
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 22 January 2025
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1495853
This article is part of the Research TopicHiding Features in Myeloid Cells: Metabolism Preference in Different Disease ModelsView all 9 articles
 Ayaka Ito1,2,3*
Ayaka Ito1,2,3* Takayoshi Suganami1,2,4,5
Takayoshi Suganami1,2,4,5Immune cells adapt their metabolism in response to their differentiation and activation status to meet the energy demands for an appropriate immune response. Recent studies have elucidated that during immune cell metabolic reprogramming, lipid metabolism, including lipid uptake, de novo lipid synthesis and fatty acid oxidation, undergoes significant alteration, resulting in dynamic changes in the quantity and quality of intracellular lipids. Given that lipids serve as an energy source and structural components of cellular membranes, they have important implications for physiological function. Myeloid cells, which are essential in bridging innate and adaptive immunity, are sensitive to these changes. Dysregulation of lipid metabolism in myeloid cells can result in immune dysfunction, chronic inflammation and impaired resolution of inflammation. Understanding the mechanism by which lipids regulate immune cell function might provide novel therapeutic insights into chronic inflammatory diseases, including metabolic diseases, autoimmune diseases and cancer. (143 words)
Chronic inflammation has been shown to be the underlying pathogenesis of various diseases. In addition to autoimmune diseases known to be caused by immune abnormalities, immune cell infiltration and aberrant cytokine production are observed in the focal points of metabolic diseases such as obesity and atherosclerosis, as well as in cancer and neurodegenerative diseases. Immune cell differentiation or activation induce metabolic reprogramming in cells, which in turn influences cellular function. This strong link between dysregulation of systemic or intracellular metabolism and immune cell dysfunction, termed immunometabolism, is an emerging field providing critical insights into the pathogenesis of chronic inflammatory diseases (1). In particular, myeloid cells including macrophages, neutrophils and dendritic cells, serve as the first line of defense against pathogens, and play a crucial role in initiating and shaping immune response. They are also involved in tissue repair. Therefore, their dysfunction triggers various pathologies.
Activated immune cells predominantly rely on glycolysis and exhibit downregulated mitochondrial respiration. Although glycolysis is inefficient in ATP production compared to oxidative phosphorylation, it has an advantage in supporting rapid and continuous ATP production because NAD+ is regenerated from NADH through the conversion of pyruvate to lactate to sustain further glycolysis. In addition, many of the intermediates of glycolysis serve as precursors for anabolic pathways, including the pentose phosphate pathway which generates NADPH and ribose 5-phosphate for nucleotide synthesis, the hexosamine pathway for glycosylation, and pathways for amino acid and lipid synthesis. Despite the downregulated mitochondrial activity under these conditions, it is not completely lost. Acetyl CoA, which is generated from pyruvate, enters the tricarboxylic acid (TCA) cycle and is converted to citric acid, which is essential for synthesis of fatty acids and triglycerides, as well as cholesterol biosynthesis. These metabolic products are crucial for cell proliferation, cytokine production, pathogen engulfment, and the presentation of antigens to T cells, thereby initiating adaptive immune responses. On the contrary, cells with low energy requirements, such as anti-inflammatory M2 macrophages, regulatory or memory cells, enhances fatty acid oxidation (FAO) and oxidative phosphorylation. This metabolic shift supports the resolution of inflammation and promotes tissue repair. In addition to FAO, glucose utilization remains crucial, as the inhibition of glycolysis prevent M2 macrophage activation (2, 3). This differential metabolic programming in different activation states underscores the crucial role of lipid metabolism in determining cell functions. Lipids are not only an important energy source, but also fundamental components of cell membranes. In this review, we summarize how lipid metabolism is differentially regulated in myeloid cells, contributing to their functions and influencing the development of various diseases.
Lipid homeostasis is regulated by the balance between endogenous biosynthesis, dietary intake, metabolism and elimination from the body. Several nuclear receptors and transcription factors, including liver X receptors (LXRs), peroxisome proliferator-activated receptors (PPARs), farnesoid X receptor (FXR) and sterol-regulatory element-binding proteins (SREBPs), respond to changes in cellular levels of endogenous lipid ligands and regulate the expression of genes involving in lipid metabolism. Both cellular and systemic cholesterol levels are tightly regulated in a reciprocal fashion by SREBP2 and LXRs. When cholesterol levels are low, SREBP2 is proteolytically cleaved and enters the nucleus to activate transcription of genes controlling cholesterol synthesis, including HMG-CoA reductase, and uptake, such as low-density lipoprotein (LDL) receptor (LDLR), whereas cleavage of SREBP2 is decreased when the cholesterol levels are high (4). Instead, LXRα and LXRβ are activated in response to elevated cholesterol levels and induce the expression of cholesterol transporters ATP-binding cassette transporter A1 and G1 (ABCA1 and ABCG1, respectively) to accomplish cholesterol efflux from peripheral cells such as macrophages. LXRs also activate the transcription of inducible degrader of LDLR (IDOL), and E3 ligase that targets LDLR for ubiquitination and lysosomal degradation, to attenuate LDL uptake by cells (5, 6) (Figure 1).
