 Niccolette Schaunaman
Niccolette Schaunaman Diana Cervantes
Diana Cervantes Deborah A. Ferrington2
Deborah A. Ferrington2 Hong Wei Chu
Hong Wei Chu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 18 March 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1501898
Introduction: Immunoproteasome (IP) is induced by pro-inflammatory stimuli such as interferon gamma to regulate inflammation and immunity. Asthma patients with airway type 2 high inflammation (e.g., IL-13) demonstrate more eosinophils and airway hyperresponsiveness (AHR) with less interferon gamma. The role of IP in regulating airway eosinophilic inflammation and AHR has not been investigated.
Methods: This study was aimed to determine how IP regulates type 2 inflammation and AHR using LMP7 (a subunit of IP) deficient mouse lungs, precision-cut lung slices (PCLS), and cultured human airway epithelial cells treated with IL-13 in the absence or presence of an IP inhibitor ONX-0914 or exogenous IP.
Results: LMP7 KO mouse lungs had significantly more IL-4Rα protein expression than the wildtype (WT) mice. Following IL-13 treatment in PCLS, LMP7 KO mice had significantly more airway contraction than WT mice, which was coupled with increased eotaxin-2 levels. IP inhibition by ONX-0914 in IL-13 treated human airway epithelial cells resulted in significantly more IL-4Rα protein expression and eotaxin-3 release. IP inhibition in human PCLS significantly increased AHR.
Conclusion: Collectively, these data demonstrated that IP promotes degradation of IL-4Rα, while inhibits type 2 inflammation and AHR. Enhancement of IP expression or activity may serve as an alternative approach to reduce the severity of type 2 inflammation and AHR.
Asthma is a chronic respiratory disease affecting approximately 25 million people, or 7.7% of all Americans (1). Of those suffering from asthma, allergic asthma is the most prevalent phenotype (2, 3). Allergic asthma is associated with wheezing, bronchoconstriction, and type 2 or allergic inflammation characterized by the presence of type 2 cytokines, such as IL-13 and IL-4 (4, 5), which induce the production of chemokines involved in the recruitment and activation of inflammatory cells such as eosinophils (6–9). Increased levels of eosinophils have been associated with increased asthma morbidity such as worsened asthma exacerbations and airways hyperresponsiveness (10–15).
The immunoproteasome (IP) is proteolytic machinery derived from the constitutive proteasome (16, 17). In the presence of pro-inflammatory stimuli such as interferon gamma (IFN-γ), IP subunits LMP2, MECL-1, and LMP7 are induced to replace β1, β2, and β5 subunits in the constitutive proteasome (16). Interestingly, in type 2 inflammation-high asthmatics, IFN-γ levels are low compared to type-2 low asthmatics (18–21), suggesting a potential deficiency of IP. IP is known for its ability to cleave peptides for antigen presentation on major histocompatibility complex (MHC) class I molecules in T cells (22, 23). In addition, IP has been shown to regulate inflammatory responses to viral infection and NF-kB signaling (22–24). The role of IP in regulating allergic inflammation remains unknown. We have recently found that IP deficient mice challenged with house dust mite and then infected with rhinovirus showed significantly more eosinophilic inflammation than their wild-type counterparts (25). The mechanism of how IP inhibits type 2 inflammation was not determined. Chen et. al., demonstrated that LMP7 deficiency enhances M2 macrophage polarization through the regulation of the IL-4 signaling pathway (26). In this study, they showed that LMP7 deficient alveolar macrophages increased STAT6 activation and IRF4 expression, which was associated with an increase in IL-4Rα, a receptor shared by both IL-4 and IL-13 (27–30). However, the authors did not address how IP regulated IL-4Rα expression.
In the present study, we hypothesize that IP reduces lung IL-4Rα levels, leading to reduction in type 2 inflammation, a key contributor to airway hyperresponsiveness. To test this hypothesis, we utilized LMP7 deficient mouse lungs or human donor lungs to generate precision-cut lung slices (PCLS), which allows us to maintain the three-dimensional structure of the lung and measure airway contraction induced by stimuli such as IL-13. Additionally, we performed cell culture experiments to examine the role of IP in human airway epithelial IL-4Rα expression and eosinophils chemokine production with or without IL-13 treatment.
Wild-type (WT) C57BL/6 mice were purchased from the Jackson Laboratories. LMP7 knockout (KO) mice on a C57BL/6 background were kindly provided by Dr. Deborah A. Ferrington at the Doheny Eye Institute, CA. All mice were bred at the National Jewish Health (NJH) biological resource center (BRC) under pathogen-free housing condition. All animal studies were reviewed and approved by the Institutional Animal Care and use Committee at NJH.
To induce mouse allergic airway inflammation, WT and LMP7 KO mice (8 – 12 weeks of age, gender matched) were intranasally sensitized with 10µg/mouse of HDM (Greer laboratories, Lenoir, NC) or 50µl of phosphate-buffered saline (PBS) on day 0 and 7. Mice were then challenged once a day for three consecutive days (day 14-16) with 10µg of HDM or 50µl PBS (25, 31, 32)). 72 hours after the last HDM challenged mice were sacrificed. The left lung was used for IL-4Rα protein quantification via western blot as well as for cytokine mRNA expression. One lobe of the right lung was fixed in 10% formalin for immunofluorescent staining.
Naïve WT and LMP7 KO mice were euthanized by intraperitoneal injection of pentobarbital sodium (Fatal-Plus) in sodium chloride. Lungs were inflated with 1.5% low-melting agarose and sliced into consecutive 250µm thickness sections using a Compresstome® VF-300 vibratome (Precisionary Instruments, Natick, MA).
The upper lobes of the right lung from eight healthy, non-smoking donors were obtained from the International Institute for the Advancement of Medicine (IIAM, Philadelphia, PA), and Donor Alliance of Colorado (Denver, CO). All donor lungs were selected based on non-smoking status and no history of lung disease/infection. The detailed donor demographic information is given in Table 1. Lungs were inflated as previously published (33) and sliced into consecutive 300µm thickness sections. The Institutional Review Board (IRB) at NJH approved our studies as meeting requirements of exempt human subject research.
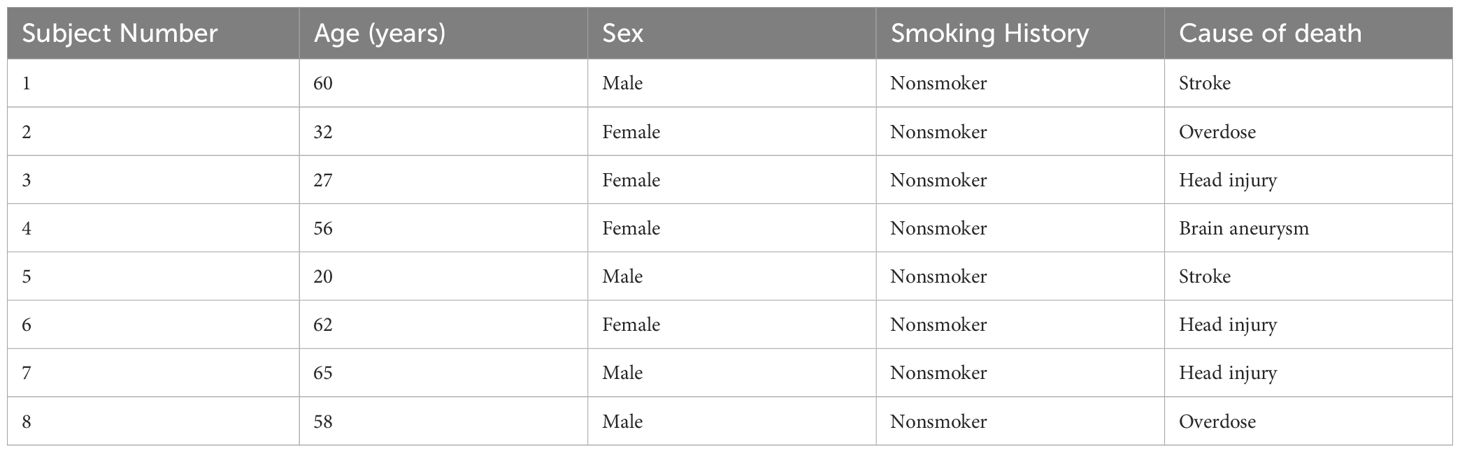
Table 1. Demographic information of human donors for the PCLS experiment.
