 Yannick Galipeau
Yannick Galipeau Curtis Cooper2
Curtis Cooper2 Marc-André Langlois
Marc-André Langlois
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 January 2025
Sec. Viral Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1509289
Few pathogens have historically been subjected to as intense scientific and clinical scrutiny as SARS-CoV-2. The genetic, immunological, and environmental factors influencing disease severity and post-infection clinical outcomes, known as correlates of immunity, remain largely undefined. Clinical outcomes of SARS-CoV-2 infection vary widely, ranging from asymptomatic cases to those with life-threatening COVID-19 symptoms. While most infected individuals return to their former health and fitness within a few weeks, some develop debilitating chronic symptoms, referred to as long-COVID. Autoimmune responses have been proposed as one of the factors influencing long-COVID and the severity of SARS-CoV-2 infection. The association between viral infections and autoimmune pathologies is not new. Viruses such as Epstein-Barr virus and cytomegalovirus, among others, have been shown to induce the production of autoantibodies and the onset of autoimmune conditions. Given the extensive literature on SARS-CoV-2, here we review current evidence on SARS-CoV-2-induced autoimmune pathologies, with a focus on autoantibodies. We closely examine mechanisms driving autoantibody production, particularly their connection with disease severity and long-COVID.
SARS-CoV-2 has profoundly impacted the lives of the global community since its emergence from Hubei province, China, in late 2019 (1, 2). This novel beta-coronavirus rapidly spread among humans and animals causing the COVID-19 pandemic. Fortunately, the prompt development of vaccines and extensive immunization efforts significantly reduced the mortality rate following infection. Nonetheless, over seven million deaths have so far been attributed to SARS-CoV-2 (3). Despite these medical advances, the incidence of breakthrough infections and vaccine hesitancy among some individuals means that SARS-CoV-2 continues to impose a significant burden on healthcare systems worldwide (4–7).
While most infected individuals return to their pre-infection health levels, some do not. These individuals continue to experience ongoing and/or new chronic symptoms for months after the initial infection (8–10). Although diagnostic guidelines remain unclear, these individuals are generally referred to as having post-acute sequelae of SARS-CoV-2, also known as long-COVID patients or long-haulers (11–13).
Another question that has stymied the scientific community is the highly heterogeneous nature of COVID-19 disease severity (14, 15). While many individuals experience mild or even asymptomatic infections (16, 17), others suffer severe infections requiring hospitalization, which can sometimes result in death (18, 19). Multiple studies have identified independent factors and co-morbidities that increase the likelihood of poor outcomes following infection, such as older age, respiratory illnesses, cardiovascular conditions, obesity, and weak antibody immunity (20, 21). However, these factors do not account for all severe clinical outcomes following infection. Given this conundrum, some have suggested that SARS-CoV-2-induced autoimmune pathologies may be a contributing factor (22–24). It has also been hypothesized that autoantibodies are a contributing factor to long-COVID (25).
The underlying factors governing the etiology and development of autoimmune responses and disease are not well understood and are believed to have roots in genetics, demographic characteristics (e.g., age and sex), and environmental factors (26–28). Viruses are considered one of several central exogenous factors able to trigger autoimmunity (26). Correlational studies have highlighted close links between certain viruses and the subsequent development of autoimmune pathologies. For example, Bjornevik et al. showed that individuals who seroconverted to Epstein-Barr virus (EBV) had a 32-fold increased risk of being diagnosed with multiple sclerosis (MS) (29). Other studies have also linked EBV infections to systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and other autoimmune pathologies (30–33). Type 1 diabetes mellitus is another example of a disease with possible linkages to viral infections and autoimmunity. Enteroviruses such as coxsackievirus and rotavirus have been linked with beta-cell autoimmunity (26, 34–36). Although multiple mechanisms have been proposed to explain viral-induced autoimmunity, the etiological link remains unclear due to the scarcity of underlying mechanistic evidence.
Here, we review the association between SARS-CoV-2 infection and autoimmune pathologies, emphasizing the production of various autoantibody classes. We present current experimental evidence on the presence and induction of these autoantibodies in the context of SARS-CoV-2 infection, and explore potential mechanisms underlying their emergence. Additionally, we address the complexities of establishing causal relationships between autoimmunity and disease processes, situating these findings within the broader context of SARS-CoV-2 disease severity and the evolving clinical picture of long-COVID.
Autoantibodies are immunoglobulins that recognize and bind to self-antigens (e.g., serum proteins, cellular proteins, phospholipids, nucleic acids) (37–39). These antibodies, which seem to defy tolerance mechanisms, have been detected in many vertebrates including sharks, fish, turtles, mice, and humans (40–43). Their ubiquitous prevalence suggests a broader role in homeostasis (44). Some propose that autoantibodies are important for immune regulation and function, clearance of apoptotic cells, transport and modulation of biologically active molecules, and other physiological processes (45). Autoantibodies can differ in isotype and binding properties. Some can bind multiple self-antigens with varying affinities and can be constituted of minimally mutated recombined V(D)J gene sequences, while others have undergone more intense somatic hypermutation resulting in higher affinity autoantibodies (46). While these antibodies can occur in healthy individuals without any obvious impact on health, environmental exposures such as viral infections can sometimes trigger the generation of pathogenic autoantibodies or increase existing autoantibodies to pathogenic levels. This section summarizes experimental evidence and describes various potentially pathogenic autoantibodies linked with SARS-CoV-2 infections (Figure 1).
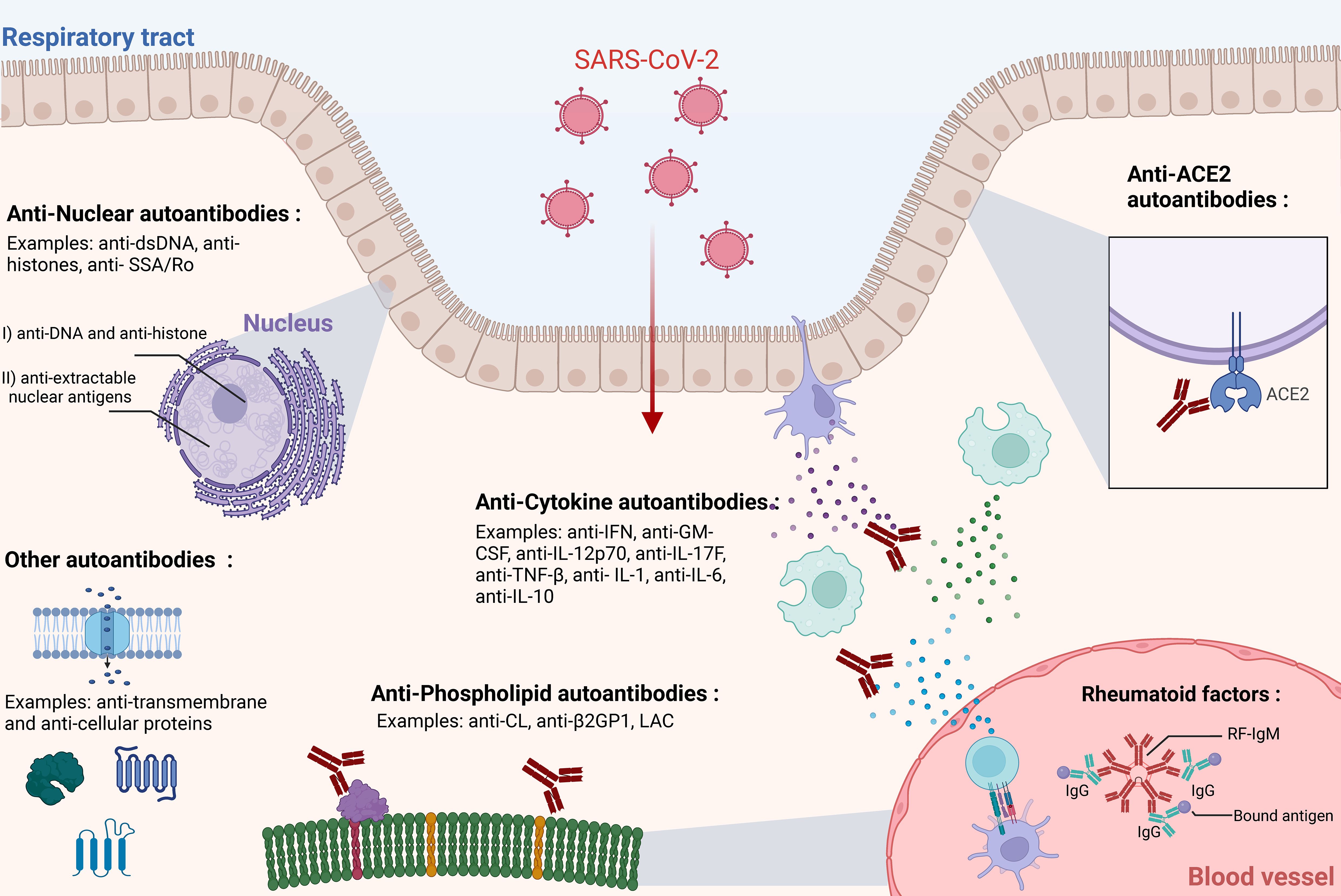
Figure 1. Types of autoantibodies associated with SARS-CoV-2 infection. Multiple studies have highlighted the association, induction, and possible roles of various types of autoantibodies in SARS-CoV-2 infection. This figure illustrates the primary categories of autoantibodies: Anti-ACE2, Anti-Cytokine Antibodies (ACAs), Antinuclear Antibodies (ANAs), and Anti-Phospholipid Antibodies (APLAs), Rheumatoid factors (RFs). Autoantibodies can generally be detected in circulation but may act in specific physiological locations (e.g., blood, respiratory tract epithelium). For simplicity, IgG antibodies are mostly depicted here, although IgM and IgA antibodies may also be present. The respiratory epithelium layer is shown as a representative site for SARS-CoV-2 infection, although the virus can infect multiple other tissues. The figure was created in https://BioRender.com.
Antinuclear antibodies (ANA) are a well-known group of autoantibodies that recognize and bind to antigens within the nuclear compartment (47–49). These autoantibodies are generally classified into two large groups: (I) antibodies targeting DNA and histones, and (II) antibodies targeting extractable nuclear antigens, including RNA-binding proteins complexed to RNA (50, 51). Examples of ANA antibodies include anti-dsDNA, anti-histones, anti-Smith antigen, anti-SSA/Ro, anti-SSB/La, anti-Scl70, and anti-Jo-1 (51). Their combinatorial presence has been used as laboratory biomarkers for several autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Sjögren’s disease, and systemic sclerosis (50, 52–54).
The prevalence of ANA autoantibodies in SARS-CoV-2 infections has been explored by multiple groups. Early in 2020, studies reported ANA seroprevalence in small cohorts of SARS-CoV-2-infected individuals that ranged from 34.5% to 54.5% (55–57). Larger cohorts estimated ANA prevalence at around 35.6% by Gazzaruso et al., around 25% by Lerma et al., and around 57.5% by Sacchi et al. (58–60). Interestingly, ANA autoantibodies cannot simply be explained by hyperglobulinemia, as they are produced in different proportions to total immunoglobulin levels in plasma (61). The notable heterogeneity in seroprevalence of ANA antibodies across studies could be explained by SARS-CoV-2 severity and cohort characteristics. For example, Taeschler et al. observed a higher trend of ANA antibodies in individuals with severe COVID-19 compared to those with milder infections (62). Despite numerous seroprevalence studies on ANA, mechanistic studies are limited. Recently, a longitudinal cohort study of 106 convalescent individuals identified ANA antibodies as a predictor of long-COVID (63). Interestingly, in another large recent study, the authors found an increased risk of an autoimmune pathology diagnosis after COVID-19 in those with detectable ANA antibodies (64). Nonetheless, it is important to note that ANA antibodies are also found in 14-27% of healthy individuals without symptomatic presentations (65–68). The low predictive value of positive antinuclear antibodies (ANA) requires careful correlation with clinical findings to support an autoimmune diagnosis. Consequently, studies linking SARS-CoV-2 infection with ANA positivity must be interpreted within the context of the study’s healthy control cohorts, the specific assays used, and the established positivity thresholds.
Antibodies recognizing various cytokines have been described in the context of viral infections prior to the COVID-19 pandemic (69, 70). Anti-cytokine antibodies (ACA) have been shown to bind a vast array of common cytokines such as IL-1, IL-6, IL-8, IL-12, GM-CSF, IFN-α, IFN-γ, and TNF-α (71–80). Interestingly, certain categories of anti-cytokine antibodies have been linked with disease. For example, anti-GM-CSF autoantibodies are believed to be responsible for idiopathic pulmonary alveolar proteinosis (PAP). While primary PAP is caused by a mutation in the GM-CSF receptor, idiopathic PAP is caused by the neutralization of GM-CSF itself (81–83). Anti-cytokine autoantibodies, particularly anti-IFN-γ, have been linked with poor clinical outcomes and increased disease severity in individuals infected with nontuberculous mycobacteria and other opportunistic infections (84–87). These antibodies can effectively neutralize interferon-gamma, leading to poor immune engagement and an overall compromised immune response (84, 88). Immune suppression by ACA has led to their inclusion in the IUIS classification of primary immunodeficiency diseases, commonly referred to as adult-onset immunodeficiency syndrome (AOID) (89, 90). Bastard et al. first reported that in a cohort of 987 individuals with severe COVID-19, 10.2% had detectable levels of autoantibodies against type I IFN. These autoantibodies were not found in individuals with mild or asymptomatic SARS-CoV-2 infections. The overall seroprevalence of these autoantibodies in healthy individuals was approximately 0.33%, underscoring a significant association with severe COVID-19 (91). These observations have been confirmed by other groups across various demographics (92–96). Anti-IFN antibodies are thought to pre-exist in certain individuals rather than being induced by infection. This highlights their potential to exacerbate common infection severity through IFN neutralization, thereby inhibiting downstream antiviral signaling pathways (91). Other types of ACA have also been identified in severe cases of SARS-CoV-2. For example, Chang et al. used a multiplex protein array to identify antibodies targeting IL-1, -6, -10, -15, -17A, -22, -21, MIP-α, and VEGF-B (61). Another group reported similar findings, including IFN-γ, GM-CSF, IL-12p70, IL-17F, and TNF-β, but only found anti-IL-12p70, anti-IL-22, and anti-IL-6 to be neutralizing. Non-neutralizing anti-cytokine antibodies can still have clinical relevance; however, their role in SARS-CoV-2 infection is not well defined. Key questions remain regarding the triggers for their induction and the persistence of ACA levels over time. Additionally, the clinical impact of most ACAs is poorly understood. In the context of long-COVID, there is currently no evidence to suggest that ACAs contribute to the condition.