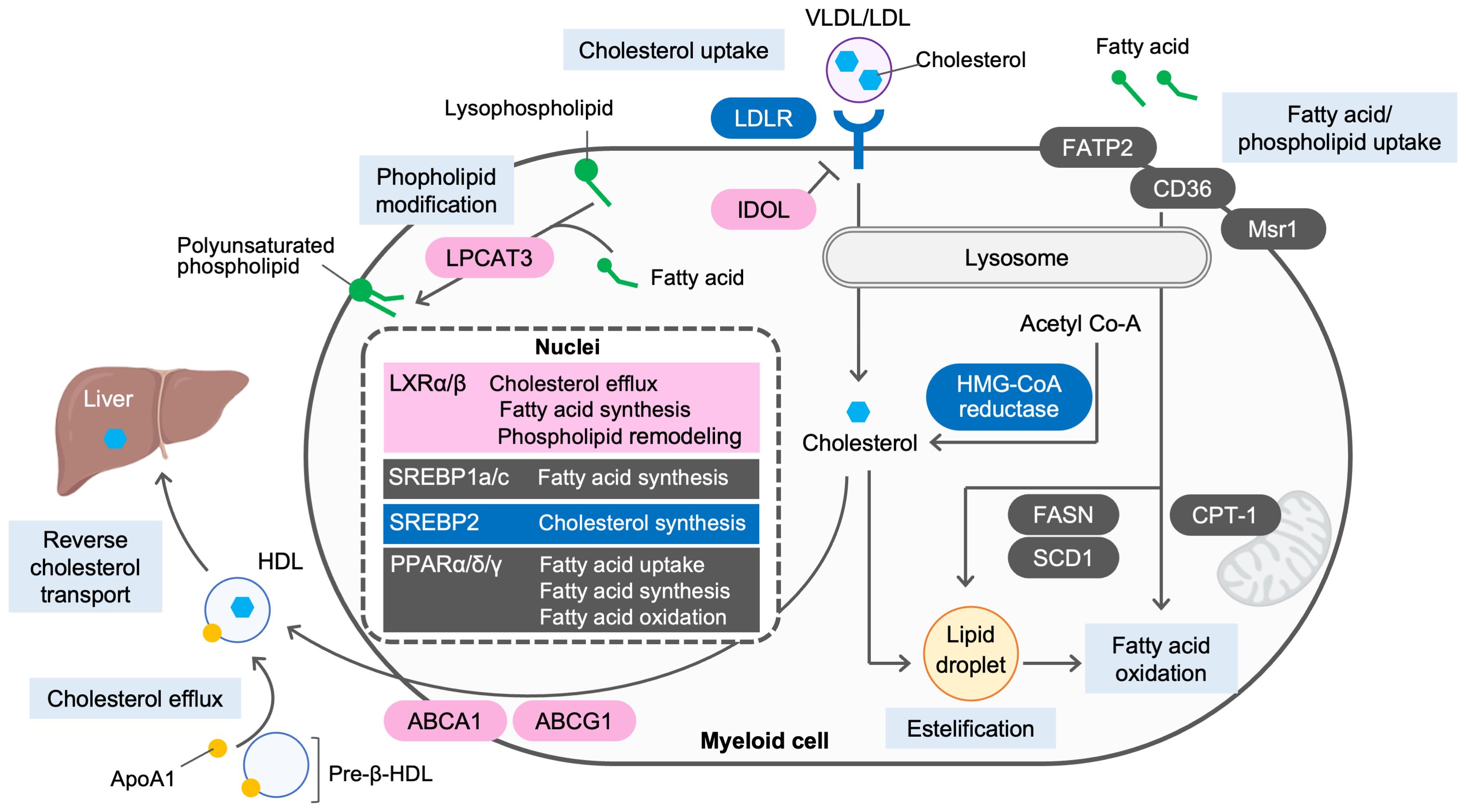
Figure 1. Lipid metabolism in myeloid cells. Lipid homeostasis is regulated by the balance between endogenous biosynthesis, dietary intake, metabolism and elimination from the body. When cholesterol levels are low, sterol regulatory element-binding protein 2 (SREBP2) is activated to synthesize cholesterol from acetyl-CoA and to facilitate cholesterol uptake via low-density lipoprotein (LDL) receptor (LDLR). When cholesterol levels are high, SREBP2 activity is decreased. Instead, activated liver X receptors (LXRs) induce ATP-binding cassette transporter A1 and G1 (ABCA1 and ABCG1, respectively) to accomplish cholesterol efflux to apolipoprotein A1 (ApoA1) or pre-β HDL and reverse cholesterol transport from periphery to the liver. LXRs also induce the expression of inducible degrader of LDLR (IDOL) to degrade LDLR and attenuate cholesterol uptake by the cells. De novo lipogenesis is regulated by LXRs, SREBP1a and SREBP1c. Activation of LXRs promotes fatty acid biosynthesis by inducing the expression of SREBP1c. As a key regulator of fatty acid biosynthesis, SREBP1a and SREBP1c induces their targets, including fatty acid synthase (FASN) and stearoyl coenzyme A desaturase 1 (SCD1). Peroxisome proliferator-activated receptors (PPARs) promotes fatty acid uptake via fatty acid transport protein 2 (FATP2) and CD36, modulate fatty acid oxidation or fatty acid synthesis by inducing their targets, such as carnitine palmitoyl transferase 1A (CPT1A), and FASN and SCD1, respectively. Lysophosphatidylcholine acyltransferase 3 (LPCAT3), an enzyme that incorporates arachidonic acid to lysophospholipids and is induced by LXR activation, modulates membrane dynamics by altering phospholipid composition.
In addition to cholesterol synthesis and efflux, de novo lipogenesis is an important component in regulating lipid homeostasis. SREBP1a and SREBP1c, key transcription activators, promote de novo lipogenesis by inducing fatty acid synthase (FASN), acetyl-CoA carboxylase (ACC) and stearoyl-coenzyme A desaturase 1 (SCD1) (7). LXRs also stimulate lipogenesis through the direct induction of SREBP1c, FASN and SCD1 (8–11). PPAR family, composed of PPARα, PPARδ and PPARγ, also modulate fatty acid uptake, synthesis and oxidation (12) (Figure 1).
