 Md. Monirul Islam1,2
Md. Monirul Islam1,2 Eizo Watanabe1*
Eizo Watanabe1* Umme Salma1Masayuki Ozaki1
Umme Salma1Masayuki Ozaki1 Takayuki Irahara1Subaru Tanabe1Ryusuke Katsuki1Dai Oishi1Naoshi Takeyama1
Takayuki Irahara1Subaru Tanabe1Ryusuke Katsuki1Dai Oishi1Naoshi Takeyama1- 1Department of Emergency and Critical Care Medicine, Aichi Medical University, Nagakute, Japan
- 2Department of Biochemistry and Biotechnology, University of Science and Technology Chittagong (USTC), Chattogram, Bangladesh
Sepsis is characterized by a concomitant early pro-inflammatory response by immune cells to an infection, and an opposing anti-inflammatory response that results in protracted immunosuppression. The primary pathological event in sepsis is widespread programmed cell death, or cellular self-sacrifice, of innate and adaptive immune cells, leading to profound immunological suppression. This severe immune dysfunction hampers effective primary pathogen clearance, thereby increasing the risk of secondary opportunistic infections, latent viral reactivation, multiple organ dysfunction, and elevated mortality. The types of cell death include apoptosis (type I programmed cell death), autophagy (type II programmed cell death), NETosis (a program for formation of neutrophil extracellular traps (NETs)) and other programmed cell deaths like pyroptosis, ferroptosis, necroptosis, each contributing to immunosuppression in distinct ways during the later phases of sepsis. Extensive apoptosis of lymphocytes, such as CD4+, CD8+ T cells, and B cells, is strongly associated with immunosuppression. Apoptosis of dendritic cells further compromises T and B cell survival and can induce T cell anergy or promote regulatory Treg cell proliferation. Moreover, delayed apoptosis and impaired neutrophil function contribute to nosocomial infections and immune dysfunction in sepsis. Interestingly, aberrant NETosis and the subsequent depletion of mature neutrophils also trigger immunosuppression, and neutrophil pyroptosis can positively regulate NETosis. The interaction between programmed cell death 1 (PD-1) or programmed cell death 1 ligand (PD-L1) plays a key role in T cell modulation and neutrophil apoptosis in sepsis. The dendritic cell growth factor, Fms-like tyrosine kinase (FLTEL), increases DC numbers, enhances CD 28 expression, attenuates PD-L1, and improves survival in sepsis. Recently, immunoadjuvant therapies have attracted attention for their potential to restore host physiological immunity and homeostasis in patients with sepsis. This review focuses on several potential immunotherapeutic agents designed to bolster suppressed innate and adaptive immune responses in the management of sepsis.
Introduction
Sepsis is characterized by a dysregulated inflammatory host response to life-threatening infection, which can trigger circulatory shock, organ dysfunction, and ultimately, death (1). The hallmark of sepsis is a simultaneous, cytokine-mediated, early excessive pro-inflammatory response to infection that contributes to the recruitment and activation of innate and adaptive immune cells to the site of infection. Progression of sepsis leads to immune cell exhaustion by continuous encounters with pathogens and inflammatory signals with increased expression of immune checkpoint molecules like PD-1 and CTLA-4. The exhausted immune cells exert a compensatory anti-inflammatory response and undergo different types of massive cell death (2). The human body manages this through a negative feedback mechanism referred to as immunosuppression, which leads to immunoparalysis involving both the innate and adaptive immune systems. This profound immune dysfunction results in poor primary pathogen clearance and enhances the risk of secondary opportunistic infections, latent viral reactivation, multiple organ dysfunction, and increased mortality (3, 4). Despite numerous clinical trials focused on mitigating hyper-inflammation by blocking pro-inflammatory mediators (5, 6), no FDA-approved treatments have been approved to date, and sepsis remains a predominant cause of death among critically ill patients in most intensive care settings worldwide (7). While advances in treatment and supportive care have reduced mortality and improved overall survival in sepsis management, researchers have yet to elucidate various immunological aspects of the syndrome or identify novel, targeted therapeutics to reverse sepsis effectively. It is well established that cell death, a conserved mechanism in multicellular organisms, plays a vital role in responding to external injuries. Dysregulation of widespread programmed immune cell death, or cellular self-sacrifice, is now recognized as the primary pathological event in sepsis, leading to significant immunological suppression (8). These cell death mechanisms include apoptosis (type I programmed cell death), autophagy (type II programmed cell death), NETosis [a program for formation of neutrophil extracellular traps (NETs)] pyroptosis, ferroptosis, and necroptosis. An inadequate immune response resulting from cell death and subsequent aggressive immunosuppression has been identified as a significant contributor to sepsis pathogenesis (9, 10). Excessive apoptosis of splenic CD4+, CD8+ T, and B cells, coupled with reduced autophagy in CD4+ T cells, has been observed in patients with sepsis, thereby accelerating acquired immunodeficiency (11). Furthermore, apoptosis of dendritic cells in sepsis also compromises the survival of T cell and B cells and can induce a state of T cell anergy or promote regulatory T cell (Treg) proliferation (12). In addition, during sepsis, the major interferon-gamma (IFN-γ)- producing natural killer (NK) cells encounter immoderate apoptosis after a reduced number is present in the circulation, thus increasing the risk of secondary infection (13). Due to the inhibition of spontaneous apoptosis, neutrophils may undergo other types of cell death, including NETosis and pyroptosis (14). Excessive NETosis, followed by the depletion of mature neutrophils, heightens the risk of nosocomial infections and immune dysfunction. Investigators are actively trying to clarify the underlying inconsistencies in innate and adaptive immunity as well as the mechanisms of immunosuppression that contribute to the long-term prognoses in sepsis. Consequently, immunoadjuvant therapies have recently attracted considerable attention in sepsis management to restore host immunity. This review focuses on the dysregulation of immune cell death patterns induced by sepsis and the subsequent disruptions to immunity. In addition, we outline current potential therapeutic interventions, including interleukin (IL)-7, IL-15, IFN-γ, granulocyte-macrophage colony stimulating factor (GM-CSF), Fms-like tyrosine kinase-3 ligand, inhibition of programmed cell death protein 1(PD-1), programmed cell death ligand 1 (PD-L1), and other cell death checkpoints, as well as future directions for sepsis management.
Mechanism of immune cell deaths in sepsis
The prime mechanism of immune cell death in sepsis is a type I programmed cell death, apoptosis. To date, three pathways of apoptosis have been reported: the extrinsic (death receptor) pathway, the intrinsic (mitochondrial) pathway, and the perforin/granzyme pathway. The three pathways intersect into the common execution pathway initiated by activating the effector enzyme cysteinyl aspartate- specific protease (caspase)-3 (15). Tumor necrosis factor (TNF)-α, high mobility group box-1 protein (HMGB1), Fas ligand (FasL), heat shock, oxygen-free radicals, nitric oxide (NO), glucocorticoids, granzymes, and TNF-alpha-induced protein eight like-2 (TIPE2) are the triggers of apoptosis, on the contrary interleukin (IL)-1, IL-6, and granulocyte colony-stimulating factor (G-CSF) are the inhibitor of apoptosis (16). The extrinsic pathway of apoptosis involves interaction between TNF family-derived extracellular death ligands, e.g., FasL, TNF-α, and the corresponding death receptors that include Fatty acid synthetase receptor, FasR, TNFR1. The ligand-receptor binding induces the recruitment of cytoplasmic adaptor protein FADD for FasL/FasR and TRADD with the recruitment of FADD and RIP in the case of TNF-α/TNFR1. Currently, caspase-8 gets activated by forming a death-inducing signaling complex (DISC) through the association of FADD and procaspase-8 (17). Caspase-8 activation leads to the downstream execution phase of apoptosis. The intrinsic pathway involves a wide range of non-receptor-mediated stimuli, and the intracellular signal generated changes the inner mitochondrial membrane. This pathway is governed by anti-apoptotic versus pro-apoptotic Bcl-2 family members. Bcl-2, Bcl-x, Bcl-XL, Bcl-XS, Bcl-w, BAG, Mcl-1, and Bfl-1/A1 are anti-apoptotic proteins, and Bcl-10, Bak, Bax, Bim, Bik, Blk, Bmf, Bad, and Bid are pro-apoptotic protein. These proteins determine whether the cell will undergo cell death or skip the death process. The intrinsic and extrinsic pathways are associated, and the molecules involved in each pathway can impact each other.
NETosis is a novel cell death program distinct from apoptosis and necrosis (14). In NETosis stimulated neutrophil release, NETs, a web-like architecture composed of a DNA backbone decorated with anti-microbial proteins like myeloperoxidase (MPO), neutrophil elastase (NE), and cathepsin G (18, 19). Although NETosis is a physiological process and essential part of the innate immune system to eliminate invading microbes, but the uncontrolled NETosis has a pathological role in numerous ways, attributed to sepsis, autoimmune, infectious, and non-infectious diseases that have attracted recent attention. NETosis occurs via two pathways: suicidal NETosis and vital NETosis. Suicidal NETosis is a lytic and slow cell death process, usually taking 2–4 h, whereas vital NETosis is a cell-death-independent non-lytic process that happens faster, within 5–60 min (20). To date, two different mechanisms, NADPH Oxidase 2 (Nox 2)-dependent and Nox 2-independent of NETosis, have been validated. Nox-dependent NETosis is triggered by inducers like PMA, LPS, and bacteria such as Pseudomonas aeruginosa, while agonists like calcium ionophores (A231128, ionomycin), uric acid crystals, certain microbes, and UV light trigger Nox-independent NETosis through the formation of different ROS, Nox-ROS and mitochondrial ROS, respectively (21). Different sets of kinases (MAPK, ERK, p38, and JNK) specific to both NETosis get activated, leading to transcriptional firing and activation of downstream pathways. In both types of NETosis ultimately nuclear membrane disintegrates, and NETs are expelled.
