 Shama Ahmad
Shama Ahmad Wesam Nasser
Wesam Nasser Aftab Ahmad
Aftab Ahmad- Anesthesiology and Perioperative Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
Airways, alveoli and the pulmonary tissues are the most vulnerable to the external environment including occasional deliberate or accidental exposure to highly toxic chemical gases. However, there are many effective protective mechanisms that maintain the integrity of the pulmonary tissues and preserve lung function. Alveolar macrophages form the first line of defense against any pathogen or chemical/reactant that crosses the airway mucociliary barrier and reaches the alveolar region. Resident alveolar macrophages are activated or circulating monocytes infiltrate the airspace to contribute towards inflammatory or reparative responses. Studies on response of alveolar macrophages to noxious stimuli are rapidly emerging and alveolar macrophage are also being sought as therapeutic target. Here such studies have been reviewed and put together for a better understanding of the role pulmonary macrophages in general and alveolar macrophage in particular play in the pathogenesis of disease caused by chemical induced acute lung injury.
1 Introduction
The respiratory system is at the frontline of constant encounter by environmental components such as non-toxic or toxic environmental gases and particulates. These can include occasional massive exposures to noxious chemical vapors or gases during accidental/occupational spills or deliberately released poisonous chemicals during terroristic attacks or wars. The type of chemical, its concentration and duration of exposure determine the extent of airway damage, edema, activation of immune cells and inflammation that may often lead to respiratory dysfunction and death. Various pulmonary cells including those of airway and alveolar epithelium, interact with and respond to such toxicants via a cascade of events that include sloughing of epithelial cells, loss of alveolar capillary membrane integrity, and subsequent activation and infiltration of immune cells. Human lungs were originally thought to be composed of about 40 different types of cells (1–4). Emerging technologies like those of single cell sequencing have transformed this information and according to currently published findings, pulmonary tissues were noted to have about 61-62 cell types which may change their identity with altered respiratory health conditions (Figure 1) (2). Pulmonary macrophages are the most abundant myeloid cells of the immune system which are the primary controllers of both innate and adaptive immunity and attain several phenotypes to respond to a wide variety of stimuli/insults including environmental pollutants, pathogens (microbes such as fungi, bacteria, virus, their products etc.) or other inhaled toxicants/threat agents (5–13).
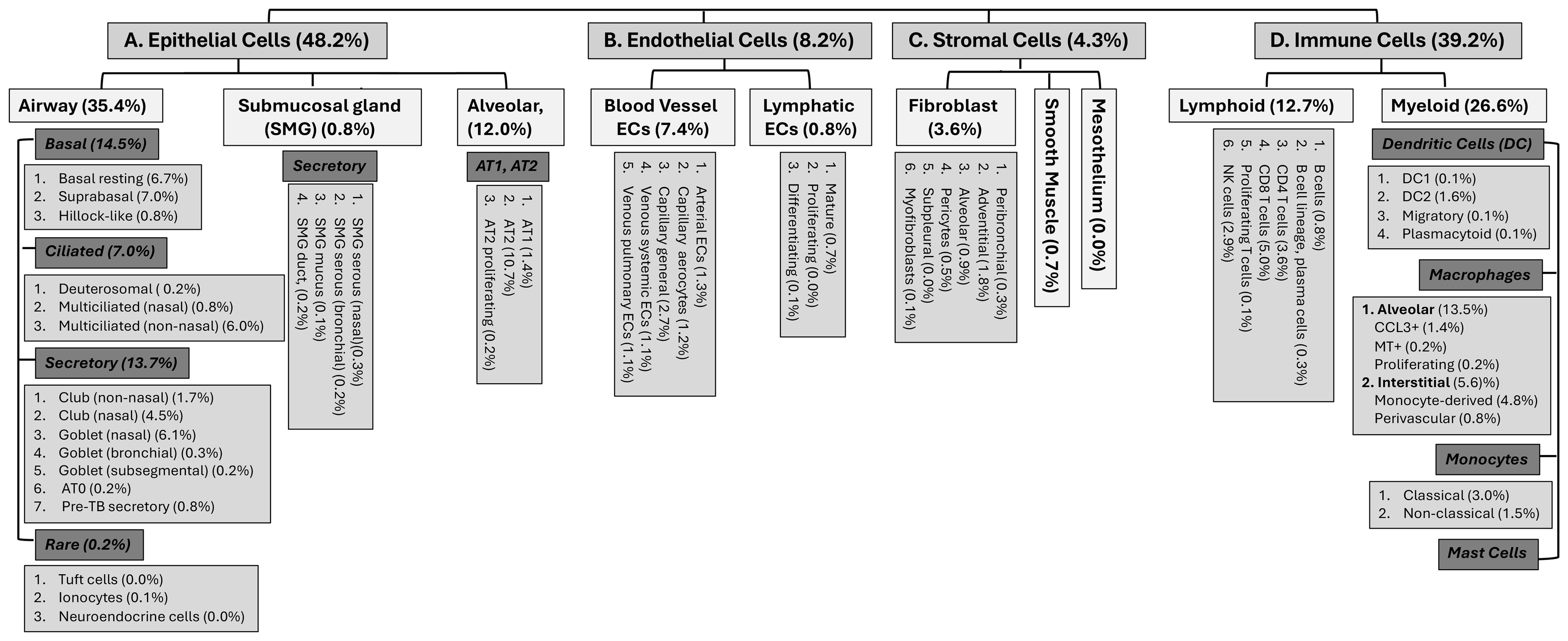
Figure 1. Different cell types and sub-types and their potential distributions in the human pulmonary tissues (2, 3).
Inhaled stimuli such as the toxic vapors or gases induce an immune response which is initiated by both macrophages and the airway epithelium, however the macrophages produce the most potent local proinflammatory response in the lungs which subsequently results in the systemic response (11, 14, 15). Previous studies have shown that the lung microenvironment is important in shaping the distinct transcriptional and epigenetic landscape of the cellular identity and function of resident macrophages (16, 17). Macrophages are the first kind of immune cells that appear during embryogenesis and are essential for early stages of organism development (18). Two types of macrophages populate the lung the a) alveolar macrophage and b) the interstitial macrophage. Alveolar macrophages (AMs) and interstitial macrophages (IMs) reside in different anatomical compartments in the lungs. IMs are monocyte derived uniform cell population that reside in the interstitium (the space between alveolar and vascular endothelium) and are often found to be associated with the airways, nerves and vessels (Figure 1). They are less well studied and have been implicated in maintenance of lung homeostasis and in prevention of immune-mediated allergic airway inflammation (19). Macrophages can sense the danger signals from their microenvironment via specific receptors such as the pattern recognition receptors. Both AMs and IMs are activated by various stimuli to polarize and form activated (M1 or M2) macrophages that determine the progression of acute lung injury (20). The nomenclature of such activated macrophage subsets is dynamic and has seen many revisions and now considered too complex with overlapping characteristics, to be defined by these two terms (21). In this review we will cover the mechanisms by which toxic inhaled chemicals and gases affect the alveolar macrophages.
1.1 Alveolar macrophages
Alveolar macrophages (AMs) have been shown via lineage tracing studies to originate from the embryonic precursors that populate the alveolar space soon after birth (22). They are the major macrophage population found attached on the epithelial surface, where they not only replicate and self-maintain but also protect the gas exchange function and barrier immunity of the lung. The high self-replicating ability, development, maintenance and function of AMs was shown to be dependent on the neutrophil derived 12-HETE and type II alveolar epithelial cell derived GM-CSF (22, 23). Once damaged or depleted, AMs can be restored by circulating monocytes that are often referred to as monocyte derived alveolar macrophages to distinguish them from the regular tissue resident AMs (9).
1.1.1 Alveolar macrophage identifying markers
Macrophage phenotypes are identified by a combination of multiple markers that enable understanding of their heterogeneity and plasticity in reference to their microenvironment (21, 24, 25). The high degree of diversity observed in the pulmonary resident macrophages (AMs and IMs) is due to their different activation states and contribution of infiltrating monocytes to their populations (Table 1) (25, 62). At steady state alveolar macrophages are maintained by epithelial GM-CSF and TGF β (63). When injury occurs, monocytes are recruited to the alveolar lumen and interstitium and develop into the activated monocyte derived macrophages that may further contribute to damage by secreting cytokines or perform reparative function. Alveolar macrophages have been shown to interact with the monocyte derived macrophages and other lymphoid cells to modulate their function (64).
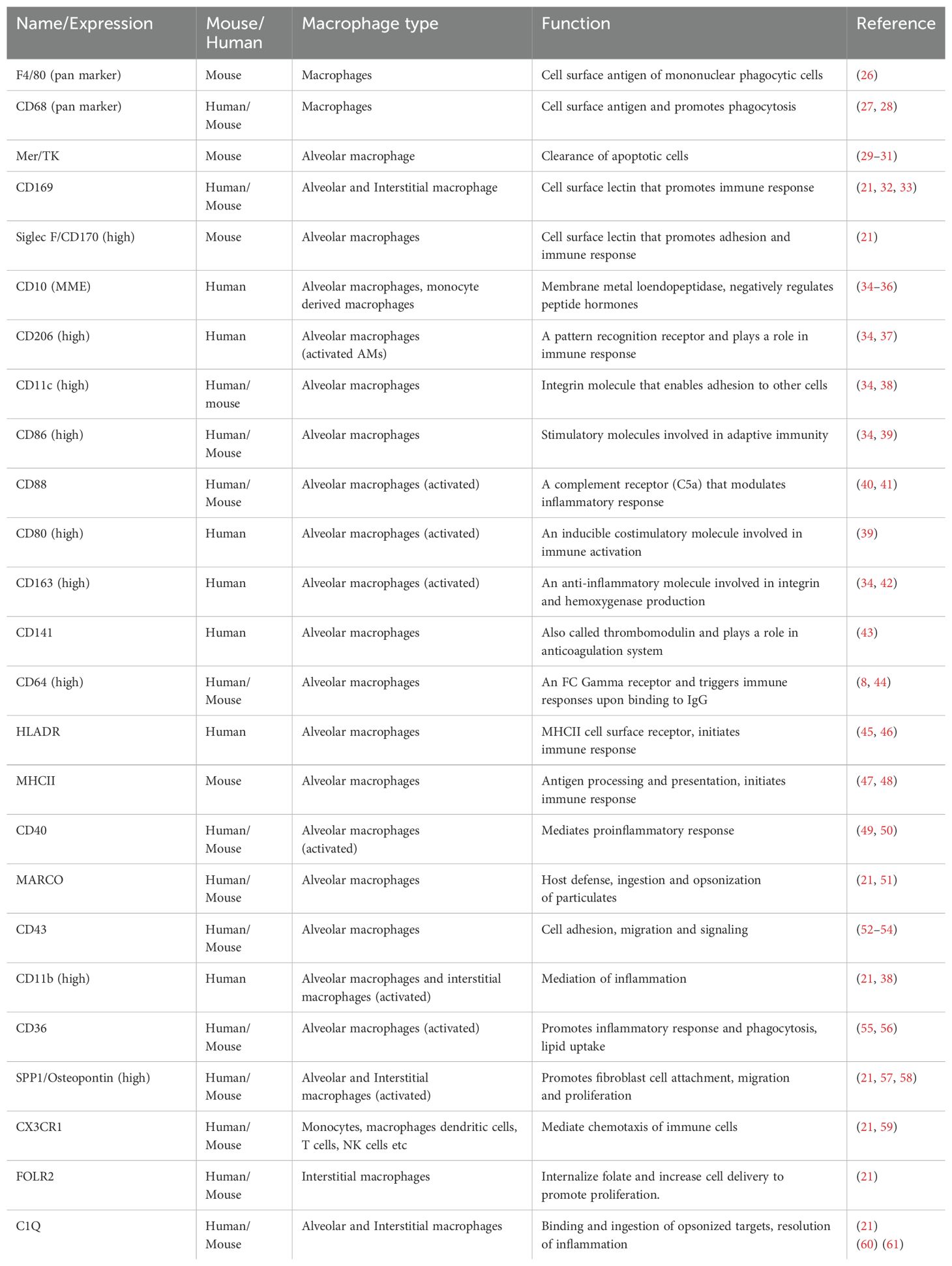
Table 1. Common pulmonary (alveolar) macrophage markers utilized for their identification.
