 Huiling Wang
Huiling Wang Peiqi Xu
Peiqi Xu Kai Yin
Kai Yin Shengjun Wang
Shengjun Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 18 February 2025
Sec. Nutritional Immunology
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1521196
This article is part of the Research TopicImmunometabolism: Exploring the Nexus of Metabolism and Immune Function in Health and DiseaseView all 3 articles
Macrophage metabolic reprogramming refers to the process by which macrophages adjust their physiological pathways to meet survival and functional demands in different immune microenvironments. This involves a range of metabolic pathways, including glycolysis, the tricarboxylic acid cycle, oxidative phosphorylation, fatty acid oxidation, and cholesterol transport. By modulating the expression and activity of key enzymes and molecules within these pathways, macrophages can make the transition between pro- and anti-inflammatory phenotypes, thereby linking metabolic reprogramming to inflammatory responses and the progression of several diseases, such as atherosclerosis, inflammatory bowel disease (IBD), and acute lung injury (ALI). N6-methyladenosine (m6A) modification has emerged as a critical regulatory mechanism during macrophage metabolic reprogramming, broadly affecting RNA stability, translation, and degradation. Therapeutic strategies targeting m6A modification can regulate the onset of metabolic diseases by influencing macrophage metabolic changes, for instance, small molecule inhibitors of methyltransferase-like 3 (METTL3) can affect glucose metabolism and inhibit IBD. This review systematically explores recent findings on the role and molecular mechanisms of m6A modification during macrophage metabolic reprogramming in human diseases and animal models, underscoring its potential as a therapeutic target for metabolic diseases.
Macrophages are a type of immune cell widely distributed across various organisms, playing a central role in immune response and homeostasis. They are found in a variety of species, ranging from invertebrates to vertebrates, and are involved in processes such as pathogen defense, tissue repair, and immune regulation. However, in mammals, including both mice and humans, macrophages exhibit a higher level of functional complexity, with their polarization states and metabolic reprogramming being particularly important in diseases such as inflammation, metabolic disorders, and cancer.
Macrophages, as one of the primary responders of the immune system in mammals, are key participants in innate immunity and serve as a bridge between innate and adaptive immune response through the antigen presentation process (1). These cells exhibit notable plasticity, with non-polarized M0 macrophages polarizing into pro-inflammatory M1 or anti-inflammatory M2 cells depending on the environmental cues, allowing them to play pivotal roles in inflammation, tissue repair, and disease progression (2). All cells require sufficient and appropriate nutrients and oxygen to maintain metabolic homeostasis. Macrophage metabolic reprogramming is an activity carried out by optimizing the physiological pathways of macrophages in response to their different metabolic demands during polarization. After polarization to the M1 or M2 phenotype, macrophages show modulated metabolic efficiency, reflected in different pathways and metabolite levels, as illustrated in Figure 1. Upon stimulation by factors such as lipopolysaccharide (LPS), interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), and oxidized low-density lipoprotein (oxLDL), macrophages are activated into the M1 phenotype and play a role in pro-inflammatory and antimicrobial responses (3). Glycolysis is the primary metabolic pathway used by M1 macrophages (4). Upon activation, these cells exhibit a Warburg-like effect (5) to meet their energy demands, shifting from reliance on oxidative phosphorylation (OXPHOS) to a more glycolytic-dependent pathway. The dependence of M1 cells on glycolysis for ATP production is due to blocks in the tricarboxylic acid cycle (TCA cycle), which limit the production of NADH and FADH2 required for the electron transport chain, thereby inhibiting OXPHOS (6). In contrast, upon stimulation by cytokines such as interleukin-4 (IL-4), interleukin-13 (IL-13), interleukin-10 (IL-10), and transforming growth factor-beta (TGF-β), macrophages are polarized into the M2 phenotype, which can be further subdivided into M2a, M2b, M2c, and M2d subtypes, contributing to anti-inflammatory responses, tissue repair, and parasitic infections (7, 8). Aerobic glucose oxidation and fatty acid oxidation (FAO) are the primary metabolic pathways used by M2 macrophages. Oxidative metabolism is also significantly enhanced in M2 cells (9, 10). Unlike the demands of energy metabolism in M1 macrophages, M2 cells maintain an intact TCA cycle, thus guaranteeing a smooth process of ATP production by OXPHOS (11, 12). Indeed the metabolic process in macrophages involves more than just changes in metabolite levels and ATP production; it also influences macrophage phenotype through the regulation of transcriptional and post-transcriptional events.
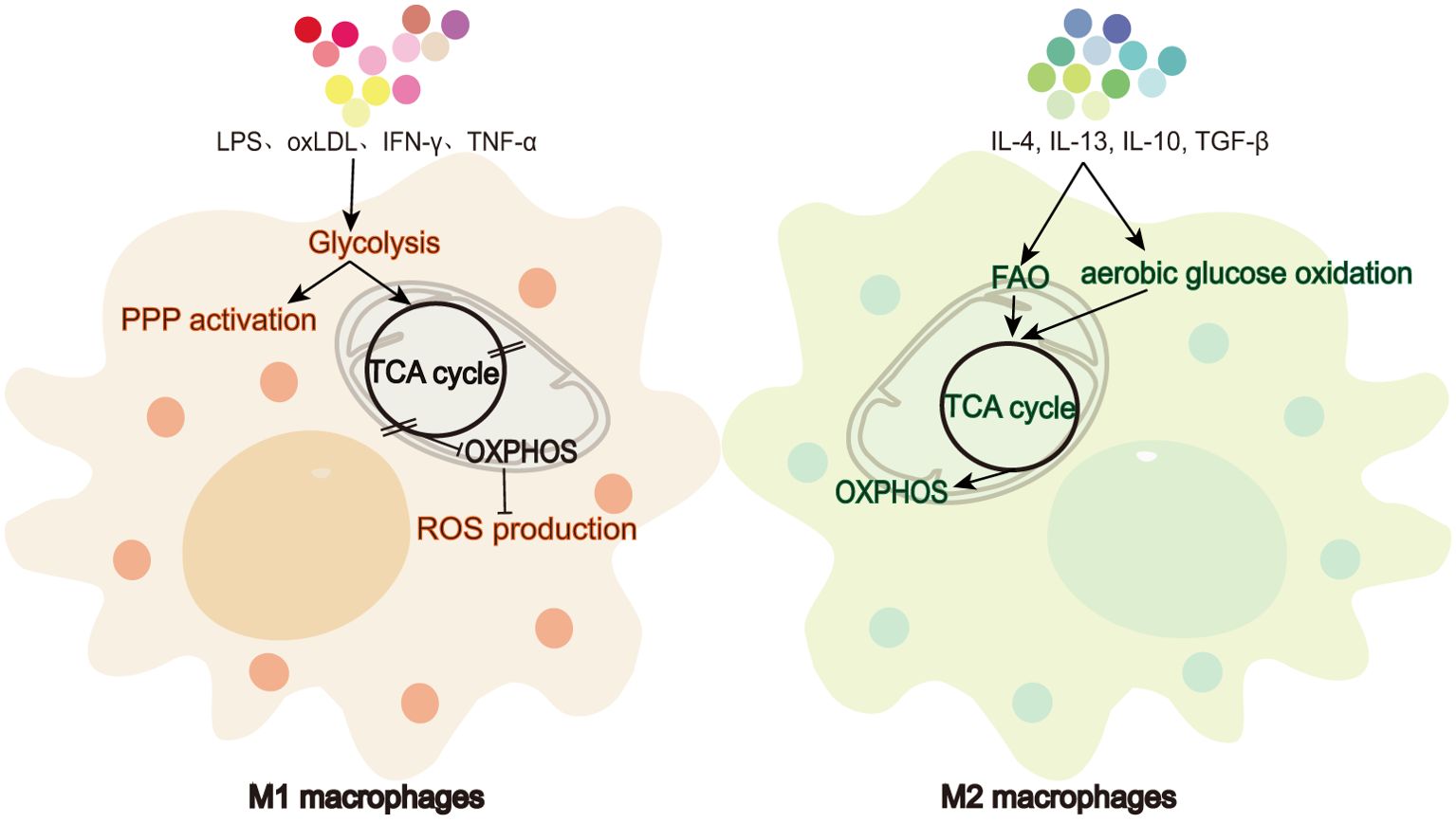
Figure 1. Metabolic differences between M1 and M2 macrophages. M1 macrophages favor glycolysis and ROS production while limiting TCA cycle and OXPHOS. In contrast, M2 macrophages rely on FAO, aerobic glucose oxidation, TCA cycle, and OXPHOS.
N6-methyladenosine (m6A) modification refers to a chemical modification in which a methyl group is added to the nitrogen at the sixth position of adenosine in RNA molecules, making it the most prevalent, abundant, and conserved post-transcriptional modification in eukaryotic cells (13). This is widely present in both mRNAs and non-coding RNAs and mainly concentrated in coding sequences (CDS), 3′ untranslated regions (3′ UTR), long introns, and near stop codons (14). RRACH sequences are consensus motifs that are recognized and methylated by RNA methyltransferases (14, 15). In mRNA, m6A modifications are reversible and can be removed by demethylases. Reader proteins recognize specific m6A sites and regulate mRNA stability, localization, translation, splicing, and transport (13), and they interact to determine the fate of mRNA. m6A modifications are involved in the physiological and pathological processes of various immune cell types, playing crucial roles in cell differentiation, development, and disease pathogenesis (16). When macrophages polarize to the M1 phenotype, the expression of methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14) is significantly upregulated, leading to m6A modification levels being markedly increase (17, 18). Recent studies indicate that m6A modifications are extensively involved in regulating macrophage phenotypes, further influencing disease progression by modulating metabolic pathways. In this paper, we are describing the role of m6A modification in macrophage metabolic reprogramming, including how this process alters metabolic disease progression, and we highlight potential opportunities for targeting m6A modifications to treat diseases.
m6A modification is a dynamic and reversible process controlled by three main types of proteins—writers, erasers, and readers (19)—as summarized in Table 1.