Pyroptosis is a caspase-1 (canonical pathway) or caspase-4/5/11(non-canonical pathway)-dependent proinflammatory programmed cell death process (22). The control form of this cell death is a part of innate immunity to actuate phagocytic immune cells and thus control pathogen infection. On the other hand, exaggerated pyroptosis results in a dysregulated host immune response and augments inflammatory injury, leading to organ dysfunction or septic shock. In the canonical pathway, intracellular pattern recognition receptors (PRRs) such as NLRP1B, NLRP3, NLRC4, recognize the stimulus signals of pathogens and activate caspase-1 protein through the association of pro-caspase-1, and adaptor protein ASC. In the non-canonical pathway, bacterial LPS directly bind and activate caspase-11/4/5 (23). At this point, gasdermin D gets activated, and pyroptosis occurs by rapid cell membrane disruption and release of proinflammatory mediators.
Autophagy, a type II programmed cell death, is an essential cellular process and a damaged protein or organelle degradation system necessary for cellular homeostasis. These regulated innate immune defense mechanisms act as cellular defense against oxidative stress and the elimination of pathogenic microorganisms and play a role in antigen presentation (24). Autophagy begins with the formation of a double-membrane vesicle called autophagosomes. Many signaling complexes and pathways participate in the initiation and maturation of the autophagy process.
Ferroptosis is a unique form of iron-dependent programmed cell death distinct from apoptosis, necrosis, and autophagy (25). In this process, lipid peroxides are generated from intracellular ROS and hydrogen peroxide (H2O2) by the action of iron and oxidize lipid membranes with polyunsaturated fatty acids (PUFAs) (26). At this stage, membrane damage begins followed by cell death. Innate and adaptive immune cells such as macrophages, T, and B cells undergo ferroptosis, reducing numbers and function. This cell death favors bacterial multiplication and dampens the body’s immune function, leading to sepsis (27).
Necroptosis, a novel form of programmed cell death, plays a significant role in the pathophysiology of sepsis. This death process is initiated by activating death receptors like TNF receptor 1 (TNFR1) (28). Then, the receptor-interacting protein kinase 1 (RIPK1) gets activated, which subsequently phosphorylates and activates RIPK3. The RIPK1-RIPK3-mixed kinase domain-like protein (MLKL) complex facilitates cell death by forming membrane pores. Staphylococcus aureus is responsible for nosocomial infection and sepsis (29). Staphylococcus aureus can also induce necroptosis of macrophages (30) and neutrophils (31) in host cells.
Role of immune checkpoint in sepsis
Immune checkpoints are specific membrane molecules and the key controllers of the immune system that balance immune homeostasis and limit excessive immune response. Immune checkpoints play a significant role in the pathophysiology of sepsis (32). Leukocytes (neutrophils, monocytes, natural killer cells, and dendritic cells) and lymphocytes (T and B cells) express checkpoint molecule PD-1 on their surface. PD-1 can interact with complementary ligand PD-L1 on the surface of antigen-presenting cells (APCs) such as monocytes, macrophages, and dendritic cells. Cell surface inhibitory immune checkpoint molecules include PD-1, PD-L1, PD-L2, cytotoxic T lymphocyte antigen-4 (CTLA-4), B and T lymphocyte attenuator (BTLA), lymphocyte activation-gene-3 (LAG-3) and T cell membrane protein-3 (TIM-3) and 2B4 (33). This review will focus on the PD-1/PD-L1 axis. During sepsis, both innate and adaptive immune cells become immunocompromised. PD-1/PD-L1 axis is involved in immune cell dysfunction and sepsis-induced immunosuppression (34). A few studies confirmed that increased PD-L1 expression on neutrophils and monocytes is linked to both pro- and anti-inflammatory cytokine levels, decreased phagocytic capacity, delayed apoptosis of neutrophils, and mortality in septic patients (34, 35). A recent study suggests that overexpression of NK cell PD-L1 is associated with increased sepsis severity (36). Increased levels of PD-1 expression in T cells have been reported to be associated with lymphopenia, T cell death, and increased mortality (37).
Sepsis-induced innate immune cell death
Sepsis markedly affects the lifespan, production, and function of the effector cells within the innate immune system, thereby disrupting homeostasis. The innate immune system, which serves as the body’s front line of defense, consists of neutrophils, monocytes and macrophages, dendritic cells, and other components. Sepsis induces marked losses of these innate immune cells through various cellular death pathways, contributing to immune suppression (Figure 1).
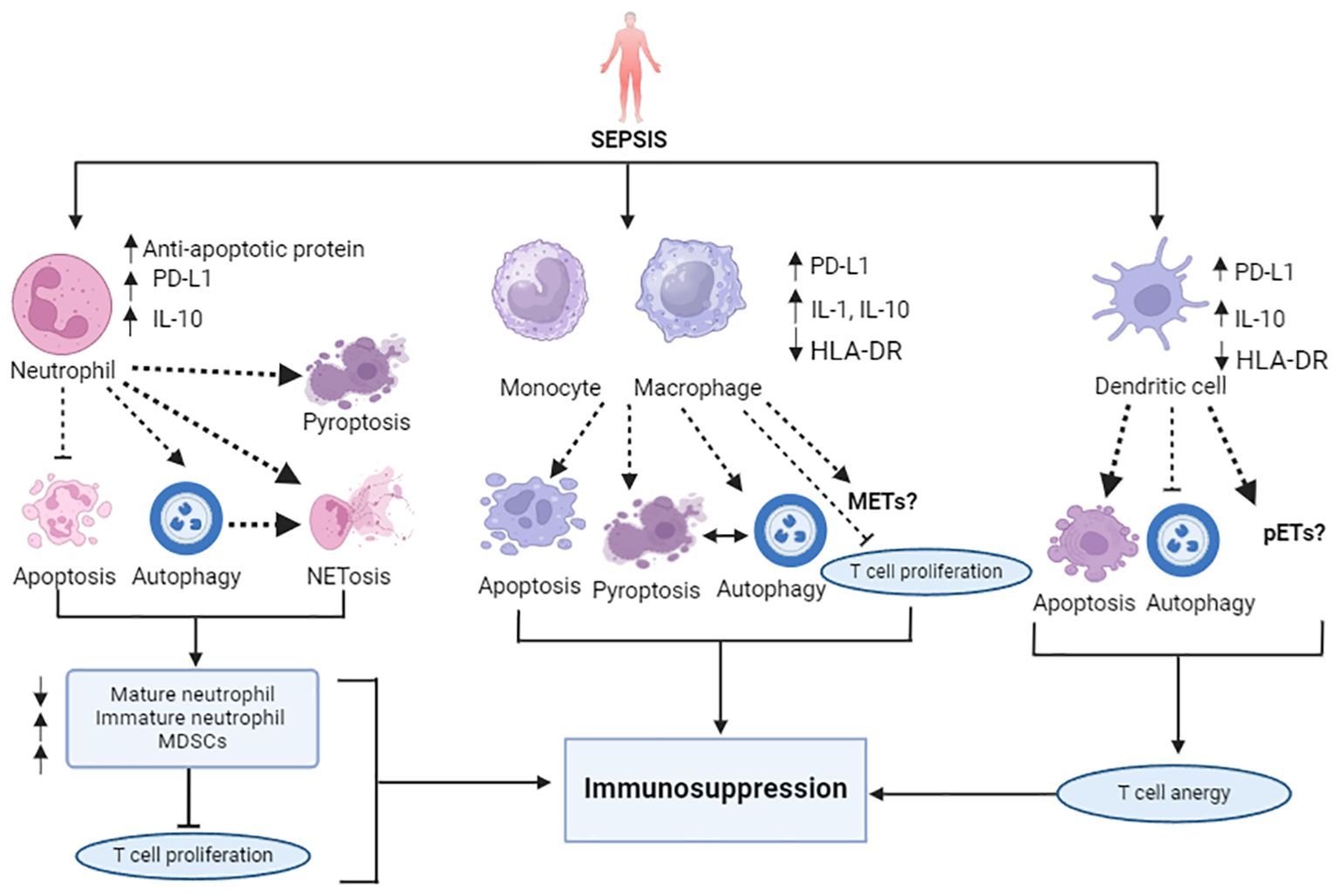
Figure 1. Overview of sepsis-attributed immunosuppression: Impairment of innate immune cell death pathways. In sepsis, alteration of cytokines (upregulated IL-1, IL-10) and reduced antigen presentation (downregulated HLA-DR) is marked. Sepsis slows neutrophil apoptosis and augments NETosis, autophagy, and pyroptosis, like cell deaths, resulting in an increase in the number of immature neutrophils, T cell proliferation inhibitory MDSCs, and the depletion of mature neutrophils. Monocytes and macrophages also encounter increased apoptosis, pyroptosis, autophagy, and NETosis-like cell death. Dendritic cells undergo extensive apoptosis, which induces a tolerogenic state. Unlike increased NETosis, DCs have less potential to undergo autophagy in sepsis. The PD-1/PD L-1 axis plays a significant role in the induction of all these cell death pathways. All these different cell death patterns contribute to immunosuppression in sepsis.
