 Anna De Lucia1
Anna De Lucia1 Lucia Mazzotti1,2
Lucia Mazzotti1,2 Anna Gaimari1,2
Anna Gaimari1,2 Matteo Zurlo1
Matteo Zurlo1 Roberta Maltoni3
Roberta Maltoni3 Claudio Cerchione4
Claudio Cerchione4 Sara Bravaccini5
Sara Bravaccini5 Angelo Delmonte6
Angelo Delmonte6 Lucio Crinò6
Lucio Crinò6 Patricia Borges de Souza1
Patricia Borges de Souza1 Luigi Pasini1*†
Luigi Pasini1*† Fabio Nicolini1
Fabio Nicolini1 Fabrizio Bianchi7
Fabrizio Bianchi7 Manel Juan8
Manel Juan8 Hugo Calderon8
Hugo Calderon8 Chiara Magnoni1,2
Chiara Magnoni1,2 Luca Gazzola1,2
Luca Gazzola1,2 Paola Ulivi9
Paola Ulivi9 Massimiliano Mazza1
Massimiliano Mazza1- 1Advanced Cellular Therapies and Rare Tumors Unit, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy
- 2Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy
- 3Healthcare Administration, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy
- 4Hematology Unit, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy
- 5Department of Medicine and Surgery, “Kore” University of Enna, Enna, Italy
- 6Medical Oncology Department, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy
- 7Unit of Cancer Biomarker, Fondazione IRCCS Casa Sollievo Della Sofferenza, San Giovanni Rotondo, FG, Italy
- 8Department of Immunology, Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 9Translational Oncology Unit, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, Italy
Over the past decades, significant progress has been made in the understanding of non-small cell lung cancer (NSCLC) biology and tumor progression mechanisms, resulting in the development of novel strategies for early detection and wide-ranging care approaches. Since their introduction, over 20 years ago, targeted therapies with tyrosine kinase inhibitors (TKIs) have revolutionized the treatment landscape for NSCLC. Nowadays, targeted therapies remain the gold standard for many patients, but still they suffer from many adverse effects, including unexpected toxicity and intrinsic acquired resistance mutations, which lead to relapse. The adoption of immune checkpoint inhibitors (ICIs) in 2015, has offered exceptional survival benefits for patients without targetable alterations. Despite this notable progress, challenges remain, as not all patients respond favorably to ICIs, and resistance to therapy can develop over time. A crucial factor influencing clinical response to immunotherapy is the tumor microenvironment (TME). The TME is pivotal in orchestrating the interactions between neoplastic cells and the immune system, influencing tumor growth and treatment outcomes. In this review, we discuss how the understanding of this intricate relationship is crucial for the success of immunotherapy and survey the current state of immunotherapy intervention, with a focus on forthcoming and promising chimeric antigen receptor (CAR) T cell therapies in NSCLC. The TME sets major obstacles for CAR-T therapies, creating conditions that suppress the immune response, inducing T cell exhaustion. To enhance treatment efficacy, specific efforts associated with CAR-T cell therapy in NSCLC, should definitely focus TME-related immunosuppression and antigen escape mechanisms, by combining CAR-T cells with immune checkpoint blockades.
Introduction
Lung cancer is a major cause of cancer-related deaths and the second most diagnosed cancer globally (1). Non-small cell lung cancer (NSCLC) accounts for 80 to 90% of all lung cancer diagnoses in the United States (2), and in Europe (3), with lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) being the two most prevalent NSCLC subtypes (4). Numerous variables play a role in the development of NSCLC. Smoking and secondhand smoke exposure are key risk factors (5). Despite remaining uncertain, marijuana and e-cigarette usage have been related to a possible risk of NSCLC (6). Additionally, environmental and professional exposure to asbestos, radon gas, air pollution, and certain pollutants are strong risk factors associated with NSCLC development (7), in addition to genetic predisposition (8).
Although surgery and chemotherapy (including neoadjuvant and/or adjuvant therapy), as well as radiotherapy, have shown improvements in prolonging overall survival (OS) in NSCLC patients, they still present with severe toxic side effects (9). In the past 20 years, targeted therapies have become the gold standard to treat NSCLC patients with actionable oncogenic alterations, including driver mutations and fusions/rearrangements (10). These patients represent only 15–20% of all NSCLC patients and, while targeted therapies are initially effective, resulting in prolonged progression-free survival (PFS) and improved OS, their efficacy is limited by the emergence of resistance mechanisms (11).
Therefore, developing new treatment approaches that impact on PFS and OS is now the primary focus in NSCLC research. In this context, targeting specific components of the tumor microenvironment (TME) in NSCLC is seen today as the Holy Grail to boost the effectiveness of established drugs and design novel anticancer agents. Drugs targeting cancer associated fibroblasts, extracellular components, immune cells, endothelial cells and the surrounding tumor vasculature have been recently approved by regulatory agencies (12, 13). A dominant trait of NSCLC is the elevated presence of tumor-specific (TSAs) and tumor-associated antigens (TAAs) on the surface of malignant cells (14). Cancer immunotherapy has the goal to overcome TME by eliciting (or re-igniting) an adaptive immune response, especially the T-cell-mediated TSA- and TAA-directed cytotoxicity against cancer cells (15). Immunotherapies include therapeutic vaccines, autologous cellular therapies, and different types of immune modulators, including checkpoint inhibitors, cytokines, T-cell agonists and adjuvants (16). In patients without a driver mutation, immunotherapy in the form of immune checkpoint inhibitors (ICIs) is currently an integral part of NSCLC treatment (17). Programmed cell death ligand-1 (PD-L1) expression is the main predictive biomarker of response to ICIs directed to PD-L1 and to its binding partner, the programmed cell death protein 1 (PD-1) (18). Normally, the five-year OS rate exceeds 25% for patients having advanced NSCLC with a PD-L1 score of 50% or higher (19, 20). This statistics is still superior to the average OS of patients treated with conventional drugs, which rarely exceed 15% (21, 22). Nevertheless, a more reliable marker of response is missing and highly wanted to improve patient stratification (23) considering that a significant fraction of patients fails to benefit from those treatments. Among investigated biomarkers, the tumor mutational burden (TMB) has recently emerged as a major predictor of immunotherapy efficacy in NSCLC (17). However, to elicit effective immune response novel immunotherapeutic approaches are needed in the so-called “cold tumors”, which are generally characterized by a decreased TMB, defective HLA class I antigen processing machinery (APM), reduced T-cell presence, and low PD-L1 expression. Two of the most advanced approaches involve adoptive cell transfer (ACT) with autologous tumor-infiltrating lymphocytes (TILs) and chimeric antigen receptor (CAR)-based therapies. In this scenario, there has been a rise in the number of clinical trials exploring the safety and efficacy of different antigen-based ACTs.
Targeted therapies
The discovery of targetable genetic alterations has radically changed the approach to NSCLC treatment. With the identification of specific cancer driver mutations, today clinicians can provide individualized therapies that lead to extremely efficient responses in some patients’ subsets when treated for example with the matched targeted tyrosine-kinase inhibitor (TKI) (24, 25).
Targeting of the epidermal growth factor receptor (EGFR) is a paradigm of this type of therapy. Mutations in the EGFR gene are prevalent in approximately ~15% of patients with NSCLC (~50% in Asians), and they play a primary role in disrupting cellular functions such as cell growth, survival, invasion, and angiogenesis. Mutations in EGFR can lead to continuous activation and independence from ligands, particularly in the ATP-binding region of the tyrosine kinase domain. Notably, common EGFR mutations, including exon 19 deletions and the L858R mutation on exon 21, have been associated with heightened sensitivity to EGFR TKIs (24, 26–29). First-generation anti-EGFR TKIs, like gefitinib (NCT01203917) (30) and erlotinib (NCT00446225) (31), have demonstrated superior response rates and longer PFS (9.2–13.1 months) than traditional cytotoxic therapies in treatment-naive patients with EGFR mutations (24, 31–37). Second-generation inhibitors, such as afatinib (NCT00949650; NCT01121393) (38) and dacomitinib (NCT01774721) (39), target EGFR but also the receptor tyrosine-protein kinase erbB-2 (HER2) and erb-4 (HER4), resulting in improved progression-free survival compared to gefitinib (24, 38–40). The most common acquired resistance to first-generation TKIs arises due to the additional EGFR mutation T790M in exon 20 (24, 41, 42); other resistance mechanisms to targeted therapy include HER2 amplification, mutations in the MET proto-oncogene receptor tyrosine kinase (MET), the B-Raf proto-oncogene serine/threonine kinase (BRAF), the ROS proto-oncogene 1 receptor tyrosine kinase (ROS1), and the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3Cclinical A) genes (24, 43). Interestingly, resistance to TKIs can also cause lung adenocarcinoma to evolve into small-cell lung cancer (SCLC): roughly, 3% to 10% of EGFR-mutated NSCLC could shift to SCLC clinical subtype (44). Third-generation EGFR inhibitors selectively target the original activating mutations, in addition to the T790M resistance mutation. Osimertinib, a representative third-generation anti-EGFR TKI, while exhibiting higher response rates and longer PFS than platinum-based chemotherapeutic treatment (45), has also shown efficacy in NSCLC patients with EGFR-T790M mutations after progression to treatment with first-generation TKIs (24, 46, 47). FLAURA clinical trial (NCT02296125) comparing osimertinib to erlotinib or gefitinib as a first-line treatment in advanced NSCLC patients with EGFR mutations revealed that osimertinib significantly improved PFS, establishing it as the preferential first-line treatment option (24, 48, 49). Acquired resistance to third-generation anti-EGFR TKIs can also occur through the C797S mutation (24, 50). Triple mutants carrying the original sensitizing mutation, plus acquired T790M and C797S mutations, show strong resistance to TKIs from all three generations (24, 51). Promising approaches for these triple mutants include allosteric inhibitors like EAI045 in tumors with the L858R-sensitizing mutation and the anaplastic lymphoma kinase (ALK) inhibitor brigatinib in tumors with exon 19 deletion. These inhibitors can be combined with cetuximab, an anti-EGFR monoclonal antibody, to target tumors harboring the triple mutant (24, 52, 53).
ALK-positive tumors represent about 4% of lung cancers, and generally appear in adenocarcinoma NSCLC of younger non-smoker patients (54). Typically, the ALK gene is rearranged with the echinoderm microtubule-associated protein-like 4 (EML4) gene, forming the EML4-ALK fusion protein (24, 55). Crizotinib, which is an oral inhibitor that targets mutations in the kinase domain of ALK (NCT00932893) (56), MET, and ROS1 (NCT00585195) (24, 57, 58), also shows effectiveness in treating NSCLCs with ALK fusion by improving 62% PFS and response rate, compared to traditional chemotherapy (24, 59–62). Second-generation ALK inhibitors like ceritinib (63), brigatinib (NCT02737501) (64), and alectinib (NCT02075840) (60) have been shown to be effective in the second-line setting after resistance to crizotinib, which is typically used as first-line treatment for ALK-mutated patients (65). As a second-line treatment, alectinib showed an objective response rate (ORR) of 45% and PFS of 8 to 12 months, and brigatinib showed an ORR of 45% to 54% with a PFS of 9.2 to 12.9 months (65). Also, clinical studies have shown higher ORR and median PFS for alectinib than crizotinib in previously untreated patients with ALK-positive NSCLC, establishing alectinib as a viable first-line option (59, 65). Resistance to ALK inhibitors can arise from various alterations in the ALK gene, such as mutations and amplification, or through the activation of alternative signaling pathways, like the EGFR and the mitogen-activated protein kinase (MAPK) cascade (66). Among all resistance mechanisms, secondary ALK mutations are the main drivers of resistance to second-generation TKIs (67). The most common ALK resistance mutation observed in patients treated with second-generation TKIs is the G1202R. This mutation confers in vitro resistance to all available ALK inhibitors except lorlatinib. Lorlatinib, a potent third-generation ALK inhibitor, is effective against most known ALK resistance mutations, and it has demonstrated efficacy in patients previously treated with up to three lines of ALK inhibitors, providing a potential treatment option for overcoming resistance (NCT01970865) (68, 69).
As mentioned before, crizotinib is an oral inhibitor that targets ALK, MET, and ROS1 tyrosine kinases. ROS1 gene can undergo rearrangements and typically fuses with the CD74 gene, which causes the receptor tyrosine kinase domain to become persistently active (70). Ceritinib and lorlatinib (NCT01970865) (69) also exhibit notable efficacy in ROS1-positive tumors (68, 71, 72). Resistance to crizotinib in cases of ROS1 rearrangements can arise through various mechanisms, including secondary mutations, most notably the G2032R mutation. Additionally, resistance may occur due to the activation of wild-type EGFR signaling or mutations in the Kirsten rat sarcoma viral proto-oncogene (KRAS) and the KIT proto-oncogene receptor tyrosine kinase (KIT) genes (73, 74).
Additional targetable gene alterations in NSCLC include BRAF and HER2 mutations, as well as rearrangements in the RET proto-oncogene (RET), and fusions involving the neurotrophic receptor tyrosine kinase (NTRK) genes 1-3 (NTRK1, NTRK2, NTRK3) (24). Roughly half of the NSCLC patients with a BRAF mutation carry the V600E activating mutation in exon 15 (75, 76). This mutation indicates sensitivity to BRAF inhibitors like vemurafenib (NCT01524978) (77) and dabrafenib, alone or combined with trametinib (NCT01336634) (78–82). Acquired resistance mechanisms to BRAF inhibitors typically include secondary BRAF alterations (e.g., splice variants), activating mutations in the mitogen-activated extracellular signal-regulated kinase (MEK) and the proto oncogene serine/threonine protein kinase CRAF, neurofibromatosis type 1 (NF1) gene loss, and sustained activation of the MAPK pathway thought bypassing signaling via other tyrosine receptor kinases like EGFR or MET (83, 84). Targeted therapies against HER2 and RET alterations have shown moderate activity compared to other targeted treatments, likely due to their dominant role as drivers of tumor growth (85–89). Table 1 summarizes the major TKIs, their targets, and associated resistance mechanisms.
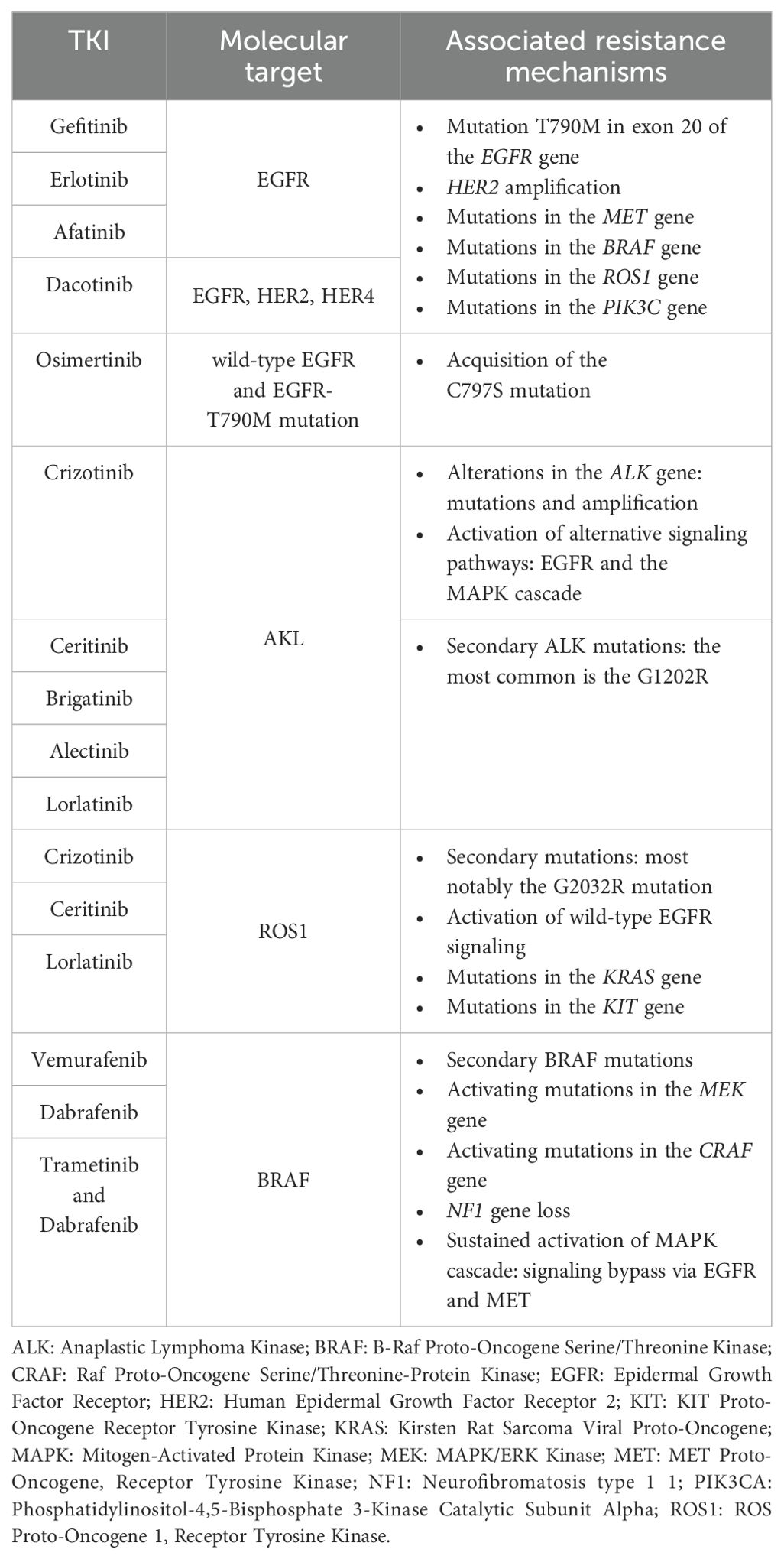
Table 1. Summary of major tyrosine kinase inhibitors (TKIs) and related therapeutic targets in non-small cell lung cancer, along with most frequent mechanisms of acquired resistance.