It has long been known that macrophages in atherosclerotic plaques produce a variety of inflammatory cytokines and chemokines when they uptake excess lipoproteins and become foam cells (13), suggesting that the quantity of intracellular lipids is associated with inflammatory responses. Indeed, macrophages activated by toll-like receptors (TLRs), induce the accumulation of cholesterol and triglycerides by promoting the uptake of lipoproteins and free-fatty acids, enhancing lipogenesis (14, 15), and inhibiting lipolysis and cholesterol efflux (15–17). Enhanced synthesis of fatty acid and phospholipid is essential for the development of cellular organelles, including mitochondria, lysosome, endoplasmic reticulum and Golgi, for cell proliferation and proper phagocytosis (18). Consistent with this, immune cells are unable to proliferate when cellular cholesterol is insufficient (19, 20). Also, mice lacking SREBP1a, which is a dominant isoform of SREBPs in macrophages, exhibit defects in lipogenesis and thereby decreased phagocytotic ability (21). These mice also show suppressed inflammasome activation and reduced IL-1β production in macrophages, making them resistant to endotoxin shock and systemic inflammatory responses induced by cecal ligation and puncture (22). TLR3 or TLR4 activation strongly induces cholesterol 25-hydroxylase (Ch25h), an enzyme that hydroxylates cholesterol to generate 25-hydroxycholesterol (25-HC) (23). Notably, 25-HC antagonizes SREBP processing, thereby suppressing inflammasome activation in LXR-independent manner, despite being one of the ligands for LXRs (24, 25). Rather, LXR target gene expression, including ABCA1 and ABCG1, and cholesterol efflux are downregulated during inflammatory activation (17, 26). Considering that Ch25h is induced by type I interferons downstream of TLR3 or TLR4, the effect of 25-HC may occur in a late phase of TLR activation, in response to suppressed cholesterol efflux and increased intracellular cholesterol levels. One possible explanation for the antagonism of 25-HC against SREBP is the increased activity of the oxysterol-metabolizing enzyme SULT2B1 and the ABCC transporter, which leads to the elimination of oxysterol ligands for LXRs, thus reducing cholesterol efflux and maintain cellular cholesterol amount (27). LXR-dependent gene expression is also crucial for attenuating inflammation and survival of macrophages and neutrophil (28, 29). In fact, systemic LXR deficiency results in increased susceptibility to inflammatory responses in atherosclerosis, Listeria monocytogenes infection and dermatitis (29–32). Intriguingly, viral infection decreases cholesterol biosynthesis while paradoxically increasing cellular cholesterol levels by upregulating cholesterol import and downregulating cholesterol efflux, leading the activation of the stimulator of interferon genes (STING) intracellular DNA sensing pathway (33). A defect in SREBP cleavage-activation protein (SCAP), a key regulator of SREBP, or the silencing of SREBP-2 leads increased production of antiviral cytokines in a STING-dependent manner (33). Given that STING protein resides in the ER, where SCAP/SREBP sense cholesterol, it is likely that STING signaling is activated in response to decreased cholesterol levels in the ER membrane.
While lipids accumulate in inflammatory cells, lipolysis is more prominent in anti-inflammatory cells. Lysosomal lipolysis and subsequent FAO, and mitochondrial oxidative phosphorylation is critical for M2 macrophage activation (14). Additionally, since M2 macrophages play an important role in anti-parasitic immunity, inhibiting lipolysis can impair the elimination of the intestinal helminth parasite H. polygyrus (14). Similarly, enhancing FAO and thereby reduction of triglyceride content in macrophages—by overexpressing PPARγ coactivator 1β (PGC1β), a key transcriptional coactivator of oxidative metabolism, or carnitine palmitoyl transferase 1A (CPT1A), the rate-limiting enzyme in mitochondrial FAO—reduced ER stress and the production of inflammatory cytokines (34, 35). On the contrary, suppression of FAO by PGC1β knockdown impairs alternative macrophage activation and rather enhances the production of inflammatory cytokines (35).
Changes in lipid quality, particularly fatty acid composition of phospholipids and cholesterol content, have been shown to determine the biophysical properties, such as fluidity and curvature, of the membrane, thereby influencing various immune cell functions, including cell proliferation, cytokine production, phagocytosis and antigen presentation (36). Wei et al. demonstrated that the deletion of FASN in macrophages resulted in impaired inflammatory responses, which is attributed to decreased membrane cholesterol levels, which in turn disrupts protein trafficking to lipid rafts (37). Correspondingly, activating ABCA1-dependent reduction of membrane cholesterol by LXRs has been shown to modulate membrane lipid organization and preventing the localization of TLR4 adaptor molecules to lipid rafts, thereby impairing downstream MAPK and NFκB signaling (38, 39). Recent comprehensive lipidomic studies have revealed that the activation of macrophages with TLR agonists reprograms cellular lipid composition. In particular, MyD88-dependent TLRs induce de novo long-chain fatty acid synthesis in SREBP1c- and SCD1/2-dependent manner, which suppresses prolonged inflammation (40–42). Consistent with this, Hsieh et al. reported that mice lacking SREBP1c exhibit accelerated clearance of Staphylococcus aureus (41). The reprograming of lipid composition by various TLRs occurs in distinct, albeit partially overlapping, manners. For instance, TRIF-dependent TLRs attenuate de novo long-chain fatty acid synthesis through autocrine type I IFN signaling. Such alterations in lipid quality occur not only at the plasma membrane but also in the endoplasmic reticulum (ER). Even a slight increase in ER cholesterol content reduces the transcriptional activity of SREBP1/2, inhibiting of fatty acid and cholesterol synthesis, while a decrease in ER cholesterol enhances SREBP activity, driving lipid synthesis (43). Lysophosphatidylcholine acyltransferase 3 (LPCAT3), an enzyme that incorporates arachidonic acid to lysophospholipids and is induced by LXR activation, has been reported to contribute to lipid synthesis by altering ER phospholipid composition. This alteration increases SREBP1c activity, thereby promoting fatty acid synthesis. Increased LPCAT3 expression and the abundance of polyunsaturated phospholipids in membranes also ameliorate ER stress in hepatocytes in the setting of obesity (44–46). These findings underscore the crucial role of lipid metabolism in immune cell function.