Neutrophils, the most abundant circulating leukocytes derived from bone marrow and primary responders to pathogen attack, typically undergo apoptosis within 24 hours of release (38). In sepsis, unlike the delayed apoptosis observed in lymphocytes, mature neutrophils exhibit several dysfunctions: release of immature neutrophils from bone marrow to circulation, reduced oxidative burst capacity, decreased cell migration, diminished complement activation, and impaired bacterial clearance. These factors contribute to the development of immune suppression and persistent inflammation, which may continue even after the disappearance of symptoms (9). This significant impairment of neutrophil functions increases the susceptibility of patients to nosocomial infections (39), ventilator-associated pneumonia (40), and other secondary infections (41). Experiments using a mouse model of sepsis have provided further support for these findings, demonstrating reduced neutrophil functions and increased risk of secondary Pseudomonas aeruginosa infection (42) and organ injury (43). Prolonged neutrophil survival is attributed to an imbalance between anti-apoptotic and pro-apoptotic signals. Notably, the activation of anti-apoptotic factors such as B-cell lymphoma-extra large (Bcl-xL), annexin A1, Bak, and myeloid cell leukemia-1 (MCL-1) is the primary cause of delayed neutrophil apoptosis (14, 44). In addition, certain neutrophil subsets (CD16hi, CD62Llow) exhibit suppressive properties by releasing large amounts of the immunosuppressive cytokine IL-10 (45), which is associated with delayed neutrophil apoptosis (46) and also suppresses T-cell proliferation (47). Furthermore, NF-kB-mediated inhibition of caspase-3 and caspase-9, along with impaired phosphorylation and inhibition of caspase-8 catalytic activity, also affects neutrophil apoptosis (48, 49). Overexpression of PD-L1 on septic neutrophils is strongly associated with delayed neutrophil apoptosis, and has been shown to drive lung injury and increase mortality in experimental sepsis, i.e., a cecal ligation and puncture (CLP) model (43). In sepsis, delayed apoptosis allows mature neutrophils to undergo other types of cell death, such as NETosis and autophagy (50, 51). Like NETosis, pyroptosis mediated by caspase-1/11, GSDMD is an essential physiological host defense mechanism. However, excessive neutrophil pyroptosis also contributes to sepsis (52). Overproduction of IL-1β and IL-18 through the classical caspase-1-dependent pathway increases the magnitude of the inflammatory response, suppresses immunity (53), and reduces survival rate (54). Consequently, marked depletion of neutrophils through various cell death pathways accelerates immunosuppression in sepsis.
Monocyte and macrophage apoptosis occurs during the progression of sepsis, potentially leading to immunosuppression and increasing host vulnerability to secondary infections or mortality. Apoptosis in monocytes may reprogram the immune system towards an anti-inflammatory, immunosuppressive response. These monocytes exhibit a reduced capacity to release pro-inflammatory cytokines, such as tumor necrosis factor (TNF), IL-1α, IL-6 and IL-12 against lipopolysaccharide (LPS) and other bacterial inducers, a phenomenon resembling ‘endotoxin tolerance’ that results in poor outcomes (55, 56). Interestingly, the same monocytes are capable of secreting significant levels of anti-inflammatory mediators, such as IL-1 receptor antagonist and IL-10, which correlate with increased rates of nosocomial infection and higher mortality (9). Also, impaired monocytes are linked to decreased antigen-specific lymphocyte proliferation (57). This endotoxin tolerance, along with increased susceptibility to nosocomial infections and elevated mortality, is associated with reduced HLA-DR expression on monocytes and macrophages, referred to as ‘anergy’ (58, 59). Moreover, reports have shown that expression of PD-L1 is increased on the monocytes of septic patients (60) and that this can be used as an independent predictor of mortality (61). In addition, studies have revealed a correlation between reduced monocyte activities and the levels of PD-1 on T lymphocytes (34). Macrophage pyroptosis also contributes to the pathology of septic disseminated intravascular coagulation (DIC) (62), with caspase-11-dependent pyroptosis playing a pivotal role in exacerbating damage and reducing survival (63, 64). Caspase-1-induced monocyte pyroptosis has also been noted in patients with post-traumatic sepsis (65). NETotic-like cell death and macrophage extracellular traps (METs) have also been observed in macrophages (66). Additionally, autophagy influences sepsis progression by affecting senescence, phagocytic capacity, and the activation of inflammatory cytokine release by macrophages (67). Thus, these excessive self-sacrificial processes may facilitate immunosuppression in sepsis.
Dendritic cells (DCs) are dynamic antigen-presenting cells (APCs) that link innate and adaptive immunity and contribute to pathogen recognition, immune response regulation, and inflammation (68, 69). Both conventional dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs) are highly susceptible to sepsis-induced apoptosis, resulting in significant depletion of DCs in patients with sepsis (70, 71). A study in mouse and other animal models of sepsis has investigated caspase-3-mediated apoptosis of DCs (16). In addition, recent reports have also confirmed the involvement of PD-1 in activating DC apoptosis (72, 73). This intense apoptosis and depletion of DCs not only increases susceptibility to nosocomial infections (74), but also diminishes their functional capabilities (75), resulting in reduced expression of CD40, CD 86, and HLA-DR, and increased section of IL-10 (76, 77). These alterations reflect the tolerogenic state of surviving DCs, which lose their ability to activate effector T cell responses, instead inducing either T cell anergy or Treg cell proliferation (12). Consequently, these immunosuppressive DCs fail to mount an immune response against subsequent bacterial challenges (78). In addition to apoptosis, pDCs can release NET-like extracellular traps (pETs) in response to bacterial infection (79). Furthermore, the loss of autophagy potential in DCs heightens the risk of sepsis (80).
Natural Killer Cells (NK cells) are innate lymphocytes that play a crucial role in coordinating innate and adaptive immune responses in sepsis and defense against pathogen attack (81). NK cells produce IFN-γ during microbial sepsis, and IFN- γ can activate macrophages (71). In sepsis, NK cells undergo extensive apoptosis, significantly decreasing their number in circulation (13). Thus, the low titer of IFN- γ increases the risk of secondary infection. Due to impaired cytokine secretion, the surviving and remaining NK cells cannot correctly induce an immune response against endotoxin. NK cells also lose cytotoxic function, which results in immune suppression (82).
Sepsis-induced adaptive immune cell death
The adaptive immune system consists of highly specialized lymphocytes, including T and B lymphocytes. These subpopulations of adaptive immune cells are also susceptible to sepsis-induced cell death. Persistent lymphopenia, a hallmark characteristic in patients with sepsis, is associated with an increased risk of nosocomial infection and a higher risk of mortality (Figure 2).
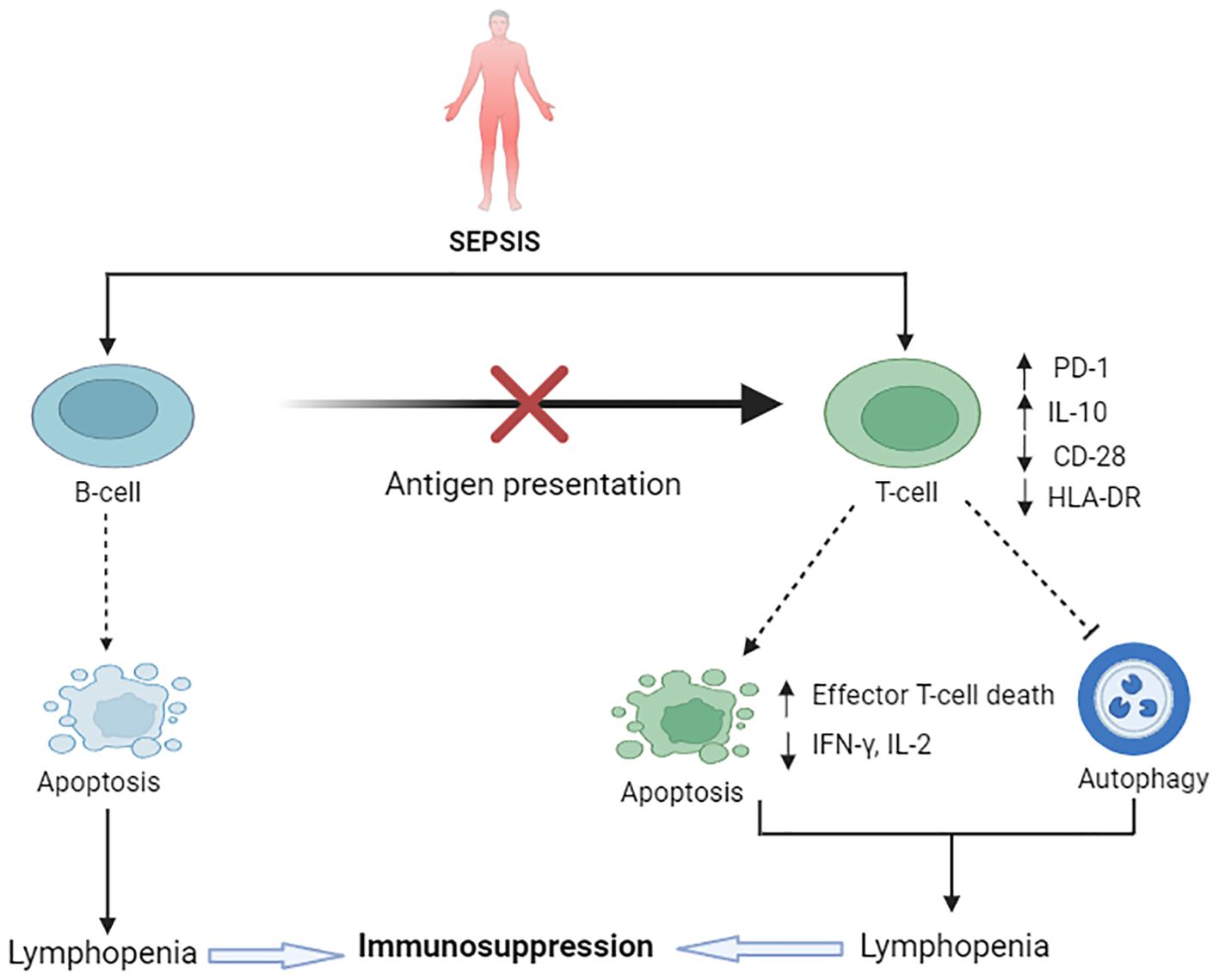
Figure 2. Sepsis-induced immunosuppression: impairment of adaptive immune cell death pathways. Upregulated IL-10 and downregulated CD8 and HLA-DR is notable in sepsis. B-cells undergo apoptosis in sepsis, resulting in a reduction of overall B-cell populations and impairment of the antigen-presenting role of B-cells. Sepsis causes excessive T-cell apoptosis, causing lymphopenia and ultimate immunosuppression. On the contrary, reduced T-cell autophagy in sepsis also contributes to immunosuppression. PD-1 and PD-L-1 play a vital role in this immunosuppression.