Intensive research has centered on the angiotensin-converting enzyme 2 (ACE2) protein, recognized as the entry receptor for both SARS-CoV-1 and SARS-CoV-2. Additionally, ACE2 serves as the entry receptor for NL63, a common human alphacoronavirus (97). Given the key role of this protein in viral entry, some hypothesized that auto-reactivity to ACE2 could contribute to severe COVID-19 symptoms (22, 98). Building on these questions, several studies examined anti-ACE2 autoantibodies in SARS-CoV-2-infected individuals. Arthur et al. found that antibodies recognizing ACE2 were detected in 93% of inpatients, and 81% of convalescent controls, but in none of the healthy controls (99). Another study found IgM antibodies to ACE2 in 27.2% of individuals with severe infections but only 3.8% of those with mild disease, similar to the control cohort (4%) (100). Additional studies have documented the presence of anti-ACE2 antibodies (101, 102). However, anti-ACE2 antibodies have not shown significant capacity to inhibit ACE2 enzymatic activity (99, 100). Interestingly, a study demonstrated the induction of anti-ACE2 antibodies following immunization with recombinant RBD in mice (101). While there is evidence that SARS-CoV-2 infection can induce anti-ACE2 autoantibodies, the topic remains controversial. For example, Chang et al. did not find elevated anti-ACE2 autoantibodies in hospitalized SARS-CoV-2 patients compared to other autoantibodies (61). In a cohort study of 464 individuals, anti-ACE2 IgG autoantibodies were detected in 10.3% of participants, IgA in 6.3%, and IgM in 18.8%, with no association found with SARS-CoV-2 vaccination or infection (103). ACE2 autoantibodies are not exclusive to SARS-CoV-2 infection, having been detected in conditions such as Parkinson’s disease, vasculopathy, and rheumatoid arthritis (104–108). The presence and function of anti-ACE2 antibodies in serum remain unclear, with mixed evidence of their association with SARS-CoV-2 infections.
Anti-phospholipid antibodies (APLAs) target phospholipids and phospholipid-binding proteins such as cardiolipin (CL), β2-glycoprotein-1 (β2GP1), and non-classical antigens like prothrombin (PT) and annexin-V (109–113). These autoantibodies are primarily associated with antiphospholipid syndrome (APS), a condition characterized by thrombotic events and pregnancy complications (113, 114). While murine models have demonstrated APLA pathogenicity in APS, the underlying etiology remains unclear (115, 116).
Historically, APLAs were first identified in fetuses with congenital syphilis in 1906 and have since been linked to infections such as HIV and HCV (117, 118). COVID-19 coagulopathy shares similarities with thrombotic events observed in both standard and catastrophic APS (119, 120). In early 2020, a case report suggested a potential role for APLAs in severe COVID-19 (121). Subsequent studies have further explored this association. For example, critically ill COVID-19 patients were reported to exhibit elevated levels of APLAs, such as IgA against CL and IgG/IgA against β2GP1 (122).
While several other studies have also highlighted the prevalence of elevated APLAs in COVID-19 patients (59, 123–125), Trahtemberg et al., however, found that APLAs associated with increased disease severity of patients with respiratory failure regardless if they were infected by SARS-CoV-2 or not (126). Reports have also correlated APLAs with thrombosis. Helms et al., for instance, identified APLAs in 87.7% of 57 SARS-CoV-2 patients with coagulation abnormalities (127). Supporting evidence from other studies has confirmed similar findings (122, 128).
Mechanistically, Zuo et al. demonstrated that IgG fractions from COVID-19 patients with APLAs induced neutrophil extracellular trap (NET) release in healthy neutrophils and promoted venous thrombosis in mice (129). However, Serrano et al. observed elevated anti-β2GP1 levels in COVID-19 patients but found no association with thrombosis (130). Additionally, some studies reported no link between APLAs and thrombosis (123, 124, 131).The relationship between COVID-19-associated coagulopathy and APLAs remains ambiguous. It is unclear whether these coagulopathies are independent of APLAs, whether APLA prevalence is specific to COVID-19, or if it is simply a feature of severe illness and hospitalization. Uncharacterized APLAs may play a role in these coagulopathies. Notably, baseline APLA levels vary with age and may be influenced by other infections or autoimmune disorders (118, 132, 133).
Rheumatoid factors (RFs) are a common class of autoantibodies that recognize and bind the Fc region of IgG immunoglobulins (Figure 1) (134, 135). RFs can be of any isotype and have been described extensively in the context of rheumatoid arthritis but have also been associated with other autoimmune conditions (e.g., SLE, Sjögren’s disease) (136–138). Notably, RFs have also been described in the context of viral infections, such as hepatitis C virus (HCV) (139, 140). Recently, RFs have been linked to COVID-19. In a cohort of 129 individuals, Xu et al. detected RFs in 20% of individuals infected with COVID-19 (141). Other studies have reported a variable RF seroprevalence among COVID-19 patients, ranging from 20% to 50% (141–144). One study found higher rates of death, ICU admission, and mechanical ventilation in COVID-19 patients that were RF-positive, suggesting a link with disease severity (142). Interestingly, another study identified novel polyreactive RFs in COVID-19, with one RF able to bind epitopes on IgG and the spike protein (145). The role of RFs in SARS-CoV-2 infection and recovery remains unclear. Previous research has suggested that RFs may neutralize virus-antibody complexes (146) and enhance viral clearance (147). Additionally, RF-positive B cells have been shown to present antigens to T cells, potentially augmenting cellular immune responses during viral infections (148).These findings raise the possibility that RFs may play a protective role during SARS-CoV-2 infection; however, further mechanistic studies are required to elucidate their exact function.
Anti-citrullinated protein antibodies (ACPAs) are a notable class of autoantibodies that recognize peptides containing citrulline, a post-translational modification of arginine (149). ACPAs are strongly associated with autoimmunity and serve as a hallmark marker of rheumatoid arthritis (150–152). Early in the COVID-19 pandemic, a case study reported a potential association between ACPAs and SARS-CoV-2 infection (153). However, subsequent studies have produced mixed results, with some supporting a connection between ACPAs and COVID-19, while others have not found a significant link (154–156).
Another group of autoantibodies relevant to SARS-CoV-2 infections are anti-neutrophil cytoplasmic antibodies (ANCAs). These autoantibodies are primarily associated with ANCA-associated vasculitis (AAV) (157). Case reports have documented instances of AAV occurring in the context of SARS-CoV-2 infection (158, 159). However, the association of both ACPAs and ANCAs with COVID-19 requires validation through studies involving larger cohorts.
Recent advancements in high-throughput multiplexed approaches have significantly enhanced our understanding of autoantibody prevalence. For instance, human exoproteome display analysis using a yeast library has facilitated the profiling of several hundred potential autoantigens. These techniques have been applied to studies of SARS-CoV-2 infection and long-COVID. Interestingly, Klein et al. utilized these methods to evaluate autoantibodies against thousands of putative self-antigens. They observed no significant differences in autoantibody levels or specific enrichment in individuals with long COVID (160). However, in severe COVID-19 cases, the same approach revealed a broad range of autoantibodies targeting lymphocyte function, cytokines, complement factors, growth factors, cell surface proteins, and more (95). Other high-throughput approaches confirmed that severe SARS-CoV-2 infections induce autoantibodies against a broad array of secreted and non-secreted proteins (61, 161, 162). These unbiased assays provide a comprehensive assessment of circulating autoantibody distributions.
Studies on the prevalence of autoantibodies across various health conditions, including viral infections such as SARS-CoV-2, are relatively common; however, the mechanisms leading to increased self-tolerance breakdown during viral infections remain unclear. In this review, we describe, explain, and discuss the most relevant mechanisms thought to drive autoantibody production and potential pathogenesis in the context of viral infections (Figure 2). These mechanisms are not mutually exclusive, and multiple pathways may contribute to the pool of circulating autoantibodies. While T cells are also crucial in the generation and maintenance of autoantibodies, their role is beyond the scope of this review.
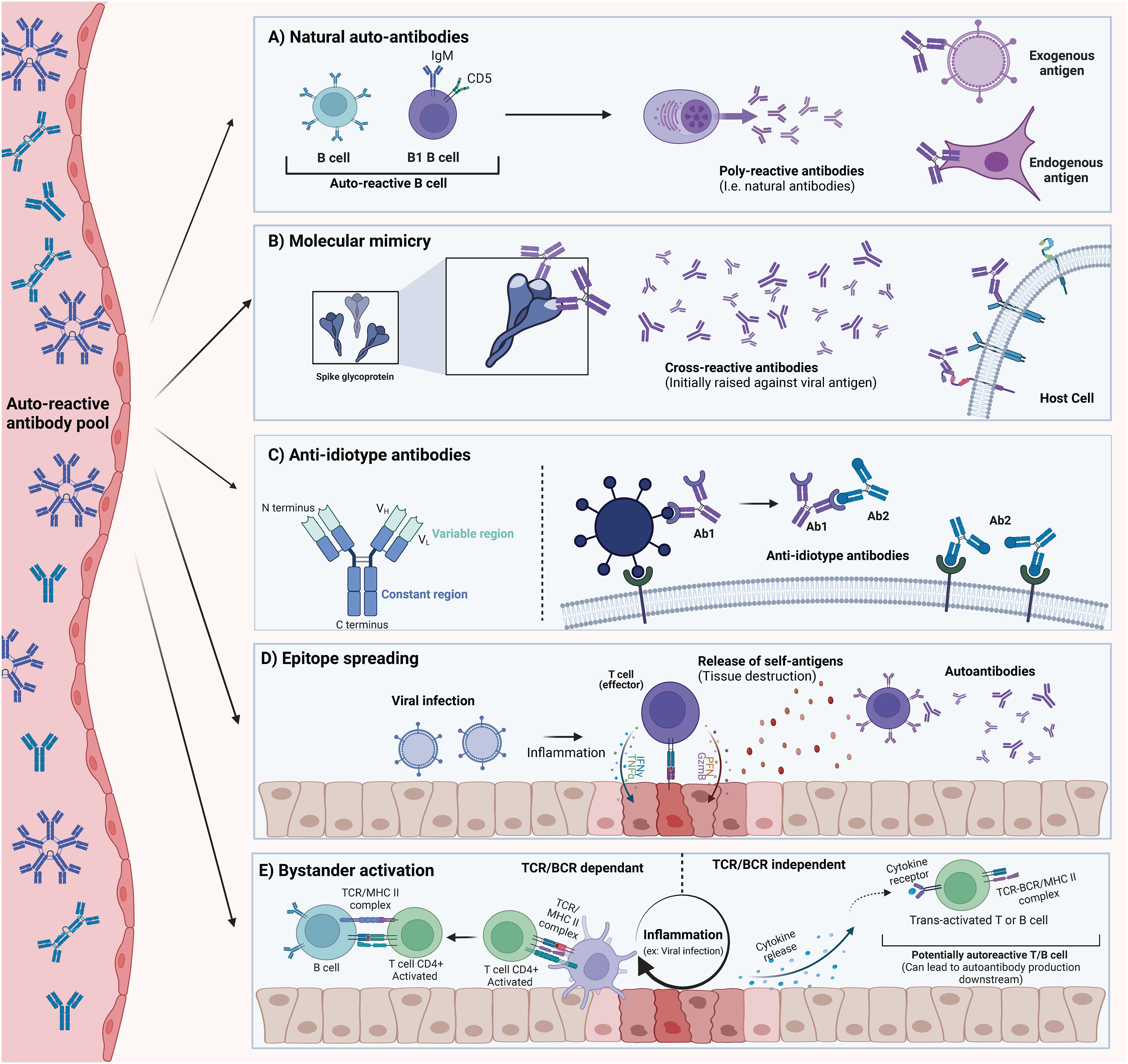
Figure 2. Description of possible mechanisms for autoantibody generation. These mechanisms should be considered as non-exclusive, as more than one mechanism can explain the pool of autoantibodies detected in SARS-CoV-2. (A) Natural Autoantibodies can be found in most vertebrates and seem to be important in normal physiological processes. They are believed to arise mainly from the B1 cell subset. These autoantibodies usually exhibit poly-reactivity to endogenous and exogenous proteins. (B) Molecular Mimicry occurs when an epitope is shared between an exogenous antigen and an endogenous protein. This can result in cross-reactive autoantibodies that bind to host-derived proteins. (C) Anti-Idiotype Autoantibodies (Ab2) are generated against the variable region of another antibody (Ab1). This antibody (Ab2) can potentially retain structural determinants of the original antigen, and in some cases, could bind its target. (D) Epitope Spreading (Intermolecular) can occur when an ongoing inflammatory process (initiated by a viral infection) results in the release of normally sequestered self-antigens. T and B cell specificity can then extend to self-antigens that would not normally be presented. (E) Bystander Activation refers to the loss of TCR/BCR engagement requirement for activation that can happen to immune cells in proximity to a highly inflammatory milieu. Other signals (such as cytokine release) decouple the need for proper engagement with innate cells and BCR/TCR-MHC interaction. This can result in the activation of auto-reactive clones. The figure was created in https://BioRender.com.
