 Luis Brieva
Luis Brieva- 1Neurology Department, Hospital Universitari Arnau de Vilanova, Lleida, Spain
- 2Medicine Department, Universitat de Lleida (UdL), Lleida, Spain
- 3Neuroimmunology Group, Institut de Recerca Biomedica de Lleida (IRBLLEIDA), Lleida, Spain
- 4Neurology Department, Hospital Universitario Son Espases, Palma de Mallorca, Spain
- 5Neurology Department, Hospital Universitario Doctor Peset, Valencia, Spain
- 6Department of Neurology, Hospital Clinico San Carlos, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos (IdISSC), Madrid, Spain
- 7Departament of Medicine, Medicine Faculty, Universidad Complutense de Madrid (UCM), Madrid, Spain
Approximately 50% diagnosed with relapsing-remitting multiple sclerosis (RRMS) transition to secondary progressive multiple sclerosis (SPMS) within 20 years following disease onset. However, early diagnosis of SPMS and effective treatment remain important clinical challenges. The lack of established diagnostic criteria often leads to delays in identifying SPMS. Also, there are limited disease-modifying therapies (DMTs) available for progressive forms of MS, and these therapies require evidence of disease activity to be initiated. This review examines the challenges in diagnosing SPMS at an early stage and summarizes the current and potential use of biomarkers of disease progression in clinical practice. We also discuss the difficulties in initiating the DMTs indicated for active SPMS (aSPMS), particularly in patients already undergoing treatment with DMTs that suppress disease activity, which may mask the presence of inflammatory activity required for the therapy switch. The article also addresses the DMTs available for both active and non-active SPMS, along with the clinical trials that supported the approval of DMTs indicated for aSPMS or relapsing MS in Europe, which includes aSPMS. We also offer insights on when discontinuing these treatments may be appropriate.
1 Introduction
Multiple Sclerosis (MS) is a chronic autoimmune disorder of the central nervous system (CNS), characterized by neuroinflammation, demyelination, and neurodegeneration. Approximately, 2.9 million individuals are affected by MS worldwide, with the average age at diagnosis being 32 years (1). MS places a huge burden on both individuals and society as a whole (2). The impact of the disease on daily living, work-related activities, and cost becomes more pronounced with the progression of disability (3, 4).
The clinical manifestations and course of MS are unpredictable and vary from person to person. Standardized definitions of MS clinical courses are important for clinicians to diagnose, monitor, and treat their patients, as well as for researchers to design studies, and interpret and compare findings. MS was initially classified into four phenotypes: relapsing-remitting MS (RRMS), primary progressive MS (PPMS), secondary progressive MS (SPMS), and progressive-relapsing MS (PRMS) (5). In the revision of 2013 by the International Advisory Committee on Clinical Trials of MS (6), patients were classified according to the presence of activity (active or non-active) and disease progression (progressing or non-progressing). These phenotypes described in 2013 are still not widely used by the entire scientific community. Moreover, the classification of MS varies between different organizations (the Food and Drug Administration [FDA], the European Medicines Agency [EMA], and the MS scientific community), which can lead to discrepancies in diagnosis and treatment (7). These phenotypes, in any case, seem to represent different stages along the MS disease continuum rather than distinct disease categories (8, 9).
In natural history studies examining untreated cohorts, about 50% of patients progressed to SPMS within 10 to 20 years following disease onset (5, 10, 11). The introduction of disease-modifying therapies (DMTs) three decades ago significantly delayed this conversion rate (12, 13). Findings from DMT-treated cohorts showed that 18% of patients progressed to SPMS after a median duration of 16.8 years from MS onset (13), which is an important reduction compared to untreated cohorts. Factors increasing the risk of conversion from RRMS to SPMS include older age at onset, smoking, higher Expanded Disability Status Scale (EDSS) at onset, a high number of early relapses, motor dysfunction, cerebellar dysfunction, and presence of lesions in the spinal cord (10, 11). From an EDSS score of 3, patients seem to progress at a similar rate, independent of their prior disease course. A study with 2054 patients (1609 relapsing/445 progressive onset) showed that disability progression in the period until irreversible EDSS 3 did not influence disability progression during the period from irreversible EDSS 3 to irreversible EDSS 6 (14).
Despite these increases in the delay of progression due to DMTs, there is an underestimation of the size of the SPMS population in certain regions (15), with the prevalence of SPMS widely varying among countries (15, 16). A study involving patients from Germany, the UK, and Sweden demonstrated that the use of a classifier, which allowed for the reclassification of RRMS/SPMS patients, increased the number of patients classified as SPMS, resulting in a more uniform prevalence of SPMS across these countries (15). Potential factors influencing SPMS prevalence include demographic and geographic factors, healthcare resources, operational definitions of SPMS, or reimbursement criteria for DMTs (16, 17).
Notably, the diagnosis of SPMS is usually made retrospectively through clinical assessment, often based on the observation of irreversible disability progression as measured by the EDSS. The delay between the detection of initial signs suggestive of progression and the confirmed diagnosis of SPMS can last up to three years (18). Despite the publication of various national consensus statements (19, 20), unified criteria for defining the onset of SPMS are still lacking. Timely SPMS diagnosis is key because it provides a window for therapeutic intervention. Early signs of MS activity, such as cognitive decline, brain atrophy, and fatigue, are often missed in routine monitoring, which usually focuses on relapses and MRI findings. Silent progression, undetected by these standard methods, contributes to disease worsening. This highlights the need to update MS management strategies, ensuring timely initiation and escalation of DMT (21).
In Europe, five DMTs are approved for relapsing MS (RMS; interferon-beta-a [IFN-β-1a], cladribine, ocrelizumab, ofatumumab, and ponesimod), which includes both RRMS and SPMS with disease activity, one is approved for RRMS and active SPMS (aSPMS; INF-β-b), and another is approved for aSPMS (siponimod). Detecting disease activity in SPMS is difficult not only due to a decrease in inflammatory activity, but also because these patients are often already under treatment with a DMT that effectively controls clinical and radiological activity. This scenario highlights the complexity of SPMS management: the need to identify disease activity to select the appropriate DMT in patients with ongoing treatments that reduces or eliminate such activity.
A narrative review was conducted for the elaboration of the article. No structured method for searching or evaluating the research literature were used. Instead, authors reviewed publications on predefined topics (diagnostic challenges in SPMS, difficulty in detecting disease activity in treated patients and disease-modifying treatments) and then shared the most relevant information of these publications and their opinions on the gaps in current knowledge, current challenges, and future directions in two structured meetings held on February 27, 2024, and April 10, 2024. In these meetings, the discussion was guided by a moderator who also summarized the contributions by the authors and shared them via email following each meeting for final review. This article aims to provide a comprehensive review and position statement on the current challenges associated with SPMS, addressing key issues such as diagnosis, the assessment of disease activity in patients undergoing DMT treatment, and the available therapeutic options for managing aSPMS.
2 Diagnostic challenges in SPMS
2.1 Diagnostic criteria
The diagnosis of SPMS is still challenging due to the lack of established clinical, imaging, immunological, or pathological criteria that clearly define this progression. Currently, the definition of SPMS by Lorscheider et al. published in 2016 is the most widely used to identify SPMS (22). This definition includes a disability progression by 1 EDSS step in patients with EDSS ≤5.5 or 0.5 EDSS steps in patients with EDSS ≥6 in the absence of a relapse, a minimum EDSS score of 4.0 and Pyramidal Functional System score of 2 and confirmed progression over at least 3 months (22). However, this definition does not capture all patients transitioning to SPMS, as some may experience a progressive course without reaching the specified EDSS threshold. In fact, some national consensus supports recognition of progression without setting a specific minimum EDSS score (19), whereas others advocate for the minimum EDSS score of 4.0 as a criterion for identifying disease progression in MS (20).
In clinical trials assessing DMTs for SPMS, different inclusion criteria have been established to select participants. In the EXPAND trial (siponimod vs placebo), patients with an EDSS score ranging from 3.0 to 6.5, who had also shown EDSS progression within the two years before the study, were included (23). Similarly, the ASCEND trial (natalizumab vs placebo) required participants to have an EDSS score between 3.0 and 6.5, but also a Multiple Sclerosis Severity Score (MSSS) of 4 or higher, and disability progression independent of relapse activity (PIRA) in the year before the study (24). In the HERCULES trial, which has evaluated the efficacy and safety of tolebrutinib compared with placebo in patients with non-relapsing SPMS (nrSPMS) (25), patients were selected if they had an EDSS from 3.0 to 6.5, had disability progression within the year before the study, and had an absence of clinical relapses for at least two years before the study. The variation in criteria among national consensus and clinical trials exemplifies the ongoing lack of standardization for defining disease progression in MS.
Although the EDSS remains the primary assessment for defining SPMS, this tool is not exempt from limitations (26). In fact, the EDSS mainly focuses on ambulation and physical disability, with cognition, vision, and upper limb function being underrepresented (21). To conduct a complete assessment of disability, the EDSS could be used with other functional assessments, such as the 9-Hole Peg Test (9HPT) and the Timed 25-Foot Walk (T25FW) (20). The Multiple Sclerosis Functional Composite (MSFC) combines the 9HPT, T25FW, and cognition (Paced Auditory Serial Addition Test; PASAT) into one score, providing a multidimensional tool for evaluating functional changes and disease progression. Cognition, which is impaired in most patients with SPMS (27), should be assessed with validated neuropsychological tests. Cognitive assessment to detect progression should be conducted with a neuropsychological evaluation. In those cases in which the administration of a full battery of neuropsychological tests is not possible, applying at least a short and validated test, such as the Symbol Digit Modalities Test (SDMT), for the screening of cognitive function is recommended (20). Electronic self-administered tools and other digital tools can be also practical to assess mobility and cognitive changes. Consistent criteria for identifying cognitive impairment in MS are also needed to improve diagnosis and monitoring disease progression (21).
2.2 Pathophysiology
The pathological mechanisms underlying disease progression in MS are complex and not fully elucidated. Both inflammation and neurodegeneration are already present at disease onset (28–32). These pathological processes coexist, to varying degrees and in different CNS locations, throughout the disease course (33, 34). The disease progression seems to involve quantitative changes in the extent of these processes rather than qualitative changes in the type of pathological activity (35, 36). Indeed, no qualitative differences are found when comparing patients with PPMS and SPMS, although there are some quantitative differences. For instance, the presence of focal and active classical white matter lesions and the global degree of inflammation are lower in PPMS compared to SPMS (37). This suggests that RRMS, PPMS and SPMS are part of a disease spectrum modulated by genetics and environmental factors (38).
The widespread neuroaxonal injury observed in progressive MS seems to be caused by the interplay of several pathological mechanisms, including compartmentalized neuroinflammation within the CNS, axonal degeneration, microglial activation, astrogliosis, oxidative stress, mitochondrial dysfunction, iron toxicity, and deficient remyelination, among others (9, 36, 39, 40). The activation of these mechanisms marks the biological onset of the disease and initiates the prodromal period (Figure 1). The dysregulation of the immune system that triggers these pathological mechanisms in the progression of MS includes chronic and elevated levels of pro-inflammatory cytokines (interleukin-1β, interleukin-6, tumor necrosis factor-alpha, interleukin-17, and tumor necrosis factor-alpha), chemokines, and autoantibodies (41).