B cells are critical components of both innate and adaptive immunity, with multifunctional roles and diverse phenotypes. Different subpopulations of B cells have specific roles in immunity. For instance, B cells that are activated by pattern recognition receptors (PRRs), referred to as innate response activator (IRA) B cells, augment the antimicrobial response to clear bacteria and induce emergency myelopoiesis by producing GM-CSF and IL-3, respectively (83, 84). Sepsis induces apoptosis in B cells and reduces the diversity of B cell subtypes (85, 86). A growing body of evidence has revealed impaired B cell functions, including diminished antigen presentation to T lymphocytes (87), imperfect interactions with bacterial products (88), and increased secretion of IL-10 (89), which collectively suppress immune responses. Antibody-producing memory B cells, extracellular signal-regulated kinase (ERK)-activation-associated B cells, and CD5+ B1 cells are more susceptible to apoptosis compared to other B cell types (85, 86). Sepsis also reduces the number of naïve B cells, triggers B cell exhaustion (90), and impairs the production of IgM (91). While there is no strong evidence directly linking B cell pyroptosis to sepsis, studies using a caspase-1 knockout, IL-1 knockout, and IL-1/IL-18 double knockout mouse models suggest that caspase-1-dependent pyroptosis delays B lymphocyte apoptosis, potentially improving macrophage phenotype and survival rates (92). Further extensive investigation is required to elucidate the relationship between lymphocyte pyroptosis and sepsis.
T cells are the primary actors of the adaptive immune system. Sepsis triggers significant apoptosis of different T cell subsets (93). Specifically, marked apoptosis and reductions in CD4+ and CD8+ T lymphocytes occur in the early phase of sepsis (82, 94). This extensive cell death leads to lymphopenia, which is associated with immunosuppression following the acute resuscitation phase of sepsis. In patients with sepsis, apoptosis of T cells occurs via both intrinsic and extrinsic pathways (95). The interaction of PD-1 with PD-L1 plays a critical pathological role in the immunosuppression observed in sepsis (96). Patients exhibit elevated PD-1 and PD-L1 expression on CD4+ T cells, decreased lymphocyte proliferation, and increased IL-10 secretion (97). In addition, stimulatory molecules such as CD28 and HLA-DR are significantly downregulated in sepsis, reflecting the host’s impaired ability to combat pathogens. Research has shown that the interaction of PD-L1 on APC with PD-1 on T cells disrupts the positive costimulatory signaling of CD28, inhibits T cell proliferation, increases immune effector cell death, reduces cytokine secretion (such as IL-2 and IFN-γ), and ultimately impairs antigen clearance (98). Moreover, a deficiency in T cell autophagy in sepsis contributes to sepsis-induced immunosuppression and increased mortality (99). Another mechanism of immune impairment in the subacute phase of sepsis is T cell anergy (Figure 1), which is characterized by the inability of lymphocytes to recognize the cognate antigen, activate, proliferate, and produce cytokines (100). Guinault et al. demonstrated that an expression pattern of the three CD8+ T cell exhaustion markers (2B4, PD-1, and CD160) was strongly associated with the mortality of patients with sepsis (101). To date, there are no reports of lymphocytes releasing NET-like structures.
Collaboration and interplay among cell death pathways in sepsis
Among the various forms of cell death, apoptosis of immune cells is a central pathophysiological event responsible for sepsis-induced immunosuppression. On one hand, an increased propensity for apoptosis among B cells, T cells, macrophages, and dendritic cells leads to a tolerogenic nature of these cells and a significant reduction in their presence in circulation. On the other hand, delayed apoptosis in neutrophils facilitates alternative forms of cell death, such as autophagy, pyroptosis, and NETosis. Experimental murine models have revealed that sepsis also compromises T cell viability and function by suppressing autophagy and accelerating apoptosis (102). A growing evidence is uncovering the cross-talk among these different cell-death pathways. Notably, there is interaction between apoptosis and autophagy; the cleavage of autophagy-related protein 5 (Atg5) by the protease calpain induces mitochondria-mediated apoptosis through its binding to the anti-apoptotic protein Bcl-xL (103). In septic CD4-Cre/Atg5f/f mice, an increase in the apoptosis of CD+ T cells has been observed, accompanied by upregulation of the pro-apoptotic gene PDCD1 and downregulation of the anti-apoptotic gene BCL2 (102). Further research has demonstrated that decreased ATG5 expression levels are correlated with the severity of sepsis progression and mortality (104), suggesting that the inhibition of autophagy promotes immune cell apoptosis and immunosuppression. Other recent studies highlighted the close association between neutrophil autophagy pathways and increased NET formation in patients with septic-DIC (23, 105). Increased neutrophil autophagy has been noted in survivors of sepsis, and autophagy in healthy neutrophils may stimulate NETs (50). Additionally, autophagy and pyroptosis are inter-connected (106), as are pyroptosis and NETosis, with each having mutual effects. In patients and a mouse model with sepsis, the use of specific inhibitors against primary actors (PAD)2 has been shown to reduce NETosis and macrophage caspase 11-dependent pyroptosis. This inhibition of caspase 11 results in decreased release of inflammatory mediators, increased macrophage counts, enhanced bacterial clearance, and improved survival (107). Targeting GSDMD and PADs could therefore be promising in sepsis therapy, as it couples pyroptosis and NETosis. The ablation of caspase-1/11 in septic mice enhances neutrophil phagocytosis and the levels of inflammatory cytokines, which reflects improved immunity (53).
Why immunotherapy?
Recent extensive research has broadened our understanding of sepsis pathophysiology, evolving from the traditional view of early inflammation-driven pathology to encompassing concurrent immunosuppression. The discussions above confirm that immunosuppression in sepsis results from the extensive loss of both innate and adaptive immune cells through apoptosis, autophagy, NETosis, pyroptosis, ferroptosis, and necroptosis. Consequently, targeting a single cell death pathway and enhancing host immunity with immunomodulatory agents represents a promising therapeutic strategy to restore impaired host defenses. The following section of this review focuses on immunomodulatory agents aimed at enhancing immune cell function by modulating cell death pathways.
Current status of clinical therapies
Interleukin-7, primarily derived from stromal cells, is an indispensable hematopoietic cytokine crucial for T cell survival, proliferation, differentiation, and effector functions. It is also an attractive immunoadjuvant therapeutic molecule targeting adaptive immune irregularities in sepsis. Clinical trials have demonstrated that IL-7 is safe and well-tolerated, without inducing toxicities such as cytokine storms or exacerbating inflammation or organ dysfunction. Moreover, IL-7 can inhibit the massive apoptosis of immune-effector cells induced by sepsis and restore the production of IFN-γ, which is essential for the host’s response to invading pathogens (108). A recent clinical trial demonstrated that IL-7 therapy successfully restored depleted CD4+ and CD8+ effector cells by threefold to fourfold in patients with sepsis (109). This pluripotent cytokine can mitigate lymphocyte apoptosis by enhancing the expression of anti-apoptotic proteins such as Bcl-2, CD-28, boosting IFN-γ levels, and increasing TCR diversity, which are typically diminished in patients with sepsis (110, 111). Consequently, recombinant IL-7 therapy not only increases the numbers of CD4+ and CD8+ T cells, but it also reduces Treg cells in circulation, decreasing morbidity and mortality. Interestingly, when IL-7 is administered alongside antiretroviral therapy in patients with HIV exhibiting lymphopenia and immune suppression, it results in reduced PD-1 expression (112). We conclude that IL-7 immunostimulatory therapy, whether used individually or in combination, may represent a promising and potentially protective treatment option for sepsis-induced immunosuppression.
Interleukin-15 promotes the proliferation of memory CD8+ T cells, stimulates dendritic cells, and enhances B cell immunoglobulin production. In a mouse model of sepsis, IL-15 has been shown to attenuate sepsis-induced apoptosis of natural killer (NK) cells, dendritic cells, and CD8+ T cells by increasing the expression of the anti-apoptotic protein Bcl-2, and decreasing the expression of pro-apoptotic proteins Bim and PUMA (113). Additionally, IL-15 enhances IFN-γ production and improves survival in the cecal ligation and puncture (CLP) sepsis model. In cancer trials, the combination of IL-15 and anti-PD-1 therapy has demonstrated reduced IL-10 production and PD-1 expression on CD8+ T cell, augmenting anti-tumor activity (114). Given that IL-15 has shown toxicity in a previous animal study (115), it is important to determine the optimal dosage when employing it as an immunotherapeutic agent in sepsis. Further clinical trials are necessary to evaluate the synergistic potential of IL-15 with other immunotherapeutic agents.
IFN-γ is a key cytokine that is essential for the activation of innate immunity, which is necessary for the clearance of microbial pathogens. However, IFN-γ production is markedly decreased in sepsis. Recombinant IFN-γ therapy in protracted Staphylococcus aureus sepsis has been shown to increase monocyte HLA-DR expression and function, as well as enhance bacterial clearance, without any adverse effects (116). Similarly, INF-γ treatment in patients with invasive fungal infections has also been demonstrated to restore HLA-DR expression on leukocytes (117). Although IFN-γ therapy offers potential benefits in patients with sepsis exhibiting immunosuppression by reviving monocyte functions associated with reduced HLA-DR expression, there are no records of it ameliorating T cell defects. Interestingly, the combined application of IFN-γ therapy with the anti-PD-1 antibody, nivolumab, in fungal sepsis has shown promising in restoring immune function and eliminating infection (118). Immunoadjuvant adjunctive IFN-γ therapy, along with IL-7 and anti-PD1/PD-L1, could be beneficial for patients with sepsis, as it has proven impacts on enhancing CD4+ and CD8+ T cell functions.