Tumor microenvironment and immunotherapy
Immune suppression within the TME
The genetic alterations that initiate and drive tumor growth not only affect the behavior of cancer cells but also shape the TME composition, by affecting both immune cells function and non-cellular components of the extracellular matrix (90).
LUAD tumors exemplify the intricate signaling networks employed by cancer cells to coerce non-malignant cells for their advantage. These types of lung cancer which are characterized by a high burden of clonal neoantigens that promote an “inflamed” TME, with an abundance of activated effector T cells, increased expression of proteins, such as the chemokine (C–X–C motif) ligand-9 (CXCL-9) and -10 (CXCL-10), involved in antigen presentation and T-cell migration (91). At the same time, an inflamed TME also expresses negative regulators of T-cell activity, like the lymphocyte-activation gene 3 (LAG-3), PD-L1, PD-1 and T cell immunoglobulin and ITIM (TIGIT). The bright side is that LUAD tumors with a high TMB and elevated PD-L1/PD-1 expression may respond well to ICI treatment (91). Also, an important cellular alteration associated with a high TMB is the loss of mismatch repair function followed by an increased microsatellite instability, the last being associated with improved responses to ICIs (92). Although tumors with microsatellite instability had shown some promising responses to ICIs (93), recent findings revealed that genome instability can fuel resistant phenotypes of tumor cells to both targeted therapy and ICI (94). Nevertheless, genetic alterations can negatively affect the TME in other ways. For instance, the inactivation of the tumor suppressor gene serine/threonine kinase 11 (STK11) in KRAS-mutated LUAD shifts the TME toward tumor infiltration by immunosuppressive neutrophils and the reduction of PD-L1 expression in cancer cells and less TILs (95).
Research on TILs has provided valuable understanding about the role of lymphocytes within the tumor stroma and their contribution to the development of an immunogenic response (96–98). In fact, a high density of T lymphocytes within the tumor bulk, including CD4+ and CD8+ cells, typically correlates with improved outcomes (97, 99). Specifically, CD8+ T cells and M1 macrophages have been associated with favorable prognosis and prolonged OS (97). A recent study focused on the association between the presence of CD8+/PD-L1+ TILs and the TMB, indicating an immunosuppressive TME, in those patients that were more likely to respond to anti-PD-1 therapy (100). To investigate more deeply the underlying mechanisms, Caushi et al. utilized single-cell transcriptomics to analyze specific TILs targeting mutation-associated neoantigens (MANAs) in NSCLC tumors from patients enrolled in a clinical trial with nivolumab alone or in combination with ipilimumab (101–103). The study revealed that MANA-specific CD8+ T cells were more abundant within the TME compared to the normal lung tissue of the same patient. Moreover, MANA-specific T cells from responsive patients exhibited an increased expression of genes associated with T cell memory, including the interleukin 7 receptor (IL7R), T-cell factor/lymphoid enhancer-binding factor 7 (TCF7), and the granzyme K (GZMK). Conversely, MANA-specific T cells from non-responsive patients predominantly expressed genes linked to T cell dysfunction, such as TOX high mobility group box family member 2 (TOX2), cytotoxic T-lymphocyte antigen 4 (CTLA4), hepatitis A virus cellular receptor 2 (HAVCR2), and the ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1) gene (102).
While some patients that are responsive to immunotherapy exhibit a tumor with a TME characterized by the presence of TILs, macrophages, and dendritic cells (DCs), other patients have tumors with a so-called “cold” TME, which is less permeated by TILs, or show an “altered” TME, where TILs are primarily found at the tumor’s edge (104). Comprehensive analyses integrating spatial histology and genetic information have shown that tumors with multiple immune cold regions are characterized by a higher risk of relapse (105). Furthermore, low TIL counts were found associated with reduced efficacy of ICI treatment and resistance to immunotherapy (105). These findings emphasize the importance of understanding the composition of the TME and spatial distribution of its components in predicting therapy response and patients’ outcome. Dysfunctional CD8+ TILs, referred to as “burned-out” (Ebo) TILs, have been identified in advanced NSCLC patients (106). Ebo TILs showed heightened proliferation and activation markers but reduced production of interferon-gamma (IFNγ). Notably, Ebo TILs expressing elevated PD-1, T-cell immunoglobulin, mucin-domain containing-3 (TIM-3), and LAG-3 were associated with resistance to anti-PD therapy in NSCLC patients (106). Furthermore, the presence of inhibitory receptors on TILs, including PD-1, TIM-3, the cytotoxic T-lymphocyte antigen 4 (CTLA-4), LAG-3, and B and T Lymphocyte associated (BTLA) receptor, associate to a progressively impaired capacity of T cells to respond to polyclonal activation (107).
Tertiary lymphoid structures in NSCLC
The so called ‘Tertiary Lymphoid Structures’ (TLS) are organized aggregates of ectopic lymphoid tissues that form within the TME, and consisting of germinal centers where cognate T cells and B cells interact to develop the anti-tumor adaptive immune response. TLS are essential to promote an antigen-specific immune response at sites of chronic inflammation, after consecutive antibody somatic hypermutation and affinity maturation happening (96, 108, 109). Several studies have associated the presence of B cells in TLS with more favorable outcomes in NSCLC (110–114). B cells have the capacity to activate and proliferate into plasma cells, which can generate tumor-specific antibodies that attack tumor cells and trigger the complement system, enhancing both antibody-dependent cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) (115, 116). In a recent study, it was demonstrated that TLS maturation is associated with major pathological response, which can be used as an independent predictor of disease free survival (DFS) in resectable neoadjuvant chemoimmunotherapy-treated NSCLC (117). The study collected formalin-fixed paraffin embedded tissues from patients with resectable NSCLC, divided in three cohorts based on treatment: naïve, neoadjuvant chemoimmunotherapy, and neoadjuvant chemotherapy. Among the three cohorts, neoadjuvant chemoimmunotherapy-treated NSCLCs showed the highest TLS maturation and abundance; both the maturation and abundance of TLS were significantly correlated with major pathological response in both the neoadjuvant chemoimmunotherapy and the chemotherapy group. Patients with high maturation and abundance of TLS exhibited better DFS in all the three cohorts. TLS maturation was also an independent predictor for DFS in the neoadjuvant chemoimmunotherapy and treatment naïve group (117).
Novel strategies and targets in NSCLC
Natural killer (NK) cells are also very important in regulating the interplay between cancer cells and the TME in NSCLC patients treated with ICIs. NK cells, specifically the non-cytotoxic CD56-bright-CD16-subset, express immunoactivation markers that accumulate in the stroma of NSCLC tumors, including the NK-specific triggering receptor (NKp44), the CD69, and the human leucocyte antigen DR (HLA-DR) protein (118, 119). Importantly, PD-1 is also expressed by NK cells (120). To optimize ICI treatment, a randomized controlled trial in NSCLC patients with positive expression of PD-L1 explored the combination of in vitro expanded allogenic NK cells with anti-PD-1 therapy. This novel approach yielded promising results, as it improved OS and PFS, as compared to anti-PD-1 therapy alone (121).
ICI resistance in NSCLC patients associates with an increased number of immunosuppressive cells, including regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAM)-M2, and neutrophils (122–124). Treg cells inhibit T cell responses and are associated with poor clinical outcomes in lung cancer patients (125). Studies have shown an increase in PD-1+ Treg cells in patients who do not respond to anti-PD-1/PD-L1 ICI, suggesting that the balance of PD-1 expression between CD8+ T cells and Treg cells in the TME can be used as a more accurate predictor of ICI therapy effectiveness rather than the expression of PD-L1 itself or the TMB (126). Hence, targeting Treg cells could potentially enhance the efficacy of ICI treatment for lung cancer (127, 128). MDSCs can induce immunosuppression through various mechanisms, including the production of molecules that hinder T cell function and interfere with T cell movement (129). MDSCs expressing specific receptors, like CD39 and CD73, have been found in NSCLC tumor tissue, being associated with disease progression (130, 131). Additionally, a 2020 study suggested an association between PD-L1 protein expression on macrophage cells and improved OS in patients treated with anti-PD-1 therapy (132).
Tumor associated neutrophils (TAN) in the TME may also contribute to immune suppression and resistance to ICI treatment in NSCLC, by mediating the suppression of Th1 and cytotoxic T lymphocytes. In particular, in these NSCLC patients, arginase-1 (ARG1)-expressing neutrophils negatively correlates with the proportion of CD8+ T cells, while ARG1-expressing granulocytic cells can lead to CD3ζ chain downregulation on T cells though L-arginine depletion and ultimately inhibit T-cell proliferation and cytokine secretion. In addition to the direct inhibition of effector T cell functions, TANs have also been implicated in regulatory T cell (Treg) recruitment (133–136) to improve T cell activation and ICI therapy response TME is also influenced by the family of vascular endothelial growth factor (VEGF) proteins and their receptors (VEGFRs). VEGF signaling plays a key role in tumor-induced angiogenesis and in promoting tumor growth in NSCLC patients (137). VEGF also influences the immune response within the TME. It can suppress the activity of antigen-presenting cells (APCs), including DCs, NK cells, and T cells. At the same time, VEGF enhances the suppressive effect of Tregs, TAMs, and MDSCs. This combination creates an immunosuppressive microenvironment that allows the tumor to evade the immune system’s surveillance. During the past years, the hypothesis that targeting VEGF could enhance the effectiveness of ICI has also been explored (137). For example, in a phase 3 study (NCT02366143) bevacizumab, an anti-VEGFA antibody, was combined with atezolizumab, an anti-PD-L1 antibody, plus chemotherapy in patients with metastatic lung cancer who had not previously received chemotherapy. This combination significantly improved OS and PFS, irrespective of their PD-L1 expression levels or EGFR or ALK genetic alteration status (138).
Lately, there has been a concerted effort to identify reliable TME-based markers to predict the effectiveness of immunotherapy for lung cancer. One notable tool is the “immunoscore” (IS), which assesses the presence of T lymphocytes within the tumor tissue (139). This digital test examines various T cell subpopulations in both the central and peripheral regions of the tumor, generating a score that ranges from IS 0 (low immune cell density) to IS 4 (high density in both areas). The IS has shown promising results in several cancer types, including NSCLC, where a higher IS score is associated with better survival outcomes (140). CD8+TILs have emerged as a potent biomarker for differentiating patients with more favorable PFS following immunotherapy with ICI (141–144). Expression of CD8 can act either as a prognostic or a predictive factor of clinical outcome: in NSCLC patients not treated by immunotherapy, high CD8A expression is associated with longer OS, while in NSCLC patients treated with anti PD1, high CD8 expression is associated with longer PFS (145). Another approach to predict the effectiveness of immunotherapy for lung cancer involves the generation of specific gene signatures to characterize the TME, aka the “immune gene signatures” (146, 147). These signatures consist of lists of genes that indicate if enriched/depleted (i.e., coherently up- or down-regulated, respectively, in a tumor sample), the presence of specific immune or stromal cell populations or describe TME-cell activation states. High-throughput technologies like microarray and RNA sequencing have facilitated the development of computational algorithms capable of predicting non-cancer cell infiltration in tumors (148–154). These algorithms generate scores that define the grade of immune and stromal cell infiltration, providing valuable insights into cancer molecular and immune characteristics and its potential impact on ICI response. As a matter of fact, the gene signature scores provide specific TME-based markers that offer valuable information on tumor heterogeneity, and enable meaningful comparisons between different tumor samples (155), showing great promise in advancing our understanding of immunotherapy response in lung cancer.
ICIs, such as monoclonal antibodies targeting CTLA-4 and antibodies against PD-1 or PD-L1, have opened new avenues for managing lung cancer (156–159). So far, the Food and Drug Administration (FDA) has approved the following ICIs: the anti-PD-1 antibodies nivolumab (NCT01642004) (160), pembrolizumab (NCT01295827) (19), and cemiplimab (NCT03409614) (161), as well as the anti-PD-L1 antibodies atezolizumab (NCT01903993) (162), durvalumab (NCT02125461) (163), avelumab (NCT02576574) (164), and sugemalimab (NCT03789604) (165, 166). Also, FDA has approved ipilimumab, an anti CTLA-4 antibody (NCT02477826) (167).
Immunotherapy has revolutionized the treatment approach for advanced NSCLC, due to the favorable safety profile of most ICIs and improved survival outcomes, which make these drugs particularly effective for patients who experience disease progression after initial cytotoxic therapy (160, 168), and a promising first-line treatment option (169).
In the first-line setting, pembrolizumab has become the standard of care for metastatic NSCLC patients with tumor expression of PD-L1 over 50%, a condition that may occur in approximately 30% of NSCLC cases (170). Pembrolizumab has significantly improved ORR, PFS, and OS compared to platinum-based cytotoxic therapy (171, 172). On the other hand, nivolumab did not show similar benefits in PFS or OS among patients with PD-L1 expression levels above 5% (171). Notably, in patients treated with nivolumab who had both high PD-L1 expression and high TMB, the objective response rate was 75%, suggesting that a predictive value for both TMB and PD-L1 expression in determining the efficacy of ICI therapy exists (169).
Even further, combination of ICI with cytotoxic chemotherapy has shown enhanced treatment outcomes in NSCLC (173). Combination of pembrolizumab with carboplatin and pemetrexed has demonstrated improved ORR and PFS compared to cytotoxic therapy alone, making it a promising first-line treatment option for advanced NSCLC patients (174). A detailed meta-analysis, including all randomized controlled trials published before February 2022, showed that the combination of ICIs with chemotherapy is much more effective in enhancing PFS, ORR, and OS in NSCLC patients (175).
Several novel immune checkpoints with promising therapeutic potential have recently been identified, among them the LAG-3, T-cell immunoglobulin and TIM-3, B7 Homolog 3 (B7-H3), and T cell immunoglobulin and TIGIT domain (176). To target LAG-3, researchers are exploring the use of a soluble dimeric recombinant LAG-3 (eftilagimod alpha, or IMP321), which stimulates DCs through the binding with the major histocompatibility complex (MHC) class II receptor, leading to sustained immune responses when combined with anti-PD-1 therapy in patients with previously untreated unresectable or metastatic NSCLC [NCT03625323 (177)]. Another approach involves bispecific antibodies (BsAbs) simultaneously targeting LAG-3 and PD-1 [NCT04140500 (178); NCT03219268 (179)]. Besides, the anti-LAG-3 antibody relatlimab (BMS-986016) has shown promising results in the phase III trial RELATIVITY-047 (NCT03470922), where treatment-naïve patients with metastatic melanoma who received nivolumab plus relatlimab demonstrated significantly longer median PFS than those who received nivolumab plus placebo (180). Considering these encouraging findings, further investigations are underway to evaluate the dual blockade in other solid tumors, including NSCLC (NCT04623775 (181).
TIM-3, known for its presence on CTL, NK, Treg, DC, and macrophages (where it promotes M2 polarization), is a critical immune checkpoint being investigated in various clinical trials for solid tumors, including NSCLC (182). Monoclonal antibodies targeting TIM-3 alone or combined with anti-PD-1 are being studied in trials such as the studies NCT03652077 (183) and NCT02608268 (184). Moreover, bispecific antibodies (BsAbs) that can simultaneously block TIM-3 and PD-1 are also explored in ongoing trials involving NSCLC patients [NCT03708328 (182, 185); NCT04931654 (186)].
The B7 homologous 3 (B7-H3), also known as CD276, is a transmembrane protein commonly expressed by cancer cells that act as an immune checkpoint, allowing cancer cells to evade immune surveillance. Researchers believe that B7-H3 expression might play a role in the resistance to anti-PD-1/PD-L1 therapy in NSCLC (187, 188). There are currently three ongoing clinical trials evaluating the use of an anti-B7-H3 antibody in combination with either anti-PD-1 or anti-CTLA-4 for advanced solid tumors, including NSCLC (NCT03729596; NCT02475213; NCT02381314) (189–191). As detailed below, novel immunotherapies targeting B7-H3 are considered very promising in enhancing the clinical response and overcoming resistance in NSCLC patients, including the use of chimeric antigen receptor (CAR)-T cells (192).
The immune receptor TIGIT is expressed by several immune cells, including CD8+ T cells, CD4+ T cells, and NK cells, and it is a candidate target of vibostolimab antibody in NSCLC (12, 193). The phase I study NCT02964013 (194) was the first to investigate safety and efficacy of vibostolimab as a monotherapy or in combination with pembrolizumab for treating advanced solid tumors, including NSCLC (12, 194). Another study, the phase II CITYSCAPE trial (NCT03563716), combined another TIGIT inhibitor, tiragolumab, with the anti-PD-L1 atezolizumab (195). The study showed that tiragolumab plus atezolizumab is a promising immunotherapy combination for the treatment of NSCLC. In particular, tiragolumab plus atezolizumab showed a considerable improvement in ORR and PFS compared with placebo plus atezolizumab in patients with chemotherapy-naive, PD-L1-positive, recurrent and metastatic NSCLC. Tiragolumab plus atezolizumab was well tolerated, with a safety profile generally similar to that of atezolizumab alone (195).
