 Shan Guo
Shan Guo Qi Zhang
Qi Zhang Xiaoyan Yin
Xiaoyan Yin Zibin Tian
Zibin Tian Xiaoyu Li
Xiaoyu Li- Department of Gastroenterology, The Affiliated Hospital of Qingdao University, Qingdao, China
CCL2, a pivotal cytokine within the chemokine family, functions by binding to its receptor CCR2. The CCL2/CCR2 signaling pathway plays a crucial role in the development of fibrosis across multiple organ systems by modulating the recruitment and activation of immune cells, which in turn influences the progression of fibrotic diseases in the liver, intestines, pancreas, heart, lungs, kidneys, and other organs. This paper introduces the biological functions of CCL2 and CCR2, highlighting their similarities and differences concerning fibrotic disorders in various organ systems, and reviews recent progress in the diagnosis and treatment of clinical fibrotic diseases linked to the CCL2/CCR2 signaling pathway. Additionally, further in-depth research is needed to explore the clinical significance of the CCL2/CCR2 axis in fibrotic conditions affecting different organs.
Introduction
Chemokines are a large family of small proteins that play a crucial role in regulating cell movement (1), particularly in guiding leukocyte migration (2), which is central to maintaining immune system balance (3). In 2000, chemokines were systematically classified into four families: CXC, CC, CX3C, and C (4). Among these, CC chemokine ligand 2 (CCL2), also known as monocyte chemotactic protein-1 (MCP-1), is a key regulator in immune response. Discovered in 1989, CCL2 is primarily anchored to endothelial cell membranes (5) and is most abundantly expressed in monocytes, macrophages, and lymphocytes (6). Other cells, such as smooth muscle cells, endothelial cells, and fibroblasts, can also secrete CCL2 (7), particularly in response to cytokines like interleukin (IL)-6, tumor necrosis factor-α (TNF-α), and transforming growth factor-β (TGF-β) (8).
CCL2 exerts its effects mainly by binding to the receptor CCR2 (9), which has the highest affinity for CCL2 due to its specific structural characteristics (10). This binding activates various downstream pathways (Figure 1), resulting in diverse chemotactic responses, particularly in monocytes. The CCL2-CCR2 interaction not only promotes cell migration but also regulates cell adhesion and macrophage chemotaxis. Upon ligand binding, CCR2 experiences conformational changes that initiate the activation of phospholipase C (PLC). This activation leads to an increased release of Ca2+ ions and the subsequent activation of protein kinase C (PKC) and phosphoinositide 3-OH kinase (PI3K). These processes further activate signaling molecules such as protein kinase B (PKB, Akt) and mitogen-activated protein kinase (MAPK). This signaling pathway is crucial for the regulation of central transduction mechanisms within the cell (11–13). Additionally, it plays a significant role in cancer progression by activating pathways such as p38-MAPK (14, 15) and PI3K/AKT/mTOR (16–18), which enhance tumor cell invasion, migration, and survival.
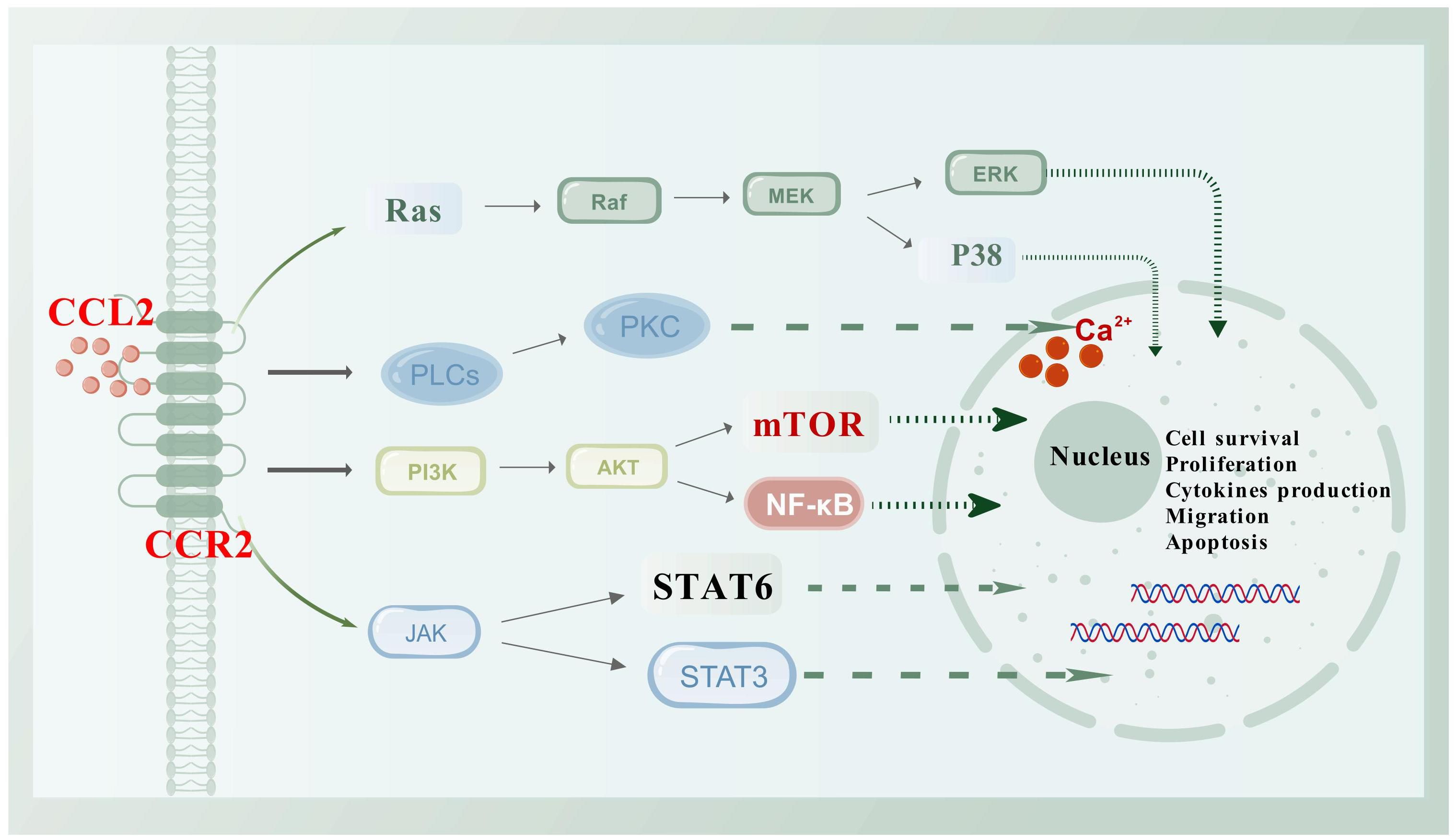
Figure 1. Schematic diagram of the CCL2/CCR2 axis and its associated signaling pathways. CCR2, a classic G protein-coupled receptor, activates a variety of downstream signaling pathways upon binding to its ligand CCL2, such as PI3K/Akt, JAK/STAT, and P38/MAPK. Activation of these pathways leads to the regulation of various transcription factors and genes involved in cell survival, proliferation, cytokine production, migration, and apoptosis. This figure was created with biogdp.com.
CCL2 is extensively involved in disease regulation, particularly in inflammation, fibrosis, and cancer. It mobilizes monocytes from the bone marrow into the bloodstream and directs their migration to sites of inflammation (19), underscoring its essential role in immune defense (20). Beyond inflammation, CCL2 contributes to fibrosis development in various organs like the liver, pancreas, and kidneys (Figure 2). It also plays a role in cancer biology, known as the “tumor chemokine” (21), aiding in tumor initiation, growth, and metastasis (22, 23). Tumor-associated macrophages secrete CCL2 (24), which aids in the progression of cancers like breast (25) and pancreatic cancers (26, 27). CCL2 expression in the tumor microenvironment, involving various cell types, highlights its significance in cancer pathology (28) and its potential as a therapeutic target.
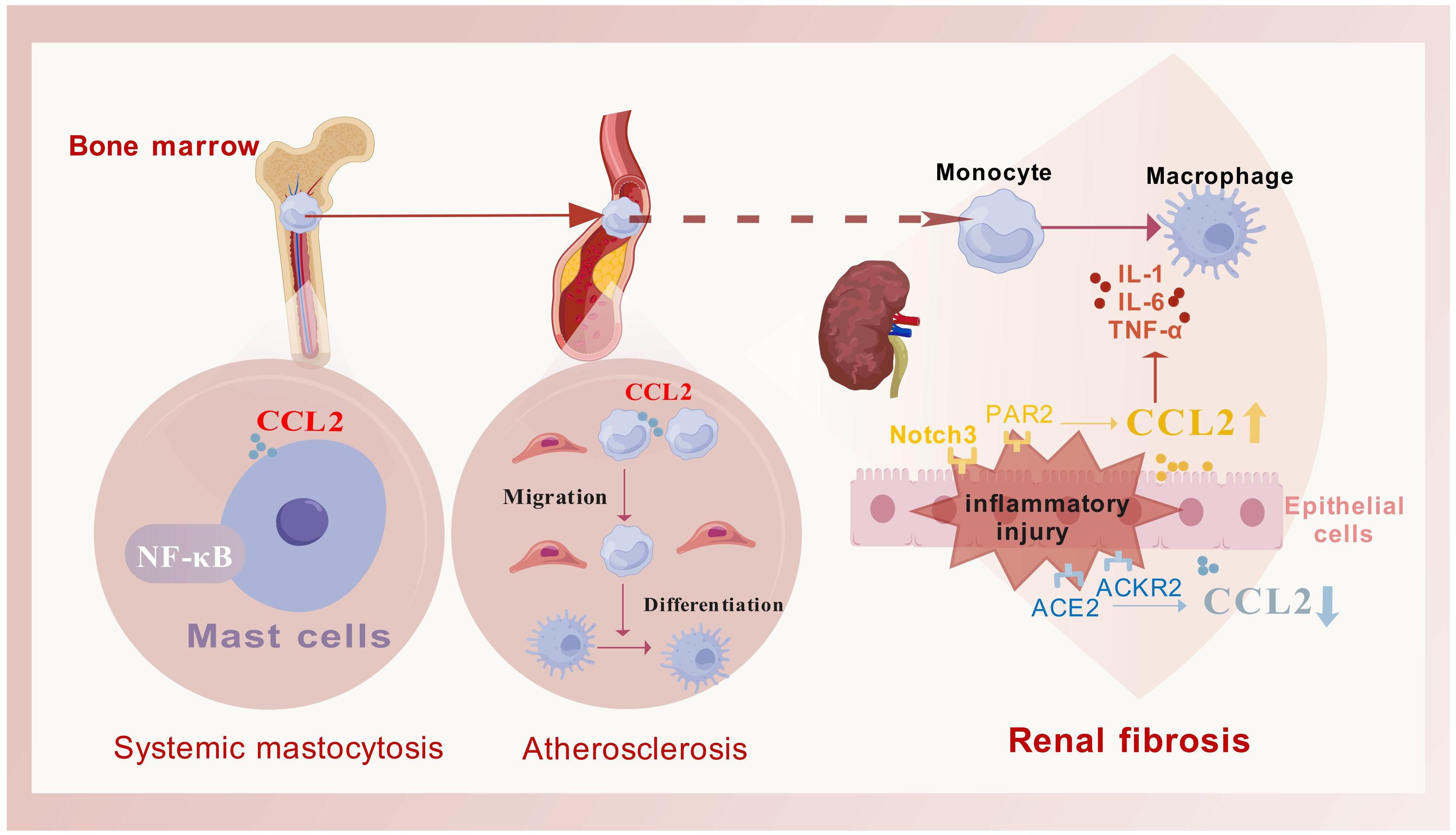
Figure 2. The primary role process of CCL2 in fibrotic diseases. CCL2 facilitates the mobilization of monocytes from the bone marrow into the bloodstream and guides their migration to targeted inflammatory fibrotic areas. Once there, these monocytes differentiate into macrophages to address tissue damage (as exemplified by atherosclerotic plaques), this process is frequently associated with the activation of the NF-κB signaling pathway (as exemplified by systemic mastocytosis). Additionally, the regulation of CCL2 secretion is often influenced by various receptors, which in turn modulate the release of inflammatory mediators (as seen in fibrosis associated with chronic kidney disease). This figure was created with biogdp.com.
Chemokine receptors have been targeted in therapeutic strategies, and recent years have seen promising results from CCL2/CCR2 antagonists in treating inflammatory and fibrotic diseases (29) and cancer immunotherapy (30, 31). This review explores the mechanisms and the diagnostic and therapeutic potentials of the CCL2/CCR2 axis in inflammatory and fibrotic diseases. These conditions are characterized by chronic inflammation leading to fibrosis, where prolonged immune responses cause excessive extracellular matrix deposition, tissue scarring, and organ dysfunction. Understanding the role of the CCL2/CCR2 axis in fibrosis will provide a basis for developing novel therapies.
Mechanisms of the CCL2/CCR2 axis in inflammatory and fibrotic diseases
CCL2 plays a pivotal role in fibrogenesis in the liver, pancreas, and intestine by attracting immune cells such as monocytes and macrophages to fibrotic areas, promoting inflammatory responses and matrix remodeling. In viral hepatitis, CCL2 helps create an immunosuppressive microenvironment that facilitates fibrosis progression and favors viral infection. In liver fibrosis, CCL2 synergizes with TGF-β to activate hepatic stellate cells (HSCs) and promote matrix deposition. In intestinal fibrosis, CCL2 works with intestinal-specific factors like IL-17. In pancreatic fibrosis, CCL2 leads to M2-like polarization of macrophages after attracting immune cells to the tissue.
Hepatic fibrosis diseases
CCL2 is crucial in liver diseases such as acute and chronic liver injury, cirrhosis, and tumor progression (32). Liver inflammation often coexists with fibrosis, driven by the recruitment and polarization of macrophages, particularly through hepatic stellate and Kupffer cell activation (33, 34). Key pathways like hedgehog (35) and TGF-β (36) are involved in these processes, contributing to conditions like non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma (HCC) (37).
NASH
NASH results from excessive fat accumulation in the liver, causing hepatocyte damage and triggering inflammation. This inflammatory state activates HSCs, leading to fibrosis, cirrhosis, or liver cancer. Studies have shown that increased expression of CCL2 is a hallmark of NASH, promoting the infiltration of monocyte-derived macrophages through CCL2-mediated chemotaxis.