Fms-like tyrosine kinase-3 ligand (Flt3L), a stem cell growth factor, acts on the class III tyrosine kinase receptor (Flt3R), which is typically expressed on hematopoietic progenitor cells and dendritic cell populations. Enhanced dendritic cell apoptosis and the subsequent impairment of T cell function are common in sepsis pathophysiology. Flt3L treatment has demonstrated effectiveness in promoting the growth and expansion of dendritic cells in both human (119) and mouse models (120). In addition, Flt3L therapy in models of burn injury and sepsis not only increases dendritic cell populations but also enhances neutrophil antimicrobial functions and improves survival (121). A recent study using a mouse model of burn injury and sepsis has shown that Flt3L treatment mitigates T cell depletion, restores CD28 expression on CD4+ and CD8+ T cells, and increases IFN-γ production by CD8+ T cells, thereby reducing organ injury markers and enhancing survival (122). Flt3L also suppresses PD-L1 expression on APCs, such as dendritic cells, macrophages and monocytes. A research group has revealed that Flt3 can mitigate oxidative stress and protect cardiomyocytes from apoptotic death through the regulation of Bcl-2 family proteins (123). Thus, Flt3 is hypothesized to reduce T cell apoptosis in sepsis. Further investigation is warranted to explore the synergistic potential of Flt3 therapy with other established therapeutics, such as IL-7, in the management of sepsis.
Granulocyte macrophage colony stimulating factor (GM-CSF), a hematopoietic growth factor, enhances the production of neutrophils and monocytes, enhances monocyte survival, and restores TNF production, thereby helping to prevent nosocomial infections and mitigate immunosuppression (124). GM-CSF therapy increases the expression of HLA-DR on monocytes, facilitates bacterial clearance, and contributes to more ventilation-free days and reduced stays in the intensive care unit (125). However, a meta-analysis by Bo et al. found no evidence supporting the routine use of G-CSF or GM-CSF in patients with sepsis (126).
PD-1 receptor system acts as a negative regulator of the immune response. In sepsis, there is an overexpression of inhibitory receptors PD-1 on B and T lymphocytes and PD-L1 and PD-L2 on epithelial cells, endothelial cells, and APCs. This overexpression leads to decreased cytokine secretion, increased apoptotic cell death, immunosuppression, and, eventually, deleterious outcomes. A high serum soluble form of PD-L1 (sPD-Ll) has also been detected in patients with sepsis, and is correlated with disease severity and poor clinical outcomes (127). The PD-1/PD-L1 axis, targeted by immune check point inhibitor antibodies, is gaining attention as an immunotherapeutic approach in sepsis due to its successful application in the treatment of infectious diseases and regression of advanced-stage cancers (128, 129). In addition, anti-PD-1/PD-L1 therapy has also been shown to increase the expression of CD28 on proliferating peripheral CD8+ T cells following treatment. Mice models and ex vivo clinical studies of patients with sepsis have shown that blockade of PD or PD-L1 plays a significant role in reversing immune defects caused by sepsis (130). Furthermore, anti-PD-L1 treatment has been shown to promote apoptosis in septic neutrophils in mice models (131). The immunosuppressive properties of septic neutrophils, monocytes, and macrophages can also be reversed by blocking either PD-1 or PD-L1 (34, 132). Additionally, treatment with anti-PD1 antibodies enhances DC survival in sepsis (72). An anti-PD-1 antibody nivolumab, i.e., immune checkpoint inhibitor has been evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics at the phase 1b (133) and the phase 1/2 study (134). Taken together, these findings suggest that targeting the PD-1/PD-L1 axis with immunoadjuvant therapy represents a promising approach to reverse sepsis-induced immunosuppression.
Autophagy has recently gained attention in the field of critical care due to its role in regulating cell apoptosis. Enhancing T cell autophagy may alleviate sepsis-induced immunosuppression by modulating apoptosis (102). Additionally, inhibiting NETosis represents another potential strategy for sepsis management. A recent study in rodent models of sepsis has shown that NET inhibition using Cl-amidine, a PAD4 inhibitor, is effective (135). Another study has demonstrated the inhibition of PAD4 and NETosis in both mice and humans using YW3-56 as an inhibitor (136). Moreover, disulfiram, an FDA-approved drug, targets GSDMD activation, blocking pyroptosis and NETosis, thereby improving survival in mice (137). Table 1 summarizes some preclinical treatments against immunosuppression in sepsis.
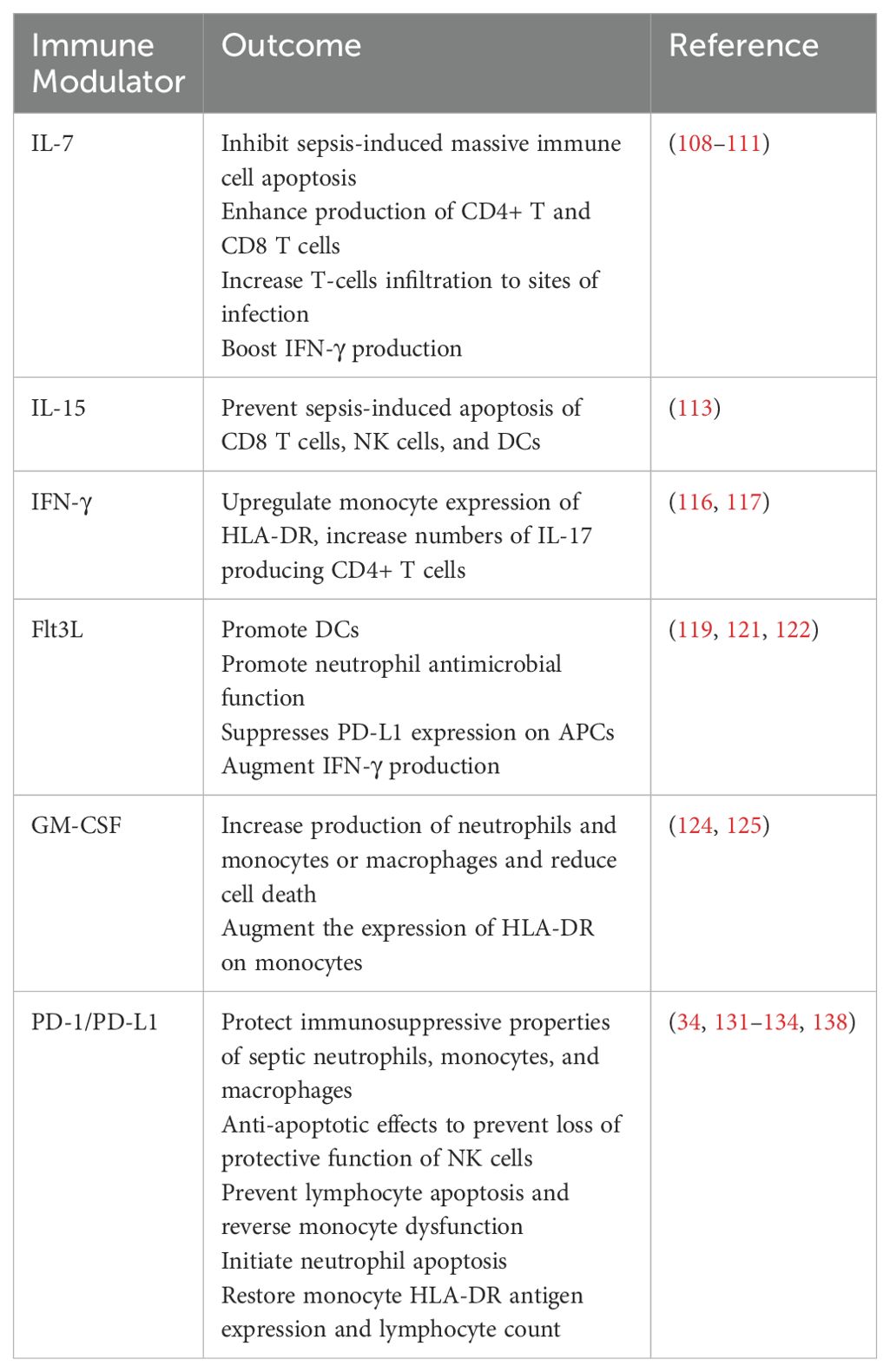
Table 1. Preclinical treatments for immunosuppression during sepsis.
Adverse reactions of immunotherapy
The main concern of immune therapy in sepsis is the risk of hyper-inflammatory response that can increase the severity of the disease, even death. An animal study reported that IL-15 immunotherapy has a toxic effect, causing liver injury and cachexia (115, 139). However, clinical trials of IL-7 and IFN-γ therapy showed no adverse reactions like cytokine storms or exacerbating inflammation and were well tolerated (108, 116). Although PD-1/PD-L1 is a promising therapy, PD-1 deficiency is also related to the occurrence of autoimmune diseases such as lupus-like syndromes, de novo type 1 diabetes, and dilated cardiomyopathy (140, 141). That is why the timing and duration of PD-1/PD-L1 blocking should be done with proper attention. In conclusion, patients’ immune status, optimal dosage, timing, and personalized approach should be considered before starting the clinical application of immunotherapy to avoid adverse reactions.