Both human and mouse slices were transferred to 24-well plates containing Dulbecco’s Modified Eagle’s Medium (DMEM, ThermoFisher Scientific, Waltham, MA) with antifungal agents and antibiotics and incubated in a humidified incubator at 37°C supplemented with 5% CO2.
24 hours after slicing, mouse PCLS were washed with 1X PBS (Fisher Scientific, Hampton, NH), and then treated with 25ng/ml of recombinant murine IL-13 (PeproTech, Cranbury, NJ). Supernatant was harvested 72 hours after IL-13 treatment, and slices underwent methacholine challenge for airway hyperresponsiveness measurement.
24 hours after slicing, human PCLS were washed with 1X PBS (Fisher Scientific, Hampton, NH), and then pre-treated with 100nM ONX-0914, an immunoproteasome inhibitor (APExBio, Houston, TX), for two hours followed by treatment with 25ng/ml of recombinant human IL-13. Supernatant was harvested 72 hours after IL-13 treatment, and slices underwent methacholine challenge for airway hyperresponsiveness measurement.
AHR measurement was performed as we previously published (34). After removing the supernatant, 500µl of warmed DMEM with antifungal agents and antibiotics was added back to PCLS. For both mouse PCLS and human PCLS, baseline images of the airways were taken, followed by adding increasing doses of methacholine (McH) for a final concentration of 1, 10, 100, and 1,000µM to each well. After 30 seconds of each dose of McH, images of airways were taken. The area (relative pixel number) within the lumen was traced using the image J freehand tool, and the percent airway constriction was calculated as (1 – post McH area/baseline area) x 100. The number of airways measured for AHR was between 3-6 per condition for each donor/mouse.
HTBE cells were processed as previously described (35) from deidentified donor lungs that were not suitable for transplantation under a protocol approved by the IRB at NJH. The selected donor lungs were obtained through the IIAM and Donor Alliance of Colorado, and all were non-smokers. The HTBE cells were isolated by enzymatic digestion from the distal region of the trachea and proximal regions of the main bronchi. Freshly isolated HTBE cells were cultured up to one week on 100mm dishes in BronchiaLife culture medium (Lifeline Cell Technology, Frederick, MD). HTBE cells were trypsinized and frozen in liquid nitrogen for future use in BronchiaLife medium supplemented with 30% Fetal Bovine Serum (FBS, SeraPrime, Fort Collins, CO), and 10% dimethyl sulfoxide.
HTBE cells from seven healthy donors were expanded on 100mm culture dishes in BronchiaLife media. Once 100% confluent, cells were trypsinized and lysed in 100µl of radioimmunoprecipitation assay buffer (RIPA). 50µg of lysed protein was incubated with either 0nM, 5nM, or 20nM of whole IP isolated from human spleens (South Bay Bio, San Jose, CA) in 100mM BTP (pH 7.5), 100mM KCL, 10mM EGTA, and 0.035% sodium dodecyl sulfate (SDS) for 5 hours at 37°C. After 5 hours of IP treatment, the mixture of cell lysate and IP was frozen immediately at -80°C for western blotting later.
HTBE cells from six healthy donors were expanded and cultured on 24-well plates in BronchiaLife media. Once 80% confluent, cells were pre-treated with 100nM ONX-0914 for two hours. After two hours, cells were treated with 2.5µg/ml of an anti-IL4Rα antibody (R&D Systems, Minneapolis, MN) or IgG control for 30 minutes. After 30 minutes, cells were treated with 10ng/ml of recombinant human IL-13 (PeptroTech, Cranbury, NJ). Cells and supernatants were harvested both 5 hours after ONX-0914 treatment and 72 hours after IL-13 treatment. 5 hours was chosen to match the incubation time of exogenous IP with airway epithelial cell lysates done above.
The doses of ONX-0914 and IL-13 were selected based on our previous publications (36–39). We chose the dose of the anti-IL4Rα antibody from our dose-response optimization study that showed the maximal reduction of eotaxin-3 levels at 2.5µg/ml in IL-13-stimulated human airway epithelial cells (data not shown).
Cells were lysed in RIPA buffer with Halt protease and phosphatase inhibitor cocktail 100x (ThermoFisher, Waltham, MA). Equal amounts of proteins were loaded and separated by SDS-PAGE, transferred onto PVDF membranes, blocked with blocking buffer, and incubated with the following primary antibodies overnight at 4°C: IL4R Polyclonal antibody (Anti-human, Proteintech Group, Inc., Rosemont, IL), LMP2 (Abcam, Cambridge, United Kingdom), LMP7 (Proteintech Group Inc. Rosemont, IL), IL-4Rα Antibody (Anti-mouse, Santa Cruz Biotechnology, Dallas, TX), and Beta-Actin antibody (Santa Cruz Biotechnology, Dallas, TX). After washes in PBS with 0.1% Tween-20, membranes were incubated with the appropriate horseradish peroxidase (HRP)-linked secondary antibodies and developed using a Fotodyne imaging system (Fotodyne Inc., Harland, WI).
Immunofluorescent (IF) staining was performed on formalin-fixed and paraffin-embedded lung tissue sections from WT and LMP7 KO mice treated with either PBS or challenged with HDM. Sections cut at 5µm were deparaffinized and immersed in pre-heated (95°C) 10mM sodium citrate buffer with 0.05% Tween 20 (pH 6.0) for 20 minutes for antigen retrieval. Slides were incubated with an anti-IL4Rα antibody (ThermoFisher Scientific, Waltham, MA) at 1:100 dilution in 0.1% Triton X-100 in PBS overnight at 4°C, and then incubated with a secondary anti-rabbit antibody conjugated with AF594 at a 1:500 dilution at room temperature for one hour. Slides were then mounted with anti-fade mounting solution with 4’,6-diamidino-2-phenylindole (DAPI) (ThermoFisher). IF staining was imaged using the Revolve microscope (Echo, San Diego, CA). Images were taken at 200x using the same overlay settings for each tissue section.
RNA was extracted from homogenized lung tissue as decribed previously (25, 31, 39, 40). Briefly, RNA was isolated using the TRIzol reagent method, and then reversely transcribed to cDNA.
Taqman Gene Expression Assay (ThermoFisher, Waltham, MA) was used to determine mRNA relative levels of IL-13 and IL-4. Target gene expression was normalized to the housekeeping gene 18S rRNA. The comparative threshold cycle method (ΔΔCt) was applied to determine the relative levels of target genes.
GraphPad PRISM version 10.0 software was used for all statistical analysis. Nonparametric data were analyzed using the Kruskal-Wallis test for multiple comparisons, and the Mann-Whitney test for two-group comparisons. A p-value <0.05 was considered statistically significant.
We have previously shown that allergen challenged IP deficient mice had significantly more eosinophils and eotaxin-2 compared to WT mice (25). To determine the possible mechanisms by which IP regulates type 2 inflammation, we measured IL-4Rα in lungs of WT and LMP7 KO mice treated with either PBS or house dust mite (HDM). HDM has not been found to regulate IL-4Rα levels (41). Similarly in our model, HDM challenge did not increase IL-4Rα levels in either strain of mice. However, LMP7 KO mice had significantly more IL-4Rα protein expression in homogenized lung tissue compared to WT mice (Figure 1A), both at baseline as well as after HDM challenge. This data supports our previous findings that LMP7 KO mice are more susceptible to type 2 inflammatory responses (e.g., eosinophil recruitment) during allergen challenges. To localize IL-4Rα in the lung, immunofluorescent staining was performed. IL-4Rα signal (Figure 1B) was observed in airway epithelium as well as in the alveolar areas which may include immune cells and structural cells. LMP7 KO mice treated with either PBS or HDM seem to have more positive IL-4Rα staining in airway epithelial cells.
