 Nagisa Nakata Arimitsu
Nagisa Nakata Arimitsu Alicja Witkowska
Alicja Witkowska Ayaka Ohashi
Ayaka Ohashi Chie Miyabe3
Chie Miyabe3 Yoshishige Miyabe
Yoshishige Miyabe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 21 March 2025
Sec. Cytokines and Soluble Mediators in Immunity
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1547256
Multiple sclerosis (MS) is a chronic autoinflammatory disease of unknown origin, involving characterized by immune cell infiltration into the target tissue, central nervous system (CNS), resulting in local and/or systemic inflammation. The symptoms vary from gait disturbance, visual impairment and learning and memory impairment and are being managed with corticosteroid and/or immunosuppressive agents. However, several patients do not respond to these treatments, which can also elevate the risk of severe infections. Therefore, there remains an ongoing need to identify new therapeutic targets. MS exhibits distinctive pathology, clinical course, and treatment responses, suggesting the importance of targeting disease site-specific immune cells to mitigate immune system-induced inflammation, rather than employing broad immunosuppression. Chemokines and chemokine receptors play a crucial role in the pathogenesis of MS by recruiting immune cells to the CNS, leading to inflammation and demyelination. Therapies targeting chemokines have shown promising results in preclinical studies and clinical trials, but more research is needed to fully understand their mechanisms and optimize their efficacy.
Multiple sclerosis (MS) is a chronic autoinflammatory disease of unknown pathogenesis characterized by immune cell infiltration into target tissue, especially the central nervous systems (CNS), leading to local and/or systemic inflammation. This infiltration results in subsequent axonal damage, demyelinating inflammation, and the formation of sclerosing plaques in brain tissue (1, 2). In general, corticosteroid and/or immunosuppressive agents are among the first line treatment for MS patients with CNS involvement. Nevertheless, some patients fail to respond to these agents, and their usage carries an elevated risk of severe infection (3, 4). With the primary objective of therapy being to reduce the frequency of relapses, limit the accumulation of persistent disability, and prevent or delay the onset of progressive disability, there remains an urgent need to identify new treatments for MS, one potential avenue being the targeting of chemokines.
Chemokines constitute a family of small chemotactic cytokines crucial for regulating leukocyte migration during inflammation. They initiate intracellular signaling cascades that drives processes such as cell polarization and adhesion. “Classical” chemokine receptors, characterized by four conserved cysteine residues arranged in CXC, CC, CX3C, and XC patterns, are categorized into four subclasses based on their structural configuration. These receptors, expressed on the surface of immune cells, bind to chemokines and activate intracellular signaling pathways that promote cell polarization, adhesion, and migration. Classical chemokine receptors function as G protein-coupled transmembrane receptors (GPCRs). Upon chemokine binding, they initiate a series of intracellular signals that facilitate cell migration from the circulation into inflamed tissues or extravascular spaces (5–7). This migration process plays a pivotal role in various biological responses, particular in immune reactions, aiding immune cells in homing to sites of inflammation or infection.
Additionally, “atypical” chemokine receptors exist to regulate chemokine levels by removing them, thus suppressing inflammation. These receptors play a vital role in chemokine biology by capturing, scavenging, or transporting chemokines, thereby modulating their signaling through classical chemokine receptors (8).
Chemokines and chemokine receptors, pivotal in regulating leukocytes migration into tissue, play a crucial role in the pathogenesis of MS. Many studies have reported their involvement in recruiting of immune cells to the CNS, leading to CNS inflammation and demyelination. Thus, targeting these molecules has emerged as a promising therapeutic strategy for MS (7, 8). Further research is essential to fully understand the mechanisms behind these therapies and optimize their efficacy, given the complex relationship between immune regulation and MS pathophysiology. Therefore, this review provides a comprehensive overview of MS immunopathology, disease progression, and current treatment approaches drawing insights from analogous mechanisms observed in animal models. Additionally, we discuss the clinical effects of drugs with CNS involvement and their potential as new targets for MS with present up-to-date opinions in this regard.
The precise triggers for immunocompetent cell activation in patients with MS remain largely unknown. However, given that MS lesions are predominantly observed in the CNS, and HLA-DRB1*15:01 alleles are major susceptibility genes for MS (9), T cells and B cells have been implicated in the pathogenesis of MS (10). In patients with MS, CSF analysis often reveals mild pleocytosis with a clear predominance of lymphocytes over neutrophils (11). These immune cells are thought to react with various myelin antigens, including myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP) (10). Recent research has shed light on the role of viral infections, particularly Epstein-Barr virus (EBV), in MS development. Longitudinal studies have shown a significant increase in MS risk following EBV infection, but not that with cytomegalovirus infection (12). Intriguingly, clinical studies have demonstrated symptomatic and objective improvement in MS patients following in vitro-expanded autologous EBV-specific T cell administrations (13). Notably, the efficacy of this therapy was associated with high EBV reactivity of the T cell product and downregulation of CSF IgG levels. Furthermore, molecular mimicry between EBV nuclear antigen (EBNA)-1 and CNS protein, a glial cell adhesion molecule (glialCAM) (Figure 1) was observed, leading to the production of a cross-reactive antibodies by pathogenic B cells (14). This finding suggests a promising clinical strategy for treating MS patients. Pathogenic roles of immune cells on MS are shown (Figure 1). During the early stages of inflammation, self-antigen activation triggers immune responses. Subsequently, helper T (Th) cells migrate into the CNS through the vasculature, initiating pathological processes (15–17) (Figure 1). The CNS has several barriers to prevent leukocyte infiltration. These barriers include the blood brain barrier (BBB) and blood-spinal cord barrier (BSCB), and the blood-cerebrospinal fluid barriers (BCSFB), which limit the entry of T cell entry into the CNS. The interaction between immune cells and endothelial cells, mediated by chemokines and adhesion molecules such as P/E selectins, Mac-1, LFA-1 (Lymphocyte function-associated antigen 1), VLA-4 (α4-β1 integrin), ICAM (Intercellular Adhesion Molecule) -1,2 and VCAM -1 (vascular cell adhesion molecule 1), is crucial for regulating leukocyte infiltration (18).
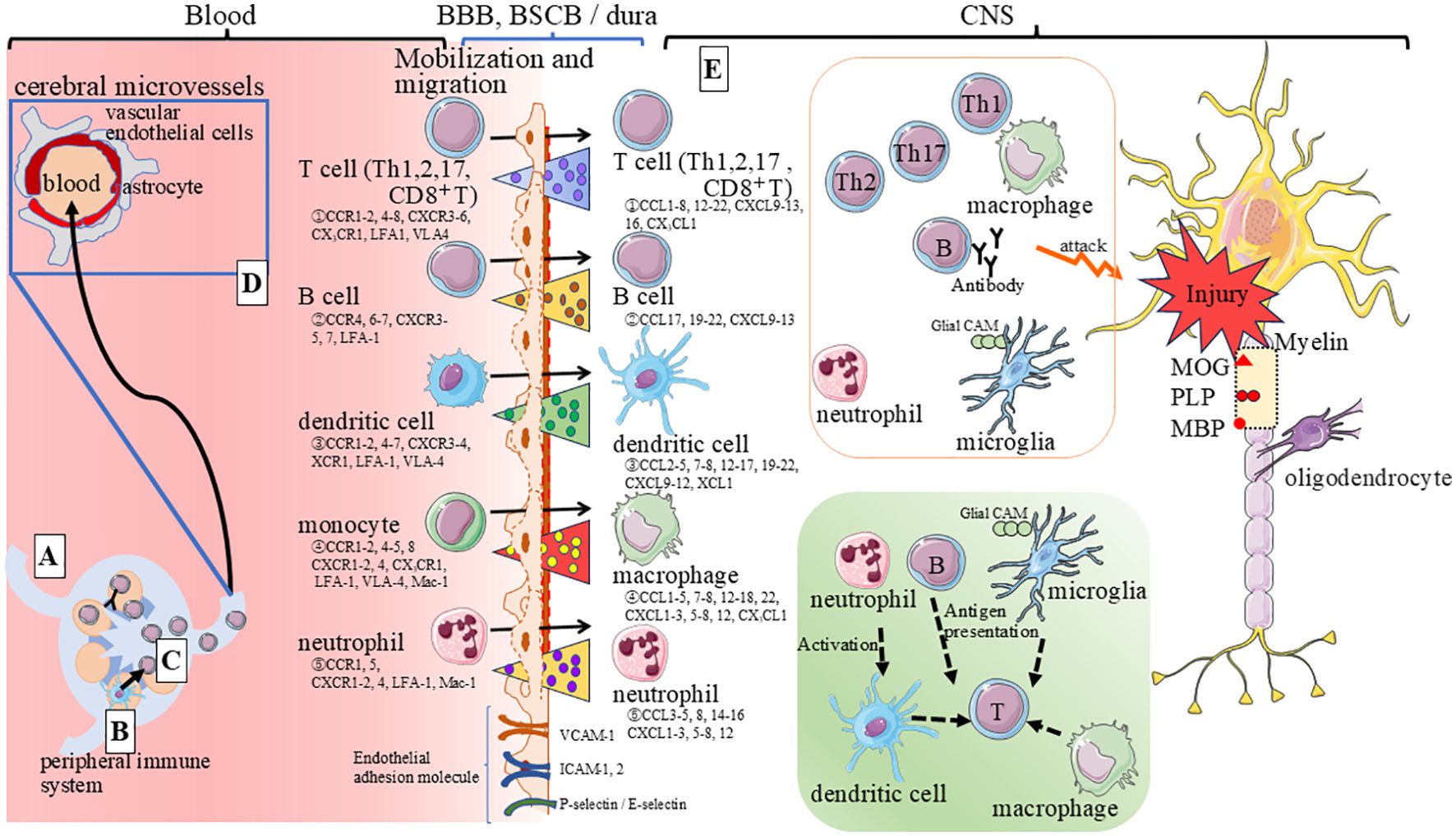