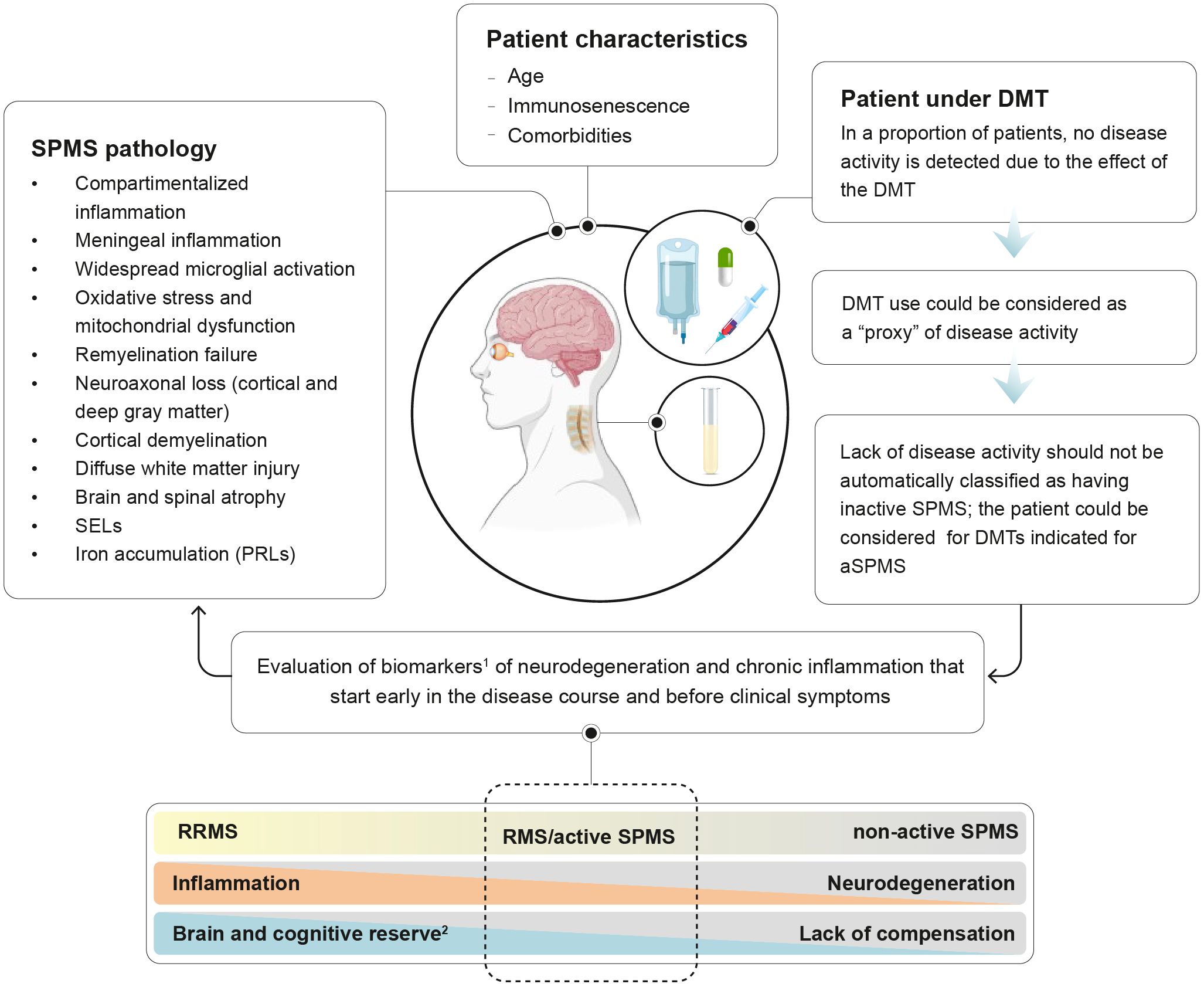
Figure 1. Detecting disease activity in treated patients transitioning to SPMS. 1biomarkers of MS progression are presented on Table 1; 2Brain reserve: structural characteristics of the brain that enable to maintain cognitive function despite brain pathology; cognitive reserve: ability to optimize performance through the differential recruitment of brain networks and alternative cognitive strategies.
Additionally, the biological process of aging superimposes the pathogenesis of progressive MS. Immunosenescence, the gradual deterioration of the immune system associated with aging, drives chronic low-grade inflammation, known as inflammaging, which is superimposed over the pathogenesis of MS (42, 43). Moreover, biological aging of glial cells has been observed to be accelerated in the brains of people with MS (44).
The balance of such mechanisms, together with repair capacity and brain and cognitive reserve influences clinical progression during the disease course. Importantly, aging is associated with a decline in brain reserve and cognitive reserve, rendering the CNS less resilient to the cumulative effects of MS pathology (45). Indeed, it has been hypothesized that loss of neurologic reserve explains the onset of progressive MS (46). In the early stages of MS, inflammation leads to brain atrophy, but symptoms are mitigated by the neurologic reserve and functional reorganization of neural networks (47). Over time, as injury from both MS and aging accumulates, the neurologic reserve and the ability to compensate diminishes. This depletion reveals the effects of subclinical MS activity and aging, presenting as progressive MS (46). Therefore, the interplay between age-related neurodegeneration, MS-specific pathological processes, and neurologic reserve explains the progression of disability observed in the advanced stages of the disease. Once the accumulation of irreversible damage surpasses the CNS ability to compensate, disability worsening emerges.
Genetics, environmental and lifestyle factors, including obesity, dysbiosis of the gut microbiota, lack of physical activity, smoking and vitamin D deficiency, play and important role in the progression of MS pathophysiology and the CNS ability to compensate (48–50).
Disability accumulation can result from relapse-associated worsening (RAW) or PIRA (progression independent of relapse activity, or the so-called “silent progression”). Studies with large patient cohorts have demonstrated that PIRA is the main driver of disability accumulation, particularly as the disease advances (29, 51–53). The occurrence of PIRA following an initial demyelinating event is a negative prognostic indicator of the disease course (53). However, the definition of PIRA is mainly based on motor worsening, assessed by the EDSS or EDSS-plus, and does not capture the worsening of other symptoms (independent of relapses), such as subtle motor impairment, cognitive slowing, early fatigability, neuropathic pain, and bowel/bladder and sexual dysfunction (39). The term smoldering-associated worsening (SAW) has been suggested to capture the pathobiological processes associated with these clinical manifestations (35, 39).
2.3 Phenotyping MS
Considering the diverse pathological processes underlying MS, several MS phenotyping methods more rooted in the biological mechanisms of MS have been proposed to provide a more comprehensive definition of MS phenotypes than the traditional classification (54–56). For instance, Pitt et al. (54) suggested extending the current classification of MS by including additional pathological processes, such as chronic perilesional inflammation, neuroaxonal degeneration, and remyelination, to improve the phenotyping of MS. Incorporating these processes could help differentiate MS phenotypes that appear clinically similar but are driven by different underlying pathological patterns (54).
MS subtypes based on pathological features using MRI have also been proposed (55). The three subtypes identified in one study using unsupervised machine learning were cortex-led, normal-appearing white matter-led, and lesion-led. Patients with the lesion-led subtype had the highest risk of confirmed disability progression (CDP) and relapse and showed positive DMT response in selected clinical trials (55).
Immune signatures in the blood have also been used as a surrogate of disease pathophysiology to explain MS endophenotypes. Using a combination of high-dimensional flow cytometry and serum proteomics, and unsupervised clustering, three distinct peripheral blood immunological endophenotypes have been recently observed. DMT response was associated with the endophenotype. Patients with endophenotype 3 treated with interferon-beta had higher disease progression and MRI activity compared with treatment with other DMTs (56).
2.4 Biomarkers
To date, no clear clinical, imaging, immunologic, or pathologic criteria exist to determine when RRMS transitions into SPMS. There is a need for tools to support early identification of SPMS. The development, validation, and implementation of biomarkers of MS progression are key not only for SPMS diagnosis, but also for monitoring the disease and the long-term treatment outcomes in these patients. To improve SPMS identification and monitoring, the updated definition of SPMS should include clinical evaluations with biomarkers in blood, cerebrospinal fluid (CSF), and imaging techniques. Current MRI assessments in clinical practice only include Gd+ lesions on T1 and new/enlarging lesions on T2, which provide limited information about disease progression. Other pathological processes and potential biomarkers, such as chronic active lesions (CAL; localized areas of compartmentalized inflammation within the CNS that occur in the absence of blood-brain barrier breakdown) and brain and spinal cord atrophy associated with MS progression, specifically with PIRA (57), are currently only being evaluated in research and have not yet been included in routine clinical practice.
Slowly expanding lesions (SELs; contiguous regions of existing T2 lesions showing local expansion) and paramagnetic rim lesions (PRLs; rim of paramagnetic material, mainly iron, that accumulates within activated microglia and macrophages at the edges of the lesion) are promising MRI measures of smoldering inflammation and CAL (58–60). SELs and PRLs are associated with PIRA and with progressive MS (59–65). A higher number, volume or proportion of SELs (66), particularly when combined with PRLs (67), is associated with more pronounced clinical progression than either lesion type alone, suggesting a compounded impact on disease severity when multiple lesion types coexist. While both SELs and PRLs represent CALs, it is unclear how these lesions are interconnected and have different associations with clinical outcomes or SPMS. Together with PRLs and SELs, 18-kDa translocator protein-positive lesions on PET is a promising candidate biomarkers of CAL (68). Further studies are warranted to confirm the role of these biomarkers in SPMS.
Advanced MRI techniques, such as quantitative susceptibility mapping (QSM), diffusion tensor imaging (DTI) and susceptibility-weighted imaging (SWI) are promising for the detection of MS pathogenesis (69–71). Iron content seems to be the primary source of QSM values in deep grey matter and at the edges of rim lesions, with the latter being associated with more severe disability (71). QSM can be acquired with standard field strength (3T) MRI scanners, which makes it a feasible option for clinical settings (72). DTI provides information on the integrity of white matter tracts, which is important in progressive MS where widespread damage occurs beyond visible lesions (73). Recently, the T1-dark rim has been proposed as a novel imaging sign identifiable in a standard 3DT1 gradient-echo inversion-recovery sequence to detect PRLs (74). CALs can be also identified by SWI (60), which makes this technique not only useful for improving MS diagnosis (70) but also for detecting progression.
On the other hand, the rate of retinal layer thinning, measured by optical coherence tomography (OCT), also correlates with disability progression and brain atrophy in MS (75–77). Retinal thinning is associated with PIRA, probably indicating neurodegenerative processes rather than focal inflammation (78). It is worth noting that, although these MRI or OCT features increase the likelihood of accumulating disability, there are still no specific neuroimaging abnormalities that can be considered a hallmark of a distinct MS phenotype, including SPMS.
