 Yaojie Kong
Yaojie Kong Yanwei Wu
Yanwei Wu- School of Medicine, Shanghai University, Shanghai, China
Chimeric Antigen Receptor (CAR)-T cell therapy has rapidly emerged as a groundbreaking approach in cancer treatment, particularly for hematologic malignancies. However, the application of CAR-T cell therapy in solid tumors remains challenging. This review summarized the development of CAR-T technologies, emphasized the challenges and solutions in CAR-T cell therapy for solid tumors. Also, key innovations were discussed including specialized CAR-T, combination therapies and the novel use of CAR-Treg, CAR-NK and CAR-M cells. Besides, CAR-based cell therapy have extended its reach beyond oncology to autoimmune disorders. We reviewed preclinical experiments and clinical trials involving CAR-T, Car-Treg and CAAR-T cell therapies in various autoimmune diseases. By highlighting these cutting-edge developments, this review underscores the transformative potential of CAR technologies in clinical practice.
1 Introduction
Cancer has long been a very threatening chronic disease. Conventional therapies, despite some relief, have notable limitations and adverse impacts on patients’ immune systems and overall health. In recent years, CAR-T cell therapy has revolutionized cancer treatment by offering personalized treatment based on the specific type of cancer and the patient’s requirements. The CAR-T cell therapy process begins with the extraction and isolation of T cells from the patient. These T cells are then genetically engineered to express the CAR, which are then able to recognize and bind to tumor antigens. After in vitro expansion of the CAR-T cells, the CAR-T cells are infused back into the patient (Figure 1).
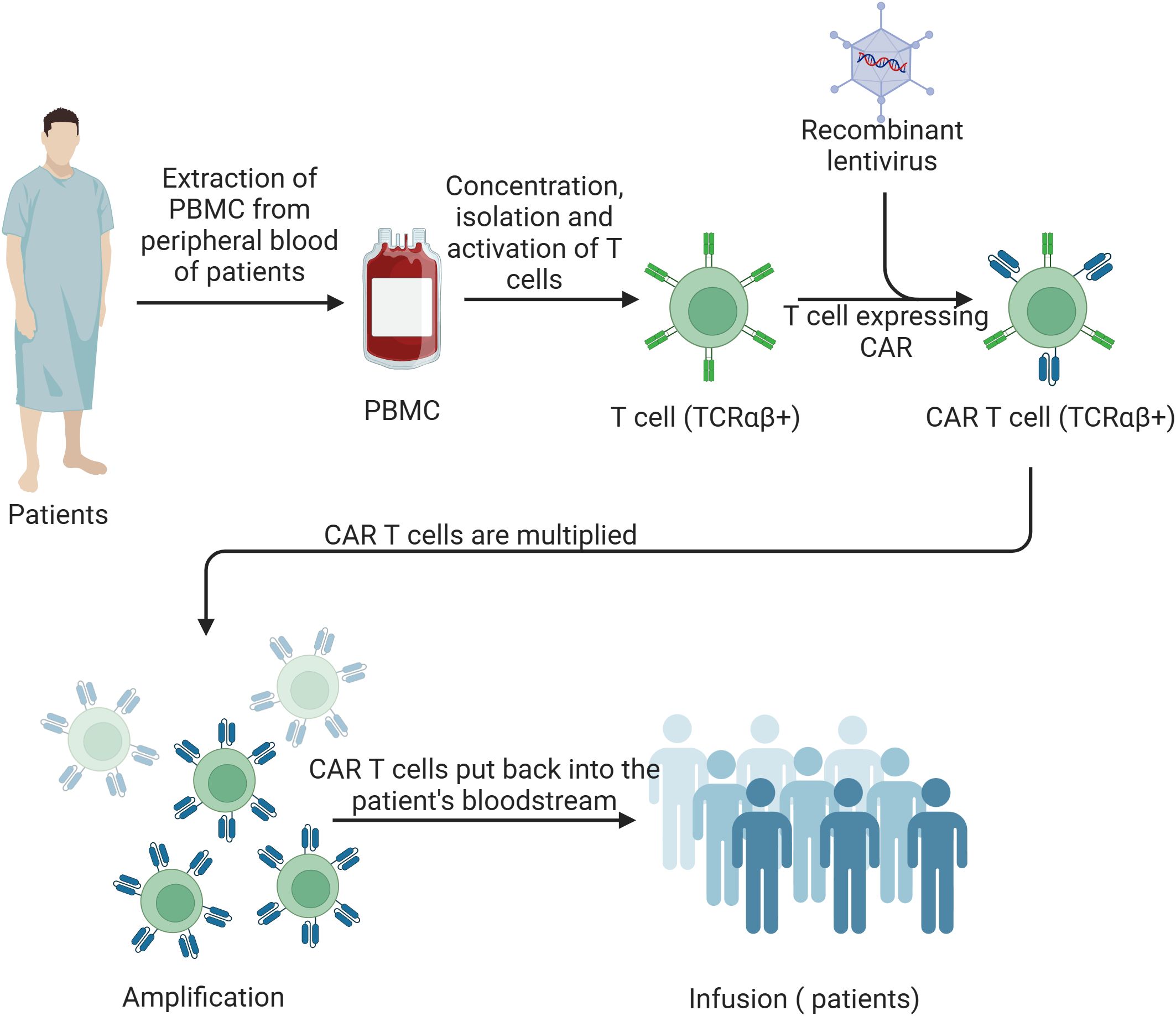
Figure 1. Schematic of CAR-T cell therapy process. Peripheral blood mononuclear cells (PBMCs) are extracted from the patient’s blood. T cells are then concentrated, isolated, and activated. These activated T cells (TCRαβ+) are transduced with a recombinant lentivirus to express the chimeric antigen receptor (CAR). The CAR T cells (TCRαβ+) are subsequently expanded and multiplied in vitro. Finally, the amplified CAR T cells are infused back into the patient’s bloodstream to target and eliminate cancer cells.
CAR-T cell therapy has unique advantages over conventional therapies, including its highly personalized and tailored nature. This customization involves analyzing the patient’s tumor to identify specific tumor-associated antigens (TAAs). Based on this analysis, the patient’s T cells are collected and genetically engineered to express CARs that specifically target these TAAs on the cancer cells. This precise targeting ensures that the therapy is specifically designed for the individual’s cancer, resulting in remarkable therapeutic efficacy (1). Furthermore, CAR-T cell therapy has shown enhanced effectiveness in addressing cancer types that are challenging to treat with more traditional cancer therapies, particularly hematological malignancies such as lymphomas, multiple myeloma, chronic lymphocytic leukemia and B-cell acute lymphoblastic leukemia (1).
CAR-T cell therapy has shown success in treating hematological tumors, but it still faces challenges in solid tumors treatment, such as therapeutic efficacy and safety. In response to these issues, studies continue to improve the design of CAR-T cells. Recent advancements in CAR-T technology have addressed some of the critical limitations of earlier generations. For instance, the development of the fourth-generation CAR-T cells has integrated inducible cytokine expression to enhance the recruitment and activation of innate immune cells and overcome the immunosuppressive tumor microenvironment, thus providing more robust and sustained anti-tumor responses compared to existing therapies. Moreover, the introduction of dual CARs and tandem CARs that can target multiple antigens simultaneously has improved the therapy’s effectiveness in overcoming tumor antigen escape, a common mechanism of resistance.
Beyond the realm of oncology, CAR-T cell therapy has shown tremendous potential for the treatment of autoimmune diseases. Currently, autoimmune conditions are predominantly managed using a wide array of immunosuppressive agents and blocking antibodies, which control the symptoms of the disease but often fall short of achieving a cure. Recent advancements highlight the potential of CAR-T cell therapy for treating autoimmune disorders like multiple sclerosis (MS) and systemic lupus erythematosus (SLE), opening the prelude of CAR-based cell therapy on the field of autoimmune diseases.
In summary, CAR-T cell therapy stands at the forefront of cancer treatment innovation, offering superior efficacy and specific advantages over traditional therapies. Continued research and development aim to expand its applicability and improve its outcomes, bringing new hope for patients with both hematologic and solid malignancies, as well as autoimmune diseases.
2 CAR structure
The foundation of CAR-T cell therapy lies in the architecture of the genetically engineered CARs. CARs are modified synthetic antigen receptors with four key structural elements: the extracellular antigen recognition domain, which encompasses a single-chain variable fragment (ScFv) and a hinge region; a transmembrane domain (TMD); and an intracellular T-cell activation domain (Figure 2). Below, we describe the roles of each of these domains and their engineering for CAR-T cell therapy in greater depth.
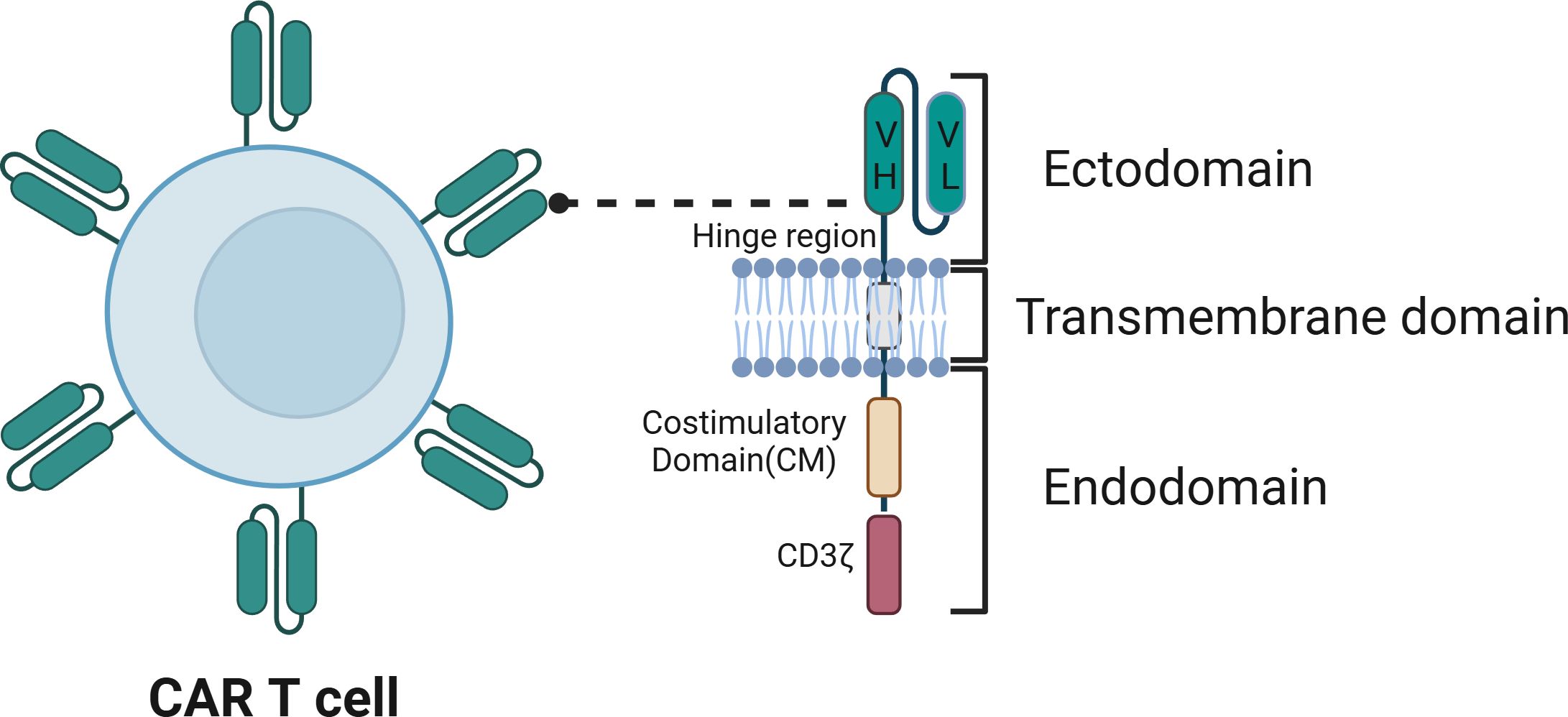
Figure 2. The structure of chimeric antigen receptor (CAR). The CAR contains an ectodomain, usually a single-chain variable fragment (scFv) from an antibody, comprising a variable heavy chain region (VH) and a variable light chain region (VL), which is responsible for recognizing the antigen. The transmembrane domain anchors the CAR to the T cell membrane. The endodomain includes signaling elements such as the CD3ζ chain, which contains ITAMs, and the costimulatory domain (CM), which enhances T cell activation, proliferation, and persistence.
2.1 The antigen recognition domain
The antigen recognition domain consists of an ScFv formed by linking the variable heavy chain (VH) and light chain (VL) of a monoclonal antibody through a linker. The primary function of the antigen recognition domain is the specific recognition of TAAs present on the surface of target cells, which subsequently facilitates the activation of the T cell bearing the CAR (2, 3).
Importantly, the recognition of target cells by CAR-T cell rely strongly on the high affinity of the ScFv component for the TAA (4–6). Therefore, the selection of appropriate ScFvs is crucial for antigen targeting. Initially, it is important to select an antibody with high affinity for the TAA and to determine its VH and VL sequences. Subsequently, CARs targeting the antigen can be constructed using ScFv sequences, after which in vitro cytotoxicity assays can be used to screen for CARs with cytotoxic functionality (5).
However, it is important to note that the affinity of the ScFv for the target antigen and its efficacy for CAR-T are not necessarily linearly correlated. In fact, excessive affinity can cause toxicity, as the CAR-T cells attack normal cells with low antigen density as well as the target tumor tissues, ultimately leading to damage to healthy tissue. Rather, researchers have found that the use of low-affinity ScFvs allows optimal CAR function (4–6). The ability to appropriately reduce the affinity of ScFvs to mitigate CAR-T cell therapy-induced tumor-associated toxicity has led researchers to speculate that there is an “affinity threshold” for CARs. This threshold represents the optimal balance where the ScFvs can effectively target tumor cells without attacking normal tissues with lower antigen density. Current research is actively exploring this concept by attempting to engineer ScFvs with affinities that fall within this ideal range. Scientists are using various screening and optimization techniques to fine-tune the affinity of ScFvs, ensuring they are strong enough to bind to tumor antigens while avoiding off-target effects on normal cells. This ongoing work aims to enhance the safety and efficacy of CAR-T cell therapies (5).
Almost all CAR constructs use ScFvs derived from monoclonal antibodies. However, the low folding stability of VH and VL could lead to aggregation or misfolding of ScFv, which may weaken the target effect and lead to exhaustion of CAR-T cells. To overcome these problems caused by conventional scFv, Xie et al. developed convenient and structurally stable CAR-T cell antigen-binding domains. This study designed a high-affinity binding protein (called “binder”) replacing typical ScFv to target tumor antigens. This binder CAR-T cells showed a stronger solid tumor treatment effect, providing a potential therapeutic benefit for CAR T cell therapy (7).
2.2 Hinge region
The hinge region connects the ScFv to the transmembrane structural domain. The hinge functions to enhance the flexibility of the ScFv, overcoming steric hindrance and facilitating the binding of the CAR to target antigens (8).
In the early, the hinge region is mainly derived from variants of immunoglobulin G (IgG), such as IgG1 and IgG4; therefore, one drawback to the use of these hinge regions is their potential interactions with the Fcγ receptor. Such interactions can lead to CAR-T cell depletion and ultimately adversely affect the durability of the therapy (4, 9). As an alternative, hinge regions from CD28 and CD8α have been adopted. In CD19 CAR-T cells, investigations have revealed that CD8α outperforms CD28 as a hinge region because CD8α stimulates a lower level of cytokine release and activation-induced cell death than CD28 (10). Moving forward, further optimization of hinge regions, including exploring other potential alternatives or modifications to CD8α, could enhance the safety and effectiveness of CAR-T therapies.
2.3 The transmembrane domain (TMD)
The TMD represents the intermediary section of the CAR, comprising a hydrophobic α-helix that spans the cellular membrane and connects the extracellular and intracellular domains. Its roles involve tethering the CAR to the cell membrane and transmitting activating signals to the intracellular domain (4, 11).
Historically, typical sources of TMDs have included CD3ζ, CD8α, CD28, and ICOS. However, the CD3ζ transmembrane structural domain is being abandoned because CARs containing this domain can form homodimers or heterodimers with the T-cell receptor (TCR), leading to strong T-cell activation in the absence of appropriate ligand binding. This uncontrolled activation can result in excessive immune responses, causing inflammation and potential damage to healthy tissues, making it a significant safety concern in CAR-T cell therapy. Conversely, CD8α and CD28 have become more frequently employed in clinical applications. Several investigations have revealed that the use of the CD8α TMD in CD19 CAR-T cells can attenuate the cell death caused by T-cell activation, and the CD8α TMD exhibits improved tolerability compared to that of CD28. In addition, the ICOS TMD has demonstrated superior efficacy and sustained antitumor activity compared to that of CD8 (10).
The TMD has attracted less attention in terms of structural innovations than other CAR domains. Nonetheless, researchers have shown that manipulating the length of the TMD can decrease CAR-T cell proliferation without compromising CAR-T cell antitumor capabilities. This characteristic offers more durable management of inflammatory cytokines and mitigation of the inflammation associated with the therapy (12). Furthermore, a recently introduced engineered TMD referred to as a programmable membrane protein (proMP) was reported to modulate the functionality of CAR receptors. As an entirely novel sequence, proMPs can form transmembrane homodimers, resulting in the generation of proCAR constructs. Compared to native CARs, ProCAR-T cells offer a more predictable functional range in vivo and significantly reduced release of inflammatory cytokines (13). Consequently, further research is warranted to explore the role and optimization of TMDs in enhancing this therapy.
