 Lejla Alic
Lejla Alic Kristina Dendinovic
Kristina Dendinovic Nikolina Papac-Milicevic
Nikolina Papac-Milicevic- 1Department of Medical Biochemistry, Faculty of Medicine, University of Sarajevo, Sarajevo, Bosnia and Herzegovina
- 2Department of Neurophysiology and Neuropharmacology, Center for Physiology and Pharmacology, Medical University of Vienna, Vienna, Austria
The complement system, a coordinator and facilitator of the innate immune response, plays an essential role in maintaining host homeostasis. It promotes clearance of pathogen- and danger-associated molecular patterns, regulates adaptive immunity, and can modify various metabolic processes such as energy expenditure, lipid metabolism, and glucose homeostasis. In this review, we will focus on the intricate interplay between complement components and lipid metabolism. More precisely, we will display how alterations in the activation and regulation of the complement system affect pathological outcome in lipid-associated diseases, such as atherosclerosis, obesity, metabolic syndrome, age-related macular degeneration, and metabolic dysfunction-associated steatotic liver disease. In addition to that, we will present and evaluate underlying complement-mediated physiological mechanisms, observed both in vitro and in vivo. Our manuscript will demonstrate the clinical significance of the complement system as a bridging figure between innate immunity and lipid homeostasis.
Introduction
Living organisms require immune homeostasis - a balance between the immune tolerance to self and immunogenicity to exogenous challenges deleterious to the host. This equilibrium is achieved through coordinated interplay between tissues with proteins and cells of the immune system. In contrast to pathogen-induced inflammation, metabolic changes caused by abnormal amounts of nutrients lead to sterile low-grade inflammation (1). This kind of inflammation can be initiated within various organs, and if not resolved by immune system action, it can drive disease development.
One of the essential energy sources, lipids, are fundamental for building cell structures, cellular signaling, and the generation of physiologically active compounds. They affect immunity in a bidirectional manner, e.g., through the balance between pro- and anti-inflammatory lipid-derived mediators modulating cellular and humoral immune response by shaping repertoires of immune cells, circulating antibodies and complement system components. At the same time, the immune system can steer lipid metabolism and determine the fate of lipid derivatives affecting the general metabolic homeostasis. Next to native lipids, modified lipids and lipoproteins are increasingly recognized as drivers in many cellular and immune processes and disease pathogenesis such as atherosclerosis, metabolic dysfunction-associated steatotic liver disease (MASLD), etc. (1–3).
Lipid-mediated pathologies comprise a broad spectrum of diseases in which abnormal lipid metabolism, signaling and storage affect various organs and systems. The most prevalent are obesity, atherosclerosis, type 2 diabetes mellitus (T2DM), age-related macular degeneration (AMD), and MASLD (4). This group of non-communicable diseases reduces the quality of life and is responsible for 20 million deaths annually (https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases). Several factors, including genetics, diet, and lifestyle contribute to these complex conditions. The diagnosis is usually established by anthropometric measurements, biochemical, genetic, liver function tests, inflammatory markers, and imaging techniques.
In this mini-review, we will report recent knowledge on the interaction between lipids, lipid metabolism within tissues and the complement system.
Complement system – an overview
The complement system, a humoral part of innate immunity, is a network of proteolytic cascades exerting its function in extracellular and intracellular fluids and on cellular surfaces. Most of its constituents are synthesized in the liver, whereas some are produced in immune or non-immune cells. It is an ancient defense system consisting of pattern recognition receptors (PRRs) and regulatory proteins, organized into three different pathways: classical (CP), lectin (LP) and alternative (AP). The CP is initiated by binding of its PRR - C1q, to antigen/antibody complexes, some pentraxins, apoptotic bodies and amyloid fibrils. Furthermore, repeating carbohydrates or acetylated residues, as well as aberrant glycocalyx patterns, engage recognition by PRRs of the LP (5–7). In contrast to that, the AP has a continuous low level of activation on self and non-self surfaces by the tick-over mechanism (8). Recently it was shown that complement can be activated in a non-canonical manner, by certain proteases from coagulation and fibrinolysis pathways (9–12). Cascades of all complement pathways conduct and control deposition of the central effector molecule C3 (13). Once deposited, and if not inactivated, C3 can guide the recruitment and formation of C5-convertase and terminal complement complex (C5b-C9) with a lytic function (8). Moreover, C3 and C5 cleavage generates anaphylatoxins that can drive chemotaxis and activation of immune cells, further propagating damage and inflammation (14, 15). Due to their strong auto-damaging potential, complement cascades are controlled by multiple regulators and inhibitors present in the fluid phase and on host cells (7, 16).
Novel evidence demonstrated that complement acts intracellularly. The intracellular complement, complosome, plays a role in cellular responses to the environment or the homeostatic balance maintenance, by regulating many cellular functions, e.g., cell metabolism, autophagy, survival, signaling, response to infections, and efferocytosis capacity (17–21).
Finally, the complement system functions are versatile: it eliminates pathogens and altered self-structures, coordinates innate and adaptive immune responses, controls tissue reorganization, instructs clearance of metabolic waste, and responds to metabolic alternations on intra- and extracellular levels (7, 17, 22).
The complement system in atherosclerosis
Atherosclerosis is a chronic inflammatory disease characterized by the deposition and oxidation of low-density lipoprotein (LDL) particles in the vessel wall, followed by immune cell infiltration, leading to the formation of fatty streaks that can progress to plaques. If untreated, plaques become larger, more fibrous, calcified and prone to rupture. According to WHO, atherosclerosis is a major cause of mortality worldwide and is responsible for most myocardial infarctions (MI), strokes, and peripheral artery disease (PAD) (23).
The relevance of the complement system in atherosclerosis was demonstrated in the 1970s (24). This was followed by detection of complement proteins C3, C1q, C4, C9, C-reactive protein (CRP), C5b-C9, CD55, CD35, C3aR1, C5aR1, factor B (FB), factor H (FH), C1-inhibitor (C1-INH), C4-binding protein (C4BP), as well as active degradation products of some, within atherosclerotic plaques (25–32). Various clinical or genome-wide association studies (GWAS), demonstrated that components of the LP and CP have both proatherogenic and atheroprotective effects. More precisely, ficolin-1 and -2, pentraxin 3 (PTX3), mannan-binding lectin serine protease 2 (MASP2) and MASP3 have predictive value towards adverse cardiovascular events (33–35). The absence of mannan-binding lectin (MBL) predisposes to atherogenesis, but there are some controversies over its serum level effect on atherosclerotic cardiovascular disease (ACVD) (36–41). C1Q has been identified as a risk gene; in advanced lesions its local production is higher, but clear mechanisms of how its serum levels affect disease outcome are unknown (42–49). Predisposition for MI or cerebrovascular episodes was seen in hereditary C2 deficiency (50). Also, elevated levels of C4 have been associated with cardiovascular disease (CVD) or diabetic stroke, independently of traditional risk factors (51–55). In contrast to CP or LP components, elevated C3 levels correlated with classical risk factors and worsened CVD outcomes (56–63). Similarly, components of the C5b-C9 complex have a strong association with pathogenicity (60, 64–69). Serum levels of C4BP can be predictive of the severity of the PAD (70, 71). Although AP is proatherogenic, there is no conclusion about the effect of CFH polymorphisms on coronary heart disease (CHD) (72–76). On the contrary, plasma concentrations of factor H-related protein 1 (FHR1) were elevated in patients with ACVD and correlated with the expression of the inflammation markers (77).
The mechanistic role of complement in atherosclerosis was explored in vitro and in vivo. High-fat diet (HFD) feeding led to elevated amounts of circulating lipids and increased levels of circulating C3, C4 and C1q (78, 79). Oxidatively modified LDL (OxLDL), located within the vessel, presents various damage-associated molecular patterns (DAMPs) on its surface, which are recognized by natural IgM antibodies, CRP, C1q, MBL, C3a, FH, FHR1, FHR3, FHR5 or scavenger receptors on macrophages (1, 2, 80–86). Due to its damage potential, if not neutralized by the immune system, OxLDL can activate endothelial cells. It alters their phenotype to procoagulatory and induces their secretion of C3a, C5a and other chemokines. This further propagates endothelial distress and activates immune response, partially through C3a/C3aR, C5a/C5aR axis, or by deposition of sublytic C5b-C9 (87–92). Consequently, monocytes are recruited to the intima, where they become foam cells (23). Macrophage uptake of OxLDL, cholesterol efflux and foam cell transformation is affected by PTX3, C3a, C1q, factor D (FD), FH and MBL (81, 84, 93–97). If the amount of engulfed lipids is too excessive, cholesterol crystals (CCs) build up and trigger NLR family pyrin domain containing 3 protein (NLRP3) activation, resulting in macrophage death. At this stage, IgM, C1q, MBL-A, MBL-C, and C3b play a protective role – guiding their removal of dying cells by macrophages, employing complement receptor 3 (CR3) and V-set and immunoglobulin domain containing 4 protein (VSIG4) receptors (98–102). Next to it, a balance between intracellular C3 activation and repression by FH controls the efferocytosis rate of lesional macrophages and affects necrotic core formation (19). The impaired clearance rate of lipid-overloaded macrophages leads to the release of DAMPs resulting in the recruitment of additional immune cells and generation of C3a and C5a, propagating inflammation, and smooth muscle cell expansion (103). This leads to the necrotic core formation filled with CCs, cellular debris, monocytes/macrophages rich in tissue factor and erythrocytes. Cholesterol crystals are recognized by C1q, C3c, ficolin-2, MBL, PTX3, and CRP. When cleared by CR3-rich monocytes, opsonized CCs activate the inflammasome in a C5a-dependent manner and enhance IL-1β secretion (104–108). Later, it was clarified that the metabolic switch required for IL-1β production by macrophages requires mitochondrial C5aR1 ligation generated by cell-intrinsic C5a (109). Furthermore, C5a makes plaques unstable and prone to rupture by affecting the senescence and death rate of smooth muscle cells (110, 111). Upon plaque rupture, released CCs activate complement and drive thrombus formation (112).
The presence of components of the C5b-C9 complex within lesions is shown to be proatherogenic in vivo. This observation was further supported by the finding that the absence of CD59, a C5b-C9 inhibitor, accelerated advanced atherosclerosis (113, 114). The previously mentioned proatherogenic effect of C5a was further confirmed by findings that inhibition of surface-expressed C5aR1 and deficiency of C5aR2 resulted in smaller lesions using animal models (111, 115–117).
The discrepancy in complement inhibition effectiveness between human trials and animal studies in atherosclerosis highlights the need for further research on the complement system function to develop novel therapeutic strategies (118–120).
The complement system in metabolic syndrome
Metabolic syndrome (MetS) represents a cluster of several disorders, including insulin resistance (IR), obesity, dyslipidemia, hypertension and hyperglycemia. It contributes to the development of ACVD and T2DM (121–124). Even though increased C3 and C4 levels have been associated with the risk of developing MetS, metabolic alterations such as IR, obesity, inflammation, and neurohormonal dysfunction are pivotal initiators of this pathogenic cascade (123, 125–130).
Insulin resistance is characterized by the loss of sensitivity to insulin within insulin-dependent tissues such as adipocytes, muscles and liver. Observational studies have shown that C4 levels are associated with the homeostasis model assessment (HOMA) index, the parameter for IR (131). Although C1qA deficiency protects from HFD-induced IR, conflicting findings for the involvement of CP have been reported (132–135). In contrast to CP, LP is predominantly protective. Independently of multiple metabolic features, MBL correlated with insulin sensitivity and its levels were low among obese individuals (136–139). Additionally, low ficolin-3 was independently associated with IR and predicted type 2 diabetes mellitus (T2DM) (140). Baseline C3 levels and level changes correlated with HOMA, multiple organ IR and T2DM independently of obesity, metabolism- and inflammation-related risk factors (131, 141–149). Mechanistically, C3 influence on IR may be linked to the activity of C3a and C5a and their receptors. Studies in mice show that lack of C3aR or C3 can increase insulin sensitivity, although human studies did not find a connection between C3a and IR or T2DM (147, 150, 151). Furthermore, in vivo data on the role of C3adesArg – acylation stimulating protein (ASP) in IR are inconsistent (152, 153). In humans, higher ASP levels correlate with increased IR through altered lipid and glucose metabolism (154). Downstream proteins of the AP have been negatively correlated with insulin sensitivity. Weight loss and treatment of IR with rosiglitazone decreased FH concentrations in plasma, although conflicting results were found in SLE patients (126, 155). Additionally, the association between properdin, FH and Bb with HOMA was observed (156). However, data on the role of properdin obtained in vivo differ from human ones, as properdin deficiency did not affect insulin-mediated glucose uptake (157). Mechanistic data confirm the relevance of FB in MetS, as Cfb-/- mice exhibit increased insulin sensitivity and decreased inflammation (158). For more downstream components, in vitro and in vivo data have shown C5aR1 to contribute to IR development (159, 160). Furthermore, the role of C5b-C9 complex in IR is still unclear. For instance, it does not correlate with IR nor influence the incidence of T2DM (147). However, in chronic heart failure, a positive correlation with HOMA-IR, fasting glucose and insulin level was seen (161).
