 Lisha Mou
Lisha Mou Zhenkun Fu1
Zhenkun Fu1 Yuxian Chen
Yuxian Chen Zuhui Pu
Zuhui Pu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 19 February 2025
Sec. Alloimmunity and Transplantation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1555310
Type 2 diabetes (T2D) is a widespread metabolic disorder marked by hyperglycemia, arising from insulin resistance and relative insulin deficiency. This review investigates the critical role of Na+/K+-ATPase (NKA), a transmembrane protein essential for maintaining cellular ion gradients, in the pathophysiology of T2D. We provide an overview of NKA’s biological functions, emphasizing its involvement in cellular signaling pathways, insulin secretion, and glucose homeostasis. The potential of NKA as a therapeutic target for T2D is analyzed, showcasing innovative strategies such as NKA activators, gene therapy, and stem cell therapy aimed at enhancing NKA activity to achieve better glycemic control. Additionally, NKA’s multifunctional role in maintaining cell viability and modulating immune responses in islet transplantation may offer potential benefits for improving transplant outcomes. By elucidating the complex interactions between NKA and T2D, this review aims to shed light on developing novel therapeutic interventions that meet the multifaceted needs of individuals suffering from this chronic condition, ultimately improving their health outcomes.
Type 2 diabetes (T2D) is the most prevalent form of diabetes (1), affecting millions of people worldwide and representing a significant global health challenge (2, 3). Despite efforts, T2D is complex to manage, and there’s a need for more effective treatments targeting its underlying causes (4). Understanding the pathophysiology of T2D is essential for developing effective therapeutic strategies (5). Among the various proteins involved in glucose metabolism and insulin signaling, Na+/K+-ATPase (NKA) plays a crucial role in maintaining ionic balance across cell membranes, which is vital for proper cellular function (6). NKA is responsible for the active transport of sodium out of cells and potassium into cells, processes that are critical for maintaining membrane potential and facilitating cellular signaling pathways (7). Dysfunction of NKA can have significant implications for insulin secretion and action, thereby contributing to T2D (5, 8). Recent studies have also linked NKA to diabetes complications like nephropathy and neuropathy (9, 10), with changes in its activity potentially exacerbating these conditions through oxidative stress and inflammation (11, 12). Additionally, NKA’s role in islet transplantation, where it may influence cell viability and immune responses, offers potential benefits for improving transplant outcomes. To develop new therapeutic approaches for T2D, it is essential to conduct in-depth research into the specific mechanisms governing NKA function and its regulation.
Structurally, NKA consists of two main subunits: the α subunit, which is responsible for the catalytic activity of the pump, and the β subunit, which aids in the proper maturation and localization of the enzyme within the membrane (13). NKA’s main role is to pump out three sodium ions and import two potassium ions per ATP molecule hydrolyzed, maintaining the cell’s membrane potential, which is vital for functions like muscle contraction and nerve signaling (13). This activity accounts for 30-40% of animal cells’ energy use, highlighting its importance in energy metabolism and electrolyte transport (14).
The regulation of NKA activity is complex and involves various mechanisms, including modulation of its expression levels, localization within the plasma membrane, enzymatic activity, and interactions with other proteins (14). NKA’s plasma membrane location is crucial for its antiport activity, with its movement between the membrane and intracellular spaces strictly regulated, although the precise mechanisms remain poorly understood (14).
NKA also functions as a signaling molecule, influencing various cellular pathways beyond ion transport. It regulates intracellular sodium and potassium concentrations, which significantly impact the flow of calcium ions and intracellular pH, thereby influencing signaling pathways critical for cell survival and function (15, 16). Additionally, NKA is linked to several metabolic pathways, including glucose transport and energy metabolism, which are essential for maintaining cellular homeostasis (14). Its interaction with signaling molecules allows it to influence pathways that regulate inflammation, cell growth, and apoptosis (17).
Research has shown that alterations in NKA activity can lead to various pathological conditions. For instance, impaired NKA function has been associated with diseases such as heart failure, where reduced NKA activity contributes to cardiac dysfunction and hypertrophy (18). Similarly, in the context of hyperuricemia, NKA signaling impairment has been linked to renal tubular injury, highlighting its role in kidney health (19, 20), Moreover, NKA’s involvement in regulating intracellular calcium levels is crucial for neuronal excitability and synaptic transmission, indicating its importance in the nervous system (6).
Overall, NKA is a multifunctional enzyme that is integral to maintaining cellular ion balance and facilitating various signaling processes. Its role extends beyond simple ion transport, influencing metabolic pathways and cellular responses to stressors. Understanding the complex regulatory mechanisms of NKA and its involvement in disease processes is essential for developing targeted therapies that can modulate its activity and improve cellular function in pathological conditions (17).