2.4 The costimulatory domain
The costimulatory domain, situated within the T-cell activation region, is pivotal for T-cell activation because it facilitates dual activation through costimulatory molecules and intracellular signaling. This dual activation is essential as it involves two key pathways: the first pathway provides the primary signal through the TCR for antigen recognition, while the second pathway, mediated by costimulatory molecules, enhances the T-cell’s response and promotes survival, proliferation, and cytokine production. Both pathways are necessary to ensure a robust and sustained immune response, preventing premature T-cell exhaustion and maximizing the effectiveness of CAR-T cell therapy (14, 15).
The most common costimulatory structural domains are CD28 and 4-1BB. CARs incorporating the CD28 costimulatory structural domain stimulate the differentiation of T cells into effector memory T cells and foster aerobic glycolysis. On the other hand, CARs containing the 4-1BB costimulatory structural domain drive the differentiation of T cells into central memory T cells, facilitating mitochondrial production, enhancing respiration, and promoting fatty acid oxidation (16, 17). Notably, CD28 exhibits faster and more robust signaling activity, while 4-1BB has a slower and more gradual profile that extends the lifespan of T cells and sustains their anticancer effects (16, 17). Furthermore, the CD28 family comprises additional costimulatory structural domains, such as ICOS, while the tumor necrosis factor receptor family includes OX40 and CD27. These additional costimulatory domains have been explored in various preclinical and clinical studies. For instance, ICOS has been tested and shown to enhance T-cell function and survival, suggesting its potential to improve CAR-T cell efficacy. Similarly, OX40 and CD27 have been investigated for their roles in enhancing T-cell proliferation and longevity. Incorporating these domains into CAR designs could further optimize the therapeutic outcomes of CAR-T cell therapy, though ongoing research is needed to fully understand their benefits and any associated risks (18).
Moreover, in recent years, the development of the pioneering in vivo gene delivery system approach, in which CAR genes are directly delivered and expressed within the patient’s body, has offered significant time and cost advantages over traditional manufacturing procedures for CAR-T cell production. Unlike conventional ex vivo methods that require T cells to be extracted, modified in the lab, and then reinfused into the patient, this in vivo system streamlines the process by modifying T cells directly within the body, reducing both production time and associated cost (19). For example, CD3-targeted lentiviral vectors (CD3-LVs) can achieve selective gene transfer into T cells specifically, thereby enabling in vivo gene delivery and T-cell expansion and allowing direct generation of CAR-T cells within the patient (20, 21). As a result, this approach has the potential to greatly simplify and accelerate the CAR-T therapy process, making it more accessible and scalable, and opening new possibilities for treating a broader range of diseases.
3 Development of CAR-T cell therapy
CAR-T cell therapy has shown encouraging results for tumor treatment. However, several limitations and safety concerns, including low persistence and the potential for inflammatory reactions, have emerged as critical considerations. Extensive research has been performed to improve the therapeutic efficacy and safety of this therapy, particularly with respect to novel approaches to CAR design. As noted above, Since the initial development of CARs in 1989, the antigen recognition domains of CARs have predominantly comprised variable antibody regions or their derived fragments, such as ScFvs. The transmembrane region has undergone minimal change. Nevertheless, the introduction of innovative costimulatory domains within CARs has been recognized as a crucial advancement (22). Based on efforts to engineer the intracellular domains of CARs, the development of CAR-T cells can be categorized into five distinct generations (Figure 3). It’s important to note that CAR-T cells from multiple generations are currently in use, each offering unique advantages despite their different limitations. Progression to newer generations does not render the earlier versions obsolete but rather expands the options available for different therapeutic needs.
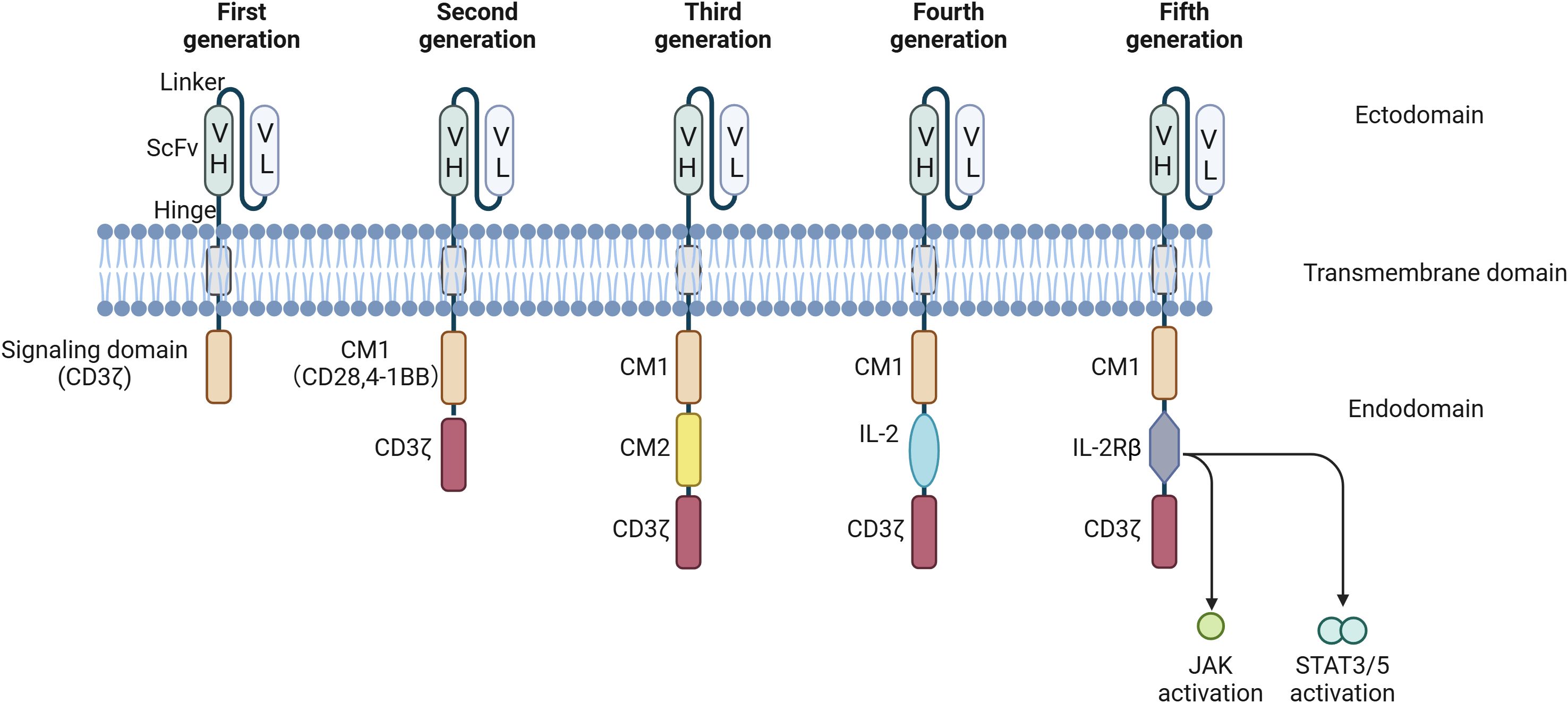
Figure 3. CAR development from the first to the fifth generation. First-generation CARs are characterized by the presence of a signaling domain originating from CD3ζ. Second-generation CARs encompass an additional co-stimulatory domain (CM1), such as CD28 or 4-1-BB. Building upon the second generation, third-generation CARs incorporate a second co-stimulatory domain (CM2). Fourth-generation CAR T cells, also known as T cells Redirected for Universal Cytokine-mediated Killing (TRUCKs), contain NFAT transcriptional elements, which can induce the expression of specific chemokines, such as IL-12, in the tumor microenvironment. Fifth-generation CARs are universal CAR-T cells. VH, variable heavy chain; VL, light chain; CM, co-stimulatory domain; IL, interleukin; CD, cluster of differentiation; NFAT, nuclear factor of activated T cells; ITAM, immunoreceptor tyrosine-based activation motify.
3.1 First-generation CAR-T cells
CARs of this generation consist of an ScFv, a hinge region, a transmembrane domain, and an intracellular signaling domain, such as CD3-ζ or FcγRI, that act as major transducers of TCR signaling and induce signaling cascades. The immunoreceptor tyrosine-based activation motif (ITAM) region of CD3ζ has been the most popular intracellular signaling domain due to the presence of three ITAMs in CD3ζ rather than one ITAM in FcϵRI (23, 24), although CAR-T cells containing the 4-1BB signaling domain demonstrate significantly better antitumor activity and persistence than those with CD3ζ (25). Leveraging the CD3ζ signaling domain, CAR-T cells of this generation trigger T-cell activation, resulting in cytotoxicity and cytokine secretion (e.g., IL-2) independent of human leukocyte antigen (HLA). Consequently, tumor cells are effectively eliminated by these CAR-T cells (26).
Nevertheless, CAR-T cells of this generation have several limitations. First, they do not produce sufficient IL-2 to support their long-term proliferation and sustain their cytotoxic activity against cancer cells, so exogenous IL-2 must be used to achieve effective tumor cell eradication (9). Second, the intracellular domains of CAR-T cells are deficient in costimulatory signals that are necessary for robust T-cell activation, limiting the efficacy of T-cell activation. Third, although these CAR-T cells exhibit conventional T-cell cytotoxicity, their proliferation is transient, and they secrete limited amounts of cytokines, ultimately leading to T-cell apoptosis and an inability to proliferate and survive long-term in vivo, greatly limiting their antitumor efficacy (27, 28).
Due to these limitations, CAR-T cells of this generation failed to achieve the expected efficacy in clinical applications. This failure propelled the development of second-generation CAR-T cell therapies aiming to enhance overall treatment efficacy by extending the in vivo persistence of CAR-T cells and augmenting their cytotoxic capabilities.
3.2 Second-generation CAR-T cells
The major advance of second-generation CAR-T cells over the first-generation therapies is the incorporation of a costimulatory domain, predominantly CD28 or 4-1BB, linked with CD3-ζ (14, 29). This modification allows T cells to receive both antibody stimulation signals and costimulatory signals (14), promoting IL-2 synthesis and facilitating T-cell activation. Second-generation CAR-T cells have been further optimized to address issues concerning T-cell proliferation, survival, cytotoxicity, and persistence (29–32).
Second-generation CAR-T cells featuring CD3ζ and costimulatory structural domains have been extensively utilized in clinical practice, with CAR-T cell products containing 4-1BB as a costimulatory molecule gradually becoming mainstream. Currently, all approved CAR-T cell therapies on the market are second-generation CAR-T products targeting hematologic malignancies. The Food and Drug Administration (FDA) has approved six CAR-T cell products, with two targeting BCMA (B-cell Maturation Antigen) (see Glossary) and four targeting CD19 (Table 1). For instance, the FDA first approved Tisagenlecleucel (Kymriah) in 2017, a CD19-targeting CAR-T cell therapy widely used for treating B-cell acute lymphoblastic leukemia and B-cell lymphoma, particularly in patients under 25 years old. Kymriah is currently approved for three indications and remains the only CAR-T cell therapy approved for both adult and pediatric populations (33). Similarly, Axicabtagene ciloleucel (Yescarta) received its first FDA approval in 2017 for the treatment of relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in adult patients and is now approved for three indications (34). In March 2021, Abecma became the first FDA-approved BCMA-targeting CAR-T cell therapy for adults with relapsed or refractory multiple myeloma after at least four prior lines of therapy, marking a significant advancement in multiple myeloma treatment (34).
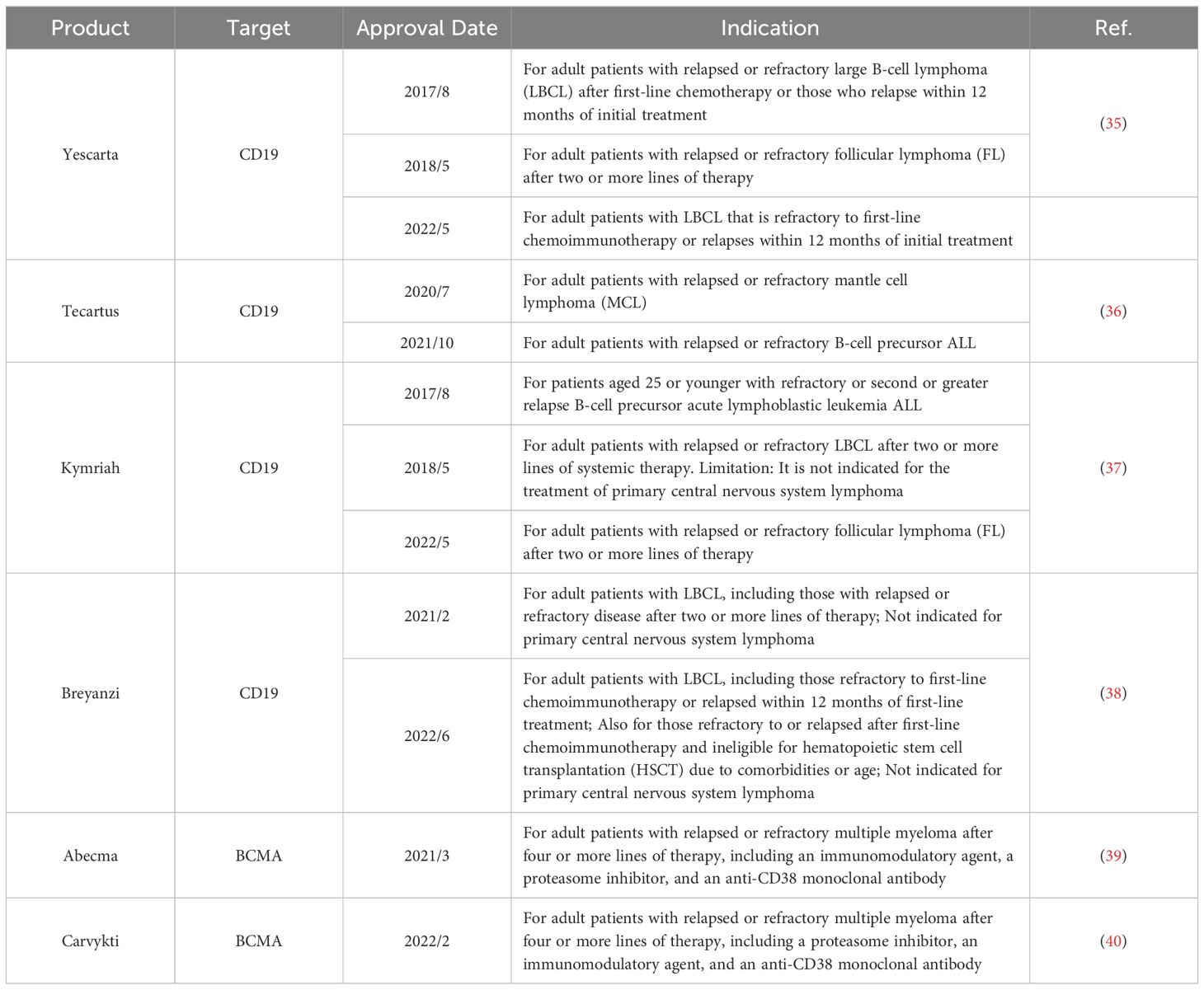
Table 1. Summary of FDA approved CAR-T products.
Several studies have demonstrated that the costimulatory molecule CD28 imparts potent activation signals, allowing T cells to rapidly attain high levels of cytotoxic activity while also contributing significantly to the generation of memory and effector cells, albeit with a shorter duration. On the other hand, the activation signals mediated by 4-1BB are more persistent, playing a vital role in regulating lymphocyte proliferation and survival and enhancing IL-2 production but with limited cytotoxic capabilities (41–43).
Unfortunately, studies have shown that a single costimulatory domain fails to resolve the issues of CAR-T cell therapy persistence and relapse. The use of retroviral vectors for gene transfer into T cells, a prevailing method for generating second-generation CAR-T cells, constrains the size of the gene fragments they can accommodate, and consequently, it is not feasible to transfect the ITAM regions of both CD28 and CD137 costimulatory molecules into T lymphocytes simultaneously. As a result, researchers have been forced to choose between achieving persistence and preventing relapse during therapy with the use of second-generation CAR-T cells.
3.3 Third-generation CAR-T cells
The development of third-generation CAR-T cells has focused on using specific transduction techniques to endow them with two or more costimulatory domains linked with CD3ζ. These approaches typically involve advanced gene delivery methods such as lentiviral vectors or transposon systems, which allow for the precise insertion of multiple costimulatory signals into the CAR-T cells. These techniques enable the enhancement of T-cell activation and persistence by integrating additional signaling domains (10, 44). CAR-T cells of the third generation are embodied by the CAR-T cell construct, which arises from the fusion of CD3ζ and multiple costimulatory domains. The viral vectors used for third-generation CAR-T cells are mostly lentiviral, which allows larger gene fragments to be carried into T lymphocytes, thus allowing CAR-T cells of this generation to contain more costimulatory structural domains, such as CD28, 4-1BB, and CD134 (OX40). At present, the most widely utilized third-generation CAR-T cell construct is the CD3ζ-CD28-4-1BB construct (44–49). The costimulatory molecules in these CAR-T cells can activate signaling pathways within T cells, including the JNK, ERK, and NF-κB pathways, thereby enhancing T-cell antitumor activity, proliferation capacity, and survival time, as well as the secretion of cytokines such as IL-2, TNF-α, and IFN-γ (27).
