 Lexin Fang
Lexin Fang Yu Song
Yu Song Jiangtao Chen1
Jiangtao Chen1 Yueping Ding
Yueping Ding- 1Department of Intensive Care Unit, The Second Affiliated Hospital of Zhejiang Chinese Medical University, Xinhua Hospital of Zhejiang Province, Hangzhou, Zhejiang, China
- 2Department of Hepatology, The Second Affiliated Hospital of Zhejiang Chinese Medical University, Xinhua Hospital of Zhejiang Province, Hangzhou, Zhejiang, China
Sepsis is often accompanied by liver injury and is associated with an increase in the number of circulating and hepatic neutrophils. In sepsis-associated liver injury, neutrophils exhibit phenotypic heterogeneity and perform both pro- and anti-inflammatory functions. Moreover, neutrophil dysfunction and neutrophil-associated immunosuppression are also involved in the pathogenesis of sepsis. Given the complex functionality of this cell type, the aim of this review was to describe the possible mechanistic role of neutrophils in sepsis-associated liver injury, with a brief introduction to neutrophil recruitment and subsequent discussion of the potential contributions of neutrophils to different subtypes of sepsis-associated liver injury.
1 Introduction
Sepsis is the main cause of death in intensive care units (ICU). It is accompanied by multi-organ dysfunction, with sepsis-associated liver injury occurring in 34–46% of patients with sepsis (1). Sepsis-associated liver injury (SALI) can generally be classified as either hypoxic hepatitis, cholestatic, hepatocellular, or severe cholangitis (2, 3), although the precise pathogenesis remains to be elucidated. Neutrophils are first-responder cells recruited to protect host organisms from infection or sterile tissue injury, and their accumulation has been observed in the livers of model animals with sepsis (4). Neutrophils clear pathogens through a variety of processes, including phagocytosis, degranulation, reactive oxygen species (ROS) production, and neutrophil extracellular traps (NETs), which consist of nuclear DNA, histones, and proteases. However, there is evidence that neutrophils are an independent predictor of SALI (5), and excessive accumulation or dysfunction of neutrophils may induce SALI (4). Therefore, the role of neutrophils is a double-edged sword. This mini-review aimed to discuss the role of circulating neutrophils as a component of innate immunity in sepsis, with a focus on possible mechanisms through which neutrophils induce liver injury in patients with the disease.
2 Recruitment of neutrophils in sepsis
2.1 Sepsis-induced release of large numbers of neutrophils into the circulation
In infectious states, cytokines such as granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor are released and subsequently activate multiple transcriptional mechanisms that promote granulopoiesis. Ioannou et al. discovered that G-CSF exposure shortens the lifespan of mature neutrophils, causing a disproportionate shift in neutrophil populations toward an immature phenotype in septic patient plasma; these changes primarily occur in the late granulocyte or maturation phase, with potential enhancement by extracellular histones (6). The same group also reported high G-CSF levels were associated with a poor prognosis, whereas the onset of sepsis was delayed when mice were pre-treated with G-CSF 24 hours before infection. In addition, G-CSF promotes neutrophil egress from bone marrow, primarily by promoting the activation of the C-X-C motif chemokine receptor (CXCR) 4/CXCR2 axis, skewing the balance towards CXCR2 (7, 8). Given the increased proportion of immature neutrophils in the general circulation, their cell counts could be useful biomarkers of sepsis, helping rule in and rule out the possibility of the disease with a certain specificity (9). One study demonstrated a differential enrichment of neutrophil subsets in patients with sepsis (10). Using flow cytometry, they roughly grouped neutrophil subsets into mature, immature, and others based on the expression of cluster of differentiation 10 (CD10). They further identified that CD10−CD177+ immature neutrophil subset showed reduced of oxidative burst capabilities as well as phagocytosis, and CD10+ mature subset with high level of programmed death ligand 1 (PD-L1) exhibited inhibition of T-cell proliferation. Meghraoui et al. reported that CD10- CD64+CD16low/- CD123+ neutrophils and CD10-CD64+PD-L1+neutrophils could facilitate the early diagnosis of sepsis (11). Moreover, Chen et al. discovered that Ly6G+Lta4h+Sort1+ neutrophil (Neu-3) levels are closely correlated with the occurrence of SALI in a mouse model (12). In recent years, research methods such as single-cell RNA sequencing (scRNA-seq) transcriptomics and proteomics have been used to analyze neutrophil heterogeneity, enriching our understanding of these cells and facilitating the exploration of their unique characteristics in sepsis.
2.2 Neutrophil migration in circulation
The ability of neutrophils to combat infection is dependent on their ability to first undergo migration to the infectious site. The phases of migration include release from the bone marrow, migration and rolling, adherence, and transmigration (13). Neutrophil migration is influenced by the concentration gradient of chemoattractant signals, and the cells respond to these signals hierarchically, mainly through the activation of G-protein-coupled receptors (GPCRs) (14). However, neutrophil chemotaxis becomes impaired and migration may even be reversed in sepsis (15). Ciupe et al. utilized mathematical models to demonstrate that the tightly regulated migratory behavior of neutrophils toward an infectious site can be altered with different concentrations of lipopolysaccharide (LPS) (16). More specifically, neutrophils treated with ultra-low doses of LPS or those exposed to LPS for extended periods might lose their ability to move up the chemotactic gradient, whereas high-dose LPS treatment enhanced their directional migration. Bao et al. reported that priming of neutrophils with LPS somewhat prevented the onset and progression of LPS-induced sepsis in a murine model (17). Impaired neutrophil chemotaxis is also associated with the internalization or desensitization of GPCRs (18). In a clinical study investigating neutrophil surface receptors, Seree-Aphinan et al. found that only the levels of CXCR2 correlated with sepsis, that a decrease in CXCR2 expression occurred in parallel with the peak of infectious activity and that this change could be used to differentiate sepsis from systemic inflammatory responses (19). In addition to neutrophil surface receptors, a large proportion of immotile neutrophils and high neutrophil mobility could each serve as an independent predictor of sepsis in patients with cirrhosis (20).