As chronic overnutrition progresses to obesity, immune cells, including macrophages, are infiltrated into adipose tissue (47). In obese adipose tissue, macrophages are activated by saturated fatty acids produced by adipocytes, shifting them towards a pro-inflammatory phenotype. This shift is associated with the development of metabolic disorders such as diabetes, dyslipidemia, and fatty liver disease, as well as an increased risk of atherosclerotic diseases (48–50). PPARγ, a sensor of fatty acid, acts as a master regulator of macrophage polarization. Genetic deletion of PPARγ in macrophages results in decreased FAO and alternative activation while increasing the production of inflammatory cytokines, making them more susceptible to obesity and impairing glucose metabolism (51). Furthermore, Ferrante and colleagues demonstrated that in obese adipose tissue, macrophages induce lysosomal biogenesis and consequently accumulate lipids within the cells, leading to an increase in macrophage number rather than macrophage activation (52). Recent single-cell transcriptomics studies demonstrate that adipose tissue macrophages are highly heterogeneous (53, 54). Among the macrophage populations in obese adipose tissue, lipid-associated macrophages (LAM) form a cluster with a distinct transcriptional signature related to lipid metabolism and phagocytosis, and their numbers increase during the progression of obesity. Trem2 null macrophages lack the majority of the LAM gene signature and mice lacking Trem2 exhibit adipocyte hypertrophy and metabolic abnormalities, suggesting that LAMs play a protective role against adipose inflammation and systemic metabolic dysregulation (53). On the other hand, scRNA-seq analysis of CD45+ cells from atherosclerotic aorta revealed that macrophages with high Trem2 expression display osteoclastic gene signature, in addition to genes related to lipid metabolism and phagocytosis, suggesting that these macrophages are associated with calcification in atherosclerotic lesion (55). The scavenging receptor CD36 is one of the genes commonly expressed in LAM in obesity, atherosclerosis and cancer (56). Mice lacking CD36 have been reported to show increased insulin sensitivity, decreased inflammation in adipose tissue and resistant to the development of atherosclerosis (57–59). Although these reports do not accurately reflect the role of CD36 in macrophages due to the use of global knockout mice, subsequent study by Moore and colleagues demonstrated that the activation of inflammasomes in macrophages by CD36 is an underlying mechanism (60). Given that cholesterol crystals are endogenous activators of inflammasomes, oxidized LDL taken up via CD36 might form crystals rather than be esterified, thereby activating inflammasomes and exacerbating atherosclerosis (61–63). These findings highlight that managing metabolic diseases through lipid regulation may not only correct systemic lipid metabolism but also improve immune cell functions to mitigate inflammation and metabolic dysregulation.
While lipogenic pathway is essential in maintaining the immune cell function, aberrant lipid accumulation can impair tumor immunity and promotes tumor progression. For instance, it has been reported that dendritic cells from tumor-bearing mice or cancer patients have increased lipid accumulation due to enhanced lipid uptake via the scavenger receptor Msr1, which subsequently impairs the ability of presentation of tumor-associated antigens to T cells, partially through triggering ER stress (64, 65). Similarly, myeloid-derived suppressor cells (MDSCs) that are pathologically activated neutrophils and monocytes with immunosuppressive activity and tumor-associated macrophages (TAM) have also been shown to have increased lipid uptake and accumulation via fatty acid transport protein 2 (FATP2) and CD36, respectively. This lipid accumulation supports their proliferation, thereby suppressing tumor immunity and contributing to tumor progression (66–68). Mechanistically, the upregulation of FATP2 increases incorporation of arachidonic acid and leading to the synthesis and secretion of prostaglandin E2 (PGE2), and PGE2 fuels tumorigenesis by expanding immunosuppressive cells and inhibiting cytotoxic immune cells (67, 69, 70). Moreover, these tumor-associated myeloid cells actively utilize FAO rather than glycolysis to meet their energy demand. Elevated FAO and mitochondrial oxidative phosphorylation result in producing reactive oxygen species and oxidized lipids, which further disrupt tumor-associated myeloid cell function and promote tumor progression (65, 71). Notably, FATP2 inhibition suppresses tumor growth and enhances the efficacy of immune checkpoint inhibitor in tumor-bearing mice (67). Additionally, lipids in tumor microenvironment serve as energy source for tumor cells. Goossens et al. reported that tumor cells promote membrane cholesterol efflux from TAM through ABCA1 and ABCG1, which in turn enhances IL-4 signaling and supports anti-inflammatory/tumor-promoting macrophage polarization (72). In agreement with this observation, myeloid-specific genetic ablation of ABCA1 and/or ABCG1 has been shown to prevent tumor growth (73, 74).
Targeting lipid metabolism has demonstrated promising anti-tumor effects in pre-clinical models, highlighting its potential for clinical applications. For instance, statins, inhibitors of HMG-CoA reductase and cholesterol synthesis, are widely used for cardiovascular diseases and are currently undergoing clinical trials for cancer treatment (75). Additionally, the inhibition of CD36 using thrombospondin analog VT1021 is being evaluated in a phase I trial for solid cancer (NCT03364400), although another thrombospondin analog, ABT-510, failed to achieve positive outcomes in a phase II trial as a monotherapy. Meanwhile, FASN inhibitor TVB-2640 is under clinical studies for various tumors, including breast cancer (NCT03179904), non-small cell lung carcinomas (NCT03808558). Phase I and II trials have already been completed for oral cancer (NCT02223247) and astrocytoma brain cancer (NCT03032484) have already been completed. The anti-tumor effects of targeting lipid metabolism may extend beyond direct impacts on myeloid cells to include the modulation of lipid metabolism in lymphocytes. Indeed, mounting evidence underscores the importance of lipid reprogramming in lymphocyte function, such as those of T cells and natural killer cells, in regulating tumor progression. While this review focuses on lipid metabolism in myeloid cells, the role of lymphocyte lipid metabolism in tumor progression has been extensively reviewed elsewhere (75–77).
