 Ziyan Huang
Ziyan Huang Miao Hao
Miao Hao Naixu Shi
Naixu Shi Xinyu Wang
Xinyu Wang Lin Yuan
Lin Yuan Haotian Yuan
Haotian Yuan Xiaofeng Wang
Xiaofeng Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 February 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1482033
Porphyromonas gingivalis (P. gingivalis) is a gram-negative bacterium and the main causative agent of periodontitis, a disease closely associated with the development of periodontal disease. The progression of periodontitis, a chronic infectious disease, is intricately linked to the inflammatory immune response. Inflammatory cytokines act on periodontal tissues via immunomodulation, resulting in the destruction of the periodontal tissue. Recent studies have established connections between periodontitis and various systemic diseases, including cardiovascular diseases, tumors, and neurodegenerative diseases. Neurodegenerative diseases are neurological disorders caused by immune system dysfunction, including Alzheimer’s and Parkinson’s diseases. One of the main characteristics of neurodegenerative diseases is an impaired inflammatory response, which mediates neuroinflammation through microglial activation. Some studies have shown an association between periodontitis and neurodegenerative diseases, with P. gingivalis as the primary culprit. P. gingivalis can cross the blood-brain barrier (BBB) or mediate neuroinflammation and injury through a variety of pathways, including the gut-brain axis, thereby affecting neuronal growth and survival and participating in the onset and progression of neurodegenerative diseases. However, comprehensive and systematic summaries of studies on the infectious origin of neurodegenerative diseases are lacking. This article reviews and summarizes the relationship between P. gingivalis and neurodegenerative diseases and its possible regulatory mechanisms. This review offers new perspectives into the understanding of neurodegenerative disease development and highlights innovative approaches for investigating and developing tailored medications for treating neurodegenerative conditions, particularly from the viewpoint of their association with P. gingivalis.
The aging population is leading to an annual increase in neurodegenerative diseases like Alzheimer’s disease (AD) and Parkinson’s diseases (PD), which poses a significant threat to human health. Currently, effective treatments strategies and drugs are lacking, suggesting that some key pathogenic mechanisms may remain undiscovered. Recent studies indicate that Porphyromonas gingivalis, the primary bacterium responsible for periodontitis, may significantly contribute to the onset and progression of neurodegenerative diseases.
Periodontitis, a common chronic inflammatory disease, affects over 700 million people worldwide and is frequently overlooked and untreated (1). Porphyromonas gingivalis (P. gingivalis) is the primary pathogenic bacterium causing periodontitis, with approximately 85.75% of subgingival plaque samples testing positive for this opportunistic bacteria (2). Typically, inflammatory factors such as C-reactive protein, IL-6 and IL-21 are significantly upregulated in serum of patients with P. gingivalis -induced periodontitis (3). This is a distinctive feature of Chronic Low-Grade Inflammatory Phenotype (CLIP), which often occurs during the aging process (4, 5). Studies have shown that periodontitis is an important risk factor for various health issues, including cardiovascular diseases, diabetes, and Parkinson’s disease (6, 7). Recent research found P. gingivalis in the brains and spinal cord of Alzheimer’s patients (8). Furthermore, study confirms that P. gingivalis infection enhances BBB permeability, and gingivally infected P. gingivalis may cause cognitive decline with periodontitis (9, 10). These findings confirm the strong link between P. gingivalis and the development of neurodegenerative diseases.
Most studies on the relationship between periodontitis, P. gingivalis periodontitis, and neurodegenerative diseases have focused primarily on clinical and epidemiological perspectives. However, there is little research on the specific regulatory mechanisms connecting the two, and the existing summaries are inadequate. This review aims to closely examine the association and potential mechanisms linking P. gingivalis and neurodegenerative diseases, including the roles of bacteria, inflammation, and immune system responses. Through this review, we hope to propose some innovative concepts to clarify the origins of neurodegenerative diseases. At the same time, we also hope to offer fresh ideas for preventing and treating these diseases.
P. gingivalis colonizes the oral epithelium and forms part of the plaque beneath the gums. It can alter the symbiotic composition of bacteria in the oral cavity, leading to ecological dysbiosis. The production of P. gingivalis biofilms is associated with the formation of bacterial plaques in gingival tissue, which further exacerbates gingival damage by other oral bacteria (11, 12).
P. gingivalis possesses several unique properties and virulence factors that affect the host (2). One notable characteristic is the shedding of outer membrane vesicles (OMVs). These vesicles contain virulence factors, particularly gingival proteases and lipopolysaccharide (LPS) (13, 14). Gingipain triggers an immune escape response by modulating inflammatory mediators and suppressing immune cell activity, including the activities of the lysine-gingipain (Kgp) and arginine-gingipain (Rgp). P. gingivalis also produces P. gingivalis lipopolysaccharide (P.g-LPS), which activates the natural host immune response (15, 16). Most strains of P. gingivalis are covered by a capsule that protects the bacteria from attack and host complement killing (17). Some studies have shown that encapsulated strains are more virulent in a mouse model of infection (18, 19). Therefore, these toxins make P. gingivalis highly pathogenic, enabling its components to enter the brain through various pathways. This entry triggers pathological reactions and contributes to the development of neurodegenerative diseases.
Based on current research, we conclude that oral infections caused by P. gingivalis can affect the brain in three ways (Figure 1). First, P. gingivalis causes local chronic inflammation and disrupts central nervous system (CNS) homeostasis through the blood-brain barrier, indirectly promoting neuroinflammation. The association of high loads of P. gingivalis with increased serum TNF-α levels suggests that P. gingivalis not only triggers the development of local inflammatory periodontitis but also leads to elevated serum levels of pro-inflammatory cytokines (20). In addition, prolonged exposure to harmful substances disrupts and increases blood-brain barrier permeability, allowing peripheral pro-inflammatory cytokines to enter the vagus nerve (21).
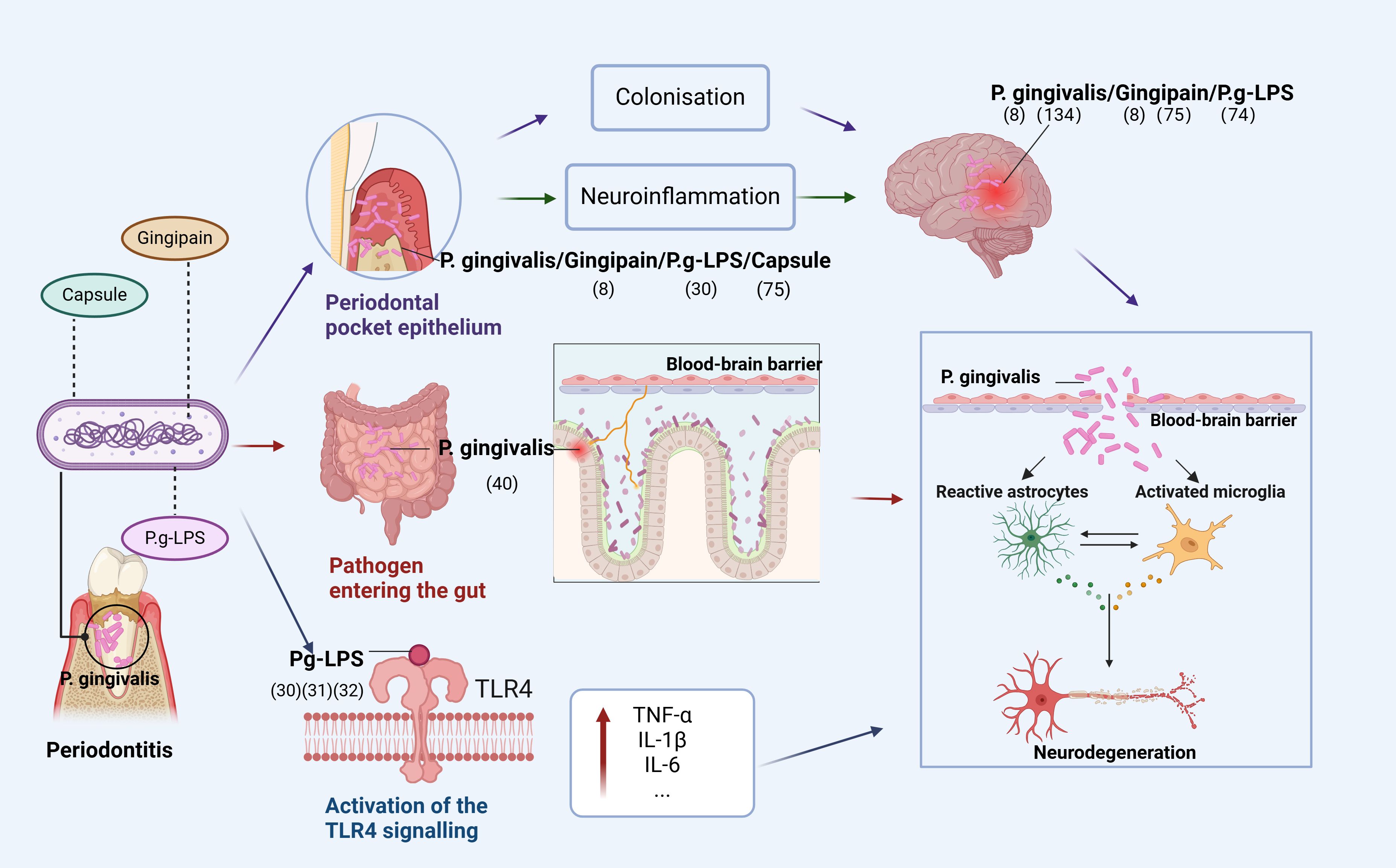
Figure 1. Pathways of P. gingivalis to the brain. The figure depicts that P. gingivalis virulence factor P.g-LPS can enter the brain through three pathways: (1) enters the epithelium of periodontal pockets to cause local inflammation and indirectly promotes neuroinflammation or directly colonize the brain; (2) disrupts the homeostatic balance of intestinal flora and enters the brain through the gut-brain axis to cause neuroinflammation; (3) activates the TLR4 signaling-inducing OS, leading to mitochondrial dysfunction and neuroinflammation (Figure was created with BioRender.com. Huang, Z. (2025) https://BioRender.com/a16s851).