Several blood and CSF biomarkers have also received great attention in the last years. Neurofilament light chain (NfL), a marker of neuroaxonal injury, and glial fibrillary acidic protein (GFAP), a marker of astrocytic activation, have shown promising results in predicting disease activity and progression (79–85). Elevated levels of serum GFAP (sGFAP) have been associated with accelerated grey matter brain volume loss and disease progression, particularly in nonactive patients (82, 85–87). However, another study revealed that, in the absence of inflammatory activity, changes in serum GFAP did not correlate with disability progression in SPMS patients (88). Elevated baseline levels of NfL in RRMS are predictive of brain and spinal cord atrophy and disability progression (89–91), though GFAP seems to have a stronger correlation with disease progression (85, 92, 93). High NfL levels also correlate with an increased number of relapses and the presence of new or enlarging T2 lesions and T1 Gd+ lesions (89, 94). Some studies have shown that NfL levels are more affected by recent relapses and high-efficacy DMTs (heDMTs) than GFAP levels (92, 93, 95). The persistence of elevated GFAP levels in patients receiving heDMTs might suggest a patient profile with a higher likelihood of disability progression. The combination of high serum NfL and GFAP has been associated with a 4- to 5-fold increased risk of confirmed disability worsening (CDW) and PIRA (86).sNfL and sGFAP could help in differentiating PIRA caused by underlying peripheral inflammation (associated with high sNfL levels) from PIRA associated with smoldering compartmentalized inflammation (observed in cases with elevated sGFAP and low sNfL levels) (96). These findings suggest that NfL and GFAP may provide complementary information about different aspects of the disease progression (96).
Other potential biomarkers of SPMS are soluble triggering receptor expressed on myeloid cells 2 (sTREM2) as a marker of microglial activation, and chitinase-3-like-1 (CHI3L1) as a marker of inflammatory activity (97).
Despite research efforts in identifying and validating these biomarkers of disease progression, their implementation in clinical practice remains limited (Table 1). Most of these biomarkers are currently used in research settings only, and their clinical feasibility is still under evaluation. There is a need to establish standardized protocols and cut-off values at an individual level for these biomarkers to be adopted in clinical practice. Current methods of measuring MS disease activity and progression often miss subtle signs of neurodegeneration, such as CAL or brain atrophy. Integrating these “hidden” indicators with biomarkers could offer a more complete picture of disease activity and allow earlier detection of progression (21). First, identification, quantification and monitoring of these biomarkers must be standardized.
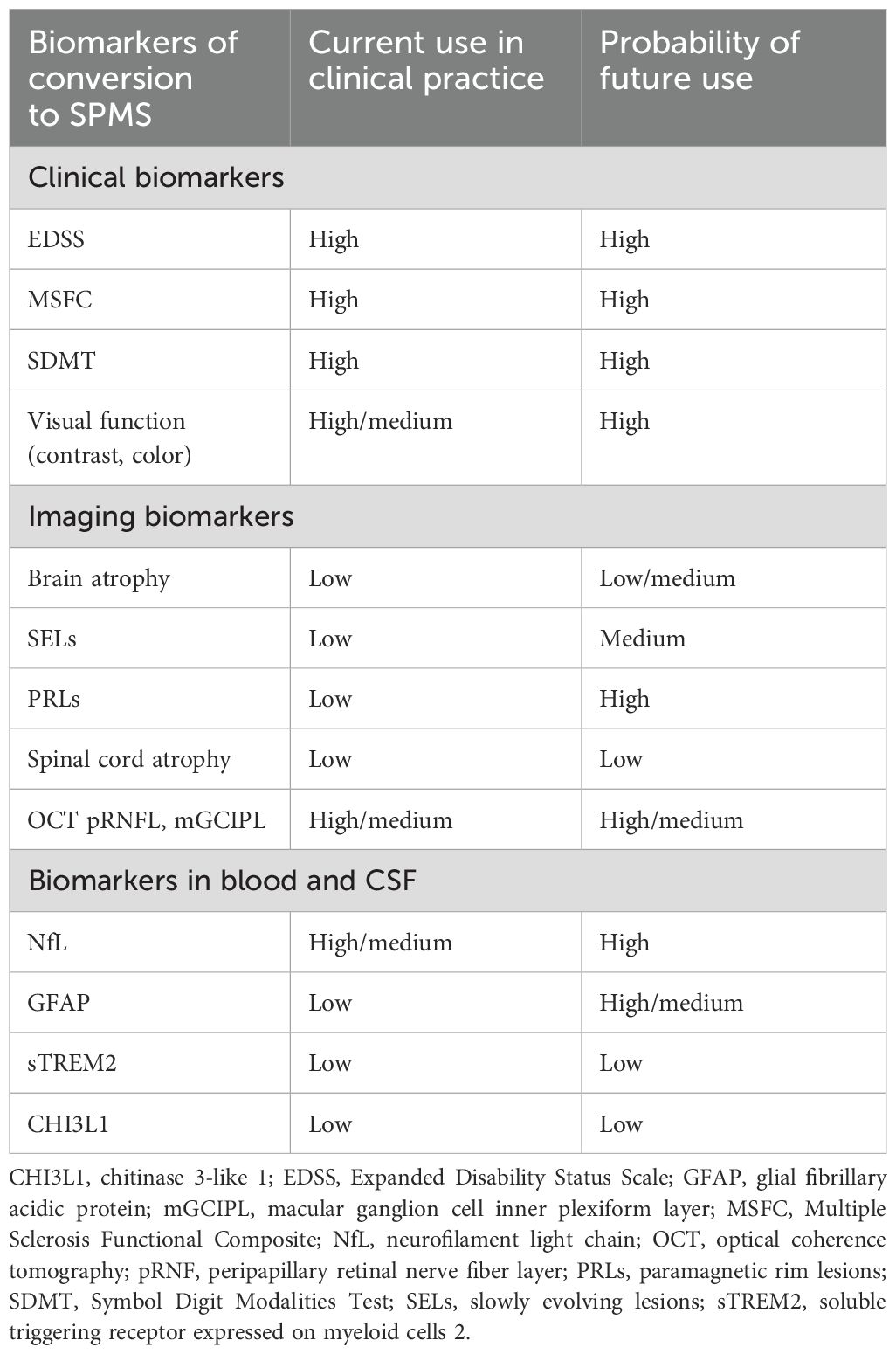
Table 1. Clinical and paraclinical biomarkers of MS progression.
3 Difficulty in detecting disease activity in treated patients
3.1 Assumption of disease activity
There is a paradox in the use of DMTs indicated for aSPMS. These treatments require patients to present disease activity evidenced by relapses or imaging features of inflammation to initiate treatment (98, 99). However, most patients with RRMS transitioning to SPMS are already treated with DMTs that effectively suppress disease activity (100, 101), making it difficult to detect the required activity to select candidates for initiating the treatments for aSPMS (98). Moreover, MRI has limited utility in detecting new lesions, particularly in cases with a high T2 lesion load, and the use of spinal MRI is not widespread. Theoretically, it could be assumed that if the DMT was discontinued, disease activity would resurge and, in some cases, a rebound effect would be observed. This assumption is based on studies showing an increase in relapses and MRI findings after DMT discontinuation in patients with SPMS (102–105), despite disease activity after discontinuation being lower in SPMS than in RRMS patients (103). A recent study further supports this assumption, demonstrating that patients 50 years and older classified as non-active MS (no evidence of relapse or MRI activity in at least two years, and treated with heDMTs for at least one year) who discontinued the heDMT had an increased probability of inflammatory activity compared with those who continued the heDMT (104).
DMTs indicated for highly active RRMS, such as natalizumab, indeed assume the presence of disease activity. That is, for patients treated with natalizumab who are at higher risk of progressive multifocal leukoencephalopathy (PML), it is recommended to consider their history of disease activity as sufficient criteria to initiate another DMT to reduce the risk of PML (106). Moreover, some European Public Assessment Reports (EPAR) consider patients under DMT as active (107, 108). For example, the EPAR for ofatumumab recognizes that patients currently receiving a DMT for controlling MS inflammatory activity can be considered as fulfilling the ‘activity’ criterion. This is based on the assumption that these patients were experiencing disease activity when the initial DMT was prescribed and that a patient whose inflammatory activity is adequately controlled by a DMT intended to control this activity might not be without activity. The EPAR states that “the fact that these patients are currently receiving a DMT for controlling the MS inflammatory activity could be considered as a ‘proxy’ of fulfilment of an ‘activity’ criterion”. Similarly, in the EPAR for ozanimod it is stated that “switching from other drugs to ozanimod should be allowed despite no clear indication of active disease, for example in patients with side effects or intolerability issues” (108). Even if an estimation of the impact of patients not meeting the ‘activity’ criterion cannot be directly drawn, it could be inferred that patients switching from their current DMT due to safety or tolerability issues could have a positive risk-benefit ratio (107). Thus, it could be argued that a patient transitioning to SPMS who is treated with a DMT and shows no signs of disease activity should not necessarily be considered as having inactive SPMS (Figure 1).
To estimate the likelihood of residual inflammatory activity, the type and duration of treatment, along with other relevant factors (duration of MS, current age, degree of disability, baseline activity before treatment, time without clinical or radiological activity) should be evaluated before concluding that the disease activity observed prior to the initiation of the DMT will remain unchanged, as this activity may have evolved (109). The interruption of a DMT, especially a heDMT, without adequately switching to another DMT, can expose the patient to a higher risk of inflammatory activity (104). Therefore, the neurologist’s judgment, together with the patient preferences, should prevail when a change to a DMT indicated for RMS or aSPMS is required for lack of safety or effectiveness reasons.
The variability in the concept of disease activity is further exemplified by the fact that certain selection criteria for recent clinical trials in nrSPMS, such as in the HERCULES trial, considered only relapses as disease activity, while radiological activity (i.e. gadolinium-enhancing lesions on T1 or new/enlarging T2 lesions) was not taken into account for selecting the patients (25). Thus, patients with disease activity on MRI months or weeks before the study could be included as a nrSPMS patient in the trial (25).
3.2 Barriers to activity detection
Detecting disease activity, even when present, is unlikely in treated-RRMS patients who begin to progress, due to several factors. On one hand, less frequent monitoring visits might be conducted in these patients. Some studies have found MRIs are conducted with low frequency in SPMS patients (109, 110). MRI seems to be a more sensitive tool to measuring disease activity than relapses, and therefore, the limited use of MRI in clinical practice might reduce the chance of detecting disease activity in SPMS, as already reported (109). Also, in routine clinical practice, spinal cord MRIs are not regularly performed, and the detection of new/enlarging lesions on T2 is complicated, particularly when there is a high T2 lesion load.
On the other hand, underreporting of relapses by these patients may occur, as they may report minimal symptoms or fail to recognize them as relevant over time (111). Also, physicians may sometimes fail to distinguish relapses from pseudo-relapses, recognize subtle changes, or attribute symptoms to other causes (112), further complicating the detection of activity and progression. Considering this context, we emphasize the importance of face-to-face monitoring in these patients. More frequent monitoring in patients diagnosed with SPMS, compared to the current standard practice, could improve the detection of disease activity. The use of spinal MRI in the follow-up of patients with worsening disability not explained by cranial MRI findings could improve the detection of active lesions, even though it is not currently performed in clinical practice (113). Fluid biomarkers, such as NfL and GFAP, hold promise for establishing progression based on underlying pathological mechanisms.
4 Disease-modifying treatments
4.1 Approved treatments
The availability of DMTs for SPMS has increased in the last few years, though options remain limited (Table 2). More than 25 years ago, IFN-β-1b was the first DMT approved for the treatment of aSPMS, based on positive results in delaying progression in patients with SPMS (114). IFN-β-1a also showed delayed progression in the PRISMS trial (115) and was approved for RMS, even if in the IFN-β-1a trial progression was a secondary endpoint and patients had RMS and a baseline EDSS of 0–5.0. Disappointingly, subsequent clinical trials of these treatments showed conflicting results, with no significant difference in time to confirmed progression between placebo-treated and IFN-β-1b- or IFN-β-1a-treated patients (99, 116–118). Post-hoc analyses showed that IFN-β-1a reduced disability progression only when patients had experienced relapses in the two prior years (118, 119). The lack of consistency between studies regarding IFN-β efficacy to delay progression has resulted in limited use of IFN-β to treat SPMS in clinical practice.