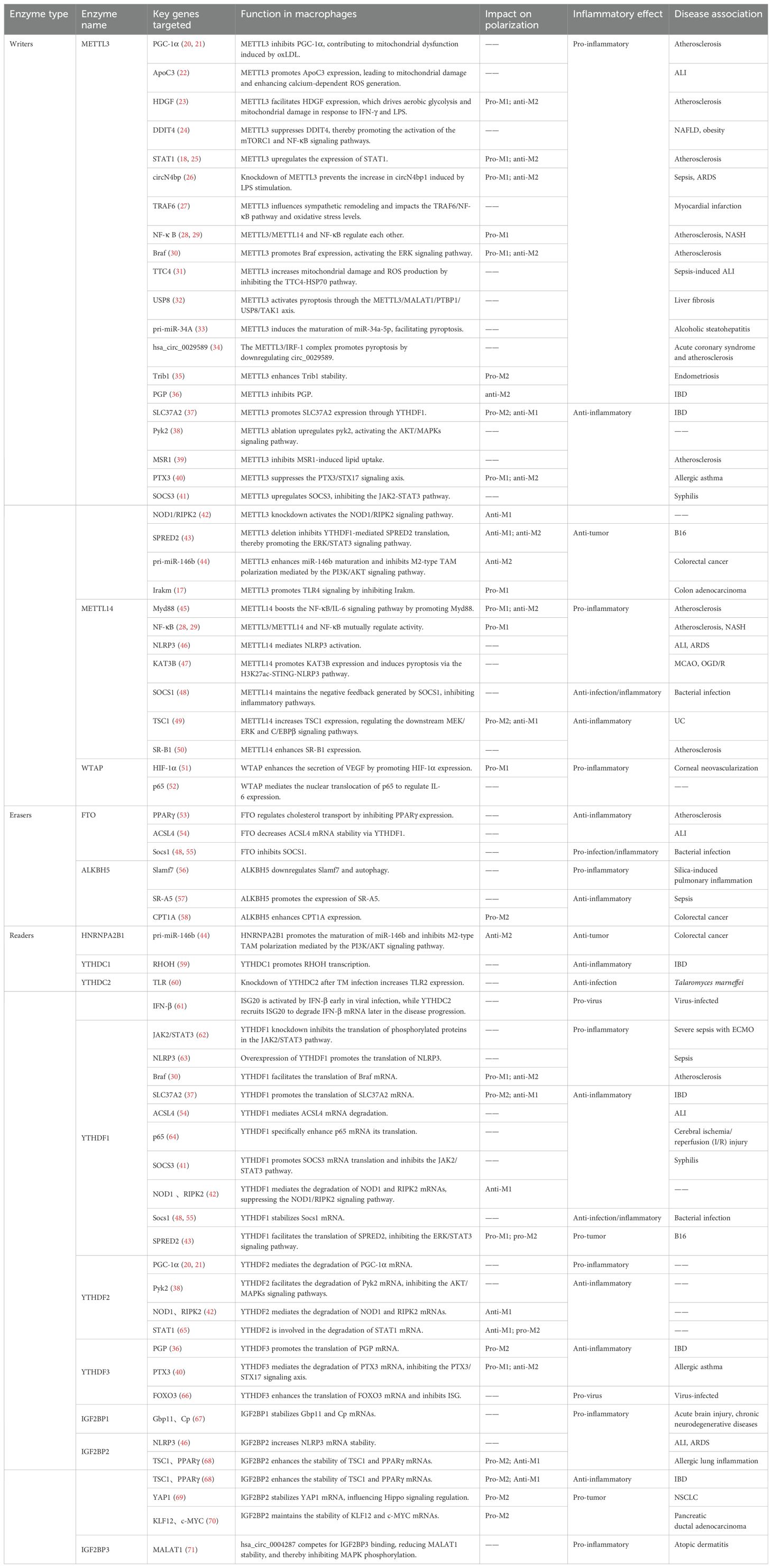
Table 1. Summary of m6A regulatory enzymes in macrophage polarization and inflammation.
m6A methyltransferases, such as METTL3, METTL14, Wilms’ tumor 1-associating protein (WTAP), methyltransferase-like 16 (METTL16), RNA-binding motif protein 15/15B (RBM15/15B), zinc finger CCCH domain-containing protein 13 (ZC3H13), HAKAI, zinc finger CCHC-type containing 4 (ZCCHC4), and vir-like m6A methyltransferase associated (VIRMA, KIAA1429), are writer proteins. METTL3, the first identified writer, is the only catalytically active subunit in the m6A methyltransferase complex (MTC) (72). In macrophages, METTL3 promotes glycolysis, mitochondrial damage, and ROS production while suppressing the TCA cycle and OXPHOS, thereby contributing to inflammatory responses. In diseases such as atherosclerosis, non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), obesity, myocardial infarction, endometriosis, sepsis, acute lung injury (ALI), and inflammatory bowel disease (IBD), METTL3 promotes glycolysis (18, 23–25, 27–29, 35), enhances mitochondrial damage and ROS production (22, 31), and suppresses the TCA cycle and OXPHOS (20, 21, 36) by regulating the m6A modifications of key genes (see Table 1 for details). Notably, M1 macrophages play a central role in driving inflammation and disease progression in conditions such as atherosclerosis, NASH, and sepsis (18, 23, 25, 26, 28–30). Also, METTL3 orchestrates these effects by reprogramming macrophage metabolism to favor M1 polarization and inflammatory responses.
The METTL3–METTL14 heterodimer, as the core of the MTC, catalyzes the majority of mRNA m6A methylation (72). Although METTL14 lacks catalytic activity, it structurally stabilizes the interaction between METTL3 and RNA substrates, enhancing the catalytic efficiency of the MTC (73). WTAP is a regulatory subunit that anchors METTL3 to METTL14 and facilitates the assembly and nuclear localization of the MTC (13). Both METTL14 and WTAP assist METTL3 in promoting M1 polarization and amplifying inflammatory responses. In diseases such as atherosclerosis, NASH and corneal neovascularization, METTL14 (28, 29, 45), and WTAP (51) enhance glycolysis, thereby promoting M1 polarization, inflammation, and disease progression.
While writers primarily enhance inflammation, they can also inhibit inflammatory processes (41, 42). In diseases such as IBD and atherosclerosis, writers can modulate the expression of solute carrier family 37 member 2 (SLC37A2), macrophage scavenger receptor 1 (MSR1), and scavenger receptor class B type 1 (SR-B1), inhibiting glycolysis and lipid uptake while promoting cholesterol efflux. This leads to anti-M1 and pro-M2 polarization, thereby suppressing macrophage inflammatory responses at the metabolic level (37, 39, 50). These findings suggest that macrophage m6A modifications are complex and multifaceted, contributing to both pro- and anti-inflammatory effects. The dual nature of this process makes it a “double-edged sword” for host immunity.
“Erasers” are m6A demethylases, including fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5). FTO was the first identified eraser (74). In diseases like atherosclerosis and ALI, FTO promotes fatty acid oxidation, inhibits lipid uptake, and accelerates cholesterol efflux, thereby facilitating M2 polarization and suppressing inflammation (53, 54, 68, 75). ALKBH5, the second identified m6A demethylase, shares a similar activity with FTO (19). It follows that m6A modifications dynamically modulate macrophage metabolic pathways in macrophages through the addition and removal of methyl groups by writers and erasers, balancing the pro-inflammatory M1 and anti-inflammatory M2 phenotypes.
“Readers” are m6A-specific reader proteins, including YTH domain containing proteins 1/2 (YTHDC1/2), heterogeneous nuclear ribonucleoproteins (hnRNPs), insulin-like growth factor 2 mRNA-binding protein 1/2/3 (IGF2BP1/2/3), and YTH domain family proteins 1/2/3 (YTHDF1/2/3). The nuclear m6A reader binds m6A-containing precursor RNAs in the nucleus and participates in RNA-selective shearing. Intranuclear m6A readers include YTHDC1 and hnRNP family members (13). YTHDC2, an RNA helicase, aids in RNA binding and influences mRNA translation or degradation (76). After being processed from the precursor transcript, mature mRNAs are further regulated by cytoplasmic m6A readers. YTHDF1 promotes mRNA translation, YTHDF2 facilitates mRNA degradation, and YTHDF3 contributes to either process (77). The IGF2BP family, which includes IGF2BP1, IGF2BP2, and IGF2BP3, stabilizes mRNA (78).
While the dynamic m6A levels are regulated by writers and erasers, m6A-modified target mRNAs are primarily regulated by readers. For example, YTHDF1 and YTHDF2 are required for the inhibitory effect of METTL3/METTL14 on suppressor of cytokine signaling 3 (SOCS3), nucleotide-binding oligomerization domain containing 1 (NOD1), and receptor interacting protein kinase 2 (RIPK2)-mediated inflammation (41, 42). These studies indicate that the role of readers in macrophage regulation is dependent on whether the target genes are pro- or anti-inflammatory. The role of readers in macrophage regulation is complex, as it depends not only on the nature of the target genes (pro- or anti-inflammatory) but also on the disease context. Even within the same macrophage mechanism, readers can exhibit different inflammatory effects depending on the disease. For example, IGF2BP2 enhances the stability of tuberous sclerosis complex 1 (TSC1) and peroxisome proliferator-activated receptor gamma (PPAR-γ) mRNAs, promoting an anti-inflammatory response in IBD but a pro-inflammatory response in allergic lung inflammation (68).
Macrophage metabolic reprogramming involves dynamic processes such as glycolysis, the TCA cycle, OXPHOS, FAO, and cholesterol transport, with the specific roles of m6A modifiers in these pathways summarized in Table 2.
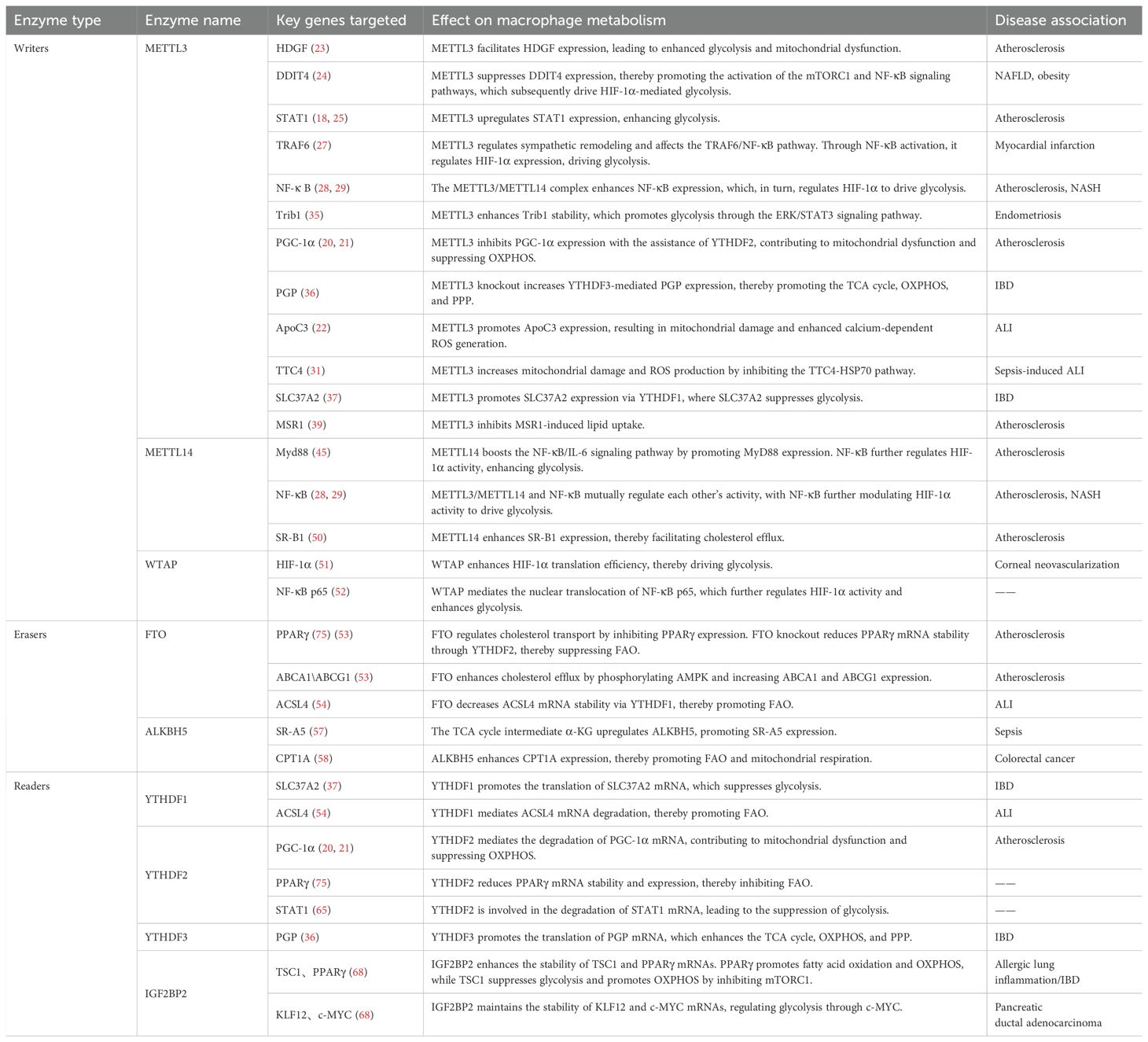
Table 2. Summary of m6A modifiers and their specific roles in macrophage metabolism.