Second, P. gingivalis enters the intestine through the mouth and can disrupt the intestinal flora, leading to an inflammatory response transmitted to the brain through the gut-brain axis (GBA). Dysregulation of the gut microbiota is strongly associated with the development of neurodegenerative diseases through the regulation of the GBA, and P. gingivalis can alter the ratio of T lymphocytes to inflammatory T cells in mesenteric lymph nodes and increase inflammatory cytokines, disrupting the gut microbiota (22–24). Gut microbiota can alter immune cells and stimulate the production of pro-inflammatory cytokines (25–27), with immune cells and inflammatory mediators subsequently entering the brain (28).
Finally, TLR4 signaling activated by P. gingivalis induces OS, leading to mitochondrial dysfunction and neuroinflammation. In both cases, it is involved in the progression of degenerative diseases due to neuroinflammation. A recent study showed that Pg-LPS acts via TLR4. Furthermore, administering Pg-LPS triggers TLR4 signals and elevates markers of dementia and neuroinflammation that have been linked to AD (29). Additional research has revealed that neuroinflammation caused by Pg-LPS is facilitated by the activation of the TLR4 signaling route (30, 31). Research indicates that neurons are capable of expressing both TLR2 and TLR4, implying that these receptors are crucial for neuroinflammatory responses (32, 33).
Neurodegenerative diseases are disorders that impact the central nervous system, such as AD, PD, multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD). These diseases are commonly associated with aging, and their occurrence rises as people get older (34). Immune responses from activated neuroglial cells is crucial in neurodegenerative diseases, particularly in the most prevalent forms: AD and PD (35). Neurodegenerative diseases target various areas of the brain, resulting in different symptoms and causes. Despite these differences, neurodegenerative diseases share common features and involve similar cellular and molecular processes.
An imbalance in the inflammatory response is a common issue in neurodegenerative diseases that leads to neuroinflammation in various parts of the brain, worsening the overall condition (36). The released mediators impact microglia and astrocytes, potentially harming neurons and the central nervous system (36–38). For instance, pyroptotic cell of microglia can lead to neuroinflammation, triggering various neurodegenerative diseases (39). Similarly, damage to astrocytes can lead to specific neuroinflammatory markers, resulting in further complications (40). Another notable feature of neurodegenerative diseases is oxidative stress. In the brains of patients with neurodegenerative diseases, elevated levels of reactive oxygen species are often seen, suggesting that oxidative damage might make the disease worse (41, 42).
Inflammation is the response of cells and tissues to injury, trauma, or infection. Research shows that there is a significant link between the brain and the immune system. Inflammation occurring in the brain is called neuroinflammation (43). Neuroinflammation is an early feature of neurodegenerative diseases. It can activate the immune system, leading to the release of pro-inflammatory cytokines, increased oxidative stress (OS), and abnormal protein deposition, all of which may harm neurons directly or indirectly (44–46). For instance, while neuroinflammation can help remove deposited Aβ, it can also generate cytotoxic substances that worsen Aβ deposition and contribute to neurodegenerative damage (47). Recent studies suggest that neuroinflammation might be a result of periodontitis (8). The immune cells in the brain mainly consist of microglia and astrocytes. Chronic inflammation in the body triggers the activation of microglia and astrocytes in the brain. For instance, when the Toll-like receptors (TLRs) in these cells get activated, they produce various pro-inflammatory cytokines, leading to neuroinflammation that causes damage or death to neurons (48–50).
Microglia are innate immune cells that participate in synaptic remodeling and defense functions, involved in homeostasis and host defense against pathogens and CNS diseases (51). Under normal conditions, microglia are highly branched and perform their sensory functions (52). With aging and chronic stress, microglia activation exhibits dystrophic morphology and an exaggerated inflammatory response (53). Although neuroinflammation is a neuroprotective mechanism, it induces neurotoxicity and is associated with neurodegeneration (51). In neurodegenerative diseases, microglia can migrate the site of injury, produce cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and accumulate pathogenic proteins that mediate neuroinflammation (49).
Astrocytes, the most abundant glial cells in the CNS, perform various functions in healthy neural tissues, including the regulation of blood flow and extracellular fluid, ionic and transmitter balance, energy supply, and synaptic function (54). Astrocytes are activated in response to pathological stimulation. During neuroinflammation, astrocytes enhance the activity of the IL-17 receptor, a crucial inflammatory agent released by effector T lymphocytes (55). The binding of IL-17 to various transmembrane receptors results in the activation of NF-κB-activating factor 1 (Act1) and the formation of signaling complexes, leading to the synthesis of pro-inflammatory cytokines, chemokines, and metalloproteinases (50).
The role of astrocytes in neuroinflammation is twofold. Astrocytes can reduce inflammation by releasing anti-inflammatory factors and promoting the production of neuroprotective factors. Also, they can exacerbate inflammation and nerve damage by releasing pro-inflammatory and neurotoxic molecules. Thus, the role of astrocytes in neuroinflammation is complex and encompasses pro-inflammatory, inhibitory, and pro-neuroprotective effects.
P. gingivalis has significant implications in the pathophysiology of neurodegenerative diseases because its virulence factors can enter the brain through three pathways, causing OS and neuroinflammation. First, toxic proteases produced by P. gingivalis, such as gingipains, may directly damage neurons in the brain, leading to the activation of microglia and astrocytes, thereby inducing neuroinflammation (56). Second, P. gingivalis infection may enter the brain through blood circulation, causing local and systemic inflammatory responses that may form positive feedback loops with oxidative stress and neurodegenerative changes (57). In addition, P. gingivalis infection may further exacerbate neuroinflammation by promoting the accumulation of misfolded proteins, such as Aβ and tau proteins (8, 58). The interaction between the abnormal accumulation of these proteins and neuroinflammation may lead to the onset and development of neurodegenerative events such as AD and PD.
AD is characterized by the progressive cognitive decline resulting from synapse degeneration and neuronal death. It is an irreversible chronic degenerative neurological disease and the most common neurodegenerative disease leading to cognitive impairment (59, 60). AD is associated with cognitive dysfunction, Aβ plaques and neurofibrillary tangles (NFTs) formed by hyperphosphorylated tau are the two hallmark pathological features of AD (61, 62). Aβ plaques are recognized by the brain as foreign bodies, triggering inflammatory and immune responses through activation of microglia and cytokine release, ultimately leading to cell death and neurodegeneration (63). And phosphorylated tau proteins contribute to AD by causing microtubule rupture, synaptic loss, and, ultimately, cognitive dysfunction (64, 65). Moreover, both Aβ and tau aggregate impair synaptic plasticity and lead to neuronal cell death (66).
Firstly, P. gingivalis could induce neuroinflammation and neurodegeneration via OS. Le Sage et al. found that at the cellular level, P.g-LPS induces oxidative stress by increasing intracellular ROS production and altering the expression of genes encoding the oxidoreductases NOX2, NOX4, iNOS, and catalase (67). Accumulation of ROS, reduced MMP expression, and increased 4-HNE protein expression in neuroblastoma cells due to P.g-LPS underscore its significance in the pathogenesis of AD (29). Furthermore, LPS from P. gingivalis increases OS in periodontal ligament fibroblasts and brain endothelial cells (68, 69).