Although clinical data indicate that these CAR-T cells show superior anticancer potency relative to that of second-generation CAR-T cells, as demonstrated by their prolonged duration and increased expansion in B-cell non-Hodgkin lymphoma patients (50, 51), the safety profile of these CAR-T cells is statistically equivalent to or even worse than that of second-generation CAR-T cells. Hence, the simple addition of costimulatory molecules within the ITAM domain does not necessarily guarantee enhanced therapeutic efficacy (52, 53).
3.4 Fourth-generation CAR-T cells
Known as precision CAR-T cell therapy, fourth-generation CAR-T cell therapy is an advanced approach that offers improved targeting and a reduced risk of tumor recurrence. Building upon the foundation of second-generation CAR-T cells, precision CAR-T cells include regulated suicide genes within CAR-T cells to control their longevity within the organism. This innovative modification aims to circumvent the adverse effects associated with cytokine storms while still precisely targeting tumor cells within the host system. These CAR-T cells are known as TRUCK or armored CAR-T cells (27, 54–56).
These CAR-T cells also harbor an element that activates the transcription of the nuclear factor of activated T cells (NFAT) gene, enabling the secretion of specific cytokines (primarily IL-12) within the TME to recruit and activate other immune cells for an immune response (57). Additionally, the secretion of these cytokines promotes T lymphocyte infiltration within tumor tissues, thereby offering therapeutic advantages for solid tumors (as opposed to hematologic malignancies) (58).
3.5 Fifth-generation CAR-T cells
Earlier generations of CAR-T cell therapies, while effective, face challenges such as immune rejection by the host and the risk of graft-versus-host disease (GVHD) when infused into patients. To address these issues, a promising strategy has emerged that involves precise genetic modifications of HLA and TCR genes in T cells obtained from healthy donors. This fifth-generation approach aims to enhance CAR-T cells by reducing the likelihood of immune rejection and preventing GVHD, making the therapy safer and more effective for a broader range of patients. Notably, the inherent advantage of this approach is that it eliminates the need for patient-specific alterations, thereby offering a promising avenue for the treatment of diverse patients with the same therapy. This innovation can benefit a broader range of patients, including those with different types of cancers or diseases and individuals who were previously excluded from CAR-T cell therapy due to compatibility issues or concerns about GVHD (59). Universal chimeric antigen receptor T cells (UCAR-T cells) are prototypical examples of fifth-generation CAR-T cells. Unlike conventional CAR-T cell therapy, which involves extracting, genetically modifying, amplifying, and transferring T cells from patients (45, 60–62), UCAR-T cell therapy relies on T cells acquired from healthy allogeneic donors. Subsequently, endogenous TCRs and HLA class I molecules are knocked out of these cells through gene editing techniques, such as ZFN, TALEN, and CRISPR-Cas9, enabling large-scale production and therapy without the constraints of individual compatibility (63).
Compared with autologous CAR-T cells, allogeneic CAR-T cells possess distinct advantages, including broad individual compatibility and potentially scalable manufacturing and treatment. However, despite their promise, current clinical trials of UCAR-T cells face certain challenges that may limit their clinical application. For instance, their CAR recognition capacity is limited, with affinity restricted to only one or two targets. Nonetheless, allogeneic CAR-T cell therapy has great potential for addressing issues related to the lengthy production cycle and high costs associated with CAR-T cell therapy, underscoring the need for extensive exploration of this approach (64–67).
The design, preparation, and application of CAR-T cells exhibit considerable heterogeneity, as their effectiveness varies depending on the tumor type. Notably, first-generation CAR-T cells have exhibited substantial therapeutic efficacy in specific hematologic malignancies but have shown limited effectiveness in targeting solid tumors. Second- and third-generation CAR-T cells have significantly enhanced the viability, effectiveness, and safety profile of CAR-T cells, thereby improving therapeutic outcomes in clinical trials. Although third-, fourth-, and fifth-generation CAR-T cell technologies are still in the developmental stage and the cost of first- and second-generation CAR-T cell treatments remains high, it is reasonably presumed that these technologies will progressively enhance the stability and effectiveness of CAR-T cells, reduce their cost, and mitigate their adverse effects (68).
4 Challenges and solutions in CAR-T cell therapy for solid tumors
CAR-T cell therapy has enabled substantial progress in the treatment of hematological tumors, and the US FDA has granted market approval for six CAR-T cell products targeting various hematological malignancie (10, 69–76). However, for solid tumors, which constitute more than 90% of all cancers, CAR-T cell therapy faces significant challenges and limitations (5, 77). The six primary obstacles faced by CAR-T cell therapy in treating solid tumors, along with the corresponding strategies proposed to address these challenges, are presented below (78).
4.1 Antigen escape
Antigen escape describes the situation in which the expression of target antigens by malignant tumor cells in the body is partially or completely inhibited. This event plays a pivotal role in the development of acquired resistance against CAR-T cells in solid tumors (79, 80).
To overcome this limitation and minimize damage to healthy tissues, researchers have explored the utilization of dual-CAR or tandem-CAR systems that target multiple antigens simultaneously (4). Several studies have reported encouraging results from the application of dual CAR-T cells that target both CD19 and CD22 to treat patients with acute lymphoblastic leukemia (ALL), reporting that this therapy is more effective than CD19 CAR-T cell therapy alone (81). Additionally, in this situation, CAR-T cells are activated upon recognizing multiple antigens, thereby reducing the likelihood of accidental activation in healthy tissues and mitigating adverse effects (82).
Neoplasm-targeting allele-sensing CAR (NASCAR), is designed to target a mutation commonly found in the cancer transformation process called clonal loss of heterozygosity (LOH). LOH is observed in 90% of human cancers and often involves tumor suppressor genes, providing a clear distinction between normal and cancer cells. Previous therapeutic approaches targeting this distinction, such as antisense oligonucleotides and siRNA, have generally been ineffective. However, programming CAR-T cells to recognize LOH can significantly enhance their precision. This is achieved by introducing both CAR and an inhibitory CAR (iCAR) into CAR-T cells, with iCAR carrying intracellular domains like PD-1 or CTLA-4. In cancer cell lines engineered with LOH using CRISPR technology, NASCAR-T cells have demonstrated strong antitumor efficacy, highlighting their potential for precise and effective tumor targeting (83).
Above, we introduced several representative strategies focusing primarily on enhancing the targeting capabilities of CAR-T cells. However, researchers are also making significant efforts to improve other aspects of CAR-T cell functionality, such as the controllability of their activity and cytotoxicity. These enhancements include the addition of drug-responsive interfaces to T cells, allowing for the precise modulation of CAR-T cell activity through external drug administration. Looking ahead, more advanced versions of CAR-T cells will likely emerge, taking into greater account the specific needs and conditions of patients (78).
4.2 T-cell exhaustion
Prolonged proliferation of CAR-T cells is a key requirement for achieving an effective antitumor response within patients (84, 85), and T-cell exhaustion therefore poses a significant hurdle in CAR-T cell therapy (86, 87). Continuous stimulation of T cells by antigens within the tumor environment renders them susceptible to exhaustion, leading to inadequate production of IL-2 (88, 89).
Recent studies have provided compelling evidence indicating that CAR-T cell expansion and persistence can be augmented through the judicious selection of costimulatory signaling molecules, such as CD28, ICOS, OX40, and 4-1BB (29, 90–92). CAR-T cells incorporating CD28 displayed greater persistence than those containing 4-1BB, thus mitigating T-cell exhaustion and accelerating the expression of effector molecules after antigenic stimulation (37, 93).
Additionally, genetic engineering methods have emerged as promising strategies to address T-cell exhaustion. For example, enhancing the expression of the cytokine IL-7 or IL-15 has been shown to promote T-cell proliferation and, when combined with the downregulation of PD-1 expression, prevent CAR-T cell exhaustion (94, 95). Furthermore, gene manipulation to induce the overexpression of mutated Fas variants within T cells has been used to effectively suppress FasL-mediated cellular apoptosis (96). Modulating the expression of inhibitory receptors, such as PD-1 and TIM-3, in CAR-T cells is another approach that has been successful in counteracting CAR-T cell exhaustion (97, 98).
Further advancements in understanding and manipulating these and other exhaustion pathways could lead to more robust and enduring CAR-T cell therapies, ultimately improving patient outcomes and expanding the applicability of this treatment across various cancers.
4.3 Off-tumor effects
A significant challenge confronting solid tumor CAR-T cell therapy is the widespread presence of TAA in normal tissues, which makes it more difficult to target the desired effects specifically to tumor cells. For instance, trastuzumab-based (anti-HER2) CAR-T cell therapy exhibited fatal cardiopulmonary toxicity in a patient diagnosed with metastatic colon cancer (47). Current approaches to address this issue include targeting posttranslational modifications that are specific to tumor cells (99, 100). Examples of these modifications include phosphorylation patterns unique to oncogenic signaling pathways, glycosylation changes that alter cell surface antigens, and acetylation modifications affecting tumor suppressor proteins. By focusing on these tumor-specific modifications, therapies can more precisely target cancer cells while sparing normal tissues. For instance, certain truncated O-glycans, such as Tn (GalNAcα1-O-Ser/Thr) and sialyl-Tn (STn) (NeuAcα2-6-GalNAcα1-O-Ser/Thr), are overexpressed in solid tumors, providing opportunities for targeted therapy. Therapies targeting these specific glycan modifications are currently in various stages of development. Some have progressed to early clinical trials, where they have demonstrated initial promise in selectively targeting cancer cells while minimizing damage to normal tissues. However, many of these approaches are still in the preclinical or experimental phase, where they are being explored for their potential to enhance specificity and efficacy in cancer treatment (99).
Another approach taken by some researchers has been to endow T cells with two engineered receptors: a conventional CAR and a chimeric costimulatory receptor (CCR). This method is related to the evolution seen in the different generations of CAR-T cell therapy. Specifically, while second and third-generation CAR-T cells incorporate single and multiple costimulatory domains directly into the CAR structure to enhance T-cell activation and persistence, the strategy of using separate CCRs represents a novel enhancement beyond these generations. This dual-receptor strategy differs from the dual activation mentioned earlier in that the CCR is designed to provide an additional, distinct costimulatory signal only upon encountering a specific antigen in the tumor microenvironment. This ensures that T cells are fully activated only when they bind both the tumor-associated antigen via the CAR and the second antigen via the CCR, thereby increasing precision and reducing the likelihood of off-target effects (101). For example, these dual-receptor T cells continuously expand and persist for longer durations in multiple myeloma and low-antigen leukemia. This phenomenon concurrently prolongs patient survival and delays cancer progression (102).
Masked CAR-T cell (mCAR-T) therapy is another strategy to reduce the off-tumor effects. It has a CAR receptor with an additional masking peptide and a protease-sensitive linker. The masking peptide covers the antigen recognition domain and is removed through protease cleavage, which is triggered by the high protease levels typically found in the tumor microenvironment. This approach enhances targeting precision, and the antitumor efficacy of mCAR-T cells is comparable to that of conventional CAR-T cells (103).
4.4 CAR-T cell trafficking
Despite the advancements in CAR-T cell therapies, targeting solid tumors remains particularly challenging. CAR-T cell delivery to solid tumors encounters significant challenges related to the tumor microenvironment (TME), tumor stroma, and disparities between the chemokine receptors expressed by CAR-T cells and the chemokines released by tumor tissue. These factors collectively impede the effective infiltration and activity of CAR-T cells within solid tumors (104).
Therefore, specific strategies are needed to enhance the delivery of CAR-T cells to solid tumors. Some approaches that have been used are localized administration to facilitate CAR-T localization at the tumor site and engineering CAR-T cells to express chemokine receptors that are compatible with tumor-derived chemokine factors (105). For example, a recent study found that compared to intravenous infusion, directly injecting CAR-T cells into the pleural cavity significantly improves therapeutic outcomes and substantially reduces the required T cell dosage. Based on these promising results, the researchers have initiated a clinical trial to evaluate the safety of locally administered mesothelin-targeted CAR-T cell therapy in patients with malignant pleural tumors (106). Moreover, studies focusing on chemokine factors have revealed that CAR-T cells targeting the tumor-associated αvβ6 integrin and expressing CXCR1/2 are effectively enriched at the tumor site (107–109). Additionally, for neuroblastoma, which exhibits high CCL2 expression, the use of CAR-T cells targeting GD2 and CCR2b has yielded promising outcomes in terms of transportation (110).
Another obstacle to effective CAR-T cell transportation stems from the vasculature in the TME (111). Tumors often exploit abnormal blood vessel formation to acquire nutrients and promote infiltration, and these abnormalities can hinder therapeutic efficacy. Consequently, specifically targeting abnormal vessels represent a promising approach for enhancing CAR-T cell penetration (112, 113). Considering the pivotal role of vascular endothelial growth factors and their receptors in angiogenic signaling pathways, studies have suggested that inhibiting these factors and receptors could be employed as an antiangiogenic therapy for treating such aberrant vessels (113). Furthermore, considering that heparan sulfate proteoglycans are major constituents of the tumor stroma, the expression of heparinase, an enzyme capable of degrading heparan sulfate proteoglycans, in CAR-T cells has been suggested as a potential strategy (114).
In summary, overcoming the challenges of CAR-T cell trafficking to solid tumors is crucial for enhancing their therapeutic effectiveness. Strategies such as modifying chemokine receptors, degrading tumor stroma components, and employing innovative delivery methods are key to improving CAR-T cell infiltration and persistence within the tumor microenvironment.
4.5 Immunosuppressive microenvironment
Other factors limiting CAR-T cell efficacy include the presence of an immunosuppressive TME with a cytokine profile (such as IL-10, TGF-β, VEGF and IL-4) that preferentially recruits immunosuppressive Treg cells, myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages(TAMs) (Figure 4) (115). Treg cells suppress cytotoxic T cell activity through various mechanisms, including the secretion of immunosuppressive cytokines, competitive consumption of IL-2, CTLA4-mediated inhibition of antigen-presenting cells (APCs), and the prevention of T cell activation (116). Similarly, MDSCs exert a strong immunosuppressive effect on effector T cells, significantly impairing CAR-T cell functionality. Clinical observations have linked low MDSC levels in patients receiving CD19 CAR-T therapy with favorable responses in lymphoma and leukemia treatment. TAMs, as the predominant immune-infiltrating cells in the TME, suppress T cell-mediated anti-tumor responses by secreting immunosuppressive cytokines, producing amino acid-depleting enzymes such as arginase 1 and indoleamine 2,3-dioxygenase (IDO), and promoting the recruitment of Treg cells. Moreover, immune checkpoints, particularly PD-1, contribute to the reduced efficacy of immunotherapy (117). Other elements of the TME, including physical barriers, hypoxia, and immunosuppressive factors, exert similar effects (118, 119). Targeting these immunosuppressive cells or molecular signals has the potential to enhance the therapeutic efficacy of CAR-T cells (78).
