 Ruifang Lin1,2
Ruifang Lin1,2 Yunfeng Yu
Yunfeng Yu Lixin Du
Lixin Du Ziyan Wang
Ziyan Wang Jiaming Wei
Jiaming Wei Zhihua Guo
Zhihua Guo- 1College of Chinese Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, China
- 2Hunan Key Laboratory of Colleges and Universities of Intelligent TCM Diagnosis and Preventive Treatment of Chronic Diseases, Hunan University of Chinese Medicine, Changsha, China
- 3First Clinical College of Chinese Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, China
- 4College of Pharmacy, Hunan University of Chinese Medicine, Changsha, Hunan, China
Heart failure (HF) has emerged as a significant global public health challenge owing to its high rates of morbidity and mortality. Activation of the NOD-like receptor protein 3 (NLRP3) inflammasome is regarded as a pivotal factor in the onset and progression of HF. Therefore, inhibiting the activation of the NLRP3 inflammasome may represent a promising therapeutic approach for preventing and treating HF. The active ingredients serve as the foundation for the therapeutic effects of traditional Chinese medicine (TCM). Recent research has revealed significant advantages of TCM active ingredients in inhibiting the activation of the NLRP3 inflammasome and enhancing cardiac structure and function in HF. The study aimed to explore the impact of NLRP3 inflammasome activation on the onset and progression of HF, and to review the current advancements in utilizing TCM active ingredients to inhibit the NLRP3 inflammasome for preventing and treating HF. This provides a novel perspective for the future development of precise intervention strategies targeting the NLRP3 inflammasome to prevent and treat HF.

Graphical Abstract.
1 Introduction
Heart failure (HF) is the terminal stage of the progression of diverse functional or organic cardiovascular diseases, characterized by impaired ventricular filling and ejection capacity, with prevalent risk factors including hypertension, myocardial infarction, and myocardial disease (1). Epidemiological studies have indicated a global total of > 64 million patients with HF, with a prevailing trend toward a younger age of onset (2, 3). HF has emerged as a significant public health issue, posing risks to human health and escalating societal burden (2). Currently, the standard pharmacological interventions for HF management include angiotensin receptor-neprilysin inhibitors, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, sodium-glucose co-transporter 2 inhibitors, beta blockers, aldosterone receptor antagonists, and diuretics (4). Although they ameliorate HF to a certain extent, uncertainties persist concerning their long-term effects, and the potential adverse events associated with long-term medication are concerning. Therefore, there is an urgent need to develop safe and effective therapeutic strategies. HF is a clinical syndrome encompassing a range of complex pathological processes, including myocardial inflammation, myocardial fibrosis, myocardial hypertrophy, impaired angiogenesis, abnormal cardiac electrical signal conduction, energy metabolism disorders, and abnormal cardiomyocyte apoptosis (5–9). The activation of the NOD-like receptor protein 3 (NLRP3) inflammasome plays a crucial role in driving these pathological changes (5–8, 10). As an intracellular multiprotein complex, persistent or excessive activation of the NLRP3 inflammasome serves as a critical driver of both the onset and progression of HF, with the extent of its activation being strongly correlated with disease severity and patient prognosis (5, 11, 12). Therefore, inhibiting the activation of the NLRP3 inflammasome holds promise as a novel breakthrough in the prevention and treatment of HF.
Traditional Chinese medicine (TCM) has been recognized as a promising therapeutic strategy for HF, owing to its ability to effectively reverse adverse cardiac remodeling, lower rehospitalization and mortality rates, and enhance the quality of life of patients (13, 14). The active ingredients of TCM are the material basis for its therapeutic effects and constitute the focal point of research on TCM. Relevant studies have indicated that the active ingredients of TCM can mitigate the onset and progression of HF by inhibiting the NLRP3 inflammasome (15–17). Thus, this study summarizes the role of NLRP3 inflammasome activation in the onset and progression of HF, as well as the current research on the use of TCM active ingredients to prevent and treat HF through targeted inhibition of the NLRP3 inflammasome, aiming to provide insights for future basic research and novel drug development.
2 NLRP3 inflammasome
2.1 Structure of the NLRP3 inflammasome
The innate immune system is the first line of defense in the human body. Innate immune cells activate inflammasome by recognizing pathogen-associated molecular patterns and damage-associated molecular patterns via pattern recognition receptors, subsequently initiating inflammatory responses (12). The NLRP3 inflammasome is the most widely and intensively studied inflammasome and is a multiprotein complex comprising the NLRP3 protein, apoptosis speck-like protein containing a caspase recruitment domain (ASC), and caspase-1 precursor (pro-caspase-1) (12) (Figure 1). NLRP3 acts as a sensor and consists of a central NACHT domain, a leucine-rich repeat (LRR) domain at the carboxyl-terminal (C-terminal), and a pyrin domain (PYD) at the amino-terminal (N-terminal). The NACHT domain primarily facilitates NLRP3 protein oligomerization and contains an adenosine triphosphatase active site, enabling the regulation of NLRP3 protein activity through adenosine triphosphate (ATP) hydrolysis (18). The LRR domain mediates protein–protein interactions and plays a crucial role in NLRP3 inflammatory signaling by recognizing and interacting with both exogenous and endogenous molecules (19). PYD recruits downstream effector signaling molecules that trigger inflammasome assembly (20). ASC functions as an adaptor with two domains: the N-terminal PYD and the C-terminal caspase activation and recruitment domain (CARD) (21). The PYD of ASC corresponds to the homotypic PYD of NLRP3 proteins, which mediates the interaction between ASC and NLRP3 proteins (20, 21). The CARD of ASC is responsible for binding pro-caspase-1 (21). Pro-caspase-1 functions as an effector and comprises three domains: the N-terminal CARD, central large catalytic subunit domain p20, and C-terminal small catalytic subunit domain p10 (22). The CARD of pro-caspase-1 is responsible for interactions with the CARD of ASC (22). Subsequently, p20 and p10 facilitate the cleavage of the interleukin-1β precursor (pro-IL-1β) and interleukin-18 precursor (pro-IL-18) into mature forms of IL-1β and IL-18.
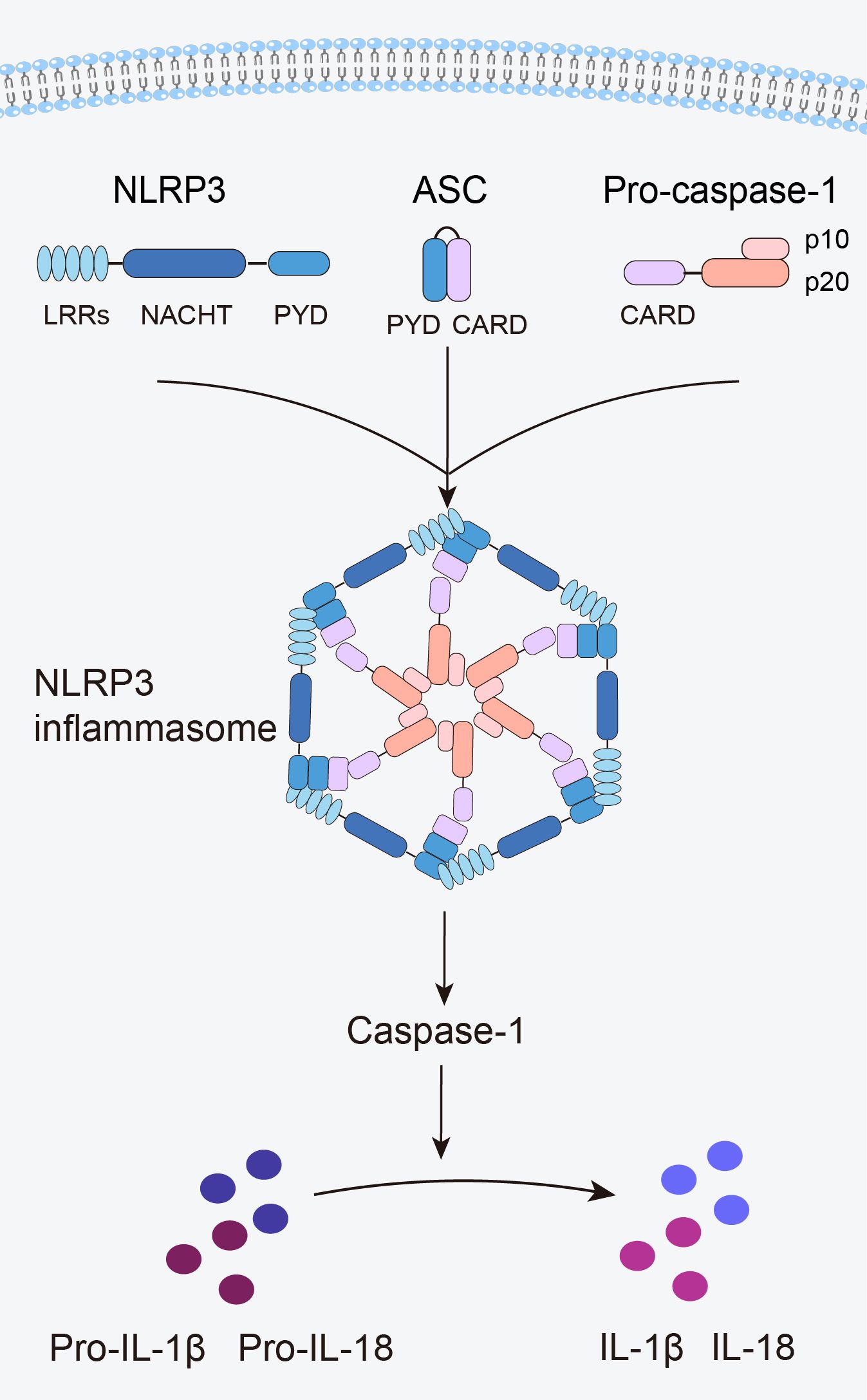
Figure 1. Structure of the NLRP3 inflammasome.
When an organism encounters an external stimulus, NLRP3 interacts with the PYD of ASC via its PYD. The CARD of ASC recruits and binds to the CARD of pro-caspase-1, triggering self-cleavage of pro-caspase-1 to yield active caspase-1. Caspase-1 cleaves pro-IL-1β and pro-IL-18 to generate mature IL-1β and IL-18, thus initiating an inflammatory response (Figure 1).
2.2 Mechanism of NLRP3 inflammasome activation
Three distinct pathways exist for NLRP3 inflammasome activation: canonical NLRP3, non-canonical NLRP3, and alternative NLRP3 inflammasome activation (23) (Figure 2).
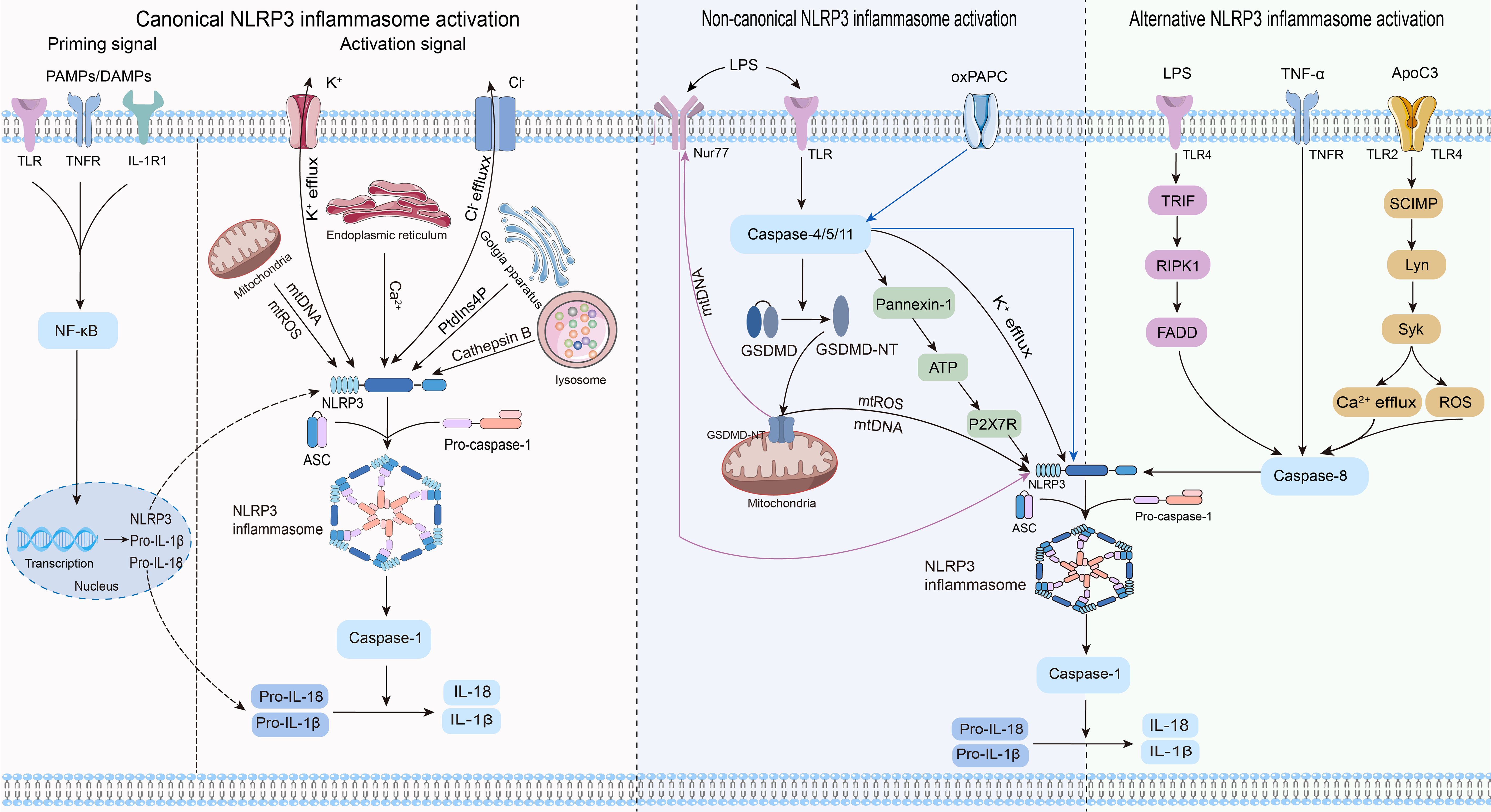
Figure 2. Three pathways and related mechanisms of NLRP3 inflammasome activation.
2.2.1 Canonical NLRP3 inflammasome activation
Canonical activation of the NLRP3 inflammasome involves two distinct processes: priming and activation (24). The priming phase involves transcriptional regulation and posttranslational modifications of NLRP3. Recognition of pathogen-associated molecular patterns or damage-associated molecular patterns by the corresponding pattern recognition receptors (25–27) triggers nuclear factor-κB (NF-κB) translocation and transcription, leading to increased expression of NLRP3, pro-IL-1β, and pro-IL-18 within the nucleus. Furthermore, priming signals trigger posttranslational modifications of NLRP3, including phosphorylation (28), ubiquitination (29), alkylation (30), S-nitrosylation (31), acetylation (32), and sumoylation (33), all of which are crucial for modulating the activation or inhibition of NLRP3. During the activation phase, NLRP3 responds to activating stimuli, subsequently initiating the assembly of the NLRP3 inflammasome, activation of caspase-1, and processing pro-IL-1β and pro-IL-18. This process ultimately produces proinflammatory cytokines IL-1β and IL-18, which are subsequently secreted into the extracellular space to trigger an inflammatory response.
Previous studies agree that the stimulus signals for NLRP3 inflammasome activation include potassium ion (K+) efflux (34), chloride ion (Cl-) efflux (35), mitochondrial dysfunction (36, 37), endoplasmic reticulum stress (38), trans-Golgi network catabolism (39), and the release of tissue protease B from damaged lysosomes (40). Remarkably, the interplay between some of these stimuli complicates the activation phase of NLRP3 (41).