CCL2 is implicated in NASH progression through several mechanisms. Li et al. (38) identified methyltransferase 3 (METTL3) as a key negative regulator, loss of Mettl3 accelerates NASH by enhancing CD36-dependent fatty acid uptake and CCL2-driven inflammation. The absence of CX3CR1 promotes a shift from M1 to M2 macrophages, slowing NASH progression. CCL2 deficiency in Cx3cr1-/- mice reduces macrophage infiltration and supports M2 dominance in the liver, alleviating NASH (39). Recent research has indicated that the activation of Notch signaling pathways in hepatocytes is crucial for the advancement of liver fibrosis linked to NASH (40). Additionally, existing literature has underscored the role of Notch signaling in hepatocytes, which further facilitates the infiltration of macrophages dependent on CCL2, thereby exacerbating fibrosis (41).
Autophagy in liver sinusoidal endothelial cells is also involved in NASH. Insufficient autophagy increases the expression of CCL2, CCL5, and CD68, exacerbating fibrosis in high-fat diet-fed mice (42). CD11c+CD206+ cells, which express high levels of CCR2, show increased CCL2 expression in NASH, correlating with disease severity. Inhibition of CCR2 reduces the infiltration of CD11b+CD11c+F4/80+ monocytes and improves liver inflammation and fibrosis in NASH models (43).
Multiple signaling pathways are implicated in steatohepatitis, with the CCL2/CCR2 axis playing a key role. Gasdermin D, involved in programmed necrosis, promotes the secretion of pro-inflammatory cytokines IL-1β and CCL2 and activates the NF-κB pathway, driving NASH progression (44). The NLRP3-IL-1β pathway also contributes to inflammation and obesity-related comorbidities. Inhibition of NLRP3 inflammasome reduces lipopolysaccharide-induced inflammation by down-regulating CCL2 mRNA levels (45). Suppressing the IL-33 signaling pathway can inhibit NASH progression by down-regulating CCL2 and α-SMA expression (46), while activating IL-19 signaling reduces HSC migration by down-regulating CCL2 expression, alleviating liver fibrosis (47).
Viral hepatitis
CCL2 plays a specific role in immune evasion during hepatitis B virus (HBV) infection. Chronic HBV infection, a major risk factor for liver diseases such as HCC, can persist for decades. HBV evades immune response by downregulating CCL2 (48), reducing inflammatory monocyte and macrophage recruitment to the liver. In chronic HBV cases complicated by cirrhosis, plasma CCL2 levels tend to decrease (49). Additionally, HBeAg can promote HSC proliferation, movement, and contraction in a macrophage-dependent manner, inducing CCL2 production that activates HSCs and worsens liver fibrosis (50).
Hepatitis C virus (HCV)-mediated hepatitis is also a significant global health issue, particularly in the United States. Studies show that HCV-infected individuals can downregulate specific microRNAs, such as miRNA-107 and miRNA-449a, to regulate CCL2 expression by targeting the interleukin-6 receptor (IL-6R) complex (51), suggesting potential therapeutic avenues. A novel pathway involving the HCV core protein interacting with gC1qR has been identified, leading to CCL2 and CXCL10 secretion in macrophages via the NF-κB signaling pathway (52).
Other hepatic fibrotic diseases
Analysis of the liver fibrosis expression dataset GSE84044 from the GEO database identified 10 key genes in the protein interaction network, including CCL2, highlighting its importance in fibrosis and inflammation (53). Glucocorticoid-induced leucine zippers (GILZ), encoded by the Tsc22d3 gene in mice, mimic glucocorticoids’ anti-inflammatory effects. Mice deficient in GILZ show increased CCL2 production and pro-inflammatory leukocyte infiltration in early liver fibrosis, accelerating its progression (54). Moreover, sphingosine kinase 1 (SPHK1) levels were significantly higher in fibrotic compared to normal human livers. SPHK1 knockout in Kupffer cells reduced CCL2 secretion, while its knockout in HSCs decreased CCR2 expression (55).
CCL2 also has a role in pediatric liver fibrosis. A recent study identified FOCAD germline recessive mutation in pediatric liver cirrhosis. In a zebrafish model with FOCAD deficiency, liver injury was accompanied by increased CCL2 expression, suggesting that targeting the CCL2/CCR2 axis could be a new approach for treating pediatric liver cirrhosis (56).
Inflammatory bowel disease
Inflammatory bowel disease (IBD) is a chronic autoimmune inflammatory disease affecting the gastrointestinal tract, with two main subtypes: ulcerative colitis (UC) and Crohn’s disease (CD). The dextran sodium sulfate (DSS)-induced colitis model, which activates the classic NF-κB signaling pathway, is widely used to study UC. In intestinal tissues, CCL2 responds to diverse signals. Inflammatory damage triggers monocytes to secrete CCL2, recruiting white blood cells or macrophages to injury sites, ultimately leading to intestinal fibrosis.
In early intestinal inflammation, Ly6c high-expressing cells markedly enhance the expression of CCL2 and CCR2 genes through the activation of STAT1 signaling, in contrast to Ly6c intermediate-expressing cells. Additionally, inhibiting the CCR2 pathway can mitigate colonic damage in models of acute colitis (57). PC3-secreted microproteins (PSMP) upregulate phosphorylated ERK levels via the CCR2 pathway (58), driving CCR2+ monocyte migration to inflamed colonic tissue. PSMP attracts Ly6chi monocytes in a CCR2-dependent manner, aided by Dectin-1 on myeloid cells (59, 60), while cobalt protoporphyrin IX (CoPP) reduces the migration of CCR2+ Ly6chi monocytes to the inflamed colon (61).
As inflammation reaches the muscle layer, Ly6c+ monocytes infiltrate, adopting a unique transcriptional state and promoting muscle inflammation. They alter the microenvironment, promoting more monocyte infiltration, which then differentiates into anti-inflammatory CD206+ macrophages through CCL2 (62). Bone marrow-derived mesenchymal stromal cells (BM-MSCs) secrete CCL2, influencing colitis development, while IL-10 in MSCs polarizes resident macrophages (63). During chronic inflammation, CCR2+ monocytes and fibrocytes infiltrate the colon, promoting fibrosis by inhibiting collagen degradation (64).
CCL2 promotes intestinal fibrosis by activating pro-inflammatory factors, notably IL-6, a process reversible with CCR2 antagonist RS102895 (65). Antisense IL-7 (IL-7-AS) accelerates inflammation by upregulating IL-6 and CCL2 (66). Poly(rC)-binding protein 1 (PCBP1) deficiency reduces CCL2 and IL-6 production in colitis macrophages (67). Additionally, in colitis models, CCR2 and CD30L expression in monocytes are positively correlated; CD30L drives monocyte homing and differentiation via the CCL2/CCR2 axis and NF-κB pathway, enhancing inflammation (68).
CCL2 is also involved in hyperoxia-induced intestinal injury. IL-17D, a member of the IL-17 family, promotes CCL2 expression in intestinal epithelial cells under hyperoxic conditions, leading to chronic intestinal inflammation (69).
Chronic pancreatitis
Chronic pancreatitis (CP) is characterized by progressive, irreversible inflammation and fibrosis, with pancreatic stellate cells (PSCs) playing a key role in this process (70). Studies in CP patients have shown that prostaglandin E2 mediates CCL2 synthesis in PSCs via TNF-α regulation, and inhibiting cyclooxygenase (COX)-2 activity can slow the progress of pancreatitis and fibrosis (71).
Research on CCL2’s role in CP progression is limited. Interestingly, alternatively activated macrophages (M2) secrete PDGF and TGF-β, which activate PSCs and promote fibrosis—a key factor in CP development (72). This contrasts with the immune microenvironment in NASH (39), highlighting the plasticity of macrophages and suggesting that CCL2 regulates immune cells differently across organs. Additionally, during chronic pancreatic inflammation, CCL2 secretion and NF-κB pathway activation persist (73).
Cardiovascular fibrotic diseases
Cardiovascular diseases remain a leading cause of morbidity and mortality worldwide (74). Recent studies highlight the inflammatory immune response as a central mechanism in their pathogenesis (75). Among the key mediators, CCL2 plays a crucial role in regulating leukocyte migration and infiltration in various cardiovascular diseases. Consequently, CCL2/CCR2 inhibitors are being explored as novel therapeutic targets for inflammation-related cardiovascular conditions (76).
Myocardial infarction
During the healing process of Myocardial infarction (MI), macrophages undergo dynamic polarization. CCL2 is differentially expressed in LPS-induced M1 and IL-4-induced M2 macrophages (77). Unlike in the liver and pancreas, CCL2 deficiency in the heart results in reduced total macrophage and M1 macrophage numbers in the infarcted area, while M2 macrophages increase (78). This suggests that CCL2 is a pivotal regulator of macrophage polarization during MI healing, both in vivo and in vitro, and it specifically promotes M1 polarization. The organ-specific and environment-dependent nature of CCL2’s role likely explains these differential effects.
The repair process following MI is critical for cardiac recovery. In response to acute myocardial ischemia-reperfusion (MIR), macrophages infiltrate damaged heart tissue and shift their polarization to manage acute inflammation and subsequent fibrotic remodeling. Studies indicate that CCL2/CCR2 signaling in macrophages facilitates the transition from acute injury to chronic fibrosis in MIR in mice through the NLRP3 inflammatory pathway and phenotypic changes (79). Furthermore, IL-34 enhances CCL2 expression by maintaining NF-κB pathway activation, aggravating cardiac remodeling and fibrosis after reperfusion injury (80).
Other cardiovascular inflammatory diseases
A meta-analysis by Georgakis et al. demonstrated that circulating CCL2 levels, synonymous with CCL2, are strongly associated with cardiovascular mortality (81). Elevated CCL2 levels in atherosclerotic plaques correlate with increased plaque vulnerability, and persistently high circulating CCL2 levels heighten the risk of adverse outcomes following plaque removal (82). Moreover, circulating CCL2 levels appear genetically influenced; higher levels are linked to a greater risk of stroke, suggesting that CCL2-targeted therapies might reduce stroke incidence (83).
CCL2 is also implicated in the regulation of circadian rhythms during atherosclerosis progression. Winter et al. (84) revealed that CCL2 is a key chemokine at atherosclerotic sites and that the chronic inflammation of large blood vessels depends on the rhythmic release of CCL2, which peaks in the early stage and declines later. This discovery opens new avenues for chronotherapy targeting the CCL2-CCR2 axis in atherosclerosis treatment.
Mast cells (MCs), known for their roles in allergy and parasitic infections, are also pivotal in inflammation and fibrosis. CCL2 acts as an activator of MCs (85). Luo et al. (86) highlighted that early MC-fibroblast interaction and the stem cell factor/mast cell/CCL2/monocyte/macrophage axis are critical for initiating myocardial fibrosis. Additionally, hypoxia, which can be both a cause and a result of heart failure, induces CCL2 expression in both right ventricle (RV) and left ventricle (LV) of mice, potentially contributing to cardiac inflammation, fibrosis, and ventricular dysfunction (87).
Pulmonary fibrotic diseases
Pulmonary inflammatory lesions tend to progress to fibrosis more consistently than in other organ systems, with CCL2 playing a significant role in this process. Pulmonary fibrosis is a multicellular phenomenon involving alveolar epithelial cells (AECs), recruited monocytes/macrophages, and fibroblasts. Several cell types, including AECs, produce CCL2, which promotes fibrosis via CCR2 activation (88). CCR2 signaling is therefore crucial for the development and progression of pulmonary fibrosis.
Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease characterized by chronic inflammation, AEC damage, and excessive deposition of extracellular matrix proteins, with myofibroblasts acting as the main effector cells (89). In mouse models of bleomycin-induced pulmonary fibrosis, transcriptional analysis has shown a significant increase in CCL2 expression (90, 91). FoxF1, an endothelial transcription factor, has also been implicated in pulmonary fibrosis. In vitro, studies reveal that FoxF1-deficient endothelial cells enhance lung fibroblast proliferation, invasion, activation, and macrophage migration by secreting chemokines such as CCL2, collectively contributing to fibrosis (92). Moreover, IL-33 treatment increases CCL2 and CXCL2 production in NFATc3+/+ macrophages, but not in NFATc3+/- mice, suggesting that NFATc3 regulates pulmonary fibrosis by regulating CCL2 and CXCL2 expression in macrophages (93).
Myofibroblasts play a central role in IPF pathogenesis. Single-cell RNA sequencing (scRNA-seq) data from IPF patients indicates that the CCL2/CCR2 axis is essential for M1 macrophage polarization (94). Pathogenesis varies among IPF subgroups, with ligand-receptor analysis suggesting a monocyte-macrophage chemotactic axis, potentially involving CCL2-CCR2, particularly in cilia-rich subgroups (95). Immunohistochemical analysis of human lung tissues has shown that activated IPF fibroblasts possess high contractile forces and produce abundant CCL2 (96), with the NF-κB signaling pathway contributing to CCL2 production and release in these fibroblasts (97). Another study on acute exacerbation of IPF found significantly higher CCL2 concentrations in bronchoalveolar lavage fluid compared to serum, indicating a localized inflammatory response (98).
Radiation-induced pulmonary fibrosis
Radiation exposure to the lungs triggers a damage response, including the release of cytokines and chemotactic mediators, signaling the recruitment of immune cells for inflammation and wound healing. A recent study has reported significantly elevated CCL2 expression in macrophages of irradiated lungs, which potentially stimulates the fibrotic phenotype in lung fibroblasts (99). Following radiation, bone marrow-derived inflammatory monocytes migrate to the lungs, where CCR2+ monocyte-derived macrophages infiltrate and may play a critical role in the development of Radiation-induced pulmonary fibrosis (RIPF) (100).
Other pulmonary fibrotic diseases
CCL2 also contributes to receptor-mediated pulmonary fibrosis progression. Adhesion G Protein-Coupled Receptor F5 (ADGRF5) is a key regulator of lung surfactant homeostasis in type II alveolar cells. Research has shown that ADGRF5 can regulate CCL2 gene expression, maintaining its potential role in lung immune homeostasis. Disruption of ADGRF5 results in airway inflammation mediated by type 2 immune response and CCL2-induced inflammation (101). In a mouse model of pulmonary fibrosis, Leucine-Rich Repeat Kinase 2 (LRRK2) expression is significantly reduced in alveolar type II epithelial (ATII) cells, and LRRK2-deficient ATII cells exhibit an enhanced ability to recruit profibrotic macrophages via the CCL2/CCR2 axis. This recruitment triggers extensive macrophage-associated profibrotic responses and progressive pulmonary fibrosis (102).