The activation of classical M1 macrophages is characterized by an upregulation of glycolysis, which is accompanied by an increased extracellular acidification rate (ECAR) and a decreased oxygen consumption rate (OCR) (79). This shift ultimately results in mitochondrial dysfunction, including reduced oxygen uptake and ATP production (80). M1 macrophages become activated by stimulating bone-marrow-derived macrophages with LPS for 24 h. This leads to a significant upregulation of the glucose transporter solute carrier family 2 member 1 (SLC2A1, also known as GLUT1) and the glycolytic enzymes, hexokinase 3 (HK3), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), phosphoglucomutase 2 (PGM2), and alpha-enolase (ENO2), along with an accumulation of the glycolytic product, lactate (81). See Figure 2 for additional glycolytic enzyme and product changes. GLUT1 is the primary glucose transporter in mouse macrophages and is upregulated 10-fold and twofold in M1 and M2 macrophages, respectively, after polarization from M0 cells (4). Macrophages use GLUT1 to take up glucose in response to LPS stimulation (82). ECAR represents the rate of glycolysis, while OCR indicates mitochondrial respiration. GLUT1 overexpression in macrophages effectively increases ECAR, reduces OCR, and promotes macrophage polarization toward the M1 inflammatory phenotype (4). All m6A-regulated mechanisms of macrophage glycolysis are summarized in Figure 2. During atherosclerosis, METTL3 regulates hepatoma-derived growth factor (HDGF) mRNA stability in macrophages through m6A modification, increasing HDGF expression that, in turn, accelerates glycolysis and enhances mitochondrial dysfunction. This is reflected in increased glucose uptake and ECAR, decreased OCR, and reduced mitochondrial membrane potential, thereby promoting M1 macrophage polarization (23).
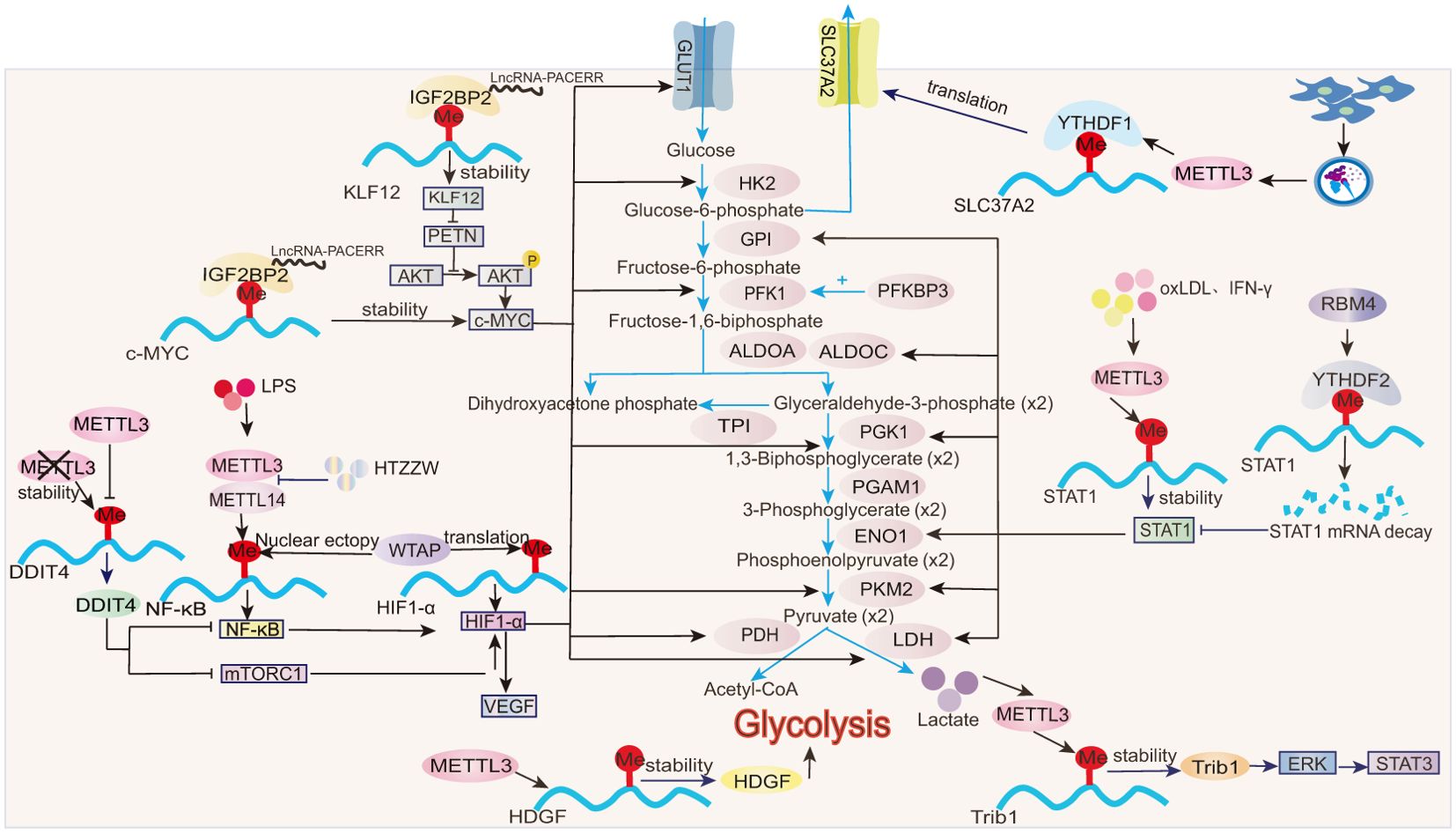
Figure 2. Role of m6A in glycolysis. In macrophages, m6A modifications influence the stability, translation, and expression of transcription factors (e.g., HIF-1α, STAT1), then modulating glycolytic enzymes (e.g., HK2, PKM2).
Signal transducer and activator of transcription 1 (STAT1) is a hallmark transcription factor involved in M1 macrophage polarization. After stimulation with IFN-γ or oxLDL, m6A modification increases significantly in macrophages. METTL3 directly mediates m6A modifications on the CDS and 3′UTR regions of STAT1 mRNA, thereby enhancing the stability, gene expression, and transcriptional activity of STAT1 (18, 25). STAT1, acting at both the transcriptional and translational levels, upregulates key glycolytic enzymes, including glucose-6-phosphate isomerase (GPI), fructose-bisphosphate aldolase A (ALDOA), fructose-bisphosphate aldolase C(ALDOC), triosephosphate isomerase (TPI), phosphoglycerate kinase 1 (PGK1), phosphoglycerate mutase 1 (PGAM1), ENO1, pyruvate kinase M2 isoform (PKM2), and lactate dehydrogenase (LDH). Furthermore, STAT1 promotes the upregulation of NADH, FADH2, and ATP synthase to meet the increased energy demands of M1 macrophages during polarization (83). In RAW264.7 macrophages, overexpression of RNA-binding motif 4 (RBM4) promotes the binding of the reader protein YTHDF2 to m6A sites on the STAT1 mRNA 3′UTR region, inducing STAT1 mRNA degradation and reducing STAT1 transcriptional activity. Consequently, the expression of glycolysis-related genes regulated by STAT1, such as ALDOA, ALDOC, and GPI, is significantly decreased (65). This suppression of glycolysis is reflected by decreased glucose uptake, reduced lactate production, and downregulated ECAR profiles (65). Notably, overexpression of RBM4 inhibits IFN-γ-induced M1 macrophage polarization without affecting IL-4-induced M2 polarization, suggesting a selective regulatory role in metabolic reprogramming (65).
IL-4-induced M2 macrophage activation is primarily dependent on mitochondrial respiratory function. However, this process is also accompanied by a moderate enhancement in glycolysis, as indicated by delayed and slight increases in ECAR (79). While glycolysis is well established as a key metabolic feature of M1 macrophage polarization, emerging evidence suggests its involvement in M2 polarization under specific contexts. For instance, in endometriosis, enhanced glycolysis is positively associated with the infiltration of M2 macrophages into lesion sites, as evidenced by the increased levels of PKM2 and lactate (35). High lactate concentrations promote the stability of tribbles pseudokinase 1 (Trib1) mRNA through METTL3-mediated m6A modifications, thereby increasing Trib1 protein expression (35). Trib1 facilitates M2 macrophage polarization through the activation of the ERK/STAT3 signaling pathway (35, 84). Collectively, glycolysis may contribute to M2 macrophage activation in some contexts (35, 65), but its precise role depends on the specific inflammatory immune environment.
Glucose-6-phosphate (G6P) is an intermediate in glycolysis. SLC37A2, a phosphate-linked G6P antiporter, exhibits the highest transcript abundance among the SLC37 family in macrophages. LPS-induced glycolysis is increased in SLC37A2 knockout macrophages, characterized by the rapid depletion of key glycolytic intermediates such as G6P and dihydroxyacetone phosphate (DHAP), alongside the increased accumulation of downstream products, including pyruvate and lactate. This metabolic shift is accompanied by elevated ECAR (85). Notably, SLC37A2 expression is significantly reduced in the colorectal tissues of IBD mouse models (37). In RAW264.7 macrophages, exosomes derived from human umbilical cord mesenchymal stem cells (hucMSC-Ex) mediate the m6A modification of SLC37A2 3′UTR mRNA through METTL3 and enhance the binding of the reader protein YTHDF1 to SLC37A2, thereby promoting SLC37A2 mRNA translation and expression (37). By downregulating glycolysis, SLC37A2 mitigates macrophage inflammatory activation, thereby alleviating intestinal inflammation in IBD (37).
Hypoxia-inducible factor 1 α (HIF-1α) is a key regulator of glycolysis (86). The rate-limiting enzyme of glycolysis, pyruvate kinase isozyme type M 2 (PKM2), is upregulated in LPS-treated macrophages, and PKM2 dimers promote HIF-1α stabilization (87). HIF-1α drives glycolysis by inducing the expression of several genes, including GLUT1, hexokinase 2 (HK2), phosphofructokinase 1 (PFK1), LDH, and pyruvate dehydrogenase (PDH) (81, 86). In corneal neovascularization, the methyltransferase, WTAP, modulates the m6A enrichment level on HIF-1α and partially affects its translation efficiency without changing its mRNA stability, thereby promoting HIF-1α protein production (51).
LPS enhances HIF-1α mRNA transcription in macrophages by inducing nuclear factor kappa B (NF-κB) activity, further amplifying HIF pathway signaling (88, 89). Beyond its role in HIF-1α translation (51), WTAP also facilitates the nuclear translocation of NF-κB p65 in tamm-horsfall protein-1 (THP-1) macrophages (52), enhancing HIF-1α stability and activity via NF-κB-dependent mechanisms (88, 89). In LPS-stimulated macrophages, NF-κB recruits the RelA subunit (NF-κB p65) to the HIF-1α promoter to regulate its transcription (89). In BMDMs treated with the hypoxia mimetic drug deferoxamine (DFX), IκB kinase β (IKKβ) is indispensable for HIF-1α accumulation (89). In the absence of IKKβ, hypoxia-driven HIF-1α expression and the transcription of its downstream targets, such as vascular endothelial growth factor (VEGF) and GLUT1, are significantly impaired (89). In LPS-stimulated liver macrophages (Kupffer cells), the NF-κB p65 subunit transcriptionally activates METTL3 and METTL14 and increases global m6A modification levels (29). The traditional Chinese herbal formula, Huatuo Zizao pill (HTZZW), mitigates atherosclerosis by attenuating NF-κB signaling. Specifically, HTZZW suppresses METTL3 and METTL14 expression in macrophages, reducing m6A modification within the NF-κB mRNA 3′-UTR and ultimately lowering NF-κB mRNA translation and protein expression (28). m6A modification influences NF-κB activity through the methyltransferases, METTL3 and METTL14, which, in turn, regulates HIF-1α through NF-κB, driving the expression of glycolytic genes and significantly promoting M1 macrophage polarization.
