 Serina Tokita
Serina Tokita Takayuki Kanaseki
Takayuki Kanaseki Toshihiko Torigoe
Toshihiko Torigoe- 1Department of Pathology, Sapporo Medical University, Sapporo, Japan
- 2Joint Research Center for Immunoproteogenomics, Sapporo Medical University, Sapporo, Japan
Somatic mutations in tumor cells give rise to mutant proteins, fragments of which are often presented by MHC and serve as neoantigens. Neoantigens are tumor-specific and not expressed in healthy tissues, making them attractive targets for T-cell-based cancer immunotherapy. On the other hand, since most somatic mutations differ from patient to patient, neoantigen-targeted immunotherapy is personalized medicine and requires their identification in each patient. Computational algorithms and machine learning methods have been developed to prioritize neoantigen candidates. In fact, since the number of clinically relevant neoantigens present in a patient is generally limited, this process is like finding a needle in a haystack. Nevertheless, MHC presentation of neoantigens is not random but follows certain rules, and the efficiency of neoantigen detection may be further improved with technological innovations. In this review, we discuss current approaches to the detection of clinically relevant neoantigens, with a focus on antigen processing and presentation.
Introduction
Mutant peptides derived from somatic mutations are often presented by MHC molecules and give rise to neoantigens (1, 2). Neoantigens are not subject to central tolerance and can therefore induce T cell responses in patients. In preclinical and clinical settings, antitumor effects of immune checkpoint blockade (ICB) are mediated by T cells recognizing neoantigens (3–5). Adoptive cell transfer of neoantigen-reactive CD4+ T cells resulted in tumor regression in a patient with epithelial tumor (6). The phase IIb study of the neoantigen vaccine in combination with ICB showed improved recurrence-free survival (RFS) and distant metastasis-free survival in patients with resected melanoma (7). Even in patients with pancreatic cancer, vaccinated patients with neoantigen reactive T cells showed prolonged RFS (8). Vaccination-induced neoantigen-reactive T cells persist for years, suggesting their contribution to long-term prevention of relapse or metastasis (9–11). These observations strongly suggest a central role for neoantigens in mediating host T cell immune responses against tumors, and manipulation of T cell responses to neoantigens may lead to success in cancer immunotherapy (12–15).
Although neoantigens are attractive targets for immunotherapy in a variety of cancers, most (if not all) neoantigens differ between patients. In addition, it is becoming clear that the frequency of immunogenic neoantigens that induce T-cell responses in patients is very low and may be limited to a few percent of somatic mutations (16–19). Thus, neoantigen-targeted immunotherapy becomes personalized medicine, requiring screening for each patient, and such a needle in a haystack must be efficiently identified from a large number of mutations. Advances in next-generation sequencing technology have enabled the identification of nonsynonymous mutations in tumor exomes and the in silico prediction of the affinity between mutant peptides and patient MHC alleles for potential neoantigens. The in silico prediction algorithm for neoantigens is being continuously improved (20, 21). However, when looking at a patient’s T cell response, the percentage of predicted sequences that do not induce a response remains high, making it a challenge to efficiently detect neoantigens by in silico prediction alone (22). In a previous report, neoantigens were predicted using different pipelines from different laboratories, but in total only about 6% of the predicted neoantigens were successfully recognized by the patient’s T cells (23). Such a high false-positive rate may be due to T cell-side factors, such as the diversity of T cell receptor (TCR) repertoires, or tumor cell-side factors, such as the complexity of intracellular antigen processing. In this review, we focus on the latter, highlighting current issues in the identification of clinically relevant neoantigens and potential solutions from an antigen processing perspective with the goal of clinical application of neoantigens.
What are clinically relevant neoantigens from an antigen presentation perspective?
Naive T cells are primed by professional antigen presenting cells (APCs) in the lymph nodes, and effector T cells subsequently migrate and recognize tumor cells. For a clinically relevant T cell immune response to occur against an antigen, the antigen must be immunogenic (capable of inducing a host T cell response) and presented on cell surfaces (naturally processed and presented by MHC). Although these two requirements are closely related, they are not necessarily the same. For example, in a vaccination setting targeting cytotoxic CD8+ T cells, administered neoantigens (or nucleotide sequences encoding neoantigens) will be taken up by APCs and may be able to induce a circulating T cell response in the peripheral blood since they are in principle foreign to the host. However, an anti-tumor effect cannot be expected if the tumor cells themselves do not endogenously present the neoantigens on their MHC. As discussed below, for various reasons, not all expressed gene products with MHC-binding properties are necessarily processed and presented to cell surface MHCs. Conversely, even if tumor cells present immunogenic neoantigens, the neoantigens may fail to elicit a spontaneous endogenous T cell response if cross-presentation of the neoantigens by professional APCs was insufficient. Therefore, clinically relevant neoantigens that elicit spontaneous T cell responses should be those presented by APCs (e.g., cross-presentation) and tumor cells (e.g., endogenous presentation).
