 Longyan Hong
Longyan Hong Xiangyu Chen1,2†
Xiangyu Chen1,2† Hao Liang
Hao Liang Yinghui Zhao
Yinghui Zhao Pengbo Guo
Pengbo Guo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 18 March 2025
Sec. Microbial Immunology
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1550968
Respiratory infectious diseases, particularly those caused by respiratory viruses, have the potential to lead to global pandemics, thereby posing significant threats to public and human health. Historically, the primary treatment for respiratory bacterial infections has been antibiotic therapy, while severe cases of respiratory viral infections have predominantly been managed by controlling inflammatory cytokine storms. Ferroptosis is a novel form of programmed cell death that is distinct from apoptosis and autophagy. In recent years, Recent studies have demonstrated that ferroptosis plays a significant regulatory role in various respiratory infectious diseases, indicating that targeting ferroptosis may represent a novel approach for the treatment of these conditions. This article summarized the toxic mechanisms underlying ferroptosis, its relationship with respiratory infectious diseases, the mechanisms of action, and current treatment strategies. Particular attentions were given to the interplay between ferroptosis and Mycobacterium tuberculosis, Epstein-Barr virus, severe acute respiratory syndrome coronavirus-2, Pseudomonas aeruginosa, dengue virus, influenza virus and herpes simplex virus type1infection. A deeper understanding of the regulatory mechanisms of ferroptosis in respiratory infections will not only advance our knowledge of infection-related pathophysiology but also provide a theoretical foundation for the development of novel therapeutic strategies. Targeting ferroptosis pathways represents a promising therapeutic approach for respiratory infections, with significant clinical and translational implications.
In 2012, Brent Stockwell, Scott Dixon, and colleagues introduced the concept of iron-dependent regulatory cell death, termed ferroptosis (1). This oxidative form of cell death is distinct from apoptosis, non-regulatory necrosis, and necroptosis (regulated necrosis) (2). Ferroptosis is primarily triggered by the accumulation of lethal lipid hydroperoxides within cellular membranes, particularly in the endoplasmic reticulum (ER), due to the failure of protective mechanisms that prevent excessive lipid peroxidation (3). Respiratory diseases pose a significant global health burden, ranking as the third leading cause of mortality in urban areas and the leading cause of death in rural regions (4, 5). Historically, the primary treatment for bacterial infections has been antibiotic therapy. However, the emergence of multiple drug-resistant strains has posed significant challenges to the effectiveness of such treatments (6). In contrast, there are limited specific antiviral medications available; most existing drugs primarily function by inhibiting viral replication or transmission within the host (7). Consequently, it is essential to elucidate the mechanisms underlying respiratory bacterial and viral infectious diseases in order to identify new insights and develop innovative therapeutic strategies. The discovery of ferroptosis has significant implications for respiratory diseases caused by certain viruses and bacteria, offering new avenues for treatment (8), particularly in targeting host-pathogen interactions to mitigate disease severity and improve clinical outcomes.
M. tuberculosis infection causes tuberculosis (TB), which ranks among the top ten causes of death worldwide (9). Prior to the COVID-19 pandemic, it was the leading cause of mortality attributable to a single infectious pathogen (10). TB is characterized as a chronic respiratory disease, manifesting a range of symptoms, including cough, hemoptysis, dyspnea, and chest pain. We summarized the role and molecular mechanisms of ferroptosis-related molecules in M. tuberculosis infection to provide novel insights and research directions for the treatment of this infection (Figure 1A).
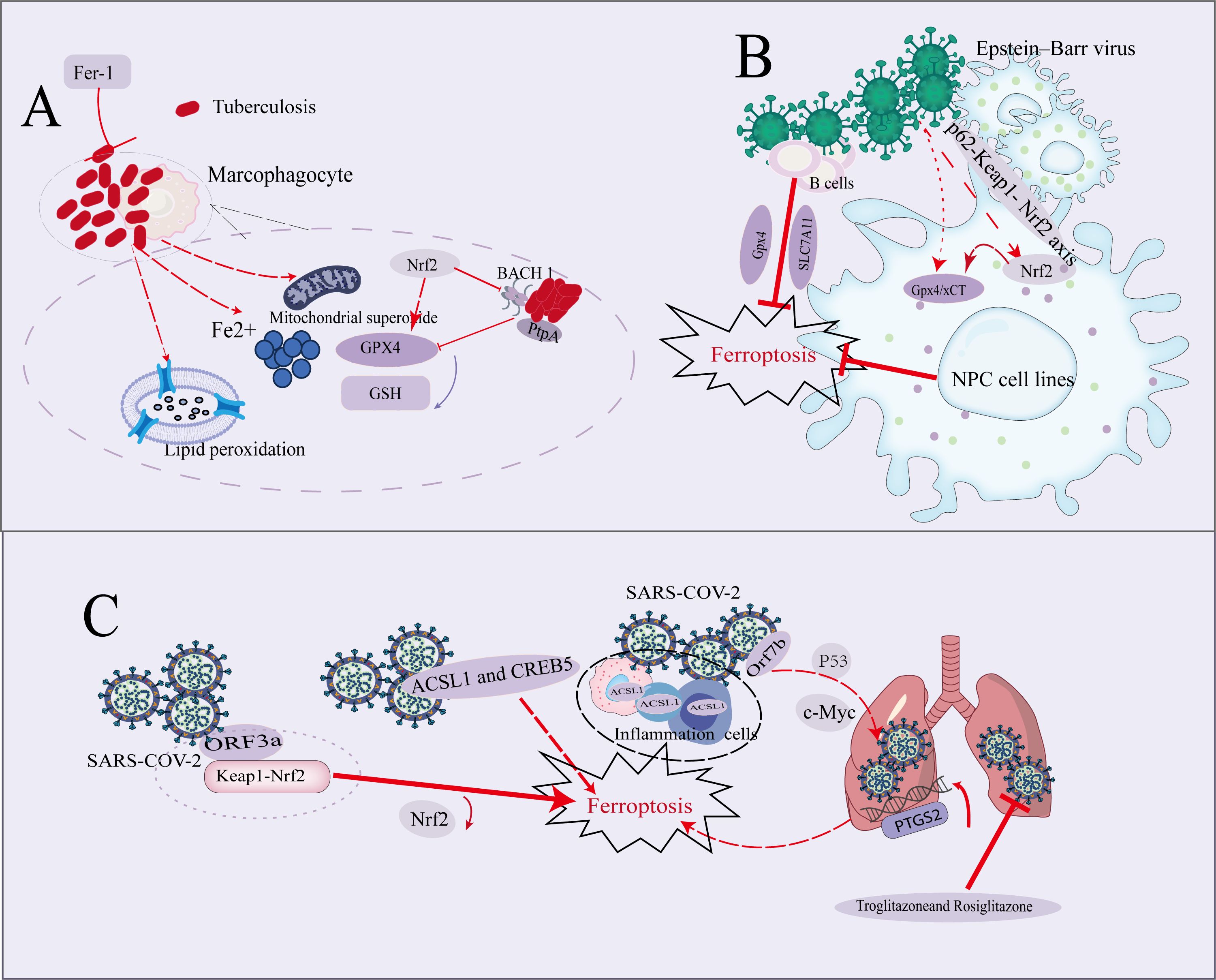
Figure 1. Overview Diagram of Ferroptosis Mechanism in Mycobacterium tuberculosis, EB Virus, and SARS-CoV-2 Infections. (A) The necrosis of macrophages triggered by M. tuberculosis is closely linked to elevated levels of intracellular iron, increased mitochondrial superoxide, and enhanced lipid peroxidation. The use of iron chelators or inhibitors of lipid peroxidation effectively averted M. tuberculosis-induced macrophage necrosis. The nuclear factor (Nrf2) played a role in inhibiting the expression of Bach1 and regulating Gpx4 to mitigate ferroptosis. Additionally, the effector molecule PtpA secreted by M. tuberculosis promoted ferroptosis, thereby increasing its pathogenicity and ability to spread. (B) This panel outlines the molecular mechanisms through which EBV infects B cells and nasopharyngeal carcinoma (NPC) cell lines. EBV infection makes B cells reliant on the activity of SLC7A11 and Gpx4 to resist ferroptosis. In NPC cell lines, EBV infection activates the p62-Keap1-NRF2 pathway, leading to enhanced Nrf2 activity. This activation further upregulates downstream target genes, such as Gpx4 and xCT, which play a critical role in reducing lipid peroxidation and inhibiting the accumulation of lipid ROS, thereby preventing ferroptosis. (C) The accessory protein Orf7b of SARS-CoV-2 promotes ferroptosis through the c-Myc signaling pathway, leading to lung damage. The expression of ferroptosis-related genes ACSL1 and CREB5 in monocytes may further promote ferroptosis by regulating the inflammatory response. In lung cells infected with SARS-CoV-2, the mRNA levels of the ferroptosis marker PTGS2 are significantly increased. Additionally, ORF3a is considered a positive regulator of ferroptosis; it binds to Keap1, enhancing Keap1 stability, promoting the interaction between Keap1 and Nrf2, and thereby accelerating the degradation of Nrf2, which in turn facilitates ferroptosis.