Obesity is a low-grade inflammatory disease defined by excessive fat accumulation in visceral and subcutaneous fat depots (BMI≥30kg/m2) (162, 163). It is driven by lifestyle, genetic, environmental and cultural factors and is considered endocrine and metabolic disease (162, 164–166). Excessive nutrient intake or low energy expenditure causes lipid accumulation, leading to adipocyte hypertrophy or hyperplasia, consequently driving inflammation that further exacerbates obesity and associated health issues (166–168). Adipose tissue produces many complement proteins including C3, FB, FH, CR1, C1q, C1r, C1s and properdin, and is the predominant source of key players in adipose tissue biology such as ASP, FD, and adiponectin (155, 169–173). In obesity, serum levels of C3, FB, FH and factor I (FI), but not FD, were elevated when compared to normal weight controls (174, 175). Similarly, analysis of BMI-discordant monozygotic obese twin pairs demonstrated that levels of FHR5, C4, C1qA, C1-INH, MASP1, FH, FI, C3 and C8 were elevated in a twin with higher BMI (176). Moreover, visceral and subcutaneous adipose tissue of a heavier twin had increased expressions of C1, C2, C3, FB, FI, properdin, FH, FHR2, C3aR, C5aR1, VISIG4, CD59, in contrast to FD and components of the C5b-C9 (177). In most rodent models of obesity, decreases in FD levels, induction of C1q, and inconsistency in overproduction and secretion of C3 or FB by adipose tissue were seen (170, 173, 178–180). Different production rate of certain complement proteins between subcutaneous and visceral fat of obese subjects, or between dissimilar stages of adipocyte maturation was reported in mice and men (172, 181–185). Within adipose tissue, by binding to the C5L2 receptor, ASP stimulates TG synthesis, increases glucose transport through GLUT1 and GLUT4, fractional free fatty acids re-esterification and inhibits lipolysis (186–194). In line with this, C3-deficient mice have a reduction in fat mass and are resistant to diet-induced obesity. Exogenous ASP administration to C3-/- animals on a standard diet led to a weight increase of a fat pad (193, 195–199). Overexpression of FB in preadipocytes boosts their lipid accumulation and maturation (185, 200). Additionally, through C3a/C3aR axis, FD regulates glucose uptake, increases TG synthesis and inhibits lipolysis (169, 172, 201–203). However, animals deficient in FD had no abnormality in development or body weight (204). Another adipokine similar to C1q is adiponectin, with an anti-inflammatory and anti-fibrotic function. It enhances insulin sensitivity and is downregulated in obesity (205–208).
Diabetes mellitus is the dysregulation of blood glucose levels due to insufficient insulin secretion by pancreatic beta cells, insensitivity of peripheral tissues to insulin, or a combination of both. Type 2 DM, an inflammatory disease, represents 90% of newly diagnosed cases. It is related to obesity and multiple metabolic disturbances, e.g., IR leading to hyperinsulinemia, beta cell exhaustion and finally insulin insufficiency (209). The complement system was shown to have critical metabolic functions within the beta cells. It affects insulin secretion, substrate and metabolite processing and regulates inflammatory processes within islets. Elevated C3 is associated with an increased risk of developing diabetes, independently of demographic, hereditary, metabolism- and inflammation-related factors (141, 147, 210). Interestingly, C3 was associated with insulin secretion, even after adjustment for insulin sensitivity index (211). Moreover, C3a, C3c and C3d correlated with T2DM, although for C3a these associations were attenuated after adjustment for confounding factors (147, 212, 213). Decreased levels of FD were observed in T2DM patients (174, 203). In one study, levels of properdin and soluble C5b-C9 were associated with a family history of T2DM, although the effect of C5b-C9 niveau was not confirmed in others (147, 156, 214, 215).
Since insulin infusion did not affect C3 expression within adipocytes, this implicated that C3 levels might affect insulin secretion in vivo (216). In line with this, increased C3 expression in T2DM pancreatic cells demonstrated that intracellular C3 has protective effects on islet beta cells in stress conditions, through interaction with ATG16L1 (18, 20). The highest expressed complement gene in human beta cells, CD59, was shown to control glucose-mediated insulin secretion (217–219). This effect is further promoted by ASP (220). On the contrary, beta cell FH suppresses insulin secretion via adrenomedullin (221). Additionally, C3a and C5b-C9 have been identified as potent inflammasome activators, suggesting their role in insulitis (222, 223). Interestingly, C3a and C5a generated by FD activity, and through the activity of their receptors, play a key role in adipose tissue-pancreas axis in murine models, by inducing insulin production and dampening beta cell death and dedifferentiation (201, 203, 224). Accordingly, C3aR and C5aR1 agonists improved glucose-dependent insulin production (225).
Dyslipidemia represents abnormal levels of lipids and lipoprotein particles in circulation and is a crucial risk factor for ACVD. It accounts for nearly 50% of deaths due to ischemic heart disease (226). Dyslipidemia is characterized by abnormal lipid levels, e.g., increased TGs, and decreased high-density lipoprotein (HDL). Many complement proteins have been shown to bind lipoprotein particles in plasma and are essential for their metabolic turnover. For instance, C3, acidic form of C4 (C4-A), basic form of C4 (C4-B), and C9 were detected on very low-density lipoprotein (VLDL) and LDL particles, while FHR3 appeared only on LDL (227, 228). Moreover, HDL was associated with C3, C4-A, C4-B, C9, vitronectin and clusterin in coronary artery disease and cholesterol ester protein transfer deficiency (229, 230). Additionally, FH, FD, properdin and MASP3 showed associations with lipoprotein particle concentration and size (228). Apolipoprotein E, a major protein component of lipoproteins, binds to FH, and C1q (97, 231, 232). In observational studies, levels of TGs, adverse lipoprotein subclass profile and enrichment in TGs in all lipoprotein subclasses are associated with higher levels of circulating C3; however, contradictory opinions were reported (129, 144, 216, 228, 233–238). Accordingly, ASP and TG levels correlate, although significance is lost when adjusted for waist/hip ratio and LDL size (195, 239). Elevated expression of C1S, C5aR1, CD59 and CD55 in subcutaneous adipose tissue was seen in patients with familial combined hyperlipidemia (240). Experimental data further underscores the significance of complement-lipid interaction. For instance, postprandial C3 and ASP secretion by adipocytes is shown to be stimulated by chylomicrons (190, 241). Additionally, male C3-/- mice (ASP deficient) display delayed postprandial clearance of TGs and increased fasting and postprandial free fatty acids levels (198). Properdin-deficient animals have increased fat accumulation on HFD, impaired postprandial TG clearance and decreased energy expenditure (157, 242).
As demonstrated above, the complement system controls insulin production and resistance, adipose tissue remodeling, and lipoprotein metabolism, consequently affecting chronic low-grade inflammation.
The complement system in metabolic dysfunction-associated steatotic liver disease
The liver, a primary site for complement protein synthesis, is particularly susceptible to complement-mediated damage (243, 244). Dysregulation of the complement system can exacerbate liver inflammation and fibrosis (245).
Metabolic dysfunction-associated steatotic liver disease (MASLD) is characterized by excess fat accumulation in hepatocytes, without significant alcohol intake and is among the most commonly diagnosed liver disorders (246, 247). A progressive and inflammatory form of MASLD, known as metabolic dysfunction-associated steatohepatitis (MASH), features hepatic steatosis, inflammation, and fibrosis (248, 249). Predisposing factors for MASLD and MASH are components of MetS, oxidative stress, and lipid peroxidation. This condition can further advance to cirrhosis and hepatocellular carcinoma (250, 251).
A positive correlation between high C3 in serum and the prevalence and severity of MASLD was demonstrated in clinical studies (252–255). In addition to this, ASP, involved in adipocyte lipid metabolism, was increased in MASLD patients (255). Histological analyses demonstrated that in 74% of patients, cleaved C3b and C4d were deposited in liver tissue, with more than 50% of C3-positive livers also showing C1q and MBL deposits, and exhibiting C5b-C9 formation. Additionally, these components were more frequently detected in MASH, demonstrating the involvement of both CP and LP in liver inflammation (256). However, in some patients, C3 activation was not associated with C1q, MBL, or C4d deposition, suggesting that the AP may also play a role in complement activation in the MASLD. This was confirmed by the positive correlation of properdin and the C3c deposits with liver inflammation. Additionally, levels of FH were downregulated in MASH subjects (257). C3a and C5a were identified to promote hepatic inflammation by attracting immune cells (258). Additionally, blocking or removing C3aR and C5aR1 might offer protection against steatosis, fibrosing MASH, inflammation, and metabolic dysfunction (17, 259–261). However, while some evidence points to C3a/C3aR and C5a/C5aR1 as a potential therapeutic approach, clinical trials targeting the complement system are still lacking, making its effectiveness uncertain.
The complement system and age-related macular degeneration
Age-related macular degeneration (AMD) is a chronic, inflammatory disease of the retina, and it is the most common cause of blindness in the elderly in developed countries (262). It is characterized by the accumulation of lipid-rich drusen between the Bruch membrane (BrM) and retinal pigment epithelium (RPE), resulting in degeneration of RPE, photoreceptors and consequently loss of vision. Joint action of aging, genetic and environmental factors play a significant role in the disease onset and progression (263).
Lipids within the retina are crucial players in the pathogenesis of AMD. Due to photo-oxidative stress, they are highly susceptible to lipid peroxidation and the generation of oxidized and reactive breakdown products (1, 264–267). These degradation products can initiate sterile inflammation and stimulate immune responses (1, 2, 266, 268–270).
Seminal studies discovered 52 variants with genome-wide association (GWA) significance, among which CFH variant rs1061170 (Y402H), and variants within ARMS2-HTR1, C2-CFB-SKVI2L and C3 had the strongest associations, confirming the key role of complement in the AMD pathogenesis (271–273). Today, Y402H is recognized as the major susceptibility variant. Heterozygous carriers of the minor C allele have a 2-4-fold, and homozygous carriers have a 3-7-fold increased risk for developing AMD (274). Moreover, it is important to note that this variant also affects the splice variant of FH, FHL-1, a main regulator of AP within BrM and RPE (275–277). Y402H decreases the binding ability of FH to heparin sulfate, CRP and malondialdehyde (MDA), thereby affecting its availability to act and regulate AP on surfaces decorated with these ligands (85, 276, 278–281). A similar aftermath was observed for the rare variant R1210C located within the C-terminal part of FH (273, 282). Deletion of CFHR3-1 genes decreases the risk of AMD development (283–285). Moreover, deleterious effects were reported for some variants within CFI, C3 and C9 (274, 286–296). On the other hand, loss-of-function variants within complement activators C2 and CFB are mostly protective (293, 297, 298).
Accumulation of oxidative damage shown by intensive MDA staining in BrM and choroid which co-stained with FH, implicated the binding of FH to MDA-adducts. Additionally, FH promoted the generation of iC3b on MDA surfaces and suppressed the proinflammatory effects of MDA in vitro. Due to the weaker binding to MDA, Y402H carriers have diminished regulation of AP and attenuated anti-MDA inflammatory properties (85). This was supported in a murine model, expressing chimeric FH, containing a human Y402H variant (299). Loss of endogenous FH in RPE cells renders them more susceptible to oxidative stress and reduces their viability (300, 301). Furthermore, we have demonstrated that FHR1 and FHR3 bind to MDA and thereby compete with FH for it, affecting FH AP regulatory function. These findings mechanistically explain the protective properties of CFHR3-1 deletion (80).
Next to AP, the relevance of locally produced C5b-C9, deposited in choriocapillaris during the photoreceptor outer segment recycling by RPE was reported (302–304). In the aging retina, recycling capabilities decrease, resulting in enhanced C5b-C9 formation, pronounced in the presence of AMD-predisposing CFH variants (303, 305). Apart from locally produced FHL-1 and C5b-C9, increased systemic C3d/C3 ratio and enhanced local C3d deposition are implicated in AMD (302, 306, 307).
Activation of the AP, dysfunctional FH, together with aging, oxidative damage and disturbed lipid metabolism, are identified as critical steps in the development and progression of AMD (7).
Concluding remarks
Experimental evidence presented in this review confirms that the functions of the complement system in lipid homeostasis are versatile (Figure 1). Certain complement proteins sense and control lipid homeostasis locally or systemically. However, in case of excessive lipid accumulation or oxidation, complement proteins, if unable to neutralize it, can also act as initiators or propagators of lipid-driven inflammation. Therefore, understanding all mechanisms involved in cross-talk between lipid metabolism and complement system should be of importance for developing better diagnostic or therapeutic approaches.