Many cluster of differentiation (CD) markers or cell surface molecules on the macrophages enable their identification within the pulmonary tissues of humans and mice (Table 1). Alveolar macrophages under normal conditions can be identified by the presence of CD11c and interstitial macrophages by CD11b on their surface (21). CD11c, CD169 and MARCO are expressed by the alveolar macrophages of both mice and humans (24, 65). Additional species-specific (human and mice) markers are utilized to identify the alveolar macrophages (Table 1). Although, many different (common or unique) macrophage markers have been identified in rodent and human tissues very little information is found for other mammals (66). In alveolar macrophage samples from healthy humans of different geographical areas, high expression of CD64, CD80, CD86, CD163 and CD206 was observed although normally these markers are associated with polarized M1/M2 macrophages (67). Some of these markers (CD206) are altered by the lung microenvironment such as surfactant protein content (68). On the other hand, some markers (e.g. MARCO) may act as target of therapy for alleviating pulmonary fibrosis (69). In general, the resident alveolar macrophages are characterized by F4/80+, CD64+, MerTK+, SiglecFhi, CD11chi, CD11blo and CD206hi markers. The monocyte derived alveolar macrophages demonstrate F4/80+, CD64+, MerTK+, SiglecF-, CD11c-, CD11bhi and CD206lo markers and is generally proinflammatory and involved with pathogen phagocytosis and cytokine production. There are two kinds of interstitial macrophage populations a) MHCIIhi Lyvelo with F4/80+, CD64+, MerTK+, SiglecF-, CD11c-, CD11bhi, CD206lo and CX3CR1hi markers that represents the antigen presenting and proinflammatory macrophage population and b) MHCIIlo Lyvehi with F4/80+, CD64+, MerTK+, SiglecF-, CD11c-, CD11bhi, CD206hi and CX3CR1lo markers, which represents the wound healing and tissue repair population (62). However, antibody specificity, origin and detection method can greatly affect the identity of the cells (70, 71).
1.1.2 Alveolar macrophages and inhaled chemical-induced acute lung injury
Alveolar macrophages are one of the first cells to encounter the inhaled chemicals. As mentioned earlier toxic vapors or gas initiate responses by both macrophages and the airway epithelial cells, however the macrophages produce the most potent local proinflammatory response in the lungs which subsequently results in the systemic response (11, 14, 15). Toxic chemicals and environmental agents may not only be affecting the macrophage function but may often lead to their decreased clearance and subsequent accumulation to cause long term consequences (72). Adverse effects of inhaled pharmaceutical agents delivered for therapy are not covered here but can be read elsewhere (73, 74). Inhaled anesthetics also modulate the macrophage function and may alter pulmonary disease outcomes (75–78). Similarly, AMs contribute to the inflammatory response induced by cigarette smoke and environmental pollutants (14, 79–81). Common toxic environmental pollutants and gases like sulfur dioxide damage and reduce AM function (82–88). Immune cell development and their responses are highly susceptible to environmental factors.
Many highly toxic chemicals such as sulfur mustard, chlorine gas, ammonia, and phosgene exert their harmful effects through direct toxicity to the pulmonary epithelium and endothelium leading to cell death, loss of barrier function, and increased permeability (89, 90). Reactive oxygen species (ROS) generated either directly by the toxic agent or because of cellular injury also play a significant role in the pathophysiology of chemical-induced lung damage (91). The imbalance between ROS and the antioxidant defenses leads to oxidative stress damaging cellular proteins, lipids, and DNA (92). Chemical exposure also triggers a robust inflammatory response, characterized by the recruitment of immune cells, including macrophages and neutrophils, to the lung (93). While initially aimed at clearing the injury, persistent inflammation can lead to further tissue damage and fibrosis (94). Macrophages play an essential role as critical regulators in the early response to chemical injury, repair, or progression towards fibrosis. When exposed to noxious chemicals, macrophages are among the first responders, releasing pro-inflammatory cytokines and chemokines that mediate the acute inflammatory phase (95). However, dysregulated activation of macrophages can further exacerbate tissue damage and lead to fibrosis through the release of various profibrotic factors (96).
Alveolar macrophages are strategically situated at the tissue-air interface where they play important role in regulating the pulmonary immune response (97). This has been demonstrated by several studies where depletion of alveolar macrophages caused increased immune response to particulate antigens (98), reduced neutrophil influx (99), decreased clearance of antigens (100) and increased lung injury (101). Depletion of macrophages on the other hand facilitated efficient absorption of therapeutic macromolecules (102) and reduced inflammatory response after acid and radiation-induced lung injury in experimental models (103, 104). Currently, studies to clarify the role of monocyte derived circulating macrophages and the resident alveolar macrophages, where monocytes/macrophages were depleted from circulation demonstrated protection from adverse lung conditions resulting from inhaled endotoxic exposures (105). Thus, AMs are the primary immune cells of the lung at steady state where their function is to dampen inflammatory response, but this immune balance can be easily disturbed by perturbations in surrounding microenvironment. In the lungs, the AMs are constantly bathed by the surfactant containing airway lining fluid layer. Inhaled noxious substances can also damage the airway lining fluid and destroy or inhibit the factors that promote macrophage function (106). The outcome of tissue responses to such toxic agents depends on the balance of the mediators produced by the normal quiescent alveolar macrophages and those from the activated alveolar macrophages exposed to the toxic compounds (107).
1.1.2.1 Inhaled halogens and phosgene
Elemental halogens such as chlorine (Cl2) and bromine (Br2) and phosgene (COCl2) are common toxic industrial chemicals and have been used historically in warfare and currently in various armed conflicts (108–111). They are highly reactive oxidizing and corrosive agents and cause acute lung injury (ALI), acute respiratory distress syndrome (ARDS) and even cardiac damage to the exposed individuals when present in high dose and prolonged duration (109, 112–122). One of the earliest case report of bronchial brushings from patients exposed accidently to chlorine demonstrated increased presence of non-pigmented alveolar macrophages 5 days after exposure (123). A dose dependent increase in bronchoalveolar lavage fluid alveolar (BALF) macrophage content was observed at 24h post chlorine exposure in mice (124). Increased inducible nitric oxide and 3-nitrotyrosine content was detected in the macrophages by these authors. Chlorine exposure causes TRPV activation which can result in formation of ROS and NO leading to peroxynitrite formation and tyrosine nitrosylation of proteins (125–127). In another study using relatively lower concentrations of chlorine the macrophage content was unaltered in the BALF up to 48h post exposure (128). These investigators did observe increase in the genes (e.g. Arg1) related to alternative macrophage activation. Damage to the airway lining fluid as demonstrated by loss of surface-active function by chlorine exposure was also demonstrated in this study (128). Chlorine exposure did not alter the number of resident macrophages and anti-inflammatory macrophages in the BALF collected 24h after exposure (129). However, the number of COX-2 or iNOS expressing proinflammatory macrophages were increased in this study. With repeated chlorine exposure a pulmonary adaptation to oxidative stress was observed which could be related to a specific alveolar macrophage population which was dependent on TGF-β and prostaglandin E2 (130). Hemeoxygenase dependent increase in pulmonary macrophages 24h after bromine exposure were recently reported (131–133). Phosgene exposure in mice reduced the BALF macrophage content when evaluated 24h after exposure (134). This could be related to the interaction of phosgene with surfactant and the role AMs play in endocytosis of dysfunctional surfactant proteins and subsequent efferocytosis of these overloaded AMs (135). Not surprisingly, inhibition of phosgene-induced AM galectin-3 production reduced alveolar epithelial cell death and lung damage (136).
1.1.2.2 Inhaled mustard vesicants
Exposure to vesicants like sulfur mustard (SM) and nitrogen mustard (NM) activate the pulmonary macrophages by ensuing inflammation which furthers the tissue damage by production of ROS and proinflammatory mediators (137–140). Dermally applied vesicants also cause increased macrophage content in the lungs of mice after exposure (141, 142). A nonsignificant decrease in BALF macrophage content after exposure to half mustard (2-chloroethyl ethyl sulfide, CEES) was reported in rats 18h after exposure (143). Inhaled nitrogen mustard caused increased inflammatory cytokine (TNFα) production and infiltration of CD11b+ macrophages in the lungs of exposed mice 3 days after exposure (144). The resident alveolar macrophages decreased upon NM treatment and were replaced by the infiltrating proinflammatory CD11b+ macrophages of M1 phenotype that matured later into profibrotic M2 macrophages suggesting a role of alveolar macrophages in the pathogenesis of NM injury (145). Treatment with anti-TNFα antibody not only reduced the macrophage infiltration in the lungs but also reduced injury, inflammation and subsequent fibrosis in this model and in similar SM model (144, 146, 147). Macrophage derived TNFα has been described as a major pathway in vesicant induced lung injury (148, 149). NM induced macrophage activation and lung injury could also be mitigated by antioxidants like N-acetyl cysteine (NAC) and surfactant protein D administration (137, 150). NAC was also protective against CEES induced lung injury in a guinea pig model (151). A role of histones, miRNA and histone acetylase and deacetylase in the phenotype switching of the alveolar macrophages was demonstrated in this model (152, 153). Transcriptional profiling of the early inflammatory phase and later profibrotic/resolution phase of alveolar macrophages from NM treated animals identified cytokine genes involved in cell migration and significant enrichment of canonical pathways related to STAT3 and NFκB signaling (154). Farnesoid X receptor (FXR) that regulates lipid homeostasis and inflammation was shown to limit alveolar macrophage inflammatory response in a mouse model of IT administered NM (155). Many studies with cutaneous or inhaled vesicant demonstrate increased HMGB1 in the BALF (121, 141, 156–159). HMGB1 causes increase in IL6 and TNF-α that switch the macrophage phenotype (160). Macrophage polarization, polarization phenotypes and their intermediates can be potential new targets to reduce inflammatory responses and tissue injury caused by inhaled vesicants and other toxic stimuli (161, 162).