Figure 1. Estimated targets of disease-modifying therapies in MS. This figure illustrates the factors involved in immune cell migration and transition from peripheral tissues to the CNS in MS. The process begins with self-antigen activation, triggering immune responses. Helper T cells then migrate into the CNS, initiating pathological processes. This infiltration is regulated by interactions between immune and endothelial cells, mediated by chemokines and adhesion molecules. Autoantigen-specific T cells activate in the periphery, migrate to the CNS, and reactivate upon encountering APCs, initiating MS-like autoimmune processes. The figure depicts the following key steps: (A) Antigen Presentation in Lymph Nodes: Dendritic cells (DCs) and macrophages, activated by exposure to self-myelin antigens (e.g., MOG, MBP, PLP), migrate to lymph nodes as APCs. (B) Immune Cell Activation: Immune cells are further activated in peripheral lymph nodes as autoantigen-specific cells. (C) Migration to the CNS: Activated myelin-specific T cells (and sometimes B cells) enter the bloodstream and migrate toward the CNS. This process is facilitated by specific adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and leukocyte function-associated antigen-1 (LFA-1). (D) Blood-Brain Barrier Disruption: Inflammatory cytokines produced by activated immune cells disrupt the BBB, BSCB, allowind immune cells to invade the CNS. This disruption is enhanced by expression of specific chemokines. Pro-inflammatory cytokines disrupt the BBB, enhancing leukocyte-endothelial adhesion. Th17 cells, known for their pro-inflammatory role, are increased in peripheral blood and CNS lesions. IL-17, produced by Th17 cells, disrupts BBB tight junctions, facilitating their transmigration across endothelial cells. Cytokines released by Th1 cells, such as IFN-γ and TNF-β, can activate macrophages, causing damage to oligodendrocytes and pathological changes in myelination. (E) Autoimmune Processes in the CNS: Upon encountering APCs presenting self-antigens in the CNS, these cells initiate MS-like autoimmune processes, such as axonal injury and myelin loss. This is accompanied by the release of chemokines that activate and mobilize other inflammatory immune cells.
Autoantigen-specific T cells are initially activated in the periphery and subsequently migrate to the CNS, where they encounter antigen-presenting cells (APCs) presenting the autoantigen. This reactivation in the CNS initiates autoimmune processes similar to MS, accompanied by the release of chemokines that activate and recruit other inflammatory immune cells.
Post-mortem examinations have frequently revealed the accumulation of both T cells and B cells in the perivascular cuffs within the meninges and brain parenchyma of MS patients (16, 19).
In these tissues, the degree of cell infiltration are often associated with demyelination and neurodegeneration (19). Exposure of endothelial cells to proinflammatory cytokines such as IFN-gamma, TNF-alpha and IL-1beta, IL-17 etc. disrupts the BBB by disorganizing cell-cell junctions, reducing the brain solute barrier, and enhancing leukocyte-endothelial adhesion (20). Notably, Th17 cells, known for their proinflammatory role, are increased in both peripheral blood (16, 21, 22) and CNS lesions of MS patients, particularly those with active disease (17). IL-17 produced by Th17 cells disrupts BBB tight junctions, facilitating their transmigration across endothelial cells in vitro and in vivo (15).
Using animal models in research offers several advantages, including their physiological and pathobiological similarities to humans, well-characterized genomes and immune responses, and appropriate experimental size. Despite these advantages, challenges persist regarding reproducibility and translating findings to human disease models. One major difference is that EAE is induced by active immunization with CNS antigens, whereas the etiology of MS appears to be a complex interplay of genetic and environmental factors.
However, animal models remain valuable tools for investigating specific research questions. For instance, in studying MS pathogenesis, mouse models such as EAE induced in C57BL/6 or SJL/J mice using MOG or PLP peptides demonstrate molecular mimicry as a disease model for MS (23). The SJL/J strain specifically models relapsing–remitting MS. Additionally, genetic susceptibility or resistance to EAE in MHC congenic mice is associated with differential cytokine production. Antigens used in rodent EAE models include spinal cord homogenate (SCH), purified myelin, myelin proteins such as MBP, PLP, and MOG, or peptides derived from these proteins. The resulting disease phenotype varies from monophasic to relapsing-remitting or chronic EAE depending on the antigens and genetic background, and can be easily examined from the time of antigen stimulation (24). MHC background largely determines disease susceptibility, while epigenetic factors influence EAE incidence, onset time, severity, neurological signs, and CNS lesion distribution (25, 26).
The expression of several chemokines and their receptors is significantly elevated in the blood and inflamed tissues of MS patients, as well as in animal models of EAE. Considering the functions of chemokines, this suggests a pivotal role for chemokines in recruiting leukocytes and initiating inflammation in MS (27–31). The temporal dynamics of chemokine expression in EAE offer valuable insights into disease progression of the disease and the identification of potential therapeutic targets. Chemokines are produced by various cell types, but the specific cells involved vary depending on the context and location in the body. In blood, chemokines are primarily produced by immune cells, endothelial cells, and fibroblasts. In CSF, they are synthesized by ependymocytes, immune cells, and interstitial cells lining the ventricles. Within the CNS, chemokines are produced by cells forming brain blood vessels, as well as lymphocytes, macrophages, and microglia, particularly in active demyelinating MS brain lesions under pathological conditions. This process enables chemokines to induce and activate leukocyte adhesion molecules locally in certain tissues, establishing a chemotactic concentration gradient and contributing to endothelial permeability and monolayer recruitment (29, 30, 32, 33).
In the EAE animal model, the disease progresses through three distinct stages: the pre-symptomatic acute phase, the onset to disease phase (peak phase), and the chronic phase. Chemokine expression typically follows this pattern:
Pre-symptomatic Acute Phase: The disease manifests approximately 8 days after EAE induction in animals. Before onset, no visible motor impairments are observed, but an increase in T cells in the blood precedes the T cell infiltration into the CNS. Temporary vascular leakage in the cortical gray matter is observed within the first several days of disease induction, initiating microglial activation, followed by continuous accumulation of dendritic cells, leading to the infiltration of myelin-specific T cells (34–36). M-CSF1 (Macrophage Colony-Stimulating Factor) is upregulated in CSF, blood and CNS tissue as early as one day post-immunization (DPI). Before the peak onset of clinical symptoms, the expression of several chemokines, including CCL2, CCL3, CCL5, CXCL1, CXCL2, CXCL10, and CXCL11 increases in the CNS of EAE animals (37–41). For example, spatial variations in chemokine concentrations, driven by differences in expression in various CNS regions and in CSF versus serum (e.g., suppression of brainstem CXCL2 expression by IFN-γ and localized variations in CCL2 expression), have been shown to influence BBB integrity and recruit neutrophils and monocytes to the CNS (29, 33, 42). The IFN-γ pathway plays a significant role in chemokine localization from the spinal cord to the brain. EAE phenotype of IFN-γ KO mice exhibit altered patterns of chemokine expression in the spinal cord and brain, with a greater tendency for inflammatory cell infiltration (43).
Onset Phase (Peak Phase): The onset phase of EAE occurs approximately 8-15 days post-immunization, during which clinical symptoms reach their peak. During this period, CCL2, CCL3, CXCL1, and CXCL10 remain elevated. Additionally, proinflammatory cytokines, such as IL-1β, IFN-γ, CCL5, CCL12, and CXCL9 are upregulated in the spinal fluid, blood, and CNS of EAE animals (44, 45).
Chronic Phase: The chronic phase of EAE occurs approximately 15-30 days post-immunization. Following the peak phase, inflammation and demyelination begin to subside, leading to partial recovery in the animals, depending on the antigen type. During this phase the expression of cytokines such as M-CSF1, CCL2, CCL3, and CCL5 decreases (29, 38, 46). This reduction is associated with the resolution of inflammation and diminished mononuclear cell infiltration into the CNS. However, nerve damage still remains and chronic neurologic symptoms persist. The chronic phase may last the animal’s entire life and results in progressive neurologic dysfunction. Persistent inflammation in the CNS can lead to further recurrence and further neurological deficits (29).