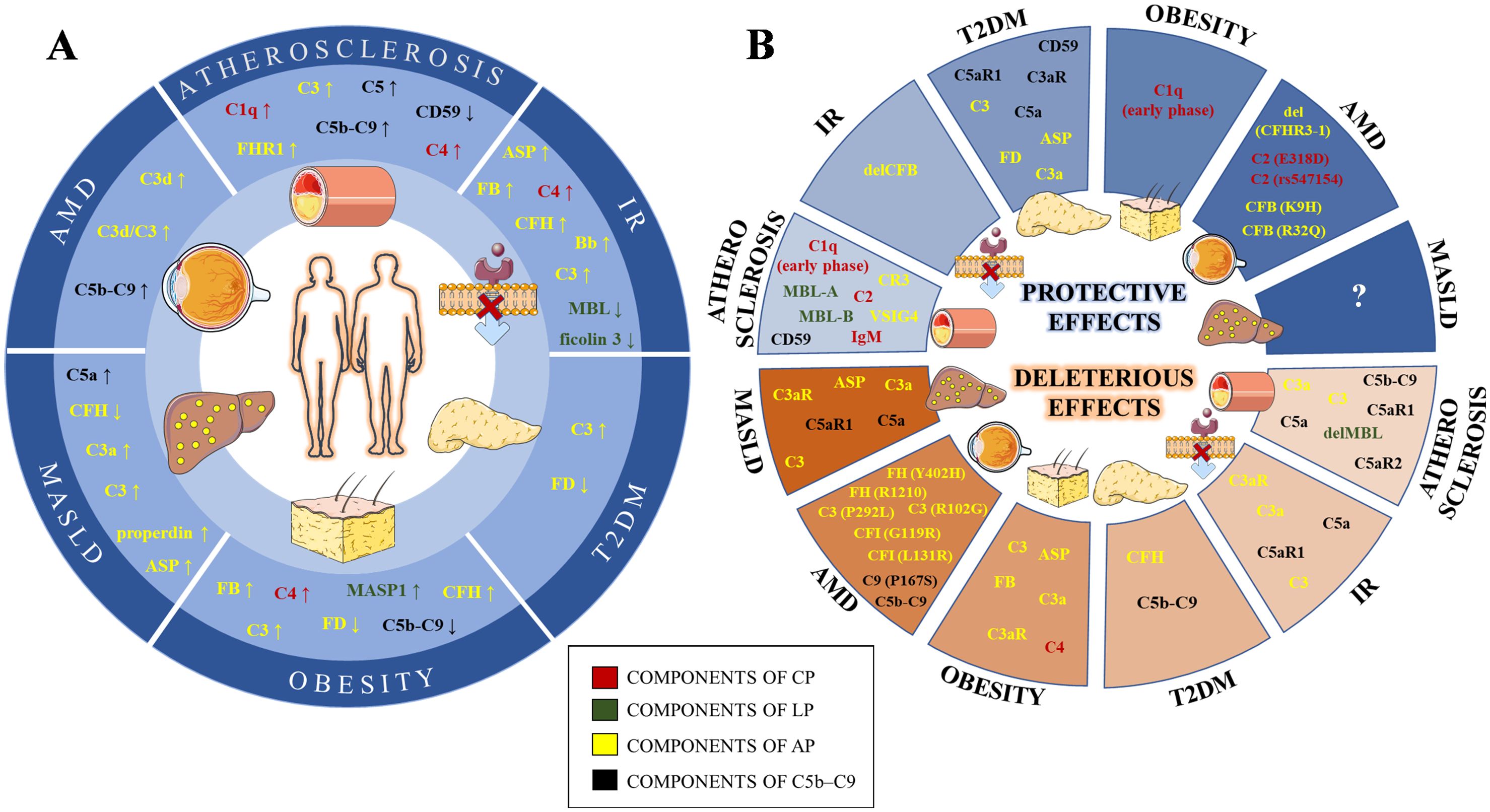
Figure 1. Schematic illustration of complement levels alterations (A) and their effects (B) in lipid-mediated pathologies. The components of the classical (CP), lectin (LP), alternative (AP) and terminal (C5b-C9) pathways are labeled in red, green, yellow and black, respectively. IR, Insulin resistance; T2DM, Type 2 diabetes mellitus; MASLD, Metabolic dysfunction-associated steatotic liver disease; AMD, Age-related macular degeneration; IgM, Immunoglobulin M; C1q, Complement Component 1q; C2, Complement Component 2; C4, Complement Component 4; MBL, Mannan binding lectin; MASP1, MBL Associated Serine Protease 1; C3, Complement Component 3; Asp, C3adesArg – acylation stimulating protein; FB, Factor B; FD, Factor D; FH, Factor H; FHR1, Factor H related protein 1; FI, Factor I; C3aR, Complement C3a Receptor; VSIG4, V-Set And Immunoglobulin Domain Containing 4; CR3, Complement Component Receptor 3; C5, Complement Component 5; C5aR1, Complement C5a Receptor 1; C5aR2, Complement C5a Receptor 2; C9, Complement Component 9; del, gene deletion; gene variants are given in the bracket after affected gene.
Author contributions
LA: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. KD: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. NP-M: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. NP-M and KD were supported by the Vienna Science and Technology Fund (WWTF LS20-081), LA by European Federation of Clinical Chemistry and Laboratory Medicine Research Grant 2024 and Ministry for Science, Higher Education and Youth of Canton Sarajevo (27-02-3533087-41/24).
Acknowledgments
The figure was created using Servier Medical Art (https://servier.com/en/brochure/servier-medical-art/).
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this paper.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16:485–97. doi: 10.1038/nri.2016.63
2. Alic L, Binder CJ, Papac-Milicevic N. The OSE complotype and its clinical potential. Front Immunol. (2022) 13:1010893. doi: 10.3389/fimmu.2022.1010893
3. Zhivaki D, Kagan JC. Innate immune detection of lipid oxidation as a threat assessment strategy. Nat Rev Immunol. (2022) 22:322–30. doi: 10.1038/s41577-021-00618-8
4. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity. (2017) 47:406–20. doi: 10.1016/j.immuni.2017.08.009
5. Zhang W, Chen Y, Pei H. C1q and central nervous system disorders. Front Immunol. (2023) 14:1145649. doi: 10.3389/fimmu.2023.1145649
6. Beltrame MH, Catarino SJ, Goeldner I, Boldt AB, de Messias-Reason IJ. The lectin pathway of complement and rheumatic heart disease. Front Pediatr. (2014) 2:148. doi: 10.3389/fped.2014.00148
7. Mastellos DC, Hajishengallis G, Lambris JD. A guide to complement biology, pathology and therapeutic opportunity. Nat Rev Immunol. (2024) 24:118–41. doi: 10.1038/s41577-023-00926-1
8. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
9. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
10. Bekassy ZD, Kristoffersson AC, Rebetz J, Tati R, Olin AI, Karpman D. Aliskiren inhibits renin-mediated complement activation. Kidney Int. (2018) 94:689–700. doi: 10.1016/j.kint.2018.04.004
11. Donado CA, Jonsson AH, Theisen E, Zhang F, Nathan A, Rupani KV, et al. Granzyme K drives a newly-intentified pathway of complement activation. bioRxiv. (2024). doi: 10.1101/2024.05.22.595315
12. Irmscher S, Doring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein cleaves C3 and activates complement. J Innate Immun. (2018) 10:94–105. doi: 10.1159/000484257
13. Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev. (2016) 274:9–15. doi: 10.1111/imr.2016.274.issue-1
14. Pandey S, Maharana J, Li XX, Woodruff TM, Shukla AK. Emerging insights into the structure and function of complement C5a receptors. Trends Biochem Sci. (2020) 45:693–705. doi: 10.1016/j.tibs.2020.04.004
15. Zarantonello A, Revel M, Grunenwald A, Roumenina LT. C3-dependent effector functions of complement. Immunol Rev. (2023) 313:120–38. doi: 10.1111/imr.v313.1
16. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. (2009) 9:729–40. doi: 10.1038/nri2620
17. King BC, Blom AM. Intracellular complement: Evidence, definitions, controversies, and solutions. Immunol Rev. (2023) 313:104–19. doi: 10.1111/imr.v313.1
18. King BC, Renstrom E, Blom AM. Intracellular cytosolic complement component C3 regulates cytoprotective autophagy in pancreatic beta cells by interaction with ATG16L1. Autophagy. (2019) 15:919–21. doi: 10.1080/15548627.2019.1580515
19. Kiss MG, Papac-Milicevic N, Porsch F, Tsiantoulas D, Hendrikx T, Takaoka M, et al. Cell-autonomous regulation of complement C3 by factor H limits macrophage efferocytosis and exacerbates atherosclerosis. Immunity. (2023) 56:1809–24.e10. doi: 10.1016/j.immuni.2023.06.026
20. Kulak K, Kuska K, Colineau L, McKay M, Maziarz K, Slaby J, et al. Intracellular C3 protects beta-cells from IL-1beta-driven cytotoxicity via interaction with Fyn-related kinase. Proc Natl Acad Sci U S A. (2024) 121:e2312621121. doi: 10.1073/pnas.2312621121
21. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
22. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
23. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
24. Geertinger P, Sorensen H. Complement as a factor in arteriosclerosis. Acta Pathol Microbiol Scand A. (1970) 78:284–8. doi: 10.1111/j.1699-0463.1970.tb03303.x
25. Hollander W, Colombo MA, Kirkpatrick B, Paddock J. Soluble proteins in the human atherosclerotic plaque. With spectral reference to immunoglobulins, C3-complement component, alpha 1-antitrypsin and alpha 2-macroglobulin. Atherosclerosis. (1979) 34:391–405. doi: 10.1016/0021-9150(79)90064-9
26. Niculescu F, Rus H. Complement activation and atherosclerosis. Mol Immunol. (1999) 36:949–55. doi: 10.1016/S0161-5890(99)00117-0
27. Niculescu F, Rus HG, Vlaicu R. Immunohistochemical localization of C5b-9, S-protein, C3d and apolipoprotein B in human arterial tissues with atherosclerosis. Atherosclerosis. (1987) 65:1–11. doi: 10.1016/0021-9150(87)90002-5
28. Oksjoki R, Kovanen PT, Pentikainen MO. Role of complement activation in atherosclerosis. Curr Opin Lipidol. (2003) 14:477–82. doi: 10.1097/00041433-200310000-00008
29. Oksjoki R, Laine P, Helske S, Vehmaan-Kreula P, Mayranpaa MI, Gasque P, et al. Receptors for the anaphylatoxins C3a and C5a are expressed in human atherosclerotic coronary plaques. Atherosclerosis. (2007) 195:90–9. doi: 10.1016/j.atherosclerosis.2006.12.016
30. Seifert PS, Hansson GK. Complement receptors and regulatory proteins in human atherosclerotic lesions. Arteriosclerosis. (1989) 9:802–11. doi: 10.1161/01.ATV.9.6.802
31. Seifert PS, Hansson GK. Decay-accelerating factor is expressed on vascular smooth muscle cells in human atherosclerotic lesions. J Clin Invest. (1989) 84:597–604. doi: 10.1172/JCI114204
32. Vlaicu R, Niculescu F, Rus HG, Cristea A. Immunohistochemical localization of the terminal C5b-9 complement complex in human aortic fibrous plaque. Atherosclerosis. (1985) 57:163–77. doi: 10.1016/0021-9150(85)90030-9
33. Carbone F, Valente A, Perego C, Bertolotto M, Pane B, Spinella G, et al. Ficolin-2 serum levels predict the occurrence of acute coronary syndrome in patients with severe carotid artery stenosis. Pharmacol Res. (2021) 166:105462. doi: 10.1016/j.phrs.2021.105462
34. Fumagalli S, Perego C, Zangari R, De Blasio D, Oggioni M, De Nigris F, et al. Lectin pathway of complement activation is associated with vulnerability of atherosclerotic plaques. Front Immunol. (2017) 8:288. doi: 10.3389/fimmu.2017.00288
35. Vengen IT, Enger TB, Videm V, Garred P. Pentraxin 3, ficolin-2 and lectin pathway associated serine protease MASP-3 as early predictors of myocardial infarction - the HUNT2 study. Sci Rep. (2017) 7:43045. doi: 10.1038/srep43045
36. Madsen HO, Videm V, Svejgaard A, Svennevig JL, Garred P. Association of mannose-binding-lectin deficiency with severe atherosclerosis. Lancet. (1998) 352:959–60. doi: 10.1016/S0140-6736(05)61513-9
37. Poppelaars F, Gaya da Costa M, Berger SP, Assa S, Meter-Arkema AH, Daha MR, et al. Strong predictive value of mannose-binding lectin levels for cardiovascular risk of hemodialysis patients. J Transl Med. (2016) 14:236. doi: 10.1186/s12967-016-0995-5
38. Vengen IT, Madsen HO, Garred P, Platou C, Vatten L, Videm V. Mannose-binding lectin deficiency is associated with myocardial infarction: the HUNT2 study in Norway. PloS One. (2012) 7:e42113. doi: 10.1371/journal.pone.0042113
39. Kieninger-Grafitsch A, Vogt S, Ribi C, Dubler D, Chizzolini C, Huynh-Do U, et al. No association of complement mannose-binding lectin deficiency with cardiovascular disease in patients with Systemic Lupus Erythematosus. Sci Rep. (2020) 10:3693. doi: 10.1038/s41598-020-60523-3
40. Saevarsdottir S, Oskarsson OO, Aspelund T, Eiriksdottir G, Vikingsdottir T, Gudnason V, et al. Mannan binding lectin as an adjunct to risk assessment for myocardial infarction in individuals with enhanced risk. J Exp Med. (2005) 201:117–25. doi: 10.1084/jem.20041431
41. Meziani S, Ferrannini G, Bjerre M, Hansen TK, Ritsinger V, Norhammar A, et al. Mannose-binding lectin does not explain the dismal prognosis after an acute coronary event in dysglycaemic patients. A report from the GAMI cohort. Cardiovasc Diabetol. (2022) 21:129. doi: 10.1186/s12933-022-01562-0
42. Bashore AC, Yan H, Xue C, Zhu LY, Kim E, Mawson T, et al. High-dimensional single-cell multimodal landscape of human carotid atherosclerosis. Arterioscler Thromb Vasc Biol. (2024) 44:930–45. doi: 10.1161/ATVBAHA.123.320524
43. Cui HK, Tang CJ, Gao Y, Li ZA, Zhang J, Li YD. An integrative analysis of single-cell and bulk transcriptome and bidirectional mendelian randomization analysis identified C1Q as a novel stimulated risk gene for Atherosclerosis. Front Immunol. (2023) 14:1289223. doi: 10.3389/fimmu.2023.1289223
44. Guo S, Mao X, Li X, Ouyang H, Gao Y, Ming L. Serum complement C1q activity is associated with obstructive coronary artery disease. Front Cardiovasc Med. (2021) 8:618173. doi: 10.3389/fcvm.2021.618173
45. Hasegawa N, Fujie S, Horii N, Uchida M, Toyama Y, Inoue K, et al. Aging-induced elevation in circulating complement C1q level is associated with arterial stiffness. Exp Gerontol. (2019) 124:110650. doi: 10.1016/j.exger.2019.110650