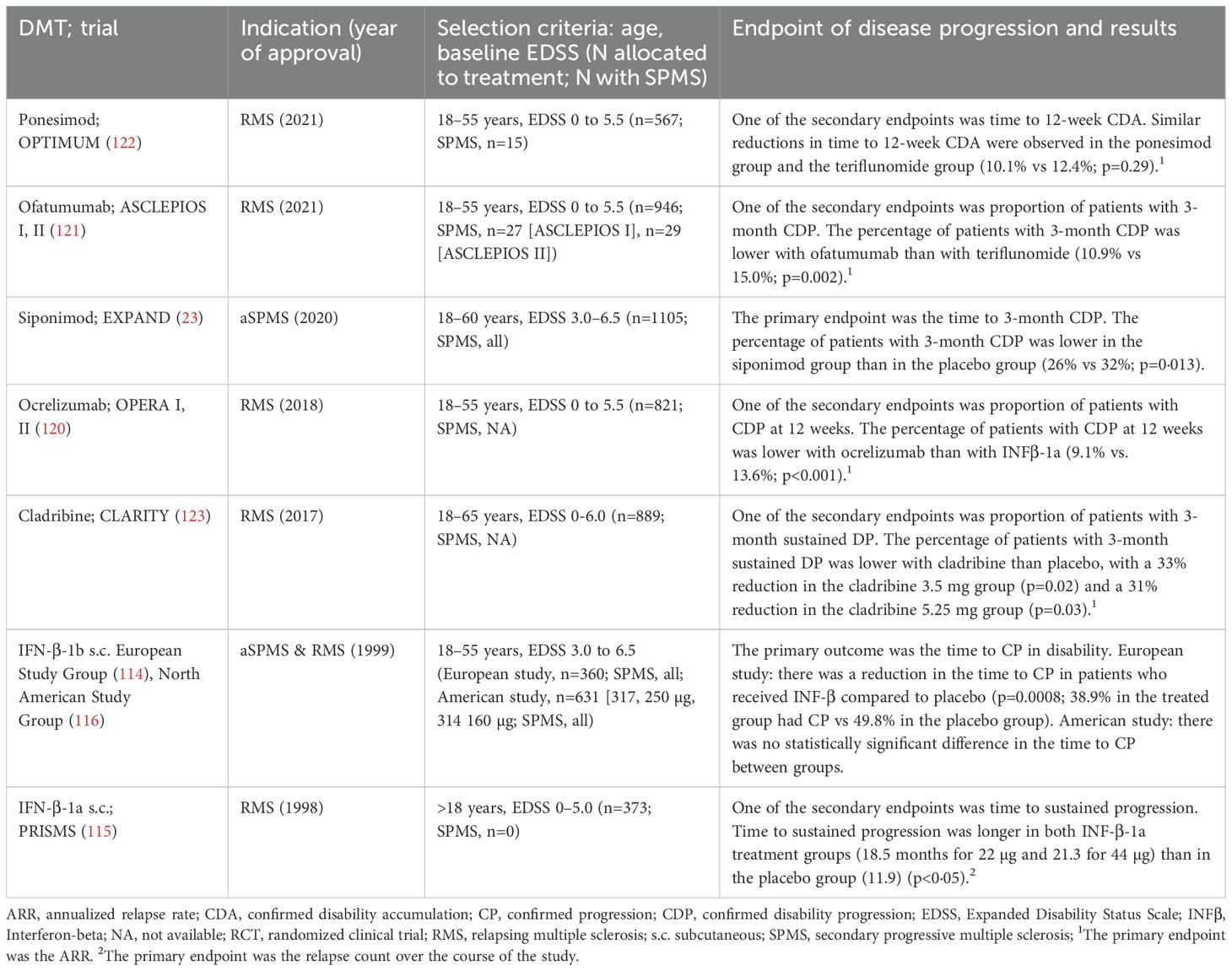
Table 2. DMTs approved for SPMS and RMS in Europe: pivotal phase III RCT.
Other DMTs, including ocrelizumab, cladribine, ofatumumab, and ponesimod have been approved for RMS. However, these approvals were mainly based on trials that were not specifically designed to assess efficacy in SPMS. These studies included patients with a baseline EDSS ranging from 0 to 5.5 (or 6.0 for cladribine) and used disease activity, measured by the annualized relapse rate, as the primary endpoint (120–123). Disability progression was a secondary outcome in these trials (120–123), and efficacy in SPMS was often inferred from post-hoc analyses of small subgroups. The approval of these DMTs, therefore, relied on the assumption that the efficacy in delaying MS progression observed in patients with RRMS and in a small number of patients with SPMS could be generalized to SPMS.
In contrast, the EXPAND study, which led to the approval of siponimod, was specifically designed to evaluate the treatment efficacy in delaying progression in SPMS patients with or without disease activity. The study included patients with a baseline EDSS of 3.0–6.5, and its primary endpoint was time to 3-month confirmed disability progression (CDP) (23). The trial demonstrated a statistically significant 21% relative reduction in the risk of 3-month CDP for patients treated with siponimod compared to those receiving placebo (23). Patients treated with siponimod also had a significantly reduced risk of 6-month CDP, less worsening in processing speed, decreased clinical and radiological disease activity, and a reduction in total brain volume loss (23). Post-hoc analysis revealed that siponimod also slowed progression of whole-brain and grey matter atrophy and improved brain tissue integrity/myelination (124). Most of these effects on brain integrity were sustained in the long-term and were more pronounced in patients who initiated siponimod earlier (124). Additionally, in a subgroup analysis of 779 patients with aSPMS, siponimod significantly reduced the risk of disability progression, cognitive decline, and MRI lesions compared to placebo (125).
In the HERCULES trial (25), eligible participants (18–60 years; EDSS: 3.0 - 6.5) were required to have no relapses in the 24 months before screening and disability progression within the 12 months before screening. The primary endpoint was time to onset of 6-month CDP. Tolebrutinib showed a statistically significant 31% risk reduction in time to 6-month CDP compared to placebo. It is worth noting that the study included nrSPMS without clinical relapses for at least 24 months, but the absence of radiological activity was not a selection criterion. Therefore, the nrSPMS population in this study allowed the inclusion of radiologically active and non-active patients.
Masitinib has showed to delayed progression (primary endpoint: EDSS change from baseline) in PPMS and nrSPMS in a phase 3 trial; a confirmatory phase 3 study is ongoing and will provide further data (126). Another ongoing study assesses the efficacy and safety of fenebrutinib compared with ocrelizumab in PPMS in disability progression (primary endpoint: time to onset of composite 12-week CDP) (127). Other clinical trials assessing the efficacy of treatments on disability progression as primary endpoint in patients with SPMS have failed to achieve positive results (128), highlighting the difficulties in developing effective treatments for SPMS. Multiple factors should be considered to improve the design of these trials (128). First, the profile of included patients should align with the mechanism of action of the drug (i.e. immunomodulation, neuroprotection, or remyelination). For example, an immunomodulation mechanism that targets inflammation would be relevant for patients with aSPMS. In contrast, for patients with nrSPMS, the treatment should target remyelination or neuroprotection rather than inflammation. Also, including a more heterogeneous profile, such as older adults and those with comorbidities, would increase the generalization of results. Second, the primary endpoint should be a sensitive measure that captures several aspects of progression, such as the EDSS-plus or other clinically meaningful composite measures; relevant biomarkers of progression and patient-reported outcomes (PROs) should also be included as secondary outcomes. Lastly, trials could be more efficient with a multi-arm design, where several active drugs are compared to the placebo arm, or a multi-stage design that allows modifying the study protocol based on interim results (128).
4.2 Discontinuation
The question of whether, and if yes, when and how, to safely discontinue DMTs in older and stable SPMS patients remains a topic of ongoing debate. Research on DMT discontinuation has yielded contradictory results about relapse rates and progression, probably due to differences in disease activity before discontinuation, age at discontinuation and treatment efficacy (129–132).
One of the most anticipated results was the findings of the DISCOMS trial (130). This trial allocated stable patients 55 years or older to either stop or to continue their DMT and had a 2-year follow-up. Those who discontinued experienced an increase in clinical or radiological disease activity (12.2%) compared with those who continued DMTs (4.7%), with no significant differences in increased disability. With an absolute difference of 7.5%, the authors were unable to reject the null hypothesis that DMT discontinuation was non-inferior to continuation. The study included a small proportion of patients treated with heDMTs (9%), and therefore, the conclusion of this trial cannot be generalized to all DMTs.
The STOP-I-SEP (NCT03653273) will provide further evidence on the impact of discontinuation on MS. The study assesses the effect of treatment discontinuation in disability progression, disease activity and health-related quality of life (HRQoL) in SPMS patients older than 50 years. The DOT-MS study (NCT04260711), which evaluated disease activity, disability and PROs in patients treated with moderate-efficacy DMTs (meDMTs; interferons, glatiramer acetate, dimethyl fumarate, or teriflunomide) was terminated early after an interim analysis revealed increased disease activity above the predefined limit in the group that discontinued treatment (133).
While we await these results, real-world evidence provides valuable data. A recent meta-analysis, which included 22 real-world studies and 2942 patients followed for 1–7 years after discontinuation, found that the risk of relapses after DMT discontinuation became negligible (i.e. < 1% per year) around the age of 60, and after either 10 years of DMT use or 8 years of stable disease (129). However, as in the DISCOMS trial, the majority of patients included in the study were treated with meDMTs. This raises the question of whether the impact of discontinuation on disease activity might depend on the efficacy of the treatments (heDMTs vs. meDMTs). A recent study using propensity-matching scores evaluated disease activity in patients 50 years and older with nonactive MS who either continued or discontinued heDMT (rituximab, ocrelizumab, natalizumab, and fingolimod). The study found that the risk of relapse was significantly higher in patients who discontinued the heDMT compared with patients who continued but varied greatly according to the DMT (104).
The integration of findings from clinical trials and real-world studies can guide clinicians in making informed decisions regarding DMT discontinuation. However, to date, there is a lack of studies specifically evaluating the discontinuation of DMTs approved for SPMS, and the long-term effects and risks of stopping these therapies are not well understood yet. When considering discontinuing treatment in a patient with SPMS, several factors must be considered, such as age, duration of MS, accumulated disability, progression rate, time of clinical/radiological stability, DMT type and safety, and comorbidities. The risk-benefit balance of discontinuing a DMT should be carefully addressed for each individual patient.
5 Discussing the progressive phase with patients
Discussing the progressive phase of MS with patients is a delicate aspect. Most patients want to know their long-term prognosis as soon as possible (134). However, predicting disease progression is difficult, as the variability in the rate of progression is high, although several prognostic factors influencing long-term progression have been identified and can help to anticipate disability (135). Also, new tools such as machine learning can be used to predict conversion to a secondary progressive course, confirmed disability accumulation, and disease severity with rapid accumulation of disability (136, 137).