Autoimmune diseases are a diverse group of disorders characterized by loss of immune tolerance with activation of both innate and adaptive immune system and development of antibodies against self-antigens. Impaired clearance of apoptotic or necrotic cells leads increased exposure of self-antigens, resulting in an activation of self-reactive lymphocytes and a break of self-tolerance. This accumulation of apoptotic cell debris also triggers TLRs and cytosolic nucleic acid sensors, which drives the production of inflammatory cytokines (78, 79). Several lines of evidence suggest a link between systemic and cellular dysregulation of lipid metabolism and the development of autoimmunity. Systemically, cholesterol efflux capacity of high-density lipoproteins (HDL) is impaired in patient with systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA), while serum HDL levels are comparable to healthy individuals (80–82). This impaired HDL function could promote lipid accumulation in myeloid cells. The phagocytosis of apoptotic cells activates LXRs, which induces their target gene Mer, a receptor tyrosine kinase that mediates phagocytosis. This positive feedback loop promotes efficient apoptotic cell clearance and suppresses autoimmunity (83). Furthermore, LXR-deficient mice exhibit defective phagocytosis of apoptotic cells, thereby developing autoantibodies (83), suggesting the importance of LXRs in autoimmunity. In fact, polymorphisms in LXR gene are reported to be associated with SLE in Koreans (84). Subsequent mechanistic analyses revealed that cholesterol accumulation in CD11c+ antigen-presenting cells and dendritic cells is a driver of systemic autoimmune disease (85–87). LXR deficiency leads to cholesterol accumulation in antigen-presenting cells, enhancing antigen presentation, T cell priming, and production of B cell activating factor (BAFF), a cytokine that supports B cell expansion and autoantibody production and plays a pivotal role in pathogenesis of SLE (86). Increased BAFF production is likely attributed to enhanced TLR signaling due to cholesterol accumulation in lipid rafts (38) Similarly, the loss of LXR targets, ABCA1 and ABCG1, in dendritic cells promotes cellular cholesterol accumulation and cytokine production through inflammasome activation and increased cell surface expression of granulocyte-macrophage colony-stimulating factor (GM-CSF), leading to autoimmunity (87). Mice lacking Ch25h also exert elevated inflammasome activity and exacerbate experimental autoimmune encephalomyelitis (24).
Interestingly, an expression quantitative trait loci analysis in myeloid cells has revealed that fatty acid desaturase genes are associated with increased susceptibility to RA (88), highlighting the significance of fatty acid metabolism in autoimmunity. For instance, the loss of 12/15-lipoxygenase, an enzyme mediates oxidation of polyunsaturated fatty acids, results in increased phagocytosis of apoptotic cells by inflammatory monocytes. This defect leads to the break of self-tolerance and the development of SLE. Mechanistically, 12/15-lipoxygenase, expressed in resident macrophages, oxidizes phosphatidylethanolamine, which sequesters a soluble ‘eat-me’ signal from inflammatory monocytes onto the plasma membrane of resident macrophages. This process inhibits aberrant phagocytosis of apoptotic cells by inflammatory monocytes (89, 90).
Dyslipidemia is frequently observed in autoimmune disease patients and is a major cause of mortality due to cardiovascular diseases. Prolonged glucocorticoid therapy can exacerbate dyslipidemia, making appropriate lipid management an important issue. Statins, widely used for lipid management, have been reported to improve the disease symptoms in RA patients (91). Preclinical studies further demonstrate that statins attenuate autoimmune pathology in models of autoimmune diseases, such as RA, encephalomyelitis, and SLE by reducing antigen presentation, suppressing cytokine production and inhibiting Th1 differentiation (92). Omega-3 polyunsaturated fatty acids, known for its triglyceride-lowering effects, have also shown anti-inflammatory effect on myeloid cells and direct impacts on B cell differentiation (93, 94). Improvements in lipid metabolism are expected to positively influence autoimmune pathology by simultaneously modulating lipid metabolism and immune cell functions.
Over the past two decades, significant progress has been made in understanding the crosstalk between lipid metabolism and inflammatory responses, as well as their collective impact on chronic inflammatory diseases (Table 1). Targeting lipid metabolism has emerged as a promising therapeutic strategy to correct immune dysfunction in these conditions. However, translating these findings into clinical applications requires a deeper understanding of lipid metabolism across diverse immune cell subsets, as the regulatory mechanisms vary depending on cell type and disease context. Another critical question that remains unanswered is whether interventions targeting lipid metabolism directly reprogram lipids within immune cells or if the effects are a consequence of alterations in systemic lipid metabolism. Future research, employing lipidomic analyses and imaging technologies, will enable us to elucidate lipid dynamics across organelles, cells, and organs. These insights will open new avenues for developing innovative strategies to address the unmet medical needs in chronic inflammatory diseases.