In addition to NF-κB, the mechanistic target of rapamycin complex 1 (mTORC1) also serves as an upstream regulator of HIF-1α, inducing the transcription of glycolytic genes such as LDH and PGK1 as well as VEGF (90). In METTL3-deficient macrophages, the stability of DNA damage-inducible transcript 4 (DDIT4) mRNA is significantly enhanced (24). DDIT4 inhibits mTORC1 and NF-κB signaling pathways, thereby reducing downstream HIF-1α transcriptional activity. This suppression reduces glycolysis and M1 macrophage activation, ultimately protecting against diet-induced nonalcoholic fatty liver disease (NAFLD) and obesity (24).
MYC oncogene (c-MYC) is also a core regulator of glycolysis (91, 92). In pancreatic ductal adenocarcinoma (PDAC), the reader IGF2BP2 interacts with LncRNA-PACERR to enhance the stability and expression of krüppel-like factor 12 (KLF12) and c-MYC mRNAs. This interaction further increases c-MYC expression through KLF12\PETN\pAKT signaling (70). c-MYC overexpression significantly increases the expression of GLUT1, SLC1A5, and the glycolytic enzymes HK2, PKM2, and LDH, promoting the M2 polarization of tumor-associated macrophages (TAMs) (70, 91, 92).
All m6A-regulated mechanisms of TCA cycle in macrophages are summarized in Figure 3. The TCA cycle remains intact in M2 macrophages (93). In colonic infiltrating macrophages from METTL3-knockout IBD mice, YTHDF3-mediated upregulation of phosphoglycolate phosphatase (PGP) mRNA and protein expression has been observed (36). PGP enhances the TCA cycle, OXPHOS, and the pentose phosphate pathway (PPP) in macrophages, while glycolysis remains unaffected (36). This is evidenced by the increased levels of TCA intermediates such as malate, citrate, and fumarate along with elevated OCR. In addition, the PPP-related genes, including glucose-6-phosphate dehydrogenase X (G6PDX), phosphogluconate dehydrogenase (PGD), and 6-phosphogluconolactonase (PGLS), are markedly upregulated, leading to an increased NADPH/NADP+ ratio. Conversely, lactate levels, ECAR, and expression of the glycolytic enzymes remain unchanged (36). The metabolic reprogramming in METTL3-deficient macrophages promotes M2 polarization, suppressing pathogenic Th1 cells and alleviating colitis (36).
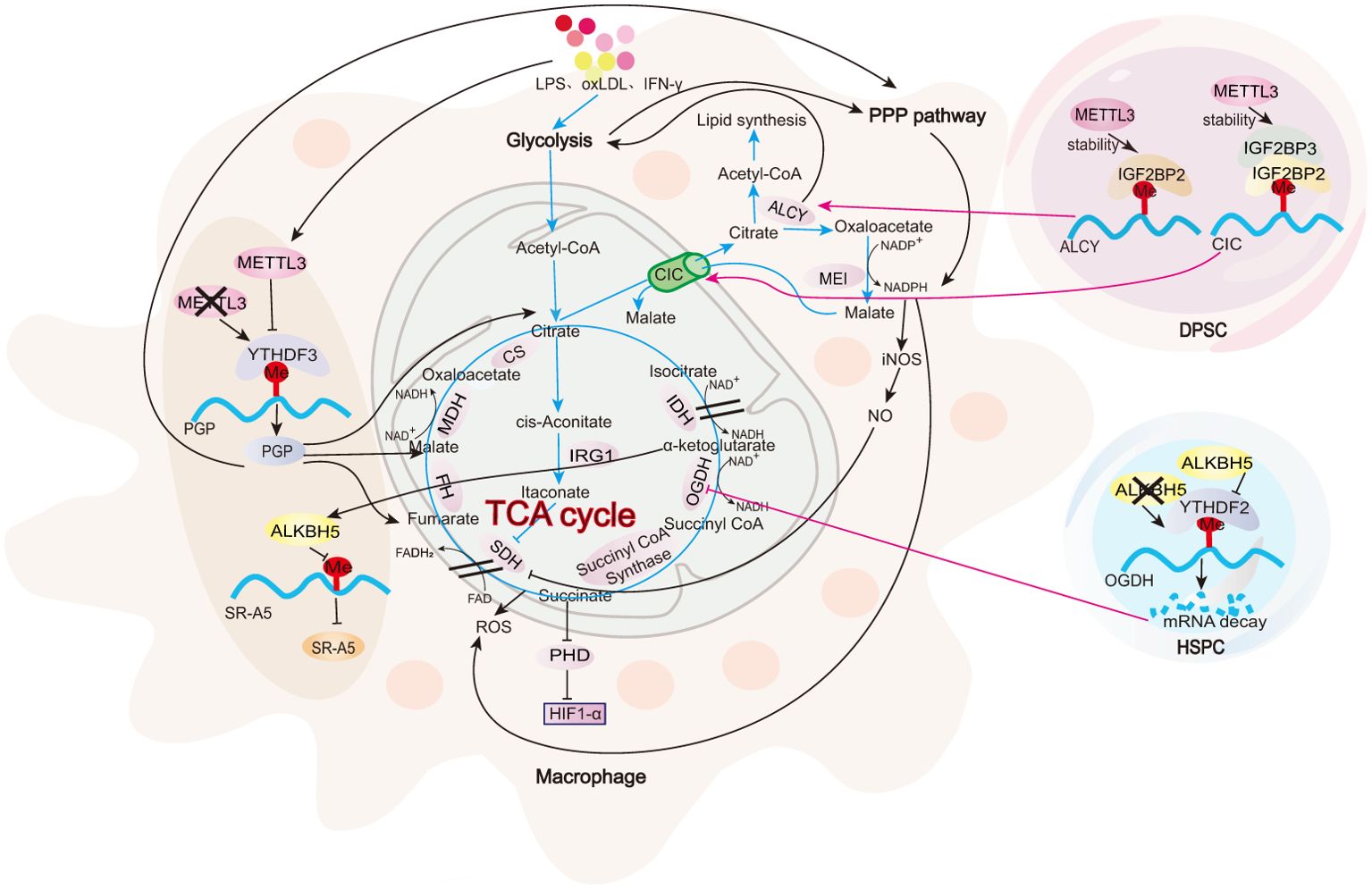
Figure 3. Role of m6A in TCA cycle. In macrophages, m6A modifications regulate PGP, which, in turn, modulates key metabolic enzymes and intermediates involved in the TCA cycle. Moreover, DPSCs and HSPCs serve as examples to illustrate the impact of m6A modifications on two critical metabolic checkpoints: citrate metabolism and succinate metabolism. Further investigation is needed to explore these checkpoints in the context of macrophages.
In M1 macrophages, alongside the upregulation of glycolysis, there is a marked reduction in the levels of key mitochondrial enzymes, such as malate dehydrogenase (MDH1) and isocitrate dehydrogenase (IDH2). This reduction, coupled with a significant depletion of isocitrate in the mitochondrial respiratory chain, indicates an impairment of the TCA cycle (81). The disruption of the TCA cycle occurs at two key metabolic points: citrate metabolism and succinate metabolism (94).
During citrate metabolism, LPS-, TNF-α-, or IFN-γ-activated M1 macrophages show a reduction in IDH expression (95), leading to isocitrate depletion in the mitochondria. At the same time, the mitochondrial citrate carrier (CIC, also known as solute carrier family 25 member 1, SLC25A1) is upregulated, facilitating the export of citrate from the mitochondria to the cytoplasm in exchange for malate. This results in the accumulation of citrate in the cytoplasm (96, 97), where ATP-citrate lyase (ACLY) converts it into acetyl-CoA and oxaloacetate (98). The increase in acetyl-CoA promotes lipid synthesis, while oxaloacetate is converted into malate by malic enzyme (ME1). This is accompanied by the reduction of NADP+ to NADPH, enhancing the production of nitric oxide (NO) and ROS (99). m6A modification at this first interruption point in the TCA cycle has been identified in dental pulp stem cells (DPSCs). In these cells, METTL3-IGF2BP2 mediates the stability of ACLY mRNA, while METTL3-IGF2BP2/3 mediates the stability of CIC mRNA (100). Additional research is required to validate this observation in macrophages.
During succinate metabolism, activated M1 macrophages show a significant upregulation of aconitate decarboxylase 1 (ACOD1, also known as IRG1) (101). ACOD1 catalyzes the decarboxylation of cis-aconitate to produce itaconate, thereby disrupting the TCA cycle (101). Itaconate inhibits succinate dehydrogenase (SDH), preventing succinate from further metabolizing into fumarate and leading to its accumulation in the cell (102). Succinate acts as a competitive inhibitor of α-ketoglutarate (α-KG), suppressing the activity of HIF-1α hydroxylase (PHD) (81) and stabilizing and activating HIF-1α, a key transcription factor for glycolysis (103). The m6A modification at this second interruption point in the TCA cycle has been observed in hematopoietic stem and progenitor cells (HSPCs). In these cells, the α-ketoglutarate dehydrogenase complex (OGDH), the rate-limiting enzyme in the conversion of α-KG to succinyl-CoA, is regulated by the m6A demethylase ALKBH5. Deletion of ALKBH5 increases m6A modifications on OGDH mRNA, which are recognized by the m6A reader protein YTHDF2. This recognition reduces OGDH mRNA stability, decreases OGDH protein levels, and consequently slows TCA cycle progression (104). In macrophages, α-KG upregulates the m6A demethylase, ALKBH5, reducing the m6A modification of scavenger receptor class A member 5 (SR-A5), enhancing SR-A5 expression, inhibiting M1 polarization, and promoting M2 polarization, thereby alleviating the inflammatory response in septic macrophages (57).
IL-4-induced M2 macrophages are characterized by enhanced oxidative metabolism, exhibiting high levels of OCR and spare respiratory capacity (SRC) (10) and primarily relying on OXPHOS and FAO (9). All m6A-regulated mechanisms of macrophage OXPHOS are summarized in Figure 4. In response to IL-4, STAT6 directly binds to the high-mobility group AT-hook 2 (HMGA2) promoter region, promoting the expression of the reader IGF2BP2 (68). IGF2BP2 enhances TSC1 and PPARγ mRNA stability and expression in an m6A modification-dependent manner (68). Among them, PPARγ promotes FAO and OXPHOS (105), while TSC1 inhibits glycolysis and induces OXPHOS (90, 106). PPARγ and TSC1 expression effectively elevates OCR and SRC production, enhancing OXPHOS and mitochondrial respiration and promoting M2 cell polarization (68). This process not only exacerbates allergic pulmonary inflammation but also has a positive role in alleviating colitis (68).