1.1.2.3 Inhaled ammonia and acids
Ammonia is a highly reactive irritant gas and a toxic industrial chemical, which quickly forms caustic ammonium hydroxide on moist surfaces. Exposure to ammonia causes burns on skin and acute respiratory tract injury, pulmonary edema and respiratory failure. The survivors have long term pulmonary complications and develop bronchiectasis, AHR, BO, COPD (163–166). Long term ammonia exposure in occupational settings causes lower airway diseases and ILD which sometimes even need lung transplantation (167). High dose ammonia inhalation can be lethal and the extent of lung injury and damage is a predictor of fatal outcome (168, 169). Intratracheal ammonia administration in animal models causes severe lung injury, respiratory acidosis and alveolar and interstitial damage (170–172). Increased infiltration of pulmonary macrophages was observed on day 7 after exposure, although neutrophils and inflammatory mediators were significantly increased at day 1 postexposure (171). Others also reported increased inflammatory cells in animals exposed to various concentration of inhaled ammonia (172, 173). Sensitivity of various mice strains to pulmonary toxicity by ammonia has also been demonstrated and many candidate genes were identified that determined the susceptibility to ammonia (174). Exposure to ammonia causes oxidative stress and increased mRNA levels of glutathione peroxidase, COX-2, iNOS, TNF-α and TGF-β (175–177). Whether the TNF-α is macrophage derived in this model is unknown but genes related to macrophage infiltration were increased upon ammonia exposure in a swine model (176).
Exposure to acids such as sulfuric acid may alter the clearance from the alveolar region by affecting the alveolar macrophage function (106, 178). HCl administration in mice causes both acute and chronic lung injury and HSP70 and HSP90 were shown to play regulatory role in causing endothelial barrier disruption and dysfunction (179–186). HCl administration increased BAL macrophages and proinflammatory cytokines like TNF-α, MCP-1 and IL-1β (187, 188). HCl administration into rabbit trachea caused increased neutrophil influx in the lungs and reduced alveolar macrophage adherence function (189). Acid induced lung injury and fibrosis was alleviated by a TLPQ-21 derivative which is an activator of alveolar macrophage function (190). Thus, alveolar macrophages are activated upon acute exposure to toxic chemicals and acids causing tissue damage and inflammation which may eventually lead to chronic effects including fibrosis.
1.1.3 Epigenetic mechanisms driving macrophage functions
Macrophages are pivotal to the innate immune system, especially within the lung environment, where they are primarily responsible for detecting, engulfing, and destroying pathogens through phagocytosis (191). They also secrete various cytokines and chemokines that mediate inflammation and recruit other immune cells to infection sites, crucial for controlling infection spread and initiating tissue repair processes (192). Macrophages display remarkable versatility in their activation; they respond to a variety of cytokines and pathogen-associated signals, which can drastically alter their behavior and function, adapting to the needs of the host defense and repair mechanisms (193). Macrophages are important in initiating immune responses through their role as antigen-presenting cells, which is critical for linking the innate and adaptive sides of the immune system (194).
Phenotypic plasticity results in macrophages that can convert from one functional phenotype to another in response to local microenvironment signals (195). This plasticity allows them to adopt various roles, from pro-inflammatory (activated, M1) phenotypes, which are essential during the initial phases of infection and inflammation, to anti-inflammatory and tissue repairing resident (M2) phenotype, crucial for resolution of inflammation and tissue healing (196). Macrophages switch between these phenotypes under the influence of environmental cues and cytokines, a process that is essential for the balanced immune response required to resolve infections while minimizing tissue damage efficiently (197). The plasticity of macrophages in function is such a key point that significantly assists in understanding their complete involvement in either disease progression or resolution within the lung. The ability of macrophages to switch phenotypes is not only essential in the resolution of different phases of diseases but also provides potential therapeutic targets through manipulating these transitions in diseases such as asthma and COPD (65, 198).
Epigenetic regulations are essential in controlling the functions of cells of innate immunity, including macrophages. Epigenetic changes to genes have the ability to change their expression without altering the sequence, significantly impacting the actions of macrophages and, thus, affecting the overall immune response (199). Genes are generally repressed by DNA methylation, which is crucial for the differentiation and functioning of cells. DNA methylation has been shown to impact the expression of specific cytokines and other proteins in macrophages. Abnormal methylation patterns are related to changes in macrophage activities, which influence the immune response to microbes and injury (200). Histone modifications, such as acetylation and methylation, can act to either promote or inhibit gene expression. Histone acetylation in macrophages is necessary for the transcriptional activation of inflammatory genes after infection (201). At the same time, the expression of these genes can be inhibited by histone deacetylases (HDCAs), enabling dynamic suppression of inflammatory responses. Histone lactylation is another fairly recent concept linking metabolic changes to epigenetic modifications in cells including macrophages (6). Histone lactylation was shown to affect the polarization of macrophages and release of lactylated DAMPS like HMGB1 from such macrophages promoted endothelial permeability and pathogenesis of sepsis (202, 203).
Non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), also play a significant role in regulating gene expression in innate immunity. miRNAs can fine-tune the immune response by targeting mRNA transcripts for degradation or inhibiting their translation. miR-155 enhances the inflammatory response by modulating pathways that affect cytokine production in macrophages (204). lncRNAs contribute to the regulation of immune gene expression by interacting with chromatin-modifying complexes, thereby influencing the epigenetic landscape of immune cells (65, 205). These miRNAs and lncRNAs are often packaged in extracellular vesicles (EVs) released from macrophages and can influence the course of injury or disease process.
1.1.4 Alveolar macrophages and epigenetic mechanisms in inhaled chemical injury
Extracellular vesicles (EVs) are cell derived membranous structures that are shed in the extracellular microenvironment that is a critical component of the epigenetic landscape where inflammatory signaling including those of the inflammatory cells/activated macrophages establish crosstalk with chromatin leading to transcription of inflammatory genes (65, 206). EVs and their cargoes are generated by multiple cell types including the alveolar macrophages after exposure to toxicants (207). The cargoes of EVs and their cargoes generated from exposure to toxic gases can potentially be transferred to other cells to promote their effects. EVs especially those from the macrophages mediate epigenetic pathways that regulate injurious and inflammatory responses of inhaled toxicants (208–211). It was demonstrated that the imbalance of histone acetylase and deacetylase contributes to lung macrophage activation following inhaled nitrogen mustard exposure (152). Additional alterations such as DNA methylation could be contributed by metabolic changes during macrophage activation by inhaled toxicants (212). Additional epigenetic mechanisms may be involved in general lung injury caused by inhaled poisonous gases like sulfur mustard (213).
1.1.5 Alveolar macrophages activating pathways and therapeutic targets for chemical induced ALI
Resident alveolar macrophages are highly influenced by their local pulmonary microenvironment which includes the airspace and the vasculature. Despite their protective roles in normal conditions, once activated during injury they play diverse roles in both initiating and driving inflammatory pathways post chemical exposure in the lung making them the ideal therapeutic target (214). Alveolar macrophages have several attributes that make them attractive and effective therapeutic targets viz; their position at the airway-tissue interface, they mediate early innate immune response, availability of inhalable products designed to target them and their long life and immune training (215, 216). Chlorine exposure increased activated proinflammatory alveolar macrophages that expressed COX-2 and iNOS (129, 217). Treatment with corticosteroids like dexamethasone or budesonide reduced inflammation and fibrosis in the lungs of chlorine exposed mice (217, 218). Chlorine-induced airway hyperreactivity was reversed by inhibition of inducible nitric oxide synthase (iNOS) which was potentially contributed by resident alveolar macrophages (124). Phosgene exposure increased inflammatory cytokines like IL-6 and impaired macrophage function and reduced viral clearance in influenza-infected rats (219, 220). Single cell RNA sequencing studies revealed that macrophages and macrophage proliferating cells were prominent clusters of cells in the BALF of chemical (phosgene) induced acute lung injury in rats (221, 222). Phosgene exposure enhanced galectin 3 expression on alveolar macrophages causing enhanced interaction with alveolar epithelial cells leading to membrane damage and death. Galectin 3 inhibition or elimination of alveolar macrophages protected the alveolar epithelial cells and reduced alveolar damage after phosgene exposure (136). Another important aspect of alveolar macrophage activation is formation of foamy or lipid laden macrophages (LLMs) (223, 224). The formation of LLMs is enhanced in inflammation where lipid accumulation compromises the macrophage function. Dysregulated lipid metabolism most commonly due to oxidative stress during lung injury contributes to LLM formation and can be an important therapeutic target. Accordingly, it was shown that antioxidants like NAC reduced LLM formation (225). Therefore, it is critical to explore the interactions of macrophages and other cells and to understand the mechanisms underlying macrophage phenotype development in order to evaluate therapies for diseases associated with acute lung injury caused by chemical exposures.
2 Discussion
Alveolar macrophages have important roles in host defense against environmental pathogens, particulates and toxic chemicals that enter through the airways into the alveolar space. Macrophages play an essential role as key regulators in the early response to chemical injury, influencing repair, or progression towards fibrosis. When exposed to noxious chemicals, macrophages are among the first responders, releasing pro-inflammatory cytokines and chemokines that mediate the acute inflammatory phase (39). However, dysregulated activation of macrophages can further exacerbate tissue damage and lead to fibrosis through the release of various profibrotic factors (96). DNA methylation, histone modification, and noncoding RNAs are some of the key epigenetic mechanisms that have been demonstrated to have a considerable impact on the macrophage’s response to chemicals. These mechansims along with others will help bridge the gap in understanding that exists in the development of chronic lung diseases resulting from chemical exposure-induced acute lung injury.
Author contributions
SA: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – original draft, Writing – review & editing. WN: Writing – original draft, Writing – review & editing. AA: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding by the CounterACT Program grants, National Institutes of Health Office of the Director (NIH OD), the National Institute of Environmental Health Sciences (NIEHS) Grants U01ES028182 (SA), U01ES033263 (SA), R21ES030525 (SA), R21ES032353 (AA), and R56ES034423 (AA&SA) are gratefully acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AM, alveolar macrophage; IM, interstitial macrophage; GM-CSF, granulocyte macrophage colony stimulating factor; TGF-β, transforming growth factor beta; CD, cluster of differentiation; 12-HETE, hydroxyeicosatetraenoic acid; MARCO, macrophage receptor with collagenous structure.