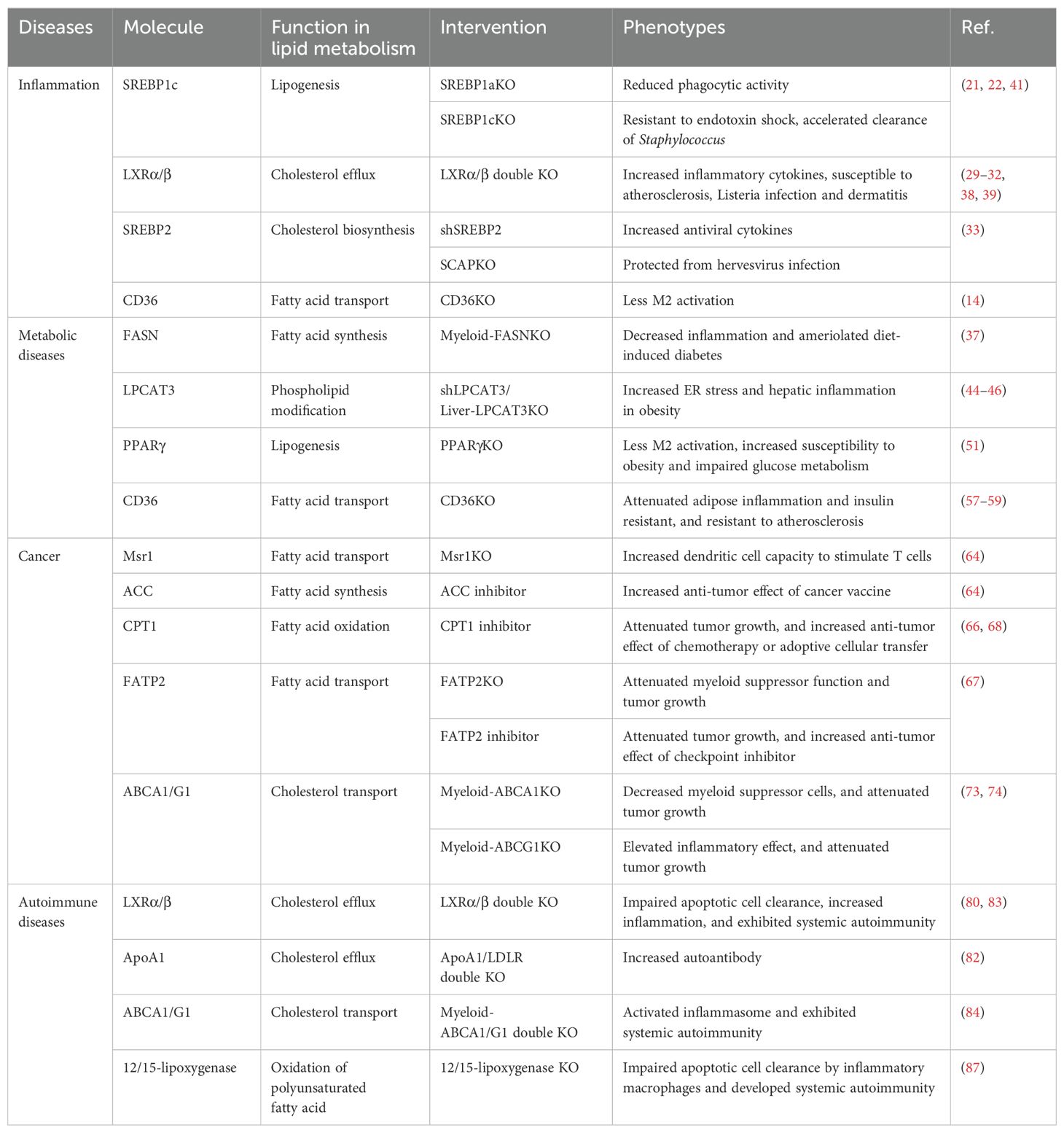
Table 1. Targeting lipid metabolism in inflammation and chronic inflammatory diseases.
AI: Conceptualization, Data curation, Funding acquisition, Investigation, Visualization, Writing – original draft, Writing – review & editing. TS: Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Grants-in Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (23K27377, 22K11700, 22K19524, 21H05625, 24KK0150), Fusion Oriented Research for Disruptive Science and Technology (JST FOREST, JPMJFR2259), and the Japan Agency for Medical Research and Development (CREST, JP24gm1210009s0106). This work was also supported by research grants from Takeda Science Foundation, Lotte Foundation, Inamori Foundation, The Hitachi Global Foundation and Kobayashi Foundation.
Figures are created with Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
2. Huang SC-C, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. (2016) 45:817–30. doi: 10.1016/j.immuni.2016.09.016
3. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife. (2016) 5:e11612. doi: 10.7554/elife.11612
4. Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. (2015) 161:161–72. doi: 10.1016/j.cell.2015.01.036
5. Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through idol-dependent ubiquitination of the LDL receptor. Science. (2009) 325:100–4. doi: 10.1126/science.1168974
6. Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L, et al. The LXR–idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab. (2014) 20:910–8. doi: 10.1016/j.cmet.2014.10.001
7. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology — divergent pathophysiology. Nat Rev Endocrinol. (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
8. Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, et al. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors*. J Biol Chem. (2002) 277:11019–25. doi: 10.1074/jbc.m111041200
9. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro J-MA, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev. (2000) 14:2819–30. doi: 10.1101/gad.844900
10. Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, et al. Role of LXRs in control of lipogenesis. Genes Dev. (2000) 14:2831–8. doi: 10.1101/gad.850400
11. Chu K, Miyazaki M, Man WC, Ntambi JM. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol Cell Biol. (2006) 26:6786–98. doi: 10.1128/mcb.00077-06
12. Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev. (2008) 18:461–7. doi: 10.1016/j.gde.2008.07.016
13. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. (2011) 145:341–55. doi: 10.1016/j.cell.2011.04.005
14. Huang SC-C, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for macrophage alternative activation. Nat Immunol. (2014) 15:846–55. doi: 10.1038/ni.2956
15. Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. (2018) 19:526–37. doi: 10.1038/s41590-018-0113-3
16. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. (2012) 92:829–39. doi: 10.1189/jlb.1111537
17. Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, et al. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. (2003) 12:805–16. doi: 10.1016/s1097-2765(03)00384-8
18. Ecker J, Liebisch G, Englmaier M, Grandl M, Robenek H, Schmitz G. Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes. Proc Natl Acad Sci. (2010) 107:7817–22. doi: 10.1073/pnas.0912059107
19. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. The sterol regulatory element binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. (2013) 14:489–99. doi: 10.1038/ni.2570
20. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. (2016) 531:651–5. doi: 10.1038/nature17412
21. Lee J-H, Phelan P, Shin M, Oh B-C, Han X, Im S-S, et al. SREBP-1a–stimulated lipid synthesis is required for macrophage phagocytosis downstream of TLR4-directed mTORC1. Proc Natl Acad Sci. (2018) 115:E12228–34. doi: 10.1073/pnas.1813458115
22. Im S-S, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
23. Park K, Scott AL. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol. (2010) 88:1081–7. doi: 10.1189/jlb.0610318
24. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. 25-Hydroxycholesterol suppresses interleukin-1–driven inflammation downstream of type I interferon. Science. (2014) 345:679–84. doi: 10.1126/science.1254790
25. Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. (2017) 171:1057–1071.e11. doi: 10.1016/j.cell.2017.09.029
26. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. (2003) 9:213–9. doi: 10.1038/nm820
27. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. (2008) 134:97–111. doi: 10.1016/j.cell.2008.04.052
28. Hong C, Kidani Y, A-Gonzalez N, Phung T, Ito A, Rong X, et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Investig. (2012) 122:337–47. doi: 10.1172/jci58393
29. Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. (2004) 119:299–309. doi: 10.1016/j.cell.2004.09.032
30. Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc Natl Acad Sci. (2002) 99:11896–901. doi: 10.1073/pnas.182199799
31. Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X, et al. Ligand activation of LXRβ reverses atherosclerosis and cellular cholesterol overload in mice lacking LXRα and apoE. J Clin Invest. (2007) 117:2337–46. doi: 10.1172/jci31909
32. Hong C, Bradley MN, Rong X, Wang X, Wagner A, Grijalva V, et al. LXRα is uniquely required for maximal reverse cholesterol transport and atheroprotection in ApoE-deficient mice. J Lipid Res. (2012) 53:1126–33. doi: 10.1194/jlr.m022061
33. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. (2015) 163:1716–29. doi: 10.1016/j.cell.2015.11.045
34. Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol-Endocrinol Metab. (2015) 308:E756–69. doi: 10.1152/ajpendo.00362.2014
35. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
36. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. (2017) 18:361–74. doi: 10.1038/nrm.2017.16
37. Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. (2016) 539:294–8. doi: 10.1038/nature20117
38. Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. (2015) 4:e08009. doi: 10.7554/elife.08009
39. Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, et al. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol[S. J Lipid Res. (2010) 51:3196–206. doi: 10.1194/jlr.m006486
40. Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, et al. A mouse macrophage lipidome* ♦. J Biol Chem. (2010) 285:39976–85. doi: 10.1074/jbc.m110.182915
41. Hsieh W-Y, Zhou QD, York AG, Williams KJ, Scumpia PO, Kronenberger EB, et al. Toll-like receptors induce signal-specific reprogramming of the macrophage lipidome. Cell Metab. (2020) 32:128–143.e5. doi: 10.1016/j.cmet.2020.05.003
42. Oishi Y, Spann NJ, Link VM, Muse ED, Strid T, Edillor C, et al. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab. (2017) 25:412–27. doi: 10.1016/j.cmet.2016.11.009
43. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. (2008) 8:512–21. doi: 10.1016/j.cmet.2008.10.008
44. Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. (2013) 18:685–97. doi: 10.1016/j.cmet.2013.10.002
45. Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol. (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
46. Rong X, Wang B, Palladino END, Vallim TQ de A, Ford DA, Tontonoz P. ER phospholipid composition modulates lipogenesis during feeding and in obesity. J Clin Invest. (2017) 127:3640–51. doi: 10.1172/jci93616
47. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Investig. (2003) 112:1796–808. doi: 10.1172/jci19246
48. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Investig. (2007) 117:175–84. doi: 10.1172/jci29881
49. Suganami T, Tanaka M, Ogawa Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr J. (2012) 59:849. doi: 10.1507/endocrj.ej12-0271
50. Chawla A, Nguyen KD, Goh YPS. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. (2011) 11:738–49. doi: 10.1038/nri3071
51. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. (2007) 447:1116–20. doi: 10.1038/nature05894
52. Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. (2013) 18:816–30. doi: 10.1016/j.cmet.2013.11.001
53. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. (2019) 178:686–698.e14. doi: 10.1016/j.cell.2019.05.054
54. Stansbury CM, Dotson GA, Pugh H, Rehemtulla A, Rajapakse I, Muir LA. A lipid-associated macrophage lineage rewires the spatial landscape of adipose tissue in early obesity. JCI Insight. (2023) 8:e171701. doi: 10.1172/jci.insight.171701
55. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/circresaha.117.312509
56. Xu R, Vujić N, Bianco V, Reinisch I, Kratky D, Krstic J, et al. Lipid-associated macrophages between aggravation and alleviation of metabolic diseases. Trends Endocrinol Metab. (2024) 35:981-995. doi: 10.1016/j.tem.2024.04.009
57. Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Investig. (2000) 105:1049–56. doi: 10.1172/jci9259
58. Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Investig. (2002) 109:1381–9. doi: 10.1172/jci14596
59. Kennedy DJ, Kuchibhotla S, Westfall KM, Silverstein RL, Morton RE, Febbraio M. A CD36-dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc Res. (2011) 89:604–13. doi: 10.1093/cvr/cvq360
60. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. (2013) 14:812–20. doi: 10.1038/ni.2639
61. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
62. Que X, Hung M-Y, Yeang C, Gonen A, Prohaska TA, Sun X, et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature. (2018) 558:301–6. doi: 10.1038/s41586-018-0198-8
63. Gioia MD, Spreafico R, Springstead JR, Mendelson MM, Joehanes R, Levy D, et al. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat Immunol. (2020) 21:42–53. doi: 10.1038/s41590-019-0539-2
64. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
65. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
66. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.cir-15-0036