Additionally, while IL-17 is known for its pro-inflammatory role in the intestine, it also contributes to parenchymal fibrosis in chronic pulmonary graft-versus-host disease (cpGVHD). Blocking IL-17A leads to reduced CCL2 expression in cpGVHD-related pulmonary fibrosis (103), though this effect is not observed in kidney diseases (104).
Renal fibrotic diseases
During kidney injury, CCL2 recruits immune cells such as macrophages and T cells to the site of injury, activating them and triggering the release of additional inflammatory mediators. These mediators not only worsen kidney damage but also promote fibrosis. Proteomic analysis of an in vitro model of renal fibrosis identified CCL2 as a key factor contributing to collagen deposition in the kidney (105).
Unilateral ureteral obstruction
Unilateral ureteral obstruction (UUO) can lead to renal interstitial fibrosis and is a potential precursor to chronic kidney disease (CKD), promoting extensive research into the underlying mechanisms. As mentioned earlier, activation of the Notch signaling pathway is known to upregulate CCL2 expression, contributing to fibrosis in organs like the liver (41). Recent animal studies have shown that Notch3 is newly expressed in damaged renal epithelium at early stages of CKD. Notably, systemic deficiency of Notch3 prevents leukocyte invasion and organ fibrosis, suggesting that targeting Notch3 may protect the epithelial epithelium and interrupt pro-inflammatory signaling, thereby alleviating kidney injury (106). Brandt et al. (107) utilized chimeric mice with Notch3 deficiency in hematopoietic cells and/or resident histiocytes to analyze renal fibrosis and inflammation following UUO. Their results indicated that a pro-inflammatory environment is characterized by upregulation of CCL2 and CCL5, which are regulated in a Notch3-dependent manner.
A recent study identified increased Twist1 expression in both the UUO mouse model and IgA nephropathy. Knockout of Twist1 in macrophages partially inhibited CCL2-mediated macrophage chemotaxis and suppressed M2 macrophage polarization, thereby mitigating fibrosis progression (108). Conversely, IL-15 has been shown to reduce CCL2 expression in the UUO mouse model, alleviating fibrosis and decreasing the likelihood of progression to CKD (109). Nicotinamide exhibits similar effects (110).
Other renal fibrotic diseases
In CKD patients, angiopoietin-1 has been found to reduce the expression of chemokine CCL2 in fibrotic renal endothelial cells. In contrast, angiopoietin-2 induces CCL2 expression in endothelial cells, which promotes macrophage infiltration and increases apoptosis in fibrotic renal endothelial cells. This process negatively impacts the renal survival rate in CKD patients (111). Consistently, angiotensin-converting enzyme 2 (ACE2), which is inversely correlated with CCL2 expression, is strongly associated with CKD and is known to limit renal fibrosis (112). Another important factor is protease-activated receptor 2 (PAR2), which is involved in renal inflammation and fibrosis. Activation of PAR2 in cultured renal tubular epithelial cells triggers extracellular signal-regulated kinase signaling and CCL2 secretion, contributing to tubulointerstitial inflammation and fibrosis (113).
Abnormal immune system activation is also implicated in CKD development. Wilkening et al. (114) analyzed glomerular CCR2 expression in focal segmental glomerulosclerosis (FSGS) and demonstrated that macrophages expressing CCR2 contribute to kidney damage and fibrosis remodeling in conditions such as glomerulonephritis and diabetic nephropathy. In contrast, atypical chemokine receptor 2 (ACKR2), also known as D6, degrades CCL2, limiting the recruitment of immune cells and myofibroblasts to renal mesenchymal cells. This degradation inhibits renal inflammation and fibrosis remodeling in glomerulonephritis (115).
Kashyap et al. (116)reported that CCL2 deficiency protects against chronic kidney damage in a mouse model of renovascular hypertension caused by renal artery stenosis (RAS). The deficiency leads to reduced monocyte infiltration and lower expression of CCR2 and CD206, suggesting that CCL2 is a key mediator of kidney injury in renovascular hypertension.
Bone marrow fibrotic diseases
Myelofibrosis (MF) is a prominent clinical feature observed in patients with myeloproliferative neoplasms (MPN). Prior research has established a correlation between the progression of MF and the aberrant expression of cytokines (117). Within the context of myeloproliferative disorders, CCL2 and its associated signaling pathways may play a critical role. CCL2 is thought to be involved in both the proliferation and differentiation of fibrosis-associated cells, thereby impacting the bone marrow microenvironment.
MPN
A recent investigation revealed that among the MPN cohort, the proportion of CCR2+ cells is significantly associated with the severity of myelofibrosis in patients, and CCR2 expression on CD34+ cells correlates with higher-risk classifications in MF and the presence of circulating blast cells (118). Additionally, polymorphisms in the CCL2 gene have been shown to influence the bone marrow microenvironment in MPN, with homozygosity for the CCL2 rs1024611 SNP linked to diminished survival in individuals with primary myelofibrosis (119). The -2518 A/G SNP of CCL2 has emerged as a potential susceptibility marker for MPN and myelofibrosis (120).
Systemic mastocytosis
As noted in the aforementioned myeloproliferative neoplasms, there is also a frequent elevation in the production of profibrotic and angiogenic cytokines in Systemic mastocytosis (SM), which contributes to alterations in the bone marrow microenvironment. A defining characteristic of SM is the abnormal accumulation of neoplastic MCs harboring the activating KIT mutation D816V within the bone marrow. Recent studies indicate that KIT D816V enhances CCL2 expression in abnormal MCs via the classical NF-κB signaling pathway. Furthermore, it has been demonstrated that CCL2 derived from MCs facilitates the migration of endothelial cells in SM, both in vitro and in vivo, thereby modifying the tumor microenvironment to favor fibrosis and angiogenesis. Serum CCL2 levels are markedly elevated in SM patients, with significantly higher concentrations observed in individuals with advanced SM compared to asymptomatic SM patients and those with cutaneous mastocytosis. Importantly, CCL2 levels exhibit only a moderate correlation with MCs infiltration in the bone marrow and instances of myelofibrosis (121).
Acute myeloid leukemia
Literature indicates that the CCL2/CCR2 axis may be implicated in cell migration in acute AML. However, plasma CCL2 levels in Acute myeloid leukemia (AML) patients are reported to be lower than those in healthy donors (122). This finding contrasts with earlier observations made by Manzur et al. (123), highlighting the need for further investigation.
Other fibrotic diseases
Fibrosis of the skin and internal organs is a hallmark of systemic sclerosis (SSc), with monocytes playing a crucial role in the skin’s inflammatory response (124). Studies have shown that CCL2 expression is significantly higher in SSc mouse models compared to normal mice (125), and CCL2 holds significant promise as a valuable biological marker for SSc (126). Additionally, knockout of STAT6 in SSc mouse models significantly reduces the expression of CCL2 and CD206, indicating that CCL2 is involved in the immune response that drives skin and organ fibrosis (127).
Experimental autoimmune orchitis (EAO) serves as an animal model for studying chronic testicular inflammation and fibrosis, replicating pathogenic changes similar to those seen in humans. During EAO, there is an increase in pro-inflammatory CCL2 expression, which coincides with leukocyte infiltration into the testicular parenchyma. Elevated levels of activin A are correlated with the severity of EAO, while high CCL2 levels mediate leukocyte transport and the recruitment of macrophages through its receptor, CCR2. These findings suggest that both CCR2 and activin A promote fibrosis in testicular inflammation by regulating macrophage function (128).
CCL2/CCR2 axis as a diagnostic marker for fibrotic diseases
Renal diseases
Urine is an easily obtainable sample that can reflect the severity of kidney disease, making it a focus for studying CCL2 levels as a diagnostic tool and for predicting prognosis. Urrego-Callejas et al. (129) included 120 systemic lupus erythematosus (SLE) patients and found that serum anti-C1Q antibody and urinary ceruloplasmin levels were associated with CCL2 activity in chronic injury. Similarly, in 125 patients with active lupus nephritis (LN), urinary CCL2 levels were positively correlated with interstitial inflammation, glomerulosclerosis, interstitial fibrosis, and tubular atrophy based on kidney biopsies (130). Standard LN therapies, like mycophenolate and rapamycin, improve glomerular sclerosis by downregulating CCL2 and reducing fibrosis-related proteins (131).
Studies have shown that excessive cisplatin administration induces renal interstitial fibrosis in C57BL/6 mice, and CCL2 serves as a marker of renal injury in this model (132). Additionally, high urinary CCL2 and low urinary epidermal growth factor (EGF) levels are associated with renal tubulointerstitial fibrosis (133), suggesting that urinary CCL2 could be a useful diagnostic marker for CKD patients.
Identifying early prognostic markers in high-risk renal transplant recipients can help optimize immunosuppressive therapy and improve outcomes. The ratio of CCL2 to creatinine (CCL2:Cr) has been shown to predict BK nephropathy; however, the diagnostic value of CCL2 for BKV infection in immunocompromised transplant recipients still needs further investigation (134).
Cardiovascular disease
Macrophages drive vasculopathy changes in Takayasu arteritis (TA) through phenotypic transformation. Peripheral CCL2 levels fluctuate at different disease stages, suggesting CCL2 as a potential biomarker for disease activity (135). In human atria, markers of leukocyte infiltration and matrix degradation indicate severe inflammation—a key factor in atrial fibrillation—linked to CCL2 (136). In a study of 131 patients undergoing aortic valve replacement, CCL2 was identified as an inflammatory marker for aortic remodeling (137). In diabetic cardiomyopathy (DCM), both serum CCL2 and myocardial CCL2 mRNA levels are elevated (138).
IBD
An interesting study of 33 children with IBD found that blood CCL2 levels were significantly higher in patients with CD compared to those with UC at all stages of the disease (139). This suggests that CCL2 could serve as a diagnostic biomarker for CD and help differentiate subtypes of pediatric IBD. However, it remains unclear if the same pattern is present in adult IBD patients. Given that both heart failure and IBD have immune-related pathogenesis, a study identified 34 genes associated with immune diseases, including CCL2, through Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (140).
IPF
As an inflammation-related gene, CCL2 may help predict the prognosis of IPF (141). While combined detection of CCL2, KL-6, and CXCL13 improves diagnostic accuracy for idiopathic interstitial pneumonia (IIP), it does not effectively differentiate among lung fibrosis diseases (142). Interestingly, serum CCL2 levels may help distinguish between IPF and fibrotic hypersensitivity pneumonitis (fHP), two conditions that are often difficult to differentiate (143).
CCL2 as a therapeutic target for fibrotic diseases
Recent research has identified CCL2 as a key therapeutic target for treating fibrotic diseases across various organs. Its significance in fibrosis pathogenesis has led to the development of diverse treatment strategies, including traditional Chinese medicines (TCM), chemical agents, and application of nanotechnology. The broad potential of targeting CCL2 underscores the need for further research to fully leverage its role in creating effective antifibrotic therapies. CCR2 antagonists show significant potential for treating fibrotic diseases by specifically blocking CCR2 receptors. These antagonists have demonstrated positive effects in various conditions, including liver, lung, and renal fibrosis. Studies suggest that CCR2 antagonists reduce inflammatory cell infiltration, inhibit profibrotic gene expression, and slow fibrosis progression by modulating relevant signaling pathways.
Liver diseases
In addition to the previously mentioned GILZ and SPHK1 that play a role in regulating CCL2 expression and thereby affecting liver fibrosis progression (54, 55), several pharmacological agents can also mitigate liver fibrosis by targeting the CCL2/CCR2 signaling pathway. Moreover, recent developments in nanotechnology have shown promise in the treatment of liver fibrosis through the modulation of CCL2 levels.
A recent investigation has introduced an innovative strategy involving the silencing of CCR2 through small interfering RNA (siCcr2) encapsulated within tetrahedron framework DNA nanostructure (tFNA) vehicle (tFNA-siCcr2). This method effectively overcomes the challenges associated with the in vivo efficacy of siCcr2 by facilitating targeted delivery to the liver, and resulting in improved therapeutic effects for liver fibrosis (144). Additionally, genetic engineering is being explored to create transgenic nano decoys that interfere with liver fibrosis. These decoys, engineered to overexpress CCR2 on their surface, can neutralize CCL2 levels. When loaded with curcumin and delivered to the liver, this combined therapy effectively reduces macrophage infiltration, offering promising therapeutic outcomes (145).
TCM formulations like Dahuang Zhizhu Pill, Tianhuang formula, and Fu-Gan-Wan have been shown to reduce the expression of CCL2 and its receptor CCR2 in the liver, particularly targeting CCR2+ macrophages (146–148). Similarly, Fuzheng Huayu (FZHY) modulates macrophage recruitment and polarization via CCL2 and CX3CL1, providing anti-inflammatory and antifibrotic effects. The Ganxianfang formula exhibits comparable benefits and may offer superior protective effects compared to FZHY (149). These findings suggest that TCM’s antifibrotic effects primarily result from inhibiting CCL2-induced macrophage recruitment to fibrotic sites, though the exact mechanisms—direct or indirect—remain unclear.Cenicriviroc (CVC) has gained attention for its therapeutic value in liver diseases. Krenkel et al. (150) conducted studies in NAFLD patients and C57BL/6 mice, demonstrating that CVC is effective and safe. CVC also enhances the therapeutic effect of fibroblast growth factor 21 on NASH, primarily by reducing liver fibrosis (151). In a mouse model of liver fibrosis induced by CCL4 and using Ccr2 knockout (Ccr2-/-) mice, Guo et al. (152) showed that CVC treatment was effective in both mice and human liver samples. Yu et al. (153) further demonstrated that CVC was effective in treating cholestatic liver injury in bile duct-ligated rats and Mdr2 (Abcb4-/-) mouse models. Additionally, Mbade et al. (154) found that CVC could prevent liver injury and steatosis in an alcoholic liver disease mouse model.