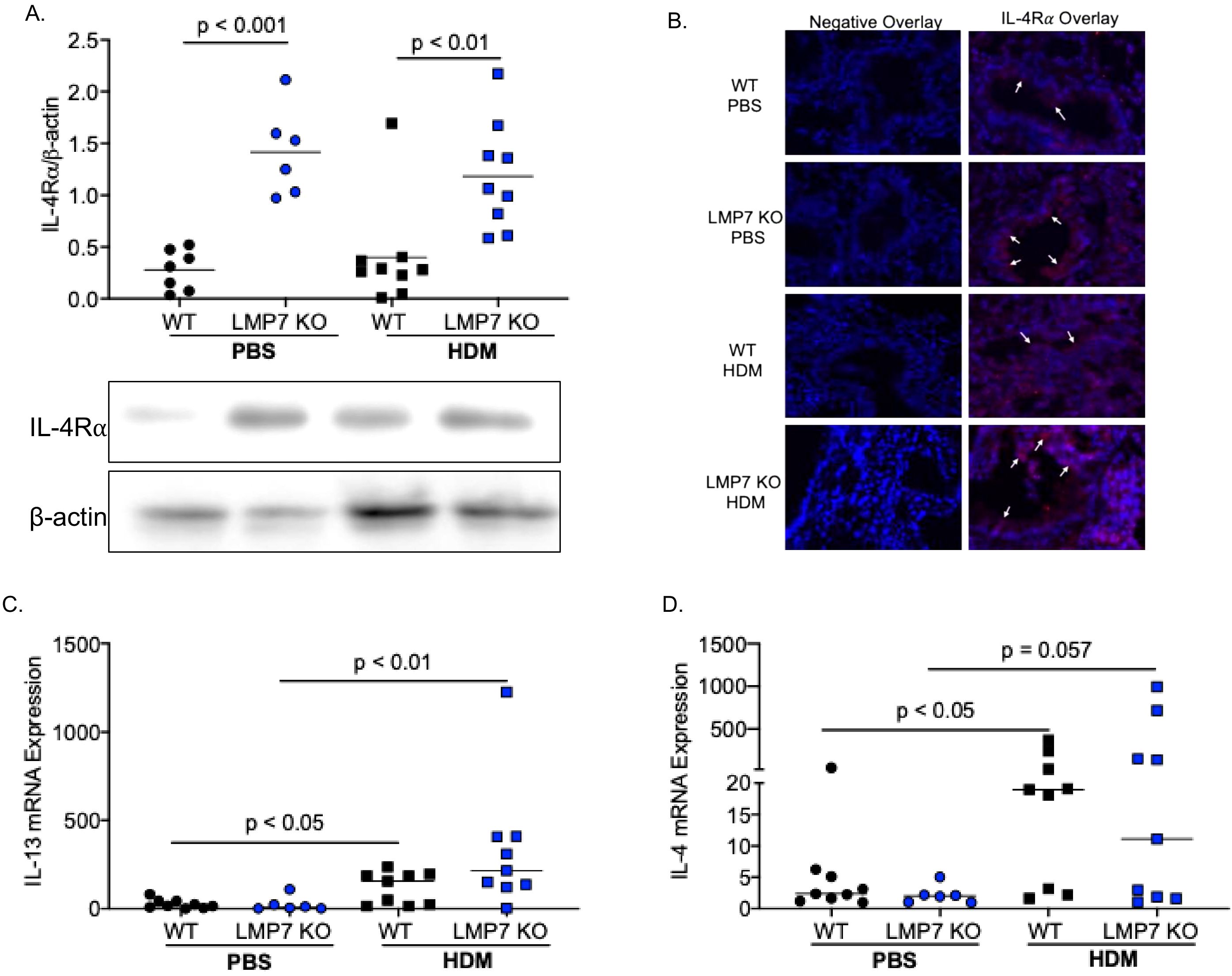
Figure 1. LMP7 deficient mice have significantly more IL-4Rα in homogenized lung tissue. (A) LMP7 KO mice treated with PBS or house dust mite (HDM) had significantly more IL-4Rα protein present in the lung tissue compred to WT mice. (B) IL-4Rα immunofluorescent staining of WT and LMP7 KO lung tissue treated with or without HDM; white arrows represent IL-4Rα staining (red color) on airway epithelial cells. HDM challenge significantly increased IL-13 (C) and IL-4 (D) mRNA levels in both WT and LMP7 KO mice, however there was not statistical difference between WT and LMP7 KO.
To determine if the increased lung eosinophil recruitment in LMP7 KO mice may be induced by heightened type 2 cytokine production, we measured mRNA expression of IL-13 and IL-4. As expected, HDM challenge significantly increased IL-13 levels in WT and LMP7 KO mice (Figure 1C) (42, 43). However, there was no significant difference in IL-13 mRNA levels between WT and LMP7 KO mice. A similar trend was seen with IL-4 mRNA expression (Figure 1D). Our data suggests that excessive eosinophilic inflammation seen in LMP7 KO mice may be attributed to the up-regulation of IL-4Rα and subsequent type 2 inflammatory response.
To determine if IP deficiency enhances AHR, a common feature associated with type 2 inflammation, WT and LMP7 KO mouse PCLS were treated with IL-13 for 72 hours after which airway lumen area was measured following methacholine challenges. Figure 2A shows a dose-dependent increase in airway contraction with each increasing dose of methacholine. As previously reported (44–48), IL-13 treatment in WT mouse PCLS significantly increased AHR compared to control treated slices (Figure 2B). LMP7 KO PCLS had significantly more airway contraction compared to WT slices at baseline, which was further enhanced after IL-13 treatment as AHR in IL-13 treated LMP7 KO PCLS was significantly greater than WT PCLS. Eotaxin-2, an eosinophil chemokine, was upregulated by IL-13 in WT and LMP7 KO PCLS compared to controls (Figure 2C). IL-13 treated LMP7 KO PCLS also had more eotaxin-2 compared to IL-13 treated WT slices.
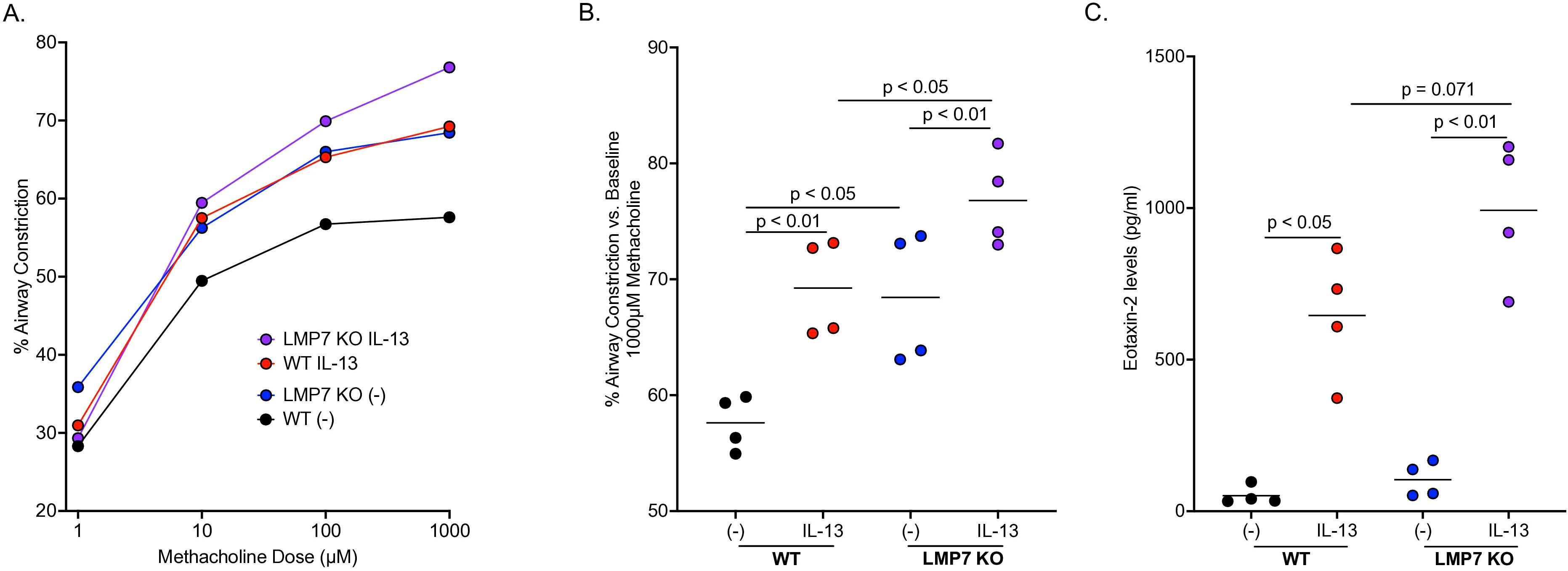
Figure 2. LMP7 deficiency enhances airway hyperresponsiveness in mouse precision-cut lung slices (PCLS) exposed to IL-13. WT and LMP7 KO mouse lungs were inflated, sliced at 250µm, and treated with or without 25ng/ml of IL-13 for 72 hours. Airway contraction with increasing doses of methacholine was measured with each symbol representing the average airway contraction of all experiments (A). LMP7 KO mice had significantly more airway contraction compared to WT mice, which was further increased by IL-13 (B). Eotaxin-2 levels were increased in PCLS by IL-13, with LMP7 KO vs. WT lung tissue having significantly higher eotaxin-2 levels (C). The symbols represent individual experiment with 3 – 8 technical replicates per experiment.