The relationship between NKA and diabetes is complex and multifaceted, involving alterations in the expression and activity of NKA that are closely linked to pancreatic function (21). In diabetes, research indicates a significant reduction in NKA activity (22, 23). The reduced activity of NKA can lead to an increase in intracellular sodium levels, which may disrupt cellular homeostasis and contribute to the dysfunction of pancreatic α-cells and β-cells.
Moreover, studies have highlighted the role of oxidative stress in T2D (24), which can further compromise NKA activity. Oxidative stress markers correlate with reduced NKA activity in various tissues (21). This suggests that the metabolic disturbances associated with T2D may be, in part, due to the inability of NKA to maintain ionic balance under oxidative conditions.
Furthermore, the relationship between NKA activity and metabolic syndrome (MS) is noteworthy, as individuals with T2D and MS exhibit significantly lower NKA activity compared to healthy controls (25). Decreased NKA activity correlates with higher oxidative stress, potentially promoting diabetes-related complications like cardiovascular disease (26). NKA dysfunction directly links to features of metabolic syndrome through several mechanisms. In obesity, reduced NKA activity leads to increased intracellular sodium levels, which can promote insulin resistance and hypertension (27–29). High sodium levels can also activate the renin-angiotensin-aldosterone system (RAAS), contributing to elevated blood pressure (29). By modulating NKA activity, it may be possible to address multiple features of metabolic syndrome, improving overall metabolic health.
The implications of NKA dysfunction extend beyond the pancreas. For instance, in diabetic peripheral neuropathy (DPN), which affects a substantial number of T2D patients, GLP-1 receptor agonist therapies have demonstrated improvements in nerve conduction and axonal excitability, potentially through the restoration of NKA function (30). This indicates that therapeutic strategies aimed at enhancing NKA activity may offer a protective effect against the neurological complications of diabetes. Additionally, the modulation of NKA activity has been implicated in the regulation of renal function in diabetes, as alterations in NKA expression and activity can lead to renal tubular dysfunction and contribute to the progression of diabetic kidney disease (31). Overall, the evidence suggests that the dysregulation of NKA is a key factor in the pathogenesis of T2D and its complications, highlighting the importance of further research into targeted therapies that could restore NKA function and improve metabolic outcomes in diabetic patients.
NKA is essential for maintaining cellular ion balance, which is crucial for the proper function of pancreatic islet cells. In β-cells, NKA helps maintain membrane potential by regulating sodium and potassium ion levels, which is vital for calcium influx and insulin release (32). Impaired NKA function can disrupt this balance, leading to reduced insulin secretion and contributing to hyperglycemia. Similarly, in α-cells, NKA’s role in establishing ion gradients is critical for glucagon secretion. Reduced NKA activity can impair glucagon secretion, leading to dysregulated blood sugar levels. This dysfunction can make α-cells less sensitive to glucose changes, resulting in excessive glucagon secretion even at low blood sugar levels, which further disrupts glucose homeostasis.
NKA’s ion transport activity also plays a significant role in cellular metabolism and energy balance (13). It accounts for 20-25% of whole-body ATP consumption, highlighting its importance in energy metabolism (33). In disease conditions, NKA dysfunction can interfere with mitochondrial function and disrupt energy production (34). Additionally, NKA interacts with metabolic signaling pathways such as the AMPK pathway, which is crucial for cellular energy sensing and regulation (19, 33). Impaired NKA function can affect AMPK activity, disrupting cellular energy balance and contributing to metabolic disorders.
NKA regulates multiple aspects of insulin secretion by affecting membrane potential and calcium influx (32). For example, normal NKA activity promotes the opening of voltage-gated calcium channels, increasing intracellular calcium levels and activating the fusion and release of insulin secretion granules (32). Additionally, NKA interacts with the cAMP-PKA signaling pathway (35). When cAMP levels rise, PKA is activated, enhancing NKA activity and promoting insulin secretion (36).
NKA dysfunction triggers cell apoptosis via multiple mechanisms (37). One key mechanism involves the accumulation of intracellular sodium, which disrupts mitochondrial function and elevates reactive oxygen species (ROS) levels (37). The increase in ROS not only damages cellular macromolecules but also activates apoptotic pathways, such as the mitochondrial and endoplasmic reticulum stress pathways (37). Additionally, NKA dysfunction alters the balance of intracellular calcium, leading to the activation of proteases like calpain (38). This activation results in structural and functional damage to the cell, ultimately inducing apoptosis. These pathways underscore the critical role of NKA in maintaining cell viability and highlight the consequences of its dysfunction in the context of metabolic diseases like T2D.