46. Kishida K, Nakagawa Y, Kobayashi H, Mazaki T, Yokoi H, Yanagi K, et al. High serum C1q-binding adiponectin levels in male patients with acute coronary syndrome. Cardiovasc Diabetol. (2014) 13:9. doi: 10.1186/1475-2840-13-9
47. Li QX, Ma XT, Shao QY, Yang ZQ, Liang J, Yang LX, et al. Association of serum complement C1q with cardiovascular outcomes among patients with acute coronary syndrome undergoing percutaneous coronary intervention. J Geriatr Cardiol. (2022) 19:949–59. doi: 10.11909/j.issn.1671-5411.2022.12.001
48. Ni XN, Yan SB, Zhang K, Sai WW, Zhang QY, Ti Y, et al. Serum complement C1q level is associated with acute coronary syndrome. Mol Immunol. (2020) 120:130–5. doi: 10.1016/j.molimm.2020.02.012
49. Sasaki S, Nishihira K, Yamashita A, Fujii T, Onoue K, Saito Y, et al. Involvement of enhanced expression of classical complement C1q in atherosclerosis progression and plaque instability: C1q as an indicator of clinical outcome. PloS One. (2022) 17:e0262413. doi: 10.1371/journal.pone.0262413
50. Jonsson G, Truedsson L, Sturfelt G, Oxelius VA, Braconier JH, Sjoholm AG. Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Med (Baltimore). (2005) 84:23–34. doi: 10.1097/01.md.0000152371.22747.1e
51. Cavusoglu E, Eng C, Chopra V, Ruwende C, Yanamadala S, Clark LT, et al. Usefulness of the serum complement component C4 as a predictor of stroke in patients with known or suspected coronary artery disease referred for coronary angiography. Am J Cardiol. (2007) 100:164–8. doi: 10.1016/j.amjcard.2007.02.075
52. Engstrom G, Hedblad B, Janzon L, Lindgarde F. Complement C3 and C4 in plasma and incidence of myocardial infarction and stroke: a population-based cohort study. Eur J Cardiovasc Prev Rehabil. (2007) 14:392–7. doi: 10.1097/01.hjr.0000244582.30421.b2
53. Zhang X, Yin J, Shao K, Yang L, Liu W, Wang Y, et al. High serum complement component C4 as a unique predictor of unfavorable outcomes in diabetic stroke. Metab Brain Dis. (2021) 36:2313–22. doi: 10.1007/s11011-021-00834-0
54. Kramer J, Rajczy K, Hegyi L, Fulop T, Mohacsi A, Mezei Z, et al. C4B*Q0 allotype as risk factor for myocardial infarction. BMJ. (1994) 309:313–4. doi: 10.1136/bmj.309.6950.313
55. Nityanand S, Hamsten A, Lithell H, Holm G, Lefvert AK. C4 null alleles and myocardial infarction. Atherosclerosis. (1999) 143:377–81. doi: 10.1016/S0021-9150(98)00329-3
56. Sorensen H, Dissing J. Association between the C3F gene and atherosclerotic vascular diseases. Hum Hered. (1975) 25:279–83. doi: 10.1159/000152736
57. Muscari A, Massarelli G, Bastagli L, Poggiopollini G, Tomassetti V, Volta U, et al. Relationship between serum C3 levels and traditional risk factors for myocardial infarction. Acta Cardiol. (1998) 53:345–54.
58. Cai G, Li L, Chen Y, Huang H, Yu L, Xu L. Complement C3 gene polymorphisms are associated with lipid levels, but not the risk of coronary artery disease: a case-control study. Lipids Health Dis. (2019) 18:217. doi: 10.1186/s12944-019-1163-8
59. El Khoudary SR, Chen X, McConnell D, Brooks MM, Billheimer J, Orchard TJ. Associations of HDL subclasses and lipid content with complement proteins over the menopause transition: The SWAN HDL ancillary study: HDL and complement proteins in women. J Clin Lipidol. (2022) 16:649–57. doi: 10.1016/j.jacl.2022.07.015
60. Hernandez-Diaz M, Rodriguez-Gonzalez D, Heras-Recuero E, Gomez-Bernal F, Quevedo-Abeledo JC, Gonzalez-Rivero AF, et al. The Relationship between the complement system and subclinical carotid atherosclerosis in patients with rheumatoid arthritis. Arthritis Res Ther. (2024) 26:127. doi: 10.1186/s13075-024-03360-3
61. Kojima C, Takei T, Ogawa T, Nitta K. Serum complement C3 predicts renal arteriolosclerosis in non-diabetic chronic kidney disease. J Atheroscler Thromb. (2012) 19:854–61. doi: 10.5551/jat.12286
62. Leban N, Jraba K, Chalghoum A, Hassine S, Elhayek D, Denden S, et al. Polymorphism of C3 complement in association with myocardial infarction in a sample of central Tunisia. Diagn Pathol. (2013) 8:93. doi: 10.1186/1746-1596-8-93
63. Zhang B, Yang N, Gao C. Is plasma C3 and C4 levels useful in young cerebral ischemic stroke patients? Associations with prognosis at 3 months. J Thromb Thrombolysis. (2015) 39:209–14. doi: 10.1007/s11239-014-1100-7
64. Aragam KG, Jiang T, Goel A, Kanoni S, Wolford BN, Atri DS, et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat Genet. (2022) 54:1803–15. doi: 10.1038/s41588-022-01233-6
65. Ge X, Xu C, Liu Y, Zhu K, Zeng H, Su J, et al. Complement activation in the arteries of patients with severe atherosclerosis. Int J Clin Exp Pathol. (2018) 11:1–9.
66. Henes JK, Groga-Bada P, Schaeffeler E, Winter S, Hack L, Zdanyte M, et al. C5 variant rs10985126 is associated with mortality in patients with symptomatic coronary artery disease. Pharmgenomics Pers Med. (2021) 14:893–903. doi: 10.2147/PGPM.S307827
67. Martinez-Lopez D, Roldan-Montero R, Garcia-Marques F, Nunez E, Jorge I, Camafeita E, et al. Complement C5 protein as a marker of subclinical atherosclerosis. J Am Coll Cardiol. (2020) 75:1926–41. doi: 10.1016/j.jacc.2020.02.058
68. Niculescu F, Niculescu T, Rus H. C5b-9 terminal complement complex assembly on apoptotic cells in human arterial wall with atherosclerosis. Exp Mol Pathol. (2004) 76:17–23. doi: 10.1016/j.yexmp.2003.10.002
69. Si W, He P, Wang Y, Fu Y, Li X, Lin X, et al. Complement complex C5b-9 levels are associated with the clinical outcomes of acute ischemic stroke and carotid plaque stability. Transl Stroke Res. (2019) 10:279–86. doi: 10.1007/s12975-018-0658-3
70. Kimoto K, Inoue T, Oku K, Mori T, Kusuda M, Handa K, et al. Relation of C4b-binding protein to athero-sclerosis of the descending thoracic aorta. Artery. (1996) 22:101–14.
71. Martin M, Gottsater A, Nilsson PM, Mollnes TE, Lindblad B, Blom AM. Complement activation and plasma levels of C4b-binding protein in critical limb ischemia patients. J Vasc Surg. (2009) 50:100–6. doi: 10.1016/j.jvs.2008.12.033
72. Nicaud V, Francomme C, Ruidavets JB, Luc G, Arveiler D, Kee F, et al. Lack of association between complement factor H polymorphisms and coronary artery disease or myocardial infarction. J Mol Med (Berl). (2007) 85:771–5. doi: 10.1007/s00109-007-0185-2
73. Pai JK, Manson JE, Rexrode KM, Albert CM, Hunter DJ, Rimm EB. Complement factor H (Y402H) polymorphism and risk of coronary heart disease in US men and women. Eur Heart J. (2007) 28:1297–303. doi: 10.1093/eurheartj/ehm090
74. Sofat R, Casas JP, Kumari M, Talmud PJ, Ireland H, Kivimaki M, et al. Genetic variation in complement factor H and risk of coronary heart disease: eight new studies and a meta-analysis of around 48,000 individuals. Atherosclerosis. (2010) 213:184–90. doi: 10.1016/j.atherosclerosis.2010.07.021
75. Volcik KA, Ballantyne CM, Braun MC, Coresh J, Mosley TH, Boerwinkle E. Association of the complement factor H Y402H polymorphism with cardiovascular disease is dependent upon hypertension status: The ARIC study. Am J Hypertens. (2008) 21:533–8. doi: 10.1038/ajh.2007.81
76. Zee RY, Diehl KA, Ridker PM. Complement factor H Y402H gene polymorphism, C-reactive protein, and risk of incident myocardial infarction, ischaemic stroke, and venous thromboembolism: a nested case-control study. Atherosclerosis. (2006) 187:332–5. doi: 10.1016/j.atherosclerosis.2005.09.009
77. Irmscher S, Zipfel SLH, Halder LD, Ivanov L, Gonzalez-Delgado A, Waldeyer C, et al. Factor H-related protein 1 (FHR-1) is associated with atherosclerotic cardiovascular disease. Sci Rep. (2021) 11:22511. doi: 10.1038/s41598-021-02011-w
78. Schepers A, de Vries MR, van Leuven CJ, Grimbergen JM, Holers VM, Daha MR, et al. Inhibition of complement component C3 reduces vein graft atherosclerosis in apolipoprotein E3-Leiden transgenic mice. Circulation. (2006) 114:2831–8. doi: 10.1161/CIRCULATIONAHA.106.619502
79. Verdeguer F, Castro C, Kubicek M, Pla D, Vila-Caballer M, Vinue A, et al. Complement regulation in murine and human hypercholesterolemia and role in the control of macrophage and smooth muscle cell proliferation. Cardiovasc Res. (2007) 76:340–50. doi: 10.1016/j.cardiores.2007.06.028
80. Alic L, Papac-Milicevic N, Czamara D, Rudnick RB, Ozsvar-Kozma M, Hartmann A, et al. A genome-wide association study identifies key modulators of complement factor H binding to malondialdehyde-epitopes. Proc Natl Acad Sci U S A. (2020) 117:9942–51. doi: 10.1073/pnas.1913970117
81. Fraser DA, Tenner AJ. Innate immune proteins C1q and mannan-binding lectin enhance clearance of atherogenic lipoproteins by human monocytes and macrophages. J Immunol. (2010) 185:3932–9. doi: 10.4049/jimmunol.1002080
82. Palinski W, Horkko S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. (1996) 98:800–14. doi: 10.1172/JCI118853
83. Rudnick RB, Chen Q, Stea ED, Hartmann A, Papac-Milicevic N, Person F, et al. FHR5 binds to laminins, uses separate C3b and surface-binding sites, and activates complement on malondialdehyde-acetaldehyde surfaces. J Immunol. (2018) 200:2280–90. doi: 10.4049/jimmunol.1701641
84. Veneskoski M, Turunen SP, Kummu O, Nissinen A, Rannikko S, Levonen AL, et al. Specific recognition of malondialdehyde and malondialdehyde acetaldehyde adducts on oxidized LDL and apoptotic cells by complement anaphylatoxin C3a. Free Radic Biol Med. (2011) 51:834–43. doi: 10.1016/j.freeradbiomed.2011.05.029
85. Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. (2011) 478:76–81. doi: 10.1038/nature10449
86. Gillotte KL, Horkko S, Witztum JL, Steinberg D. Oxidized phospholipids, linked to apolipoprotein B of oxidized LDL, are ligands for macrophage scavenger receptors. J Lipid Res. (2000) 41:824–33. doi: 10.1016/S0022-2275(20)32391-9
87. Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, et al. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. (1994) 94:1147–55. doi: 10.1172/JCI117430
88. Kilgore KS, Schmid E, Shanley TP, Flory CM, Maheswari V, Tramontini NL, et al. Sublytic concentrations of the membrane attack complex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1 through nuclear factor-kappa B activation. Am J Pathol. (1997) 150:2019–31.