Chemokine ligands and receptors are considered potential risk factors for MS, with their expression linked to clinical disease activity and severity. Inhibiting the chemokine system has been shown to suppress MS inflammation and potentially prevent the onset of MS (Tables 1a, b). Consequently, controlling immune cell recruitment to the CNS has become a major therapeutic target.
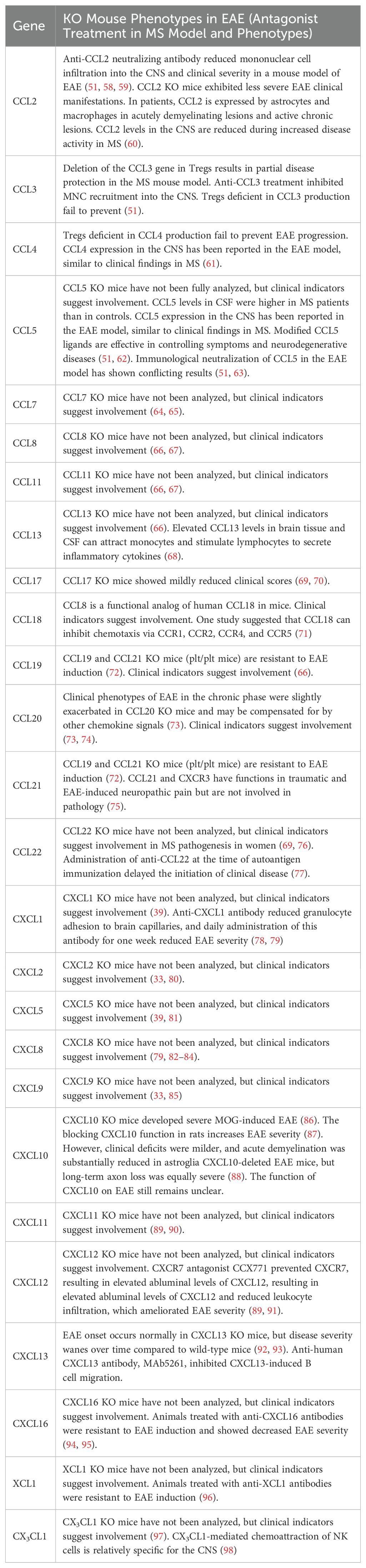
Table 1a. Chemokine targets for MS and phenotypes of chemokine ligand gene-modified mice in MS models.
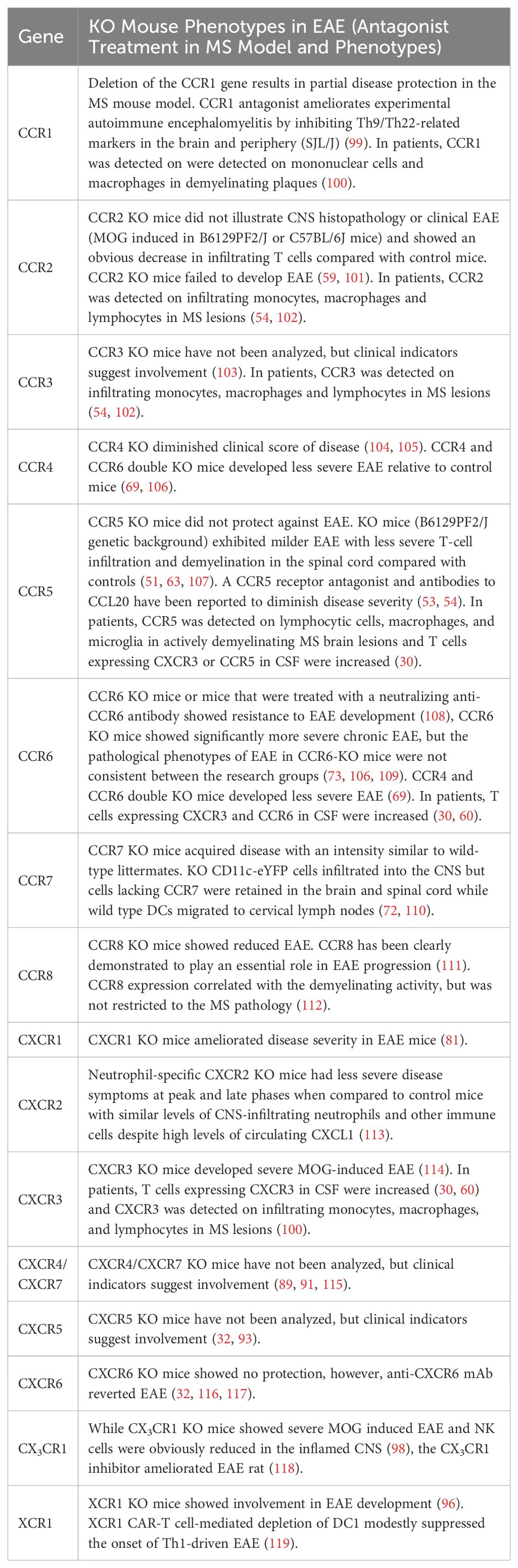
Table 1b. The phenotypes of chemokine receptors gene-modified mice in MS models.
Furthermore, due to functional overlap between chemokine systems, inhibiting a single chemokine may not be sufficient to completely suppress leukocyte migration. Therefore, targeting multiple chemokines and/or their receptors in combination may offer a more effective approach for treating human MS (5, 6).
The study of chemokines can 1) involve an influx of inflammatory cells from blood vessels, but since this occurs before the onset of symptoms, the timing and site-specific control of activity is possible from the outset, 2) provide potential lead compounds for MS therapy, 3) lead to a deeper understanding of MS pathogenesis, and 4) can be used therapeutically. The anti-inflammatory effect may be due to inhibition of chemotaxis, thereby preventing leukocyte influx into affected tissues. In addition, since there are multiple chemokines for each receptor, it is necessary to verify whether it is effective to inhibit them collectively or ligand-specifically. Chemokine activity can be controlled by inhibition against the chemokine itself or against the chemokine receptor, using peptide antagonists, neutralizing antibodies, and small non-peptide antagonists.
Immune neutralization of some chemokines [e.g., CCL3 acting on both CCR5 and CCR1, multiple chemokines sharing a single receptor, etc (47)] generally involves interactions with a variety of chemokine-receptors, so details of tissue localization and time-specific effects need to be identified. From both therapeutic and mechanistic perspectives, it is meaningful to administer selective small-molecule agents to block the effects of chemokine receptors on acute EAE and MS during the induction phase of the autoimmune response. Table 1c describes chemokines and receptors with potential therapeutic effects. We would like to discuss effective chemokine modulation for treatment by describing those that are affected by disease transition and protein/gene expression, those that are suspected to be involved in chemokines in gene-deficient animals, and those that are affected by chemokines in drugs already used in clinical practice.