M. tuberculosis-induced necrotic cell death was associated with increased levels of intracellular iron and mitochondrial superoxide, as well as lipid peroxidation (11). Using an in vitro model of M. tuberculosis-induced macrophage death, researchers observed that necrosis exhibited classic characteristics associated with ferroptosis. The necrotic cell death of macrophages infected with M. tuberculosis was linked to decreased expression levels of GSH and Gpx4 (12, 13) (Figure 1A).
Iron chelation or inhibition of lipid peroxidation could prevent macrophage necrosis induced by M. tuberculosis. Fer-1 is widely recognized as a potent inhibitor of ferroptosis due to its highly effective inhibition of lipid peroxidation, partly attributed to its antioxidant activity (1). Treatment with Fer-1 significantly reduced lung weight and calculated relative lung mass in infected mice (Figure 1A). Histological examination confirmed that this inhibition of lipid peroxidation correlates with a reduction in granulomatous inflammation.
By comparing tuberculosis patients with healthy individuals, it was observed a decrease in the levels of Gpx4/GSH and an increase in lipid peroxidation among TB patients (10, 14).One potential strategy is to target upstream signaling components that positively regulate Gpx4 expression and/or activity in glutathione metabolism. A key component in this process was nuclear factor erythroid 2-related factor 2 (Nrf2), which not only regulated Gpx4 expression but also influenced various other antioxidant molecules (Figure 1A). However, this approach requires extensive clinical trials for further validation (10, 15–17).
In addition, some antioxidants, including vitamin E, were shown to inhibit ferroptosis, and studies indicated that adjunctive use of vitamin E with conventional antibiotic therapy improved TB treatment outcomes (13, 18). Furthermore, supplementing selenium was shown to enhance Gpx4 expression and activity, thereby inhibiting ferroptosis (12). Notably, selenium, as a nutritional supplement, was demonstrated to improve TB treatment outcomes (13).
PtpA is an effector secreted by M. tuberculosis. PtpA triggered ferroptosis, thereby enhancing the pathogenicity and transmission of M. tuberculosis. It interacted with the host RanGDP through its Cys 11 site to enter the host cell nucleus. Once inside, nuclear PtpA promoted the asymmetric dimethylation of histone H3 at arginine 2 (H3 R2 me2a) by targeting protein arginine methyltransferase 6 (PRMT 6), resulting in the inhibition of Gpx4 expression and ultimately inducing ferroptosis, which facilitates M. tuberculosis pathogenicity and spread (Figure 1A) (19–21).
The exacerbation of lipid peroxidation is a consequence of uncontrolled oxidative stress, which is linked to iron deposition and necrotic cell death (22–24).Bach 1 is a transcription factor that inhibits a variety of antioxidant genes. Notably, BACH 1 mRNA expression is increased in patients who develop active TB following M. tuberculosis infection. Importantly, it was observed that iron sepsis-related genes associated with antioxidant properties were upregulated in various bone marrow cell populations in M. tuberculosis-infected Bach 1 −/− mice, suggesting a role for Bach 1 in regulating M. tuberculosis-induced necrosis in vivo. Under conditions of Bach 1 deficiency, complete ablation of cell necrosis was not observed in either in vivo or in vitro settings, suggesting that Bach 1 deficiency and its role in cell necrosis coexist.
Nrf2 is the primary upstream regulator of the host antioxidant response (including GSH metabolism), and inhibiting its repressive factor Bach might provide an alternative strategy to enhance the expression of enzymes and their cofactors, which were crucial for mitigating lipid peroxidation-mediated tissue death (25). Discovering and developing other biologically acceptable Bach 1 inhibitors represented an effective strategy for addressing various diseases, particularly those where lipid peroxidation-mediated cell necrosis is recognized as a significant mechanism of pathogenesis (1, 23, 24, 26).
EBV is a double-stranded DNA virus that establishes lifelong latency in the host following primary infection (27). EBV infection has been closely associated with various B-cell malignancies (28–31). Among these, the malignancy most strongly associated with EBV infection has been undifferentiated nasopharyngeal carcinoma (NPC) (32). Although chemotherapy and radiotherapy are the primary treatment modalities for NPC, significant challenges remained due to chemotherapy resistance. Nevertheless, there had been considerable advancements in understanding the pathogenesis and exploring treatment options for the role of ferroptosis EBV infection (33).
EBV activated lipid metabolism, transforming B cells into immortalized lymphoblastic cell lines (LCLs), which served as a model for post-transplant lymphoproliferative disorders and promoted lipid peroxidation in primary B cells. Anti-EBV therapy remains a significant unmet medical need, especially for immunocompromised patients. Although antiviral drugs approved for other herpesviruses have been tested for EBV-associated diseases, their results have been disappointing (31). Studies showed that dipyridamole (DIP), a drug with good safety and broad pharmacological properties, effectively inhibited EBV reactivation in B cell lines (34). Additionally, research found that different stages of EBV transformation generate varying levels of lipid-derived reactive oxygen species (ROS) byproducts. In the early stages of infection (Burkitt-like hyperproliferation phase), EBV induces lipid metabolism and ROS production, making B cells dependent on cystine import mediated by SLC7A11 and the activity of Gpx4 to resist ferroptosis (Figure 1B). Blocking these pathways, such as using erastin to inhibit SLC7A11 or ML-210 to inhibit Gpx4, significantly induces cell death (35). This phenomenon suggested that ferroptosis could be a potential therapeutic strategy for preventing or treating certain EBV-associated lymphomas (35).
By suppressing Gpx4 expression, EBV promoted the escape of ferroptosis and the restoration of redox homeostasis, thereby contributing to chemotherapy resistance in NPC. EBV infection led to the upregulation of p62, which subsequently activated Nrf2 in NPC cell lines through the p62-Keap1- Nrf2 axis. This activation resulted in the upregulation of downstream effectors xCT and Gpx4, effectively reducing lipid ROS accumulation in tumor cells and protecting them from ferroptosis (Figure 1B). Gene knockout of Gpx4 or treatment with low-dose Gpx4-targeted inhibitors could significantly diminish chemotherapy resistance in EBV-positive NPC cell lines, presenting a potential therapeutic target for treating chemotherapy-resistant tumors (35–37).
SARS-CoV-2 was a novel coronavirus identified as the causative agent of the global epidemic of coronavirus disease 2019 (COVID-19) (38–41). This virus belonged to the coronavirus family and was a highly transmissible airborne infectious pathogen (41). COVID-19 primarily presented as a respiratory disease that manifested as acute upper and/or lower respiratory syndrome of varying severity. The most common symptoms included fever, cough, acute lung injury, septic shock, and acute respiratory distress syndrome (ARDS) (42, 43). Additionally, various other symptoms had been reported in COVID-19 patients, such as loss of taste or smell, headache, muscle or body pain, nausea or vomiting, and diarrhea (44, 45). (The relationship between SARS-CoV-2 infection and the ferroptosis mechanism is illustrated in Figure 1C as a schematic diagram).
The seven main cell types identified in COVID-19 patients, which are affected peripherally by SARS-CoV-2, include monocytes, NK cells, platelets, CD34+ pre-B cells, T cells, B cells, and HSC-G-CSF cells. The ferroptosis-related genes ACSL1 and CREB5 were predominantly expressed in monocytes, HSC-G-CSF cells, and CD34+ pre-B cells (46, 47). The esterification of acyl-CoA synthase long-chain member 4 (ACSL4) allowed polyunsaturated fatty acids (PUFAs) to disrupt cell membranes during the execution phase (48). Recently, ACSL1 had been identified as a promoter of ferroptosis (47) (Figure 1C), withACSL1-induced gallic acid (ESA) triggered ferroptosis. In contrast to typical ferroptosis inducers such as the Gpx4 inhibitor ML160 and the FSP1 inhibitor iFSP1, the ACSL1-dependent ferroptosis induced by ESA shows significant differences (49). The high expression of five ferroptosis-related genes (such as ACSL1 and CREB5) in monocytes of COVID-19 patients may affect the inflammatory response, which could further influence the development of stroke, particularly in strokes triggered by COVID-19 infection (50, 51).Targeted ferroptosis may serve as a potential therapeutic approach for managing excessive inflammation induced by coronavirus infection (52).