References
1. Cardoso WV, Whitsett JA. Resident cellular components of the lung: developmental aspects. Proc Am Thorac Soc. (2008) 5:767–71. doi: 10.1513/pats.200803-026HR
2. Sikkema L, Ramirez-Suastegui C, Strobl DC, Gillett TE, Zappia L, Madissoon E, et al. An integrated cell atlas of the lung in health and disease. Nat Med. (2023) 29:1563–77. doi: 10.1038/s41591-023-02327-2
3. Ahmad S, Ahmad A. Epithelial regeneration and lung stem cells. In: Koval M, Sidhaye VK, editors. Lung epithelial biology in the pathogenesis of pulmonary disease. Academic Press, Boston, MA (2017).
4. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. (2020) 587:619–25. doi: 10.1038/s41586-020-2922-4
5. Lohmann-Matthes ML, Steinmüller C, Franke-Ullmann G. Pulmonary macrophages. Eur Respir J. (1994) 7:1678–89. doi: 10.1183/09031936.94.07091678
6. Wei Y, Guo H, Chen S, Tang XX. Regulation of macrophage activation by lactylation in lung disease. Front Immunol. (2024) 15:1427739. doi: 10.3389/fimmu.2024.1427739
7. Hetzel M, Ackermann M, Lachmann N. Beyond "Big eaters": the versatile role of alveolar macrophages in health and disease. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22073308
8. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology. (2020) 160:126–38. doi: 10.1111/imm.v160.2
9. Mass E, Nimmerjahn F, Kierdorf K, Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol. (2023) 23:563–79. doi: 10.1038/s41577-023-00848-y
10. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
11. Tapak M, Sadeghi S, Ghazanfari T, Mosaffa N. Chemical exposure and alveolar macrophages responses: the role of pulmonary defense mechanism in inhalation injuries. BMJ Open Respir Res. (2023) 10. doi: 10.1136/bmjresp-2022-001589
12. Laskin DL, Malaviya R, Laskin JD. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol Sci. (2019) 168:287–301. doi: 10.1093/toxsci/kfy309
13. Kulle A, Thanabalasuriar A, Cohen TS, Szydlowska M. Resident macrophages of the lung and liver: The guardians of our tissues. Front Immunol. (2022) 13:1029085. doi: 10.3389/fimmu.2022.1029085
14. Hiraiwa K, van Eeden SF. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediators Inflammation. (2013) 2013:619523. doi: 10.1155/2013/619523
15. Hogg JC, van Eeden S. Pulmonary and systemic response to atmospheric pollution. Respirology. (2009) 14:336–46. doi: 10.1111/j.1440-1843.2009.01497.x
16. Hey J, Paulsen M, Toth R, Weichenhan D, Butz S, Schatterny J, et al. Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat Commun. (2021) 12:6520. doi: 10.1038/s41467-021-26777-9
17. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
18. Chitu V, Stanley ER. Regulation of embryonic and postnatal development by the CSF-1 receptor. Curr Top Dev Biol. (2017) 123:229–75. doi: 10.1016/bs.ctdb.2016.10.004
19. Schyns J, Bureau F, Marichal T. Lung interstitial macrophages: past, present, and future. J Immunol Res. (2018) 2018:5160794. doi: 10.1155/2018/5160794
20. Guan T, Zhou X, Zhou W, Lin H. Regulatory T cell and macrophage crosstalk in acute lung injury: future perspectives. Cell Death Discovery. (2023) 9:9. doi: 10.1038/s41420-023-01310-7
21. Bain CC, MacDonald AS. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol. (2022) 15:223–34. doi: 10.1038/s41385-021-00480-w
22. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
23. Pernet E, Sun S, Sarden N, Gona S, Nguyen A, Khan N, et al. Neonatal imprinting of alveolar macrophages via neutrophil-derived 12-HETE. Nature. (2023) 614:530–38. doi: 10.1038/s41586-022-05660-7
24. Wei Q, Deng Y, Yang Q, Zhan A, Wang L. The markers to delineate different phenotypes of macrophages related to metabolic disorders. Front Immunol. (2023) 14:1084636. doi: 10.3389/fimmu.2023.1084636
25. Li X, Mara AB, Musial SC, Kolling FW, Gibbings SL, Gerebtsov N, et al. Coordinated chemokine expression defines macrophage subsets across tissues. Nat Immunol. (2024) 25:1110–22. doi: 10.1038/s41590-024-01826-9
26. Nibbering PH, Leijh PC, van Furth R. Quantitative immunocytochemical characterization of mononuclear phagocytes. I. Monoblasts, promonocytes, monocytes, and peritoneal and alveolar macrophages. Cell Immunol. (1987) 105:374–85. doi: 10.1016/0008-8749(87)90085-2
27. Gordon S, Lawson L, Rabinowitz S, Crocker PR, Morris L, Perry VH. Antigen markers of macrophage differentiation in murine tissues. Curr Top Microbiol Immunol. (1992) 181:1–37.
28. Pulford KA, Sipos A, Cordell JL, Stross WP, Mason DY. Distribution of the CD68 macrophage/myeloid associated antigen. Int Immunol. (1990) 2:973–80. doi: 10.1093/intimm/2.10.973
29. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol. (2016) 38:409–23. doi: 10.1007/s00281-016-0555-3
30. Haczku A. Cell-corpse clearance after lung damage: the essential role of merTK-mediated alveolar macrophage efferocytosis. Am J Respir Cell Mol Biol. (2024) 70:433–34. doi: 10.1165/rcmb.2024-0108ED
31. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. (2001) 411:207–11. doi: 10.1038/35075603
32. Ural BB, Yeung ST, Damani-Yokota P, Devlin JC, de Vries M, Vera-Licona P, et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.aax8756
33. Liu Y, Xia Y, Qiu CH. Functions of CD169 positive macrophages in human diseases (Review). BioMed Rep. (2021) 14:26. doi: 10.3892/br.2020.1402
34. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. (2022) 55:1564–80. doi: 10.1016/j.immuni.2022.08.010
35. Huang X, He C, Lin G, Lu L, Xing K, Hua X, et al. Induced CD10 expression during monocyte-to-macrophage differentiation identifies a unique subset of macrophages in pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun. (2020) 524:1064–71. doi: 10.1016/j.bbrc.2020.02.042
36. Wang S, Xiao Y, An X, Luo L, Gong K, Yu D. A comprehensive review of the literature on CD10: its function, clinical application, and prospects. Front Pharmacol. (2024) 15:1336310. doi: 10.3389/fphar.2024.1336310
37. Kim GD, Lim EY, Shin HS. Macrophage polarization and functions in pathogenesis of chronic obstructive pulmonary disease. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25115631
38. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. (2013) 49:503–10. doi: 10.1165/rcmb.2013-0086MA
39. Camp B, Jorde I, Sittel F, Pausder A, Jeron A, Bruder D, et al. Comprehensive analysis of lung macrophages and dendritic cells in two murine models of allergic airway inflammation reveals model- and subset-specific accumulation and phenotypic alterations. Front Immunol. (2024) 15:1374670. doi: 10.3389/fimmu.2024.1374670
40. Leach SM, Gibbings SL, Tewari AD, Atif SM, Vestal B, Danhorn T, et al. Human and mouse transcriptome profiling identifies cross-species homology in pulmonary and lymph node mononuclear phagocytes. Cell Rep. (2020) 33:108337. doi: 10.1016/j.celrep.2020.108337
41. Crane JW, Baiquni GP, Sullivan RKP, Lee JD, Sah P, Taylor SM, et al. The C5a anaphylatoxin receptor CD88 is expressed in presynaptic terminals of hippocampal mossy fibres. J Neuroinflamm. (2009) 6:34. doi: 10.1186/1742-2094-6-34
42. Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal. (2013) 18:2352–63. doi: 10.1089/ars.2012.4834
43. Desch AN, Gibbings SL, Goyal R, Kolde R, Bednarek J, Bruno T, et al. Flow cytometric analysis of mononuclear phagocytes in nondiseased human lung and lung-draining lymph nodes. Am J Respir Crit Care Med. (2016) 193:614–26. doi: 10.1164/rccm.201507-1376OC
44. Akinrinmade OA, Chetty S, Daramola AK, Islam MU, Thepen T, Barth S. CD64: an attractive immunotherapeutic target for M1-type macrophage mediated chronic inflammatory diseases. Biomedicines. (2017) 5. doi: 10.3390/biomedicines5030056
45. Bratke K, Weise M, Stoll P, Virchow JC, Lommatzsch M. Flow cytometry as an alternative to microscopy for the differentiation of BAL fluid leukocytes. Chest. (2024). doi: 10.1016/j.chest.2024.03.037
46. Ikegami H, Yamasaki K, Ogawa M, Nemoto K, Akata K, Noguchi S, et al. Reduced phagocytic activity of human alveolar macrophages infected with Mycobacterium avium complex. J Infect Chemother. (2022) 28:1506–12. doi: 10.1016/j.jiac.2022.07.018
47. Draijer C, Florez-Sampedro L, Reker-Smit C, Post E, van Dijk F, Melgert BN. Explaining the polarized macrophage pool during murine allergic lung inflammation. Front Immunol. (2022) 13:1056477. doi: 10.3389/fimmu.2022.1056477
48. Vo T, Saini Y. Case report: Mafb promoter activity may define the alveolar macrophage dichotomy. Front Immunol. (2022) 13:1050494. doi: 10.3389/fimmu.2022.1050494
49. Liu P-S, Chen Y-T, Li X, Hsueh P-C, Tzeng S-F, Chen H, et al. CD40 signal rewires fatty acid and glutamine metabolism for stimulating macrophage anti-tumorigenic functions. Nat Immunol. (2023) 24:452–62. doi: 10.1038/s41590-023-01430-3
50. Dong L, Wang S, Chen M, Li H, Bi W. The activation of macrophage and upregulation of CD40 costimulatory molecule in lipopolysaccharide-induced acute lung injury. J BioMed Biotechnol. (2008) 2008:852571. doi: 10.1155/bmri.v2008.1