89. Lozada C, Levin RI, Huie M, Hirschhorn R, Naime D, Whitlow M, et al. Identification of C1q as the heat-labile serum cofactor required for immune complexes to stimulate endothelial expression of the adhesion molecules E-selectin and intercellular and vascular cell adhesion molecules 1. Proc Natl Acad Sci U S A. (1995) 92:8378–82. doi: 10.1073/pnas.92.18.8378
90. Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. (2003) 17:1003–14. doi: 10.1096/fj.02-0737com
91. Hertle E, van Greevenbroek MM, Arts IC, van der Kallen CJ, Geijselaers SL, Feskens EJ, et al. Distinct associations of complement C3a and its precursor C3 with atherosclerosis and cardiovascular disease. The CODAM study. Thromb Haemost. (2014) 111:1102–11. doi: 10.1160/TH13-10-0831
92. Miller YI, Shyy JY. Context-dependent role of oxidized lipids and lipoproteins in inflammation. Trends Endocrinol Metab. (2017) 28:143–52. doi: 10.1016/j.tem.2016.11.002
93. Liu W, Jiang J, Yan D, Li D, Li W, Ma Y, et al. Pentraxin 3 promotes oxLDL uptake and inhibits cholesterol efflux from macrophage-derived foam cells. Exp Mol Pathol. (2014) 96:292–9. doi: 10.1016/j.yexmp.2014.03.007
94. Pulanco MC, Cosman J, Ho MM, Huynh J, Fing K, Turcu J, et al. Complement protein C1q enhances macrophage foam cell survival and efferocytosis. J Immunol. (2017) 198:472–80. doi: 10.4049/jimmunol.1601445
95. Duan Y, Zhang X, Zhang X, Lin J, Shu X, Man W, et al. Inhibition of macrophage-derived foam cells by Adipsin attenuates progression of atherosclerosis. Biochim Biophys Acta Mol Basis Dis. (2022) 1868:166533. doi: 10.1016/j.bbadis.2022.166533
96. Liu L, Chan M, Yu L, Wang W, Qiang L. Adipsin deficiency does not impact atherosclerosis development in Ldlr(-/-) mice. Am J Physiol Endocrinol Metab. (2021) 320:E87–92. doi: 10.1152/ajpendo.00440.2020
97. Nissila E, Hakala P, Leskinen K, Roig A, Syed S, Van Kessel KPM, et al. Complement factor H and apolipoprotein E participate in regulation of inflammation in THP-1 macrophages. Front Immunol. (2018) 9:2701. doi: 10.3389/fimmu.2018.02701
98. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. (1998) 188:2313–20. doi: 10.1084/jem.188.12.2313
99. Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. (2007) 170:416–26. doi: 10.2353/ajpath.2007.060406
100. Lewis MJ, Malik TH, Ehrenstein MR, Boyle JJ, Botto M, Haskard DO. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. (2009) 120:417–26. doi: 10.1161/CIRCULATIONAHA.109.868158
101. Matthijsen RA, de Winther MP, Kuipers D, van der Made I, Weber C, Herias MV, et al. Macrophage-specific expression of mannose-binding lectin controls atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. (2009) 119:2188–95. doi: 10.1161/CIRCULATIONAHA.108.830661
102. Helmy KY, Katschke KJ Jr., Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. (2006) 124:915–27. doi: 10.1016/j.cell.2005.12.039
103. Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci U S A. (2020) 117:15818–26. doi: 10.1073/pnas.2006348117
104. Niyonzima N, Halvorsen B, Sporsheim B, Garred P, Aukrust P, Mollnes TE, et al. Complement activation by cholesterol crystals triggers a subsequent cytokine response. Mol Immunol. (2017) 84:43–50. doi: 10.1016/j.molimm.2016.09.019
105. Pilely K, Rosbjerg A, Genster N, Gal P, Pal G, Halvorsen B, et al. Cholesterol crystals activate the lectin complement pathway via ficolin-2 and mannose-binding lectin: implications for the progression of atherosclerosis. J Immunol. (2016) 196:5064–74. doi: 10.4049/jimmunol.1502595
106. Pilely K, Fumagalli S, Rosbjerg A, Genster N, Skjoedt MO, Perego C, et al. C-reactive protein binds to cholesterol crystals and co-localizes with the terminal complement complex in human atherosclerotic plaques. Front Immunol. (2017) 8:1040. doi: 10.3389/fimmu.2017.01040
107. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. (2014) 192:2837–45. doi: 10.4049/jimmunol.1302484
108. Niyonzima N, Samstad EO, Aune MH, Ryan L, Bakke SS, Rokstad AM, et al. Reconstituted high-density lipoprotein attenuates cholesterol crystal-induced inflammatory responses by reducing complement activation. J Immunol. (2015) 195:257–64. doi: 10.4049/jimmunol.1403044
109. Niyonzima N, Rahman J, Kunz N, West EE, Freiwald T, Desai JV, et al. Mitochondrial C5aR1 activity in macrophages controls IL-1beta production underlying sterile inflammation. Sci Immunol. (2021) 6:eabf2489. doi: 10.1126/sciimmunol.abf2489
110. Herman AB, Tsitsipatis D, Anerillas C, Mazan-Mamczarz K, Carr AE, Gregg JM, et al. DPP4 inhibition impairs senohemostasis to improve plaque stability in atherosclerotic mice. J Clin Invest. (2023) 133:e165933. doi: 10.1172/JCI165933
111. Wezel A, de Vries MR, Lagraauw HM, Foks AC, Kuiper J, Quax PH, et al. Complement factor C5a induces atherosclerotic plaque disruptions. J Cell Mol Med. (2014) 18:2020–30. doi: 10.1111/jcmm.2014.18.issue-10
112. Gravastrand CS, Steinkjer B, Halvorsen B, Landsem A, Skjelland M, Jacobsen EA, et al. Cholesterol crystals induce coagulation activation through complement-dependent expression of monocytic tissue factor. J Immunol. (2019) 203:853–63. doi: 10.4049/jimmunol.1900503
113. Lewis RD, Jackson CL, Morgan BP, Hughes TR. The membrane attack complex of complement drives the progression of atherosclerosis in apolipoprotein E knockout mice. Mol Immunol. (2010) 47:1098–105. doi: 10.1016/j.molimm.2009.10.035
114. Wu G, Hu W, Shahsafaei A, Song W, Dobarro M, Sukhova GK, et al. Complement regulator CD59 protects against atherosclerosis by restricting the formation of complement membrane attack complex. Circ Res. (2009) 104:550–8. doi: 10.1161/CIRCRESAHA.108.191361
115. Selle J, Asare Y, Kohncke J, Alampour-Rajabi S, Shagdarsuren G, Klos A, et al. Atheroprotective role of C5ar2 deficiency in apolipoprotein E-deficient mice. Thromb Haemost. (2015) 114:848–58. doi: 10.1160/TH14-12-1075
116. Shagdarsuren E, Bidzhekov K, Mause SF, Simsekyilmaz S, Polakowski T, Hawlisch H, et al. C5a receptor targeting in neointima formation after arterial injury in atherosclerosis-prone mice. Circulation. (2010) 122:1026–36. doi: 10.1161/CIRCULATIONAHA.110.954370
117. Manthey HD, Thomas AC, Shiels IA, Zernecke A, Woodruff TM, Rolfe B, et al. Complement C5a inhibition reduces atherosclerosis in ApoE-/- mice. FASEB J. (2011) 25:2447–55. doi: 10.1096/fj.10-174284
118. Emmens RW, Wouters D, Zeerleder S, van Ham SM, Niessen HWM, Krijnen PAJ. On the value of therapeutic interventions targeting the complement system in acute myocardial infarction. Transl Res. (2017) 182:103–22. doi: 10.1016/j.trsl.2016.10.005
119. Kiss MG, Binder CJ. The multifaceted impact of complement on atherosclerosis. Atherosclerosis. (2022) 351:29–40. doi: 10.1016/j.atherosclerosis.2022.03.014
120. Maffia P, Mauro C, Case A, Kemper C. Canonical and non-canonical roles of complement in atherosclerosis. Nat Rev Cardiol. (2024) 21:743–61. doi: 10.1038/s41569-024-01016-y
121. Denisenko YK, Kytikova OY, Novgorodtseva TP, Antonyuk MV, Gvozdenko TA, Kantur TA. Lipid-induced mechanisms of metabolic syndrome. J Obes. (2020) 2020:5762395. doi: 10.1155/2020/5762395
122. Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. (2000) 106:171–6. doi: 10.1172/JCI10583
123. Fahed G, Aoun L, Bou Zerdan M, Allam S, Bou Zerdan M, Bouferraa Y, et al. Metabolic syndrome: updates on pathophysiology and management in 2021. Int J Mol Sci. (2022) 23:786. doi: 10.3390/ijms23020786
124. Li M, Chi X, Wang Y, Setrerrahmane S, Xie W, Xu H. Trends in insulin resistance: insights into mechanisms and therapeutic strategy. Signal Transduct Target Ther. (2022) 7:216. doi: 10.1038/s41392-022-01073-0
125. Ajjan R, Carter AM, Somani R, Kain K, Grant PJ. Ethnic differences in cardiovascular risk factors in healthy Caucasian and South Asian individuals with the metabolic syndrome. J Thromb Haemost. (2007) 5:754–60. doi: 10.1111/j.1538-7836.2007.02434.x
126. Garcia-Gonzalez M, Gomez-Bernal F, Quevedo-Abeledo JC, Fernandez-Cladera Y, Gonzalez-Rivero AF, Lopez-Mejias R, et al. The complement system is linked to insulin resistance in patients with systemic lupus erythematosus. Clin Exp Rheumatol. (2024) 42:115–21. doi: 10.55563/clinexprheumatol/t2k0hn
127. Liu Z, Tang Q, Wen J, Tang Y, Huang D, Huang Y, et al. Elevated serum complement factors 3 and 4 are strong inflammatory markers of the metabolic syndrome development: a longitudinal cohort study. Sci Rep. (2016) 6:18713. doi: 10.1038/srep18713
128. Meng G, Zhu Q, Shao J, Zhang Q, Liu L, Wu H, et al. Comparing the diagnostic ability of inflammatory markers in metabolic syndrome. Clin Chim Acta. (2017) 475:1–6. doi: 10.1016/j.cca.2017.09.023
129. Nilsson B, Hamad OA, Ahlstrom H, Kullberg J, Johansson L, Lindhagen L, et al. C3 and C4 are strongly related to adipose tissue variables and cardiovascular risk factors. Eur J Clin Invest. (2014) 44:587–96. doi: 10.1111/eci.2014.44.issue-6
130. Onat A, Hergenc G, Can G, Kaya Z, Yuksel H. Serum complement C3: a determinant of cardiometabolic risk, additive to the metabolic syndrome, in middle-aged population. Metabolism. (2010) 59:628–34. doi: 10.1016/j.metabol.2009.09.006
131. Bratti LOS, do Carmo IAR, Vilela TF, Wopereis S, de Moraes ACR, Borba BGM, et al. Complement component 3 (C3) as a biomarker for insulin resistance after bariatric surgery. Clin Biochem. (2017) 50:529–32. doi: 10.1016/j.clinbiochem.2017.02.006
132. Hillian AD, McMullen MR, Sebastian BM, Roychowdhury S, Kashyap SR, Schauer PR, et al. Mice lacking C1q are protected from high fat diet-induced hepatic insulin resistance and impaired glucose homeostasis. J Biol Chem. (2013) 288:22565–75. doi: 10.1074/jbc.M113.465674
133. Peake PW, Kriketos AD, Campbell LV, Shen Y, Charlesworth JA. The metabolism of isoforms of human adiponectin: studies in human subjects and in experimental animals. Eur J Endocrinol. (2005) 153:409–17. doi: 10.1530/eje.1.01978
134. Hertle E, Stehouwer CD, van Greevenbroek MM. The complement system in human cardiometabolic disease. Mol Immunol. (2014) 61:135–48. doi: 10.1016/j.molimm.2014.06.031
135. Moreno-Navarrete JM, Fernandez-Real JM. The complement system is dysfunctional in metabolic disease: Evidences in plasma and adipose tissue from obese and insulin resistant subjects. Semin Cell Dev Biol. (2019) 85:164–72. doi: 10.1016/j.semcdb.2017.10.025
136. Fernandez-Real JM, Straczkowski M, Vendrell J, Soriguer F, Perez Del Pulgar S, Gallart L, et al. Protection from inflammatory disease in insulin resistance: the role of mannan-binding lectin. Diabetologia. (2006) 49:2402–11. doi: 10.1007/s00125-006-0381-6
137. Kowalska I, Fernandez-Real JM, Straczkowski M, Kozlowska A, Adamska A, Ortega F, et al. Insulin resistance is associated with decreased circulating mannan-binding lectin concentrations in women with polycystic ovary syndrome. Diabetes Care. (2008) 31:e20. doi: 10.2337/dc07-1872
138. Stienstra R, Dijk W, van Beek L, Jansen H, Heemskerk M, Houtkooper RH, et al. Mannose-binding lectin is required for the effective clearance of apoptotic cells by adipose tissue macrophages during obesity. Diabetes. (2014) 63:4143–53. doi: 10.2337/db14-0256
139. Manco M, Fernandez-Real JM, Equitani F, Vendrell J, Valera Mora ME, Nanni G, et al. Effect of massive weight loss on inflammatory adipocytokines and the innate immune system in morbidly obese women. J Clin Endocrinol Metab. (2007) 92:483–90. doi: 10.1210/jc.2006-0960
140. Chen H, Lu J, Chen X, Yu H, Zhang L, Bao Y, et al. Low serum levels of the innate immune component ficolin-3 is associated with insulin resistance and predicts the development of type 2 diabetes. J Mol Cell Biol. (2012) 4:256–7. doi: 10.1093/jmcb/mjs032
141. Engstrom G, Hedblad B, Eriksson KF, Janzon L, Lindgarde F. Complement C3 is a risk factor for the development of diabetes: a population-based cohort study. Diabetes. (2005) 54:570–5. doi: 10.2337/diabetes.54.2.570