2.3 Neutrophil recruitment to the liver
The mechanisms underlying neutrophil recruitment to liver sinusoids differ from the classical recruitment cascade and appear independent of processes involving integrins and rolling, which have been extensively reviewed elsewhere (21). Neutrophils with high CXCR2 expression levels are recruited to the liver and guided along the concentration gradient of chemoattractant signals later in sepsis. In cases of endothelial barrier damage, immature neutrophils with low CXCR2 expression move to the liver for disposal through diapedesis (22). However, in comparison to septic mice who are fed high-fat and normal diets, neutrophil accumulation in the liver was shown to be unaffected by CXCR2, and obese mice exhibited a higher survival rate (23). Infections caused by different pathogens may induce differential neutrophil recruitment to the liver (24). For example, formyl peptide receptor signaling may act as an initial chemotactic signal during Listeria monocytogenes infection (25). In a septic model involving Staphylococcus aureus infection, heparan sulfate (HS) binding proteins were shown to be significantly enriched on the surface of the hepatic vasculature and were involved in modulating neutrophil recruitment (26). Reducing endothelial HS sulfation can selectively attenuate hepatic neutrophil infiltration and tissue damage. Pioneering neutrophils in the liver lead to swarming behavior via self-amplifying chemotactic signals, resulting in tissue damage (27, 28), with leukotriene B4 (LTB4) playing an important role in this process. Yu et al. demonstrated, in a cecal ligation and puncture (CLP)-induced murine model, that SALI and neutrophil infiltration could be ameliorated by inhibiting 5-lipoxygenase/LTB4 through caffeic acid administration (29). Neutrophil transmigration to the liver parenchyma occurs more rapidly compared to the migration rates toward other organs due to discontinuities in the hepatic sinusoidal capillaries (30), and neutrophil recruitment to the liver sinusoids is dependent on organ-specific adhesion mechanisms. Neutrophils adhere to the sinusoidal endothelium in the liver predominantly through interactions between a cluster of differentiation 44 (CD44) and hyaluronan (HA) as well as between dipeptidyl peptidase 1 (DPEP-1) and its ligands (31). Notably, the CD44–HA interaction is weak (32), although it is enhanced by the modification of heavy chains during sepsis, with the levels of heavy chain Itih3 in hepatic tissue samples increasing significantly (33). Furthermore, toll-like receptor 4 (TLR4) signaling affects serum-derived HA-associated protein, promoting the direct adherence of neutrophils to the sinusoidal endothelium of the liver partially through HA–CD44 interactions (34). McDonald et al. discovered that blocking HA–CD44 interactions using an anti-CD44 antibody significantly ameliorated LPS-induced hepatic injury (32). In addition, neutrophils promote their own extravasation through the endothelium into inflamed tissue by expressing macrophage-1 antigen (MAC-1) and very late antigen 4, which binds to intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1, respectively, on hepatic sinusoidal endothelial cells (21). It has been shown that LPS induces neutrophil priming, as reflected by increased ICAM-1 expression; this change is associated with enhanced neutrophil infiltration into the liver and tissue injury (35). Xiao et al. found that reducing hepatic neutrophil transmigration and hepatocyte injury through the administration of neutrophil membrane-mimicking nanodecoys in an endotoxemia mouse model may be linked to decreased endothelial ICAM-1 expression (36).
3 Effects of neutrophils on the liver during sepsis
3.1 Protective functions of neutrophils
Neutrophils act as a first line of defense against infections; once they enter the liver, they initiate various antimicrobial activities by secreting protein hydrolases and ROS. In the liver, neutrophils have a weak ability to entrap circulating bacteria; however, this ability is enhanced by the release of NETs (37). GPCRs modulate ROS levels to mediate the release of NETs in the early stages of disease (38). Neutrophils might tune their response according to a microbe’s size, as large pathogens promote the upregulated expression of interleukin (IL)-1β and neutrophil recruitment by triggering extracellular ROS release, while also inducing neutrophils to selectively release NETs (39, 40). Shao et al. reported that ‘targeted nuclear degranulation’ that a portion of the CD44 expressed on the surface of neutrophils moved to the nucleus after neutrophil activation was delayed to limit the rapid formation of NETs in response to strong stimulation in mouse models (41). Moreover, Oliveira-Costa et al. proposed the “innate triad” model, which involves interactions between neutrophils, platelets, and macrophages to enhance pathogen clearance (42).
Neutrophils are also associated with the resolution of liver inflammation and tissue repair. The restorative effects of neutrophils have been investigated in a murine model of toxic liver injury (43). Neutrophils may help induce a phenotypic switch in macrophages toward a pro-restorative profile through microRNA-223 or ROS, thereby resolving inflammation (44, 45). In addition, microRNA-223 is known to regulate neutrophil elastase (NE) enrichment to protect the liver from LPS-induced injury (46).
3.2 Neutrophil-driven hyperinflammation in liver injury
Overexuberant recruitment or the uncontrolled activation of neutrophils may lead to an overwhelming pro-inflammatory condition that is associated with multi-organ injury. An abundant neutrophil population has been observed in mouse livers during sepsis (30); however, it remains uncertain whether their lifespan in the liver can be extended or constantly replenished de novo from bone marrow. In recent years, granulocyte–monocyte progenitors have been identified to undergo release into the circulatory system during sepsis, and cell division has been reported in neutrophils (47). Therefore, further studies are required to investigate whether numerous neutrophils in the liver partially originate from peripheral neutrophil proliferation before altering their function. Several studies have utilized scRNA-seq to analyze the phenotype and function of hepatic neutrophils in SALI (12, 48). Chen et al. identified three distinct hepatic neutrophil subsets in septic mouse models (12) and demonstrated that the proportion of pro-inflammatory subsets continued to increase in a time-dependent manner. TLR plays an important role in the activation of neutrophils, driving a shift toward pro-inflammatory responses. One study also confirmed that microRNA-let-7b regulates neutrophil function by inhibiting the TLR4/nuclear factor kappa-B (NF-κB) pathway while also attenuating hepatic inflammation in septic mice, with the data showing decreased gene expression of tumor necrosis factor (TNF)-α and IL-8 in neutrophils (49). He et al. demonstrated that neutrophils identify pathogen-associated molecular patterns (PAMPs) via TLR2, which actives NF-κB signaling and the subsequent release of pro-inflammatory factors (48), and that artesunate treatment could reverse the increase in the proportion of pro-inflammatory subsets by inhibiting TLR2 expression while alleviating sepsis-induced liver injury in mouse models. Moreover, these proinflammatory neutrophils may be associated with dysregulated cytoplasmic Ca2+ concentrations and enhanced membrane depolarization and glycolytic metabolism (50, 51).