In addition to inducing neuroinflammation through OS, P. gingivalis can directly cause neuroinflammation. Animal studies have shown that P.g-LPS-induced neuroinflammation leads to cognitive impairment in C57BL/6 mice and that P.g-LPS significantly activates astrocytes and microglia and upregulates the TLR4/NF-κB signaling pathway (70). Hu et al. found that periodontitis caused by P.g-LPS exacerbates neuroinflammation by stimulating TLR4 and the NF-κB signaling pathway, which have been linked to learning and memory deficits in Sprague-Dawley rats (71). P. gingivalis OMV, which carries high levels of gingipain, may play a significant role in AD (72). Research have demonstrated that LPS derived from P. gingivalis OMV activates glial cells and induces brain inflammation. It is also linked to the expression of AD markers, such as Aβ and NFTs (73). In addition, the capsules of P. gingivalis play a central role in chronic inflammatory responses and cognitive deficits caused by short-term oral infections. More toxic capsules are likely to induce AD-like pathology and accelerate the pathogenic process (74).
At the same time, the pathogenic factors of P. gingivalis can also lead to the development of pathological features associated with AD, such as influencing Aβ accumulation and tau protein function. Animal studies corroborate that oral P. gingivalis infection in mice leads to brain colonization and enhances the generation of the amyloid plaque element Aβ1-42 (8). Similarly, Ryra et al. reported, using a rat model, that P.g-LPS induces an increase in serum levels of Aβ peptide (75). Tang et al. demonstrated that rats infected with P. gingivalis exhibit robust tau phosphorylation at the Thr181 and Thr231 loci linked to AD, and these loci are abundant in activated astrocytes (76).
At the cellular level, it has been established that prolonged contact with P.g-LPS led to the buildup of Aβ in the brains of mice of middle age. The exposure further led to the peripheral build-up of Aβ in inflammatory monocytes and macrophages (58). Gingipains, another toxic product of P. gingivalis, are associated with tau phosphorylation and tau cleavage (77, 78). Dominy et al. suggested that the origin of tau in the brains of patients with AD could stem from the transneuronal spread of P. gingivalis, Gingipain may also play a role in the adaptive elevation of tau protein synthesis in patients with AD (8).
In summary, P. gingivalis has been linked to the pathogenesis of AD through multiple mechanisms, including neuroinflammation, oxidative stress, Aβ accumulation, and interference with tau protein function, as summarized in Table 1. These findings suggest that P. gingivalis may contribute to the development of AD through different pathways, whether at the cellular or animal, level, thus providing potential targets for future therapeutic strategies.
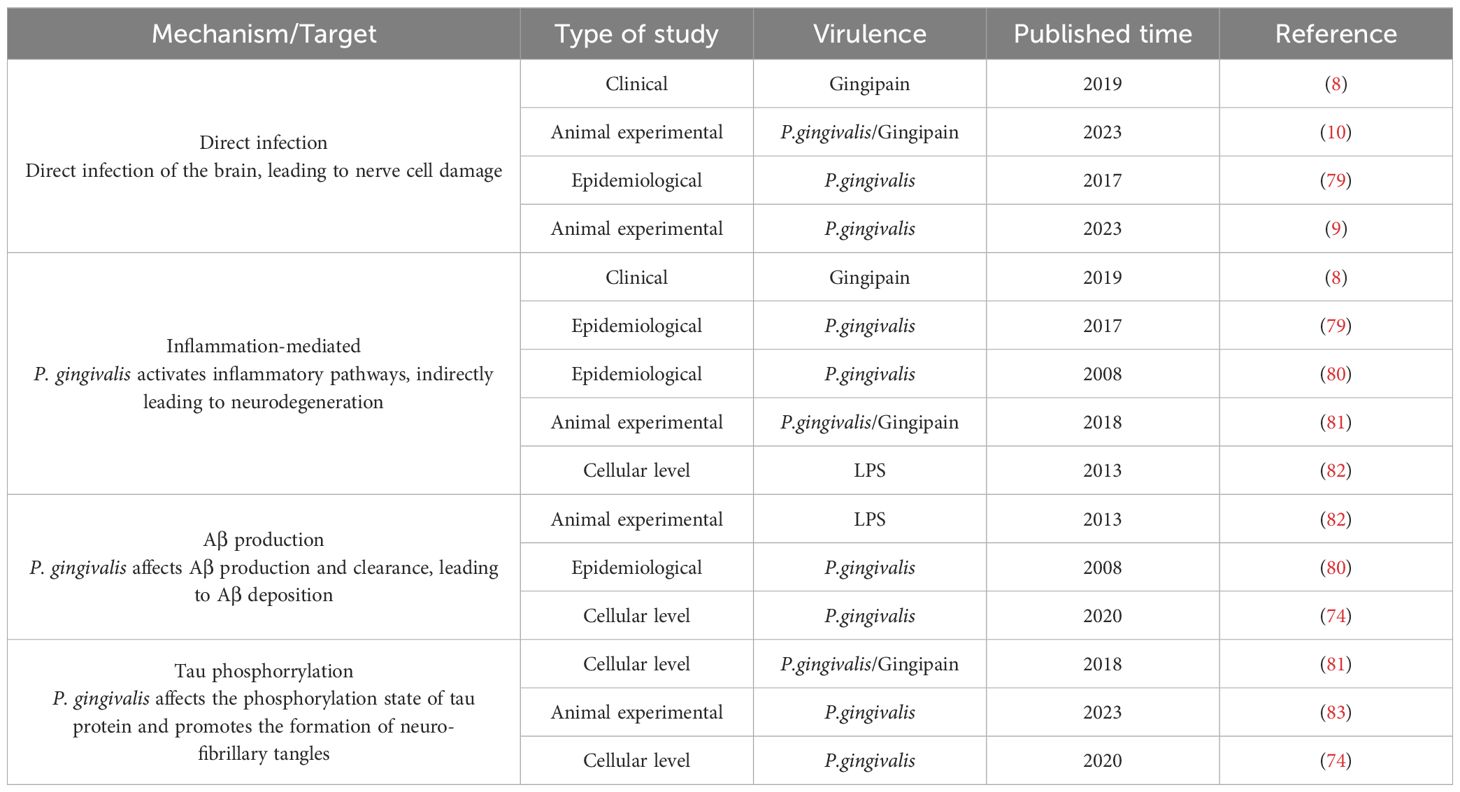
Table 1. P. gingivalis affects the development of AD.
PD is the second most prevalent neurodegenerative disorder that results from the death of dopaminergic nerve cells in the substantia nigra pars compacta (SNPC) (84, 85). It leads to movement disorders such as tremors, bradykinesia, and cognitive impairment (86). Pathologically, PD involves alpha-synuclein (α-Syn) misfolding, neuroinflammation, and mitochondrial dysfunction (87, 88). In recent years, periodontitis, a common slow inflammatory disease, has been associated with the risk of PD (89, 90). Most previous associations between the two diseases have been based on PD-induced dyskinesias, which may lead to the progression of periodontal disease (90). And P. gingivalis is a major periodontal pathogen that induces intestinal dysbiosis (91, 92).
Our previous study employed bioconfidence data mining to demonstrate that periodontitis is a high-risk causative factor of PD, and our results suggest that P. gingivalis, the main causative agent of periodontitis, can contribute to the development of PD (93). Previous studies have also detected P. gingivalis major virulence factors such as gingipain R1 and P.g-LPS in the blood of PD patients (94, 95). A recent study suggests that gingipains from P.gingivalis may accumulate in the SNPC of the human brain (96). An animal study confirmed that P. gingivalis reduces dopaminergic neurons in SNPC of mice with the leucine-rich repeat kinase 2 (LRRK2) R144G mutation, which is associated with late-onset PD (97, 98).
P. gingivalis may affect the onset and development of PD primarily through two mechanisms: OS and neuroinflammation. This mechanism was confirmed in various studies on animal models, including a report by La Vitola et al., which found that LPS from Escherichia coli (E. coli) induces neuroinflammation and enhances α-Syn toxicity along with cognitive impairment (99). Similarly, a previous study showed that P.g-LPS hindered spatial learning and memory in the Morris Water Maze (MWM) test, whereas the effects of the two LPS types were not significantly different (70). The findings imply that P.g-LPS, notwithstanding its structural variances, might possess a mechanism akin to Escherichia coli LPS that leads to the intensification of harmful impacts of α-Syn and cognitive deficits.
There is currently no direct evidence that P. gingivalis contributes to the development of PD via oxidative stress. However, studies indicate that P.g-LPS induces oxidative stress, leading to mitochondrial dysfunction and neuroinflammation in SH-SY5Y cells (29). In addition, a positron emission tomography (PET) study of patients with PD have shown that a widespread presence of activated microglia (100). Interestingly, this response does not correlate with clinical severity, suggesting it may occur early in the disease. The mechanism by which microglia are involved in PD may be similar to that seen in AD (51). Microglia internalize and degrade the proteinα-Syn. If this process fails, extracellular α-Syn accumulates, similar to Aβ (101). Microglia gather around α-Syn deposits and display pro-inflammatory properties base on receptors that also bind Aβ (102, 103). Therefore, we hypothesized that P. gingivalis may contribute to PD through oxidative stress and neuroinflammation.