51. Kanno S, Hirano S, Sakamoto T, Furuyama A, Takase H, Kato H, et al. Scavenger receptor MARCO contributes to cellular internalization of exosomes by dynamin-dependent endocytosis and macropinocytosis. Sci Rep. (2020) 10:21795. doi: 10.1038/s41598-020-78464-2
52. Randhawa AK, Ziltener HJ, Merzaban JS, Stokes RW. CD43 is required for optimal growth inhibition of Mycobacterium tuberculosis in macrophages and in mice. J Immunol. (2005) 175:1805–12. doi: 10.4049/jimmunol.175.3.1805
53. Venosa A, Malaviya R, Gow AJ, Hall L, Laskin JD, Laskin DL. Protective role of spleen-derived macrophages in lung inflammation, injury, and fibrosis induced by nitrogen mustard. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L1487–98. doi: 10.1152/ajplung.00276.2015
54. Hume PS, Gibbings SL, Jakubzick CV, Tuder RM, Curran-Everett D, Henson PM, et al. Localization of macrophages in the human lung via design-based stereology. Am J Respir Crit Care Med. (2020) 201:1209–17. doi: 10.1164/rccm.201911-2105OC
55. Smith LC, Gow AJ, Abramova E, Vayas K, Guo C, Noto J, et al. Role of PPARgamma in dyslipidemia and altered pulmonary functioning in mice following ozone exposure. Toxicol Sci. (2023) 194:109–19. doi: 10.1093/toxsci/kfad048
56. Sun S, Yao Y, Huang C, Xu H, Zhao Y, Wang Y, et al. CD36 regulates LPS-induced acute lung injury by promoting macrophages M1 polarization. Cell Immunol. (2022) 372:104475. doi: 10.1016/j.cellimm.2021.104475
57. Moos PJ, Cheminant JR, Cowman S, Noll J, Wang Q, Musci T, et al. Spatial and phenotypic heterogeneity of resident and monocyte-derived macrophages during inflammatory exacerbations leading to pulmonary fibrosis. Front Immunol. (2024) 15:1425466. doi: 10.3389/fimmu.2024.1425466
58. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. (2019) 54. doi: 10.1183/13993003.02441-2018
59. Lee M, Lee Y, Song J, Lee J, Chang SY. Tissue-specific role of CX(3)CR1 expressing immune cells and their relationships with human disease. Immune Netw. (2018) 18:e5. doi: 10.4110/in.2018.18.e5
60. Ogawa T, Shichino S, Ueha S, Bando K, Matsushima K. Profibrotic properties of C1q+ interstitial macrophages in silica-induced pulmonary fibrosis in mice. Biochem Biophys Res Commun. (2022) 599:113–19. doi: 10.1016/j.bbrc.2022.02.037
61. Xu-Vanpala S, Deerhake ME, Wheaton JD, Parker ME, Juvvadi PR, MacIver N, et al. Functional heterogeneity of alveolar macrophage population based on expression of CXCL2. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.aba7350
62. Hou F, Xiao K, Tang L, Xie L. Diversity of macrophages in lung homeostasis and diseases. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.753940
63. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. (2014) 506:503–6. doi: 10.1038/nature12902
64. Liu X, Boyer MA, Holmgren AM, Shin S. Legionella-infected macrophages engage the alveolar epithelium to metabolically reprogram myeloid cells and promote antibacterial inflammation. Cell Host Microbe. (2020) 28:683–98.e6. doi: 10.1016/j.chom.2020.07.019
65. Ahmad S, Zhang XL, Ahmad A. Epigenetic regulation of pulmonary inflammation. Semin Cell Dev Biol. (2024) 154:346–54. doi: 10.1016/j.semcdb.2023.05.003
66. Nikovics K, Favier AL. Macrophage identification in situ. Biomedicines. (2021) 9. doi: 10.3390/biomedicines9101393
67. Mitsi E, Kamngona R, Rylance J, Solórzano C, Reiné JJ, Mwandumba HC, et al. Human alveolar macrophages predominately express combined classical M1 and M2 surface markers in steady state. Respir Res. (2018) 19:66. doi: 10.1186/s12931-018-0777-0
68. Beharka AA, Gaynor CD, Kang BK, Voelker DR, McCormack FX, Schlesinger LS. Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J Immunol. (2002) 169:3565–73. doi: 10.4049/jimmunol.169.7.3565
69. Chang M, Li N, Zhou Q, Yan Y, Xu W, Zhao Y, et al. The inhibition of MARCO by PolyG alleviates pulmonary fibrosis via regulating mitochondrial function in a silicotic rat model. Environ Toxicol. (2024) 39:3808–19. doi: 10.1002/tox.24241
70. Cummings RD. The mannose receptor ligands and the macrophage glycome. Curr Opin Struct Biol. (2022) 75:102394. doi: 10.1016/j.sbi.2022.102394
71. Sicherre E, Favier AL, Riccobono D, Nikovics K. Non-specific binding, a limitation of the immunofluorescence method to study macrophages in situ. Genes (Basel). (2021) 12. doi: 10.3390/genes12050649
72. Bowden DH, Loosli CG. The alveolar macrophage and its role in toxicology. CRC Crit Rev Toxicol. (1973) 2:95–124. doi: 10.1080/10408447309163832
73. Hoffman E, Urbano L, Martin A, Mahendran R, Patel A, Murnane D, et al. Profiling alveolar macrophage responses to inhaled compounds using in vitro high content image analysis. Toxicol Appl Pharmacol. (2023) 474. doi: 10.1016/j.taap.2023.116608
74. Nikula KJ, McCartney JE, McGovern T, Miller GK, Odin M, Pino MV, et al. STP position paper: interpreting the significance of increased alveolar macrophages in rodents following inhalation of pharmaceutical materials. Toxicologic Pathol. (2013) 42:472–86. doi: 10.1177/0192623313507003
75. Hoidal JR, White JG, Repine JE. Impairment of human alveolar macrophage oxygen consumption, and superoxide anion production by local anesthetics used in bronchoscopy. Chest. (1979) 75:243–6. doi: 10.1378/chest.75.2.243
76. Kotani N, Hashimoto H, Sessler DI, Kikuchi A, Suzuki A, Takahashi S, et al. Intraoperative modulation of alveolar macrophage function during isoflurane and propofol anesthesia. Anesthesiology. (1998) 89:1125–32. doi: 10.1097/00000542-199811000-00012
77. Oliveira TB, Braga CL, Battaglini D, Pelosi P, Rocco PRM, Silva PL, et al. Comparison between sevoflurane and propofol on immunomodulation in an in vitro model of sepsis. Front Med (Lausanne). (2023) 10:1225179. doi: 10.3389/fmed.2023.1225179
78. Gerber TJ, Fehr VCO, Oliveira SDS, Hu G, Dull R, Bonini MG, et al. Sevoflurane promotes bactericidal properties of macrophages through enhanced inducible nitric oxide synthase expression in male mice. Anesthesiology. (2019) 131:1301–15. doi: 10.1097/ALN.0000000000002992
79. Wohnhaas CT, Bassler K, Watson CK, Shen Y, Leparc GG, Tilp C, et al. Monocyte-derived alveolar macrophages are key drivers of smoke-induced lung inflammation and tissue remodeling. Front Immunol. (2024) 15:1325090. doi: 10.3389/fimmu.2024.1325090
80. Li CH, Tsai ML, Chiou HC, Lin YC, Liao WT, Hung CH. Role of macrophages in air pollution exposure related asthma. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232012337
81. Barbosa-de-Oliveira MC, Oliveira-Melo P, Goncalves da Silva MH, Santos da Silva F, Andrade Carvalho da Silva F, Silva de Araujo BV, et al. Modulation of alveolar macrophage activity by eugenol attenuates cigarette-smoke-induced acute lung injury in mice. Antioxidants (Basel). (2023) 12. doi: 10.3390/antiox12061258
82. Costa-Beber LC, Guma F. The macrophage senescence hypothesis: the role of poor heat shock response in pulmonary inflammation and endothelial dysfunction following chronic exposure to air pollution. Inflammation Res. (2022) 71:1433–48. doi: 10.1007/s00011-022-01647-2
83. Guttenberg MA, Vose AT, Tighe RM. Role of innate immune system in environmental lung diseases. Curr Allergy Asthma Rep. (2021) 21:34. doi: 10.1007/s11882-021-01011-0
84. Lawal AO. Diesel exhaust particles and the induction of macrophage activation and dysfunction. Inflammation. (2018) 41:356–63. doi: 10.1007/s10753-017-0682-6
85. Patial S, Saini Y. Lung macrophages: current understanding of their roles in Ozone-induced lung diseases. Crit Rev Toxicol. (2020) 50:310–23. doi: 10.1080/10408444.2020.1762537
86. Skornik WA, Brain JD. Effect of sulfur dioxide on pulmonary macrophage endocytosis at rest and during exercise. Am Rev Respir Dis. (1990) 142:655–9. doi: 10.1164/ajrccm/142.3.655
87. Knorst MM, Kienast K, Groß S, Fries B, Müller-Quernheim J, Ferlinz R. Chemotactic response of human alveolar macrophages and blood monocytes elicited by exposure to sulfur dioxide. Res Exp Med. (1996) 196:127–35. doi: 10.1007/BF02576834
88. Belchamber KBR, Donnelly LE. Macrophage dysfunction in respiratory disease. Results Probl Cell Differ. (2017) 62:299–313.
89. Winder C. The toxicology of chlorine. Environ Res. (2001) 85:105–14. doi: 10.1006/enrs.2000.4110
90. Ghanei M, Harandi AA. Long term consequences from exposure to sulfur mustard: a review. Inhal Toxicol. (2007) 19:451–6. doi: 10.1080/08958370601174990
91. Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. (2006) 28:219–42. doi: 10.1183/09031936.06.00053805
92. Kirkham P, Rahman I. Oxidative stress in asthma and COPD: antioxidants as a therapeutic strategy. Pharmacol Ther. (2006) 111:476–94. doi: 10.1016/j.pharmthera.2005.10.015
93. Moss M, Guidot DM, Wong-Lambertina M, Ten Hoor T, Perez RL, Brown LA. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med. (2000) 161:414–9. doi: 10.1164/ajrccm.161.2.9905002
94. Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. (2005) 172:417–22. doi: 10.1164/rccm.200501-017PP
95. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: A new therapeutic pathway in fibrosing lung disease? Trends Mol Med. (2016) 22:303–16.
96. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.v214:2
97. Sabatel C, Bureau F. The innate immune brakes of the lung. Front Immunol. (2023) 14:1111298. doi: 10.3389/fimmu.2023.1111298
98. Thepen T, Van Rooijen N, Kraal G. Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J Exp Med. (1989) 170:499–509. doi: 10.1084/jem.170.2.499
99. Hashimoto S, Pittet JF, Hong K, Folkesson H, Bagby G, Kobzik L, et al. Depletion of alveolar macrophages decreases neutrophil chemotaxis to Pseudomonas airspace infections. Am J Physiol. (1996) 270:L819–28. doi: 10.1152/ajplung.1996.270.5.L819
100. Guttenberg MA, Vose AT, Birukova A, Lewars K, Cumming RI, Albright MC, et al. Tissue-resident alveolar macrophages reduce O(3)-induced inflammation via MerTK mediated efferocytosis. bioRxiv. (2023). doi: 10.1101/2023.11.06.565865
101. Nakamura T, Abu-Dahab R, Menger MD, Schäfer U, Vollmar B, Wada H, et al. Depletion of alveolar macrophages by clodronate-liposomes aggravates ischemia-reperfusion injury of the lung. J Heart Lung Transplant. (2005) 24:38–45. doi: 10.1016/j.healun.2003.10.007
102. Lombry C, Edwards DA, Préat V, Vanbever R. Alveolar macrophages are a primary barrier to pulmonary absorption of macromolecules. Am J Physiol Lung Cell Mol Physiol. (2004) 286:L1002–8. doi: 10.1152/ajplung.00260.2003
103. Beck-Schimmer B, Rosenberger DS, Neff SB, Jamnicki M, Suter D, Fuhrer T, et al. Pulmonary aspiration: new therapeutic approaches in the experimental model. Anesthesiology. (2005) 103:556–66. doi: 10.1097/00000542-200509000-00019
104. Song G, Cai F, Liu L, Xu Z, Peng Y, Yang Z, et al. Liposomal sodium clodronate mitigates radiation-induced lung injury through macrophage depletion. Transl Oncol. (2024) 47:102029. doi: 10.1016/j.tranon.2024.102029
105. Schwab AD, Wyatt TA, Moravec G, Thiele GM, Nelson AJ, Gleason A, et al. Targeting transitioning lung monocytes/macrophages as treatment strategies in lung disease related to environmental exposures. Respir Res. (2024) 25:157. doi: 10.1186/s12931-024-02804-3
106. Gardner DE. Alterations in macrophage functions by environmental chemicals. Environ Health Perspect. (1984) 55:343–58. doi: 10.1289/ehp.8455343
107. Laskin DL. Macrophages and inflammatory mediators in chemical toxicity: A battle of forces. Chem Res Toxicol. (2009) 22:1376–85. doi: 10.1021/tx900086v
108. Marzec J, Nadadur S. Countermeasures against pulmonary threat agents. J Pharmacol Exp Ther. (2024) 388:560–67. doi: 10.1124/jpet.123.001822
109. Addis DR, Aggarwal S, Lazrak A, Jilling T, Matalon S. Halogen-induced chemical injury to the mammalian cardiopulmonary systems. Physiol (Bethesda). (2021) 36:272–91. doi: 10.1152/physiol.00004.2021
110. Zellner T, Eyer F. Choking agents and chlorine gas - History, pathophysiology, clinical effects and treatment. Toxicol Lett. (2020) 320:73–9. doi: 10.1016/j.toxlet.2019.12.005
111. Achanta S, Jordt SE. Toxic effects of chlorine gas and potential treatments: a literature review. Toxicol Mech Methods. (2021) 31:244–56. doi: 10.1080/15376516.2019.1669244
112. Ahmad S, Ahmad A, Hendry-Hofer TB, Loader JE, Claycomb WC, Mozziconacci O, et al. Sarcoendoplasmic reticulum Ca(2+) ATPase. A critical target in chlorine inhalation-induced cardiotoxicity. Am J Respir Cell Mol Biol. (2015) 52:492–502. doi: 10.1165/rcmb.2014-0005OC
113. Ahmad S, Ahmad A, Neeves KB, Hendry-Hofer T, Loader JE, White CW, et al. In vitro cell culture model for toxic inhaled chemical testing. J Vis Exp. (2014). doi: 10.3791/51539
114. Ahmad S, Masjoan Juncos JX, Ahmad A, Zaky A, Wei CC, Bradley WE, et al. Bromine inhalation mimics ischemia-reperfusion cardiomyocyte injury and calpain activation in rats. Am J Physiol Heart Circ Physiol. (2019) 316:H212–H23. doi: 10.1152/ajpheart.00652.2017
115. Juncos JXM, Shakil S, Ahmad A, Aishah D, Morgan CJ, DellItalia LJ, et al. Circulating and tissue biomarkers as predictors of bromine gas inhalation. Ann N Y Acad Sci. (2020) 1480:104–15. doi: 10.1111/nyas.v1480.1
116. Masjoan Juncos JX, Nadeem F, Shakil S, El-Husari M, Zafar I, Louch WE, et al. Myocardial SERCA2 protects against cardiac damage and dysfunction caused by inhaled bromine. J Pharmacol Exp Ther. (2024) 390:146–58. doi: 10.1124/jpet.123.002084
117. Masjoan Juncos JX, Shakil S, Ahmad A, Mariappan N, Zafar I, Bradley WE, et al. Sex differences in cardiopulmonary effects of acute bromine exposure. Toxicol Res (Camb). (2021) 10:1064–73. doi: 10.1093/toxres/tfab079
118. Shakil S, Masjoan Juncos JX, Mariappan N, Zafar I, Amudhan A, Amudhan A, et al. Behavioral and neuronal effects of inhaled bromine gas: oxidative brain stem damage. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22126316
119. Zaky A, Ahmad A, DellItalia LJ, Jahromi L, Reisenberg LA, Matalon S, et al. Inhaled matters of the heart. Cardiovasc Regener Med. (2015) 2.
120. Zaky A, Bradley WE, Lazrak A, Zafar I, Doran S, Ahmad A, et al. Chlorine inhalation-induced myocardial depression and failure. Physiol Rep. (2015) 3. doi: 10.14814/phy2.12439
121. Mariappan N, Husain M, Zafar I, Singh V, Smithson KG, Crowe DR, et al. Extracellular nucleic acid scavenging rescues rats from sulfur mustard analog-induced lung injury and mortality. Arch Toxicol. (2020) 94:1321–34. doi: 10.1007/s00204-020-02699-1
122. Zhou T, Song WF, Shang Y, Yao SL, Matalon S. Halogen inhalation-induced lung injury and acute respiratory distress syndrome. Chin Med J (Engl). (2018) 131:1214–19. doi: 10.4103/0366-6999.231515
123. Shroff CP, Khade MV, Srinivasan M. Respiratory cytopathology in chlorine gas toxicity: a study in 28 subjects. Diagn Cytopathol. (1988) 4:28–32. doi: 10.1002/dc.2840040108
124. Martin JG, Campbell HR, Iijima H, Gautrin D, Malo JL, Eidelman DH, et al. Chlorine-induced injury to the airways in mice. Am J Respir Crit Care Med. (2003) 168:568–74. doi: 10.1164/rccm.200201-021OC
125. Morty RE, Kuebler WM. TRPV4: an exciting new target to promote alveolocapillary barrier function. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L817–21. doi: 10.1152/ajplung.00254.2014
126. Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, et al. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L158–72. doi: 10.1152/ajplung.00065.2014
127. Vermillion MS, Saari N, Bray M, Nelson AM, Bullard RL, Rudolph K, et al. Effect of TRPV4 antagonist GSK2798745 on chlorine gas-induced acute lung injury in a swine model. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25073949
128. Massa CB, Scott P, Abramova E, Gardner C, Laskin DL, Gow AJ. Acute chlorine gas exposure produces transient inflammation and a progressive alteration in surfactant composition with accompanying mechanical dysfunction. Toxicol Appl Pharmacol. (2014) 278:53–64. doi: 10.1016/j.taap.2014.02.006
129. Malaviya R, Gardner CR, Rancourt RC, Smith LC, Abramova EV, Vayas KN, et al. Lung injury and oxidative stress induced by inhaled chlorine in mice is associated with proinflammatory activation of macrophages and altered bioenergetics. Toxicol Appl Pharmacol. (2023) 461:116388. doi: 10.1016/j.taap.2023.116388
130. Allard B, Panariti A, Pernet E, Downey J, Ano S, Dembele M, et al. Tolerogenic signaling of alveolar macrophages induces lung adaptation to oxidative injury. J Allergy Clin Immunol. (2019) 144:945–61.e9. doi: 10.1016/j.jaci.2019.07.015
131. Aggarwal S, Lam A, Bolisetty S, Carlisle MA, Traylor A, Agarwal A, et al. Heme attenuation ameliorates irritant gas inhalation-induced acute lung injury. Antioxid Redox Signal. (2016) 24:99–112. doi: 10.1089/ars.2015.6347
132. Ahmad I, Molyvdas A, Jian MY, Zhou T, Traylor AM, Cui H, et al. AICAR decreases acute lung injury by phosphorylating AMPK and upregulating heme oxygenase-1. Eur Respir J. (2021) 58. doi: 10.1183/13993003.03694-2020
133. Addis DR, Lambert JA, Ren C, Doran S, Aggarwal S, Jilling T, et al. Vascular endothelial growth factor-121 administration mitigates halogen inhalation-induced pulmonary injury and fetal growth restriction in pregnant mice. J Am Heart Assoc. (2020) 9:e013238. doi: 10.1161/JAHA.119.013238
134. Aggarwal S, Jilling T, Doran S, Ahmad I, Eagen JE, Gu S, et al. Phosgene inhalation causes hemolysis and acute lung injury. Toxicol Lett. (2019) 312:204–13. doi: 10.1016/j.toxlet.2019.04.019
135. Pauluhn J. Phosgene inhalation toxicity: Update on mechanisms and mechanism-based treatment strategies. Toxicology. (2021) 450:152682. doi: 10.1016/j.tox.2021.152682
136. Memet O, Cao C, Hu H, Dun Y, Bao X, Liu F, et al. Galectin-3 inhibition ameliorates alveolar epithelial cell pyroptosis in phosgene-induced acute lung injury. Int Immunopharmacol. (2024) 132:111965. doi: 10.1016/j.intimp.2024.111965
137. Malaviya R, Meshanni JA, Sunil VR, Venosa A, Guo C, Abramova EV, et al. Role of macrophage bioenergetics in N-acetylcysteine-mediated mitigation of lung injury and oxidative stress induced by nitrogen mustard. Toxicol Appl Pharmacol. (2024) 485:116908. doi: 10.1016/j.taap.2024.116908
138. Malaviya R, Sunil VR, Venosa A, Vayas KN, Businaro R, Heck DE, et al. Macrophages and inflammatory mediators in pulmonary injury induced by mustard vesicants. Ann N Y Acad Sci. (2016) 1374:168–75. doi: 10.1111/nyas.2016.1374.issue-1
139. Sadeghi S, Tapak M, Ghazanfari T, Mosaffa N. A review of Sulfur Mustard-induced pulmonary immunopathology: An Alveolar Macrophage Approach. Toxicol Lett. (2020) 333:115–29. doi: 10.1016/j.toxlet.2020.07.035
140. Gao X, Anderson DR, Brown AW, Lin H, Amnuaysirikul J, Chua AL, et al. Pathological studies on the protective effect of a macrolide antibiotic, roxithromycin, against sulfur mustard inhalation toxicity in a rat model. Toxicologic Pathol. (2011) 39:1056–64. doi: 10.1177/0192623311422079