It is important to note that the safety profile of these ICIs in combination may differ from when ICIs are coupled to chemotherapy, particularly regarding immune-related adverse events. Plus, despite initial encouraging results in certain patients, many lung tumors exhibit intrinsic resistance to immunotherapies. Hence, the next challenge will lie in identifying rational combinations that can enhance treatment responses and delay the onset of resistance (196).
Latest immunotherapy-based strategies for NSCLC
To elicit an effective immune response in so-called ‘immune desert tumors’, which are devoid of lymphocyte infiltration, little TMB, and low PD-L1 expression, the main challenge is to attract effector T cells to the TME and present them with the tumor antigens. Among those novel approaches, the two most promising include the adoptive cell transfer (ACT), based on autologous T cells derived from TILs, and CAR-T therapies (197). However, identifying NSCLC-specific or unique cell surface antigens is necessary for the exploitation of CAR-T approach. TAAs that are frequently found to be overexpressed in NSCLC include the Mucin 1 (MUC-1), the carcinoembryonic antigen (CEA), the New York esophageal squamous cell carcinoma 1 (NY-ESO), and the melanoma-associated antigen 3 (MAGE-A3) (198–200). The problem is that these antigens are also commonly expressed in normal lung cells. As a consequence, these TAAs are potentially not very much immunogenic and can be also used by the tumor to develop immune tolerance, which results in reduced responsiveness to ICIs (201).
On the other hand, TSAs are unique to cancer cells and result from nonsynonymous somatic mutations. These TSAs represent ideal targets for cellular immunotherapy (202, 203). NSCLC, like other tumors with high TMB, have been shown to possess a significant number of TSAs arising from various somatic mutations, including driver genes like tumor protein 53 (TP53), Kristen Rat sarcoma viral oncogene homolog (KRAS), cyclin-dependent kinase inhibitor 2A (CDKN2A), AT-Rich Interaction Domain 1A (ARID1A), Neurogenic locus notch homolog protein 1 (NOTCH1), myelocytomatosis oncogene (MYC), SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4 (SMARCA4), and retinoblastoma 1 (RB1) (204, 205). TILs can recognize these neoantigens, and their density has been linked to a more favorable prognosis, higher cytotoxic T lymphocyte (CTL) content, and increased benefit from ICI (206, 207). Despite the inherent challenges, recent technological advancements, such as the MANA Expansion of Specific T cells platform, has already been used to detect and monitor peripheral and intratumoral MANA-specific T cell responses in NSCLC patients with acquired resistance to checkpoint blockade (208). Notably, CTLs that are specifically directed against peptides derived from oncogenic driver mutations, such as TP53 R248L and BRAF N581I (208, 209), have been detected, offering potential avenues for new targeted immunotherapies in lung cancer.
Cancer vaccines may augment the body’s T cell and B cell response against TAA or TSA. There are different mechanisms to stimulate the immune system and generate an effective anti-tumor response (210). One common approach involves the use of DCs collected from a patient’s blood, loaded with TSAs derived from the tumor, and then administered back to the patient. These DCs migrate to lymphoid organs, where they interact with host immune cells, including T- and B- cells, leading to their activation and enhancing of the immune response against tumor cells (211). A different strategy entails using whole-cell preparations obtained from cancer cells that have undergone either inactivation or genetic modification. The inactivated cancer cells are recognized by the host immune system, triggering an immediate nonspecific inflammatory response (212). Induced pluripotent stem cells (iPSCs) derived from primary fibroblasts exhibit genetic and transcriptomic similarities with cancer tissues, encompassing numerous cancer-associated genes as well as over 100 TAAs and TSAs, which are protein markers detectable by the immune system (213, 214). Recently, research in mice has explored the use of iPSCs as a source of tumor- and patient-specific antigens to direct the immune system to target cancer (213). Kooreman further developed the idea of an iPSC-based anticancer vaccine by combining irradiated autologous mice iPSCs with the Toll-like receptor 9 (TLR9) agonist, CpG oligodeoxynucleotide. Using autologous iPSCs helped reduce immune reactions caused by MHC mismatches, while the addition of the CpG adjuvant enhanced the activation of antigen-presenting cells like dendritic cells (213, 215). In a humanized mouse model of lung cancer, vaccination with iPSCs and CpG led to an increase in splenic APCs, cytotoxic T cells, circulating effector/memory CD4+ and CD8+ T cells, and tumor-infiltrating CD8+ T cells, while reducing regulatory T cells (Tregs). This immune response contributed to effective tumor growth suppression, with tumor antigen-specific T cells playing a pivotal role. This was demonstrated by the protective effect seen after transferring spleen T cells from vaccinated mice to unvaccinated ones. The immunity triggered by iPSCs was thought to stem from shared gene expression patterns between iPSCs and lung adenocarcinoma stem cells (213, 216). With an alternative approach, viral or bacterial -based cancer vaccines are used to directly activate the immune response against TSAs and TAAs: the antigens are either expressed by a virus or bacteria, delivered to infect the host cells, which process the antigen and present it to activate the T cell response (217).
Numerous clinical trials are currently investigating different vaccines targeting specific NSCLC antigens like MAGE-A3, CEA, mesothelin, KRAS proto-oncogene (KRAS), New York esophageal squamous cell carcinoma 1 (NY-ESO-1), and telomerase (TERT), as well as immunomodulatory enzymes such as indoleamine 2,3-dioxygenase (IDO) and arginase-1, in lung cancer patients (199, 218). Some of these cancer vaccines are combined with ICIs in phase I/II studies (NCT04908111, NCT02879760, NCT03562871, NCT05202561, NCT04117087, NCT01935154; NCT03689192, NCT03970746, NCT02187848) (219–227).
Importantly, the success of COVID-19 messenger RNA (mRNA) vaccines has shown how effective this type of vaccine can be, how fast it can reach a clinical stage and the ease of production at global scale (228). This approach involves the use of synthetic mRNA sequences, either alone or in combination with other molecules, which encode proteins found in cancer cells. The expression of these proteins triggers immune reactions targeting tumor antigens, such as the production of antibodies and cytotoxic T cells (229). Early studies, like the NCT00004604 trial conducted at the beginning of 2000s to evaluate mostly safety and dose-limiting toxicities of mRNA-based vaccines using DCs, proved quite ineffective in terms of clinical response and disease progression (230). At present, an upgraded form of these vaccines is being introduced, showing improved efficacy and tolerability. For example, the mRNA-based vaccine CV9201, tested in the NCT00923312 phase I/IIa trial, showed promising results for advanced NSCLC patients (231). Also, the CV9202 mRNA-based vaccine was used in combination with radiation therapy in stage-IV NSCLC patients in the NCT01915524 study showing increased antigen-specific immune responses, and stable disease achieved in 46.2% of patients (232). On balance, while mRNA vaccines offer substantial benefits over conventional options thanks to their high efficacy, minimized toxicity, accelerated manufacturing, and reliable administration, there are some potential limitations that could overshadow their widespread use (233). For example, these constraints include lack of stability and reduced translation rates upon delivery into target cells, mostly due to inadequate methylation of the mRNA, or small impurities in the preparation process (234). Intrinsic immunogenicity of the construct can also impair the stability of the mRNA vaccine, and ultimately cause decreased translation. Increasing the capping efficiency and preventing de-capping through the incorporation of modified nucleosides or pseudouridine can actually avoid recognition by the innate immune system and prevent mRNA destruction (233). Inefficiency of in vivo delivery in target tissues, including the lungs, is another major issue that limits the potential of mRNA vaccines and requires further optimization of the vectors (viral, non-viral, cell-based, lipid vesicles). A robust targeting of mRNA has been achieved in pulmonary tissues, for example, by the use of nanoparticles (235). Furthermore, the specificity of the mRNA-encoded polypeptide, although tailored to generate only the specific antigen of interest, may not only limit the efficacy of inducing a robust immune response in the cancer tissue but can also result in serious side effects, typically including uncontrolled inflammation and anaphylaxis (236).
The YL202/BNT326 is a novel antibody-drug conjugate ADC designed by BioNTech to target HER3, combining an anti-HER3 monoclonal antibody with a topoisomerase I inhibitor, YL0010014, linked via a tripeptide linker. The results of the phase I trial conducted on advanced or metastatic non-small cell lung cancer (NSCLC) patients bearing EGFR-activating mutations, as well as HR-positive, HER2-negative breast cancer (BC) that had previously received third-generation tyrosine kinase inhibitors (TKIs) or CDK4/6 inhibitors and at least one line of chemotherapy were recently presented at the American Society of Clinical Oncology (ASCO) 2024 Annual Meeting held in Chicago. Six different doses of YL202/BNT326 were tested, using a dose-escalation approach, followed by additional dosing in selected cohorts. Safety and tolerability were the primary endpoints, with secondary endpoints including pharmacokinetics and efficacy. In terms of efficacy, 46 patients were evaluable for tumor response. At doses levels 3 to 5, the overall response rate (ORR) was 41.0%, and the disease control rate (DCR) was 94.9%. In breast cancer patients, the ORR reached 54.5%, with an impressive DCR of 100% (237).
ATMPs for lung cancer: from ACTs to CAR-Ts
Another version of antigen-specific immunotherapy is the adoptive T cell transfer (ACT) of lymphocytes that exhibit antitumor activity. The concept of ACT involves ex vivo activation of the patient’s own immune cells, before transferring them back to the patient to recognize and eliminate cancer cells (197). ACT therapies encompass different methods, including adoptive transfer of TILs or genetically engineered T cells with retargeted specificity, such as affinity-enhanced αβ-T-cell-receptor (TCR) and CAR (197). Compared to vaccine-based strategies, ACT provides patients with pre-activated effector cells, eliminating the need for T cell priming in patients who have compromised immune systems or have developed immune tolerance to tumor antigens (238). In current strategies for targeting advanced NSCLC, ACT is employed with engineered T cells directed against specific TAA, like NY-ESO-1/LAGE-1, often in combination with ICI (NCT03709706) (239). ACT approaches using TCR show some challenges, however, since they may be susceptible to tumor escape due to immunoediting processes, through which tumor clones develop mechanisms to evade antigen presentation, such as loss of antigenicity and/or loss of immunogenicity. Loss of antigenicity can occur either through the acquisition of defects in the antigen processing and presentation, or through the loss of immunogenic tumor antigens: both mechanisms lead to a lack of immunogenic peptides presented in the context of a peptide/MHC complex (240). Malignant cells also can earn additional immunosuppressive properties, such as expression of PD-L1 or secretion of suppressive cytokines (240). To address this issue in particular, CAR-T cells have been developed as an alternative technology to redirect T cell specificity by recognizing intact cell surface proteins, and so bypassing MHC-mediated antigen presentation (241). CARs are engineered receptors formed by three parts: an extracellular antigen recognition domain, usually a single-chain fragment variant (scFv), a transmembrane domain, and an intracellular T cell activation domain (242). All surface-expressed target molecules represent a potential CAR-triggering epitope, so that the genetic modification of T cells with CARs combines the specificity of antibody-like recognition with the cytotoxic activation of T cells (243).
First, construction of a CAR relies on the identification of a suitable antibody that can effectively target a cell surface molecule of interest (244). Carbohydrates and glycolipid antigens on cancer cells are also suitable targets, since they can be recognized by CARs engineered with the antigen-recognition domain derived from monoclonal antibodies (245). The simplest level of CAR structure contains an extracellular domain, connected through a hinge to the transmembrane domain, and an intracellular signaling domain (246). The CAR ectodomain is designed to specifically recognize an antigen on the cancer-cell membrane. Upon engagement, the receptor triggers downstream signaling, resulting in CAR-T cell activation. This leads to a complex network of events comprising transcription factor expression, cell proliferation, survival, and cytokine release which culminates in the execution of a cytotoxic program against the target cell (246).
A hinge sequence is used to connect the ectodomain to the transmembrane (TM) region of the CAR. The hinge region’s length is a crucial parameter in CAR design, since it can modify the flexibility of the scFv and its ability to interact with hidden or distant epitopes on the antigen. On the other end, the specific makeup of the hinge-scFv moiety can be detrimental to CAR efficacy by inducing an unwanted tonic signal of the CAR triggered also in the absence of the antigen, resulting in T cell exhaustion (246). The transmembrane domain serves as an anchor of the CAR to the T cell membrane. Although this domain can also be relevant for CAR-T cell function influencing CAR expression level, stability, signaling, and dimerization with endogenous signaling molecules (247–250). Most transmembrane domains are derived from natural proteins including CD3ζ, CD4, CD8α, or CD28. Endodomain, which transmits the binding signal from the tumor antigen into the T cell, has received most of its attention over the years, resulting in multiple CAR generations. In fact, since the initial development of CARs in 1989, CAR-T constructs can be divided into five generations according to the structure of the endodomain (250, 251). The first-generation CAR design is formed only by Fcγ (the γ-chain from FcϵRI) or CD3ζ (ζ- ζ-chain of the TcR complex) intracellular domain. The durability and persistence of these first generation CARs was not robust because they produced limited amounts of interleukin-2 (IL-2), rendering those CAR T cell dependent on exogenous administration of IL-2 (251). Hence, costimulatory domains were added to the CAR constructs to create the second-generation CARs, which achieved to fully activate T cell proliferation and induce persistent cytotoxicity. Specifically, the insertion in the endodomain of CD28 or 4-1BB was enough to block apoptotic signaling via adequate IL-2 synthesis and complete stimulation of T cells (252). The costimulatory domains CD28 and 4-1BB differ in their functional and metabolic profiles. CARs with CD28 domains differentiate into effector memory T cells and primarily use aerobic glycolysis, while CARs possessing the 4-1BB domain differentiate into central memory T cells and display increased mitochondrial biogenesis and oxidative metabolism (250, 252–254). Thanks to these coreceptors, which yield enhanced persistence, decreased differentiation and exhaustion, prolific expansion, increased cytotoxicity, second generation of CAR constructs have proven to be more effective, compared to first generation CARs, which only included the CD3ζ sequence (252, 253). To further increase the cytokine production and cytotoxicity against tumor cells, a third generation of CARs was designed by adding an extra intracellular signaling sequence in the costimulatory domain such as CD134 or CD137 (251, 252). However, different studies led to ambiguous performance results of third-generation CAR-T use, indicating that clinical outcomes were not always improved compared to the second generation. Moreover, third-generation CAR-T cells have been reported to worsen T cell exhaustion or activation-induced cell death (AICD) (255, 256). A fourth-generation CARs followed based on the second-generation constructs but with the addition of an ILs expression cassette, resulting in the so-called T cells redirected for universal cytokine-mediated killing (TRUCKs), which exhibited remarkable efficacy against different solid tumor types (257). Specifically, IL-12 enhances the response of innate and adaptive immune cells, IFN-γ secretion and the expression of granzyme B and perforin by T cells and NK cells, and suppresses tumor-induced T-regulatory (T-reg) cell proliferation (258, 259). Finally, a fifth-generation CAR-T cells were created to avoid host immune rejection or graft-vs.-host disease against transplanted CAR-T cells (260, 261). These advanced CARs are based on the second-generation of constructs, but they contain a truncated cytoplasmic IL-2 receptor β-chain domain with a binding site for the transcription factor STAT3: the antigen-specific activation of this receptor simultaneously triggers TCR (through the CD3ζ domains), costimulatory (CD28 domain), and cytokine (JAK– STAT3/5) signaling (262).
So far, various NSCLC-associated antigens have been selected as potential candidates for CAR-T cell therapy (263). For instance, CAR-T cells directed against CEA and MUC1, two TSA that are highly expressed in lung cancer, are being evaluated for safety and efficacy in the NCT02349724 (264), NCT04348643 (265), NCT03525782 (266), NCT02587689 (267), NCT05239143 (268) clinical trials (9, 269, 270). Alternatively, the infiltration of CAR-T cells into the tumor is being improved by expressing the C-X-C chemokine receptor type 5 (CXCR5). Also known as CD185, it is the only known receptor for the chemokine ligand 13 (CXCL13), which is abundant in the TME of many NSCLC tumors (271). CAR-T cells engineered to express CXCR5 are now being studied in the trial NCT05060796 (272) for enhanced lymphocytes’ infiltration in the tumor and activation against CXCL13-expressing tumor cell (9, 273–275). Overexpression of EGFR, resulting from EGFR gene amplification and/or mutations, has been detected in a variety of human cancers, including over 60% of NSCLC. This overexpression is linked to tumor recurrence, the formation of new blood vessels, and metastasis (276). The extracellular domain of EGFR present on the surface of tumor cells forms a highly immunogenic and tumor-specific epitope, making it a promising target for CAR-T therapy in NSCLC. Recombinant anti-EGFR CAR-T cells have been developed with specific cytolytic activity against EGFR-positive tumor cells (277). Co-incubation of EGFR-positive tumor cells with anti-EGFR CAR-T cells resulted in the release of high levels of cytokines such as IL-2, IL-4, IL-10, TNF-α, and interferon (IFN)-γ, within 24 hours (276, 278). In vivo experiments have shown that these CAR-T cells can proliferate against NSCLC and are found in high proportions among CD8+ cytotoxic T-lymphocyte populations (278). Clinical trials are underway to assess the efficacy and safety of anti-EGFR CAR-T cells in treating advanced NSCLC patients with EGFR-positive tumors (279). For example, a phase I clinical trial at Sun Yat-sen University evaluated anti-EGFR CAR-T cells, modified for the expression of CXCR5, in EGFR-positive patients with advanced NSCLC: among 11 patients being assessed receiving different doses, two showed a partial response, and five remained stable for eight months (280). Another phase I clinical study (NCT03182816) investigated anti EGFR CAR-T cell therapy in NSCLC patients. This trial revealed that EGFR-CAR T cell therapy was feasible and safe in treatment of EGFR-positive advanced relapsed/refractory NSCLC patients, with a progression-free survival of 7.13 months (281).
