 Leiyi Yang1†
Leiyi Yang1† Ruibing Guo1†
Ruibing Guo1† Hongjiang Liu1†
Hongjiang Liu1† Bo Chen1Changpei Li1Ruiting Liu1Shuyi Liao1
Bo Chen1Changpei Li1Ruiting Liu1Shuyi Liao1 Qibing Xie1*
Qibing Xie1* Geng Yin2*
Geng Yin2*- 1Department of Rheumatology and Immunology, West China Hospital, Sichuan University, Chengdu, China
- 2Health Management Center, General Practice Medical Center, West China Hospital, Sichuan University, Chengdu, China
Antiphospholipid syndrome (APS) is an autoimmune disease characterized by the occurrence of thrombotic or obstetrical events in patients with persistent antiphospholipid antibodies (aPL). Thrombotic events, the primary pathological hallmarks and clinical manifestations, are among the leading causes of mortality in APS. Our understanding of the mechanism underlying APS-related thrombosis has significantly advanced in recent years. The presence of aPL, particularly anti-β2-glycoprotein I (anti-β2GPI) antibodies, is a major driver of thrombosis. The proposed pathophysiological mechanisms of aPL-mediated pro-thrombotic events can be broadly categorized into three types: disruption of anticoagulant reactions and fibrinolysis, interference with coagulation cascade cells, and complement activation. A triggering ‘second hit’ is typically necessary to initiate thrombosis. The development of animal models of APS has further refined our understanding of the role of aPL in thrombosis. In this review, we focused on the role of β2GPI-dependent aPL in thrombosis of thrombotic APS.
1 Introduction
Antiphospholipid syndrome (APS), first described in the early 1980s (1), is defined by the presence of antiphospholipid antibodies (aPL) in patients with thrombotic complications and/or adverse pregnancy outcomes (2). APS is one of the most common causes of acquired thrombophilia, affecting both arterial and venous circulation, particularly in young people (3, 4). Thrombotic events are the most frequent clinical manifestations and the leading causes of mortality in APS (4). The annual incidence and prevalence of APS in adults are 1-2 and 40-50 per 100,000 individuals, respectively (5). The 10-year survival rate for patients with APS has been reported to be 90.7% (4). Less than 1% of APS patients may develop catastrophic APS, characterized by life-threatening microvascular thrombosis in at least three organs, with a mortality rate of up to 50% (6), typically occurring within one week (7). Emerging evidence highlights significant sex-related differences in APS pathogenesis, with a striking 5:1 female predominance (8). In thrombotic APS, women tend to present venous thrombosis at a younger age while men manifest arterial events later in life (9). This happens because estrogen and progesterone work together in specific ways. First, estrogen induces a prothrombotic environment through various effects on the hemostatic pathways (10). Second, progesterone activates platelets through glucocorticoid receptor signaling, increasing the likelihood of venous thrombosis when progesterone levels are high (11). Women with APS are at substantially elevated risk for pregnancy-related complications, with pooled analyses demonstrating 12.3-fold increased odds of severe preeclampsia, 9.1-fold risk of fetal loss, and 6.8-fold higher perinatal mortality compared to the general obstetric population (12).
APS can present either as a primary condition (primary APS) (13) or in association with other systemic autoimmune diseases, most notably systemic lupus erythematosus (SLE), in which case it sometimes referred to as secondary APS (8, 14). However, the expert committee on APS advises against using the term ‘secondary APS’ (2). This is primarily because there are no significant differences in the clinical consequences between primary APS and so-called secondary APS (14, 15). The 2023 ACR/EULAR classification criteria emphasize a phenotype-based classification framework over etiological associations (16). It is now generally accepted that APS manifests in two main clinical variants: thrombotic APS and obstetric APS (17, 18). In addition to the presence of aPL, thrombotic APS is characterized by clinical features related to venous, arterial, or microvascular thrombosis, while obstetric APS is distinguished by pregnancy complications (17, 19). Over the past 30 years, our understanding of the mechanism of APS thrombosis has evolved significantly. However, the exact mechanisms are still not fully understood, which may explain why current treatment relies mainly on anticoagulants (17). Although anticoagulants are somewhat effective in preventing aPL-associated thrombosis (especially venous), they exhibit limited therapeutic impact on the microvascular manifestations of APS that affect the heart, kidneys, skin, and brain (20). Despite existing treatments, mainly including oral anticoagulants and/or anti-aggregation agents, patients with APS continue to experience considerable morbidity and mortality (4). Therefore, it is imperative to intensify efforts to develop therapeutic strategies to prevent these critical complications. Recently, Meroni et al. proposed that thrombotic and obstetric APS may represent two distinct diseases mediated by the same antibodies (18).
The aPL, including anticardiolipin (aCL), lupus anticoagulant (LA) and anti-β2-glycoprotein (anti-β2GPI) antibodies, recognize plasma proteins that bind avidly to anionic phospholipid surfaces, with β2GPI being the primary target (2, 21). While there is general consensus that aPL detected in patients with APS mediate thrombosis, the underlying pathophysiological mechanisms remain under debate (17, 22–24). Herein, we review current evidence on the role of β2GPI-dependent aPL in thrombosis of thrombotic APS. Our aim is to enhance understanding of these mechanisms, potentially illuminating future therapeutic targets and strategies to prevent thrombotic events.
2 Murine models of APS thrombosis
Animal models are crucial for investigating the mechanisms of aPL-induced thrombogenesis. In the 1990s, Pierangeli et al. developed the femoral vein pinch model, a mouse model for studying aPL-induced venous thrombosis in vivo (25–27). In this model, the appropriate dose of IgG-APS was injected intraperitoneally (i.p.) at 0 and 48h. The surgical procedure was performed 72 hours after the first injection. The model enabled the study of thrombus size and growth dynamics in the vein using a transilluminator equipped with digital video analysis (25–27). Several research groups have adopted this mouse model to investigate mechanisms of aPL-mediated thrombosis (28–31). Then, Jankowski’s laboratory adapted a murine model of arterial thrombosis induced by a photochemical reaction (32, 33). In this model, thrombosis was induced in the left carotid artery using filtered green light irradiation combined with the fluorescent dye rose-bengal in mice or hamsters (32, 33). Purified human β2GPI mAbs or IgG from APS patients were infused 15 minutes prior to photochemical vessel injury. Thrombus formation was continuously monitored by a transilluminator mounted on the artery and quantitatively analyzed with image processing software (32, 33). Furie and colleagues later utilized a laser-induced arteriole injury model in mice and applied intravital microscopy to image thrombus formation in real time within the microcirculation (34, 35). Using the laser-induced thrombosis model and intravital microscopy, the same group investigated the in vivo roles of platelets and endothelial cells in anti-β2GPI antibody/β2GPI complex-mediated thrombosis (36, 37). Seshan et al. developed a mouse model of thrombotic microangiopathy that replicated the early-stage pathophysiology of thrombotic microangiopathy induced by aPL in APS patients (38). This model used smaller amounts of aPL-IgG and enabled the investigation of mechanisms underlying renal vascular thrombosis (38). In addition, two research groups employed a ferric chloride-induced thrombosis model to explore the molecular and cellular events mediated by aPL in vascular thrombosis in vivo (39, 40).
Deep vein thrombosis represents the most frequently occurring form of thrombosis in APS (4). Manukyan and colleagues recently introduced a novel mouse model of flow restriction-induced thrombosis, termed stenosis, which simulates the clinical features of deep vein thrombosis in humans (41, 42). In this model, a spacer was positioned around the exposed inferior vena cava (IVC), and a permanent ligature was tightened below the left renal vein to create narrowing. The wire was then removed to prevent complete vessel occlusion. Antibodies were administered one hour before inducing flow restriction (41). Building on this thrombosis model, Knight’s group developed a murine model of APS-induced thrombosis via flow restriction or stenosis of the IVC (43, 44). However, most existing murine models of APS-related thrombosis have been confined to microscopic vascular beds. To better replicate large-vein thrombosis, Knight and colleagues recently developed an electrolytic IVC model of aPL-accelerated thrombosis (45, 46). A 30-gauge silver-coated copper wire was attached to a 25-gauge needle, which was inserted into the exposed IVC. A mild direct current of 250 μA was applied for 15 minutes. The mouse was then treated with 500 μg of aPL-IgG and allowed to recover. Thrombus size and content were assessed after 24 hours (46).
Patients with APS continuously produce aPL, leading to vascular thrombosis in both small and large vessels across venous and arterial beds (17, 47). However, existing animal models have demonstrated that a single, temporary aPL injection is insufficient to induce a pro-thrombotic phenotype. These models are often restricted to studying thrombosis in only one type of blood vessel. Furthermore, the aPL dosage, injection intervals, and choice of stimulating factor vary widely among these models.
3 Antiphospholipid antibodies and β2-glycoprotein I
The aPL included in the 2006 Sydney classification criteria are LA, IgG and/or IgM aCL antibodies, and anti-β2GPI antibody of IgG and/or IgM isotype (2). A diagnosis of APS requires persistently positive aPL (detected on two occasions ≥12 weeks apart) and at least one of two clinical manifestations: vascular thrombosis or pregnancy morbidity (2, 16). However, some individuals exhibit a clinical profile highly suggestive of APS but consistently test negative for ‘criteria’ aPL (48). The term ‘seronegative APS’ has been proposed to describe this subset of patients (49, 50). Seronegative APS patients are not entirely without autoantibodies but instead present with ‘extra-criteria’ autoantibodies. Notably, the clinical validity of these non-criteria antibodies varies significantly: while anti-phosphatidylserine/prothrombin (aPS/PT) and anti-β2GPI-domain I antibodies show strong mechanistic evidence and are under consideration for inclusion in future classification criteria (51–53), others such as anti-annexin antibodies and anti-phosphatidylethanolamine lack robust clinical validation (51). It is likely that all subpopulations of ‘criteria’ or ‘extra-criteria’ autoantibodies in patients with APS can influence their hemostatic balance. Nonetheless, it is widely accepted that APS-related thrombosis is driven by aPL, with antibodies against β2GPI being the most significant (17).
Initially, it was thought that aPL could directly recognize anionic phospholipids. However, some studies conducted in the 1990s confirmed that these antibodies do not bind phospholipids directly but instead interact with phospholipids via the plasma protein β2GPI (21, 54, 55). In APS, phospholipid-bound β2GPI is the primary target of aPL, and anti-β2GPI antibodies are thought to play a central role in the mechanisms of thrombosis (17, 24, 56). Multiple studies using animal models have demonstrated that anti-β2GPI antibodies or β2GPI-dependent antibodies play important roles in inducing thrombosis (33, 41, 57). Further research, which separated the heterogeneous aPL population from APS patients into different subpopulations, showed that anti-β2GPI antibodies significantly enhanced the thrombotic response in a mouse model (36).
β2GPI, also known as apolipoprotein H, is a 50-kDa phospholipid-binding glycoprotein present in plasma at a concentration of approximately 200 μg/mL (58). The primary function of β2GPI remains largely unknown, although it has been reported to have roles in anticoagulant activity, antiangiogenic activity, complement regulation, and other physiological process (59–62). β2GPI exists in several conformations, including J-elongated, S-twisted, and O-circular, with the J conformation likely being predominant under physiological conditions (63). Normally, β2GPI circulates in a circular form, but in the presence of elevated aPL or exposed anionic phospholipids on cell membranes, it adopts an open conformation, which may contribute to the pathogenesis of APS and thrombosis (62). However, the mechanisms underlying these conformational changes remain unclear. Structurally, β2GPI is composed of 326 amino acids arranged into five homologous domains, with domain V containing a unique lysine cluster and a C-terminal loop (64–66). The specific structure of domain V forms a binding site for negatively charged phospholipids, such as cardiolipin and phosphatidylserine (67, 68). When β2GPI binds to the surface of anionic phospholipids, it exposes a hidden epitope that is recognized by aPL in APS (69–71). These aPL do not recognize β2GPI in solution and only bind to domain I of β2GPI which has undergone a conformational change (70, 71).
4 Potential mechanisms of aPL-mediated thrombosis
The possible pathogenesis of thrombosis mediated by β2GPI-dependent aPL includes (1) disruption of fluid-phase coagulation by interfering with protein C, antithrombin, annexin A5, and fibrinolysis (2), impairment of coagulation cascade cell functions by interacting with monocytes, endothelial cells, neutrophils, and platelets (3), and complement activation. It is now widely accepted that a ‘second hit’ is necessary to trigger thrombotic events (17, 47, 72, 73).
4.1 Disruptions of fluid-phase coagulation
4.1.1 Inhibition of the protein C pathway
Protein C is an important vitamin K-dependent anticoagulant that becomes activated when thrombin binds to thrombomodulin. Activated protein C (APC) plays crucial anticoagulant and antithrombotic roles by binding to and inactivating the procoagulant factors Va and VIIIa (74). Researchers discovered that the activation of protein C and the function of APC are inhibited by purified immunoglobulin fractions from patients with APS (75, 76). APL can disrupt the protein C system in several ways, including inhibiting the assembly of the protein C complex, interfering with protein C activation, and blocking thrombin formation (77–79). Murine monoclonal anti-β2GPI antibodies, in the presence of β2GPI, have been demonstrated to inhibit the anticoagulant activity of APC in vitro (80). In addition, anti-β2GPI antibodies and β2GPI-dependent LA can induce APC resistance, increasing the risk of venous thromboembolism in patients with APS (81, 82). These studies indicate that protein C dysfunction caused by aPL is primarily mediated by β2GPI. Autoantibodies directed against protein C have been detected in the serum APS patients, and they show a significant correlation with APC resistance and thrombosis in these individuals (83).