There is some evidence that the oxidative burst capacity of neutrophils is altered in SALI. Sustained neutrophil activation induces oxidative bursts by activating the GPCR/phospholipase C (PLC)/Ca2+ signaling pathway (14). Wang et al. reported that increased oxidative stress might result from activation of CXCR2 and its downstream target protein kinase C in neutrophils, with the process being regulated by CXCL2 on macrophage extracellular vesicles isolated from mouse (52). ROS activate the formation of NOD-like receptor 3 (NLRP3) via thioredoxin-interacting protein activation, subsequently resulting in IL-1β release into the extracellular space, causing an excessive inflammatory response (53). Moreover, NLRP3 triggers caspase-1-mediated endothelial pyroptosis, increasing hepatic vascular permeability and the likelihood of mortality (54). NETs exert a protective function by limiting bacterial spread; however, NET production or imbalances in clearance can also induce thrombosis, disseminated intravascular coagulation, and tissue damage (55). NET expression in neutrophils is upregulated in a time-dependent manner in septic liver tissues, far surpassing the levels in other areas of the microcirculation (56, 57). Hsieh et al. reported that histone H4 might trigger a sustained elevation of intracellular Ca2+ levels in human neutrophils, thereby inducing hydrogen peroxide production and degranulation through the G protein/phosphoinositide 3-kinase (PI3K) pathway to promote inflammatory responses (58). Levels of calgranulins S100A8/S100A9 become elevated in the blood of patients with sepsis and may be produced from neutrophil movement (59). Hepatic neutrophils with a hyperactivated phenotype exhibit high expression levels of S100A8/S100A9 in septic models (12), which may damage the liver by binding to receptors of advanced glycation endproducts (RAGE) and subsequently activating NF-κB signaling to promote TNF-α expression, providing evidence of its role as a damage-associated molecular pattern (DAMP) (4). Zhang et al. reported that S100A9 can disrupt mitochondrial respiratory chain functionality in the hepatic tissues of septic mice (60). S100A9 knockout ameliorated liver injury in these animals by inhibiting protein kinase B (Akt) and 5′adenosine monophosphate-activated protein kinase-activated mitochondrial metabolism.
3.3 Neutrophil-associated immunosuppression in liver injury
Although hyperactivated neutrophil responses lead to SALI, these cells exhibit impaired antimicrobial functions, and neutrophil-associated immunosuppression is reduced, implying an increased susceptibility to secondary infections. In a study involving septic mice in which neutrophilic gasdermin D (GSDMD) was specifically knocked out, no significant change in the degree of neutrophilic NET formation was observed, and this deletion led to more severe liver damage (61). This suggests that neutrophil-specific GSDMD may regulate the bactericidal activity of neutrophils and that impaired regulation is unrelated to NETs. Taylor et al. demonstrated that neutrophils exhibit substantially diminished functional capacity, including impairment of oxidative burst capabilities and phagocytosis in patients with SALI (62). The energetic state of neutrophils governs their function. In comparison to patients with sepsis and healthy volunteers, the lactic acid levels were lower in the septic patients, as evidenced by a study in which sustained LPS stimulation inhibited neutrophil phagocytosis through the decreased production of lactates and the inhibition of glycolysis via the PI3K/Akt/hypoxia-inducible factor (HIF)-1α pathway (63). A study identified an important role of monocyte chemotactic protein-induced protein-1 (MCPIP-1) in reducing the oxidative burst of neutrophils through degradation of cold-inducible RNA in mice with infectious hepatic disease in which neutrophilic MCPIP-1 expression becomes elevated (64).
Impaired antibacterial activity in neutrophils can also be attributed to their hyporesponsiveness to pathogens. Neutrophilic susceptibility to pathogens is also reduced by high levels of pro-inflammatory cytokines, soluble receptors, and endotoxins (65). The sustained inflammatory stimulation may be associated with tolerance development that affects TLR signaling or the upregulation of inhibitors of TLR signaling (66). Conversely, however, DAMP expression levels remain persistently elevated in LPS-challenged model piglets, which share similar pattern recognition receptors (PRRs) on neutrophils as well as microbial-associated molecular patterns, leading to impaired pathogen recognition (67, 68). Recent studies have identified a subset of low-density neutrophils (LDNs), including immature neutrophils and myeloid-derived suppressor cells with immunosuppressive characteristics, and there is evidence that some LDNs are degranulated from high-density neutrophils (HDNs) (69, 70). While these LDNs exhibit limited phagocytic abilities in a CLP mouse model, they are more actively engaged in the formation of NETs, which promote naïve CD4+ T cell differentiation into regulatory T cells (Tregs) while enhancing their immunosuppressive function (71). Neutrophil-derived immunosuppressive cells have been reported to undergo expansion in the liver of septic mice and are associated with T-cell dysfunction (72). Human neutrophils produce MAC-1 and PD-L1, which exert immunosuppressive effects that include T-lymphocyte apoptosis and the inhibition of T-cell activation and proliferation (73, 74). In addition, neutrophils in patients with sepsis produce large amounts of immunosuppressive cytokines, such as IL-10 (10).
3.4 The role of neutrophils in the gut–liver axis
Intestinal dysbiosis and disruption of the intestinal barrier induce intestinal bacteria and their metabolites translocation that causes the inflammatory response. Intestinal dysbiosis was associated with severe liver injury in septic models (75). Generally, intestinal pathogens and their products can go through the circulation, portal, and biliary to the liver and are processed by the liver (76). A review analyzing changes in neutrophil intracellular bacterial communities at different stages of sepsis found that the alterations in neutrophil-specific microbiomes were similar to intestinal microbiome composition (77). Intestinal epithelial cells and hepatocytes can produce LPS-binding protein, which enhances LPS transfer, binds to the membrane CD14 on neutrophils, and consequently promotes the inflammatory response (76). Liu et al. discovered that gut-derived bacteria and LPS promote the formation of NETs in the liver via TLR4 in a mice model (78). Recent studies have discovered that the intestinal microbiome can also regulate intrahepatic neutrophil infiltration Using in vivo imaging to track neutrophil movement in mice with Staphylococcus aureus infection, D-lactate-producing gut microbiota prime hepatic endothelial cells to upregulate DPEP-1 expression (79). Collectively, these studies suggest that targeted restoration of axis equilibrium to combat gut dysbiosis in SALI may prevent excessive neutrophil recruitment.