67. Veglia F, Tyurin VA, Blasi M, Leo AD, Kossenkov A, Donthireddy L, et al. Fatty acid transporter 2 reprograms neutrophils in cancer. Nature. (2019) 569:73–8. doi: 10.1038/s41586-019-1118-2
68. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. (2020) 80:1438–50. doi: 10.1158/0008-5472.can-19-2994
69. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. (2015) 162:1257–70. doi: 10.1016/j.cell.2015.08.015
70. Morotti M, Grimm AJ, Hope HC, Arnaud M, Desbuisson M, Rayroux N, et al. PGE2 inhibits TIL expansion by disrupting IL-2 signalling and mitochondrial function. Nature. (2024) 629:426–34. doi: 10.1038/s41586-024-07352-w
71. Cao W, Ramakrishnan R, Tuyrin VA, Veglia F, Condamine T, Amoscato A, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol. (2014) 192:2920–31. doi: 10.4049/jimmunol.1302801
72. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. (2019) 29:1376–1389.e4. doi: 10.1016/j.cmet.2019.02.016
73. Zamanian-Daryoush M, Lindner DJ, DiDonato JA, Wagner M, Buffa J, Rayman P, et al. Myeloid-specific genetic ablation of ATP-binding cassette transporter ABCA1 is protective against cancer. Oncotarget. (2017) 8:71965–80. doi: 10.18632/oncotarget.18666
74. Sag D, Cekic C, Wu R, Linden J, Hedrick CC. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat Commun. (2015) 6:6354. doi: 10.1038/ncomms7354
75. Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt S-M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev Cell. (2021) 56:1363–93. doi: 10.1016/j.devcel.2021.04.013
76. Howie D, Bokum AT, Necula AS, Cobbold SP, Waldmann H. The role of lipid metabolism in T lymphocyte differentiation and survival. Front Immunol. (2018) 8:1949. doi: 10.3389/fimmu.2017.01949
77. Lim SA, Su W, Chapman NM, Chi H. Lipid metabolism in T cell signaling and function. Nat Chem Biol. (2022) 18:470–81. doi: 10.1038/s41589-022-01017-3
78. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. (2016) 2:16039. doi: 10.1038/nrdp.2016.39
79. Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
80. Charles-Schoeman C, Lee YY, Grijalva V, Amjadi S, FitzGerald J, Ranganath VK, et al. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann Rheum Dis. (2012) 71:1157. doi: 10.1136/annrheumdis-2011-200493
81. Ronda N, Favari E, Borghi MO, Ingegnoli F, Gerosa M, Chighizola C, et al. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. (2014) 73:609. doi: 10.1136/annrheumdis-2012-202914
82. Liu Y, Kaplan MJ. Cardiovascular disease in systemic lupus erythematosus. Curr Opin Rheumatol. (2018) 30:441–8. doi: 10.1097/bor.0000000000000528
83. A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. (2009) 31:245–58. doi: 10.1016/j.immuni.2009.06.018
84. Jeon J-Y, Nam J-Y, Kim H-A, Park Y-B, Bae S-C, Suh C-H. Liver X receptors alpha gene (NR1H3) promoter polymorphisms are associated with systemic lupus erythematosus in Koreans. Arthritis Res Ther. (2014) 16:R112. doi: 10.1186/ar4563
85. Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arter Thromb Vasc Biol. (2009) 29:843–9. doi: 10.1161/atvbaha.108.183442
86. Ito A, Hong C, Oka K, Salazar JV, Diehl C, Witztum JL, et al. Cholesterol accumulation in CD11c+ Immune cells is a causal and targetable factor in autoimmune disease. Immunity. (2016) 45:1311–26. doi: 10.1016/j.immuni.2016.11.008
87. Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. (2017) 25:1294–1304.e6. doi: 10.1016/j.cmet.2017.04.005
88. Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. (2014) 344:519–23. doi: 10.1126/science.1249547
89. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. (2009) 9:353–63. doi: 10.1038/nri2545
90. Uderhardt S, Herrmann M, Oskolkova OV, Aschermann S, Bicker W, Ipseiz N, et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity. (2012) 36:834–46. doi: 10.1016/j.immuni.2012.03.010
91. Robinson G, Pineda-Torra I, Ciurtin C, Jury EC. Lipid metabolism in autoimmune rheumatic disease: implications for modern and conventional therapies. J Clin Investig. (2022) 132:e148552. doi: 10.1172/jci148552
92. Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol. (2006) 6:358–70. doi: 10.1038/nri1839
93. Li X, Bi X, Wang S, Zhang Z, Li F, Zhao AZ. Therapeutic potential of ω-3 polyunsaturated fatty acids in human autoimmune diseases. Front Immunol. (2019) 10:2241. doi: 10.3389/fimmu.2019.02241
Keywords: lipid metabolism, immunometabolism, autoimmunity, cancer, metabolic disease
Citation: Ito A and Suganami T (2025) Lipid metabolism in myeloid cell function and chronic inflammatory diseases. Front. Immunol. 15:1495853. doi: 10.3389/fimmu.2024.1495853
Received: 13 September 2024; Accepted: 30 December 2024;
Published: 22 January 2025.
Edited by:
Duoyao Cao, Cedars Sinai Medical Center, United StatesReviewed by:
Andy Ruiz, National Institute of Respiratory Diseases-Mexico (INER), MexicoCopyright © 2025 Ito and Suganami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ayaka Ito, YWl0b0ByaWVtLm5hZ295YS11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.