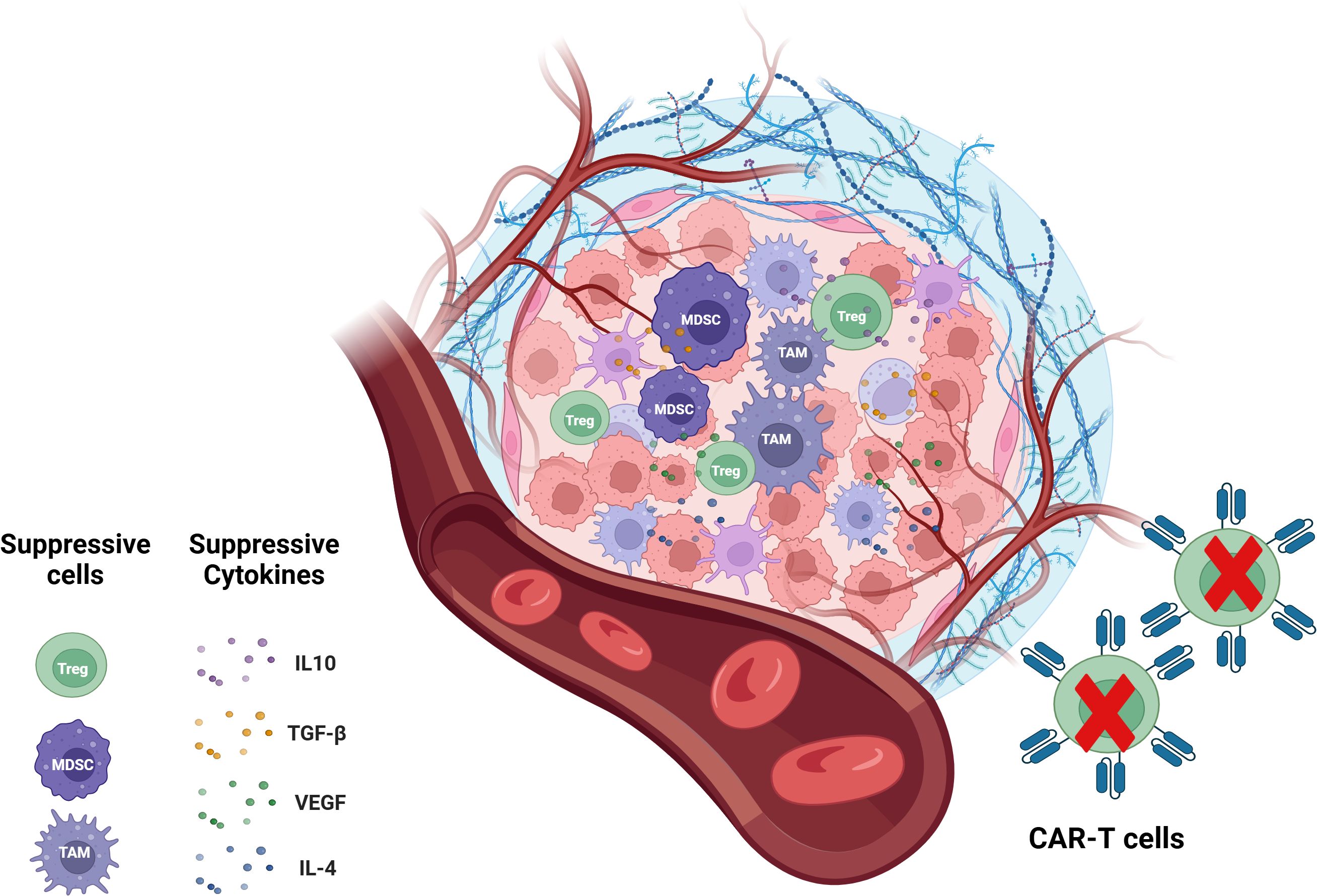
Figure 4. CAR-T cells in immunosuppressive TME. The immunosuppressive TME is a key factor that limits the efficacy of CAR-T cell therapy. The immunosuppressive tumor microenvironment is contributed by immunosuppressive cells including Treg cells, MDSCs, TAMs and immunosuppressive molecules like IL-10, TGF-0, IL-4 and VEGF. TME, tumor microenvironment; MDSCs, myeloid-derived suppressor cells; TAMs, tumor-associated macrophages.
Numerous strategies have been proposed to address these challenges. First, engineered synthetic receptors have been developed to counteract suppressive cytokines, with notable targets including TGF-β and IL-4. One such innovation is the synthetic TGF-β dominant-negative receptor (DNR), a modified version of TGF-β receptor II (TGF-βRII) that lacks the cytoplasmic signaling domain. These TGF-β DNRs inhibit native TGF-β signaling by forming non-functional ligand-receptor complexes, effectively serving as a molecular sink to sequester TGF-β (120). The incorporation of immune checkpoint blockade agents into CAR-T cells also promotes their long-term persistence, leading to promising outcomes (121). Furthermore, gene-editing technologies such as CRISPR/Cas9 can be utilized to disrupt PD-1 expression, further enhancing CAR-T cell persistence (98). Designing CAR-T cells to resist immune inhibitory factors can also result in prolonged persistence, contributing to their sustained presence and efficacy in the TME (122).
4.6 CAR-T cell toxicity
A notable obstacle to the widespread adoption of CAR-T cell therapy is the toxicity associated with these approaches including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) (36). CAR-T cell activation and cytokine secretion depend on the binding of the antigen-binding domain to its corresponding target antigen and reaching a specific activation threshold. This threshold refers to the amount of antigen engagement needed to sufficiently activate the CAR-T cells. Typically, it involves a certain number of antigen receptors being bound on the surface of CAR-T cells. When this threshold is met, it triggers a robust immune response. However, if the number of bound receptors or the intensity of activation signals exceeds this threshold, it can lead to excessive cytokine release and potentially cause toxicity, such as CRS or off-target effects on healthy tissues. Consequently, there has been a comprehensive exploration of modifying the CAR construct as a potential strategy to mitigate such toxicity (123).
One effective approach involves reducing the affinity of the antigen recognition domain, which consequently requires higher levels of target antigens on tumor cells to trigger T-cell activation. This alteration allows for the relative sparing of healthy cells that express lower levels of the targeted antigens (124). Another strategy to mitigate toxicity involves modifying the hinge and transmembrane regions of CAR constructs. These modifications are designed to optimize the flexibility and stability of the CAR structure, which can influence the strength and duration of the activation signal. By fine-tuning these regions, the CAR-T cells can achieve a more controlled and balanced activation response. This helps in regulating the amount of cytokine secretion, preventing excessive immune responses and reducing the risk of toxicity. Notably, modifying the CD8-α hinge and transmembrane region in anti-CD19 CAR-T cell therapy significantly reduced cytokine release (12).
Therefore, effectively managing CAR-T cell toxicity is essential to ensure the safety and success of these therapies. Current strategies focus on refining CAR designs to better regulate activation and cytokine release, thereby reducing adverse effects.
5 Innovations in CAR-T cell therapy
5.1 Specialized CAR-T cells
Several efforts to address the limitations arising from the adverse effects of CAR-T cell therapy have led to the development of CAR-T cells with specialized functionalities (125). For example, “self-driving” CARs are characterized by the expression of chemokine receptors that can detect and respond to the production of chemokines by tumor cells targeted for destruction. This mechanism enables the efficient trafficking of CAR-T cells to tumor sites, thereby augmenting their antitumor efficacy (45). For example, CAR-T cells carrying the tumor antigen GD2 enhance the migration of CAR-T cells to CCL2 by expressing the chemokine receptor CCR2b (126).
In the dual-CAR strategy, described above, genetic engineering techniques enable T cells to express two distinct CARs. This allows the targeting of multiple antigens simultaneously, potentially reducing patient resistance to CAR-T cell therapy and enhancing its overall effectiveness (4). Recent experimental evidence suggests that patients with ALL treated with CAR-T cells specifically targeting CD19 may experience loss or downregulation of the CD19 antigen (127, 128). However, the utilization of dual CAR-T cells that target both CD19 and CD22 has proven effective in addressing this challenge (81).
Inhibitory CARs (I-CARs) are CARs that can suppress T-cell responses. These CAR-T cells express two different CARs, one conventional and the other carrying an I-CAR. The I-CAR acts as a self-regulating switch that inhibits T-cell activation when it specifically recognizes and binds to antigens that are present exclusively or predominantly in healthy tissues. This design ensures that CAR-T cells function only in the absence of I-CAR stimulation, preventing damage to healthy tissues. Examples include I-CARs based on CTLA-4 or PD-1, which selectively limit the cytotoxicity induced by endogenous T-cell receptors or the activation of chimeric receptors (129).
Tan-CARs are characterized by the presence of two ScFv structural domains, each capable of recognizing a different target. This configuration increases the likelihood of CAR-T cell activation. However, it also carries the risk of causing damage to healthy tissues (125).
The concept of “off-switch” CARs emerged in response to the severe adverse effects associated with CAR-T cells, such as CRS. These CAR-T cells are designed such that specific inducers can be used to halt their excessive toxicity and induce depletion, thereby reducing or preventing adverse effects (130). For instance, CD20 CAR-T cells can be depleted through the administration of rituximab (131). However, one limitation of this antibody-mediated “off-switch” approach is that it acts on a relatively long timescale; that is, the rate of depletion of the undesired cells is slow (132). Moreover, alternative methods have been developed, including the use of dasatinib, inducible Cas9, and protease-based small molecule-assisted shutdown CARs (SMASh-CARs) (133, 134). In addition to “off-switch CARs”, there are also “on-switch CARs” that remain inactive under normal circumstances and are activated only upon stimulation by exogenous molecules, thus enabling the regulation of the timing and location of CAR-T cell activity (135).
In conclusion, continued research and optimization of these specialized CAR-T cells will be crucial to maximize their therapeutic potential and extend their application to a broader range of cancers and patient populations.
5.2 Combination therapies: CAR-T-based approaches in conjunction with other treatments
While CAR-T cells have demonstrated promising efficacy, achieving complete eradication of solid tumors using CAR-T cell therapy alone is difficult (136). However, some researchers have found that CAR-T cell therapy can significantly enhance the antitumor effects of other treatment modalities. Combination therapies that have been tested in conjunction with CAR-T cells to date include synergistic approaches that activate the patient’s endogenous T cells (137), as well as chemotherapy, radiation therapy, nanoparticles (NPs), oncolytic viruses (OVs), immune checkpoint blockade (ICB) and mRNA vaccine techniques (138).
The PD-L1/PD-1 signaling pathway is a tumor hallmark, with the TME inducing the expression of PD-L1 on tumor cells, and blocking the PD-1 receptor can deactivate CAR-T cells. The binding of PD-L1 on tumor cells to PD-1 on CAR-T cells inhibits the antitumor effects of CAR-T cells (139, 140). Hence, integrating ICB into CAR-T cells by blocking the PD-1/PD-L1 signaling pathway, such as by combining CAR-T cell therapy with anti-PD-1/PD-L1 agents (121, 141, 142), has shown potential in prolonging the persistence of CAR-T cell therapy and increasing the ratio of CD8+/CD4+ T cells within the TME (121, 143–145). Clinically, combination therapy targeting both PD-1 and CD19 has yielded favorable treatment outcomes and persistence in patients with hematologic malignancies (146).
Moreover, blocking the PD-1 signaling pathway in combination with CAR-T cell therapy has significantly improved the efficacy of CAR-T treatment of solid tumors (78). Recent studies have shown that combining CAR-T cell therapy with pembrolizumab (a PD-1 inhibitor) substantially increases the longevity of chimeric antigen receptor (CAR) cells in a murine model of metastatic melanoma (147, 148). Additionally, another study revealed that CAR-T cells secreting an anti-PD-1 scFv significantly enhanced CAR-T cell persistence and prevented the toxicity associated with systemic checkpoint inhibition (149, 150). Furthermore, employing CRISPR-Cas9 to knock out the PD-1 gene in CAR-T cells enhances CAR-T cell function, prolongs persistence, and reduces cytotoxicity, although it falls short in inducing T-cell infiltration (36, 151, 152).
Other gene editing techniques can also be applied to the modification and manufacturing of CAR-T cells. Studies have shown that CRISPR/Cas9 knockout of the TCR in tumor cells enhances T-cell potency (153) and reduces the risk of GVHD. Additionally, CRISPR/Cas9 can be used in conjunction with viral vectors to achieve targeted gene integration at specific sites (154). This precise integration can enhance the efficiency and safety of CAR-T cell therapies by ensuring that the CAR gene is inserted into a known and controlled location within the genome. However, some studies have indicated that this method may inadvertently shorten the lifespan of the CAR-T cell response. This unexpected outcome could be related to the disruption of genomic stability or the altered expression of nearby genes caused by the integration process, rather than the specific gene that was knocked in. As a result, while CRISPR/Cas9 offers a powerful tool for precise gene editing, it is essential to further investigate and mitigate these potential impacts to fully harness its benefits in CAR-T cell therapy (155). In terms of manufacturing, CRISPR/Cas9-based methods are characterized by low toxicity and high efficiency.
OVs are particularly interesting as a combination therapy because themselves exhibit antitumor activity (156, 157), and moreover, combining CAR-T cell therapy with OVs provides a more effective targeting of solid tumors, leading to enhanced antitumor effects (158). The OV genome can also be modified to express tumor necrosis factor-alpha or IL-2, which has been shown to increase the quantity of CAR-T cells in the TME and thereby enhance antitumor efficacy (159, 160).
Various nanotechnologies promise to enhance cancer immunotherapy by facilitating the delivery of immune-modulating drugs. In particular, NPs can serve as carriers for CAR-T cells into the TME, preventing their inhibition by TMICs and other immunosuppressive factors (115, 161–163).
The remarkable success of the SARS-CoV-2 mRNA vaccine has spurred extensive efforts to broaden the mRNA vaccine platform for the treatment of other conditions. Recent research has demonstrated that modified mRNAs encoding a CAR designed against fibroblast activation protein (FAP) (a marker of activated fibroblasts) encapsulated in targeted LNPs can be intravenously injected to induce the generation of functionally engineered T cells in vivo, offering potential for treating cardiac injury (164). These mRNAs typically encode for CARs or TCRs, which can reprogram T cells to target specific antigens associated with disease, thereby enhancing their therapeutic potential (165–167). Furthermore, numerous experiments have shown the potential of this method for treating solid tumors. Recent findings from a pioneering phase 1/2 basket trial investigated a claudin-6 (CLDN6)-directed CAR-T cell therapy for patients with CLDN6 tumors with the use of CARVac, an innovative antigen delivery mechanism that leverages an mRNA vaccine to bolster CAR-T cell amplification and persistence (168). This strategy is attractive because it enables CAR-T cell therapy to be performed directly in vivo, that is, without removing T cells from the body for editing, which saves considerable time and money. Additionally, the ability to promptly and precisely adjust the mRNA sequence and dosage to suit various tumor types and variants offers clear practical advantages. For example, it avoids prolonged CAR-T cell activity and the resulting amplification effects, thus reducing the risk of severe side effects such as CRS and neurotoxicity. Maintaining CAR-T cell efficacy and durability through multiple sequential injections addresses challenges such as immunosuppression and drug resistance within the TME. Moreover, synergistic enhancement of CAR-T cell antitumor efficacy can be achieved by combining or coadministering various mRNA types or drugs. Overall, the integration of mRNA technology with CAR-T cell approaches has potential for ‘off-the-shelf’ universal therapies, offering a scalable and affordable solution in oncology and for other diseases, such as autoimmune disorders, cardiovascular diseases, infectious diseases, and certain genetic conditions
5.3 Novel strategies: CAR-Tregs, CAR-NK cells, and CAR-M cells
Recent advances have expanded the scope of immunotherapy beyond CAR-T cells alone. Innovations in cellular immunotherapy have propelled the expansion of the core CAR strategy to different cell types, such as Tregs, NK cells, and macrophages (M cells), to enhance their efficacy and broaden the therapeutic landscape. The resulting therapies hold great potential for treating cancer and immune-related disorders. These innovative approaches are described in detail below.
5.3.1 Advantages and challenges of CAR-Treg therapy
Treg cells possess potent immunoregulatory functions and promote immune tolerance, making Treg cell therapy desirable for inducing tolerance to allogeneic tissues and cells in organ and hematopoietic stem cell transplantation (169–175). Treg cells can be used for the management of GVHD (176, 177), type 1 diabetes (T1D) (178), and other autoimmune diseases.
Despite the potential of Treg cell therapy, it has several limitations. Treg cell activation can lead to bystander suppression, in which non-targeted immune responses are suppressed, potentially reducing the effectiveness of the overall immune response. Moreover, the therapeutic effects are suboptimal when an insufficient number of Treg cells are present. In certain immune environments, nonspecific Tregs can be converted into proinflammatory Th17 cells, and this treatment approach may also induce nonspecific immune suppression (170, 171). Specific Treg cell therapies, such as TCR-Tregs, have been developed to overcome these limitations. However, the reliance of TCR-Tregs on MHC for antigen recognition limits the effectiveness of this approach. The engineering of Treg cells to express CARs offers advantages such as independence from MHC, lack of HLA restrictions, lower dependence on IL-2, stable phenotype and function, and improved specificity and suppressive capacity through coreceptor signaling requirements (170, 171, 179).
CAR-Tregs can recognize specific antigens and induce their activation and proliferation. Subsequently, the CAR-Tregs directly inactivate APCs, preventing antigen presentation to T cells. Additionally, CAR-Tregs can directly impede T-cell activation by producing inhibitory cytokines such as TGF-β, IL-10, and IL-35. Upon activation, CAR-Tregs can curb rejection responses by releasing granule enzymes and perforin to eliminate cytotoxic T cells (180).
The process of manufacturing CAR-Tregs is similar to that of CAR-T cells and therefore faces similar challenges, such as high cost and complexity (Table 2) (181). Currently, there are two methods for obtaining CAR-Treg cells. Isolation from PBMCs is the most commonly used method, but the relatively low abundance of Tregs in the peripheral blood limits the suppressive capacity of Tregs obtained in this way (182). Another method involves engineering and modifying CD4+ or CD3+ conventional T cells by introducing CARs and further introducing FoxP3 cDNA to induce the differentiation of these cells into Tregs. This approach overcomes the limitations of low Treg levels in the peripheral blood and the lack of FoxP3 expression in endogenous Tregs (180). Transfecting T cells with FoxP3 can also induce regulatory activity, specifically enhancing their ability to suppress immune responses and maintain immune tolerance (183). In CAR-Treg manufacturing, CAR genes are mainly delivered using viral vectors such as lentiviruses (175) or adenoviruses (184), although nonviral methods such as the Sleeping Beauty system (185) and CRISPR-Cas9 technology (186) have also been employed. Cell sorting is primarily performed using magnetic-activated cell sorting (MACS), but this technique may result in insufficient purity and low Treg cell recovery rates. Although FACS ensures higher purity and recovery rates, as well as more precise separation of Treg cells, the relatively small quantity of cells that can be produced and slow processing speed are drawbacks of this approach (187, 188). Currently, activation is achieved through stimulation with magnetic beads coated with anti-CD3/CD28 antibodies and IL-2. Notably, during in vitro expansion, the mTOR inhibitor rapamycin is often added to deplete effector T cells (Teffs) to maintain Treg stability (189).