Natural autoantibodies are commonly occurring, low-specificity, and polyreactive antibodies found in all vertebrates (44, 163–165). These antibodies can occur independently of antigenic encounters and are able to recognize both endogenous and exogenous antigens (43, 166, 167). Networks of natural antibodies have been proposed to be remnants of evolution prior to the appearance of specific adaptive immune responses (163, 168). The presence of natural autoantibodies raises critical questions about our understanding of self-tolerance mechanisms. Central and peripheral tolerance mechanisms during B cell development are designed to eliminate autoreactive clones. For instance, in the bone marrow, negative selection of immature B cells upon recognition of self-antigens can lead to B cell receptor (BCR) editing (169). In addition, auto-reactive immature B cells that are unable to rescue a self-reactive BCR may undergo apoptosis by a mechanism known as clonal deletion (170). Despite this, a large number of self-reactive clones persist in the periphery where other tolerance checkpoints are in place such as B cell anergy (171). Nevertheless, the immune system appears capable of maintaining a repertoire of B cell receptors that are polyreactive and can secrete polyreactive immunoglobulins. These are believed to be secreted primarily by B1-cells co-expressing CD5, although other subsets have been reported (172–175). Given this, it is crucial to consider that some of the autoantibodies detected during and after SARS-CoV-2 infection may be natural autoantibodies or could result from the expansion or upregulation of polyreactive B cells already present in circulation due to inflammation or immune system activation during infection. A better characterization of the affinities and rate of somatic hypermutation, as well as identifying B-cell subsets able to generate such autoantibodies is critical to understand if natural antibodies play a key role in SARS-CoV-2 related autoantibodies.
Molecular mimicry is one of the most widely recognized mechanisms associated with autoimmunity. In its simplest form, it refers to the phenomenon where certain exogenous antigens, such as viral proteins, including those from SARS-CoV-2, share common epitopes with endogenous antigens (26, 176–178). Antibodies raised against these regions of high homology can in some cases lead to cross-reactive antibodies that can be pathogenic to the host (179–181). In 1962, the work and observations of Kaplan et al. showed that antibodies against group A streptococci can cross-react with cardiac tissues, leading to the death of an individual due to rheumatic pancarditis caused by a streptococcal infection (182). In the following years, multiple lines of evidence led to the acceptance of molecular mimicry as a mechanism explaining cross-reactive antibodies following infection. In fact, molecular mimicry has been observed with various viruses such as Influenza, Zika, Epstein-Barr virus (EBV), and others (183–189). For instance, mimicry between EBV nuclear antigen 1 (EBNA1) and the central nervous system protein glial cell adhesion molecule (GlialCAM) has been shown to generate cross-reactive antibodies, which are detected in approximately 25% of multiple sclerosis (MS) patients (190).
Recent computational analyses of structural homology between SARS-CoV-2 antigens and the human proteome have identified several shared epitopes. For example, Kanduc et al. identified several linear hexapeptide epitopes that displayed conserved homology between SARS-CoV-2 proteins and 460 human proteins (191). Other groups, using slightly different approaches, have also reported several SARS-CoV-2 proteins containing regions that can be referred to as “molecular mimicry hot-spots” (192–194). Interestingly, immunization of mice with the receptor-binding domain (RBD) protein, as demonstrated by Lai et al. generated antibodies capable of binding to ACE2, suggesting the presence of cross-reactive epitopes (101). Validation of these findings from animal models in larger cohorts of vaccinated individuals remains to be conducted. Importantly, molecular mimicry is not a binary “all-or-nothing” model; genetic factors, such as HLA haplotypes, and environmental factors, such as infections, can influence antigen processing and peptide presentation (195).
Anti-idiotype antibodies are not often discussed in the context of autoimmunity. However, these types of antibodies remain interesting as they could explain, at least partially, some of the autoantibodies detected during SARS-CoV-2 infections. The concept of anti-idiotype antibodies, first introduced by Niels Jerne in 1974, suggests that antibodies generated in response to infection (Ab1) possess an immunogenic component within their variable regions that constitutes the idiotype. This region could then be recognized as an antigen, and as such, new antibodies (Ab2) generated against this region would structurally mimic the original antigen (196). This can hypothetically have ramifications whereby anti-idiotype Ab2 antibodies could bind membrane-bound proteins of the host, form immune complexes, and potentially drive pathogenic effects (197, 198). In the context of the SARS-CoV-2 spike protein, one can hypothesize that antibodies against the spike protein could represent the Ab1, and newly generated Ab2 antibodies would be able to bind Ab1 antibodies, but also potentially the ACE2 protein. Interestingly, anti-idiotype antibodies have been explored as possible vaccine candidates for infectious diseases and cancer therapies (199–203). Despite this intriguing hypothesis, at the time of writing this review, no experimental evidence has been reported to support this mechanism in the context of SARS-CoV-2.
Epitope spreading (ES) can be described as the broadening of reactive lymphocytes to other antigen/epitopes (204, 205). Epitope spreading can be subdivided into intramolecular ES when reactive lymphocytes are able to react with cryptic, non-presented, non-available, or sub-dominant epitopes (206, 207). In contrast, ES can also occur through the diversification of reactive T and B lymphocytes toward antigens distinct from the initially presented antigen that triggered their expansion, a phenomenon commonly referred to as intermolecular ES. Intermolecular ES is frequently discussed in contexts of tissue damage, whether caused by direct trauma or by tissue destruction through phagocytic and inflammatory mechanisms (208, 209). Several factors influence the magnitude of ES, some of which include the type and intensity of the primary inflammatory process and the magnitude of the tissue damage (210). It is also important to consider that ES can be a normal feature of immunity, allowing for more efficient and diverse adaptive responses (208). While ES has been associated with multiple viruses, whether ES contributes to SARS-CoV-2-related autoantibodies remains inconclusive (26, 211). However, tissue damage has been extensively described in severe COVID-19 (212, 213). As such, it is reasonable to suggest that ES is a valid possibility that could explain the generation of some of the autoantibodies identified following SARS-CoV-2 infections.
Bystander activation has been proposed as a mechanism that may explain the activation of auto-reactive lymphocytes independently of their BCR/TCR specificity (214, 215). This antigen-independent activation relies on several co-stimulatory signals that decouple the requirement of BCR/TCR signaling with their specific antigen (214, 216). Some of these signaling mediators include ligands (co-signaling receptors, pathogen-associated molecular patterns), cytokines, and chemokines (214, 217–221). These signaling mediators can all occur during infection, resulting in a localized pro-inflammatory environment that can trigger bystander activation of nearby lymphocytes. For example, during primary HIV infection, CD8+ T cells against influenza, EBV, and CMV show markers of activation and expansion in some individuals, despite the absence of cognate antigens (219). Several autoimmune disorders have been associated with bystander activation such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Grave’s disease, and Hashimoto’s thyroiditis (222–225). While direct evidence of bystander activation in SARS-CoV-2 infections is limited, it is well known that severe infections have been correlated with an overall release of pro-inflammatory cytokines (226, 227), which have the potential to initiate bystander activation in some individuals.
In addition to the mechanisms discussed in this review, several other processes may contribute to the generation of autoantibodies. These include direct infection of lymphocytes, the activity of superantigens, and the breakdown of immune tolerance mechanisms (228, 229). It is also important to recognize that polyreactive antibody-secreting cells can positively contribute to antibody-mediated immune responses during infection. For instance, in the context of influenza infections, polyreactive monoclonal antibodies (mAbs) have demonstrated increased binding breadth to antigenically drifted and shifted influenza A virus (IAV) antigens (230). Similarly, in HIV infections, two well-characterized broadly neutralizing mAbs were found to be polyreactive and cross-reactive to cardiolipin (231).
Autoantibodies have also been linked to the relief of inflammatory pathologies, suggesting possible regulatory functions. It is important to consider that autoantibodies may not all be pathogenic, and some may possibly be important features of normal physiology. For example, Sjöwall et al. showed that in patients with systemic lupus erythematosus (SLE) a reduction of TNFα autoantibodies was linked with disease exacerbation (78). Similarly, another group showed that autoantibodies against type 1 IFN in SLE correlated with lower levels of IFN bioactivity and reduced downstream IFN pathways, which correlated with a lower disease score (77). It is also important to consider the regulatory role of antibodies in the treatment of numerous autoimmune or inflammatory conditions. For example, Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, vasculitis, immune thrombocytopenic purpura, and several others are often treated with intravenous immunoglobulins (IVIG) (232–234). Anti-idiotype interactions, inhibition of complement deposition, saturation of the neonatal Fc receptor (FcRn) involved in antibody recycling, and competitive blockade of activating Fc gamma receptors are all non-exclusive mechanisms proposed to explain the effects of intravenous immunoglobulin (IVIG) (233, 235). These examples highlight that antibodies, including autoantibodies, can be important in controlling aberrant or excessive inflammatory processes. Whether autoantibodies associated with SARS-CoV-2 are of regulatory importance in viral infections or post-infection physiological processes remains unclear but represents an interesting proposition.
Research on autoantibodies in individuals infected with or recovering from SARS-CoV-2 has primarily focused on those with severe COVID-19, aiming to elucidate their role in adverse outcomes. As a result, most studies have reported autoantibody presence in hospitalized patients with severe disease, leaving limited data on those with mild or asymptomatic infections. The definitive relationship between autoantibody production and disease severity remains under investigation.
A recent study stratified participants by disease severity, including healthy controls, and found an increased prevalence of autoantibodies—particularly those targeting cardiolipin, claudin, and platelet glycoproteins—with escalating disease severity and advancing age (236). Mechanistically, severe SARS-CoV-2 infections may result in a temporary loss of T cell tolerance. Woodruff et al. demonstrated that severe COVID-19 is associated with the expansion of antibody-secreting B cell populations with low somatic hypermutation, which contract upon recovery, indicating a transient period of reduced selection pressure (229). Similar findings have been observed in acute respiratory distress syndrome caused by bacterial pneumonia, suggesting this phenomenon may represent a physiological response to severe pulmonary infections (229). Recent data from Jaycox et al. suggest that the increase in autoantibodies seen in SARS-CoV-2 infections is not a feature of mRNA vaccinations, suggesting that severe infection may well be the cause (237). While anecdotal reports and small cohort studies have identified autoantibodies after vaccination (238, 239), vaccination remains protective against the development of autoimmune diseases (240, 241).
The underlying mechanisms driving autoantibody production during SARS-CoV-2 infection remain unclear. It is hypothesized that factors such as the intensity of the immune response, inflammation (e.g., cytokine storm), or viral proteins may contribute to their development. Variability in symptom profiles and infection characteristics, such as viral variants, may also influence autoantibody production.
Interestingly, one study highlighted that autoantibodies are not unique to SARS-CoV-2 but are a common feature in critically ill patients with non-SARS-CoV-2 respiratory infections (242).Anti-cytokine autoantibodies were more prevalent in critically ill patients with non-SARS-CoV-2 infections compared to non-infected critically ill individuals (242). Although baseline levels of anti-cytokine autoantibodies (ACAs) in SARS-CoV-2 patients remain understudied, ACAs are recognized as potential risk factors for severe disease. Future longitudinal studies are needed to quantify the risk of severe COVID-19 associated with pre-existing ACAs and to monitor whether specific ACAs are induced during infection.
The COVID-19 pandemic has highlighted the established link between viral infections and autoantibodies with a remarkable volume of literature emerging on this topic. Reaching robust conclusions about the causative factors behind autoantibody production and their physiological roles is challenging due to the wide diversity of study designs (e.g., cross-sectional, longitudinal, retrospective, case reports), laboratory assay methodologies, and the varying characteristics of SARS-CoV-2 infections and clinical outcomes (e.g., disease severity, hospitalization, viral variants, therapeutic interventions, vaccination status, and co-morbidities) (Figure 3).