In contrast, it is unknown whether this principle applies to MHC-II neoantigens. The significance of MHC-II neoantigen presentation on tumor cells is likely to depend on the phenotype of the neoantigen-reactive CD4+ T cells. For helper T cells that exert their antitumor effects indirectly through the production of effector cytokines, by helping to prime CD8+ T cells, or by activating myeloid-derived cells, MHC-II neoantigen presentation on tumor cells would not be mandatory, and in fact many tumor cells of epithelial origin lack surface MHC-II expression (24–26). Meanwhile, cytotoxic CD4+ T cells capable of lysing tumor cells require MHC-II neoantigen presentation on tumor cells (27, 28). Paradoxically, however, MHC-II neoantigen presentation on tumor cells can also inhibit antitumor effects, possibly by inducing neoantigen-reactive regulatory CD4+ T cells in the tumor microenvironment (29). Thus, clinically relevant neoantigens that may lead to therapeutic effects must be presented by MHC; while the case of MHC-I neoantigens is straightforward, the case of MHC-II neoantigens requires further classification and validation with respect to CD4+ T cell differentiation types.
Endogenous antigen processing and MHC class I presentation
The cell surface repertoire of peptide-MHC-I complex (pMHC-I) is formed by the intracellular antigen presentation pathway consisting of a multi-step process involving the antigen processing machinery (APM) (Figure 1) (30–32). The proteasomes degrade proteins in the cytoplasm and yield protein fragments, which serve as precursors of MHC-binding peptides. APCs and tumor cells differ in the composition of proteasome subunits, with APCs expressing the immunoproteasomes and tumor cells of epithelial origin expressing the immunoproteasomes only under IFNγ stimulation. In melanoma patients treated with ICB, higher expression of the immunoproteasomes correlates with improved prognosis, possibly suggesting their influence on neoantigen production with clinical significance (33). The peptide precursors, which are yet too long for MHC binding, are then transported into the endoplasmic reticulum (ER) by the transporters associated with antigen processing (TAP). An ER-resident aminopeptidase, ERAAP (or ERAP1), is present in the ER and trims the amino acids from the N-terminus of the precursors to yield short peptides with optimal lengths for MHC binding (34). Hence, the C-terminal end of peptides is completed in the cytoplasm (by the proteasomes), while the N-terminal end is completed in the ER (by ERAAP). In the endoplasmic reticulum, empty MHC-I molecules associate with TAP, tapasin, ERp57, and calreticulin to form a structure called the peptide loading complex (PLC), which exchanges candidate peptides to form pMHC-I (35). ERAAP and PLC are thought to generate and select peptides that fit into MHC-I molecules, thereby providing stable pMHC-I on the cell surface.