Mesothelin (MSLN) is another promising CAR target, and assessment of safety and feasibility of anti-MSLN CAR-T cell therapy in the clinical trial NCT02414269 (282, 283) conducted at the Memorial Sloan Kettering Cancer Center is under way. The study focuses on anti-MSLN CAR combined with inducible caspase 9-M28z (iCasp9M28z) suicide system expression and shows promise as a targeted therapy (282). However, the phase I/II trial NCT01583686 led by the US National Cancer Institute (NCI) and assessing anti-MSLN CAR-T cell therapy for patients with MSLN-positive metastatic lung cancer was discontinued due to slow and insufficient patient accrual (284). Nevertheless, there is ongoing interest in the potential of intravenous administration of mRNA-engineered T cells to express anti-MSLN CAR temporarily (285).
Researchers have also developed ROR1-specific CAR-T cells using lentiviral vectors encoding ROR1, scFv/4-1BB/CD3ζ, and truncated EGFR molecules. This engineered CAR-T cell approach effectively eliminated ROR1-positive tumor cells in 3D tumors established from A549 (a non–small cell lung cancer) cell lines on a biological scaffold and an intact basement membrane (286). To evaluate the safety and anti-tumor effects of autologous anti-ROR1 CAR-T cells, a phase I clinical study (NCT02706392) conducted by the Fred Hutchinson Cancer Research Center involved patients with advanced, ROR1-positive, stage IV NSCLC (287). Furthermore, exciting findings from Wallstabe et al. demonstrated the effectiveness of anti-ROR1 CAR-T cells in eliminating both NSCLC and TNBC cells, as shown in organoid tumor models (286) This evidence highlights the potential of anti-ROR1 CAR-T cell therapy as a promising and innovative strategy for treating NSCLC, providing new hope for patients in need of additional treatment options.
Although CAR-T immunotherapy for solid tumors like NSCLC is still in its infancy and has faced limited achievements, it still holds great potential to manage cancers at an advanced stage (288). Importantly, some aspects contribute more than others to hinder CAR-T therapy efficacy in solid tumors:
1. On-target/off-tumor toxicity: CAR-T cells may inadvertently target healthy cells expressing the same antigen as cancer cells, leading to unintended side effects and toxicities (289).
2. Neurological toxicity: some patients may experience neurologic complications due to the activation of CAR-T cells in the central nervous system (289).
3. Cytokine release syndrome (CRS): CAR-T cells can trigger an excessive release of cytokines, causing systemic inflammation and potentially life-threatening complications. CRS is characterized by releasing various inflammatory cytokines upon T cell activation and antigen recognition. This immune response can surge pro-inflammatory molecules like TNF-α, C-reactive protein, IL-2, IL-6, IL-8, and IFN-γ. Consequently, patients may experience fever, fatigue, loss of appetite, hypotension, and, in severe cases, multi-organ dysfunction or even sudden death due to the cytokine storm (289, 290). Early detection and effective management of CRS are vital to safeguard patient well-being during CAR-T cell therapy. Substantial evidence supports using IL-6 pathway inhibitors like tocilizumab or siltuximab as effective treatments for CRS (291, 292). Additionally, infliximab, a TNF-α inhibitor, is a viable option for managing cytokine-related complications (290). By carefully monitoring and promptly intervening, healthcare professionals can minimize the impact of CRS and enhance the overall safety and success of CAR-T cell therapy for patients.
4. The lack of reliable TSA: identifying antigens unique to cancer cells is challenging in solid tumors, reducing the specificity of CAR-T cell therapy.
5. Immunosuppressive TME: solid tumors often create an immune-suppressive environment, hindering the efficacy of CAR-T cells (293). The tumor-associated stroma may form a stumbling block against the entry of T cells, which are already increasingly dimmed by dysregulation of adhesion molecules, aberrant tumor-related vasculature, and mismatching of chemokines and their receptors. Furthermore, TME is characterized by restricted nutrient availability, acidosis, and local hypoxia.
6. Low levels of lymphocytes infiltration within tumor tissue: CAR-T cells may struggle to penetrate and accumulate within the tumor, limiting their ability to target cancer cells effectively.
7. Tumor antigen escape: cancer cells can downregulate or lose the target antigen, evading recognition and destruction by CAR-T cells. A strategy to enhance the function of CAR-T can be reached by targeting TAAs while treating with anti-PD-1 blocking antibodies in combination (294). The co-application of prostate stem cell antigen (PSCA)-targeted and mucin 1 (MUC1)-targeted CAR-T cells effectively eradicated cancer cells in individuals diagnosed with PSCA and MUC1 positive non-small cell lung cancer (294).
Addressing these barriers is crucial to unlock CAR-cell therapy to its full potential and improve patient outcomes. Research efforts are ongoing to overcome these challenges and advance the application of CAR-T cell therapy in the context of solid malignancies. Enhancing CAR-T cell infiltration into the TME is a critical challenge in solid tumors, including NSCLC. Specific strategies include structurally altering CAR-T cells combined with targeted therapy, radiotherapy, or chemotherapy (295). Chemotherapy has the ability to alter TME and enhance the effectiveness of CAR-T cell therapy. In particular, neoadjuvant therapies, have shown the ability to enhance the effectiveness of GD2 CAR-T cells by decreasing MDSCs in the tumor (295). In a phase I clinical trial, combined therapy involving CAR-T cells along with paclitaxel and cyclophosphamide revealed clinical improvements in 21 of the 28 patients who had previously experienced unsuccessful paclitaxel therapy (296). Murty et al. in 2020 demonstrated in a glioblastoma model that radiotherapy enabled the swift movement of CAR-T cells from the vascular system into the TME, as well as their amplification within the TME, leading to enhanced and prolonged immune responses (297). In order to increase the CAR-T infiltration into the TME, researchers are actively exploring novel approaches. In particular, dual-receptor CAR-T cells have been designed to improve specificity while enhancing T-cell activation and persistence in the TME. These constructs are engineered to recognize two different TAAs simultaneously. By targeting multiple antigens, dual-receptor CAR-T cells can mitigate antigen escape and improve infiltration through stronger, more sustained signaling (298). A second strategy focuses on optimizing the delivery methods for CAR-T cells, directly injecting CAR-T cells into or near the tumor site. Intratumoral delivery reduces systemic exposure, potentially minimizing off-tumor toxicity and enhancing CAR-T cell concentrations at the target site. This strategy has shown promise in preclinical models of glioblastoma and pancreatic cancer and is currently under investigation in lung cancer (299, 300). To further enhance T-cell migration, researchers are modifying CAR-T cells to express chemokine receptors to exploit the chemokine profile of the TME as chemoattractant. For example, expressing CXCR2 allows CAR-T cells to home more effectively to tumors secreting chemokines like CXCL1 and CXCL8. Similar modifications, such as introducing CXCR5 to target CXCL13-rich NSCLC tumors, have shown significant improvements in infiltration and therapeutic outcomes (273, 301, 302). Additionally, combining CAR-T cell therapy with immune checkpoint inhibitors, such as anti-PD-1 or anti-CTLA-4 antibodies, has shown promise in preclinical and early-phase clinical trials for NSCLC. This combination approach can help overcome immune suppression and enhance T-cell function (303, 304). The application and testing of novel strategies through forthcoming clinical trials will be key to assess their real benefits for the broadest population of NSCLC patients.
Discussion
NSCLC, the predominant form of lung cancer, is a major contributor to cancer-related deaths (4). Over time, however, there have been substantial advancements in the treatment of NSCLC, particularly due to the emergence of targeted therapies that have revolutionized the field toward individualized medical care. Undoubtedly, TKIs have brought about a revolutionary shift in the treatment of individuals with specific genetic mutations (305). Within the realm of targeted therapy, EGFR stands as a quintessential model: activating mutations found in the EGFR gene are widespread among a significant portion of individuals afflicted with NSCLC, and can be effectively targeted by TKIs such as erlotinib, gefitinib, and osimertinib, which have markedly improved PSF and OS of many patients (306). Similarly, ALK rearrangements respond well to alectinib and lorlatinib, while MET, BRAF, and ROS1 mutations are targeted by crizotinib, vemurafenib, and dabrafenib (307).
Regrettably, the group of individuals that respond well to TKIs accounts for just 15–20% of all NSCLC patients, and although targeted treatments show promise at first, leading to longer PFS and improved OS, life expectancy is hindered by the development of pharmacological resistance mechanisms for most patients (11). Consequently, the main goal in NSCLC investigations is the design and testing of innovative therapies that impact PFS and OS, taking into account the major role played by the TME in sustaining therapeutic resistance and tumor progression (12). Understanding the tumor TME is key, as it has a substantial impact on how effective these treatments are. Importantly, the presence of neoantigens, TAAs, TSAs, and TILs can define an “inflamed” TME in NSCLC, underscoring its significance in amplifying the immune system’s ability to target tumors by enhancing the effectiveness of neoantigen-based immunotherapies (271). Additionally, the presence of TLS within the TME - organized aggregates of immune cells resembling germinal centers - can further enhance, refine and sustain local and systemic immune responses. TLS have been associated with better prognoses and may play a crucial role in the success of immunotherapies (308).
Immunotherapy has become a cornerstone in NSCLC treatment, with ICIs leading the charge, specifically in those patients without any driver mutations (17). ICIs such as nivolumab and pembrolizumab, which target PD-1, and ipilimumab, targeting CTLA-4, have dramatically improved survival rates for patients with advanced NSCLC (309). The success of these therapies has spurred the development of new ICIs targeting other checkpoint molecules like LAG-3, TIM-3, and TIGIT (310). These novel ICIs aim to overcome resistance mechanisms and further potentiate the immune response against cancer, highlighting the dynamic and expanding landscape of immunotherapy in NSCLC. Yet, a considerable number of patients do not benefit from immunotherapies. In particular, the failure of standard immunotherapy is a frequent issue in the context of a ‘cold’ TME with a lack of neoantigens, diminished T cell numbers, and minimal PD-L1/PD-1 expression (23).
The exploration of novel immunotherapeutic approaches is ongoing, with TSA-directed strategies at the forefront. Vaccines targeting TSAs derived from NSCLC common drivers, such as MAGE-A3, CEA, KRAS, NY-ESO-1, and TERT, show great promise in several early trials (311). These new strategies are designed to boost the immune response of the body to target cancer cells in a precise manner and to defeat the immune escape mechanisms triggered by tumors. ACT-based therapies are gaining momentum in the panorama of clinical experimentation. ACT involves infusing patients with T cells able to recognize and attack cancer cells. ACT includes TCR and CAR-T cell therapy, which features genetically modified T cells to express receptors specific to intracellular or membrane associated tumor antigens, respectively. These therapies represent a significant leap forward in the quest for effective and durable cancer treatments (312). CAR-T cell therapy, in particular, has garnered attention for its potential in treating NSCLC. Over successive generations, CAR-T cells have been refined to enhance their efficacy and safety, with modifications aimed at improving persistence, reducing toxicity, and boosting antitumor activity (313). While early results are promising, challenges remain, particularly in managing on-target, off-tumor effects and ensuring sustained responses. Nevertheless, the potential of CAR-T cell therapy to significantly impact NSCLC treatment is undeniable specially in vivo transient expression of mRNA in mRNA-engineered T cells, which is a highly promising approach (314). This method eliminates the need to isolate patient cells, by simplifying the production process and allowing for a purely off-the-shelf solution. Another promising format relies on TRUCKs, allowing cytokines and other immunological molecules to be delivered locally at the tumor site to restrain TME immunosuppressive cues and elicit immune responses previously kept in check. Indeed, ACTs are also suited to be combined with ICIs to boost their own activity in difficult contests, promote epitope spreading across APCs and engage preexisting adaptive and innate immunity against tumor cells. In conclusion, understanding the complexities of the TME and leveraging novel treatment strategies that exploit the ability of immune cells to recognize TAAs/TSAs is crucial to improve patient outcomes. Boosted by this primary goal, the treatment landscape for NSCLC is rapidly advancing toward the integration of targeted therapies and innovative immunotherapeutic approaches, while new clinical trials are increasingly looking into how ACT and CAR-T cell therapy can influence NSCLC by targeting diverse surface antigens. In order to guide the reader through the complex scenario of NSCLC therapy options, we provide a brief yet informative summary indicating the reference trials that allowed drug registration in Figure 1. As research continues to unravel the intricacies of NSCLC and its microenvironment, the promise of achieving more effective and durable responses and a cure becomes increasingly attainable. In this review, we adeptly merge the literature and highlight the critical hurdles that must be tackled in order to impact effectively on patients’ health, with a specific emphasis on the complexities associated with non-small cell lung cancer, by implementing novel and smarter immunotherapy approaches into clinical practice. In Figure 2 we have summarized the relationships occurring between the tumor microenvironment (TME) and the associated mechanisms of resistance, indicating the approaches currently under clinical testing to overcome resistance in NSCLC patients.
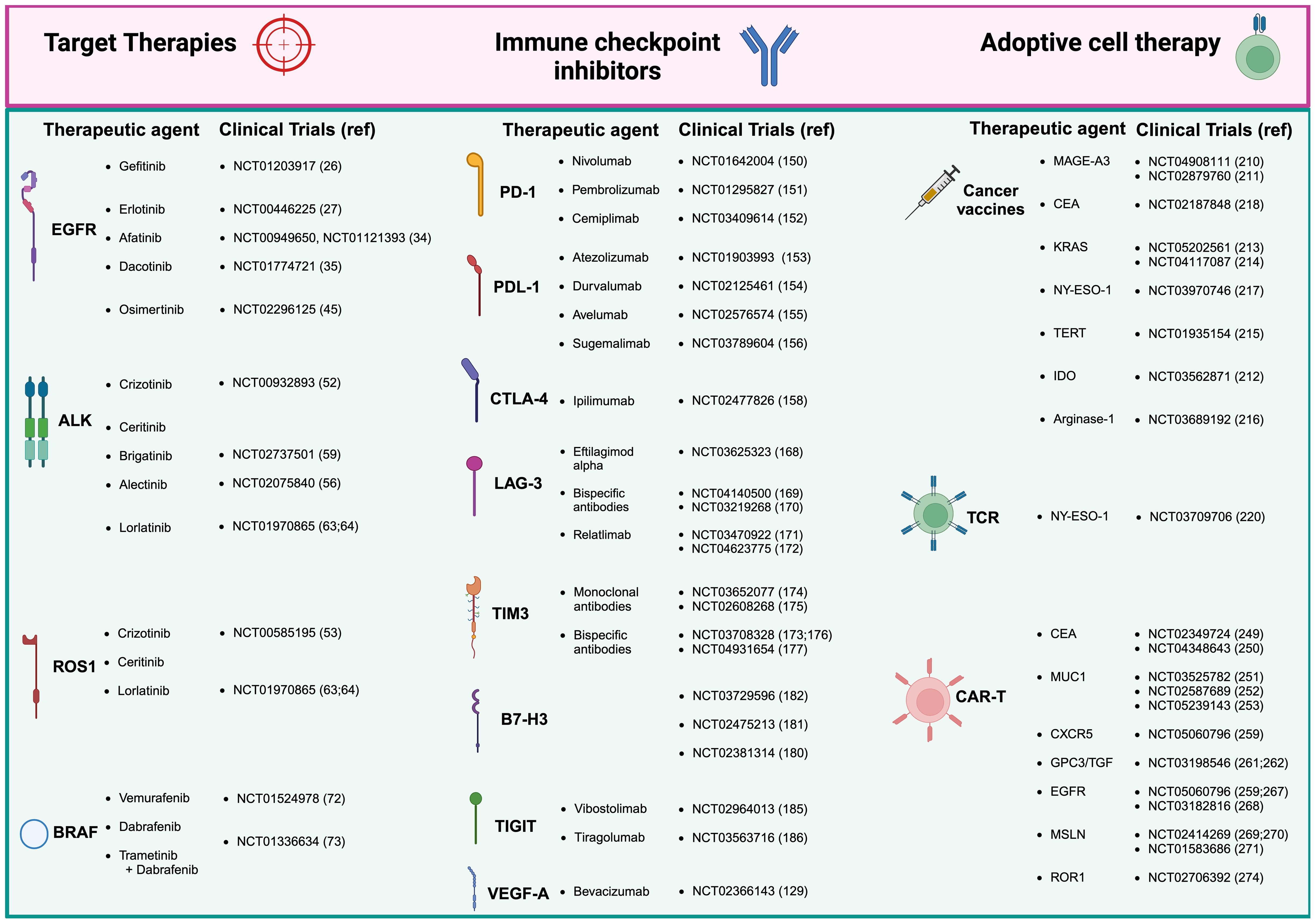
Figure 1. Therapeutic approaches and clinical trials for non-small cell lung cancer (NSCLC). Current treatment of NSCLC range mainly in three areas: tyrosine kinase inhibitors (TKIs), which remain the gold standard for many patients; immune checkpoint inhibitors (ICIs), which have offered exceptional survival benefits to selected patients; adoptive cell therapy, representing the latest frontier in immunotherapy. Adoptive cell therapy divides then into three sub-areas of therapeutic interventions: cancer vaccines, T cell receptor (TCR) based therapy, and the most innovative antigen receptor chimeric (CAR) T cell therapies, which has shown great promise in NSCLC. Created in BioRender. Mazza, M. (2025) https://BioRender.com/o50k263.