2.2.2 Non-canonical NLRP3 inflammasome activation
Non-canonical activation of the NLRP3 inflammasome primarily relies on the mediation of human caspase-4/5 or mouse caspase-11. CARDs of caspase-4/5/11 directly recognize lipopolysaccharides (LPS) from gram-negative bacteria, prompting their oligomerization of caspase-4/5/11 (42–44). This leads to the cleavage of GSDMD into its active form, GSDMD-NT, which in turn induces pyroptosis by creating pores in the cytoplasmic membrane (42–44). Notably, while inducing pyroptosis, caspase-4/5/11 do not directly cleave pro-IL-1β (45, 46). They are required to indirectly promote the cleavage of pro-IL-1β and the release of IL-1β through NLRP3-dependent activation of caspase-1 (45, 46). Research has revealed that caspase-4/5/11 can trigger the release of mitochondrial reactive oxygen species (mtROS) and mitochondrial DNA (mtDNA) by enhancing the pore-forming capability of GSDMD in the mitochondria, thereby contributing to the activation of the NLRP3 inflammasome (47, 48). The orphan receptor Nur77 is activated upon binding to LPS and mtDNA (48). Subsequently, Nur77 interacts with NLRP3, triggering activation of the NLRP3 inflammasome (48). After LPS stimulation, caspase-4/5/11 trigger intracellular K+ efflux, one of the pathways by which caspase-4/5/11 mediates NLRP3 inflammasome activation (46, 49). Furthermore, the activated caspase-11 triggers the cleavage of pannexin-1 channels, leading to the release of ATP into the extracellular environment (50). Subsequently, the P2X7 receptor (P2X7R) responds to extracellular ATP, triggering the assembly of NLRP3 inflammasome and the release of IL-1β (50). The scaffold structural domain of pro-caspase-11 facilitates the activation of NLRP3 through interaction with the LRRs and PYD of NLRP3 (51). Intriguingly, this activation is mediated by the co-induction of live gram-negative bacterial mRNA and LPS (51). In addition to LPS, oxidized phospholipids (oxPAPC) serve as endogenous ligands for caspase-11 (52). It induces the oligomerization of caspase-11 by binding to its catalytic domain in dendritic cells, thereby promoting the assembly of the NLRP3 inflammasome and inflammation (52). Nevertheless, indications suggest that oxPAPC exerts an anti-inflammatory effect as it can diminish the non-canonical activation of the macrophage NLRP3 inflammasome and dampen the inflammatory response through competitive binding of LPS to caspase-4 and caspase-11 (53). Consequently, further investigations are warranted to explore the potential divergent effects of oxPAPCs on non-canonical NLRP3 inflammasome activation across different cell types.
2.2.3 Alternative NLRP3 inflammasome activation
In contrast to the previously mentioned activation pathways, the alternative activation pathway requires only one step to activate the NLRP3 inflammasome and lacks the features of canonical and non-canonical NLRP3 inflammasome activation, such as K+ efflux, pyroptosis, or pyroptosome formation (54). This activation pathway exhibits species specificity and has been identified exclusively in human and porcine monocytes (54). Research revealed that toll-like receptor (TLR) 4 in human monocytes triggered the NLRP3 inflammasome through the TRIF/RIPK1/FADD/caspase-8 signaling pathway upon stimulation by LPS, eliminating the need for a secondary signal for mediation (54). Tumor necrosis factor-α (TNF-α), a closely associated cytokine in psoriasis, selectively induces the initiation of the NLRP3 inflammasome through the TNFR/caspase-8 pathway even without an initial signal (55). Apolipoprotein C3 (ApoC3), an endogenous mediator, selectively triggers activation of the NLRP3 inflammasome in human monocytes (56). This process involves the formation of heterodimers between TLR2 and TLR4, initiating a pathway dependent on SCIMP/Lyn/Syk for calcium influx and ROS production, leading to caspase-8 activation and ultimately triggering activation of the NLRP3 inflammasome (56).
3 Role of NLRP3 inflammasome activation in the onset and progression of HF
Upon systematically reviewing studies on NLRP3 inflammasome activation in HF, we discovered that its activation promotes the onset and progression of HF by exacerbating multiple crucial pathophysiological processes. These pathological changes include myocardial inflammatory injury, adverse myocardial fibrosis, pathological myocardial hypertrophy, inhibited angiogenesis, abnormal cardiac electrical signal conduction, disturbed cardiac energy metabolism, and abnormal cardiomyocyte apoptosis (Table 1). Among these processes, myocardial inflammatory injury stands as a central nexus, where chronic inflammation not only directly harms the myocardium but also has the potential to initiate a cascade of events that worsen other pathological alterations in HF. Notably, the activation of the NLRP3 inflammasome involves the modulation of multiple signaling pathways, which are pivotal in mediating the aforementioned pathological processes (Figure 3).
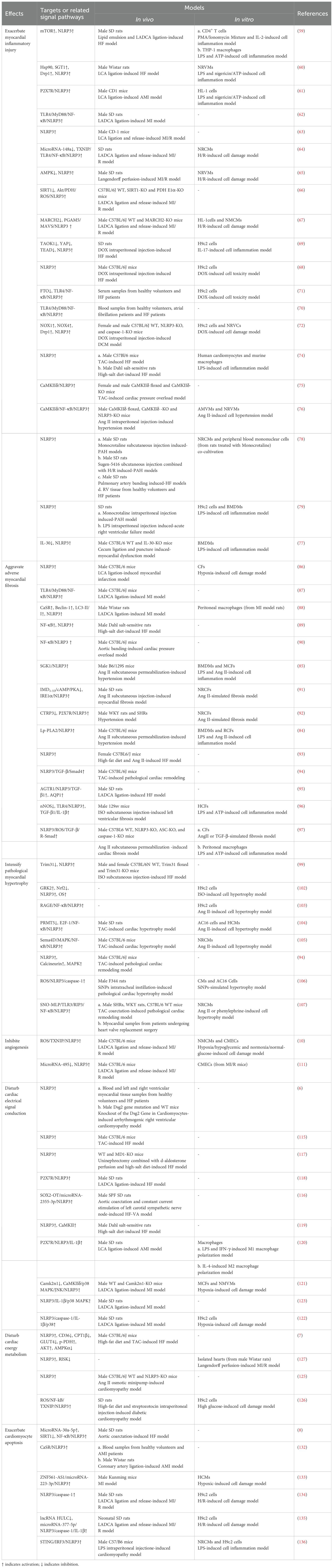
Table 1. Role of NLRP3 inflammasome activation in the onset and progression of HF.
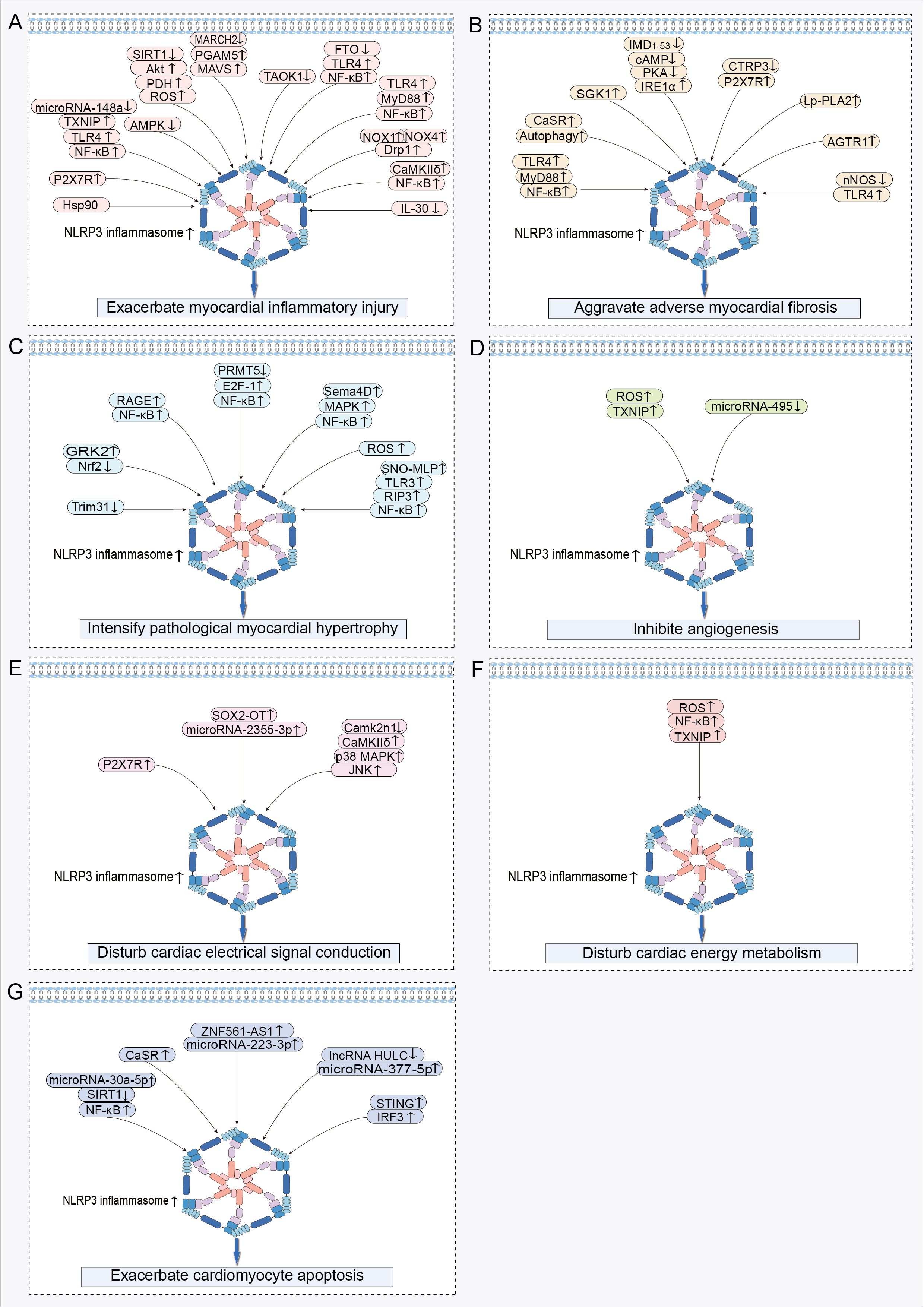
Figure 3. Signaling pathways regulating NLRP3 inflammasome activation in HF: (A) Signaling pathway exacerbating myocardial inflammatory injury. (B) Signaling pathway aggravating adverse myocardial fibrosis. (C) Signaling pathway intensifying pathological myocardial hypertrophy. (D) Signaling pathway inhibiting angiogenesis. (E) Signaling pathway disturbing cardiac electrical signal conduction. (F) Signaling pathway disturbing cardiac energy metabolism. (G) Signaling pathway exacerbating cardiomyocyte apoptosis. ↑ indicates activation; ↓ indicates inhibition.
3.1 Exacerbate myocardial inflammatory injury
An appropriate inflammatory response serves as a protective mechanism that eliminates harmful stimuli and repairs damaged tissues (57). However, excessive or prolonged inflammation escalates the risk of cardiac dysfunction and adverse cardiac remodeling (57, 58).
In the cardiac tissues of HF rats, increased NLRP3-positive spots, caspase-1 shear activation, and elevated levels of mature IL-1β were accompanied by a heightened inflammatory response (59, 60). These findings suggest that the NLRP3 inflammasome plays a contributory role in the development of myocardial inflammation in HF (59, 60). During acute myocardial infarction (AMI), dying heart myocytes initiate the assembly of the NLRP3 inflammasome by activating P2X7R via ATP release (61). This process amplifies cardiac inflammation, leads to further loss of functional myocardium, and even results in HF (61). Following myocardial infarction (MI), myocardial injury triggers the activation of the NLRP3 inflammasome, which exacerbates the myocardial inflammatory response, leading to enlargement of the infarct and worsening of cardiac dysfunction (62). Nicorandil pretreatment decreased NLRP3 inflammasome activation by inhibiting the TLR4/myeloid differentiation primary response protein 88 (MyD88)/NF-κB pathway, thereby alleviating the detrimental effects of MI on the heart (62). Activation of the NLRP3 inflammasome plays a crucial role in promoting myocardial ischemia/reperfusion (MI/R) injury (63). MI/R injury results in decreased microRNA-148a expression in myocardial cells, which increases the expression of thioredoxin-interacting protein (TXNIP) (64). Subsequently, TXNIP activates the TLR4/NF-κB/NLRP3 signaling pathway, promoting the release of inflammatory factors IL-1β and IL-18, thereby increasing inflammatory cell death in myocardial cells, leading to more extensive myocardial damage and worsening of cardiac function (64). Furthermore, studies have revealed that signaling pathways including adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) (65), silent information regulator of transcription (SIRT) 1/serine/threonine protein kinase B (Akt)/pyruvate dehydrogenase (PDH)/ROS (66), and E3 ubiquitin ligase membrane-associated RING finger protein 2 (MARCH2)/phosphoglycerate mutase 5 (PGAM5)/mitochondrial anti-viral-signaling protein (MAVS) (67) contribute to the exacerbation of cardiac inflammatory injury by activating the NLRP3 inflammasome, thereby exacerbating adverse cardiac outcomes caused by MI/R.
Activation of the NLRP3 inflammasome is a pivotal factor contributing to increased inflammatory damage in the non-ischemic myocardium. Activation of the NLRP3 inflammasome led to increased cardiomyocyte pyroptosis and reduced proliferative capacity in the doxorubicin (DOX)-induced HF model, collectively exacerbating the pathogenic progression of HF (68, 69). Mechanistically, activation of the TLR4/MyD88/NF-κB signaling pathway serves as an upstream event that triggers the activation of the NLRP3 inflammasome, thereby exacerbating cardiomyocyte pyroptosis and myocardial inflammation in DOX-induced HF (70, 71). Myocardial tissues from patients with dilated cardiomyopathy (DCM) exhibit aberrant NLRP3 inflammasome activation and pronounced pyroptosis, which are correlated with diminished cardiac function (72). In a DOX-induced DCM mouse model, DOX triggered the hyperactivation of the NLRP3 inflammasome by upregulating NOX1 and NOX4 expression and activating dynamin-related protein 1 (Drp1)-dependent mitochondrial fragmentation (72). This process exacerbates cardiomyocyte pyroptosis and contributes to the progression of cardiac dysfunction (72). Macrophages play a pivotal role in the regulation of cardiac inflammation (73). In the HF state, the activation of the NLRP3 inflammasome in myocardial tissue promotes macrophage infiltration into the heart (74). Mechanistic study has demonstrated that cardiomyocytes activate NLRP3 through the calmodulin-regulated kinase δ (CaMKIIδ) signaling pathway, promoting the release of pro-inflammatory cytokines IL-1β, IL-18, and IL-6, as well as the production of monocyte chemotactic protein-1 (MCP-1) and macrophage inflammatory protein 1α (75). These factors synergistically promote macrophage migration to myocardial tissue, thereby further amplifying cardiac inflammation (75). Furthermore, activation of myocardium-specific CaMKIIδ can also mediate the activation of the NLRP3 inflammasome through the NF-κB pathway, leading to increased macrophage recruitment to the damaged myocardium and exacerbating the cardiac inflammatory cascade (76). Interestingly, macrophages demonstrate two pro-inflammatory effects, pyroptosis and pro-inflammatory polarization, upon recruitment to the heart (77–79). In pulmonary arterial hypertension (PAH)-induced right ventricular failure, there was a significant increase in the number of macrophages within the right ventricle, accompanied by an elevated expression of the NLRP3 inflammasome in these macrophages (78). The elevated expression of the NLRP3 inflammasome not only promoted macrophage pyroptosis, but also drove macrophages toward a pro-inflammatory M1-type phenotype (78). This shift exacerbated the inflammatory response in the right ventricle, contributing to further deterioration of right ventricular dysfunction (78). Additionally, cardiomyocyte NLRP3-dependent pyroptosis further stimulates macrophage polarization toward a pro-inflammatory M1 phenotype in myocardial tissues through the release of pro-inflammatory cytokines and MCP-1 (79). Similarly, in sepsis-induced cardiac inflammatory injury and dysfunction, macrophage pyroptosis in cardiac tissues, along with the polarization of Ly6Chigh macrophages, is positively regulated by NLRP3 complex activation (77). These studies confirm that activation of the NLRP3 inflammasome exacerbates the cardiac inflammatory cascade by promoting macrophage recruitment to the heart and stimulating macrophage pyroptosis and pro-inflammatory polarization.