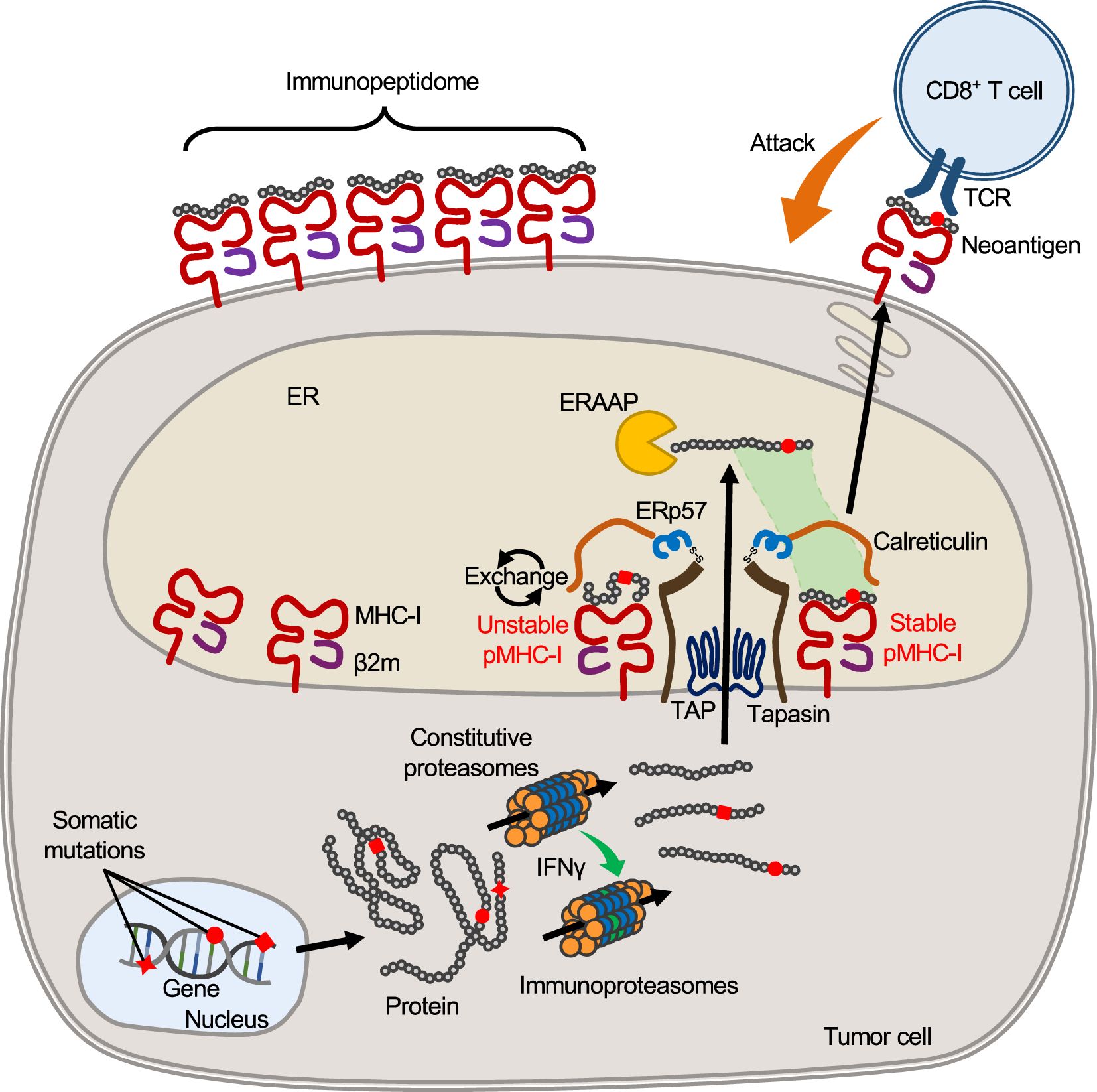
Figure 1. MHC-I antigen processing and presentation pathway. In tumor cells, mutant proteins with substituted amino acids are translated from transcripts with nonsynonymous somatic mutations and, like other proteins, are processed and fragmented by proteasomes in the cytoplasm. Upon IFNγ stimulation, proteasomes are transformed into immunoproteasomes composed of different subunits, yielding protein fragments of different lengths that serve as precursors of MHC-bound peptides. Such peptide precursors are then transported to the ER through the TAP. Inside the ER, an aminopeptidase, ERAAP (ERAP1), trims amino acids from the N-terminus of the precursors, yielding peptides of optimal length for binding to host MHC-I. At the same time, empty MHC-I forms the PLC with β2m, tapasin, calreticulin, and ERp57, which removes and exchanges unstable peptides bound to MHC-I. Stabilized pMHC-I is released from the PLC and transported to the cell surface to form the surface pMHC-I repertoire. When peptides with mutated amino acids are presented, they may serve as neoantigens that elicit a T cell response because they are foreign to the host.
It should be noted that, in theory, cells are not capable of presenting all protein fragments. Since the number of MHC-I molecules (on the order of 105) is limited compared to the number of protein fragments produced in the cytosol, the proportion of intracellular peptides that are ultimately presented by surface MHCs is thought to be very small (36). The actual number of different peptides would be much smaller, since peptides with high gene expression levels are likely to account for multiple copies of MHC-I molecules. Peptides are selected mainly due to their ability to bind to the host MHC-I molecule, but there are other limitations besides MHC binding. For example, ERAAP cannot trim proline, so sequences a few amino acids downstream of proline are unlikely to be presented by MHC-I, even if they have appropriate MHC-I binding motifs (37–39). Thus, not only peptide sequences but also surrounding sequences influence the efficiency of antigen processing. Furthermore, APM expression is not always constant across cells. Loss of APM expression, which is often observed in tumor cells, is likely to influence pMHC-I repertoire formation (40, 41). Thus, the surface MHC-I peptide repertoire is elaborately regulated through endogenous antigen processing pathway; however, the repertoire can be influenced by multiple factors, such as APM expression and competition among candidate peptides, and this complexity may preclude accurate prediction of the peptides presented on the cell surface.