4 The role of neutrophils in different types of SALI
4.1 Neutrophils in sepsis-associated cholestatic liver injury
Sepsis-associated cholestasis is a clinical phenotype of SALI. A study that evaluated liver samples from patients with sepsis revealed that ductular cholestasis holds diagnostic value for identifying the disease, with a sensitivity of 68% and a specificity of 45% (3). Furthermore, hepatic neutrophilic infiltration has been observed, although there was no significant difference in the rates of portal and lobular neutrophilic inflammation in patients with and without sepsis (3). Accumulation of neutrophils around the bile ducts has been observed in an acute biliary epithelial cell mouse model (80). However, impaired LPS excretion by hepatocytes has been observed in bile salt efflux pump knockout mice; in these animals, LPS was not excreted through bile acids and was a direct cause of further infiltration of neutrophils and inflammatory mediators (81). Sepsis-associated cholestasis may be associated with impaired bile excretion rather than an increase in bile synthesis (82). Wu et al. reported a reduction in the direct IL-1β/IL-18-mediated neutrophilic damage to transporter proteins, which resulted in the restoration of tubular transporter protein and sepsis-induced hyper-bileacidaemia in NLPR3 knockout mice (83). In addition, septic bile duct-associated neutrophil subsets exist in an exhausted state, and the biological function is characterized by reduced neutrophil migration and phagocytosis, making it difficult to control infections and exacerbating septic shock development (84). Finally, a recent study demonstrated that tuft cells are negatively associated with biliary inflammation and microbiome-dependent neutrophilic infiltration, and biliary inflammation increased in tuft cell-deficient mice of cholestasis (85).
4.2 Neutrophils in sepsis-associated hypoxic liver injury
In sepsis, the liver increases its capacity for oxygen extraction; however, decreased hepatic perfusion and impaired oxygen utilization can still result in hepatocyte death (86). In addition, increased oxidative stress in septic livers is somewhat indicative of oxygen depletion, which ultimately leads to hypoxic liver injury. Neutrophils can work actively under hypoxic conditions and can sense oxygen tension; a series of responses triggered by stimulation is succeeded by increased degranulation, reduced ROS production, and prolonged survival (87, 88). However, continuous LPS stimulation has been shown to reduce glycolysis in neutrophils through the HIF-α pathway, thereby affecting their phagocytic and migratory functions (63). Recruited neutrophils accumulate in the sinusoids, blocking the lumen and exacerbating ischemia and hypoxia (89). Boufenzer reported that hypoxic conditions activate triggering receptors expressed on myeloid cell-1 (TREM-1), which synergizes with TLR4 to induce intracellular calcium currents and ROS production, thereby enhancing NET release by human and murine neutrophils (90). NETs impair hepatic microcirculation and further exacerbate hypoxia (91); in turn, ischemia and hypoxia lead to neutrophil recruitment to the liver in large numbers. The detrimental cycle of increased neutrophil accumulation and hypoxia can continue and accelerate liver injury progression.
5 Discussion
Neutrophils can be regarded as either a blessing or a curse, depending on their differential functionality in SALI. Proteases, ROS, and NETs released from neutrophils are involved in pathogens clearance and liver injury resolution, but they also act as pro-inflammatory mediators leading to liver injury. This may be influenced by time, concentration, and environment. This dual role of neutrophils deepens the difficulty of exploring their therapeutic role in SALI. By studying neutrophil heterogeneity, we have gained insight into the functions of different neutrophil subsets (Table 1), and helped develop targeted therapies against neutrophils and their associated components. Animal studies found that pro-inflammatory neutrophils are progressively increased during the progression of SALI, but the mechanism remains unclear. Reversing the proportion of pro-inflammatory neutrophils may be one of the future research directions.
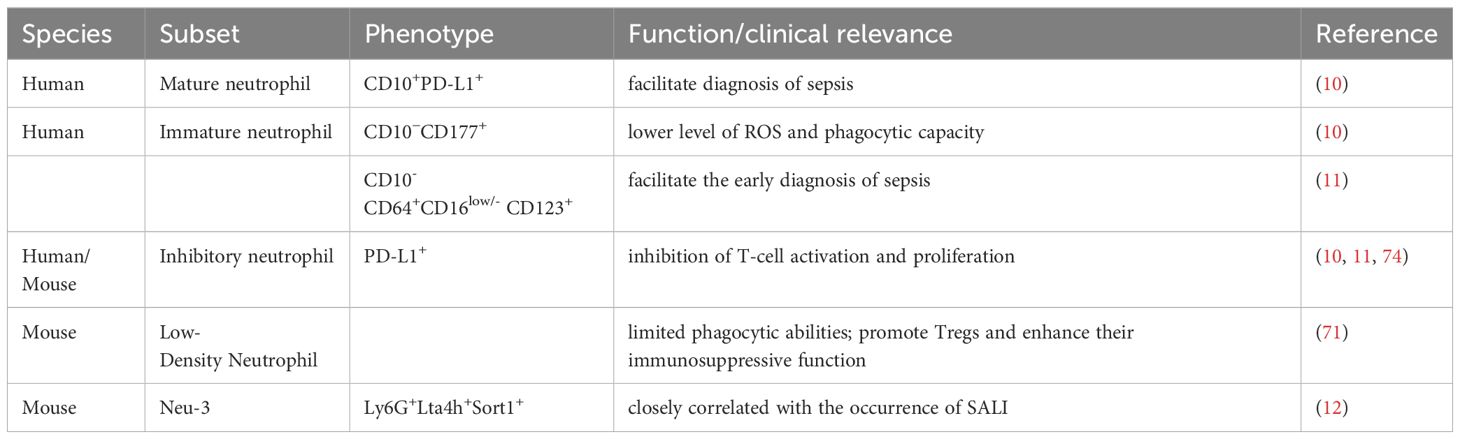
Table 1. The characteristics of neutrophil subsets in sepsis.