NKA is involved in multiple signaling pathways in immune regulation. For example, NKA regulates the activity of the nuclear factor κB (NF-κB) signaling pathway by affecting intracellular sodium and calcium concentrations (39). NF-κB plays a key role in the activation, proliferation, and cytokine secretion of immune cells (40). Normal NKA activity inhibits the overactivation of the NF-κB signaling pathway, reducing the production of pro-inflammatory cytokines and alleviating inflammatory responses (39).
Oxidative stress is a central mechanistic factor in the dysfunction of NKA and the pathogenesis of T2D (24, 41). Elevated levels of oxidative markers such as malondialdehyde (MDA), protein carbonyls, and 8-hydroxy-2’-deoxyguanosine (8-OHdG) are directly linked to T2D (42). MDA, a product of lipid peroxidation, indicates damage to cell membranes and organelles, disrupting cellular homeostasis (43). Protein carbonyls reflect protein oxidation, which can alter protein structure and function, impairing NKA’s catalytic activity (44). 8-OHdG, a marker of DNA damage, suggests genomic instability and can affect the expression and function of NKA. These oxidative markers contribute to metabolic dysfunction by impairing insulin signaling, reducing β-cell function, and promoting insulin resistance (45).
Given the fundamental role of NKA in regulating insulin secretion and action, NKA is considered a promising therapeutic target for improving pancreatic function and insulin sensitivity in T2D patients. Research indicates that modulating NKA activity can lead to enhanced glucose homeostasis and improved metabolic outcomes (22, 46). For instance, specific antibodies targeting the NKA may stimulate its activity, leading to cardioprotective effects and improved metabolic profiles in models of chronic kidney disease and heart failure (47). Furthermore, the use of NKA activators has been associated with increased osteogenic differentiation in mesenchymal stem cells, suggesting a broader therapeutic potential for NKA modulation beyond metabolic diseases (48).
Recent investigations have also highlighted the significance of NKA in various cellular processes, including its role as a signaling molecule (6). NKA interacts with several proteins, influencing pathways that regulate cellular metabolism and proliferation (49). For example, the G protein-coupled receptor GPR35 has been shown to enhance NKA’s ion transport and signaling activity, which may have implications for T2D treatment (50). Additionally, the identification of endogenous negative regulators of NKA, such as inositol pyrophosphate 5-InsP7, suggests that manipulating these regulatory pathways could provide new strategies for enhancing NKA function in T2D (15).
Overall, the modulation of NKA presents a multifaceted approach to treating T2D and potentially other related conditions. The ongoing research into NKA’s role in cellular signaling, its interactions with various proteins, and its implications in metabolic and oncological contexts highlights its potential as a versatile therapeutic target. Continued exploration of NKA’s mechanisms and regulatory pathways will be essential in developing effective strategies to harness its therapeutic potential in clinical settings.
Recent studies have highlighted the importance of NKA in the context of T2D and its potential as a therapeutic target. NKA dysfunction is linked to insulin resistance, β-cell failure, and increased oxidative stress, all of which contribute to the pathophysiology of T2D. Although there are no reports of direct applications of NKA activators in the treatment of T2D, existing findings suggest that modulating NKA activity may offer new insights for improving insulin secretion and restoring glucose homeostasis. Future research may further explore the potential applications of NKA activators in the treatment of T2D. Novel approaches including NKA activators, gene therapy, and stem cell therapy, can improve insulin secretion, and restore glucose homeostasis. These approaches offer novel therapeutic strategies that complement existing antidiabetic drugs and immunomodulatory therapies, providing a more comprehensive and updated perspective on the treatment of T2D.
NKA activators enhance NKA activity, which is crucial for cellular ion homeostasis and insulin secretion. The mechanism involves increasing NKA activity, which helps maintain intracellular sodium and potassium balance and promotes insulin release from pancreatic beta cells. This is beneficial for T2D patients with impaired insulin secretion. Specific NKA activators may improve glycemic control and insulin sensitivity, reducing blood glucose levels. However, potential side effects and off-target effects require careful monitoring and dosage control. NKA activators offer a novel therapeutic strategy for T2D.
Gene therapy, especially using CRISPR/Cas9, targets genetic factors in T2D (51). It can edit genes associated with NKA function to restore normal activity, enhancing insulin secretion and glucose homeostasis. This is relevant in T2D, where ion transport dysregulation is a significant issue. Gene editing can correct mutations impairing NKA function, improving glucose responses and insulin sensitivity. Gene therapy can also deliver genes encoding beneficial proteins to enhance beta-cell function (52). Despite challenges like delivery methods and off-target effects, ongoing research explores its feasibility and efficacy, offering personalized and durable solutions for T2D.