To further establish a role for IP during type 2 inflammation in human airway epithelial cells, IL-4Rα protein levels were measured five hours after HTBE cells were exposed to IL-13 with and without ONX-0914. ONX-0914 in IL-13 treated cells significantly increased IL-4Rα levels compared to IL-13 treatment alone (Figure 3A). Eotaxin-3, and eosinophil chemokine upregulated by IL-13, was increased by IL-13 at 72 hours (Figure 3B). ONX-0914 in IL-13-stimulated cells significantly enhanced eotaxin-3 production. Importantly, when cells were treated with an anti-IL4Rα antibody in the presence of ONX-0914 and IL-13, eotaxin-3 levels were completely inhibited. Our data suggests that IP inhibition increases IL-4Rα protein expression, which is critical to eotaxin production.
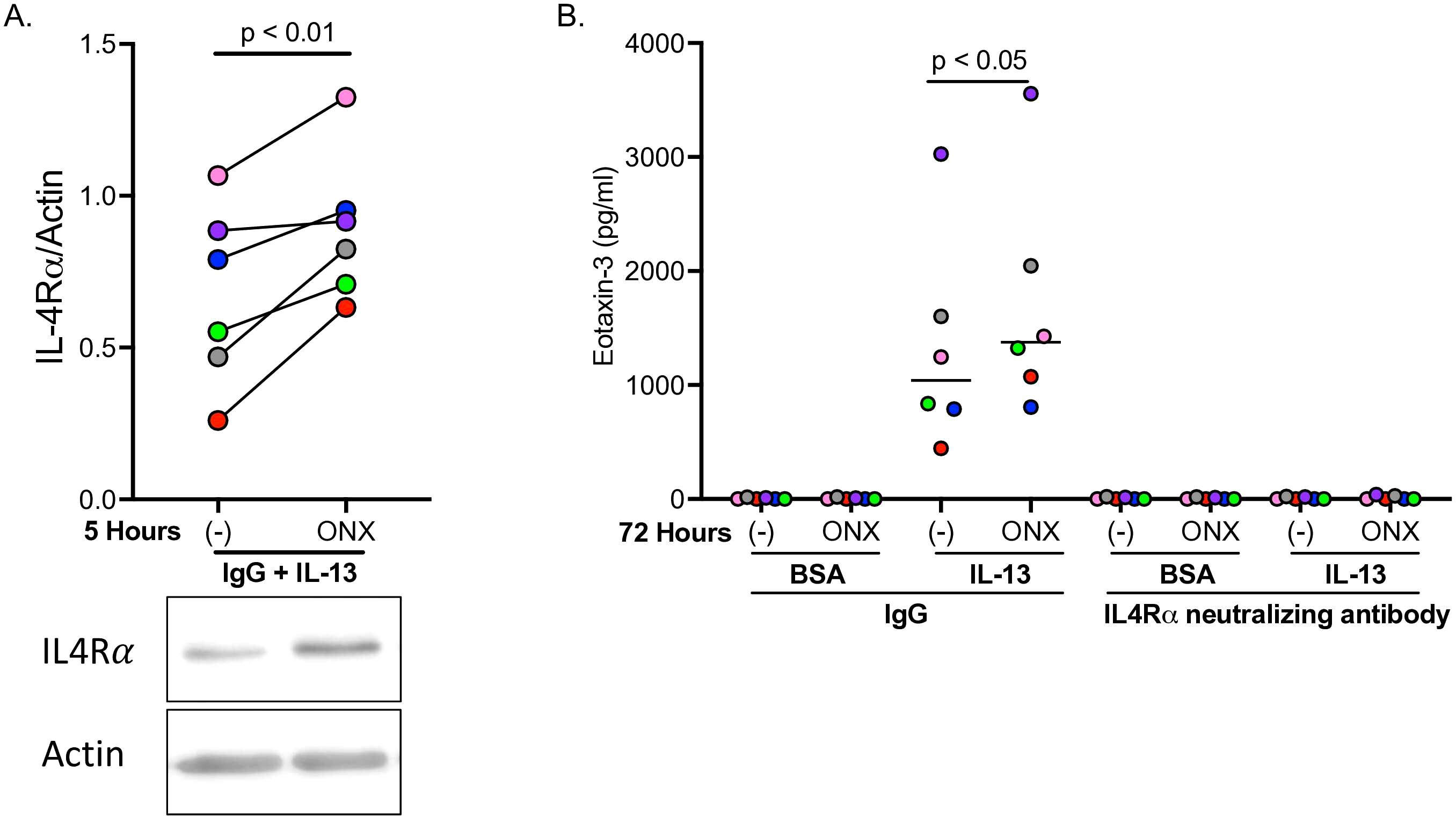
Figure 3. Inhibition of immunoproteasome (IP) increases IL-4Rα and eotaxin-3 in IL-13-stimulated human airway epithelial cells. Tracheobronchial epithelial cells from n=6 healthy donors were pre-treated with immunoproteasome LMP7 inhibitor ONX-0914, then given anti-IL4Rα antibody followed by IL-13 treatment. IP inhibition in IL-13 treated cells significantly upregulated IL-4Rα protein levels five hours after ONX-0914 treatment (A), as well as significantly increased eotaxin-3 levels after 72 hours (B). Anti-IL4Rα antibody significantly inhibited IL-13-induced eotaxin-3.
To determine the direct effect of IP on IL-4Rα protein content, IP isolated from human spleen was incubated with naïve HTBE cells from seven healthy donors for five hours at 37°C. Accordingly, LMP2 and LMP7 (subunits of IP) protein levels increased in a dose-dependent manner following incubation with exogenous IP at 5nM and 20nM (Figures 4A, B). While 5nM IP trended to decrease IL-4Rα protein levels, 20nM IP significantly decreased IL-4Rα levels compared to control and the 5nM IP concentration (Figure 4C). Representative western blot images for IL-4Rα, LMP2, and LMP7 protein expression can be seen in Figure 4D.