MS is a progressive disorder of the central nervous system, characterized by invasion by immune cells, detachment of myelin sheaths, growth of reactive glial cells, and damage to neural axons. This sequence results in sensory, motor, and cognitive impairments (104). Although the exact cause of MS recurrence remains unclear, it is believed to stem from an autoimmune inflammatory condition in which environmental and genetic elements trigger CNS antigens, such as myelin basic protein, to be addressed by the immune system (105).
The correlation between MS and P. gingivalis is currently being investigated, although there are no published articles describing the role of P. gingivalis in the pathogenesis of MS. As one of the most prevalent neurodegenerative diseases, MS is also associated with neuroinflammation. Lucchinetti et al. showed that acute MS leads to astrocyte and microglial activation and occasionally to oligodendrocyte apoptosis (106).
Some relevant experiments have sought to demonstrate this link, including a report by Moreno et al. aimed to investigate whether systemic inflammatory stimuli exacerbate axonal damage. Through the use of rat models of autoimmune encephalomyelitis, the researchers showed that microglia activation leads to an increase in carbon monoxide synthase, IL-1β, and axonal damage (107). The findings confirmed a relationship between peripheral inflammation and neurodegeneration in a rodent model (91). In another study, Polak et al. engineered an MS mouse model by infecting it with P. gingivalis. Their experiment showed that tail weakness and paralysis in the extremities developed while exacerbating MS pathology and increasing lymphocyte proliferation (108). These findings suggest that P. gingivalis can contribute to the exacerbation of MS pathology by causing an inflammatory response.
ALS is the most common motor neuron (MN) disease, with an average age of onset of 50-65 years (109). Neuroinflammatory processes induced by microglia and astrocytes appear to play an important role in ALS pathology. Reactive astrogliosis occurs under pathological conditions such as ALS, shifting these cells from a ‘neuroprotective’ to a ‘neurodegenerative’ role (110).
In ALS, astrocyte activation is associated with motor neuron degeneration, which promotes inflammation and OS. In the early stages of disease, astrocytes provide neuroprotection. As the disease progresses, activated astrocytes promote a neurotoxic environment. This occurs either through microglial activation processes or through compounds released by motor neuron, ultimately resulting in motor neuron death (111). Microglia also play a key role in ALS, with M2 microglia being neuroprotective and M1 microglia being toxic. Studies in animal indicate that as the ALS progresses in fALS mice, the number of ‘neuroprotective’ M2 microglia increases. However, at later stage, these microglia transition to ‘neurotoxic’ M1 microglia (112–115). Inflammatory cytokines released by astrocytes and microglia may promote glutamatergic excitotoxicity, thereby linking neuroinflammation to excitotoxic cell death. When critical thresholds are reached, reactive astrocytes and microglia may trigger irreversible pathological processes that subsequently lead to non-cell-autonomous death of motor neurons in ALS patients (116).
Despite the absence of P. g-LPS, research has shown that microglia in ALS models can be stimulated by various LPS sources. This triggers a transition from protective to pro-inflammatory conditions, resulting in the production of IL-12, TNFα, NO, superoxide anion, and peroxynitrite. This process causes motor neuron deterioration and exacerbates the disease in mice models of ALS. Additionally, these molecules enhance the interaction between extracellular glutamate and its receptor on motor neurons, causing increased calcium influx into the cells and triggering cellular death (117–119). In ALS, the motor regions of the CNS may be affected by neuroinflammation and OS, evidenced by the activation of reactive astrocytes and microglia, moderate invasion of peripheral immune cells, and increased levels of inflammatory mediators (120). Animal models primarily show unusual growth of astrocytes and the presence of inflammatory indicators such as cyclooxygenase-2, inducible NOS, and neuronal NOS (121). Clinical studies have found that astrocytes in the spinal cords of patients with ALS are cytotoxic to MN in culture (122). Cytokines, such as G-CSF, IL2, IL15, IL17, MCP-1 MIP1α, TNFα, and VEGF found in the cerebrospinal fluid of individuals with ALS were found to be unusually elevated (123). Furthermore, patients with ALS exhibit increased IL-6 levels in exosomes derived from astrocytes, indicating a potential role of CNS-derived exosomes in uncovering neuroinflammation in patients with ALS and a direct relationship with the rate of disease progression (124).
Moreover, in ALS, OS may play a role in the deterioration of neuromuscular junctions. Mouse models show enhanced sensitivity of nerve endings to ROS, which may lead to the degeneration of presynapses at neuromuscular junctions. Concurrently, excessive activation of excitatory amino acids leads to the irregular release of acetylcholinesterase, diminishing acetylcholine levels in the synaptic gap and potentially resulting in diminished muscle strength in patients with ALS. These initial malfunctions, coupled with diminished inflammation and nutritional aid, potentially culminate in neurodegenerative disorders (125).
Although no direct evidence exists that P. gingivalis is involved in the pathogenesis of ALS, we can conclude that neuroinflammation and OS are intertwined mechanisms involved in the pathophysiology of ALS. Although further research is needed to determine whether P. gingivalis influences ALS through mechanisms of neuroinflammation and OS, P. gingivalis can trigger neuroinflammation and OS, and we, therefore, hypothesize that it is involved in the development of ALS through neuroinflammation and OS.
Apart from the associations between P. gingivalis and AD, PD, and MS, which has been summarized above, no studies on the effects of P. gingivalis on other neurodegenerative diseases, such as ALS, have been reported. However, the unifying clinical feature or disease phenotype of these disorders is neuroinflammation. P. gingivalis not only induces neuroinflammation directly but also mediates neuroinflammation through the oral-intestinal-brain axis.
The oral cavity is the starting site of the digestive tract, and humans ingest approximately 1.5 × 1012 oral bacteria daily from swallowed saliva (126), and P. gingivalis of oral origin can induce dysbiosis of the intestinal flora (127, 128). For example, a clinical study found altered gut microbiota in patients with periodontitis (129). Furthermore, in a previous study, we found that periodontitis-associated periodontal pathogens disrupt the gut microbiota, exacerbate the systemic immune response, and worsen colitis (130). According to Wang et al., oral microbes can have an impact on the gut, and P. gingivalis was detected in the feces of patients with colorectal cancer (131).
All these studies suggest that oral pathogenic bacteria can affect the central nervous system via the Gut-brain axis, suggesting the existence of an oral-intestinal-brain axis. A previous study also defined the presence of the Oral-gut-brain axis (132). It has been shown that P. gingivalis affects neuroinflammation by influencing intestinal ecology by increasing the number of inflammatory T and B lymphocytes, thereby inducing neuroinflammation (133, 134). Dysbiosis of the gut microbiota reduces the production of short-chain fatty acids, which are linked to inflammatory responses (135, 136). Another study indicated that the gut microbiota affects both microglial maturation and normal function and that dysregulation of the microbiota can lead to neuroinflammation (137). Feng et al. found that oral administration of P. gingivalis reduces intestinal permeability and elevates IL-17a levels in the peripheral blood of R1441G mice, potentially linked to neuronal demise and neuroinflammation (97). Thus, the aforementioned studies suggest that P. gingivalis influences neuroinflammation via the oral-intestina-brain axis, which may be closely related to the pathogenesis of several neurodegenerative diseases.
Currently, there is convincing evidence that oral P. gingivalis may influence neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. Here, we summarize the relationship between the two diseases (Figure 2).