3.2 Aggravate adverse myocardial fibrosis
Myocardial fibrosis is characterized by abnormal proliferation and differentiation of cardiac fibroblasts (CFs) and excessive accumulation and abnormal distribution of the extracellular matrix (ECM). Myocardial fibrosis is a critical reparative response aimed at maintaining cardiac integrity after myocardial injury (80). However, excessive myocardial fibrosis results in diminished myocardial compliance and cardiac diastolic and systolic dysfunction, serving as a pivotal pathological foundation for the onset and progression of HF (81, 82).
Myofibroblasts play a crucial role as mediator cells in the progression of myocardial fibrosis (83). They induce cardiac fibrous scar formation and dysfunction by synthesizing significant quantities of ECM and collagen, secreting pro-fibrotic cytokines, and expressing α-smooth muscle actin (α-SMA) (83). IL-1β was identified as a key mediator in promoting the proliferation and differentiation of CFs into myofibroblasts, indicating that NLRP3 inflammasome activation is an important factor mediating the progression of myocardial fibrosis (84, 85). MI leads to a notable upregulation in the expression of fibrotic markers in myocardial tissues, including collagen I, collagen III, and α-SMA (86). Importantly, the degree of NLRP3 inflammasome activation is positively correlated with the severity of myocardial fibrosis (86). Myocardial ischemia triggered the activation of the TLR4/MyD88/NF-κB signaling pathway, which facilitated the assembly and activation of the NLRP3 inflammasome, thereby exacerbating cardiac inflammation (87). The progression of inflammation enhances fibrosis, resulting in increased cardiac stiffness and reduced cardiac pumping function (87). The expression of calcium-sensitive receptor (CaSR) is elevated in myocardial tissue following MI (88). CaSR exacerbates both inflammation and fibrosis post-MI by activating the autophagy/NLRP3 inflammasome pathway (88).
In pressure overload-induced HF, the activation of the NLRP3 inflammasome was identified as a critical factor driving the progression of myocardial fibrosis (89). Mechanistically, chronic stress overload initiates the activation of the NF-κB/NLRP3 inflammasome pathway (90). This pathway amplifies the aberrant activation of cardiac fibroblasts and promotes the over-synthesis of collagen associated with fibrosis, thereby fueling the adverse progression of cardiac fibrosis (90). Administration of angiotensin II (Ang II) induced myocardial fibrosis in mice, as indicated by the excessive deposition of collagen fibers, elevated expression levels of transforming growth factor-β (TGF-β) and connective tissue growth factor, along with NLRP3 inflammasome activation in cardiac tissues and heightened IL-1β secretion (85). Notably, treatment with MCC950 successfully reversed these pathological alterations (85). Inositol-requiring enzyme 1α (IRE1α) acted as a sensor of endoplasmic reticulum stress, capable of triggering NLRP3 inflammasome activation, thereby exacerbating the progression of myocardial fibrosis (91). The endogenous cardiovascular protective peptide, intermedin1-53 (IMD1-53), had the ability to reduce the expression of IRE1α through the activation of the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathway, leading to the inhibition of NLRP3 inflammasome activation and mitigation of Ang II-induced cardiac fibrosis (91). Reduced expression of C1q/TNF-related protein-3 (CTRP3) in myocardial tissue is linked to the advancement of cardiac fibrosis (92). The restoration of CTRP3 expression ameliorated Ang II-induced myocardial fibrosis by inhibiting the P2X7R/NLRP3 inflammasome pathway to reduce α-SMA, collagen I/III, and matrix metallopeptidase (MMP) 2/9 expression (92).
TGF-β is a central signaling pathway in the promotion of fibrosis. In HF, activation of the NLRP3 inflammasome is considered a significant factor in the upregulation of TGF-β gene expression in the cardiac tissue (93). Specifically, the activated NLRP3 inflammasome promotes the advancement of cardiac fibrosis by triggering the TGF-β/Smad4 signaling pathway to enhance the expression levels of collagen type I, collagen type III, MMP-2, MMP-9, and α-SMA (94). Further study has demonstrated that Ang II receptor type 1 (AGTR1) accelerates myocardial fibrosis progression by activating the NLRP3 inflammasome and enhancing the production of TGF-β1 (95). Concurrently, activation of the TLR4 receptor also triggers NLRP3 inflammasome activation, initiating a signaling cascade that enhances the pro-fibrotic effects of the TGF-β1/IL-1β axis, promotes cardiac myofibroblast differentiation, increases interstitial collagen deposition, and ultimately exacerbates fibrosis (96). Additionally, a study revealed the mitochondrial localization of NLRP3 in CFs and demonstrated that NLRP3 is involved in the development of cardiac fibrosis by enhancing mitochondrial ROS production, promoting activation of the TGF-β/R-Smad pathway, and facilitating CF differentiation (97).
3.3 Intensify pathological myocardial hypertrophy
Pathological myocardial hypertrophy is an adaptive response of the heart to prolonged pressure or increased volume overload. Nevertheless, as myocardial hypertrophy advances to a certain level, it can exert significant adverse effects on the cardiac structure and function, thereby increasing the risk of HF (98).
In the context of cardiac remodeling, the activation of the NLRP3 inflammasome promotes not only cardiac inflammation and fibrosis but also aggravates pathological myocardial hypertrophy, consequently exacerbating symptoms of HF (99). Elevated levels of G protein-coupled receptor kinase 2 (GRK2) were identified in hypertrophied myocardial tissue (100, 101). Mechanistically, GRK2 promotes the activation of the NLRP3 inflammasome and induces oxidative stress (OS) by downregulating the expression of nuclear factor erythroid-2-related factor 2 (Nrf2), thereby exacerbating isoproterenol (ISO)-induced pathological cardiac hypertrophy (102). The receptor for advanced glycation endproducts (RAGE) participated in Ang II-induced pathological cardiomyocyte hypertrophy by activating the NF-κB/NLRP3/IL-1β pathway (103). Under pressure overload, there is a reduction in protein arginine methyltransferase 5 (PRMT5) expression in hypertrophic myocardial tissues. Low PRMT5 expression triggered the activation of the E2F-1/NF-κB signaling pathway, leading to the activation of the NLRP3 inflammasome that promotes maladaptive cardiac hypertrophy induced by transverse aortic constriction (TAC) or Ang II (104). The overexpression of Sema4D contributed to pressure overload-induced cardiac hypertrophy (105). It promotes the assembly and activation of NLRP3 complexes by activating the mitogen-activated protein kinase (MAPK)/NF-κB signaling pathway, thereby exacerbating TAC-induced pathological cardiac hypertrophy and dysfunction (105). Elevated levels of calcineurin and MAPK phosphorylation were observed in the TAC surgery group of pressure-overloaded mice (94). In contrast, MCC950 ameliorates pathological cardiac hypertrophy and enhances cardiac function by inhibiting calcineurin expression and the MAPK signaling pathway (94). Silica nanoparticles (SiNPs) contributed to the exacerbation of cardiac hypertrophy (106). SiNPs worsen myocardial hypertrophy by inducing cardiomyocyte pyroptosis via activation of the ROS/NLRP3/caspase-1 signaling pathway (106). Transfection of cardiomyocytes with si-NLRP3 or the caspase-1 inhibitor VX-765 limited SiNP-induced pathological cardiac hypertrophy (106). S-nitrosylated muscle LIM protein (SNO-MLP) expression is markedly elevated in patients and animals with myocardial hypertrophy (107). This upregulation primarily facilitated the interaction between TLR3 and receptor-interacting protein kinase 3 (RIP3), thus initiating activation of the NF-κB/NLRP3 inflammasome pathway, ultimately fostering the progression of myocardial hypertrophy (107).
3.4 Inhibite angiogenesis
Angiogenesis generates new blood vessels from existing capillaries or capillary post-veins. When the heart is exposed to ischemic and hypoxic stimuli, angiogenesis enhances its blood supply, thereby mitigating damage and preserving cardiac function resulting from ischemia and hypoxia (108, 109). However, the progression of cardiac pathological remodeling inhibits angiogenesis, resulting in decreased microvascular density and ultimately leading to HF (109).
Coronary microvessel rarefaction and decreased blood flow reserve have been identified as the primary drivers of diastolic dysfunction in patients with HF with a preserved ejection fraction (HFpEF) (9). Moreover, decreased cardiac microvascular density is intricately linked to NLRP3 inflammasome activation (110). Phosphorylation of microfibrillar-associated protein 4 (MAP4) downregulates the expression of angiogenic markers, such as CD31, CD34, VEGFA, VEGFR2, ANG2, and TIE2 (110). Mechanistically, MAP4 inhibited angiogenesis via NLRP3 inflammasome activation, leading to reduced cardiac microvessel density (110). Endothelial cells (ECs) serve as primary effector cells in cardiac angiogenesis, and any damage to or aberrant apoptosis of these cells significantly affects their capacity for cardiac angiogenesis. During MI/R injury, microvascular endothelial cells (CMECs) mediated interactions between TXNIP and NLRP3 by generating excessive ROS (10). This action subsequently escalates the activation level of the NLRP3 inflammasome, exacerbating damage to cardiac microvascular endothelial cells (10). In ischemia-reperfused myocardial tissues, there was a reduction in microRNA-495 expression, which facilitates the activation of the NLRP3 inflammasome, worsening inflammatory damage and apoptosis in CMECs (111). Conversely, elevating the expression of microRNA-495 or suppressing the NLRP3 gene decreases apoptosis and enhances the proliferation of CMECs by shifting the cell population from the G0/G1 phase to the S phase (111). This observation implies that the suppression of NLRP3 inflammasome activation may facilitate the repair and angiogenesis of cardiac microvessels. SIRT3 deficiency resulted in diminished expression of hypoxia-inducible factor-2α, VEGF, and angiopoietin-1, leading to decreased angiogenesis and subsequently causing coronary microvessel rarefaction and cardiac diastolic dysfunction (112). Trimethylamine N-oxide (TMAO) induces vascular inflammation by suppressing SIRT3 expression and superoxide dismutase 2 (SOD2) activity in endothelial cells, subsequently triggering mtROS/NLRP3 inflammasome signaling (113). Therefore, SIRT3 deficiency may impede coronary microvascular angiogenesis by activating the NLRP3 inflammasome.
3.5 Disturb cardiac electrical signal conduction
Ventricular arrhythmias (VAs) are common triggers and causes of death in HF (114). The cardiac electrical conduction system is crucial for maintaining normal heart function, and conduction abnormalities are the underlying precursors of arrhythmias.
Numerous studies have established that the activation of the NLRP3 inflammasome is a key factor in disrupting the electrical signaling in the heart and inducing malignant arrhythmias, particularly in the context of HF (6, 115–119). The activation of the NLRP3 inflammasome not only enhances myocardial inflammatory responses but also promotes the development of cardiac hypertrophy and fibrosis, creating a pro-arrhythmic environment (115). Additionally, NLRP3 inflammasome activation results in changes to myocyte ion channel functions, including a reduced expression of ion channel proteins such as Kv4.2, KChIP2, and Cav1.2, which affect the timing and morphology of cardiac action potentials and contribute to the development and maintenance of arrhythmias (115). Simultaneously, sympathetic nervous hyperactivity contributes to an increased susceptibility to HF-related ventricular arrhythmias due to NLRP3 inflammasome activation (117, 118). In particular, the activation of the NLRP3 inflammasome exacerbates cardiac sympathetic hyperactivity by promoting the release of the proinflammatory cytokine IL-1β, and this inflammatory-neural interaction results in altered electrophysiological properties of the heart, such as prolongation of the action potential duration and shortening of the effective refractory period, which increases the risk of ventricular arrhythmias (117, 118). In the myocardial tissues of rats with HF-related ventricular arrhythmias (VAs-HF), there was a notable increase in the expression of SOX2-overlapping transcript (SOX2-OT) and NLRP3 (116). Furthermore, silencing of the SOX2-OT gene reduced NLRP3 inflammasome activation levels by regulating microRNA-2355-3p, thus alleviating HF symptoms and diminishing VAs (116). In HFpEF, the activation of the NLRP3 inflammasome facilitates the development of atrial fibrillation through the promotion of atrial fibrosis, by prolonging the atrial action potential duration, increasing the dispersion of action potential duration, and activating inflammation-associated signaling pathways (119). Following MI, P2X7R facilitates the upregulation of nerve growth factor, tyrosine hydroxylase, and growth-associated protein 43 by mediating the activation of the NLRP3/IL-1β pathway, thereby fostering sympathetic sprouting (120). This cascade leads to altered cardiac electrophysiological characteristics and an increased susceptibility to arrhythmias (120). After MI, the expression of Camk2n1 is markedly reduced in the infarct border zone, leading to the activation of the CaMKIIδ/p38 MAPK/C-Jun N-terminal kinase (JNK)/NLRP3 inflammasome signaling pathway (121). This exacerbates myocardial fibrosis and increases the vulnerability to premature ventricular contractions, tachycardia, and ventricular fibrillation (121). Connexin 43 (Cx43) is a key regulator of cardiac electrical signal conduction (122, 123). The activation of the NLRP3 inflammasome within the myocardial infarct zone diminishes the expression of Cx43 in myocardial tissue, resulting in compromised intercellular communication and heightened vulnerability to VAs (122, 123). Conversely, restoring the expression of Cx43 in the heart by inhibiting the NLRP3/IL-1β/p38 MAPK pathway helps enhance cardiac conduction function and decrease the heart’s susceptibility to VAs (122, 123).
3.6 Disturb cardiac energy metabolism
The heart, as an organ with high energy and oxygen demands, relies on homeostasis of its energy metabolism as the foundational mechanism for maintaining the stability of the cardiac tissue structure and internal environment (124). The myocardial energy metabolism relies heavily on mitochondrial oxidative phosphorylation. When mitochondria are damaged, myocardial energy substrate utilization is altered, leading to decreased cardiac energy production and lactic acid build-up, which in turn affects cardiac energy metabolism and cardiomyocyte survival and accelerates the malignant progression of HF (124).
A complex interplay exists between NLRP3 inflammasome activation and myocardial energy metabolism disruption. Mitochondrial dysfunction is the trigger for the activation of the NLRP3 inflammasome (36, 37), while the activation of the NLRP3 inflammasome further impairs mitochondrial function and homeostasis (7, 125–127). In an obesity-associated HF model, overactivation of the NLRP3 inflammasome results in an imbalance between cardiac energy supply and demand, as evidenced by decreased fatty acid utilization and increased reliance on glycolysis and glucose oxidation in cardiomyocytes, thereby triggering cardiac metabolic reprogramming (7). This metabolic transition was concomitant with the downregulation of genes associated with mitochondrial energy transfer and respiratory pathways, consequently intensifying the advancement of HF (7). During MI/R injury, the inhibition of the NLRP3 inflammasome activates the reperfusion injury salvage kinase (RISK) pathway, subsequently enhancing the expression of markers associated with mitochondrial biogenesis and energy metabolism, such as mitochondrial transcription factor A, nuclear respiratory factor-1, and mitochondrial creatine kinase (127). These findings suggest an association between disturbed myocardial energy metabolism and the formation of the NLRP3 inflammasome complex during MI/R injury, indicating that inhibition of NLRP3 inflammasome activation contributes to the improvement of cardiac energy metabolism, thereby enhancing the resistance of cardiomyocytes to ischemic and hypoxic injury (127). In the Ang II-induced cardiomyopathy model, increased NLRP3 inflammasome activity was accompanied by decreased mtDNA copy number, reduced ATP synthase activity, increased ROS production, as well as mitochondrial structural alterations, including swelling, disordered matrix organization, and fragmentation (125). The knockdown of the NLRP3 gene mitigated Ang II-induced mitochondrial structural and functional damage, as well as alleviated cardiac dysfunction (125). In rats with diabetic cardiomyopathy, cardiomyocyte mitochondria exhibit swelling and matrix disorders, along with activation of the NLRP3 inflammasome (126). Silencing of the NLRP3 gene aided in restoring mitochondrial structure and reducing glycogenolysis and lipid accumulation in cardiomyocytes, suggesting an enhancement in cardiomyocyte energy metabolism (126).