4.1.2 Inhibition of antithrombin activity
Antithrombin is the primary inhibitor of thrombin, as well as factors IXa and Xa. As early as the 1980s, it was reported that an APS patient with recurrent thrombosis had normal levels of antithrombin antigen but reduced functional activity (84). APL can inhibit the cofactor activity of the heparin/antithrombin III complex and interfere with the formation of antithrombin III-thrombin complexes, thereby promoting thrombosis in patients with APS (85, 86). Two studies have demonstrated that injecting anti-prothrombin autoantibodies into mice can induce a pro-thrombotic phenotype, which may be linked to the inhibition of antithrombin activity (86, 87).
4.1.3 Disruption of annexin A5 anticoagulant shield
Annexin A5 is a protein that binds to anionic phospholipids with high affinity. It forms a protective crystal shield on vascular cells, inhibiting phospholipid-dependent coagulation reactions. Rand et al. were the first to report that aPL reduce the levels of annexin A5 and promote plasma coagulation on vascular endothelial cells. This finding suggested a potential mechanism for thrombosis (88). They hypothesized that aPL might increase resistance to the anticoagulant effects of annexin A5. Based on this hypothesis, Rand and colleagues developed a method to detect what they termed ‘annexin A5 resistance’ in plasma, which they confirmed in several populations of patients with APS (89). Subsequently, an in vitro study by the same group showed that anti-β2GPI antibodies, in complex with β2GPI, can disrupt annexin A5’s anticoagulant shield (90). This disruption exposes procoagulant phosphatidylserine, increasing the risk of thrombosis. Hydroxychloroquine has been found to inhibit the ability of β2GPI immune complexes to disrupt the protective annexin A5 barrier on the surface of vascular endothelial cells (91). This provides new evidence for the therapeutic potential of this old antimalarial drug in APS patients.
4.1.4 Insufficient fibrinolysis
Fibrinolysis is the process by which fibrin, formed during blood coagulation, is broken down and liquefied. Fibrinolysis is a crucial anticoagulant process in vivo. Musiał J et al. discovered that fibrin clots in thrombotic APS patients are composed of thin fibers and small pores. This structure makes the clots firmer, quicker to form, and slower to degrade compared to those in similar VTE cases (92). The altered fibrin structure is more resistant to lysis, which negatively impacts anticoagulation. Annexin A2 acts as a receptor for β2GPI and tissue plasminogen activator (tPA), playing a significant role in the process of fibrinolysis (93). Studies have demonstrated that patients with APS have autoantibodies against the Annexin A2, with high titers of these antibodies being significantly correlated with thrombosis (94, 95), as they inhibit tPA-dependent plasmin generation. Additionally, anti-β2GPI antibodies in APS patients can neutralize the capacity of β2GPI to enhance tPA activity, thereby further inhibiting fibrinolysis (96).
4.2 Cell-mediated events
Vascular inflammation is another critical mechanism of thrombosis in APS. Various types of cells are involved in the inflammatory process, including endothelial cells, monocytes, and neutrophils. Anti-β2GPI antibodies bind to membrane-bound β2GPI, triggering intracellular signaling and thereby promoting inflammation (Figure 1).
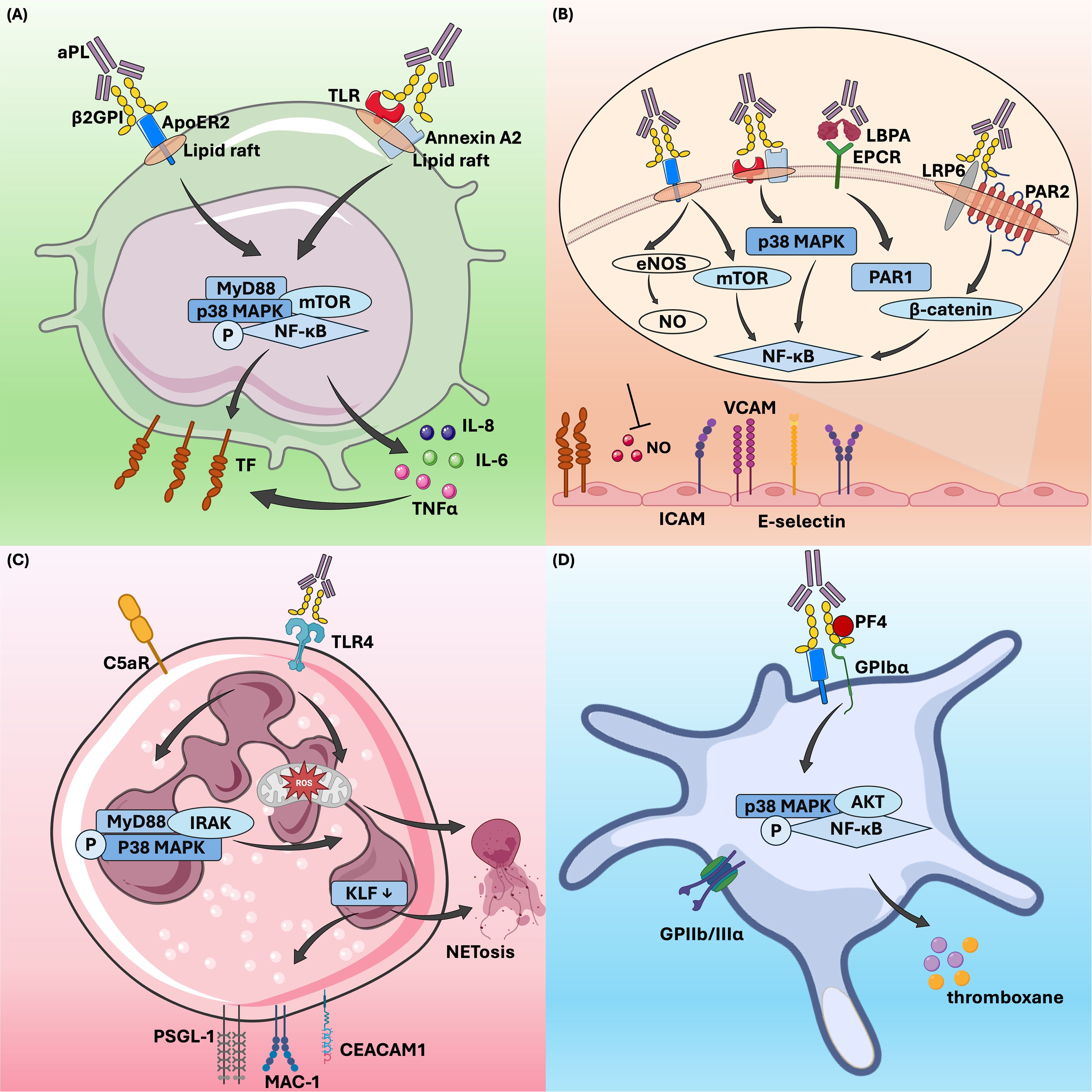
Figure 1. β2GPI-dependent aPL effects on cells in thrombotic antiphospholipid syndrome. β2GPI-dependent aPL activates monocytes (A), endothelial cells (B), neutrophils (C), and platelets (D) through various signaling pathways, thereby regulating downstream cellular activities and contributing to thrombosis in APS. aPL, antiphospholipid antibody;β2GPI, β2-glycoprotein I; ApoER2, apolipoprotein E receptor 2; TLR, toll-like receptor; MyD88, myeloid differentiation primary response gene 88; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa B; AKT, protein kinase B; TF, tissue factor; TNFα, tumor necrosis factor-alpha; LBPA, lysobisphosphatidic acid; EPCR, endothelial protein C receptor; LRP6, LDL receptor-related protein 6; PAR1, protease-activated receptor 1;eNOS, endothelial nitric oxide synthase; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; ROS, reactive oxygen species; NETs, neutrophil extracellular traps; KLF, krüppel-like factor 2; PSGL-1, P-selectin glycoprotein ligand 1; IRAK, interleukin-1 receptor-associated kinase; MAC-1, macrophage-1 antigen; CEACAM1, carcinoembryonic antigen related cell adhesion molecule 1; PF4, platelet factor 4; GPIIbIIIα, glycoprotein IIIbIIIα.
4.2.1 On endothelial cells: expression of adhesion molecules and procoagulant substances
Quiescent endothelial cells are crucial for maintaining blood flow by expressing anticoagulant proteins and generating an actively antithrombotic surface within blood vessels (97). However, when endothelial cells are disrupted, their membranes can shift from an anticoagulant surface to a procoagulant phenotype. This change is mainly characterized by the induction of tissue factor (TF), plasminogen activator inhibitors, and the synthesis of specific binding sites for coagulation factors (97). TF is a single-stranded transmembrane glycoprotein that acts as a key initiator of the blood coagulation cascade by binding to factor VIIa (98, 99). Under normal conditions, TF is not expressed by intravascular cells but can be induced in monocytes and endothelial cells in response to nonphysiological or pathophysiological stimulation (100, 101). Numerous studies have shown that aPL, especially anti-β2GPI antibodies, can activate endothelial cells, promoting thrombosis in APS (102–104). Endothelial cells activated by aPL display increased expression of adhesion molecules, such as E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 (102, 105).
The induction of TF expression in endothelial cells by antiphospholipid sera was reported in 1993 (106). Subsequent in vitro studies have confirmed that aPL can induce the expression of TF in endothelial cells (107, 108). Additionally, aPL-activated endothelial cells contribute to a prothrombotic state through mechanisms such as releasing microparticles with proinflammatory and procoagulant properties (109, 110), producing proinflammatory cytokines (111), and reducing levels of endothelial cell-derived nitric oxide (112).
There are multiple pathways through which activated endothelial cells mediate the prethrombotic state in APS. Currently, it is accepted that the binding of β2GPI -antibody complexes to Annexin A2 or toll-like receptor (TLR) on endothelial cell surfaces triggers the activation of the p38 mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-κB) signaling pathways, which in turn promote the expression of procoagulant substances (29, 113, 114). Annexin A2 has been identified as a significant aPL receptor on endothelial cell membranes and is essential in endothelial cell activation and thrombosis in APS (95, 115). Research suggested that Annexin A2 might be part of a larger aPL receptor complex on endothelial cells, potentially forming co-receptors with TLR2 or TLR4 to mediate this activation (116, 117). In contrast, Annexin V, another member of the annexin family, acts protectively by blocking aPL from binding to phospholipids on the cell membrane. These antibodies cannot interact with endothelial cells unless Annexin V is interrupted (118). Endothelial protein C receptor (EPCR) serves as a receptor for β2GPI/anti-β2GPI antibody complexes in APS, playing crucial roles in anticoagulation and placental development. In APS, anti-EPCR antibodies can inhibit protein C activation, thereby increasing the risk of fetal loss and thrombotic events (119, 120). Binding of aPL to EPCR accelerates the endocytosis of the EPCR- lysobisphosphatidic acid complex, which leads to thrombin-induced and protease-activated receptor 1-mediated endothelial cell activation (121, 122). Furthermore, different EPCR haplotypes, particularly the H1 haplotype, can influence APS symptoms, modulating the risk of arterial thrombosis (123). Apolipoprotein E receptor 2 (ApoER2), also known as LDL receptor-related protein 8 (LRP8), is a member of the low-density lipoprotein receptor family and plays a crucial role in the pathogenic mechanism of aPL. Compared to ApoER2+/+ mice, ApoER2-/- mice exhibited prolonged vascular occlusion times induced by aPL, along with significant reductions in aPL-induced vascular TF activity, thrombosis formation, and monocyte activation (30). ApoER2 also participates in the inhibition of endothelial cell migration and regeneration by aPL. In cell experiments conducted by Ulrich V. et al., siRNA knockdown of ApoER2 in endothelial cells restored migration capacity, which had been suppressed by aPL (124). In APS, ApoER2 facilitates thrombosis by serving as a scaffold for the assembly of protein phosphatase 2A, leading to antagonism of endothelial nitric oxide synthase (eNOS) (125). This mechanism involves the recruitment of Disabled-2 and Src homology domain-containing transforming protein 1 to ApoER2, which activates protein phosphatase 2A and promotes the dephosphorylation of protein kinase B (AKT) and eNOS, ultimately contributing to aPL-induced thrombosis (40).
4.2.2 On monocytes: induction of tissue factor
Monocytes contribute to thrombosis in APS primarily through the expression of TF (126). Kornberg et al. demonstrated that murine monoclonal aCL can induce TF expression in monocytes and enhance TF procoagulant activity (127). Later, Guadrado et al. found that monocytes from APS patients with thrombosis showed significantly increased TF mRNA expression and TF-related procoagulant activity compared to those without thrombosis (128). Subsequent studies confirmed that human monoclonal aCL and IgG from patients with APS promote TF expression and boost TF activity in monocytes (129, 130). Extensive research has since established that aPL, especially anti-β2GPI antibodies, can induce TF expression on monocytes when they form β2GPI/anti-β2GPI immune complexes (131–134). These complexes interact with various cell surface receptors, such as phosphatidylserine, ApoER2, and Annexin A2, as well as coreceptors like TLRs (135). These interactions activate signaling pathways, including mitogen-activated protein kinase kinase 1 (MEK-1)/extracellular regulated protein kinases (ERK), p38 MAPK, mammalian target of rapamycin (mTOR), and NF-κB, predominantly through the TLR4-myeloid differentiation primary response 88 (MyD88) pathway (131–134, 136–138). Furthermore, the β2GPI/anti-β2GPI complexes upregulate inflammatory cytokines through the TLR/MyD88 and NF-κB pathways, leading to increase TF expression in monocytes (134, 139). In vitro studies demonstrated that monocytes from healthy donors incubated with monoclonal aPL or affinity-purified anti-β2GPI antibodies showed significantly higher secretion of tumor necrosis factor-alpha (TNFα) compared to those incubated with control IgG, further amplifying TF expression (134, 140, 141).