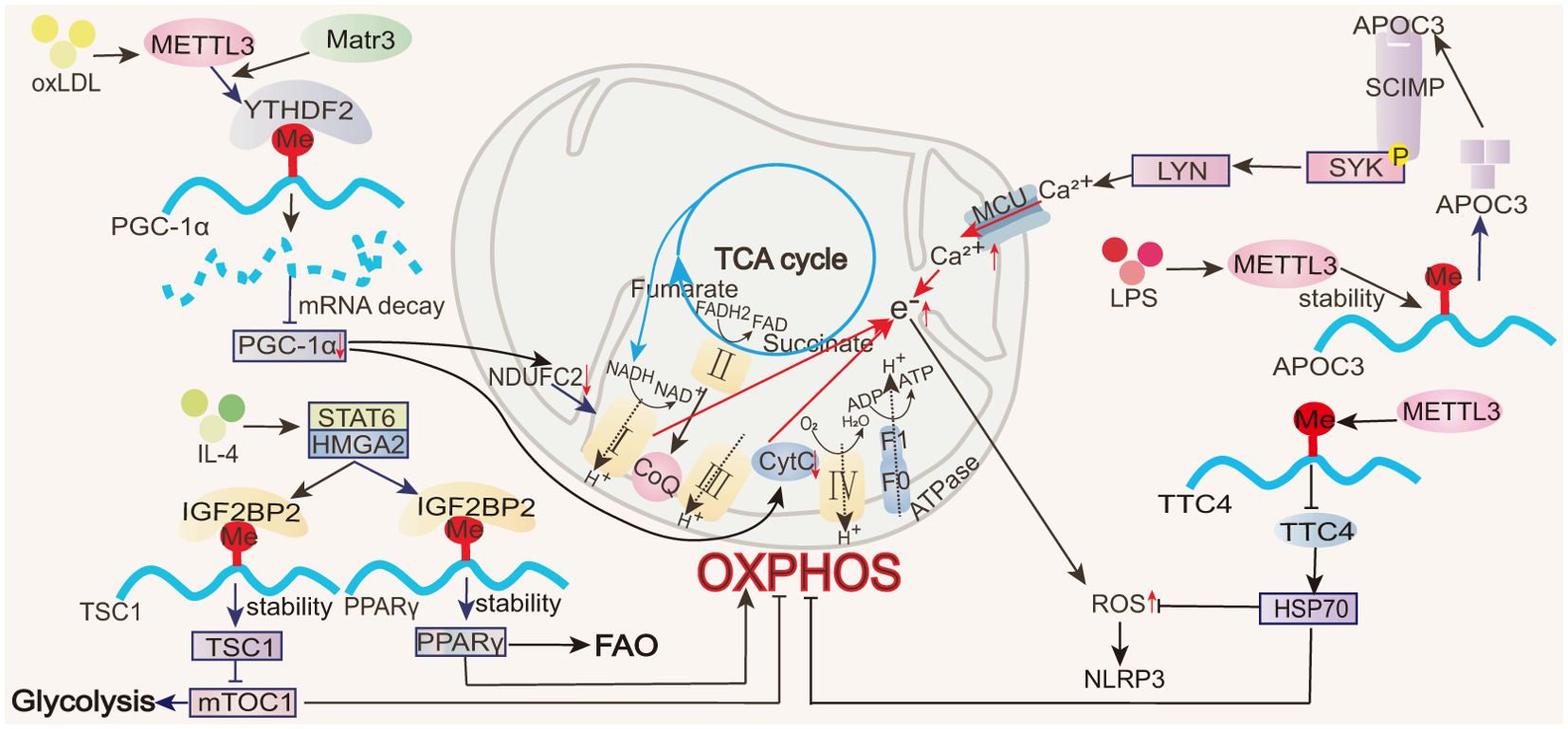
Figure 4. Role of m6A in OXPHOS. In macrophages, m6A modifications regulate transcription factors (e.g., PGC-1α, PPARγ), thereby affecting the electron transport chain in OXPHOS and influencing ROS production.
NADH and FADH2, generated during the TCA cycle, are critical electron donors for OXPHOS. During M1 macrophage activation, TCA cycle disruption leads to succinate accumulation, causing reverse electron transfer (RET) in the mitochondria. This increases electron leakage in the electron transport chain, reducing OXPHOS efficiency and generating ROS (107). Excessive ROS can impair mitochondrial function, favoring a shift from OXPHOS to glycolysis for ATP production. During ALI, METTL3 modulates the stability and expression of apolipoprotein C3 (ApoC3) mRNA in macrophages. ApoC3, through the SCIMP-SYK signaling pathway, promotes calcium influx, damages the mitochondrial membrane potential, impairs OXPHOS, generates ROS and activates the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome (22). METTL3 also suppresses the stability and expression of tetratricopeptide repeat domain 4 (TTC4) 3’-UTR mRNA in ALI macrophages, inhibiting downstream heat shock protein 70 (HSP70) induction, increasing mitochondrial damage, and activating the ROS/NLRP3 signaling pathway (31).
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is an important regulator of OXPHOS, binding to various transcription factors and nuclear hormone receptors to support mitochondrial biogenesis and oxidative metabolism. This includes the activation of nuclear respiratory factor 1 (NRF-1), estrogen-related receptor alpha (ERRα), yin yang 1 (YY1), and myocyte enhancer factor 2C (MEF2C), which act on the mitochondrial respiratory chain, and PPARα and PPARγ, which control FAO (108). During oxLDL-induced monocyte inflammation, METTL3 mediates m6A modifications in the coding region of PGC-1α. YTHDF2 specifically recognizes these m6A modifications and promotes the degradation of PGC-1α mRNA, reducing the PGC-1α levels (20, 21). The reduction in PGC-1α further inhibits the expression of the nuclear-encoded mitochondrial respiratory chain proteins, cytochrome c somatic (CYCS), and NADH: ubiquinone oxidoreductase core subunit C2 (NDUFC2), thereby suppressing OXPHOS, increasing the accumulation of cellular and mitochondrial ROS, reducing OCR, and impairing mitochondrial function (20, 21). Matr3 participates in the formation of the METTL3–METTL14 complex. While Matr3 overexpression does not directly affect the level of m6A in polyadenylated RNA, this protein can assist METTL3 in increasing the m6A modifications of PGC-1α mRNA (20, 21).
All m6A-regulated mechanisms of FAO in macrophages are summarized in Figure 5. Oxidative metabolism in M2 macrophages primarily depends on FAO and OXPHOS (9). The rate-limiting step of FAO is mediated by carnitine palmitoyltransferase 1A (CPTIA), which converts long-chain fatty acids into acyl-carnitine, facilitating its transport into the mitochondrial matrix (109) and making CPT1A the most critical transport enzyme for FAO (110). FAO and ATP levels increase significantly when THP-1 macrophages are co-cultured with colorectal cancer (CRC) cells. This is accompanied by a notable increase in OCR and a decrease in ECAR, indicating that fatty acid metabolism and mitochondrial respiration are increased in the macrophages, inducing TAM M2 polarization and promoting tumor growth (58). Notably, ALKBH5 mediates a reduction in the m6A modification of CPT1A, increasing CPT1A mRNA stability and expression and thus regulating the FAO pathway involved in M2 polarization (58).
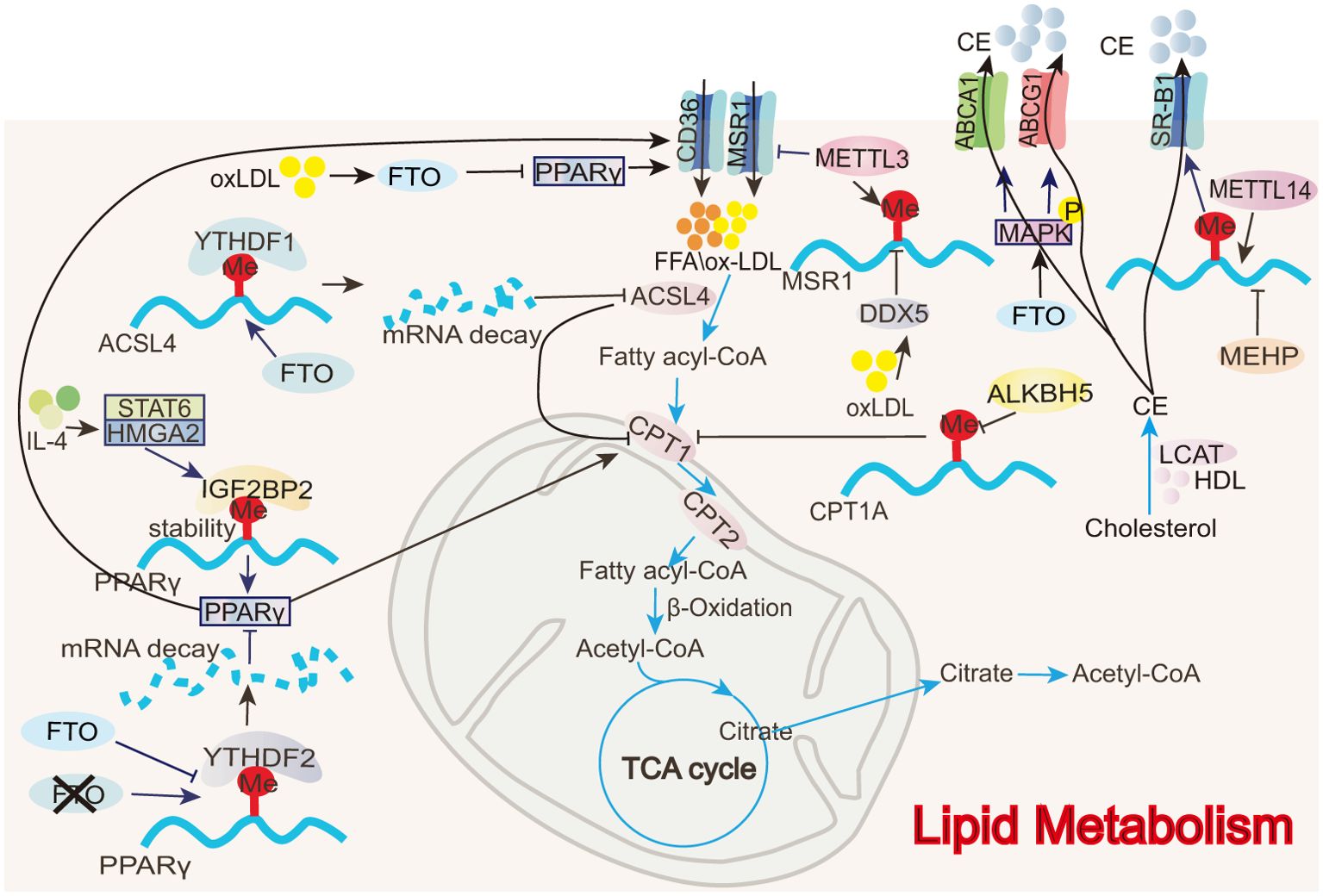
Figure 5. Role of m6A in FAO and cholesterol transport. In macrophages, m6A modifications directly or indirectly regulate FAO-related enzymes (e.g., ACSL4, CPT1) as well as lipid transport proteins (e.g., ABCA1, ABCG1).
Acyl-CoA synthetase long-chain family member 4 (ACSL4) is involved in polyunsaturated fatty acid metabolism, promoting lipid peroxide production during fatty acid oxidation (111). Inhibiting ACSL4 enhances mitochondrial respiration and fatty acid oxidation, as evidenced by increased OCR, mitochondrial membrane potential, ATP production, and a higher expression of genes, including PPARα, PGC1α, CPT1A, acetyl-CoA carboxylase 2 (ACC2), and acyl-CoA dehydrogenase long-chain (ACADL), associated with mitochondrial fatty acid oxidation (111). During ALI, FTO reduces the stability of ACSL4 mRNA via YTHDF1, interrupting poly-unsaturated fatty acid metabolism, decreasing lipid peroxide production, and promoting FAO to suppress macrophage inflammation and inhibit disease progression (54). The FTO inhibitor FB23-2 exacerbates lung injury and inflammation in ALI mice (54).