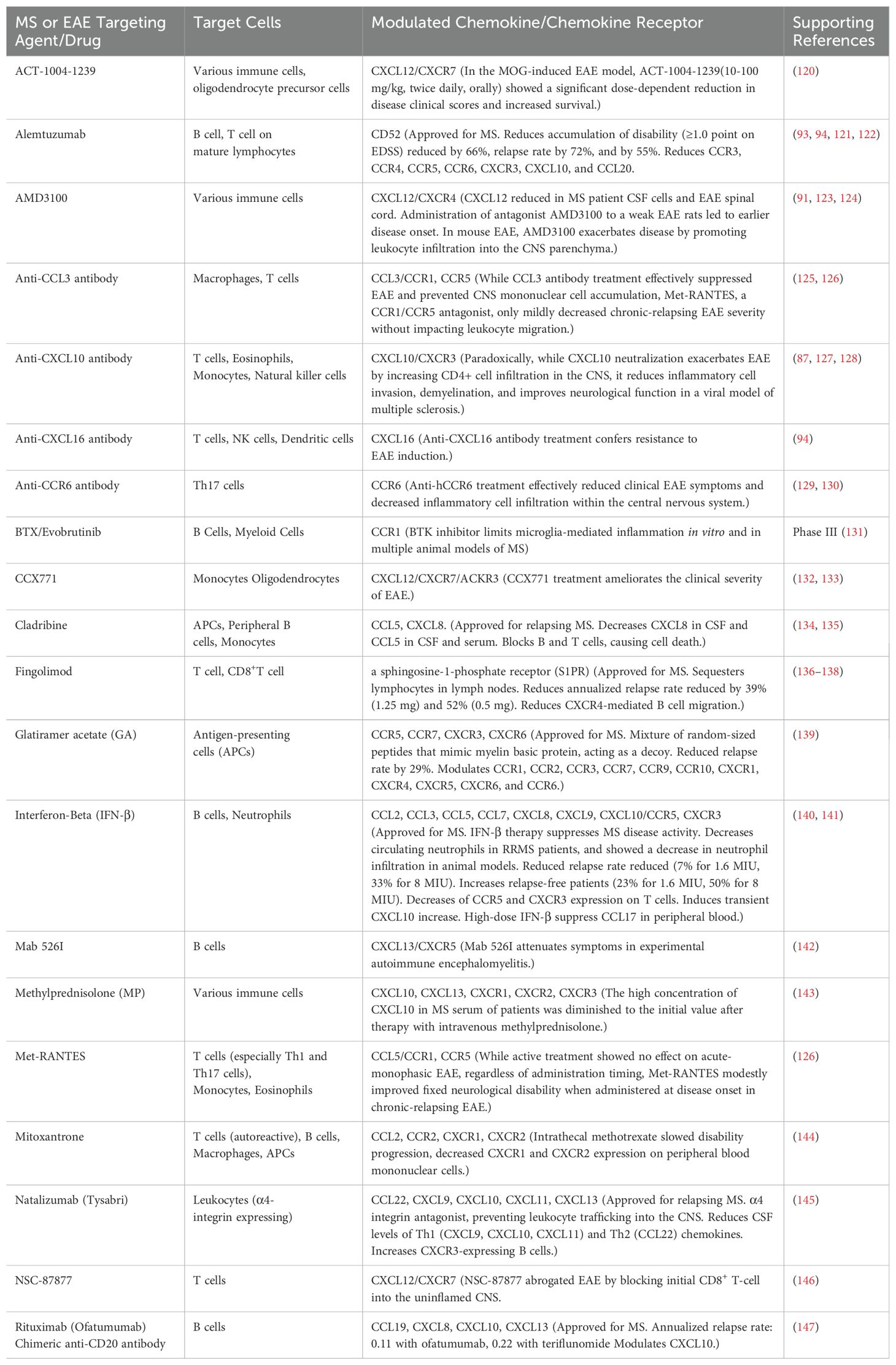
Table 1c. The chemokine as the target for therapy of MS.
• CCR1 Antagonists
CCX354 is an orally administered, highly potent, and selective antagonist of the CCR1 receptor. In clinical studies, it has shown good tolerance and exhibited a linear dose-exposure relationship, with a half-life of approximately 7 hours at a 300-mg dose. Effective blockade of inflammatory cell infiltration into tissues requires maintaining ≥90% CCR1 inhibition on blood leukocytes at all times (48, 49). CCX354 was evaluated in two clinical trials for rheumatoid arthritis, where it demonstrated a favorable safety and tolerability profile, as well as clinical activity. The response rate at week 12 was 39% in the placebo group and 56% in the 200 mg once-daily group (p=0.01). To date, there is a lack of specific data on the use of CCX354 in MS. However, other CCR1 antagonists, such as CCX721, avacopan (CCX168), BMS-817399, and ASK-8007, show promise in addressing MS-related neuroinflammation (50). Notably, elevated CCR1 levels have been observed in cerebrospinal fluid (CSF) during the early and acute stages of demyelination episodes in MS. In an animal model of EAE, CCR1 expression was significantly reduced during clinical remission, correlating with decreased spinal cord inflammation and demyelination (51). This evidence supports the exploration of CCR1 antagonists as a potential therapeutic approach for MS.
• CCR2 Antagonists
CCX872, a CCR2 antagonist, targets the G-protein-coupled receptor CCR2, which is expressed on monocytes and macrophages and plays a crucial role in their migration and infiltration into tissues. Its anti-inflammatory potential was demonstrated using CX3CR1GFP/+CCR2RFP/+ reporter mice, where treatment with CCX872 effectively reduced the accumulation of peripheral macrophages, inhibited the neurotoxic polarization of these cells, and suppressed the expression of NOX2, a superoxide-generating enzyme that produces reactive oxygen species (ROS), within a day post-injury. Importantly, this intervention prevented traumatic brain injury (TBI)-induced deficits in hippocampal-dependent learning and memory observed 28 days after injury (52). Given the evidence that CCR2 is expressed on microglia and macrophages in chronic active MS lesions and on perivascular mononuclear cells in both white matter lesions and unaffected cortex, CCX872 presents a particularly promising candidate for further investigation in MS. However, if therapeutic intervention is considered, multiple targets must be addressed. This is because EAE in CCR2 knockout (KO) mice led to the replacement of neutrophils by monocytes, ultimately resulting in demyelination (53).
Dual Antagonists
• CCR2/CCR5
BMS-813160 is a dual antagonist of CCR2/CCR5 activation presenting with an excellent human liver microsome stability, oral bioavailability and low clearance in mouse, dog and monkey. It may inhibit inflammatory processes, angiogenesis, tumor cell migration, tumor cell proliferation and invasion and was clinically tested in this regard. Simpson et al.'s study, chronic active MS lesions were found to be associated with CCR2, CCR3, and CCR5, particularly in foamy macrophages and activated microglia, with CCR2 and CCR5 also being prevalent in infiltrating lymphocytes (54, 55). Given these findings, BMS-813160 might be a promising candidate for evaluation in MS treatment.
• CCR1/3 and CCR2/5
UCB 35625 and its enantiomer J113863 are known for their strong affinity towards CCR1 and CCR3, making them promising candidates for modulating immune responses. In preclinical studies using EAE model, daily administration of J113863 at a dose of 10 mg/kg for 12 days resulted in a significant reduction of pro-inflammatory CD4+GM-CSF+ and CD4+IL-6+ cells, while simultaneously increasing the levels of anti-inflammatory CD4+IL-27+ and CD4+IL-10+ cells in the spleen. Moreover, J113863 treatment was shown to suppress the mRNA and protein expression of pro-inflammatory cytokines GM-CSF and IL-6 in brain tissues, while enhancing the expression of anti-inflammatory cytokines IL-10 and IL-27. These findings suggest that CCR1 antagonists could play a valuable role in reducing neuroinflammation in MS. Interestingly, while UCB 35625 and J113863 primarily target CCR1 and CCR3, they also demonstrate low-affinity interactions with CCR2 and CCR5. Depending on the specific receptor, the enantiomer, and the signaling pathway involved, these compounds may act as antagonists, partial agonists, or full agonists. This versatility in function underscores the potential utility of these compounds in addressing the complex immune dysregulation observed in MS (56, 57).
This review synthesizes research on the role of chemokines in inflammatory diseases, with a particular focus on MS, and outlines recommended therapies and promising targets based on the disease mechanisms presented in Figure 1. Several chemokines and their receptors are implicated in MS pathophysiology, with by some being directly targeted or indirectly modulated by immunomodulatory agents and drugs (Table 1c). The distinctive pathology, clinical course, and treatment responses of MS underscore the importance of targeting disease site-specific immune cells to mitigate immune system-induced inflammation, rather than relying on broad immunosuppression. The pleiotropic nature of the chemokine system presents challenges for effective targeting with selective antibodies or receptor blockers. Furthermore, general chemokine blockade carries the risk of compromising host defenses, and discrepancies between findings in EAE models and species-specific differences in chemokine responsiveness necessitate careful consideration for clinical translation to human MS patients. These insights are valuable for developing novel therapeutic approaches in inflammatory diseases, especially in inhibiting the mobilization of pathogenic lymphoid cells within lesions and contribute to advancing therapeutic strategies for MS.
NA: Funding acquisition, Writing – original draft, Writing – review & editing. AW: Supervision, Writing – review & editing. AO: Supervision, Writing – review & editing. CM: Supervision, Writing – review & editing. YM: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. The work of NA is supported by the Japanese Society for the Promotion of Science (JSPS) KAKENHI (grant number 21K07379); Terumo life science foundation. The work of YM is supported by the Japanese Society for the Promotion of Science (JSPS) KAKENHI (grant number JP22K08531); Kato Memorial Bioscience Foundation; The Uehara Memorial Foundation; The Naito Foundation; The Mochida Memorial Foundation for Medical and Pharmaceutical Research; The JCR Grant for Promoting Research for FRONTIER and The Mitsubishi Foundation.