Stem cell therapy provides insulin-producing cells to restore pancreatic function in T2D (53). The differentiation of stem cells into pancreatic beta cells addresses the loss of insulin secretion (54). Advances in stem cell research have enabled the generation of functional pancreatic cells from PSCs, which can secrete insulin in response to glucose, potentially reversing hyperglycemia (55, 56). Adult pancreatic stem cells also show promise in regenerating damaged tissue and restoring insulin production (57). However, challenges include generating sufficient functional beta cells, managing tumorigenesis risk, and post-transplantation immunosuppression. Ongoing research aims to optimize differentiation protocols and explore combination therapies to improve T2D outcomes. Stem cell therapy is a promising regenerative approach to address the root causes of T2D.
This review has highlighted the multifaceted role of NKA in T2D, emphasizing its importance in cellular ion homeostasis, insulin secretion, and immune responses (Figure 1). The key points addressed include the biological functions of NKA, its involvement in T2D pathophysiology, and the potential of NKA as a therapeutic target. Areas warranting further investigation involve the development of more specific NKA modulators with fewer side effects and the application of experimental models to explore the clinical feasibility of targeting NKA. Future research should focus on refining NKA activators, gene therapy, and stem cell therapy to enhance their safety and efficacy. The development of novel experimental models, such as humanized mouse models and iPSC-derived islets, will be crucial for preclinical testing and translational research. In conclusion, modulating NKA activity offers promising therapeutic strategies for T2D, with the potential to improve glycemic control and enhance β-cell function. These strategies, if developed and applied effectively, could significantly impact the management of T2D and related complications, and may also contribute to improving outcomes in islet transplantation.
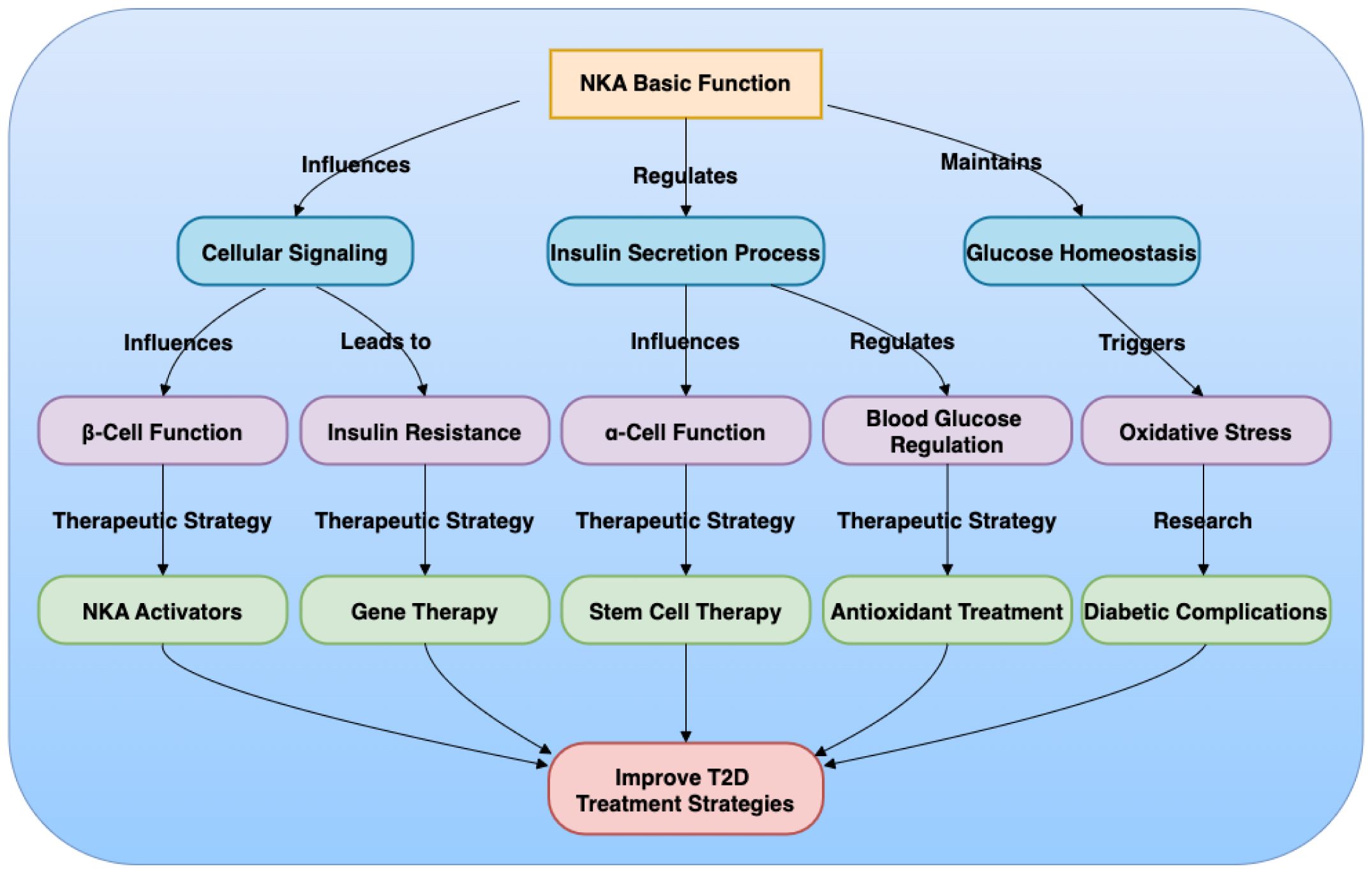
Figure 1. Mechanism of Na+/K+-ATPase (NKA) in Type 2 Diabetes (T2D).
