 Bingtao Zhang1
Bingtao Zhang1 Yan Zou1
Yan Zou1 Zixuan Yuan2Kun Jiang1
Zixuan Yuan2Kun Jiang1 Zhaoxiang Zhang1Shujuan Chen1Xiaoming Zhou1
Zhaoxiang Zhang1Shujuan Chen1Xiaoming Zhou1 Qi Wu1
Qi Wu1 Xin Zhang1*
Xin Zhang1*- 1Department of Neurosurgery, Nanjing Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, China
- 2Department of Neurosurgery, Jiangsu Province Hospital of Chinese Medicine, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, China
Cardiovascular and cerebrovascular diseases have surpassed cancer as significant global health challenges, which mainly include atherosclerosis, myocardial infarction, hemorrhagic stroke and ischemia stroke. The inflammatory response immediately following these diseases profoundly impacts patient prognosis and recovery. Efficient resolution of inflammation is crucial not only for halting the inflammatory process but also for restoring tissue homeostasis. Efferocytosis, the phagocytic clearance of dying cells by phagocytes, especially microglia and macrophages, plays a critical role in this resolution process. Upon tissue injury, phagocytes are recruited to the site of damage where they engulf and clear dying cells through efferocytosis. Efferocytosis suppresses the secretion of pro-inflammatory cytokines, stimulates the production of anti-inflammatory cytokines, modulates the phenotype of microglia and macrophages, accelerates the resolution of inflammation, and promotes tissue repair. It involves three main stages: recognition, engulfment, and degradation of dying cells. Optimal removal of apoptotic cargo by phagocytes requires finely tuned machinery and associated modifications. Key molecules in efferocytosis, such as ‘Find-me signals’, ‘Eat-me signals’, and ‘Don’t eat-me signals’, have been shown to enhance efferocytosis following cardiovascular and cerebrovascular diseases. Moreover, various additional molecules, pathways, and mitochondrial metabolic processes have been identified to enhance prognosis and outcomes via efferocytosis in diverse experimental models. Impaired efferocytosis can lead to inflammation-associated pathologies and prolonged recovery periods. Therefore, this review consolidates current understanding of efferocytosis mechanisms and its application in cardiovascular and cerebrovascular diseases, proposing future research directions.
1 Introduction
Cell death and turnover are continuous processes essential for maintaining physiological equilibrium across all human organs. Even under healthy conditions, the human body undergoes the turnover of over one million cells per second primarily through programmed cell death, known as apoptosis. Efferocytosis, the phagocytic removal and recycling of dying cells by specialized phagocytes at specific sites, is pivotal in this cellular clearance process (1). Furthermore, efferocytosis assumes critical significance in various pathophysiological contexts such as acute myocardial infarction and transient middle cerebral artery occlusion, prevalent in cardiovascular and cerebrovascular diseases (2–4). Upon injury, extensive cell death leads to the release of damage-associated molecular patterns (DAMPs), triggering localized inflammatory responses (5, 6). Concurrently, dying cells release signals that recruit phagocytes (7) to the injury site, facilitating the clearance of dying cells and DAMPs to mitigate inflammation and restore homeostasis (8). Following the engulfment and recycling of dying cells, phagocytes contribute to the resolution of inflammation by releasing anti-inflammatory cytokines while dampening the secretion of pro-inflammatory cytokines. This underscores efferocytosis as a pivotal process in regulating inflammatory responses and facilitating tissue repair (9, 10).
Cardiovascular and cerebrovascular diseases, such as stroke and atherosclerosis, represent significant global health challenges characterized by altered blood flow dynamics and vascular morphological changes, coupled with inflammation. In this context, efferocytosis has emerged as a focal point in contemporary research aimed at understanding its mechanistic underpinnings and its impact on the pathophysiology of cardiovascular and cerebrovascular diseases (11, 12). Therefore, this review consolidates current insights into the mechanisms of efferocytosis and its implications for cardiovascular and cerebrovascular diseases.