YM received grant/research support from Chugai Pharmaceutical Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Compston A, Coles A. Multiple sclerosis. Lancet. (2008) 372:1502–17. doi: 10.1016/S0140-6736(08)61620-7
2. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. (1998) 338:278–85. doi: 10.1056/NEJM199801293380502
3. Coerver EME, Fung WH, de Beukelaar J, Bouvy WH, Canta LR, Gerlach OHH, et al. Discontinuation of first-line disease-modifying therapy in patients with stable multiple sclerosis: the DOT-MS randomized clinical trial. JAMA Neurol. (2025) 82:123–31. doi: 10.1001/jamaneurol.2024.4164
4. Dargahi N, Katsara M, Tselios T, Androutsou ME, de Courten M, Matsoukas J, et al. Multiple sclerosis: immunopathology and treatment update. Brain Sci. (2017) 7. doi: 10.3390/brainsci7070078
5. Dyer DP. Understanding the mechanisms that facilitate specificity, not redundancy, of chemokine-mediated leukocyte recruitment. Immunology. (2020) 160:336–44. doi: 10.1111/imm.13200
6. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. (2018) 285:2944–71. doi: 10.1111/febs.14466
7. Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. (2002) 2:106–15. doi: 10.1038/nri722
8. Xu H, Lin S, Zhou Z, Li D, Zhang X, Yu M, et al. New genetic and epigenetic insights into the chemokine system: the latest discoveries aiding progression toward precision medicine. Cell Mol Immunol. (2023) 20:739–76. doi: 10.1038/s41423-023-01032-x
9. Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. Lancet Neurol. (2014) 13:700–9. doi: 10.1016/S1474-4422(14)70041-9
10. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. (2016) 15:198–209. doi: 10.1016/S1474-4422(15)00334-8
11. Berek K, Bsteh G, Auer M, Di Pauli F, Zinganell A, Berger T, et al. Cerebrospinal fluid findings in 541 patients with clinically isolated syndrome and multiple sclerosis: A monocentric study. Front Immunol. (2021) 12:675307. doi: 10.3389/fimmu.2021.675307
12. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. (2022) 375:296–301. doi: 10.1126/science.abj8222
13. Pender MP, Csurhes PA, Smith C, Douglas NL, Neller MA, Matthews KK, et al. Epstein-Barr virus-specific T cell therapy for progressive multiple sclerosis. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.124714
14. Lanz TV, Brewer RC, Ho PP, Moon JS, Jude KM, Fernandez D, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. (2022) 603:321–7. doi: 10.1038/s41586-022-04432-7
15. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. (2007) 13:1173–5. doi: 10.1038/nm1651
16. Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. (2009) 66:390–402. doi: 10.1002/ana.21748
17. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. (2008) 172:146–55. doi: 10.2353/ajpath.2008.070690
18. Kadry H, Noorani B, Cucullo L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. (2020) 17:69. doi: 10.1186/s12987-020-00230-3
19. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132(Pt. (2009) 5):1175–89. doi: 10.1093/brain/awp070
20. Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. (2003) 9:540–9. doi: 10.1191/1352458503ms965oa
21. Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. (1999) 5:101–4. doi: 10.1177/135245859900500206
22. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. (2009) 65:499–509. doi: 10.1002/ana.21652
23. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. (2006) 177:566–73. doi: 10.4049/jimmunol.177.1.566
24. Miller SD, Karpus WJ. Experimental autoimmune encephalomyelitis in the mouse. Curr Protoc Immunol Chapter. (2007) 15:15.1.1–15.1.18. doi: 10.1002/0471142735.im1501s77
25. deLuca LE, Pikor NB, O’Leary J, Galicia-Rosas G, Ward LA, Defreitas D, et al. Substrain differences reveal novel disease-modifying gene candidates that alter the clinical course of a rodent model of multiple sclerosis. J Immunol. (2010) 184:3174–85. doi: 10.4049/jimmunol.0902881
26. Sobel RA. Genetic and epigenetic influence on EAE phenotypes induced with different encephalitogenic peptides. J Neuroimmunol. (2000) 108:45–52. doi: 10.1016/s0165-5728(99)00270-2
27. Berek K, Bauer A, Rudzki D, Auer M, Barket R, Zinganell A, et al. Immune profiling in multiple sclerosis: a single-center study of 65 cytokines, chemokines, and related molecules in cerebrospinal fluid and serum. Front Immunol. (2023) 14:1200146. doi: 10.3389/fimmu.2023.1200146
28. Lepennetier G, Hracsko Z, Unger M, Van Griensven M, Grummel V, Krumbholz M, et al. Cytokine and immune cell profiling in the cerebrospinal fluid of patients with neuro-inflammatory diseases. J Neuroinflamm. (2019) 16:219. doi: 10.1186/s12974-019-1601-6
29. Borjini N, Fernández M, Giardino L, Calzà L. Cytokine and chemokine alterations in tissue, CSF, and plasma in early presymptomatic phase of experimental allergic encephalomyelitis (EAE), in a rat model of multiple sclerosis. J Neuroinflamm. (2016) 13:291. doi: 10.1186/s12974-016-0757-6
30. Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. (1999) 103:807–15. doi: 10.1172/JCI5150
31. Kothur K, Wienholt L, Tantsis EM, Earl J, Bandodkar S, Prelog K, et al. B cell, Th17, and neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PLoS One. (2016) 11:e0149411. doi: 10.1371/journal.pone.0149411
32. Schropp V, Chunder R, Dietel B, Tacke S, Kuerten S. The presence of cerebellar B cell aggregates is associated with a specific chemokine profile in the cerebrospinal fluid in a mouse model of multiple sclerosis. J Neuroinflamm. (2023) 20:18. doi: 10.1186/s12974-023-02695-z
33. Stoolman JS, Duncker PC, Huber AK, Giles DA, Washnock-Schmid JM, Soulika AM, et al. An IFNγ/CXCL2 regulatory pathway determines lesion localization during EAE. J Neuroinflamm. (2018) 15:208. doi: 10.1186/s12974-018-1237-y
34. Xin L, Madarasz A, Ivan DC, Weber F, Aleandri S, Luciani P, et al. Impairment of spinal CSF flow precedes immune cell infiltration in an active EAE model. J Neuroinflamm. (2024) 21:272. doi: 10.1186/s12974-024-03247-9
35. Wimmer I, Scharler C, Zrzavy T, Kadowaki T, Mödlagl V, Rojc K, et al. Microglia pre-activation and neurodegeneration precipitate neuroinflammation without exacerbating tissue injury in experimental autoimmune encephalomyelitis. Acta Neuropathol Commun. (2019) 7:14. doi: 10.1186/s40478-019-0667-9
36. Bartholomäus I, Kawakami N, Odoardi F, Schläger C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. (2009) 462:94–8. doi: 10.1038/nature08478
37. Shrestha B, Ge S, Pachter JS. Resolution of central nervous system astrocytic and endothelial sources of CCL2 gene expression during evolving neuroinflammation. Fluids Barriers CNS. (2014) 11:6. doi: 10.1186/2045-8118-11-6
38. Chui R, Dorovini-Zis K. Regulation of CCL2 and CCL3 expression in human brain endothelial cells by cytokines and lipopolysaccharide. J Neuroinflamm. (2010) 7:1. doi: 10.1186/1742-2094-7-1
39. Rumble JM, Huber AK, Krishnamoorthy G, Srinivasan A, Giles DA, Zhang X, et al. Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med. (2015) 212:23–35. doi: 10.1084/jem.20141015
40. Godiska R, Chantry D, Dietsch GN, Gray PW. Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol. (1995) 58:167–76. doi: 10.1016/0165-5728(95)00008-p
41. Fife BT, Kennedy KJ, Paniagua MC, Lukacs NW, Kunkel SL, Luster AD, et al. CXCL10 (IFN-gamma-inducible protein-10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. (2001) 166:7617–24. doi: 10.4049/jimmunol.166.12.7617
42. Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. (2005) 25:593–606. doi: 10.1038/sj.jcbfm.9600055
43. Stoolman JS, Duncker PC, Huber AK, Segal BM. Site-specific chemokine expression regulates central nervous system inflammation and determines clinical phenotype in autoimmune encephalomyelitis. J Immunol. (2014) 193:564–70. doi: 10.4049/jimmunol.1400825