3.7 Exacerbate cardiomyocyte apoptosis
Cardiomyocyte apoptosis is a type of programmed cell death that is genetically regulated (128). Cardiomyocytes, which are primary cardiac cells, are responsible for contraction (129). Excessive apoptosis of cardiomyocytes is a significant contributor to the structural alterations and functional deterioration of the heart. Moreover, it is a crucial driver of HF onset and progression (130, 131).
During the pathological progression of HF, the overactivation of NLRP3 exerts a pro-apoptotic effect on cardiomyocytes (8). Mechanistically, microRNA-30a-5p activates the NF-κB/NLRP3 signaling cascade by targeting SIRT1, thereby exacerbating cardiomyocyte apoptosis (8). CaSR expression is markedly elevated in the neutrophils of patients and rats with AMI (132). This upregulation facilitated NLRP3 inflammasome activation, release of IL-1β through the PLC-IP3 pathway, and calcium release from the endoplasmic reticulum (132). IL-1β interacted with the IL-1 receptor on cardiomyocytes, leading to an increase in Bax expression and caspase-3 cleavage, while decreasing Bcl2 expression, thereby effectively promoting cardiomyocyte apoptosis (132). In the myocardial tissue of MI mice, the expression of the long noncoding RNA zinc finger protein 561 antisense RNA 1 (ZNF561-AS1) is significantly upregulated (133). This upregulation leads to the inhibition of cardiomyocyte proliferation and augmentation of cardiomyocyte apoptosis via activation of the microRNA-223-3p/NLRP3 inflammasome pathway (133). During MI/R injury, activation of the NLRP3 inflammasome results in increased cardiomyocyte apoptosis through the upregulation of Bax protein expression and downregulation of Bcl2 expression (134). In myocardial tissues injured by ischemia reperfusion, the expression of long noncoding RNA highly up-regulated in liver cancer (lncRNA HULC) is downregulated (135). The decrease in lncRNA HULC expression results in heightened microRNA-377-5p activity, triggering the NLRP3/caspase-1/IL-1β signaling pathway (135). This cascade amplifies caspase-3 and cleaved-caspase-3 expression, ultimately worsening cardiomyocyte apoptosis (135). In a mouse model of cardiomyopathy, STING activation triggers the activation of the NLRP3 inflammasome by enhancing the phosphorylation and intranuclear translocation of IRF3 (136). This process elevates the ratios of Bax/Bcl-2 and C-Caspase3/T-Caspase3, leading to an increase in cardiomyocyte apoptosis (136).
4 TCM active ingredients in preventing and treating HF by inhibiting the NLRP3 inflammasome
Active ingredients are fundamental to the efficacy of TCM. Existing studies have revealed that active ingredients in TCM exert positive regulatory effects on key pathological processes of HF by inhibiting the NLRP3 inflammasome. In particular, these active ingredients are effective in ameliorating myocardial inflammation, adverse myocardial fibrosis, pathological myocardial hypertrophy, angiogenesis, cardiac electrical signal conduction, cardiac energy metabolism, and reducing abnormal cardiomyocyte apoptosis (Table 2). Further analysis revealed that these active ingredients, with the potential to prevent and treat HF, are primarily found in flavonoids and their glycosides, terpenes and their glycosides, phenolic acids, quinones, and phenylpropanoids (Table 2).
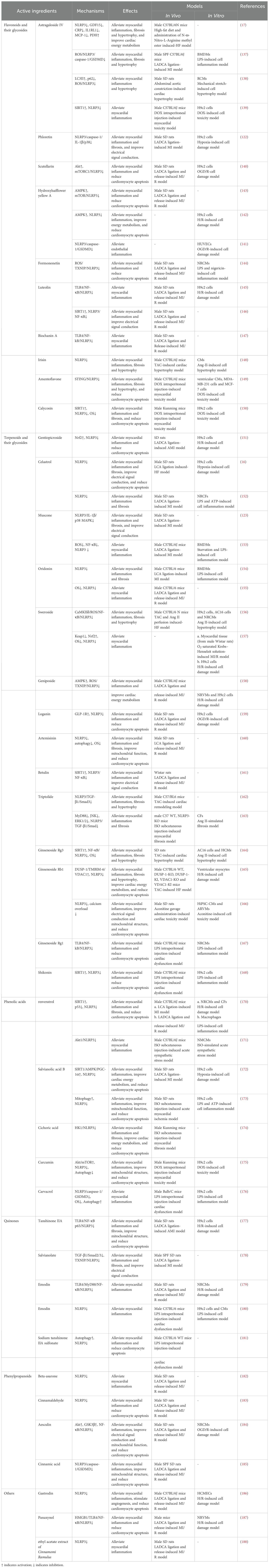
Table 2. Mechanisms of active ingredients in TCM regulating the NLRP3 inflammasome in HF.
4.1 Flavonoids and their glycosides
Astragaloside IV (AS-IV) demonstrates significant therapeutic potential for HFpEF (17). Specifically, AS-IV intervention markedly decreased NLRP3, IL-1β, and caspase-1 levels in the myocardium of HFpEF mice, with this reduction of biomarkers significantly linked to the amelioration of myocardial inflammation and enhancement of cardiac function (17). Additionally, AS-IV exerted a beneficial effect on maintaining cardiac metabolic homeostasis in HFpEF by optimizing cardiac glycolipid metabolism, enhancing mitochondrial function, and regulating energy metabolic pathways (17). AS-IV also effectively alleviated cardiac remodeling caused by MI (137). By inhibiting the ROS/caspase-1/GSDMD signaling pathway, As-IV reduces cardiomyocyte pyroptosis and lowers the expression levels of collagen I, collagen III, α-SMA, and fibronectin (137). This process effectively reduces post-MI cardiac fibrosis and hypertrophy, consequently enhancing the heart function (137). As-IV exerts protective effects against pressure overload-induced cardiac dysfunction (138). Through the upregulation of LC3II levels and inhibition of p62 expression, As-IV activated autophagy, subsequently inhibiting the ROS/NLRP3 inflammasome pathway and reducing the expression levels of IL-1β and IL-18 (138). This action effectively alleviates pressure overload-induced myocardial hypertrophy (138). In addition, As-IV mitigates DOX-induced myocardial toxicity (139). It exerts cardioprotective effects by reversing the DOX-induced downregulation of SIRT1 protein expression, upregulation of NLRP3 expression, and reduction in cardiomyocyte pyroptosis (139). Phloretin mitigates the electrical remodeling process in the heart post-MI (122). By inhibiting the NLRP3/caspase-1/IL-1β pathway, it diminished p38 phosphorylation, facilitating the restoration of Cx43 expression and mitigating cardiac electrical remodeling post-MI, consequently lowering cardiac susceptibility to VAs and the occurrence of HF (122). Furthermore, Phloretin also decreased the expression of fibrotic markers including collagen 1, collagen 3, TGF-β, and α-SMA post-MI by suppressing inflammatory responses orchestrated by NLRP3 inflammasome activation, consequently alleviating detrimental cardiac remodeling (122). The cardioprotective effects of scutellarin are mediated by its regulation of the Akt/mTORC1/NLRP3 signaling pathway (140). More precisely, scutellarin inhibits mTORC phosphorylation by upregulating Akt expression (140). This action subsequently diminishes the activation of the NLRP3 inflammasome, thus mitigating inflammatory injury and dysfunction in the heart induced by MI/R (140). Hydroxylsafflower yellow A (HSYA) was recognized for its ability to mitigate myocardial ischemia and hypoxic injury (141–143). In MI/R injury, HSYA suppressed the NLRP3 inflammasome by modulating the AMPK/mTOR signaling pathway, thereby reducing myocardial infarct size and decreasing cardiomyocyte apoptosis, ultimately improving heart function (143). In an H/R-induced H9c2 cell study, the AMPK inhibitor compound C nullified the suppressive impact of HSYA on NLRP3 inflammasome activation, as demonstrated by elevated levels of NLRP3, caspase-1, and IL-1β expression (142). This observation further corroborates that the inhibition of the AMPK/NLRP3 inflammasome signaling pathway is an important mechanism in the anti-MI/R injury effect of HSYA (142). In a study on oxygen-glucose deprivation/reoxygenation (OGD/R)-induced HUVECs, NLRP3 inflammasome-mediated pyroptosis was heightened (141). Treatment with HSYA mitigated pyroptosis by inhibiting the NLRP3/caspase-1/GSDMD pathway, thereby mitigating inflammatory damage to HUVECs resulting from OGD/R (141). Formononetin can alleviate MI/R injury (144). It restricts the activation of the NLRP3 inflammasome by diminishing the release of ROS, suppressing the expression of TXNIP, and attenuating the interaction between TXNIP and NLRP3, thereby decreasing the secretion of proinflammatory factors and cardiomyocyte apoptosis (144). Luteolin similarly demonstrated the potential to alleviate MI/R injury, and this protective attribute was associated with its suppression of the TLR4/NF-κB/NLRP3 inflammasome pathway (145). Luteolin downregulates the expression of TLR4, MyD88, and NF-κB in a dose-dependent manner to inhibit NLRP3 inflammasome activation, consequently diminishing myocardial infarct size and enhancing left ventricular function (145). Intriguingly, another study identified the SIRT1/NLRP3/NF-κB signaling pathway as the primary regulatory mechanism by which luteolin alleviates MI/R damage (146). These findings suggest that luteolin may exert cardioprotective effects by inhibiting the NLRP3 inflammasome through multiple molecular signaling pathways. Biochanin A alleviates the cardiac inflammatory response and reduces the infarcted myocardial area resulting from MI/R (147). Its cardioprotective effect was intricately linked to its inhibition of the TLR4/NF-κB/NLRP3 signaling pathway (147). By inhibiting NLRP3 inflammasome activation, irisin effectively restrained the expression of GSDMD-N and IL-1β, thereby mitigating the detrimental effects of pressure overload on the heart such as myocardial inflammation, fibrosis, and hypertrophy (148). By inhibiting the STING/NLRP3 signaling pathway, amentoflavone mitigates cardiomyocyte pyroptosis and cardiac inflammation, consequently ameliorating DOX-induced heart damage and functional impairment (149). Calycosin also shows promise for the treatment of myocardial toxicity (150). Mechanistically, it inhibited NLRP3 inflammasome activation by upregulating SIRT1 expression, thereby reducing cardiac inflammatory infiltration, myocardial fibrosis, and cardiomyocyte apoptosis, ultimately mitigating DOX-induced cardiac injury (150).
4.2 Terpenoids and their glycosides
AMI triggered intense inflammatory responses and oxidative stress (OS) (151). Gentiopicroside mitigates cardiac inflammatory responses, OS, and cardiomyocyte apoptosis induced by AMI by regulating the Nrf2/NLRP3 signaling pathway, thereby safeguarding cardiac function (151). In the pathological progression of chronic HF, Celastrol improves cardiac electrophysiological stability, upregulates Cx43 and ion channel expression, and reduces myocardial fibrosis and inflammatory responses by inhibiting the NLRP3/caspase-1/IL-1β signaling pathway, ultimately reducing susceptibility to ventricular fibrillation (16). Following MI, a notable increase was observed in macrophage and neutrophil infiltration of myocardial tissues alongside a significant upregulation in the expression of pro-fibrotic proteins such as collagen I, collagen III, and α-SMA (152). Celastrol mitigates these pathological alterations by inhibiting the NLRP3 inflammasome (152). Muscone exhibits a promising therapeutic potential against MI (123). It diminishes ventricular inflammation and fibrosis, while decreasing vulnerability to VAs via the upregulation of Cx43 expression in the infarct border zone (123). These effects were associated with its inhibitory impact on the NLRP3/IL-1β/p38 MAPK pathway (123). Furthermore, Muscone mitigated the macrophage-driven cardiac inflammatory response by suppressing NF-κB expression and NLRP3 inflammasome activation in myocardial macrophages, leading to enhanced cardiac function and increased survival rates in mice post-MI (153). Oridonin can alleviate cardiac remodeling post-MI (154). By inhibiting the NLRP3 inflammasome, it reduced the expression of fibrosis markers, including collagen-I, collagen-III, collagen-IV, and α-SMA, thereby alleviating myocardial fibrosis and cardiac dysfunction following MI (154). Moreover, pretreatment with oridonin suppressed the overactivation of OS and NLRP3 inflammasome, consequently mitigating cardiac pathological alterations induced by ischemia reperfusion, including the alleviation of myocardial inflammatory damage and reduction of infarct size (155). Sweroside inhibits the ROS-mediated NF-κB/NLRP3 inflammasome pathway in cardiomyocytes by directly binding to CaMKIIδ, alleviating myocardial inflammation and adverse cardiac remodeling, thereby improving HF induced by pressure overload (156). Sweroside also exerts protective effects on ischemia reperfusion myocardium (157). Its intervention alleviates myocardial inflammatory damage and reduces the size of the infarcted area, helping to alleviate cardiac dysfunction caused by MI/R (157). This effect is primarily due to the inhibition of NLRP3 inflammasome-mediated pyroptosis (157). Geniposide has therapeutic potential for alleviating MI/R injury (158). It inhibits the ROS/TXNIP/NLRP3 inflammasome pathway by activating the AMPK signaling pathway (158). This process efficiently suppresses cardiac inflammation, enhances myocardial energy metabolism, and ultimately reduces the damage inflicted on the myocardium by ischemia reperfusion (158). The glucagon-like peptide-1 receptor (GLP-1R)/NLRP3 pathway plays a pivotal role in mediating the cardioprotective effects of loganins (159). MI/R induces a notable decline in GLP-1R expression within the myocardial tissue, which promotes the formation of the NLRP3 inflammasome and pyroptosis, exacerbating myocardial damage and cardiomyocyte apoptosis (159). Conversely, treatment with loganin alleviates these pathological changes (159). Artemisinin pretreatment mitigates MI/R-induced myocardial inflammation, cardiomyocyte apoptosis, and myocardial fibrosis primarily by inhibiting the NLRP3 inflammasome (160). Betulin attenuated the cardiac inflammatory response, decreased myocardial infarct size, and enhanced cardiac electrical signaling by modulating the SIRT1/NLRP3/NF-κB signaling pathway. This action ultimately helps mitigate the cardiac pathological damage induced by MI/R (161). Tretinoin has proven advantageous in alleviating negative cardiac repercussions induced by pressure overload (162). Mechanistically, tretinoin impeded the TGF-β1/Smad3 pathway by dampening the activation of the NLRP3 inflammasome, which in turn attenuated TAC-induced myocardial fibrosis and hypertrophy and improved cardiac function (162). Another study demonstrated that the mechanism by which tretinoin mitigates myocardial fibrosis involves the inhibition of the NLRP3 inflammasome. By diminishing MyD88-mediated JNK and ERK1/2 activity, tretinoin suppressed the NLRP3 inflammasome, subsequently inhibiting the TGF-β1/Smad signaling pathway (163). This cascade of events aids in reducing ECM deposition caused by pressure overload, thus exerting an anti-myocardial fibrotic effect (163). Ginsenoside Rg3 inhibited pathological myocardial hypertrophy induced by pressure overload (164). It achieved its anti-inflammatory and antioxidant effects by regulating the SIRT1/NF-κB/NLRP3 inflammasome signaling pathway, thereby reducing myocardial fibrosis and hypertrophy (164). Ginsenoside Rb1 attenuated HF induced by MI/R injury by targeting the DUSP-1/TMBIM-6/VDAC1 pathway, regulating intestinal microbiota homeostasis and the equilibrium of the mitochondrial quality control network, as well as suppressing the NLRP3-mediated inflammatory response and pyroptosis (165). Cardiac injury is a common adverse effect of aconitine. Following aconitine intervention, there is an increase in the expression of NLRP3-dependent pyroptosis-related proteins in myocardial tissue, accompanied by disruptions in electrophysiology, significant myocardial apoptosis, and cardiac dysfunction (166). Ginsenoside Rb1 effectively ameliorated aconitine-induced cardiac pathological alterations (167). Ginsenoside Rg1 effectively mitigated LPS-induced cardiotoxicity by reducing cardiac inflammation and cardiomyocyte apoptosis (167). This was achieved by lowering the Bax/Bcl2 ratio and the quantity of TUNEL-positive cells in myocardial tissues via the inhibition of the TLR4/NF-κB/NLRP3 pathway (167). Shikonin also attenuates LPS-induced cardiac dysfunction by inhibiting the NLRP3 inflammasome through upregulation of the SIRT1 pathway (168). This process reduces the release of inflammatory factors and macrophage infiltration into cardiac tissues, thereby alleviating LPS-induced myocardial injury and improving cardiac function (168).