Conclusions and future directions
Immune cells employ mechanisms such as autophagy in B cells and T cells, as well as NETosis and pyroptosis in neutrophils and macrophages, initially to protect the host. However, when overactivated, these protective effects can become detrimental. Dysregulated immune cell death, including apoptosis, autophagy, NETosis, and pyroptosis, along with impaired immune status, contributes significantly to the pathophysiology of sepsis. To effectively address sepsis, it is imperative to explore other types of cell death and their underlying mechanisms. In addition, elucidating the cross-talk among apoptosis, autophagy, pyroptosis, and NETosis is necessary. These cell death processes augment inflammation, deplete immune cells, and lead to immunosuppression. Targeting, closely monitoring, and regulating these cell death mechanisms could offer a promising approach to treating patients with sepsis, ultimately improving survival. In this review, we have highlighted numerous immunoadjuvant therapeutic agents that possess significant potential to enhance suppressed immunity in sepsis. While immunotherapy represents a promising strategy against sepsis, the broad variations in immune status among patients must be carefully considered for clinical applications. Biomarker-guided stratification and a personalized approach for each patient are imperative. Furthermore, combination therapies may offer a higher success rate in countering immunosuppressive sepsis in the future.
Author contributions
MMI: Writing – original draft, Writing – review & editing. EW: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. US: Writing – review & editing. MO: Writing – review & editing. TI: Writing – review & editing. ST: Writing – review & editing. RK: Writing – review & editing. DO: Writing – review & editing. NT: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
3. Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, Bauer M, et al, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. (2011) 15:R183. doi: 10.1186/cc10332
4. Kethireddy S, Kumar A. Mortality due to septic shock following early, appropriate antibiotic therapy: can we do better?*. Crit Care Med. (2012) 40:2228–9. doi: 10.1097/CCM.0b013e318256bb99
5. Cohen J, Opal S, Calandra T. Sepsis studies need new direction. Lancet Infect Dis. (2012) 12:503–5. doi: 10.1016/S1473-3099(12)70136-6
6. Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA. (2011) 306:2614–5. doi: 10.1001/jama.2011.1853
7. Fleischmann C, Thomas-Rueddel DO, Hartmann M, Hartog CS, Welte T, Heublein S, et al. Hospital incidence and mortality rates of sepsis. Dtsch Arztebl Int. (2016) 113:159–66. doi: 10.3238/arztebl.2016.0159
8. Wen X, Xie B, Yuan S, Zhang J. The “Self-sacrifice” of immuneCells in sepsis. Front Immunol. (2022) 13:833479. doi: 10.3389/fimmu.2022.833479
9. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. (2016) 126:23–31. doi: 10.1172/JCI82224
10. Patil NK, Bohannon JK, Sherwood ER. Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol Res. (2016) 111:688–702. doi: 10.1016/j.phrs.2016.07.019
11. Watanabe E, Thampy LK, Hotchkiss RS. Immunoadjuvant therapy in sepsis: novel strategies for immunosuppressive sepsis coming down the pike. Acute Med Surg. (2018) 5:309–15. doi: 10.1002/ams2.2018.5.issue-4
12. Faivre V, Lukaszewicz AC, Alves A, Charron D, Payen D, Haziot A. Human monocytes differentiate into dendritic cells subsets that induce anergic and regulatory T cells in sepsis. PloS One. (2012) 7:e47209. doi: 10.1371/journal.pone.0047209
13. Wesselkamper SC, Eppert BL, Motz GT, Lau GW, Hassett DJ, Borchers MT. NKG2D is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J Immunol. (2008) 181:5481–9. doi: 10.4049/jimmunol.181.8.5481
14. Zhu CL, Wang Y, Liu Q, Li HR, Yu CM, Li P, et al. Dysregulation of neutrophil death in sepsis. Front Immunol. (2022) 13:963955. doi: 10.3389/fimmu.2022.963955
15. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
16. Luan YY, Yao YM, Xiao XZ, Sheng ZY. Insights into the apoptotic death of immune cells in sepsis. J Interferon Cytokine Res. (2015) 35:17–22. doi: 10.1089/jir.2014.0069
17. Hattori Y, Takano K, Teramae H, Yamamoto S, Yokoo H, Matsuda N. Insights into sepsis therapeutic design based on the apoptotic death pathway. J Pharmacol Sci. (2010) 114:354–65. doi: 10.1254/jphs.10R04CR
18. Islam MM, Takeyama N. Inorganic arsenic administration suppresses human neutrophil function in vitro. Hum Exp Toxicol. (2021) 40:725–34. doi: 10.1177/0960327120966040
19. Islam MM, Salma U, Irahara T, Watanabe E, Takeyama N. Quantifying myeloperoxidase-DNA and neutrophil elastase-DNA complexes from neutrophil extracellular traps by using a modified sandwich ELISA. J Vis Exp. (2023) 195. doi: 10.3791/64644
20. Ravindran M, Khan MA, Palaniyar N. Neutrophil extracellular trap formation: physiology, pathology, and pharmacology. Biomolecules. (2019) 9(8):365. doi: 10.3390/biom9080365
21. Islam MM, Takeyama N. Role of neutrophil extracellular traps in health and disease pathophysiology: recent insights and advances. Int J Mol Sci. (2023) 24(21):15805. doi: 10.3390/ijms242115805
22. Xia D, Wang S, Yao R, Han Y, Zheng L, He P, et al. Pyroptosis in sepsis: Comprehensive analysis of research hotspots and core genes in 2022. Front Mol Biosci. (2022) 9:955991. doi: 10.3389/fmolb.2022.955991
23. Zheng X, Chen W, Gong F, Chen Y, Chen E. The role and mechanism of pyroptosis and potential therapeutic targets in sepsis: A review. Front Immunol. (2021) 12:711939. doi: 10.3389/fimmu.2021.711939
24. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
25. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523
26. Yang J, Yan C, Chen S, Li M, Miao Y, Ma X, et al. The possible mechanisms of ferroptosis in sepsis-associated acquired weakness. Front Physiol. (2024) 15:1380992. doi: 10.3389/fphys.2024.1380992
27. Xl L, Gy Z, R G, N C. Ferroptosis in sepsis: The mechanism, the role and the therapeutic potential. Front Immunol. (2022) 13:956361. doi: 10.3389/fimmu.2022.956361
28. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. (2019) 20:19–33. doi: 10.1038/s41583-018-0093-1
29. Murray RJ. Recognition and management of Staphylococcus aureus toxin-mediated disease. Intern Med J. (2005) 35 Suppl 2:S106–19. doi: 10.1111/j.1444-0903.2005.00984.x
30. Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PloS Pathog. (2015) 11:e1004820. doi: 10.1371/journal.ppat.1004820
31. Zhou Y, Niu C, Ma B, Xue X, Li Z, Chen Z, et al. Inhibiting PSMalpha-induced neutrophil necroptosis protects mice with MRSA pneumonia by blocking the agr system. Cell Death Dis. (2018) 9:362. doi: 10.1038/s41419-018-0398-z
32. McBride MA, Patil TK, Bohannon JK, Hernandez A, Sherwood ER, Patil NK. Immune checkpoints: novel therapeutic targets to attenuate sepsis-induced immunosuppression. Front Immunol. (2020) 11:624272. doi: 10.3389/fimmu.2020.624272
33. Patil NK, Guo Y, Luan L, Sherwood ER. Targeting immune cell checkpoints during sepsis. Int J Mol Sci. (2017) 18(11):2413. doi: 10.3390/ijms18112413
34. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol. (2016) 100:1239–54. doi: 10.1189/jlb.4HI0616-255R
35. Huang X, Chen Y, Chung CS, Yuan Z, Monaghan SF, Wang F, et al. Identification of B7-H1 as a novel mediator of the innate immune/proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J Immunol. (2014) 192:1091–9. doi: 10.4049/jimmunol.1302252
36. Jiang W, Li X, Wen M, Liu X, Wang K, Wang Q, et al. Increased percentage of PD-L1(+) natural killer cells predicts poor prognosis in sepsis patients: a prospective observational cohort study. Crit Care. (2020) 24:617. doi: 10.1186/s13054-020-03329-z
37. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care. (2014) 18:R3. doi: 10.1186/cc13176
38. Rosales C. Neutrophils at the crossroads of innate and adaptive immunity. J Leukoc Biol. (2020) 108:377–96. doi: 10.1002/JLB.4MIR0220-574RR
39. Stephan F, Yang K, Tankovic J, Soussy CJ, Dhonneur G, Duvaldestin P, et al. Impairment of polymorphonuclear neutrophil functions precedes nosocomial infections in critically ill patients. Crit Care Med. (2002) 30:315–22. doi: 10.1097/00003246-200202000-00009
40. Wilkinson TS, Conway Morris A, Kefala K, O'Kane CM, Moore NR, Booth NA, et al. Ventilator-associated pneumonia is characterized by excessive release of neutrophil proteases in the lung. Chest. (2012) 142:1425–32. doi: 10.1378/chest.11-3273
41. Morris AC, Brittan M, Wilkinson TS, McAuley DF, Antonelli J, McCulloch C, et al. C5a-mediated neutrophil dysfunction is RhoA-dependent and predicts infection in critically ill patients. Blood. (2011) 117:5178–88. doi: 10.1182/blood-2010-08-304667
42. Delano MJ, Thayer T, Gabrilovich S, Kelly-Scumpia KM, Winfield RD, Scumpia PO, et al. Sepsis induces early alterations in innate immunity that impact mortality to secondary infection. J Immunol. (2011) 186:195–202. doi: 10.4049/jimmunol.1002104
43. Wang JF, Wang YP, Xie J, Zhao ZZ, Gupta S, Guo Y, et al. Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood. (2021) 138:806–10. doi: 10.1182/blood.2020009417