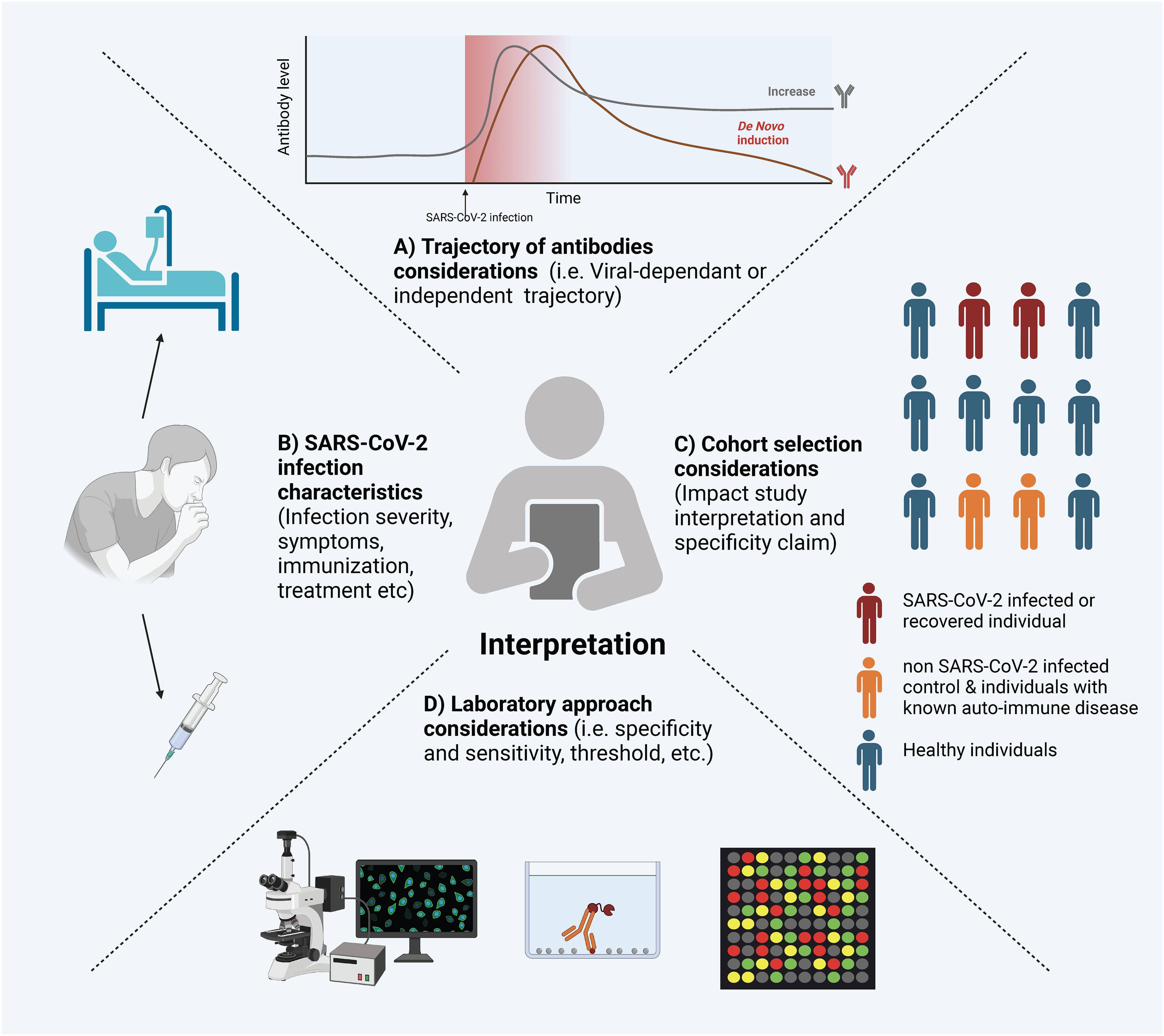
Figure 3. Considerations for Interpreting Experimental Evidence of Autoantibodies Following SARS-CoV-2 Infection. Several factors can influence the interpretation of SARS-CoV-2 induced autoantibodies. Here is a visual representation of a non-exclusive list of factors: (A) The Trajectory of Autoantibodies: Understanding whether autoantibodies are present chronically, induced independently of SARS-CoV-2, or induced by SARS-CoV-2 infection. (B) SARS-CoV-2 Infection Characteristics: Factors such as infection severity, treatment, and immunization status can significantly influence the presence and levels of autoantibodies. (C) Cohort Selection: The selection of cohorts, including characteristics like age, sex, underlying health conditions, and control groups, can have a substantial impact on the interpretation of results. (D) Laboratory Considerations: The experimental methods employed can significantly influence the sensitivity and specificity of autoantibody detection. The figure was created in https://BioRender.com.
The wide range of validated and in-house assays used to measure autoantibodies results in variability in reporting and interpretation, which often depends on the specific method employed. Taking ANA detection as an example, it has traditionally been performed using indirect immunofluorescence (IIF), which is considered the gold standard method (243). However, the development of alternative methods, such as solid-phase assays and bead-based multiplex platforms, has introduced challenges in standardizing results across different techniques (244). Beyond the challenges of standardization, each experimental system has distinct sensitivity and specificity profiles. While most assays used in clinical settings have been validated, in-house assays are inherently more flexible but their performance more heterogeneous. Therefore, it is essential to carefully select the assay type based on the specific research questions being asked. Furthermore, the choice of thresholds for seroprevalence measurements should be considered when comparing findings across different studies.
The presence of autoreactive antibodies during and after SARS-CoV-2 infection is of significant clinical interest, but whether these antibodies are directly induced by the virus remains unclear. For example, Lebedin et al. demonstrated that some autoantibodies associated with COVID-19 are non-specific, polyreactive IgG. These polyreactive antibodies can interfere with the accurate quantification of target-specific and functionally relevant autoantibodies, which may have greater clinical importance (245). Additionally, the profiles of these autoantibodies before, during, and after infection are becoming increasingly well-characterized, enabling the proposal of distinct autoantibody trajectories (Figure 4).
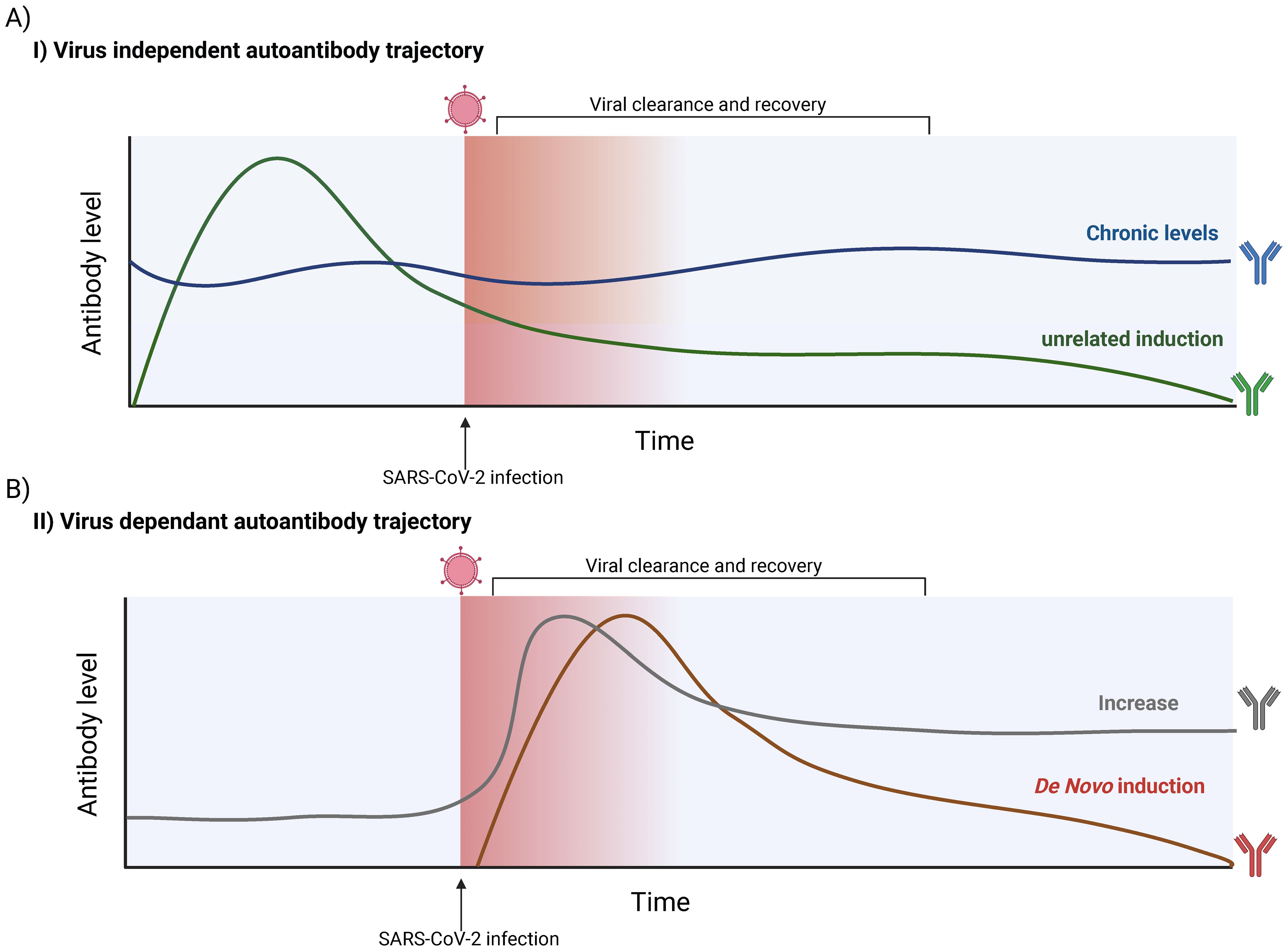
Figure 4. Possible SARS-CoV-2-dependant and -independent trajectories of autoantibody production. (A) Hypothetical trajectories of virus-independent autoantibodies. Autoantibody levels are not influenced by SARS-CoV-2 infection. Such antibodies are generally already present but can vary in level. A chronically present autoantibody (Blue) can remain relatively stable over time, with slight biological fluctuations. A SARS-CoV-2-independent induction can also occur (Green), in which antibody levels can rapidly increase and possibly decay over time. (B) Hypothetical trajectories virus-dependent autoantibodies detected in individuals. The presence and/or quantity of an autoantibody is influenced by SARS-CoV-2 infection. It is possible to see an increase in the titer of an autoantibody already detected in the infected host (Grey). This trajectory contrasts with a de novo induction, where SARS-CoV-2 infection leads to the induction of an autoantibody that was not in circulation prior to the infection (Red). These features of possible trajectories need to be considered when interpreting experimental evidence. The figure was created in https://BioRender.com.
Observational studies and those with limited control cohorts often cannot establish whether SARS-CoV-2 infection directly triggers autoantibody production. Given the heterogeneity of autoantibodies observed across various diseases, it is plausible that some findings may be unrelated to SARS-CoV-2 infection (246). For cases where a link between SARS-CoV-2 and autoantibodies has been demonstrated, a key question remains: are these autoantibodies induced de novo, or are pre-existing antibodies elevated to higher titers? For instance, Bastard et al. showed that autoantibodies targeting interferons (IFNs), associated with severe COVID-19, were present prior to infection but did not cause symptoms (91). This indicates that such autoantibodies may preexist in predisposed individuals. Further longitudinal studies that assess baseline autoantibody levels are needed to clarify their trajectories over time and elucidate their origins.
It is important to understand that the presence of autoreactive antibodies does not automatically indicate a causal relationship with the onset of autoimmune diseases. Notably, conditions associated with autoimmunity, such as myocarditis, arthritis, vasculitis, and encephalitis, have been reported following SARS-CoV-2 infection (247, 248). Retrospective studies in unvaccinated individuals have confirmed that SARS-CoV-2 infection significantly increases the risk of developing autoimmune disorders (240, 247). One study reported a 42.6% higher likelihood of autoimmune disease in individuals previously infected with SARS-CoV-2 (249). However, no definitive mechanistic studies have linked these conditions to autoantibodies, making it difficult to establish causality.
Despite this, the detection of autoantibodies raises important questions about their clinical significance and their potential utility as biomarkers for disease. Future research should focus on disentangling the pathogenic and non-pathogenic roles of these autoantibodies to better understand their implications in post-infection outcomes.
Autoantibodies have been extensively reported in association with SARS-CoV-2 infections, reinforcing the notion that viral infections, are important environmental factors that can induce de novo autoantibody production or amplify existing autoantibody levels. However, there remains a significant gap in our understanding of the precise mechanisms by which these autoantibodies are generated following infection and, more specifically, whether and how they contribute to pathology in the host. It is also plausible that some autoantibodies contribute to regulatory physiological processes during infection or recovery, highlighting their multiple complex roles beyond their known pathogenic effects. Moreover, there is a scarcity of studies exploring therapeutic interventions, such as plasmapheresis or immunosuppressants, specifically targeting these autoantibodies. These gaps in our knowledge represent both challenges and valuable research opportunities. The findings related to SARS-CoV-2 and its association to autoantibodies underscore the complex and intertwined relationship between viral infections and autoimmunity, inviting further exploration into their clinical significance and therapeutic potential.
YG: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. CC: Writing – original draft, Writing – review & editing. M-AL: Conceptualization, Investigation, Supervision, Writing – original draft, Writing – review & editing, Funding acquisition.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported in part by a COVID-19 Rapid Response grant by the Canadian Institute of Health Research (CIHR) (VR2-172722) and by a grant supplement by the Canadian Immunity Task Force (CITF) to M-AL. YG holds a Canadian Institute of Health Research (CIHR) Frederick Banting and Charles Best graduate scholarship (CGS-Doctoral) (476885). M-AL holds the Faculty of Medicine Chair of Excellence in Pandemic Viruses and Preparedness Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that Generative AI was used in the creation of this manuscript. It was used as a tool for English editing purposes only.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
3. WHO. COVID-19 dashboard geneva (2023). Available online at: https://covid19.who.int/ (Accessed November 14, 2024).
4. Myers LC, Liu VX. The COVID-19 pandemic strikes again and again and again. JAMA Network Open. (2022) 5:e221760–e. doi: 10.1001/jamanetworkopen.2022.1760
5. Blumenthal D, Fowler EJ, Abrams M, Collins SR. Covid-19 — Implications for the health care system. New Engl J Med. (2020) 383:1483–8. doi: 10.1056/NEJMsb2021088
7. Duong D, Vogel L. Canada’s public health system is stretched “dangerously thin. Can Med Assoc J. (2022) 194:E57–E. doi: 10.1503/cmaj.1095983
8. Peghin M, Palese A, Venturini M, De Martino M, Gerussi V, Graziano E, et al. Post-COVID-19 symptoms 6 months after acute infection among hospitalized and non-hospitalized patients. Clin Microbiol Infection. (2021) 27:1507–13. doi: 10.1016/j.cmi.2021.05.033
9. Augustin M, Schommers P, Stecher M, Dewald F, Gieselmann L, Gruell H, et al. Post-COVID syndrome in non-hospitalised patients with COVID-19: a longitudinal prospective cohort study. Lancet Regional Health - Europe. (2021) 6:100122. doi: 10.1016/j.lanepe.2021.100122
10. Fernández-de-las-Peñas C, Palacios-Ceña D, Gómez-Mayordomo V, Florencio LL, Cuadrado ML, Plaza-Manzano G, et al. Prevalence of post-COVID-19 symptoms in hospitalized and non-hospitalized COVID-19 survivors: A systematic review and meta-analysis. Eur J Internal Med. (2021) 92:55–70. doi: 10.1016/j.ejim.2021.06.009
11. Crook H, Raza S, Nowell J, Young M, Edison P. Long covid—mechanisms, risk factors, and management. BMJ. (2021) 374::n1648. doi: 10.1136/bmj.n1648
12. Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. (2023) 133–46. doi: 10.1038/s41579-022-00846-2
13. Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. (2021) 12. doi: 10.3389/fmicb.2021.698169
14. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): A review. Jama. (2020) 324:782–93. doi: 10.1001/jama.2020.12839
15. Lamers MM, Haagmans BL. SARS-coV-2 pathogenesis. Nat Rev Microbiol. (2022) 20:270–84. doi: 10.1038/s41579-022-00713-0
16. Oran DP, Topol EJ. The proportion of SARS-coV-2 infections that are asymptomatic. Ann Internal Med. (2021) 174:655–62. doi: 10.7326/M20-6976
17. Sah P, Fitzpatrick MC, Zimmer CF, Abdollahi E, Juden-Kelly L, Moghadas SM, et al. Asymptomatic SARS-CoV-2 infection: A systematic review and meta-analysis. Proc Natl Acad Sci. (2021) 118:e2109229118. doi: 10.1073/pnas.2109229118
18. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
19. Goh KJ, Choong MC, Cheong EH, Kalimuddin S, Duu Wen S, Phua GC, et al. Rapid progression to acute respiratory distress syndrome: review of current understanding of critical illness from coronavirus disease 2019 (COVID-19) infection. Ann Acad Med Singap. (2020) 49:108–18. doi: 10.47102/annals-acadmedsg.