4.3 Phenolic acids
Resveratrol shows promise in HF treatment (169). After a 3-month treatment with resveratrol, patients with systolic HF experienced significant reductions in IL-1, IL-6, NT-proBNP, galectin-3, total cholesterol, and low-density lipoprotein cholesterol levels, along with substantial enhancements in cardiac function and quality of life (169). Resveratrol can mitigate MI/R injury (170). It mitigates cardiac inflammation, fibrosis, and apoptosis by modulating the SIRT1/p53 signaling pathway and inhibiting the NLRP3 inflammasome, thereby ameliorating MI/R-induced cardiac dysfunction (170). Furthermore, resveratrol reduced acute sympathetic stress-induced cardiac inflammation by inhibiting the Akt1/NLRP3 inflammasome pathway (171). Salvianolic acid B inhibited the activation of the NLRP3 inflammasome by regulating the SIRT1/AMPK/PGC-1α signaling pathway (172). This action leads to decreased cardiac inflammation, mitigation of mitochondrial dysfunction, and a reduction in cardiomyocyte apoptosis, ultimately exerting a cardioprotective effect in ischemic and hypoxic conditions (172). Salvianolic acid B can also ameliorate myocardial inflammation and enhance mitochondrial function by promoting mitochondrial autophagy and inhibiting the NLRP3 inflammasome, thus alleviating ISO-induced acute myocardial ischemic injury (173). Chicoric acid effectively mitigates the detrimental effects of cardiac overload (174). Specifically, it reduced ISO-induced cardiac inflammation, fibrosis, apoptosis, and mitochondrial structural damage by inhibiting the hexokinase 1(HK1)/NLRP3 inflammasome signaling pathway (174). When exposed to DOX, cardiomyocytes exhibit impaired contractile function (175). Curcumin activates the AKT/mTOR pathway, leading to a reduction in DOX-induced pyroptosis and autophagy, thereby contributing to the alleviation of cardiomyocyt\e apoptosis and cardiac dysfunction (175). Carvacrol is beneficial in attenuating LPS-induced cardiac dysfunction, and its protective effect against myocardial injury is linked to the inhibition of pyroptosis mediated by the NLRP3/caspase1/GSDMD pathway (176).
4.4 Quinones
After AMI, the expression of TLR4, NF-κB p65, NLRP3, IL-1β, and IL-18 increased in cardiac tissue (177). These changes lead to adverse cardiac effects such as myocardial inflammation, fibrosis, cardiomyocyte apoptosis, and cardiac dysfunction (177). Tanshinone IIA mitigated these adverse changes by inhibiting the TLR4/NF-κB p65/NLRP3 inflammasome signaling pathway, thus enhancing cardiac structure and restoring left ventricular function (177). After MI, treatment with salvianolate ameliorated interstitial fibrosis in the atria, decreased the susceptibility of the heart to atrial fibrillation, and reduced the duration of atrial fibrillation (178). Salvianolate’s cardioprotective effect was attributed to its capacity to reduce collagen deposition and attenuate inflammatory responses by inhibiting the TGF-β1/Smad2/3 and TXNIP/NLRP3 inflammasome signaling pathways (178). Emodin decreased the expression of GSDMD-NT and IL-1β by inhibiting the TLR4/MyD88/NF-κB/NLRP3 inflammasome signaling pathway, thereby mitigating myocardial inflammatory injury induced by MI/R (179). Furthermore, Emodin potentially ameliorated LPS-induced cardiac injury and dysfunction (180). This is primarily achieved by inhibiting the NLRP3 inflammasome, decreasing the levels of inflammatory cytokines, and inducing cardiomyocyte pyroptosis (180). Sodium tanshinone IIA sulfonate has demonstrated potential for the treatment of sepsis-induced myocardial dysfunction (181). In mice with LPS-induced cardiomyopathy, sodium tanshinone IIA sulfonate mitigates myocardial inflammation and enhances cardiac function by promoting autophagy and inhibiting NLRP3 inflammasome activation, leading to increased survival rates (181).
4.5 Phenylpropanoids
Beta-asarone reduces cardiac inflammation and diminishes the size of MI by inhibiting the NLRP3 inflammasome, thus enhancing cardiac recovery after ischemia reperfusion (182). The cardioprotective effect of cinnamaldehyde also depends on its inhibitory effects on the NLRP3 inflammasome (183). Pretreatment with Cinnamaldehyde attenuated cardiomyocyte pyroptosis and the number of TUNEL-positive cells by suppressing the expression of NLRP3, ASC, pro-caspase-1, caspase-1, and GSDMD, as well as the release of IL-18 and IL-1β, thereby alleviating MI/R injury (183). Aesculin also confers protective effects against ischemia reperfusion in the myocardium (184). It hindered the activation of the NF-κB/NLRP3 inflammasome signaling pathway by enhancing Akt and GSK3β expression, leading to reduced cardiac inflammation, enhanced mitochondrial function, reduced cardiomyocyte apoptosis, and decreased vulnerability to ventricular arrhythmias, ultimately enhancing cardiac function (184). Cinnamic acid alleviates MI/R injury by reducing the infarct size, preventing myocardial cell apoptosis, and improving cardiac diastolic function (185). The cardioprotective effects of cinnamic acid have been attributed to the suppression of NLRP3 inflammasome activation-induced pyroptosis (185).
4.6 Other active ingredients
In addition to the aforementioned compounds, gastrodin (186), panaxynol (187), and the ethyl acetate extract of cinnamomi ramulus (188) had been identified as having the potential to ameliorate symptoms and enhance the prognosis of HF by inhibiting the NLRP3 inflammasome. By inhibiting NLRP3/caspase-1 signaling, Gastrodin reduced the production of IL-1β (186). This mechanism alleviates the inflammatory responses in the heart and microvasculature, reduces myocardial apoptosis, and promotes capillary formation, thereby offering protection against myocardial and cardiac microvascular damage induced by MI/R (186). Panaxynol exhibits anti-inflammatory and anti-apoptotic properties (187). It demonstrated beneficial effects in mitigating MI/R injury by suppressing the HMGB1/TLR4/NF-κB/NLRP3 inflammasome signaling pathway, leading to a significant reduction in MI size and enhancement of cardiac function (187). Cinnamomi Ramulus also has the potential to ameliorate adverse cardiac outcomes resulting from MI/R (188). It attenuates cardiac inflammation and enhances cardiac function by reducing NLRP3 inflammasome activation and pyroptosis, thereby exerting cardioprotective effects (188).
5 Conclusions and prospects
The activation of the NLRP3 inflammasome is a complex process involving three distinct pathways: canonical, non-canonical, and alternative NLRP3 inflammasome activation. These activated pathways adversely affect cardiac function by promoting the progression of various pathological processes, including the exacerbation of myocardial inflammatory injury, adverse myocardial fibrosis, pathological myocardial hypertrophy, and abnormal cardiomyocyte apoptosis; inhibition of angiogenesis; and disruption of cardiac electrical signaling and energy metabolism. These factors synergistically accelerate the onset and progression of HF. In the prevention and treatment of HF, the active ingredients of TCM demonstrate significant potential. They inhibit the NLRP3 inflammasome through multiple pathways, effectively attenuating the aforementioned pathological changes and thereby improving both the structure and function of the heart. Furthermore, we found that these active ingredients are primarily concentrated in flavonoids and their glycosides, terpenes and their glycosides, phenolic acids, quinones, and phenylpropanoids. Based on these findings, we posit that there is both theoretical value and clinical significance in reviewing studies on TCM active ingredients for preventing and treating HF, with a focus on the inhibition of the NLRP3 inflammasome. This endeavor aims to lay the foundation for future research and the development of novel therapeutic agents.
Despite some progress in investigating the inhibition of the NLRP3 inflammasome by the active ingredients of TCM for the prevention and treatment of HF, various limitations and challenges persist. First, some studies lack in-depth exploration of the upstream signaling molecules that regulate the NLRP3 inflammasome. It remains unclear which specific signaling pathways the active components of TCM utilize to inhibit the NLRP3 inflammasome. Second, active ingredients of TCM may engage multiple signaling pathways to suppress the NLRP3 inflammasome, such as Astragaloside IV, Luteolin, and Resveratrol. However, there remains a shortage of comprehensive investigations into the interplay between molecular signals. Third, most current research is confined to animal and cell experiments and lacks robust clinical studies that offer evidential support, particularly high-quality randomized controlled trials. Fourth, the etiology of HF is multifaceted and includes myocardial ischemia, cardiac overload, and myocardial toxicity. Further research is necessary to verify whether there are variations in the mechanism of action and the effects of the same active ingredients in TCM on HF with different etiologies. Fifth, although the active ingredients of TCM exhibit minimal side effects and low drug resistance, HF is characterized by a protracted course of illness that may require extended dosing periods. Therefore, the safety profiles of the active ingredients in Chinese medicines require evaluation through meticulously designed clinical studies.
Considering the aforementioned limitations and challenges, future studies should conduct multidimensional validations using HF models derived from various etiological sources both in vitro and in vivo. Simultaneously, emphasis should be placed on exploring upstream signaling molecules that suppress the NLRP3 inflammasome and investigating the interactions among diverse molecular mechanisms. Furthermore, after verifying the efficacy and safety of the active ingredients in TCM in basic research, clinical trials should be conducted to assess the therapeutic potential of these components in preventing and treating HF, thereby enabling the translation of research findings into clinical applications.
Author contributions
RL: Conceptualization, Investigation, Software, Visualization, Writing – original draft. YY: Investigation, Writing – review & editing. LD: Visualization, Writing – review & editing. ZD: Writing – review & editing. ZW: Writing – review & editing. JW: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. ZG: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82174343, 82405372), Key Subject of Scientific Research Projects of Traditional Chinese Medicine in Hunan Province (A2023017), Hunan Provincial Health Commission Research Project (D202303017861), Key Research and Development Plan of Hunan Province (2022SK2012), Changsha Science and Technology Bureau Project (kq2402175), Scientific Research Project of Hunan University of Chinese Medicine (Z2023XJYB05), Graduate Innovation Project of Hunan University of Chinese Medicine (2024CX036, 2024CX008, 2024CX019).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1520482/full#supplementary-material
Glossary
Akt: serine/threonine protein kinase B
AMI: acute myocardial infarction
AMPK: adenosine 5’-monophosphate (AMP)-activated protein kinase
AMVMs: adult mouse ventricular myocytes
Ang II: angiotensin II
ASC: apoptosis speck-like protein containing a caspase recruitment domain
As-IV: Astragaloside IV
α-SMA: α-smooth muscle actin
ATP: adenosine triphosphate
BMDMs: bone marrow-derived macrophages
CARD: caspase activation and recruitment domain
CFs: cardiac fibroblasts
CMECs: cardiac microvascular endothelial cells
H/R: hypoxia/reoxygenation
Cx43: Connexin 43
DCM: dilated cardiomyopathy
DOX: Doxorubicin
ECM: Extracellular matrix
ECs: endothelial cells
GSDMD: gasderminD
GSDMD-NT: GSDMD N-terminal
HCMs: human cardiomyocytes
HF: heart failure
HFpEF: heart failure with preserved ejection fraction
HK1: hexokinase 1
HUVECs: human umbilical vein endothelial cells
IFN‐γ: interferon-gamma
IL: interleukin
ISO: isoproterenol
KI: knock-in
KO: knockout
K+: potassium ion
LADCA: left anterior descending coronary artery
LCA: left coronary artery
LPS: lipopolysaccharide
LRR: leucine rich repeat
MAPK: mitogen-activated protein kinase
MCFs: mouse cardiac fibroblasts
MCP-1: monocyte chemoattractant protein-1
MI: myocardial infarction
MI/R: myocardial ischemia/reperfusion
mtROS: mitochondrial reactive oxygen species
mtDNA: mitochondrial DNA
MyD88: myeloid differentiation primary response protein 88
NF-κB: nuclear factor-κB
NLRP3: NOD-like receptor protein 3
NMCMs: neonatal mouse cardiomyocytes
NMVMs: neonatal mouse ventricular cardiomyocytes
NRCFs: neonatal rat cardiac fibroblasts
NRCMs: neonatal rat cardiomyocytes;NRVMs, neonatal rat ventricular myocytes
OGD/R: oxygen-glucose deprivation/reoxygenation
OS: oxidative stress
pro-caspase-1: caspase-1 precursor
PYD: pyrin domain
P2X7R: P2X7 receptor
RV: right ventricular
SD: sprague dawley
SHRs: spontaneously hypertensive rats
SIRT: silent information regulator of transcription
TAC: transverse aortic constriction
TGF-β: transforming growth factor-β
TLR: toll-like receptor
TNF-α: tumor necrosis factor-α;VAs, ventricular arrhythmias
WKY: Wistar-Kyoto
WT: wild type.