There is no consensus on the best time to introduce discussions with patients about MS progression. Some neurologists prefer to address the issue early to facilitate informed decision-making and planning. In contrast, others delay the conversation out of concern that the conversation may increase anxiety and delay decisions regarding treatment (138). One of the main difficulties in starting these discussions has been the lack of effective treatments in reducing disease progression. However, in the last few years, the approval of DMTs for aSPMS has provided an option to treat these patients (23, 124, 125). The positive results from clinical trials of other treatments in delaying progression (25, 126) give hope that additional DMTs that modify the disease course and delay progression will be available.
Something important to consider when addressing the progressive phase with patients is the fact that most patients have probably already come across the concept of disease progression when searching for information online about MS (139). Patients can now easily access medical and scientific information by using artificial intelligence (AI)-powered large language models. These tools have transformed information retrieval, as they provide instant, context-specific explanations of medical concepts in a human-like manner (140). Furthermore, online patient communities and social media platforms create spaces where patients can share their experiences (141). As a result, physician–patient communication must evolve to match the increasing accessibility of medical knowledge.
Neurologists should be prepared to engage with patients and caregivers in discussions that clarify, validate, and contextualize the information gathered from AI tools and digital platforms. Effective communication—characterized by empathy, validation of emotions, clarity, and active listening—plays a key role in managing patients. A recent study evaluated the preferences of patients with MS towards responses to frequently asked health-related questions provided by either neurologists or by ChatGPT. Patients, who were unaware of who generated the response, perceived ChatGPT responses as more empathetic compared to responses from neurologists, although patients with higher levels of education showed lower satisfaction towards the responses created by ChatGPT (142). In fact, empathy has been reported by older patients with MS as an area for improvement among neurologists (143). Therefore, improving soft skills, especially oral communication skills, is more important than ever when discussing sensitive topics with patients, such as disease progression. Including the family in the conversation and involving the patient in shared decision-making of DMT and symptomatic treatment are also relevant for increasing treatment satisfaction (144, 145).
A key strategy in discussing progression is having an ongoing conversation rather than a one-time disclosure. Instead of presenting progression as an inevitable outcome, neurologists could emphasize that progression is a gradual process, often subtle and that proactive management—including lifestyle modifications, symptomatic treatment, and rehabilitation strategies—can improve long-term outcomes. Patients’ emotional responses to information about MS are shaped by the way their providers communicate prognosis and treatment expectations. One of the most important factors influencing patients’ satisfaction with the MS diagnosis is adequate emotional support (146). By acknowledging patient concerns while providing reassurance that their care plan is adaptable, neurologists can help patients engage with their treatment strategy without feeling a sense of impending loss.
Discussing MS progression requires a patient-centered, stepwise approach that evolves over time. While the initial focus should be on controlling disease activity and maximizing HRQoL, patients should also be prepared for the possibility of progression. When discussing progression, neurologists should also clarify that disease progression is not only defined by motor decline but can include cognitive, sensory, and functional changes, which have an impact on HRQoL (147), even in the absence of relapses or MRI lesions (148).
Patients often notice these subtle changes before they are clinically confirmed, and thus, neurologists must listen carefully and integrate these patient-reported symptoms into their assessments. There is no standard guideline on when to address progression, and therefore, neurologists should tailor these discussions to individual patients, considering their level of health literacy, emotional readiness, and personal concerns.
6 Concluding remarks
Optimizing the management of SPMS requires a comprehensive approach. First, there is a critical need to establish a unified definition of SPMS that captures the early stages of disease progression, where the introduction of DMTs may be most beneficial. This definition should incorporate an understanding of the underlying pathological processes and be supported by biomarkers that can be routinely applied in clinical practice.
Given that neurodegenerative changes begin early in the disease course and contribute to long-term disability, these processes should be identified as soon as possible. Current clinical measures alone may not fully capture disease activity and the underlying neurodegeneration that may be masked by compensatory mechanisms during the early stages of MS. Emerging biomarkers, such as SELs and GFAP, hold promise for detecting early neurodegenerative changes in the near future.
Furthermore, the initiation of DMTs with proven efficacy in reducing progression should be used as soon as the patient transitions to SPMS. The difficulties in identifying SPMS in patients currently treated with DMTs that reduce disease activity should not prevent the decision to start DMTs indicated for aSPMS. The use of current DMTs could be considered an assumption of ongoing clinical activity.
To support evidence-based decisions in MS management, clinical trials for MS treatments should include more patients with SPMS. Also, discussing MS progression with patients requires a patient-centered, stepwise approach, balancing transparency, emotional support, and shared decision-making. By addressing these issues, we can improve long-term outcomes for patients with SPMS and provide more effective, safe and personalized care.
Author contributions
LB: Conceptualization, Writing – original draft, Writing – review & editing. CC: Conceptualization, Writing – original draft, Writing – review & editing. LL: Conceptualization, Writing – original draft, Writing – review & editing. CO: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors declare that this manuscript received funding from Novartis Farmaceútica S.A for the development of this manuscript. The funder was not involved in the literature review, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
We would like to express our gratitude to Laura Prieto del Val from Evidenze Health Spain S.L.U. for her support as a medical writer during the meetings to revise the content for this article and during elaboration of this article.
Conflict of interest
LB has received funding for his group’s research projects or in the form of fees for lectures, tutoring, and assistance in attending conferences from Bayer, Biogen, Roche, Merck, Novartis, Allmirall, BMS, Janssen, Sanofi and Neuraxpharm. CC has received fees for participating as a speaker in meetings, courses, advisories, monographs by Teva, Biogen, Sanofi, Merck, Bristol-Myers, Roche, Celgene, Novartis, Sandoz. LL has received fees for participating in advisories, scientific and educational activities from Almirall, Bayer, Biogen, Bristol-Myers, Sanofi-Genzyme, Merck, Novartis, UCB Pharma, Roche, and Teva. And CO-G has received fees for lectures and consultations from Biogen Idec, Celgene, Sanofi-Genzyme, Novartis, BMS, Jannsen, Roche, Merck, Viatris, Neuraxpharm and Teva.The authors declare that this study received funding from Novartis. The funder was not involved in the study design, collection, analysis, interpretationof data, the writing of this article or the decision to submit it for publication.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. MS international federation. Epidemiology (2024). Available online at: https://www.atlasofms.org/map/global/epidemiology/number-of-people-with-ms (Accessed June 10, 2024).
2. Oreja-Guevara C, Kobelt G, Berg J, Capsa D, Eriksson J. New insights into the burden and costs of multiple sclerosis in Europe: Results for Spain. Multiple Sclerosis J. (2017) 23:166–78. doi: 10.1177/1352458517708672
3. Battaglia MA, Bezzini D, Cecchini I, Cordioli C, Fiorentino F, Manacorda T, et al. Patients with multiple sclerosis: a burden and cost of illness study. J Neurol. (2022) 269:5127–35. doi: 10.1007/s00415-022-11169-w
4. Oreja-Guevara C, Meca-Lallana JE, Díaz-Díaz J, Ara J-R, Hernández Pérez MÁ, Gracia Gil J, et al. Clinical characteristics and impact on patient-reported outcomes and quality of life of people with ambulatory secondary progressive multiple sclerosis: DISCOVER study. Multiple Sclerosis Related Disord. (2024) 90:105787. doi: 10.1016/j.msard.2024.105787
5. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. (1996) 46:907–11. doi: 10.1212/wnl.46.4.907
6. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. (2014) 83:278–86. doi: 10.1212/wnl.0000000000000560
7. Korsukewitz C, Wiendl H. Emerging trends and challenges in multiple sclerosis in Europe: rethinking classification and addressing COVID-19 impact. Lancet Regional Health Europe. (2024) 44. doi: 10.1016/j.lanepe.2024.101017
8. Cree BAC, Arnold DL, Chataway J, Chitnis T, Fox RJ, Pozo Ramajo A, et al. Secondary progressive multiple sclerosis: new insights. Neurology. (2021) 97:378–88. doi: 10.1212/WNL.0000000000012323
9. Kuhlmann T, Moccia M, Coetzee T, Cohen JA, Correale J, Graves J, et al. Multiple sclerosis progression: time for a new mechanism-driven framework. Lancet Neurol. (2023) 22:78–88. doi: 10.1016/s1474-4422(22)00289-7
10. Scalfari A, Neuhaus A, Daumer M, Muraro PA, Ebers GC. Onset of secondary progressive phase and long-term evolution of multiple sclerosis. J Neurol Neurosurg Psychiatry. (2014) 85:67–75. doi: 10.1136/jnnp-2012-304333
11. Barzegar M, Najdaghi S, Afshari-Safavi A, Nehzat N, Mirmosayyeb O, Shaygannejad V. Early predictors of conversion to secondary progressive multiple sclerosis. Multiple Sclerosis Related Disord. (2021) 54:103115. doi: 10.1016/j.msard.2021.103115
12. Tedeholm H, Piehl F, Lycke J, Link J, Stawiarz L, Burman J, et al. Effectiveness of first generation disease-modifying therapy to prevent conversion to secondary progressive multiple sclerosis. Mult Scler Relat Disord. (2022) 68:104220. doi: 10.1016/j.msard.2022.104220
13. Cree BA, Gourraud PA, Oksenberg JR, Bevan C, Crabtree-Hartman E, Gelfand JM, et al. Long-term evolution of multiple sclerosis disability in the treatment era. Ann Neurol. (2016) 80:499–510. doi: 10.1002/ana.24747
14. Leray E, Yaouanq J, Le Page E, Coustans M, Laplaud D, Oger J, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. (2010) 133:1900–13. doi: 10.1093/brain/awq076
15. Forsberg L, Stahmann A, Middleton R, Ellenberger D, Rodgers J, Nicholas R, et al. Comparison of the proportions of Secondary Progressive Multiple Sclerosis between three registries within the SPMS Research Collaboration Network (3977). Neurology. (2020) 94:3977. doi: 10.1212/WNL.94.15_supplement.3977
16. Vasanthaprasad V, Khurana V, Vadapalle S, Palace J, Adlard N. Systematic literature review and meta-analysis of the prevalence of secondary progressive multiple sclerosis in the USA, Europe, Canada, Australia, and Brazil. BMC Neurol. (2022) 22:301. doi: 10.1186/s12883-022-02820-0
17. Sharmin S, Roos I, Simpson-Yap S, Malpas C, Sánchez MM, Ozakbas S, et al. The risk of secondary progressive multiple sclerosis is geographically determined but modifiable. Brain. (2023) 146:4633–44. doi: 10.1093/brain/awad218
18. Katz Sand I, Krieger S, Farrell C, Miller AE. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult Scler. (2014) 20:1654–7. doi: 10.1177/1352458514521517
19. Ciron J, Gueguen A, Al Khedr A, Bourre B, Clavelou P, Defer G, et al. Secondary progressive multiple sclerosis: A national consensus paper on diagnostic criteria. Rev Neurol. (2022) 178:1098–104. doi: 10.1016/j.neurol.2022.07.004
20. Meca-Lallana JE, Casanova B, Rodríguez-Antigüedad A, Eichau S, Izquierdo G, Durán C, et al. Consensus on early detection of disease progression in patients with multiple sclerosis. Front Neurol. (2022) 13:931014. doi: 10.3389/fneur.2022.931014
21. Ontaneda D, Chitnis T, Rammohan K, Obeidat AZ. Identification and management of subclinical disease activity in early multiple sclerosis: a review. J Neurol. (2024) 271:1497–514. doi: 10.1007/s00415-023-12021-5
22. Lorscheider J, Buzzard K, Jokubaitis V, Spelman T, Havrdova E, Horakova D, et al. Defining secondary progressive multiple sclerosis. Brain. (2016) 139:2395–405. doi: 10.1093/brain/aww173
23. Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomized, phase 3 study. Lancet. (2018) 391:1263–73. doi: 10.1016/S0140-6736(18)30475-6
24. Kapoor R, Ho PR, Campbell N, Chang I, Deykin A, Forrestal F, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomized, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. (2018) 17:405–15. doi: 10.1016/s1474-4422(18)30069-3
25. Fox RJ, Bar-Or A, Traboulsee A, Oreja-Guevara C, Giovannoni G, Vermersch P. Efficacy and safety of tolebrutinib versus placebo in non-relapsing secondary progressive multiple sclerosis: results from the Phase 3 HERCULES Trial. Multiple Sclerosis and Related Disorders. (2024) 92.