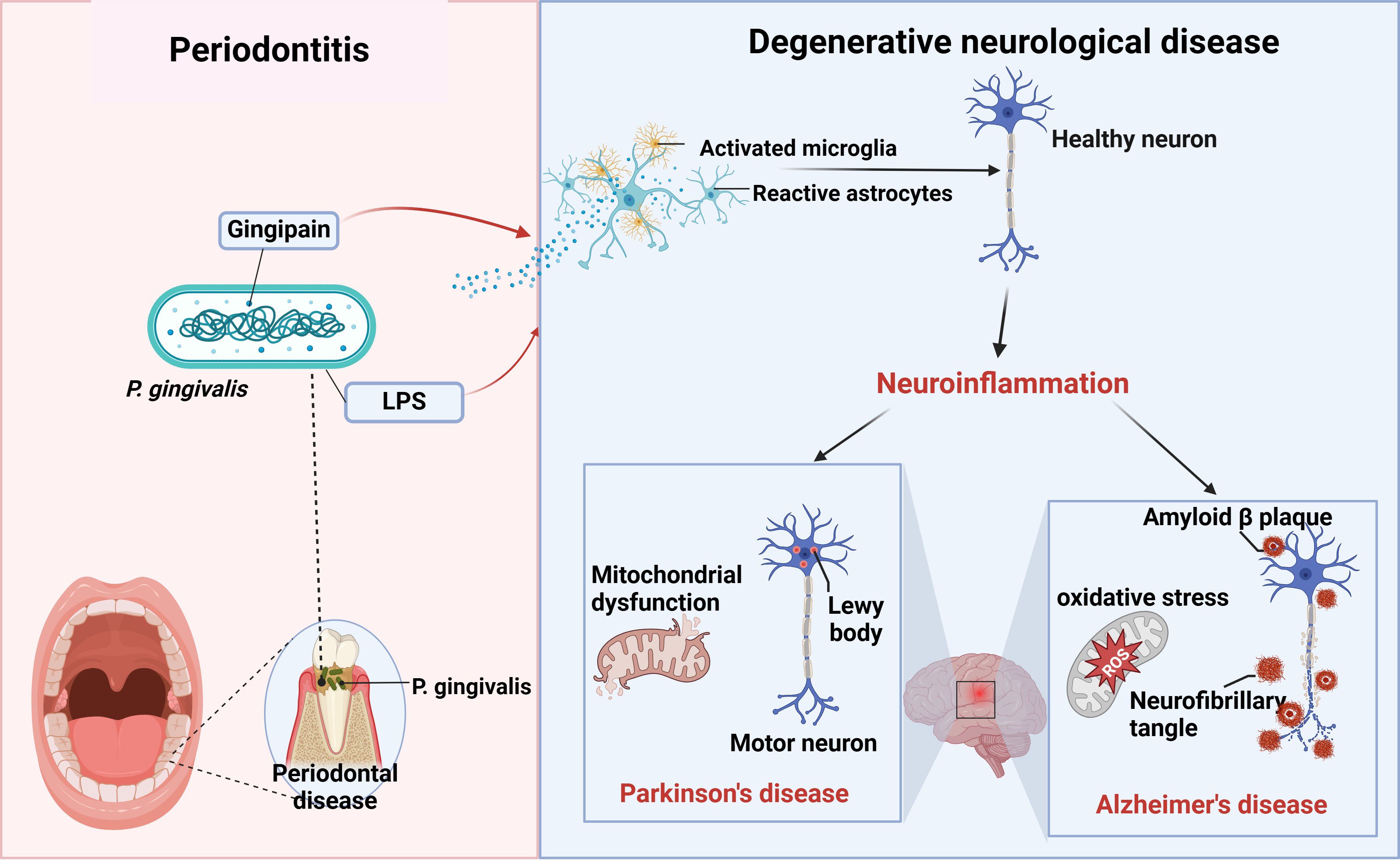
Figure 2. P. gingivalis and neurodegenerative diseases. Periodontitis triggers neuroinflammation through the production of virulence factors P.g-LPS and gingipain by P. gingivalis, which activate the brain’s immune cells, astrocytes and microglia, causing alterations in healthy neurons that ultimately lead to neurodegenerative diseases such as AD and PD (figure was created with BioRender.com. Huang, Z. (2025) https://BioRender.com/a16s851).
P. gingivalis is the main causative agent of periodontitis, inducing both periodontal inflammation and systemic chronic inflammation, as well as neuroinflammation. Recent studies indicate that P. gingivalis is not only relevant to periodontitis, but also directly induces AD through neuroinflammation and oxidative stress, suggesting its involvement in the development of neurodegenerative diseases. This review systematically summarizes the literature regarding P. gingivalis’s role in the development of neurodegenerative diseases through neuroinflammation and further analyzes the underlying mechanisms involved.
Neurodegenerative diseases, such as AD, may be associated with infections. T Numerous studies have confirmed that pathogenic microorganisms, including human immunodeficiency virus (HIV) and specific herpes viruses like herpes simplex virus (HSV), are linked to neurodegenerative disorders. HIV infection can lead to AIDS-associated dementia, while HSV infection may increase the risk of AD (138, 139). Additionally, certain bacteria, such as Streptococcus pneumoniae, are linked to neurodegenerative diseases. For example, infections with Streptococcus pneumoniae can result in meningitis, potentially causing lasting neurological harm and cognitive deterioration (140). Additionally, gut microbes can influence AD through the gut-brain axis. A study found notable differences in the gut bacterial community structure between AD model mice and their age-matched wild-type counterparts. AD mice have significantly lower abundance of members of the phyla Thick-walled Bacteria, Micrococcus wartyi, Aspergillus, and Actinobacteria, and increased abundance of members of the phyla Synechococcus and Teneribacteria (141); these changes may lead to TNF-mediated gastrointestinal inflammation, which can increase the risk of AD (142). This suggests that shaping the composition of the gut microbiota may influence the progression of AD. These findings indicate that the mechanisms of neurodegenerative diseases are complex and, therefore, studies from the point of view of pathogenic microorganisms, especially oral flora, are of great importance.
Porphyromonas gingivalis impacts host’s immune function and triggers inflammatory, which may lead to various immune diseases beyond neurodegenerative disorders. Recent studies suggest a link between P. gingivalis and several immune disorders, including autoimmune and inflammatory conditions. In autoimmune disorders, P. gingivalis can trigger immune responses through molecular mimicry, leading to the production of autoantibodies. Research shows that a particular peptide (Pep19) from P. gingivalis heat shock protein 60 reacts significantly in the serum of individuals with autoimmune disorders. This finding suggests P. gingivalis may play a role in the emergence and progression of these diseases (143). Furthermore, P. gingivalis is associated with inflammatory disorders atherosclerosis, diabetes, and rheumatoid arthritis. Frequently associated with persistent inflammatory reactions, these conditions can worsen due to P. gingivalis by enhancing the inflammatory response regulation (143).
Investigating the connection between P. gingivalis and neuroinflammation is an emerging area of research, especially regarding neurodegenerative disorders like AD. Many studies have suggested ways in which P. gingivalis is connected to neuroinflammation. However, this research is still in its early stages, requiring more experimental and clinical studies to validate these links and investigate possible treatment options. In conclusion, studying the relationship between P. gingivalis and neurodegenerative conditions deepens our understanding of these diseases and raises public awareness of periodontitis. Improving the prevention and treatment of periodontitis may help reduce the onset and progression of neurodegenerative diseases. The intricate connection between Porphyromonas gingivalis and neurodegenerative diseases requires further investigation to establish a scientific foundation for their prevention and treatment.
ZH: Writing – original draft. MH: Writing – review & editing. NS: Writing – review & editing. XyW: Investigation, Writing – review & editing. LY: Validation, Writing – review & editing. HY: Validation, Writing – review & editing. XfW: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by Natural Science Foundation of Jilin Province (grant no. YDZJ202401450ZYTS), Special Project for Health Research Talents in Jilin Province (grant no.2024SCZ23) and the Chunlei project of China-Japan Union Hospital of Jilin University (grant no. 2024CL07).