References
2. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano G, Coats A. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. (2023) 118:3272–87. doi: 10.1093/cvr/cvac013
3. Nedkoff L, Weber C. Heart failure: not just a disease of the elderly. Heart. (2022) 108:249–50. doi: 10.1136/heartjnl-2021-320273
4. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. AHA/ACC/HFSA guideline for the management of heart failure: A report of the american college of cardiology/american heart association joint committee on clinical practice guidelines. J Am Coll Cardiol. (2022) 79:e263–421. doi: 10.1016/j.jacc.2021.12.012
5. Cheng X, Zhao H, Wen X, Li G, Guo S, Zhang D. NLRP3-inflammasome inhibition by MCC950 attenuates cardiac and pulmonary artery remodelling in heart failure with preserved ejection fraction. Life Sci. (2023) 333:122185. doi: 10.1016/j.lfs.2023.122185
6. Fu M, Hua X, Shu S, Xu X, Zhang H, Peng Z, et al. Single-cell RNA sequencing in donor and end-stage heart failure patients identifies NLRP3 as a therapeutic target for arrhythmogenic right ventricular cardiomyopathy. BMC Med. (2024) 22:11. doi: 10.1186/s12916-023-03232-8
7. Wang M, Zhao M, Yu J, Xu Y, Zhang J, Liu J, et al. MCC950, a selective NLRP3 inhibitor, attenuates adverse cardiac remodeling following heart failure through improving the cardiometabolic dysfunction in obese mice. Front Cardiovasc Med. (2022) 9:727474. doi: 10.3389/fcvm.2022.727474
8. Wu YX, Xu RY, Jiang L, Chen XY, Xiao XJ. MicroRNA-30a-5p promotes chronic heart failure in rats by targeting sirtuin-1 to activate the nuclear factor-kappaB/NOD-like receptor 3 signaling pathway. Cardiovasc Drugs Ther. (2023) 37:1065–76. doi: 10.1007/s10557-021-07304-w
9. Zeng H, Chen JX. Microvascular rarefaction and heart failure with preserved ejection fraction. Front Cardiovasc Med. (2019) 6:15. doi: 10.3389/fcvm.2019.00015
10. Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C, et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. (2014) 109:415. doi: 10.1007/s00395-014-0415-z
11. Fan Y, Li C, Fu L, Yuan P, Ge M. To investigate the correlation of serum NLRP3 level with endothelial function, severity of disease and prognosis in elderly patients with chronic heart failure. Minerva Med. (2024) 115:1–3. doi: 10.23736/S0026-4806.24.09322-4
12. Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. (2007) 19:615–22. doi: 10.1016/j.coi.2007.09.002
13. Wang A, Zhao W, Yan K, Huang P, Zhang H, Zhang Z, et al. Mechanisms and efficacy of traditional chinese medicine in heart failure. Front Pharmacol. (2022) 13:810587. doi: 10.3389/fphar.2022.810587
14. Fan Y, Yang Z, Wang L, Liu Y, Song Y, Liu Y, et al. Traditional Chinese medicine for heart failure with preserved ejection fraction: clinical evidence and potential mechanisms. Front Pharmacol. (2023) 14:1154167. doi: 10.3389/fphar.2023.1154167
15. Fang HY, Zhao XN, Zhang M, Ma YY, Huang JL, Zhou P. Beneficial effects of flavonoids on cardiovascular diseases by influencing NLRP3 inflammasome. Inflammopharmacology. (2023) 31:1715–29. doi: 10.1007/s10787-023-01249-2
16. Tan W, Cheng S, Qiu Q, Huang J, Xie M, Song L, et al. Celastrol exerts antiarrhythmic effects in chronic heart failure via NLRP3/Caspase-1/IL-1beta signaling pathway. BioMed Pharmacother. (2024) 177:117121. doi: 10.1016/j.biopha.2024.117121
17. Wang X, Chen X, Wang Y, He X, Li L, Wang X, et al. Astragaloside IV alleviates inflammation and improves myocardial metabolism in heart failure mice with preserved ejection fraction. Front Pharmacol. (2024) 15:1467132. doi: 10.3389/fphar.2024.1467132
18. Xu H, Chen J, Chen P, Li W, Shao J, Hong S, et al. Costunolide covalently targets NACHT domain of NLRP3 to inhibit inflammasome activation and alleviate NLRP3-driven inflammatory diseases. Acta Pharm Sin B. (2023) 13:678–93. doi: 10.1016/j.apsb.2022.09.014
19. Rahman T, Nagar A, Duffy EB, Okuda K, Silverman N, Harton JA. NLRP3 sensing of diverse inflammatory stimuli requires distinct structural features. Front Immunol. (2020) 11:1828. doi: 10.3389/fimmu.2020.01828
20. Hochheiser IV, Behrmann H, Hagelueken G, Rodriguez-Alcazar JF, Kopp A, Latz E, et al. Directionality of PYD filament growth determined by the transition of NLRP3 nucleation seeds to ASC elongation. Sci Adv. (2022) 8:eabn7583. doi: 10.1126/sciadv.abn7583
21. Nambayan R, Sandin SI, Quint DA, Satyadi DM, de Alba E. The inflammasome adapter ASC assembles into filaments with integral participation of its two Death Domains. J Biol Chem. (2019) 294:439–52. doi: 10.1074/jbc.RA118.004407
22. Segovia JA, Tsai SY, Chang TH, Shil NK, Weintraub ST, Short JD, et al. Nedd8 regulates inflammasome-dependent caspase-1 activation. Mol Cell Biol. (2015) 35:582–97. doi: 10.1128/MCB.00775-14
23. Seok JK, Kang HC, Cho YY, Lee HS, Lee JY. Regulation of the NLRP3 inflammasome by post-translational modifications and small molecules. Front Immunol. (2020) 11:618231. doi: 10.3389/fimmu.2020.618231
24. Toldo S, Mezzaroma E, McGeough MD, Pena CA, Marchetti C, Sonnino C, et al. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc Res. (2015) 105:203–12. doi: 10.1093/cvr/cvu259
25. Li Y, Xia Y, Yin S, Wan F, Hu J, Kou L, et al. Targeting microglial alpha-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in parkinson's disease. Front Immunol. (2021) 12:719807. doi: 10.3389/fimmu.2021.719807
26. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J Am Coll Cardiol. (2018) 71:875–86. doi: 10.1016/j.jacc.2017.12.037
27. McGeough MD, Wree A, Inzaugarat ME, Haimovich A, Johnson CD, Pena CA, et al. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J Clin Invest. (2017) 127:4488–97. doi: 10.1172/JCI90699
28. Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell. (2017) 68:185–97. doi: 10.1016/j.molcel.2017.08.017
29. Xu T, Yu W, Fang H, Wang Z, Chi Z, Guo X, et al. Ubiquitination of NLRP3 by gp78/Insig-1 restrains NLRP3 inflammasome activation. Cell Death Differ. (2022) 29:1582–95. doi: 10.1038/s41418-022-00947-8
30. Kim H, Kwon S, Sun X, Akther M, Han J, Kim T, et al. Vanillylacetone attenuates NLRP3 inflammasome mediated immune responses in murine bone marrow derived macrophages via NLRP3 alkylation. J Funct Foods. (2020) 64:103655. doi: 10.1016/j.jff.2019.103655
31. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol. (2013) 14:52–60. doi: 10.1038/ni.2474
32. Zhang L, Gai Y, Liu Y, Meng D, Zeng Y, Luo Y, et al. Tau induces inflammasome activation and microgliosis through acetylating NLRP3. Clin Transl Med. (2024) 14:e1623. doi: 10.1002/ctm2.1623
33. Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun. (2018) 9:3001. doi: 10.1038/s41467-018-05321-2
34. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
35. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. (2017) 8:202. doi: 10.1038/s41467-017-00227-x
36. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–25. doi: 10.1038/nature09663
37. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
38. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U.S.A. (2012) 109:11282–87. doi: 10.1073/pnas.1117765109
39. Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. (2018) 564:71–6. doi: 10.1038/s41586-018-0761-3
40. Wang Y, Jia L, Shen J, Wang Y, Fu Z, Su SA, et al. Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PloS Pathog. (2018) 14:e1006872. doi: 10.1371/journal.ppat.1006872
41. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
42. Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med. (2018) 215:2279–88. doi: 10.1084/jem.20180589
43. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–65. doi: 10.1038/nature15514
44. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
45. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
46. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol. (2015) 45:2911–17. doi: 10.1002/eji.201545523
47. Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, et al. Shiga toxin/lipopolysaccharide activates caspase-4 and gasdermin D to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep. (2018) 25:1525–36. doi: 10.1016/j.celrep.2018.09.071
48. Zhu F, Ma J, Li W, Liu Q, Qin X, Qian Y, et al. The orphan receptor Nur77 binds cytoplasmic LPS to activate the non-canonical NLRP3 inflammasome. Immunity. (2023) 56:753–67. doi: 10.1016/j.immuni.2023.03.003
49. Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. (2015) 45:2927–36. doi: 10.1002/eji.201545772
50. Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
51. Moretti J, Jia B, Hutchins Z, Roy S, Yip H, Wu J, et al. Caspase-11 interaction with NLRP3 potentiates the noncanonical activation of the NLRP3 inflammasome. Nat Immunol. (2022) 23:705–17. doi: 10.1038/s41590-022-01192-4
52. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. (2016) 352:1232–36. doi: 10.1126/science.aaf3036
53. Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun. (2018) 9:996. doi: 10.1038/s41467-018-03409-3
54. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
55. Verma D, Fekri SZ, Sigurdardottir G, Bivik EC, Sandin C, Enerback C. Enhanced inflammasome activity in patients with psoriasis promotes systemic inflammation. J Invest Dermatol. (2021) 141:586–95. doi: 10.1016/j.jid.2020.07.012
56. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. (2020) 21:30–41. doi: 10.1038/s41590-019-0548-1
57. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11:255–65. doi: 10.1038/nrcardio.2014.28
58. Westman PC, Lipinski MJ, Luger D, Waksman R, Bonow RO, Wu E, et al. Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. (2016) 67:2050–60. doi: 10.1016/j.jacc.2016.01.073
59. Lan T, Zeng Q, Zhu Y, Zheng G, Chen K, Jiang W, et al. Xin-Li formula attenuates heart failure induced by a combination of hyperlipidemia and myocardial infarction in rats via Treg immunomodulation and NLRP3 inflammasome inhibition. J Tradit Complement Med. (2023) 13:441–53. doi: 10.1016/j.jtcme.2023.03.009
60. Marunouchi T, Iguchi A, Shindo A, Shimbo N, Yano E, Tanonaka K. Involvement of Hsp90 in NLRP3 inflammasome activation in the failing heart following myocardial infarction in rats. Biochem Pharmacol. (2023) 212:115547. doi: 10.1016/j.bcp.2023.115547
61. Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U.S.A. (2011) 108:19725–30. doi: 10.1073/pnas.1108586108
62. Chen F, Chen ZQ, Zhong GL, Zhu JJ. Nicorandil inhibits TLR4/MyD88/NF-kappaB/NLRP3 signaling pathway to reduce pyroptosis in rats with myocardial infarction. Exp Biol Med (Maywood). (2021) 246:1938–47. doi: 10.1177/15353702211013444
63. Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol. (2016) 209:215–20. doi: 10.1016/j.ijcard.2016.02.043
64. Dai Y, Wang S, Chang S, Ren D, Shali S, Li C, et al. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-kappaB/NLRP3 inflammasome signaling pathway. J Mol Cell Cardiol. (2020) 142:65–79. doi: 10.1016/j.yjmcc.2020.02.007
65. Zhang J, Huang L, Shi X, Yang L, Hua F, Ma J, et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging (Albany Ny). (2020) 12:24270–87. doi: 10.18632/aging.202143
66. Han Y, Sun W, Ren D, Zhang J, He Z, Fedorova J, et al. SIRT1 agonism modulates cardiac NLRP3 inflammasome through pyruvate dehydrogenase during ischemia and reperfusion. Redox Biol. (2020) 34:101538. doi: 10.1016/j.redox.2020.101538
67. Liu S, Bi Y, Han T, Li YE, Wang Q, Wu NN, et al. The E3 ubiquitin ligase MARCH2 protects against myocardial ischemia-reperfusion injury through inhibiting pyroptosis via negative regulation of PGAM5/MAVS/NLRP3 axis. Cell Discovery. (2024) 10:24. doi: 10.1038/s41421-023-00622-3
68. Zhang L, Li Y, Fan CD, Jiang YH, Sheng LS, Song XY, et al. Chinese medicinal formula Fu Xin decoction against chronic heart failure by inhibiting the NLRP3/caspase-1/GSDMD pyroptotic pathway. BioMed Pharmacother. (2024) 174:116548. doi: 10.1016/j.biopha.2024.116548
69. Zhou J, Wu C, Zhao M. TAOK1-mediated regulation of the YAP/TEAD pathway as a potential therapeutic target in heart failure. PloS One. (2024) 19:e308619. doi: 10.1371/journal.pone.0308619
70. Ping Z, Fangfang T, Yuliang Z, Xinyong C, Lang H, Fan H, et al. Oxidative stress and pyroptosis in doxorubicin-induced heart failure and atrial fibrillation. Oxid Med Cell Longev. (2023) 2023:4938287. doi: 10.1155/2023/4938287
71. Tu W, Huang X, Liu S, Zhan Y, Cai X, Shao L. The m(6)A demethylase fat mass and obesity-associated protein mitigates pyroptosis and inflammation in doxorubicin-induced heart failure via the toll-like receptor 4/NF-kappaB pathway. Cardiovasc Diagn Ther. (2024) 14:158–73. doi: 10.21037/cdt-23-326
72. Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. (2020) 34:101523. doi: 10.1016/j.redox.2020.101523
73. Lafuse WP, Wozniak DJ, Rajaram M. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells. (2020) 10:51. doi: 10.3390/cells10010051
74. Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, Darwesh AM, et al. Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (Nucleotide-binding domain-like receptor protein 3) inflammasome activation in heart failure. Circ Heart Fail. (2020) 13:e6277. doi: 10.1161/CIRCHEARTFAILURE.119.006277
75. Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by ca(2+)/calmodulin-dependent protein kinase II delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation. (2018) 138:2530–44. doi: 10.1161/CIRCULATIONAHA.118.034621
76. Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller BJ. CaMKIIdelta-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight. (2018) 3:e97054. doi: 10.1172/jci.insight.97054
77. Zhao M, Zheng Z, Zhang P, Xu Y, Zhang J, Peng S, et al. IL-30 protects against sepsis-induced myocardial dysfunction by inhibiting pro-inflammatory macrophage polarization and pyroptosis. Iscience. (2023) 26:107544. doi: 10.1016/j.isci.2023.107544