Neutrophils are first-responder cells recruited to infection. In recent years, progress has been made in the use of neutrophils as carriers in the development of drugs to treat SALI; however, because of the difficulty of obtaining liver specimens from septic patients, this research is still in the laboratory stage. Developing animal models that more simulate patients with SALI could help study potential treatments of SALI.
Author contributions
LF: Writing – original draft, Writing – review & editing. YS: Writing – review & editing. JC: Writing – review & editing. YD: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang Z, Tan X-J, Shi H-Q, Zhang H, Li J-B, Liao X-L. Bibliometric study of sepsis-associated liver injury from 2000 to 2023. World J Gastroenterol. (2024) 30:3609. doi: 10.3748/wjg.v30.i30.3609
2. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. (2017) 14:55–66. doi: 10.1038/nrgastro.2016.168
3. Bsirini C, Katerji R, Lee EJ, Gonzalez RS. Liver histology in septic patients: is it all about ductular cholestasis? Arch Pathol Lab Med. (2022) 146:1329–37. doi: 10.5858/arpa.2021-0190-OA
4. Weinhage T, Wirth T, Schütz P, Becker P, Lueken A, Skryabin BV, et al. The receptor for advanced glycation endproducts (Rage) contributes to severe inflammatory liver injury in mice. Front Immunol. (2020) 11:1157. doi: 10.3389/fimmu.2020.01157
5. Xie T, Xin Q, Cao X, Chen R, Ren H, Liu C, et al. Clinical characteristics and construction of a predictive model for patients with sepsis related liver injury. Clin Chim Acta. (2022) 537:80–6. doi: 10.1016/j.cca.2022.10.004
6. Ioannou M, Hoving D, Aramburu IV, Temkin MI, De Vasconcelos NM, Tsourouktsoglou TD, et al. Microbe capture by splenic macrophages triggers sepsis via T cell-death-dependent neutrophil lifespan shortening. Nat Commun. (2022) 13:4658. doi: 10.1038/s41467-022-32320-1
7. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. Cxcr2 and cxcr4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. (2010) 120:2423–31. doi: 10.1172/jci41649
8. Kim HK, de la Luz Sierra M, Williams CK, Gulino AV, Tosato G. G-csf down-regulation of cxcr4 expression identified as a mechanism for mobilization of myeloid cells. Blood. (2006) 108:812–20. doi: 10.1182/blood-2005-10-4162
9. Rubio I, Osuchowski MF, Shankar-Hari M, Skirecki T, Winkler MS, Lachmann G, et al. Current gaps in sepsis immunology: new opportunities for translational research. Lancet Infect Dis. (2019) 19:e422–e36. doi: 10.1016/s1473-3099(19)30567-5
10. Parthasarathy U, Kuang Y, Thakur G, Hogan JD, Wyche TP, Norton JE Jr., et al. Distinct subsets of neutrophils crosstalk with cytokines and metabolites in patients with sepsis. iScience. (2023) 26:105948. doi: 10.1016/j.isci.2023.105948
11. Meghraoui-Kheddar A, Chousterman BG, Guillou N, Barone SM, Granjeaud S, Vallet H, et al. Two new neutrophil subsets define a discriminating sepsis signature. Am J Respir Crit Care Med. (2022) 205:46–59. doi: 10.1164/rccm.202104-1027OC
12. Chen G, Ren C, Xiao Y, Wang Y, Yao R, Wang Q, et al. Time-resolved single-cell transcriptomics reveals the landscape and dynamics of hepatic cells in sepsis-induced acute liver dysfunction. JHEP Rep. (2023) 5:100718. doi: 10.1016/j.jhepr.2023.100718
13. Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. (2002) 159:91–102. doi: 10.1083/jcb.200202114
14. Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol. (2020) 17:433–50. doi: 10.1038/s41423-020-0412-0
15. Bruserud Ø, Mosevoll KA, Bruserud Ø, Reikvam H, Wendelbo Ø. The regulation of neutrophil migration in patients with sepsis: the complexity of the molecular mechanisms and their modulation in sepsis and the heterogeneity of sepsis patients. Cells. (2023) 12:1003. doi: 10.3390/cells12071003
16. Ciupe SM, Boribong BP, Kadelka S, Jones CN. Bistable mathematical model of neutrophil migratory patterns after lps-induced epigenetic reprogramming. Front Genet. (2021) 12:633963. doi: 10.3389/fgene.2021.633963
17. Bao W, Xing H, Cao S, Long X, Liu H, Ma J, et al. Neutrophils restrain sepsis associated coagulopathy via extracellular vesicles carrying superoxide dismutase 2 in a murine model of lipopolysaccharide induced sepsis. Nat Commun. (2022) 13:4583. doi: 10.1038/s41467-022-32325-w
18. Wang Y, Zhu CL, Li P, Liu Q, Li HR, Yu CM, et al. The role of G protein-coupled receptor in neutrophil dysfunction during sepsis-induced acute respiratory distress syndrome. Front Immunol. (2023) 14:1112196. doi: 10.3389/fimmu.2023.1112196
19. Seree-Aphinan C, Vichitkunakorn P, Navakanitworakul R, Khwannimit B. Distinguishing sepsis from infection by neutrophil dysfunction: A promising role of cxcr2 surface level. Front Immunol. (2020) 11:608696. doi: 10.3389/fimmu.2020.608696
20. Langer MM, Sichelschmidt S, Bauschen A, Bornemann L, Guckenbiehl S, Gunzer M, et al. Pathological neutrophil migration predicts adverse outcomes in hospitalized patients with liver cirrhosis. Liver Int. (2023) 43:896–905. doi: 10.1111/liv.15486
21. Lee W-Y, Kubes P. Leukocyte adhesion in the liver: distinct adhesion paradigm from other organs. J Hepatol. (2008) 48:504–12. doi: 10.1016/j.jhep.2007.12.005