LM: Writing – original draft, Writing – review & editing, Funding acquisition. XW: Writing – review & editing, Funding acquisition. ZP: Writing – review & editing, Funding acquisition. ZF: Writing – original draft, Writing – review & editing. TW: Writing – review & editing. YC: Writing – review & editing. ZL: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported in part by the Shenzhen Science and Technology Program (JCYJ20220818102001003, JCYJ20230807115107015, GJHZ20240218114714027 and GCZX2015043017281705) and Team-based Medical Science Research Program (2024YZZ04).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Taylor R. Understanding the cause of type 2 diabetes. Lancet Diabetes Endocrinol. (2024) 12:664–73. doi: 10.1016/S2213-8587(24)00157-8
2. Zhou B, Rayner AW, Gregg EW, Sheffer KE, Carrillo-Larco RM, Bennett JE, et al. Worldwide trends in diabetes prevalence and treatment from 1990 to 2022: a pooled analysis of 1108 population-representative studies with 141 million participants. Lancet. (2024) 404:2077–93. doi: 10.1016/S0140-6736(24)02317-1
3. Ogurtsova K, Guariguata L, Barengo NC, Ruiz PL-D, Sacre JW, Karuranga S, et al. IDF diabetes Atlas: Global estimates of undiagnosed diabetes in adults for 2021. Diabetes Res Clin Pract. (2022) 183:109118. doi: 10.1016/j.diabres.2021.109118
4. Huang Y-J, Chen C, Yang H-C. AI-enhanced integration of genetic and medical imaging data for risk assessment of Type 2 diabetes. Nat Commun. (2024) 15:4230. doi: 10.1038/s41467-024-48618-1
5. Vague P, Coste TC, Jannot MF, Raccah D, Tsimaratos M. C-peptide, Na+,K+-ATPase, and diabetes. J Diabetes Res. (2004) 5:37–50. doi: 10.1080/15438600490424514
6. Contreras RG, Torres-Carrillo A, Flores-Maldonado C, Shoshani L, Ponce A. Na+/K+-ATPase: more than an electrogenic pump. IJMS. (2024) 25:6122. doi: 10.3390/ijms25116122
7. Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front Physiol. (2017) 8:371. doi: 10.3389/fphys.2017.00371
8. Wen X-P, Wan Q-Q. Regulatory effect of insulin on the structure, function and metabolism of Na+/K+−ATPase (Review). Exp Ther Med. (2021) 22:1243. doi: 10.3892/etm.2021.10678
9. Scarpini E, Bianchi R, Moggio M, Sciacco M, Fiori MG, Scarlato G. Decrease of nerve Na+,K+-ATPase activity in the pathogenesis of human diabetic neuropathy. J Neurol Sci. (1993) 120:159–67. doi: 10.1016/0022-510X(93)90268-4
10. Shahid SM, Mahboob T. Electrolytes and NA(+)-K(+)-ATPase: potential risk factors for the development of diabetic nephropathy. Pak J Pharm Sci. (2008) 21:172–9.