The figure was created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, et al. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. (2015) 86:611–22. doi: 10.1902/jop.2015.140520
2. How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. (2016) 7:53. doi: 10.3389/fmicb.2016.00053
3. Sun XJ, Meng HX, Shi D, Xu L, Zhang L, Chen ZB, et al. Elevation of C-reactive protein and interleukin-6 in plasma of patients with aggressive periodontitis. J Periodontal Res. (2009) 44(3):311–6. doi: 10.1111/j.1600-0765.2008.01131.x
4. Chen Y, Liu S, Leng SX. Chronic low-grade inflammatory phenotype (CLIP) and senescent immune dysregulation. Clin Ther. (2019) 41:400–9. doi: 10.1016/j.clinthera.2019.02.001
5. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
6. Carter CJ, France J, Crean S, Singhrao SK. The porphyromonas gingivalis/host interactome shows enrichment in GWASdb genes related to alzheimer’s disease, diabetes and cardiovascular diseases. Front Aging Neurosci. (2017) 9:408. doi: 10.3389/fnagi.2017.00408
7. Olsen I, Taubman MA, Singhrao SK. Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. J Oral Microbiol. (2016) 8:33029. doi: 10.3402/jom.v8.33029
8. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) 5:eaau3333. doi: 10.1126/sciadv.aau3333
9. Ma X, Shin Y-J, Yoo J-W, Park H-S, Kim D-H. Extracellular vesicles derived from Porphyromonas gingivalis induce trigeminal nerve-mediated cognitive impairment. J Adv Res. (2023) 54:293–303. doi: 10.1016/j.jare.2023.02.006
10. Lei S, Li J, Yu J, Li F, Pan Y, Chen X, et al. Porphyromonas gingivalis bacteremia increases the permeability of the blood-brain barrier via the Mfsd2a/Caveolin-1 mediated transcytosis pathway. Int J Oral Sci. (2023) 15:3. doi: 10.1038/s41368-022-00215-y
11. Olsen I, Lambris JD, Hajishengallis G. Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J Oral Microbiol. (2017) 9:1340085. doi: 10.1080/20002297.2017.1340085
12. Fiorillo L, Cervino G, Laino L, D’Amico C, Mauceri R, Tozum TF, et al. Porphyromonas gingivalis, periodontal and systemic implications: A systematic review. Dent J (Basel). (2019) 7:114. doi: 10.3390/dj7040114
13. Okamura H, Hirota K, Yoshida K, Weng Y, He Y, Shiotsu N, et al. Outer membrane vesicles of Porphyromonas gingivalis: Novel communication tool and strategy. Jpn Dent Sci Rev. (2021) 57:138–46. doi: 10.1016/j.jdsr.2021.07.003
14. Veith PD, Chen Y-Y, Gorasia DG, Chen D, Glew MD, O’Brien-Simpson NM, et al. Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J Proteome Res. (2014) 13:2420–32. doi: 10.1021/pr401227e
15. Karkowska-Kuleta J, Surowiec M, Gogol M, Koziel J, Potempa B, Potempa J, et al. Peptidylarginine Deiminase of Porphyromonas gingivalis Modulates the Interactions between Candida albicans Biofilm and Human Plasminogen and High-Molecular-Mass Kininogen. Int J Mol Sci. (2020) 21:2495. doi: 10.3390/ijms21072495
16. Konig MF, Paracha AS, Moni M, Bingham CO, Andrade F. Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann Rheum Dis. (2015) 74:2054–61. doi: 10.1136/annrheumdis-2014-205385
17. Lunar Silva I, Cascales E. Molecular strategies underlying porphyromonas gingivalis virulence. J Mol Biol. (2021) 433:166836. doi: 10.1016/j.jmb.2021.166836
18. Laine ML, van Winkelhoff AJ. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol Immunol. (1998) 13:322–5. doi: 10.1111/j.1399-302x.1998.tb00714.x
19. Singh A, Wyant T, Anaya-Bergman C, Aduse-Opoku J, Brunner J, Laine ML, et al. The capsule of Porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect Immun. (2011) 79:4533–42. doi: 10.1128/IAI.05016-11
20. Andrukhov O, Ulm C, Reischl H, Nguyen PQ, Matejka M, Rausch-Fan X. Serum cytokine levels in periodontitis patients in relation to the bacterial load. J Periodontol. (2011) 82:885–92. doi: 10.1902/jop.2010.100425
21. Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. (2011) 130:226–38. doi: 10.1016/j.pharmthera.2011.01.014
22. Stefano GB, Pilonis N, Ptacek R, Raboch J, Vnukova M, Kream RM. Gut, microbiome, and brain regulatory axis: relevance to neurodegenerative and psychiatric disorders. Cell Mol Neurobiol. (2018) 38:1197–206. doi: 10.1007/s10571-018-0589-2
23. Cryan JF, O’Riordan KJ, Sandhu K, Peterson V, Dinan TG. The gut microbiome in neurological disorders. Lancet Neurol. (2020) 19:179–94. doi: 10.1016/S1474-4422(19)30356-4
24. Zhu F, Li C, Chu F, Tian X, Zhu J. Target dysbiosis of gut microbes as a future therapeutic manipulation in alzheimer’s disease. Front Aging Neurosci. (2020) 12:544235. doi: 10.3389/fnagi.2020.544235
25. Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, et al. Circulating and tissue-resident CD4+ T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology. (2017) 153:1320–1337.e16. doi: 10.1053/j.gastro.2017.07.047
26. McCoy KD, Ronchi F, Geuking MB. Host-microbiota interactions and adaptive immunity. Immunol Rev. (2017) 279:63–9. doi: 10.1111/imr.12575
27. Zhao Q, Elson CO. Adaptive immune education by gut microbiota antigens. Immunology. (2018) 154:28–37. doi: 10.1111/imm.12896
28. Campos-Acuña J, Elgueta D, Pacheco R. T-cell-driven inflammation as a mediator of the gut-brain axis involved in parkinson’s disease. Front Immunol. (2019) 10:239. doi: 10.3389/fimmu.2019.00239
29. Verma A, Azhar G, Zhang X, Patyal P, Kc G, Sharma S, et al. P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. Int J Mol Sci. (2023) 24:950. doi: 10.3390/ijms24020950
30. Pandur E, Varga E, Tamási K, Pap R, Nagy J, Sipos K. Effect of inflammatory mediators lipopolysaccharide and lipoteichoic acid on iron metabolism of differentiated SH-SY5Y cells alters in the presence of BV-2 microglia. Int J Mol Sci. (2018) 20:17. doi: 10.3390/ijms20010017
31. Nativel B, Couret D, Giraud P, Meilhac O, d’Hellencourt CL, Viranaïcken W, et al. Porphyromonas gingivalis lipopolysaccharides act exclusively through TLR4 with a resilience between mouse and human. Sci Rep. (2017) 7:15789. doi: 10.1038/s41598-017-16190-y
32. Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. (2007) 9:1081–8. doi: 10.1038/ncb1629
33. Acosta C, Davies A. Bacterial lipopolysaccharide regulates nociceptin expression in sensory neurons. J Neurosci Res. (2008) 86:1077–86. doi: 10.1002/jnr.21565
34. Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. (2005) 115:1449–57. doi: 10.1172/JCI24761
35. Hussain R, Zubair H, Pursell S, Shahab M. Neurodegenerative diseases: regenerative mechanisms and novel therapeutic approaches. Brain Sci. (2018) 8:E177. doi: 10.3390/brainsci8090177
36. Kwon HS, Koh S-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. (2020) 9:42. doi: 10.1186/s40035-020-00221-2
37. Isik S, Yeman Kiyak B, Akbayir R, Seyhali R, Arpaci T. Microglia mediated neuroinflammation in parkinson’s disease. Cells. (2023) 12:1012. doi: 10.3390/cells12071012
38. Wendimu MY, Hooks SB. Microglia phenotypes in aging and neurodegenerative diseases. Cells. (2022) 11:2091. doi: 10.3390/cells11132091
39. Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. (2019) 11:e10248. doi: 10.15252/emmm.201810248
40. Franklin H, Clarke BE, Patani R. Astrocytes and microglia in neurodegenerative diseases: Lessons from human in vitro models. Prog Neurobiol. (2021) 200:101973. doi: 10.1016/j.pneurobio.2020.101973
41. Liu Z, Zhou T, Ziegler AC, Dimitrion P, Zuo L. Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid Med Cell Longev. (2017) 2017:2525967. doi: 10.1155/2017/2525967
42. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. (2013) 3:461–91. doi: 10.3233/JPD-130230
43. Dhapola R, Hota SS, Sarma P, Bhattacharyya A, Medhi B, Reddy DH. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology. (2021) 29:1669–81. doi: 10.1007/s10787-021-00889-6
44. Zhao W, Spiers JG, Vassileff N, Khadka A, Jaehne EJ, van den Buuse M, et al. microRNA-146a modulates behavioural activity, neuroinflammation, and oxidative stress in adult mice. Mol Cell Neurosci. (2023) 124:103820. doi: 10.1016/j.mcn.2023.103820
45. Mander P, Brown GC. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J Neuroinflamm. (2005) 2:20. doi: 10.1186/1742-2094-2-20
46. Spiers JG, Chen H-JC, Bourgognon J-M, Steinert JR. Dysregulation of stress systems and nitric oxide signaling underlies neuronal dysfunction in Alzheimer’s disease. Free Radic Biol Med. (2019) 134:468–83. doi: 10.1016/j.freeradbiomed.2019.01.025
47. Ahmad MH, Fatima M, Mondal AC. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J Clin Neurosci. (2019) 59:6–11. doi: 10.1016/j.jocn.2018.10.034
48. Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. (2016) 37:608–20. doi: 10.1016/j.it.2016.06.006
49. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in alzheimer’s disease. Lancet Neurol. (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