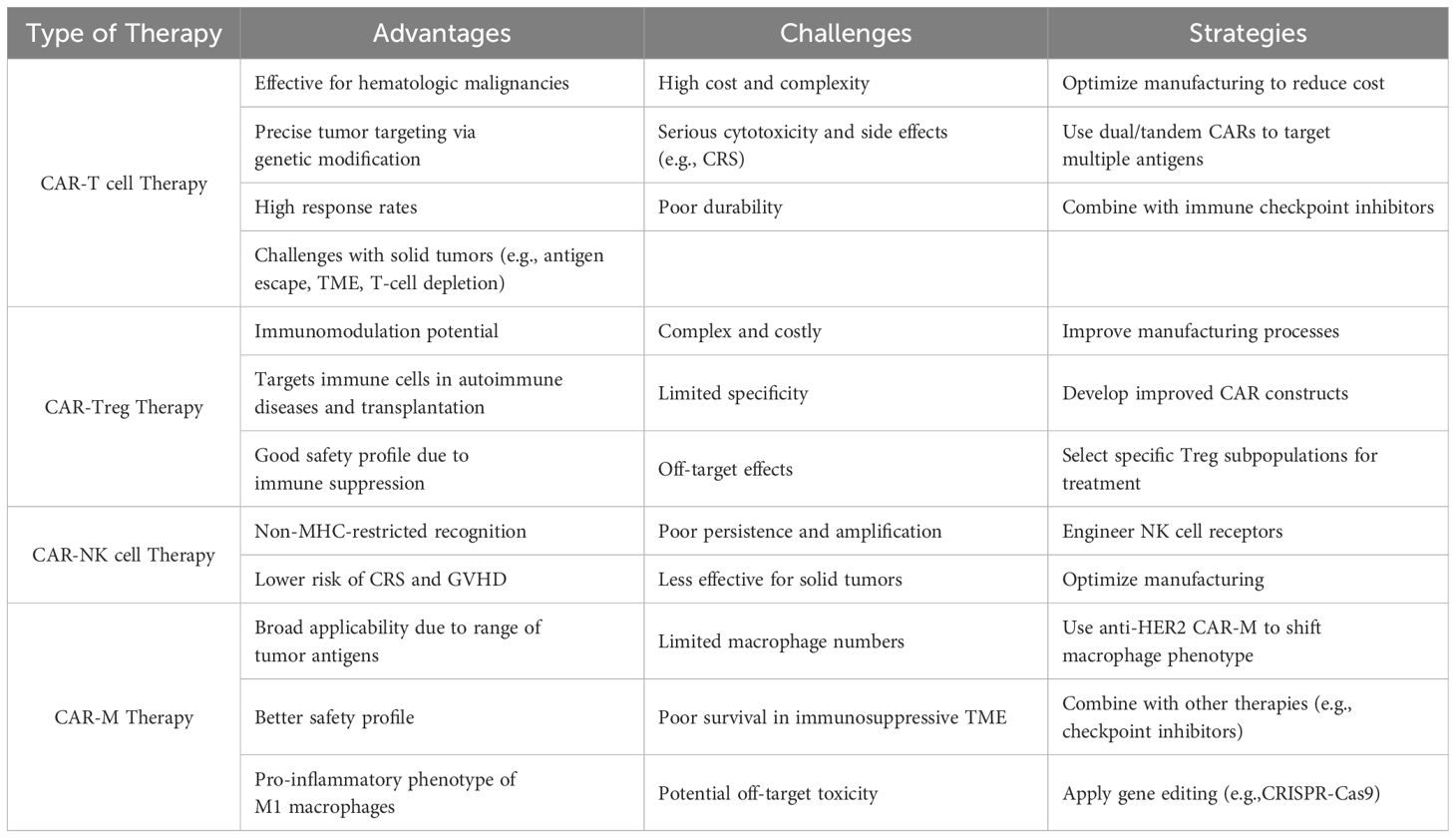
Table 2. Advantages and challenges of CAR-T cell, CAR-Treg, CAR-NK cell, and CAR-M therapies.
5.3.2 Advantages and challenges of CAR-NK therapy
NK cells are innate cytotoxic immune cells that can mount immune responses against foreign cells (190, 191) and recognize malignant cells through various signals from different cell surface receptors (192). In the context of cancer immunotherapy, NK cells are appealing as they intrinsically regulate their own cytotoxic activity toward normal cells and avoid “on-target, off-tumor” toxicity by employing killer cell immunoglobulin-like receptors (KIRs) for self-recognition (193–195). In contrast to CAR-T cells, which can only specifically recognize cells with tumor-associated target antigens, CAR-NK cells can kill cancer cells that do not express target antigens (196).
Furthermore, NK cells exhibit non-MHC-restricted recognition, infiltrative properties within tumor tissues, and fewer side effects, such as CRS and GVHD, among other advantages. Consequently, the distinct characteristics of NK cells position them as a potential alternative to T cells, and the fusion of CAR technology with NK cells has established the groundwork for CAR-NK therapy (Table 2) (138).
CAR-NK cells can effectively eliminate tumors through a combination of CAR-dependent and NK cell receptor-dependent mechanisms, resembling the mechanisms observed in CAR-T cell antitumor therapy (138). This allows CAR-NK cells to effectively target cancer cells for eradication. CAR-NK cells can also be activated by CAR-independent pathways, thereby broadening their versatility in tumor eradication (197, 198). For instance, antibody-dependent cellular cytotoxicity (ADCC) mediated by CD16 can be utilized by CAR-NK cells to eliminate tumor cells (199). These mechanisms show the multifaceted capabilities of CAR-NK cells in combating tumors and illustrate their promise for therapeutic applications.
As previously mentioned, allogeneic CAR-T cell therapy carries a risk of GVHD, a condition where donor immune cells attack the recipient’s tissues and organs. However, allogeneic CAR-NK cell transplantation carries a lower risk of GVHD than does CAR-T cell therapy, and potential complications such as CRS and ICANS are avoided, making CAR-NK cells a potential alternative for immunotherapy (200). Consequently, CAR-NK cell-based therapies have demonstrated improved clinical safety profiles and have addressed the problems associated with immune-mediated adverse events commonly associated with allogeneic CAR-T cell therapies.
Allogeneic NK cells are frequently favored for cellular immunotherapy, and they can be derived from induced pluripotent stem cells (iPSCs), peripheral blood (PB), and umbilical cord blood (UCB), among other sources (201). Among these, UCB has the capacity to generate NK cells in high quantities (202), facilitating the production of readily available CAR-NK cells for patient administration when needed (203). This approach offers notable advantages, such as safety, convenience, rapidity, and cost-effectiveness. However, a significant drawback is that UCB must be collected at birth, limiting its availability for current patients. Alternative approaches, such as deriving NK cells from iPSCs or peripheral blood, are being explored to address this limitation (204).
Indeed, while CAR-NK-cell therapy offers several advantages over CAR-T cell therapy, it also has several limitations similar to those faced by CAR-T cell therapy, particularly in the context of treating solid tumors. One of the significant challenges is that TME components such as immune cells, stromal cells, and extracellular matrix can hinder CAR-NK cell activity (205). To address these limitations, ongoing research has focused on developing strategies to enhance CAR-NK cell function within the TME, such as combination approaches involving immune checkpoint inhibitors, cytokine support, or genetic modifications like enhancing expression of cytokine receptors, incorporating co-stimulatory domains, or knocking out inhibitory receptors To address these limitations, ongoing research has focused on developing strategies to enhance CAR-NK cell function within the TME, such as combination approaches involving immune checkpoint inhibitors, cytokine support, or genetic modifications like enhancing expression of cytokine receptors, incorporating co-stimulatory domains, or knocking out inhibitory receptors. Additionally, advancements in nanotechnology and engineering approaches have been applied to optimize CAR-NK cell delivery and improve CAR-NK persistence and efficacy in solid tumor environments (206).
Additionally, the transduction, in vitro expansion, and in vivo maintenance of NK cells pose greater challenges than those of infiltrating T cells, creating a hurdle for achieving long-term persistence and sustained antitumor effects. In particular, the transient nature of NK cell function and the shorter lifespans of these cells in vivo can impact the durability of CAR-NK therapy (207). Ongoing research efforts to address these limitations include the optimization of transduction techniques and the development of methods for more effective in vitro expansion and enhancing NK cell survival and persistence in vivo.
Due to the promising attributes of CAR-NK cell treatment, there has been notable progress within this domain in recent years. Numerous researchers have proposed the fabrication of CAR vectors that specifically target NK cells to enhance the efficacy of tumor therapy. Significantly, the inclusion of costimulatory domains that are specific to NK cells, such as 2B4 and DNAX activation protein 10 or 12 (DAP-10 or DAP-12), has been shown to induce increased cytotoxicity and the production of IFN-γ (208). Furthermore, the integration of transgenes that encode activating cytokines, such as IL-21, which can stimulate the growth and viability of NK cells, can extend the longevity of CAR-NK cells (207). For instance, the introduction of CXCR3 receptors into the activation signaling domain of NK cells facilitates their migration to tumor sites, thereby bolstering their therapeutic efficacy against solid tumors (209, 210). In addition, the use of novel viral transduction enhancers, such as polyethyleneimine, can enhance the transduction efficiency of NK cells, thus amplifying their anticancer effects (211).
5.3.3 Advantages and challenges of CAR-M-cell therapy
In addition to NK cells, macrophages have garnered significant attention as promising cells for cancer therapy. Their efficacy stems from their capacity to engulf tumor antigens and secrete anti-inflammatory factors, along with other anticancer mechanisms (Table 2) (212).
TAMs can assume two phenotypes, known as M1 and M2 (213). M1 macrophages release interleukin-12 (IL-12), which activates the cytotoxic activity of NK cells and stimulates Th1 and CD8+ T cells, thus facilitating the eradication of tumors (214–216). Moreover, M1 macrophages can function as APCs for tumor antigens, thereby contributing to their antitumor effects (217). Conversely, M2 macrophages promote tumor growth (218) and typically impede the immune-mediated clearance of tumor cells (219). Macrophages can be derived from different sources, such as UCB, bone marrow (BM), and iPSCs. Each source has its own advantages and disadvantages. UCB provides a readily available source with high proliferation potential, but its collection is limited to the time of birth, making it unavailable for current patients. BM-derived macrophages are more accessible for adult patients and have a well-established use in clinical settings, but their collection is invasive and can be painful. iPSCs offer the advantage of being able to generate large quantities of macrophages with specific desired properties, including genetic modifications for enhanced anticancer effects. Macrophages derived from iPSCs that are engineered to express chimeric antigen receptors (CAR-iMacs) retain their innate immune functions and can shift from an M2 phenotype to an M1 phenotype upon encountering antigens, exerting potent anticancer effects. However, iPSC-derived macrophages require extensive laboratory work and are cost-intensive. The choice of source is crucial as it impacts the practicality, scalability, and effectiveness of macrophage-based therapies, with iPSCs providing a particularly flexible and potent option for engineering macrophages with enhanced therapeutic properties (220).
CAR-M therapy and CAR-NK cell therapy share several characteristics, including a reduced likelihood of inducing GVHD, that make them promising alternatives for allogeneic cell therapy. Furthermore, the remarkable capacity of macrophages to remodel the extracellular matrix (ECM) enables their infiltration into the immunosuppressive TME (221). Consequently, CAR-M therapy has a significant advantage over CAR-T cell and CAR-NK-cell therapies. Another noteworthy aspect of CAR-M therapy is its ability to elicit antitumor responses through both the innate and adaptive immune systems (222).
Nevertheless, CAR-M therapy has certain limitations, including a constrained macrophage quantity, limited effectiveness of CAR transduction, and compromised survival and persistence within immunosuppressive TMEs (138). Additionally, since macrophages serve as a primary source of cytokines, the administration of CAR-M treatment may induce CRS (223). Furthermore, the systemic circulation of macrophages may result in off-target effects (224).
The strategies that have been pursued to improve CAR-M therapy include viral and nonviral engineering approaches. Research has shown the effectiveness of modified lentiviral particles encoding the Vpx protein for proficiently transferring transgenes into myeloid cells (225). Additionally, anti-HER2 CAR-M therapy has been shown to induce a phenotypic shift from M2 to M1 macrophages, thereby augmenting antitumor efficacy (226).
6 The expanded application of CAR-based cell therapy in autoimmune diseases
Although CAR-T cell therapy is predominantly used for oncology, recent studies have indicated its potential application in the treatment of autoimmune diseases such as SLE, colitis, and common aspergillosis (57, 227–230). Despite the significant progress that has been made in the treatment of autoimmune diseases since the beginning of this century, the current interventions (primarily immunosuppressive agents and blocking antibodies) can control the disease but often fail to achieve a cure, as they still inadequately control the underlying autoimmune processes. Achieving the therapeutic goal of long-term remission in patients with autoimmune diseases remains challenging. CAR therapies offer a promising approach for controlling autoimmune diseases by specifically targeting and modulating the immune cells responsible for the pathological immune response. By engineering T cells to express CAR that recognize autoantigens or immune cell markers, CAR-based therapies can selectively eliminate or suppress autoreactive immune cells, thereby restoring immune balance and preventing tissue damage. To date, three CAR-based cell therapy strategies for treating autoimmune diseases have been reported: CAR-T cell therapy, CAR-Treg therapy and Chimeric autoantibody receptor (CAAR)-T cell therapy.
6.1 CAR-T cell therapy for autoimmune diseases
Following the success of CAR-T cell therapy for cancer, researchers have explored its potential in treating autoimmune diseases. Excessive B-cell activation leads to autoantibody production, driving autoimmune disorders. CAR-T cell targeting of autoreactive B cells aims to reduce autoantibodies and suppress autoimmunity. This innovative approach offers treatment promise across autoimmune disorders, enhancing patient outcomes and quality of life.
6.1.1 Systemic lupus erythematosus
SLE is a chronic autoimmune disease characterized by the immune system attacking the body’s own tissues, leading to widespread inflammation and tissue damage in organs such as the skin, joints, kidneys, and brain. B cells play a crucial role in SLE by producing autoantibodies that form immune complexes, which contribute to the inflammatory process and tissue damage. Given the role of B-cell hyperactivation in SLE, targeting B cells is a promising approach for SLE treatment (231). CAR-T cell therapy has potential for enabling selective B-cell targeting (232). Recent studies have highlighted the efficacy of CAR-T cell therapy in SLE, particularly when directed against the CD19 antigen. CD19 CAR-T cell infusion in chronic lymphocytic leukemia and acute myeloid leukemia patients leads to CD19+ B-cell reduction, autoantibody suppression, and lupus nephritis reversal. Subsequent work has focused on constructing CD19 CARs with internal costimulatory domains such as CD28 or 4-1BB, which, when employed in SLE patients, demonstrate similar therapeutic effectiveness and even preventive effects. Notably, CAR-T cells integrating the 4-1BB costimulatory domain outperform those with CD28 in terms of treatment outcomes (23, 210, 211, 233–236). As a result, CD19 CAR-T cell therapy has proven to be efficacious in controlling CD19+ B cells (25, 37, 93, 230, 231, 237, 238). A latest follow-up research evaluated 8 patients with severe SLE who received a single infusion of CD19 CAR-T cells 2 years ago. As a result, all the patients with SLE had Definition of Remission in SLE (DORIS) remission and Immunosuppressive therapy was completely stopped in all the patients, providing rationale for further controlled clinical trials (239).
Despite the notable adverse events linked to CAR-T cell therapy, such as CRS and ICANS (36), it is worth noting that SLE patients, who have a lower B cell burden than patients with B-cell malignancies, do not exhibit high-grade CRS following CAR-T cell therapy. Furthermore, the incidence of ICANS in SLE patients receiving CAR-T cell therapy is lower than that in non-SLE patients (233, 240–243). As a result, CAR-T cell therapy represents a viable approach for achieving sustained B-cell depletion, highlighting the significance of CD19-targeting CAR-T cells as a valuable therapeutic option for managing SLE.
In addition, B cell maturation antigen (BCMA)-based CAR T cell therapies also have been applied to various B cell-mediated autoimmune diseases including systemic lupus erythematosu, promoting the field of CAR-T cell therapy in autoimmune diseases rapidly evolve (244).
6.1.2 Rheumatoid arthritis
B lymphocyte function in the etiology of RA is similar to that in SLE. Recent investigations have shown the promise of employing inert UCAR-T cells that are engineered target fluorescein isothiocyanate (FITC) epitopes to alleviate RA. These CAR-T constructs consist of one end capable of connecting to CAR-T cells through FITC and the other end capable of binding to self-reactive B cells via antigen peptides, connected with a linker molecules. By simply substituting the antigenic terminus of the linker compound according to the patient’s antibody profile, a tailored strategy can be developed to specifically target and eradicate distinct B lymphocyte populations, resulting in prolonged and efficacious enhancement of RA outcomes (245).
6.2 Application of CAR-Tregs in autoimmune diseases
Originally utilized for GVHD treatment (246), Treg-based therapy aims to reinstate immune tolerance in the context of autoimmune diseases (188). Since its original application, the amalgamation of Treg cells with CAR technology has been considered to have considerable potential in addressing diverse autoimmune conditions, facilitating the extensive utilization of CAR-Tregs (247, 248). Clinical trials commonly employ manipulation techniques involving CD4+ FOXP3+ Treg cells (249).