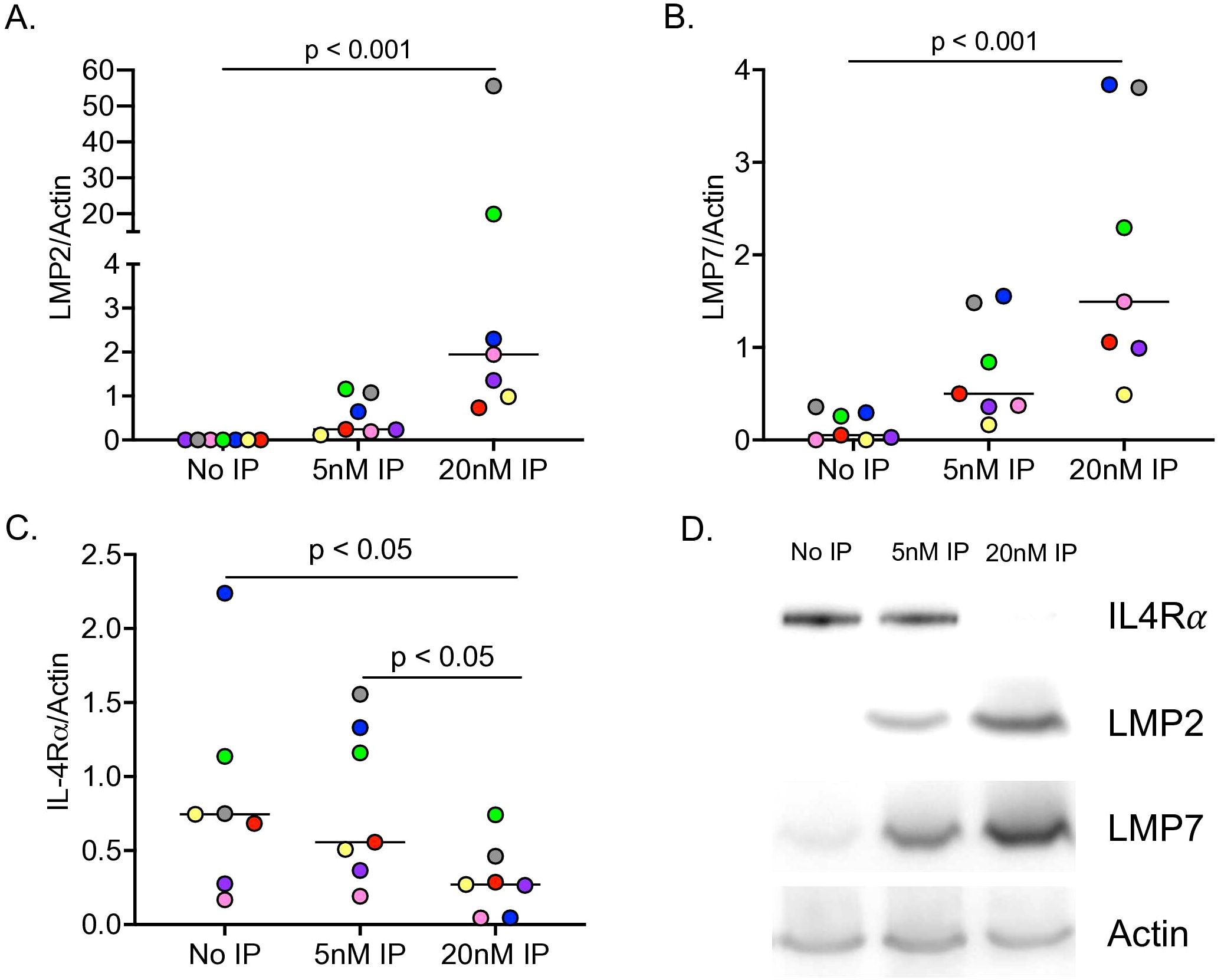
Figure 4. The immunoproteasome (IP) degrades IL-4Rα in human airway epithelial cell lysates. Tracheobronchial epithelial cells from n=7 healthy donors were incubated with 5nM and 20nM of exogenous IP for 5 hours at 37°C. IP it increased LMP2 (A) and LMP7 (B) protein expression, while it decreased IL-4Rα protein expression in a dose-dependent manner (C). Representative western image of IP degrading IL-4Rα and increasing IP subunits (D).
As we observed the enhanced AHR in IP deficient mouse PCLS even without IL-13 treatment, we tested if IP deficiency in human distal lungs increases AHR. PCLS from eight healthy donors were treated with or without ONX-0914 for 72 hours before measuring AHR. IP inhibition by ONX-0914 significantly increased airway contraction as compared to control slices (Figures 5A, B).
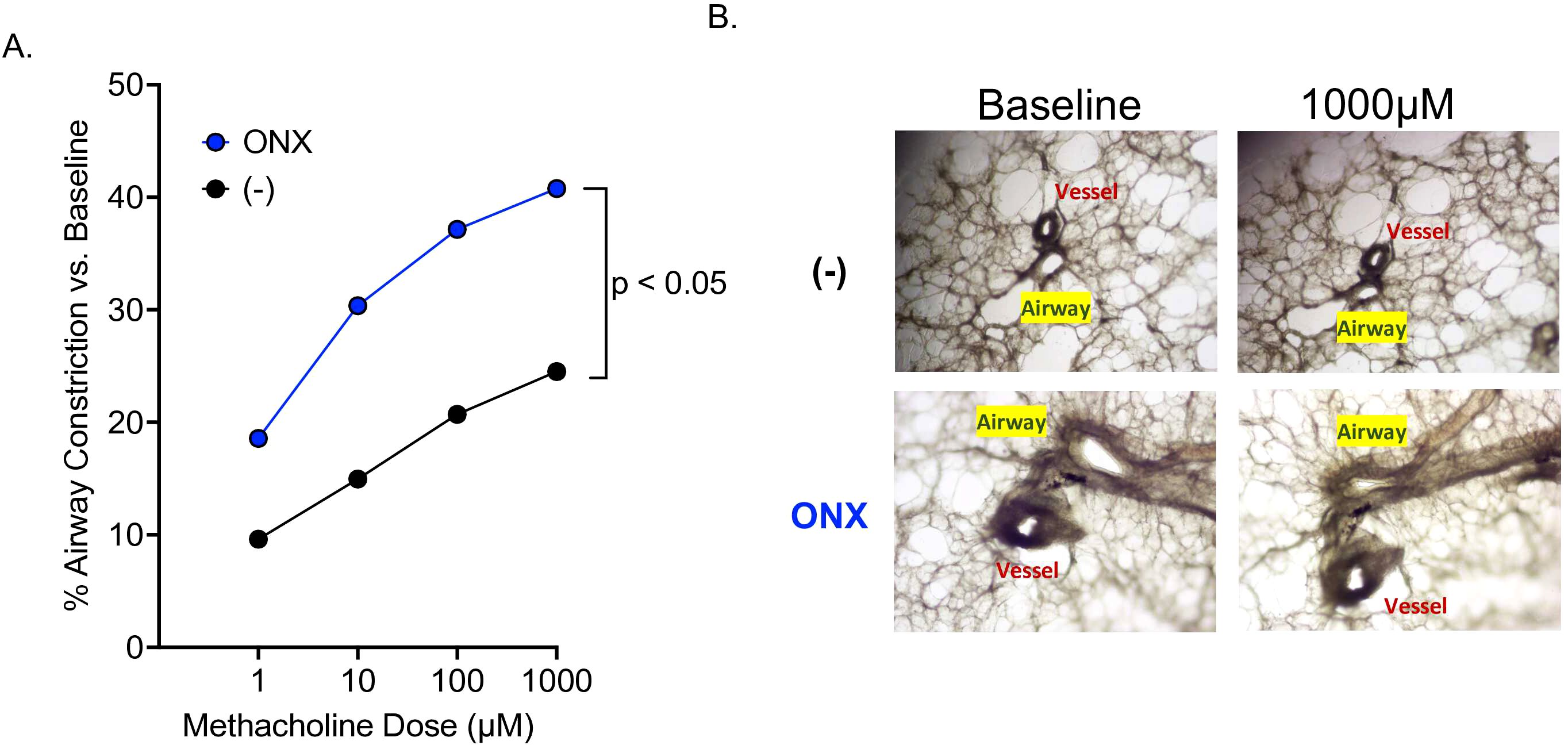
Figure 5. IP inhibition in human PCLS significantly increases airway hyperresponsiveness. Lung from n=8 healthy human lung donors were inflated with low melting agarose, cored, sliced to 300µM, and treated with or without ONX-0914. (A) Airway contraction data. (B) Representative images of airway contraction in PCLS.