11. Puljko B, Stojanović M, Ilic K, Kalanj-Bognar S, Mlinac-Jerkovic K. Start me up: how can surrounding gangliosides affect sodium-potassium ATPase activity and steer towards pathological ion imbalance in neurons? Biomedicines. (2022) 10:1518. doi: 10.3390/biomedicines10071518
12. Vrbjar N, Jasenovec T, Kollarova M, Snurikova D, Chomova M, Radosinska D, et al. Na,K-ATPase kinetics and oxidative stress in kidneys of Zucker diabetic fatty (fa/fa) rats depending on the diabetes severity—Comparison with lean (fa/+) and wistar rats. Biology. (2022) 11:1519. doi: 10.3390/biology11101519
13. Huang S, Dong W, Lin X, Bian J. Na+/K+-ATPase: ion pump, signal transducer, or cytoprotective protein, and novel biological functions. Neural Regener Res. (2024) 19:2684–97. doi: 10.4103/NRR.NRR-D-23-01175
14. Hodeify R, Kreydiyyeh S, Zaid LMJ. Identified and potential internalization signals involved in trafficking and regulation of Na+/K+ ATPase activity. Mol Cell Biochem. (2024) 479:1583–98. doi: 10.1007/s11010-023-04831-y
15. Chin AC, Gao Z, Riley AM, Furkert D, Wittwer C, Dutta A, et al. The inositol pyrophosphate 5-InsP 7 drives sodium-potassium pump degradation by relieving an autoinhibitory domain of PI3K p85α. Sci Adv. (2020) 6:eabb8542. doi: 10.1126/sciadv.abb8542
16. Nguyen PT, Deisl C, Fine M, Tippetts TS, Uchikawa E, Bai X, et al. Structural basis for gating mechanism of the human sodium-potassium pump. Nat Commun. (2022) 13:5293. doi: 10.1038/s41467-022-32990-x
17. Ren Y, Anderson AT, Meyer G, Lauber KM, Gallucci JC, Douglas Kinghorn A. Digoxin and its Na+/K+-ATPase-targeted actions on cardiovascular diseases and cancer. Bioorg Medicinal Chem. (2024) 114:117939. doi: 10.1016/j.bmc.2024.117939
18. Yan X, Li M, Lan P, Xun M, Zhang Y, Shi J, et al. Regulation of Na+-K+-ATPase leads to disturbances of isoproterenol-induced cardiac dysfunction via interference of Ca2+-dependent cardiac metabolism. Clin Sci (Lond). (2024) 138:23–42. doi: 10.1042/CS20231039
19. Xiao J, Zhu S, Guan H, Zheng Y, Li F, Zhang X, et al. AMPK alleviates high uric acid-induced Na+-K+-ATPase signaling impairment and cell injury in renal tubules. Exp Mol Med. (2019) 51:1–14. doi: 10.1038/s12276-019-0254-y
20. Zheng J, Lan P, Li M, Kang M, Xun M, Ma X, et al. Anti–Na+/K+-ATPase DR antibody attenuates UUO-induced renal fibrosis through inhibition of Na+/K+-ATPase α1–dependent HMGB1 release. Int Immunopharmacol. (2023) 116:109826. doi: 10.1016/j.intimp.2023.109826
21. Radosinska D, Gaal Kovalcikova A, Gardlik R, Chomova M, Snurikova D, Radosinska J, et al. Oxidative Stress Markers and Na,K-ATPase Enzyme Kinetics Are Altered in the Cerebellum of Zucker Diabetic Fatty fa/fa Rats: A Comparison with Lean fa/+ and Wistar Rats. Biology. (2024) 13:759. doi: 10.3390/biology13100759
22. Sun H-J, Cao L, Zhu M-Y, Wu Z-Y, Shen C-Y, Nie X-W, et al. DR-region of Na+/K+-ATPase is a target to ameliorate hepatic insulin resistance in obese diabetic mice. Theranostics. (2020) 10:6149–66. doi: 10.7150/thno.46053
23. Shi Q, Zeng J, Dong Y, Xu KY, Shi Q, Zeng J, et al. Concurrent impairment of (Na++K+)-ATPase activity in multi-organ of type-1 diabetic NOD mice. J Diabetes Its Complicat. (2013) 27:29–33. doi: 10.1016/j.jdiacomp.2012.07.004
24. Caturano A, D’Angelo M, Mormone A, Russo V, Mollica MP, Salvatore T, et al. Oxidative stress in type 2 diabetes: impacts from pathogenesis to lifestyle modifications. CIMB. (2023) 45:6651–66. doi: 10.3390/cimb45080420
25. Najafi A, Pourfarzam M, Zadhoush F. Oxidant/antioxidant status in Type-2 diabetes mellitus patients with metabolic syndrome. J Res Med Sci. (2021) 26:6. doi: 10.4103/jrms.JRMS_249_20
26. Fedorova OV, Fadeev AV, Grigorova YN, Marshall CA, Zernetkina V, Kolodkin NI, et al. Cardiotonic steroids induce vascular fibrosis via pressure-independent mechanism in NaCl-loaded diabetic rats. J Cardiovasc Pharmacol. (2019) 74:436–42. doi: 10.1097/FJC.0000000000000730