142. Martinez-Gomez D, Eisenmann JC, Warnberg J, Gomez-Martinez S, Veses A, Veiga OL, et al. Associations of physical activity, cardiorespiratory fitness and fatness with low-grade inflammation in adolescents: the AFINOS Study. Int J Obes (Lond). (2010) 34:1501–7. doi: 10.1038/ijo.2010.114
143. Phillips CM, Kesse-Guyot E, Ahluwalia N, McManus R, Hercberg S, Lairon D, et al. Dietary fat, abdominal obesity and smoking modulate the relationship between plasma complement component 3 concentrations and metabolic syndrome risk. Atherosclerosis. (2012) 220:513–9. doi: 10.1016/j.atherosclerosis.2011.11.007
144. Volp AC, Barbosa KB, Bressan J. Triacylglycerols and body fat mass are possible independent predictors of C3 in apparently healthy young Brazilian adults. Nutrition. (2012) 28:544–50. doi: 10.1016/j.nut.2011.08.014
145. Muscari A, Antonelli S, Bianchi G, Cavrini G, Dapporto S, Ligabue A, et al. Serum C3 is a stronger inflammatory marker of insulin resistance than C-reactive protein, leukocyte count, and erythrocyte sedimentation rate: comparison study in an elderly population. Diabetes Care. (2007) 30:2362–8. doi: 10.2337/dc07-0637
146. van Greevenbroek MM, Jacobs M, van der Kallen CJ, Vermeulen VM, Jansen EH, Schalkwijk CG, et al. The cross-sectional association between insulin resistance and circulating complement C3 is partly explained by plasma alanine aminotransferase, independent of central obesity and general inflammation (the CODAM study). Eur J Clin Invest. (2011) 41:372–9. doi: 10.1111/j.1365-2362.2010.02418.x
147. Wlazlo N, van Greevenbroek MM, Ferreira I, Feskens EJ, van der Kallen CJ, Schalkwijk CG, et al. Complement factor 3 is associated with insulin resistance and with incident type 2 diabetes over a 7-year follow-up period: the CODAM Study. Diabetes Care. (2014) 37:1900–9. doi: 10.2337/dc13-2804
148. Yang Y, Lu HL, Zhang J, Yu HY, Wang HW, Zhang MX, et al. Relationships among acylation stimulating protein, adiponectin and complement C3 in lean vs obese type 2 diabetes. Int J Obes (Lond). (2006) 30:439–46. doi: 10.1038/sj.ijo.0803173
149. Hernandez-Mijares A, Jarabo-Bueno MM, Lopez-Ruiz A, Sola-Izquierdo E, Morillas-Arino C, Martinez-Triguero ML. Levels of C3 in patients with severe, morbid and extreme obesity: its relationship to insulin resistance and different cardiovascular risk factors. Int J Obes (Lond). (2007) 31:927–32. doi: 10.1038/sj.ijo.0803543
150. Mamane Y, Chung Chan C, Lavallee G, Morin N, Xu LJ, Huang J, et al. The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes. (2009) 58:2006–17. doi: 10.2337/db09-0323
151. Xia Z, Sniderman AD, Cianflone K. Acylation-stimulating protein (ASP) deficiency induces obesity resistance and increased energy expenditure in ob/ob mice. J Biol Chem. (2002) 277:45874–9. doi: 10.1074/jbc.M207281200
152. Fisette A, Poursharifi P, Oikonomopoulou K, Munkonda MN, Lapointe M, Cianflone K. Paradoxical glucose-sensitizing yet proinflammatory effects of acute ASP administration in mice. Mediators Inflamm. (2013) 2013:713284. doi: 10.1155/2013/713284
153. Munkonda MN, Lapointe M, Miegueu P, Roy C, Gauvreau D, Richard D, et al. Recombinant acylation stimulating protein administration to C3-/- mice increases insulin resistance via adipocyte inflammatory mechanisms. PloS One. (2012) 7:e46883. doi: 10.1371/journal.pone.0046883
154. Wamba PC, Mi J, Zhao XY, Zhang MX, Wen Y, Cheng H, et al. Acylation stimulating protein but not complement C3 associates with metabolic syndrome components in Chinese children and adolescents. Eur J Endocrinol. (2008) 159:781–90. doi: 10.1530/EJE-08-0467
155. Moreno-Navarrete JM, Martinez-Barricarte R, Catalan V, Sabater M, Gomez-Ambrosi J, Ortega FJ, et al. Complement factor H is expressed in adipose tissue in association with insulin resistance. Diabetes. (2010) 59:200–9. doi: 10.2337/db09-0700
156. Somani R, Richardson VR, Standeven KF, Grant PJ, Carter AM. Elevated properdin and enhanced complement activation in first-degree relatives of South Asian subjects with type 2 diabetes. Diabetes Care. (2012) 35:894–9. doi: 10.2337/dc11-1483
157. Gauvreau D, Roy C, Tom FQ, Lu H, Miegueu P, Richard D, et al. A new effector of lipid metabolism: complement factor properdin. Mol Immunol. (2012) 51:73–81. doi: 10.1016/j.molimm.2012.02.110
158. Coan PM, Barrier M, Alfazema N, Carter RN, Marion de Proce S, Dopico XC, et al. Complement factor B is a determinant of both metabolic and cardiovascular features of metabolic syndrome. Hypertension. (2017) 70:624–33. doi: 10.1161/HYPERTENSIONAHA.117.09242
159. Phieler J, Chung KJ, Chatzigeorgiou A, Klotzsche-von Ameln A, Garcia-Martin R, Sprott D, et al. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol. (2013) 191:4367–74. doi: 10.4049/jimmunol.1300038
160. Shah DS, McNeilly AD, McCrimmon RJ, Hundal HS. The C5aR1 complement receptor: A novel immunomodulator of insulin action in skeletal muscle. Cell Signal. (2024) 113:110944. doi: 10.1016/j.cellsig.2023.110944
161. Bjerre M, Kistorp C, Hansen TK, Faber J, Lip GY, Hildebrandt P, et al. Complement activation, endothelial dysfunction, insulin resistance and chronic heart failure. Scand Cardiovasc J. (2010) 44:260–6. doi: 10.3109/14017431.2010.484506
162. Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. (2019) 20:242–58. doi: 10.1038/s41580-018-0093-z
163. Global BMIMC, Di Angelantonio E, Bhupathiraju Sh N, Wormser D, Gao P, Kaptoge S, et al. Body-mass index and all-cause mortality: individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet. (2016) 388:776–86. doi: 10.1016/S0140-6736(16)30175-1
164. Bluher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. (2019) 15:288–98. doi: 10.1038/s41574-019-0176-8
165. Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet. (2022) 23:120–33. doi: 10.1038/s41576-021-00414-z
166. Valenzuela PL, Carrera-Bastos P, Castillo-Garcia A, Lieberman DE, Santos-Lozano A, Lucia A. Obesity and the risk of cardiometabolic diseases. Nat Rev Cardiol. (2023) 20:475–94. doi: 10.1038/s41569-023-00847-5
167. Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. (2021) 320:C375–C91. doi: 10.1152/ajpcell.00379.2020
168. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. (2017) 13:633–43. doi: 10.1038/nrendo.2017.90
169. Cook KS, Groves DL, Min HY, Spiegelman BM. A developmentally regulated mRNA from 3T3 adipocytes encodes a novel serine protease homologue. Proc Natl Acad Sci U S A. (1985) 82:6480–4. doi: 10.1073/pnas.82.19.6480
170. Choy LN, Rosen BS, Spiegelman BM. Adipsin and an endogenous pathway of complement from adipose cells. J Biol Chem. (1992) 267:12736–41. doi: 10.1016/S0021-9258(18)42338-1
171. Cianflone K, Maslowska M. Differentiation-induced production of ASP in human adipocytes. Eur J Clin Invest. (1995) 25:817–25. doi: 10.1111/j.1365-2362.1995.tb01690.x
172. White RT, Damm D, Hancock N, Rosen BS, Lowell BB, Usher P, et al. Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. J Biol Chem. (1992) 267:9210–3. doi: 10.1016/S0021-9258(19)50409-4
173. Zhang J, Wright W, Bernlohr DA, Cushman SW, Chen X. Alterations of the classic pathway of complement in adipose tissue of obesity and insulin resistance. Am J Physiol Endocrinol Metab. (2007) 292:E1433–40. doi: 10.1152/ajpendo.00664.2006
174. Pomeroy C, Mitchell J, Eckert E, Raymond N, Crosby R, Dalmasso AP. Effect of body weight and caloric restriction on serum complement proteins, including Factor D/adipsin: studies in anorexia nervosa and obesity. Clin Exp Immunol. (1997) 108:507–15. doi: 10.1046/j.1365-2249.1997.3921287.x
175. Wei JN, Li HY, Sung FC, Lin CC, Chiang CC, Carter AM, et al. Obesity and clustering of cardiovascular disease risk factors are associated with elevated plasma complement C3 in children and adolescents. Pediatr Diabetes. (2012) 13:476–83. doi: 10.1111/j.1399-5448.2012.00864.x
176. Sahebekhtiari N, Saraswat M, Joenvaara S, Jokinen R, Lovric A, Kaye S, et al. Plasma proteomics analysis reveals dysregulation of complement proteins and inflammation in acquired obesity-A study on rare BMI-discordant monozygotic twin pairs. Proteomics Clin Appl. (2019) 13:e1800173. doi: 10.1002/prca.201800173
177. Kaye S, Lokki AI, Hanttu A, Nissila E, Heinonen S, Hakkarainen A, et al. Upregulation of early and downregulation of terminal pathway complement genes in subcutaneous adipose tissue and adipocytes in acquired obesity. Front Immunol. (2017) 8:545. doi: 10.3389/fimmu.2017.00545
178. Flier JS, Lowell B, Napolitano A, Usher P, Rosen B, Cook KS, et al. Adipsin: regulation and dysregulation in obesity and other metabolic states. Recent Prog Horm Res. (1989) 45:567–80. doi: 10.1016/B978-0-12-571145-6.50017-0
179. Platt KA, Claffey KP, Wilkison WO, Spiegelman BM, Ross SR. Independent regulation of adipose tissue-specificity and obesity response of the adipsin promoter in transgenic mice. J Biol Chem. (1994) 269:28558–62. doi: 10.1016/S0021-9258(19)61941-1
180. Plubell DL, Wilmarth PA, Zhao Y, Fenton AM, Minnier J, Reddy AP, et al. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol Cell Proteomics. (2017) 16:873–90. doi: 10.1074/mcp.M116.065524
181. Dusserre E, Moulin P, Vidal H. Differences in mRNA expression of the proteins secreted by the adipocytes in human subcutaneous and visceral adipose tissues. Biochim Biophys Acta. (2000) 1500:88–96. doi: 10.1016/S0925-4439(99)00091-5
182. Gabrielsson BG, Johansson JM, Lonn M, Jernas M, Olbers T, Peltonen M, et al. High expression of complement components in omental adipose tissue in obese men. Obes Res. (2003) 11:699–708. doi: 10.1038/oby.2003.100
183. Lowell BB, Napolitano A, Usher P, Dulloo AG, Rosen BS, Spiegelman BM, et al. Reduced adipsin expression in murine obesity: effect of age and treatment with the sympathomimetic-thermogenic drug mixture ephedrine and caffeine. Endocrinology. (1990) 126:1514–20. doi: 10.1210/endo-126-3-1514
184. Milek M, Moulla Y, Kern M, Stroh C, Dietrich A, Schon MR, et al. Adipsin serum concentrations and adipose tissue expression in people with obesity and type 2 diabetes. Int J Mol Sci. (2022) 23:2222. doi: 10.3390/ijms23042222
185. Peake PW, O’Grady S, Pussell BA, Charlesworth JA. Detection and quantification of the control proteins of the alternative pathway of complement in 3T3-L1 adipocytes. Eur J Clin Invest. (1997) 27:922–7. doi: 10.1046/j.1365-2362.1997.2090759.x
186. Baldo A, Sniderman AD, St-Luce S, Avramoglu RK, Maslowska M, Hoang B, et al. The adipsin-acylation stimulating protein system and regulation of intracellular triglyceride synthesis. J Clin Invest. (1993) 92:1543–7. doi: 10.1172/JCI116733
187. Cianflone K, Roncari DA, Maslowska M, Baldo A, Forden J, Sniderman AD. Adipsin/acylation stimulating protein system in human adipocytes: regulation of triacylglycerol synthesis. Biochemistry. (1994) 33:9489–95. doi: 10.1021/bi00198a014
188. Cianflone KM, Sniderman AD, Walsh MJ, Vu HT, Gagnon J, Rodriguez MA. Purification and characterization of acylation stimulating protein. J Biol Chem. (1989) 264:426–30. doi: 10.1016/S0021-9258(17)31275-9
189. Kalant D, MacLaren R, Cui W, Samanta R, Monk PN, Laporte SA, et al. C5L2 is a functional receptor for acylation-stimulating protein. J Biol Chem. (2005) 280:23936–44. doi: 10.1074/jbc.M406921200
190. Maslowska M, Sniderman AD, Germinario R, Cianflone K. ASP stimulates glucose transport in cultured human adipocytes. Int J Obes Relat Metab Disord. (1997) 21:261–6. doi: 10.1038/sj.ijo.0800396
191. Murray I, Parker RA, Kirchgessner TG, Tran J, Zhang ZJ, Westerlund J, et al. Functional bioactive recombinant acylation stimulating protein is distinct from C3a anaphylatoxin. J Lipid Res. (1997) 38:2492–501. doi: 10.1016/S0022-2275(20)30034-1