This is the first report studying the mechanism of regulation of type 2 inflammation by IP, and the impact of IP deficiency on airway hyperresponsiveness. Our results suggest that degradation of IL-4Rα and reduced IL-13-induced eosinophil chemokine production. In addition, IP deficiency, specifically LMP7 deficiency, contributed to airway contraction in the absence or presence of IL-13 stimulation.
The role of IP in allergic or type 2 inflammation remains controversial. Several research groups have reported differing roles of IP on eosinophilic inflammation depending on types of allergens given. Oliveri et. al., found that mice challenged with ovalbumin and an IP inhibitor had less eosinophils present than vehicle control mice. However, when challenged with house dust mite (HDM) and an IP inhibitor, eosinophil levels were not different from vehicle control mice (49). Another group found similar results when using LMP7 deficient mice and a high dose of HDM (50). Lack of mechanistic studies into how IP directly regulates allergic, or type 2 inflammation may in part explain the above controversial or inconsistent findings. In this study, we clearly demonstrated that IP regulates IL-4Rα protein expression in human airway epithelial cells as IP inhibitor treatment in IL-13-stimulated human airway epithelial cells increased IL-4Rα and eotaxin-3 levels. Moreover, this was evident in our LMP7 deficient mouse model where IL-4Rα levels were increased in LMP7 deficient lung tissue. This increase was not coupled with an increase in type 2 cytokines IL-13 or IL-4 in mice, suggesting that IP does not appear to up-regulate the expression or production of typical type 2 cytokines (e.g., IL-4 and IL-13). Instead, IP may inhibit the signaling of type 2 cytokines through IL-4Rα, such as eotaxin-2 production as we previously reported. Eotaxin-2 production is highly dependent on IL-4/IL-13 and IL-4Rα signaling and is critical to lung eosinophil recruitment and inflammation (51, 52).
Many biologics have been developed to combat type 2 inflammation and allergic asthma, with IL-4Rα being one of the main targets, since both IL-13 and IL-4 utilize it as a receptor (27–30). Our data shows that IP inhibition in human epithelial cells treated with an anti-IL-4Rα neutralizing antibody abolished IL-13-induced eotaxin-3 levels. While little is known about IL-4Rα regulation by IP or proteasome, Wei et. al., found that ubiquitination-mediated proteasomal degradation of IL-4Rα is important in controlling airway inflammation (53). The IP is proteolytic machinery that has been classically considered to play a role in antigen presentation (22, 23) by cleaving different peptides to initiate adaptive immune responses (24, 54). However, multiple studies support a more general role for IP in responding to stress (55–59). Here, we have improved our understanding of how IP may regulate type 2 inflammation by showing the inhibitory role of IP in regulating IL-4Rα content. Increased IL-4Rα expression in airway epithelium has been reported in asthma, particularly atopic/allergic asthma (60). Although genetics and other factors may contribute to increased IL-4Rα in asthma, investigating the role of IP is critical for enhancing our understanding of this important mechanism. Given that IFN-γ induces IP and IFN-γ levels are low in type 2 inflammation-high asthmatics (18–21), it is possible that IP deficiency may exist in type 2 inflammation-high asthmatics. This speculation needs to be confirmed in future work, but reduced IP may offer a new mechanism for type 2 inflammation. Severe chronic obstructive pulmonary disease (COPD) has been associated with a decrease in IP levels (61, 62). Interestingly, impaired IFN signaling has been reported in COPD patients with viral infection (63). Thus, it appears that IFN supplementation may be appropriate in asthma or COPD patients with IP deficiency to attenuate type 2 inflammation.
Airway obstruction is a salient feature of asthma, but the role of IP in airway obstruction or AHR has not been tested. By leveraging the PCLS model where the three-dimensional structure of the lung is maintained, we found that LMP7 deficiency enhanced AHR, in the absence or especially in the presence of IL-13. Even without IL-13 stimulation or at the baseline, LMP7 KO mouse PCLS had significantly more airway contraction than WT slices. This was mirrored in our human PCLS model where IP inhibition by ONX-0914 resulted in significantly more airway contraction. How IP contributes to AHR remains to be determined. One explanation could be related to IL-4Rα-mediated signaling such as smooth muscle contractility and mucus production in the presence of IL-13 stimulation. We still do not know why IP deficiency alone (no IL-13 stimulation) increases AHR. Whether IP deficiency in the lung increased the activity (e.g., release of acetylcholine) of parasympathetic nerves and smooth muscle contraction warrants further investigation. Calcium signaling is critical to airway smooth muscle contraction (64). Proteasome inhibitor may contribute to disruption of intracellular calcium homeostasis (65). Whether IP deficiency at the baseline affects calcium signaling and subsequently enhance AHR at the baseline may need to be tested.
One limitation of our study is that while IP reduces airway epithelial IL-4Rα content, the exact mechanisms (direct vs. indirect) were not determined. Further studies could investigate the direct mechanisms by determining if immunoproteasome components (e.g., LMP7) interact with IL-4Ra and ubiquitinate it, and target IL-4R1 for ubiquitination-mediated proteasomal degradation. Regarding indirect mechanisms, we could test if IP deficiency decreases the expression of other mediators involved in IL-4Rα degradation, including STIP1 homology and U-Box containing protein 1 (STUB1), a chaperone-dependent E3 ubiquitin ligase. In a previous study, STUB1 was shown to promote IL-4Rα degradation (53). Additionally, our study only examined IP-mediated IL-4Rα under IL-13. Adding IL-4, another type 2 cytokine that shares IL-4Rα as a receptor, would further our understanding of IP in the regulation of type 2 inflammatory responses. Another limitation is that we did not use a mouse asthma model to determine the role of IP in AHR. Nonetheless, the PCLS model allowed us to accurately measure airway contraction. Finally, future studies are needed to determine if IP expression and/or activity exist in asthmatics with type 2 inflammation, but not with type 1 inflammation.
By utilizing IP deficient mouse lungs and PCLS models, we have demonstrated that immunoproteasome is a negative regulator of IL-4Rα expression and type 2 inflammation, as well as airway hyperresponsiveness. Enhancement of IP expression or function may be therapeutically beneficial to reduce the severity of asthma in a subset of patients who present with high levels of type 2 inflammation in the airways.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by The Institutional Review Board at National Jewish Health. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from International Institute for the Advancement of Medicine or Donor Alliance of Colorado. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The animal study was approved by Institutional Animal and use Committee at National Jewish Health. The study was conducted in accordance with the local legislation and institutional requirements.
NS: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. DC: Data curation, Methodology, Validation, Writing – review & editing. DF: Resources, Writing – review & editing, Funding acquisition. HC: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the NIH (grants: R01AI150082, R01AI152504, R01AI161296, and U19AI125357 to HWC) and the Stephen J. Ryan-Arnold and Mabel Beckman Foundation (to DAF). These funding sources were not involved in the preparation of data or the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript. References of IP response to stress- these articles are from my lab, but you can do a quick search for other authors to add. The literature is quite robust.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Moorman JE, Akinbami LJ, Bailey CM, Zahran HS, King ME, Johnson CA, et al. National surveillance of asthma: United States, 2001-2010. Vital Health Stat 3. (2012) 35:1–58.
2. Akar-Ghibril N, Casale T, Custovic A, Phipatanakul W. Allergic endotypes and phenotypes of asthma. J Allergy Clin Immunol Pract. (2020) 8:429–40. doi: 10.1016/j.jaip.2019.11.008
3. Schatz M, Rosenwasser L. The allergic asthma phenotype. J Allergy Clin Immunol Pract. (2014) 2:645–8. doi: 10.1016/j.jaip.2014.09.004
4. Behnke O, Ammitzboll T, Jessen H, Klokker M, Nilausen K, Tranum-Jensen J, et al. Non-specific binding of protein-stabilized gold sols as a source of error in immunocytochemistry. Eur J Cell Biol. (1986) 41:326–38.