Diffuse alveolar injury (DAD) was a significant pathological feature observed in autopsy reports of COVID-19 patients, characterized by extensive cell death and lung injury indicative of disease exposure (53–56). Previous study investigated Orf 7b promoted ferroptosis through the upregulation of c-Myc. Researches had indicated that c-Myc could inhibit ferroptosis, and it was established that P53 suppressed c-Myc via a histone deacetylation mechanism (57–59). Thus, Orf 7b-mediated P53 inhibition might represent a potential mechanism for c-Myc activation, providing a foundation for future investigations (Figure 1C).
PTGS2 is a biomarker associated with ferroptosis. In lung cells infected with SARS-CoV-2, the mRNA level of PTGS2 significantly increased (60)(Figure 1C). In contrast, the transcription levels of ferroptosis regulatory factors—such as solute carrier family 7member 11 (SLC7A11), ferritin light chain (FTL), ferritin heavy chain 1 (FTH1), NAD(P)H quinone dehydrogenase 1 (NQO1), heme oxygenase-1 (HO-1), and Gpx4—in lung tissue significantly decreased following SARS-CoV-2 infection. These findings indicated a substantial association between SARS-CoV-2 infection and ferroptosis. Among the seven accessory proteins, cells overexpressing SARS-CoV-2 ORF3a exhibited the highest levels of lipid ROS induced by RSL3. Both erastin and RSL3 promoted significant cell death and increased lipid ROS levels. Evaluating whether ORF3a expression renders cells sensitive to erastin and RSL3-induced ferroptosis revealed that the inhibition of cell growth and lipid ROS formation induced by erastin and RSL3 increased significantly in a dose-dependent manner. This effect could be completely reversed by iron-induced apoptosis inhibitors (e.g., DFO, Fer-1), but not by apoptosis or necrosis inhibitors. These results indicated that SARS-CoV-2 ORF3a acted as a positive regulator of ferroptosis in cells.
Research had identified that the Nrf2- ARE pathway regulated ferroptosis mediated by ORF3a, with the iron-inhibiting gene driven by Nrf2 being suppressed in SARS-CoV-2 infected cells, making them more susceptible to ferroptosis. The Keap1- Nrf2 axis was known to prevent ferroptosis. ORF3a directly bind to Keap1, enhancing its stability and promoting the interaction between Keap1 and Nrf2. This interaction facilitated the degradation of Nrf2, thereby alleviating the inhibition of ferroptosis (Figure 1C). Notably, knockdown of Keap1 partially rescued lipid ROS levels in cells overexpressing ORF3a, thus continuing to prevent iron-induced apoptosis (61). Two types of ferroptosis inhibitors, troglitazone and rosiglitazone, had been shown to reduce viral production of coronaviruses, including SARS-CoV-2 (62) (Figure 1C).
P. aeruginosa is an obligate aerobic bacterium capable of infecting any tissue or organ in the human body, particularly when the immune system is compromised. Following infection of the respiratory tract, pneumonia may develop, predominantly in patients with impaired pulmonary immune function, such as those with cystic fibrosis or those who have undergone tracheal intubation. In severe cases, the infection could lead to complications such as heart failure and emphysema. Currently, pharmacological therapy is the primary treatment approach in clinical practice, often requiring prolonged duration (63–66). Recent studies had demonstrated that ferroptosis played a crucial regulatory role in P. aeruginosa infections. These findings offered novel insights for the prevention and treatment of P. aeruginosa infections in the future (67).
Dar et al. reported the expression of lipoxygenase (pLoxA) in P. aeruginosa, the host’s arachidonic acid phosphatidylethanolamine was oxidized, triggering ferroptosis in human bronchial epithelial cells (Figure 2A). Idebenone, an effective antioxidant compound and inhibitor of TMEM 16A, had been approved for use in treating Duchenne muscular dystrophy and Leber hereditary optic neuropathy (68–71). Furthermore, it could mitigate lipid peroxidation and cell death in the lungs of cystic fibrosis patients. Additionally, the direct inhibition of P. aeruginosa-induced cell death in CF lungs and other forms of bacterial pneumonia by a new generation of ferroptosis (UAMC-3203) inhibitors might significantly reduce lung inflammation (72) (Figure 2A).
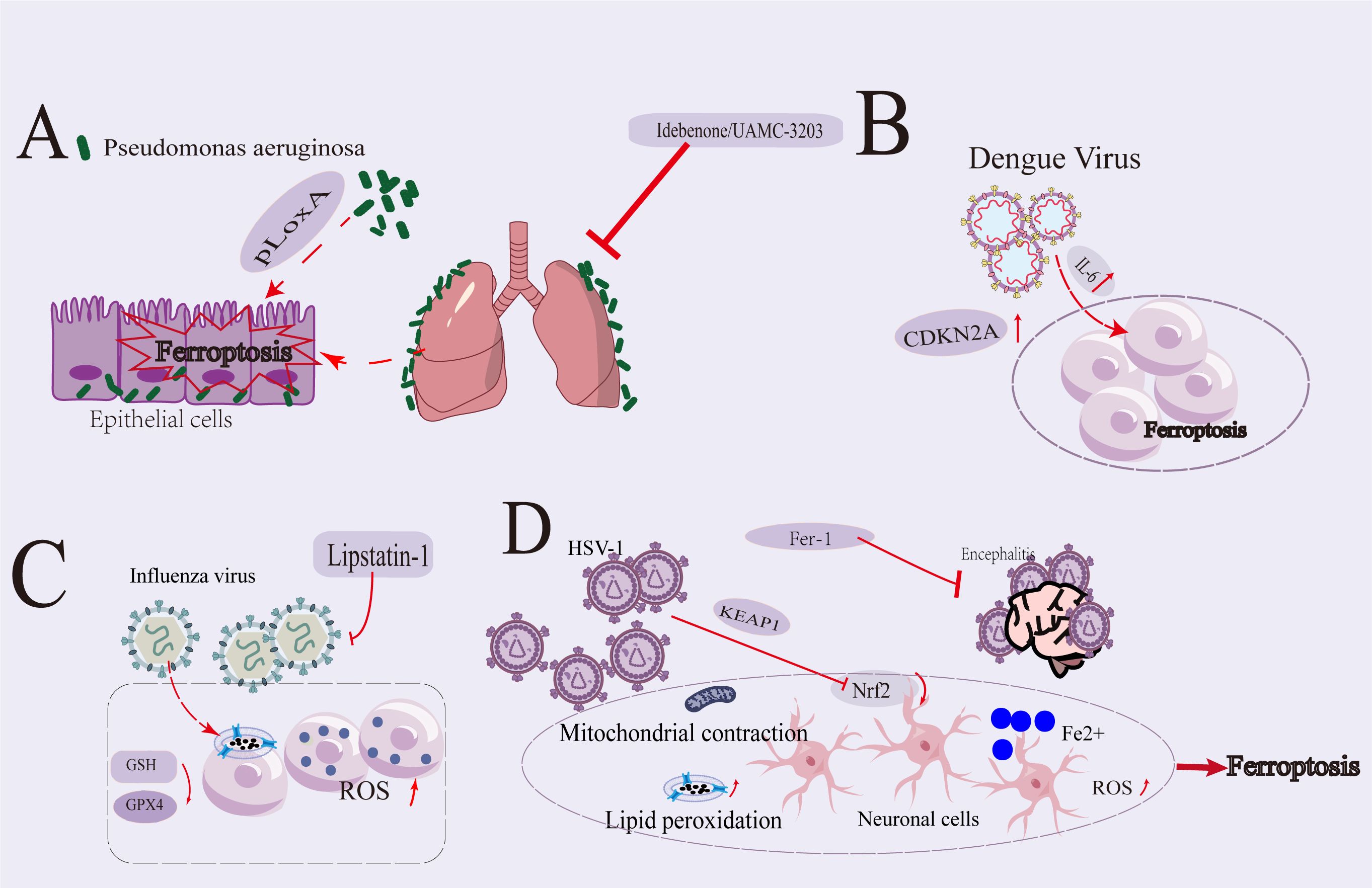
Figure 2. Overview Diagram of the Role of Ferroptosis Mechanism in Pseudomonas aeruginosa, Dengue Virus, Influenza Virus, and Herpes Simplex Virus 1 Infections. (A) P. aeruginosa triggered ferroptosis in human bronchial epithelial cells through the secretion of lipoxygenase (pLoxA). Inhibitors of iron-mediated cell death significantly mitigated cellular apoptosis in the lungs affected by cystic fibrosis and in inflammatory responses associated with bacterial pneumonia. (B) During DENV infection, IL-6, functioning as a pro-inflammatory cytokine, facilitated ferroptosis through the induction of ROS-mediated lipid peroxidation and the perturbation of iron homeostasis. Furthermore, there was an upregulation of CDKN2A expression in HepG2 cell lines infected with DENV. (C) The influenza virus impeded its own replication by increasing the accumulation of lipid peroxidation metabolites and lipid ROS, along with the depletion of intracellular GSH and the downregulation of Gpx4. Lipstatin-1 had the capacity to inhibit the intracellular propagation of the virus. (D) HSV-1 precipitated ferroptosis in both cellular and organismal models. HSV-1 augmented the KEAP1-mediated proteasomal degradation of Nrf2, thereby facilitating ferroptosis. The ferroptosis triggered by HSV-1 infection was typified by iron accumulation, ROS buildup, GSH depletion, lipid peroxidation, and mitochondrial shrinkage. Additionally, ferroptosis induced by HSV-1 plays a significant role in the development of viral encephalitis in mice. Inhibition of ferroptosis by Fer-1 alleviates this process.