2 Mechanisms of efferocytosis
Efferocytosis plays a pivotal role in tissue repair and homeostasis by orchestrating the recognition, engulfment, and digestion of dying cells (Figure 1) (13). Optimal uptake of apoptotic cargo by phagocytes necessitates a finely tuned phagosomal machinery and requisite modifications. Following the recognition of apoptotic cells (ACs), phagocytes undergo a series of morphological and functional adaptations. These include the suppression of pro-inflammatory cytokine secretion, potentiation of anti-inflammatory cytokine production, acceleration of inflammation resolution, and facilitation of tissue repair processes. Efficient clearance of dying cells not only resolves inflammation but also contributes decisively to the restoration of tissue and organ homeostasis (14). Consequently, enhancing any of these facets presents potential avenues to improve efferocytosis and mitigate inflammatory responses. Subsequent sections will delve into the molecular intricacies and therapeutic strategies targeting each phase of efferocytosis.
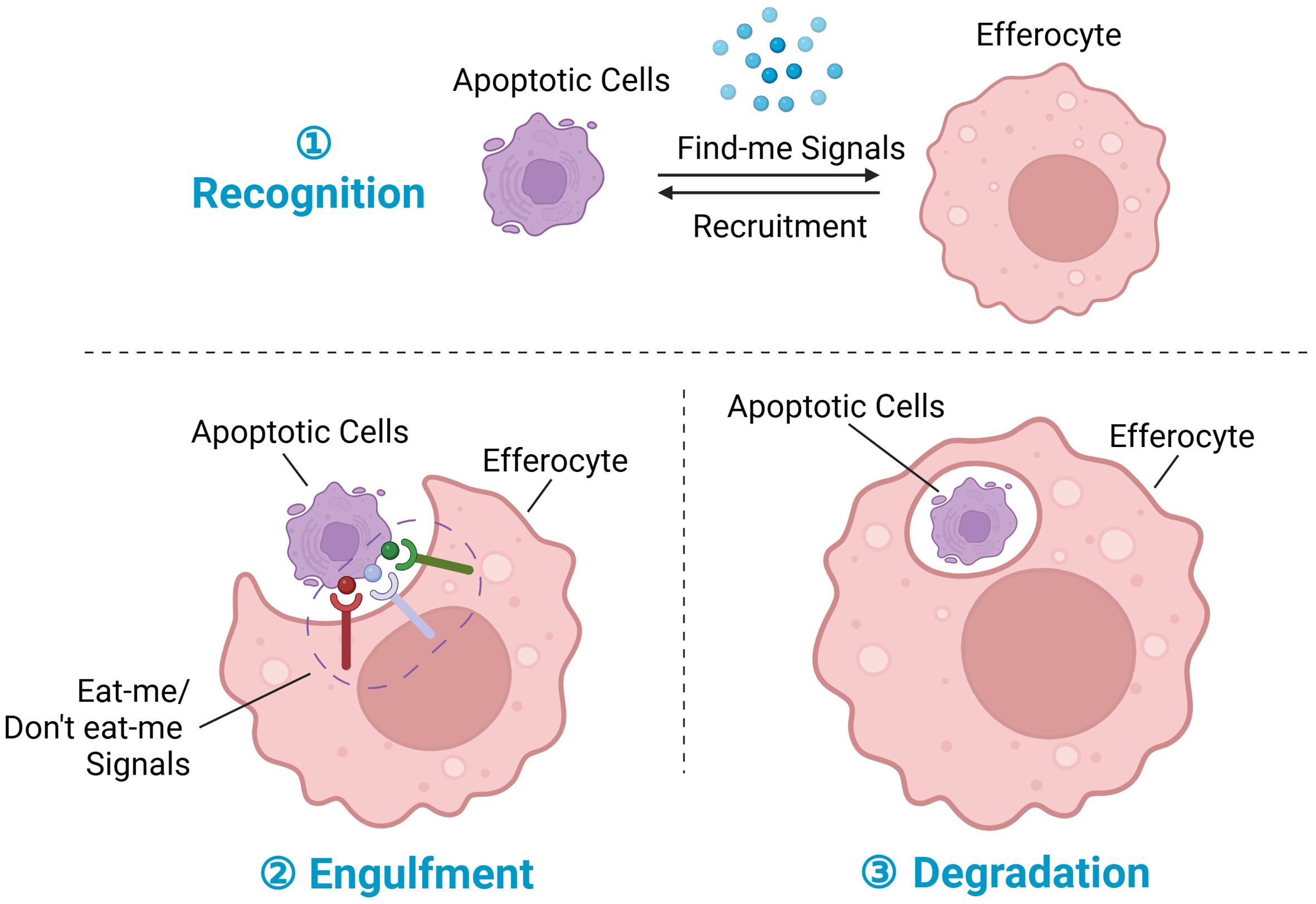
Figure 1. Steps in efferocytosis. ① Recognition phase: Following tissue injury, cells at the injury site undergo various programmed cell death pathways, notably apoptosis, ferroptosis, pyroptosis, and necroptosis, with apoptosis being the most prominent. During apoptosis, dying cells release chemokines termed “find-me” signals, which serve to recruit efferocytes to the injury site. ② Engulfment phase: Membrane proteins on dying cells interact with corresponding receptors on phagocytes, facilitating the regulation of efferocytosis; these membrane proteins are collectively referred to as “eat-me” signals or “don’t eat-me” signals. ③ Degradation phase: Upon recognition and engulfment of dying cells, the process of subsequent degradation commences. This process involves the maturation of the phagosome, its transformation into a phagolysosome, and the subsequent breakdown and digestion of the engulfed dying cells. Created with BioRender.com.
2.1 Recognition of dying cells: “Find-me” signals and “Eat-me” signals