50. Y Q, C L, J H, Cz A, Mf G, D J-W, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. (2007) 8:247–256. doi: 10.1038/ni1439
51. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
52. Boche D, Perry VH, Nicoll J a. R. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. (2013) 39:3–18. doi: 10.1111/nan.12011
53. A N, Jf S, Jp G. Microglia priming with aging and stress. Neuropsychopharmacology. (2017) 42:318–33. doi: 10.1038/npp.2016.185
54. Acosta MT, Gioia GA, Silva AJ. Neurofibromatosis type 1: new insights into neurocognitive issues. Curr Neurol Neurosci Rep. (2006) 6:136–43. doi: 10.1007/s11910-996-0036-5
55. Colombo E, Di Dario M, Capitolo E, Chaabane L, Newcombe J, Martino G, et al. Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann Neurol. (2014) 76:325–37. doi: 10.1002/ana.24217
56. Hu Y, Li H, Zhang J, Zhang X, Xia X, Qiu C, et al. Periodontitis induced by P. gingivalis-LPS is associated with neuroinflammation and learning and memory impairment in sprague-dawley rats. Front Neurosci. (2020) 14:658. doi: 10.3389/fnins.2020.00658
57. Ding Y, Ren J, Yu H, Yu W, Zhou Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing. (2018) 15:6. doi: 10.1186/s12979-017-0110-7
58. Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, et al. Porphyromonas gingivalis infection induces amyloid-β Accumulation in monocytes/macrophages. J Alzheimers Dis. (2019) 72:479–94. doi: 10.3233/JAD-190298
59. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. (2013) 61:349–60. doi: 10.1002/glia.22437
61. Canter RG, Penney J, Tsai L-H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature. (2016) 539:187–96. doi: 10.1038/nature20412
62. Edwards FA. A unifying hypothesis for alzheimer’s disease: from plaques to neurodegeneration. Trends Neurosci. (2019) 42:310–22. doi: 10.1016/j.tins.2019.03.003
63. Barage SH, Sonawane KD. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. (2015) 52:1–18. doi: 10.1016/j.npep.2015.06.008
64. Giraldo E, Lloret A, Fuchsberger T, Viña J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. (2014) 2:873–7. doi: 10.1016/j.redox.2014.03.002
65. Bulat N, Widmann C. Caspase substrates and neurodegenerative diseases. Brain Res Bull. (2009) 80:251–67. doi: 10.1016/j.brainresbull.2009.07.007
66. Begcevic I, Brinc D, Brown M, Martinez-Morillo E, Goldhardt O, Grimmer T, et al. Brain-related proteins as potential CSF biomarkers of Alzheimer’s disease: A targeted mass spectrometry approach. J Proteomics. (2018) 182:12–20. doi: 10.1016/j.jprot.2018.04.027
67. Le Sage F, Meilhac O, Gonthier M-P. Porphyromonas gingivalis lipopolysaccharide induces pro-inflammatory adipokine secretion and oxidative stress by regulating Toll-like receptor-mediated signaling pathways and redox enzymes in adipocytes. Mol Cell Endocrinol. (2017) 446:102–10. doi: 10.1016/j.mce.2017.02.022
68. Gölz L, Memmert S, Rath-Deschner B, Jäger A, Appel T, Baumgarten G, et al. LPS from P. gingivalis and hypoxia increases oxidative stress in periodontal ligament fibroblasts and contributes to periodontitis. Mediators Inflammation. (2014) 2014:986264. doi: 10.1155/2014/986264
69. Charoensaensuk V, Chen Y-C, Lin Y-H, Ou K-L, Yang L-Y, Lu D-Y. Porphyromonas gingivalis Induces Proinflammatory Cytokine Expression Leading to Apoptotic Death through the Oxidative Stress/NF-κB Pathway in Brain Endothelial Cells. Cells. (2021) 10:3033. doi: 10.3390/cells10113033
70. Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflamm. (2018) 15:37. doi: 10.1186/s12974-017-1052-x
71. Blasco-Baque V, Garidou L, Pomié C, Escoula Q, Loubieres P, Le-Gall-David S, et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut. (2017) 66:872–85. doi: 10.1136/gutjnl-2015-309897
72. Zhang Z, Liu D, Liu S, Zhang S, Pan Y. The role of porphyromonas gingivalis outer membrane vesicles in periodontal disease and related systemic diseases. Front Cell Infect Microbiol. (2020) 10:585917. doi: 10.3389/fcimb.2020.585917
73. Singhrao SK, Olsen I. Are porphyromonas gingivalis outer membrane vesicles microbullets for sporadic alzheimer’s disease manifestation? J Alzheimers Dis Rep. (2018) 2:219–28. doi: 10.3233/ADR-180080
74. Díaz-Zúñiga J, More J, Melgar-Rodríguez S, Jiménez-Unión M, Villalobos-Orchard F, Muñoz-Manríquez C, et al. Alzheimer’s disease-like pathology triggered by porphyromonas gingivalis in wild type rats is serotype dependent. Front Immunol. (2020) 11:588036. doi: 10.3389/fimmu.2020.588036
75. Leira Y, Rodríguez-Yáñez M, Arias S, López-Dequidt I, Campos F, Sobrino T, et al. Periodontitis is associated with systemic inflammation and vascular endothelial dysfunction in patients with lacunar infarct. J Periodontol. (2019) 90:465–74. doi: 10.1002/JPER.18-0560
76. Tang Z, Liang D, Cheng M, Su X, Liu R, Zhang Y, et al. Effects of porphyromonas gingivalis and its underlying mechanisms on alzheimer-like tau hyperphosphorylation in sprague-dawley rats. J Mol Neurosci. (2021) 71:89–100. doi: 10.1007/s12031-020-01629-1
77. Chu J, Lauretti E, Praticò D. Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3β kinase: implications for Alzheimer’s disease. Mol Psychiatry. (2017) 22:1002–8. doi: 10.1038/mp.2016.214
78. Sandhu P, Naeem MM, Lu C, Kumarathasan P, Gomes J, Basak A. Ser422 phosphorylation blocks human Tau cleavage by caspase-3: Biochemical implications to Alzheimer’s Disease. Bioorg Med Chem Lett. (2017) 27:642–52. doi: 10.1016/j.bmcl.2016.11.087
79. Chen C-K, Wu Y-T, Chang Y-C. Periodontal inflammatory disease is associated with the risk of Parkinson’s disease: a population-based retrospective matched-cohort study. PeerJ. (2017) 5:e3647. doi: 10.7717/peerj.3647
80. Kamer AR, Craig RG, Dasanayake AP, Brys M, Glodzik-Sobanska L, de Leon MJ. Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimers Dement. (2008) 4:242–50. doi: 10.1016/j.jalz.2007.08.004
81. V I, Pk Z, Sj G, Pt T, Me R, K L, et al. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PloS One. (2018) 13:1–24. doi: 10.1371/journal.pone.0204941
82. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. (2013) 36:665–77. doi: 10.3233/JAD-121918
83. Wang RP-H, Huang J, Chan KWY, Leung WK, Goto T, Ho Y-S, et al. IL-1β and TNF-α play an important role in modulating the risk of periodontitis and Alzheimer’s disease. J Neuroinflamm. (2023) 20:71. doi: 10.1186/s12974-023-02747-4
84. MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. (2006) 52:587–93. doi: 10.1016/j.neuron.2006.10.008
85. Nl K, S J, Jm L, N P, P A-S, Jl H, et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain: J Neurol. (2005) 128:2786–96. doi: 10.1093/brain/awh667
86. Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. (2012) 2:a009258. doi: 10.1101/cshperspect.a009258
87. Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, et al. Integrating pathways of Parkinson’s disease in a molecular interaction map. Mol Neurobiol. (2014) 49:88–102. doi: 10.1007/s12035-013-8489-4
88. Titova N, Padmakumar C, Lewis SJG, Chaudhuri KR. Parkinson’s: a syndrome rather than a disease? J Neural Transm (Vienna). (2017) 124:907–14. doi: 10.1007/s00702-016-1667-6
89. Kaur T, Uppoor A, Naik D. Parkinson’s disease and periodontitis - the missing link? A review. Gerodontology. (2016) 33:434–8. doi: 10.1111/ger.12188
90. Hanaoka A, Kashihara K. Increased frequencies of caries, periodontal disease and tooth loss in patients with Parkinson’s disease. J Clin Neurosci. (2009) 16:1279–82. doi: 10.1016/j.jocn.2008.12.027
91. Nakajima M, Arimatsu K, Kato T, Matsuda Y, Minagawa T, Takahashi N, et al. Oral administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PloS One. (2015) 10:e0134234. doi: 10.1371/journal.pone.0134234
92. Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. (2014) 4:4828. doi: 10.1038/srep04828
93. Wang X, Shi N, Wu B, Yuan L, Chen J, Ye C, et al. Bioinformatics analysis of gene expression profile and functional analysis in periodontitis and Parkinson’s disease. Front Aging Neurosci. (2022) 14:1029637. doi: 10.3389/fnagi.2022.1029637
94. Olsen I, Kell DB, Pretorius E. Is Porphyromonas gingivalis involved in Parkinson’s disease? Eur J Clin Microbiol Infect Dis. (2020) 39:2013–8. doi: 10.1007/s10096-020-03944-2
95. Adams B, Nunes JM, Page MJ, Roberts T, Carr J, Nell TA, et al. Parkinson’s disease: A systemic inflammatory disease accompanied by bacterial inflammagens. Front Aging Neurosci. (2019) 11:210. doi: 10.3389/fnagi.2019.00210
96. Ermini F, Low VF, Song JJ, Tan AYS, Faull RLM, Dragunow M, et al. Ultrastructural localization of Porphyromonas gingivalis gingipains in the substantia nigra of Parkinson’s disease brains. NPJ Parkinsons Dis. (2024) 10:90. doi: 10.1038/s41531-024-00705-2