Bile duct diseases
The CCL2/CCR2 axis’s chemotactic effect on monocytes and macrophages is a key driver of primary sclerosing cholangitis (PSC) progression. Studies have shown a positive correlation between serum CCL2 levels and fibrosis severity in primary biliary cholangitis (PBC) patients (155). In acute sclerosing cholangitis mouse models treated with the apoptosis-inhibiting BV6 inhibitor, both CVC treatment and CCR2 deletion were found to reduce macrophage accumulation, liver damage, and biliary fibrosis (156). Similarly, Reuveni et al. (157) observed improvements in primary biliary cholangitis models using Cx3cr1gfp/+ and Ccr2-/-Cx3cr1gfp/+ mice following intraperitoneal CVC injection.
Intestinal diseases
In the treatment of IBD, agonists or inhibitors targeting specific factors or receptors have been developed. For example, SARI, commonly used as a colon cancer inhibitor, has been found to increase CCL2 production when deficient, whereas knocking out CCR2 can block this effect, thereby reducing colitis symptoms (158). Dimethyl itaconate decreases CCL2 production in epithelial cells, reduces macrophage recruitment to the tumor microenvironment, alleviates the hyperinflammatory state of UC, and lowers the risk of colitis-associated cancer (159).
Several other drugs directly target the CCL2/CCR2 axis. Luteolin and homoharringtonine both exhibit strong anti-inflammatory effects by inhibiting NF-κB signaling and reducing CCL2 production in colonic tissues (160, 161). Nobiletin reduces CCL2 expression and collagen deposition in colitis-induced mice (162), while berberine and geniposide show similar effects in chronic colitis (163, 164).
Pulmonary diseases
Excessive inflammation in silicosis is triggered by silica exposure. Research has shown that the Caspase-1 inhibitor VX-765 reduces the infiltration of inflammatory M1 alveolar macrophages and decreases CCL2 expression, thereby mitigating the inflammatory response (165). TAS-115, a novel polytyrosine kinase inhibitor, has been found to inhibit the phosphorylation of the macrophage colony-stimulating factor receptor c-FMS in mouse bone marrow-derived macrophages, both in vivo and in vitro. This inhibition reduces CCL2 production, highlighting CCL2 as a key molecule in pulmonary fibrosis development (166).
Pancreatic diseases
Oxidative stress is a major pathway in pancreatitis pathogenesis. Hydroxytyrosol has anti-inflammatory and antioxidant effects, reducing CCL2 release in pancreatic and colon tissues (167). Additionally, the classic TCM formulation known as Dahuang Chaihu Decoction has been shown to lower the levels of CCL2 in the pancreas, which in turn reduces macrophage infiltration and fibrosis associated with CP (168).
Renal diseases
In a glomerulosclerosis model, the use of a CCR2 antagonist (CCR2A) can inhibit renal macrophage accumulation and enhance the effects of conventional treatments like angiotensin-converting enzyme inhibitors (ACEi) (169). CKD often leads to complications such as salt-sensitive (SS) hypertension. Daily intraperitoneal administration of a CCR2 antagonist (RS 102895) during active disease stages has been shown to reduce renal leukocyte infiltration, kidney injury, and hypertension incidence (170). Additionally, bladder outlet obstruction (BOO) causes bladder remodeling, affecting its structure and function. Wang et al. (171) demonstrated that treatment with a CCR2 antagonist (RS504393) may offer a therapeutic strategy for managing bladder remodeling in such conditions.
Clinical applications of CCL2/CCR2 antagonists
In recent years, a considerable number of clinical studies focusing on adult patients have been conducted to explore the effects of CCL2/CCR2 antagonists (Figure 3). Currently, significant advancements have been made in clinical research concerning the treatment of NASH-related liver fibrosis using CVC, with clinical trials advancing to Phase III. It is widely acknowledged that the medication shows good tolerability, with a safety profile similar to that of a placebo (172). Initial results from ongoing trials indicate that CVC may offer additional benefits for patients suffering from advanced fibrosis (173, 174). However, the combination of CVC with Tropifexor did not yield improved therapeutic efficacy (175). Although the Phase III clinical trial demonstrated good therapeutic efficacy, it still lacks histological data from clinical studies to confirm its efficacy in the treatment of liver fibrosis (176).
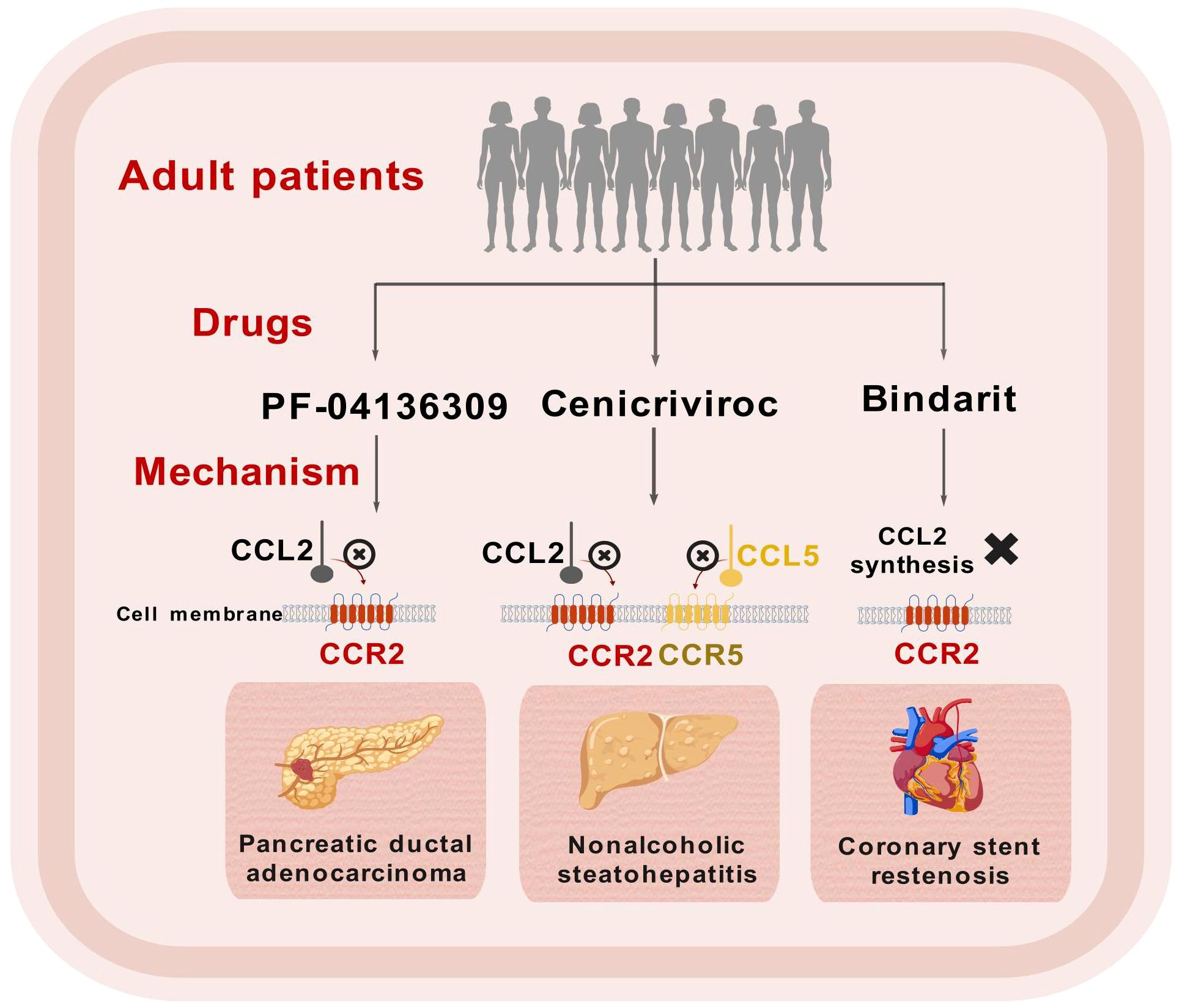
Figure 3. Schematic representation of clinical trials for CCL2/CCR2 inhibitors. This figure was created with biogdp.com.
Primary sclerosing cholangitis (PSC) is a chronic cholestatic condition that can result in liver fibrosis. A multicenter clinical trial conducted with adult patients diagnosed with PSC revealed that after 24 weeks of treatment with CVC, there was a moderate decrease in the surrogate marker of serum alkaline phosphatase (ALP), and the therapy was well tolerated by the participants (177).
In addition to fibrotic diseases, there has been notable advancement in clinical trials involving CCL2/CCR2 antagonists for other conditions, including pancreatic ductal adenocarcinoma (PDAC) (178, 179) and coronary stent restenosis (180). Future exploration of larger clinical studies in these domains is warranted (Table 1).
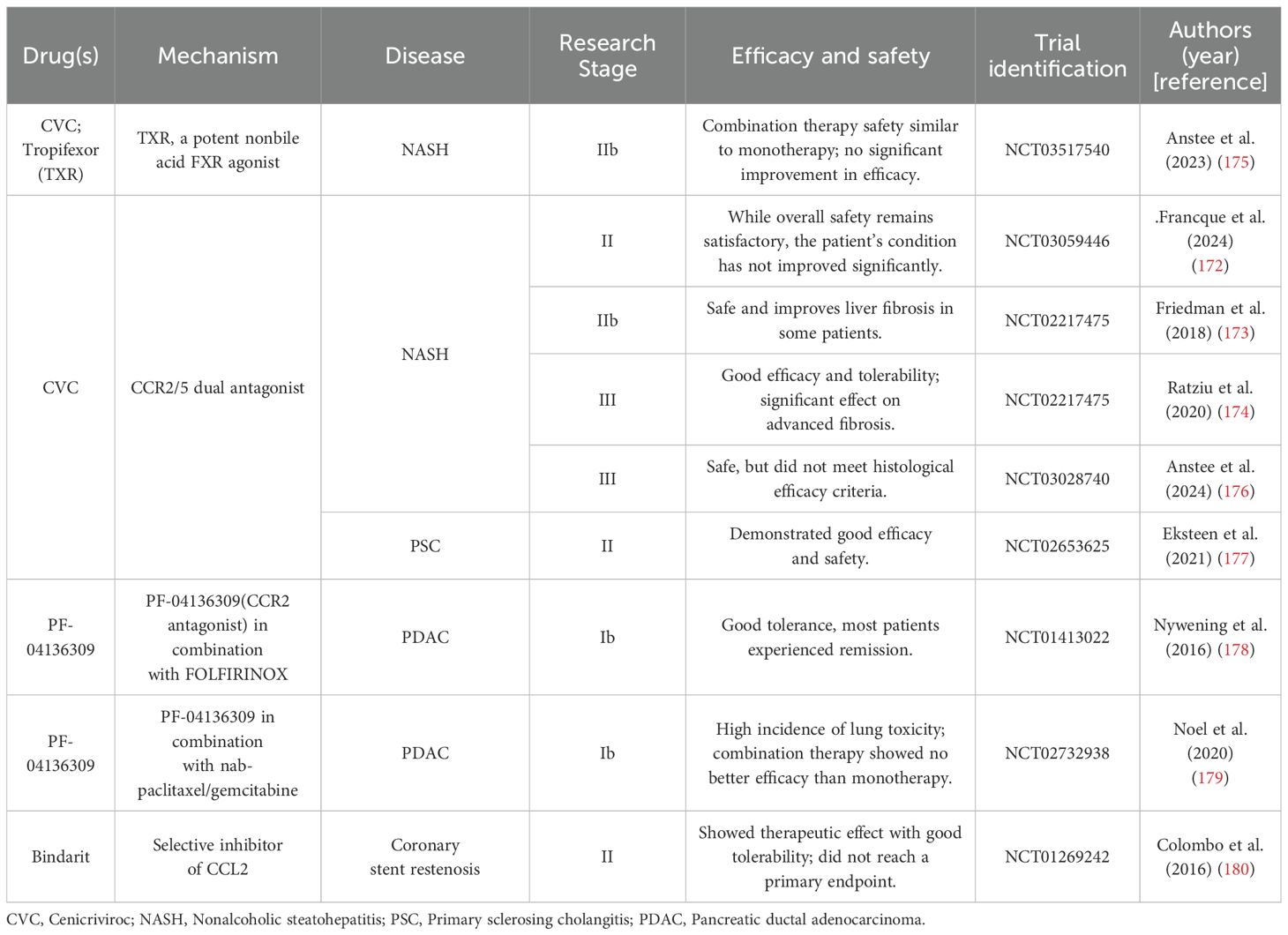
Table 1. Clinical trials of CCR2/CCL2 antagonists in treating inflammatory and fibrotic diseases.
Conclusions and future perspectives
Recent research underscores the crucial role of the CCL2/CCR2 axis in the development and progression of inflammatory and fibrotic diseases across various organs (Table 2). While the mechanisms by which CCL2 mediates cell interactions differ slightly between organs (Figure 4), its primary role in most fibrotic conditions is to recruit macrophages to sites of tissue damage. An exception is viral hepatitis, where CCL2 expression is downregulated in liver cells, contributing to an immunosuppressive environment that allows viral persistence, a key early step in the disease’s pathogenesis.

Table 2. The role of CCL2/CCR2 axis in organ fibrosis.