44. Hidaka Y, Inaba Y, Matsuda K, Itoh M, Kaneyama T, Nakazawa Y, et al. Cytokine production profiles in chronic relapsing-remitting experimental autoimmune encephalomyelitis: IFN-γ and TNF-α are important participants in the first attack but not in the relapse. J Neurol Sci. (2014) 340:117–22. doi: 10.1016/j.jns.2014.02.039
45. McColl SR, Mahalingam S, Staykova M, Tylaska LA, Fisher KE, Strick CA, et al. Expression of rat I-TAC/CXCL11/SCYA11 during central nervous system inflammation: comparison with other CXCR3 ligands. Lab Invest. (2004) 84:1418–29. doi: 10.1038/labinvest.3700155
46. Hulkower K, Brosnan CF, Aquino DA, Cammer W, Kulshrestha S, Guida MP, et al. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J Immunol. (1993) 150:2525–33. doi: 10.4049/jimmunol.150.6.2525
47. Dyer DP, Medina-Ruiz L, Bartolini R, Schuette F, Hughes CE, Pallas K, et al. Chemokine receptor redundancy and specificity are context dependent. Immunity. (2019) 50:378–389.e5. doi: 10.1016/j.immuni.2019.01.009
48. Tak PP, Balanescu A, Tseluyko V, Bojin S, Drescher E, Dairaghi D, et al. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: CARAT-2, a randomised, placebo controlled clinical trial. Ann Rheum Dis. (2013) 72:337–44. doi: 10.1136/annrheumdis-2011-201605
49. Dairaghi DJ, Zhang P, Wang Y, Seitz LC, Johnson DA, Miao S, et al. Pharmacokinetic and pharmacodynamic evaluation of the novel CCR1 antagonist CCX354 in healthy human subjects: implications for selection of clinical dose. Clin Pharmacol Ther. (2011) 89:726–34. doi: 10.1038/clpt.2011.33
50. Shu J, Ren Y, Tan W, Wei W, Zhang L, Chang J. Identification of potential drug targets for vascular dementia and carotid plaques by analyzing underlying molecular signatures shared by them. Front Aging Neurosci. (2022) 14:967146. doi: 10.3389/fnagi.2022.967146
51. Eltayeb S, Sunnemark D, Berg AL, Nordvall G, Malmberg A, Lassmann H, et al. Effector stage CC chemokine receptor-1 selective antagonism reduces multiple sclerosis-like rat disease. J Neuroimmunol. (2003) 142:75–85. doi: 10.1016/s0165-5728(03)00264-9
52. Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, et al. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. (2015) 35:748–60. doi: 10.1523/JNEUROSCI.2405-14.2015
53. Gaupp S, Pitt D, Kuziel WA, Cannella B, Raine CS. Experimental autoimmune encephalomyelitis (EAE) in CCR2(-/-) mice: susceptibility in multiple strains. Am J Pathol. (2003) 162:139–50. doi: 10.1016/S0002-9440(10)63805-9
54. Simpson J, Rezaie P, Newcombe J, Cuzner ML, Male D, Woodroofe MN. Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol. (2000) 108:192–200. doi: 10.1016/s0165-5728(00)00274-5
55. Cherney RJ, Anjanappa P, Selvakumar K, Batt DG, Brown GD, Rose AV, et al. BMS-813160: A potent CCR2 and CCR5 dual antagonist selected as a clinical candidate. ACS Med Chem Lett. (2021) 12:1753–8. doi: 10.1021/acsmedchemlett.1c00373
56. Ansari MA, Nadeem A, Attia SM, Bakheet SA, Shahid M, Rehman MU, et al. CCR1 antagonist J-113863 corrects the imbalance of pro- and anti-inflammatory cytokines in a SJL/J mouse model of relapsing-remitting multiple sclerosis. Immunobiology. (2022) 227:152245. doi: 10.1016/j.imbio.2022.152245
57. Corbisier J, Huszagh A, Galés C, Parmentier M, Springael JY. Partial agonist and biased signaling properties of the synthetic enantiomers J113863/UCB35625 at chemokine receptors CCR2 and CCR5. J Biol Chem. (2017) 292:575–84. doi: 10.1074/jbc.M116.757559
58. Karpus WJ, Kennedy KJ, Fife BT, Bennett JL, Dal Canto MC, Kunkel SL, et al. Anti-CCL2 treatment inhibits Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Neurovirol. (2006) 12:251–61. doi: 10.1080/13550280600873819
59. Bose S, Cho J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch Pharm Res. (2013) 36:1039–50. doi: 10.1007/s12272-013-0161-z
60. van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain. (2018) 141:1334–49. doi: 10.1093/brain/awy069
61. Ronin E, Pouchy C, Khosravi M, Hilaire M, Grégoire S, Casrouge A, et al. Tissue-restricted control of established central nervous system autoimmunity by TNF receptor 2-expressing Treg cells. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2014043118
62. Zang YC, Samanta AK, Halder JB, Hong J, Tejada-Simon MV, Rivera VM, et al. Aberrant T cell migration toward RANTES and MIP-1 alpha in patients with multiple sclerosis. Overexpression of chemokine receptor CCR5. Brain. (2000) 123:1874–82. doi: 10.1093/brain/123.9.1874
63. Rottman JB, Slavin AJ, Silva R, Weiner HL, Gerard CG, Hancock WW. Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur J Immunol. (2000) 30:2372–7. doi: 10.1002/1521-4141(2000)30:8<2372::AID-IMMU2372>3.0.CO;2-D
64. Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. (2001) 193:713–26. doi: 10.1084/jem.193.6.713
65. Zhang Y, Han JJ, Liang XY, Zhao L, Zhang F, Rasouli J, et al. miR-23b suppresses leukocyte migration and pathogenesis of experimental autoimmune encephalomyelitis by targeting CCL7. Mol Ther. (2018) 26:582–92. doi: 10.1016/j.ymthe.2017.11.013
66. Cui LY, Chu SF, Chen NH. The role of chemokines and chemokine receptors in multiple sclerosis. Int Immunopharmacol. (2020) 83:106314. doi: 10.1016/j.intimp.2020.106314
67. Banisor I, Leist TP, Kalman B. Involvement of beta-chemokines in the development of inflammatory demyelination. J Neuroinflamm. (2005) 2:7. doi: 10.1186/1742-2094-2-7
68. Pigard N, Elovaara I, Kuusisto H, Paalavuo R, Dastidar P, Zimmermann K, et al. Therapeutic activities of intravenous immunoglobulins in multiple sclerosis involve modulation of chemokine expression. J Neuroimmunol. (2009) 209:114–20. doi: 10.1016/j.jneuroim.2009.01.014
69. Scheu S, Ali S, Ruland C, Arolt V, Alferink J. The C-C chemokines CCL17 and CCL22 and their receptor CCR4 in CNS autoimmunity. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18112306
70. Ruland C, Renken H, Kuzmanov I, Fattahi Mehr A, Schwarte K, Cerina M, et al. Chemokine CCL17 is expressed by dendritic cells in the CNS during experimental autoimmune encephalomyelitis and promotes pathogenesis of disease. Brain Behav Immun. (2017) 66:382–93. doi: 10.1016/j.bbi.2017.06.010
71. Krohn SC, Bonvin P, Proudfoot AE. CCL18 exhibits a regulatory role through inhibition of receptor and glycosaminoglycan binding. PLoS One. (2013) 8:e72321. doi: 10.1371/journal.pone.0072321
72. Kuwabara T, Ishikawa F, Yasuda T, Aritomi K, Nakano H, Tanaka Y, et al. CCR7 ligands are required for development of experimental autoimmune encephalomyelitis through generating IL-23-dependent Th17 cells. J Immunol. (2009) 183:2513–21. doi: 10.4049/jimmunol.0800729
73. Sachi N, Kamiyama N, Saechue B, Ozaka S, Dewayani A, Ariki S, et al. CCL20/CCR6 chemokine signaling is not essential for pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochem Biophys Res Commun. (2023) 641:123–31. doi: 10.1016/j.bbrc.2022.11.088
74. El Sharkawi FZ, Ali SA, Hegazy MI, Atya HB. The combined effect of IL-17F and CCL20 gene polymorphism in susceptibility to multiple sclerosis in Egypt. Gene. (2019) 685:164–9. doi: 10.1016/j.gene.2018.11.006
75. Schmitz K, Pickert G, Wijnvoord N, Häussler A, Tegeder I. Dichotomy of CCL21 and CXCR3 in nerve injury-evoked and autoimmunity-evoked hyperalgesia. Brain Behav Immun. (2013) 32:186–200. doi: 10.1016/j.bbi.2013.04.011
76. Jafarzadeh A, Ebrahimi HA, Bagherzadeh S, Zarkesh F, Iranmanesh F, Najafzadeh A, et al. Lower serum levels of Th2-related chemokine CCL22 in women patients with multiple sclerosis: a comparison between patients and healthy women. Inflammation. (2014) 37:604–10. doi: 10.1007/s10753-013-9775-z
77. Dogan RN, Long N, Forde E, Dennis K, Kohm AP, Miller SD, et al. CCL22 regulates experimental autoimmune encephalomyelitis by controlling inflammatory macrophage accumulation and effector function. J Leukoc Biol. (2011) 89:93–104. doi: 10.1189/jlb.0810442
