-derived suppressor cells and tumor growth in mice. J Immunother Cancer. (2020) 8(1):e000478. doi: 10.1136/jitc-2019-000478
90. Gobbo J, Marcion G, Cordonnier M, Dias AMM, Pernet N, Hammann A, et al. Restoring anticancer immune response by targeting tumor-derived exosomes with a HSP70 peptide aptamer. JNCI J Natl Cancer Inst. (2016) 108:djv330. doi: 10.1093/jnci/djv330
91. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin J-P, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. (2010) 120:457–71. doi: 10.1172/JCI40483
92. Z L, L C, G Z, S W, E X, J T, et al. Loss of MNX1 sensitizes tumors to cytotoxic T cells by degradation of PD-L1 mRNA. Adv Sci Weinh Baden-Wurtt Ger. (2025) 12(12):e2403077. doi: 10.1002/advs.202403077
93. Xu X, Xie T, Zhou M, Sun Y, Wang F, Tian Y, et al. Hsc70 promotes anti-tumor immunity by targeting PD-L1 for lysosomal degradation. Nat Commun. (2024) 15:4237. doi: 10.1038/s41467-024-48597-3
94. Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J Biol Chem. (2018) 293:5414–24. doi: 10.1074/jbc.R117.818237
95. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. (2011) 20:131–9. doi: 10.1016/j.devcel.2010.12.003
96. Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Høyer-Hansen M, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. (2004) 200:425–35. doi: 10.1084/jem.20040531
97. R R, Al K, Zr I PJ, Dm B. Similarities and differences of hsp70, hsc70, grp78 and mortalin as cancer biomarkers and drug targets. Cells. (2021) 10(11):2996. doi: 10.3390/cells10112996
98. N W, J L, L L, L Y, L D, C S, et al. MerTK+ macrophages promote melanoma progression and immunotherapy resistance through AhR-ALKAL1 activation. Sci Adv. (2024) 10(40):eado8366. doi: 10.1126/sciadv.ado8366
99. Guo D, Chen Y, Wang S, Yu L, Shen Y, Zhong H, et al. Exosomes from heat-stressed tumour cells inhibit tumour growth by converting regulatory T cells to Th17 cells via IL-6. Immunology. (2018) 154:132–43. doi: 10.1111/imm.12874
100. Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. (2005) 65:5238–47. doi: 10.1158/0008-5472.CAN-04-3804
101. Hosseini R, Sarvnaz H, Arabpour M, Ramshe SM, Asef-Kabiri L, Yousefi H, et al. Cancer exosomes and natural killer cells dysfunction: biological roles, clinical significance and implications for immunotherapy. Mol Cancer. (2022) 21:15. doi: 10.1186/s12943-021-01492-7
102. Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. (2006) 31:164–72. doi: 10.1016/j.tibs.2006.01.006
103. Xu F, Lin D, Jiang W, Meng L, Xu Y, Wang C, et al. HSP70 inhibitor VER155008 suppresses pheochromocytoma cell and xenograft growth by inhibition of PI3K/AKT/mTOR and MEK/ERK pathways. Int J Clin Exp Pathol. (2019) 12:2585–94.