6.2.1 cGVHD
CAR-Treg technology has the ability to produce allogeneic antigen-specific Tregs (175). This approach facilitates the use of CAR-Tregs to treat autoimmune diseases.
HLA disparities are common in transplantation and can lead to graft incompatibility and immune-mediated rejection when donor-recipient HLA mismatch occurs (250). Studies have demonstrated that targeting HLA-A2 via CAR and FOXP3 transduction enables the generation of HLA-A2-specific HLA-A2-CAR-Tregs. Activated HLA-A2 CAR-Tregs effectively suppress skin graft rejection in humanized mice with immune reconstitution (175). Moreover, they outperform polyclonal Treg cells in inhibiting delayed-type hypersensitivity (DTH) reactions to allogeneic antigens, completely preventing cytotoxicity against allogeneic human skin grafts (234). Second-generation HLA-A2-CAR-Tregs designed by another research team to have enhanced suppressive potential upon specific HLA-A2 activation effectively alleviated alloimmune-induced skin damage without inducing cytotoxicity (251, 252).
CAR-Treg therapy holds promising prospects for the treatment of GVHD. Sangamo Therapeutics initiated the first clinical trial (NCT04817774) of CAR-Tregs, known as TX200. TX200 was specifically developed to mitigate immune-mediated rejection in kidney transplantation with HLA-A2 mismatch, particularly in patients with end-stage renal disease. In March 2022, TX200 was administered to a patient for the first time as part of the phase 1/2 STEADFAST clinical study (253). Additionally, Quell Therapeutics is currently conducting a clinical trial (NCT05234190) of QEL-001, a self-targeting CAR-T regulatory cell therapy that specifically targets HLA-A2, to mitigate transplant rejection in patients after liver transplantation.
6.2.2 Inflammatory bowel disease (IBD)
IBDs, clinically consisting of ulcerative colitis (UC) and Crohn’s disease (CD), are chronic inflammatory disorders of the gastrointestinal tract (254). In a 2,4,6-trinitrophenol (TNP)-induced IBD mice model, the accumulation of TNP-CAR-Treg cells was observed at the inflamed colonic sites upon activation. These cells effectively suppressed the activity of Teffs and significantly improved colitis, representing an initial preclinical exploration of CAR-Treg therapy (227). The induction of the IL-23 receptor (IL-23R) plays a significant role in CD (255). Hence, IL-23R-CAR-Treg cells, which incorporate CD28 as a costimulatory domain, were used to enhance the management of CD (256).
6.2.3 T1D
T1D is a chronic autoimmune disorder, characterized by the autoimmune destruction of pancreatic β cells leading to insulin deficiency (257, 258). The current therapeutic options for T1D include insulin injections, dietary management, and exercise (259). Changes in Treg cell number and function have been observed and documented in the context of T1D (260). Notably, an increased prevalence of IFN-γ+ FoxP3+ (Th1-like) Treg cells with reduced immunosuppressive activity has been observed in the peripheral blood of individuals affected by this condition (261). Consequently, the continuous infusion of Treg cells has been suggested as a potential strategy to mitigate T1D (262–264). However, the long-term delivery of Treg cells poses challenges due to the intricate and costly manufacturing process involved in the large-scale production of these cells (181).
Preclinical studies utilizing mouse models have successfully demonstrated effective control of T1D through the use of FoxP3-engineered islet-specific T cells (265). Expanding on this discovery, researchers are currently considering the application of CAR-Treg cells targeting distinct structural domains for the treatment of other forms of diabetes. One approach involves redirecting T effector cells to Tregs by employing FoxP3 engineering while simultaneously utilizing CAR technology to specifically target T cells to insulin, thereby reducing autoimmune attacks on pancreatic beta cells and preserving insulin production in patients with diabetes. The resulting insulin-specific CAR-Treg cells exhibit enduring functionality and sustained effects (266). Moreover, by incorporating a FITC-binding domain into the CAR structure, antigen-specific monoclonal CAR-Treg cells can be designed to selectively target desired antigens using FITC-conjugated corresponding antibodies. This innovative strategy holds promise for addressing immune rejection associated with allogeneic islet transplantation and ultimately ameliorating T1D (267).
6.2.4 Multiple sclerosis
The recognition of self-antigens by autoreactive T cells targeting myelin epitopes contributes to the pathogenesis of MS (268). Individuals with MS exhibit increases in the populations of Th1-like Treg cells, which exhibit impaired suppressive function. The generation of these cells can be regulated by the PI3K/AKT/FOXO signaling pathway, and inhibiting this pathway therefore has the potential to decrease the quantity of Th1-like Treg cells and restore their suppressive capabilities (269, 270). By utilizing a lentiviral vector system, CD4+ T cells were modified to express a CAR targeting myelin oligodendrocyte glycoprotein (MOG), as well as the FoxP3 gene, facilitating the desired expression in the cells. In murine experiments involving MOG-CAR-Treg cells, these cells migrated to the central nervous system, resulting in decreased levels of IL-12 and IFN-γ, thereby leading to long-term suppression of experimental autoimmune encephalomyelitis (183).
Overall, CAR-T and CAR-Treg therapies show significant potential for the treatment of multiple sclerosis. For example, researchers have utilized KYV-101, a fully humanized CD19 CAR-T cell therapy developed by Kyverna, to treat patients with multiple sclerosis. Treatment data indicate that this therapy exhibits tolerable safety and promising efficacy in progressive MS, including patients for whom conventional antibody-mediated B-cell depletion fails (271). By precisely targeting immune cells and mitigating pathological immune responses, these therapies are expected to alleviate symptoms and enhance the quality of life for patients.
6.2.5 Rheumatoid arthritis
An increased presence of Th17-like Treg cells (Treg cells with impaired suppressive function) has been observed in the inflamed joints of RA patients and may be associated with persistent arthritis (272, 273). CD4+ T cells were modified to express a CAR targeting type II collagen and the FoxP3 gene. These CAR-Treg cells effectively suppressed ovalbumin (OVA)-induced arthritis by inhibiting the immune response to OVA (274). Furthermore, antigen-specific CAR-Tregs targeting citrullinated vimentin (CV) can be engineered for the treatment of RA. CV is a key autoantigen implicated in the pathogenesis of RA, as it is commonly found in the synovial tissue of patients and is associated with the autoimmune response driving the disease. By specifically targeting CV, CAR-Tregs can potentially modulate this immune response, reducing the inflammation and joint damage characteristic of RA (275).
6.2.6 Other autoimmune diseases
CAR-Treg therapy has the potential to treat a variety of autoimmune diseases in addition to those mentioned above. For example, autoimmune liver disease (AILD) is characterized by persistent immune-mediated inflammation of the liver, loss of immune tolerance to hepatocytes and bile duct epithelial cells, elevated serum total IgG, and the presence of circulating autoantibodies (235). Currently, there is no curative treatment for AILD, and patients need lifelong medication to manage liver inflammation and prevent bile duct injury (236). Two AILD subtypes, PBC and AIH type 2, are characterized by distinct self-antigens, making CAR-Treg therapy targeting these antigens a promising approach for treatment. CAR-Treg technology for AILD treatment is still in the clinical research phase (NCT05234190). This phase I/II single-arm, open-label clinical trial targets the HLA-A2 antigen. When these CAR-Tregs recognize the HLA-A2 antigen on the transplanted liver, they become activated and work to induce and maintain immune tolerance to the transplanted organ.
Although CAR-Treg therapy shows potential for treating autoimmune diseases, clinical investigation of such applications is still in its infancy, several obstacles remain to be addressed, such as CAR-Treg depletion. Nonetheless, preliminary evidence suggests their potential as a future therapeutic choice. Future research should aim to evaluate the safety, effectiveness, and long-term impacts of CAR-Treg therapies and to fully scope their potential in managing autoimmune diseases (276).
6.3 Application of CAAR-T-cell therapy in autoimmune diseases
Another recent area of exploration within the field of CAR-T cell and CAR-Treg therapy for autoimmune disease treatment involves modifying CAR-T cells to proficiently eliminate self-reactive B lymphocytes. Of particular interest is a novel engineered T-cell construct, termed the chimeric autoantibody receptor (CAAR). In contrast to CAR-T cells, CAAR-T cells possess an extracellular domain derived from autoantigen. This unique feature enables CAAR-T cells to identify and eradicate self-reactive B-cell receptor (BCR)-expressing B lymphocytes, granting them the ability to specifically target this population (229). Although the investigation of CAAR-T cells is still in its initial phases, it represents an enticing strategy for treating autoimmune diseases.
Pemphigus vulgaris (PV) is a severe blistering disease affecting the skin and mucous membranes caused by autoantibodies targeting desmoglein proteins (277). Therefore, the presence of anti-desmoglein BCR on memory B cells contributes to PV pathogenesis (278, 279). Recent studies have demonstrated that cell-based therapies targeting anti-desmoglein 3 (Dsg3) antibodies can be employed for treating PV. Compared with CD19 CAR-T cells, chimeric CAAR-T cells with Dsg3 extracellular domains have shown superior and more durable therapeutic effects. However, they are still associated with several adverse effects, such as CRS (280). Despite being in its nascent phase, CAAR-T-cell technology has exhibited promise in preclinical investigations and initial clinical trials. Further research endeavors and clinical studies will facilitate a comprehensive assessment and refinement of the therapeutic efficacy, safety, and long-term sustainability of this technology.
7 Discussion
CAR-T cell therapy has revolutionized the landscape of cancer treatment, demonstrating remarkable efficacy in hematologic malignancies and showing promising potential in autoimmune diseases. However, significant challenges remain, particularly in the treatment of solid tumors. The evolution of CAR-T technology, from first-generation to fifth-generation CARs, highlights ongoing efforts to enhance efficacy, safety, and specificity. To overcome these challenges, current research is focusing on discovering new antigen targets, optimizing CAR designs, and developing multitargeting strategies. Future research still needs to focus on overcoming the immunosuppressive tumor microenvironment, improving CAR-T cell persistence, and minimizing off-target effects. Ultimately, the continued refinement and expansion of CAR-T cell therapy hold great promise for transforming oncological and immunological treatments, offering new hope for patients with challenging diseases.
Author contributions
YK: Conceptualization, Formal analysis, Writing – original draft, Writing – review & editing. JL: Formal analysis, Investigation, Validation, Writing – review & editing. XZ: Formal analysis, Investigation, Validation, Writing – review & editing. YW: Conceptualization, Formal analysis, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. LC: Conceptualization, Funding acquisition, Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Major Program of the National Natural Science Foundation of China (No. 82293635), the National Key Research and Development Program of China (No. 2023YFC2306401) and the National Natural Science Foundation of China (No. 82173823 and No. 82471823).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
BCMA: (B-cell Maturation Antigen) A protein found on the surface of multiple myeloma cells. It is a common target for CAR-T cell therapies aimed at treating multiple myeloma
CAR: (Chimeric Antigen Receptor)A genetically engineered receptor introduced into T cells to help them recognize and attack cancer cells. CAR-T cell therapy is a breakthrough in cancer immunotherapy
CRISPR/Cas9: A gene-editing technology used to modify genes within organisms. In CAR-T therapies, it is used to improve the efficacy and persistence of T cells by knocking out genes like PD-1
CRS: (Cytokine Release Syndrome) A potentially life-threatening side effect of CAR-T cell therapy characterized by a severe immune response, where large numbers of cytokines are released into the blood
GVHD: (Graft-versus-Host Disease) A condition where transplanted immune cells (like CAR-T cells) attack the recipient's tissues, causing severe complications. Allogeneic CAR-T therapies aim to mitigate this risk. ICANS (Immune Effector Cell-Associated Neurotoxicity Syndrome), A neurological side effect associated with CAR-T cell therapies, causing symptoms ranging from confusion to seizures
IL: (Interleukin) A group of cytokines (immune system proteins) that play various roles in regulating immune responses. In CAR-T therapies, IL-12 is often used to enhance the anti-tumor activity of the modified T cells
MHC: (Major Histocompatibility Complex) A set of proteins displayed on cell surfaces that help the immune system recognize foreign substances. CAR-T cells work independently of MHC, allowing them to target tumor cells more broadly
NK Cells: (Natural Killer Cells) A type of immune cell involved in killing virus-infected and tumor cells. CAR-NK cell therapies represent an alternative to CAR-T cell therapies with fewer side effects like GVHD
PD-1: (Programmed Cell Death Protein 1) A checkpoint protein on T cells that regulates immune response by preventing the activation of T cells, thus reducing their ability to attack cancer cells. Blocking PD-1 enhances the effectiveness of CAR-T therapies
TME: (Tumor Microenvironment) The environment around a tumor, including various cells, blood vessels, and signaling molecules. The TME can inhibit CAR-T cell efficacy, making it a significant barrier to successful immunotherapy
References
1. Zhao Z, Chen Y, Francisco NM, Zhang Y, Wu M. The application of CAR-T cell therapy in hematological Malignancies: advantages and challenges. Acta Pharm Sin B. (2018) 8:539–51. doi: 10.1016/j.apsb.2018.03.001
2. Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J, et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. (2014) 4:3571. doi: 10.1038/srep03571
3. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. (2019) 18:125. doi: 10.1186/s12943-019-1057-4
4. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
5. Duan Y, Chen R, Huang Y, Meng X, Chen J, Liao C, et al. Tuning the ignition of CAR: optimizing the affinity of scFv to improve CAR-T therapy. Cell Mol Life Sci. (2021) 79:14. doi: 10.1007/s00018-021-04089-x
6. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. (2004) 173:7647–53. doi: 10.4049/jimmunol.173.12.7647
7. Xia Z, Jin Q, Long Z, He Y, Liu F, Sun C, et al. Targeting overexpressed antigens in glioblastoma via CAR T cells with computationally designed high-affinity protein binders. Nat BioMed Eng. (2024) 8:1634–50. doi: 10.1038/s41551-024-01258-8
8. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
9. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. biomark Res. (2017) 5:22. doi: 10.1186/s40364-017-0102-y
10. Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. (2020) 13:86. doi: 10.1186/s13045-020-00910-5
11. Guedan S, Calderon H, Posey AD Jr., Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. (2019) 12:145–56. doi: 10.1016/j.omtm.2018.12.009
12. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
13. Elazar A, Chandler NJ, Davey AS, Weinstein JY, Nguyen JV, Trenker R, et al. De novo-designed transmembrane domains tune engineered receptor functions. Elife. (2022) 11:e75660. doi: 10.7554/eLife.75660
14. Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. (2002) 20:70–5. doi: 10.1038/nbt0102-70
15. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. (2014) 257:107–26. doi: 10.1111/imr.2013.257.issue-1
16. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:712. doi: 10.1016/j.immuni.2016.02.023
17. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
18. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol Rev. (2019) 290:127–47. doi: 10.1111/imr.v290.1
19. Hamilton JR, Chen E, Perez BS, Sandoval Espinoza CR, Kang MH, Trinidad M, et al. In vivo human T cell engineering with enveloped delivery vehicles. Nat Biotechnol. (2024) 42:1684–92. doi: 10.1038/s41587-023-02085-z
20. DeWitt MA, Corn JE, Carroll D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. (2017) 121-122:9–15. doi: 10.1016/j.ymeth.2017.04.003
21. Frank AM, Braun AH, Scheib L, Agarwal S, Schneider IC, Fusil F, et al. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. (2020) 4:5702–15. doi: 10.1182/bloodadvances.2020002229
22. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
23. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U.S.A. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
24. Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. (2009) 21:215–23. doi: 10.1016/j.coi.2009.02.009
25. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
26. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U.S.A. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
27. Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. (2017) 10:78. doi: 10.1186/s13045-017-0444-9
28. Heuser C, Hombach A, Lösch C, Manista K, Abken H. T-cell activation by recombinant immunoreceptors: impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. (2003) 10:1408–19. doi: 10.1038/sj.gt.3302023
29. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
30. Bretscher PA, two-step A. two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U.S.A. (1999) 96:185–90. doi: 10.1073/pnas.96.1.185
31. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. (1998) 161:2791–7. doi: 10.4049/jimmunol.161.6.2791
32. Jenkins MK, Burrell E, Ashwell JD. Antigen presentation by resting B cells. Effectiveness at inducing T cell proliferation is determined by costimulatory signals, not T cell receptor occupancy. J Immunol. (1990) 144:1585–90. doi: 10.4049/jimmunol.144.5.1585
33. Awasthi R, Maier HJ, Zhang J, Lim S. Kymriah® (tisagenlecleucel) - An overview of the clinical development journey of the first approved CAR-T therapy. Hum Vaccin Immunother. (2023) 19:2210046. doi: 10.1080/21645515.2023.2210046
34. Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. (2022) 386:640–54. doi: 10.1056/NEJMoa2116133
35. AlDallal SM. Yescarta: A new era for non-hodgkin lymphoma patients. Cureus. (2020) 12:e11504. doi: 10.7759/cureus.11504
36. Lu J, Jiang G. The journey of CAR-T therapy in hematological Malignancies. Mol Cancer. (2022) 21:194. doi: 10.1186/s12943-022-01663-0
37. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
38. García-Sancho AM, Cabero A, Gutiérrez NC. Treatment of Relapsed or Refractory Diffuse Large B-Cell Lymphoma: New Approved Options. J Clin Med. (2023) 13:70. doi: 10.3390/jcm13010070
39. Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
40. Ciltacabtagene autoleucel (Carvykti) for multiple myeloma. Med Lett Drugs Ther. (2022) 64:e188–9.
41. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. (2003) 3:939–51. doi: 10.1038/nri1248
42. Hu L, Charwudzi A, Li Q, Zhu W, Tao Q, Xiong S, et al. Anti-CD19 CAR-T cell therapy bridge to HSCT decreases the relapse rate and improves the long-term survival of R/R B-ALL patients: a systematic review and meta-analysis. Ann Hematol. (2021) 100:1003–12. doi: 10.1007/s00277-021-04451-w
43. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
44. Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. (2005) 12:933–41. doi: 10.1016/j.ymthe.2005.04.016
45. Poorebrahim M, Sadeghi S, Fakhr E, Abazari MF, Poortahmasebi V, Kheirollahi A, et al. Production of CAR T-cells by GMP-grade lentiviral vectors: latest advances and future prospects. Crit Rev Clin Lab Sci. (2019) 56:393–419. doi: 10.1080/10408363.2019.1633512
46. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. (2010) 18:413–20. doi: 10.1038/mt.2009.210
47. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
48. Tang XY, Sun Y, Zhang A, Hu GL, Cao W, Wang DH, et al. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: a non-randomised, open-label phase I trial protocol. BMJ Open. (2016) 6:e013904. doi: 10.1136/bmjopen-2016-013904
49. Enblad G, Karlsson H, Gammelgård G, Wenthe J, Lövgren T, Amini RM, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T Cells for lymphoma and leukemia. Clin Cancer Res. (2018) 24:6185–94. doi: 10.1158/1078-0432.CCR-18-0426
50. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
51. Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-hodgkin’s lymphomas. Mol Ther. (2018) 26:2727–37. doi: 10.1016/j.ymthe.2018.09.009
52. Künkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, et al. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol Res. (2015) 3:368–79. doi: 10.1158/2326-6066.CIR-14-0200
53. Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation. Mol Ther. (2013) 21:2268–77. doi: 10.1038/mt.2013.192
54. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
55. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
56. Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. (2007) 13:1440–9. doi: 10.1038/nm1676
57. Malaer JD, Marrufo AM, Mathew PA. 2B4 (CD244, SLAMF4) and CS1 (CD319, SLAMF7) in systemic lupus erythematosus and cancer. Clin Immunol. (2019) 204:50–6. doi: 10.1016/j.clim.2018.10.009
58. Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current challenges and emerging opportunities of CAR-T cell therapies. J Control Release. (2020) 319:246–61. doi: 10.1016/j.jconrel.2019.12.047
59. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
60. Smith JW. Apheresis techniques and cellular immunomodulation. Ther Apher. (1997) 1:203–6. doi: 10.1111/j.1744-9987.1997.tb00137.x
61. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev. (2019) 290:60–84. doi: 10.1111/imr.v290.1
62. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. (2018) 53:164–81. doi: 10.1016/j.copbio.2018.01.025
63. Lin H, Cheng J, Mu W, Zhou J, Zhu L. Advances in universal CAR-T cell therapy. Front Immunol. (2021) 12:744823. doi: 10.3389/fimmu.2021.744823
64. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. [amp]]lsquo;Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
65. DiNofia AM, Grupp SA. Will allogeneic CAR T cells for CD19(+) Malignancies take autologous CAR T cells ‘off the shelf’? Nat Rev Clin Oncol. (2021) 18:195–6. doi: 10.1038/s41571-021-00485-1
66. Köhl U, Arsenieva S, Holzinger A, Abken H. CAR T cells in trials: recent achievements and challenges that remain in the production of modified T cells for clinical applications. Hum Gene Ther. (2018) 29:559–68. doi: 10.1089/hum.2017.254
67. Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. (2018) 33:547–62. doi: 10.1016/j.ccell.2018.03.012
68. Asmamaw Dejenie T, Tiruneh GMM, Dessie Terefe G, Tadele Admasu F, Wale Tesega W, Chekol Abebe E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum Vaccin Immunother. (2022) 18:2114254. doi: 10.1080/21645515.2022.2114254
69. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. (2019) 20:31–42. doi: 10.1016/S1470-2045(18)30864-7
70. Vairy S, Garcia JL, Teira P, Bittencourt H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des Devel Ther. (2018) 12:3885–98. doi: 10.2147/DDDT.S138765
71. O’Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin Cancer Res. (2019) 25:1142–6. doi: 10.1158/1078-0432.CCR-18-2035
72. Anderson LD Jr. Idecabtagene vicleucel (ide-cel) CAR T-cell therapy for relapsed and refractory multiple myeloma. Future Oncol. (2022) 18:277–89. doi: 10.2217/fon-2021-1090
73. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
74. Iacoboni G, Rejeski K, Villacampa G, van Doesum JA, Chiappella A, Bonifazi F, et al. Real-world evidence of brexucabtagene autoleucel for the treatment of relapsed or refractory mantle cell lymphoma. Blood Adv. (2022) 6:3606–10. doi: 10.1182/bloodadvances.2021006922
75. Mullard A. FDA approves second BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discovery. (2022) 21:249. doi: 10.1038/d41573-022-00048-8
76. Chen YJ, Abila B, Mostafa Kamel Y. CAR-T: what is next? Cancers (Basel). (2023) 15:663. doi: 10.3390/cancers15030663
77. Hay KA, Turtle CJ. Chimeric antigen receptor (CAR) T cells: lessons learned from targeting of CD19 in B-cell Malignancies. Drugs. (2017) 77:237–45. doi: 10.1007/s40265-017-0690-8
78. Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song SX, Ma CQ. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol Res. (2022) 175:106036. doi: 10.1016/j.phrs.2021.106036
79. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. (2019) 568:112–6. doi: 10.1038/s41586-019-1054-1
80. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
81. Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell Malignancies: a phase 1 trial. Nat Med. (2021) 27:1419–31. doi: 10.1038/s41591-021-01436-0
82. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
83. Hwang MS, Mog BJ, Douglass J, Pearlman AH, Hsiue EH, Paul S, et al. Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc Natl Acad Sci U.S.A. (2021) 118:e2022410118. doi: 10.1073/pnas.2022410118
84. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
85. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
86. Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological Malignancies. J Hematol Oncol. (2018) 11:91. doi: 10.1186/s13045-018-0629-x
87. Kumar J, Kumar R, Kumar Singh A, Tsakem EL, Kathania M, Riese MJ, et al. Deletion of Cbl-b inhibits CD8(+) T-cell exhaustion and promotes CAR T-cell function. J Immunother Cancer. (2021) 9:e001688. doi: 10.1136/jitc-2020-001688
88. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
89. Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T cell, exhaustion, and tumor immunity. Immunol Med. (2020) 43:1–9. doi: 10.1080/25785826.2019.1698261
90. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. (2004) 172:104–13. doi: 10.4049/jimmunol.172.1.104
91. Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. (2011) 71:4617–27. doi: 10.1158/0008-5472.CAN-11-0422
92. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. (2019) 8:e1049. doi: 10.1002/cti2.2019.8.issue-5
93. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
94. Duan D, Wang K, Wei C, Feng D, Liu Y, He Q, et al. The BCMA-targeted fourth-generation CAR-T cells secreting IL-7 and CCL19 for therapy of refractory/recurrent multiple myeloma. Front Immunol. (2021) 12:609421. doi: 10.3389/fimmu.2021.609421
95. He C, Zhou Y, Li Z, Farooq MA, Ajmal I, Zhang H, et al. Co-expression of IL-7 improves NKG2D-based CAR T cell therapy on prostate cancer by enhancing the expansion and inhibiting the apoptosis and exhaustion. Cancers (Basel). (2020) 12(7):1969. doi: 10.3390/cancers12071969
96. Yamamoto TN, Lee PH, Vodnala SK, Gurusamy D, Kishton RJ, Yu Z, et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J Clin Invest. (2019) 129:1551–65. doi: 10.1172/JCI121491
97. Ping Y, Li F, Nan S, Zhang D, Shi X, Shan J, et al. Augmenting the effectiveness of CAR-T cells by enhanced self-delivery of PD-1-neutralizing scFv. Front Cell Dev Biol. (2020) 8:803. doi: 10.3389/fcell.2020.00803
98. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
99. Steentoft C, Migliorini D, King TR, Mandel U, June CH, Posey AD Jr. Glycan-directed CAR-T cells. Glycobiology. (2018) 28:656–69. doi: 10.1093/glycob/cwy008
100. Lindo L, Wilkinson LH, Hay KA. Befriending the hostile tumor microenvironment in CAR T-cell therapy. Front Immunol. (2020) 11:618387. doi: 10.3389/fimmu.2020.618387
101. Katsarou A, Sjöstrand M, Naik J, Mansilla-Soto J, Kefala D, Kladis G, et al. Combining a CAR and a chimeric costimulatory receptor enhances T cell sensitivity to low antigen density and promotes persistence. Sci Transl Med. (2021) 13:eabh1962. doi: 10.1126/scitranslmed.abh1962
102. Liao Q, Mao Y, He H, Ding X, Zhang X, Xu J. PD-L1 chimeric costimulatory receptor improves the efficacy of CAR-T cells for PD-L1-positive solid tumors and reduces toxicity in vivo. biomark Res. (2020) 8:57. doi: 10.1186/s40364-020-00237-w
103. Han X, Bryson PD, Zhao Y, Cinay GE, Li S, Guo Y, et al. Masked chimeric antigen receptor for tumor-specific activation. Mol Ther. (2017) 25:274–84. doi: 10.1016/j.ymthe.2016.10.011
104. Yan ZX, Li L, Wang W, OuYang BS, Cheng S, Wang L, et al. Clinical efficacy and tumor microenvironment influence in a dose-escalation study of anti-CD19 chimeric antigen receptor T cells in refractory B-cell non-hodgkin’s lymphoma. Clin Cancer Res. (2019) 25:6995–7003. doi: 10.1158/1078-0432.CCR-19-0101
105. Ren B, Cui M, Yang G, Wang H, Feng M, You L, et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer. (2018) 17:108. doi: 10.1186/s12943-018-0858-1
106. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
107. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-cells targeting the integrin αnte and co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid Malignancies. Cancers (Basel). (2019) 11(5):674. doi: 10.3390/cancers11050674
108. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. (2020) 50:712–24. doi: 10.1002/eji.201948457
109. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
110. Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front Immunol. (2021) 12:628906. doi: 10.3389/fimmu.2021.628906
111. Zhao Z, Xiao X, Saw PE, Wu W, Huang H, Chen J, et al. Chimeric antigen receptor T cells in solid tumors: a war against the tumor microenvironment. Sci China Life Sci. (2020) 63:180–205. doi: 10.1007/s11427-019-9665-8
112. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of Malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. (2013) 11:187. doi: 10.1186/1479-5876-11-187
113. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. (2005) 307:58–62. doi: 10.1126/science.1104819
114. Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. (2016) 59:340–8. doi: 10.1007/s11427-016-5027-4
115. Yong SB, Chung JY, Song Y, Kim J, Ra S, Kim YH. Non-viral nano-immunotherapeutics targeting tumor microenvironmental immune cells. Biomaterials. (2019) 219:119401. doi: 10.1016/j.biomaterials.2019.119401
116. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T&xa0;cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther - Oncolytics. (2022) 25:69–77. doi: 10.1016/j.omto.2022.03.009
117. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
118. Zhang SY, Song XY, Li Y, Ye LL, Zhou Q, Yang WB. Tumor-associated macrophages: A promising target for a cancer immunotherapeutic strategy. Pharmacol Res. (2020) 161:105111. doi: 10.1016/j.phrs.2020.105111
119. Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. (2022) 21:184. doi: 10.1186/s12943-022-01657-y
120. Bradley JD, Hu C, Komaki RR, Masters GA, Blumenschein GR, Schild SE, et al. Long-term results of NRG oncology RTOG 0617: standard- versus high-dose chemoradiotherapy with or without cetuximab for unresectable stage III non-small-cell lung cancer. J Clin oncology: Off J Am Soc Clin Oncol. (2020) 38:706–14. doi: 10.1200/JCO.19.01162
121. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. (2019) 36:471–82. doi: 10.1016/j.ccell.2019.09.006
122. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-n receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
123. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
124. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned erbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. (2015) 75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159
125. Santamaria-Alza Y, Vasquez G. Are chimeric antigen receptor T cells (CAR-T cells) the future in immunotherapy for autoimmune diseases? Inflammation Res. (2021) 70:651–63. doi: 10.1007/s00011-021-01470-1
126. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
127. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discovery. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
128. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
129. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
130. Jan M, Scarfò I, Larson RC, Walker A, Schmidts A, Guirguis AA, et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci Transl Med. (2019) 11(499):eaau5907. doi: 10.1126/scitranslmed.aau5907
131. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
132. Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. (2005) 105:4247–54. doi: 10.1182/blood-2004-11-4564
133. Juillerat A, Tkach D, Busser BW, Temburni S, Valton J, Duclert A, et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. (2019) 19:44. doi: 10.1186/s12896-019-0537-3
134. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. (2019) 11(499):eaau5907. doi: 10.1126/scitranslmed.aau5907
135. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. (2015) 350:aab4077. doi: 10.1126/science.aab4077
136. Lin YJ, Mashouf LA, Lim M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front Immunol. (2022) 13:817296. doi: 10.3389/fimmu.2022.817296
137. Ramello MC, Haura EB, Abate-Daga D. CAR-T cells and combination therapies: What’s next in the immunotherapy revolution? Pharmacol Res. (2018) 129:194–203. doi: 10.1016/j.phrs.2017.11.035
138. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22:20. doi: 10.1186/s12943-023-01723-z
139. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292
140. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. (2013) 21:2087–101. doi: 10.1038/mt.2013.185
141. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. (2017) 129:1039–41. doi: 10.1182/blood-2016-09-738245
142. Gomez S, Tabernacki T, Kobyra J, Roberts P, Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin Cancer Biol. (2020) 65:99–113. doi: 10.1016/j.semcancer.2019.12.019
143. Lotfinejad P, Kazemi T, Mokhtarzadeh A, Shanehbandi D, Jadidi Niaragh F, Safaei S, et al. PD-1/PD-L1 axis importance and tumor microenvironment immune cells. Life Sci. (2020) 259:118297. doi: 10.1016/j.lfs.2020.118297
144. Shergold AL, Millar R, Nibbs RJB. Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol Res. (2019) 145:104258. doi: 10.1016/j.phrs.2019.104258
145. Lind H, Gameiro SR, Jochems C, Donahue RN, Strauss J, Gulley JM, et al. Dual targeting of TGF-e and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: status of preclinical and clinical advances. J Immunother Cancer. (2020) 8(1):e000433. doi: 10.1136/jitc-2019-000433
146. Palomba ML, Qualls D, Monette S, Sethi S, Dogan A, Roshal M, et al. CD19-directed chimeric antigen receptor T cell therapy in Waldenström macroglobulinemia: a preclinical model and initial clinical experience. J Immunother Cancer. (2022) 10(2):e004128. doi: 10.1136/jitc-2021-004128
147. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
148. Song W, Zhang M. Use of CAR-T cell therapy, PD-1 blockade, and their combination for the treatment of hematological Malignancies. Clin Immunol. (2020) 214:108382. doi: 10.1016/j.clim.2020.108382
149. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
150. Zhu H, You Y, Shen Z, Shi L. EGFRvIII-CAR-T cells with PD-1 knockout have improved anti-glioma activity. Pathol Oncol Res. (2020) 26:2135–41. doi: 10.1007/s12253-019-00759-1
151. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
152. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:737. doi: 10.1038/s41598-017-00462-8
153. Safarzadeh Kozani P, Shokrgozar MA, Evazalipour M, Roudkenar MH. CRISPR/Cas9-medaited knockout of endogenous T-cell receptor in Jurkat cells and generation of NY-ESO-1-specific T cells: An in vitro study. Int Immunopharmacol. (2022) 110:109055. doi: 10.1016/j.intimp.2022.109055
154. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) 367(6481):eaba7365. doi: 10.1126/science.aba7365
155. Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. (2020) 136:1407–18. doi: 10.1182/blood.2020005185
156. Shi T, Song X, Wang Y, Liu F, Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front Immunol. (2020) 11:683. doi: 10.3389/fimmu.2020.00683
157. Marelli G, Howells A, Lemoine NR, Wang Y. Oncolytic viral therapy and the immune system: A double-edged sword against cancer. Front Immunol. (2018) 9:866. doi: 10.3389/fimmu.2018.00866
158. Rosewell Shaw A, Suzuki M. Oncolytic viruses partner with T-cell therapy for solid tumor treatment. Front Immunol. (2018) 9:2103. doi: 10.3389/fimmu.2018.02103
159. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. (2018) 3(7):e99573. doi: 10.1172/jci.insight.99573
160. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. (2014) 74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697
161. Yang S, Gao H. Nanoparticles for modulating tumor microenvironment to improve drug delivery and tumor therapy. Pharmacol Res. (2017) 126:97–108. doi: 10.1016/j.phrs.2017.05.004