It was found that P. aeruginosa significantly exacerbated the damage caused by TBI, further confirming that the pLoxA-rich vesicles secreted by P. aeruginosa triggered ferroptosis in epithelial cells (66). Additionally, selectively targeting pLoxA might represent a promising therapeutic strategy aimed at reducing the high mortality rate associated with TBI combined with P. aeruginosa infection (73, 74).
Dengue fever (DF), a global arthropod-borne viral disease, is caused by infection with the DENV, primarily transmitted through mosquito bites (75, 76). DENV infection induced a characteristic pathology in humans, manifesting as vascular system dysfunction. However, the pathogenesis of DENV remained incompletely elucidated, as many viruses exploited host responses to facilitate their own replication (77). Recent studies had indicated a potential relationship between iron dysregulation and DENV infection (78). (Figure 2B illustrates the potential relationship between dengue virus and ferroptosis).
Li et al. identified and validated nine key genes associated with iron metabolism and DENV infection (HSPA5, CAV1, HRAS, PTGS2, JUN, IL6, ATF3, XBP1, and CDKN2A). IL-6, PTGS2, ATF3, and XBP1 were categorized as markers of ferroptosis and might signify the presence of ferroptosis during DENV infection. JUN is regarded as a suppressor gene of ferroptosis, while HRAS, NRAS, ATF3, and CDKN2A were considered driver genes that might promote ferroptosis. DENV infection had been shown to increase IL-6 production, which was associated with the pathogenesis of severe DENV disease. Reports indicated that IL-6, a pro-inflammatory cytokine, promoted ferroptosis by inducing cellular ROS-dependent lipid peroxidation and disrupting iron homeostasis (Figure 2B). These findings suggested that IL-6 played a crucial role in the pathogenesis of DENV infection by regulating ferroptosis (79–83).
CDKN2A, also known as ARF, stimulated ROS-induced ferroptosis in a p53-independent manner. Previous researches suggested that IL-6, ATF3, XBP1, and CDKN2A were closely associated with iron dysregulation and viral infection, providing a foundational basis for our hypothesis that these genes may be involved in the pathogenesis of DENV infection through the regulation of host cell iron homeostasis. The potential relationship between ferroptosis and DENV infection was elucidated based on a microarray gene expression dataset from a public database using bioinformatics analysis of an exojunction cell culture model (78, 84, 85). This finding provided a basis for further research on the specific regulatory role of ferroptosis in the pathogenesis of dengue fever and laid the foundation for the development of new therapeutic strategies.
Influenza virus is classified into three types: A, B, and C. The acute respiratory infection caused by the influenza virus is known as influenza. Influenza A and B viruses frequently led to seasonal epidemics, resulting in localized outbreaks, while influenza A viruses have the potential to cause a global pandemic. Recent studies had indicated a significant relationship between influenza virus infection and ferroptosis (86–89).
Influenza virus infection led to excessive accumulation of intracellular ROS, and the replication of influenza A virus is influenced by the REDOX(Reduction-Oxidation) state of the host cell, including GSH level (90). Increasing evidence suggested a correlation between the intracellular replication of influenza virus and cell iron level (91). The influenza virus induced ferroptosis by accumulating lipid ROS, depleting GSH, and downregulating Gpx4 expression (Figure 2C). Hemagglutinin (HA) induced ferroptosis via ferritinophagy, leading to lipid peroxidation and impairing the MAVS (mitochondrial antiviral signaling)-mediated type I interferon response, ultimately promoting viral replication (92). Additionally, Lipstatin-1 inhibited the intracellular replication of the virus, indicating that the replication of influenza viruses is reliant on cellular iron levels (14, 89–91).
HSV-1 is an enveloped double-stranded DNA virus that belongs to the genus Herpes Simplex virus within the Herpesviridae family. It is a ubiquitous and highly contagious pathogen that primarily infects humans. HSV-1 primarily causes contact infections through the oral or ocular mucosa, can infiltrate the nervous system, and establishes latent infections via complex immune evasion mechanisms (14, 93, 94).
The study conducted by Xu et al. demonstrated that HSV-1 could induce ferroptosis in both in vitro and in vivo. Specifically, HSV-1 enhanced KEAP1-dependent Nrf2 degradation, which contributed to ferroptosis. (Figure 2D illustrates the potential relationship between HSV-1 infection and ferroptosis.) Furthermore, exogenous GSH inhibited HSV-1 replication by interfering with the later stages of the virus lifecycle, without affecting cellular metabolism (95).
Findings suggested that HSV-1-induced ferroptosis was crucial to the neuro-pathogenesis of the virus. Evidence showed that HSV-1 infection led to ferroptosis characterized by iron overload, ROS, GSH depletion, lipid peroxidation, and mitochondrial contraction in cultured astrocytes, microglial cells, and mouse brains (Figure 2D). Additionally, HSV-1 infection promoted the ubiquitination and degradation of KEAP1-dependent Nrf2, resulting in a significant decrease in the expression levels of downstream anti-iron deposition genes, thereby disrupting cellular redox homeostasis and promoting iron accumulation (1, 96).
The interplay between HSV-1 infection and ferroptosis provided new insights into the physiological impact of ferroptosis on the pathogenesis of HSV-1 infection and encephalitis (Figure 2D). Furthermore, the inhibition of ferroptosis showed promise as a therapeutic strategy against HSV-1-induced encephalitis, highlighting its potential as an effective immunotherapy approach for treating HSV-1 infections and associated encephalitis (97).
Ferroptosis is a type of regulated cell death distinct from apoptosis and necrosis. It is associated with amino acid metabolism, iron metabolism, lipid metabolism, and mitochondrial activity (2). Ferroptosis is implicated in processes such as inflammation, oxidative stress, and lipid peroxidation. Additionally, it is associated with vascular diseases, including Alzheimer’s disease, stroke, and ischemia-reperfusion injury (98).
Numerous studies had demonstrated a close relationship between ferroptosis and respiratory pathogenic microbial infection (78, 99–105). Tuberculosis, caused by infection with M. tuberculosis, remains a significant global public health issue (106). The current lack of safe and effective adult tuberculosis vaccines, the need for prolonged and adequate antibiotic treatment, the increase in global population, and the concentration of populations in underdeveloped areas have all contributed to the increased challenges in the prevention and treatment of M. tuberculosis infection (107, 108). This challenge has spurred significant interest in developing new strategies to target M. tuberculosis infection. EBV is the first identified virus associated with cancer. However, the precise mechanisms by which EBV influences the progression of malignancies in related tumors remain unclear, posing challenges for the prevention and treatment of EBV-associated cancers (109, 110). SARS-CoV-2 caused a global pandemic at the end of 2019 (111). Despite the emergence and administration of multiple vaccines against SARS-CoV-2, infections continue to occur. Cytokine storms are a significant factor contributing to mortality in severe cases of SARS-CoV-2 infection (112, 113). The emergence of multidrug-resistant strains of P. aeruginosa infections, particularly among ICU patients, presents significant challenges to treatment (114). Dengue fever is highly prevalent in tropical and subtropical regions, particularly in Africa, where mosquito-borne fatalities are common (115). The influenza A virus has caused numerous global pandemics, resulting in tens of millions of deaths. Despite the use of vaccines against certain strains of the virus, its multiple serotypes and rapid mutation rate continue to pose significant challenges for prevention, control, and treatment (116, 117). This remains a particularly pressing issue in many countries, especially Japan. HSV-1 spreads rapidly and has a high prevalence among humans. In addition to causing oral herpes, it can lead to severe diseases affecting the central nervous system and circulatory system, which are significant factors in mortality (118, 119).Consequently, the investigation of ferroptosis inhibitors offered new avenues for the treatment of these respiratory infectious diseases, which was summarized in Table 1.