27. Ertuglu LA, Elijovich F, Laffer CL, Kirabo A. Salt-sensitivity of blood pressure and insulin resistance. Front Physiol. (2021) 12:793924. doi: 10.3389/fphys.2021.793924
28. Wu Q, Burley G, Li L, Lin S, Shi Y, Wu Q, et al. The role of dietary salt in metabolism and energy balance: Insights beyond cardiovascular disease. Diabetes Obes Metab. (2023) 25:1147–61. doi: 10.1111/dom.14980
29. Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME, Hall JE, et al. Obesity, kidney dysfunction and hypertension: mechanistic links. Nat Rev Nephrol. (2019) 15:367–85. doi: 10.1038/s41581-019-0145-4
30. Dhanapalaratnam R, Issar T, Poynten AM, Milner K-L, Kwai NCG, Krishnan AV. Impact of glucagon-like peptide-1 receptor agonists on axonal function in diabetic peripheral neuropathy. J Neurophysiol. (2025) 133:14–21. doi: 10.1152/jn.00228.2024
31. Silva-Aguiar RP, Teixeira DE, Peres RAS, Alves SAS, Novaes-Fernandes C, Dias WB, et al. O-Linked GlcNAcylation mediates the inhibition of proximal tubule (Na++K+)ATPase activity in the early stage of diabetes mellitus. Biochim Biophys Acta (BBA) Gen Subj. (2023) 1867:130466. doi: 10.1016/j.bbagen.2023.130466
32. Dickerson MT, Dadi PK, Zaborska KE, Nakhe AY, Schaub CM, Dobson JR, et al. Gi/o protein-coupled receptor inhibition of beta-cell electrical excitability and insulin secretion depends on Na+/K+ ATPase activation. Nat Commun. (2022) 13:6461. doi: 10.1038/s41467-022-34166-z
33. Pirkmajer S, Petrič M, Chibalin AV, Pirkmajer S, Petrič M, Chibalin AV, et al. The role of AMPK in regulation of Na+,K+-ATPase in skeletal muscle: does the gauge always plug the sink? J Muscle Res Cell Motil. (2021) 42:77–97. doi: 10.1007/s10974-020-09594-3
34. Dhalla NS, Elimban V, Adameova AD. Role of Na+-K+ ATPase alterations in the development of heart failure. IJMS. (2024) 25:10807. doi: 10.3390/ijms251910807
35. Poulsen H, Nissen P, Mouritsen OG, Khandelia H, Poulsen H, Nissen P, et al. (PKA) phosphorylation of Na+/K+-ATPase opens intracellular C-terminal water pathway leading to third Na+-binding site in molecular dynamics simulations. J Biol Chem. (2012) 287:15959–65. doi: 10.1074/jbc.M112.340406
36. Stožer A, Paradiž Leitgeb E, Pohorec V, Dolenšek J, Križančić Bombek L, Gosak M, et al. The role of cAMP in beta cell stimulus–secretion and intercellular coupling. Cells. (2021) 10:1658. doi: 10.3390/cells10071658
37. Felippe-Gonçalves-de-Albuquerque C, Ribeiro Silva A, Ignácio da Silva C, Caire-Castro-Faria-Neto H, Burth P. Na/K pump and beyond: Na/K-ATPase as a modulator of apoptosis and autophagy. Molecules. (2017) 22:578. doi: 10.3390/molecules22040578
38. Rodrigues T, Piccirillo S, Magi S, Preziuso A, dos Santos Ramos V, Serfilippi T, et al. Control of Ca2+ and metabolic homeostasis by the Na+/Ca2+ exchangers (NCXs) in health and disease. Biochem Pharmacol. (2022) 203:115163. doi: 10.1016/j.bcp.2022.115163
39. Zhang J, Chang J, Beg MA, Huang W, Zhao Y, Dai W, et al. Na/K-ATPase suppresses LPS-induced pro-inflammatory signaling through Lyn. iScience. (2022) 25:104963. doi: 10.1016/j.isci.2022.104963
40. Guo Q, Jin Y, Chen X, Ye X, Shen X, Lin M, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Sig Transduct Target Ther. (2024) 9:53. doi: 10.1038/s41392-024-01757-9
41. de Melo AD, Freire VAF, Diogo ÍL, Santos H de L, Barbosa LA, de Carvalho LED, et al. Antioxidant therapy reduces oxidative stress, restores Na,K-ATPase function and induces neuroprotection in rodent models of seizure and epilepsy: A systematic review and meta-analysis. Antioxidants. (2023) 12:1397. doi: 10.3390/antiox12071397
42. Goriuc A, Cojocaru K-A, Luchian I, Ursu R-G, Butnaru O, Foia L. Using 8-hydroxy-2′-deoxiguanosine (8-OHdG) as a reliable biomarker for assessing periodontal disease associated with diabetes. IJMS. (2024) 25:1425. doi: 10.3390/ijms25031425