The expression of TF is also regulated by tissue factor pathway inhibitor (TFPI), which inhibits factors VIIa and Xa. One study revealed that anti-β2GPI antibodies suppress TFPI activity, thereby enhancing factor Xa generation (142). Notably, recent findings showed that aPL dissociated TFPI from monocytes, increasing the risk of thrombosis in APS patients (142). Furthermore, protease-activated receptors (PARs), which are triggered by thrombin or factor X, contribute to the production of pro-inflammatory cytokine (135). It was reported that the expression of PARs was elevated in monocytes from APS patients, and inhibition of PAR2 prevents aPL-induced TF expression (143).
4.2.3 On neutrophil: increased release of neutrophil extracellular traps
Neutrophils are the most abundant leukocyte subsets in human peripheral blood and play an important role in innate immune against the invasion of various pathogenic microbes (144). They defend against external pathogens through multiple mechanisms, including the release of neutrophil extracellular traps (NETs), which are meshwork substances composed of DNA, histones, and antibacterial proteins (145). The release of NETs is the most striking phase in a unique cell death process called NETosis, distinct from apoptosis and necrosis (146). NET formation is a double-edged sword, playing a role in the pathogenesis of inflammatory and autoimmune disorders (147, 148).
Compared to the extensive research on the aforementioned cell types, the interactions between aPL and neutrophils have been less thoroughly investigated. Initially, only two studies demonstrated that aPL could directly activate neutrophils (149, 150), a process that may be amplified by complement C5, thereby promoting blood coagulation (151). With the growing recognition of the connection between NETs and thrombosis in non-autoimmune contexts, increasing attention has been given to the role of neutrophils in APS-related thrombosis (152, 153). Leffler et al. discovered that APS sera exhibited an impaired ability to degrade NETs, a dysfunction associated with specific clinical features (154). In recent years, Knight’s group has conducted several pivotal studies examining the role of neutrophils in APS-related thrombosis. In a 2015 study, they identified elevated levels of NETs in the circulation of APS patients and demonstrated that anti-β2GPI IgG could promote NET release in vitro (155). Moreover, these aPL-stimulated NETs were shown to facilitate thrombin generation in vitro (155), introducing a novel mechanism of thrombosis in patients with APS (156). Knight and his team later confirmed the in vivo relevance of NETs in thrombosis using a mouse model of APS. In this model, they observed that APS-associated thrombi were enriched with NETs and that NET-disrupting treatments could prevent APS IgG-mediated thrombosis (43). To further understand the mechanism of neutrophil hyperactivity in APS, they conducted a transcriptome analysis of APS neutrophils. This analysis revealed a proinflammatory gene expression signature, with overexpressed genes linked to interferon signaling, cellular defense, and intercellular adhesion (44). Among the upregulated genes, they identified a notable group of leukocyte immunoglobulin-like receptor (LILR) genes, which included all activating members of the LILR family: LILRA1, LILRA2, LILRA3, LILRA4, LILRA5, and LILRA6 (44). The role of these LILR members in regulating thromboinflammation in APS remains unclear. More recently, Knight’s group has showed that neutrophils in APS exhibit increased adhesive potential, lowering the threshold for NETosis and elevating the risk of thrombotic events (157). Additionally, a specific neutrophil subgroup known as low-density granulocytes, which is elevated in APS, exhibits a higher propensity for NET release (158). Mechanistically, anti-β2GPI antibodies promote NETs formation in a time- and concentration-dependent manner by forming complexes with β2GPI, activating TLR4, triggering ROS production, and inducing NET release via the MyD88-IRAKs-MAPKs pathway (159). Targeting NET release, for instance, by activating adenosine receptor, has been shown to reduce thrombosis in APS mouse models (46).
In APS, neutrophils exhibit an increased tendency to interact with endothelial cells, driven by the upregulation of adhesion molecules induced by aPL. The loss of the transcription factor Krüppel-like factor 2 (KLF2) is a key factor that transforms neutrophils into a prothrombotic state, enhancing their migration, adhesion, and release of factors like P-selectin glycoprotein ligand 1 (PSGL-1), which facilitates binding to the endothelium (160). Neutrophils in APS exhibit lower KLF2 levels, resulting in increased clustering of PSGL-1, elevated NET formation, and higher TF activity. Additionally, these neutrophils also upregulate other adhesion proteins, including CD64, carcinoembryonic antigen related cell adhesion molecule-1, β2-glycoprotein, and activated macrophage-1 antigen, all of which contribute to their heightened thrombotic potential (157).
4.2.4 On platelets: enhanced platelet activation/aggregation
Platelets are essential for physiological hemostasis, as they aggregate and adhere to injured vessel walls, become activated, and release granule, promoting blood clotting and thrombus formation. Thrombocytopenia is a common clinical feature in patients with APS, leading to the hypothesis that aPL may bind to platelets, causing aggregation and thrombosis. The proposed causes of thrombocytopenia include platelet destruction by autoantibodies targeting platelet glycoproteins, as well as platelet activation and depletion initiated by aPL (161). Studies have confirmed that platelet activation in patients with APS is primarily linked to β2GPI-dependent antiphospholipid antibodies (33, 162, 163). The binding of anti-β2GP auto-antibody/β2GPI immune complex to ApoER2 and glycoprotein Ibα can promote platelet activation (164–167). These receptors cross-link with anti-β2GPI, activating platelets and promoting the release of thromboxane A2. This process neutralizes the inhibitory effect of β2GPI on von Willebrand factor, thereby enhancing platelet adhesion and aggregation (166–168). Platelet factor 4 (PF4) is a specific protein released by activated platelets that promotes thrombosis. In patients with APS, β2GPI can form a stable complex with PF4. The binding of anti-β2GPI antibody to the complex triggers p38 MAPK phosphorylation and induces the production of thromboxane B2 (169). In a mouse model using fluorescently labeled β2GPI and anti-β2GPI autoantibodies, Proulle et al. found that platelets, rather than endothelial cells, were the first effector cells activated by the anti-β2GPI antibody/β2GPI complex (37). Moreover, the anti-β2GPI antibody/β2GPI complex enhances platelet activation, and the secretion from activated platelets promotes endothelial cell activation (37). These findings underscore the critical role of platelets in the pathogenesis of APS.
4.2.5 Procoagulant signaling in plasma membrane microdomains: focus on lipid rafts and heparanase
Although soluble coagulation factors and intracellular signaling pathways are well understood in APS pathogenesis, recent study showed that plasma membrane microdomains play an important role in regulating prothrombotic events. Specifically, lipid rafts and heparanase, despite working through different mechanisms, worsens the risk of thrombosis in APS.
Lipid rafts are believed to play an important role in the development of APS. They are small (10-200 nm) non-homogeneous regions of the membrane, rich in glycosphingolipids and cholesterol (170). In APS, studies have shown that anti-β2GPI antibodies bind to their target antigens, such as β2GPI, annexin A2, TLR2, and TLR4, within lipid rafts in the plasma membrane of monocytes or endothelial cells (133). Biochemical analyses revealed that β2GPI exists in a dimeric form within lipid rafts. This suggests that β2GPI can only interact with lipid rafts after it dimerizes, possibly due to conformational changes (171). Further studies showed that lipid rafts play a key role in the signaling pathway triggered by anti-β2GPI antibodies. These antibodies activate IRAK and NF-κB through lipid rafts, leading to the release of TNF-α and TF, which contribute to a proinflammatory and procoagulant state (172, 173). Additionally, it has been found that anti-β2GPI antibodies activate LRP6 and LRP8/ApoER2 signaling in endothelial cells through lipid rafts (173, 174). Riitano et al. used a raft-disrupting agent called methyl-β-cyclodextrin (MβCD) to further study the role of lipid rafts. The results revealed that anti-β2GPI antibodies induce TF expression in endothelial cells through the LRP8/ApoER2 signaling pathway, which requires intact lipid rafts for efficient signal transduction (174). These findings underline the importance of lipid rafts in APS and suggest potential targets for therapeutic intervention in APS.
Heparanase is the only known enzyme that cleaves heparan sulfate side chains, contributing to inflammatory disorders by aiding vascular endothelial cell migration and immune cell activation (175). It affects coagulation by upregulating TF expression and interacting with TFPI, increasing coagulation activity (176). Heparanase also acts as a cofactor for TF, promoting factor Xa production (177). In APS, inhibiting heparanase, such as RDS3337, prevents TF expression and platelet aggregation (175), suggesting that targeting heparanase could be a potential treatment for APS-related prothrombotic conditions.
4.3 Complement activation
Given the established interactions between the complement and the coagulation systems, complement components may be directly involved in thrombosis (178). Supporting this view, injecting IgG aPL purified from APS patients into complement C6-/- rats does not induce thrombosis in the mesenteric blood vessels but does cause blood clots in normal rats (57). A subsequent study replicated these findings in C6 knockout mice (179). Activation of complement components C3 and C5 can enhance aPL-mediated thrombosis and activate endothelial cells (28). APL-induced complement activation generates downstream C5a, which recruits and activates neutrophils, resulting in TF expression (180, 181). These animal experiments confirmed the critical role of complement in aPL-mediated thrombosis. Several studies have shown significantly elevated levels of complement activation products (fragments Bb and C3a-desArg) in patients with APS, which correlate with increased aPL titers (182, 183). Nevertheless, the underlying mechanisms driving complement activation in patients with APS are not yet fully understood. Recent study indicated that reduced levels of complement factor H (FH), a key regulatory factor, may contribute to complement activation in APS (184). A prior study used ELISA to detected FH autoantibodies in APS patients from Serbian and Italian cohorts, suggesting these autoantibodies may contribute to reduced FH levels (185). In 2016, a case report provided evidence that complement directly impacted human thrombosis. The report described an APS patient undergo femoral artery bypass surgery for vascular thrombosis, with complement deposition observed on the endothelium and vascular wall at the thrombotic site (186). Treatment with a humanized anti-C5a monoclonal antibody (eculizumab) effectively prevented thrombosis following the surgery (186). Furthermore, FXa and thrombin can activate the complement pathway by cleaving C3 and C5. However, β2GPI inhibits complement activation by modulating the activities of thrombin and FXa (187, 188). This interplay between coagulation factors and β2GPI may indicate a potential mechanism for complement activation.
Complement activation is considered a significant factor in early pregnancy loss (189, 190). Shamonki et al. conducted an immunohistochemical analysis using antibodies against C4d, C3b, and C5b-9 on placental tissue from APS patients and controls, highlighting complement’s crucial role in APS-related fetal tissue damage (191). Elevated levels of Bb and sC5b-9 detected in early pregnancy have been strongly associated with negative pregnancy outcomes in patients with aPL (192). Studies indicated that inhibiting the complement cascade with C3 convertase inhibitors or blocking C5a receptor interactions could mitigate the harmful effects of aPL in early pregnancy (193). Further evidence showed that heparins alleviated early pregnancy complications mainly by inhibiting aPL-induced complement activation, rather than through their anticoagulant properties (194). Genetic mutations that cause immune dysregulation are associated with complement-mediated diseases (195), and approximately 60% of catastrophic APS patients have germline variants in complement regulatory genes (196).
5 Two-hit model
Thrombotic events are infrequent, even with the persistent presence of aPL. Moreover, aPL alone do not seem to induce thrombotic phenotypes (197). The ‘two hit’ hypothesis, proposed in 2001, explains these clinical and experimental observations (198). The presence of aPL (the first hit) increases the risk of thrombophilia, while clotting occurs when an additional procoagulant condition (second hit) is present (Figure 2) (198). The ‘two-hit’ model was later validated using a photochemically induced thrombosis model in hamsters (33). A low dose of bacterial lipopolysaccharide was necessary for β2GPI-dependent aPL to trigger thrombosis in rat mesenteric microcirculation (57). Consistent with this finding, infection might act as a second hit, amplifying the thrombophilic effect of aPL (199).