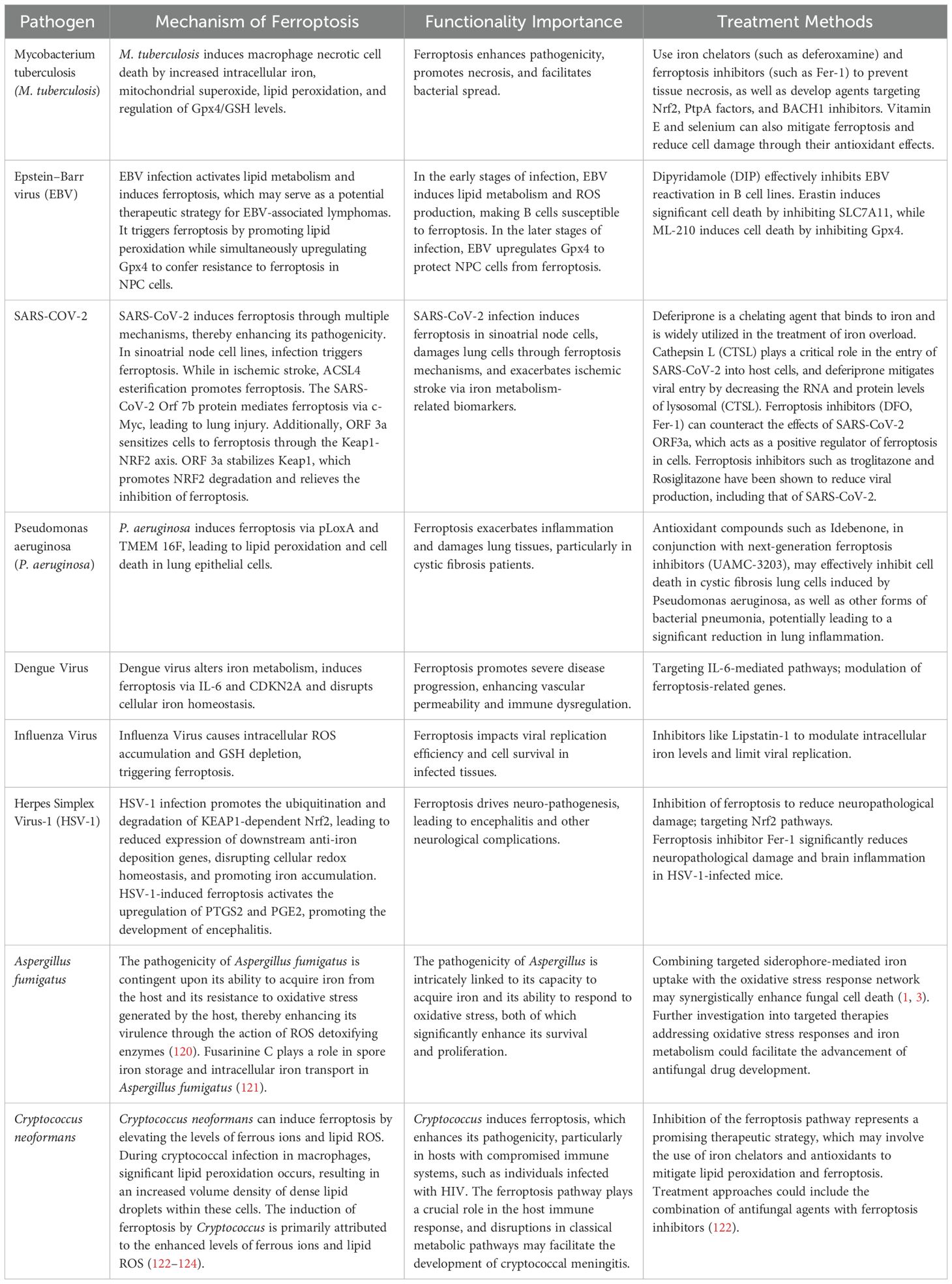
Table 1. Ferroptosis mechanisms and therapeutic strategies in respiratory infectious diseases.
Inhibiting ferroptosis had emerged as a promising new direction for the treatment of respiratory diseases, leading to the development of various ferroptosis inhibitors, such as deferoxamine, troglitazone, and Rosiglitazone (125–127). However, despite the promising results of these ferroptosis and iron-induced apoptosis inhibitors (such as DFO, Fer-1, and UAMC-3203) in cellular and animal models, they still face certain challenges in clinical applications (128). For instance, selectivity and targeting may pose problems, as these inhibitors could affect other physiological processes, leading to adverse reactions. Additionally, ferroptosis and iron-induced apoptosis pathways may vary across different cell types and disease states, limiting the widespread application of these molecules. Moreover, issues related to drug stability, solubility, and pharmacokinetics still need to be optimized to ensure the safety and efficacy of their clinical use.
Given the complexity of the human respiratory microbiome, the interactions between different microorganisms are intricate, and the biological effects and mechanisms of various pathogenic microorganisms differ. The precise relationship between ferroptosis and the immune system remains inadequately elucidated. Consequently, the application of ferroptosis in treating respiratory infectious diseases is still in its nascent stages. Future research should delve deeper into the relationship between ferroptosis and the respiratory microbiome to determine whether ferroptosis affects the survival and pathogenicity of other bacteria or viruses. It is also essential to identify any infection-specific ferroptosis markers, thereby providing a more robust theoretical foundation for utilizing ferroptosis as a novel approach in preventing and treating respiratory infections.
LH: Writing – original draft, Writing – review & editing. XC: Writing – original draft, Writing – review & editing. YL: Writing – original draft, Writing – review & editing. HL: Conceptualization, Writing – review & editing. YZ: Formal Analysis, Writing – review & editing. PG: Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Natural Science Youth Foundation of Shandong Province (No. ZR2022QH039) and National Natural Science Youth Foundation (No. 82402627).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
2. Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discovery. (2021) 7:193. doi: 10.1038/s41420-021-00579-w
3. Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol Cancer research: MCR. (2018) 16:1073–6. doi: 10.1158/1541-7786.Mcr-18-0055
4. Wisnivesky J, De-Torres JP. The global burden of pulmonary diseases: most prevalent problems and opportunities for improvement. Ann Global Health. (2019) 85. doi: 10.5334/aogh.2411
5. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: A systematic analysis for the global burden of disease study 2017. Lancet Respir Med. (2020) 8:585–96. doi: 10.1016/s2213-2600(20)30105-3
6. Chinemerem Nwobodo D, Ugwu MC, Oliseloke Anie C, Al-Ouqaili MTS, Chinedu Ikem J, Victor Chigozie U, et al. Antibiotic resistance: the challenges and some emerging strategies for tackling a global menace. J Clin Lab Anal. (2022) 36:e24655. doi: 10.1002/jcla.24655
7. Tompa DR, Immanuel A, Srikanth S, Kadhirvel S. Trends and strategies to combat viral infections: A review on fda approved antiviral drugs. Int J Biol Macromolecules. (2021) 172:524–41. doi: 10.1016/j.ijbiomac.2021.01.076
8. Wu T, Ji M, Li T, Luo L. The molecular and metabolic landscape of ferroptosis in respiratory diseases: pharmacological aspects. J Pharm Anal. (2025) 15:101050. doi: 10.1016/j.jpha.2024.101050
9. Miggiano R, Rizzi M, Ferraris DM. Mycobacterium tuberculosis pathogenesis, infection prevention and treatment. Pathog (Basel Switzerland). (2020) 9. doi: 10.3390/pathogens9050385
10. Amaral EP, Foreman TW, Namasivayam S, Hilligan KL, Kauffman KD, Barbosa Bomfim CC, et al. Gpx4 regulates cellular necrosis and host resistance in mycobacterium tuberculosis infection. J Exp Med. (2022) 219. doi: 10.1084/jem.20220504
11. Wang J, Cao H, Xie Y, Xu Z, Li Y, Luo H. Mycobacterium tuberculosis infection induces a novel type of cell death: ferroptosis. Biomedicine Pharmacotherapy. (2024) 177:117030. doi: 10.1016/j.biopha.2024.117030
12. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by gpx4 is required to prevent hydroperoxide-induced ferroptosis. Cell. (2018) 172:409–22.e21. doi: 10.1016/j.cell.2017.11.048
13. Seyedrezazadeh E, Ostadrahimi A, Mahboob S, Assadi Y, Ghaemmagami J, Pourmogaddam M. Effect of vitamin E and selenium supplementation on oxidative stress status in pulmonary tuberculosis patients. Respirology (Carlton Vic). (2008) 13:294–8. doi: 10.1111/j.1440-1843.2007.01200.x
14. Amaral EP, Namasivayam S, Queiroz ATL, Fukutani E, Hilligan KL, Aberman K, et al. Bach1 promotes tissue necrosis and mycobacterium tuberculosis susceptibility. Nat Microbiol. (2024) 9:120–35. doi: 10.1038/s41564-023-01523-7
15. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. (2022) 185:2401–21. doi: 10.1016/j.cell.2022.06.003
16. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discovery. (2022) 8:43. doi: 10.1038/s41420-021-00807-3
17. Nishizawa H, Yamanaka M, Igarashi K. Ferroptosis: regulation by competition between nrf2 and bach1 and propagation of the death signal. FEBS J. (2023) 290:1688–704. doi: 10.1111/febs.16382
18. Hernández J, Garibay-Escobar A, Mendoza-Mendoza A, Pinelli-Saavedra A, Valenzuela O. Effect of exogenous vitamin E on proliferation and cytokine production in peripheral blood mononuclear cells from patients with tuberculosis. Br J Nutr. (2008) 99:224–9. doi: 10.1017/s0007114507795302
19. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death differentiation. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
20. Qiang L, Zhang Y, Lei Z, Lu Z, Tan S, Ge P, et al. A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nat Commun. (2023) 14:1430. doi: 10.1038/s41467-023-37148-x
21. Shi Z, Naowarojna N, Pan Z, Zou Y. Multifaceted mechanisms mediating cystine starvation-induced ferroptosis. Nat Commun. (2021) 12:4792. doi: 10.1038/s41467-021-25159-5
22. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
23. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
24. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. (2020) 30:478–90. doi: 10.1016/j.tcb.2020.02.009
25. Cazalla E, Cuadrado A, García-Yagüe ÁJ. Role of the transcription factor nrf2 in maintaining the integrity of the blood-brain barrier. Fluids Barriers CNS. (2024) 21:93. doi: 10.1186/s12987-024-00599-5
26. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar macrophages generate a noncanonical nrf2-driven transcriptional response to mycobacterium tuberculosis in vivo. Sci Immunol. (2019) 4. doi: 10.1126/sciimmunol.aaw6693