26. Cohen M, Bresch S, Thommel Rocchi O, Morain E, Benoit J, Levraut M, et al. Should we still only rely on EDSS to evaluate disability in multiple sclerosis patients? A study of inter and intra rater reliability. Multiple Sclerosis Related Disord. (2021) 54:103144. doi: 10.1016/j.msard.2021.103144
27. Planche V, Gibelin M, Cregut D, Pereira B, Clavelou P. Cognitive impairment in a population-based study of patients with multiple sclerosis: differences between late relapsing-remitting, secondary progressive and primary progressive multiple sclerosis. Eur J Neurol. (2016) 23:282–9. doi: 10.1111/ene.12715
28. Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. (1997) 120:393–9. doi: 10.1093/brain/120.3.393
29. Kappos L, Wolinsky JS, Giovannoni G, Arnold DL, Wang Q, Bernasconi C, et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. (2020) 77:1132–40. doi: 10.1001/jamaneurol.2020.1568
30. Bonzano L, Bove M, Sormani MP, Stromillo ML, Giorgio A, Amato MP, et al. Subclinical motor impairment assessed with an engineered glove correlates with magnetic resonance imaging tissue damage in radiologically isolated syndrome. Eur J Neurol. (2019) 26:162–7. doi: 10.1111/ene.13789
31. Eshaghi A, Marinescu RV, Young AL, Firth NC, Prados F, Jorge Cardoso M, et al. Progression of regional grey matter atrophy in multiple sclerosis. Brain. (2018) 141:1665–77. doi: 10.1093/brain/awy088
32. Cagol A, Benkert P, Melie-Garcia L, Schaedelin SA, Leber S, Tsagkas C, et al. Association of spinal cord atrophy and brain paramagnetic rim lesions with progression independent of relapse activity in people with MS. Neurology. (2024) 102:e207768. doi: 10.1212/WNL.0000000000207768
33. Haider L, Zrzavy T, Hametner S, Höftberger R, Bagnato F, Grabner G, et al. The topography of demyelination and neurodegeneration in the multiple sclerosis brain. Brain. (2016) 139:807–15. doi: 10.1093/brain/awv398
34. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. (2009) 132:1175–89. doi: 10.1093/brain/awp070
35. Giovannoni G, Popescu V, Wuerfel J, Hellwig K, Iacobaeus E, Jensen MB, et al. Smoldering multiple sclerosis: the ‘real MS’. Ther Adv Neurol Disord. (2022) 15:17562864211066751. doi: 10.1177/17562864211066751
36. Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol. (2022) 18:40–55. doi: 10.1038/s41582-021-00581-x
37. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol. (2019) 9:3116. doi: 10.3389/fimmu.2018.03116
38. Antel J, Antel S, Caramanos Z, Arnold DL, Kuhlmann T. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol. (2012) 123:627–38. doi: 10.1007/s00401-012-0953-0
39. Scalfari A, Traboulsee A, Oh J, Airas L, Bittner S, Calabrese M, et al. Smoldering-associated worsening in multiple sclerosis: an international consensus statement on definition, biology, clinical implications, and future directions. Ann Neurol. (2024) 96(5):826–45. doi: 10.1002/ana.27034
40. Popescu BF, Frischer JM, Webb SM, Tham M, Adiele RC, Robinson CA, et al. Pathogenic implications of distinct patterns of iron and zinc in chronic MS lesions. Acta Neuropathol. (2017) 134:45–64. doi: 10.1007/s00401-017-1696-8
41. Khan Z, Mehan S, Gupta GD, Narula AS. Immune system dysregulation in the progression of multiple sclerosis: molecular insights and therapeutic implications. Neuroscience. (2024) 548:9–26. doi: 10.1016/j.neuroscience.2024.04.004
42. Thakolwiboon S, Mills EA, Yang J, Doty J, Belkin MI, Cho T, et al. Immunosenescence and multiple sclerosis: inflammaging for prognosis and therapeutic consideration. Front Aging. (2023) 4:1234572. doi: 10.3389/fragi.2023.1234572
43. Perdaens O, van Pesch V. Molecular mechanisms of immunosenescene and inflammaging: relevance to the immunopathogenesis and treatment of multiple sclerosis. Front Neurol. (2022) 12:811518. doi: 10.3389/fneur.2021.811518
44. Kular L, Klose D, Urdánoz-Casado A, Ewing E, Planell N, Gomez-Cabrero D, et al. Epigenetic clock indicates accelerated aging in glial cells of progressive multiple sclerosis patients. Front Aging Neurosci. (2022) 14:926468. doi: 10.3389/fnagi.2022.926468
45. Sumowski JF, Rocca MA, Leavitt VM, Riccitelli G, Comi G, DeLuca J, et al. Brain reserve and cognitive reserve in multiple sclerosis: what you’ve got and how you use it. Neurology. (2013) 80:2186–93. doi: 10.1212/WNL.0b013e318296e98b
46. Vollmer TL, Nair KV, Williams IM, Alvarez E. Multiple sclerosis phenotypes as a continuum. Neurol Clin Pract. (2021) 11:342–51. doi: 10.1212/CPJ.0000000000001045
47. Chard DT, Alahmadi AAS, Audoin B, Charalambous T, Enzinger C, Hulst HE, et al. Mind the gap: from neurons to networks to outcomes in multiple sclerosis. Nat Rev Neurol. (2021) 17:173–84. doi: 10.1038/s41582-020-00439-8
48. Hecker M, Bühring J, Fitzner B, Rommer PS, Zettl UK. Genetic, environmental and lifestyle determinants of accelerated telomere attrition as contributors to risk and severity of multiple sclerosis. Biomolecules. (2021) 11(10):1510. doi: 10.3390/biom11101510
49. Harroud A, Stridh P, McCauley JL, Saarela J, van den Bosch AMR, Engelenburg HJ, et al. Locus for severity implicates CNS resilience in progression of multiple sclerosis. Nature. (2023) 619:323–31. doi: 10.1038/s41586-023-06250-x
50. Zhou X, Baumann R, Gao X, Mendoza M, Singh S, Katz Sand I, et al. Gut microbiome of multiple sclerosis patients and paired household healthy controls reveal associations with disease risk and course. Cell. (2022) 185:3467–3486.e16. doi: 10.1016/j.cell.2022.08.021
51. Lublin FD, Häring DA, Ganjgahi H, Ocampo A, Hatami F, Čuklina J, et al. How patients with multiple sclerosis acquire disability. Brain. (2022) 145:3147–61. doi: 10.1093/brain/awac016
52. Cree BAC, Hollenbach JA, Bove R, Kirkish G, Sacco S, Caverzasi E, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. (2019) 85:653–66. doi: 10.1002/ana.25463
53. Tur C, Carbonell-Mirabent P, Cobo-Calvo Á, Otero-Romero S, Arrambide G, Midaglia L, et al. Association of early progression independent of relapse activity with long-term disability after a first demyelinating event in multiple sclerosis. JAMA Neurol. (2023) 80:151–60. doi: 10.1001/jamaneurol.2022.4655
54. Pitt D, Lo CH, Gauthier SA, Hickman RA, Longbrake E, Airas LM, et al. Toward precision phenotyping of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2022) 9(6):e200025. doi: 10.1212/nxi.0000000000200025
55. Eshaghi A, Young AL, Wijeratne PA, Prados F, Arnold DL, Narayanan S, et al. Identifying multiple sclerosis subtypes using unsupervised machine learning and MRI data. Nat Commun. (2021) 12:2078. doi: 10.1038/s41467-021-22265-2
56. Gross CC, Schulte-Mecklenbeck A, Steinberg OV, Wirth T, Lauks S, Bittner S, et al. Multiple sclerosis endophenotypes identified by high-dimensional blood signatures are associated with distinct disease trajectories. Sci Trans Med. (2024) 16:eade8560. doi: 10.1126/scitranslmed.ade8560
57. Ciccarelli O, Barkhof F, Calabrese M, De Stefano N, Eshaghi A, Filippi M, et al. Using the progression independent of relapse activity framework to unveil the pathobiological foundations of multiple sclerosis. Neurology. (2024) 103:e209444. doi: 10.1212/WNL.0000000000209444
58. Kim S, Lee EK, Song CJ, Sohn E. Iron rim lesions as a specific and prognostic biomarker of multiple sclerosis: 3T-based susceptibility-weighted imaging. Diagnostics. (2023) 13:1866. doi: 10.3390/diagnostics13111866
59. Kolb H, Al-Louzi O, Beck ES, Sati P, Absinta M, Reich DS. From pathology to MRI and back: Clinically relevant biomarkers of multiple sclerosis lesions. NeuroImage: Clin. (2022) 36:103194. doi: 10.1016/j.nicl.2022.103194
60. Bagnato F, Sati P, Hemond CC, Elliott C, Gauthier SA, Harrison DM, et al. Imaging chronic active lesions in multiple sclerosis: a consensus statement. Brain. (2024) 147(9):2913–33. doi: 10.1093/brain/awae013
61. Beynon V, George IC, Elliott C, Arnold DL, Ke J, Chen H, et al. Chronic lesion activity and disability progression in secondary progressive multiple sclerosis. BMJ Neurol Open. (2022) 4:e000240. doi: 10.1136/bmjno-2021-000240
62. Absinta M, Dal-Bianco A. Slowly expanding lesions are a marker of progressive MS – Yes. Multiple Sclerosis J. (2021) 27:1679–81. doi: 10.1177/13524585211013748
63. Ghione E, Bergsland N, Dwyer MG, Hagemeier J, Jakimovski D, Paunkoski I, et al. Brain atrophy is associated with disability progression in patients with MS followed in a clinical routine. AJNR Am J Neuroradiol. (2018) 39:2237–42. doi: 10.3174/ajnr.A5876
64. Bischof A, Papinutto N, Keshavan A, Rajesh A, Kirkish G, Zhang X, et al. Spinal cord atrophy predicts progressive disease in relapsing multiple sclerosis. Ann Neurol. (2022) 91:268–81. doi: 10.1002/ana.26281
65. Krajnc N, Hofer L, Föttinger F, Dal-Bianco A, Leutmezer F, Kornek B, et al. Paramagnetic rim lesions are associated with inner retinal layer thinning and progression independent of relapse activity in multiple sclerosis. Eur J Neurol. (2025) 32:e16529. doi: 10.1111/ene.16529
66. Preziosa P, Pagani E, Meani A, Moiola L, Rodegher M, Filippi M, et al. Slowly expanding lesions predict 9-year multiple sclerosis disease progression. Neurol Neuroimmunol Neuroinflamm. (2022) 9(2):e1139. doi: 10.1212/nxi.0000000000001139
67. Calvi A, Clarke MA, Prados F, Chard D, Ciccarelli O, Alberich M, et al. Relationship between paramagnetic rim lesions and slowly expanding lesions in multiple sclerosis. Mult Scler. (2023) 29:352–62. doi: 10.1177/13524585221141964
68. Polvinen E, Matilainen M, Nylund M, Sucksdorff M, Airas LM. TSPO-detectable chronic active lesions predict disease progression in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200133. doi: 10.1212/NXI.0000000000200133