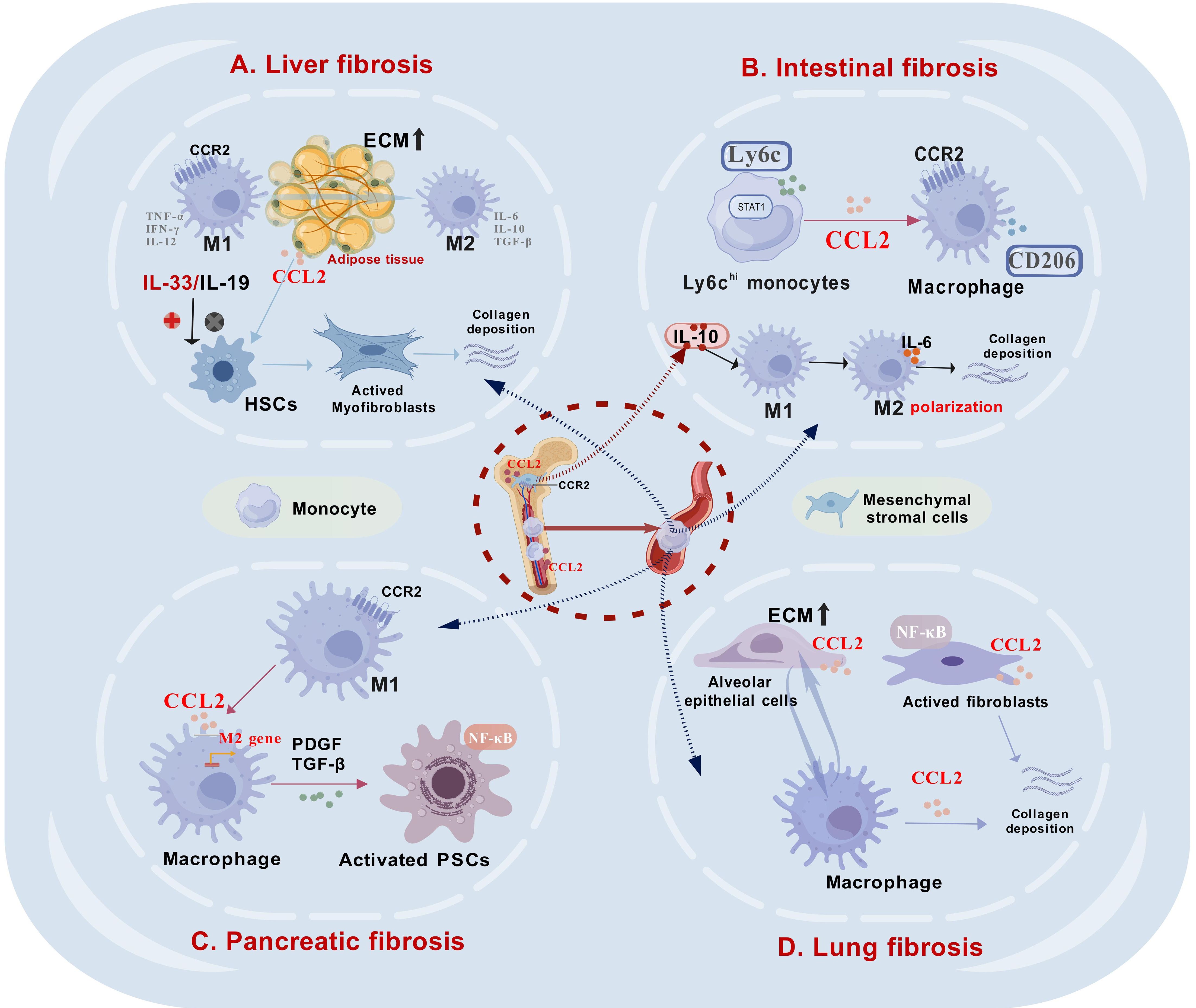
Figure 4. The role of CCL2 in mediating intercellular interactions within fibrotic tissues in various organs. (A). In liver fibrosis, CCL2 plays a critical role by recruiting macrophages to the fibrotic regions and facilitating their M2 polarization, which subsequently activates hepatic stellate cells (HSCs) and promotes collagen deposition. (B). In intestinal fibrosis, Ly6Chi monocytes are predominant, and an increase in their proportion significantly enhances the expression of CCL2. Bone marrow-derived mesenchymal stromal cells (BM-MSCs) can release IL-10, which facilitates the M2 polarization of intestinal macrophages, subsequently contributing to the development of fibrosis. (C). In pancreatic fibrosis, M2 macrophages secrete pro-fibrotic factors that activate pancreatic stellate cells (PSCs), a process similar to that in liver fibrosis. (D). In lung fibrosis, alveolar epithelial cells (AECs) exhibit increased CCL2 production, facilitating macrophage recruitment to fibrotic regions. Additionally, activated fibroblasts release significant amounts of CCL2 via NF-κB signaling, contributing to collagen accumulation. This figure was created with biogdp.com.
In pancreatic fibrosis, the role of the CCL2/CCR2 axis is less understood. It remains unclear whether CCL2 is the primary chemokine driving macrophage infiltration in the pancreas or which genes trigger its activation during fibrosis. Recent evidence suggests that SPHK1 is critical in promoting fibrosis in CP models (181). Given SPHK1’s regulatory effect on CCL2 in liver fibrosis, it is worth investigating if a similar mechanism occurs in SPHK1-induced pancreatic fibrosis.
Another area needing exploration is the role of CCL2 in pain associated with fibrotic diseases. Emerging studies have linked CCL2 to pancreatic tumor growth, neural invasion, and pancreatic cancer-related pain (182, 183). Since pain is a common, debilitating symptom in many inflammatory and fibrotic conditions, such as CP (184), understanding how CCL2 contributes to neural invasion and pain mechanisms could reveal new research and treatment avenues.
The therapeutic potential of targeting the CCL2/CCR2 axis is promising. Various strategies, including natural compounds, chemical agents, and traditional Chinese medicine, have been explored to modulate this pathway in treating fibrotic diseases, highlighting its clinical translation potential. Beyond inflammatory and fibrotic conditions, targeting CCL2 could also enhance cancer treatment, particularly immunotherapy in cancers such as esophageal squamous cell carcinoma (185, 186) and breast cancer (187). Circulating CCL2 levels may also serve as biomarkers for predicting the onset, progression, and prognosis of various cancers, including liver and prostate cancers. Further research is needed to determine if CCL2 has similar predictive value in other cancers and its potential to improve chemotherapy outcomes.
Clinical trials of CCL2/CCR2 antagonists are ongoing for various diseases. In conclusion, recent studies investigating the role of CCR2 antagonists in treating fibrotic diseases have shown promising safety profiles, with the majority of participants reporting significant symptom relief. However, there is an urgent necessity for more sophisticated assessment methodologies to rigorously evaluate the effectiveness of CCL2/CCR2 inhibitors in various fibrotic conditions. Notwithstanding these challenges, CCL2 is anticipated to undergo further evaluation as a viable therapeutic target for a broader spectrum of organ-related diseases, potentially providing patients with innovative and effective treatment alternatives.
Author contributions
SG: Writing – original draft, Conceptualization, Software, Visualization. QZ: Conceptualization, Writing – review & editing. YG: Formal Analysis, Writing – review & editing. XY: Software, Writing – review & editing. PZ: Software, Writing – review & editing. TM: Methodology, Writing – review & editing. ZT: Supervision, Writing – review & editing. XL: Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. National Natural Science Foundation of China (Grant No.: 82270676); Taishan Young Scholars Program of Shandong Province (NO. tsqn202306395); 2023 Qingdao Technology Benefiting Demonstration Project (Grant No.: 23–2-8-smjk-8-nsh); 2021 Shandong Province Graduate Education and Teaching Reform Research Project (SDYJG21110); Qingdao Chinese Medicine Technology Project (Grant No.: 2021-zyym26).
Conflict of interest
The authors declare that there are no commercial or financial relationships that could be construed as a potential conflict of interest in the conduct of this research.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. (2018) 285:2944–71. doi: 10.1111/febs.2018.285.issue-16
2. Ono SJ, Nakamura T, Miyazaki D, Ohbayashi M, Dawson M, Toda M. Chemokines: roles in leukocyte development, trafficking, and effector function. J Allergy Clin Immunol. (2003) 111:1185–99. doi: 10.1067/mai.2003.1594
3. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. (2015) 7(5):a016303. doi: 10.1101/cshperspect.a016303
4. Murphy PM, Baggiolini M, Charo IF, Hébert CA, Horuk R, Matsushima K, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. (2000) 52:145–76.
5. Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med. (1989) 169:1485–90. doi: 10.1084/jem.169.4.1485
6. Van Coillie E, Van Damme J, Opdenakker G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev. (1999) 10:61–86. doi: 10.1016/S1359-6101(99)00005-2
7. Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J Biol Chem. (1991) 266:9912–8. doi: 10.1016/S0021-9258(18)92905-4
8. Gschwandtner M, Derler R, Midwood KS. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol. (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
9. Huma ZE, Sanchez J, Lim HD, Bridgford JL, Huang C, Parker BJ, et al. Key determinants of selective binding and activation by the monocyte chemoattractant proteins at the chemokine receptor CCR2. Sci Signal. (2017) 10(480):eaai8529. doi: 10.1126/scisignal.aai8529
10. Bhusal RP, Foster SR, Stone MJ. Structural basis of chemokine and receptor interactions: Key regulators of leukocyte recruitment in inflammatory responses. Protein Sci. (2020) 29:420–32. doi: 10.1002/pro.v29.2
11. Ren J, Wang Q, Morgan S, Si Y, Ravichander A, Dou C, et al. Protein kinase C-δ (PKCδ) regulates proinflammatory chemokine expression through cytosolic interaction with the NF-κB subunit p65 in vascular smooth muscle cells. J Biol Chem. (2014) 289:9013–26. doi: 10.1074/jbc.M113.515957
12. Chiu HY, Sun KH, Chen SY, Wang HH, Lee MY, Tsou YC, et al. Autocrine CCL2 promotes cell migration and invasion via PKC activation and tyrosine phosphorylation of paxillin in bladder cancer cells. Cytokine. (2012) 59:423–32. doi: 10.1016/j.cyto.2012.04.017
13. Jiménez-Sainz MC, Fast B, Mayor F Jr., Aragay AM. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol Pharmacol. (2003) 64:773–82. doi: 10.1124/mol.64.3.773
14. Hao Q, Vadgama JV, Wang P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun Signal. (2020) 18:82. doi: 10.1186/s12964-020-00589-8
15. Arefieva TI, Kukhtina NB, Antonova OA, Krasnikova TL. MCP-1-stimulated chemotaxis of monocytic and endothelial cells is dependent on activation of different signaling cascades. Cytokine. (2005) 31:439–46. doi: 10.1016/j.cyto.2005.06.016
16. Yasui H, Kajiyama H, Tamauchi S, Suzuki S, Peng Y, Yoshikawa N, et al. CCL2 secreted from cancer-associated mesothelial cells promotes peritoneal metastasis of ovarian cancer cells through the P38-MAPK pathway. Clin Exp Metastasis. (2020) 37:145–58. doi: 10.1007/s10585-019-09993-y
17. Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem. (2008) 283:25057–73. doi: 10.1074/jbc.M801073200
18. Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y, et al. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. (2020) 111:47–58. doi: 10.1111/cas.14230
19. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. (2006) 354:610–21. doi: 10.1056/NEJMra052723
20. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
21. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
22. Morein D, Erlichman N, Ben-Baruch A. Beyond cell motility: the expanding roles of chemokines and their receptors in Malignancy. Front Immunol. (2020) 11:952. doi: 10.3389/fimmu.2020.00952
23. Xu M, Wang Y, Xia R, Wei Y, Wei X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. (2021) 54:e13115. doi: 10.1111/cpr.13115
24. Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity. (2021) 54:859–74. doi: 10.1016/j.immuni.2021.01.012
25. Lee S, Lee E, Ko E, Ham M, Lee HM, Kim ES, et al. Tumor-associated macrophages secrete CCL2 and induce the invasive phenotype of human breast epithelial cells through upregulation of ERO1-α and MMP-9. Cancer Lett. (2018) 437:25–34. doi: 10.1016/j.canlet.2018.08.025
26. Hou P, Kapoor A, Zhang Q, Li J, Wu CJ, Li J, et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discovery. (2020) 10:1058–77. doi: 10.1158/2159-8290.CD-19-0597
27. Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H, et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discovery. (2020) 10:608–25. doi: 10.1158/2159-8290.CD-19-0297
28. Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. (2018) 15:335–45. doi: 10.1038/cmi.2017.135
29. Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. (2002) 2:106–15. doi: 10.1038/nri722
30. Vilgelm AE, Richmond A. Chemokines modulate immune surveillance in tumorigenesis, metastasis, and response to immunotherapy. Front Immunol. (2019) 10:333. doi: 10.3389/fimmu.2019.00333
31. Saxena S, Singh RK. Chemokines orchestrate tumor cells and the microenvironment to achieve metastatic heterogeneity. Cancer Metastasis Rev. (2021) 40:447–76. doi: 10.1007/s10555-021-09970-6
32. Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. (2014) 147:577–594.e1. doi: 10.1053/j.gastro.2014.06.043
33. Li S, Zhou B, Xue M, Zhu J, Tong G, Fan J, et al. Macrophage-specific FGF12 promotes liver fibrosis progression in mice. Hepatology. (2023) 77:816–33. doi: 10.1002/hep.32640
34. Chen Y, Wang J, Zhou N, Fang Q, Cai H, Du Z, et al. Protozoan-derived cytokine-transgenic macrophages reverse hepatic fibrosis. Adv Sci (Weinh). (2024) 11:e2308750. doi: 10.1002/advs.202308750
35. Guillen-Sacoto MJ, Martinez AF, Abe Y, Kruszka P, Weiss K, Everson JL, et al. Human germline hedgehog pathway mutations predispose to fatty liver. J Hepatol. (2017) 67:809–17. doi: 10.1016/j.jhep.2017.06.008
36. Giannelli G, Chieti A, Cigliano A, Mancarella S, Dituri F. TGF-β as multifaceted orchestrator in HCC progression: signaling, EMT, immune microenvironment, and novel therapeutic perspectives. Semin Liver Dis. (2018) 39:053–69. doi: 10.1055/s-0038-1676121
37. Zhuang H, Cao G, Kou C, Liu T. CCL2/CCR2 axis induces hepatocellular carcinoma invasion and epithelial-mesenchymal transition in vitro through activation of the Hedgehog pathway. Oncol Rep. (2018) 39:21–30. doi: 10.3892/or.2017.6069
38. Li X, Yuan B, Lu M, Wang Y, Ding N, Liu C, et al. The methyltransferase METTL3 negatively regulates nonalcoholic steatohepatitis (NASH) progression. Nat Commun. (2021) 12:7213. doi: 10.1038/s41467-021-27539-3
39. Ni Y, Zhuge F, Ni L, Nagata N, Yamashita T, Mukaida N, et al. CX3CL1/CX3CR1 interaction protects against lipotoxicity-induced nonalcoholic steatohepatitis by regulating macrophage migration and M1/M2 status. Metabolism. (2022) 136:155272. doi: 10.1016/j.metabol.2022.155272
40. Yu J, Zhu C, Wang X, Kim K, Bartolome A, Dongiovanni P, et al. Hepatocyte TLR4 triggers inter-hepatocyte Jagged1/Notch signaling to determine NASH-induced fibrosis. Sci Transl Med. (2021) 13(599):eabe1692. doi: 10.1126/scitranslmed.abe1692
41. Kang J, Postigo-Fernandez J, Kim K, Zhu C, Yu J, Meroni M, et al. Notch-mediated hepatocyte MCP-1 secretion causes liver fibrosis. JCI Insight. (2023) 8:e165369. doi: 10.1172/jci.insight.165369
42. Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion AC, et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. (2020) 72:528–38. doi: 10.1016/j.jhep.2019.10.028
43. Parker R, Weston CJ, Miao Z, Corbett C, Armstrong MJ, Ertl L, et al. CC chemokine receptor 2 promotes recruitment of myeloid cells associated with insulin resistance in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. (2018) 314:G483–93. doi: 10.1152/ajpgi.00213.2017
44. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol. (2018) 68:773–82. doi: 10.1016/j.jhep.2017.11.040
45. Unamuno X, Gómez-Ambrosi J, Ramírez B, Rodríguez A, Becerril S, Valentí V, et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell Mol Immunol. (2021) 18:1045–57. doi: 10.1038/s41423-019-0296-z
46. Wakamatsu S, Jojima T, Hashiguchi M, Kishi H, Niitani T, Sakurai S, et al. Inhibition of IL-33 signaling ameliorate hepatic fibrosis with decreasing MCP-1 in a mouse model of diabetes and non-alcoholic steatohepatitis; comparison for luseogliflozin, an SGLT2 inhibitor. J Diabetes Complications. (2024) 38:108650. doi: 10.1016/j.jdiacomp.2023.108650
47. Ono N, Fujita T, Miki M, Nishiyama K, Izawa T, Aoyama T, et al. Interleukin-19 gene-deficient mice promote liver fibrosis via enhanced TGF-β Signaling, and the interleukin-19-CCL2 axis is important in the direction of liver fibrosis. Biomedicines. (2023) 11(7):2064. doi: 10.3390/biomedicines11072064
48. Duriez M, Mandouri Y, Lekbaby B, Wang H, Schnuriger A, Redelsperger F, et al. Alternative splicing of hepatitis B virus: A novel virus/host interaction altering liver immunity. J Hepatol. (2017) 67:687–99. doi: 10.1016/j.jhep.2017.05.025
49. Barathan M, Riazalhosseini B, Iyadorai T, Vellasamy KM, Vadivelu J, Chang LY, et al. Comparative expression of pro-inflammatory and apoptotic biosignatures in chronic HBV-infected patients with and without liver cirrhosis. Microb Pathog. (2021) 161:105231. doi: 10.1016/j.micpath.2021.105231
50. Xie X, Lv H, Liu C, Su X, Yu Z, Song S, et al. HBeAg mediates inflammatory functions of macrophages by TLR2 contributing to hepatic fibrosis. BMC Med. (2021) 19:247. doi: 10.1186/s12916-021-02085-3
51. Sarma NJ, Tiriveedhi V, Crippin JS, Chapman WC, Mohanakumar T, Williams B. Hepatitis C virus-induced changes in microRNA 107 (miRNA-107) and miRNA-449a modulate CCL2 by targeting the interleukin-6 receptor complex in hepatitis. J Virol. (2014) 88:3733–43. doi: 10.1128/JVI.03060-13
52. Song X, Gao X, Wang Y, Raja R, Zhang Y, Yang S, et al. HCV core protein induces chemokine CCL2 and CXCL10 expression through NF-κB signaling pathway in macrophages. Front Immunol. (2021) 12:654998. doi: 10.3389/fimmu.2021.654998
53. Bai YM, Liang S, Zhou B. Revealing immune infiltrate characteristics and potential immune-related genes in hepatic fibrosis: based on bioinformatics, transcriptomics and q-PCR experiments. Front Immunol. (2023) 14:1133543. doi: 10.3389/fimmu.2023.1133543
54. Flamini S, Sergeev P, Viana de Barros Z, Mello T, Biagioli M, Paglialunga M, et al. Glucocorticoid-induced leucine zipper regulates liver fibrosis by suppressing CCL2-mediated leukocyte recruitment. Cell Death Dis. (2021) 12:421. doi: 10.1038/s41419-021-03704-w
55. Lan T, Li C, Yang G, Sun Y, Zhuang L, Ou Y, et al. Sphingosine kinase 1 promotes liver fibrosis by preventing miR-19b-3p-mediated inhibition of CCR2. Hepatology. (2018) 68:1070–86. doi: 10.1002/hep.29885
56. Moreno Traspas R, Teoh TS, Wong PM, Maier M, Chia CY, Lay K, et al. Loss of FOCAD, operating via the SKI messenger RNA surveillance pathway, causes a pediatric syndrome with liver cirrhosis. Nat Genet. (2022) 54:1214–26. doi: 10.1038/s41588-022-01120-0
57. Kii S, Kitamura H, Hashimoto S, Ikeo K, Ichikawa N, Yoshida T, et al. STAT1-mediated induction of Ly6c-expressing macrophages are involved in the pathogenesis of an acute colitis model. Inflammation Res. (2022) 71:1079–94. doi: 10.1007/s00011-022-01620-z
58. Pei X, Sun Q, Zhang Y, Wang P, Peng X, Guo C, et al. PC3-secreted microprotein is a novel chemoattractant protein and functions as a high-affinity ligand for CC chemokine receptor 2. J Immunol. (2014) 192:1878–86. doi: 10.4049/jimmunol.1300758
59. Pei X, Zheng D, She S, Ma J, Guo C, Mo X, et al. The PSMP-CCR2 interactions trigger monocyte/macrophage-dependent colitis. Sci Rep. (2017) 7:5107. doi: 10.1038/s41598-017-05255-7
60. Rahabi M, Jacquemin G, Prat M, Meunier E, AlaEddine M, Bertrand B, et al. Divergent roles for macrophage C-type lectin receptors, dectin-1 and mannose receptors, in the intestinal inflammatory response. Cell Rep. (2020) 30:4386–4398.e5. doi: 10.1016/j.celrep.2020.03.018
61. Schaefer REM, Callahan RC, Atif SM, Orlicky DJ, Cartwright IM, Fontenot AP, et al. Disruption of monocyte-macrophage differentiation and trafficking by a heme analog during active inflammation. Mucosal Immunol. (2022) 15:244–56. doi: 10.1038/s41385-021-00474-8
62. Stakenborg M, Abdurahiman S, De Simone V, Goverse G, Stakenborg N, van Baarle L, et al. Enteric glial cells favor accumulation of anti-inflammatory macrophages during the resolution of muscularis inflammation. Mucosal Immunol. (2022) 15:1296–308. doi: 10.1038/s41385-022-00563-2
63. Giri J, Das R, Nylen E, Chinnadurai R, Galipeau J. CCL2 and CXCL12 derived from mesenchymal stromal cells cooperatively polarize IL-10+ Tissue macrophages to mitigate gut injury. Cell Rep. (2020) 30:1923–1934.e4. doi: 10.1016/j.celrep.2020.01.047
64. Kuroda N, Masuya M, Tawara I, Tsuboi J, Yoneda M, Nishikawa K, et al. Infiltrating CCR2(+) monocytes and their progenies, fibrocytes, contribute to colon fibrosis by inhibiting collagen degradation through the production of TIMP-1. Sci Rep. (2019) 9:8568. doi: 10.1038/s41598-019-45012-6
65. Cao Q, Lin Y, Yue C, Wang Y, Quan F, Cui X, et al. IL-6 deficiency promotes colitis by recruiting Ly6C(hi) monocytes into inflamed colon tissues in a CCL2-CCR2-dependent manner. Eur J Pharmacol. (2021) 904:174165. doi: 10.1016/j.ejphar.2021.174165
66. Liu X, Lu Y, Zhu J, Liu M, Xie M, Ye M, et al. A long noncoding RNA, antisense IL-7, promotes inflammatory gene transcription through facilitating histone acetylation and switch/sucrose nonfermentable chromatin remodeling. J Immunol. (2019) 203:1548–59. doi: 10.4049/jimmunol.1900256
67. Yang X, Yabe-Wada T, Han J, Saito F, Ogasawara C, Yamada S, et al. PCBP1 acts as a regulator of CCL2 expression in macrophages to induce recruitment of monocyte-derived macrophages into the inflamed colon. Int Immunol. (2023) 35:287–99. doi: 10.1093/intimm/dxad003
68. Mei C, Meng F, Wang X, Yan S, Zheng Q, Zhang X, et al. CD30L is involved in the regulation of the inflammatory response through inducing homing and differentiation of monocytes via CCL2/CCR2 axis and NF-κB pathway in mice with colitis. Int Immunopharmacol. (2022) 110:108934. doi: 10.1016/j.intimp.2022.108934
69. Li T, Liu Y, Yu X, Wang P, Sun S, Liu D. IL-17D affects the chemokines and chemokine receptors of intestinal epithelial cells under hyperoxia. Int Immunopharmacol. (2022) 113:109386. doi: 10.1016/j.intimp.2022.109386
70. Mews P, Phillips P, Fahmy R, Korsten M, Pirola R, Wilson J, et al. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut. (2002) 50:535–41. doi: 10.1136/gut.50.4.535
71. Sun LK, Reding T, Bain M, Heikenwalder M, Bimmler D, Graf R. Prostaglandin E2 modulates TNF-alpha-induced MCP-1 synthesis in pancreatic acinar cells in a PKA-dependent manner. Am J Physiol Gastrointest Liver Physiol. (2007) 293:G1196–204. doi: 10.1152/ajpgi.00330.2007
72. Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ, et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. (2015) 6:7158. doi: 10.1038/ncomms8158
73. Wu N, Xu XF, Xin JQ, Fan JW, Wei YY, Peng QX, et al. The effects of nuclear factor-kappa B in pancreatic stellate cells on inflammation and fibrosis of chronic pancreatitis. J Cell Mol Med. (2021) 25:2213–27. doi: 10.1111/jcmm.16213
74. Vos T, Lim SS, Abbafati C, Abbas KM, Abbasi M, Abbasifard M, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. (2020) 396:1204–22. doi: 10.1016/S0140-6736(20)30925-9
75. Liberale L, Badimon L, Montecucco F, Lüscher TF, Libby P, Camici GG. Inflammation, aging, and cardiovascular disease: JACC review topic of the week. J Am Coll Cardiol. (2022) 79:837–47. doi: 10.1016/j.jacc.2021.12.017
76. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. (2011) 17:1410–22. doi: 10.1038/nm.2538
77. Chen L, Pan D, Zhang Y, Zhang E, Ma L. C-C motif chemokine 2 regulates macrophage polarization and contributes to myocardial infarction healing. J Interferon Cytokine Res. (2024) 44:68–79. doi: 10.1089/jir.2023.0132
78. Cheng Y, Luo D, Zhao Y, Rong J. N-Propargyl caffeate amide (PACA) prevents cardiac fibrosis in experimental myocardial infarction by promoting pro-resolving macrophage polarization. Aging (Albany NY). (2020) 12:5384–98. doi: 10.18632/aging.102959
79. Shen SC, Xu J, Cheng C, Xiang XJ, Hong BY, Zhang M, et al. Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching. Acta Pharmacol Sin. (2024) 45:959–74. doi: 10.1038/s41401-023-01222-3
80. Zhuang L, Zong X, Yang Q, Fan Q, Tao R. Interleukin-34-NF-κB signaling aggravates myocardial ischemic/reperfusion injury by facilitating macrophage recruitment and polarization. EBioMedicine. (2023) 95:104744. doi: 10.1016/j.ebiom.2023.104744
81. Georgakis MK, de Lemos JA, Ayers C, Wang B, Björkbacka H, Pana TA, et al. Association of circulating monocyte chemoattractant protein-1 levels with cardiovascular mortality: A meta-analysis of population-based studies. JAMA Cardiol. (2021) 6:587–92. doi: 10.1001/jamacardio.2020.5392
82. Georgakis MK, van der Laan SW, Asare Y, Mekke JM, Haitjema S, Schoneveld AH, et al. Monocyte-chemoattractant protein-1 levels in human atherosclerotic lesions associate with plaque vulnerability. Arterioscler Thromb Vasc Biol. (2021) 41:2038–48. doi: 10.1161/ATVBAHA.121.316091
83. Georgakis MK, Gill D, Rannikmäe K, Traylor M. Genetically determined levels of circulating cytokines and risk of stroke. Circulation. (2019) 139:256–68. doi: 10.1161/CIRCULATIONAHA.118.035905
84. Winter C, Silvestre-Roig C, Ortega-Gomez A, Lemnitzer P, Poelman H, Schumski A, et al. Chrono-pharmacological targeting of the CCL2-CCR2 axis ameliorates atherosclerosis. Cell Metab. (2018) 28:175–182.e5. doi: 10.1016/j.cmet.2018.05.002
85. Singh S, Anshita D, Ravichandiran V. MCP-1: Function, regulation, and involvement in disease. Int Immunopharmacol. (2021) 101:107598. doi: 10.1016/j.intimp.2021.107598
86. Luo Y, Zhang H, Yu J, Wei L, Li M, Xu W. Stem cell factor/mast cell/CCL2/monocyte/macrophage axis promotes Coxsackievirus B3 myocarditis and cardiac fibrosis by increasing Ly6C(high) monocyte influx and fibrogenic mediators production. Immunology. (2022) 167:590–605. doi: 10.1111/imm.13556
87. Chouvarine P, Legchenko E, Geldner J, Riehle C, Hansmann G. Hypoxia drives cardiac miRNAs and inflammation in the right and left ventricle. J Mol Med (Berl). (2019) 97:1427–38. doi: 10.1007/s00109-019-01817-6
88. Yang J, Agarwal M, Ling S, Teitz-Tennenbaum S, Zemans RL, Osterholzer JJ, et al. Diverse injury pathways induce alveolar epithelial cell CCL2/12, which promotes lung fibrosis. Am J Respir Cell Mol Biol. (2020) 62:622–32. doi: 10.1165/rcmb.2019-0297OC
89. Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. (2007) 132:1311–21. doi: 10.1378/chest.06-2568
90. Zhao T, Wu X, Zhao X, Yao K, Li X, Ni J. Identification and validation of chemokine system-related genes in idiopathic pulmonary fibrosis. Front Immunol. (2023) 14:1159856. doi: 10.3389/fimmu.2023.1159856
91. Hult EM, Gurczynski SJ, O'Dwyer DN, Zemans RL, Rasky A, Wang Y, et al. Myeloid- and epithelial-derived heparin-binding epidermal growth factor-like growth factor promotes pulmonary fibrosis. Am J Respir Cell Mol Biol. (2022) 67:641–53. doi: 10.1165/rcmb.2022-0174OC