20. Ng WH, Tipih T, Makoah NA, Vermeulen JG, Goedhals D, Sempa JB, et al. Comorbidities in SARS-coV-2 patients: a systematic review and meta-analysis. mBio. (2021) 12. doi: 10.1128/mBio.03647-20
21. Mason KE, Maudsley G, McHale P, Pennington A, Day J, Barr B. Age-adjusted associations between comorbidity and outcomes of COVID-19: A review of the evidence from the early stages of the pandemic. Front Public Health. (2021) 9. doi: 10.3389/fpubh.2021.584182
22. McMillan P, Dexhiemer T, Neubig RR, Uhal BD. COVID-19—A theory of autoimmunity against ACE-2 explained. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.582166
23. Mobasheri L, Nasirpour MH, Masoumi E, Azarnaminy AF, Jafari M, Esmaeili S-A. SARS-CoV-2 triggering autoimmune diseases. Cytokine. (2022) 154:155873. doi: 10.1016/j.cyto.2022.155873
24. Kocivnik N. Velnar T. A Rev Pertaining to SARS-CoV-2 Autoimmune Diseases: What Is Connection? Life. (2022) 12:1918. doi: 10.3390/life12111918
25. Anaya J-M, Herrán M, Beltrán S, Rojas M. Is post-COVID syndrome an autoimmune disease? Expert Rev Clin Immunol. (2022) 18:653–66. doi: 10.1080/1744666X.2022.2085561
26. Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM. Viruses and autoimmunity: A review on the potential interaction and molecular mechanisms. Viruses. (2019) 11. doi: 10.3390/v11080762
27. Arleevskaya MI, Manukyan G, Inoue R, Aminov R. Editorial: microbial and environmental factors in autoimmune and inflammatory diseases. Front Immunol. (2017) 8:243. doi: 10.3389/fimmu.2017.00243
28. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. (2015) 125:2228–33. doi: 10.1172/JCI78088
29. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. (2022) 375:296–301. doi: 10.1126/science.abj8222
30. Moon UY, Park SJ, Oh ST, Kim WU, Park SH, Lee SH, et al. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Res Ther. (2004) 6:R295–302. doi: 10.1186/ar1181
31. Draborg AH, Jørgensen JM, Müller H, Nielsen CT, Jacobsen S, Iversen LV, et al. Epstein-Barr virus early antigen diffuse (EBV-EA/D)-directed immunoglobulin A antibodies in systemic lupus erythematosus patients. Scand J Rheumatol. (2012) 41:280–9. doi: 10.3109/03009742.2012.665944
32. Draborg AH, Duus K, Houen G. Epstein-Barr virus and systemic lupus erythematosus. Clin Dev Immunol. (2012) 2012:370516. doi: 10.1155/2012/370516
33. Dostál C, Newkirk MM, Duffy KN, Palecková A, Bosák V, Cerná M, et al. Herpes viruses in multicase families with rheumatoid arthritis and systemic lupus erythematosus. Ann N Y Acad Sci. (1997) 815:334–7. doi: 10.1111/j.1749-6632.1997.tb52078.x
34. Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, et al. Coxsackievirus B1 is associated with induction of β-cell autoimmunity that portends type 1 diabetes. Diabetes. (2014) 63:446–55. doi: 10.2337/db13-0619
35. Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. Bmj. (2011) 342:d35. doi: 10.1136/bmj.d35
36. Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, et al. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes. (2000) 49:1319–24. doi: 10.2337/diabetes.49.8.1319
37. Elkon K, Casali P. Nature and functions of autoantibodies. Nat Clin Pract Rheumatol. (2008) 4:491–8. doi: 10.1038/ncprheum0895
38. Lutz HU, Binder CJ, Kaveri S. Naturally occurring auto-antibodies in homeostasis and disease. Trends Immunol. (2009) 30:43–51. doi: 10.1016/j.it.2008.10.002
39. Casali P, Schettino EW. Structure and function of natural antibodies. In: Immunology of silicones. Springer Berlin Heidelberg, Berlin, Heidelberg (1996).
40. Adelman MK, Schluter SF, Marchalonis JJ. The natural antibody repertoire of sharks and humans recognizes the potential universe of antigens. Protein J. (2004) 23:103–18. doi: 10.1023/B:JOPC.0000020077.73751.76
41. Gonzalez R, Charlemagne J, Mahana W, Avrameas S. Specificity of natural serum antibodies present in phylogenetically distinct fish species. Immunology. (1988) 63:31–6.
42. Hunter KW, duPré SA, Sharp T, Sandmeier FC, Tracy CR. Western blot can distinguish natural and acquired antibodies to Mycoplasma agassizii in the desert tortoise (Gopherus agassizii). J Microbiological Methods. (2008) 75:464–71. doi: 10.1016/j.mimet.2008.07.022
43. Haury M, Sundblad A, Grandien A, Barreau C, Coutinho A, Nobrega A. The repertoire of serum IgM in normal mice is largely independent of external antigenic contact. Eur J Immunol. (1997) 27:1557–63. doi: 10.1002/eji.1830270635
44. Avrameas S. Natural autoantibodies: from ‘horror autotoxicus’ to ‘gnothi seauton’. Immunol Today. (1991) 12:154–9. doi: 10.1016/S0167-5699(05)80045-3
45. Siloşi I, Siloşi CA, Boldeanu MV, Cojocaru M, Biciuşcă V, Avrămescu CS, et al. The role of autoantibodies in health and disease. Rom J Morphol Embryol. (2016) 57:633–8.
46. Detanico T, St Clair JB, Aviszus K, Kirchenbaum G, Guo W, Wysocki LJ. Somatic mutagenesis in autoimmunity. Autoimmunity. (2013) 46:102–14. doi: 10.3109/08916934.2012.757597
47. Li H, Zheng Y, Chen L, Lin S. High titers of antinuclear antibody and the presence of multiple autoantibodies are highly suggestive of systemic lupus erythematosus. Sci Rep. (2022) 12:1687. doi: 10.1038/s41598-022-05807-6
48. Holborow EJ, Weir DM, Johnson GD. A serum factor in lupus erythematosus with affinity for tissue nuclei. Br Med J. (1957) 2:732–4. doi: 10.1136/bmj.2.5047.732
49. Irure-Ventura J, López-Hoyos M. The past, present, and future in antinuclear antibodies (ANA). Diagnostics (Basel). (2022) 12. doi: 10.3390/diagnostics12030647
50. Pisetsky DS. Antinuclear antibody testing — misunderstood or misbegotten? Nat Rev Rheumatol. (2017) 13:495–502. doi: 10.1038/nrrheum.2017.74
51. Kumar Y, Bhatia A, Minz RW. Antinuclear antibodies and their detection methods in diagnosis of connective tissue diseases: a journey revisited. Diagn Pathol. (2009) 4:1. doi: 10.1186/1746-1596-4-1
52. Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. (2009) 8:632–7. doi: 10.1016/j.autrev.2009.02.010
53. Hoffman IE, Peene I, Meheus L, Huizinga TW, Cebecauer L, Isenberg D, et al. Specific antinuclear antibodies are associated with clinical features in systemic lupus erythematosus. Ann Rheum Dis. (2004) 63:1155–8. doi: 10.1136/ard.2003.013417
54. Gunawardena H. The clinical features of myositis-associated autoantibodies: a review. Clin Rev Allergy Immunol. (2017) 52:45–57. doi: 10.1007/s12016-015-8513-8
55. Gagiannis D, Steinestel J, Hackenbroch C, Schreiner B, Hannemann M, Bloch W, et al. Clinical, serological, and histopathological similarities between severe COVID-19 and acute exacerbation of connective tissue disease-associated interstitial lung disease (CTD-ILD). Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.587517
56. Zhou Y, Han T, Chen J, Hou C, Hua L, He S, et al. Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin Transl Sci. (2020) 13:1077–86. doi: 10.1111/cts.12805
57. Vlachoyiannopoulos PG, Magira E, Alexopoulos H, Jahaj E, Theophilopoulou K, Kotanidou A, et al. Autoantibodies related to systemic autoimmune rheumatic diseases in severely ill patients with COVID-19. Ann Rheumatic Diseases. (2020) 79:1661–3. doi: 10.1136/annrheumdis-2020-218009
58. Lerma LA, Chaudhary A, Bryan A, Morishima C, Wener MH, Fink SL. Prevalence of autoantibody responses in acute coronavirus disease 2019 (COVID-19). J Transl Autoimmun. (2020) 3:100073. doi: 10.1016/j.jtauto.2020.100073
59. Gazzaruso C, Carlo Stella N, Mariani G, Nai C, Coppola A, Naldani D, et al. High prevalence of antinuclear antibodies and lupus anticoagulant in patients hospitalized for SARS-CoV2 pneumonia. Clin Rheumatol. (2020) 39:2095–7. doi: 10.1007/s10067-020-05180-7
60. Sacchi MC, Tamiazzo S, Stobbione P, Agatea L, De Gaspari P, Stecca A, et al. SARS-CoV-2 infection as a trigger of autoimmune response. Clin Transl Sci. (2021) 14:898–907. doi: 10.1111/cts.12953
61. Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun. (2021) 12:5417. doi: 10.1038/s41467-021-25509-3
62. Taeschler P, Cervia C, Zurbuchen Y, Hasler S, Pou C, Tan Z, et al. Autoantibodies in COVID-19 correlate with antiviral humoral responses and distinct immune signatures. Allergy. (2022) 77:2415–30. doi: 10.1111/all.15302
63. Son K, Jamil R, Chowdhury A, Mukherjee M, Venegas C, Miyasaki K, et al. Circulating anti-nuclear autoantibodies in COVID-19 survivors predict long COVID symptoms. Eur Respir J. (2023) 61. doi: 10.1183/13993003.00970-2022
64. Hileman CO, Malakooti SK, Patil N, Singer NG, McComsey GA. New-onset autoimmune disease after COVID-19. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1337406
65. Watanabe A, Kodera M, Sugiura K, Usuda T, Tan EM, Takasaki Y, et al. Anti-DFS70 antibodies in 597 healthy hospital workers. Arthritis Rheumatism. (2004) 50:892–900. doi: 10.1002/art.20096
66. Wandstrat AE, Carr-Johnson F, Branch V, Gray H, Fairhurst A-M, Reimold A, et al. Autoantibody profiling to identify individuals at risk for systemic lupus erythematosus. J Autoimmunity. (2006) 27:153–60. doi: 10.1016/j.jaut.2006.09.001
67. Li X, Liu X, Cui J, Song W, Liang Y, Hu Y, et al. Epidemiological survey of antinuclear antibodies in healthy population and analysis of clinical characteristics of positive population. J Clin Lab Analysis. (2019) 33:e22965. doi: 10.1002/jcla.22965
68. Grygiel-Górniak B, Rogacka N, Puszczewicz M. Antinuclear antibodies in healthy people and non-rheumatic diseases - diagnostic and clinical implications. Reumatologia. (2018) 56:243–8. doi: 10.5114/reum.2018.77976
69. Knight V, Merkel PA, O'Sullivan MD. Anticytokine autoantibodies: association with infection and immune dysregulation. Antibodies (Basel). (2016) 5. doi: 10.3390/antib5010003
70. Browne SK, Holland SM. Anticytokine autoantibodies in infectious diseases: pathogenesis and mechanisms. Lancet Infect Dis. (2010) 10:875–85. doi: 10.1016/S1473-3099(10)70196-1
71. Mizutani H, Ohmoto Y, Kupper TS, Shimizu M. Endogenous neutralizing anti-IL-1 alpha autoantibodies in inflammatory skin diseases: possible natural inhibitor for over expressed epidermal IL-1. J Dermatol Sci. (1998) 20:63–71. doi: 10.1016/s0923-1811(98)00074-7
72. Takemura H, Suzuki H, Yoshizaki K, Ogata A, Yuhara T, Akama T, et al. Anti-interleukin-6 autoantibodies in rheumatic diseases. Increased frequency sera patients systemic sclerosis. Arthritis Rheumatol. (1992) 35:940–3. doi: 10.1002/art.1780350814
73. Fudala R, Krupa A, Stankowska D, Allen TC, Kurdowska AK. Anti-interleukin-8 autoantibody:interleukin-8 immune complexes in acute lung injury/acute respiratory distress syndrome. Clin Sci (Lond). (2008) 114:403–12. doi: 10.1042/CS20070272
74. Meager A, Vincent A, Newsom-Davis J, Willcox N. Spontaneous neutralising antibodies to interferon–alpha and interleukin-12 in thymoma-associated autoimmune disease. Lancet. (1997) 350:1596–7. doi: 10.1016/S0140-6736(05)64012-3
75. Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. (2007) 356:567–79. doi: 10.1056/NEJMoa062505
76. Czaja CA, Merkel PA, Chan ED, Lenz LL, Wolf ML, Alam R, et al. Rituximab as successful adjunct treatment in a patient with disseminated nontuberculous mycobacterial infection due to acquired anti-interferon-γ autoantibody. Clin Infect Dis. (2014) 58:e115–8. doi: 10.1093/cid/cit809
77. Morimoto AM, Flesher DT, Yang J, Wolslegel K, Wang X, Brady A, et al. Association of endogenous anti-interferon-α autoantibodies with decreased interferon-pathway and disease activity in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2011) 63:2407–15. doi: 10.1002/art.30399
78. Sjöwall C, Ernerudh J, Bengtsson AA, Sturfelt G, Skogh T. Reduced anti-TNFalpha autoantibody levels coincide with flare in systemic lupus erythematosus. J Autoimmun. (2004) 22:315–23. doi: 10.1016/j.jaut.2004.02.003