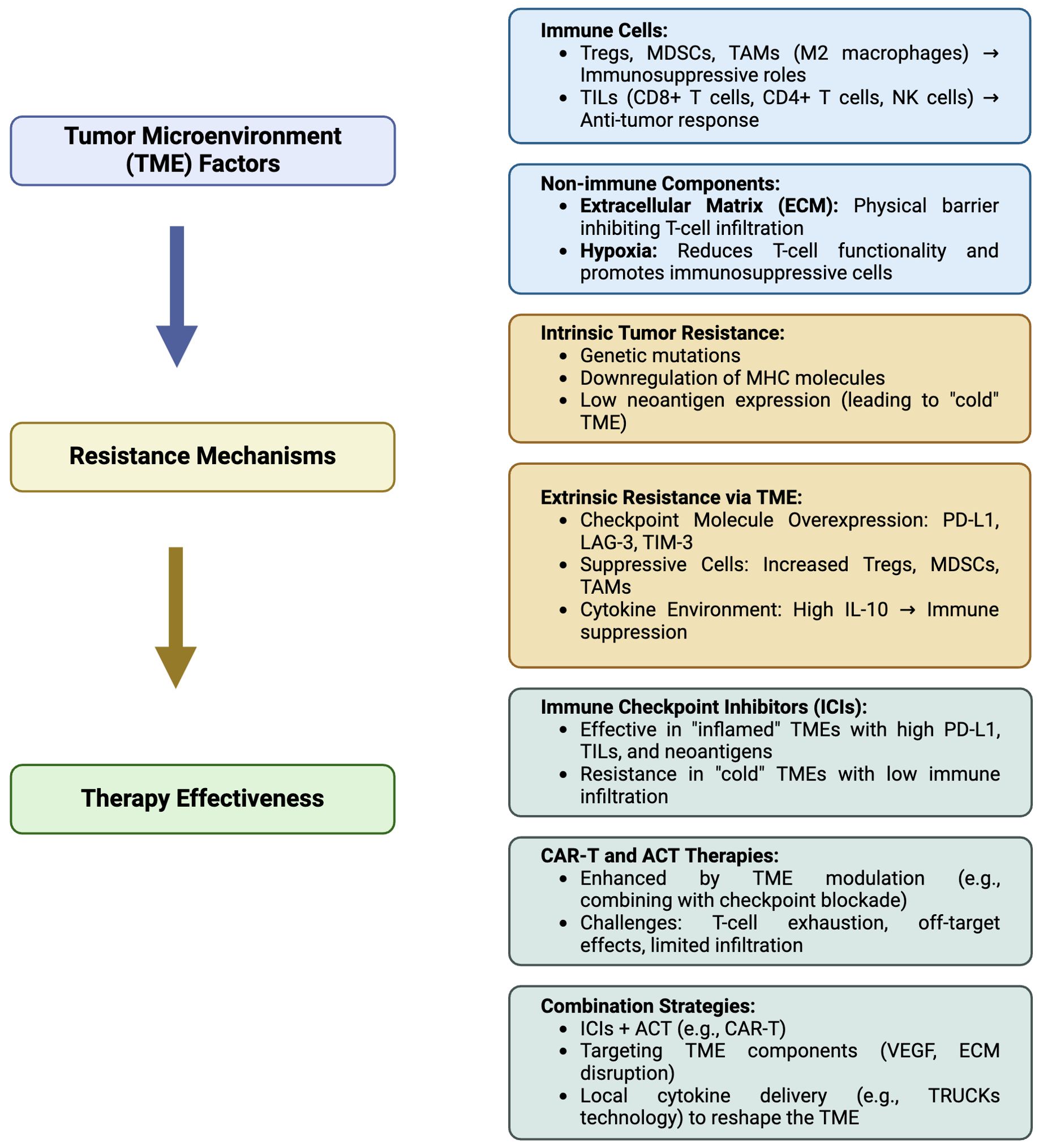
Figure 2. Summarizing flowchart of the relationship between the tumor microenvironment (TME) and associated resistance mechanisms, affecting immunotherapy of non-small cell lung cancer (NSCLC). Tregs, Regulatory T cells; MDSC, Myeloid-derived suppressor cells; TAMs, Tumor-associated macrophages (TAM)-M2; TILs, Tumor-infiltrating lymphocytes; NK, natural killer; MHC, Major histocompatibility complex; PD-L1, Programmed cell death ligand-1; LAG-3, Lymphocyte-activation gene 3; TIM-3, T-cell immunoglobulin, mucin-domain containing-3; IL-10 interleukin-10; ACT, adoptive T cell transfer; CAR-T, Chimeric antigen receptor (CAR)-T cells; VEGF, vascular endothelial growth factor; TRUCKs, T cells redirected for universal cytokine-mediated killing. Created in BioRender. Mazza, M. (2025) https://BioRender.com/m08u912.
Author contributions
ADL: Conceptualization, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. LM: Methodology, Visualization, Writing – review & editing. AG: Methodology, Visualization, Writing – review & editing. MZ: Methodology, Writing – review & editing. RM: Methodology, Writing – review & editing. CC: Methodology, Writing – review & editing. SB: Methodology, Project administration, Writing – original draft. AD: Writing – review & editing. LC: Writing – review & editing. PBS: Visualization, Writing – review & editing. LP: Conceptualization, Validation, Writing – original draft, Writing – review & editing. FN: Conceptualization, Project administration, Supervision, Writing – original draft. FB: Methodology, Writing – review & editing. MJ: Writing – review & editing. HC: Writing – review & editing. CM: Visualization, Writing – review & editing. LG: Visualization, Writing – review & editing. PU: Writing – review & editing. MM: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Italian Ministry of Health within the Ricerca Corrente Reti ACC - CAR-T project: RCR-2023-23684268.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roces B. Patients with advanced lung cancer: quality of life and perception of dyspnea. (Dissertations). University of San Diego (2014) 471. doi: 10.22371/07.2014.023
3. Jezdic S, Planchard D, Novello S, Peters S, Califano R, Douillard JY, et al. Non-small-cell lung cancer: A guide for patients(2019). Available online at: https://www.esmo.org/content/download/7252/143219/1/EN-Non-Small-Cell-Lung-Cancer-Guide-for-Patients.pdf (Accessed January 2025).
4. Wang W, Liu H, Li G. What’s the difference between lung adenocarcinoma and lung squamous cell carcinoma? Evidence from a retrospective analysis in a cohort of Chinese patients. Front Endocrinol. (2022) 13:947443. doi: 10.3389/fendo.2022.947443
5. Health. NC for CDP and HP (US) O on S and. The Health Consequences of Smoking—50 Years of Progress (2014). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK179276/.
6. Shehata SA, Toraih EA, Ismail EA, Hagras AM, Elmorsy E, Fawzy MS. Vaping, environmental toxicants exposure, and lung cancer risk. Cancers. (2023) 15:4525. doi: 10.3390/cancers15184525
7. Yang P. Lung cancer in never smokers. Semin Respir Crit Care Med. (2011) 32:010–21. doi: 10.1055/s-0031-1272865
8. Wang J, Liu Q, Yuan S, Xie W, Liu Y, Xiang Y, et al. Genetic predisposition to lung cancer: comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci Rep. (2017) 7:08371. doi: 10.1038/s41598-017-07737-0
9. Lahiri A, Maji A, Potdar PD, Singh N, Parikh P, Bisht B, et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol Cancer. (2023) 22:40. doi: 10.1186/s12943-023-01740-y
10. Li S, de Camargo Correia GS, Wang J, Manochakian R, Zhao Y, Lou Y. Emerging targeted therapies in advanced non-small-cell lung cancer. Cancers. (2023) 15:2899. doi: 10.3390/cancers15112899
11. Mayekar MK, Bivona TG. Current landscape of targeted therapy in lung cancer. Clin Pharmacol Ther. (2017) 102:757–64. doi: 10.1002/cpt.v102.5
12. Wang L, Jia Q, Chu Q, Zhu B. Targeting tumor microenvironment for non-small cell lung cancer immunotherapy. Chin Méd J Pulm Crit Care Med. (2023) 1:18–29. Available online at: https://www.sciencedirect.com/science/article/pii/S2772558823000014 (Accessed January 2025).
13. Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer. (2019) 19:9–31. doi: 10.1038/s41568-018-0081-9
14. Wang L, Jia Q, Chu Q, Zhu B. Targeting tumor microenvironment for non-small cell lung cancer immunotherapy (2023). Available online at: https://www.sciencedirect.com/science/article/pii/S2772558823000014 (Accessed January 2025).
15. Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. (2003) 21:807–39. doi: 10.1146/annurev.immunol.21.120601.141135
16. Gupta SL, Basu S, Soni V, Jaiswal RK. Immunotherapy: an alternative promising therapeutic approach against cancers. Mol Biol Rep. (2022) 49:9903–13. doi: 10.1007/s11033-022-07525-8
17. Mamdani H, Matosevic S, Khalid AB, Durm G, Jalal SI. Immunotherapy in lung cancer: current landscape and future directions. Front Immunol. (2022) 13:823618. doi: 10.3389/fimmu.2022.823618
18. Gutic B, Bozanovic T, Mandic A, Dugalic S, Todorovic J, Stanisavljevic D, et al. Programmed cell death-1 and its ligands: Current knowledge and possibilities in immunotherapy. Clinics. (2023) 78:100177. doi: 10.1016/j.clinsp.2023.100177
19. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian A, Eder JP, et al. Pembrolizumab for the treatment of non–Small-Cell lung cancer. N Engl J Med. (2015) 372:2018–28. doi: 10.1056/nejmoa1501824
20. Velcheti V, Rai P, Kao YH, Chirovsky D, Nunes AT, Liu SV. 5-year real-world outcomes with frontline pembrolizumab monotherapy in PD-L1 expression ≥ 50% Advanced NSCLC. Clin Lung Cancer. (2024) 25:502–508.e3. doi: 10.1016/j.cllc.2024.05.002
21. Gijtenbeek RGP, Damhuis RAM, van der Wekken AJ, Hendriks LEL, Groen HJM, van Geffen WH. Overall survival in advanced epidermal growth factor receptor mutated non-small cell lung cancer using different tyrosine kinase inhibitors in The Netherlands: a retrospective, nationwide registry study. Lancet Reg Heal - Eur. (2023) 27:100592. doi: 10.1016/j.lanepe.2023.100592
22. Chi SA, Yu H, Choi YL, Park S, Sun JM, Lee SH, et al. Trends in survival rates of non–small cell lung cancer with use of molecular testing and targeted therapy in Korea, 2010-2020. JAMA Netw Open. (2023) 6:e232002. doi: 10.1001/jamanetworkopen.2023.2002
23. Wang DR, Wu XL, Sun YL. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct Target Ther (2022) 7:331. doi: 10.1038/s41392-022-01136-2
24. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. (2018) 553:446–54. doi: 10.1038/nature25183
25. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. (2014) 311:1998–2006. doi: 10.1001/jama.2014.3741
26. Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol. (2014) 6:a020768. doi: 10.1101/cshperspect.a020768
27. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med. (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
28. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. (2004) 304:1497–500. doi: 10.1126/science.1099314
29. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nat Rev Clin Oncol. (2010) 7:493–507. doi: 10.1038/nrclinonc.2010.97
30. Douillard JY, Ostoros G, Cobo M, Ciuleanu T, McCormack R, Webster A, et al. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer. (2014) 110:55–62. doi: 10.1038/bjc.2013.721
31. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. (2012) 13:239–46. doi: 10.1016/S1470-2045(11)70393-X
32. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. (2011) 12:735–42. doi: 10.1016/S1470-2045(11)70184-X
33. Mok T,S, Yi-Long W, Sumitra T, Chih-Hsin Y, Da-Tong C, Nagahiro S, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. (2009) 361:947–57. doi: 10.1056/NEJMoa0810699
34. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. (2010) 11:121–8. doi: 10.1016/S1470-2045(09)70364-X
35. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N Engl J Med. (2010) 362:2380–8. doi: 10.1056/NEJMoa0909530
36. Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non–small-cell lung cancer in asia (IPASS). J Clin Oncol. (2011) 29:2866–74. doi: 10.1200/JCO.2010.33.4235
37. Wu Y, Du B, Lv C, Ji X, Lan Y, Yao N, et al. First-generation EGFR-TKI plus bevacizumab and chemotherapy for advanced EGFR-mutated non-squamous non-small-cell lung cancer: a retrospective study. Ann Med. (2023) 55:2243967. doi: 10.1080/07853890.2023.2243967
38. Yang JCH, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. (2015) 16:141–51. doi: 10.1016/S1470-2045(14)71173-8
39. Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. (2017) 18:1454–66. doi: 10.1016/S1470-2045(17)30608-3
40. Paz-Ares L, Tan EH, O’Byrne K, Zhang L, Hirsh V, Boyer M, et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial. Ann Oncol. (2017) ;28:270–7. doi: 10.1093/annonc/mdw611
41. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. (2011) 3:75ra26. doi: 10.1126/scitranslmed.3002003
42. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med. (2005) 352:786–92. doi: 10.1056/NEJMoa044238
43. Okabe T, Okamoto I, Tamura K, Terashima M, Yoshida T, Satoh T, et al. Differential constitutive activation of the epidermal growth factor receptor in non–small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res. (2007) 67:2046–53. doi: 10.1158/0008-5472.CAN-06-3339
44. Xu J, Xu L, Wang B, Kong W, Chen Y, Yu Z. Outcomes in patients with lung adenocarcinoma with transformation to small cell lung cancer after EGFR tyrosine kinase inhibitors resistance: A systematic review and pooled analysis. Front Oncol. (2022) 11:766148. doi: 10.3389/fonc.2021.766148
45. Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med. (2017) 376:629–40. doi: 10.1056/NEJMoa1612674
46. Janne PA, Yang JC-H, Dong-Wan K, David P, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor–resistant non–small-cell lung cancer. N Engl J Med. (2015) 372:1689–99. doi: 10.1056/nejmoa1411817
47. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. (2014) 11:473–81. doi: 10.1038/nrclinonc.2014.104
48. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N Engl J Med. (2017) 378:113–25. doi: 10.1056/nejmoa1713137
49. Viray H, Piper-Vallillo AJ, Widick P, Academia E, Shea M, Rangachari D, et al. A real-world study of patient characteristics and clinical outcomes in EGFR mutated lung cancer treated with first-line osimertinib: expanding the FLAURA trial results into routine clinical practice. Cancers. (2024) 16:1079. doi: 10.3390/cancers16061079
50. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat Med. (2015) 21:560–2. doi: 10.1038/nm.3854
51. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. (2015) 21:3924–33. doi: 10.1158/1078-0432.CCR-15-0560
52. Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, et al. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat Commun. (2017) 8:14768. doi: 10.1038/ncomms14768
53. Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. (2016) 534:129–32. doi: 10.1038/nature17960
54. Zheng ZR, Ku HY, Chen KC, Chiang CJ, Wang CL, Chen CY, et al. Association of smoking and ALK tyrosine-kinase inhibitors on overall survival in treatment-naïve ALK-positive advanced lung adenocarcinoma. Front Oncol. (2023) 13:1063695. doi: 10.3389/fonc.2023.1063695
55. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature. (2007) 448:561–6. doi: 10.1038/nature05945
56. Shaw TA, Dong-Wan K, Kazuhiko N, Takashi S, Lucio C, Myung-Ju A, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. (2013) 368:2385–94. doi: 10.1056/NEJMoa1214886
57. Shaw AT, Riely GJ, Bang YJ, Kim DW, Camidge DR, Solomon BJ, et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol. (2019) 30:1121–6. doi: 10.1093/annonc/mdz131
58. Kwak EL, Yung-Jue B, Ross Camidge D, Shaw AT, Solomon B, Maki R, et al. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med. (2010) 363:1693–703. doi: 10.1056/NEJMoa1006448
59. Soria JC, Tan DSW, Chiari R, Wu YL, Paz-Ares L, Wolf J, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet. (2017) 389:917–29. doi: 10.1016/S0140-6736(17)30123-X
60. Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N Engl J Med. (2017) 377:829–38. doi: 10.1056/NEJMoa1704795
61. Hida T, Nokihara H, Kondo M, Kim YH, Azuma K, Seto T, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. Lancet. (2017) 390:29–39. doi: 10.1016/S0140-6736(17)30565-2
62. Cruz BD, Barbosa MM, Torres LL, Azevedo PS, Silva VEA, Godman B, et al. Crizotinib versus conventional chemotherapy in first-line treatment for ALK-positive non-small cell lung cancer: A systematic review and meta-analysis. Oncol Ther. (2021) 9:505–24. doi: 10.1007/s40487-021-00155-3
63. Shaw AT, Dong-Wan K, Mehra R, Tan DSW, Felip E, Chow LQM, et al. Ceritinib in ALK-rearranged non–small-cell lung cancer. N Engl J Med. (2014) 370:1189–97. doi: 10.1056/NEJMoa1311107
64. Camidge DR, Kim HR, Ahn MJ, Yang JCH, Han JY, Hochmair MJ, et al. Brigatinib versus crizotinib in advanced ALK inhibitor–naive ALK-positive non–small cell lung cancer: second interim analysis of the phase III ALTA-1L trial. J Clin Oncol. (2020) 38:3592–603. doi: 10.1200/JCO.20.00505
65. Beardslee T, Lawson J. Alectinib and brigatinib: new second-generation ALK inhibitors for the treatment of non-small cell lung cancer. J Adv Pr Oncol. (2018) 9:94–101.