78. Al-Qazazi R, Lima P, Prisco SZ, Potus F, Dasgupta A, Chen KH, et al. Macrophage-NLRP3 activation promotes right ventricle failure in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2022) 206:608–24. doi: 10.1164/rccm.202110-2274OC
79. Guo L, Qin G, Cao Y, Yang Y, Dai S, Wang L, et al. Regulation of the immune microenvironment by an NLRP3 inhibitor contributes to attenuation of acute right ventricular failure in rats with pulmonary arterial hypertension. J Inflammation Res. (2021) 14:5699–711. doi: 10.2147/JIR.S336964
80. Su Y, Shi D, Xia G, Liu Y, Xu L, Dao L, et al. Carbonic Anhydrase 3 is required for cardiac repair post myocardial infarction via Smad7-Smad2/3 signaling pathway. Int J Biol Sci. (2024) 20:1796–814. doi: 10.7150/ijbs.91396
81. Gulati A, Japp AG, Raza S, Halliday BP, Jones DA, Newsome S, et al. Absence of myocardial fibrosis predicts favorable long-term survival in new-onset heart failure. Circ Cardiovasc Imaging. (2018) 11:e7722. doi: 10.1161/CIRCIMAGING.118.007722
82. Roy C, Slimani A, de Meester C, Amzulescu M, Pasquet A, Vancraeynest D, et al. Associations and prognostic significance of diffuse myocardial fibrosis by cardiovascular magnetic resonance in heart failure with preserved ejection fraction. J Cardiovasc Magn Reson. (2018) 20:55. doi: 10.1186/s12968-018-0477-4
83. Liu M, Lopez DJAB, Cheng K. Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies. Adv Drug Delivery Rev. (2021) 173:504–19. doi: 10.1016/j.addr.2021.03.021
84. Lv SL, Zeng ZF, Gan WQ, Wang WQ, Li TG, Hou YF, et al. Lp-PLA2 inhibition prevents Ang II-induced cardiac inflammation and fibrosis by blocking macrophage NLRP3 inflammasome activation. Acta Pharmacol Sin. (2021) 42:2016–32. doi: 10.1038/s41401-021-00703-7
85. Gan W, Ren J, Li T, Lv S, Li C, Liu Z, et al. The SGK1 inhibitor EMD638683, prevents Angiotensin II-induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1–10. doi: 10.1016/j.bbadis.2017.10.001
86. Gao R, Shi H, Chang S, Gao Y, Li X, Lv C, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunopharmacol. (2019) 74:105575. doi: 10.1016/j.intimp.2019.04.022
87. Zhang X, Qu H, Yang T, Liu Q, Zhao D, Liu W, et al. LuQi formula ameliorates myocardial fibrosis by suppressing TLR4/myD88/NF-kappaB pathway and NLRP3 inflammasome activation in mice with myocardial infarction. Evid Based Complement Alternat Med. (2022) 2022:5867987. doi: 10.1155/2022/5867987
88. Liu W, Sun J, Guo Y, Liu N, Ding X, Zhang X, et al. Calhex231 ameliorates myocardial fibrosis post myocardial infarction in rats through the autophagy-NLRP3 inflammasome pathway in macrophages. J Cell Mol Med. (2020) 24:13440–53. doi: 10.1111/jcmm.15969
89. Shen S, Duan J, Hu J, Qi Y, Kang L, Wang K, et al. Colchicine alleviates inflammation and improves diastolic dysfunction in heart failure rats with preserved ejection fraction. Eur J Pharmacol. (2022) 929:175126. doi: 10.1016/j.ejphar.2022.175126
90. Li X, Zhu Q, Wang Q, Zhang Q, Zheng Y, Wang L, et al. Protection of Sacubitril/Valsartan against Pathological Cardiac Remodeling by Inhibiting the NLRP3 Inflammasome after Relief of Pressure Overload in Mice. Cardiovasc Drugs Ther. (2020) 34:629–40. doi: 10.1007/s10557-020-06995-x
91. Zhang LS, Zhang JS, Hou YL, Lu WW, Ni XQ, Lin F, et al. Intermedin(1-53) inhibits NLRP3 inflammasome activation by targeting IRE1alpha in cardiac fibrosis. Inflammation. (2022) 45:1568–84. doi: 10.1007/s10753-022-01642-z
92. Liu N, Gong Z, Li Y, Xu Y, Guo Y, Chen W, et al. CTRP3 inhibits myocardial fibrosis through the P2X7R-NLRP3 inflammasome pathway in SHR rats. J Hypertens. (2024) 42:315–28. doi: 10.1097/HJH.0000000000003591
93. Li S, Withaar C, Rodrigues PG, Zijlstra SN, de Boer RA, Sillje H, et al. The NLRP3-inflammasome inhibitor MCC950 improves cardiac function in a HFpEF mouse model. BioMed Pharmacother. (2024) 181:117711. doi: 10.1016/j.biopha.2024.117711
94. Zhao M, Zhang J, Xu Y, Liu J, Ye J, Wang Z, et al. Selective inhibition of NLRP3 inflammasome reverses pressure overload-induced pathological cardiac remodeling by attenuating hypertrophy, fibrosis, and inflammation. Int Immunopharmacol. (2021) 99:108046. doi: 10.1016/j.intimp.2021.108046
95. Wei XH, Liu WJ, Jiang W, Lan TH, Pan H, Ma MY, et al. XinLi formula, a traditional Chinese decoction, alleviates chronic heart failure via regulating the interaction of AGTR1 and AQP1. Phytomedicine. (2023) 113:154722. doi: 10.1016/j.phymed.2023.154722
96. Caceres FT, Gaspari TA, Samuel CS, Pinar AA. Serelaxin inhibits the profibrotic TGF-beta1/IL-1beta axis by targeting TLR-4 and the NLRP3 inflammasome in cardiac myofibroblasts. FASEB J. (2019) 33:14717–33. doi: 10.1096/fj.201901079RR
97. Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JJ, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem. (2014) 289:19571–84. doi: 10.1074/jbc.M114.550624
98. Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. (2014) 114:565–71. doi: 10.1161/CIRCRESAHA.114.300507
99. Duan F, Li H, Lu B, Wang X, Xu X. Loss of trim31 worsens cardiac remodeling in a mouse model of heart failure by enhancing the activation of the NLRP3 inflammasome. Inflammation. (2024) 47:1–13. doi: 10.1007/s10753-024-02217-w
100. Schlegel P, Reinkober J, Meinhardt E, Tscheschner H, Gao E, Schumacher SM, et al. G protein-coupled receptor kinase 2 promotes cardiac hypertrophy. PloS One. (2017) 12:e182110. doi: 10.1371/journal.pone.0182110
101. Sun X, Zhou M, Wen G, Huang Y, Wu J, Peng L, et al. Paroxetine attenuates cardiac hypertrophy via blocking GRK2 and ADRB1 interaction in hypertension. J Am Heart Assoc. (2021) 10:e16364. doi: 10.1161/JAHA.120.016364
102. Liu J, Li X, Ding L, Li W, Niu X, Gao D. GRK2 participation in cardiac hypertrophy induced by isoproterenol through the regulation of Nrf2 signaling and the promotion of NLRP3 inflammasome and oxidative stress. Int Immunopharmacol. (2023) 117:109957. doi: 10.1016/j.intimp.2023.109957
103. Lim S, Lee ME, Jeong J, Lee J, Cho S, Seo M, et al. sRAGE attenuates angiotensin II-induced cardiomyocyte hypertrophy by inhibiting RAGE-NFkappaB-NLRP3 activation. Inflammation Res. (2018) 67:691–701. doi: 10.1007/s00011-018-1160-9
104. Zhang L, Wang H, Li W. PRMT5 up-regulation improves myocardial hypertrophy by mediating E2F-1/NF-kappaB/NLRP3 pathway. Prev Med. (2023) 172:107553. doi: 10.1016/j.ypmed.2023.107553
105. Wu B, Xu C, Xu C, Qiu L, Gao JX, Li M, et al. Inhibition of Sema4D attenuates pressure overload-induced pathological myocardial hypertrophy via the MAPK/NF-kappaB/NLRP3 pathways. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:166944. doi: 10.1016/j.bbadis.2023.166944
106. Wang F, Liang Q, Ma Y, Sun M, Li T, Lin L, et al. Silica nanoparticles induce pyroptosis and cardiac hypertrophy via ROS/NLRP3/Caspase-1 pathway. Free Radic Biol Med. (2022) 182:171–81. doi: 10.1016/j.freeradbiomed.2022.02.027
107. Tang X, Pan L, Zhao S, Dai F, Chao M, Jiang H, et al. SNO-MLP (S-nitrosylation of muscle LIM protein) facilitates myocardial hypertrophy through TLR3 (Toll-like receptor 3)-mediated RIP3 (Receptor-interacting protein kinase 3) and NLRP3 (NOD-like receptor pyrin domain containing 3) inflammasome activation. Circulation. (2020) 141:984–1000. doi: 10.1161/CIRCULATIONAHA.119.042336
108. Ferraro B, Leoni G, Hinkel R, Ormanns S, Paulin N, Ortega-Gomez A, et al. Pro-angiogenic macrophage phenotype to promote myocardial repair. J Am Coll Cardiol. (2019) 73:2990–3002. doi: 10.1016/j.jacc.2019.03.503
109. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. (2005) 115:2108–18. doi: 10.1172/JCI24682
110. Feng YH, Li LF, Zhang Q, Zhang JH, Huang Y, Lv YL, et al. Microtubule associated protein 4 (MAP4) phosphorylation reduces cardiac microvascular density through NLRP3-related pyroptosis. Cell Death Discovery. (2021) 7:213. doi: 10.1038/s41420-021-00606-w
111. Zhou T, Xiang DK, Li SN, Yang LH, Gao LF, Feng C. MicroRNA-495 ameliorates cardiac microvascular endothelial cell injury and inflammatory reaction by suppressing the NLRP3 inflammasome signaling pathway. Cell Physiol Biochem. (2018) 49:798–815. doi: 10.1159/000493042
112. He X, Zeng H, Chen ST, Roman RJ, Aschner JL, Didion S, et al. Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J Mol Cell Cardiol. (2017) 112:104–13. doi: 10.1016/j.yjmcc.2017.09.007
113. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. (2017) 6:e006347. doi: 10.1161/JAHA.117.006347
114. Cho JH, Zhang R, Aynaszyan S, Holm K, Goldhaber JI, Marban E, et al. Ventricular arrhythmias underlie sudden death in rats with heart failure and preserved ejection fraction. Circ Arrhythm Electrophysiol. (2018) 11:e6452. doi: 10.1161/CIRCEP.118.006452
115. Jiang X, Yang F, Ou D, Huang L, Li H, Lang M. MCC950 ameliorates ventricular arrhythmia vulnerability induced by heart failure. Bioengineered. (2022) 13:8593–604. doi: 10.1080/21655979.2022.2053813
116. Liang Y, Wang B, Huang H, Wang M, Wu Q, Zhao Y, et al. Silenced SOX2-OT alleviates ventricular arrhythmia associated with heart failure by inhibiting NLRP3 expression via regulating miR-2355-3p. Immun Inflammation Dis. (2021) 9:255–64. doi: 10.1002/iid3.388
117. Yang HJ, Kong B, Shuai W, Zhang JJ, Huang H. Knockout of MD1 contributes to sympathetic hyperactivity and exacerbates ventricular arrhythmias following heart failure with preserved ejection fraction via NLRP3 inflammasome activation. Exp Physiol. (2020) 105:966–78. doi: 10.1113/EP088390
118. Yang H, Hu Y, Kong B, Zhou Y, Shuai W. Low-intensity pulsed ultrasound treatment mitigates ventricular arrhythmias via inhibiting microglia-mediated neuroinflammation in heart failure rat model. Int Immunopharmacol. (2024) 126:111317. doi: 10.1016/j.intimp.2023.111317
119. Yang H, Zhu J, Fu H, Shuai W. Dapansutrile ameliorates atrial inflammation and vulnerability to atrial fibrillation in HFpEF rats. Heart Lung Circ. (2024) 33:65–77. doi: 10.1016/j.hlc.2023.09.017
120. Yin J, Wang Y, Hu H, Li X, Xue M, Cheng W, et al. P2X(7) receptor inhibition attenuated sympathetic nerve sprouting after myocardial infarction via the NLRP3/IL-1beta pathway. J Cell Mol Med. (2017) 21:2695–710. doi: 10.1111/jcmm.13185
121. Wei Z, Fei Y, Wang Q, Hou J, Cai X, Yang Y, et al. Loss of Camk2n1 aggravates cardiac remodeling and Malignant ventricular arrhythmia after myocardial infarction in mice via NLRP3 inflammasome activation. Free Radic Biol Med. (2021) 167:243–57. doi: 10.1016/j.freeradbiomed.2021.03.014
122. Li B, Xu L, Liu J, Zhou M, Jiang X. Phloretin ameliorates heart function after myocardial infarction via NLRP3/Caspase-1/IL-1beta signaling. BioMed Pharmacother. (2023) 165:115083. doi: 10.1016/j.biopha.2023.115083
123. Yang S, Bi Y, Wei Y, Li W, Liu J, Mao T, et al. Muscone attenuates susceptibility to ventricular arrhythmia by inhibiting NLRP3 inflammasome activation in rats after myocardial infarction. J Biochem Mol Toxicol. (2023) 37:e23458. doi: 10.1002/jbt.23458
124. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. (2021) 128:1487–513. doi: 10.1161/CIRCRESAHA.121.318241
125. Chen Y, Zeng M, Zhang Y, Guo H, Ding W, Sun T. Nlrp3 deficiency alleviates angiotensin II-induced cardiomyopathy by inhibiting mitochondrial dysfunction. Oxid Med Cell Longev. (2021) 2021:6679100. doi: 10.1155/2021/6679100
126. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PloS One. (2014) 9:e104771. doi: 10.1371/journal.pone.0104771
127. Mastrocola R, Penna C, Tullio F, Femmino S, Nigro D, Chiazza F, et al. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid Med Cell Longev. (2016) 2016:5271251. doi: 10.1155/2016/5271251
128. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26:239–57. doi: 10.1038/bjc.1972.33
129. Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. (2020) 588:466–72. doi: 10.1038/s41586-020-2797-4
130. Chunhacha P, Pinkaew D, Sinthujaroen P, Bowles DE, Fujise K. Fortilin inhibits p53, halts cardiomyocyte apoptosis, and protects the heart against heart failure. Cell Death Discovery. (2021) 7:310. doi: 10.1038/s41420-021-00692-w
131. Wu QQ, Yao Q, Hu TT, Wan Y, Xie QW, Zhao JH, et al. Tax1 banding protein 1 exacerbates heart failure in mice by activating ITCH-P73-BNIP3-mediated cardiomyocyte apoptosis. Acta Pharmacol Sin. (2022) 43:2562–72. doi: 10.1038/s41401-022-00950-2
132. Ren Z, Yang K, Zhao M, Liu W, Zhang X, Chi J, et al. Calcium-sensing receptor on neutrophil promotes myocardial apoptosis and fibrosis after acute myocardial infarction via NLRP3 inflammasome activation. Can J Cardiol. (2020) 36:893–905. doi: 10.1016/j.cjca.2019.09.026
133. Li X, Long J, Zong L, Zhang C, Yang Z, Guo S. ZNF561-AS1 regulates cell proliferation and apoptosis in myocardial infarction through miR-223-3p/NLRP3 axis. Cell Transplant. (2022) 31:73831512. doi: 10.1177/09636897221077928
134. Zhang H, Huang C, Zhang D, Zhu Y. Pioglitazone protects against hypoxia-induced cardiomyocyte apoptosis through inhibiting NLRP3/caspase-1 pathway in vivo and in vitro. Int Heart J. (2022) 63:893–903. doi: 10.1536/ihj.21-404
135. Liang H, Li F, Li H, Wang R, Du M. Overexpression of lncRNA HULC Attenuates Myocardial Ischemia/reperfusion Injury in Rat Models and Apoptosis of Hypoxia/reoxygenation Cardiomyocytes via Targeting miR-377-5p through NLRP3/Caspase−1/IL−1beta Signaling Pathway Inhibition. Immunol Invest. (2021) 50:925–38. doi: 10.1080/08820139.2020.1791178
136. Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. (2019) 24:101215. doi: 10.1016/j.redox.2019.101215
137. Zhang X, Qu H, Yang T, Liu Q, Zhou H. Astragaloside IV attenuate MI-induced myocardial fibrosis and cardiac remodeling by inhibiting ROS/caspase-1/GSDMD signaling pathway. Cell Cycle. (2022) 21:2309–22. doi: 10.1080/15384101.2022.2093598
138. Zhang T, Wang H, Lu M, Zhao K, Yin J, Liu Y, et al. Astragaloside IV prevents myocardial hypertrophy induced by mechanical stress by activating autophagy and reducing inflammation. Am J Transl Res. (2020) 12:5332–42.