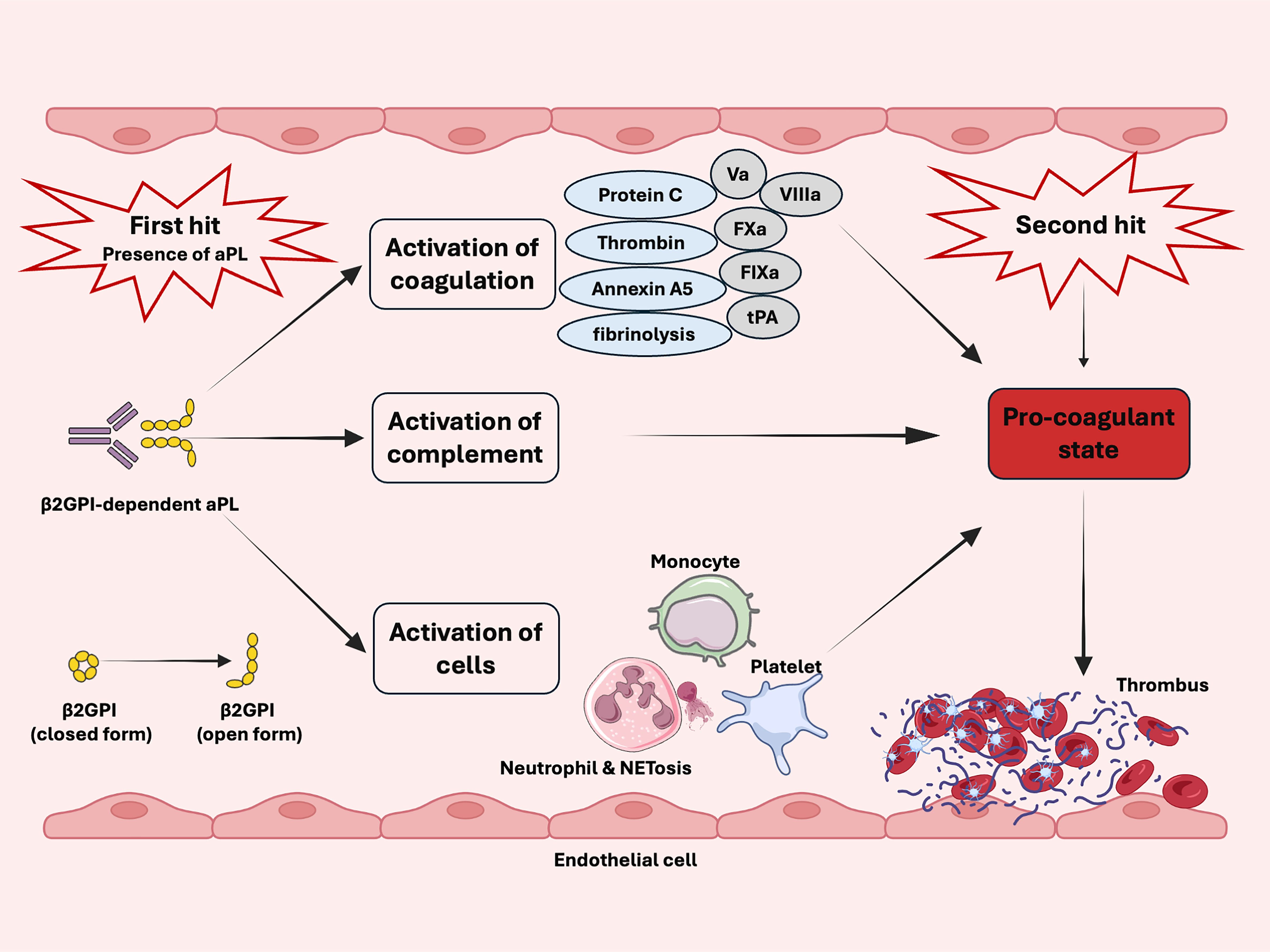
Figure 2. Schematic diagram of two-hit model in antiphospholipid syndrome. The presence of aPL (first hit) and the activation of the coagulation system, complement, monocytes, endothelial cells, neutrophils, and platelets increase the risk of thrombosis. Clotting occurs in the presence of additional procoagulant condition (second hit).
The presence of aPL is recognized as necessary but insufficient from thrombosis in APS while an additional ‘second hit’ is required (18, 22, 23, 47, 72, 200). The second hit often includes but not limited to inflammatory responses, mechanical trauma, immobility, venous stasis and estrogen-containing contraceptive (22, 47, 72). In addition, cardiovascular risk factors, such as arterial hypertension, diabetes, obesity, smoking, and hyperlipidemia, further increase the risk of thrombosis (72, 200).
6 Current and potential drugs for APS
The management of thrombotic APS focuses on preventing thrombotic events and addressing the multifaceted pathogenesis of the disease. Current strategies emphasize anticoagulation as the cornerstone of therapy, targeting the hypercoagulable state driven by aPL. Standard regimens typically include vitamin K antagonists and low-molecular-weight heparin (LMWH), with close monitoring of the international normalized ratio (INR) to ensure an appropriate balance between thromboprophylaxis and the risk of bleeding. Immunomodulation plays a critical role in suppressing autoimmune-driven pathways, particularly in patients with concurrent SLE or refractory cases. Table 1 summarizes the primary therapeutic agents for preventing thrombotic events in APS.
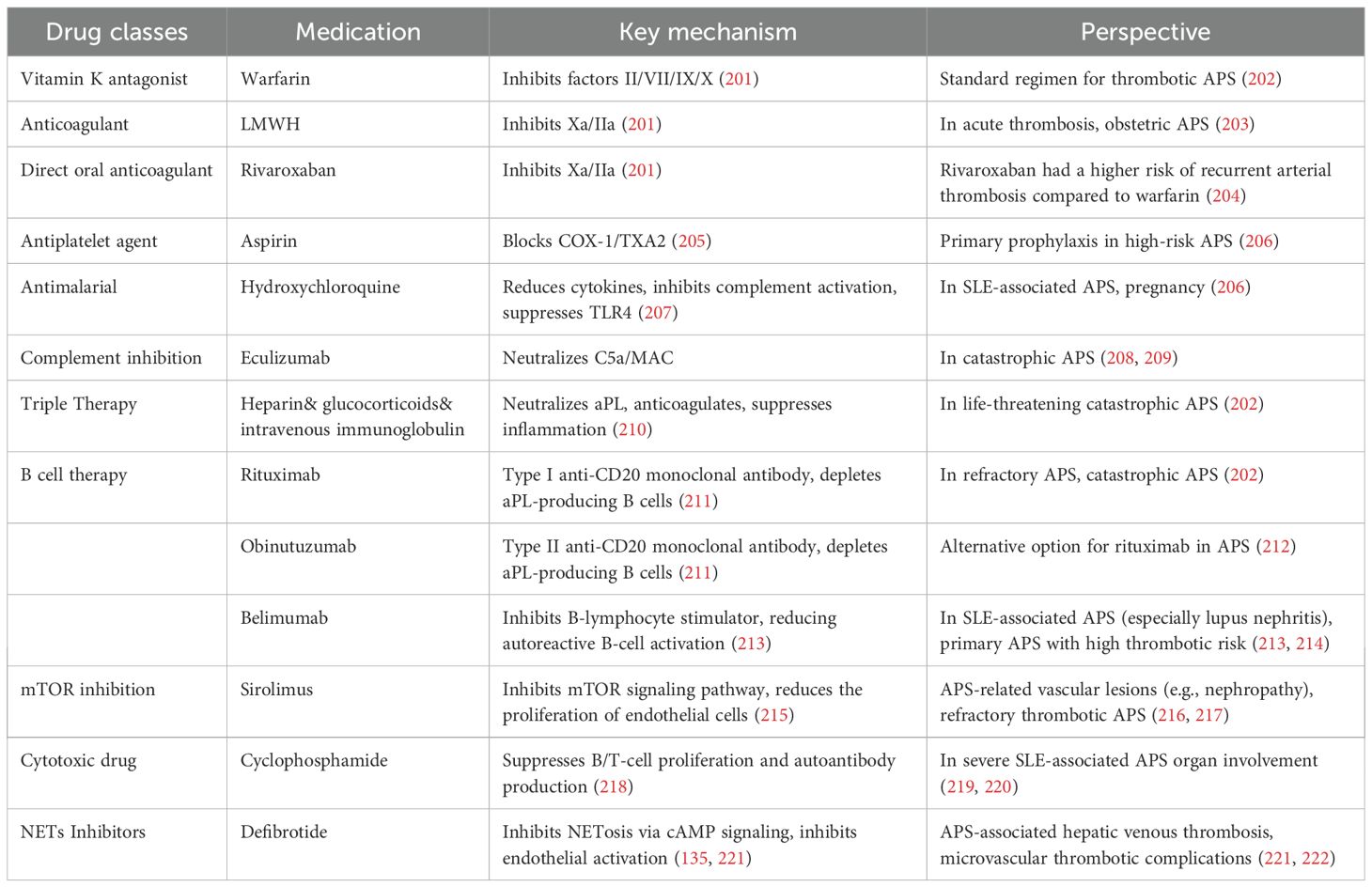
Table 1. Drugs used in the management of APS.
While anticoagulants remain foundational, their inability to prevent non-thrombotic complications underlines the need for potential therapies. Potential approaches aim to tackle non-thrombotic complications and specific pathogenic mechanisms that lead diseases. Among these strategies, statins have shown dual benefits by not only improving endothelial function through lipid-lowering effects but also suppressing aPL-induced pro-inflammatory signaling (e.g., NF-κB and MAPK pathways), thereby reducing TF expression and monocyte activation (223). Direct targeting of coagulation activation is exemplified by anti-TF molecules (e.g., ALT-836), which inhibit thrombin generation by neutralizing TF upregulated in monocytes (224). In addition, interventions against NETs, such as DNase-mediated degradation, PAD4 inhibition, and histone toxicity blockade, aim to mitigate NETs-driven thromboinflammation and placental damage (225). To disrupt the core antigen-antibody interaction in APS, anti-β2GPI domain I monoclonal antibodies competitively block pathogenic aPL binding to β2GPI, preventing its engagement with cellular receptors like ApoER2 and subsequent complement activation (226, 227). Further upstream, lipid raft-targeting agents (e.g., MβCD) destabilize membrane microdomains essential for β2GPI anchoring, thereby suppressing platelet activation and complement deposition (173). Beyond thrombotic pathways, immune modulation is being explored through probiotics and vitamin D3, which restore gut microbiota balance, regulate Th17/Treg dynamics, and suppress autoreactive B-cell responses (202, 228, 229). Collectively, these therapies exemplify a paradigm shift from broad anticoagulation to mechanistically precise interventions targeting aPL-mediated thrombosis, autoantibody pathogenicity, and immune dysregulation.
7 Conclusion and future directions
Despite the strong association between aPL and thrombosis, the exact mechanisms underlying aPL-mediated thrombotic events remain unclear. Current evidence suggests that aPL triggers activation of endothelial cells, monocytes, neutrophils, and platelets. This activation, coupled with the disruption of natural anticoagulant and fibrinolytic pathways, leads to a pro-thrombotic state in patients with thrombotic APS. An additional triggering event, or ‘second hit,’ is ultimately necessary to initiate thrombus formation.
Despite extensive research efforts, several crucial questions remain unanswered. One unresolved question is which cell type serves as the primary target for aPL. Some studies argue that endothelial cells play a key role in APS-associated thrombosis (104), while others downplay the role of the endothelium and emphasize the importance of platelets (37). Recent insights suggest that anti-inflammatory treatments targeting NETosis may be more effective than conventional anticoagulation therapy in reducing thrombosis (230). It is also possible that all of these cell types contribute, either directly or indirectly, through the release of prothrombotic microparticles (231, 232). While it is well-established that a ‘second hit,’ such as mechanical trauma combined with antibody binding to β2GPI on the endothelium, plays a key role in initiating clot formation at specific sites (18), it remains obscure how other types of ‘second hit’ trigger thrombosis. In addition, β2GPI, the primary antibody target, is a complex protein with an unclear physiological role (62). Understanding the precise function of β2GPI could be crucial for unraveling the mechanism of thrombosis. Finally, a consensus on the optimal animal model that accurately replicates APS pathophysiology is urgently needed to improve in vivo experiments. Advances in APS research will deepen our understanding of the underlying mechanisms and contribute to future treatments.
Author contributions
LY: Visualization, Writing – original draft, Writing – review & editing. RG: Writing – original draft, Writing – review & editing. HL: Writing – original draft, Writing – review & editing. BC: Visualization, Writing – review & editing. CL: Writing – review & editing. RL: Writing – review & editing. SL: Visualization, Writing – review & editing. QX: Writing – review & editing. GY: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study is supported by Sichuan Province Science and Technology Program (2024YFFK0349, 2024YFFK0062), the Postdoctor Research Fund of West China Hospital, Sichuan University (2024HXBH084), China Postdoctoral Science Foundation (2024M752245), and Clinical Research Incubation Project of West China Hospital (2021HXFH018).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. (2006) 4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x
3. Giannakopoulos B, Passam F, Ioannou Y, Krilis SA. How we diagnose the antiphospholipid syndrome. Blood. (2009) 113:985–94. doi: 10.1182/blood-2007-12-129627
4. Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, de Ramón E, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. (2015) 74:1011–8. doi: 10.1136/annrheumdis-2013-204838
5. Dabit JY, Valenzuela-Almada MO, Vallejo-Ramos S, Duarte-García A. Epidemiology of antiphospholipid syndrome in the general population. Curr Rheumatol Rep. (2022) 23:85. doi: 10.1007/s11926-021-01038-2
6. Rodríguez-Pintó I, Moitinho M, Santacreu I, Shoenfeld Y, Erkan D, Espinosa G, et al. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun Rev. (2016) 15:1120–4. doi: 10.1016/j.autrev.2016.09.010
7. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol. (2016) 28:218–27. doi: 10.1097/BOR.0000000000000269
8. Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheumatol. (2002) 46:1019–27. doi: 10.1002/art.10187
9. Moschetti L, Dal Pozzolo L, Le Guern V, Morel N, Yelnik CM, Lambert M, et al. Gender differences in primary antiphospholipid syndrome with vascular manifestations in 433 patients from four European centres. Clin Exp Rheumatol. (2022) 40 Suppl 134:19–26. doi: 10.55563/clinexprheumatol/9royri
10. Abou-Ismail MY, Citla D Sridhar, Nayak L. Estrogen and thrombosis: A bench to bedside review. Thromb Res. (2020) 192:40–51. doi: 10.1016/j.thromres.2020.05.008
11. Bukhari S, Fatima S, Barakat AF, Fogerty AE, Weinberg I, Elgendy IY. Venous thromboembolism during pregnancy and postpartum period. Eur J Intern Med. (2022) 97:8–17. doi: 10.1016/j.ejim.2021.12.013
12. Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. (2010) 376:1498–509. doi: 10.1016/S0140-6736(10)60709-X