104. Hong S-K, Starenki D, Johnson OT, Gestwicki JE, Park J-I. Analogs of the heat shock protein 70 inhibitor MKT-077 suppress medullary thyroid carcinoma cells. Int J Mol Sci. (2022) 23:1063. doi: 10.3390/ijms23031063
105. Starenki D, Park JI. Selective mitochondrial uptake of MKT-077 can suppress medullary thyroid carcinoma cell survival in vitro and in vivo. Endocrinol Metab Seoul Korea. (2015) 30:593–603. doi: 10.3803/EnM.2015.30.4.593
106. Song T, Zhang H, Zhao Q, Hu Z, Wang Z, Song Y, et al. Small molecule inhibitor targeting the Hsp70-Bim protein–protein interaction in estrogen receptor-positive breast cancer overcomes tamoxifen resistance. Breast Cancer Res. (2024) 26:33. doi: 10.1186/s13058-024-01790-0
107. Song T, Guo Y, Xue Z, Guo Z, Wang Z, Lin D, et al. Small-molecule inhibitor targeting the Hsp70-Bim protein-protein interaction in CML cells overcomes BCR-ABL-independent TKI resistance. Leukemia. (2021) 35:2862–74. doi: 10.1038/s41375-021-01283-5
108. Zhang H, Song T, Wang Z, Laura Bonnette U, Guo Y, Wang H, et al. Bcr-Abl drives the formation of Hsp70/Bim PPI to stabilize oncogenic clients and prevent cells from undergoing apoptosis. Biochem Pharmacol. (2022) 198:114964. doi: 10.1016/j.bcp.2022.114964
109. Skorupan N, Ahmad MI, Steinberg SM, Trepel JB, Cridebring D, Han H, et al. A phase II trial of the super-enhancer inhibitor Minnelide™ in advanced refractory adenosquamous carcinoma of the pancreas. Future Oncol Lond Engl. (2022) 18:2475–81. doi: 10.2217/fon-2021-1609
110. Lan Y-H, Li Y-G, Liang Z-W, Chen M, Peng M-L, Tang L, et al. A DNA vaccine against chimeric AFP enhanced by HSP70 suppresses growth of hepatocellular carcinoma. Cancer Immunol Immunother CII. (2007) 56:1009–16. doi: 10.1007/s00262-006-0254-3
111. University of Connecticut. A Feasibility and Toxicity Study of Vaccination With HSP70 for the Treatment of Chronic Myelogenous Leukemia in the Chronic Phase. [Clinical trial registration]. clinicaltrials.gov (2005). Available online at: https://clinicaltrials.gov/study/NCT00027144 (Accessed March 25, 2025).
112. Li Z, Qiao Y, Liu B, Laska EJ, Chakravarthi P, Kulko JM, et al. Combination of imatinib mesylate with autologous leukocyte-derived heat shock protein and chronic myelogenous leukemia. Clin Cancer Res Off J Am Assoc Cancer Res. (2005) 11:4460–8. doi: 10.1158/1078-0432.CCR-05-0250
113. Memorial Sloan Kettering Cancer Center. Vaccination of Melanoma Patients With Tyrosinase YMD and gp100 IMD-Javelin Fusion Peptides&x2F;HSP70 Complexes: A Trial Comparing the Immunogenicity of Three Doses. [Clinical trial registration]. clinicaltrials.gov (2013). Available online at: https://clinicaltrials.gov/study/NCT00005633 (Accessed March 25, 2025).
114. Li X, Shao H, Taylor IR, Gestwicki JE. Targeting allosteric control mechanisms in heat shock protein 70 (Hsp70). Curr Top Med Chem. (2016) 16:2729–40. doi: 10.2174/1568026616666160413140911
115. Schlecht R, Scholz SR, Dahmen H, Wegener A, Sirrenberg C, Musil D, et al. Functional analysis of Hsp70 inhibitors. PloS One. (2013) 8:e78443. doi: 10.1371/journal.pone.0078443
116. Aj M, Ds W, H B, Jb M, P D, T S, et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol. (2010) 66(3):535–45. doi: 10.1007/s00280-009-1194-3
117. Massey AJ. ATPases as drug targets: insights from heat shock proteins 70 and 90. J Med Chem. (2010) 53:7280–6. doi: 10.1021/jm100342z
118. Modica-Napolitano JS, Koya K, Weisberg E, Brunelli BT, Li Y, Chen LB. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. (1996) 56:544–50.
119. Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ERP. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol. (2011) 411:614–32. doi: 10.1016/j.jmb.2011.06.003
120. Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, et al. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. (2000) 60:6818–21.