27. Hoover K, Higginbotham K. Epstein-barr virus. In: Statpearls. StatPearls Publishing LLC, Treasure Island (FL (2025).
28. Wen KW, Wang L, Menke JR, Damania B. Cancers associated with human gammaherpesviruses. FEBS J. (2022) 289:7631–69. doi: 10.1111/febs.16206
29. Münz C. Latency and lytic replication in epstein-barr virus-associated oncogenesis. Nat Rev Microbiol. (2019) 17:691–700. doi: 10.1038/s41579-019-0249-7
30. Pei Y, Lewis AE, Robertson ES. Current progress in ebv-associated B-cell lymphomas. Adv Exp Med Biol. (2017) 1018:57–74. doi: 10.1007/978-981-10-5765-6_5
31. Andrei G, Trompet E, Snoeck R. Novel therapeutics for epstein⁻Barr virus. Molecules (Basel Switzerland). (2019) 24. doi: 10.3390/molecules24050997
32. Tsao SW, Tsang CM, Lo KW. Epstein-barr virus infection and nasopharyngeal carcinoma. Philos Trans R Soc London Ser B Biol Sci. (2017) 372. doi: 10.1098/rstb.2016.0270
33. Esmaeili A, Awasthi P, Tabaee S. Beyond immortality: epstein-barr virus and the intricate dance of programmed cell death in cancer development. Cancer Treat Res Commun. (2025) 43:100880. doi: 10.1016/j.ctarc.2025.100880
34. Thomé MP, Borde C, Larsen AK, Henriques JAP, Lenz G, Escargueil AE, et al. Dipyridamole as a new drug to prevent epstein-barr virus reactivation. Antiviral Res. (2019) 172:104615. doi: 10.1016/j.antiviral.2019.104615
35. Burton EM, Voyer J, Gewurz BE. Epstein-barr virus latency programs dynamically sensitize B cells to ferroptosis. Proc Natl Acad Sci United States America. (2022) 119:e2118300119. doi: 10.1073/pnas.2118300119
36. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a cancer dependency map. Cell. (2017) 170:564–76.e16. doi: 10.1016/j.cell.2017.06.010
37. Chiba S, Itoh Y, Sekine S, Yokoyama S. Structural basis for the major role of O-phosphoseryl-trna kinase in the uga-specific encoding of selenocysteine. Mol Cell. (2010) 39:410–20. doi: 10.1016/j.molcel.2010.07.018
38. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
39. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
40. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. New Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
41. Pal M, Berhanu G, Desalegn C, Kandi V. Severe acute respiratory syndrome coronavirus-2 (Sars-cov-2): an update. Cureus. (2020) 12:e7423. doi: 10.7759/cureus.7423
42. Alam MS, Czajkowsky DM. Sars-cov-2 infection and oxidative stress: pathophysiological insight into thrombosis and therapeutic opportunities. Cytokine Growth factor Rev. (2022) 63:44–57. doi: 10.1016/j.cytogfr.2021.11.001
43. Yegiazaryan A, Abnousian A, Alexander LJ, Badaoui A, Flaig B, Sheren N, et al. Recent developments in the understanding of immunity, pathogenesis and management of covid-19. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23169297
44. Chavda VP, Kapadia C, Soni S, Prajapati R, Chauhan SC, Yallapu MM, et al. A global picture: therapeutic perspectives for covid-19. Immunotherapy. (2022) 14:351–71. doi: 10.2217/imt-2021-0168
45. Long B, Carius BM, Chavez S, Liang SY, Brady WJ, Koyfman A, et al. Clinical update on covid-19 for the emergency clinician: presentation and evaluation. Am J Emergency Med. (2022) 54:46–57. doi: 10.1016/j.ajem.2022.01.028
46. Zhao X, Liang Q, Li H, Jing Z, Pei D. Single-cell rna sequencing and multiple bioinformatics methods to identify the immunity and ferroptosis-related biomarkers of sars-cov-2 infections to ischemic stroke. Aging. (2023) 15:8237–57. doi: 10.18632/aging.204966
47. Qiu Y, Cao Y, Cao W, Jia Y, Lu N. The application of ferroptosis in diseases. Pharmacol Res. (2020) 159:104919. doi: 10.1016/j.phrs.2020.104919
48. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. Cd8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via acsl4. Cancer Cell. (2022) 40:365–78.e6. doi: 10.1016/j.ccell.2022.02.003
49. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
50. Knoll R, Schultze JL, Schulte-Schrepping J. Monocytes and macrophages in covid-19. Front Immunol. (2021) 12:720109. doi: 10.3389/fimmu.2021.720109
51. Merad M, Martin JC. Pathological inflammation in patients with covid-19: A key role for monocytes and macrophages. Nat Rev Immunol. (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
52. Xia H, Zhang Z, You F. Inhibiting acsl1-related ferroptosis restrains murine coronavirus infection. Viruses. (2021) 13. doi: 10.3390/v13122383
53. Upadhya S, Rehman J, Malik AB, Chen S. Mechanisms of lung injury induced by sars-cov-2 infection. Physiol (Bethesda Md). (2022) 37:88–100. doi: 10.1152/physiol.00033.2021
54. Borczuk AC, Salvatore SP, Seshan SV, Patel SS, Bussel JB, Mostyka M, et al. Covid-19 pulmonary pathology: A multi-institutional autopsy cohort from Italy and new york city. Modern pathology: an Off J United States Can Acad Pathology Inc. (2020) 33:2156–68. doi: 10.1038/s41379-020-00661-1
55. Martines RB, Ritter JM, Matkovic E, Gary J, Bollweg BC, Bullock H, et al. Pathology and pathogenesis of sars-cov-2 associated with fatal coronavirus disease, United States. Emerging Infect Dis. (2020) 26:2005–15. doi: 10.3201/eid2609.202095
56. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (Covid-19) through postmortem core biopsies. Modern pathology: an Off J United States Can Acad Pathology Inc. (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
57. Ratan RR. The chemical biology of ferroptosis in the central nervous system. Cell Chem Biol. (2020) 27:479–98. doi: 10.1016/j.chembiol.2020.03.007
58. Jin Y, Qiu J, Lu X, Li G. C-myc inhibited ferroptosis and promoted immune evasion in ovarian cancer cells through ncoa4 mediated ferritin autophagy. Cells. (2022) 11. doi: 10.3390/cells11244127
59. Ho JS, Ma W, Mao DY, Benchimol S. P53-dependent transcriptional repression of C-myc is required for G1 cell cycle arrest. Mol Cell Biol. (2005) 25:7423–31. doi: 10.1128/mcb.25.17.7423-7431.2005
60. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by gpx4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
61. Liu L, Du J, Yang S, Zheng B, Shen J, Huang J, et al. Sars-cov-2 orf3a sensitizes cells to ferroptosis via keap1-nrf2 axis. Redox Biol. (2023) 63:102752. doi: 10.1016/j.redox.2023.102752
62. Kung YA, Chiang HJ, Li ML, Gong YN, Chiu HP, Hung CT, et al. Acyl-coenzyme a synthetase long-chain family member 4 is involved in viral replication organelle formation and facilitates virus replication via ferroptosis. mBio. (2022) 13:e0271721. doi: 10.1128/mbio.02717-21
63. Flitter BA, Hvorecny KL, Ono E, Eddens T, Yang J, Kwak DH, et al. Pseudomonas aeruginosa sabotages the generation of host proresolving lipid mediators. Proc Natl Acad Sci United States America. (2017) 114:136–41. doi: 10.1073/pnas.1610242114
64. Alverdy J, Holbrook C, Rocha F, Seiden L, Wu RL, Musch M, et al. Gut-derived sepsis occurs when the right pathogen with the right virulence genes meets the right host: evidence for in vivo virulence expression in pseudomonas aeruginosa. Ann Surg. (2000) 232:480–9. doi: 10.1097/00000658-200010000-00003