13. Alarcón-Segovia D, Sanchez-Guerrero J. Primary antiphospholipid syndrome. J Rheumatol. (1989) 16:482–8.
14. Vianna JL, Khamashta MA, Ordi-Ros J, Font J, Cervera R, Lopez-Soto A, et al. Comparison of the primary and secondary antiphospholipid syndrome: a European Multicenter Study of 114 patients. Am J Med. (1994) 96:3–9. doi: 10.1016/0002-9343(94)90108-2
15. Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. (2002) 346:752–63. doi: 10.1056/NEJMra002974
16. Barbhaiya M, Zuily S, Naden R, Hendry A, Manneville F, Amigo MC, et al. ACR/EULAR antiphospholipid syndrome classification criteria. Ann Rheum Dis. (2023) 82:1258–70. doi: 10.1136/ard-2023-224609
17. Garcia D, Erkan D. Diagnosis and management of the antiphospholipid syndrome. N Engl J Med. (2018) 378:2010–21. doi: 10.1056/NEJMra1705454
18. Meroni PL, Borghi MO, Grossi C, Chighizola CB, Durigutto P, Tedesco F. Obstetric and vascular antiphospholipid syndrome: same antibodies but different diseases? Nat Rev Rheumatol. (2018) 14:433–40. doi: 10.1038/s41584-018-0032-6
19. Cervera R. Antiphospholipid syndrome. Thromb Res. (2017) 151 Suppl 1:S43–s7. doi: 10.1016/S0049-3848(17)30066-X
20. Abreu MM, Danowski A, Wahl DG, Amigo MC, Tektonidou M, Pacheco MS, et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid Antibodies Technical Task Force Report on Antiphospholipid Syndrome Clinical Features. Autoimmun Rev. (2015) 14:401–14. doi: 10.1016/j.autrev.2015.01.002
21. McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: beta 2-glycoprotein I (apolipoprotein H). Proc Natl Acad Sci USA. (1990) 87:4120–4. doi: 10.1073/pnas.87.11.4120
22. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. (2011) 7:330–9. doi: 10.1038/nrrheum.2011.52
23. Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. (2013) 368:1033–44. doi: 10.1056/NEJMra1112830
24. de Groot PG, Urbanus RT. The significance of autoantibodies against β2-glycoprotein I. Blood. (2012) 120:266–74. doi: 10.1182/blood-2012-03-378646
25. Pierangeli SS, Barker JH, Stikovac D, Ackerman D, Anderson G, Barquinero J, et al. Effect of human IgG antiphospholipid antibodies on an in vivo thrombosis model in mice. Thromb Haemost. (1994) 71:670–4. doi: 10.1055/s-0038-1642501
26. Pierangeli SS, Liu XW, Barker JH, Anderson G, Harris EN. Induction of thrombosis in a mouse model by IgG, IgM and IgA immunoglobulins from patients with the antiphospholipid syndrome. Thromb Haemost. (1995) 74:1361–7. doi: 10.1055/s-0038-1649940
27. Pierangeli SS, Colden-Stanfield M, Liu X, Barker JH, Anderson GL, Harris EN. Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation. (1999) 99:1997–2002. doi: 10.1161/01.CIR.99.15.1997
28. Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheumatol. (2005) 52:2120–4. doi: 10.1002/art.21157
29. Pierangeli SS, Vega-Ostertag ME, Raschi E, Liu X, Romay-Penabad Z, De Micheli V, et al. Toll-like receptor and antiphospholipid mediated thrombosis: in vivo studies. Ann Rheum Dis. (2007) 66:1327–33. doi: 10.1136/ard.2006.065037
30. Romay-Penabad Z, Aguilar-Valenzuela R, Urbanus RT, Derksen RH, Pennings MT, Papalardo E, et al. Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood. (2011) 117:1408–14. doi: 10.1182/blood-2010-07-299099
31. Pericleous C, Ruiz-Limón P, Romay-Penabad Z, Marín AC, Garza-Garcia A, Murfitt L, et al. Proof-of-concept study demonstrating the pathogenicity of affinity-purified IgG antibodies directed to domain I of β2-glycoprotein I in a mouse model of anti-phospholipid antibody-induced thrombosis. Rheumatol (Oxford). (2015) 54:722–7. doi: 10.1093/rheumatology/keu360
32. Kawasaki T, Kaida T, Arnout J, Vermylen J, Hoylaerts MF. A new animal model of thrombophilia confirms that high plasma factor VIII levels are thrombogenic. Thromb Haemost. (1999) 81:306–11. doi: 10.1055/s-0037-1614471
33. Jankowski M, Vreys I, Wittevrongel C, Boon D, Vermylen J, Hoylaerts MF, et al. Thrombogenicity of beta 2-glycoprotein I-dependent antiphospholipid antibodies in a photochemically induced thrombosis model in the hamster. Blood. (2003) 101:157–62. doi: 10.1182/blood-2002-05-1310
34. Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. (2002) 8:1175–81. doi: 10.1038/nm782
35. Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. (2008) 118:1123–31. doi: 10.1172/JCI34134
36. Arad A, Proulle V, Furie RA, Furie BC, Furie B. beta(2)-Glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood. (2011) 117:3453–9. doi: 10.1182/blood-2010-08-300715
37. Proulle V, Furie RA, Merrill-Skoloff G, Furie BC, Furie B. Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood. (2014) 124:611–22. doi: 10.1182/blood-2014-02-554980
38. Seshan SV, Franzke CW, Redecha P, Monestier M, Mackman N, Girardi G. Role of tissue factor in a mouse model of thrombotic microangiopathy induced by antiphospholipid antibodies. Blood. (2009) 114:1675–83. doi: 10.1182/blood-2009-01-199117
39. Laplante P, Fuentes R, Salem D, Subang R, Gillis MA, Hachem A, et al. Antiphospholipid antibody-mediated effects in an arterial model of thrombosis are dependent on Toll-like receptor 4. Lupus. (2016) 25:162–76. doi: 10.1177/0961203315603146
40. Sacharidou A, Chambliss KL, Ulrich V, Salmon JE, Shen YM, Herz J, et al. Antiphospholipid antibodies induce thrombosis by PP2A activation via apoER2-Dab2-SHC1 complex formation in endothelium. Blood. (2018) 131:2097–110. doi: 10.1182/blood-2017-11-814681
41. Manukyan D, Müller-Calleja N, Jäckel S, Luchmann K, Mönnikes R, Kiouptsi K, et al. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J Thromb Haemost. (2016) 14:1011–20. doi: 10.1111/jth.13263
42. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
43. Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. (2017) 69:655–67. doi: 10.1002/art.39938
44. Knight JS, Meng H, Coit P, Yalavarthi S, Sule G, Gandhi AA, et al. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight. (2017) 2:e93897. doi: 10.1172/jci.insight.93897
45. Palmer OR, Shaydakov ME, Rainey JP, Lawrence DA, Greve JM, Diaz JA. Update on the electrolytic IVC model for pre-clinical studies of venous thrombosis. Res Pract Thromb Haemost. (2018) 2:266–73. doi: 10.1002/rth2.12074
46. Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun. (2019) 10:1916. doi: 10.1038/s41467-019-09801-x
47. Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, et al. Antiphospholipid syndrome. Nat Rev Dis Primers. (2018) 4:17103. doi: 10.1038/nrdp.2017.103
48. Rodriguez-Garcia JL, Bertolaccini ML, Cuadrado MJ, Sanna G, Ateka-Barrutia O, Khamashta MA. Clinical manifestations of antiphospholipid syndrome (APS) with and without antiphospholipid antibodies (the so-called ‘seronegative APS’). Ann Rheum Dis. (2012) 71:242–4. doi: 10.1136/annrheumdis-2011-200614
49. Hughes GR, Khamashta MA. Seronegative antiphospholipid syndrome. Ann Rheum Dis. (2003) 62:1127. doi: 10.1136/ard.2003.006163
51. Zohoury N, Bertolaccini ML, Rodriguez-Garcia JL, Shums Z, Ateka-Barrutia O, Sorice M, et al. Closing the serological gap in the antiphospholipid syndrome: the value of “Non-criteria” Antiphospholipid antibodies. J Rheumatol. (2017) 44:1597–602. doi: 10.3899/jrheum.170044
52. Zhang S, Zhang F, Li Y. Should aPS/PT Be Incorporated into the Routine Serological Tests in the Diagnosis of Antiphospholipid Syndrome? J Rheumatol. (2019) 46:114–6. doi: 10.3899/jrheum.171402
53. Sciascia S, Khamashta MA, Bertolaccini ML. New tests to detect antiphospholipid antibodies: antiprothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT) antibodies. Curr Rheumatol Rep. (2014) 16:415. doi: 10.1007/s11926-014-0415-x
54. Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, van-Breda-Vriesman PJ, et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. (1990) 335:1544–7. doi: 10.1016/0140-6736(90)91374-J
55. Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K, Koike T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet. (1990) 336:177–8. doi: 10.1016/0140-6736(90)91697-9
56. Bai A. [amp]]beta;2-glycoprotein I and its antibodies involve in the pathogenesis of the antiphospholipid syndrome. Immunol Lett. (2017) 186:15–9. doi: 10.1016/j.imlet.2017.03.013
57. Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. (2005) 106:2340–6. doi: 10.1182/blood-2005-03-1319
58. Yasuda S, Atsumi T, Ieko M, Koike T. Beta2-glycoprotein I, anti-beta2-glycoprotein I, and fibrinolysis. Thromb Res. (2004) 114:461–5. doi: 10.1016/j.thromres.2004.07.013
59. Schousboe I. beta 2-Glycoprotein I: a plasma inhibitor of the contact activation of the intrinsic blood coagulation pathway. Blood. (1985) 66:1086–91. doi: 10.1182/blood.V66.5.1086.1086
60. Nakagawa H, Yasuda S, Matsuura E, Kobayashi K, Ieko M, Kataoka H, et al. Nicked {beta}2-glycoprotein I binds angiostatin 4.5 (plasminogen kringle 1-5) and attenuates its antiangiogenic property. Blood. (2009) 114:2553–9. doi: 10.1182/blood-2008-12-190629
61. Gropp K, Weber N, Reuter M, Micklisch S, Kopka I, Hallström T, et al. [amp]]beta;₂-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. (2011) 118:2774–83. doi: 10.1182/blood-2011-02-339564
62. de Groot PG, Meijers JC. [amp]]beta;(2) -Glycoprotein I: evolution, structure and function. J Thromb Haemost. (2011) 9:1275–84. doi: 10.1111/j.1538-7836.2011.04327.x
63. Ruben E, Planer W, Chinnaraj M, Chen Z, Zuo X, Pengo V, et al. The J-elongated conformation of β2-glycoprotein I predominates in solution: implications for our understanding of antiphospholipid syndrome. J Biol Chem. (2020) 295:10794–806. doi: 10.1074/jbc.RA120.013939
64. Bouma B, de Groot PG, van den Elsen JM, Ravelli RB, Schouten A, Simmelink MJ, et al. Adhesion mechanism of human beta(2)-glycoprotein I to phospholipids based on its crystal structure. EMBO J. (1999) 18:5166–74. doi: 10.1093/emboj/18.19.5166
65. Schwarzenbacher R, Zeth K, Diederichs K, Gries A, Kostner GM, Laggner P, et al. Crystal structure of human beta2-glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. EMBO J. (1999) 18:6228–39. doi: 10.1093/emboj/18.22.6228
66. Lozier J, Takahashi N, Putnam FW. Complete amino acid sequence of human plasma beta 2-glycoprotein I. Proc Natl Acad Sci U S A. (1984) 81:3640–4. doi: 10.1073/pnas.81.12.3640
67. Mehdi H, Naqvi A, Kamboh MI. A hydrophobic sequence at position 313-316 (Leu-Ala-Phe-Trp) in the fifth domain of apolipoprotein H (beta2-glycoprotein I) is crucial for cardiolipin binding. Eur J Biochem. (2000) 267:1770–6. doi: 10.1046/j.1432-1327.2000.01174.x
68. Hamdan R, Maiti SN, Schroit AJ. Interaction of beta2-glycoprotein 1 with phosphatidylserine-containing membranes: ligand-dependent conformational alterations initiate bivalent binding. Biochemistry. (2007) 46:10612–20. doi: 10.1021/bi700621j
69. Iverson GM, Victoria EJ, Marquis DM. Anti-beta2 glycoprotein I (beta2GPI) autoantibodies recognize an epitope on the first domain of beta2GPI. Proc Natl Acad Sci U S A. (1998) 95:15542–6. doi: 10.1073/pnas.95.26.15542
70. de Laat B, Derksen RH, van Lummel M, Pennings MT, de Groot PG. Pathogenic anti-beta2-glycoprotein I antibodies recognize domain I of beta2-glycoprotein I only after a conformational change. Blood. (2006) 107:1916–24. doi: 10.1182/blood-2005-05-1943
71. Yamaguchi Y, Seta N, Kaburaki J, Kobayashi K, Matsuura E, Kuwana M. Excessive exposure to anionic surfaces maintains autoantibody response to beta(2)-glycoprotein I in patients with antiphospholipid syndrome. Blood. (2007) 110:4312–8. doi: 10.1182/blood-2007-07-100008
72. Sciascia S, Amigo MC, Roccatello D, Khamashta M. Diagnosing antiphospholipid syndrome: ‘extra-criteria’ manifestations and technical advances. Nat Rev Rheumatol. (2017) 13:548–60. doi: 10.1038/nrrheum.2017.124
73. Li J, Zhu X, Feng J. Role of β2-glycoprotein I in the pathogenesis of the antiphospholipid syndrome. Rheumatol Autoimmunity. (2023) 3:131–9. doi: 10.1002/rai2.12072
74. Esmon CT, Gu JM, Xu J, Qu D, Stearns-Kurosawa DJ, Kurosawa S. Regulation and functions of the protein C anticoagulant pathway. Haematologica. (1999) 84:363–8.