Landscape of MHC class I presented peptides
Landscapes of peptides displayed by MHC can now be explored by immunopeptidomics or MHC ligandome analysis, in which MHCs are extracted directly from cell lysates and the bound peptides are eluted and comprehensively sequenced by mass spectrometry (MS). The increased sensitivity of MS allows the sequencing of thousands of MHC-I and MHC-II peptides, separately or simultaneously, per sample (42). Sequencing results from a variety of normal and tumor tissue types have been collected and used as training data to improve in silico algorithms for predicting MHC-presented peptides (43). The recovery of naturally presented peptides reveals the nature of antigen processing and presentation. The repertoire of source genes providing an immunopeptidome is not ubiquitous but limited, with only about 60% of expressed protein-coding genes (exon regions) yielding MHC-I represented peptides (44). This proportion is likely to vary further as translation and MHC-I presentation of peptides is now known to occur outside the exons or from unconventional open reading frames (45–47). Furthermore, peptides with higher source gene expression or abundant proteins are more likely to be presented to MHC (38, 48). The distribution of MHC-presented peptide sites within a protein sequence is not also uniform, but rather skewed toward certain sites, often forming “hotspots” (44, 49).Because MHC anchors often contain hydrophobic amino acids, peptide sequences tend to concentrate in transmembrane regions where hydrophobic residues are unevenly distributed (50). The presence of such hotspots clearly indicates that the peptides presented by MHC are not randomly selected from a given protein fragments, but that their selection follows certain biological rules.
Identification of neoantigens presented by MHC class I or II
As mentioned above, neoantigens with immunogenicity and a higher probability of being presented by MHC need to be prioritized for clinical applications, such as use as vaccines or efficacy biomarkers for ICB. An efficient approach to identify immunogenic neoantigens would be to use the TCRs of clonally expanded tumor-infiltrating T cells (TILs) as a screening probe. Since TILs are already primed and migrating into the tumor, neoantigens recognized by such TCRs should naturally be presented to MHC. High-throughput and sensitive screening platforms have been developed and reported (51–53). Meanwhile, a reliable way to identify MHC-presented peptides would be immunopeptidomics using MS. This approach allows comprehensive identification of MHC-presented neoantigens directly from tissue samples, including solid tumors of epithelial origin (54–57). While this approach may have a lower detection sensitivity due to technical limitations of MS, this approach may be useful for identifying both immunodominant and subdominant neoantigens, the latter of which do not spontaneously induce host T-cell responses and thus may be missed by TIL probes. These MS-detected neoantigens tend to be immunogenic and recognized by patient TILs, potentially suggesting a link between immunogenicity and the level of natural MHC presentation.
On the other hand, both approaches require a specific type of biomaterial, TILs or frozen tumor tissue, which are not always available for every patient in clinical settings and require organized logistics. To overcome this limitation, we propose to exploit the nature of antigen processing and use wild-type surrogate immunopeptidome data instead of tumor immunopeptidomes (Figure 2). The majority of immunopeptidomes are matched between different individuals carrying the same MHC type, which may be consistent with the presence of hotspots for MHC presentation in protein sequences.In fact, even among different individuals, about 70% of presenting peptides overlap when MHC types match in the same organ (57, 58). Even between different organs, about 60% of the immunopeptidomes match in both mouse and human, which could be due to the sharing of highly expressed transcripts between different organs (59). Furthermore, MHC presentation of neoantigens is often accompanied by presentation of their wild-type counterparts, as demonstrated by immunopeptidomics (33, 54, 57, 60–63). Neoantigens accompanied with wild-type MHC presentation or those whose wild-type counterparts are already registered in public databases as MHC-presented peptides tend to be immunogenic (64). Therefore, it may be possible to predict naturally presented neoantigens using MHC-matched surrogate wild-type immunopeptidome data in combination with somatic mutation data from the patient’s tumor. We have recently reported an approach called neoantigen selection using surrogate immunopeptidomes (NESSIE), which has efficiently identified immunogenic neoantigens in colorectal and endometrial cancer patients compared to conventional in silico prediction (65). Since this approach analyzes surrogate (e.g. blood) immunopeptidomes but not tumor immunopeptidomes, frozen tumor samples are not required and can be widely used in clinical practice.