92. Bian F, Lan YW, Zhao S, Deng Z, Shukla S, Acharya A, et al. Lung endothelial cells regulate pulmonary fibrosis through FOXF1/R-Ras signaling. Nat Commun. (2023) 14:2560. doi: 10.1038/s41467-023-38177-2
93. Nie Y, Zhai X, Li J, Sun A, Che H, Christman JW, et al. NFATc3 promotes pulmonary inflammation and fibrosis by regulating production of CCL2 and CXCL2 in macrophages. Aging Dis. (2023) 14:1441–57. doi: 10.14336/AD.2022.1202
94. Li C, Feng X, Li S, He X, Luo Z, Cheng X, et al. Tetrahedral DNA loaded siCCR2 restrains M1 macrophage polarization to ameliorate pulmonary fibrosis in chemoradiation-induced murine model. Mol Ther. (2024) 32:766–82. doi: 10.1016/j.ymthe.2024.01.022
95. Karman J, Wang J, Bodea C, Cao S, Levesque MC. Lung gene expression and single cell analyses reveal two subsets of idiopathic pulmonary fibrosis (IPF) patients associated with different pathogenic mechanisms. PloS One. (2021) 16:e0248889. doi: 10.1371/journal.pone.0248889
96. Walsh SMW, Worrell JC, Fabre A, Hinz B, Kane R, Keane MP. Novel differences in gene expression and functional capabilities of myofibroblast populations in idiopathic pulmonary fibrosis. Am J Physiol-Lung C. (2018) 315:L697–710. doi: 10.1152/ajplung.00543.2017
97. Hadjicharalambous MR, Roux BT, Feghali-Bostwick CA, Murray LA, Clarke DL, Lindsay MA. Long non-coding RNAs are central regulators of the IL-1β-induced inflammatory response in normal and idiopathic pulmonary lung fibroblasts. Front Immunol. (2018) 9:2906. doi: 10.3389/fimmu.2018.02906
98. Gui X, Qiu X, Tian Y, Xie M, Li H, Gao Y, et al. Prognostic value of IFN-γ, sCD163, CCL2 and CXCL10 involved in acute exacerbation of idiopathic pulmonary fibrosis. Int Immunopharmacol. (2019) 70:208–15. doi: 10.1016/j.intimp.2019.02.039
99. Su L, Dong Y, Wang Y, Wang Y, Guan B, Lu Y, et al. Potential role of senescent macrophages in radiation-induced pulmonary fibrosis. Cell Death Dis. (2021) 12:527. doi: 10.1038/s41419-021-03811-8
100. Groves AM, Johnston CJ, Williams JP, Finkelstein JN. Role of infiltrating monocytes in the development of radiation-induced pulmonary fibrosis. Radiat Res. (2018) 189:300–11. doi: 10.1667/RR14874.1
101. Kubo F, Ariestanti DM, Oki S, Fukuzawa T, Demizu R, Sato T, et al. Loss of the adhesion G-protein coupled receptor ADGRF5 in mice induces airway inflammation and the expression of CCL2 in lung endothelial cells. Respir Res. (2019) 20:11. doi: 10.1186/s12931-019-0973-6
102. Tian Y, Lv J, Su Z, Wu T, Li X, Hu X, et al. LRRK2 plays essential roles in maintaining lung homeostasis and preventing the development of pulmonary fibrosis. Proc Natl Acad Sci U.S.A. (2021) 118(35):e2106685118. doi: 10.1073/pnas.2106685118
103. Martinu T, McManigle WC, Kelly FL, Nelson ME, Sun J, Zhang HL, et al. IL-17A contributes to lung fibrosis in a model of chronic pulmonary graft-versus-host disease. Transplantation. (2019) 103:2264–74. doi: 10.1097/TP.0000000000002837
104. Rosendahl A, Kabiri R, Bode M, Cai A, Klinge S, Ehmke H, et al. Adaptive immunity and IL-17A are not involved in the progression of chronic kidney disease after 5/6 nephrectomy in mice. Br J Pharmacol. (2019) 176:2002–14. doi: 10.1111/bph.14509
105. Zhou S, Yin X, Mayr M, Noor M, Hylands PJ, Xu Q. Proteomic landscape of TGF-β1-induced fibrogenesis in renal fibroblasts. Sci Rep. (2020) 10:19054. doi: 10.1038/s41598-020-75989-4
106. Kavvadas P, Keuylian Z, Prakoura N, Placier S, Dorison A, Chadjichristos CE, et al. Notch3 orchestrates epithelial and inflammatory responses to promote acute kidney injury. Kidney Int. (2018) 94:126–38. doi: 10.1016/j.kint.2018.01.031
107. Brandt S, Ballhause TM, Bernhardt A, Becker A, Salaru D, Le-Deffge HM, et al. Fibrosis and immune cell infiltration are separate events regulated by cell-specific receptor notch3 expression. J Am Soc Nephrol. (2020) 31:2589–608. doi: 10.1681/ASN.2019121289
108. Wu Q, Sun S, Wei L, Liu M, Liu H, Liu T, et al. Twist1 regulates macrophage plasticity to promote renal fibrosis through galectin-3. Cell Mol Life Sci. (2022) 79:137. doi: 10.1007/s00018-022-04137-0
109. Devocelle A, Lecru L, Ferlicot S, Bessede T, Candelier JJ, Giron-Michel J, et al. IL-15 prevents renal fibrosis by inhibiting collagen synthesis: A new pathway in chronic kidney disease? Int J Mol Sci. (2021) 22(21):11698. doi: 10.3390/ijms222111698
110. Zheng M, Cai J, Liu Z, Shu S, Wang Y, Tang C, et al. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J Cell Mol Med. (2019) 23(6):3995–4004. doi: 10.1111/jcmm.14285
111. Chang FC, Liu CH, Luo AJ, Tao-Min Huang T, Tsai MH, Chen YJ, et al. Angiopoietin-2 inhibition attenuates kidney fibrosis by hindering chemokine C-C motif ligand 2 expression and apoptosis of endothelial cells. Kidney Int. (2022) 102(4):780–97. doi: 10.1016/j.kint.2022.06.026
112. Maksimowski N, Williams VR, Scholey JW. Kidney ACE2 expression: Implications for chronic kidney disease. PloS One. (2020) 15:e0241534. doi: 10.1371/journal.pone.0241534
113. Ma FY, Han Y, Ozols E, Chew P, Vesey DA, Gobe GC, et al. Protease-activated receptor 2 does not contribute to renal inflammation or fibrosis in the obstructed kidney. Nephrol (Carlton). (2019) 24:983–91. doi: 10.1111/nep.13635
114. Wilkening A, Krappe J, Mühe AM, Lindenmeyer MT, Eltrich N, Luckow B, et al. C-C chemokine receptor type 2 mediates glomerular injury and interstitial fibrosis in focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2020) 35:227–39. doi: 10.1093/ndt/gfy380
115. Bideak A, Blaut A, Hoppe JM, Müller MB, Federico G, Eltrich N, et al. The atypical chemokine receptor 2 limits renal inflammation and fibrosis in murine progressive immune complex glomerulonephritis. Kidney Int. (2018) 93:826–41. doi: 10.1016/j.kint.2017.11.013
116. Kashyap S, Osman M, Ferguson CM, Nath MC, Roy B, Lien KR, et al. Ccl2 deficiency protects against chronic renal injury in murine renovascular hypertension. Sci Rep. (2018) 8:8598. doi: 10.1038/s41598-018-26870-y
117. Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. (2013) 24:133–45. doi: 10.1016/j.cytogfr.2013.01.004
118. Pozzi G, Carubbi C, Gobbi G, Tagliaferri S, Mirandola P, Vitale M, et al. Tracking fibrosis in myeloproliferative neoplasms by CCR2 expression on CD34(+) cells. Front Oncol. (2022) 12:980379. doi: 10.3389/fonc.2022.980379
119. Masselli E, Carubbi C, Pozzi G, Percesepe A, Campanelli R, Villani L, et al. Impact of the rs1024611 polymorphism of CCL2 on the pathophysiology and outcome of primary myelofibrosis. Cancers (Basel). (2021) 13(11):2552. doi: 10.3390/cancers13112552
120. Masselli E, Carubbi C, Cambò B, Pozzi G, Gobbi G, Mirandola P, et al. The -2518 A/G polymorphism of the monocyte chemoattractant protein-1 as a candidate genetic predisposition factor for secondary myelofibrosis and biomarker of disease severity. Leukemia. (2018) 32:2266–70. doi: 10.1038/s41375-018-0088-y
121. Greiner G, Witzeneder N, Berger A, Schmetterer K, Eisenwort G, Schiefer AI, et al. CCL2 is a KIT D816V-dependent modulator of the bone marrow microenvironment in systemic mastocytosis. Blood. (2017) 129:371–82. doi: 10.1182/blood-2016-09-739003
122. Macanas-Pirard P, Quezada T, Navarrete L, Broekhuizen R, Leisewitz A, Nervi B, et al. The CCL2/CCR2 axis affects transmigration and proliferation but not resistance to chemotherapy of acute myeloid leukemia cells. PloS One. (2017) 12:e0168888. doi: 10.1371/journal.pone.0168888
123. Mazur G, Wróbel T, Butrym A, Kapelko-Słowik K, Poreba R, Kuliczkowski K. Increased monocyte chemoattractant protein 1 (MCP-1/CCL-2) serum level in acute myeloid leukemia. Neoplasma. (2007) 54:285–9.
124. Haub J, Roehrig N, Uhrin P, Schabbauer G, Eulberg D, Melchior F, et al. Intervention of inflammatory monocyte activity limits dermal fibrosis. J Invest Dermatol. (2019) 139:2144–53. doi: 10.1016/j.jid.2019.04.006
125. Park MJ, Park Y, Choi JW, Baek JA, Jeong HY, Na HS, et al. Establishment of a humanized animal model of systemic sclerosis in which T helper-17 cells from patients with systemic sclerosis infiltrate and cause fibrosis in the lungs and skin. Exp Mol Med. (2022) 54:1577–85. doi: 10.1038/s12276-022-00860-7
126. Jin J, Liu Y, Tang Q, Yan X, Jiang M, Zhao X, et al. Bioinformatics-integrated screening of systemic sclerosis-specific expressed markers to identify therapeutic targets. Front Immunol. (2023) 14:1125183. doi: 10.3389/fimmu.2023.1125183
127. Huang J, Puente H, Wareing NE, Wu M, Mayes MD, Karmouty-Quintana H, et al. STAT6 suppression prevents bleomycin-induced dermal fibrosis. FASEB J. (2023) 37:e22761. doi: 10.1096/fj.202200994R
128. Peng W, Kepsch A, Kracht TO, Hasan H, Wijayarathna R, Wahle E, et al. Activin A and CCR2 regulate macrophage function in testicular fibrosis caused by experimental autoimmune orchitis. Cell Mol Life Sci. (2022) 79:602. doi: 10.1007/s00018-022-04632-4
129. Urrego-Callejas T, Álvarez SS, Arias LF, Reyes BO, Vanegas-García AL, González LA, et al. Urinary levels of ceruloplasmin and monocyte chemoattractant protein-1 correlate with extra-capillary proliferation and chronic damage in patients with lupus nephritis. Clin Rheumatol. (2021) 40:1853–9. doi: 10.1007/s10067-020-05454-0
130. Pérez-Arias AA, Méndez-Pérez RA, Cruz C, Zavala-Miranda MF, Romero-Diaz J, Márquez-Macedo SE, et al. The first-year course of urine MCP-1 and its association with response to treatment and long-term kidney prognosis in lupus nephritis. Clin Rheumatol. (2023) 42:83–92. doi: 10.1007/s10067-022-06373-y
131. Zhang C, Chan CCY, Cheung KF, Chau MKM, Yap DYH, Ma MKM, et al. Effect of mycophenolate and rapamycin on renal fibrosis in lupus nephritis. Clin Sci (Lond). (2019) 133:1721–44. doi: 10.1042/CS20190536
132. Sears SM, Sharp CN, Krueger A, Oropilla GB, Saforo D, Doll MA, et al. C57BL/6 mice require a higher dose of cisplatin to induce renal fibrosis and CCL2 correlates with cisplatin-induced kidney injury. Am J Physiol Renal Physiol. (2020) 319:F674–85. doi: 10.1152/ajprenal.00196.2020
133. Chanrat E, Worawichawong S, Radinahamed P, Sathirapongsasuti N, Nongnuch A, Assanatham M, et al. Urine epidermal growth factor, monocyte chemoattractant protein-1 or their ratio as predictors of complete remission in primary glomerulonephritis. Cytokine. (2018) 104:1–7. doi: 10.1016/j.cyto.2018.01.015
134. Gniewkiewicz MS, Czerwińska M, Gozdowska J, Czerwińska K, Sadowska A, Dęborska-Materkowska D, et al. Urinary levels of CCL2 and CXCL10 chemokines as potential biomarkers of ongoing pathological processes in kidney allograft: an association with BK virus nephropathy. Pol Arch Intern Med. (2019) 129:592–7. doi: 10.20452/pamw.14926
135. Kong X, Xu M, Cui X, Ma L, Cheng H, Hou J, et al. Potential role of macrophage phenotypes and CCL2 in the pathogenesis of takayasu arteritis. Front Immunol. (2021) 12:646516. doi: 10.3389/fimmu.2021.646516
136. Aguiar CM, Gawdat K, Legere S, Marshall J, Hassan A, Kienesberger PC, et al. Fibrosis independent atrial fibrillation in older patients is driven by substrate leukocyte infiltration: diagnostic and prognostic implications to patients undergoing cardiac surgery. J Transl Med. (2019) 17:413. doi: 10.1186/s12967-019-02162-5
137. Sequeira Gross TM, Lindner D, Ojeda FM, Neumann J, Grewal N, Kuntze T, et al. Comparison of microstructural alterations in the proximal aorta between aortic stenosis and regurgitation. J Thorac Cardiovasc Surg. (2021) 162:1684–95. doi: 10.1016/j.jtcvs.2020.03.002
138. Wang L, Wu H, Deng Y, Zhang S, Wei Q, Yang Q, et al. FTZ ameliorates diabetic cardiomyopathy by inhibiting inflammation and cardiac fibrosis in the streptozotocin-induced model. Evidence-Based Complementary Altern Med. (2021) 2021:1–16. doi: 10.1155/2021/5582567
139. Ott A, Tutdibi E, Goedicke-Fritz S, Schöpe J, Zemlin M, Nourkami-Tutdibi N. Serum cytokines MCP-1 and GCS-F as potential biomarkers in pediatric inflammatory bowel disease. PloS One. (2023) 18:e0288147. doi: 10.1371/journal.pone.0288147