162. Mi Y, Hagan CT4, Vincent BG, Wang AZ. Emerging nano-/microapproaches for cancer immunotherapy. Adv Sci (Weinh). (2019) 6:1801847. doi: 10.1002/advs.201801847
163. Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. (2020) 20:1578–89. doi: 10.1021/acs.nanolett.9b04246
164. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science. (2022) 375(6576):91–6. doi: 10.1126/science.abm059
165. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science. (2022) 375:91–6. doi: 10.1126/science.abm0594
166. Gao TA, Chen YY. T cells to fix a broken heart. Science. (2022) 375:23–4. doi: 10.1126/science.abn0851
167. Chehrazi-Raffle A, Budde LE, Pal SK. Boosting CAR T cells with anti-tumor mRNA vaccines. Nat Med. (2023) 29:2711–2. doi: 10.1038/s41591-023-02623-x
168. Deng Z, Tian Y, Song J, An G, Yang P. mRNA vaccines: the dawn of a new era of cancer immunotherapy. Front Immunol. (2022) 13:887125. doi: 10.3389/fimmu.2022.887125
169. Elkord E. Thymus-derived, peripherally derived, and in vitro-induced T regulatory cells. Front Immunol. (2014) 5:17. doi: 10.3389/fimmu.2014.00017
170. McGovern JL, Wright GP, Stauss HJ. Engineering specificity and function of therapeutic regulatory T cells. Front Immunol. (2017) 8:1517. doi: 10.3389/fimmu.2017.01517
171. Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, et al. Chimeric antigen receptor (CAR) Treg: A promising approach to inducing immunological tolerance. Front Immunol. (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
172. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
173. Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology. (2008) 124:13–22. doi: 10.1111/j.1365-2567.2008.02813.x
174. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
175. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
176. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
177. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. (2016) 127:1044–51. doi: 10.1182/blood-2015-06-653667
178. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
179. Wing JB, Sakaguchi S. TCR diversity and Treg cells, sometimes more is more. Eur J Immunol. (2011) 41:3097–100. doi: 10.1002/eji.201142115
180. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T regulatory (CAR-Treg) cells: engineering and applications. Biomedicines. (2022) 10(2):287. doi: 10.3390/biomedicines10020287
181. Abou-El-Enein M, Römhild A, Kaiser D, Beier C, Bauer G, Volk HD, et al. Good Manufacturing Practices (GMP) manufacturing of advanced therapy medicinal products: a novel tailored model for optimizing performance and estimating costs. Cytotherapy. (2013) 15:362–83. doi: 10.1016/j.jcyt.2012.09.006
182. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. (2006) 203:1693–700. doi: 10.1084/jem.20060468
183. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflamm. (2012) 9:112. doi: 10.1186/1742-2094-9-112
184. Dai X, Park JJ, Du Y, Kim HR, Wang G, Errami Y, et al. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat Methods. (2019) 16:247–54. doi: 10.1038/s41592-019-0329-7
185. Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia. (2017) 31:186–94. doi: 10.1038/leu.2016.180
186. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
187. Arroyo Hornero R, Betts GJ, Sawitzki B, Vogt K, Harden PN, Wood KJ. CD45RA distinguishes CD4+CD25+CD127-/low TSDR demethylated regulatory T cell subpopulations with differential stability and susceptibility to tacrolimus-mediated inhibition of suppression. Transplantation. (2017) 101:302–9. doi: 10.1097/TP.0000000000001278
188. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discovery. (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
189. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant. (2017) 17:2945–54. doi: 10.1111/ajt.14415
190. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
191. Kiessling R, Klein E, Pross H, Wigzell H. Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. (1975) 5:117–21. doi: 10.1002/eji.1830050209
192. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-like receptors (KIRs): their role in NK cell modulation and developments leading to their clinical exploitation. Front Immunol. (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
193. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med. (2005) 202:1001–12. doi: 10.1084/jem.20051143
194. Gunesch JT, Angelo LS, Mahapatra S, Deering RP, Kowalko JE, Sleiman P, et al. Genome-wide analyses and functional profiling of human NK cell lines. Mol Immunol. (2019) 115:64–75. doi: 10.1016/j.molimm.2018.07.015
195. Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res. (2015) 62:341–56. doi: 10.1007/s12026-015-8664-y
196. Khawar MB, Sun H. CAR-NK cells: from natural basis to design for kill. Front Immunol. (2021) 12:707542. doi: 10.3389/fimmu.2021.707542
197. Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol. (2015) 12:292–302. doi: 10.1038/cmi.2014.91
198. Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol Sin. (2015) 36:1191–9. doi: 10.1038/aps.2015.41
199. Wu J, Mishra HK, Walcheck B. Role of ADAM17 as a regulatory checkpoint of CD16A in NK cells and as a potential target for cancer immunotherapy. J Leukoc Biol. (2019) 105:1297–303. doi: 10.1002/JLB.2MR1218-501R
200. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
201. Fang F, Xie S, Chen M, Li Y, Yue J, Ma J, et al. Advances in NK cell production. Cell Mol Immunol. (2022) 19:460–81. doi: 10.1038/s41423-021-00808-3
202. Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical cord blood natural killer cells, their characteristics, and potential clinical applications. Front Immunol. (2017) 8:329. doi: 10.3389/fimmu.2017.00329
203. Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers (Basel). (2019) 11(6):769. doi: 10.3390/cancers11060769
204. Albinger N, Hartmann J, Ullrich E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. (2021) 28:513–27. doi: 10.1038/s41434-021-00246-w
205. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ Jr., Guedan S. CAR-T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. (2020) 11:1109. doi: 10.3389/fimmu.2020.01109
206. Pang Z, Wang Z, Li F, Feng C, Mu X. Current progress of CAR-NK therapy in cancer treatment. Cancers (Basel). (2022) 14(17):4318. doi: 10.3390/cancers14174318
207. Meazza R, Azzarone B, Orengo AM, Ferrini S. Role of common-gamma chain cytokines in NK cell development and function: perspectives for immunotherapy. J BioMed Biotechnol. (2011) 2011:861920. doi: 10.1155/bmri.v2011.1
208. Wrona E, Borowiec M, Potemski P. CAR-NK cells in the treatment of solid tumors. Int J Mol Sci. (2021) 22(11):5899. doi: 10.3390/ijms22115899
209. Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1yificedddcs glioblastoma. J Immunother. (2015) 38:197–210. doi: 10.1097/CJI.0000000000000082
210. Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother. (2015) 64:225–35. doi: 10.1007/s00262-014-1629-5
211. Davis HE, Morgan JR, Yarmush ML. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys Chem. (2002) 97:159–72. doi: 10.1016/S0301-4622(02)00057-1
212. Dolgin E. Cancer-eating immune cells kitted out with CARs. Nat Biotechnol. (2020) 38:509–11. doi: 10.1038/s41587-020-0520-5
213. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. (2020) 10:188. doi: 10.3389/fonc.2020.00188
214. Jeannin P, Paolini L, Adam C, Delneste Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. (2018) 285:680–99. doi: 10.1111/febs.2018.285.issue-4
215. Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflammation. (2013) 2013:697972. doi: 10.1155/2013/697972
216. Bruckmeier M, Kuehnl A, Culmes M, Pelisek J, Eckstein HH. Impact of oxLDL and LPS on C-type natriuretic peptide system is different between THP-1 cells and human peripheral blood monocytic cells. Cell Physiol Biochem. (2012) 30:199–209. doi: 10.1159/000339044
217. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. (2021) 591:131–6. doi: 10.1038/s41586-021-03218-7
218. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.v233.9
219. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
220. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. (2020) 13:153. doi: 10.1186/s13045-020-00983-2
221. Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. (2017) 10:58. doi: 10.1186/s13045-017-0430-2
222. Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Núñez NG, et al. Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell. (2022) 185:1189–1207.e25. doi: 10.1016/j.cell.2022.02.021
223. Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp Hematol Oncol. (2020) 9:15. doi: 10.1186/s40164-020-00171-5
224. Ying W, Cheruku PS, Bazer FW, Safe SH, Zhou B. Investigation of macrophage polarization using bone marrow derived macrophages. J Vis Exp. (2013) 23(76):50323. doi: 10.3791/50323-v
225. Bobadilla S, Sunseri N, Landau NR. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. (2013) 20:514–20. doi: 10.1038/gt.2012.61
226. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J BioMed Sci. (2019) 26:78. doi: 10.1186/s12929-019-0568-z
227. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology. (2008) 134:2014–24. doi: 10.1053/j.gastro.2008.02.060
228. Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology. (2009) 136:1721–31. doi: 10.1053/j.gastro.2009.01.049
229. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
230. Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med. (2019) 11(482):eaav1648. doi: 10.1126/scitranslmed.aav1648
231. Jin X, Xu Q, Pu C, Zhu K, Lu C, Jiang Y, et al. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell Mol Immunol. (2021) 18:1896–903. doi: 10.1038/s41423-020-0472-1
232. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
233. Parra Sánchez AR, Voskuyl AE, van Vollenhoven RF. Treat-to-target in systemic lupus erythematosus: advancing towards its implementation. Nat Rev Rheumatol. (2022) 18:146–57. doi: 10.1038/s41584-021-00739-3
234. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant. (2017) 17:917–30. doi: 10.1111/ajt.14175
235. Hennes EM, Zeniya M, Czaja AJ, Parja A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. (2008) 48:169–76. doi: 10.1002/hep.22322
236. Richardson N, Wootton GE, Bozward AG, Oo YH. Challenges and opportunities in achieving effective regulatory T cell therapy in autoimmune liver disease. Semin Immunopathol. (2022) 44:461–74. doi: 10.1007/s00281-022-00940-w
237. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
238. Park JH, Rivière I, Gonen M, Wang X, Sénécha B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
239. Müller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Völkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - A case series with follow-up. N Engl J Med. (2024) 390:687–700. doi: 10.1056/NEJMoa2308917
240. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. (2010) 184:1840–8. doi: 10.4049/jimmunol.0902592
241. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. (2016) 165:551–65. doi: 10.1016/j.cell.2016.03.008
242. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
243. Bethge WA, Martus P, Schmitt M, Holtick U, Subklewe M, von Tresckow B, et al. GLA/DRST real-world outcome analysis of CAR T-cell therapies for large B-cell lymphoma in Germany. Blood. (2022) 140:349–58. doi: 10.1182/blood.2021015209
244. Schett G, Müller F, Taubmann J, Mackensen A, Wang W, Furie RA, et al. Advancements and challenges in CAR T cell therapy in autoimmune diseases. Nat Rev Rheumatol. (2024) 20:531–44. doi: 10.1038/s41584-024-01139-z
245. Zhang B, Wang Y, Yuan Y, Sun J, Liu L, Huang D, et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann Rheum Dis. (2021) 80:176–84. doi: 10.1136/annrheumdis-2020-217844
246. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
247. Fritsche E, Volk HD, Reinke P, Abou-El-Enein M. Toward an optimized process for clinical manufacturing of CAR-Treg cell therapy. Trends Biotechnol. (2020) 38:1099–112. doi: 10.1016/j.tibtech.2019.12.009
248. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol. (2019) 10:43. doi: 10.3389/fimmu.2019.00043
249. Qu G, Chen J, Li Y, Yuan Y, Liang R, Li B. Current status and perspectives of regulatory T cell-based therapy. J Genet Genomics. (2022) 49:599–611. doi: 10.1016/j.jgg.2022.05.005
250. Crux NB, Elahi S. Human leukocyte antigen (HLA) and immune regulation: how do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections? Front Immunol. (2017) 8:832. doi: 10.3389/fimmu.2017.00832
251. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
252. Bézie S, Charreau B, Vimond N, Lasselin J, Gérard N, Nerrière-Daguin V, et al. Human CD8+ Tregs expressing a MHC-specific CAR display enhanced suppression of human skin rejection and GVHD in NSG mice. Blood Adv. (2019) 3:3522–38. doi: 10.1182/bloodadvances.2019000411
253. Schreeb K, Culme-Seymour E, Ridha E, Dumont C, Atkinson G, Hsu B, et al. Study design: human leukocyte antigen class I molecule A(leculeemour,c antigen receptor regulatory T cells in renal transplantation. Kidney Int Rep. (2022) 7:1258–67. doi: 10.1016/j.ekir.2022.03.030
254. Seyedian SS, Nokhostin F, Malamir MD. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J Med Life. (2019) 12:113–22. doi: 10.25122/jml-2018-0075
255. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. (2006) 314:1461–3. doi: 10.1126/science.1135245
256. Powers AC. Type 1 diabetes mellitus: much progress, many opportunities. J Clin Invest. (2021) 131(8):e142242. doi: 10.1172/JCI142242
257. Gillespie KM. Type 1 diabetes: pathogenesis and prevention. Cmaj. (2006) 175:165–70. doi: 10.1503/cmaj.060244
258. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. (2018) 391:2449–62. doi: 10.1016/S0140-6736(18)31320-5
259. Beck RW, Bergenstal RM, Laffel LM, Pickup JC. Advances in technology for management of type 1 diabetes. Lancet. (2019) 394:1265–73. doi: 10.1016/S0140-6736(19)31142-0
260. Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev. (2011) 10:744–55. doi: 10.1016/j.autrev.2011.05.004
261. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol. (2011) 186:3918–26. doi: 10.4049/jimmunol.1003099
262. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. (2005) 54:92–9. doi: 10.2337/diabetes.54.1.92
263. Glisic-Milosavljevic S, Wang T, Koppen M, Kramer J, Ehlenbach S, Waukau J, et al. Dynamic changes in CD4+ CD25+(high) T cell apoptosis after the diagnosis of type 1 diabetes. Clin Exp Immunol. (2007) 150:75–82. doi: 10.1111/j.1365-2249.2007.03475.x
264. Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol. (2005) 175:3053–9. doi: 10.4049/jimmunol.175.5.3053
265. Jaeckel E, von Boehmer H, Manns MP. Antigen-specific FoxP3-transduced T-cells can control established type 1 diabetes. Diabetes. (2005) 54:306–10. doi: 10.2337/diabetes.54.2.306
266. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun. (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
267. Pierini A, Iliopoulou BP, Peiris H, Pérez-Cruz M, Baker J, Hsu K, et al. T cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight. (2017) 2(20):e92865. doi: 10.1172/jci.insight.92865
268. Compston A, Coles A. Multiple sclerosis. Lancet. (2008) 372:1502–17. doi: 10.1016/S0140-6736(08)61620-7
269. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. (2011) 17:673–5. doi: 10.1038/nm.2389
270. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. (2012) 491:554–9. doi: 10.1038/nature11581
271. Fischbach F, Richter J, Pfeffer LK, Fehse B, Berger SC, Reinhardt S, et al. CD19-targeted chimeric antigen receptor T cell therapy in two patients with multiple sclerosis. Med. (2024) 5:550–558.e2. doi: 10.1016/j.medj.2024.03.002
272. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. doi: 10.1038/nm.3432
273. van Roon JA, Hartgring SA, van der Wurff-Jacobs KM, Bijlsma JW, Lafeber FP. Numbers of CD25+Foxp3+ T cells that lack the IL-7 receptor are increased intra-articularly and have impaired suppressive function in RA patients. Rheumatol (Oxford). (2010) 49:2084–9. doi: 10.1093/rheumatology/keq237
274. Sun G, Hou Y, Gong W, Liu S, Li J, Yuan Y, et al. Adoptive induced antigen-specific treg cells reverse inflammation in collagen-induced arthritis mouse model. Inflammation. (2018) 41:485–95. doi: 10.1007/s10753-017-0704-4
275. Xiao Q, Li X, Li Y, Wu Z, Xu C, Chen Z, et al. Biological drug and drug delivery-mediated immunotherapy. Acta Pharm Sin B. (2021) 11:941–60. doi: 10.1016/j.apsb.2020.12.018
276. Riet T, Chmielewski M. Regulatory CAR-T cells in autoimmune diseases: Progress and current challenges. Front Immunol. (2022) 13:934343. doi: 10.3389/fimmu.2022.934343
277. Malik AM, Tupchong S, Huang S, Are A, Hsu S, Motaparthi K. An updated review of pemphigus diseases. Medicina (Kaunas). (2021) 57(10):1080. doi: 10.3390/medicina57101080
278. Lunardon L, Tsai KJ, Propert KJ, Fett N, Stanley JR, Werth VP, et al. Adjuvant rituximab therapy of pemphigus: a single-center experience with 31 patients. Arch Dermatol. (2012) 148:1031–6. doi: 10.1001/archdermatol.2012.1522
279. Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. (2008) 128:2850–8. doi: 10.1038/jid.2008.172
Keywords: CAR-T cell therapy, solid tumors, autoimmune diseases, dual-CAR strategy, CAR-Treg
Citation: Kong Y, Li J, Zhao X, Wu Y and Chen L (2025) CAR-T cell therapy: developments, challenges and expanded applications from cancer to autoimmunity. Front. Immunol. 15:1519671. doi: 10.3389/fimmu.2024.1519671
Received: 30 October 2024; Accepted: 17 December 2024;
Published: 09 January 2025.
Edited by:
Wenxue Ma, University of California, San Diego, United StatesReviewed by:
Subhash Kumar Tripathi, Seattle Children’s Research Institute, United StatesHuifei Zheng, City of Hope National Medical Center, United States
Copyright © 2025 Kong, Li, Zhao, Wu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Chen, bGNoZW4xQHNodS5lZHUuY24=; Yanwei Wu, d3V5YW53ZWlAc2h1LmVkdS5jbg==