79. Strunz B, Maucourant C, Mehta A, Wan H, Du L, Sun D, et al. Type I interferon autoantibodies correlate with cellular immune alterations in severe COVID-19. J Infect Diseases. (2024) 230:e318–e26. doi: 10.1093/infdis/jiae036
80. Saheb Sharif-Askari N, Saheb Sharif-Askari F, Bayram OS, Hafezi S, Alsayed HAH, Kasim F, et al. Salivary autoantibodies to type I IFNs: Mirror plasma levels, predispose to severe COVID-19, and enhance feasibility for clinical screening. Heart Lung. (2024) 66:31–6. doi: 10.1016/j.hrtlng.2024.03.005
81. Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. (1958) 258:1123–42. doi: 10.1056/NEJM195806052582301
82. Uchida K, Nakata K, Trapnell BC, Terakawa T, Hamano E, Mikami A, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood. (2004) 103:1089–98. doi: 10.1182/blood-2003-05-1565
83. Kitamura T, Tanaka N, Watanabe J, Uchida, Kanegasaki S, Yamada Y, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. (1999) 190:875–80. doi: 10.1084/jem.190.6.875
84. Höflich C, Sabat R, Rosseau S, Temmesfeld B, Slevogt H, Döcke WD, et al. Naturally occurring anti-IFN-gamma autoantibody and severe infections with Mycobacterium cheloneae and Burkholderia cocovenenans. Blood. (2004) 103:673–5. doi: 10.1182/blood-2003-04-1065
85. Döffinger R, Helbert MR, Barcenas-Morales G, Yang K, Dupuis S, Ceron-Gutierrez L, et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin Infect Dis. (2004) 38:e10–4. doi: 10.1086/380453
86. Patel SY, Ding L, Brown MR, Lantz L, Gay T, Cohen S, et al. Anti-IFN-gamma autoantibodies in disseminated nontuberculous mycobacterial infections. J Immunol. (2005) 175:4769–76. doi: 10.4049/jimmunol.175.7.4769
87. Ishii T, Tamura A, Matsui H, Nagai H, Akagawa S, Hebisawa A, et al. Disseminated Mycobacterium avium complex infection in a patient carrying autoantibody to interferon-γ. J Infect Chemother. (2013) 19:1152–7. doi: 10.1007/s10156-013-0572-2
88. Kampmann B, Hemingway C, Stephens A, Davidson R, Goodsall A, Anderson S, et al. Acquired predisposition to mycobacterial disease due to autoantibodies to IFN-gamma. J Clin Invest. (2005) 115:2480–8. doi: 10.1172/JCI19316
89. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. (2014) 5:162. doi: 10.3389/fimmu.2014.00162
90. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
91. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. (2020) 370:eabd4585. doi: 10.1126/science.abd4585
92. Goncalves D, Mezidi M, Bastard P, Perret M, Saker K, Fabien N, et al. Antibodies against type I interferon: detection and association with severe clinical outcome in COVID-19 patients. Clin Trans Immunol. (2021) 10:e1327. doi: 10.1002/cti2.v10.8
93. Troya J, Bastard P, Planas-Serra L, Ryan P, Ruiz M, de Carranza M, et al. Neutralizing autoantibodies to type I IFNs in >10% of patients with severe COVID-19 pneumonia hospitalized in madrid, Spain. J Clin Immunol. (2021) 41:914–22. doi: 10.1007/s10875-021-01036-0
94. Koning R, Bastard P, Casanova J-L, Brouwer MC, van de Beek D, van Agtmael M, et al. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med. (2021) 47:704–6. doi: 10.1007/s00134-021-06392-4
95. Wang EY, Mao T, Klein J, Dai Y, Huck JD, Jaycox JR, et al. Diverse functional autoantibodies in patients with COVID-19. Nature. (2021) 595:283–8. doi: 10.1038/s41586-021-03631-y
96. Solanich X, Rigo-Bonnin R, Gumucio V-D, Bastard P, Rosain J, Philippot Q, et al. Pre-existing autoantibodies neutralizing high concentrations of type I interferons in almost 10% of COVID-19 patients admitted to intensive care in barcelona. J Clin Immunol. (2021) 41:1733–44. doi: 10.1007/s10875-021-01136-x
97. Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pöhlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A. (2005) 102:7988–93. doi: 10.1073/pnas.0409465102
98. Townsend A. Autoimmunity to ACE2 as a possible cause of tissue inflammation in Covid-19. Med Hypotheses. (2020) 144:110043. doi: 10.1016/j.mehy.2020.110043
99. Arthur JM, Forrest JC, Boehme KW, Kennedy JL, Owens S, Herzog C, et al. Development of ACE2 autoantibodies after SARS-CoV-2 infection. PloS One. (2021) 16:e0257016. doi: 10.1371/journal.pone.0257016
100. Casciola-Rosen L, Thiemann DR, Andrade F, Trejo-Zambrano MI, Leonard EK, Spangler JB, et al. IgM anti-ACE2 autoantibodies in severe COVID-19 activate complement and perturb vascular endothelial function. JCI Insight. (2022) 7. doi: 10.1172/jci.insight.158362
101. Lai Y-C, Cheng Y-W, Chao C-H, Chang Y-Y, Chen C-D, Tsai W-J, et al. Antigenic cross-reactivity between SARS-coV-2 S1-RBD and its receptor ACE2. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.868724
102. Rodriguez-Perez AI, Labandeira CM, Pedrosa MA, Valenzuela R, Suarez-Quintanilla JA, Cortes-Ayaso M, et al. Autoantibodies against ACE2 and angiotensin type-1 receptors increase severity of COVID-19. J Autoimmun. (2021) 122:102683. doi: 10.1016/j.jaut.2021.102683
103. Galipeau Y, Castonguay N, McCluskie PS, Sonoda MT, Keeshan A, Collins E, et al. Autoantibodies targeting angiotensin converting enzyme 2 are prevalent and not induced by SARS-coV-2 infection. medRxiv. (2024). 2024.11.18.24317488. doi: 10.1101/2024.11.18.24317488
104. Khajeh Pour S, Scoville C, Tavernier SS, Aghazadeh-Habashi A. Plasma angiotensin peptides as biomarkers of rheumatoid arthritis are correlated with anti-ACE2 auto-antibodies level and disease intensity. Inflammopharmacology. (2022) 30:1295–302. doi: 10.1007/s10787-022-01008-9
105. Labandeira CM, Pedrosa MA, Quijano A, Valenzuela R, Garrido-Gil P, Sanchez-Andrade M, et al. Angiotensin type-1 receptor and ACE2 autoantibodies in Parkinson´s disease. NPJ Parkinson's Disease. (2022) 8:76. doi: 10.1038/s41531-022-00340-9
106. Takahashi Y, Haga S, Ishizaka Y, Mimori A. Autoantibodies to angiotensin-converting enzyme 2 in patients with connective tissue diseases. Arthritis Res Ther. (2010) 12:R85. doi: 10.1186/ar3012
107. Miziołek B, Sieńczyk M, Grzywa R, Łupicka-Słowik A, Kucharz E, Kotyla P, et al. The prevalence and role of functional autoantibodies to angiotensin-converting-enzyme-2 in patients with systemic sclerosis. Autoimmunity. (2021) 54:181–6. doi: 10.1080/08916934.2021.1916915
108. Mecoli CA, Yoshida A, Paik JJ, Lin CT, Danoff S, Hanaoka H, et al. Presence and implications of anti-angiotensin converting enzyme-2 immunoglobulin M antibodies in anti-melanoma-differentiation-associated 5 dermatomyositis. ACR Open Rheumatol. (2022) 4:457–63. doi: 10.1002/acr2.11423
109. Cristiano A, Fortunati V, Cherubini F, Bernardini S, Nuccetelli M. Anti-phospholipids antibodies and immune complexes in COVID-19 patients: a putative role in disease course for anti-annexin-V antibodies. Clin Rheumatol. (2021) 40:2939–45. doi: 10.1007/s10067-021-05580-3
110. Deegan MJ. Anti-phospholipid antibodies. Am J Clin Pathology. (1992) 98:390–1. doi: 10.1093/ajcp/98.4.390
111. Shoenfeld Y, Twig G, Katz U, Sherer Y. Autoantibody explosion in antiphospholipid syndrome. J Autoimmunity. (2008) 30:74–83. doi: 10.1016/j.jaut.2007.11.011
112. Rauch J, D'Agnillo P, Subang R, Levine JS. Anti-phospholipid antibodies (aPL) and apoptosis: prothrombin-dependent aPL as a paradigm for phospholipid-dependent interactions with apoptotic cells. Thromb Res. (2004) 114:371–82. doi: 10.1016/j.thromres.2004.08.007
113. Mackworth-Young CG. Antiphospholipid syndrome: multiple mechanisms. Clin Exp Immunol. (2004) 136:393–401. doi: 10.1111/j.1365-2249.2004.02497.x
114. Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. New Engl J Med. (2002) 346:752–63. doi: 10.1056/NEJMra002974
115. Branch DW, Dudley DJ, Mitchell MD, Creighton KA, Abbott TM, Hammond EH, et al. Immunoglobulin G fractions from patients with antiphospholipid antibodies cause fetal death in BALB/c mice: A model for autoimmune fetal loss. Am J Obstetrics Gynecology. (1990) 163:210–6. doi: 10.1016/S0002-9378(11)90700-5
116. Mason AN, Mageed RA, Mackworth-Young CG. The effects of a human IgM monoclonal anticardiolipin antibody on pregnancy in a transgenic mouse model. Lupus. (2001) 10:289–94. doi: 10.1191/096120301680416986
117. Wassermann A, Neisser A, Bruck C. Eine serodiagnostische Reaktion bei Syphilis. Dtsch Med Wochenschr. (1906) 32:745–6. doi: 10.1055/s-0028-1142018
118. Uthman IW, Gharavi AE. Viral infections and antiphospholipid antibodies. Semin Arthritis Rheumatism. (2002) 31:256–63. doi: 10.1053/sarh.2002.28303
119. Cervera R, Rodríguez-Pintó I, Espinosa G. The diagnosis and clinical management of the catastrophic antiphospholipid syndrome: A comprehensive review. J Autoimmun. (2018) 92:1–11. doi: 10.1016/j.jaut.2018.05.007
120. Serrano M, Espinosa G, Serrano A, Cervera R. Antigens and antibodies of the antiphospholipid syndrome as new allies in the pathogenesis of COVID-19 coagulopathy. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23094946
121. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with covid-19. N Engl J Med. (2020) 382:e38. doi: 10.1056/NEJMc2007575
122. Xiao M, Zhang Y, Zhang S, Qin X, Xia P, Cao W, et al. Antiphospholipid antibodies in critically ill patients with COVID-19. Arthritis Rheumatol. (2020) 72:1998–2004. doi: 10.1002/art.v72.12
123. Bowles L, Platton S, Yartey N, Dave M, Lee K, Hart DP, et al. Lupus anticoagulant and abnormal coagulation tests in patients with covid-19. N Engl J Med. (2020) 383:288–90. doi: 10.1056/NEJMc2013656
124. Siguret V, Voicu S, Neuwirth M, Delrue M, Gayat E, Stépanian A, et al. Are antiphospholipid antibodies associated with thrombotic complications in critically ill COVID-19 patients? Thromb Res. (2020) 195:74–6. doi: 10.1016/j.thromres.2020.07.016
125. Devreese KMJ, Linskens EA, Benoit D, Peperstraete H. Antiphospholipid antibodies in patients with COVID&x2010;19: A relevant observation? J Thromb Haemostasis. (2020) 18:2191–201. doi: 10.1111/jth.14994
126. Trahtemberg U, Rottapel R, Dos Santos CC, Slutsky AS, Baker A, Fritzler MJ. Anticardiolipin and other antiphospholipid antibodies in critically ill COVID-19 positive and negative patients. Ann Rheum Dis. (2021) 80:1236–40. doi: 10.1136/annrheumdis-2021-220206
127. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. (2020) 46:1089–98. doi: 10.1007/s00134-020-06062-x
128. Gil-Etayo FJ, Garcinuño S, Lalueza A, Díaz-Simón R, García-Reyne A, Pleguezuelo DE, et al. Anti-phospholipid antibodies and COVID-19 thrombosis: A co-star, not a supporting actor. Biomedicines. (2021) 9. doi: 10.3390/biomedicines9080899
129. Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.abd3876
130. Serrano M, Espinosa G, Lalueza A, Bravo-Gallego LY, Diaz-Simón R, Garcinuño S, et al. Beta-2-glycoprotein-I deficiency could precipitate an antiphospholipid syndrome-like prothrombotic situation in patients with coronavirus disease 2019. ACR Open Rheumatol. (2021) 3:267–76. doi: 10.1002/acr2.11245
131. Previtali G, Seghezzi M, Moioli V, Sonzogni A, Cerutti L, Marozzi R, et al. The pathogenesis of thromboembolic disease in covid-19 patients: Could be a catastrophic antiphospholipid syndrome? Thromb Res. (2020) 194:192–4. doi: 10.1016/j.thromres.2020.06.042
132. Meroni PL, Borghi MO. Antiphospholipid antibodies and COVID-19 thrombotic vasculopathy: one swallow does not make a summer. Ann Rheumatic Diseases. (2021) 80:1105–7. doi: 10.1136/annrheumdis-2021-220520
133. Manukyan D, Rossmann H, Schulz A, Zeller T, Pfeiffer N, Binder H, et al. Distribution of antiphospholipid antibodies in a large population-based German cohort. Clin Chem Lab Med (CCLM). (2016) 54:1663–70. doi: 10.1515/cclm-2016-0014
134. Pike RM, Sulkin SE, Coggeshall HC. Serological reactions in rheumatoid arthritis: I. Factors affecting the agglutination of sensitized sheep erythrocytes in rheumatoid-arthritis serum1. J Immunol. (1949) 63:441–6. doi: 10.4049/jimmunol.63.4.441