43. Zheng Y, Sun J, Luo Z, Li Y, Huang Y, Zheng Y, et al. Emerging mechanisms of lipid peroxidation in regulated cell death and its physiological implications. Cell Death Dis. (2024) 15:859. doi: 10.1038/s41419-024-07244-x
44. Kryvenko V, Wessendorf M, Morty RE, Herold S, Seeger W, Vagin O, et al. Hypercapnia impairs Na,K-ATPase function by inducing endoplasmic reticulum retention of the β-subunit of the enzyme in alveolar epithelial cells. IJMS. (2020) 21:1467. doi: 10.3390/ijms21041467
45. Eguchi N, Vaziri ND, Dafoe DC, Ichii H. The role of oxidative stress in pancreatic β Cell dysfunction in diabetes. IJMS. (2021) 22:1509. doi: 10.3390/ijms22041509
46. Michaels AM, Zoccarato A, Hoare Z, Firth G, Chung YJ, Kuchel PW, et al. Disrupting Na+ ion homeostasis and Na+/K+ ATPase activity in breast cancer cells directly modulates glycolysis in vitro and in vivo. Cancer Metab. (2024) 12:15. doi: 10.1186/s40170-024-00343-5
47. Zheng J, Lan P, Meng X, Kang M-C, Huang X, Yan X. Na+/K+-ATPase DR region antibody ameliorated cardiac hypertrophy and fibrosis in rats with 5/6 nephrectomy. Exp Biol Med (Maywood). (2022) 247:1785–94. doi: 10.1177/15353702221108910
48. Tang Z, Chen S, Ni Y, Zhao R, Zhu X, Yang X, et al. Role of Na+, K+-ATPase ion pump in osteoinduction. Acta Biomaterialia. (2021) 129:293–308. doi: 10.1016/j.actbio.2021.05.026
49. Cui X, Xie Z. Protein interaction and Na/K-ATPase-mediated signal transduction. Molecules. (2017) 22:990. doi: 10.3390/molecules22060990
50. Schneditz G, Elias JE, Pagano E, Zaeem Cader M, Saveljeva S, Long K, et al. GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump. Sci Signal. (2019) 12:eaau9048. doi: 10.1126/scisignal.aau9048
51. Lotfi M, Butler AE, Sukhorukov VN, Sahebkar A. Application of CRISPR-Cas9 technology in diabetes research. Diabetes Med. (2024) 41:e15240. doi: 10.1111/dme.15240
52. So WY, Han W. Gene therapy targeting key beta cell regulators as a potential intervention for diabetes. EMBO Mol Med. (2024) 16:1490–4. doi: 10.1038/s44321-024-00089-z
53. Grattoni A, Korbutt G, Tomei AA, García AJ, Pepper AR, Stabler C, et al. Harnessing cellular therapeutics for type 1 diabetes mellitus: progress, challenges, and the road ahead. Nat Rev Endocrinol. (2025) 21:14–30. doi: 10.1038/s41574-024-01029-0
54. Wu J, Li T, Guo M, Ji J, Meng X, Fu T, et al. Treating a type 2 diabetic patient with impaired pancreatic islet function by personalized endoderm stem cell-derived islet tissue. Cell Discovery. (2024) 10:45. doi: 10.1038/s41421-024-00662-3
55. Ramzy A, Thompson DM, Ward-Hartstonge KA, Ivison S, Cook L, Garcia RV, et al. Implanted pluripotent stem-cell-derived pancreatic endoderm cells secrete glucose-responsive C-peptide in patients with type 1 diabetes. Cell Stem Cell. (2021) 28:2047–2061.e5. doi: 10.1016/j.stem.2021.10.003
56. Wang S, Du Y, Zhang B, Meng G, Liu Z, Liew SY, et al. Transplantation of chemically induced pluripotent stem-cell-derived islets under abdominal anterior rectus sheath in a type 1 diabetes patient. Cell. (2024) 187:6152–6164.e18. doi: 10.1016/j.cell.2024.09.004
Keywords: diabetes, type 2 diabetes, sodium-potassium ATPase, islet, β-cells, therapeutic target, gene therapy, stem cell therapy
Citation: Mou L, Fu Z, Wang TB, Chen Y, Luo Z, Wang X and Pu Z (2025) Na+/K+-ATPase: a multifunctional target in type 2 diabetes and pancreatic islets. Front. Immunol. 16:1555310. doi: 10.3389/fimmu.2025.1555310
Received: 04 January 2025; Accepted: 21 January 2025;
Published: 19 February 2025.
Edited by:
Yi Wang, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, ChinaCopyright © 2025 Mou, Fu, Wang, Chen, Luo, Wang and Pu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisha Mou, bGlzaGFtb3VAZW1haWwuc3p1LmVkdS5jbg==; Xinyu Wang, d3h5aG9yc2VAMTYzLmNvbQ==; Zuhui Pu, enVodWlwdUBlbWFpbC5zenUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.