97. Feng Y-K, Wu Q-L, Peng Y-W, Liang F-Y, You H-J, Feng Y-W, et al. Oral P. gingivalis impairs gut permeability and mediates immune responses associated with neurodegeneration in LRRK2 R1441G mice. J Neuroinflamm. (2020) 17:347. doi: 10.1186/s12974-020-02027-5
98. Vinagre-Aragón A, Campo-Caballero D, Mondragón-Rezola E, Pardina-Vilella L, Hernandez Eguiazu H, Gorostidi A, et al. A more homogeneous phenotype in parkinson’s disease related to R1441G mutation in the LRRK2 gene. Front Neurol. (2021) 12:635396. doi: 10.3389/fneur.2021.635396
99. La Vitola P, Balducci C, Baroni M, Artioli L, Santamaria G, Castiglioni M, et al. Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson’s models. Neuropathol Appl Neurobiol. (2021) 47:43–60. doi: 10.1111/nan.12644
100. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. (2006) 21:404–12. doi: 10.1016/j.nbd.2005.08.002
101. Halliday GM, Stevens CH. Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord. (2011) 26:6–17. doi: 10.1002/mds.23455
102. Croisier E, Moran LB, Dexter DT, Pearce RKB, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflamm. (2005) 2:14. doi: 10.1186/1742-2094-2-14
103. Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. (2013) 4:1562. doi: 10.1038/ncomms2534
104. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
105. Li X, Kiprowska M, Kansara T, Kansara P, Li P. Neuroinflammation: A distal consequence of periodontitis. J Dent Res. (2022) 101:1441–9. doi: 10.1177/00220345221102084
106. C L, W B, J P, B S, M R, H L. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q
107. Moreno B, Jukes J-P, Vergara-Irigaray N, Errea O, Villoslada P, Perry VH, et al. Systemic inflammation induces axon injury during brain inflammation. Ann Neurol. (2011) 70:932–42. doi: 10.1002/ana.22550
108. Polak D, Shmueli A, Brenner T, Shapira L. Oral infection with P. gingivalis exacerbates autoimmune encephalomyelitis. J Periodontol. (2018) 89:1461–6. doi: 10.1002/JPER.17-0531
109. Couratier P, Corcia P, Lautrette G, Nicol M, Preux PM, Marin B. Epidemiology of amyotrophic lateral sclerosis: A review of literature. Rev Neurol (Paris). (2016) 172(1):37–45. doi: 10.1016/j.neurol.2015.11.002
110. Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, et al. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. (2004) 89:464–73. doi: 10.1111/j.1471-4159.2004.02357.x
111. Yamanaka K, Komine O. The multi-dimensional roles of astrocytes in ALS. Neurosci Res. (2018) 126:31–8. doi: 10.1016/j.neures.2017.09.011
112. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. (2012) 237:147–52. doi: 10.1016/j.expneurol.2012.06.011
113. Ekdahl CT. Microglial activation - tuning and pruning adult neurogenesis. Front Pharmacol. (2012) 3:41. doi: 10.3389/fphar.2012.00041
114. Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol. (2013) 8:888–99. doi: 10.1007/s11481-013-9489-x
115. Sargsyan SA, Monk PN, Shaw PJ. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia. (2005) 51:241–53. doi: 10.1002/glia.20210
116. Lee J, Hyeon SJ, Im H, Ryu H, Kim Y, Ryu H. Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp Neurobiol. (2016) 25:233–40. doi: 10.5607/en.2016.25.5.233
117. Nguyen MD, D’Aigle T, Gowing G, Julien J-P, Rivest S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci. (2004) 24:1340–9. doi: 10.1523/JNEUROSCI.4786-03.2004
118. Weydt P, Yuen EC, Ransom BR, Möller T. Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia. (2004) 48:179–82. doi: 10.1002/glia.20062
119. Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, Julien J-P, et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. (2010) 58:231–43. doi: 10.1002/glia.20919
120. Liu J, Wang F. Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front Immunol. (2017) 8:1005. doi: 10.3389/fimmu.2017.01005
121. Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics. (2010) 7:471–81. doi: 10.1016/j.nurt.2010.05.012
122. Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. (2011) 29:824–8. doi: 10.1038/nbt.1957
123. Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal fluid inflammatory cytokine aberrations in alzheimer’s disease, parkinson’s disease and amyotrophic lateral sclerosis: A systematic review and meta-analysis. Front Immunol. (2018) 9:2122. doi: 10.3389/fimmu.2018.02122
124. Chen Y, Xia K, Chen L, Fan D. Increased interleukin-6 levels in the astrocyte-derived exosomes of sporadic amyotrophic lateral sclerosis patients. Front Neurosci. (2019) 13:574. doi: 10.3389/fnins.2019.00574
125. Pollari E, Goldsteins G, Bart G, Koistinaho J, Giniatullin R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front Cell Neurosci. (2014) 8:131. doi: 10.3389/fncel.2014.00131
126. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PloS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
127. Kato T, Yamazaki K, Nakajima M, Date Y, Kikuchi J, Hase K, et al. Oral administration of porphyromonas gingivalis alters the gut microbiome and serum metabolome. mSphere. (2018) 3:e00460–18. doi: 10.1128/mSphere.00460-18
128. Ohtsu A, Takeuchi Y, Katagiri S, Suda W, Maekawa S, Shiba T, et al. Influence of Porphyromonas gingivalis in gut microbiota of streptozotocin-induced diabetic mice. Oral Dis. (2019) 25:868–80. doi: 10.1111/odi.13044
129. Lourenςo TGB, Spencer SJ, Alm EJ, Colombo APV. Defining the gut microbiota in individuals with periodontal diseases: an exploratory study. J Oral Microbiol. (2018) 10:1487741. doi: 10.1080/20002297.2018.1487741
130. Qian J, Lu J, Huang Y, Wang M, Chen B, Bao J, et al. Periodontitis salivary microbiota worsens colitis. J Dent Res. (2022) 101:559–68. doi: 10.1177/00220345211049781
131. Wang X, Jia Y, Wen L, Mu W, Wu X, Liu T, et al. Porphyromonas gingivalis promotes colorectal carcinoma by activating the hematopoietic NLRP3 inflammasome. Cancer Res. (2021) 81:2745–59. doi: 10.1158/0008-5472.CAN-20-3827
132. Sansores-España LD, Melgar-Rodríguez S, Olivares-Sagredo K, Cafferata EA, Martínez-Aguilar VM, Vernal R, et al. Oral-gut-brain axis in experimental models of periodontitis: associating gut dysbiosis with neurodegenerative diseases. Front Aging. (2021) 2:781582. doi: 10.3389/fragi.2021.781582
133. Carlessi AS, Borba LA, Zugno AI, Quevedo J, Réus GZ. Gut microbiota-brain axis in depression: The role of neuroinflammation. Eur J Neurosci. (2021) 53:222–35. doi: 10.1111/ejn.14631
134. Suganya K, Koo B-S. Gut-brain axis: role of gut microbiota on neurological disorders and how probiotics/prebiotics beneficially modulate microbial and immune pathways to improve brain functions. Int J Mol Sci. (2020) 21:E7551. doi: 10.3390/ijms21207551
135. Lach G, Schellekens H, Dinan TG, Cryan JF. Anxiety, depression, and the microbiome: A role for gut peptides. Neurotherapeutics. (2018) 15:36–59. doi: 10.1007/s13311-017-0585-0
136. Gudi R, Suber J, Brown R, Johnson BM, Vasu C. Pretreatment with yeast-derived complex dietary polysaccharides suppresses gut inflammation, alters the microbiota composition, and increases immune regulatory short-chain fatty acid production in C57BL/6 mice. J Nutr. (2020) 150:1291–302. doi: 10.1093/jn/nxz328
137. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
138. Kodidela S, Gerth K, Haque S, Gong Y, Ismael S, Singh A, et al. Extracellular vesicles: A possible link between HIV and alzheimer’s disease-like pathology in HIV subjects? Cells. (2019) 8:968. doi: 10.3390/cells8090968
139. Albaret MA, Textoris J, Dalzon B, Lambert J, Linard M, Helmer C, et al. HSV-1 cellular model reveals links between aggresome formation and early step of Alzheimer’s disease. Transl Psychiatry. (2023) 13:86. doi: 10.1038/s41398-023-02376-8
140. Farmen K, Tofiño-Vian M, Iovino F. Neuronal damage and neuroinflammation, a bridge between bacterial meningitis and neurodegenerative diseases. Front Cell Neurosci. (2021) 15:680858. doi: 10.3389/fncel.2021.680858
141. Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep. (2017) 7:41802. doi: 10.1038/srep41802
142. Schaubeck M, Clavel T, Calasan J, Lagkouvardos I, Haange SB, Jehmlich N, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. (2016) 65:225–37. doi: 10.1136/gutjnl-2015-309333
Keywords: Porphyromonas gingivalis, neurodegenerative diseases, periodontitis, Alzheimer’s disease, Parkinson’s disease, neuroinflammation
Citation: Huang Z, Hao M, Shi N, Wang X, Yuan L, Yuan H and Wang X (2025) Porphyromonas gingivalis: a potential trigger of neurodegenerative disease. Front. Immunol. 16:1482033. doi: 10.3389/fimmu.2025.1482033
Received: 17 August 2024; Accepted: 27 January 2025;
Published: 14 February 2025.
Edited by:
Hugo Caire Castro-Faria-Neto, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Jaime Díaz-Zúñiga, University of Chile, ChileCopyright © 2025 Huang, Hao, Shi, Wang, Yuan, Yuan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Wang, d2FuZ3hpYW9mZW5nQGpsdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.