78. Roy M, Richard JF, Dumas A, Vallières L. CXCL1 can be regulated by IL-6 and promotes granulocyte adhesion to brain capillaries during bacterial toxin exposure and encephalomyelitis. J Neuroinflamm. (2012) 9:18. doi: 10.1186/1742-2094-9-18
79. Shi X, Wan Y, Wang N, Xiang J, Wang T, Yang X, et al. Selection of a picomolar antibody that targets CXCR2-mediated neutrophil activation and alleviates EAE symptoms. Nat Commun. (2021) 12:2547. doi: 10.1038/s41467-021-22810-z
80. Tsutsui M, Hirase R, Miyamura S, Nagayasu K, Nakagawa T, Mori Y, et al. TRPM2 exacerbates central nervous system inflammation in experimental autoimmune encephalomyelitis by increasing production of CXCL2 chemokines. J Neurosci. (2018) 38:8484–95. doi: 10.1523/JNEUROSCI.2203-17.2018
81. Zhuang W, Zhou J, Zhong L, Lv J, Zhong X, Liu G, et al. CXCR1 drives the pathogenesis of EAE and ARDS via boosting dendritic cells-dependent inflammation. Cell Death Dis. (2023) 14:608. doi: 10.1038/s41419-023-06126-y
82. Ahn JJ, Abu-Rub M, Miller RH. B cells in neuroinflammation: new perspectives and mechanistic insights. Cells. (2021) 10. doi: 10.3390/cells10071605
83. Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab. (2010) 30:459–73. doi: 10.1038/jcbfm.2009.240
84. Uzawa A, Mori M, Hayakawa S, Masuda S, Nomura F, Kuwabara S. Expression of chemokine receptors on peripheral blood lymphocytes in multiple sclerosis and neuromyelitis optica. BMC Neurol. (2010) 10:113. doi: 10.1186/1471-2377-10-113
85. Watson AES, Goodkey K, Footz T, Voronova A. Regulation of CNS precursor function by neuronal chemokines. Neurosci Lett. (2020) 715:134533. doi: 10.1016/j.neulet.2019.134533
86. Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, et al. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol. (2004) 172:550–9. doi: 10.4049/jimmunol.172.1.550
87. Narumi S, Kaburaki T, Yoneyama H, Iwamura H, Kobayashi Y, Matsushima K. Neutralization of IFN-inducible protein 10/CXCL10 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. (2002) 32:1784–91. doi: 10.1002/1521-4141(200206)32:6<1784::AID-IMMU1784>3.0.CO;2-R
88. Mills Ko E, Ma JH, Guo F, Miers L, Lee E, Bannerman P, et al. Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. J Neuroinflamm. (2014) 11:105. doi: 10.1186/1742-2094-11-105
89. Kohler RE, Comerford I, Townley S, Haylock-Jacobs S, Clark-Lewis I, McColl SR. Antagonism of the chemokine receptors CXCR3 and CXCR4 reduces the pathology of experimental autoimmune encephalomyelitis. Brain Pathol. (2008) 18:504–16. doi: 10.1111/j.1750-3639.2008.00154.x
90. Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, et al. CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J Clin Invest. (2014) 124:2009–22. doi: 10.1172/JCI71951
91. McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol. (2006) 177:8053–64. doi: 10.4049/jimmunol.177.11.8053
92. Huber AK, Irani DN. Targeting CXCL13 during neuroinflammation. Adv Neuroimmune Biol. (2015) 6:1–8. doi: 10.3233/NIB-150101
93. Bagaeva LV, Rao P, Powers JM, Segal BM. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol. (2006) 176:7676–85. doi: 10.4049/jimmunol.176.12.7676
94. Fukumoto N, Shimaoka T, Fujimura H, Sakoda S, Tanaka M, Kita T, et al. Critical roles of CXC chemokine ligand 16/scavenger receptor that binds phosphatidylserine and oxidized lipoprotein in the pathogenesis of both acute and adoptive transfer experimental autoimmune encephalomyelitis. J Immunol. (2004) 173:1620–7. doi: 10.4049/jimmunol.173.3.1620
95. Holmøy T, Løken-Amsrud KI, Bakke SJ, Beiske AG, Bjerve KS, Hovdal H, et al. Inflammation markers in multiple sclerosis: CXCL16 reflects and may also predict disease activity. PLoS One. (2013) 8:e75021. doi: 10.1371/journal.pone.0075021
96. Matsumoto N, Kon S, Nakatsuru T, Miyashita T, Inui K, Saitoh K, et al. A novel α9 integrin ligand, XCL1/lymphotactin, is involved in the development of murine models of autoimmune diseases. J Immunol. (2017) 199:82–90. doi: 10.4049/jimmunol.1601329
97. Zhu W, Acosta C, MacNeil B, Cortes C, Intrater H, Gong Y, et al. Elevated expression of fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) in the dorsal root ganglia and spinal cord in experimental autoimmune encephalomyelitis: implications in multiple sclerosis-induced neuropathic pain. BioMed Res Int. (2013) 2013:480702. doi: 10.1155/2013/480702
98. Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. (2006) 20:896–905. doi: 10.1096/fj.05-5465com
99. Al-Mazroua HA, Nadeem A, Ansari MA, Attia SM, Bakheet SA, Albekairi TH, et al. CCR1 antagonist ameliorates experimental autoimmune encephalomyelitis by inhibition of Th9/Th22-related markers in the brain and periphery. Mol Immunol. (2022) 144:127–37. doi: 10.1016/j.molimm.2022.02.017
100. Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A. (1999) 96:6873–8. doi: 10.1073/pnas.96.12.6873
101. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. (2000) 192:899–905. doi: 10.1084/jem.192.6.899
102. Trebst C, Ransohoff RM. Investigating chemokines and chemokine receptors in patients with multiple sclerosis: opportunities and challenges. Arch Neurol. (2001) 58:1975–80. doi: 10.1001/archneur.58.12.1975
103. Shou J, Peng J, Zhao Z, Huang X, Li H, Li L, et al. CCL26 and CCR3 are associated with the acute inflammatory response in the CNS in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2019) 333:576967. doi: 10.1016/j.jneuroim.2019.576967
104. Poppensieker K, Otte DM, Schürmann B, Limmer A, Dresing P, Drews E, et al. CC chemokine receptor 4 is required for experimental autoimmune encephalomyelitis by regulating GM-CSF and IL-23 production in dendritic cells. Proc Natl Acad Sci U S A. (2012) 109:3897–902. doi: 10.1073/pnas.1114153109
105. Forde EA, Dogan RN, Karpus WJ. CCR4 contributes to the pathogenesis of experimental autoimmune encephalomyelitis by regulating inflammatory macrophage function. J Neuroimmunol. (2011) 236:17–26. doi: 10.1016/j.jneuroim.2011.04.008
106. Moriguchi K, Miyamoto K, Tanaka N, Yoshie O, Kusunoki S. The importance of CCR4 and CCR6 in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2013) 257:53–8. doi: 10.1016/j.jneuroim.2013.02.002
107. Gu SM, Park MH, Yun HM, Han SB, Oh KW, Son DJ, et al. CCR5 knockout suppresses experimental autoimmune encephalomyelitis in C57BL/6 mice. Oncotarget. (2016) 7:15382–93. doi: 10.18632/oncotarget.8097
108. Liston A, Kohler RE, Townley S, Haylock-Jacobs S, Comerford I, Caon AC, et al. Inhibition of CCR6 function reduces the severity of experimental autoimmune encephalomyelitis via effects on the priming phase of the immune response. J Immunol. (2009) 182:3121–30. doi: 10.4049/jimmunol.0713169
109. Elhofy A, Depaolo RW, Lira SA, Lukacs NW, Karpus WJ. Mice deficient for CCR6 fail to control chronic experimental autoimmune encephalomyelitis. J Neuroimmunol. (2009) 213:91–9. doi: 10.1016/j.jneuroim.2009.05.011
110. Clarkson BD, Walker A, Harris MG, Rayasam A, Hsu M, Sandor M, et al. CCR7 deficient inflammatory Dendritic Cells are retained in the Central Nervous System. Sci Rep. (2017) 7:42856. doi: 10.1038/srep42856
111. Chensue SW, Lukacs NW, Yang TY, Shang X, Frait KA, Kunkel SL, et al. Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J Exp Med. (2001) 193:573–84. doi: 10.1084/jem.193.5.573
112. Trebst C, Staugaitis SM, Kivisäkk P, Mahad D, Cathcart MK, Tucky B, et al. CC chemokine receptor 8 in the central nervous system is associated with phagocytic macrophages. Am J Pathol. (2003) 162:427–38. doi: 10.1016/S0002-9440(10)63837-0
113. Khaw YM, Cunningham C, Tierney A, Sivaguru M, Inoue M. Neutrophil-selective deletion of Cxcr2 protects against CNS neurodegeneration in a mouse model of multiple sclerosis. J Neuroinflamm. (2020) 17:49. doi: 10.1186/s12974-020-1730-y
114. Lalor SJ, Segal BM. Th1-mediated experimental autoimmune encephalomyelitis is CXCR3 independent. Eur J Immunol. (2013) 43:2866–74. doi: 10.1002/eji.201343499