Peroxisome proliferator-activated receptor gamma (PPARγ) enhances fatty acid uptake by upregulating CD36 expression and accelerates fatty acid entry into the mitochondria by promoting CPT1 activity, thereby increasing FAO (105). m6A modifications regulate PPARγ expression, modulating the FAO process and impacting M2 macrophage polarization. In BMDMs, IGF2BP2 recognizes the m6A modification on PPARγ mRNA and enhances its stability and expression, thus promoting M2 polarization (68). In contrast, an FTO gene knockout reduces PPARγ mRNA stability and expression through YTHDF2 involvement, thereby hindering macrophage M2 polarization (75).
All m6A-regulated mechanisms of cholesterol transport in macrophages are summarized in Figure 5. CD36 is a transmembrane transport protein widely expressed in various tissues, including adipose tissue, heart, and skeletal muscle, and it is responsible for the uptake and transport of fatty acids and cholesterol (53, 112). Low-density lipoprotein (LDL) is a lipoprotein particle that carries cholesterol into peripheral tissue cells. Upon oxidization, LDL forms oxidized low-density lipoprotein (oxLDL) (113). CD36 facilitates the uptake of oxLDL by macrophages, leading to the formation of foam cells, which can induce inflammatory responses and contribute to atherosclerosis (53). In RAW 264.7 macrophages, the eraser FTO inhibits PPARγ expression, suppressing CD36 uptake of oxLDL and preventing foam cell formation (53). Like CD36, MSR1 is a scavenger receptor that can also take up oxLDL and contribute to foam cell formation (39). oxLDL promotes dead-box helicase 5 (DDX5) expression in THP-1 macrophages. DDX5 then inhibits METTL3 methyltransferase activity, reducing m6A modifications and thus stabilizing MSR1 mRNA, increasing MSR1 expression, and inducing lipid uptake and atherosclerosis (39).
Reverse cholesterol transport (RCT) is the process by which cholesterol from lipid-laden peripheral cells is transported to the liver via plasma high-density lipoprotein (HDL) and subsequently excreted from the body through bile acids (114). Unlike lipid uptake, RCT can inhibit the formation of macrophage foam cells and the progression of atherosclerosis. RCT can be mediated by three pathways: simple diffusion, scavenger receptor B1 (SR-B1)-mediated facilitated diffusion, and efflux mediated by ATP-binding cassette transporter A1 (ABCA1) or G1 (ABCG1) (114). In mono- (2-ethylhexyl) phthalate (MEHP)-exposed RAW 264.7 macrophages, m6A RNA methylation is significantly reduced. MEHP inhibits SR-B1 expression by decreasing METTL14 expression, suppressing cholesterol efflux from macrophages, and accelerating atherosclerosis (50). In contrast, FTO increases ABCA1 and ABCG1 expression by phosphorylating AMPK, thereby enhancing cholesterol efflux and hindering disease progression (53).
Recently, m6A modification has played a significant role in the characterization of macrophage metabolic reprogramming and has been identified as an important therapeutic target for macrophage-related inflammatory diseases. Drugs that directly affect m6A modification or the activity of m6A-modifying proteins are used to modulate macrophage activation, polarization, and inflammatory responses. For example, Hua Tuo Zai Zao Wan (HTZZW) reduces the m6A levels in macrophages by inhibiting METTL3 and METTL14, leading to destabilization and downregulation of NF-κB mRNA, which suppresses macrophage polarization toward the M1 phenotype, effectively alleviating atherosclerosis (28). Coptisine (COP) increases macrophage m6A methylation by upregulating METTL14, enhancing TSC1 mRNA stability, inhibiting M1 polarization, and promoting M2 polarization, effectively alleviating ulcerative colitis (49). Astragalus mongholicus polysaccharides (APS) counteract the LPS-induced elevation of m6A levels in macrophages by inhibiting WTAP. This prevents WTAP-mediated p65 nuclear translocation, reducing IL-6 expression and alleviating macrophage-mediated inflammation (52).
Moreover, several specially designed small-molecule inhibitors, which exhibit good specificity and regulatory properties, are easy to synthesize and modify, showing promising translational potential in macrophage-related inflammatory diseases. For instance, the METTL3 inhibitor STM2457 suppresses the inflammatory response of M1 macrophages by inhibiting the LPS-induced NF-κB signaling pathway, thereby reducing the incidence of bone marrow inflammation in mice (115). The METTL3 inhibitors F039-0002 and 7460-0250 specifically inhibit METTL3 activity in macrophages, regulating glucose metabolic reprogramming and significantly alleviating intestinal inflammation (36). In lung macrophages (MH-S), treatment with the METTL3 inhibitor 3-deazaadenosine (DAA) nearly restores the elevated levels of circN4bp1 induced by LPS stimulation to normal, inhibiting M1 macrophage activation and improving the prognosis of septic patients (26). Conversely, the FTO inhibitor FB23-2 exacerbates the inflammatory response, lung injury, and iron dysregulation in ALI mice (54). Other METTL3 inhibitors include UZH1a and UZH1b (116), RM3, and RSM3 (117); other FTO inhibitors include CS1 and CS2 (118), R-2HG (119), and MA (120); and other ALKBH5 inhibitors include IOX3 (121), Ena15 and Ena21 (122), Cpd 20m (123), and DDO-2728 (124) have also been developed. The efficacy of these inhibitors in treating macrophage-related inflammatory diseases requires further investigation.
While small molecule inhibitors have a high potential for clinical translation, their long-term safety and side effects require additional validation. Nanoparticle (NP) drug delivery systems may help to enhance small molecule inhibitor specificity and safety. For example, red blood cell microvesicles can deliver STM2457 to activated monocytes, inhibiting NF-κB signaling-specific inflammation. This method is used to treat monocyte inflammation and fibrosis related to cardiac remodeling associated with device implantation (125).
miRNAs and siRNAs are also delivered by NPs to alter macrophage activation and polarization. For instance, miR-1208 in hucMSCs-EVs regulates the m6A levels of NLRP3 mRNA, preventing the activation of the NLRP3 inflammasome and inhibiting pro-inflammatory factors in macrophages, thereby slowing the progression of knee osteoarthritis (126). NPs can also encapsulate METTL3, METTL14, SPRED2 mRNA, or IRAKM siRNA, specifically targeting TAMs and reprogramming them from an M2 to an M1 phenotype, thus remodeling the tumor microenvironment (TME) (127).
This article reviews recent findings on the role of m6A modification in macrophage metabolic reprogramming. In macrophages stimulated with LPS, oxLDL, or other pro-inflammatory factors, metabolic stress can induce abnormal m6A methylation and alter the expression of m6A-regulating proteins. Conversely, m6A modification can directly impact key enzymes, transporters, and transcription factors in various metabolic pathways, influencing macrophage polarization and the progression of inflammation. However, m6A-related studies have predominantly focused on modulating metabolic enzymes indirectly via transcription factors, such as HIF-1α involved in glycolysis. This highlights the need for more research directly targeting specific metabolic enzymes. In particular, studies on how m6A regulates the two breakpoints in the TCA cycle are also limited, indicating that several mechanisms remain to be elucidated.
To address these gaps, future research should prioritize uncovering how m6A modification directly regulates specific metabolic enzymes in macrophages, including key players in glycolysis, lipid metabolism, OXPHOS, and the TCA cycle. Furthermore, studying the interplay between m6A modification and the cellular microenvironment, including factors like hypoxia and nutrient availability, will enhance our understanding of m6A’s broader regulatory roles. Finally, developing new m6A-targeting small-molecule inhibitors will offer a promising strategy for therapeutic interventions in macrophage-driven inflammatory diseases.
HW: Writing – original draft. PX: Writing – review & editing. KY: Writing – review & editing. SW: Writing – review & editing, Conceptualization, Supervision.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Research Project of the Jiangsu Commission of Health (grant no. K2023062), Gynecology Project of Zhenjiang Traditional Chinese Medicine (SS2021006), and Jiangsu Provincial Medical Key Discipline Cultivation Unit (grant no. JSDW202241).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sheu KM, Hoffmann A. Functional hallmarks of healthy macrophage responses: their regulatory basis and disease relevance. Annu Rev Immunol. (2022) 40:295–321. doi: 10.1146/annurev-immunol-101320-031555
2. Oishi Y, Manabe I. Macrophages in inflammation, repair and regeneration. Int Immunol. (2018) 30:511–28. doi: 10.1093/intimm/dxy054
3. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
4. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: GLUCOSE TRANSPORTER 1 (GLUT1)-MEDIATED GLUCOSE METABOLISM DRIVES A PROINFLAMMATORY PHENOTYPE *. J Biol Chem. (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
5. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
6. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
7. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. (1992) 176:287–92. doi: 10.1084/jem.176.1.287
8. Pérez S, Rius-Pérez S. Macrophage polarization and reprogramming in acute inflammation: A redox perspective. Antioxidants (Basel). (2022) 11:1394. doi: 10.3390/antiox11071394
9. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. (2017) 169:381–405. doi: 10.1016/j.cell.2017.04.001
10. Van den Bossche J, Baardman J, de Winther MP. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J Vis Exp. (2015) 105:53424. doi: 10.3791/53424-v
11. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
12. Angajala A, Lim S, Phillips JB, Kim JH, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol. (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
13. Fang Z, Mei W, Qu C, Lu J, Shang L, Cao F, et al. Role of m6A writers, erasers and readers in cancer. Exp Hematol Oncol. (2022) 11:45. doi: 10.1186/s40164-022-00298-7
14. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
15. Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. (2017) 33:319–42. doi: 10.1146/annurev-cellbio-100616-060758
16. Shulman Z, Stern-Ginossar N. The RNA modification N(6)-methyladenosine as a novel regulator of the immune system. Nat Immunol. (2020) 21:501–12. doi: 10.1038/s41590-020-0650-4
17. Tong J, Wang X, Liu Y, Ren X, Wang A, Chen Z, et al. Pooled CRISPR screening identifies m(6)A as a positive regulator of macrophage activation. Sci Adv. (2021) 7:eabd4742. doi: 10.1126/sciadv.abd4742
18. Liu Y, Liu Z, Tang H, Shen Y, Gong Z, Xie N, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am J Physiol Cell Physiol. (2019) 317:C762–c775. doi: 10.1152/ajpcell.00212.2019
19. Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. (2018) 28:616–24. doi: 10.1038/s41422-018-0040-8
20. Zhang X, Li X, Jia H, An G, Ni J. The m(6)A methyltransferase METTL3 modifies PGC-1α mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J Biol Chem. (2021) 297:101058. doi: 10.1016/j.jbc.2021.101058
21. Sun Z, Chen W, Wang Z, Wang S, Zan J, Zheng L, et al. Matr3 reshapes m6A modification complex to alleviate macrophage inflammation during atherosclerosis. Clin Immunol. (2022) 245:109176. doi: 10.1016/j.clim.2022.109176
22. Pu Z, Wang W, Xie H, Wang W. Apolipoprotein C3 (ApoC3) facilitates NLRP3 mediated pyroptosis of macrophages through mitochondrial damage by accelerating of the interaction between SCIMP and SYK pathway in acute lung injury. Int Immunopharmacol. (2024) 128:111537. doi: 10.1016/j.intimp.2024.111537
23. Zheng L, Chen X, Yin Q, Gu J, Chen J, Chen M, et al. RNA-m6A modification of HDGF mediated by Mettl3 aggravates the progression of atherosclerosis by regulating macrophages polarization via energy metabolism reprogramming. Biochem Biophys Res Commun. (2022) 635:120–7. doi: 10.1016/j.bbrc.2022.10.032
24. Qin Y, Li B, Arumugam S, Lu Q, Mankash SM, Li J, et al. m(6)A mRNA methylation-directed myeloid cell activation controls progression of NAFLD and obesity. Cell Rep. (2021) 37:109968. doi: 10.1016/j.celrep.2021.109968
25. Li Z, Xu Q, Huangfu N, Chen X, Zhu J. Mettl3 promotes oxLDL-mediated inflammation through activating STAT1 signaling. J Clin Lab Anal. (2022) 36:e24019. doi: 10.1002/jcla.24019
26. Zhao D, Wang C, Liu X, Liu N, Zhuang S, Zhang Q, et al. CircN4bp1 Facilitates Sepsis-Induced Acute Respiratory Distress Syndrome through Mediating Macrophage Polarization via the miR-138-5p/EZH2 Axis. Mediators Inflammation. (2021) 2021:7858746. doi: 10.1155/2021/7858746
27. Qi L, Wang Y, Hu H, Li P, Hu H, Li Y, et al. m(6)A methyltransferase METTL3 participated in sympathetic neural remodeling post-MI via the TRAF6/NF-κB pathway and ROS production. J Mol Cell Cardiol. (2022) 170:87–99. doi: 10.1016/j.yjmcc.2022.06.004
28. Yu Z, Zheng X, Wang C, Chen C, Ning N, Peng D, et al. The traditional chinese medicine hua tuo zai zao wan alleviates atherosclerosis by deactivation of inflammatory macrophages. Evid Based Complement Alternat Med. (2022) 2022:2200662. doi: 10.1155/2022/2200662
29. Feng Y, Dong H, Sun B, Hu Y, Yang Y, Jia Y, et al. METTL3/METTL14 transactivation and m(6)A-dependent TGF-β1 translation in activated kupffer cells. Cell Mol Gastroenterol Hepatol. (2021) 12:839–56. doi: 10.1016/j.jcmgh.2021.05.007
30. Li Q, Yu L, Gao A, Ren R, Zhang J, Cao L, et al. METTL3 (Methyltransferase like 3)-dependent N6-methyladenosine modification on braf mRNA promotes macrophage inflammatory response and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2023) 43:755–73. doi: 10.1161/ATVBAHA.122.318451
31. Chen P, Liu J, Sun K, Wang L, Li X, Li X, et al. Methylation of TTC4 interaction with HSP70 inhibits pyroptosis in macrophages of sepsis-induced lung injury by NLRP3 inflammation. Am J Cancer Res. (2023) 13:5122–37.