139. Tian W, Zhang P, Yang L, Song P, Zhao J, Wang H, et al. Astragaloside IV alleviates doxorubicin-induced cardiotoxicity by inhibiting cardiomyocyte pyroptosis through the SIRT1/NLRP3 pathway. Am J Chin Med. (2024) 52:453–69. doi: 10.1142/S0192415X24500198
140. Xu LJ, Chen RC, Ma XY, Zhu Y, Sun GB, Sun XB. Scutellarin protects against myocardial ischemia-reperfusion injury by suppressing NLRP3 inflammasome activation. Phytomedicine. (2020) 68:153169. doi: 10.1016/j.phymed.2020.153169
141. Guo F, Han X, You Y, Xu SJ, Zhang YH, Chen YY, et al. Hydroxysafflor yellow A inhibits pyroptosis and protecting HUVECs from OGD/R via NLRP3/caspase-1/GSDMD pathway. Chin J Integr Med. (2024) 30:1027–34. doi: 10.1007/s11655-023-3716-y
142. Ye JX, Wang M, Wang RY, Liu HT, Qi YD, Fu JH, et al. Hydroxysafflor yellow A inhibits hypoxia/reoxygenation-induced cardiomyocyte injury via regulating the AMPK/NLRP3 inflammasome pathway. Int Immunopharmacol. (2020) 82:106316. doi: 10.1016/j.intimp.2020.106316
143. Ye J, Lu S, Wang M, Ge W, Liu H, Qi Y, et al. Hydroxysafflor yellow A protects against myocardial ischemia/reperfusion injury via suppressing NLRP3 inflammasome and activating autophagy. Front Pharmacol. (2020) 11:1170. doi: 10.3389/fphar.2020.01170
144. Wang DS, Yan LY, Yang DZ, Lyu Y, Fang LH, Wang SB, et al. Formononetin ameliorates myocardial ischemia/reperfusion injury in rats by suppressing the ROS-TXNIP-NLRP3 pathway. Biochem Biophys Res Commun. (2020) 525:759–66. doi: 10.1016/j.bbrc.2020.02.147
145. Zhang X, Du Q, Yang Y, Wang J, Dou S, Liu C, et al. The protective effect of Luteolin on myocardial ischemia/reperfusion (I/R) injury through TLR4/NF-kappaB/NLRP3 inflammasome pathway. BioMed Pharmacother. (2017) 91:1042–52. doi: 10.1016/j.biopha.2017.05.033
146. Zhao L, Zhou Z, Zhu C, Fu Z, Yu D. Luteolin alleviates myocardial ischemia reperfusion injury in rats via Siti1/NLRP3/NF-kappaB pathway. Int Immunopharmacol. (2020) 85:106680. doi: 10.1016/j.intimp.2020.106680
147. Bai Y, Li Z, Liu W, Gao D, Liu M, Zhang P. Biochanin A attenuates myocardial ischemia/reperfusion injury through the TLR4/NF-kappaB/NLRP3 signaling pathway. Acta Cir Bras. (2019) 34:e201901104. doi: 10.1590/s0102-865020190110000004
148. Yue R, Zheng Z, Luo Y, Wang X, Lv M, Qin D, et al. NLRP3-mediated pyroptosis aggravates pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction in mice: cardioprotective role of irisin. Cell Death Discovery. (2021) 7:50. doi: 10.1038/s41420-021-00434-y
149. Fang G, Li X, Yang F, Huang T, Qiu C, Peng K, et al. Amentoflavone mitigates doxorubicin-induced cardiotoxicity by suppressing cardiomyocyte pyroptosis and inflammation through inhibition of the STING/NLRP3 signalling pathway. Phytomedicine. (2023) 117:154922. doi: 10.1016/j.phymed.2023.154922
150. Zhai J, Tao L, Zhang S, Gao H, Zhang Y, Sun J, et al. Calycosin ameliorates doxorubicin-induced cardiotoxicity by suppressing oxidative stress and inflammation via the sirtuin 1-NOD-like receptor protein 3 pathway. Phytother Res. (2020) 34:649–59. doi: 10.1002/ptr.6557
151. Li F, Zhu H, Chang Z, Li Y. Gentiopicroside alleviates acute myocardial infarction injury in rats by disrupting Nrf2/NLRP3 signaling. Exp Biol Med (Maywood). (2023) 248:1254–66. doi: 10.1177/15353702231199076
152. Fan J, Ren M, Chen W, Wang H, He Y. Celastrol relieves myocardial infarction-induced cardiac fibrosis by inhibiting NLRP3 inflammasomes in rats. Int Immunopharmacol. (2023) 121:110511. doi: 10.1016/j.intimp.2023.110511
153. Du Y, Gu X, Meng H, Aa N, Liu S, Peng C, et al. Muscone improves cardiac function in mice after myocardial infarction by alleviating cardiac macrophage-mediated chronic inflammation through inhibition of NF-kappaB and NLRP3 inflammasome. Am J Transl Res. (2018) 10:4235–46.
154. Gao RF, Li X, Xiang HY, Yang H, Lv CY, Sun XL, et al. The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int Immunopharmacol. (2021) 90:107133. doi: 10.1016/j.intimp.2020.107133
155. Lu C, Chen C, Chen A, Wu Y, Wen J, Huang F, et al. Oridonin attenuates myocardial ischemia/reperfusion injury via downregulating oxidative stress and NLRP3 inflammasome pathway in mice. Evid Based Complement Alternat Med. (2020) 2020:7395187. doi: 10.1155/2020/7395187
156. Wang D, Yu X, Gao K, Li F, Li X, Pu H, et al. Sweroside alleviates pressure overload-induced heart failure through targeting CaMKIIdelta to inhibit ROS-mediated NF-kappaB/NLRP3 in cardiomyocytes. Redox Biol. (2024) 74:103223. doi: 10.1016/j.redox.2024.103223
157. Li J, Zhao C, Zhu Q, Wang Y, Li G, Li X, et al. Sweroside protects against myocardial ischemia-reperfusion injury by inhibiting oxidative stress and pyroptosis partially via modulation of the keap1/nrf2 axis. Front Cardiovasc Med. (2021) 8:650368. doi: 10.3389/fcvm.2021.650368
158. Li H, Yang DH, Zhang Y, Zheng F, Gao F, Sun J, et al. Geniposide suppresses NLRP3 inflammasome-mediated pyroptosis via the AMPK signaling pathway to mitigate myocardial ischemia/reperfusion injury. Chin Med. (2022) 17:73. doi: 10.1186/s13020-022-00616-5
159. Li W, Fan P, Wang X, Tang H. Loganin alleviates myocardial ischemia-reperfusion injury through GLP-1R/NLRP3-mediated pyroptosis pathway. Environ Toxicol. (2023) 38:2730–40. doi: 10.1002/tox.23908
160. Wang F, Gao Q, Yang J, Wang C, Cao J, Sun J, et al. Artemisinin suppresses myocardial ischemia-reperfusion injury via NLRP3 inflammasome mechanism. Mol Cell Biochem. (2020) 474:171–80. doi: 10.1007/s11010-020-03842-3
161. Yu C, Cai X, Liu X, Liu J, Zhu N. Betulin alleviates myocardial ischemia-reperfusion injury in rats via regulating the siti1/NLRP3/NF-kappaB signaling pathway. Inflammation. (2021) 44:1096–107. doi: 10.1007/s10753-020-01405-8
162. Li R, Lu K, Wang Y, Chen M, Zhang F, Shen H, et al. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem Biophys Res Commun. (2017) 485:69–75. doi: 10.1016/j.bbrc.2017.02.021
163. Pan XC, Liu Y, Cen YY, Xiong YL, Li JM, Ding YY, et al. Dual role of triptolide in interrupting the NLRP3 inflammasome pathway to attenuate cardiac fibrosis. Int J Mol Sci. (2019) 20:360. doi: 10.3390/ijms20020360
164. Ren B, Feng J, Yang N, Guo Y, Chen C, Qin Q. Ginsenoside Rg3 attenuates angiotensin II-induced myocardial hypertrophy through repressing NLRP3 inflammasome and oxidative stress via modulating SIRT1/NF-kappaB pathway. Int Immunopharmacol. (2021) 98:107841. doi: 10.1016/j.intimp.2021.107841
165. Pu X, Zhang Q, Liu J, Wang Y, Guan X, Wu Q, et al. Ginsenoside Rb1 ameliorates heart failure through DUSP-1-TMBIM-6-mediated mitochondrial quality control and gut flora interactions. Phytomedicine. (2024) 132:155880. doi: 10.1016/j.phymed.2024.155880
166. Wang M, Wang R, Sun H, Sun G, Sun X. Ginsenoside Rb1 ameliorates cardiotoxicity triggered by aconitine via inhibiting calcium overload and pyroptosis. Phytomedicine. (2021) 83:153468. doi: 10.1016/j.phymed.2021.153468
167. Luo M, Yan D, Sun Q, Tao J, Xu L, Sun H, et al. Ginsenoside Rg1 attenuates cardiomyocyte apoptosis and inflammation via the TLR4/NF-kB/NLRP3 pathway. J Cell Biochem. (2020) 121:2994–3004. doi: 10.1002/jcb.29556
168. Guo T, Jiang ZB, Tong ZY, Zhou Y, Chai XP, Xiao XZ. Shikonin ameliorates LPS-induced cardiac dysfunction by SIRT1-dependent inhibition of NLRP3 inflammasome. Front Physiol. (2020) 11:570441. doi: 10.3389/fphys.2020.570441
169. Gal R, Deres L, Horvath O, Eros K, Sandor B, Urban P, et al. Resveratrol improves heart function by moderating inflammatory processes in patients with systolic heart failure. Antioxidants (Basel). (2020) 9:1108. doi: 10.3390/antiox9111108
170. Feng H, Mou SQ, Li WJ, Zhang N, Zhou ZY, Ding W, et al. Resveratrol inhibits ischemia-induced myocardial senescence signals and NLRP3 inflammasome activation. Oxid Med Cell Longev. (2020) 2020:2647807. doi: 10.1155/2020/2647807
171. Wang R, Wang Y, Wu J, Guo Y, Xiao H, Zhang Y, et al. Resveratrol targets AKT1 to inhibit inflammasome activation in cardiomyocytes under acute sympathetic stress. Front Pharmacol. (2022) 13:818127. doi: 10.3389/fphar.2022.818127
172. Li Q, Zuo Z, Pan Y, Zhang Q, Xu L, Jiang B. Salvianolic acid B alleviates myocardial ischemia injury by suppressing NLRP3 inflammasome activation via SIRT1-AMPK-PGC-1alpha signaling pathway. Cardiovasc Toxicol. (2022) 22:842–57. doi: 10.1007/s12012-022-09760-8
173. Hu Y, Wang X, Li Q, Pan Y, Xu L. Salvianolic acid B alleviates myocardial ischemic injury by promoting mitophagy and inhibiting activation of the NLRP3 inflammasome. Mol Med Rep. (2020) 22:5199–208. doi: 10.3892/mmr.2020.11589
174. Cheng X, Zhang Y, Guo H, Li X, Wang Y, Song Y, et al. Cichoric acid improves isoproterenol-induced myocardial fibrosis via inhibition of HK1/NLRP3 inflammasome-mediated signaling pathways by reducing oxidative stress, inflammation, and apoptosis. Food Sci Nutr. (2024) 12:180–91. doi: 10.1002/fsn3.3758
175. Yu W, Qin X, Zhang Y, Qiu P, Wang L, Zha W, et al. Curcumin suppresses doxorubicin-induced cardiomyocyte pyroptosis via a PI3K/Akt/mTOR-dependent manner. Cardiovasc Diagn Ther. (2020) 10:752–69. doi: 10.21037/cdt-19-707
176. Joshi S, Kundu S, Priya VV, Kulhari U, Mugale MN, Sahu BD. Anti-inflammatory activity of carvacrol protects the heart from lipopolysaccharide-induced cardiac dysfunction by inhibiting pyroptosis via NLRP3/Caspase1/Gasdermin D signaling axis. Life Sci. (2023) 324:121743. doi: 10.1016/j.lfs.2023.121743
177. Chai R, Ye Z, Xue W, Shi S, Wei Y, Hu Y, et al. Tanshinone IIA inhibits cardiomyocyte pyroptosis through TLR4/NF-kappaB p65 pathway after acute myocardial infarction. Front Cell Dev Biol. (2023) 11:1252942. doi: 10.3389/fcell.2023.1252942
178. Qiu H, Liu W, Lan T, Pan W, Chen X, Wu H, et al. Salvianolate reduces atrial fibrillation through suppressing atrial interstitial fibrosis by inhibiting TGF-beta1/Smad2/3 and TXNIP/NLRP3 inflammasome signaling pathways in post-MI rats. Phytomedicine. (2018) 51:255–65. doi: 10.1016/j.phymed.2018.09.238
179. Ye B, Chen X, Dai S, Han J, Liang X, Lin S, et al. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des Devel Ther. (2019) 13:975–90. doi: 10.2147/DDDT.S195412
180. Dai S, Ye B, Chen L, Hong G, Zhao G, Lu Z. Emodin alleviates LPS-induced myocardial injury through inhibition of NLRP3 inflammasome activation. Phytother Res. (2021) 35:5203–13. doi: 10.1002/ptr.7191
181. Chen P, An Q, Huang Y, Zhang M, Mao S. Prevention of endotoxin-induced cardiomyopathy using sodium tanshinone IIA sulfonate: Involvement of augmented autophagy and NLRP3 inflammasome suppression. Eur J Pharmacol. (2021) 909:174438. doi: 10.1016/j.ejphar.2021.174438
182. Xiao B, Huang X, Wang Q, Wu Y. Beta-asarone alleviates myocardial ischemia-reperfusion injury by inhibiting inflammatory response and NLRP3 inflammasome mediated pyroptosis. Biol Pharm Bull. (2020) 43:1046–51. doi: 10.1248/bpb.b19-00926
183. Luan F, Lei Z, Peng X, Chen L, Peng L, Liu Y, et al. Cardioprotective effect of cinnamaldehyde pretreatment on ischemia/ reperfusion injury via inhibiting NLRP3 inflammasome activation and gasdermin D mediated cardiomyocyte pyroptosis. Chem Biol Interact. (2022) 368:110245. doi: 10.1016/j.cbi.2022.110245
184. Xu XN, Jiang Y, Yan LY, Yin SY, Wang YH, Wang SB, et al. Aesculin suppresses the NLRP3 inflammasome-mediated pyroptosis via the Akt/GSK3beta/NF-kappaB pathway to mitigate myocardial ischemia/reperfusion injury. Phytomedicine. (2021) 92:153687. doi: 10.1016/j.phymed.2021.153687
185. Luan F, Rao Z, Peng L, Lei Z, Zeng J, Peng X, et al. Cinnamic acid preserves against myocardial ischemia/reperfusion injury via suppression of NLRP3/Caspase-1/GSDMD signaling pathway. Phytomedicine. (2022) 100:154047. doi: 10.1016/j.phymed.2022.154047
186. Sun W, Lu H, Lyu L, Yang P, Lin Z, Li L, et al. Gastrodin ameliorates microvascular reperfusion injury-induced pyroptosis by regulating the NLRP3/caspase-1 pathway. J Physiol Biochem. (2019) 75:531–47. doi: 10.1007/s13105-019-00702-7
187. Ding HS, Huang Y, Qu JF, Wang YJ, Huang ZY, Wang FY, et al. Panaxynol ameliorates cardiac ischemia/reperfusion injury by suppressing NLRP3-induced pyroptosis and apoptosis via HMGB1/TLR4/NF-kappaB axis. Int Immunopharmacol. (2023) 121:110222. doi: 10.1016/j.intimp.2023.110222
188. Peng L, Lei Z, Rao Z, Yang R, Zheng L, Fan Y, et al. Cardioprotective activity of ethyl acetate extract of Cinnamomi Ramulus against myocardial ischemia/reperfusion injury in rats via inhibiting NLRP3 inflammasome activation and pyroptosis. Phytomedicine. (2021) 93:153798. doi: 10.1016/j.phymed.2021.153798
Keywords: NLRP3, inflammasome, heart failure, active ingredient, traditional Chinese medicine
Citation: Lin R, Yu Y, Du L, Ding Z, Wang Z, Wei J and Guo Z (2025) Active ingredients of traditional Chinese medicine inhibit NOD-like receptor protein 3 inflammasome: a novel strategy for preventing and treating heart failure. Front. Immunol. 16:1520482. doi: 10.3389/fimmu.2025.1520482
Received: 31 October 2024; Accepted: 06 January 2025;
Published: 24 January 2025.
Edited by:
Xing Zhang, National University of Singapore, SingaporeReviewed by:
Youhua Wang, Shanghai University of Traditional Chinese Medicine, ChinaXing Luo, Harbin Medical University, China
Copyright © 2025 Lin, Yu, Du, Ding, Wang, Wei and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaming Wei, MDA0OTE2QGhudWNtLmVkdS5jbg==; Zhihua Guo, MDA0Mjk0QGhudWNtLmVkdS5jbg==