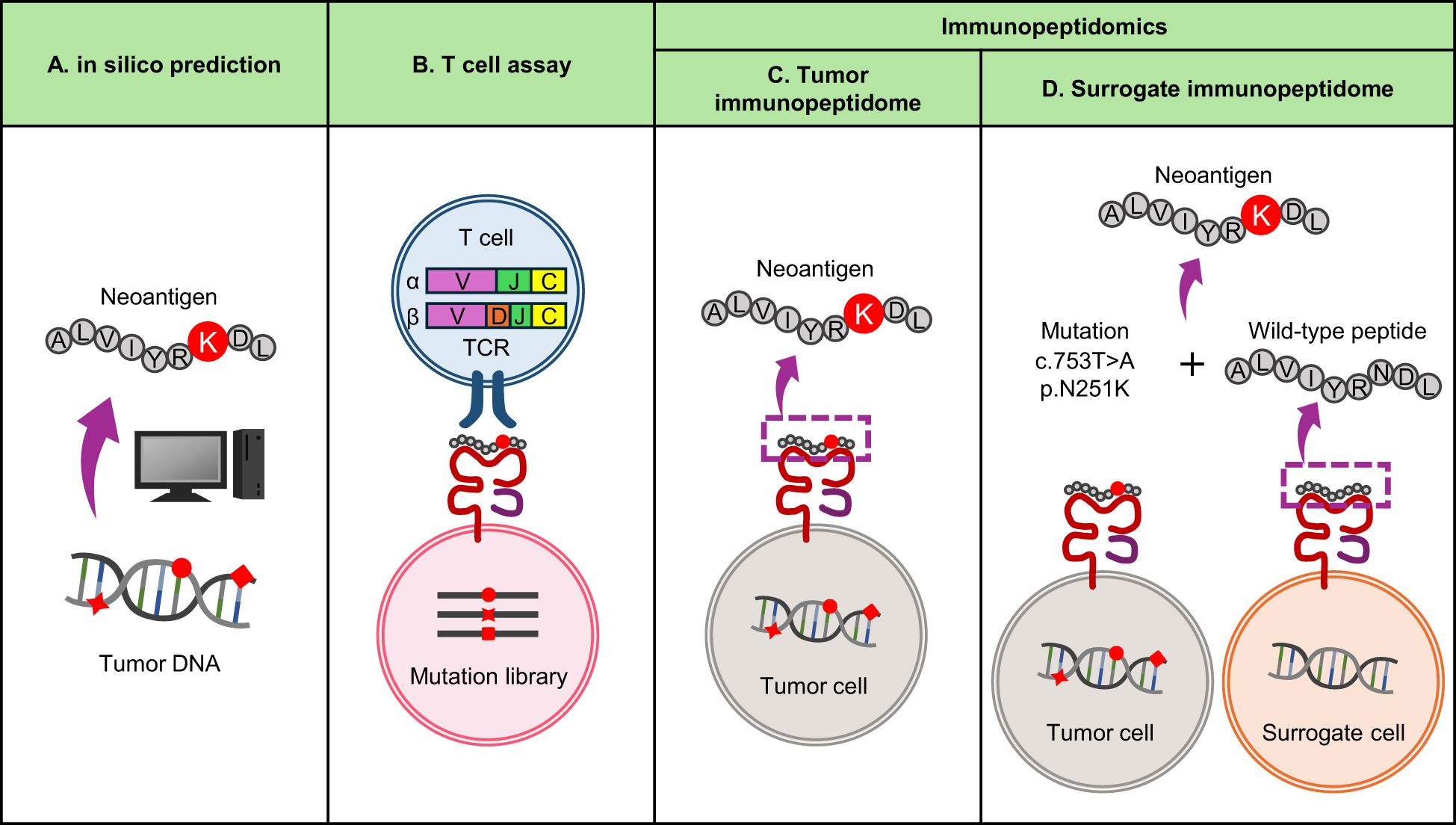
Figure 2. Current approaches to neoantigen identification. (A) In silico prediction of neoantigens from genomic data. It is widely used because it can quickly identify neoantigens based on tumor mutation and MHC data. The sensitivity is considered high, but the false positive rate is also high and the specificity is considered low. (B) T-cell assay using TIL TCRs as screening probes. A reliable approach to identify immunogenic neoantigens with high specificity, but time consuming and labor intensive. (C, D) Immunopeptidomics using MS. A reliable approach to identify neoantigens naturally presented by MHC, but time-consuming and labor-intensive. Sensitivity depends on sample quality and MS performance. (C) Conventional immunopeptidomics screening tumor immunopeptidomes. (D) Alternative immunopeptidomics screening surrogate (i.e. blood) immunopeptidomes for wild-type counterparts of neoantigens.