135. Ingegnoli F, Castelli R, Gualtierotti R. Rheumatoid factors: clinical applications. Dis Markers. (2013) 35:726598. doi: 10.1155/2013/726598
136. Witte T, Hartung K, Sachse C, Matthias T, Fricke M, Kalden JR, et al. Rheumatoid factors in systemic lupus erythematosus: association with clinical and laboratory parameters. SLE study Group Rheumatol Int. (2000) 19:107–11. doi: 10.1007/s002960050112
137. Maślińska M, Mańczak M, Kwiatkowska B, Ramsperger V, Shen L, Suresh L. IgA immunoglobulin isotype of rheumatoid factor in primary Sjögren's syndrome. Rheumatol Int. (2021) 41:643–9. doi: 10.1007/s00296-020-04782-3
138. Moore S, Ruska K, Peters L, Olsen NJ. Associations of IgA and IgA-rheumatoid factor with disease features in patients with rheumatoid arthritis. Immunol Invest. (1994) 23:355–65. doi: 10.3109/08820139409066831
139. Banks SE, Riley TR 3rd, Naides SJ. Musculoskeletal complaints and serum autoantibodies associated with chronic hepatitis C and nonalcoholic fatty liver disease. Dig Dis Sci. (2007) 52:1177–82. doi: 10.1007/s10620-006-9109-7
140. Charles ED, Orloff MI, Nishiuchi E, Marukian S, Rice CM, Dustin LB. Somatic hypermutations confer rheumatoid factor activity in hepatitis C virus-associated mixed cryoglobulinemia. Arthritis Rheumatol. (2013) 65:2430–40. doi: 10.1002/art.38041
141. Xu C, Fan J, Luo Y, Zhao Z, Tang P, Yang G, et al. Prevalence and characteristics of rheumatoid-associated autoantibodies in patients with COVID-19. J Inflammation Res. (2021) 14:3123–8. doi: 10.2147/JIR.S312090
142. Jeong H, Baek AR, Park SW, Kim T, Choo EJ, Jeon CH. Rheumatoid factor is associated with severe COVID-19. Int J Rheumatic Diseases. (2023) 26:850–61. doi: 10.1111/1756-185X.14647
143. Elghali M, Bannour I, Touil I, Changuel M, Brahem Y, Jaoued O, et al. Increased Rheumatoid Factor production in patients with severe COVID-19. Diagn Microbiol Infect Dis. (2024) 109:116284. doi: 10.1016/j.diagmicrobio.2024.116284
144. Anaya JM, Monsalve DM, Rojas M, Rodríguez Y, Montoya-García N, Mancera-Navarro LM, et al. Latent rheumatic, thyroid and phospholipid autoimmunity in hospitalized patients with COVID-19. J Transl Autoimmun. (2021) 4:100091. doi: 10.1016/j.jtauto.2021.100091
145. Amjadi MF, Parker MH, Adyniec RR, Zheng Z, Robbins AM, Bashar SJ, et al. Novel and unique rheumatoid factors cross-react with viral epitopes in COVID-19. J Autoimmun. (2024) 142:103132. doi: 10.1016/j.jaut.2023.103132
146. Ashe WK, Daniels CA, Scott GS, Notkins AL. Interaction of rheumatoid factor with infectious herpes simplex virus-antibody complexes. Science. (1971) 172:176–7. doi: 10.1126/science.172.3979.176
147. Van Snick JL, Van Roost E, Markowetz B, Cambiaso CL, Masson PL. Enhancement by IgM rheumatoid factor of in vitro ingestion by macrophages and in vivo clearance of aggregated IgG or antigen-antibody complexes. Eur J Immunol. (1978) 8:279–85. doi: 10.1002/eji.1830080412
148. Roosnek E, Lanzavecchia A. Efficient and selective presentation of antigen-antibody complexes by rheumatoid factor B cells. J Exp Med. (1991) 173:487–9. doi: 10.1084/jem.173.2.487
149. Alghamdi M, Alasmari D, Assiri A, Mattar E, Aljaddawi AA, Alattas SG, et al. An overview of the intrinsic role of citrullination in autoimmune disorders. J Immunol Res. (2019) 2019:7592851. doi: 10.1155/2019/7592851
150. Girbal-Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, et al. The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol. (1999) 162:585–94. doi: 10.4049/jimmunol.162.1.585
151. Rantapää-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheumatol. (2003) 48:2741–9. doi: 10.1002/art.v48:10
152. Willemze A, Trouw LA, Toes REM, Huizinga TWJ. The influence of ACPA status and characteristics on the course of RA. Nat Rev Rheumatol. (2012) 8:144–52. doi: 10.1038/nrrheum.2011.204
153. Perrot L, Hemon M, Busnel JM, Muis-Pistor O, Picard C, Zandotti C, et al. First flare of ACPA-positive rheumatoid arthritis after SARS-CoV-2 infection. Lancet Rheumatol. (2021) 3:e6–8. doi: 10.1016/S2665-9913(20)30396-9
154. Derksen VFAM, Kissel T, Lamers-Karnebeek FBG, van der Bijl AE, Venhuizen AC, Huizinga TWJ, et al. Onset of rheumatoid arthritis after COVID-19: coincidence or connected? Ann Rheumatic Dis. (2021) 80:1096–8. doi: 10.1136/annrheumdis-2021-219859
155. Ursini F, Ruscitti P, D'Angelo S, Cacciapaglia F, De Angelis R, Campochiaro C, et al. Broad clinical spectrum of SARS-CoV-2-associated inflammatory joint disease in adults: a report of 35 cases from the COVID-19 & Autoimmune Systemic Disease Italian study group. Ann Rheumatic Diseases. (2021) 80:1498–501. doi: 10.1136/annrheumdis-2021-220606
156. Lingel H, Meltendorf S, Billing U, Thurm C, Vogel K, Majer C, et al. Unique autoantibody prevalence in long-term recovered SARS-CoV-2-infected individuals. J Autoimmun. (2021) 122:102682. doi: 10.1016/j.jaut.2021.102682
157. Kitching AR, Anders H-J, Basu N, Brouwer E, Gordon J, Jayne DR, et al. ANCA-associated vasculitis. Nat Rev Dis Primers. (2020) 6:71. doi: 10.1038/s41572-020-0204-y
158. Selvaraj V, Moustafa A, Dapaah-Afriyie K, Birkenbach MP. COVID-19-induced granulomatosis with polyangiitis. BMJ Case Rep. (2021) 14:e242142. doi: 10.1136/bcr-2021-242142
159. Uppal NN, Kello N, Shah HH, Khanin Y, De Oleo IR, Epstein E, et al. De novo ANCA-associated vasculitis with glomerulonephritis in COVID-19. Kidney Int Rep. (2020) 5:2079–83. doi: 10.1016/j.ekir.2020.08.012
160. Klein J, Wood J, Jaycox JR, Dhodapkar RM, Lu P, Gehlhausen JR, et al. Distinguishing features of long COVID identified through immune profiling. Nature. (2023) 623:139–48. doi: 10.1038/s41586-023-06651-y
161. Credle JJ, Gunn J, Sangkhapreecha P, Monaco DR, Zheng XA, Tsai H-J, et al. Unbiased discovery of autoantibodies associated with severe COVID-19 via genome-scale self-assembled DNA-barcoded protein libraries. Nat Biomed Engineering. (2022) 6:992–1003. doi: 10.1038/s41551-022-00925-y
162. Matsuda KM, Kawase Y, Iwadoh K, Kurano M, Yatomi Y, Okamoto K, et al. Proteome-wide autoantibody screening and holistic autoantigenomic analysis unveil COVID-19 signature of autoantibody landscape. medRxiv. (2024). 2024.06.07.24308592. doi: 10.1101/2024.06.07.24308592
163. Avrameas S, Alexopoulos H, Moutsopoulos HM. Natural autoantibodies: an undersugn hero of the immune system and autoimmune disorders—A point of view. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.01320
164. Boes M. Role of natural and immune IgM antibodies in immune responses. Mol Immunol. (2000) 37:1141–9. doi: 10.1016/S0161-5890(01)00025-6
165. Lee W, Gaca JG, Edwards LA, Braedehoeft SJ, Parker W, Davis RD. Binding of polyreactive antibodies to self versus foreign antigens. Immunobiology. (2002) 205:95–107. doi: 10.1078/0171-2985-00113
166. Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. (1995) 7:812–8. doi: 10.1016/0952-7915(95)80053-0
167. Maddur MS, Lacroix-Desmazes S, Dimitrov JD, Kazatchkine MD, Bayry J, Kaveri SV. Natural antibodies: from first-line defense against pathogens to perpetual immune homeostasis. Clin Rev Allergy Immunol. (2020) 58:213–28. doi: 10.1007/s12016-019-08746-9
168. Stewart J. Immunoglobulins did not arise in evolution to fight infection. Immunol Today. (1992) 13:396–9. doi: 10.1016/0167-5699(92)90088-O
169. Duong BH, Ota T, Aoki-Ota M, Cooper AB, Ait-Azzouzene D, Vela JL, et al. Negative selection by igM superantigen defines a B cell central tolerance compartment and reveals mutations allowing escape. J Immunol. (2011) 187:5596–605. doi: 10.4049/jimmunol.1102479
170. Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. (2017) 17:281–94. doi: 10.1038/nri.2017.19
171. Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol. (2007) 7:633–43. doi: 10.1038/nri2133
172. Baumgarth N. B-1 cell heterogeneity and the regulation of natural and antigen-induced igM production. Front Immunol. (2016) 7. doi: 10.3389/fimmu.2016.00324
173. Lalor PA. An evolutionarily-conserved role for murine ly-1 B cells in protection against bacterial infections. Autoimmunity. (1991) 10:71–6. doi: 10.3109/08916939108997150
174. Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. (2011) 11:34–46. doi: 10.1038/nri2901
175. Zhou ZH, Notkins AL. Polyreactive antigen-binding B (PAB-) cells are widely distributed and the PAB population consists of both B-1+ and B-1- phenotypes. Clin Exp Immunol. (2004) 137:88–100. doi: 10.1111/j.1365-2249.2004.02511.x
176. Rojas M, Restrepo-Jiménez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, et al. Molecular mimicry and autoimmunity. J Autoimmunity. (2018) 95:100–23. doi: 10.1016/j.jaut.2018.10.012
177. Kohm AP, Fuller KG, Miller SD. Mimicking the way to autoimmunity: an evolving theory of sequence and structural homology. Trends Microbiol. (2003) 11:101–5. doi: 10.1016/S0966-842X(03)00006-4
178. Kanduc D, Stufano A, Lucchese G, Kusalik A. Massive peptide sharing between viral and human proteomes. Peptides. (2008) 29:1755–66. doi: 10.1016/j.peptides.2008.05.022
179. Agmon-Levin N, Blank M, Paz Z, Shoenfeld Y. Molecular mimicry in systemic lupus erythematosus. Lupus. (2009) 18:1181–5. doi: 10.1177/0961203309346653
180. Oldstone MB. Molecular mimicry and immune-mediated diseases. FASEB J. (1998) 12:1255–65. doi: 10.1096/fasebj.12.13.1255
181. Wildner G, Diedrichs-Möhring M. Molecular mimicry and uveitis. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.580636
182. Kaplan M, Meyeserian M. An immunological cross-reaction between group-A streptococcal cells and human heart tissue. Lancet. (1962) 279:706–10. doi: 10.1016/S0140-6736(62)91653-7
183. He D, Zhang H, Xiao J, Zhang X, Xie M, Pan D, et al. Molecular and clinical relationship between live-attenuated Japanese encephalitis vaccination and childhood onset myasthenia gravis. Ann Neurol. (2018) 84:386–400. doi: 10.1002/ana.25267
184. Lucchese G, Kanduc D. Zika virus and autoimmunity: From microcephaly to Guillain-Barré syndrome, and beyond. Autoimmun Rev. (2016) 15:801–8. doi: 10.1016/j.autrev.2016.03.020
185. Lindsey JW. Antibodies to the Epstein-Barr virus proteins BFRF3 and BRRF2 cross-react with human proteins. J Neuroimmunol. (2017) 310:131–4. doi: 10.1016/j.jneuroim.2017.07.013
186. Robinson WH, Steinman L. Epstein-Barr virus and multiple sclerosis. Science. (2022) 375:264–5. doi: 10.1126/science.abm7930
187. Anaya J-M, Ramirez-Santana C, Salgado-Castaneda I, Chang C, Ansari A, Gershwin ME. Zika virus and neurologic autoimmunity: the putative role of gangliosides. BMC Med. (2016) 14:49. doi: 10.1186/s12916-016-0601-y
188. Luo G, Ambati A, Lin L, Bonvalet M, Partinen M, Ji X, et al. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc Natl Acad Sci. (2018) 115:E12323–E32. doi: 10.1073/pnas.1818150116
189. Zhao Z-S, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. (1998) 279:1344–7. doi: 10.1126/science.279.5355.1344
190. Lanz TV, Brewer RC, Ho PP, Moon JS, Jude KM, Fernandez D, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. (2022) 603:321–7. doi: 10.1038/s41586-022-04432-7
191. Kanduc D. From anti-SARS-CoV-2 immune responses to COVID-19 via molecular mimicry Antibodies (Basel). (2020) 9. doi: 10.3390/antib9030033
192. Khavinson V, Terekhov A, Kormilets D, Maryanovich A. Homology between SARS CoV-2 and human proteins. Sci Rep. (2021) 11:17199. doi: 10.1038/s41598-021-96233-7
193. O'Donoghue SI, Schafferhans A, Sikta N, Stolte C, Kaur S, Ho BK, et al. SARS-CoV-2 structural coverage map reveals viral protein assembly, mimicry, and hijacking mechanisms. Mol Syst Biol. (2021) 17:e10079. doi: 10.15252/msb.202010079
194. Nunez-Castilla J, Stebliankin V, Baral P, Balbin CA, Sobhan M, Cickovski T, et al. Potential autoimmunity resulting from molecular mimicry between SARS-coV-2 spike and human proteins. Viruses. (2022) 14. doi: 10.3390/v14071415