75. Malia RG, Kitchen S, Greaves M, Preston FE. Inhibition of activated protein C and its cofactor protein S by antiphospholipid antibodies. Br J Haematol. (1990) 76:101–7. doi: 10.1111/j.1365-2141.1990.tb07843.x
76. Borrell M, Sala N, de Castellarnau C, Lopez S, Gari M, Fontcuberta J. Immunoglobulin fractions isolated from patients with antiphospholipid antibodies prevent the inactivation of factor Va by activated protein C on human endothelial cells. Thromb Haemost. (1992) 68:268–72. doi: 10.1055/s-0038-1656363
77. de Groot PG, Horbach DA, Derksen RH. Protein C and other cofactors involved in the binding of antiphospholipid antibodies: relation to the pathogenesis of thrombosis. Lupus. (1996) 5:488–93. doi: 10.1177/096120339600500532
78. Oosting JD, Preissner KT, Derksen RH, de Groot PG. Autoantibodies directed against the epidermal growth factor-like domains of thrombomodulin inhibit protein C activation in vitro. Br J Haematol. (1993) 85:761–8. doi: 10.1111/j.1365-2141.1993.tb03220.x
79. Mori T, Takeya H, Nishioka J, Gabazza EC, Suzuki K. beta 2-Glycoprotein I modulates the anticoagulant activity of activated protein C on the phospholipid surface. Thromb Haemost. (1996) 75:49–55. doi: 10.1055/s-0038-1650220
80. Safa O, Esmon CT, Esmon NL. Inhibition of APC anticoagulant activity on oxidized phospholipid by anti-{beta}2-glycoprotein I monoclonal antibodies. Blood. (2005) 106:1629–35. doi: 10.1182/blood-2005-01-0404
81. Nojima J, Kuratsune H, Suehisa E, Iwatani Y, Kanakura Y. Acquired activated protein C resistance associated with IgG antibodies against beta2-glycoprotein I and prothrombin as a strong risk factor for venous thromboembolism. Clin Chem. (2005) 51:545–52. doi: 10.1373/clinchem.2004.043414
82. de Laat B, Eckmann CM, van Schagen M, Meijer AB, Mertens K, van Mourik JA. Correlation between the potency of a beta2-glycoprotein I-dependent lupus anticoagulant and the level of resistance to activated protein C. Blood Coagul Fibrinolysis. (2008) 19:757–64. doi: 10.1097/MBC.0b013e32830f1b85
83. Arachchillage DR, Efthymiou M, Mackie IJ, Lawrie AS, Machin SJ, Cohen H. Anti-protein C antibodies are associated with resistance to endogenous protein C activation and a severe thrombotic phenotype in antiphospholipid syndrome. J Thromb Haemost. (2014) 12:1801–9. doi: 10.1111/jth.12722
84. Cosgriff TM, Martin BA. Low functional and high antigenic antithrombin III level in a patient with the lupus anticoagulant and recurrent thrombosis. Arthritis rheumatism. (1981) 24:94–6. doi: 10.1002/art.1780240115
85. Chamley LW, McKay EJ, Pattison NS. Inhibition of heparin/antithrombin III cofactor activity by anticardiolipin antibodies: a mechanism for thrombosis. Thromb Res. (1993) 71:103–11. doi: 10.1016/0049-3848(93)90176-O
86. Shibata S, Harpel PC, Gharavi A, Rand J, Fillit H. Autoantibodies to heparin from patients with antiphospholipid antibody syndrome inhibit formation of antithrombin III-thrombin complexes. Blood. (1994) 83:2532–40. doi: 10.1182/blood.V83.9.2532.2532
87. Vega-Ostertag M, Liu X, Kwan-Ki H, Chen P, Pierangeli S. A human monoclonal antiprothrombin antibody is thrombogenic in vivo and upregulates expression of tissue factor and E-selectin on endothelial cells. Br J Haematol. (2006) 135:214–9. doi: 10.1111/j.1365-2141.2006.06283.x
88. Rand JH, Wu XX, Andree HA, Lockwood CJ, Guller S, Scher J, et al. Pregnancy loss in the antiphospholipid-antibody syndrome–a possible thrombogenic mechanism. New Engl J Med. (1997) 337:154–60. doi: 10.1056/NEJM199707173370303
89. Rand JH, Wu XX, Lapinski R, van Heerde WL, Reutelingsperger CP, Chen PP, et al. Detection of antibody-mediated reduction of annexin A5 anticoagulant activity in plasmas of patients with the antiphospholipid syndrome. Blood. (2004) 104:2783–90. doi: 10.1182/blood-2004-01-0203
90. de Laat B, Wu XX, van Lummel M, Derksen RH, de Groot PG, Rand JH. Correlation between antiphospholipid antibodies that recognize domain I of beta2-glycoprotein I and a reduction in the anticoagulant activity of annexin A5. Blood. (2007) 109:1490–4. doi: 10.1182/blood-2006-07-030148
91. Rand JH, Wu XX, Quinn AS, Ashton AW, Chen PP, Hathcock JJ, et al. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug. Blood. (2010) 115:2292–9. doi: 10.1182/blood-2009-04-213520
92. Musiał J, Padjas A, Iwaniec T, Celińska-Löwenhoff M, Undas A. Altered fibrin clot structure/function in patients with antiphospholipid syndrome: association with thrombotic manifestation. Thromb Haemostasis. (2017) 112:287–96. doi: 10.1160/TH13-11-0980
93. Ling Q, Jacovina AT, Deora A, Febbraio M, Simantov R, Silverstein RL, et al. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest. (2004) 113:38–48. doi: 10.1172/JCI19684
94. Cesarman-Maus G, Ríos-Luna NP, Deora AB, Huang B, Villa R, Cravioto Mdel C, et al. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood. (2006) 107:4375–82. doi: 10.1182/blood-2005-07-2636
95. Romay-Penabad Z, Montiel-Manzano MG, Shilagard T, Papalardo E, Vargas G, Deora AB, et al. Annexin A2 is involved in antiphospholipid antibody-mediated pathogenic effects in vitro and in vivo. Blood. (2009) 114:3074–83. doi: 10.1182/blood-2008-11-188698
96. Bu C, Gao L, Xie W, Zhang J, He Y, Cai G, et al. [amp]]beta;2-glycoprotein i is a cofactor for tissue plasminogen activator–mediated plasminogen activation. Arthritis Rheumatism. (2009) 60:559–68. doi: 10.1002/art.24262
97. Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. (1998) 91:3527–61. doi: 10.1182/blood.V91.10.3527
98. Morrissey JH, Fakhrai H, Edgington TS. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. (1987) 50:129–35. doi: 10.1016/0092-8674(87)90669-6
99. Banner DW, D’Arcy A, Chène C, Winkler FK, Guha A, Konigsberg WH, et al. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. (1996) 380:41–6. doi: 10.1038/380041a0
100. Mackman N. Regulation of the tissue factor gene. FASEB J. (1995) 9:883–9. doi: 10.1096/fasebj.9.10.7615158
101. Østerud B, Bjørklid E. Sources of tissue factor. Semin Thromb Hemost. (2006) 32:11–23. doi: 10.1055/s-2006-933336
102. Simantov R, LaSala JM, Lo SK, Gharavi AE, Sammaritano LR, Salmon JE, et al. Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest. (1995) 96:2211–9. doi: 10.1172/JCI118276
103. Del Papa N, Guidali L, Sala A, Buccellati C, Khamashta MA, Ichikawa K, et al. Endothelial cells as target for antiphospholipid antibodies. Human polyclonal and monoclonal anti-beta 2-glycoprotein I antibodies react in vitro with endothelial cells through adherent beta 2-glycoprotein I and induce endothelial activation. Arthritis Rheumatol. (1997) 40:551–61. doi: 10.1002/art.1780400322
104. Del Papa N, Raschi E, Catelli L, Khamashta MA, Ichikawa K, Tincani A, et al. Endothelial cells as a target for antiphospholipid antibodies: role of anti-beta 2 glycoprotein I antibodies. Am J Reprod Immunol. (1997) 38:212–7. doi: 10.1111/j.1600-0897.1997.tb00301.x
105. Espinola RG, Liu X, Colden-Stanfield M, Hall J, Harris EN, Pierangeli SS. E-Selectin mediates pathogenic effects of antiphospholipid antibodies. J Thromb Haemost. (2003) 1:843–8. doi: 10.1046/j.1538-7836.2003.00119.x
106. Branch DW, Rodgers GM. Induction of endothelial cell tissue factor activity by sera from patients with antiphospholipid syndrome: a possible mechanism of thrombosis. Am J Obstet Gynecol. (1993) 168:206–10. doi: 10.1016/S0002-9378(12)90915-1
107. Ferrara DE, Swerlick R, Casper K, Meroni PL, Vega-Ostertag ME, Harris EN, et al. Fluvastatin inhibits up-regulation of tissue factor expression by antiphospholipid antibodies on endothelial cells. J Thromb Haemost. (2004) 2:1558–63. doi: 10.1111/j.1538-7836.2004.00896.x
108. López-Pedrera C, Buendía P, Barbarroja N, Siendones E, Velasco F, Cuadrado MJ. Antiphospholipid-mediated thrombosis: interplay between anticardiolipin antibodies and vascular cells. Clin Appl Thromb Hemost. (2006) 12:41–5. doi: 10.1177/107602960601200107
109. Betapudi V, Lominadze G, Hsi L, Willard B, Wu M, McCrae KR. Anti-beta2GPI antibodies stimulate endothelial cell microparticle release via a nonmuscle myosin II motor protein-dependent pathway. Blood. (2013) 122:3808–17. doi: 10.1182/blood-2013-03-490318
110. Wu M, Barnard J, Kundu S, McCrae KR. A novel pathway of cellular activation mediated by antiphospholipid antibody-induced extracellular vesicles. J Thromb Haemost. (2015) 13:1928–40. doi: 10.1111/jth.13072
111. Hamid C, Norgate K, D’Cruz DP, Khamashta MA, Arno M, Pearson JD, et al. Anti-beta2GPI-antibody-induced endothelial cell gene expression profiling reveals induction of novel pro-inflammatory genes potentially involved in primary antiphospholipid syndrome. Ann Rheum Dis. (2007) 66:1000–7. doi: 10.1136/ard.2006.063909
112. Mineo C. Inhibition of nitric oxide and antiphospholipid antibody-mediated thrombosis. Curr Rheumatol Rep. (2013) 15:324. doi: 10.1007/s11926-013-0324-4
113. Montiel-Manzano G, Romay-Penabad Z, Papalardo de Martínez E, Meillon-García LA, García-Latorre E, Reyes-Maldonado E, et al. In vivo effects of an inhibitor of nuclear factor-kappa B on thrombogenic properties of antiphospholipid antibodies. Ann N Y Acad Sci. (2007) 1108:540–53. doi: 10.1196/annals.1422.057
114. Vega-Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheumatol. (2005) 52:1545–54. doi: 10.1002/art.21009
115. Zhang J, McCrae KR. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood. (2005) 105:1964–9. doi: 10.1182/blood-2004-05-1708
116. Allen KL, Fonseca FV, Betapudi V, Willard B, Zhang J, McCrae KR. A novel pathway for human endothelial cell activation by antiphospholipid/anti-β2 glycoprotein I antibodies. Blood. (2012) 119:884–93. doi: 10.1182/blood-2011-03-344671
117. Raschi E, Testoni C, Bosisio D, Borghi MO, Koike T, Mantovani A, et al. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood. (2003) 101:3495–500. doi: 10.1182/blood-2002-08-2349
118. Feng W, Qiao J, Tan Y, Liu Q, Wang Q, Yang B, et al. Interaction of antiphospholipid antibodies with endothelial cells in antiphospholipid syndrome. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1361519
119. Gandrille S. Endothelial cell protein C receptor and the risk of venous thrombosis. Haematologica. (2008) 93:812–6. doi: 10.3324/haematol.13243
120. Hurtado V, Montes R, Gris JC, Bertolaccini ML, Alonso A, Martínez-González MA, et al. Autoantibodies against EPCR are found in antiphospholipid syndrome and are a risk factor for fetal death. Blood. (2004) 104:1369–74. doi: 10.1182/blood-2004-03-0793
121. Müller-Calleja N, Hollerbach A, Royce J, Ritter S, Pedrosa D, Madhusudhan T, et al. Lipid presentation by the protein C receptor links coagulation with autoimmunity. Science. (2021) 371:eabc0956. doi: 10.1126/science.abc0956
122. Chu CQ. The pivotal role of endothelial protein C receptor for antiphospholipid antibody-mediated pathologies. Rheumatol (Oxford). (2022) 61:883–5. doi: 10.1093/rheumatology/keab620
123. Plasín-Rodríguez MA, Rodríguez-Pintó I, Patricio P, Monteagudo J, Cervera R, Reverter JC, et al. The H1 haplotype of the endothelial protein C receptor protects against arterial thrombosis in patients with antiphospholipid syndrome. Thromb Res. (2018) 169:128–34. doi: 10.1016/j.thromres.2018.07.006
124. Ulrich V, Konaniah ES, Lee WR, Khadka S, Shen YM, Herz J, et al. Antiphospholipid antibodies attenuate endothelial repair and promote neointima formation in mice. J Am Heart Assoc. (2014) 3:e001369. doi: 10.1161/JAHA.114.001369
125. Ramesh S, Morrell CN, Tarango C, Thomas GD, Yuhanna IS, Girardi G, et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J Clin Invest. (2011) 121:120–31. doi: 10.1172/JCI39828