In contrast, there are specific types of neoantigens that cannot be detected by NESSIE. As sources of clinically relevant, immunogenic MHC-I neoantigens, SNVs account for 98.2% (56/57) and indels for 1.8% (1/57), with SNV-derived neoantigens being by far the most common (19). Of these, indel-derived neoantigens cannot be detected by NESSIE due to the lack of wild-type counterparts. SNV-derived neoantigens are more complicated. If the mutated amino acid were not an MHC-binding anchor, NESSIE would detect the neoantigen. Even if an MHC-binding anchor was substituted (anchor-type neoantigen), NESSIE would detect the neoantigen unless MHC presentation of its wild-type peptide was completely absent. The percentage of anchor-type neoantigens associated with zero (undetectable) wild-type MHC presentation is unclear. Although there may be limitations as this approach relies on the detection of wild-type counterparts, it is conceptually novel and the sensitivity and specificity of this type of approach compared to conventional methods should be further investigated.
Conclusion
Neoantigens are truly tumor-specific, a key link between T cells and tumor cells, and thus serve as attractive targets for immunotherapy. However, it is clear that the prioritization of clinically relevant neoantigens remains a challenge and a bottleneck in their clinical application. This challenge would be overcome by technological advances such as the rapid and comprehensive screening of antigens recognized by the TIL repertoire, the development of highly sensitive immunopeptidomics independent of sample quality, and the development of in silico prediction technology based on learning from the results obtained. In particular, although difficult to quantify unambiguously, certain rules exist in the selection of antigens by the antigen processing and presentation pathway, and we consider that their successful application will further improve the efficacy and lead to the success of personalized immunotherapy.
Author contributions
ST: Conceptualization, Data curation, Funding acquisition, Investigation, Visualization, Writing – original draft, Writing – review & editing. TK: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. TT: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Japan Agency for Medical Research and Development (AMED) Grant to TK (JP24ama221315), Japan Society for the Promotion of Science (JSPS) Grant to TK (20K21528 and 24K02252), JSPS Grant to ST (24K10110), and AMED Grant to TT (JP24ama221328). This work was also supported by MEXT Promotion of Distinctive Joint Research Center Program (JPMXP0723833150).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
2. Schumacher TN, Scheper W, Kvistborg P. Cancer neoantigens. Annu Rev Immunol. (2019) 37:173–200. doi: 10.1146/annurev-immunol-042617-053402
3. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. (2014) 515:577–81. doi: 10.1038/nature13988
4. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
5. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
6. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
7. Weber JS, Carlino MS, Khattak A, Meniawy T, Ansstas G, Taylor MH, et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet. (2024) 403:632–44. doi: 10.1016/S0140-6736(23)02268-7
8. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. (2023) 618:144–50. doi: 10.1038/s41586-023-06063-y
9. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222–6. doi: 10.1038/nature23003
10. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
11. Hu Z, Leet DE, Allesoe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. (2021) 27:515–25. doi: 10.1038/s41591-020-01206-4
12. Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science. (2018) 359:1355–60. doi: 10.1126/science.aar7112
13. Vormehr M, Tureci O, Sahin U. Harnessing tumor mutations for truly individualized cancer vaccines. Annu Rev Med. (2019) 70:395–407. doi: 10.1146/annurev-med-042617-101816
14. Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat Med. (2019) 25:1488–99. doi: 10.1038/s41591-019-0596-y
15. Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. (2021) 18:215–29. doi: 10.1038/s41571-020-00460-2
16. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. (2015) 350:1387–90. doi: 10.1126/science.aad1253
17. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. (2015) 21:81–5. doi: 10.1038/nm.3773
18. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. (2016) 22:433–8. doi: 10.1038/nm.4051
19. Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discovery. (2019) 9:1022–35. doi: 10.1158/2159-8290.CD-18-1494
20. Fotakis G, Trajanoski Z, Rieder D. Computational cancer neoantigen prediction: current status and recent advances. Immunooncol Technol. (2021) 12:100052. doi: 10.1016/j.iotech.2021.100052
21. Lang F, Schrors B, Lower M, Tureci O, Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat Rev Drug Discovery. (2022) 21:261–82. doi: 10.1038/s41573-023-00873-5
22. Vitiello A, Zanetti M. Neoantigen prediction and the need for validation. Nat Biotechnol. (2017) 35:815–7. doi: 10.1038/nbt.3932
23. Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. (2020) 183:818–34.e13. doi: 10.1016/j.cell.2020.09.015
24. Poncette L, Bluhm J, Blankenstein T. The role of CD4 T cells in rejection of solid tumors. Curr Opin Immunol. (2022) 74:18–24. doi: 10.1016/j.coi.2021.09.005
25. Speiser DE, Chijioke O, Schaeuble K, Munz C. CD4(+) T cells in cancer. Nat Cancer. (2023) 4:317–29. doi: 10.1038/s43018-023-00521-2
26. Kruse B, Buzzai AC, Shridhar N, Braun AD, Gellert S, Knauth K, et al. CD4(+) T cell-induced inflammatory cell death controls immune-evasive tumours. Nature. (2023) 618:1033–40. doi: 10.1038/s41586-023-06199-x
27. Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell. (2020) 181:1612–1625 e1613. doi: 10.1016/j.cell.2020.05.017
28. Cachot A, Bilous M, Liu YC, Li X, Saillard M, Cenerenti M, et al. Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci Adv. (2021) 7:eabe3348. doi: 10.1126/sciadv.abe3348
29. Oliveira G, Stromhaug K, Cieri N, Iorgulescu JB, Klaeger S, Wolff JO, et al. Landscape of helper and regulatory antitumour CD4(+) T cells in melanoma. Nature. (2022) 605:532–8. doi: 10.1038/s41586-022-04682-5
30. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. (2011) 11:823–36. doi: 10.1038/nri3084
31. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
32. Pishesha N, Harmand TJ, Ploegh HL. A guide to antigen processing and presentation. Nat Rev Immunol. (2022) 22:751–64. doi: 10.1038/s41577-022-00707-2
33. Kalaora S, Lee JS, Barnea E, Levy R, Greenberg P, Alon M, et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat Commun. (2020) 11:896. doi: 10.1038/s41467-020-14639-9
34. Kanaseki T, Blanchard N, Hammer GE, Gonzalez F, Shastri N. ERAAP synergizes with MHC class I molecules to make the final cut in the antigenic peptide precursors in the endoplasmic reticulum. Immunity. (2006) 25:795–806. doi: 10.1016/j.immuni.2006.09.012
35. Elliott T, Williams A. The optimization of peptide cargo bound to MHC class I molecules by the peptide-loading complex. Immunol Rev. (2005) 207:89–99. doi: 10.1111/j.0105-2896.2005.00311.x
36. Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. (2016) 37:724–37. doi: 10.1016/j.it.2016.08.010
37. Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. (2002) 419:480–3. doi: 10.1038/nature01074
38. Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. (2017) 46:315–26. doi: 10.1016/j.immuni.2017.02.007
39. Hongo A, Kanaseki T, Tokita S, Kochin V, Miyamoto S, Hashino Y, et al. Upstream position of proline defines peptide-HLA class I repertoire formation and CD8(+) T cell responses. J Immunol. (2019) 202:2849–55. doi: 10.4049/jimmunol.1900029
40. Hammer GE, Kanaseki T, Shastri N. The final touches make perfect the peptide-MHC class I repertoire. Immunity. (2007) 26:397–406. doi: 10.1016/j.immuni.2007.04.003
41. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. (2021) 21:298–312. doi: 10.1038/s41568-021-00339-z
42. Purcell AW, Ramarathinam SH, Ternette N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat Protoc. (2019) 14:1687–707. doi: 10.1038/s41596-019-0133-y
43. Creech AL, Ting YS, Goulding SP, Sauld JFK, Barthelme D, Rooney MS, et al. The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction. Proteomics. (2018) 18:e1700259. doi: 10.1002/pmic.201700259
44. Pearson H, Daouda T, Granados DP, Durette C, Bonneil E, Courcelles M, et al. MHC class I-associated peptides derive from selective regions of the human genome. J Clin Invest. (2016) 126:4690–701. doi: 10.1172/JCI88590
45. Laumont CM, Daouda T, Laverdure JB, Bonneil E, Caron-Lizotte O, Laumont MP, et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun. (2016) 7:10238. doi: 10.1038/ncomms10238
46. Chong C, Muller M, Pak H, Harnett D, Huber F, Grun D, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun. (2020) 11:1293. doi: 10.1038/s41467-020-14968-9