5. Andrea M, Susanna B, Francesca N, Enrico M, Alessandra V. The emerging role of type 2 inflammation in asthma. Expert Rev Clin Immunol. (2021) 17:63–71. doi: 10.1080/1744666X.2020.1860755
6. Rothenberg ME, Wen T, Shik D, Cole ET, Mingler MM, Munitz A. IL-13 receptor alpha1 differentially regulates aeroallergen-induced lung responses. J Immunol. (2011) 187:4873–80. doi: 10.4049/jimmunol.1004159
7. Munitz A, Brandt EB, Mingler M, Finkelman FD, Rothenberg ME. Distinct roles for IL-13 and IL-4 via IL-13 receptor alpha1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A. (2008) 105:7240–5. doi: 10.1073/pnas.0802465105
8. Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. (2008) 8:384–92. doi: 10.2174/156652408785161032
9. Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. (1998) 282:2258–61. doi: 10.1126/science.282.5397.2258
10. Kume H, Hojo M, Hashimoto N. Eosinophil inflammation and hyperresponsiveness in the airways as phenotypes of COPD, and usefulness of inhaled glucocorticosteroids. Front Pharmacol. (2019) 10:765. doi: 10.3389/fphar.2019.00765
11. Littlefield LC. Therapeutic drug monitoring in ambulatory pediatrics. J Pediatr Health Care. (1987) 1:113–6. doi: 10.1016/0891-5245(87)90182-9
12. Price D, Wilson AM, Chisholm A, Rigazio A, Burden A, Thomas M, et al. Predicting frequent asthma exacerbations using blood eosinophil count and other patient data routinely available in clinical practice. J Asthma Allergy. (2016) 9:1–12. doi: 10.2147/JAA.S97973
13. Talini D, Novelli F, Bacci E, Bartoli M, Cianchetti S, Costa F, et al. Sputum eosinophilia is a determinant of FEV1 decline in occupational asthma: results of an observational study. BMJ Open. (2015) 5:e005748. doi: 10.1136/bmjopen-2014-005748
14. Matsuda H, Nakano S, Shirakura R, Matsuwaka R, Ohkubo N, Ohtani M, et al. Surgery for aortic arch aneurysm with selective cerebral perfusion and hypothermic cardiopulmonary bypass. Circulation. (1989) 80:I243–8.
15. Katz LE, Gleich GJ, Hartley BF, Yancey SW, Ortega HG. Blood eosinophil count is a useful biomarker to identify patients with severe eosinophilic asthma. Ann Am Thorac Soc. (2014) 11:531–6. doi: 10.1513/AnnalsATS.201310-354OC
16. Nathan JA, Spinnenhirn V, Schmidtke G, Basler M, Groettrup M, Goldberg AL. Immuno- and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. Cell. (2013) 152:1184–94. doi: 10.1016/j.cell.2013.01.037
17. Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol. (1999) 17:739–79. doi: 10.1146/annurev.immunol.17.1.739
18. Luo W, Hu J, Xu W, Dong J. Distinct spatial and temporal roles for Th1, Th2, and Th17 cells in asthma. Front Immunol. (2022) 13:974066. doi: 10.3389/fimmu.2022.974066
19. Leonard C, Tormey V, Burke C, Poulter LW. Allergen-induced cytokine production in atopic disease and its relationship to disease severity. Am J Respir Cell Mol Biol. (1997) 17:368–75. doi: 10.1165/ajrcmb.17.3.2797
20. Fallon PG, Schwartz C. The high and lows of type 2 asthma and mouse models. J Allergy Clin Immunol. (2020) 145:496–8. doi: 10.1016/j.jaci.2019.11.031
21. Teixeira LK, Fonseca BP, Barboza BA, Viola JP. The role of interferon-gamma on immune and allergic responses. Mem Inst Oswaldo Cruz. (2005) 100 Suppl 1:137–44. doi: 10.1590/s0074-02762005000900024
22. Basler M, Kirk CJ, Groettrup M. The immunoproteasome in antigen processing and other immunological functions. Curr Opin Immunol. (2013) 25:74–80. doi: 10.1016/j.coi.2012.11.004
23. Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci. (2012) 109:75–112. doi: 10.1016/B978-0-12-397863-9.00003-1
24. Dimasuay KG, Schaunaman N, Berg B, Cervantes D, Kruger E, Heppner FL, et al. Airway epithelial immunoproteasome subunit LMP7 protects against rhinovirus infection. Sci Rep. (2022) 12:14507. doi: 10.1038/s41598-022-18807-3
25. Schaunaman N, Nichols T, Cervantes D, Hartsoe P, Ferrington DA, Chu HW. The effect of a TLR3 agonist on airway allergic inflammation and viral infection in immunoproteasome-deficient mice. Viruses. (2024) 16:1384. doi: 10.3390/v16091384
26. Chen S, Kammerl IE, Vosyka O, Baumann T, Yu Y, Wu Y, et al. Immunoproteasome dysfunction augments alternative polarization of alveolar macrophages. Cell Death Differ. (2016) 23:1026–37. doi: 10.1038/cdd.2016.3
27. Keegan AD, Leonard WJ, Zhu J. Recent advances in understanding the role of IL-4 signaling. Fac Rev. (2021) 10:71. doi: 10.12703/r/10-71
28. Aman MJ, Tayebi N, Obiri NI, Puri RK, Modi WS, Leonard WJ. cDNA cloning and characterization of the human interleukin 13 receptor alpha chain. J Biol Chem. (1996) 271:29265–70. doi: 10.1074/jbc.271.46.29265
29. Russell SM, Keegan AD, Harada N, Nakamura Y, Noguchi M, Leland P, et al. Interleukin-2 receptor gamma chain: a functional component of the interleukin-4 receptor. Science. (1993) 262:1880–3. doi: 10.1126/science.8266078
30. Kondo M, Takeshita T, Ishii N, Nakamura M, Watanabe S, Arai K, et al. Sharing of the interleukin-2 (IL-2) receptor gamma chain between receptors for IL-2 and IL-4. Science. (1993) 262:1874–7. doi: 10.1126/science.8266076
31. Nouri HR, Schaunaman N, Kraft M, Li L, Numata M, Chu HW. Tollip deficiency exaggerates airway type 2 inflammation in mice exposed to allergen and influenza A virus: role of the ATP/IL-33 signaling axis. Front Immunol. (2023) 14:1304758. doi: 10.3389/fimmu.2023.1304758
32. Dimasuay KG, Schaunaman N, Martin RJ, Pavelka N, Kolakowski C, Gottlieb RA, et al. Parkin, an E3 ubiquitin ligase, enhances airway mitochondrial DNA release and inflammation. Thorax. (2020) 75:717–24. doi: 10.1136/thoraxjnl-2019-214158
33. Agraval H, Crue T, Schaunaman N, Numata M, Day BJ, Chu HW. Electronic cigarette exposure increases the severity of influenza a virus infection via TRAIL dysregulation in human precision-cut lung slices. Int J Mol Sci. (2023) 24:4295. doi: 10.3390/ijms24054295
34. Cervantes D, Schaunaman N, Downey GP, Chu HW, Day BJ. Desert particulate matter from Afghanistan increases airway obstruction in human distal lungs exposed to type 2 cytokine IL-13. Front Med (Lausanne). (2023) 10:1177665. doi: 10.3389/fmed.2023.1177665
35. Wu Q, Jiang D, Smith S, Thaikoottathil J, Martin RJ, Bowler RP, et al. IL-13 dampens human airway epithelial innate immunity through induction of IL-1 receptor-associated kinase M. J Allergy Clin Immunol. (2012) 129:825–33 e2. doi: 10.1016/j.jaci.2011.10.043
36. Schaunaman N, Dimasuay KG, Kraft M, Chu HW. Tollip interaction with STAT3: a novel mechanism to regulate human airway epithelial responses to type 2 cytokines. Respir Res. (2022) 23:31. doi: 10.1186/s12931-022-01941-x
37. Dakhama A, Al Mubarak R, Pavelka N, Voelker D, Seibold M, Ledford JG, et al. Tollip inhibits ST2 signaling in airway epithelial cells exposed to type 2 cytokines and rhinovirus. J Innate Immun. (2020) 12:103–15. doi: 10.1159/000497072