192. Ohno M, Hirata T, Enomoto M, Araki T, Ishimaru H, Takahashi TA. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Mol Immunol. (2000) 37:407–12. doi: 10.1016/S0161-5890(00)00067-5
193. Paglialunga S, Fisette A, Yan Y, Deshaies Y, Brouillette JF, Pekna M, et al. Acylation-stimulating protein deficiency and altered adipose tissue in alternative complement pathway knockout mice. Am J Physiol Endocrinol Metab. (2008) 294:E521–9. doi: 10.1152/ajpendo.00590.2007
194. Van Harmelen V, Reynisdottir S, Cianflone K, Degerman E, Hoffstedt J, Nilsell K, et al. Mechanisms involved in the regulation of free fatty acid release from isolated human fat cells by acylation-stimulating protein and insulin. J Biol Chem. (1999) 274:18243–51. doi: 10.1074/jbc.274.26.18243
195. Cianflone K, Xia Z, Chen LY. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim Biophys Acta. (2003) 1609:127–43. doi: 10.1016/S0005-2736(02)00686-7
196. Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, et al. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology. (1999) 42:135–49. doi: 10.1016/S0162-3109(99)00021-1
197. Murray I, Havel PJ, Sniderman AD, Cianflone K. Reduced body weight, adipose tissue, and leptin levels despite increased energy intake in female mice lacking acylation-stimulating protein. Endocrinology. (2000) 141:1041–9. doi: 10.1210/endo.141.3.7364
198. Murray I, Sniderman AD, Havel PJ, Cianflone K. Acylation stimulating protein (ASP) deficiency alters postprandial and adipose tissue metabolism in male mice. J Biol Chem. (1999) 274:36219–25. doi: 10.1074/jbc.274.51.36219
199. Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. (1995) 92:11490–4. doi: 10.1073/pnas.92.25.11490
200. Matsunaga H, Iwashita M, Shinjo T, Yamashita A, Tsuruta M, Nagasaka S, et al. Adipose tissue complement factor B promotes adipocyte maturation. Biochem Biophys Res Commun. (2018) 495:740–8. doi: 10.1016/j.bbrc.2017.11.069
201. Gomez-Banoy N, Guseh JS, Li G, Rubio-Navarro A, Chen T, Poirier B, et al. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat Med. (2019) 25:1739–47. doi: 10.1038/s41591-019-0610-4
202. Lesavre PH, Hugli TE, Esser AF, Muller-Eberhard HJ. The alternative pathway C3/C5 convertase: chemical basis of factor B activation. J Immunol. (1979) 123:529–34. doi: 10.4049/jimmunol.123.2.529
203. Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, et al. Adipsin is an adipokine that improves beta cell function in diabetes. Cell. (2014) 158:41–53. doi: 10.1016/j.cell.2014.06.005
204. Xu Y, Ma M, Ippolito GC, Schroeder HW Jr., Carroll MC, Volanakis JE. Complement activation in factor D-deficient mice. Proc Natl Acad Sci U S A. (2001) 98:14577–82. doi: 10.1073/pnas.261428398
205. Combs TP, Wagner JA, Berger J, Doebber T, Wang WJ, Zhang BB, et al. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology. (2002) 143:998–1007. doi: 10.1210/endo.143.3.8662
206. Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. (2002) 8:731–7. doi: 10.1038/nm724
207. Marangoni RG, Masui Y, Fang F, Korman B, Lord G, Lee J, et al. Adiponectin is an endogenous anti-fibrotic mediator and therapeutic target. Sci Rep. (2017) 7:4397. doi: 10.1038/s41598-017-04162-1
208. Silha JV, Krsek M, Skrha JV, Sucharda P, Nyomba BL, Murphy LJ. Plasma resistin, adiponectin and leptin levels in lean and obese subjects: correlations with insulin resistance. Eur J Endocrinol. (2003) 149:331–5. doi: 10.1530/eje.0.1490331
209. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
210. Raynor LA, Pankow JS, Duncan BB, Schmidt MI, Hoogeveen RC, Pereira MA, et al. Novel risk factors and the prediction of type 2 diabetes in the Atherosclerosis Risk in Communities (ARIC) study. Diabetes Care. (2013) 36:70–6. doi: 10.2337/dc12-0609
211. Fiorentino TV, Hribal ML, Andreozzi F, Perticone M, Sciacqua A, Perticone F, et al. Plasma complement C3 levels are associated with insulin secretion independently of adiposity measures in non-diabetic individuals. Nutr Metab Cardiovasc Dis. (2015) 25:510–7. doi: 10.1016/j.numecd.2015.02.007
212. Figueredo A, Ibarra JL, Bagazgoitia J, Rodriguez A, Molino AM, Fernandez-Cruz A, et al. Plasma C3d levels and ischemic heart disease in type II diabetes. Diabetes Care. (1993) 16:445–9. doi: 10.2337/diacare.16.2.445
213. Kramer U, Herder C, Sugiri D, Strassburger K, Schikowski T, Ranft U, et al. Traffic-related air pollution and incident type 2 diabetes: results from the SALIA cohort study. Environ Health Perspect. (2010) 118:1273–9. doi: 10.1289/ehp.0901689
214. Fujita T, Hemmi S, Kajiwara M, Yabuki M, Fuke Y, Satomura A, et al. Complement-mediated chronic inflammation is associated with diabetic microvascular complication. Diabetes Metab Res Rev. (2013) 29:220–6. doi: 10.1002/dmrr.v29.3
215. Siezenga MA, Chandie Shaw PK, van der Geest RN, Mollnes TE, Daha MR, Rabelink TJ, et al. Enhanced complement activation is part of the unfavourable cardiovascular risk profile in South Asians. Clin Exp Immunol. (2009) 157:98–103. doi: 10.1111/j.1365-2249.2009.03959.x
216. Koistinen HA, Vidal H, Karonen SL, Dusserre E, Vallier P, Koivisto VA, et al. Plasma acylation stimulating protein concentration and subcutaneous adipose tissue C3 mRNA expression in nondiabetic and type 2 diabetic men. Arterioscler Thromb Vasc Biol. (2001) 21:1034–9. doi: 10.1161/01.ATV.21.6.1034
217. Golec E, Ekstrom A, Noga M, Omar-Hmeadi M, Lund PE, Villoutreix BO, et al. Alternative splicing encodes functional intracellular CD59 isoforms that mediate insulin secretion and are down-regulated in diabetic islets. Proc Natl Acad Sci U S A. (2022) 119:e2120083119. doi: 10.1073/pnas.2120083119
218. Krus U, King BC, Nagaraj V, Gandasi NR, Sjolander J, Buda P, et al. The complement inhibitor CD59 regulates insulin secretion by modulating exocytotic events. Cell Metab. (2014) 19:883–90. doi: 10.1016/j.cmet.2014.03.001
219. Golec E, Rosberg R, Zhang E, Renstrom E, Blom AM, King BC. A cryptic non-GPI-anchored cytosolic isoform of CD59 controls insulin exocytosis in pancreatic beta-cells by interaction with SNARE proteins. FASEB J. (2019) 33:12425–34. doi: 10.1096/fj.201901007R
220. Ahren B, Havel PJ, Pacini G, Cianflone K. Acylation stimulating protein stimulates insulin secretion. Int J Obes Relat Metab Disord. (2003) 27:1037–43. doi: 10.1038/sj.ijo.0802369
221. Martinez A, Pio R, Lopez J, Cuttitta F. Expression of the adrenomedullin binding protein, complement factor H, in the pancreas and its physiological impact on insulin secretion. J Endocrinol. (2001) 170:503–11. doi: 10.1677/joe.0.1700503
222. Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1beta release. J Immunol. (2013) 191:1006–10. doi: 10.4049/jimmunol.1300489
223. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
224. Kitagawa K, Rosen BS, Spiegelman BM, Lienhard GE, Tanner LI. Insulin stimulates the acute release of adipsin from 3T3-L1 adipocytes. Biochim Biophys Acta. (1989) 1014:83–9. doi: 10.1016/0167-4889(89)90244-9
225. Atanes P, Ruz-Maldonado I, Pingitore A, Hawkes R, Liu B, Zhao M, et al. C3aR and C5aR1 act as key regulators of human and mouse beta-cell function. Cell Mol Life Sci. (2018) 75:715–26. doi: 10.1007/s00018-017-2655-1
226. Moini JAR, Miller C, Samsam M. Perspective on economics and obesity. Global Health Complications Obes. (2020), 411–23. doi: 10.1016/B978-0-12-819751-6.00018-9
227. Dashty M, Motazacker MM, Levels J, de Vries M, Mahmoudi M, Peppelenbosch MP, et al. Proteome of human plasma very low-density lipoprotein and low-density lipoprotein exhibits a link with coagulation and lipid metabolism. Thromb Haemost. (2014) 111:518–30. doi: 10.1160/TH13-02-0178
228. Xin Y, Hertle E, van der Kallen CJH, Vogelzangs N, Arts ICW, Schalkwijk CG, et al. C3 and alternative pathway components are associated with an adverse lipoprotein subclass profile: The CODAM study. J Clin Lipidol. (2021) 15:311–9. doi: 10.1016/j.jacl.2021.01.011
229. Okada T, Ohama T, Takafuji K, Kanno K, Matsuda H, Sairyo M, et al. Shotgun proteomic analysis reveals proteome alterations in HDL of patients with cholesteryl ester transfer protein deficiency. J Clin Lipidol. (2019) 13:317–25. doi: 10.1016/j.jacl.2019.01.002
230. Vaisar T, Pennathur S, Green PS, Gharib SA, Hoofnagle AN, Cheung MC, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. (2007) 117:746–56. doi: 10.1172/JCI26206
231. Haapasalo K, van Kessel K, Nissila E, Metso J, Johansson T, Miettinen S, et al. Complement factor H binds to human serum apolipoprotein E and mediates complement regulation on high density lipoprotein particles. J Biol Chem. (2015) 290:28977–87. doi: 10.1074/jbc.M115.669226
232. Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. (2019) 25:496–506. doi: 10.1038/s41591-018-0336-8
233. Charlesworth JA, Peake PW, Campbell LV, Pussell BA, O’Grady S, Tzilopoulos T. The influence of oral lipid loads on acylation stimulating protein (ASP) in healthy volunteers. Int J Obes Relat Metab Disord. (1998) 22:1096–102. doi: 10.1038/sj.ijo.0800733
234. Foghmar C, Brons C, Pilely K, Vaag A, Garred P. Complement factors C4 and C3 are down regulated in response to short term overfeeding in healthy young men. Sci Rep. (2017) 7:1235. doi: 10.1038/s41598-017-01382-3
235. Halkes CJ, van Dijk H, de Jaegere PP, Plokker HW, van der Helm Y, Erkelens DW, et al. Postprandial increase of complement component 3 in normolipidemic patients with coronary artery disease: effects of expanded-dose simvastatin. Arterioscler Thromb Vasc Biol. (2001) 21:1526–30. doi: 10.1161/hq0901.095276
236. Meijssen S, van Dijk H, Verseyden C, Erkelens DW, Cabezas MC. Delayed and exaggerated postprandial complement component 3 response in familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. (2002) 22:811–6. doi: 10.1161/01.ATV.0000014079.98335.72
237. Onat A, Uzunlar B, Hergenc G, Yazici M, Sari I, Uyarel H, et al. Cross-sectional study of complement C3 as a coronary risk factor among men and women. Clin Sci (Lond). (2005) 108:129–35. doi: 10.1042/CS20040198
238. van Oostrom AJ, Alipour A, Plokker TW, Sniderman AD, Cabezas MC. The metabolic syndrome in relation to complement component 3 and postprandial lipemia in patients from an outpatient lipid clinic and healthy volunteers. Atherosclerosis. (2007) 190:167–73. doi: 10.1016/j.atherosclerosis.2006.01.009
239. Saleh J, Wahab RA, Farhan H, Al-Amri I, Cianflone K. Plasma levels of acylation-stimulating protein are strongly predicted by waist/hip ratio and correlate with decreased LDL size in men. ISRN Obes. (2013) 2013:342802. doi: 10.1155/2013/342802
240. van Greevenbroek MM, Ghosh S, van der Kallen CJ, Brouwers MC, Schalkwijk CG, Stehouwer CD. Up-regulation of the complement system in subcutaneous adipocytes from nonobese, hypertriglyceridemic subjects is associated with adipocyte insulin resistance. J Clin Endocrinol Metab. (2012) 97:4742–52. doi: 10.1210/jc.2012-2539
241. Fujita T, Fujioka T, Murakami T, Satomura A, Fuke Y, Matsumoto K. Chylomicron accelerates C3 tick-over by regulating the role of factor H, leading to overproduction of acylation stimulating protein. J Clin Lab Anal. (2007) 21:14–23. doi: 10.1002/jcla.20158
242. Wang Y, Van Oort MM, Yao M, van der Horst DJ, Rodenburg KW. Insulin and chromium picolinate induce translocation of CD36 to the plasma membrane through different signaling pathways in 3T3-L1 adipocytes, and with a differential functionality of the CD36. Biol Trace Elem Res. (2011) 142:735–47. doi: 10.1007/s12011-010-8809-8
243. Alper CA, Johnson AM, Birtch AG, Moore FD. Human C’3: evidence for the liver as the primary site of synthesis. Science. (1969) 163:286–8. doi: 10.1126/science.163.3864.286
245. Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A, et al. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet. (2005) 37:835–43. doi: 10.1038/ng1599