44. Wesche DE, Lomas-Neira JL, Perl M, Chung CS, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. (2005) 78:325–37. doi: 10.1189/jlb.0105017
45. Kasten KR, Muenzer JT, Caldwell CC. Neutrophils are significant producers of IL-10 during sepsis. Biochem Biophys Res Commun. (2010) 393:28–31. doi: 10.1016/j.bbrc.2010.01.066
46. Keel M, Ungethüm U, Steckholzer U, Niederer E, Hartung T, Trentz O, et al. Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood. (1997) 90:3356–63. doi: 10.1182/blood.V90.9.3356
47. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. (2012) 122:327–36. doi: 10.1172/JCI57990
48. Taneja R, Parodo J, Jia SH, Kapus A, Rotstein OD, Marshall JC. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. (2004) 32:1460–9. doi: 10.1097/01.CCM.0000129975.26905.77
49. Gupta S, Lee CM, Wang JF, Parodo J, Jia SH, Hu J, et al. Heat-shock protein-90 prolongs septic neutrophil survival by protecting c-Src kinase and caspase-8 from proteasomal degradation. J Leukoc Biol. (2018) 103:933–44. doi: 10.1002/JLB.4A0816-354R
50. Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med. (2017) 196:577–89. doi: 10.1164/rccm.201603-0596OC
51. Martinod K, Fuchs TA, Zitomersky NL, Wong SL, Demers M, Gallant M, et al. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood. (2015) 125:1948–56. doi: 10.1182/blood-2014-07-587709
52. Liu L, Sun B. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. (2019) 76:2031–42. doi: 10.1007/s00018-019-03060-1
53. Gentile LF, Cuenca AL, Cuenca AG, Nacionales DC, Ungaro R, Efron PA, et al. Improved emergency myelopoiesis and survival in neonatal sepsis by caspase-1/11 ablation. Immunology. (2015) 145:300–11. doi: 10.1111/imm.2015.145.issue-2
54. Yang M, Fang JT, Zhang NS, Qin LJ, Zhuang YY, Wang WW, et al. Caspase-1-inhibitor AC-YVAD-CMK inhibits pyroptosis and ameliorates acute kidney injury in a model of sepsis. BioMed Res Int 2021. (2021) p:6636621. doi: 10.1155/2021/6636621
55. Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. (2006) 10:233. doi: 10.1186/cc5055
56. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. (2009) 30:475–87. doi: 10.1016/j.it.2009.07.009
57. Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol. (2006) 177:6540–7. doi: 10.4049/jimmunol.177.9.6540
58. Monneret G, Finck ME, Venet F, Debard AL, Bohé J, Bienvenu J, et al. Monitoring immune dysfunctions in the septic patient: a new skin for the old ceremony. Mol Med. (2008) 14:64–78. doi: 10.2119/2007-00102.Monneret
59. Monneret G, Finck ME, Venet F, Debard AL, Bohé J, Bienvenu J, et al. The anti-inflammatory response dominates after septic shock: association of low monocyte HLA-DR expression and high interleukin-10 concentration. Immunol Lett. (2004) 95:193–8. doi: 10.1016/j.imlet.2004.07.009
60. Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J, et al. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care. (2011) 15:R70. doi: 10.1186/cc10059
61. Shao R, Fang Y, Yu H, Zhao L, Jiang Z, Li CS. Monocyte programmed death ligand-1 expression after 3-4 days of sepsis is associated with risk stratification and mortality in septic patients: a prospective cohort study. Crit Care. (2016) 20:124. doi: 10.1186/s13054-016-1301-x
62. Luo L, Xu M, Liao D, Deng J, Mei H, Hu Y. PECAM-1 protects against DIC by dampening inflammatory responses via inhibiting macrophage pyroptosis and restoring vascular barrier integrity. Transl Res. (2020) 222:1–16. doi: 10.1016/j.trsl.2020.04.005
63. Salvamoser R, Brinkmann K, O'Reilly LA, Whitehead L, Strasser A, Herold MJ. Characterisation of mice lacking the inflammatory caspases-1/11/12 reveals no contribution of caspase-12 to cell death and sepsis. Cell Death Differ. (2019) 26:1124–37. doi: 10.1038/s41418-018-0188-2
64. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. (2018) 24:97–108.e4. doi: 10.1016/j.chom.2018.05.009
65. Wang YC, Liu QX, Liu T, Xu XE, Gao W, Bai XJ, et al. Caspase-1-dependent pyroptosis of peripheral blood mononuclear cells predicts the development of sepsis in severe trauma patients: A prospective observational study. Med (Baltimore). (2018) 97:e9859. doi: 10.1097/MD.0000000000009859
66. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
67. Lee JP, Foote A, Fan H, Peral de Castro C, Lang T, Jones SA, et al. Loss of autophagy enhances MIF/macrophage migration inhibitory factor release by macrophages. Autophagy. (2016) 12:907–16. doi: 10.1080/15548627.2016.1164358
68. Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. (2019) 10:782. doi: 10.1038/s41419-019-2015-1
69. Baratin M, Foray C, Demaria O, Habbeddine M, Pollet E, Maurizio J, et al. Homeostatic NF-κB signaling in steady-state migratory dendritic cells regulates immune homeostasis and tolerance. Immunity. (2015) 42:627–39. doi: 10.1016/j.immuni.2015.03.003
70. Dreschler K, Bratke K, Petermann S, Thamm P, Kuepper M, Virchow JC, et al. Altered phenotype of blood dendritic cells in patients with acute pneumonia. Respiration. (2012) 83:209–17. doi: 10.1159/000328406
71. Strother RK, Danahy DB, Kotov DI, Kucaba TA, Zacharias ZR, Griffith TS, et al. Polymicrobial sepsis diminishes dendritic cell numbers and function directly contributing to impaired primary CD8 T cell responses In vivo. J Immunol. (2016) 197:4301–11. doi: 10.4049/jimmunol.1601463
72. Park SJ, Namkoong H, Doh J, Choi JC, Yang BG, Park Y, et al. Negative role of inducible PD-1 on survival of activated dendritic cells. J Leukoc Biol. (2014) 95:621–9. doi: 10.1189/jlb.0813443
73. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. (2017) 549:106–10. doi: 10.1038/nature23669
74. Grimaldi D, Louis S, Pène F, Sirgo G, Rousseau C, Claessens YE, et al. Profound and persistent decrease of circulating dendritic cells is associated with ICU-acquired infection in patients with septic shock. Intensive Care Med. (2011) 37:1438–46. doi: 10.1007/s00134-011-2306-1
75. Fan X, Liu Z, Jin H, Yan J, Liang HP. Alterations of dendritic cells in sepsis: featured role in immunoparalysis. BioMed Res Int 2015. (2015) p:903720. doi: 10.1155/2015/903720
76. Pastille E, Didovic S, Brauckmann D, Rani M, Agrawal H, Schade FU, et al. Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J Immunol. (2011) 186:977–86. doi: 10.4049/jimmunol.1001147
77. Poehlmann H, Schefold JC, Zuckermann-Becker H, Volk HD, Meisel C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: a prospective observational analysis. Crit Care. (2009) 13:R119. doi: 10.1186/cc7969
78. Wu DD, Li T, Ji XY. Dendritic cells in sepsis: pathological alterations and therapeutic implications. J Immunol Res. (2017) 2017:3591248. doi: 10.1155/2017/3591248
79. Loures FV, Röhm M, Lee CK, Santos E, Wang JP, Specht CA, et al. Recognition of Aspergillus fumigatus hyphae by human plasmacytoid dendritic cells is mediated by dectin-2 and results in formation of extracellular traps. PloS Pathog. (2015) 11:e1004643. doi: 10.1371/journal.ppat.1004643
80. Weindel CG, Richey LJ, Mehta AJ, Shah M, Huber BT. Autophagy in dendritic cells and B cells is critical for the inflammatory state of TLR7-mediated autoimmunity. J Immunol. (2017) 198:1081–92. doi: 10.4049/jimmunol.1601307
81. Guo Y, Patil NK, Luan L, Bohannon JK, Sherwood ER. The biology of natural killer cells during sepsis. Immunology. (2018) 153:190–202. doi: 10.1111/imm.2018.153.issue-2
82. Hotchkiss RS, insley KW, Swanson PE, Schmieg RE Jr, Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. (2001) 166:6952–63. doi: 10.4049/jimmunol.166.11.6952
83. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science. (2012) 335:597–601. doi: 10.1126/science.1215173
84. Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science. (2015) 347:1260–5. doi: 10.1126/science.aaa4268
85. Shankar-Hari M, Fear D, Lavender P, Mare T, Beale R, Swanson C, et al. Activation-associated accelerated apoptosis of memory B cells in critically ill patients with sepsis. Crit Care Med. (2017) 45:875–82. doi: 10.1097/CCM.0000000000002380
86. Monserrat J, de Pablo R, Diaz-Martín D, Rodríguez-Zapata M, de la Hera A, Prieto A, et al. Early alterations of B cells in patients with septic shock. Crit Care. (2013) 17:R105. doi: 10.1186/cc12750
87. Vaughan AT, Roghanian A, Cragg MS. B cells–masters of the immunoverse. Int J Biochem Cell Biol. (2011) 43:280–5. doi: 10.1016/j.biocel.2010.12.005
88. Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol. (2012) 12:282–94. doi: 10.1038/nri3190
89. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. (2002) 3:944–50. doi: 10.1038/ni833
90. Suzuki K, Inoue S, Kametani Y, Komori Y, Chiba S, Sato T, et al. Reduced immunocompetent B cells and increased secondary infection in elderly patients with severe sepsis. Shock. (2016) 46:270–8. doi: 10.1097/SHK.0000000000000619
91. Giamarellos-Bourboulis EJ, Apostolidou E, Lada M, Perdios I, Gatselis NK, Tsangaris I, et al. Kinetics of circulating immunoglobulin M in sepsis: relationship with final outcome. Crit Care. (2013) 17:R247. doi: 10.1186/cc13073
92. Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT, et al. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am J Respir Crit Care Med. (2006) 174:1003–10. doi: 10.1164/rccm.200604-546OC