141. Zafar I, Manzoor S, Mariappan N, Ahmad S, Athar M, Antony V, et al. A murine model of vesicant-induced acute lung injury. J Pharmacol Exp Ther. (2024) 388:568–75. doi: 10.1124/jpet.123.001780
142. Feng Y, Xu Q, Yang Y, Shi W, Meng W, Zhang H, et al. The therapeutic effects of bone marrow-derived mesenchymal stromal cells in the acute lung injury induced by sulfur mustard. Stem Cell Res Ther. (2019) 10:90. doi: 10.1186/s13287-019-1189-x
143. ONeill HC, White CW, Veress LA, Hendry-Hofer TB, Loader JE, Min E, et al. Treatment with the catalytic metalloporphyrin AEOL 10150 reduces inflammation and oxidative stress due to inhalation of the sulfur mustard analog 2-chloroethyl ethyl sulfide. Free Radic Biol Med. (2010) 48:1188–96. doi: 10.1016/j.freeradbiomed.2010.01.039
144. Malaviya R, Sunil VR, Venosa A, Verissimo VL, Cervelli JA, Vayas KN, et al. Attenuation of nitrogen mustard-induced pulmonary injury and fibrosis by anti-tumor necrosis factor-α Antibody. Toxicol Sci. (2015) 148:71–88. doi: 10.1093/toxsci/kfv161
145. Venosa A, Malaviya R, Choi H, Gow AJ, Laskin JD, Laskin DL. Characterization of distinct macrophage subpopulations during nitrogen mustard-induced lung injury and fibrosis. Am J Respir Cell Mol Biol. (2016) 54:436–46. doi: 10.1165/rcmb.2015-0120OC
146. Malaviya R, Bellomo A, Abramova E, Croutch CR, Roseman J, Tuttle R, et al. Pulmonary injury and oxidative stress in rats induced by inhaled sulfur mustard is ameliorated by anti-tumor necrosis factor-α antibody. Toxicol Appl Pharmacol. (2021) 428:115677. doi: 10.1016/j.taap.2021.115677
147. Murray A, Gow AJ, Venosa A, Andres J, Malaviya R, Adler D, et al. Assessment of mustard vesicant lung injury and anti-TNF-α efficacy in rodents using live-animal imaging. Ann N Y Acad Sci. (2020) 1480:246–56. doi: 10.1111/nyas.v1480.1
148. Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFα in pulmonary pathophysiology. Respir Res. (2006) 7:1–9. doi: 10.1186/1465-9921-7-125
149. Mukhopadhyay S, Mukherjee S, Smith M, Das SK. Activation of MAPK/AP-1 signaling pathway in lung injury induced by 2-chloroethyl ethyl sulfide, a mustard gas analog. Toxicol Lett. (2008) 181:112–17. doi: 10.1016/j.toxlet.2008.07.005
150. Sunil VR, Vayas KN, Cervelli JA, Ebramova EV, Gow AJ, Goedken M, et al. Protective role of surfactant protein-D against lung injury and oxidative stress induced by nitrogen mustard. Toxicol Sci. (2018) 166:108–22. doi: 10.1093/toxsci/kfy188
151. Das SK, Mukherjee S, Smith MG, Chatterjee D. Prophylactic protection by N-acetylcysteine against the pulmonary injury induced by 2-chloroethyl ethyl sulfide, a mustard analogue. J Biochem Mol Toxicol. (2003) 17:177–84. doi: 10.1002/jbt.10076
152. Venosa A, Gow JG, Hall L, Malaviya R, Gow AJ, Laskin JD, et al. Regulation of nitrogen mustard-induced lung macrophage activation by valproic acid, a histone deacetylase inhibitor. Toxicol Sci. (2017) 157:222–34. doi: 10.1093/toxsci/kfx032
153. Venosa A, Smith LC, Gow AJ, Zarbl H, Laskin JD, Laskin DL. Macrophage activation in the lung during the progression of nitrogen mustard induced injury is associated with histone modifications and altered miRNA expression. Toxicol Appl Pharmacol. (2021) 423:115569. doi: 10.1016/j.taap.2021.115569
154. Smith LC, Venosa A, Gow AJ, Laskin JD, Laskin DL. Transcriptional profiling of lung macrophages during pulmonary injury induced by nitrogen mustard. Ann N Y Acad Sci. (2020) 1480:146–54. doi: 10.1111/nyas.v1480.1
155. Murray A, Banota T, Guo GL, Smith LC, Meshanni JA, Lee J, et al. Farnesoid X receptor regulates lung macrophage activation and injury following nitrogen mustard exposure. Toxicol Appl Pharmacol. (2022) 454:116208. doi: 10.1016/j.taap.2022.116208
156. Manzoor S, Mariappan N, Zafar I, Wei CC, Ahmad A, Surolia R, et al. Cutaneous lewisite exposure causes acute lung injury. Ann N Y Acad Sci. (2020) 1479:210–22. doi: 10.1111/nyas.v1479.1
157. Mariappan N, Zafar I, Robichaud A, Wei CC, Shakil S, Ahmad A, et al. Pulmonary pathogenesis in a murine model of inhaled arsenical exposure. Arch Toxicol. (2023) 97:1847–58. doi: 10.1007/s00204-023-03503-6
158. Rana T, Ahmad A, Zafar I, Mariappan N, Chandrashekar DS, Hamid T, et al. MicroRNA-mediated inflammation and coagulation effects in rats exposed to an inhaled analog of sulfur mustard. Ann N Y Acad Sci. (2020) 1479:148–58. doi: 10.1111/nyas.v1479.1
159. Surolia R, Li FJ, Dsouza K, Zeng H, Singh P, Stephens C, et al. Cutaneous exposure to arsenicals is associated with development of constrictive bronchiolitis in mice. Am J Respir Cell Mol Biol. (2023) 68:485–97. doi: 10.1165/rcmb.2022-0321MA
160. Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduction Targeted Ther. (2023) 8:207. doi: 10.1038/s41392-023-01452-1
161. Wei L-y, Jiang A-q, Jiang R, Duan S-y, Xu X, Su Z-d, et al. Protective effects of recombinant 53-kDa protein of Trichinella spiralis on acute lung injury in mice via alleviating lung pyroptosis by promoting M2 macrophage polarization. Innate Immun. (2021) 27:313–23. doi: 10.1177/17534259211013397
162. Ge Z, Chen Y, Ma L, Hu F, Xie L. Macrophage polarization and its impact on idiopathic pulmonary fibrosis. Front Immunol. (2024) 15:1444964. doi: 10.3389/fimmu.2024.1444964
163. Saeed O, Boyer NL, Pamplin JC, Driscoll IR, DellaVolpe J, Cannon J, et al. Inhalation injury and toxic industrial chemical exposure. Military Med. (2018) 183:130–32. doi: 10.1093/milmed/usy073
164. Pangeni RP, Timilsina B, Oli PR, Khadka S, Regmi PR. A multidisciplinary approach to accidental inhalational ammonia injury: A case report. Ann Med Surg. (2022) 82:104741. doi: 10.1016/j.amsu.2022.104741
165. Brooks SM, Weiss MA, Bernstein IL. Reactive airways dysfunction syndrome (RADS). Persistent asthma syndrome after high level irritant exposures. Chest. (1985) 88:376–84. doi: 10.1378/chest.88.3.376
166. de la Hoz RE, Schlueter DP, Rom WN. Chronic lung disease secondary to ammonia inhalation injury: a report on three cases. Am J Ind Med. (1996) 29:209–14. doi: 10.1002/(SICI)1097-0274(199602)29:2<209::AID-AJIM12>3.0.CO;2-7
167. Brautbar N, Wu M, Richter E. Chronic ammonia inhalation and interstitial pulmonary fibrosis: A case report and review of the literature. Arch Environ Health. (2003) 58:592–6. doi: 10.3200/AEOH.58.9.592-596
168. Gueye L, Samb A, Ciss M, Ndoye O, Mbengue-Gaye A, Cisse F. Ammonia-gas poisoning: respiratory troubles evaluated by functional exploration. Dakar Med. (2001) 46:8–11.
169. Mulder JS, van der Zalm HO. Fatal case of ammonia poisoning. Tijdschr Sociale Geneeskunde. (1967) 45:458–60.
170. Elfsmark L, Ågren L, Akfur C, Wigenstam E, Bergström U, Jonasson S. Comparisons of acute inflammatory responses of nose-only inhalation and intratracheal instillation of ammonia in rats. Inhalation Toxicol. (2019) 31:107–18. doi: 10.1080/08958378.2019.1606367
171. Elfsmark L, Ågren L, Akfur C, Jonasson S. Ammonia exposure by intratracheal instillation causes severe and deteriorating lung injury and vascular effects in mice. Inhalation Toxicol. (2022) 34:145–58. doi: 10.1080/08958378.2022.2064566
172. Perkins MW, Wong B, Tressler J, Rodriguez A, Sherman K, Andres J, et al. Adverse respiratory effects in rats following inhalation exposure to ammonia: respiratory dynamics and histopathology. Inhalation Toxicol. (2017) 29:32–41. doi: 10.1080/08958378.2016.1277571
173. Perkins MW, Wong B, Tressler J, Coggins A, Rodriguez A, Devorak J, et al. Assessment of inhaled acute ammonia-induced lung injury in rats. Inhalation Toxicol. (2016) 28:71–9. doi: 10.3109/08958378.2015.1136715
174. Bein K, Ganguly K, Martin TM, Concel VJ, Brant KA, Di YPP, et al. Genetic determinants of ammonia-induced acute lung injury in mice. Am J Physiol Lung Cell Mol Physiol. (2021) 320:L41–l62. doi: 10.1152/ajplung.00276.2020
175. Chen D, Ning F, Zhang J, Tang Y, Teng X. NF-κB pathway took part in the development of apoptosis mediated by miR-15a and oxidative stress via mitochondrial pathway in ammonia-treated chicken splenic lymphocytes. Sci Total Environ. (2020) 729:139017. doi: 10.1016/j.scitotenv.2020.139017
176. Li D, Shen L, Zhang D, Wang X, Wang Q, Qin W, et al. Ammonia-induced oxidative stress triggered proinflammatory response and apoptosis in pig lungs. J Environ Sci. (2023) 126:683–96. doi: 10.1016/j.jes.2022.05.005
177. Tang S, Liu Z, Deng F, Xu Y, Zhong R, Chen L, et al. Ammonia exposure-triggered redox imbalance with the occurrence of inflammatory response, cell apoptosis, and the circadian clock disturbance leads to lung injury in growing pigs. Air Quality Atmosphere Health. (2024). doi: 10.1007/s11869-024-01607-1
178. Schlesinger RB. Effects of inhaled acids on respiratory tract defense mechanisms. Environ Health Perspect. (1985) 63:25–38. doi: 10.1289/ehp.856325
179. Colunga Biancatelli RML, Solopov P, Dimitropoulou C, Catravas JD. Age-dependent chronic lung injury and pulmonary fibrosis following single exposure to hydrochloric acid. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22168833
180. Colunga Biancatelli RML, Solopov P, Dimitropoulou C, Gregory B, Day T, Catravas JD. The heat shock protein 90 inhibitor, AT13387, protects the alveolo-capillary barrier and prevents HCl-induced chronic lung injury and pulmonary fibrosis. Cells. (2022) 11. doi: 10.3390/cells11061046
181. Colunga Biancatelli RML, Solopov P, Gregory B, Catravas JD. The HSP90 inhibitor, AUY-922, protects and repairs human lung microvascular endothelial cells from hydrochloric acid-induced endothelial barrier dysfunction. Cells. (2021) 10. doi: 10.3390/cells10061489
182. Colunga Biancatelli RML, Solopov PA, Day T, Gregory B, Osei-Nkansah M, Dimitropoulou C, et al. HSP70 is a critical regulator of HSP90 inhibitors effectiveness in preventing HCl-induced chronic lung injury and pulmonary fibrosis. Int J Mol Sci. (2024) 25.