69. Zhang S, Nguyen TD, Hurtado Rúa SM, Kaunzner UW, Pandya S, Kovanlikaya I, et al. Quantitative susceptibility mapping of time-dependent susceptibility changes in multiple sclerosis lesions. AJNR Am J Neuroradiol. (2019) 40:987–93. doi: 10.3174/ajnr.A6071
70. Clarke MA, Pareto D, Pessini-Ferreira L, Arrambide G, Alberich M, Crescenzo F, et al. Value of 3T susceptibility-weighted imaging in the diagnosis of multiple sclerosis. AJNR Am J Neuroradiol. (2020) 41:1001–8. doi: 10.3174/ajnr.A6547
71. Voon CC, Wiltgen T, Wiestler B, Schlaeger S, Mühlau M. Quantitative susceptibility mapping in multiple sclerosis: A systematic review and meta-analysis. NeuroImage Clin. (2024) 42:25. doi: 10.1016/j.nicl.2024.103598
72. Deh K, Nguyen TD, Eskreis-Winkler S, Prince MR, Spincemaille P, Gauthier S, et al. Reproducibility of quantitative susceptibility mapping in the brain at two field strengths from two vendors. J Magn Reson Imaging. (2015) 42:1592–600. doi: 10.1002/jmri.24943
73. Klistorner A, Wang C, Yiannikas C, Parratt J, Dwyer M, Barton J, et al. Evidence of progressive tissue loss in the core of chronic MS lesions: A longitudinal DTI study. NeuroImage Clin. (2018) 17:1028–35. doi: 10.1016/j.nicl.2017.12.010
74. Naval-Baudin P, Pons-Escoda A, Castillo-Pinar A, Martínez-Zalacaín I, Arroyo-Pereiro P, Flores-Casaperalta S, et al. The T1-dark-rim: A novel imaging sign for detecting smoldering inflammation in multiple sclerosis. Eur J Radiol. (2024) 173:111358. doi: 10.1016/j.ejrad.2024.111358
75. El Ayoubi NK, Sabbagh HM, Bou Rjeily N, Hannoun S, Khoury SJ. Rate of retinal layer thinning as a biomarker for conversion to progressive disease in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2022) 9(6):e200030. doi: 10.1212/nxi.0000000000200030
76. Bsteh G, Berek K, Hegen H, Altmann P, Wurth S, Auer M, et al. Macular ganglion cell-inner plexiform layer thinning as a biomarker of disability progression in relapsing multiple sclerosis. Mult Scler. (2021) 27:684–94. doi: 10.1177/1352458520935724
77. Pietroboni AM, Dell’Arti L, Caprioli M, Scarioni M, Carandini T, Arighi A, et al. The loss of macular ganglion cells begins from the early stages of disease and correlates with brain atrophy in multiple sclerosis patients. Multiple Sclerosis J. (2019) 25:31–8. doi: 10.1177/1352458517740214
78. Bsteh G, Hegen H, Altmann P, Auer M, Berek K, Pauli FD, et al. Retinal layer thinning is reflecting disability progression independent of relapse activity in multiple sclerosis. Mult Scler J Exp Transl Clin. (2020) 6:2055217320966344. doi: 10.1177/2055217320966344
79. Leppert D, Kropshofer H, Häring DA, Dahlke F, Patil A, Meinert R, et al. Blood neurofilament light in progressive multiple sclerosis: post hoc analysis of 2 randomized controlled trials. Neurology. (2022) 98:e2120–31. doi: 10.1212/wnl.0000000000200258
80. Thebault S, Abdoli M, Fereshtehnejad SM, Tessier D, Tabard-Cossa V, Freedman MS. Serum neurofilament light chain predicts long term clinical outcomes in multiple sclerosis. Sci Rep. (2020) 10:10381. doi: 10.1038/s41598-020-67504-6
81. Comabella M, Sastre-Garriga J, Carbonell-Mirabent P, Fissolo N, Tur C, Malhotra S, et al. Serum neurofilament light chain levels predict long-term disability progression in patients with progressive multiple sclerosis. J Neurol Neurosurg Psychiatry. (2022), jnnp-2022-329020. doi: 10.1136/jnnp-2022-329020
82. Barro C, Healy BC, Liu Y, Saxena S, Paul A, Polgar-Turcsanyi M, et al. and nfL levels differentiate subsequent progression and disease activity in patients with progressive multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2023) 10(1):e200052. doi: 10.1212/nxi.0000000000200052
83. Högel H, Rissanen E, Barro C, Matilainen M, Nylund M, Kuhle J, et al. Serum glial fibrillary acidic protein correlates with multiple sclerosis disease severity. Mult Scler. (2020) 26:210–9. doi: 10.1177/1352458518819380
84. Abdelhak A, Huss A, Kassubek J, Tumani H, Otto M. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci Rep. (2018) 8:14798. doi: 10.1038/s41598-018-33158-8
85. Cross AH, Gelfand JM, Thebault S, Bennett JL, von Büdingen HC, Cameron B, et al. Emerging cerebrospinal fluid biomarkers of disease activity and progression in multiple sclerosis. JAMA Neurol. (2024) 81:373–83. doi: 10.1001/jamaneurol.2024.0017
86. Meier S, Willemse EAJ, Schaedelin S, Oechtering J, Lorscheider J, Melie-Garcia L, et al. Serum glial fibrillary acidic protein compared with neurofilament light chain as a biomarker for disease progression in multiple sclerosis. JAMA Neurol. (2023) 80:287–97. doi: 10.1001/jamaneurol.2022.5250
87. Abdelhak A, Hottenrott T, Morenas-Rodríguez E, Suárez-Calvet M, Zettl UK, Haass C, et al. Glial activation markers in CSF and serum from patients with primary progressive multiple sclerosis: potential of serum GFAP as disease severity marker? Front Neurol. (2019) 10:280. doi: 10.3389/fneur.2019.00280
88. Jiang X, Shen C, Teunissen CE, Wessels M, Zetterberg H, Giovannoni G, et al. Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation. Mult Scler. (2023) 29:1070–9. doi: 10.1177/13524585231176732
89. Häring DA, Kropshofer H, Kappos L, Cohen JA, Shah A, Meinert R, et al. Long-term prognostic value of longitudinal measurements of blood neurofilament levels. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e856. doi: 10.1212/NXI.0000000000000856
90. Kartau M, Melkas S, Kartau J, Arola A, Laakso H, Pitkänen J, et al. Neurofilament light level correlates with brain atrophy, and cognitive and motor performance. Front Aging Neurosci. (2022) 14:939155. doi: 10.3389/fnagi.2022.939155
91. Barro C, Benkert P, Disanto G, Tsagkas C, Amann M, Naegelin Y, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain. (2018) 141:2382–91. doi: 10.1093/brain/awy154
92. Kuhle J. (2023)., in: New promising biomarkers: GFAP, IgM, complement … In: The 9th Joint ECTRIMS-ACTRIMS Meeting 2023, Milan, Italy.
93. Benkert P. (2023)., in: Serum glial fibrillary acidic protein (GFAP) is a longitudinal indicator of disease progression in MS while neurofilament light chain (NfL) associates with therapy response in patients under B-cell depleting therapy. I. The 9th Joint ECTRIMS-ACTRIMS Meeting, Milan, Italy.
94. Kuhle J, Kropshofer H, Haering DA, Kundu U, Meinert R, Barro C, et al. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology. (2019) 92:e1007–15. doi: 10.1212/wnl.0000000000007032
95. Sainz-Amo R, Rodero Romero A, Monreal E, Chico García JL, Fernández Velasco JI, Villarrubia N, et al. Effect of alemtuzumab over sNfL and sGFAP levels in multiple sclerosis. Front Immunol. (2024) 15:1454474. doi: 10.3389/fimmu.2024.1454474
96. Monreal E, Fernández-Velasco JI, Álvarez-Lafuente R, Sainz de la Maza S, García-Sánchez MI, Llufriu S, et al. Serum biomarkers at disease onset for personalized therapy in multiple sclerosis. Brain. (2024) 147:4084–93. doi: 10.1093/brain/awae260
97. Krajnc N, Bsteh G, Berger T. Clinical and paraclinical biomarkers and the hitches to assess conversion to secondary progressive multiple sclerosis: A systematic review. Front Neurol. (2021) 12:666868. doi: 10.3389/fneur.2021.666868
98. European Medicines Agency. Mayzent (siponimod) Summary of Product Characteristics (2019). Available online at: https://www.ema.europa.eu/en/documents/product-information/mayzent-epar-product-information_en.pdf (Accessed June 12, 2024).
99. European Medicines Agency. Betaferon (interferon-beta) Summary of Product Characteristics (2007).
100. Bossart J, Kamm CP, Kaufmann M, Stanikić M, Puhan MA, Kesselring J, et al. Real-world disease-modifying therapy usage in persons with relapsing-remitting multiple sclerosis: Cross-sectional data from the Swiss Multiple Sclerosis Registry. Multiple Sclerosis Related Disord. (2022) 60:103706. doi: 10.1016/j.msard.2022.103706
101. Papukchieva S, Stratil AS, Kahn M, Neß NH, Hollnagel-Schmitz M, Gerencser V, et al. Shifting from the treat-to-target to the early highly effective treatment approach in patients with multiple sclerosis - real-world evidence from Germany. Ther Adv Neurol Disord. (2024) 17:17562864241237857. doi: 10.1177/17562864241237857
102. Berkovich R. Clinical and MRI outcomes after stopping or switching disease-modifying therapy in stable MS patients: a case series report. Multiple Sclerosis Related Disord. (2017) 17:123–7. doi: 10.1016/j.msard.2017.07.007
103. Gisela Z, Carla P, Josefina B, Tomas I, Lucia B, Pappolla A, et al. Disease activity after discontinuation of disease-modifying therapies in patients with multiple sclerosis in Argentina: data from the nationwide registry RelevarEM. Neurol Res. (2023) 45:112–7. doi: 10.1080/01616412.2022.2124792
104. Jouvenot G, Courbon G, Lefort M, Rollot F, Casey R, Le Page E, et al. High-efficacy therapy discontinuation vs continuation in patients 50 years and older with nonactive MS. JAMA Neurol. (2024) 81(5):490–8. doi: 10.1001/jamaneurol.2024.0395
105. Fagius J, Feresiadou A, Larsson E-M, Burman J. Discontinuation of disease modifying treatments in middle aged multiple sclerosis patients. First line drugs vs natalizumab. Multiple Sclerosis Related Disord. (2017) 12:82–7. doi: 10.1016/j.msard.2017.01.009
106. Giovannoni G, Marta M, Davis A, Turner B, Gnanapavan S, Schmierer K. Switching patients at high risk of PML from natalizumab to another disease-modifying therapy. Pract Neurol. (2016) 16:389–93. doi: 10.1136/practneurol-2015-001355
107. European Medicines Agency. Kesimpta® EPAR assessment report (2021). Available online at: https://www.ema.europa.eu/en/documents/assessment-report/kesimpta-epar-public-assessment-report_en.pdf (Accessed June 12, 2024).