22. Liu N, Bauer M, Press AT. The immunological function of cxcr2 in the liver during sepsis. J Inflammation. (2022) 19:1–13. doi: 10.1186/s12950-022-00321-y
23. Cichon I, Ortmann W, Santocki M, Opydo-Chanek M, Kolaczkowska E. Scrutinizing mechanisms of the ‘Obesity paradox in sepsis’: obesity is accompanied by diminished formation of neutrophil extracellular traps (Nets) due to restricted neutrophil–platelet interactions. Cells. (2021) 10:384. doi: 10.3390/cells10020384
24. Margraf A, Ley K, Zarbock A. Neutrophil recruitment: from model systems to tissue-specific patterns. Trends Immunol. (2019) 40:613–34. doi: 10.1016/j.it.2019.04.010
25. Witter AR, Okunnu BM, Berg RE. The essential role of neutrophils during infection with the intracellular bacterial pathogen listeria monocytogenes. J Immunol. (2016) 197:1557–65. doi: 10.4049/jimmunol.1600599
26. Golden GJ, Toledo AG, Marki A, Sorrentino JT, Morris C, Riley RJ, et al. Endothelial heparan sulfate mediates hepatic neutrophil trafficking and injury during staphylococcus aureus sepsis. mBio. (2021) 12:e0118121. doi: 10.1128/mBio.01181-21
27. Bartneck M, Wang J. Therapeutic targeting of neutrophil granulocytes in inflammatory liver disease. Front Immunol. (2019) 10:2257. doi: 10.3389/fimmu.2019.02257
28. Irimia D. Neutrophil swarms are more than the accumulation of cells. Microbiol Insights. (2020) 13:1178636120978272. doi: 10.1177/1178636120978272
29. Yu CM, Wang Y, Ren SC, Liu ZL, Zhu CL, Liu Q, et al. Caffeic acid modulates activation of neutrophils and attenuates sepsis-induced organ injury by inhibiting 5-lox/ltb4 pathway. Int Immunopharmacol. (2023) 125:111143. doi: 10.1016/j.intimp.2023.111143
30. Ahn SY, Maeng YS, Kim YR, Choe YH, Hwang HS, Hyun YM. In vivo monitoring of dynamic interaction between neutrophil and human umbilical cord blood-derived mesenchymal stem cell in mouse liver during sepsis. Stem Cell Res Ther. (2020) 11:44. doi: 10.1186/s13287-020-1559-4
31. Choudhury SR, Babes L, Rahn JJ, Ahn BY, Goring KR, King JC, et al. Dipeptidase-1 is an adhesion receptor for neutrophil recruitment in lungs and liver. Cell. (2019) 178:1205–21.e17. doi: 10.1016/j.cell.2019.07.017
32. McDonald B, McAvoy EF, Lam F, Gill V, de la Motte C, Savani RC, et al. Interaction of cd44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med. (2008) 205:915–27. doi: 10.1084/jem.20071765
33. Toledo AG, Golden G, Campos AR, Cuello H, Sorrentino J, Lewis N, et al. Proteomic atlas of organ vasculopathies triggered by staphylococcus aureus sepsis. Nat Commun. (2019) 10:4656. doi: 10.1038/s41467-019-12672-x
34. McDonald B, Jenne CN, Zhuo L, Kimata K, Kubes P. Kupffer cells and activation of endothelial tlr4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G797–806. doi: 10.1152/ajpgi.00058.2013
35. Zhang Y, Lin R, Pradhan K, Geng S, Li L. Innate priming of neutrophils potentiates systemic multiorgan injury. Immunohorizons. (2020) 4:392–401. doi: 10.4049/immunohorizons.2000039
36. Xiao Y, Ren C, Chen G, Shang P, Song X, You G, et al. Neutrophil membrane-mimicking nanodecoys with intrinsic anti-inflammatory properties alleviate sepsis-induced acute liver injury and lethality in a mouse endotoxemia model. Mater Today Bio. (2022) 14:100244. doi: 10.1016/j.mtbio.2022.100244
37. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. (2012) 12:324–33. doi: 10.1016/j.chom.2012.06.011
38. Guo W, Gong Q, Zong X, Wu D, Li Y, Xiao H, et al. Gpr109a controls neutrophil extracellular traps formation and improve early sepsis by regulating ros/pad4/cit-H3 signal axis. Exp Hematol Oncol. (2023) 12:15. doi: 10.1186/s40164-023-00376-4
39. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. (2014) 15:1017–25. doi: 10.1038/ni.2987
40. Yam AO, Chtanova T. Imaging the neutrophil: intravital microscopy provides a dynamic view of neutrophil functions in host immunity. Cell Immunol. (2020) 350:103898. doi: 10.1016/j.cellimm.2019.01.003
41. Shao Y, Li L, Liu L, Yang Y, Huang J, Ji D, et al. Cd44/erm/F-actin complex mediates targeted nuclear degranulation and excessive neutrophil extracellular trap formation during sepsis. J Cell Mol Med. (2022) 26:2089–103. doi: 10.1111/jcmm.17231
42. Oliveira-Costa KM, Menezes GB, Paula Neto HA. Neutrophil accumulation within tissues: A damage X healing dichotomy. BioMed Pharmacother. (2022) 145:112422. doi: 10.1016/j.biopha.2021.112422
43. Chauhan A, Sheriff L, Hussain MT, Webb GJ, Patten DA, Shepherd EL, et al. The platelet receptor clec-2 blocks neutrophil mediated hepatic recovery in acetaminophen induced acute liver failure. Nat Commun. (2020) 11:1939. doi: 10.1038/s41467-020-15584-3
44. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ros to orchestrate liver repair. Nat Commun. (2019) 10:1–14. doi: 10.1038/s41467-019-09046-8
45. Wang X, He Y, Mackowiak B, Gao B. Micrornas as regulators, biomarkers and therapeutic targets in liver diseases. Gut. (2021) 70:784–95. doi: 10.1136/gutjnl-2020-322526
46. Ye D, Yao J, Du W, Chen C, Yang Y, Yan K, et al. Neutrophil extracellular traps mediate acute liver failure in regulation of mir-223/neutrophil elastase signaling in mice. Cell Mol Gastroenterol Hepatol. (2022) 14:587–607. doi: 10.1016/j.jcmgh.2022.05.012
47. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The neutrophil. Immunity. (2021) 54:1377–91. doi: 10.1016/j.immuni.2021.06.006
48. He XL, Chen JY, Feng YL, Song P, Wong YK, Xie LL, et al. Single-cell rna sequencing deciphers the mechanism of sepsis-induced liver injury and the therapeutic effects of artesunate. Acta Pharmacol Sin. (2023) 44:1–14. doi: 10.1038/s41401-022-00938-y
49. Chen B, Han J, Chen S, Xie R, Yang J, Zhou T, et al. Microlet-7b regulates neutrophil function and dampens neutrophilic inflammation by suppressing the canonical tlr4/nf-Kb pathway. Front Immunol. (2021) 12:653344. doi: 10.3389/fimmu.2021.653344
50. Robledo-Avila FH, Ruiz-Rosado JD, Brockman KL, Partida-Sanchez S. The trpm2 ion channel regulates inflammatory functions of neutrophils during listeria monocytogenes infection. Front Immunol. (2020) 11:97. doi: 10.3389/fimmu.2020.00097
51. Liu D, Sun W, Zhang D, Yu Z, Qin W, Liu Y, et al. Long noncoding rna gsec promotes neutrophil inflammatory activation by supporting pfkfb3-involved glycolytic metabolism in sepsis. Cell Death Dis. (2021) 12:1157. doi: 10.1038/s41419-021-04428-7
52. Wang G, Huang W, Wang S, Wang J, Cui W, Zhang W, et al. Macrophagic extracellular vesicle cxcl2 recruits and activates the neutrophil cxcr2/pkc/nox4 axis in sepsis. J Immunol. (2021) 207:2118–28. doi: 10.4049/jimmunol.2100229
53. Li Z, Liu T, Feng Y, Tong Y, Jia Y, Wang C, et al. Pparγ Alleviates sepsis-induced liver injury by inhibiting hepatocyte pyroptosis via inhibition of the ros/txnip/nlrp3 signaling pathway. Oxid Med Cell Longev. (2022) 2022:1269747. doi: 10.1155/2022/1269747
54. Chen Q, Yang Y, Pan Y, Shen L, Zhang Y, Zheng F, et al. Human neutrophil defensins disrupt liver interendothelial junctions and aggravate sepsis. Mediators Inflammation. (2022) 2022:7659282. doi: 10.1155/2022/7659282
55. Kiwit A, Lu Y, Lenz M, Knopf J, Mohr C, Ledermann Y, et al. The dual role of neutrophil extracellular traps (Nets) in sepsis and ischemia-reperfusion injury: comparative analysis across murine models. Int J Mol Sci. (2024) 25:3787. doi: 10.3390/ijms25073787
56. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. (2018) 15:206–21. doi: 10.1038/nrgastro.2017.183
57. Sun X, Wu J, Liu L, Chen Y, Tang Y, Liu S, et al. Transcriptional switch of hepatocytes initiates macrophage recruitment and T-cell suppression in endotoxemia. J Hepatol. (2022) 77:436–52. doi: 10.1016/j.jhep.2022.02.028
58. Hsieh IN, Deluna X, White MR, Hartshorn KL. Histone H4 directly stimulates neutrophil activation through membrane permeabilization. J Leukoc Biol. (2021) 109:763–75. doi: 10.1002/jlb.3a0620-342r
59. Marki A, Buscher K, Lorenzini C, Meyer M, Saigusa R, Fan Z, et al. Elongated neutrophil-derived structures are blood-borne microparticles formed by rolling neutrophils during sepsis. J Exp Med. (2021) 218. doi: 10.1084/jem.20200551
60. Zhang Y, Wu F, Teng F, Guo S, Li H. Deficiency of S100a9 alleviates sepsis-induced acute liver injury through regulating akt-ampk-dependent mitochondrial energy metabolism. Int J Mol Sci. (2023) 24:2112. doi: 10.3390/ijms24032112
61. Liu F, Ghimire L, Balasubramanian A, Hsu AY, Zhang Z, Yu H, et al. Neutrophil-specific depletion of gasdermin D does not protect against murine sepsis. Blood. (2023) 141:550–4. doi: 10.1182/blood.2022016931
62. Taylor NJ, Nishtala A, Manakkat Vijay GK, Abeles RD, Auzinger G, Bernal W, et al. Circulating neutrophil dysfunction in acute liver failure. Hepatology. (2013) 57:1142–52. doi: 10.1002/hep.26102
63. Pan T, Sun S, Chen Y, Tian R, Chen E, Tan R, et al. Immune effects of pi3k/akt/hif-1α-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit Care. (2022) 26:29. doi: 10.1186/s13054-022-03893-6
64. Lin J, Lu Z, Li G, Zhang C, Lu H, Gao S, et al. Mcpip-1-mediated immunosuppression of neutrophils exacerbates acute bacterial peritonitis and liver injury. J Innate Immun. (2022) 15:262–82. doi: 10.1159/000526784
65. Liu K, Wang FS, Xu R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cell Mol Immunol. (2021) 18:38–44. doi: 10.1038/s41423-020-00560-0
66. Shen X, Cao K, Zhao Y, Du J. Targeting neutrophils in sepsis: from mechanism to translation. Front Pharmacol. (2021) 12:644270. doi: 10.3389/fphar.2021.644270
67. Leliefeld PH, Wessels CM, Leenen LP, Koenderman L, Pillay J. The role of neutrophils in immune dysfunction during severe inflammation. Crit Care. (2016) 20:73. doi: 10.1186/s13054-016-1250-4
68. Xu Q, Guo J, Li X, Wang Y, Wang D, Xiao K, et al. Necroptosis underlies hepatic damage in a piglet model of lipopolysaccharide-induced sepsis. Front Immunol. (2021) 12:633830. doi: 10.3389/fimmu.2021.633830
69. Sun R, Huang J, Yang Y, Liu L, Shao Y, Li L, et al. Dysfunction of low-density neutrophils in peripheral circulation in patients with sepsis. Sci Rep. (2022) 12:685. doi: 10.1038/s41598-021-04682-x
70. Cho Y, Bukong TN, Tornai D, Babuta M, Vlachos IS, Kanata E, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. (2023) 78:28–44. doi: 10.1016/j.jhep.2022.08.029
71. Shi Y, Wu D, Wang Y, Shao Y, Zeng F, Zhou D, et al. Treg and neutrophil extracellular trap interaction contributes to the development of immunosuppression in sepsis. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.180132