93. Monserrat J, de Pablo R, Reyes E, Díaz D, Barcenilla H, Zapata MR, et al. Clinical relevance of the severe abnormalities of the T cell compartment in septic shock patients. Crit Care. (2009) 13:R26. doi: 10.1186/cc7731
94. Luperto M, Zafrani L. T cell dysregulation in inflammatory diseases in ICU. Intensive Care Med Exp. (2022) 10:43. doi: 10.1186/s40635-022-00471-6
95. Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol. (2005) 174:5110–8. doi: 10.4049/jimmunol.174.8.5110
96. Hotchkiss RS, Moldawer LL. Parallels between cancer and infectious disease. N Engl J Med. (2014) 371:380–3. doi: 10.1056/NEJMcibr1404664
97. Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care. (2011) 15:R99. doi: 10.1186/cc10112
98. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292
99. Lin CW, Lo S, Hsu C, Hsieh CH, Chang YF, Hou BS, et al. T-cell autophagy deficiency increases mortality and suppresses immune responses after sepsis. PloS One. (2014) 9:e102066. doi: 10.1371/journal.pone.0102066
100. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
101. Guinault D, Nicolau-Travers ML, Silva S, Cointault O, Daniau B, Del Bello A, et al. Expression of exhaustion markers on CD8+ T-cell patterns predict outcomes in septic patients admitted to the ICU. Crit Care Med. (2021) 49:1513–23. doi: 10.1097/CCM.0000000000005047
102. Oami T, Watanabe E, Hatano M, Sunahara S, Fujimura L, Sakamoto A, et al. Suppression of T cell autophagy results in decreased viability and function of T cells through accelerated apoptosis in a murine sepsis model. Crit Care Med. (2017) 45:e77–85. doi: 10.1097/CCM.0000000000002016
103. Luo S, Rubinsztein DC. Atg5 and Bcl-2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. (2007) 14:1247–50. doi: 10.1038/sj.cdd.4402149
104. Shao Y, Chen F, Chen Y, Zhang W, Lin Y, Cai Y, et al. Association between genetic polymorphisms in the autophagy-related 5 gene promoter and the risk of sepsis. Sci Rep. (2017) 7:9399. doi: 10.1038/s41598-017-09978-5
105. Mao JY, Zhang JH, Cheng W, Chen JW, Cui N. Effects of neutrophil extracellular traps in patients with septic coagulopathy and their interaction with autophagy. Front Immunol. (2021) 12:757041. doi: 10.3389/fimmu.2021.757041
106. Pu Q, Gan C, Li R, Li Y, Tan S, Li X, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in. J Immunol. (2017) 198:3205–13. doi: 10.4049/jimmunol.1601196
107. Tian Y, Qu S, Alam HB, Williams AM, Wu Z, Deng Q, et al. Peptidylarginine deiminase 2 has potential as both a biomarker and therapeutic target of sepsis. JCI Insight. (2020) 5(20):e138873. doi: 10.1172/jci.insight.138873
108. Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. (2011) 11:330–42. doi: 10.1038/nri2970
109. Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. (2018) 3(5):e98960. doi: 10.1172/jci.insight.98960
110. Venet F, Foray AP, Villars-Méchin A, Malcus C, Poitevin-Later F, Lepape A, et al. IL-7 restores lymphocyte functions in septic patients. J Immunol. (2012) 189:5073–81. doi: 10.4049/jimmunol.1202062
111. Perales MA, Goldberg JD, Yuan J, Koehne G, Lechner L, Papadopoulos EB, et al. Recombinant human interleukin-7 (CYT107) promotes T-cell recovery after allogeneic stem cell transplantation. Blood. (2012) 120:4882–91. doi: 10.1182/blood-2012-06-437236
112. Lévy Y, Sereti I, Tambussi G, Routy JP, Lelièvre JD, Delfraissy JF, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis. (2012) 55:291–300. doi: 10.1093/cid/cis383
113. Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. (2010) 184:1401–9. doi: 10.4049/jimmunol.0902307
114. Yu P, Steel JC, Zhang M, Morris JC, Waldmann TA. Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin Cancer Res. (2010) 16:6019–28. doi: 10.1158/1078-0432.CCR-10-1966
115. Guo J, Liang Y, Xue D, Shen J, Cai Y, Zhu J, et al. Tumor-conditional IL-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res. (2021) 31:1190–8. doi: 10.1038/s41422-021-00543-4
116. Nalos M, Santner-Nanan B, Parnell G, Tang B, McLean AS, Nanan R. Immune effects of interferon gamma in persistent staphylococcal sepsis. Am J Respir Crit Care Med. (2012) 185:110–2. doi: 10.1164/ajrccm.185.1.110
117. Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, et al. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect Dis. (2014) 14:166. doi: 10.1186/1471-2334-14-166
118. Grimaldi D, Pradier O, Hotchkiss RS, Vincent JL, et al. Nivolumab plus interferon-γ in the treatment of intractable mucormycosis. Lancet Infect Dis. (2017) 17:18. doi: 10.1016/S1473-3099(16)30541-2
119. Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, Hoek J, et al. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood. (2000) 96:878–84. doi: 10.1182/blood.V96.3.878
120. Ding Y, Wilkinson A, Idris A, Fancke B, O'Keeffe M, Khalil D, et al. FLT3-ligand treatment of humanized mice results in the generation of large numbers of CD141+ and CD1c+ dendritic cells in vivo. J Immunol. (2014) 192:1982–9. doi: 10.4049/jimmunol.1302391
121. Bohannon J, Cui W, Sherwood E, Toliver-Kinsky T. Dendritic cell modification of neutrophil responses to infection after burn injury. J Immunol. (2010) 185:2847–53. doi: 10.4049/jimmunol.0903619
122. Patil NK, Bohannon JK, Luan L, Guo Y, Fensterheim B, Hernandez A, et al. Flt3 ligand treatment attenuates T cell dysfunction and improves survival in a murine model of burn wound sepsis. Shock. (2017) 47:40–51. doi: 10.1097/SHK.0000000000000688
123. Pfister O, Lorenz V, Oikonomopoulos A, Xu L, Häuselmann SP, Mbah C, et al. FLT3 activation improves post-myocardial infarction remodeling involving a cytoprotective effect on cardiomyocytes. J Am Coll Cardiol. (2014) 63:1011–9. doi: 10.1016/j.jacc.2013.08.1647
124. Hall MW, Knatz NL, Vetterly C, Tomarello S, Wewers MD, Volk HD, et al. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med. (2011) 37:525–32. doi: 10.1007/s00134-010-2088-x
125. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. (2009) 180:640–8. doi: 10.1164/rccm.200903-0363OC
126. Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care. (2011) 15:R58. doi: 10.1186/cc10031
127. Liu M, Zhang X, Chen H, Wang G, Zhang J, Dong P, et al. Serum sPD-L1, upregulated in sepsis, may reflect disease severity and clinical outcomes in septic patients. Scand J Immunol. (2017) 85:66–72. doi: 10.1111/sji.2017.85.issue-1
128. Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. (2017) 47:765–79. doi: 10.1002/eji.201646875
129. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. (2015) 125:3335–7. doi: 10.1172/JCI83871
130. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. (2017) 127:2930–40. doi: 10.1172/JCI91190
131. Wang JF, Li JB, Zhao YJ, Yi WJ, Bian JJ, Wan XJ, et al. Up-regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis-induced immunosuppression: an animal study and a prospective case-control study. Anesthesiology. (2015) 122:852–63. doi: 10.1097/ALN.0000000000000525
132. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. (2010) 14:R220. doi: 10.1186/cc9354
133. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med. (2019) 45:1360–71. doi: 10.1007/s00134-019-05704-z
134. Watanabe E, Nishida O, Kakihana Y, Odani M, Okamura T, Harada T, et al. Pharmacokinetics, pharmacodynamics, and safety of nivolumab in patients with sepsis-induced immunosuppression: A multicenter, open-label phase 1/2 study. Shock. (2020) 53:686–94. doi: 10.1097/SHK.0000000000001443
135. Biron BM, Chung CS, O'Brien XM, Chen Y, Reichner JS, Ayala A. Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. (2017) 9:22–32. doi: 10.1159/000448808
136. Yao H, Cao G, Liu Z, Zhao Y, Yan Z, Wang S, et al. Inhibition of netosis with PAD inhibitor attenuates endotoxin shock induced systemic inflammation. Int J Mol Sci. (2022) 23(21):13264. doi: 10.3390/ijms232113264
137. Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. (2021) 138:2702–13. doi: 10.1182/blood.2021011525
138. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. (2017) 127:2930–40. doi: 10.1172/JCI91190
139. Guo Y, Luan L, Rabacal W, Bohannon JK, Fensterheim BA, Hernandez A, et al. IL-15 superagonist-mediated immunotoxicity: role of NK cells and IFN-gamma. J Immunol. (2015) 195:2353–64. doi: 10.4049/jimmunol.1500300
140. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
Keywords: apoptosis, autophagy, NETosis, pyroptosis, ferroptosis, necroptosis cell death, immunosuppression, sepsis
Citation: Islam MM, Watanabe E, Salma U, Ozaki M, Irahara T, Tanabe S, Katsuki R, Oishi D and Takeyama N (2024) Immunoadjuvant therapy in the regulation of cell death in sepsis: recent advances and future directions. Front. Immunol. 15:1493214. doi: 10.3389/fimmu.2024.1493214
Received: 08 September 2024; Accepted: 18 November 2024;
Published: 10 December 2024.
Edited by:
Pengpeng Zhang, Nanjing Medical University, ChinaReviewed by:
Cong Zhang, The First People’s Hospital of Foshan, ChinaChenglong Zhu, Second Military Medical University, China
Copyright © 2024 Islam, Watanabe, Salma, Ozaki, Irahara, Tanabe, Katsuki, Oishi and Takeyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eizo Watanabe, ZWl6b3dAYWljaGktbWVkLXUuYWMuanA=