121. Petit T, Izbicka E, Lawrence RA, Nalin C, Weitman SD, Von Hoff DD. Activity of MKT 077, a rhodacyanine dye, against human tumor colony-forming units. Anticancer Drugs. (1999) 10:309–15. doi: 10.1097/00001813-199903000-00010
122. Huryn DM, Brodsky JL, Brummond KM, Chambers PG, Eyer B, Ireland AW, et al. Chemical methodology as a source of small-molecule checkpoint inhibitors and heat shock protein 70 (Hsp70) modulators. Proc Natl Acad Sci U.S.A. (2011) 108:6757–62. doi: 10.1073/pnas.1015251108
123. Wang X-P, Wang Q-X, Lin H-P, Xu B, Zhao Q, Chen K. Recombinant heat shock protein 70 functional peptide and alpha-fetoprotein epitope peptide vaccine elicits specific anti-tumor immunity. Oncotarget. (2016) 7:71274–84. doi: 10.18632/oncotarget.12464
124. Tn M, N M, S B, V S, A S, S V, et al. Triptolide induces the expression of miR-142-3p: a negative regulator of heat shock protein 70 and pancreatic cancer cell proliferation. Mol Cancer Ther. (2013) 12(7):1266–75. doi: 10.1158/1535-7163.MCT-12-1231
125. Phillips PA, Dudeja V, McCarroll JA, Borja-Cacho D, Dawra RK, Grizzle WE, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. (2007) 67:9407–16. doi: 10.1158/0008-5472.CAN-07-1077
126. A O, G B, R C, Sj S, K M, S B, et al. Triptolide abrogates growth of colon cancer and induces cell cycle arrest by inhibiting transcriptional activation of E2F. Lab Investig J Tech Methods Pathol. (2015) 95(6):648–59. doi: 10.1038/labinvest.2015.46
127. S B, V T, V S, Tn M, Ak S, S S. Minnelide reduces tumor burden in preclinical models of osteosarcoma. Cancer Lett. (2013) 335(2):412–20. doi: 10.1016/j.canlet.2013.02.050
128. Jacobson BA, Chen EZ, Tang S, Belgum HS, McCauley JA, Evenson KA, et al. Triptolide and its prodrug minnelide suppress Hsp70 and inhibit in vivo growth in a xenograft model of mesothelioma. Genes Cancer. (2015) 6:144–52. doi: 10.18632/genesandcancer.55
129. Chugh R, Sangwan V, Patil SP, Dudeja V, Dawra RK, Banerjee S, et al. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci Transl Med. (2012) 4:156ra139. doi: 10.1126/scitranslmed.3004334
130. Li M, Li S, Guo Y, Hu P, Shi J. Magnetothermal-activated gene editing strategy for enhanced tumor cell apoptosis. J Nanobiotechnol. (2024) 22:450. doi: 10.1186/s12951-024-02734-8
131. Theivanthiran B, Yarla N, Haykal T, Nguyen Y-V, Cao L, Ferreira M, et al. Tumor-intrinsic NLRP3-HSP70-TLR4 axis drives premetastatic niche development and hyperprogression during anti-PD-1 immunotherapy. Sci Transl Med. (2022) 14:eabq7019. doi: 10.1126/scitranslmed.abq7019
Keywords: HSP70, immune regulation, tumor immunity, molecular mechanisms, therapeutic strategies
Citation: Zhang B and Qi R (2025) The dual-function of HSP70 in immune response and tumor immunity: from molecular regulation to therapeutic innovations. Front. Immunol. 16:1587414. doi: 10.3389/fimmu.2025.1587414
Received: 04 March 2025; Accepted: 27 March 2025;
Published: 14 April 2025.
Edited by:
Ming Xian Chang, Chinese Academy of Sciences (CAS), ChinaCopyright © 2025 Zhang and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruiqun Qi, xiaoqiliumin@163.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.