126. Boles J, Mackman N. Role of tissue factor in thrombosis in antiphospholipid antibody syndrome. Lupus. (2010) 19:370–8. doi: 10.1177/0961203309360810
127. Kornberg A, Blank M, Kaufman S, Shoenfeld Y. Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J Immunol (Baltimore Md: 1950). (1994) 153:1328–32. doi: 10.4049/jimmunol.153.3.1328
128. Cuadrado MJ, López-Pedrera C, Khamashta MA, Camps MT, Tinahones F, Torres A, et al. Thrombosis in primary antiphospholipid syndrome: a pivotal role for monocyte tissue factor expression. Arthritis rheumatism. (1997) 40:834–41. doi: 10.1002/art.1780400509
129. Reverter JC, Tàssies D, Font J, Khamashta MA, Ichikawa K, Cervera R, et al. Effects of human monoclonal anticardiolipin antibodies on platelet function and on tissue factor expression on monocytes. Arthritis rheumatism. (1998) 41:1420–7. doi: 10.1002/1529-0131(199808)41:8<1420::AID-ART11>3.0.CO;2-U
130. Zhou H, Wolberg AS, Roubey RA. Characterization of monocyte tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood. (2004) 104:2353–8. doi: 10.1182/blood-2004-01-0145
131. Bohgaki M, Atsumi T, Yamashita Y, Yasuda S, Sakai Y, Furusaki A, et al. The p38 mitogen-activated protein kinase (MAPK) pathway mediates induction of the tissue factor gene in monocytes stimulated with human monoclonal anti-beta2Glycoprotein I antibodies. Int Immunol. (2004) 16:1633–41. doi: 10.1093/intimm/dxh166
132. López-Pedrera C, Buendía P, Cuadrado MJ, Siendones E, Aguirre MA, Barbarroja N, et al. Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappaB/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis Rheumatol. (2006) 54:301–11. doi: 10.1002/art.21549
133. Sorice M, Longo A, Capozzi A, Garofalo T, Misasi R, Alessandri C, et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheumatol. (2007) 56:2687–97. doi: 10.1002/art.22802
134. Xie H, Zhou H, Wang H, Chen D, Xia L, Wang T, et al. Anti-β(2)GPI/β(2)GPI induced TF and TNF-α expression in monocytes involving both TLR4/MyD88 and TLR4/TRIF signaling pathways. Mol Immunol. (2013) 53:246–54. doi: 10.1016/j.molimm.2012.08.012
135. Salet DM, Bekkering S, Middeldorp S, van den Hoogen LL. Targeting thromboinflammation in antiphospholipid syndrome. J Thromb Haemost. (2023) 21:744–57. doi: 10.1016/j.jtha.2022.12.002
136. Lambrianides A, Carroll CJ, Pierangeli SS, Pericleous C, Branch W, Rice J, et al. Effects of polyclonal IgG derived from patients with different clinical types of the antiphospholipid syndrome on monocyte signaling pathways. J Immunol. (2010) 184:6622–8. doi: 10.4049/jimmunol.0902765
137. Zhou H, Yan Y, Xu G, Zhou B, Wen H, Guo D, et al. Toll-like receptor (TLR)-4 mediates anti-beta2GPI/beta2GPI-induced tissue factor expression in THP-1 cells. Clin Exp Immunol. (2011) 163:189–98. doi: 10.1111/j.1365-2249.2010.04291.x
138. Xia L, Zhou H, Wang T, Xie Y, Wang T, Wang X, et al. Activation of mTOR is involved in anti-β(2)GPI/β(2)GPI-induced expression of tissue factor and IL-8 in monocytes. Thromb Res. (2017) 157:103–10. doi: 10.1016/j.thromres.2017.05.023
139. Brandt KJ, Fickentscher C, Boehlen F, Kruithof EK, de Moerloose P. NF-κB is activated from endosomal compartments in antiphospholipid antibodies-treated human monocytes. J Thromb Haemost. (2014) 12:779–91. doi: 10.1111/jth.12536
140. Colasanti T, Alessandri C, Capozzi A, Sorice M, Delunardo F, Longo A, et al. Autoantibodies specific to a peptide of β2-glycoprotein I cross-react with TLR4, inducing a proinflammatory phenotype in endothelial cells and monocytes. Blood. (2012) 120:3360–70. doi: 10.1182/blood-2011-09-378851
141. Zhou H, Sheng L, Wang H, Xie H, Mu Y, Wang T, et al. Anti-β2GPI/β2GPI stimulates activation of THP-1 cells through TLR4/MD-2/MyD88 and NF-κB signaling pathways. Thromb Res. (2013) 132:742–9. doi: 10.1016/j.thromres.2013.09.039
142. Salemink I, Blezer R, Willems GM, Galli M, Bevers E, Lindhout T. Antibodies to beta2-glycoprotein I associated with antiphospholipid syndrome suppress the inhibitory activity of tissue factor pathway inhibitor. Thromb Haemost. (2000) 84:653–6. doi: 10.1055/s-0037-1614082
143. López-Pedrera C, Aguirre MA, Buendía P, Barbarroja N, Ruiz-Limón P, Collantes-Estevez E, et al. Differential expression of protease-activated receptors in monocytes from patients with primary antiphospholipid syndrome. Arthritis Rheumatol. (2010) 62:869–77. doi: 10.1002/art.27299
144. Kobayashi SD, DeLeo FR. Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med. (2009) 1:309–33. doi: 10.1002/wsbm.v1:3
145. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
146. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
147. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. (2012) 189:2689–95. doi: 10.4049/jimmunol.1201719
148. Grayson PC, Kaplan MJ. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J Leukoc Biol. (2016) 99:253–64. doi: 10.1189/jlb.5BT0615-247R
149. Arvieux J, Jacob MC, Roussel B, Bensa JC, Colomb MG. Neutrophil activation by anti-beta 2 glycoprotein I monoclonal antibodies via Fc gamma receptor II. J Leukoc Biol. (1995) 57:387–94. doi: 10.1002/jlb.57.3.387
150. Gladigau G, Haselmayer P, Scharrer I, Munder M, Prinz N, Lackner K, et al. A role for Toll-like receptor mediated signals in neutrophils in the pathogenesis of the anti-phospholipid syndrome. PloS One. (2012) 7:e42176. doi: 10.1371/journal.pone.0042176
151. Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. (2003) 112:1644–54. doi: 10.1172/JCI200318817
152. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
153. Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. (2012) 32:1777–83. doi: 10.1161/ATVBAHA.111.242859
154. Leffler J, Stojanovich L, Shoenfeld Y, Bogdanovic G, Hesselstrand R, Blom AM. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol. (2014) 32:66–70.
155. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez-Álvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. (2015) 67:2990–3003. doi: 10.1002/art.v67.11
156. Duarte JH. Connective tissue diseases: Neutrophil extracellular traps–a mechanism of thrombosis in patients with antiphospholipid syndrome? Nat Rev Rheumatol. (2015) 11:444. doi: 10.1038/nrrheum.2015.96
157. Sule G, Kelley WJ, Gockman K, Yalavarthi S, Vreede AP, Banka AL, et al. Increased adhesive potential of antiphospholipid syndrome neutrophils mediated by β2 integrin mac-1. Arthritis Rheumatol. (2020) 72:114–24. doi: 10.1002/art.41057
158. Mauracher L-M, Krall M, Roiß J, Hell L, Koder S, Hofbauer TM, et al. Neutrophil subpopulations and their activation potential in patients with antiphospholipid syndrome and healthy individuals. Rheumatology. (2021) 60:1687–99. doi: 10.1093/rheumatology/keaa532
159. Zha C, Zhang W, Gao F, Xu J, Jia R, Cai J, et al. Anti-β(2)GPI/β(2)GPI induces neutrophil extracellular traps formation to promote thrombogenesis via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology. (2018) 138:140–50. doi: 10.1016/j.neuropharm.2018.06.001
160. Nayak L, Sweet DR, Thomas A, Lapping SD, Kalikasingh K, Madera A, et al. A targetable pathway in neutrophils mitigates both arterial and venous thrombosis. Sci Transl Med. (2022) 14:eabj7465. doi: 10.1126/scitranslmed.abj7465
161. Mittal P, Quattrocchi G, Tohidi-Esfahani I, Sayar Z, Chandratheva A, Cohen H. Antiphospholipid syndrome, antiphospholipid antibodies, and stroke. Int J Stroke. (2023) 18:383–91. doi: 10.1177/17474930221150349
162. Font J, Espinosa G, Tassies D, Pino M, Khamashta MA, Gallart T, et al. Effects of beta2-glycoprotein I and monoclonal anticardiolipin antibodies in platelet interaction with subendothelium under flow conditions. Arthritis Rheumatol. (2002) 46:3283–9. doi: 10.1002/art.v46:12
163. Vega-Ostertag M, Harris EN, Pierangeli SS. Intracellular events in platelet activation induced by antiphospholipid antibodies in the presence of low doses of thrombin. Arthritis Rheumatol. (2004) 50:2911–9. doi: 10.1002/art.20434
164. Lutters BC, Derksen RH, Tekelenburg WL, Lenting PJ, Arnout J, de Groot PG. Dimers of beta 2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2’. J Biol Chem. (2003) 278:33831–8. doi: 10.1074/jbc.M212655200
165. van Lummel M, Pennings MT, Derksen RH, Urbanus RT, Lutters BC, Kaldenhoven N, et al. The binding site in {beta}2-glycoprotein I for ApoER2’ on platelets is located in domain V. J Biol Chem. (2005) 280:36729–36. doi: 10.1074/jbc.M504172200
166. Hulstein JJ, Lenting PJ, de Laat B, Derksen RH, Fijnheer R, de Groot PG. beta2-Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood. (2007) 110:1483–91. doi: 10.1182/blood-2006-10-053199
167. Urbanus RT, Pennings MT, Derksen RH, de Groot PG. Platelet activation by dimeric beta2-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2’. J Thromb Haemost. (2008) 6:1405–12. doi: 10.1111/j.1538-7836.2008.03021.x
168. Shi T, Giannakopoulos B, Yan X, Yu P, Berndt MC, Andrews RK, et al. Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheumatol. (2006) 54:2558–67. doi: 10.1002/art.21968
169. Sikara MP, Routsias JG, Samiotaki M, Panayotou G, Moutsopoulos HM, Vlachoyiannopoulos PG. [amp]]beta;2 Glycoprotein I ({beta}2GPI) binds platelet factor 4 (PF4): implications for the pathogenesis of antiphospholipid syndrome. Blood. (2010) 115:713–23. doi: 10.1182/blood-2009-03-206367
170. Ouweneel AB, Thomas MJ, Sorci-Thomas MG. The ins and outs of lipid rafts: functions in intracellular cholesterol homeostasis, microparticles, and cell membranes: Thematic Review Series: Biology of Lipid Rafts. J Lipid Res. (2020) 61:676–86. doi: 10.1194/jlr.TR119000383
171. Capozzi A, Manganelli V, Riitano G, Caissutti D, Longo A, Garofalo T, et al. Advances in the pathophysiology of thrombosis in antiphospholipid syndrome: molecular mechanisms and signaling through lipid rafts. J Clin Med. (2023) 12:891. doi: 10.3390/jcm12030891
172. Misasi R, Longo A, Recalchi S, Caissutti D, Riitano G, Manganelli V, et al. Molecular mechanisms of “Antiphospholipid antibodies” and their paradoxical role in the pathogenesis of “Seronegative APS. Int J Mol Sci. (2020) 21:8411. doi: 10.3390/ijms21218411
173. Riitano G, Capozzi A, Recalchi S, Caissutti D, Longo A, Mattei V, et al. Anti-β2-GPI antibodies induce endothelial cell expression of tissue factor by LRP6 signal transduction pathway involving lipid rafts. Cells. (2022) 11:1288. doi: 10.3390/cells11081288
174. Riitano G, Capozzi A, Recalchi S, Augusto M, Conti F, Misasi R, et al. Role of lipid rafts on LRP8 signaling triggered by anti-β2-GPI antibodies in endothelial cells. . Biomedicines. (2023) 11:3135. doi: 10.3390/biomedicines11123135
175. Mackman N, Archibald SJ, Hisada Y. Effect of heparanase inhibitor on tissue factor overexpression in platelets and endothelial cells induced by anti-β2-GPI antibodies: Comment from Mackman et al. J Thromb Haemost. (2022) 20:260–1. doi: 10.1111/jth.15557
176. Nadir Y. Heparanase and coagulation-new insights. Rambam Maimonides Med J. (2014) 5:e0031. doi: 10.5041/RMMJ.20769172
177. Nadir Y, Brenner B, Zetser A, Ilan N, Shafat I, Zcharia E, et al. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. (2006) 4:2443–51. doi: 10.1111/j.1538-7836.2006.02212.x
178. Oikonomopoulou K, Ricklin D, Ward PA, Lambris JD. Interactions between coagulation and complement–their role in inflammation. Semin Immunopathol. (2012) 34:151–65. doi: 10.1007/s00281-011-0280-x
179. Carrera-Marin A, Romay-Penabad Z, Papalardo E, Reyes-Maldonado E, Garcia-Latorre E, Vargas G, et al. C6 knock-out mice are protected from thrombophilia mediated by antiphospholipid antibodies. Lupus. (2012) 21:1497–505. doi: 10.1177/0961203312458839
180. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. (2006) 177:4794–802. doi: 10.4049/jimmunol.177.7.4794
181. Redecha P, Tilley R, Tencati M, Salmon JE, Kirchhofer D, Mackman N, et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. (2007) 110:2423–31. doi: 10.1182/blood-2007-01-070631
182. Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, et al. Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis. (2009) 68:1030–5. doi: 10.1136/ard.2008.090670
183. Breen KA, Seed P, Parmar K, Moore GW, Stuart-Smith SE, Hunt BJ. Complement activation in patients with isolated antiphospholipid antibodies or primary antiphospholipid syndrome. Thromb Haemost. (2012) 107:423–9. doi: 10.1160/TH11-08-0554
184. Nakamura H, Oku K, Ogata Y, Ohmura K, Yoshida Y, Kitano E, et al. Alternative pathway activation due to low level of complement factor H in primary antiphospholipid syndrome. Thromb Res. (2018) 164:63–8. doi: 10.1016/j.thromres.2018.02.142
185. Foltyn Zadura A, Memon AA, Stojanovich L, Perricone C, Conti F, Valesini G, et al. Factor H autoantibodies in patients with antiphospholipid syndrome and thrombosis. J Rheumatol. (2015) 42:1786–93. doi: 10.3899/jrheum.150185
186. Meroni PL, Macor P, Durigutto P, De Maso L, Gerosa M, Ferraresso M, et al. Complement activation in antiphospholipid syndrome and its inhibition to prevent rethrombosis after arterial surgery. Blood. (2016) 127:365–7. doi: 10.1182/blood-2015-09-672139
187. McDonnell T, Wincup C, Buchholz I, Pericleous C, Giles I, Ripoll V, et al. The role of beta-2-glycoprotein I in health and disease associating structure with function: More than just APS. Blood Rev. (2020) 39:100610. doi: 10.1016/j.blre.2019.100610
188. Shi W, Chong BH, Hogg PJ, Chesterman CN. Anticardiolipin antibodies block the inhibition by beta 2-glycoprotein I of the factor Xa generating activity of platelets. Thromb Haemost. (1993) 70:342–5. doi: 10.1055/s-0038-1649577
189. Arachchillage DRJ, Pericleous C. Evolution of antiphospholipid syndrome. Semin Thromb Hemostasis. (2023) 49:295–304. doi: 10.1055/s-0042-1760333
190. Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. (2002) 195:211–20. doi: 10.1084/jem.200116116
191. Shamonki JM, Salmon JE, Hyjek E, Baergen RN. Excessive complement activation is associated with placental injury in patients with antiphospholipid antibodies. Am J Obstet Gynecol. (2007) 196:167.e1–5. doi: 10.1016/j.ajog.2006.10.879
192. Kim MY, Guerra MM, Kaplowitz E, Laskin CA, Petri M, Branch DW, et al. Complement activation predicts adverse pregnancy outcome in patients with systemic lupus erythematosus and/or antiphospholipid antibodies. Ann Rheum Dis. (2018) 77:549–55. doi: 10.1136/annrheumdis-2017-212224
193. Salmon JE, Girardi G, Holers VM. Activation of complement mediates antiphospholipid antibody-induced pregnancy loss. Lupus. (2003) 12:535–8. doi: 10.1191/0961203303lu397oa
194. Girardi G, Redecha P, Salmon JE. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med. (2004) 10:1222–6. doi: 10.1038/nm1121
195. Chaturvedi S, Braunstein EM, Brodsky RA. Antiphospholipid syndrome: Complement activation, complement gene mutations, and therapeutic implications. J Thromb Haemostasis. (2021) 19:607–16. doi: 10.1111/jth.15082
196. Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. (2020) 135:239–51. doi: 10.1182/blood.2019003863
197. Pierangeli SS, Gharavi AE, Harris EN. Experimental thrombosis and antiphospholipid antibodies: new insights. J Autoimmun. (2000) 15:241–7. doi: 10.1006/jaut.2000.0420
198. Meroni PL, Riboldi P. Pathogenic mechanisms mediating antiphospholipid syndrome. Curr Opin Rheumatol. (2001) 13:377–82. doi: 10.1097/00002281-200109000-00006
199. Shoenfeld Y, Blank M, Cervera R, Font J, Raschi E, Meroni PL. Infectious origin of the antiphospholipid syndrome. Ann Rheum Dis. (2006) 65:2–6. doi: 10.1136/ard.2005.045443
200. Noureldine MHA, Nour-Eldine W, Khamashta MA, Uthman I. Insights into the diagnosis and pathogenesis of the antiphospholipid syndrome. Semin Arthritis Rheumatol. (2019) 48:860–6. doi: 10.1016/j.semarthrit.2018.08.004
201. Gailani D, Gruber A. Targeting factor XI and factor XIa to prevent thrombosis. Blood. (2024) 143:1465–75. doi: 10.1182/blood.2023020722
202. Knight JS, Branch DW, Ortel TL. Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. Bmj. (2023) 380:e069717. doi: 10.1136/bmj-2021-069717
203. Cohen H, Cuadrado MJ, Erkan D, Duarte-Garcia A, Isenberg DA, Knight JS, et al. 16th international congress on antiphospholipid antibodies task force report on antiphospholipid syndrome treatment trends. Lupus. (2020) 29:1571–93. doi: 10.1177/0961203320950461
204. Dufrost V, Wahl D, Zuily S. Direct oral anticoagulants in antiphospholipid syndrome: Meta-analysis of randomized controlled trials. Autoimmun Rev. (2021) 20:102711. doi: 10.1016/j.autrev.2020.102711
205. Lucotti S, Cerutti C, Soyer M, Gil-Bernabé AM, Gomes AL, Allen PD, et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J Clin Invest. (2019) 129:1845–62. doi: 10.1172/JCI121985
206. Knight JS, Erkan D. Rethinking antiphospholipid syndrome to guide future management and research. Nat Rev Rheumatol. (2024) 20:377–88. doi: 10.1038/s41584-024-01110-y
207. Signorelli F, Balbi GGM, Domingues V, Levy RA. New and upcoming treatments in antiphospholipid syndrome: A comprehensive review. Pharmacol Res. (2018) 133:108–20. doi: 10.1016/j.phrs.2018.04.012
208. Oulego-Erroz I, Martínez-Sáenz-de-Jubera J, Ocaña-Alcober C, Regueras-Santos L, Ferrero-De-la-Mano L, Martínez-Badás JP. Pediatric catastrophic antiphospholipid syndrome successfully treated with eculizumab. Am J Respir Crit Care Med. (2021) 203:640–2. doi: 10.1164/rccm.202009-3489LE
209. López-Benjume B, Rodríguez-Pintó I, Amigo MC, Erkan D, Shoenfeld Y, Cervera R, et al. Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the “CAPS Registry. Autoimmun Rev. (2022) 21:103055. doi: 10.1016/j.autrev.2022.103055
210. Bitsadze V, Yakubova F, Khizroeva J, Lazarchuk A, Salnikova P, Vorobev A, et al. Catastrophic antiphospholipid syndrome. Int J Mol Sci. (2024) 25:668. doi: 10.3390/ijms25010668
211. Freeman CL, Sehn LH. A tale of two antibodies: obinutuzumab versus rituximab. Br J Haematol. (2018) 182:29–45. doi: 10.1111/bjh.2018.182.issue-1
212. Kvacskay P, Merkt W, Günther J, Blank N, Lorenz HM. Obinutuzumab in connective tissue diseases after former rituximab-non-response: a case series. Ann Rheum Dis. (2022) 81:744–6. doi: 10.1136/annrheumdis-2021-221756
213. Sciascia S, Rubini E, Radin M, Cecchi I, Rossi D, Roccatello D. Anticardiolipin and anti-beta 2 glycoprotein-I antibodies disappearance in patients with systemic lupus erythematosus and antiphospholipid syndrome while on belimumab. Ann Rheum Dis. (2018) 77:1694–5. doi: 10.1136/annrheumdis-2018-213496
214. Yazici A, Yazirli B, Erkan D. Belimumab in primary antiphospholipid syndrome. Lupus. (2017) 26:1123–4. doi: 10.1177/0961203316682102
215. Connor DE, Gerbelli J, Chew AN, Cooley-Andrade O, Goonawardhana D, Cheung K, et al. Sirolimus and propranolol inhibit endothelial proliferation while detergent sclerosants induce endothelial activation, microparticle release and apoptosis in vitro. Phlebology. (2020) 35:566–75. doi: 10.1177/0268355520913384
216. Zhang D, Sun F, Ye S. Successful treatment of sirolimus in a Chinese patient with refractory LN and APS: a case report. Ther Adv Musculoskelet Dis. (2022) 14:1759720x221079253. doi: 10.1177/1759720X221079253
217. de Azevedo FVA, Maia DG, de Carvalho JF, Rodrigues CEM. Renal involvement in antiphospholipid syndrome. Rheumatol Int. (2018) 38:1777–89. doi: 10.1007/s00296-018-4040-2
218. Radojcic V, Luznik L. Mechanism of action of posttransplantation cyclophosphamide: more than meets the eye. J Clin Invest. (2019) 129:2189–91. doi: 10.1172/JCI128710
219. Pul S, Gökçe İ, Bodur ED, Güven S, Çiçek N, Sak M, et al. Catastrophic antiphospholipid syndrome accompanied by complement regulatory gene mutation. Turk J Pediatr. (2023) 65:330–7. doi: 10.24953/turkjped.2022.288
220. Bayraktar UD, Erkan D, Bucciarelli S, Espinosa G, Asherson R. The clinical spectrum of catastrophic antiphospholipid syndrome in the absence and presence of lupus. J Rheumatol. (2007) 34:346–52.
221. Burcoglu-O’Ral A, Erkan D, Asherson R. Treatment of catastrophic antiphospholipid syndrome with defibrotide, a proposed vascular endothelial cell modulator. J Rheumatol. (2002) 29:2006–11.
222. Ali RA, Estes SK, Gandhi AA, Yalavarthi S, Hoy CK, Shi H, et al. Defibrotide inhibits antiphospholipid antibody-mediated neutrophil extracellular trap formation and venous thrombosis. Arthritis Rheumatol. (2022) 74:902–7. doi: 10.1002/art.42017
223. Rodziewicz M, D’Cruz DP. An update on the management of antiphospholipid syndrome. Ther Adv Musculoskelet Dis. (2020) 12:1759720x20910855. doi: 10.1177/1759720X20910855
224. Jiao JA, Kelly AB, Marzec UM, Nieves E, Acevedo J, Burkhardt M, et al. Inhibition of acute vascular thrombosis in chimpanzees by an anti-human tissue factor antibody targeting the factor X binding site. Thromb Haemost. (2010) 103:224–33. doi: 10.1160/TH09-06-0400
225. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
226. Agostinis C, Durigutto P, Sblattero D, Borghi MO, Grossi C, Guida F, et al. A non-complement-fixing antibody to β2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood. (2014) 123:3478–87. doi: 10.1182/blood-2013-11-537704
227. Shemer A, Willis R, Gonzalez EB, Romay-Penabad Z, Shovman O, Shoenfeld Y, et al. Oral administration of Domain-I of beta-2glycoprotein-I induces immunological tolerance in experimental murine antiphospholipid syndrome. J Autoimmun. (2019) 99:98–103. doi: 10.1016/j.jaut.2019.02.002
228. Ruff WE, Dehner C, Kim WJ, Pagovich O, Aguiar CL, Yu AT, et al. Pathogenic autoreactive T and B cells cross-react with mimotopes expressed by a common human gut commensal to trigger autoimmunity. Cell Host Microbe. (2019) 26:100–13.e8. doi: 10.1016/j.chom.2019.05.003
229. Gysler SM, Mulla MJ, Stuhlman M, Sfakianaki AK, Paidas MJ, Stanwood NL, et al. Vitamin D reverses aPL-induced inflammation and LMWH-induced sFlt-1 release by human trophoblast. Am J Reprod Immunol. (2015) 73:242–50. doi: 10.1111/aji.2015.73.issue-3
230. Collison J. Preventing NETosis to reduce thrombosis. Nat Rev Rheumatol. (2019) 15:317. doi: 10.1038/s41584-019-0234-6
231. Chaturvedi S, Alluri R, McCrae KR. Extracellular vesicles in the antiphospholipid syndrome. Semin Thromb Hemost. (2018) 44:493–504. doi: 10.1055/s-0037-1599081
Keywords: antiphospholipid syndrome, antiphospholipid antibody, β2-glycoprotein I, thrombotic event, two hit model
Citation: Yang L, Guo R, Liu H, Chen B, Li C, Liu R, Liao S, Xie Q and Yin G (2025) Mechanism of antiphospholipid antibody-mediated thrombosis in antiphospholipid syndrome. Front. Immunol. 16:1527554. doi: 10.3389/fimmu.2025.1527554
Received: 13 November 2024; Accepted: 25 February 2025;
Published: 13 March 2025.
Edited by:
Shruti Chaturvedi, Johns Hopkins Medicine, United StatesReviewed by:
Danieli Castro Oliveira De Andrade, University of São Paulo, BrazilGloria Riitano, Sapienza University of Rome, Italy
Koshy Nithin Thomas, Christian Medical College, India
Tina Garofalo, Sapienza University of Rome, Italy
Copyright © 2025 Yang, Guo, Liu, Chen, Li, Liu, Liao, Xie and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geng Yin, eWluZ2VuZzE5NzVAMTYzLmNvbQ==; Qibing Xie, eGllcWliaW5nMTk3MUAxNjMuY29t
†These authors have contributed equally to this work and share first authorship