72. Luo M, Wang H, Liu K, Liu M, Tan S, Zhu Y, et al. Il-1r1 blockade attenuates liver injury through inhibiting the recruitment of myeloid-derived suppressor cells in sepsis. Biochem Biophys Res Commun. (2022) 620:21–8. doi: 10.1016/j.bbrc.2022.06.038
73. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through mac-1. J Clin Invest. (2012) 122:327–36. doi: 10.1172/jci57990
74. Qi X, Yu Y, Sun R, Huang J, Liu L, Yang Y, et al. Identification and characterization of neutrophil heterogeneity in sepsis. Crit Care. (2021) 25:50. doi: 10.1186/s13054-021-03481-0
75. Liu Z, Li N, Fang H, Chen X, Guo Y, Gong S, et al. Enteric dysbiosis is associated with sepsis in patients. FASEB J. (2019) 33:12299–310. doi: 10.1096/fj.201900398RR
76. Zhang X, Liu H, Hashimoto K, Yuan S, Zhang J. The gut-liver axis in sepsis: interaction mechanisms and therapeutic potential. Crit Care. (2022) 26:213. doi: 10.1186/s13054-022-04090-1
77. Wang C, Li Q, Tang C, Zhao X, He Q, Tang X, et al. Characterization of the blood and neutrophil-specific microbiomes and exploration of potential bacterial biomarkers for sepsis in surgical patients. Immun Inflammation Dis. (2021) 9:1343–57. doi: 10.1002/iid3.v9.4
78. Liu Y, Zhang X, Chen S, Wang J, Yu S, Li Y, et al. Gut-derived lipopolysaccharide promotes alcoholic hepatosteatosis and subsequent hepatocellular carcinoma by stimulating neutrophil extracellular traps through toll-like receptor 4. Clin Mol Hepatol. (2022) 28:522–39. doi: 10.3350/cmh.2022.0039
79. Zucoloto AZ, Schlechte J, Ignacio A, Thomson CA, Pyke S, Yu IL, et al. Vascular traffic control of neutrophil recruitment to the liver by microbiota-endothelium crosstalk. Cell Rep. (2023) 42:112507. doi: 10.1016/j.celrep.2023.112507
80. Guillot A, Guerri L, Feng D, Kim SJ, Ahmed YA, Paloczi J, et al. Bile acid–activated macrophages promote biliary epithelial cell proliferation through integrin Avβ6 upregulation following liver injury. J Clin Invest. (2021) 131. doi: 10.1172/jci132305
81. Remetic J, Ghallab A, Hobloss Z, Brackhagen L, Hassan R, Myllys M, et al. Loss of bile salt export pump aggravates lipopolysaccharide-induced liver injury in mice due to impaired hepatic endotoxin clearance. Hepatology. (2022) 75:1095–109. doi: 10.1002/hep.32289
82. Zhu CL, Wang Y, Liu Q, Li HR, Yu CM, Li P, et al. Dysregulation of neutrophil death in sepsis. Front Immunol. (2022) 13:963955. doi: 10.3389/fimmu.2022.963955
83. Wu Y, Ren J, Zhou B, Ding C, Chen J, Wang G, et al. Gene silencing of non-obese diabetic receptor family (Nlrp3) protects against the sepsis-induced hyper-bile acidaemia in a rat model. Clin Exp Immunol. (2015) 179:277–93. doi: 10.1111/cei.12457
84. Zhang H, Wang N, Xu Y, Pei M, Zheng Y. Comparative analysis of peripheral blood immunoinflammatory landscapes in patients with acute cholangitis and its secondary septic shock using single-cell rna sequencing. Biochem Biophys Res Commun. (2023) 683:149121. doi: 10.1016/j.bbrc.2023.149121
85. O’Leary CE, Sbierski-Kind J, Kotas ME, Wagner JC, Liang HE, Schroeder AW, et al. Bile acid-sensitive tuft cells regulate biliary neutrophil influx. Sci Immunol. (2022) 7:eabj1080. doi: 10.1126/sciimmunol.abj1080
86. Liu S, Kohler A, Langer R, Jakob MO, Salm L, Blank A, et al. Hepatic blood flow regulation but not oxygen extraction capability is impaired in prolonged experimental abdominal sepsis. Am J Physiol Gastrointest Liver Physiol. (2022) 323:G348–g61. doi: 10.1152/ajpgi.00109.2022
87. Curi R, Levada-Pires AC, Silva EBD, Poma SO, Zambonatto RF, Domenech P, et al. The critical role of cell metabolism for essential neutrophil functions. Cell Physiol Biochem. (2020) 54:629–47. doi: 10.33594/000000245
88. Lodge KM, Cowburn AS, Li W, Condliffe AM. The impact of hypoxia on neutrophil degranulation and consequences for the host. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21041183
89. Beyer D, Hoff J, Sommerfeld O, Zipprich A, Gaßler N, Press AT. The liver in sepsis: molecular mechanism of liver failure and their potential for clinical translation. Mol Med. (2022) 28:84. doi: 10.1186/s10020-022-00510-8
90. Boufenzer A, Carrasco K, Jolly L, Brustolin B, Di-Pillo E, Derive M, et al. Potentiation of nets release is novel characteristic of trem-1 activation and the pharmacological inhibition of trem-1 could prevent from the deleterious consequences of nets release in sepsis. Cell Mol Immunol. (2021) 18:452–60. doi: 10.1038/s41423-020-00591-7
Keywords: immunosuppression, inflammation, liver injury, neutrophil, sepsis
Citation: Fang L, Song Y, Chen J and Ding Y (2025) The dual role of neutrophils in sepsis-associated liver injury. Front. Immunol. 16:1538282. doi: 10.3389/fimmu.2025.1538282
Received: 02 December 2024; Accepted: 17 February 2025;
Published: 28 February 2025.
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Theocharis Konstantinidis, Democritus University of Thrace, GreeceMathieu-Benoit Voisin, Queen Mary University of London, United Kingdom
Copyright © 2025 Fang, Song, Chen and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yueping Ding, RGluZ3lwMDQyNEB6Y211LmVkdS5jbg==