115. Byun DJ, Lee J, Ko K, Hyun YM. NLRP3 exacerbates EAE severity through ROS-dependent NET formation in the mouse brain. Cell Commun Signal. (2024) 22:96. doi: 10.1186/s12964-023-01447-z
116. Kim JV, Jiang N, Tadokoro CE, Liu L, Ransohoff RM, Lafaille JJ, et al. Two-photon laser scanning microscopy imaging of intact spinal cord and cerebral cortex reveals requirement for CXCR6 and neuroinflammation in immune cell infiltration of cortical injury sites. J Immunol Methods. (2010) 352:89–100. doi: 10.1016/j.jim.2009.09.007
117. Hou L, Rao DA, Yuki K, Cooley J, Henderson LA, Jonsson AH, et al. SerpinB1 controls encephalitogenic T helper cells in neuroinflammation. Proc Natl Acad Sci U.S.A. (2019) 116:20635–43. doi: 10.1073/pnas.1905762116
118. Ridderstad Wollberg A, Ericsson-Dahlstrand A, Juréus A, Ekerot P, Simon S, Nilsson M, et al. Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic-relapsing rat model for multiple sclerosis. Proc Natl Acad Sci U.S.A. (2014) 111:5409–14. doi: 10.1073/pnas.1316510111
119. Moorman CD, Yu S, Briseno CG, Phee H, Sahoo A, Ramrakhiani A, et al. CAR-T cells and CAR-Tregs targeting conventional type-1 dendritic cell suppress experimental autoimmune encephalomyelitis. Front Immunol. (2023) 14:1235222. doi: 10.3389/fimmu.2023.1235222
120. Pouzol L, Baumlin N, Sassi A, Tunis M, Marrie J, Vezzali E, et al. ACT-1004-1239, a first-in-class CXCR7 antagonist with both immunomodulatory and promyelinating effects for the treatment of inflammatory demyelinating diseases. FASEB J. (2021) 35:e21431. doi: 10.1096/fj.202002465R
121. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. (2012) 380:1819–28. doi: 10.1016/S0140-6736(12)61769-3
122. Coles AJ, Compston DA, Selmaj KW, Lake SL, Moran S, Margolin DH, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. (2008) 359:1786–801. doi: 10.1056/NEJMoa0802670
123. Mueller AM, Yoon BH, Sadiq SA. Inhibition of hyaluronan synthesis protects against central nervous system (CNS) autoimmunity and increases CXCL12 expression in the inflamed CNS. J Biol Chem. (2014) 289:22888–99. doi: 10.1074/jbc.M114.559583
124. Miljković D, Stanojević Z, Momcilović M, Odoardi F, Flügel A, Mostarica-Stojković M. CXCL12 expression within the CNS contributes to the resistance against experimental autoimmune encephalomyelitis in Albino Oxford rats. Immunobiology. (2011) 216:979–87. doi: 10.1016/j.imbio.2011.03.013
125. Liang M, Mallari C, Rosser M, Ng HP, May K, Monahan S, et al. Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J Biol Chem. (2000) 275:19000–8. doi: 10.1074/jbc.M001222200
126. Matsui M, Weaver J, Proudfoot AE, Wujek JR, Wei T, Richer E, et al. Treatment of experimental autoimmune encephalomyelitis with the chemokine receptor antagonist Met-RANTES. J Neuroimmunol. (2002) 128:16–22. doi: 10.1016/s0165-5728(02)00121-2
127. Welsh N, Disano K, Linzey M, Pike SC, Smith AD, Pachner AR, et al. CXCL10/IgG1 axis in multiple sclerosis as a potential predictive biomarker of disease activity. Neurol Neuroimmunol Neuroinflamm. (2024) 11:e200200. doi: 10.1212/NXI.0000000000200200
128. Liu MT, Keirstead HS, Lane TE. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J Immunol. (2001) 167:4091–7. doi: 10.4049/jimmunol.167.7.4091
129. Hou L, Yuki K. CCR6 and CXCR6 identify the Th17 cells with cytotoxicity in experimental autoimmune encephalomyelitis. Front Immunol. (2022) 13:819224. doi: 10.3389/fimmu.2022.819224
130. Robert R, Ang C, Sun G, Juglair L, Lim EX, Mason LJ, et al. Essential role for CCR6 in certain inflammatory diseases demonstrated using specific antagonist and knockin mice. JCI Insight. (2017) 2. doi: 10.1172/jci.insight.94821
131. Piasecka-Stryczynska K, Rejdak K, Dyroff M, Hyvert Y, Holmberg K, Mandel M, et al. Concentration of evobrutinib, a BTK inhibitor, in cerebrospinal fluid during treatment of patients with relapsing multiple sclerosis in a phase 2 study. Mult Scler Relat Disord. (2021) 51:103001. doi: 10.1016/j.msard.2021.103001
132. Williams JL, Patel JR, Daniels BP, Klein RS. Targeting CXCR7/ACKR3 as a therapeutic strategy to promote remyelination in the adult central nervous system. J Exp Med. (2014) 211:791–9. doi: 10.1084/jem.20131224
133. Cruz-Orengo L, Chen YJ, Kim JH, Dorsey D, Song SK, Klein RS. CXCR7 antagonism prevents axonal injury during experimental autoimmune encephalomyelitis as revealed by in vivo axial diffusivity. J Neuroinflamm. (2011) 8:170. doi: 10.1186/1742-2094-8-170
134. Hermann R, Karlsson MO, Novakovic AM, Terranova N, Fluck M, Munafo A. The clinical pharmacology of cladribine tablets for the treatment of relapsing multiple sclerosis. Clin Pharmacokinet. (2019) 58:283–97. doi: 10.1007/s40262-018-0695-9
135. Holm Hansen R, von Essen MR, Reith Mahler M, Cobanovic S, Sellebjerg F. Sustained effects on immune cell subsets and autoreactivity in multiple sclerosis patients treated with oral cladribine. Front Immunol. (2024) 15:1327672. doi: 10.3389/fimmu.2024.1327672
136. Blumenfeld-Kan S, Staun-Ram E, Miller A. Fingolimod reduces CXCR4-mediated B cell migration and induces regulatory B cells-mediated anti-inflammatory immune repertoire. Mult Scler Relat Disord. (2019) 34:29–37. doi: 10.1016/j.msard.2019.06.016
137. Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. (2010) 362:387–401. doi: 10.1056/NEJMoa0909494
138. Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. (2010) 362:402–15. doi: 10.1056/NEJMoa0907839
139. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. (1995) 45:1268–76. doi: 10.1212/wnl.45.7.1268
140. The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. (1993) 43:655–61. doi: 10.1212/wnl.43.4.655
141. Zang YC, Halder JB, Samanta AK, Hong J, Rivera VM, Zhang JZ. Regulation of chemokine receptor CCR5 and production of RANTES and MIP-1alpha by interferon-beta. J Neuroimmunol. (2001) 112:174–80. doi: 10.1016/s0165-5728(00)00397-0
142. Klimatcheva E, Pandina T, Reilly C, Torno S, Bussler H, Scrivens M, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. (2015) 16:6. doi: 10.1186/s12865-015-0068-1
143. Michałowska-Wender G, Losy J, Szczuciński A, Biernacka-Łukanty J, Wender M. Effect of methylprednisolone treatment on expression of sPECAM-1 and CXCL10 chemokine in serum of MS patients. Pharmacol Rep. (2006) 58:920–3.
144. Dhaiban S, Al-Ani M, Elemam NM, Maghazachi AA. Targeting chemokines and chemokine receptors in multiple sclerosis and experimental autoimmune encephalomyelitis. J Inflammation Res. (2020) 13:619–33. doi: 10.2147/JIR.S270872
145. Mellergård J, Edström M, Vrethem M, Ernerudh J, Dahle C. Natalizumab treatment in multiple sclerosis: marked decline of chemokines and cytokines in cerebrospinal fluid. Mult Scler. (2010) 16:208–17. doi: 10.1177/1352458509355068
146. Luo Q, Sun Y, Gong FY, Liu W, Zheng W, Shen Y, et al. Blocking initial infiltration of pioneer CD8(+) T-cells into the CNS via inhibition of SHP-2 ameliorates experimental autoimmune encephalomyelitis in mice. Br J Pharmacol. (2014) 171:1706–21. doi: 10.1111/bph.12565
Keywords: multiple sclerosis (MS), experimental autoimmune encephalomyelitis (EAE), chemokines, CNS (central nervous system), immune cells, therapeutic targets
Citation: Arimitsu NN, Witkowska A, Ohashi A, Miyabe C and Miyabe Y (2025) Chemokines as therapeutic targets for multiple sclerosis: a spatial and chronological perspective. Front. Immunol. 16:1547256. doi: 10.3389/fimmu.2025.1547256
Received: 18 December 2024; Accepted: 04 March 2025;
Published: 21 March 2025.
Edited by:
Olga Barreiro, Harvard Medical School, United StatesReviewed by:
Ju Liu, First Affiliated Hospital of Zhengzhou University, ChinaCopyright © 2025 Arimitsu, Witkowska, Ohashi, Miyabe and Miyabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshishige Miyabe, eW9zaGlzaGlnZS5taXlhYmVAbWFyaWFubmEtdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.