140. Luo X, Wang R, Zhang X, Wen X, Deng S, Xie W. Identification CCL2,CXCR2,S100A9 of the immune-related gene markers and immune infiltration characteristics of inflammatory bowel disease and heart failure via bioinformatics analysis and machine learning. Front Cardiovasc Med. (2023) 10:1268675. doi: 10.3389/fcvm.2023.1268675
141. Yin YQ, Peng F, Situ HJ, Xie JL, Tan L, Wei J, et al. Construction of prediction model of inflammation related genes in idiopathic pulmonary fibrosis and its correlation with immune microenvironment. Front Immunol. (2022) 13:1010345. doi: 10.3389/fimmu.2022.1010345
142. Xue M, Guo Z, Cai C, Sun B, Wang H. Evaluation of the diagnostic efficacies of serological markers KL-6, SP-A, SP-D, CCL2, and CXCL13 in idiopathic interstitial pneumonia. Respiration. (2019) 98:534–45. doi: 10.1159/000503689
143. d’Alessandro M, Bergantini L, Cameli P, Lanzarone N, Perillo F, Perrone A, et al. BAL and serum multiplex lipid profiling in idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Life Sci. (2020) 256:117995. doi: 10.1016/j.lfs.2020.117995
144. Tian T, Zhao C, Li S, Huang Z, Guo Y, Dai W, et al. Liver-targeted delivery of small interfering RNA of C-C chemokine receptor 2 with tetrahedral framework nucleic acid attenuates liver cirrhosis. ACS Appl Mater Interfaces. (2023) 15:10492–505. doi: 10.1021/acsami.2c22579
145. Du Y, Ding H, Chen Y, Gao B, Mao Z, Wang W, et al. A genetically engineered biomimetic nanodecoy for the treatment of liver fibrosis. Adv Sci (Weinh). (2024) 11(40):e2405026. doi: 10.1002/advs.202405026
146. Lan T, Chen B, Hu X, Cao J, Chen S, Ding X, et al. Tianhuang formula ameliorates liver fibrosis by inhibiting CCL2-CCR2 axis and MAPK/NF-κB signaling pathway. J Ethnopharmacol. (2024) 321:117516. doi: 10.1016/j.jep.2023.117516
147. Chen C, Yao X, Xu Y, Zhang Q, Wang H, Zhao L, et al. Dahuang Zhechong Pill suppresses colorectal cancer liver metastasis via ameliorating exosomal CCL2 primed pre-metastatic niche. J Ethnopharmacol. (2019) 238:111878. doi: 10.1016/j.jep.2019.111878
148. Shi H, Duan X, Dong J, Tao Y, Lei Y. RNA-seq combined network pharmacology reveals that Fu-Gan-Wan (FGW) inhibits liver fibrosis via NF-κB/CCL2/CCR2 and lipid peroxidation via Nrf2/HMOX1 signaling pathway. J Ethnopharmacol. (2024) 326:117963. doi: 10.1016/j.jep.2024.117963
149. Liu Z, Xiang H, Xiang D, Xiao S, Xiang H, Xiao J, et al. Revealing potential anti-fibrotic mechanism of Ganxianfang formula based on RNA sequence. Chin Med. (2022) 17:23. doi: 10.1186/s13020-022-00579-7
150. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. (2018) 67:1270–83. doi: 10.1002/hep.29544
151. Puengel T, Lefere S, Hundertmark J, Kohlhepp M, Penners C, Van de Velde F, et al. Combined therapy with a CCR2/CCR5 antagonist and FGF21 analogue synergizes in ameliorating steatohepatitis and fibrosis. Int J Mol Sci. (2022) 23(12):6696. doi: 10.3390/ijms23126696
152. Guo Y, Zhao C, Dai W, Wang B, Lai E, Xiao Y, et al. C-C motif chemokine receptor 2 inhibition reduces liver fibrosis by restoring the immune cell landscape. Int J Biol Sci. (2023) 19:2572–87. doi: 10.7150/ijbs.83530
153. Yu D, Cai SY, Mennone A, Vig P, Boyer JL. Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int. (2018) 38:1128–38. doi: 10.1111/liv.13698
154. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology. (2019) 69:1105–21. doi: 10.1002/hep.30249
155. Bauer A, Rawa T. Circulating monocyte chemoattractant protein-1 (MCP-1) in patients with primary biliary cholangitis. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25021333
156. Guicciardi ME, Trussoni CE, Krishnan A, Bronk SF, Lorenzo Pisarello MJ, O'Hara SP, et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol. (2018) 69:676–86. doi: 10.1016/j.jhep.2018.05.018
157. Reuveni D, Gore Y, Leung PSC, Lichter Y, Moshkovits I, Kaminitz A, et al. The critical role of chemokine (C-C motif) receptor 2-positive monocytes in autoimmune cholangitis. Front Immunol. (2018) 9:1852. doi: 10.3389/fimmu.2018.01852
158. Dai L, Liu Y, Cheng L, Wang H, Lin Y, Shi G, et al. SARI attenuates colon inflammation by promoting STAT1 degradation in intestinal epithelial cells. Mucosal Immunol. (2019) 12:1130–40. doi: 10.1038/s41385-019-0178-9
159. Wang Q, Li XL, Mei Y, Ye JC, Fan W, Cheng GH, et al. The anti-inflammatory drug dimethyl itaconate protects against colitis-associated colorectal cancer. J Mol Med (Berl). (2020) 98:1457–66. doi: 10.1007/s00109-020-01963-2
160. Xue L, Jin X, Ji T, Li R, Zhuge X, Xu F, et al. Luteolin ameliorates DSS-induced colitis in mice via suppressing macrophage activation and chemotaxis. Int Immunopharmacol. (2023) 124:110996. doi: 10.1016/j.intimp.2023.110996
161. Liu J, Shi L, Huang W, Zheng Z, Huang X, Su Y. Homoharringtonine attenuates dextran sulfate sodium-induced colitis by inhibiting NF-κB signaling. Mediators Inflammation. (2022) 2022:3441357. doi: 10.1155/2022/3441357
162. Hagenlocher Y, Gommeringer S, Held A, Feilhauer K, Köninger J, Bischoff SC, et al. Nobiletin acts anti-inflammatory on murine IL-10(-/-) colitis and human intestinal fibroblasts. Eur J Nutr. (2019) 58:1391–401. doi: 10.1007/s00394-018-1661-x
163. Zhou Z, Lin Y, Gao L, Yang Z, Wang S, Wu B. Circadian pharmacological effects of berberine on chronic colitis in mice: Role of the clock component Rev-erbα. Biochem Pharmacol. (2020) 172:113773. doi: 10.1016/j.bcp.2019.113773
164. Yu Y, Bian Y, Shi JX, Gu Y, Yuan DP, Yu B, et al. Geniposide promotes splenic Treg differentiation to alleviate colonic inflammation and intestinal barrier injury in ulcerative colitis mice. Bioengineered. (2022) 13:14616–31. doi: 10.1080/21655979.2022.2092678
165. Tao H, Zhao H, Mo A, Shao L, Ge D, Liu J, et al. VX-765 attenuates silica-induced lung inflammatory injury and fibrosis by modulating alveolar macrophages pyroptosis in mice. Ecotoxicol Environ Saf. (2023) 249:114359. doi: 10.1016/j.ecoenv.2022.114359
166. Koyama KG, Goto H, Morizumi S, Kagawa K, Nishimura H, Sato S, et al. The tyrosine kinase inhibitor TAS-115 attenuates bleomycin-induced lung fibrosis in mice. Am J Resp Cell Mol. (2019) 60:478–87. doi: 10.1165/rcmb.2018-0098OC
167. Fusco R, Cordaro M, Siracusa R, D'Amico R, Genovese T, Gugliandolo E, et al. Biochemical evaluation of the antioxidant effects of hydroxytyrosol on pancreatitis-associated gut injury. Antioxidants (Basel). (2020) 9(9):781. doi: 10.3390/antiox9090781
168. Duan LF, Xu XF, Zhu LJ, Liu F, Zhang XQ, Wu N, et al. Dachaihu decoction ameliorates pancreatic fibrosis by inhibiting macrophage infiltration in chronic pancreatitis. World J Gastroenterol. (2017) 23:7242–52. doi: 10.3748/wjg.v23.i40.7242
169. Tesch GHP, Pullen N, Jesson MI. Combined inhibition of CCR2 and ACE provides added protection against progression of diabetic nephropathy in Nos3-deficient mice. Am J Physiol-Renal. (2019) 317:F1439–49. doi: 10.1152/ajprenal.00340.2019
170. Alsheikh AJ, Dasinger JH, Abais-Battad JM, Fehrenbach DJ. CCL2 mediates early renal leukocyte infiltration during salt-sensitive hypertension. Am J Physiol-Renal. (2020) 318:F982–93. doi: 10.1152/ajprenal.00521.2019
171. Wang W, Ai J, Liao B, Xiao K, Lin L, Chen H, et al. The roles of MCP-1/CCR2 mediated macrophage recruitment and polarization in bladder outlet obstruction (BOO) induced bladder remodeling. Int Immunopharmacol. (2021) 99:107947. doi: 10.1016/j.intimp.2021.107947
172. Francque SM, Hodge A, Boursier J, Younes ZH, Rodriguez-Araujo G, Park GS, et al. Phase 2, open-label, rollover study of cenicriviroc for liver fibrosis associated with metabolic dysfunction-associated steatohepatitis. Hepatol Commun. (2024) 8(2):e0335. doi: 10.1097/HC9.0000000000000335
173. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. (2018) 67:1754–67. doi: 10.1002/hep.29477
174. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. (2020) 72:892–905. doi: 10.1002/hep.31108
175. Anstee QM, Lucas KJ, Francque S, Abdelmalek MF, Sanyal AJ, Ratziu V, et al. Tropifexor plus cenicriviroc combination versus monotherapy in nonalcoholic steatohepatitis: Results from the phase 2b TANDEM study. Hepatology. (2023) 78:1223–39. doi: 10.1097/HEP.0000000000000439
176. Anstee QM, Neuschwander-Tetri BA, Wai-Sun Wong V, Abdelmalek MF, Rodriguez-Araujo G, Landgren H, et al. Cenicriviroc lacked efficacy to treat liver fibrosis in nonalcoholic steatohepatitis: AURORA phase III randomized study. Clin Gastroenterol Hepatol. (2024) 22:124–134.e1. doi: 10.1016/j.cgh.2023.04.003
177. Eksteen B, Bowlus CL, Montano-Loza AJ, Lefebvre E, Fischer L, Vig P, et al. Efficacy and safety of cenicriviroc in patients with primary sclerosing cholangitis: PERSEUS study. Hepatol Commun. (2021) 5:478–90. doi: 10.1002/hep4.1619
178. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
179. Noel M, O'Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. (2020) 38:800–11. doi: 10.1007/s10637-019-00830-3
180. Colombo A, Basavarajaiah S, Limbruno U, Picchi A, Lettieri C, Valgimigli M, et al. A double-blind randomised study to evaluate the efficacy and safety of bindarit in preventing coronary stent restenosis. EuroIntervention. (2016) 12:e1385–94. doi: 10.4244/EIJY15M12_03
181. Wang D, Han S, Lv G, Hu Y, Zhuo W, Zeng Z, et al. Pancreatic acinar cells-derived sphingosine-1-phosphate contributes to fibrosis of chronic pancreatitis via inducing autophagy and activation of pancreatic stellate cells. Gastroenterology. (2023) 165:1488–1504.e20. doi: 10.1053/j.gastro.2023.08.029
182. Pausch TM, Aue E, Wirsik NM, Freire Valls A, Shen Y, Radhakrishnan P, et al. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the CXCL8 and the CCL2 axes. Sci Rep. (2020) 10:5420. doi: 10.1038/s41598-020-62416-x
183. Wang X, Istvanffy R, Ye L, Teller S, Laschinger M, Diakopoulos KN, et al. Phenotype screens of murine pancreatic cancer identify a Tgf-α-Ccl2-paxillin axis driving human-like neural invasion. J Clin Invest. (2023) 133(21):e166333. doi: 10.1172/JCI166333
184. Kempeneers MA, Issa Y, Verdonk RC, Bruno M, Fockens P, van Goor H, et al. Pain patterns in chronic pancreatitis: a nationwide longitudinal cohort study. Gut. (2021) 70:1724–33. doi: 10.1136/gutjnl-2020-322117
185. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. (2020) 19:41. doi: 10.1186/s12943-020-01165-x
186. Higashino N, Koma YI, Hosono M, Takase N, Okamoto M, Kodaira H, et al. Fibroblast activation protein-positive fibroblasts promote tumor progression through secretion of CCL2 and interleukin-6 in esophageal squamous cell carcinoma. Lab Invest. (2019) 99:777–92. doi: 10.1038/s41374-018-0185-6
187. Brummer G, Fang W, Smart C, Zinda B, Alissa N, Berkland C, et al. CCR2 signaling in breast carcinoma cells promotes tumor growth and invasion by promoting CCL2 and suppressing CD154 effects on the angiogenic and immune microenvironments. Oncogene. (2020) 39:2275–89. doi: 10.1038/s41388-019-1141-7
Keywords: CCL2, CCR2, inflammation, fibrosis, immune regulation, clinical diagnosis and treatment
Citation: Guo S, Zhang Q, Guo Y, Yin X, Zhang P, Mao T, Tian Z and Li X (2025) The role and therapeutic targeting of the CCL2/CCR2 signaling axis in inflammatory and fibrotic diseases. Front. Immunol. 15:1497026. doi: 10.3389/fimmu.2024.1497026
Received: 16 September 2024; Accepted: 11 December 2024;
Published: 09 January 2025.
Edited by:
Jian Zheng, University of Louisville, United StatesReviewed by:
Luis Castro-Sánchez, University of Colima, MexicoFederico Diaz-Gonzalez, University of La Laguna, Spain
Andrey Elchaninov, Avtsyn Research Institute of Human Morphology of FSBI “Petrovsky National Research Centre of Surgery”, Russia
Shrabanti Chowdhury, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2025 Guo, Zhang, Guo, Yin, Zhang, Mao, Tian and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyu Li, bGl4aWFveXUwNUAxNjMuY29t