246. Chan WK, Chuah KH, Rajaram RB, Lim LL, Ratnasingam J, Vethakkan SR. Metabolic dysfunction-associated steatotic liver disease (MASLD): A state-of-the-art review. J Obes Metab Syndr. (2023) 32:197–213. doi: 10.7570/jomes23052
247. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
248. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology. (2018) 67:328–57. doi: 10.1002/hep.29367
249. Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology. (2015) 149:1784–93. doi: 10.1053/j.gastro.2015.08.011
250. Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. (2010) 53:372–84. doi: 10.1016/j.jhep.2010.04.008
251. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. (2013) 10:627–36. doi: 10.1038/nrgastro.2013.149
252. Jia Q, Li C, Xia Y, Zhang Q, Wu H, Du H, et al. Association between complement C3 and prevalence of fatty liver disease in an adult population: a cross-sectional study from the Tianjin Chronic Low-Grade Systemic Inflammation and Health (TCLSIHealth) cohort study. PloS One. (2015) 10:e0122026. doi: 10.1371/journal.pone.0122026
253. Ursini F, Russo E, Mauro D, Abenavoli L, Ammerata G, Serrao A, et al. Complement C3 and fatty liver disease in Rheumatoid arthritis patients: a cross-sectional study. Eur J Clin Invest. (2017) 47:728–35. doi: 10.1111/eci.2017.47.issue-10
254. Xu C, Chen Y, Xu L, Miao M, Li Y, Yu C. Serum complement C3 levels are associated with nonalcoholic fatty liver disease independently of metabolic features in Chinese population. Sci Rep. (2016) 6:23279. doi: 10.1038/srep23279
255. Yesilova Z, Ozata M, Oktenli C, Bagci S, Ozcan A, Sanisoglu SY, et al. Increased acylation stimulating protein concentrations in nonalcoholic fatty liver disease are associated with insulin resistance. Am J Gastroenterol. (2005) 100:842–9. doi: 10.1111/j.1572-0241.2005.40838.x
256. Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, et al. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology. (2009) 50:1809–17. doi: 10.1002/hep.23228
257. Segers FM, Verdam FJ, de Jonge C, Boonen B, Driessen A, Shiri-Sverdlov R, et al. Complement alternative pathway activation in human nonalcoholic steatohepatitis. PloS One. (2014) 9:e110053. doi: 10.1371/journal.pone.0110053
258. Guo Z, Fan X, Yao J, Tomlinson S, Yuan G, He S. The role of complement in nonalcoholic fatty liver disease. Front Immunol. (2022) 13:1017467. doi: 10.3389/fimmu.2022.1017467
259. Bavia L, Cogliati B, Dettoni JB, Ferreira Alves VA, Isaac L. The complement component C5 promotes liver steatosis and inflammation in murine non-alcoholic liver disease model. Immunol Lett. (2016) 177:53–61. doi: 10.1016/j.imlet.2016.07.014
260. Jiang K, Lu S, Li D, Liu M, Jin H, Lei B, et al. Blockade of C5aR1 alleviates liver inflammation and fibrosis in a mouse model of NASH by regulating TLR4 signaling and macrophage polarization. J Gastroenterol. (2023) 58:894–907. doi: 10.1007/s00535-023-02002-w
261. Lim J, Iyer A, Suen JY, Seow V, Reid RC, Brown L, et al. C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J. (2013) 27:822–31. doi: 10.1096/fj.12-220582
262. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. (2014) 2:e106–16. doi: 10.1016/S2214-109X(13)70145-1
263. Fleckenstein M, Keenan TDL, Guymer RH, Chakravarthy U, Schmitz-Valckenberg S, Klaver CC, et al. Age-related macular degeneration. Nat Rev Dis Primers. (2021) 7:31. doi: 10.1038/s41572-021-00265-2
264. Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. (2000) 45:115–34. doi: 10.1016/S0039-6257(00)00140-5
265. Catala A. An overview of lipid peroxidation with emphasis in outer segments of photoreceptors and the chemiluminescence assay. Int J Biochem Cell Biol. (2006) 38:1482–95. doi: 10.1016/j.biocel.2006.02.010
266. Papac-Milicevic N, Busch CJ, Binder CJ. Malondialdehyde epitopes as targets of immunity and the implications for atherosclerosis. Adv Immunol. (2016) 131:1–59. doi: 10.1016/bs.ai.2016.02.001
267. Wiegand RD, Giusto NM, Rapp LM, Anderson RE. Evidence for rod outer segment lipid peroxidation following constant illumination of the rat retina. Invest Ophthalmol Vis Sci. (1983) 24:1433–5.
268. Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. (2004) 200:1359–70. doi: 10.1084/jem.20031763
269. Curcio CA. Complementing apolipoprotein secretion by cultured retinal pigment epithelium. Proc Natl Acad Sci U S A. (2011) 108:18569–70. doi: 10.1073/pnas.1115497108
270. Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. (1999) 103:117–28. doi: 10.1172/JCI4533
271. Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. (2005) 308:421–4. doi: 10.1126/science.1110189
272. Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. (2005) 308:419–21. doi: 10.1126/science.1110359
273. Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. (2016) 48:134–43. doi: 10.1038/ng.3448
274. Geerlings MJ, de Jong EK, den Hollander AI. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol Immunol. (2017) 84:65–76. doi: 10.1016/j.molimm.2016.11.016
275. Clark SJ, McHarg S, Tilakaratna V, Brace N, Bishop PN. Bruch’s membrane compartmentalizes complement regulation in the eye with implications for therapeutic design in age-related macular degeneration. Front Immunol. (2017) 8:1778. doi: 10.3389/fimmu.2017.01778
276. Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol. (2013) 190:2049–57. doi: 10.4049/jimmunol.1201751
277. Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. Identification of factor H-like protein 1 as the predominant complement regulator in Bruch’s membrane: implications for age-related macular degeneration. J Immunol. (2014) 193:4962–70. doi: 10.4049/jimmunol.1401613
278. Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, et al. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem. (2006) 281:24713–20. doi: 10.1074/jbc.M605083200
279. Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, et al. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J Biol Chem. (2010) 285:30192–202. doi: 10.1074/jbc.M110.103986
280. Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. (2007) 178:3831–6. doi: 10.4049/jimmunol.178.6.3831
281. Molins B, Fuentes-Prior P, Adan A, Anton R, Arostegui JI, Yague J, et al. Complement factor H binding of monomeric C-reactive protein downregulates proinflammatory activity and is impaired with at risk polymorphic CFH variants. Sci Rep. (2016) 6:22889. doi: 10.1038/srep22889
282. Raychaudhuri S, Iartchouk O, Chin K, Tan PL, Tai AK, Ripke S, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet. (2011) 43:1232–6. doi: 10.1038/ng.976
283. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. (2005) 102:7227–32. doi: 10.1073/pnas.0501536102
284. Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med. (2006) 38:592–604. doi: 10.1080/07853890601097030
285. Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. (2006) 38:1173–7. doi: 10.1038/ng1890
286. de Jong S, Gagliardi G, Garanto A, de Breuk A, Lechanteur YTE, Katti S, et al. Implications of genetic variation in the complement system in age-related macular degeneration. Prog Retin Eye Res. (2021) 84:100952. doi: 10.1016/j.preteyeres.2021.100952
287. Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. (2009) 17:100–4. doi: 10.1038/ejhg.2008.140
288. Hallam TM, Marchbank KJ, Harris CL, Osmond C, Shuttleworth VG, Griffiths H, et al. Rare genetic variants in complement factor I lead to low FI plasma levels resulting in increased risk of age-related macular degeneration. Invest Ophthalmol Vis Sci. (2020) 61:18. doi: 10.1167/iovs.61.6.18
289. Java A, Baciu P, Widjajahakim R, Sung YJ, Yang J, Kavanagh D, et al. Functional analysis of rare genetic variants in complement factor I (CFI) using a serum-based assay in advanced age-related macular degeneration. Transl Vis Sci Technol. (2020) 9:37. doi: 10.1167/tvst.9.9.37
290. Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. (2007) 39:1200–1. doi: 10.1038/ng2131
291. McMahon O, Hallam TM, Patel S, Harris CL, Menny A, Zelek WM, et al. The rare C9 P167S risk variant for age-related macular degeneration increases polymerization of the terminal component of the complement cascade. Hum Mol Genet. (2021) 30:1188–99. doi: 10.1093/hmg/ddab086
292. Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. (2013) 45:1366–70. doi: 10.1038/ng.2741
293. Shughoury A, Sevgi DD, Ciulla TA. Molecular genetic mechanisms in age-related macular degeneration. Genes (Basel). (2022) 13:1233. doi: 10.3390/genes13071233
294. van de Ven JP, Nilsson SC, Tan PL, Buitendijk GH, Ristau T, Mohlin FC, et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat Genet. (2013) 45:813–7. doi: 10.1038/ng.2640
295. Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. (2007) 357:553–61. doi: 10.1056/NEJMoa072618
296. Zhang J, Li S, Hu S, Yu J, Xiang Y. Association between genetic variation of complement C3 and the susceptibility to advanced age-related macular degeneration: a meta-analysis. BMC Ophthalmol. (2018) 18:274. doi: 10.1186/s12886-018-0945-5
297. Sun C, Zhao M, Li X. CFB/C2 gene polymorphisms and risk of age-related macular degeneration: a systematic review and meta-analysis. Curr Eye Res. (2012) 37:259–71. doi: 10.3109/02713683.2011.635401
298. Thakkinstian A, McEvoy M, Chakravarthy U, Chakrabarti S, McKay GJ, Ryu E, et al. The association between complement component 2/complement factor B polymorphisms and age-related macular degeneration: a HuGE review and meta-analysis. Am J Epidemiol. (2012) 176:361–72. doi: 10.1093/aje/kws031
299. Aredo B, Li T, Chen X, Zhang K, Wang CX, Gou D, et al. A chimeric Cfh transgene leads to increased retinal oxidative stress, inflammation, and accumulation of activated subretinal microglia in mice. Invest Ophthalmol Vis Sci. (2015) 56:3427–40. doi: 10.1167/iovs.14-16089
300. Armento A, Honisch S, Panagiotakopoulou V, Sonntag I, Jacob A, Bolz S, et al. Loss of Complement Factor H impairs antioxidant capacity and energy metabolism of human RPE cells. Sci Rep. (2020) 10:10320. doi: 10.1038/s41598-020-67292-z
301. Borras C, Canonica J, Jorieux S, Abache T, El Sanharawi M, Klein C, et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci Rep. (2019) 9:13873. doi: 10.1038/s41598-019-50420-9
302. Armento A, Ueffing M, Clark SJ. The complement system in age-related macular degeneration. Cell Mol Life Sci. (2021) 78:4487–505. doi: 10.1007/s00018-021-03796-9
303. Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. (2014) 184:3142–53. doi: 10.1016/j.ajpath.2014.07.017
304. Pauly D, Agarwal D, Dana N, Schafer N, Biber J, Wunderlich KA, et al. Cell-type-specific complement expression in the healthy and diseased retina. Cell Rep. (2019) 29:2835–48 e4. doi: 10.1016/j.celrep.2019.10.084
305. Mullins RF, Dewald AD, Streb LM, Wang K, Kuehn MH, Stone EM. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp Eye Res. (2011) 93:565–7. doi: 10.1016/j.exer.2011.06.015
306. Heesterbeek TJ, Lechanteur YTE, Lores-Motta L, Schick T, Daha MR, Altay L, et al. Complement activation levels are related to disease stage in AMD. Invest Ophthalmol Vis Sci. (2020) 61:18. doi: 10.1167/iovs.61.3.18
Keywords: innate immunity, complement, lipid metabolism, lipid-mediated pathologies, complement system
Citation: Alic L, Dendinovic K and Papac-Milicevic N (2024) The complement system in lipid-mediated pathologies. Front. Immunol. 15:1511886. doi: 10.3389/fimmu.2024.1511886
Received: 15 October 2024; Accepted: 31 October 2024;
Published: 20 November 2024.
Edited by:
Marcin Okrój, Intercollegiate Faculty of Biotechnology of University of Gdańsk and Medical University of Gdańsk, PolandReviewed by:
Utpal Sengupta, The Leprosy Mission Trust India, IndiaCopyright © 2024 Alic, Dendinovic and Papac-Milicevic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolina Papac-Milicevic, bmlrb2xpbmEucGFwYWMtbWlsaWNldmljQG1lZHVuaXdpZW4uYWMuYXQ=