108. European Medicines Agency. Zeposia® EPAR assessment report (2020). Available online at: https://www.ema.europa.eu/en/documents/assessment-report/zeposia-epar-public-assessment-report_en.pdf (Accessed June 14, 2024).
109. Giovannoni G, Houchen E, Sobisek L, Karu H, Ryan S, Jones E, et al. MRI activity versus relapses as markers of disease activity in SPMS: data from adelphi real-world MS disease specific program and the phase 3 EXPAND study (S14.010). Neurology. (2022) 98:3783. doi: 10.1212/WNL.98.18_supplement.3783
110. Frahm N, Ellenberger D, Fneish F, Christoph K, Warnke C, Zettl UK, et al. Characteristics of secondary progressive multiple sclerosis: Disease activity and provision of care in Germany – A registry-based/multicentric cohort study. Multiple Sclerosis Related Disord. (2021) 56:103281. doi: 10.1016/j.msard.2021.103281
111. Duddy M, Lee M, Pearson O, Nikfekr E, Chaudhuri A, Percival F, et al. The UK patient experience of relapse in Multiple Sclerosis treated with first disease modifying therapies. Mult Scler Relat Disord. (2014) 3:450–6. doi: 10.1016/j.msard.2014.02.006
112. Cameron E, Rog D, McDonnell G, Overell J, Pearson O, French DP. Factors influencing multiple sclerosis disease-modifying treatment prescribing decisions in the United Kingdom: A qualitative interview study. Mult Scler Relat Disord. (2019) 27:378–82. doi: 10.1016/j.msard.2018.11.023
113. Wattjes MP, Ciccarelli O, Reich DS, Banwell B, de Stefano N, Enzinger C, et al. MAGNIMS&x2013;CMSC&x2013;NAIMS consensus recommendations on the use of MRI in patients with multiple sclerosis. Lancet Neurol. (2021) 20:653–70. doi: 10.1016/S1474-4422(21)00095-8
114. Kappos L. Placebo-controlled multicenter randomized trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. Lancet. (1998) 352:1491–7. doi: 10.1016/S0140-6736(98)10039-9
115. PRISMS Study Group. Randomized double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. (1998) 352:1498–504.
116. Panitch H, Miller A, Paty D, Weinshenker B. Interferon beta-1b in secondary progressive MS: results from a 3-year controlled study. Neurology. (2004) 63:1788–95. doi: 10.1212/01.wnl.0000146958.77317.3e
117. SPECTRIMS Study Group. Randomized controlled trial of interferon- beta-1a in secondary progressive MS: Clinical results. Neurology. (2001) 56:1496–504. doi: 10.1212/wnl.56.11.1496
118. European Medicines Agency. Rebif (interferon-beta-1a) Summary of Product Characteristics (2009).
119. Group* SPECToRI-b-aiMS. Randomized controlled trial of interferon- beta-1a in secondary progressive MS. Neurology. (2001) 56:1496–504. doi: 10.1212/WNL.56.11.1496
120. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. (2017) 376:221–34. doi: 10.1056/NEJMoa1601277
121. Hauser SL, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. (2020) 383:546–57. doi: 10.1056/NEJMoa1917246
122. Kappos L, Fox RJ, Burcklen M, Freedman MS, Havrdová EK, Hennessy B, et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study: A randomized clinical trial. JAMA Neurol. (2021) 78:558–67. doi: 10.1001/jamaneurol.2021.0405
123. Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sørensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. (2010) 362:416–26. doi: 10.1056/NEJMoa0902533
124. Arnold DL, Kappos L, Vermersch P, Gold R, Bar-Or A, Giovannoni G, et al. Long-term effect of siponimod on MRI outcomes in SPMS: analyses from the EXPAND study up to 5 years (2217). Neurology. (2021) 96:2217. doi: 10.1212/WNL.96.15_supplement.2217
125. Gold R, Piani-Meier D, Kappos L, Bar-Or A, Vermersch P, Giovannoni G, et al. Siponimod vs placebo in active secondary progressive multiple sclerosis: a post hoc analysis from the phase 3 EXPAND study. J Neurol. (2022) 269:5093–104. doi: 10.1007/s00415-022-11166-z
126. Vermersch P, Brieva-Ruiz L, Fox RJ, Paul F, Ramio-Torrenta L, Schwab M, et al. Efficacy and safety of masitinib in progressive forms of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1148. doi: 10.1212/NXI.0000000000001148
127. Roche. A Study to Evaluate the Efficacy and Safety of Fenebrutinib Compared With Ocrelizumab in Adult Participants With Primary Progressive Multiple Sclerosis (FENtrepid) [Internet]. ClinicalTrials.gov identifier: NCT04544449. Available online at: https://clinicaltrials.gov/ct2/show/NCT04544449 (Accessed June 14, 2024).
128. Chataway J, Williams T, Li V, Marrie RA, Ontaneda D, Fox RJ. Clinical trials for progressive multiple sclerosis: progress, new lessons learned, and remaining challenges. Lancet Neurol. (2024) 23:277–301. doi: 10.1016/S1474-4422(24)00027-9
129. Prosperini L, Haggiag S, Ruggieri S, Tortorella C, Gasperini C. Stopping disease-modifying treatments in multiple sclerosis: A systematic review and meta-analysis of real-world studies. CNS Drugs. (2023) 37:915–27. doi: 10.1007/s40263-023-01038-z
130. Corboy JR, Fox RJ, Kister I, Cutter GR, Morgan CJ, Seale R, et al. Risk of new disease activity in patients with multiple sclerosis who continue or discontinue disease-modifying therapies (DISCOMS): a multicenter, randomized, single-blind, phase 4, non-inferiority trial. Lancet Neurol. (2023) 22:568–77. doi: 10.1016/S1474-4422(23)00154-0
131. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. (2017) 8:577. doi: 10.3389/fneur.2017.00577
132. Zhang Y, Gonzalez Caldito N, Shirani A, Salter A, Cutter G, Culpepper W 2nd, et al. Aging and efficacy of disease-modifying therapies in multiple sclerosis: a meta-analysis of clinical trials. Ther Adv Neurol Disord. (2020) 13:1756286420969016. doi: 10.1177/1756286420969016
133. Coerver E, Fung WH, De Beukelaar J, Bouvy W, Canta L, Gerlach O, et al. (2023)., in: Discontinuation of first-line disease-modifying therapy in stable multiple sclerosis (DOT-MS): an early terminated multicenter randomized controlled trial. 9th Joint ECTRIMS-ACTRIMS Meeting, Milan, Italy.
134. Dennison L, Brown M, Kirby S, Galea I. Do people with multiple sclerosis want to know their prognosis? A UK nationwide study. PloS One. (2018) 13:e0193407. doi: 10.1371/journal.pone.0193407
135. Lomer NB, Asalemi KA, Saberi A, Sarlak K. Predictors of multiple sclerosis progression: A systematic review of conventional magnetic resonance imaging studies. PloS One. (2024) 19:e0300415. doi: 10.1371/journal.pone.0300415
136. Pinto MF, Oliveira H, Batista S, Cruz L, Pinto M, Correia I, et al. Prediction of disease progression and outcomes in multiple sclerosis with machine learning. Sci Rep. (2020) 10:21038. doi: 10.1038/s41598-020-78212-6
137. Andorra M, Freire A, Zubizarreta I, de Rosbo NK, Bos SD, Rinas M, et al. Predicting disease severity in multiple sclerosis using multimodal data and machine learning. J Neurol. (2024) 271:1133–49. doi: 10.1007/s00415-023-12132-z
138. Shosha E, Burton JM. Discussing the potential for progression with patients newly diagnosed with multiple sclerosis: When, how, and why? Multiple Sclerosis Related Disord. (2022) 68:104230. doi: 10.1016/j.msard.2022.104230
139. Berhanu D, Leal Rato M, Canhoto AI, Vieira da Cunha J, Geraldes R. Online information search by people with Multiple Sclerosis: A systematic review. Multiple Sclerosis Related Disord. (2023) 79:105032. doi: 10.1016/j.msard.2023.105032
140. Shah NH, Entwistle D, Pfeffer MA. Creation and adoption of large language models in medicine. JAMA. (2023) 330:866–9. doi: 10.1001/jama.2023.14217
141. Della Rosa S, Sen F. Health topics on facebook groups: content analysis of posts in multiple sclerosis communities. Interact J Med Res. (2019) 8:e10146. doi: 10.2196/10146
142. Maida E, Moccia M, Palladino R, Borriello G, Affinito G, Clerico M, et al. ChatGPT vs. neurologists: a cross-sectional study investigating preference, satisfaction ratings and perceived empathy in responses among people living with multiple sclerosis. J Neurol. (2024) 271:4057–66. doi: 10.1007/s00415-024-12328-x
143. Stanikić M, Gille F, Schlomberg J, Daniore P, Kägi S, Chan A, et al. Exploring the relationship between neurologists and older persons with multiple sclerosis through the lens of social support theory. Multiple Sclerosis J Experimental Trans Clin. (2024) 10:20552173241281458. doi: 10.1177/20552173241281458
144. Ben-Zacharia AB, Lee JM, Kahle JS, Lord B. Shared decision-making in multiple sclerosis physical symptomatic care: a systematic review. Ther Adv Chronic Dis. (2023) 14:20406223231172920. doi: 10.1177/20406223231172920
145. Alonso R, Carnero Contentti E, Graña M, Linares R, Lopez P, Mainella C, et al. Shared decision making in the treatment of multiple sclerosis: A consensus based on Delphi methodology. Multiple Sclerosis Related Disord. (2023) 70:104465. doi: 10.1016/j.msard.2022.104465
146. Messina MJ, Dalla Costa G, Rodegher M, Moiola L, Colombo B, Comi G, et al. The communication of multiple sclerosis diagnosis: the patients’ Perspective. Mult Scler Int. (2015) 2015:353828. doi: 10.1155/2015/353828
147. Højsgaard Chow H, Schreiber K, Magyari M, Ammitzbøll C, Börnsen L, Romme Christensen J, et al. Progressive multiple sclerosis, cognitive function, and quality of life. Brain Behav. (2018) 8:e00875. doi: 10.1002/brb3.875
Keywords: multiple sclerosis, secondary progressive multiple sclerosis, disease activity, silent progression, smouldering disease, multiple sclerosis treatment, disease-modifying treatments
Citation: Brieva L, Calles C, Landete L and Oreja-Guevara C (2025) Current challenges in secondary progressive multiple sclerosis: diagnosis, activity detection and treatment. Front. Immunol. 16:1543649. doi: 10.3389/fimmu.2025.1543649
Received: 11 December 2024; Accepted: 25 February 2025;
Published: 21 March 2025.
Edited by:
Francesco Patti, University of Catania, ItalyReviewed by:
Reza Rahmanzadeh, TheUltra.ai, SwitzerlandEslam Shosha, McMaster University, Canada
Paolo Preziosa, Vita-Salute San Raffaele University, Italy
Copyright © 2025 Brieva, Calles, Landete and Oreja-Guevara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celia Oreja-Guevara, orejacbn@gmail.com
†These authors have contributed equally to this work
‡ORCID: Celia Oreja-Guevara, orcid.org/0000-0002-9221-5716
Luis Brieva, orcid.org/0000-0002-5871-6189