183. Solopov P, Colunga Biancatelli RML, Dimitropoulou C, Catravas JD. Dietary phytoestrogens ameliorate hydrochloric acid-induced chronic lung injury and pulmonary fibrosis in mice. Nutrients. (2021) 13. doi: 10.3390/nu13103599
184. Solopov P, Colunga Biancatelli RML, Dimitropoulou C, Catravas JD. Sex-related differences in murine models of chemically induced pulmonary fibrosis. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22115909
185. Solopov PA, Colunga Biancatelli RML, Dimitropolou C, Day T, Catravas JD. Optimizing antidotal treatment with the oral HSP90 inhibitor TAS-116 against hydrochloric acid-induced pulmonary fibrosis in mice. Front Pharmacol. (2022) 13:1034464. doi: 10.3389/fphar.2022.1034464
186. Marinova M, Solopov P, Dimitropoulou C, Colunga Biancatelli RML, Catravas JD. Acute exposure of mice to hydrochloric acid leads to the development of chronic lung injury and pulmonary fibrosis. Inhal Toxicol. (2019) 31:147–60. doi: 10.1080/08958378.2019.1624895
187. Aschner Y, Correll KA, Beke KM, Foster DG, Roybal HM, Nelson MR, et al. PTPα promotes fibroproliferative responses after acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2022) 323:L69–l83. doi: 10.1152/ajplung.00436.2021
188. Puig F, Herrero R, Guillamat-Prats R, Gómez MN, Tijero J, Chimenti L, et al. A new experimental model of acid-and endotoxin-induced acute lung injury in rats. Am J Physiology-Lung Cell Mol Physiol. (2016) 311:L229–L37. doi: 10.1152/ajplung.00390.2015
189. Shellito J, Murphy S. The effect of experimental acid aspiration on alveolar macrophage function in rabbits. Am Rev Respir Dis. (1980) 122:551–60.
190. Zambelli V, Rizzi L, Delvecchio P, Bresciani E, Molteni L, Meanti R, et al. JMV5656, a short synthetic derivative of TLQP-21, alleviates acid-induced lung injury and fibrosis in mice. Pulm Pharmacol Ther. (2020) 62:101916. doi: 10.1016/j.pupt.2020.101916
191. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. (2011) 11:750–61. doi: 10.1038/nri3088
192. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
193. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
194. Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr Drug Targets Inflammation Allergy. (2005) 4:281–6. doi: 10.2174/1568010054022024
195. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. (2005) 23:344–6. doi: 10.1016/j.immuni.2005.10.001
196. Chawla A. Control of macrophage activation and function by PPARs. Circ Res. (2010) 106:1559–69. doi: 10.1161/CIRCRESAHA.110.216523
197. Locati M, Mantovani A, Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol. (2013) 120:163–84. doi: 10.1016/B978-0-12-417028-5.00006-5
198. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. (2019) 129:2619–28. doi: 10.1172/JCI124615
199. Natoli G. Maintaining cell identity through global control of genomic organization. Immunity. (2010) 33:12–24. doi: 10.1016/j.immuni.2010.07.006
200. Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol. (2019) 19:417–32. doi: 10.1038/s41577-019-0151-6
201. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. (2007) 447:972–8. doi: 10.1038/nature05836
202. Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. (2022) 29:133–46. doi: 10.1038/s41418-021-00841-9
203. Irizarry-Caro RA, McDaniel MM, Overcast GR, Jain VG, Troutman TD, Pasare C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc Natl Acad Sci U.S.A. (2020) 117:30628–38.
204. OConnell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. (2012) 30:295–312. doi: 10.1146/annurev-immunol-020711-075013
205. Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. (2013) 341:789–92. doi: 10.1126/science.1240925
206. Zhou H, Hu S, Yan W. Extracellular vesicles as modifiers of epigenomic profiles. Trends Genet. (2024). doi: 10.1016/j.tig.2024.05.005
207. Korkmaz A, Tan DX, Reiter RJ. Acute and delayed sulfur mustard toxicity; novel mechanisms and future studies. Interdiscip Toxicol. (2008) 1:22–6. doi: 10.2478/v10102-010-0027-x
208. Tang D, Cao F, Yan C, Fang K, Ma J, Gao L, et al. Extracellular vesicle/macrophage axis: potential targets for inflammatory disease intervention. Front Immunol. (2022) 13:705472. doi: 10.3389/fimmu.2022.705472
209. Andres J, Smith LC, Murray A, Jin Y, Businaro R, Laskin JD, et al. Role of extracellular vesicles in cell-cell communication and inflammation following exposure to pulmonary toxicants. Cytokine Growth Factor Rev. (2020) 51:12–8. doi: 10.1016/j.cytogfr.2019.12.001
210. Carnino JM, Lee H, Smith LC, Sunil VR, Rancourt RC, Vayas K, et al. Microvesicle-derived miRNAs regulate proinflammatory macrophage activation in the lung following ozone exposure. Toxicol Sci. (2022) 187:162–74. doi: 10.1093/toxsci/kfac025
211. Wang Y, Zhao M, Liu S, Guo J, Lu Y, Cheng J, et al. Macrophage-derived extracellular vesicles: diverse mediators of pathology and therapeutics in multiple diseases. Cell Death Dis. (2020) 11:924. doi: 10.1038/s41419-020-03127-z
212. Armstrong DA, Chen Y, Dessaint JA, Aridgides DS, Channon JY, Mellinger DL, et al. DNA methylation changes in regional lung macrophages are associated with metabolic differences. Immunohorizons. (2019) 3:274–81. doi: 10.4049/immunohorizons.1900042
213. Imani S, Panahi Y, Salimian J, Fu J, Ghanei M. Epigenetic: A missing paradigm in cellular and molecular pathways of sulfur mustard lung: a prospective and comparative study. Iran J Basic Med Sci. (2015) 18:723–36.
214. Pervizaj-Oruqaj L, Ferrero MR, Matt U, Herold S. The guardians of pulmonary harmony: alveolar macrophages orchestrating the symphony of lung inflammation and tissue homeostasis. Eur Respir Rev. (2024) 33. doi: 10.1183/16000617.0263-2023
215. Lim PN, Cervantes MM, Pham LK, Rothchild AC. Alveolar macrophages: novel therapeutic targets for respiratory diseases. Expert Rev Mol Med. (2021) 23:e18. doi: 10.1017/erm.2021.21
216. Vichare R, Janjic JM. Macrophage-Targeted nanomedicines for ARDS/ALI: promise and potential. Inflammation. (2022) 45:2124–41. doi: 10.1007/s10753-022-01692-3
217. Musah S, Chen J, Schlueter C, Humphrey DM Jr., Stocke K, Hoyle MI, et al. Inhibition of chlorine-induced airway fibrosis by budesonide. Toxicol Appl Pharmacol. (2019) 363:11–21. doi: 10.1016/j.taap.2018.08.024
218. Jonasson S, Wigenstam E, Koch B, Bucht A. Early treatment of chlorine-induced airway hyperresponsiveness and inflammation with corticosteroids. Toxicol Appl Pharmacol. (2013) 271:168–74. doi: 10.1016/j.taap.2013.04.037
219. Ehrlich JP, Burleson GR. Enhanced and prolonged pulmonary influenza virus infection following phosgene inhalation. J Toxicol Environ Health. (1991) 34:259–73. doi: 10.1080/15287399109531565
220. Sciuto AM, Clapp DL, Hess ZA, Moran TS. The temporal profile of cytokines in the bronchoalveolar lavage fluid in mice exposed to the industrial gas phosgene. Inhal Toxicol. (2003) 15:687–700. doi: 10.1080/08958370390197254
221. Cao C, Memete O, Shao Y, Zhang L, Liu F, Dun Y, et al. Single-cell RNA-sequencing reveals epithelial cell signature of multiple subtypes in chemically induced acute lung injury. Int J Mol Sci. (2022) 24. doi: 10.3390/ijms24010277
222. Cao C, Memet O, Liu F, Hu H, Zhang L, Jin H, et al. Transcriptional characterization of bronchoalveolar lavage fluid reveals immune microenvironment alterations in chemically induced acute lung injury. J Inflammation Res. (2023) 16:2129–47. doi: 10.2147/JIR.S407580
223. Zhu Y, Choi D, Somanath PR, Zhang D. Lipid-Laden macrophages in pulmonary diseases. Cells. (2024) 13:889. doi: 10.3390/cells13110889
224. Stevenson ER, Smith LC, Wilkinson ML, Lee SJ, Gow AJ. Etiology of lipid-laden macrophages in the lung. Int Immunopharmacol. (2023) 123:110719. doi: 10.1016/j.intimp.2023.110719
Keywords: macrophage, lung, inhaled, chemical, halogen, epigenetics
Citation: Ahmad S, Nasser W and Ahmad A (2024) Epigenetic mechanisms of alveolar macrophage activation in chemical-induced acute lung injury. Front. Immunol. 15:1488913. doi: 10.3389/fimmu.2024.1488913
Received: 30 August 2024; Accepted: 15 October 2024;
Published: 08 November 2024.
Edited by:
Xiao Xiao Tang, First Affiliated Hospital of Guangzhou Medical University, ChinaReviewed by:
Yi Wang, Huazhong University of Science and Technology, ChinaLeah Renee Reznikov, University of Florida, United States
Copyright © 2024 Ahmad, Nasser and Ahmad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shama Ahmad, c2hhbWFhaGFtZEB1YWJtYy5lZHU=