32. Shu B, Zhou YX, Li H, Zhang RZ, He C, Yang X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death Discovery. (2021) 7:368. doi: 10.1038/s41420-021-00756-x
33. Pan XS, Li BW, Wang LL, Li N, Lin HM, Zhang J, et al. Kupffer cell pyroptosis mediated by METTL3 contributes to the progression of alcoholic steatohepatitis. FASEB J. (2023) 37:e22965. doi: 10.1096/fj.202300059RR
34. Guo M, Yan R, Ji Q, Yao H, Sun M, Duan L, et al. IFN regulatory Factor-1 induced macrophage pyroptosis by modulating m6A modification of circ_0029589 in patients with acute coronary syndrome. Int Immunopharmacol. (2020) 86:106800. doi: 10.1016/j.intimp.2020.106800
35. Gou Y, Wang H, Wang T, Wang H, Wang B, Jiao N, et al. Ectopic endometriotic stromal cells-derived lactate induces M2 macrophage polarization via Mettl3/Trib1/ERK/STAT3 signalling pathway in endometriosis. Immunology. (2023) 168:389–402. doi: 10.1111/imm.v168.3
36. Yin H, Ju Z, Zhang X, Zuo W, Yang Y, Zheng M, et al. Inhibition of METTL3 in macrophages provides protection against intestinal inflammation. Cell Mol Immunol. (2024) 21:589–603. doi: 10.1038/s41423-024-01156-8
37. Xu X, Peng J, Wang N, Ocansey DKW, Zhang X, Mao F. hucMSC-Ex alleviates inflammatory bowel disease in mice by enhancing M2-type macrophage polarization via the METTL3-Slc37a2-YTHDF1 axis. Cell Biol Toxicol. (2024) 40:74. doi: 10.1007/s10565-024-09921-1
38. Cai Y, Yu R, Zhang Z, Li D, Yi B, Feng Z, et al. Mettl3/Ythdf2 regulate macrophage inflammation and ROS generation by controlling Pyk2 mRNA stability. Immunol Lett. (2023) 264:64–73. doi: 10.1016/j.imlet.2023.11.004
39. Zhao W, Wang Z, Sun Z, He Y, Jian D, Hu X, et al. RNA helicase DDX5 participates in oxLDL-induced macrophage scavenger receptor 1 expression by suppressing mRNA degradation. Exp Cell Res. (2018) 366:114–20. doi: 10.1016/j.yexcr.2018.03.003
40. Han X, Liu L, Huang S, Xiao W, Gao Y, Zhou W, et al. RNA m(6)A methylation modulates airway inflammation in allergic asthma via PTX3-dependent macrophage homeostasis. Nat Commun. (2023) 14:7328. doi: 10.1038/s41467-023-43219-w
41. Li Z, Teng M, Jiang Y, Zhang L, Luo X, Liao Y, et al. YTHDF1 Negatively Regulates Treponema pallidum-Induced Inflammation in THP-1 Macrophages by Promoting SOCS3 Translation in an m6A-Dependent Manner. Front Immunol. (2022) 13:857727. doi: 10.3389/fimmu.2022.857727
42. Cai Y, Yu R, Kong Y, Feng Z, Xu Q. METTL3 regulates LPS-induced inflammatory response via the NOD1 signaling pathway. Cell Signal. (2022) 93:110283. doi: 10.1016/j.cellsig.2022.110283
43. Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. (2021) 12:1394. doi: 10.1038/s41467-021-21514-8
44. He S, Song W, Cui S, Li J, Jiang Y, Chen X, et al. Modulation of miR-146b by N6-methyladenosine modification remodels tumor-associated macrophages and enhances anti-PD-1 therapy in colorectal cancer. Cell Oncol (Dordr). (2023) 46:1731–46. doi: 10.1007/s13402-023-00839-0
45. Zheng Y, Li Y, Ran X, Wang D, Zheng X, Zhang M, et al. Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell Mol Life Sci. (2022) 79:311. doi: 10.1007/s00018-022-04331-0
46. Cao F, Chen G, Xu Y, Wang X, Tang X, Zhang W, et al. METTL14 contributes to acute lung injury by stabilizing NLRP3 expression in an IGF2BP2-dependent manner. Cell Death Dis. (2024) 15:43. doi: 10.1038/s41419-023-06407-6
47. Li Y, Li J, Yu Q, Ji L, Peng B. METTL14 regulates microglia/macrophage polarization and NLRP3 inflammasome activation after ischemic stroke by the KAT3B-STING axis. Neurobiol Dis. (2023) 185:106253. doi: 10.1016/j.nbd.2023.106253
48. Du J, Liao W, Liu W, Deb DK, He L, Hsu PJ, et al. N(6)-adenosine methylation of socs1 mRNA is required to sustain the negative feedback control of macrophage activation. Dev Cell. (2020) 55:737–753.e7. doi: 10.1016/j.devcel.2020.10.023
49. Zhao M, Li P, Qiao D, Hua S, Yue Q, Dai Y, et al. N6-methyladenosine modification of TSC1 mRNA contributes to macrophage polarization regulated by Coptisine in DSS-induced ulcerative colitis. Phytomedicine. (2024) 122:155153. doi: 10.1016/j.phymed.2023.155153
50. Park MH, Jeong E, Choudhury M. Mono-(2-Ethylhexyl)phthalate Regulates Cholesterol Efflux via MicroRNAs Regulated m(6)A RNA Methylation. Chem Res Toxicol. (2020) 33:461–9. doi: 10.1021/acs.chemrestox.9b00367
51. Bai Y, Jiao X, Hu J, Xue W, Zhou Z, Wang W. WTAP promotes macrophage recruitment and increases VEGF secretion via N6-methyladenosine modification in corneal neovascularization. Biochim Biophys Acta Mol Basis Dis. (2023) 1869:166708. doi: 10.1016/j.bbadis.2023.166708
52. Long H, Lin H, Zheng P, Hou L, Zhang M, Lin S, et al. WTAP mediates the anti-inflammatory effect of Astragalus mongholicus polysaccharide on THP-1 macrophages. Front Pharmacol. (2022) 13:1023878. doi: 10.3389/fphar.2022.1023878
53. Mo C, Yang M, Han X, Li J, Gao G, Tai H, et al. Fat mass and obesity-associated protein attenuates lipid accumulation in macrophage foam cells and alleviates atherosclerosis in apolipoprotein E-deficient mice. J Hypertens. (2017) 35:810–21. doi: 10.1097/HJH.0000000000001255
54. Zhao Y, Ding W, Cai Y, Li Q, Zhang W, Bai Y, et al. The m(6)A eraser FTO suppresses ferroptosis via mediating ACSL4 in LPS-induced macrophage inflammation. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167354. doi: 10.1016/j.bbadis.2024.167354
55. Hu Z, Li Y, Yuan W, Jin L, Leung WK, Zhang C, et al. N6-methyladenosine of Socs1 modulates macrophage inflammatory response in different stiffness environments. Int J Biol Sci. (2022) 18:5753–69. doi: 10.7150/ijbs.74196
56. Yin H, Gu P, Xie Y, You X, Zhang Y, Yao Y, et al. ALKBH5 mediates silica particles-induced pulmonary inflammation through increased m(6)A modification of Slamf7 and autophagy dysfunction. J Hazard Mater. (2024) 462:132736. doi: 10.1016/j.jhazmat.2023.132736
57. Liang G, Hu JY, Liu RJ, Chao YP, Hu YF, Zheng H, et al. [amp]]alpha;-Ketoglutarate plays an inflammatory inhibitory role by regulating scavenger receptor class a expression through N6-methyladenine methylation during sepsis. Eur J Immunol. (2024) 54:e2350655. doi: 10.1002/eji.202350655
58. Sun M, Yue Y, Wang X, Feng H, Qin Y, Chen M, et al. ALKBH5-mediated upregulation of CPT1A promotes macrophage fatty acid metabolism and M2 macrophage polarization, facilitating Malignant progression of colorectal cancer. Exp Cell Res. (2024) 437:113994. doi: 10.1016/j.yexcr.2024.113994
59. Ge X, Xue G, Ding Y, Li R, Hu K, Xu T, et al. The loss of YTHDC1 in gut macrophages exacerbates inflammatory bowel disease. Adv Sci (Weinh). (2023) 10:e2205620. doi: 10.1002/advs.202205620
60. Chu J, Zheng R, Chen H, Chen Y, Lin Y, Li J, et al. Dynamic m(6) A profiles reveal the role of YTHDC2-TLR2 signaling axis in Talaromyces marneffei infection. J Med Virol. (2024) 96:e29466. doi: 10.1002/jmv.29466
61. Zhu J, Liu S, Fang J, Cui Z, Wang B, Wang Y, et al. Enzymolysis-based RNA pull-down identifies YTHDC2 as an inhibitor of antiviral innate response. Cell Rep. (2023) 42:113192. doi: 10.1016/j.celrep.2023.113192
62. Xing Y, Cheng D, Shi C, Shen Z. The protective role of YTHDF1-knock down macrophages on the immune paralysis of severe sepsis rats with ECMO. Microvasc Res. (2021) 137:104178. doi: 10.1016/j.mvr.2021.104178
63. Hao WY, Lou Y, Hu GY, Qian CY, Liang WR, Zhao J, et al. RNA m6A reader YTHDF1 facilitates inflammation via enhancing NLRP3 translation. Biochem Biophys Res Commun. (2022) 616:76–81. doi: 10.1016/j.bbrc.2022.05.076
64. Zheng L, Tang X, Lu M, Sun S, Xie S, Cai J, et al. microRNA-421-3p prevents inflammatory response in cerebral ischemia/reperfusion injury through targeting m6A Reader YTHDF1 to inhibit p65 mRNA translation. Int Immunopharmacol. (2020) 88:106937. doi: 10.1016/j.intimp.2020.106937
65. Huangfu N, Zheng W, Xu Z, Wang S, Wang Y, Cheng J, et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int Immunopharmacol. (2020) 83:106432. doi: 10.1016/j.intimp.2020.106432
66. Zhang Y, Wang X, Zhang X, Wang J, Ma Y, Zhang L, et al. RNA-binding protein YTHDF3 suppresses interferon-dependent antiviral responses by promoting FOXO3 translation. Proc Natl Acad Sci U.S.A. (2019) 116:976–81. doi: 10.1073/pnas.1812536116
67. Ding L, Wu H, Wang Y, Li Y, Liang Z, Xia X, et al. m6A reader igf2bp1 regulates the inflammatory responses of microglia by stabilizing gbp11 and cp mRNAs. Front Immunol. (2022) 13:872252. doi: 10.3389/fimmu.2022.872252
68. Wang X, Ji Y, Feng P, Liu R, Li G, Zheng J, et al. The m6A reader IGF2BP2 regulates macrophage phenotypic activation and inflammatory diseases by stabilizing TSC1 and PPARγ. Adv Sci (Weinh). (2021) 8:2100209. doi: 10.1002/advs.202100209
69. Liu X, Xiong H, Lu M, Liu B, Hu C, Liu P. Trans-3, 5, 4’-trimethoxystilbene restrains non-small-cell lung carcinoma progression via suppressing M2 polarization through inhibition of m6A modified circPACRGL-mediated Hippo signaling. Phytomedicine. (2024) 126:155436. doi: 10.1016/j.phymed.2024.155436
70. Liu Y, Shi M, He X, Cao Y, Liu P, Li F, et al. LncRNA-PACERR induces pro-tumour macrophages via interacting with miR-671-3p and m6A-reader IGF2BP2 in pancreatic ductal adenocarcinoma. J Hematol Oncol. (2022) 15:52. doi: 10.1186/s13045-022-01272-w