38. Francisco D, Wang Y, Conway M, Hurbon AN, Dy ABC, Addison KJ, et al. Surfactant protein-A protects against IL-13-induced inflammation in asthma. J Immunol. (2020) 204:2829–39. doi: 10.4049/jimmunol.1901227
39. Dimasuay KG, Sanchez A, Schaefer N, Polanco J, Ferrington DA, Chu HW. Immunoproteasomes as a novel antiviral mechanism in rhinovirus-infected airways. Clin Sci (Lond). (2018) 132:1711–23. doi: 10.1042/CS20180337
40. Schaunaman N, Dimasuay KG, Cervantes D, Li L, Numata M, Kraft M, et al. Tollip inhibits IL-33 release and inflammation in influenza A virus-infected mouse airways. J Innate Immun. (2023) 15:67–77. doi: 10.1159/000525315
41. Hadebe S, Khumalo J, Mangali S, Mthembu N, Ndlovu H, Scibiorek M, et al. Deletion of IL-4Ralpha signaling on B cells limits hyperresponsiveness depending on antigen load. J Allergy Clin Immunol. (2021) 148:99–109 e5. doi: 10.1016/j.jaci.2020.12.635
42. Gregory LG, Causton B, Murdoch JR, Mathie SA, O’Donnell V, Thomas CP, et al. Inhaled house dust mite induces pulmonary T helper 2 cytokine production. Clin Exp Allergy. (2009) 39:1597–610. doi: 10.1111/j.1365-2222.2009.03302.x
43. Jang YH, Choi JK, Jin M, Choi YA, Ryoo ZY, Lee HS, et al. House Dust Mite Increases pro-Th2 Cytokines IL-25 and IL-33 via the Activation of TLR1/6 Signaling. J Invest Dermatol. (2017) 137:2354–61. doi: 10.1016/j.jid.2017.03.042
44. Cooper PR, Lamb R, Day ND, Branigan PJ, Kajekar R, San Mateo L, et al. TLR3 activation stimulates cytokine secretion without altering agonist-induced human small airway contraction or relaxation. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L530–7. doi: 10.1152/ajplung.00133.2009
45. Danov O, Jimenez Delgado SM, Obernolte H, Seehase S, Dehmel S, Braubach P, et al. Human lung tissue provides highly relevant data about efficacy of new anti-asthmatic drugs. PloS One. (2018) 13:e0207767. doi: 10.1371/journal.pone.0207767
46. Jiang H, Xie Y, Abel PW, Toews ML, Townley RG, Casale TB, et al. Targeting phosphoinositide 3-kinase gamma in airway smooth muscle cells to suppress interleukin-13-induced mouse airway hyperresponsiveness. J Pharmacol Exp Ther. (2012) 342:305–11. doi: 10.1124/jpet.111.189704
47. Kellner J, Gamarra F, Welsch U, Jorres RA, Huber RM, Bergner A. IL-13Ralpha2 reverses the effects of IL-13 and IL-4 on bronchial reactivity and acetylcholine-induced Ca+ signaling. Int Arch Allergy Immunol. (2007) 142:199–210. doi: 10.1159/000097022
48. Li G, Cohen JA, Martines C, Ram-Mohan S, Brain JD, Krishnan R, et al. Preserving airway smooth muscle contraction in precision-cut lung slices. Sci Rep. (2020) 10:6480. doi: 10.1038/s41598-020-63225-y
49. Oliveri F, Basler M, Rao TN, Fehling HJ, Groettrup M. Immunoproteasome inhibition reduces the T helper 2 response in mouse models of allergic airway inflammation. Front Immunol. (2022) 13:870720. doi: 10.3389/fimmu.2022.870720
50. Volkov A, Hagner S, Loser S, Alnahas S, Raifer H, Hellhund A, et al. beta5i subunit deficiency of the immunoproteasome leads to reduced Th2 response in OVA induced acute asthma. PloS One. (2013) 8:e60565. doi: 10.1371/journal.pone.0060565
51. Watanabe K, Jose PJ, Rankin SM. Eotaxin-2 generation is differentially regulated by lipopolysaccharide and IL-4 in monocytes and macrophages. J Immunol. (2002) 168:1911–8. doi: 10.4049/jimmunol.168.4.1911
52. Menzies-Gow A, Ying S, Sabroe I, Stubbs VL, Soler D, Williams TJ, et al. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J Immunol. (2002) 169:2712–8. doi: 10.4049/jimmunol.169.5.2712
53. Wei Q, Sha Y, Bhattacharya A, Abdel Fattah E, Bonilla D, Jyothula SS, et al. Regulation of IL-4 receptor signaling by STUB1 in lung inflammation. Am J Respir Crit Care Med. (2014) 189:16–29. doi: 10.1164/rccm.201305-0874OC
54. Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med. (2009) 15:781–7. doi: 10.1038/nm.1978
55. Hussong SA, Roehrich H, Kapphahn RJ, Maldonado M, Pardue MT, Ferrington DA. A novel role for the immunoproteasome in retinal function. Invest Ophthalmol Vis Sci. (2011) 52:714–23. doi: 10.1167/iovs.10-6032
56. Ferrington DA, Hussong SA, Roehrich H, Kapphahn RJ, Kavanaugh SM, Heuss ND, et al. Immunoproteasome responds to injury in the retina and brain. J Neurochem. (2008) 106:158–69. doi: 10.1111/j.1471-4159.2008.05345.x
57. Hussong SA, Kapphahn RJ, Phillips SL, Maldonado M, Ferrington DA. Immunoproteasome deficiency alters retinal proteasome’s response to stress. J Neurochem. (2010) 113:1481–90. doi: 10.1111/j.1471-4159.2010.06688.x
58. Schuld NJ, Hussong SA, Kapphahn RJ, Lehmann U, Roehrich H, Rageh AA, et al. Immunoproteasome deficiency protects in the retina after optic nerve crush. PloS One. (2015) 10:e0126768. doi: 10.1371/journal.pone.0126768
59. Petersen A, Zetterberg M. The immunoproteasome in human lens epithelial cells during oxidative stress. Invest Ophthalmol Vis Sci. (2016) 57:5038–45. doi: 10.1167/iovs.16-19536
60. Kotsimbos TC, Ghaffar O, Minshall EM, Humbert M, Durham SR, Pfister R, et al. Expression of the IL-4 receptor alpha-subunit is increased in bronchial biopsy specimens from atopic and nonatopic asthmatic subjects. J Allergy Clin Immunol. (1998) 102:859–66. doi: 10.1016/S0091-6749(98)70029-6
61. Kammerl IE, Dann A, Mossina A, Brech D, Lukas C, Vosyka O, et al. Impairment of immunoproteasome function by cigarette smoke and in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2016) 193:1230–41. doi: 10.1164/rccm.201506-1122OC
62. Baker TA, Bach H, Gamelli RL, Love RB, Majetschak M. Proteasomes in lungs from organ donors and patients with end-stage pulmonary diseases. Physiol Res. (2014) 63:311–9. doi: 10.33549/physiolres
63. Mehta M, Paudel KR, Shukla SD, Shastri MD, Singh SK, Gulati M, et al. Interferon therapy for preventing COPD exacerbations. EXCLI J. (2020) 19:1477–80. doi: 10.17179/excli2020-2997
64. Jude JA, Wylam ME, Walseth TF, Kannan MS. Calcium signaling in airway smooth muscle. Proc Am Thorac Soc. (2008) 5:15–22. doi: 10.1513/pats.200704-047VS
Keywords: immunoproteasome, IL-4RA, type 2 inflammation, precision-cut lung slices, airway hyperresponsiveness
Citation: Schaunaman N, Cervantes D, Ferrington DA and Chu HW (2025) Degradation of IL-4Ralpha by Immunoproteasome: implication in airway type 2 inflammation and hyperresponsiveness. Front. Immunol. 16:1501898. doi: 10.3389/fimmu.2025.1501898
Received: 25 September 2024; Accepted: 03 March 2025;
Published: 18 March 2025.
Edited by:
Markus Bosmann, Boston University, United StatesReviewed by:
Georges Doumet Helou, Université Paris Cité, FranceCopyright © 2025 Schaunaman, Cervantes, Ferrington and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Wei Chu, Y2h1aHdAbmpoZWFsdGgub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.