47. Kikuchi Y, Tokita S, Hirama T, Kochin V, Nakatsugawa M, Shinkawa T, et al. CD8(+) T-cell immune surveillance against a tumor antigen encoded by the oncogenic long noncoding RNA PVT1. Cancer Immunol Res. (2021) 9:1342–53. doi: 10.1158/2326-6066.CIR-20-0964
48. Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics. (2015) 14:658–73. doi: 10.1074/mcp.M114.042812
49. Muller M, Gfeller D, Coukos G, Bassani-Sternberg M. 'Hotspots' of antigen presentation revealed by human leukocyte antigen ligandomics for neoantigen prioritization. Front Immunol. (2017) 8:1367. doi: 10.3389/fimmu.2017.01367
50. Bianchi F, Textor J. G. van den Bogaart, Transmembrane Helices Are an Overlooked Source of Major Histocompatibility Complex Class I Epitopes. Front Immunol. (2017) 8:1118. doi: 10.3389/fimmu.2017.01118
51. Arnaud M, Chiffelle J, Genolet R, Navarro Rodrigo B, Perez MAS, Huber F, et al. Sensitive identification of neoantigens and cognate TCRs in human solid tumors. Nat Biotechnol. (2022) 40:656–60. doi: 10.1038/s41587-021-01072-6
52. Cattaneo CM, Battaglia T, Urbanus J, Moravec Z, Voogd R, de Groot R, et al. Identification of patient-specific CD4(+) and CD8(+) T cell neoantigens through HLA-unbiased genetic screens. Nat Biotechnol. (2023) 41:783–7. doi: 10.1038/s41587-022-01547-0
53. Moravec Z, Zhao Y, Voogd R, Cook DR, Kinrot S, Capra B, et al. Discovery of tumor-reactive T cell receptors by massively parallel library synthesis and screening. Nat Biotechnol. (2024). doi: 10.1038/s41587-024-02210-6
54. Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. (2016) 7:13404. doi: 10.1038/ncomms13404
55. Kalaora S, Wolf Y, Feferman T, Barnea E, Greenstein E, Reshef D, et al. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in melanoma. Cancer Discovery. (2018) 8:1366–75. doi: 10.1158/2159-8290.CD-17-1418
56. Newey A, Griffiths B, Michaux J, Pak HS, Stevenson BJ, Woolston A, et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J immunotherapy Cancer. (2019) 7:309. doi: 10.1186/s40425-019-0769-8
57. Hirama T, Tokita S, Nakatsugawa M, Murata K, Nannya Y, Matsuo K, et al. Proteogenomic identification of an immunogenic HLA class I neoantigen in mismatch repair-deficient colorectal cancer tissue. JCI Insight. (2021) 6:e146356. doi: 10.1172/jci.insight.146356
58. Granados DP, Sriranganadane D, Daouda T, Zieger A, Laumont CM, Caron-Lizotte O, et al. Impact of genomic polymorphisms on the repertoire of human MHC class I-associated peptides. Nat Commun. (2014) 5:3600. doi: 10.1038/ncomms4600
59. Kubiniok P, Marcu A, Bichmann L, Kuchenbecker L, Schuster H, Hamelin DJ, et al. Understanding the constitutive presentation of MHC class I immunopeptidomes in primary tissues. iScience. (2022) 25:103768. doi: 10.1016/j.isci.2022.103768
60. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. (2014) 515:572–6. doi: 10.1038/nature14001
61. Kochin V, Kanaseki T, Tokita S, Miyamoto S, Shionoya Y, Kikuchi Y, et al. HLA-A24 ligandome analysis of colon and lung cancer cells identifies a novel cancer-testis antigen and a neoantigen that elicits specific and strong CTL responses. Oncoimmunology. (2017) 6:e1293214. doi: 10.1080/2162402X.2017.1293214
62. Ebrahimi-Nik H, Michaux J, Corwin WL, Keller GL, Shcheglova T, Pak H, et al. Mass spectrometry driven exploration reveals nuances of neoepitope-driven tumor rejection. JCI Insight. (2019) 5:e129152. doi: 10.1172/jci.insight.129152
63. Minegishi Y, et al, Kiyotani K, et al, Nemoto K, et al, Inoue Y, et al, Haga Y, et al, Fujii R, et al. Differential ion mobility mass spectrometry in immunopeptidomics identifies neoantigens carrying colorectal cancer driver mutations. Commun Biol. (2022) 5:831. doi: 10.1038/s42003-022-03807-w
64. Muller M, Huber F, Arnaud M, Kraemer AI, Altimiras ER, Michaux J, et al. Machine learning methods and harmonized datasets improve immunogenic neoantigen prediction. Immunity. (2023) 56:2650-163.e6. doi: 10.1016/j.immuni.2023.09.002
Keywords: neoantigen, MHC, antigen processing and presentation, personalized medicine, cancer vaccine
Citation: Tokita S, Kanaseki T and Torigoe T (2024) Neoantigen prioritization based on antigen processing and presentation. Front. Immunol. 15:1487378. doi: 10.3389/fimmu.2024.1487378
Received: 28 August 2024; Accepted: 21 October 2024;
Published: 06 November 2024.
Edited by:
Mihaela Angelova, The Francis Crick Institute, United KingdomReviewed by:
Richard A. Schäfer, Northwestern University, United StatesCarmen Aguilar-Gurrieri, IrsiCaixa, Spain
Copyright © 2024 Tokita, Kanaseki and Torigoe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takayuki Kanaseki, a2FuYXNla2lAc2FwbWVkLmFjLmpw