65. Zaborina O, Kohler JE, Wang Y, Bethel C, Shevchenko O, Wu L, et al. Identification of multi-drug resistant pseudomonas aeruginosa clinical isolates that are highly disruptive to the intestinal epithelial barrier. Ann Clin Microbiol antimicrobials. (2006) 5:14. doi: 10.1186/1476-0711-5-14
66. Dar HH, Tyurina YY, Mikulska-Ruminska K, Shrivastava I, Ting HC, Tyurin VA, et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest. (2018) 128:4639–53. doi: 10.1172/jci99490
67. Kirienko NV, Kirienko DR, Larkins-Ford J, Wählby C, Ruvkun G, Ausubel FM. Pseudomonas aeruginosa disrupts caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe. (2013) 13:406–16. doi: 10.1016/j.chom.2013.03.003
68. Schreiber R, Ousingsawat J, Wanitchakool P, Sirianant L, Benedetto R, Reiss K, et al. Regulation of tmem16a/ano1 and tmem16f/ano6 ion currents and phospholipid scrambling by ca(2+) and plasma membrane lipid. J Physiol. (2018) 596:217–29. doi: 10.1113/jp275175
69. Schreiber R, Buchholz B, Kraus A, Schley G, Scholz J, Ousingsawat J, et al. Lipid peroxidation drives renal cyst growth in vitro through activation of tmem16a. J Am Soc Nephrology: JASN. (2019) 30:228–42. doi: 10.1681/asn.2018010039
70. Seo Y, Park J, Kim M, Lee HK, Kim JH, Jeong JH, et al. Inhibition of ano1/tmem16a chloride channel by idebenone and its cytotoxicity to cancer cell lines. PloS One. (2015) 10:e0133656. doi: 10.1371/journal.pone.0133656
71. Lyseng-Williamson KA. Idebenone: A review in leber’s hereditary optic neuropathy. Drugs. (2016) 76:805–13. doi: 10.1007/s40265-016-0574-3
72. Devisscher L, Van Coillie S, Hofmans S, Van Rompaey D, Goossens K, Meul E, et al. Discovery of novel, drug-like ferroptosis inhibitors with in vivo efficacy. J medicinal Chem. (2018) 61:10126–40. doi: 10.1021/acs.jmedchem.8b01299
73. Liu J, Liu C, Yue J. Radiotherapy and the gut microbiome: facts and fiction. Radiat Oncol (London England). (2021) 16:9. doi: 10.1186/s13014-020-01735-9
74. Pham TA, Lawley TD. Emerging insights on intestinal dysbiosis during bacterial infections. Curr Opin Microbiol. (2014) 17:67–74. doi: 10.1016/j.mib.2013.12.002
75. Han F, Li S, Yang Y, Bai Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered. (2021) 12:5279–88. doi: 10.1080/21655979.2021.1964158
76. Rathore AP, Mantri CK, Aman SA, Syenina A, Ooi J, Jagaraj CJ, et al. Dengue virus-elicited tryptase induces endothelial permeability and shock. J Clin Invest. (2019) 129:4180–93. doi: 10.1172/jci128426
77. Hardy Céline SC, Wegman Adam D, Waldran Mitchell J, Chan Gary C, Waickman Adam T. Conventional and antibody-enhanced denv infection of human macrophages induces differential immunotranscriptomic profiles. J Virol. (2025), e01962–24. doi: 10.1128/jvi.01962-24
78. Li J, Yan X, Li B, Huang L, Wang X, He B, et al. Identification and validation of ferroptosis-related genes in patients infected with dengue virus: implication in the pathogenesis of denv. Virus Genes. (2023) 59:377–90. doi: 10.1007/s11262-023-01985-1
79. Meuren LM, Prestes EB, Papa MP, de Carvalho LRP, Mustafá YM, da Costa LS, et al. Infection of endothelial cells by dengue virus induces ros production by different sources affecting virus replication, cellular activation, death and vascular permeability. Front Immunol. (2022) 13:810376. doi: 10.3389/fimmu.2022.810376
80. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. Atf3 promotes erastin-induced ferroptosis by suppressing system xc(). Cell Death differentiation. (2020) 27:662–75. doi: 10.1038/s41418-019-0380-z
81. Fu D, Wang C, Yu L, Yu R. Induction of ferroptosis by atf3 elevation alleviates cisplatin resistance in gastric cancer by restraining nrf2/keap1/xct signaling. Cell Mol Biol Lett. (2021) 26:26. doi: 10.1186/s11658-021-00271-y
82. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. (2014) 3:e02523. doi: 10.7554/eLife.02523
83. Lew QJ, Chu KL, Lee J, Koh PL, Rajasegaran V, Teo JY, et al. Pcaf interacts with xbp-1s and mediates xbp-1s-dependent transcription. Nucleic Acids Res. (2011) 39:429–39. doi: 10.1093/nar/gkq785
84. Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, et al. Nrf2 is a major target of arf in P53-independent tumor suppression. Mol Cell. (2017) 68:224–32.e4. doi: 10.1016/j.molcel.2017.09.009
85. Hale BG, Albrecht RA, García-Sastre A. Innate immune evasion strategies of influenza viruses. Future Microbiol. (2010) 5:23–41. doi: 10.2217/fmb.09.108
86. Cheng J, Tao J, Li B, Shi Y, Liu H. Swine influenza virus triggers ferroptosis in A549 cells to enhance virus replication. Virol J. (2022) 19:104. doi: 10.1186/s12985-022-01825-y
87. Wang H, Wan K, Shi X. Recent advances in nanozyme research. Advanced materials (Deerfield Beach Fla). (2019) 31:e1805368. doi: 10.1002/adma.201805368
88. Winterbourn CC. Toxicity of iron and hydrogen peroxide: the fenton reaction. Toxicol Lett. (1995) 82-83:969–74. doi: 10.1016/0378-4274(95)03532-x
89. Kan X, Yin Y, Song C, Tan L, Qiu X, Liao Y, et al. Newcastle-disease-virus-induced ferroptosis through nutrient deprivation and ferritinophagy in tumor cells. iScience. (2021) 24:102837. doi: 10.1016/j.isci.2021.102837
90. Miao X, Yin Y, Chen Y, Bi W, Yin Y, Chen S, et al. Bidirectionally regulating viral and cellular ferroptosis with metastable iron sulfide against influenza virus. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2023) 10:e2206869. doi: 10.1002/advs.202206869
91. Nencioni L, Iuvara A, Aquilano K, Ciriolo MR, Cozzolino F, Rotilio G, et al. Influenza a virus replication is dependent on an antioxidant pathway that involves gsh and bcl-2. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2003) 17:758–60. doi: 10.1096/fj.02-0508fje
92. Frensing T, Kupke SY, Bachmann M, Fritzsche S, Gallo-Ramirez LE, Reichl U. Influenza virus intracellular replication dynamics, release kinetics, and particle morphology during propagation in mdck cells. Appl Microbiol Biotechnol. (2016) 100:7181–92. doi: 10.1007/s00253-016-7542-4
93. Yin Y, Favoreel HW. Herpesviruses and the type iii interferon system. Virologica Sin. (2021) 36:577–87. doi: 10.1007/s12250-020-00330-2
94. Bradshaw MJ, Venkatesan A. Herpes simplex virus-1 encephalitis in adults: pathophysiology, diagnosis, and management. Neurotherapeutics: J Am Soc Exp Neurother. (2016) 13:493–508. doi: 10.1007/s13311-016-0433-7
95. Palamara AT, Perno CF, Ciriolo MR, Dini L, Balestra E, D’Agostini C, et al. Evidence for antiviral activity of glutathione: in vitro inhibition of herpes simplex virus type 1 replication. Antiviral Res. (1995) 27:237–53. doi: 10.1016/0166-3542(95)00008-a
96. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of gsh and gpx4. Free Radical Biol Med. (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