66. Karachaliou N, Rosell R. Systemic treatment in EGFR-ALK NSCLC patients: second line therapy and beyond. Cancer Biol Med. (2014) 11:173–81. doi: 10.1586/14737140.2014.896210
67. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discovery. (2016) 6:1118–33. doi: 10.1158/2159-8290.CD-16-0596
68. Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. (2017) 18:1590–9. doi: 10.1016/S1470-2045(17)30680-0
69. Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. (2018) 19:1654–67. doi: 10.1016/S1470-2045(18)30649-1
70. Facchinetti F, Rossi G, Bria E, Soria JC, Besse B, Minari R, et al. Oncogene addiction in non-small cell lung cancer: Focus on ROS1 inhibition. Cancer Treat Rev. (2017) 55:83–95. doi: 10.1016/j.ctrv.2017.02.010
71. Shaw AT, Ou SHI, Yung-Jue B, Ross Cd JSB, Ravi S, Salgia R, et al. Crizotinib in ROS1-rearranged non–small-cell lung cancer. N Engl J Med. (2014) 371:1963–71. doi: 10.1056/NEJMoa1406766
72. Lim SM, Kim HR, Lee JS, Lee KH, Lee YG, Min YJ, et al. Open-label, multicenter, phase II study of ceritinib in patients with non–small-cell lung cancer harboring ROS1 rearrangement. J Clin Oncol. (2017) 35:2613–8. doi: 10.1200/JCO.2016.71.3701
73. Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PloS One. (2013) 8:e82236. doi: 10.1371/journal.pone.0082236
74. Awad MM, Ryohei K, Michele M, Wei L, Ya-Li D, Alexei B, et al. Acquired resistance to crizotinib from a mutation in CD74–ROS1. N Engl J Med. (2013) 368:2395–401. doi: 10.1056/NEJMoa1215530
75. Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non–small cell lung cancer. Clin Cancer Res. (2013) 19:4532–40. doi: 10.1158/1078-0432.CCR-13-0657
76. Marchetti A, Felicioni L, Malatesta S, Sciarrotta MG, Guetti L, Chella A, et al. Clinical features and outcome of patients with non–small-cell lung cancer harboring BRAF mutations. J Clin Oncol. (2011) 29:3574–9. doi: 10.1200/JCO.2011.35.9638
77. Subbiah V, Gervais R, Riely G, Hollebecque A, Blay JY, Felip E, et al. Efficacy of vemurafenib in patients with non–small-cell lung cancer with BRAF V600 mutation: an open-label, single-arm cohort of the histology-independent VE-BASKET study. JCO Precis Oncol. (2019) 3:PO.18.00266. doi: 10.1200/PO.18.00266
78. Planchard D, Besse B, Groen HJM, Hashemi SMS, Mazieres J, Kim TM, et al. Phase 2 study of dabrafenib plus trametinib in patients with BRAF V600E-mutant metastatic NSCLC: updated 5-year survival rates and genomic analysis. J Thorac Oncol. (2022) 17:103–15. doi: 10.1016/j.jtho.2021.08.011
79. Han J, Liu Y, Yang S, Wu X, Li H, Wang Q. MEK inhibitors for the treatment of non-small cell lung cancer. J Hematol Oncol. (2021) 14:1. doi: 10.1186/s13045-020-01025-7
80. Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. (2016) 17:984–93. doi: 10.1016/S1470-2045(16)30146-2
81. Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, et al. Dabrafenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. (2016) 17:642–50. doi: 10.1016/S1470-2045(16)00077-2
82. Hyman DM, Igor P, Subbiah V, Faris JE, Ian C, Jean-Yves B, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. (2015) 373:726–36. doi: 10.1056/NEJMoa1502309
83. Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. (2013) 31:1767–74. doi: 10.1200/JCO.2012.44.7888
84. Zhong J, Yan W, Wang C, Liu W, Lin X, Zou Z, et al. BRAF inhibitor resistance in melanoma: mechanisms and alternative therapeutic strategies. Curr Treat Options Oncol. (2022) 23:1503–21. doi: 10.1007/s11864-022-01006-7
85. Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. (2013) 31:1997–2003. doi: 10.1200/JCO.2012.45.6095
86. Mazières J, Barlesi F, Filleron T, Besse B, Monnet I, Beau-Faller M, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Ann Oncol. (2016) 27:281–6. doi: 10.1093/annonc/mdv573
87. Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. (2012) 18:375–7. doi: 10.1038/nm.2644
88. Gautschi O, Milia J, Filleron T, Wolf J, Carbone DP, Owen D, et al. Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J Clin Oncol. (2017) 35:1403–10. doi: 10.1200/JCO.2016.70.9352
89. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. (2016) 17:1653–60. doi: 10.1016/S1470-2045(16)30562-9
90. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. (2020) 18:59. doi: 10.1186/s12964-020-0530-4
91. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1126/science.aaf1490
92. Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: the rosetta stone of therapy resistance. Cancer Cell. (2020) 37:471–84. doi: 10.1016/j.ccell.2020.03.007
93. Le DT, Uram JN, Wang H, Bartlett B, Kemberling H, Eyring A, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
94. Wu W, Liu Y, Zeng S, Han Y, Shen H. Intratumor heterogeneity: the hidden barrier to immunotherapy against MSI tumors from the perspective of IFN-γ signaling and tumor-infiltrating lymphocytes. J Hematol Oncol. (2021) 14:160. doi: 10.1186/s13045-021-01166-3
95. Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. (2016) 76:999–1008. doi: 10.1158/0008-5472.CAN-15-1439
96. Genova C, Dellepiane C, Carrega P, Sommariva S, Ferlazzo G, Pronzato P, et al. Therapeutic implications of tumor microenvironment in lung cancer: focus on immune checkpoint blockade. Front Immunol. (2022) 12:799455. doi: 10.3389/fimmu.2021.799455
97. Fridman WH, Zitvogel L, Sautès–Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
98. Brambilla E, Teuff GL, Marguet S, Lantuejoul S, Dunant A, Graziano S, et al. Prognostic effect of tumor lymphocytic infiltration in resectable non–small-cell lung cancer. J Clin Oncol. (2016) 34:1223–30. doi: 10.1200/JCO.2015.63.0970
99. Al-Shibli KI, Donnem T, Al-Saad S, Persson M, Bremnes RM, Busund LT. Prognostic effect of epithelial and stromal lymphocyte infiltration in non–small cell lung cancer. Clin Cancer Res. (2008) 14:5220–7. doi: 10.1158/1078-0432.CCR-08-0133
100. Li F, Li C, Cai X, Xie Z, Zhou L, Cheng B, et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine. (2021) 41:101134. doi: 10.1016/j.eclinm.2021.101134
101. Zhang L, Chen Y, Wang H, Xu Z, Wang Y, Li S, et al. Massive PD-L1 and CD8 double positive TILs characterize an immunosuppressive microenvironment with high mutational burden in lung cancer. J Immunother Cancer. (2021) 9:e002356. doi: 10.1136/jitc-2021-002356
102. Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EHC, et al. Author Correction: Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature. (2021) 598:E1–1. doi: 10.1038/s41586-021-03893-6
103. Bristol-Myers Squibb. Neoadjuvant Nivolumab, or Nivolumab in Combination With Ipilimumab. In: Resectable Non-Small-Cell Lung Cancer. Johns Hopkins: Sidney Kimmel Comprehensive Cancer Center (2014). Available at: https://clinicaltrials.gov/study/NCT02259621.
104. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discovery. (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
105. AbdulJabbar K, Raza SEA, Rosenthal R, Jamal-Hanjani M, Veeriah S, Akarca A, et al. Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nat Med. (2020) 26:1054–62. doi: 10.1038/s41591-020-0900-x
106. Sanmamed MF, Nie X, Desai SS, Villaroel-Espindola F, Badri T, Zhao D, et al. A burned-out CD8+ T-cell subset expands in the tumor microenvironment and curbs cancer immunotherapy. Cancer Discovery. (2021) 11:1700–15. doi: 10.1158/2159-8290.CD-20-0962
107. Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res. (2015) 3:1344–55. doi: 10.1158/2326-6066.CIR-15-0097
108. Chen J, Chen J, Wang L. Tertiary lymphoid structures as unique constructions associated with the organization, education, and function of tumor-infiltrating immunocytes. J Zhejiang Univ-Sci B. (2022) 23:812–22. doi: 10.1631/jzus.B2200174
109. Jacquelot N, Tellier J, Nutt Sl, Belz Gt. Tertiary lymphoid structures and B lymphocytes in cancer prognosis and response to immunotherapies. OncoImmunology. (2021) 10:1900508. doi: 10.1080/2162402X.2021.1900508
110. Vanhersecke L, Brunet M, Guégan JP, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. (2021) 2:794–802. doi: 10.1038/s43018-021-00232-6
111. Tang J, Ramis-Cabrer D, Curull V, Wang X, Mateu-Jiménez M, Pijuan L, et al. B cells and tertiary lymphoid structures influence survival in lung cancer patients with resectable tumors. Cancers. (2020) 12:2644. doi: 10.3390/cancers12092644
112. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. (2019) 19:307–25. doi: 10.1038/s41568-019-0144-6
113. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
114. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non–small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. (2008) 26:4410–7. doi: 10.1200/JCO.2007.15.0284
115. Germain C, Devi-Marulkar P, Knockaert S, Biton J, Kaplon H, Letaïef L, et al. Tertiary lymphoid structure-B cells narrow regulatory T cells impact in lung cancer patients. Front Immunol. (2021) 12:626776. doi: 10.3389/fimmu.2021.626776
116. Zhang Y, Xu M, Ren Y, Ba Y, Liu S, Zuo A, et al. Tertiary lymphoid structural heterogeneity determines tumour immunity and prospects for clinical application. Mol Cancer. (2024) 23:75. doi: 10.1186/s12943-024-01980-6
117. Sun X, Liu W, Sun L, Mo H, Feng Y, Wu X, et al. Maturation and abundance of tertiary lymphoid structures are associated with the efficacy of neoadjuvant chemoimmunotherapy in resectable non-small cell lung cancer. J Immunother Cancer. (2022) 10:e005531. doi: 10.1136/jitc-2022-005531
118. Russick J, Joubert PE, Gillard-Bocquet M, Torset C, Meylan M, Petitprez F, et al. Natural killer cells in the human lung tumor microenvironment display immune inhibitory functions. J Immunother Cancer. (2020) 8:e001054. doi: 10.1136/jitc-2020-001054
119. Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56brightCD16– cells and display an impaired capability to kill tumor cells. Cancer. (2008) 112:863–75. doi: 10.1002/cncr.v112:4
120. Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The immune checkpoint PD-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers. (2020) 12:3285. doi: 10.3390/cancers12113285
121. Lin M, Luo H, Liang S, Chen J, Liu A, Niu L, et al. Pembrolizumab plus allogeneic NK cells in advanced non-small cell lung cancer patients. J Clin Investig. (2020) 130:2560–9. doi: 10.1172/JCI132712
122. Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
123. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
124. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. (2002) 3:991–8. doi: 10.1038/ni1102-991
125. Principe DR, Chiec L, Mohindra NA, Munshi HG. Regulatory T-cells as an emerging barrier to immune checkpoint inhibition in lung cancer. Front Oncol. (2021) 11:684098. doi: 10.3389/fonc.2021.684098
126. Casey M, Nakamura K. The cancer-immunity cycle in multiple myeloma. ImmunoTargets Ther. (2021) 10:247–60. doi: 10.2147/ITT.S305432
127. Wu SP, Liao RQ, Tu HY, Wang WJ, Dong ZY, Huang SM, et al. Stromal PD-L1–Positive Regulatory T cells and PD-1–Positive CD8-Positive T cells Define the Response of Different Subsets of Non–Small Cell Lung Cancer to PD-1/PD-L1 Blockade Immunotherapy. J Thorac Oncol. (2018) 13:521–32. doi: 10.1016/j.jtho.2017.11.132
128. Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. (2020) 21:1346–58. doi: 10.1038/s41590-020-0769-3
129. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
130. Umansky V, Shevchenko I, Bazhin AV, Utikal J. Extracellular adenosine metabolism in immune cells in melanoma. Cancer Immunol Immunother. (2014) 63:1073–80. doi: 10.1007/s00262-014-1553-8
131. Sui H, Dongye S, Liu X, Xu X, Wang L, Jin CQ, et al. Immunotherapy of targeting MDSCs in tumor microenvironment. Front Immunol. (2022) 13:990463. doi: 10.3389/fimmu.2022.990463
132. Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res. (2020) 26:970–7. doi: 10.1158/1078-0432.CCR-19-1040
133. Aloe C, Wang H, Vlahos R, Irving L, Steinfort D, Bozinovski S. Emerging and multifaceted role of neutrophils in lung cancer. Transl Lung Cancer Res. (2021) 10:2806–18. doi: 10.21037/tlcr-20-760
134. Akbay EA, Koyama S, Liu Y, Dries R, Bufe LE, Silkes M, et al. Interleukin-17A promotes lung tumor progression through neutrophil attraction to tumor sites and mediating resistance to PD-1 blockade. J Thorac Oncol. (2017) 12:1268–79. doi: 10.1016/j.jtho.2017.04.017
135. Miret JJ, Kirschmeier P, Koyama S, Zhu M, Li YY, Naito Y, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J Immunother Cancer. (2019) 7:32. doi: 10.1186/s40425-019-0504-5
136. Kargl J, Busch SE, Yang GHY, Kim KH, Hanke ML, Metz HE, et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat Commun. (2017) 8:14381. doi: 10.1038/ncomms14381
137. Ren S, Xiong X, You H, Shen J, Zhou P. The combination of immune checkpoint blockade and angiogenesis inhibitors in the treatment of advanced non-small cell lung cancer. Front Immunol. (2021) 12:689132. doi: 10.3389/fimmu.2021.689132
138. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
139. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagés C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. (2006) 313:1960–4. doi: 10.1126/science.1129139
140. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of Malignant tumours. J Pathol. (2014) 232:199–209. doi: 10.1002/path.2014.232.issue-2
141. Zhao Z, Zhao D, Xia J, Wang Y, Wang B. Immunoscore predicts survival in early-stage lung adenocarcinoma patients. Front Oncol. (2020) 10:691. doi: 10.3389/fonc.2020.00691
142. Zeng Z, Yang F, Wang Y, Zhao H, Wei F, Zhang P, et al. Significantly different immunological score in lung adenocarcinoma and squamous cell carcinoma and a proposal for a new immune staging system. OncoImmunology. (2020) 9:1828538. doi: 10.1080/2162402X.2020.1828538
143. Feng W, Li Y, Shen L, Zhang Q, Cai XW, Zhu ZF, et al. Clinical impact of the tumor immune microenvironment in completely resected stage IIIA(N2) non-small cell lung cancer based on an immunological score approach. Ther Adv Méd Oncol. (2021) 13:1758835920984975. doi: 10.1177/1758835920984975
144. Boscolo A, Fortarezza F, Lunardi F, Comacchio G, Urso L, Frega S, et al. Combined immunoscore for prognostic stratification of early stage non-small-cell lung cancer. Front Oncol. (2020) 10:564915. doi: 10.3389/fonc.2020.564915
145. Fumet JD, Richard C, Ledys F, Klopfenstein Q, Joubert P, Routy B, et al. Correction: Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br J Cancer. (2019) 121:283–3. doi: 10.1038/s41416-019-0512-8
146. Chen D, Wang Y, Zhang X, Ding Q, Wang X, Xue Y, et al. Characterization of tumor microenvironment in lung adenocarcinoma identifies immune signatures to predict clinical outcomes and therapeutic responses. Front Oncol. (2021) 11:581030. doi: 10.3389/fonc.2021.581030
147. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. (2013) 39:782–95. doi: 10.1016/j.immuni.2013.10.003
148. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. (2013) 4:2612. doi: 10.1038/ncomms3612
149. Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, Fridman WH, et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics. (2019) 35:i436–45. doi: 10.1093/bioinformatics/btz363
150. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. (2015) 12:453–7. doi: 10.1038/nmeth.3337
151. Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. (2017) 77:e108–10. doi: 10.1158/1538-7445.AM2017-108
152. Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. (2016) 17:174. doi: 10.1186/s13059-016-1028-7
153. Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. (2016) 17:218. doi: 10.1186/s13059-016-1070-5
154. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. (2017) 18:220. doi: 10.1186/s13059-017-1349-1
155. Pirrotta S, Masatti L, Corrà A, Pedrini F, Esposito G, Martini P, et al. signifinder enables the identification of tumor cell states and cancer expression signatures in bulk, single-cell and spatial transcriptomic data. bioRxiv. (2023) 10:2023.03.07.530940. doi: 10.1101/2023.03.07.530940
156. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
157. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
158. Stephen HF, O’day SJ, McDermott DFMD, Weber RWMD, Sosman JAMD, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
159. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
160. Brahmer J, Reckamp KL, Paul B, Lucio C, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
161. Gogishvili M, Melkadze T, Makharadze T, Giorgadze D, Dvorkin M, Penkov K, et al. Cemiplimab plus chemotherapy versus chemotherapy alone in non-small cell lung cancer: a randomized, controlled, double-blind phase 3 trial. Nat Med. (2022) 28:2374–80. doi: 10.1038/s41591-022-01977-y
162. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. (2016) 387:1837–46. doi: 10.1016/S0140-6736(16)00587-0
163. Paz-Ares L, Villegas A, Daniel D, Baz DV, Murakami S, Hui R, et al. PACIFIC: A double-blind, placebo-controlled phase III study of durvalumab after chemoradiation therapy (CRT) in patients with stage III, locally advanced, unresectable NSCLC. Ann Oncol. (2017) 28:v634. doi: 10.1093/annonc/mdx440.049
164. Reck M, Barlesi F, Yang JCH, Westeel V, Felip E, Özgüroğlu M, et al. Avelumab versus platinum-based doublet chemotherapy as first-line treatment for patients with high-expression programmed death-ligand 1–positive metastatic NSCLC: primary analysis from the phase 3 JAVELIN lung 100 trial. J Thorac Oncol. (2024) 19:297–313. doi: 10.1016/j.jtho.2023.09.1445
165. Zhou C, Wang Z, Sun M, Cao L, Ma Z, Wu R, et al. Interim survival analysis of the randomized phase III GEMSTONE-302 trial: sugemalimab or placebo plus chemotherapy as first-line treatment for metastatic NSCLC. Nat Cancer. (2023) 4:860–71. doi: 10.1038/s43018-023-00578-z
166. Tan S, Day D, Nicholls SJ, Segelov E. Immune checkpoint inhibitor therapy in oncology current uses and future directions: JACC: cardioOncology state-of-the-art review. JACC: CardioOncol. (2022) 4:579–97. doi: 10.1016/j.jaccao.2022.09.004
167. Hellmann MD, Paz-Ares L, Caro RB, Zurawski B, Kim SW, Costa EC, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
168. Hossein B, Luis PA, Leora H, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med. (2015) 373:1627–39. doi: 10.1056/nejmoa1507643
169. Herbst RS, Sznol M. Diminished but not dead: chemotherapy for the treatment of NSCLC. Lancet Oncol. (2016) 17:1464–5. doi: 10.1016/S1470-2045(16)30524-1
170. Majem M, Cobo M, Isla D, Marquez-Medina D, Rodriguez-Abreu D, Casal-Rubio J, et al. PD-(L)1 inhibitors as monotherapy for the first-line treatment of non-small-cell lung cancer patients with high PD-L1 expression: A network meta-analysis. J Clin Med. (2021) 10:1365. doi: 10.3390/jcm10071365
171. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
172. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. N Engl J Med. (2017) 376:2415–26. doi: 10.1056/NEJMoa1613493
173. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. (2013) 39:74–88. doi: 10.1016/j.immuni.2013.06.014
174. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. (2016) 17:1497–508. doi: 10.1016/S1470-2045(16)30498-3
175. Kang J, Zhang J, Tian Z, Xu Y, Li J, Li M. The efficacy and safety of immune-checkpoint inhibitors plus chemotherapy versus chemotherapy for non-small cell lung cancer: An updated systematic review and meta-analysis. PloS One. (2024) 19:e0276318. doi: 10.1371/journal.pone.0276318
176. Borgeaud M, Sandoval J, Obeid M, Banna G, Michielin O, Addeo A, et al. Novel targets for immune-checkpoint inhibition in cancer. Cancer Treat Rev. (2023) 120:102614. doi: 10.1016/j.ctrv.2023.102614
177. Felip E, Majem M, Doger B, Clay TD, Carcereny E, Bondarenko I, et al. A phase II study (TACTI-002) in first-line metastatic non–small cell lung carcinoma investigating eftilagimod alpha (soluble LAG-3 protein) and pembrolizumab: Updated results from a PD-L1 unselected population. J Clin Oncol. (2022) 40:9003–3. doi: 10.1200/JCO.2022.40.16_suppl.9003
178. ClinicalTrials.Gov. Dose Escalation Study of a PD1-LAG3 Bispecific Antibody in Patients With Advanced and/or Metastatic Solid Tumors. 2024. Dose Escalation Study of a PD1-LAG3 Bispecific Antibody in Patients With Advanced and/or Metastatic Solid Tumors. Available online at: https://clinicaltrials.gov/study/NCT04140500 (Accessed January 2025).
179. Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV, et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med. (2023) 29:2814–24. doi: 10.1038/s41591-023-02593-0
180. Lipson EJ, Tawbi HAH, SChadendorf D, Ascierto PA, Matamala L, Gutiérrez EC, et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J Clin Oncol. (2021) 39:9503–3. doi: 10.1200/JCO.2021.39.15_suppl.9503
181. Morgensztern D, Chaudhry A, Iannotti N, Acevedo A, Balaburski G, Balogh A, et al. 1359TiP RELATIVITY-104: First-line relatlimab (RELA) + nivolumab (NIVO) with chemotherapy vs nivo with chemotherapy in stage IV or recurrent non-small cell lung cancer (NSCLC): A phase II, randomized, double-blind study. Ann Oncol. (2021) 32:S1030. doi: 10.1016/j.annonc.2021.08.1960
182. Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. (2020) 8:e000911. doi: 10.1136/jitc-2020-000911
183. Gutierrez ME, Tang SC, Powderly JD, Balmanoukian AS, Janik J, Hoyle P, et al. 730MO First-in-human phase I study of INCAGN02390, a TIM-3 monoclonal antibody antagonist in patients with advanced Malignancies. Abstract Book ESMO Congress. (2022) 9. Available online at: https://www.sciencedirect.com/science/article/pii/S0923753422027077 (Accessed January 2025).
184. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/ib clinical trial of sabatolimab, an anti–TIM-3 antibody, alone and in combination with spartalizumab, an anti–PD-1 antibody, in advanced solid tumors. Clin Cancer Res. (2021) 27:3620–9. doi: 10.1158/1078-0432.CCR-20-4746
185. Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, et al. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. (2019) 154:21–31. doi: 10.1016/j.ymeth.2018.11.008
186. Besse B, Italiano A, Cousin S, Ruiter G, Felip E, Alvarez EC, et al. 1313MO Safety and preliminary efficacy of AZD7789, a bispecific antibody targeting PD-1 and TIM-3, in patients (pts) with stage IIIB–IV non-small-cell lung cancer (NSCLC) with previous anti-PD-(L)1 therapy. Abstract Book ESMO Congress. (2023) 34:S755. Available online at: https://www.sciencedirect.com/science/article/pii/S0923753423031848 (Accessed January 2025).
187. Yonesaka K, Haratani K, Takamura S, Sakai H, Kato R, Takegawa N, et al. B7-H3 negatively modulates CTL-mediated cancer immunity. Clin Cancer Res. (2018) 24:2653–64. doi: 10.1158/1078-0432.CCR-17-2852
188. Liu J, Yang S, Cao B, Zhou G, Zhang F, Wang Y, et al. Targeting B7-H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. J Hematol Oncol. (2021) 14:21. doi: 10.1186/s13045-020-01024-8
189. Urba W, Chmielowski B, Loo D, Baughman J, Chen F, Moore P, et al. A Phase I, open-label, dose escalation study of MGA271 in combination with ipilimumab in patients with B7-H3-expressing melanoma, squamous cell cancer of the head and neck or non-small cell lung cancer. J Immunother Cancer. (2015) 3:P176. doi: 10.1186/2051-1426-3-S2-P176
190. Aggarwal C, Prawira A, Antonia S, Rahma O, Tolcher A, Cohen RB, et al. Dual checkpoint targeting of B7-H3 and PD-1 with enoblituzumab and pembrolizumab in advanced solid tumors: interim results from a multicenter phase I/II trial. J Immunother Cancer. (2022) 10:e004424. doi: 10.1136/jitc-2021-004424
191. Jang S, Powderly JD, Spira AI, Bakkacha O, Loo D, Bohac GC, et al. Phase 1 dose escalation study of MGC018, an anti-B7-H3 antibody-drug conjugate (ADC), in patients with advanced solid tumors. J Clin Oncol. (2021) 39:2631–1. doi: 10.1200/JCO.2021.39.15_suppl.2631
192. Ventin M, Cattaneo G, Maggs L, Jia J, Arya S, Ferrone S, et al. B7-H3-targeted CAR T cell activity is enhanced by radiotherapy in solid cancers. Front Oncol. (2023) 13:1193963. doi: 10.3389/fonc.2023.1193963
193. Manieri NA, Chiang EY, Grogan JL. TIGIT: A key inhibitor of the cancer immunity cycle. Trends Immunol. (2017) 38:20–8. doi: 10.1016/j.it.2016.10.002
194. Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer☆. Ann Oncol. (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
195. Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. (2022) 23:781–92. doi: 10.1016/S1470-2045(22)00226-1
196. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
197. Du S, Yan J, Xue Y, Zhong Y, Dong Y. Adoptive cell therapy for cancer treatment. Exploration. (2023) 3:20210058. doi: 10.1002/EXP.20210058
198. Raina D, Kosugi M, Ahmad R, Panchamoorthy G, Rajabi H, Alam M, et al. Dependence on the MUC1-C oncoprotein in non–small cell lung cancer cells. Mol Cancer Ther. (2011) 10:806–16. doi: 10.1158/1535-7163.MCT-10-1050
199. Grah JJ, Katalinic D, Juretic A, Santek F, Samarzija M. Clinical significance of immunohistochemical expression of cancer/testis tumor-associated antigens (MAGE-A1, MAGE-A3/4, NY-ESO-1) in patients with non-small cell lung cancer. Tumori J. (2013) 100:60–8. doi: 10.1177/1430.15817
200. Ford CH, Stokes HJ, Newman CE. Carcinoembryonic antigen and prognosis after radical surgery for lung cancer: Immunocytochemical localization and serum levels. Br J Cancer. (1981) 44:145–53. doi: 10.1038/bjc.1981.164
201. Lao Y, Shen D, Zhang W, He R, Jiang M. Immune checkpoint inhibitors in cancer therapy—How to overcome drug resistance? Cancers. (2022) 14:3575. doi: 10.3390/cancers14153575
202. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–192.e5. doi: 10.1016/j.stem.2018.06.002
203. Kelderman S, Kvistborg P. Tumor antigens in human cancer control. Biochim Biophys Acta (BBA) - Rev Cancer. (2016) 1865:83–9. doi: 10.1016/j.bbcan.2015.10.004
204. Forde PM, Chaft JE, Pardoll DM. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. (2018) 379:e14. doi: 10.1056/NEJMoa1716078
205. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non–small cell lung cancer. Cancer Discovery. (2017) 7:264–76. doi: 10.1158/2159-8290.CD-16-0828
206. Rizvi NA, Solange P. Immunotherapy for unresectable stage III non–small-cell lung cancer. N Engl J Med. (2017) 377:1986–8. doi: 10.1056/NEJMe1711430
207. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. (2014) 24:743–50. doi: 10.1101/gr.165985.113
208. Danilova L, Anagnostou V, Caushi JX, Sidhom JW, Guo H, Chan HY, et al. The mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay: A sensitive platform for monitoring antitumor immunity. Cancer Immunol Res. (2018) 6:888–99. doi: 10.1158/2326-6066.CIR-18-0129
209. Smith KN, Llosa NJ, Cottrell TR, Siegel N, Fan H, Suri P, et al. Persistent mutant oncogene specific T cells in two patients benefitting from anti-PD-1. J Immunother Cancer. (2019) 7:40. doi: 10.1186/s40425-018-0492-x
210. Kaczmarek M, Poznańska J, Fechner F, Michalska N, Paszkowska S, Napierała A, et al. Cancer vaccine therapeutics: limitations and effectiveness—A literature review. Cells. (2023) 12:2159. doi: 10.3390/cells12172159
211. Prete AD, Salvi V, Soriani A, Laffranchi M, Sozio F, Bosisio D, et al. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol Immunol. (2023) 20:432–47. doi: 10.1038/s41423-023-00990-6
212. Zhang X, Cui H, Zhang W, Li Z, Gao J. Engineered tumor cell-derived vaccines against cancer: The art of combating poison with poison. Bioact Mater. (2023) 22:491–517. doi: 10.1016/j.bioactmat.2022.10.016
213. De Jesus BB, Neves BM, Ferreira M, Nóbrega-Pereira S. Strategies for cancer immunotherapy using induced pluripotency stem cells-based vaccines. Cancers. (2020) 12:3581. doi: 10.3390/cancers12123581
214. Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, et al. Drug delivery with carbon nanotubes for in vivo cancer treatment. arXiv. (2008) 68:6652–60. doi: 10.1158/0008-5472.CAN-08-1468
215. Kooreman NG, Kim Y, de Almeida PE, Termglinchan V, Diecke S, Shao NY, et al. Autologous iPSC-based vaccines elicit anti-tumor responses in vivo. Cell Stem Cell. (2018) 22:501–513.e7. doi: 10.1016/j.stem.2018.01.016
216. Wang J, Shao L, Wu L, Ma W, Zheng Y, Hu C, et al. Expression levels of a gene signature in hiPSC associated with lung adenocarcinoma stem cells and its capability in eliciting specific antitumor immune-response in a humanized mice model. Thorac Cancer. (2020) 11:1603–12. doi: 10.1111/1759-7714.13440
217. Fan T, Zhang M, Yang J, Zhu Z, Cao W, Dong C. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther. (2023) 8:450. doi: 10.1038/s41392-023-01674-3
218. Oliveres H, Caglevic C, Passiglia F, Taverna S, Smits E, Rolfo C. Vaccine and immune cell therapy in non-small cell lung cancer. J Thorac Dis. (2018) 1:S1602–14. doi: 10.21037/jtd.2018.05.134
219. Blackhall F, Moliner L, Samson A, Franks K, Karydis I, Joseph-Pietras D, et al. 1502TiP A phase I/IIa trial of ChAdOx1 and MVA vaccines against MAGE-A3 and NY-ESO-1. Abstract Book ESMO Congress. (2023) 34:S846. https://www.sciencedirect.com/science/article/pii/S0923753423033707 (Accessed January 2025).
220. Pol JG, Acuna SA, Yadollahi B, Tang N, Stephenson KB, Atherton MJ, et al. Preclinical evaluation of a MAGE-A3 vaccination utilizing the oncolytic Maraba virus currently in first-in-human trials. OncoImmunology. (2019) 8:e1512329. doi: 10.1080/2162402X.2018.1512329
221. Dey S, Sutanto-Ward E, Kopp KL, DuHadaway J, Mondal A, Ghaban D, et al. Peptide vaccination directed against IDO1-expressing immune cells elicits CD8+ and CD4+ T-cell-mediated antitumor immunity and enhanced anti-PD1 responses. J Immunother Cancer. (2020) 8:e000605. doi: 10.1136/jitc-2020-000605
222. Kiousi E, Lyraraki V, Mardiki GL, Stachika N, Damianou AK, Malainou CP, et al. Progress and challenges of messenger RNA vaccines in the therapeutics of NSCLC. Cancers. (2023) 15:5589. doi: 10.3390/cancers15235589
223. Haldar SD, Heumann TR, Berg M, Ferguson A, Lim SJ, Wang H, et al. A phase I study of a mutant KRAS-targeted long peptide vaccine combined with ipilimumab/nivolumab in resected pancreatic cancer and MMR-proficient metastatic colorectal cancer. J Clin Oncol. (2023) 41:TPS814–4. doi: 10.1200/JCO.2023.41.4_suppl.TPS814
224. Gridelli C, Ciuleanu T, Domine M, Szczesna A, Bover I, Cobo M, et al. Clinical activity of a htert (vx-001) cancer vaccine as post-chemotherapy maintenance immunotherapy in patients with stage IV non-small cell lung cancer: final results of a randomised phase 2 clinical trial. Br J Cancer. (2020) 122:1461–6. doi: 10.1038/s41416-020-0785-y
225. Lorentzen CL, Martinenaite E, Kjeldsen JW, Holmstroem RB, Mørk SK, Pedersen AW, et al. Arginase-1 targeting peptide vaccine in patients with metastatic solid tumors – A phase I trial. Front Immunol. (2022) 13:1023023. doi: 10.3389/fimmu.2022.1023023
226. Vansteenkiste JF, Demedts I, Pons-Tostivint E, Biesma B, Borm F, Colinet B, et al. 1176P Open-label, dose escalation, phase I/II study to assess safety, tolerability, immunogenicity and preliminary clinical activity of the therapeutic cancer vaccine PDC*lung01 with or without anti-programmed death-1 (PD-1) treatment in patients with non-small cell lung cancer (NSCLC). Ann Oncol. (2022) 33:S1086. doi: 10.1016/j.annonc.2022.07.1299
227. Ricordel C, Barlesi F, Cousin S, Cho BC, Calvo E, Kim TM, et al. Safety and efficacy of tusamitamab ravtansine (SAR408701) in long-term treated patients with nonsquamous non–small cell lung cancer (NSQ NSCLC) expressing carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5). J Clin Oncol. (2022) 40:9039–9. doi: 10.1200/JCO.2022.40.16_suppl.9039
228. Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer. (2021) 20:41. doi: 10.1186/s12943-021-01335-5
229. Park JW, Lagniton PNP, Liu Y, Xu RH. mRNA vaccines for COVID-19: what, why and how. Int J Biol Sci. (2021) 17:1446–60. doi: 10.7150/ijbs.59233
230. Morse MA, Nair SK, Mosca PJ, Hobeika AC, Clay TM, Deng Y, et al. Immunotherapy with autologous, human dendritic cells transfected with carcinoembryonic antigen mRNA. Cancer Invest. (2003) 21:341–9. doi: 10.1081/CNV-120018224
231. Sebastian M, Schröder A, Scheel B, Hong HS, Muth A, Von Boehmer L, et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol Immunother. (2019) 68:799–812. doi: 10.1007/s00262-019-02315-x
232. Gandhi L, Ramirez KA, Schwarzenberger P, Ricciardi T, Macri MJ, Ryan A, et al. Phase 1/2 study of mRNA vaccine therapy + durvalumab (durva) ± tremelimumab (treme) in patients with metastatic non-small cell lung cancer (NSCLC). J Clin Oncol. (2018) 36:TPS9107–TPS9107. doi: 10.1200/JCO.2018.36.15_suppl.TPS9107
233. Duan LJ, Wang Q, Zhang C, Yang DX, Zhang XY. Potentialities and challenges of mRNA vaccine in cancer immunotherapy. Front Immunol. (2022) 13:923647. doi: 10.3389/fimmu.2022.923647
234. Foster JB, Choudhari N, Perazzelli J, Storm J, Hofmann TJ, Jain P, et al. Purification of mRNA encoding chimeric antigen receptor is critical for generation of a robust T-cell response. Hum Gene Ther. (2019) 30:168–78. doi: 10.1089/hum.2018.145
235. Ongun M, Lokras AG, Baghel S, Shi Z, Schmidt ST, Franzyk H, et al. Lipid nanoparticles for local delivery of mRNA to the respiratory tract: Effect of PEG-lipid content and administration route. Eur J Pharm Biopharm. (2024) 198:114266. doi: 10.1016/j.ejpb.2024.114266
236. Ali T, Mujawar S, Sowmya AV, Saldanha D, Chaudhury S. Dangers of mRNA vaccines. Ind Psychiatry J. (2021) 30:S291–3. doi: 10.4103/0972-6748.328833
237. Cheng Y, Zhang X, Liu J, Huang Z, Lv D, Zhang Y, et al. YL202/BNT326, a HER3-targeted ADC, in patients with locally advanced or metastatic non-small cell lung cancer and breast cancer: Preliminary results from a first-in-human phase I trial. J Clin Oncol. (2024) 42:3034–4. doi: 10.1200/JCO.2024.42.16_suppl.3034
238. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol Rev. (2019) 290:127–47. doi: 10.1111/imr.v290.1
239. Reckamp KL, Akerley W, Calvo E, Clarke J, Edelman MJ, He K, et al. (2019). 1590TiP - Safety, tolerability and activity of autologous T-cells with enhanced T-cell receptors specific to NY ESO 1/LAGE 1a (GSK3377794) alone, or in combination with pembrolizumab, in advanced non-small cell lung cancer: A phase Ib/IIa randomised pilot study, in: Abstract Book of the 44th ESMO Congress (ESMO 2019), Barcelona, Spain, 27 September – 1 October 2019, (2019) 30:v657–8. Available at: https://www.sciencedirect.com/science/article/pii/S0923753419597976 (Accessed January 2025).
240. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
241. Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol. (2018) 18:605–16. doi: 10.1038/s41577-018-0042-2
242. Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. (2005) 23:1257–68. doi: 10.1038/nbt1127
243. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
244. Met Ö, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. (2019) 41:49–58. doi: 10.1007/s00281-018-0703-z
245. Zhao L, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front Immunol. (2019) 10:2250. doi: 10.3389/fimmu.2019.02250
246. Nair R, Westin J. Immunotherapy. Adv Exp Med Biol. (2020) 1244:215–33. doi: 10.1007/978-3-030-41008-7_10
247. Zhang T, Wu MR, Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. (2012) 189:2290–9. doi: 10.4049/jimmunol.1103495
248. Guedan S, Posey AD, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. (2018) 3:e96976. doi: 10.1172/jci.insight.96976
249. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3ζ Transmembrane Domain Is Dependent upon Incorporation of the Receptor into the Endogenous TCR/CD3 Complex. J Immunol. (2010) 184:6938–49. doi: 10.4049/jimmunol.0901766
250. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
251. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. biomark Res. (2017) 5:22. doi: 10.1186/s40364-017-0102-y
252. Tokarew N, Ogonek J, Endres S, Von-Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
253. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. (2015) 3:356–67. doi: 10.1158/2326-6066.CIR-14-0186
254. Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. (2017) 25:2452–65. doi: 10.1016/j.ymthe.2017.07.013
255. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther - Methods Clin Dev. (2019) 12:145–56. doi: 10.1016/j.omtm.2018.12.009
256. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
257. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. (2014) 257:83–90. doi: 10.1111/imr.2013.257.issue-1
258. Cao X, Leonard K, Collins LI, Cai SF, Mayer JC, Payton JE, et al. Interleukin 12 stimulates IFN-γ–mediated inhibition of tumor-induced regulatory T-cell proliferation and enhances tumor clearance. Cancer Res. (2009) 69:8700–9. doi: 10.1158/0008-5472.CAN-09-1145
259. Ferlazzo G, Pack M, Thomas D, Paludan C, Schmid D, Strowig T, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci. (2004) 101:16606–11. doi: 10.1073/pnas.0407522101
260. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. (2018) 11:132. doi: 10.1186/s13045-018-0677-2
261. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
262. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat Med. (2018) 24:352–9. doi: 10.1038/nm.4478
263. Chen L, Chen F, Li J, Pu Y, Yang C, Wang Y, et al. CAR-T cell therapy for lung cancer: Potential and perspective. Thorac Cancer. (2022) 13:889–99. doi: 10.1111/1759-7714.14375
264. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ Metastatic colorectal cancers. Mol Ther. (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
265. Yang Z, ClinicalTrials.Gov. Safety and Efficacy of CEA-Targeted CAR-T Therapy for Relapsed/Refractory CEA+ Cancer (2020). Available online at: https://clinicaltrials.gov/study/NCT04348643 (Accessed January 2025).
266. Lin Y, Chen S, Zhong S, An H, Yin H, McGowan E. 35O Phase I clinical trial of PD-1 knockout anti-MUC1 CAR-T cells in the treatment of patients with non-small cell lung cancer. Ann Oncol. (2019) 30:xi12. doi: 10.1093/annonc/mdz448
267. Yang L, ClinicalTrials.Gov. Phase I/II Study of Anti-Mucin1 (MUC1) CAR T Cells for Patients With MUC1+ Advanced Refractory Solid Tumor. NCI-Supported Clinical Trials (2016). Available online at: https://clinicaltrials.gov/study/NCT02587689 (Accessed January 2025).
268. Henry J, Oh D, Eskew J, Baranda J, Rivera IIR, Dumbrava E, et al. 728 Phase 1 study of P-MUC1C-ALLO1 allogeneic CAR-T cells in patients with epithelial-derived cancers. J Immunotherap Cancer. (2022) 10:A761–1. doi: 10.1136/jitc-2022-SITC2022.0728
269. Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. OncoImmunology. (2017) 6:e1284722. doi: 10.1080/2162402X.2017.1284722
270. Saltos A, Khalil F, Smith M, Li J, Schell M, Antonia SJ, et al. Clinical associations of mucin 1 in human lung cancer and precancerous lesions. Oncotarget. (2018) 9:35666–75. doi: 10.18632/oncotarget.26278
271. Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. (2023) 8:9. doi: 10.1038/s41392-022-01270-x
272. Zhang Z, ClinicalTrials.gov. Study of CXCR5 Modified EGFR Targeted CAR-T Cells for Advanced NSCLC (2024). Available online at: https://clinicaltrials.gov/study/NCT05060796contacts-and-locations (Accessed January 2025).
273. Li G, Guo J, Zheng Y, Ding W, Han Z, Qin L, et al. CXCR5 guides migration and tumor eradication of anti-EGFR chimeric antigen receptor T cells. Mol Ther - Oncol. (2021) 22:507–17. doi: 10.1016/j.omto.2021.07.003
274. Li F, Chen H, Wang D. Silencing of CD276 suppresses lung cancer progression by regulating integrin signaling. J Thorac Dis. (2020) 12:2137–45. doi: 10.21037/jtd.2020.04.41
275. Zhang C, Hao X. Prognostic significance of CD276 in non-small cell lung cancer. Open Med. (2019) 14:805–12. doi: 10.1515/med-2019-0076
276. Ke EE, Wu YL. EGFR as a pharmacological target in EGFR-mutant non-small-cell lung cancer: where do we stand now? Trends Pharmacol Sci. (2016) 37:887–903. doi: 10.1016/j.tips.2016.09.003
277. Li H, Huang Y, Jiang DQ, Cui LZ, He Z, Wang C, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. (2018) 9:177. doi: 10.1038/s41419-017-0238-6
278. Qu J, Mei Q, Chen L, Zhou J. Chimeric antigen receptor (CAR)-T-cell therapy in non-small-cell lung cancer (NSCLC): current status and future perspectives. Cancer Immunol Immunother. (2021) 70:619–31. doi: 10.1007/s00262-020-02735-0
279. Ma HY, Das J, Prendergast C, Jong DD, Braumuller B, Paily J, et al. Advances in CAR T cell therapy for non-small cell lung cancer. Curr Issues Mol Biol. (2023) 45:9019–38. doi: 10.3390/cimb45110566
280. Ma Y, ClinicalTrials.gov. Study of CXCR5 Modified EGFE Chimeric Antigen Receptor Autologous T Cells in EGFR- Positive Patients With Advanced Non-small Cell Lung Cancer (2020). Available online at: https://clinicaltrials.gov/study/NCT04153799contacts-and-locations (Accessed January 2025).
281. Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W, et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol. (2021) 147:3725–34. doi: 10.1007/s00432-021-03613-7
282. Castelletti L, Yeo D, Van Zandwijk N, Rasko JEJ. Anti-Mesothelin CAR T cell therapy for Malignant mesothelioma. biomark Res. (2021) 9:11. doi: 10.1186/s40364-021-00264-1
283. Adusumilli PS, Zauderer MG, Rivière I, Solomon SB, Rusch VW, O’Cearbhaill RE, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with Malignant pleural disease, in combination with the anti–PD-1 agent pembrolizumabRegional CAR T-cell therapy for mesothelioma. Cancer Discovery. (2021) 11:2748–63. doi: 10.1158/2159-8290.CD-21-0407
284. Zhai X, Mao L, Wu M, Liu J, Yu S. Challenges of anti-mesothelin CAR-T-cell therapy. Cancers. (2023) 15:1357. doi: 10.3390/cancers15051357
285. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid Malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
286. Wallstabe L, Göttlich C, Nelke LC, Kühnemundt J, Schwarz T, Nerreter T, et al. ROR1-CAR T-cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight. (2019) 4:e126345. doi: 10.1172/jci.insight.126345
287. Osorio-Rodríguez DA, Camacho BA, Ramírez-Segura C. Anti-ROR1 CAR-T cells: Architecture and performance. Front Med. (2023) 10:1121020. doi: 10.3389/fmed.2023.1121020
288. Fucà G, Reppel L, Landoni E, Savoldo B, Dotti G. Enhancing chimeric antigen receptor T-cell efficacy in solid tumors. Clin Cancer Res. (2020) 26:2444–51. doi: 10.1158/1078-0432.CCR-19-1835
289. Zhang Y, Qin D, Shou AC, Liu Y, Wang Y, Zhou L. Exploring CAR-T cell therapy side effects: mechanisms and management strategies. J Clin Med. (2023) 12:6124. doi: 10.3390/jcm12196124
290. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
291. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
292. Chen F, Teachey DT, Pequignot E, Frey N, Porter D, Maude SL, et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J Immunol Methods. (2016) 434:1–8. doi: 10.1016/j.jim.2016.03.005
293. Liu Z, Zhou Z, Dang Q, Xu H, Lv J, Li H, et al. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics. (2022) 12:6273–90. doi: 10.7150/thno.76854
294. Wang A, Lv T, Song Y. Tandem CAR-T cells targeting MUC1 and PSCA combined with anti-PD-1 antibody exhibit potent preclinical activity against non-small cell lung cancer. Cell Immunol. (2023) 391–392:104760. https://www.sciencedirect.com/science/article/pii/S0008874923000990 (Accessed January 2025).
295. Chen T, Wang M, Chen Y, Liu Y. Current challenges and therapeutic advances of CAR-T cell therapy for solid tumors. Cancer Cell Int. (2024) 24:133. doi: 10.1186/s12935-024-03315-3
296. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. (2022) 28:1189–98. doi: 10.1038/s41591-022-01800-8
297. Murty S, Haile ST, Beinat C, Aalipour A, Alam IS, Murty T, et al. Intravital imaging reveals synergistic effect of CAR T-cells and radiation therapy in a preclinical immunocompetent glioblastoma model. OncoImmunology. (2020) 9:1757360. doi: 10.1080/2162402X.2020.1757360
298. Hirabayashi K, Du H, Xu Y, Shou P, Zhou X, Fucá G, et al. Dual-targeting CAR-T cells with optimal co-stimulation and metabolic fitness enhance antitumor activity and prevent escape in solid tumors. Nat Cancer. (2021) 2:904–18. doi: 10.1038/s43018-021-00244-2
299. Zhou Z, Tao C, Li J, Tang JCO, Chan ASC, Zhou Y. Chimeric antigen receptor T cells applied to solid tumors. Front Immunol. (2022) 13. https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.984864 (Accessed January 2025).
300. Aalipour A, Boeuf FL, Tang M, Murty S, Simonetta F, Lozano AX, et al. Viral delivery of CAR targets to solid tumors enables effective cell therapy. Mol ther - oncolytics. Mol Ther Oncolytics. (2020) 17:232–40. doi: 10.1016/j.omto.2020.03.018
301. Foeng J, Comerford I, McColl SR. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep Med. (2022) 3:100543. doi: 10.1016/j.xcrm.2022.100543
302. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
303. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. (2019) 36:471–82. doi: 10.1016/j.ccell.2019.09.006
304. Lv Y, Luo X, Xie Z, Qiu J, Yang J, Deng Y, et al. Prospects and challenges of CAR-T cell therapy combined with ICIs. Front Oncol. (2024) 14:1368732. doi: 10.3389/fonc.2024.1368732
305. Attili I, Corvaja C, Spitaleri G, Signore ED, Aliaga PT, Passaro A, et al. New generations of tyrosine kinase inhibitors in treating NSCLC with oncogene addiction: strengths and limitations. Cancers. (2023) 15:5079. doi: 10.3390/cancers15205079
306. Fu K, Xie F, Wang F, Fu L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. J Hematol Oncol. (2022) 15:173. doi: 10.1186/s13045-022-01391-4
307. McLaughlin J, Berkman J, Nana-Sinkam P. Targeted therapies in non-small cell lung cancer: present and future. Fac Rev. (2023) 12:22. doi: 10.12703/r/12-22
308. Sato Y, Silina K, Van Den BM, Hirahara K, Yanagita M. The roles of tertiary lymphoid structures in chronic diseases. Nat Rev Nephrol. (2023) 19:525–37. doi: 10.1038/s41581-023-00706-z
309. Olivares-Hernández A, González del Portillo E, Tamayo-Velasco Á, Figuero-Pérez L, Zhilina-Zhilina S, Fonseca-Sánchez E, et al. Immune checkpoint inhibitors in non-small cell lung cancer: from current perspectives to future treatments—a systematic review. Ann Transl Med. (2023) 11:354–4. doi: 10.21037/atm-22-4218
310. Lu C, Tan Y. Promising immunotherapy targets: TIM3, LAG3, and TIGIT joined the party(2024). Available online at: https://www.sciencedirect.com/science/article/pii/S2950329924000158 (Accessed January 2025).
311. Fan T, Zhang M, Yang J, Zhu Z, Cao W, Dong C. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther. (2023) 8:450. doi: 10.1038/s41392-023-01674-3
312. Uscanga-Palomeque AC, Chávez-Escamilla AK, Alvizo-Báez CA, Saavedra-Alonso S, Terrazas-Armendáriz LD, Tamez-Guerra RS, et al. CAR-T cell therapy: from the shop to cancer therapy. Int J Mol Sci. (2023) 24:15688. doi: 10.3390/ijms242115688
313. Wang JY, Wang L. CAR-T cell therapy: Where are we now, and where are we heading? Blood Sci. (2023) 5:237–48. doi: 10.1097/bs9.0000000000000173
Keywords: non-small cell lung cancer (NSCLC), tumor microenvironment (TME), tyrosine kinase inhibitors (TKIs), immune checkpoint inhibitors (ICIs), chimeric antigen receptor (CAR) T cell therapy
Citation: De Lucia A, Mazzotti L, Gaimari A, Zurlo M, Maltoni R, Cerchione C, Bravaccini S, Delmonte A, Crinò L, Borges de Souza P, Pasini L, Nicolini F, Bianchi F, Juan M, Calderon H, Magnoni C, Gazzola L, Ulivi P and Mazza M (2025) Non-small cell lung cancer and the tumor microenvironment: making headway from targeted therapies to advanced immunotherapy. Front. Immunol. 16:1515748. doi: 10.3389/fimmu.2025.1515748
Received: 23 October 2024; Accepted: 20 January 2025;
Published: 10 February 2025.
Edited by:
S. Paul Gao, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Patrick C. Gedeon, Brigham and Women’s Hospital and Harvard Medical School, United StatesSijia Zhang, Heidelberg University, Germany
Copyright © 2025 De Lucia, Mazzotti, Gaimari, Zurlo, Maltoni, Cerchione, Bravaccini, Delmonte, Crinò, Borges de Souza, Pasini, Nicolini, Bianchi, Juan, Calderon, Magnoni, Gazzola, Ulivi and Mazza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luigi Pasini, bHVpZ2kucGFzaW5pQGlyc3QuZW1yLml0
†Present address: Luigi Pasini, IRCCS Istituto Romagnolo per lo Studio dei Tumori (IRST) “Dino Amadori”, Meldola, FC, Italy