195. Liblau R, Gautam AM. HLA. molecular mimicry and multiple sclerosis. Rev Immunogenet. (2000) 2:95–104.
196. Jerne NK. Towards a network theory of the immune system. Ann Immunol (Paris). (1974) 125c:373–89.
197. Murphy WJ, Longo DL. A possible role for anti-idiotype antibodies in SARS-coV-2 infection and vaccination. New Engl J Med. (2021) 386:394–6. doi: 10.1056/NEJMcibr2113694
198. Shukla AK, Misra S. Clinical implications of anti-idiotype antibodies in COVID-19. J Basic Clin Physiol Pharmacol. (2022) 33:727–33. doi: 10.1515/jbcpp-2022-0123
199. Burdette S, Schwartz RS. Idiotypes and idiotypic networks. New Engl J Med. (1987) 317:219–24. doi: 10.1056/NEJM198707233170407
200. Lanier LL, Babcock GF, Raybourne RB, Arnold LW, Warner NL, Haughton G. Mechanism of B cell lymphoma immunotherapy with passive xenogeneic anti-idiotype serum. J Immunol. (1980) 125:1730–6. doi: 10.4049/jimmunol.125.4.1730
201. Hamblin T, Cattan A, Glennie M, MacKenzie M, Stevenson F, Watts H, et al. Initial experience in treating human lymphoma with a chimeric univalent derivative of monoclonal anti-idiotype antibody. Blood. (1987) 69:790–7. doi: 10.1182/blood.V69.3.790.790
202. Bona C, Moran T. Idiotype vaccines. Annales l'Institut Pasteur / Immunologie. (1985) 136:299–312. doi: 10.1016/S0769-2625(85)80002-7
203. Sharpe AH, Gaulton GN, McDade KK, Fields BN, Greene MI. Syngeneic monoclonal antiidiotype can induce cellular immunity to reovirus. J Exp Med. (1984) 160:1195–205. doi: 10.1084/jem.160.4.1195
204. Powell AM, Black MM. Epitope spreading: protection from pathogens, but propagation of autoimmunity? Clin Exp Dermatol. (2001) 26:427–33. doi: 10.1046/j.1365-2230.2001.00852.x
205. Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. (1992) 358:155–7. doi: 10.1038/358155a0
206. Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med. (2003) 197:1501–10. doi: 10.1084/jem.20022031
207. Didona D, Di Zenzo G. Humoral epitope spreading in autoimmune bullous diseases. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.00779
208. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. (2002) 2:85–95. doi: 10.1038/nri724
209. Chan LS, Vanderlugt CJ, Hashimoto T, Nishikawa T, Zone JJ, Black MM, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. (1998) 110:103–9. doi: 10.1046/j.1523-1747.1998.00107.x
210. Cornaby C, Gibbons L, Mayhew V, Sloan CS, Welling A, Poole BD. B cell epitope spreading: Mechanisms and contribution to autoimmune diseases. Immunol Letters. (2015) 163:56–68. doi: 10.1016/j.imlet.2014.11.001
211. Miller SD, Katz-Levy Y, Neville KL, Vanderlugt CL. Virus-induced autoimmunity: Epitope spreading to myelin autoepitopes in theiler's virus infection of the central nervous system. Adv Virus Res 56: Acad Press;. (2001) p:199–217. doi: 10.1016/S0065-3527(01)56008-X
212. D’Agnillo F, Walters K-A, Xiao Y, Sheng Z-M, Scherler K, Park J, et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci Trans Med. (2021) 13:eabj7790. doi: 10.1126/scitranslmed.abj7790
213. Sauter JL, Baine MK, Butnor KJ, Buonocore DJ, Chang JC, Jungbluth AA, et al. Insights into pathogenesis of fatal COVID-19 pneumonia from histopathology with immunohistochemical and viral RNA studies. Histopathology. (2020) 77:915–25. doi: 10.1111/his.14201
214. Pacheco Y, Acosta-Ampudia Y, Monsalve DM, Chang C, Gershwin ME, Anaya J-M. Bystander activation and autoimmunity. J Autoimmunity. (2019) 103:102301. doi: 10.1016/j.jaut.2019.06.012
215. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. (1996) 272:1947–50. doi: 10.1126/science.272.5270.1947
216. Lee H, Jeong S, Shin E-C. Significance of bystander T cell activation in microbial infection. Nat Immunol. (2022) 23:13–22. doi: 10.1038/s41590-021-00985-3
217. Di Genova G, Savelyeva N, Suchacki A, Thirdborough SM, Stevenson FK. Bystander stimulation of activated CD4+ T cells of unrelated specificity following a booster vaccination with tetanus toxoid. Eur J Immunol. (2010) 40:976–85. doi: 10.1002/eji.200940017
218. Eberl GR, Brawand P, MacDonald HR. Selective bystander proliferation of memory CD4+ and CD8+ T cells upon NK T or T cell activation. J Immunol. (2000) 165:4305–11. doi: 10.4049/jimmunol.165.8.4305
219. Doisne J-M, Urrutia A, Lacabaratz-Porret C, Cc G, Meyer L, Chaix M-L, et al. Cytomegalovirus, and influenza virus are activated during primary HIV infection1. J Immunol. (2004) 173:2410–8. doi: 10.4049/jimmunol.173.4.2410
220. Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, et al. Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep. (2013) 3:701–8. doi: 10.1016/j.celrep.2013.02.020
221. Kim T-S, Shin E-C. The activation of bystander CD8+ T cells and their roles in viral infection. Exp Mol Med. (2019) 51:1–9. doi: 10.1038/s12276-019-0316-1
222. Pan Q, Liu Z, Liao S, Ye L, Lu X, Chen X, et al. Current mechanistic insights into the role of infection in systemic lupus erythematosus. Biomedicine Pharmacotherapy. (2019) 117:109122. doi: 10.1016/j.biopha.2019.109122
223. Quaglia M, Merlotti G, De Andrea M, Borgogna C, Cantaluppi V. Viral infections and systemic lupus erythematosus: new players in an old story. Viruses. (2021) 13. doi: 10.3390/v13020277
224. Kobayashi M, Yasui N, Ishimaru N, Arakaki R, Hayashi Y. Development of autoimmune arthritis with aging via bystander T cell activation in the mouse model of Sjögren's syndrome. Arthritis Rheumatol. (2004) 50:3974–84. doi: 10.1002/art.v50:12
225. McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocrine Rev. (2014) 35:59–105. doi: 10.1210/er.2013-1055
226. Montazersaheb S, Hosseiniyan Khatibi SM, Hejazi MS, Tarhriz V, Farjami A, Ghasemian Sorbeni F, et al. COVID-19 infection: an overview on cytokine storm and related interventions. Virol J. (2022) 19:92. doi: 10.1186/s12985-022-01814-1
227. Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee J-Y, Plociennikowska A, et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun Biol. (2022) 5:45. doi: 10.1038/s42003-021-02983-5
228. Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest. (2001) 108:1097–104. doi: 10.1172/JCI200114235
229. Woodruff MC, Ramonell RP, Haddad NS, Anam FA, Rudolph ME, Walker TA, et al. Dysregulated naive B cells and de novo autoreactivity in severe COVID-19. Nature. (2022) 611:139–47. doi: 10.1038/s41586-022-05273-0
230. Guthmiller JJ, Lan LY, Fernández-Quintero ML, Han J, Utset HA, Bitar DJ, et al. Polyreactive Broadly Neutralizing B cells Are Selected to Provide Defense against Pandemic Threat Influenza Viruses. Immunity. (2020) 53:1230–44.e5. doi: 10.1016/j.immuni.2020.10.005
231. Haynes BF, Fleming J, St. Clair EW, Katinger H, Stiegler G, Kunert R, et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. (2005) 308:1906–8. doi: 10.1126/science.1111781
232. Jolles S, Sewell WA, Misbah SA. Clinical uses of intravenous immunoglobulin. Clin Exp Immunol. (2005) 142:1–11. doi: 10.1111/j.1365-2249.2005.02834.x
233. Berger M, McCallus DE, Lin CS. Rapid and reversible responses to IVIG in autoimmune neuromuscular diseases suggest mechanisms of action involving competition with functionally important autoantibodies. J Peripher Nerv Syst. (2013) 18:275–96. doi: 10.1111/jns5.2013.18.issue-4
234. Siragam V, Crow AR, Brinc D, Song S, Freedman J, Lazarus AH. Intravenous immunoglobulin ameliorates ITP via activating Fcγ receptors on dendritic cells. Nat Med. (2006) 12:688–92. doi: 10.1038/nm1416
235. Ben Mkaddem S, Benhamou M, Monteiro RC. Understanding fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.00811
236. Fonseca DLM, Filgueiras IS, Marques AHC, Vojdani E, Halpert G, Ostrinski Y, et al. Severe COVID-19 patients exhibit elevated levels of autoantibodies targeting cardiolipin and platelet glycoprotein with age: a systems biology approach. NPJ Aging. (2023) 9:21. doi: 10.1038/s41514-023-00118-0
237. Jaycox JR, Lucas C, Yildirim I, Dai Y, Wang EY, Monteiro V, et al. SARS-CoV-2 mRNA vaccines decouple anti-viral immunity from humoral autoimmunity. Nat Commun. (2023) 14:1299. doi: 10.1038/s41467-023-36686-8
238. Sacchi MC, Pelazza C, Bertolotti M, Agatea L, De Gaspari P, Tamiazzo S, et al. The onset of de novo autoantibodies in healthcare workers after mRNA based anti-SARS-CoV-2 vaccines: a single centre prospective follow-up study. Autoimmunity. (2023) 56:2229072. doi: 10.1080/08916934.2023.2229072
239. Ning W, Xu W, Cong X, Fan H, Gilkeson G, Wu X, et al. COVID-19 mRNA vaccine BNT162b2 induces autoantibodies against type I interferons in a healthy woman. J Autoimmun. (2022) 132:102896. doi: 10.1016/j.jaut.2022.102896
240. Peng K, Li X, Yang D, Chan SCW, Zhou J, Wan EYF, et al. Risk of autoimmune diseases following COVID-19 and the potential protective effect from vaccination: a population-based cohort study. EClinicalMedicine. (2023) 63:102154. doi: 10.1016/j.eclinm.2023.102154
241. Olivieri B, Betterle C, Zanoni G. Vaccinations and autoimmune diseases. Vaccines (Basel). (2021) 9. doi: 10.3390/vaccines9080815
242. Feng A, Yang EY, Moore AR, Dhingra S, Chang SE, Yin X, et al. Autoantibodies are highly prevalent in non–SARS-CoV-2 respiratory infections and critical illness. JCI Insight. (2023) 8. doi: 10.1172/jci.insight.163150
243. Meroni PL, Schur PH. ANA screening: an old test with new recommendations. Ann Rheumatic Diseases. (2010) 69:1420–2. doi: 10.1136/ard.2009.127100
244. Mahler M. Lack of standardisation of ANA and implications for drug development and precision medicine. Ann Rheumatic Diseases. (2019) 78:e33–e. doi: 10.1136/annrheumdis-2018-213374
245. Lebedin M, García CV, Spatt L, Ratswohl C, Thibeault C, Ostendorf L, et al. Discriminating promiscuous from target-specific autoantibodies in COVID-19. Eur J Immunol. (2023) 53:e2250210. doi: 10.1002/eji.202250210
246. Shome M, Chung Y, Chavan R, Park JG, Qiu J, LaBaer J. Serum autoantibodyome reveals that healthy individuals share common autoantibodies. Cell Rep. (2022) 39:110873. doi: 10.1016/j.celrep.2022.110873
247. Chang R, Yen-Ting Chen T, Wang S-I, Hung Y-M, Chen H-Y, Wei C-CJ. Risk of autoimmune diseases in patients with COVID-19: a retrospective cohort study. eClinicalMedicine. (2023) 56. doi: 10.1016/j.eclinm.2022.101783
248. Machhi J, Herskovitz J, Senan AM, Dutta D, Nath B, Oleynikov MD, et al. The natural history, pathobiology, and clinical manifestations of SARS-coV-2 infections. J Neuroimmune Pharmacol. (2020) 15:359–86. doi: 10.1007/s11481-020-09944-5
Keywords: SARS-CoV-2, COVID-19, autoantibodies, ACE2, autoimmunity, long-COVID, post-acute sequelae
Citation: Galipeau Y, Cooper C and Langlois M-A (2025) Autoantibodies in COVID-19: implications for disease severity and clinical outcomes. Front. Immunol. 15:1509289. doi: 10.3389/fimmu.2024.1509289
Received: 10 October 2024; Accepted: 13 December 2024;
Published: 06 January 2025.
Edited by:
Pei-Hui Wang, Shandong University, ChinaReviewed by:
Yaping Sun, Yale University, United StatesCopyright © 2025 Galipeau, Cooper and Langlois. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc-André Langlois, bGFuZ2xvaXNAdW90dGF3YS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.