97. Xu XQ, Xu T, Ji W, Wang C, Ren Y, Xiong X, et al. Herpes simplex virus 1-induced ferroptosis contributes to viral encephalitis. mBio. (2023) 14:e0237022. doi: 10.1128/mbio.02370-22
98. Li J, Cao F, Yin H-l, Huang Z-j, Lin Z-t, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
99. Zheng Y, Huang Y, Xu Y, Sang L, Liu X, Li Y. Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discovery. (2023) 9:91. doi: 10.1038/s41420-023-01369-2
100. Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, et al. A major role for ferroptosis in mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med. (2019) 216:556–70. doi: 10.1084/jem.20181776
101. He S, Luo C, Shi F, Zhou J, Shang L. The emerging role of ferroptosis in ebv-associated cancer: implications for cancer therapy. Biology. (2024) 13. doi: 10.3390/biology13070543
102. Habib HM, Ibrahim S, Zaim A, Ibrahim WH. The role of iron in the pathogenesis of covid-19 and possible treatment with lactoferrin and other iron chelators. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2021) 136:111228. doi: 10.1016/j.biopha.2021.111228
103. Ousingsawat J, Schreiber R, Gulbins E, Kamler M, Kunzelmann KP. Aeruginosa induced lipid peroxidation causes ferroptotic cell death in airways. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol. (2021) 55:590–604. doi: 10.33594/000000437
104. Liu C, Wu X, Bing X, Qi W, Zhu F, Guo N, et al. H1n1 influenza virus infection through nrf2-keap1-gclc pathway induces ferroptosis in nasal mucosal epithelial cells. Free Radical Biol Med. (2023) 204:226–42. doi: 10.1016/j.freeradbiomed.2023.05.004
105. Qian S, Zhang D, Li R, Sha X, Lu S, Pan L, et al. Downregulation of fcrn promotes ferroptosis in herpes simplex virus-1-induced lung injury. Cell Mol Life sciences: CMLS. (2025) 82:36. doi: 10.1007/s00018-024-05555-y
106. Villar-Hernández R, Ghodousi A, Konstantynovska O, Duarte R, Lange C, Raviglione M. Tuberculosis: current challenges and beyond. Breathe (Sheffield England). (2023) 19:220166. doi: 10.1183/20734735.0166-2022
107. Hokey DA, Ginsberg A. The current state of tuberculosis vaccines. Hum Vaccines immunotherapeutics. (2013) 9:2142–6. doi: 10.4161/hv.25427
108. Zhuang L, Ye Z, Li L, Yang L, Gong W. Next-generation tb vaccines: progress, challenges, and prospects. Vaccines. (2023) 11. doi: 10.3390/vaccines11081304
109. Zhang B, Choi IK. Facts and hopes in the relationship of ebv with cancer immunity and immunotherapy. Clin Cancer research: an Off J Am Assoc Cancer Res. (2022) 28:4363–9. doi: 10.1158/1078-0432.Ccr-21-3408
110. Yu H, Robertson ES. Epstein-barr virus history and pathogenesis. Viruses. (2023) 15. doi: 10.3390/v15030714
111. Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of sars-cov-2 and covid-19. Nat Rev Microbiol. (2021) 19:141–54. doi: 10.1038/s41579-020-00459-7
112. Muralidar S, Ambi SV, Sekaran S, Krishnan UM. The emergence of covid-19 as a global pandemic: understanding the epidemiology, immune response and potential therapeutic targets of sars-cov-2. Biochimie. (2020) 179:85–100. doi: 10.1016/j.biochi.2020.09.018
113. Jiang Y, Rubin L, Peng T, Liu L, Xing X, Lazarovici P, et al. Cytokine storm in covid-19: from viral infection to immune responses, diagnosis and therapy. Int J Biol Sci. (2022) 18:459–72. doi: 10.7150/ijbs.59272
114. Pang Z, Raudonis R, Glick BR, Lin T-J, Cheng Z. Antibiotic resistance in pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv. (2019) 37:177–92. doi: 10.1016/j.bioteChadv.2018.11.013
115. da-Silva-Voorham JM, Tami A, Juliana AE, Rodenhuis-Zybert IA, Wilschut JC, Smit JM. Dengue: A growing risk to travellers to tropical and sub-tropical regions. Nederlands tijdschrift voor geneeskunde. (2009) 153:A778.
116. Shao W, Li X, Goraya MU, Wang S, Chen JL. Evolution of influenza a virus by mutation and re-assortment. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18081650
117. Low ZY, Wong KH, Wen Yip AJ, Choo WS. The convergent evolution of influenza a virus: implications, therapeutic strategies and what we need to know. Curr Res Microbial Sci. (2023) 5:100202. doi: 10.1016/j.crmicr.2023.100202
118. Su D, Han L, Shi C, Li Y, Qian S, Feng Z, et al. An updated review of hsv-1 infection-associated diseases and treatment, vaccine development, and vector therapy application. Virulence. (2024) 15:2425744. doi: 10.1080/21505594.2024.2425744
119. Rathbun MM, Szpara ML. Chapter two - a holistic perspective on herpes simplex virus (Hsv) ecology and evolution. In: Kielian M, Mettenleiter TC, Roossinck MJ, editors. Advances in Virus Research, vol. 110 . Academic Press (2021). p. 27–57.
120. Brandon M, Howard B, Lawrence C, Laubenbacher R. Iron acquisition and oxidative stress response in aspergillus fumigatus. BMC Syst Biol. (2015) 9:19. doi: 10.1186/s12918-015-0163-1
121. Happacher I, Aguiar M, Alilou M, Abt B, Baltussen TJH, Decristoforo C, et al. The siderophore ferricrocin mediates iron acquisition in aspergillus fumigatus. Microbiol Spectr. (2023) 11:e0049623. doi: 10.1128/spectrum.00496-23
122. Xu X, Lin D, Tu S, Gao S, Shao A, Sheng J. Is ferroptosis a future direction in exploring cryptococcal meningitis? Front Immunol. (2021) 12:598601. doi: 10.3389/fimmu.2021.598601
123. Gao J, Wang Q, Tang Y-D, Zhai J, Hu W, Zheng C. When ferroptosis meets pathogenic infections. Trends Microbiol. (2023) 31:468–79. doi: 10.1016/j.tim.2022.11.006
124. Zafar H, Altamirano S, Ballou ER, Nielsen K. A titanic drug resistance threat in cryptococcus neoformans. Curr Opin Microbiol. (2019) 52:158–64. doi: 10.1016/j.mib.2019.11.001
125. Jiao T, Chen Y, Sun H, Yang L. Targeting ferroptosis as a potential prevention and treatment strategy for aging-related diseases. Pharmacol Res. (2024) 208:107370. doi: 10.1016/j.phrs.2024.107370
126. Xie S, Zhang H, Cheng G, Wang B, Li Y, Xing X, et al. Deferoxamine alleviates ferroptosis in seawater immersion combined with traumatic brain injury. Exp Neurol. (2024) 382:114977. doi: 10.1016/j.expneurol.2024.114977
127. Wang Y, Zhang M, Bi R, Su Y, Quan F, Lin Y, et al. Acsl4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. (2022) 51:102262. doi: 10.1016/j.redox.2022.102262
Keywords: ferroptosis, role, respiratory infection, molecular mechanism, therapeutic approach
Citation: Hong L, Chen X, Liu Y, Liang H, Zhao Y and Guo P (2025) The relationship between ferroptosis and respiratory infectious diseases: a novel landscape for therapeutic approach. Front. Immunol. 16:1550968. doi: 10.3389/fimmu.2025.1550968
Received: 24 December 2024; Accepted: 05 March 2025;
Published: 18 March 2025.
Edited by:
Aijaz Ahmad, University of Pittsburgh, United StatesReviewed by:
Rohit Patel, Harvard Medical School, United StatesCopyright © 2025 Hong, Chen, Liu, Liang, Zhao and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengbo Guo, ODc3MjczNjIxQHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.