71. Yang L, Fu J, Han X, Zhang C, Xia L, Zhu R, et al. Hsa_circ_0004287 inhibits macrophage-mediated inflammation in an N(6)-methyladenosine-dependent manner in atopic dermatitis and psoriasis. J Allergy Clin Immunol. (2022) 149:2021–33. doi: 10.1016/j.jaci.2021.11.024
72. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. (2016) 534:575–8. doi: 10.1038/nature18298
73. Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of mettl3 and mettl14 methyltransferases. Mol Cell. (2016) 63:306–17. doi: 10.1016/j.molcel.2016.05.041
74. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
75. Gu X, Zhang Y, Li D, Cai H, Cai L, Xu Q. N6-methyladenosine demethylase FTO promotes M1 and M2 macrophage activation. Cell Signal. (2020) 69:109553. doi: 10.1016/j.cellsig.2020.109553
76. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian. spermatogenesis. Cell Res. (2017) 27:1115–27. doi: 10.1038/cr.2017.99
77. Zaccara S, Jaffrey SR. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell. (2020) 181:1582–1595.e18. doi: 10.1016/j.cell.2020.05.012
78. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
79. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. (2012) 15:813–26. doi: 10.1016/j.cmet.2012.04.023
81. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
82. Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun. (1996) 64:108–12. doi: 10.1128/iai.64.1.108-112.1996
83. Pitroda SP, Wakim BT, Sood RF, Beveridge MG, Beckett MA, MacDermed DM, et al. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. (2009) 7:68. doi: 10.1186/1741-7015-7-68
84. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. (2018) 17:428–38. doi: 10.1080/15384101.2018.1444305
85. Wang Z, Zhao Q, Nie Y, Yu Y, Misra BB, Zabalawi M, et al. Solute carrier family 37 member 2 (SLC37A2) negatively regulates murine macrophage inflammation by controlling glycolysis. iScience. (2020) 23:101125. doi: 10.1016/j.isci.2020.101125
86. Corcoran SE, O’Neill LA. HIF1α and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/JCI84431
87. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
88. Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, et al. An intact canonical NF-κB pathway is required for inflammatory gene expression in response to hypoxia. J Immunol. (2011) 186:1091–6. doi: 10.4049/jimmunol.1002256
89. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. (2008) 453:807–11. doi: 10.1038/nature06905
90. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
91. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discovery. (2015) 5:1024–39. doi: 10.1158/2159-8290.CD-15-0507
92. Tateishi K, Iafrate AJ, Ho Q, Curry WT, Batchelor TT, Flaherty KT, et al. Myc-driven glycolysis is a therapeutic target in glioblastoma. Clin Cancer Res. (2016) 22:4452–65. doi: 10.1158/1078-0432.CCR-15-2274
93. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (Going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
94. Ryan DG, O’Neill LAJ. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. (2017) 591:2992–3006. doi: 10.1002/feb2.2017.591.issue-19
95. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
96. Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochem J. (2011) 438:433–6. doi: 10.1042/BJ20111275
97. Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim Biophys Acta. (2014) 1839:1217–25. doi: 10.1016/j.bbagrm.2014.07.013
98. Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. (2004) 447:689–709. doi: 10.1007/s00424-003-1099-7
99. Iacobazzi V, Infantino V, Castegna A, Menga A, Palmieri EM, Convertini P, et al. Mitochondrial carriers in inflammation induced by bacterial endotoxin and cytokines. Biol Chem. (2017) 398:303–17. doi: 10.1515/hsz-2016-0260
100. Cai W, Ji Y, Han L, Zhang J, Ni Y, Cheng Y, et al. METTL3-dependent glycolysis regulates dental pulp stem cell differentiation. J Dent Res. (2022) 101:580–9. doi: 10.1177/00220345211051594
101. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U.S.A. (2013) 110:7820–5. doi: 10.1073/pnas.1218599110
102. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556:113–7. doi: 10.1038/nature25986
103. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. (2011) 145:732–44. doi: 10.1016/j.cell.2011.03.054
104. Gao Y, Zimmer JT, Vasic R, Liu C, Gbyli R, Zheng SJ, et al. ALKBH5 modulates hematopoietic stem and progenitor cell energy metabolism through m(6)A modification-mediated RNA stability control. Cell Rep. (2023) 42:113163. doi: 10.1016/j.celrep.2023.113163
105. Miao Y, Zhang C, Yang L, Zeng X, Hu Y, Xue X, et al. The activation of PPARγ enhances Treg responses through up-regulating CD36/CPT1-mediated fatty acid oxidation and subsequent N-glycan branching of TβRII/IL-2Rα. Cell Commun Signal. (2022) 20:48. doi: 10.1186/s12964-022-00849-9
106. Morita M, Gravel SP, Chénard V, Sikström K, Zheng L, Alain T, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. (2013) 18:698–711. doi: 10.1016/j.cmet.2013.10.001
107. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–470.e13. doi: 10.1016/j.cell.2016.08.064
108. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. (2011) 1813:1269–78. doi: 10.1016/j.bbamcr.2010.09.019
109. Qu Q, Zeng F, Liu X, Wang QJ, Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. (2016) 7:e2226–6. doi: 10.1038/cddis.2016.132
110. Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis. (2010) 33:469–77. doi: 10.1007/s10545-010-9061-2
111. Duan J, Wang Z, Duan R, Yang C, Zhao R, Feng Q, et al. Therapeutic targeting of hepatic ACSL4 ameliorates NASH in mice. Hepatology. (2022) 75:140–53. doi: 10.1002/hep.32148
112. Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. (2009) 2:re3. doi: 10.1126/scisignal.272re3
113. Parthasarathy S, Rankin SM. Role of oxidized low density lipoprotein in atherogenesis. Prog Lipid Res. (1992) 31:127–43. doi: 10.1016/0163-7827(92)90006-5
114. Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. (2012) 125:1905–19. doi: 10.1161/CIRCULATIONAHA.111.066589
115. Hu CY, Jiao Y. A METTL3 inhibitor alleviates the onset of osteomyelitis in a mouse model by targeting myD88. Jpn J Infect Dis. (2023) 76:191–6. doi: 10.7883/yoken.JJID.2022.454
116. Bedi RK, Huang D, Eberle SA, Wiedmer L, Śledź P, Caflisch A. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. (2020) 15:744–8. doi: 10.1002/cmdc.202000011
117. Li Z, Feng Y, Han H, Jiang X, Chen W, Ma X, et al. A stapled peptide inhibitor targeting the binding interface of N6-adenosine-methyltransferase subunits METTL3 and METTL14 for cancer therapy. Angew Chem Int Ed Engl. (2024) 63:e202402611. doi: 10.1002/anie.202402611
118. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. (2020) 38:79–96.e11. doi: 10.1016/j.ccell.2020.04.017
119. Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell. (2021) 81:922–939.e9. doi: 10.1016/j.molcel.2020.12.026
120. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. (2015) 43:373–84. doi: 10.1093/nar/gku1276
121. Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, et al. Structure of human RNA N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. (2014) 42:4741–54. doi: 10.1093/nar/gku085
122. Takahashi H, Hase H, Yoshida T, Tashiro J, Hirade Y, Kitae K, et al. Discovery of two novel ALKBH5 selective inhibitors that exhibit uncompetitive or competitive type and suppress the growth activity of glioblastoma multiforme. Chem Biol Drug Des. (2022) 100:1–12. doi: 10.1111/cbdd.v100.1
123. Jiang X, Yan F, Geng Y, Cheng X, Zhang S, Zhao T, et al. ALKBH5 deficiency attenuates oxygen-glucose deprivation-induced injury in mouse brain microvascular endothelial cells in an m6A dependent manner. Exp Neurol. (2024) 380:114910. doi: 10.1016/j.expneurol.2024.114910
124. Wang YZ, Li HY, Zhang Y, Jiang RX, Xu J, Gu J, et al. Discovery of pyrazolo[1,5-a]pyrimidine derivative as a novel and selective ALKBH5 inhibitor for the treatment of AML. J Med Chem. (2023) 66:15944–59. doi: 10.1021/acs.jmedchem.3c01374
125. Li J, Wei L, Hu K, He Y, Gong G, Liu Q, et al. Deciphering m(6)A methylation in monocyte-mediated cardiac fibrosis and monocyte-hitchhiked erythrocyte microvesicle biohybrid therapy. Theranostics. (2024) 14:3486–508. doi: 10.7150/thno.95664
126. Zhou H, Shen X, Yan C, Xiong W, Ma Z, Tan Z, et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate osteoarthritis of the knee in mice model by interacting with METTL3 to reduce m6A of NLRP3 in macrophage. Stem Cell Res Ther. (2022) 13:322. doi: 10.1186/s13287-022-03005-9
Keywords: M1/M2 macrophage reprogramming, glycolysis, tricarboxylic acid cycle, oxidative phosphorylation, fatty acid oxidation, cholesterol transport
Citation: Wang H, Xu P, Yin K and Wang S (2025) The role of m6A modification during macrophage metabolic reprogramming in human diseases and animal models. Front. Immunol. 16:1521196. doi: 10.3389/fimmu.2025.1521196
Received: 01 November 2024; Accepted: 28 January 2025;
Published: 18 February 2025.
Edited by:
Eric Henrique Roma, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Byron Morales-Lange, Norwegian University of Life Sciences, NorwayCopyright © 2025 Wang, Xu, Yin and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengjun Wang, c2p3anNAdWpzLmVkdS5jbg==; Peiqi Xu, cGVpcWl4dTA2MDRAMTYzLmNvbQ==; Kai Yin, anN5aW5rYWlAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.