Following tissue injury, cells at the injury site undergo various programmed cell death pathways, notably apoptosis, ferroptosis, pyroptosis, and necroptosis, with apoptosis being the most prominent (15). During these programmed cell death pathways, dying cells release chemokines termed “Find-me” signals, which serve to attract phagocytes to the injury site. Additionally, membrane proteins on dying cells interact with corresponding receptors on phagocytes, facilitating the regulation of efferocytosis; these membrane proteins are collectively referred to as “Eat-me” signals (14).
After injury and apoptosis, cellular membranes undergo rupture and release soluble signals that function in dual capacities: firstly, as chemokines directing phagocytes towards apoptotic cells; and secondly, in priming phagocytes for engulfment by modulating their cytoskeletal dynamics and enhancing the expression of engulfment receptors and digestive machinery. These “find-me” signals encompass chemokines (e.g., CX3CL1 (16) also known as fractalkine), lipids [e.g., LPC (17)], and nucleotides (e.g., ATP and UTP). During early apoptosis, apoptotic cells release CX3CL1, which binds to CX3CR1 on phagocytes and guides them to the periphery of apoptotic cells (1). LPC, an initial “find-me” signal, binds to G-protein-coupled receptor G2A to induce efferocytosis, with ATP-binding cassette transporter 1 (ABCA1) expressed by apoptotic cells also facilitating LPC release (18). Sphingosine 1-phosphate (S1P), synthesized in a caspase-dependent manner, is abundantly released post-apoptosis and engages S1P receptors on phagocytes to promote efferocytosis (19, 20). Both LPC and S1P serve as apoptosis-specific “Find-me” signals. During apoptosis, caspase-3 cleavage activates calcium-independent phospholipase A2, which synthesizes LPC from phosphatidylcholine. Additionally, some apoptotic cells upregulate S1P mitogen-activated protein kinases SPK1 and SPK2, phosphorylating sphingosine to produce S1P (Figure 2) (21). Binding of S1P to S1P receptor (S1PR) on macrophages fosters an autocrine signaling loop involving hypoxia-inducible factor-1α (HIF-1α), erythropoietin (EPO), EPO receptor, and peroxisome proliferator-activated receptor-α (PPARα), which upregulates receptors for dying cells (22). Nucleotides (ATP/UTP) also act as chemokines, promoting recruitment of phagocytes via binding to P2X or P2Y receptors following cleavage of the C-terminal tail of Panx1 of plasma membrane Pannexin-1 (Panx1) channels by caspase 3/7 during apoptosis (6, 23). These released nucleotides “prime” phagocytes for engulfment by inducing expression of binding and engulfment receptors such as CD11b and α5β3 integrin (24).
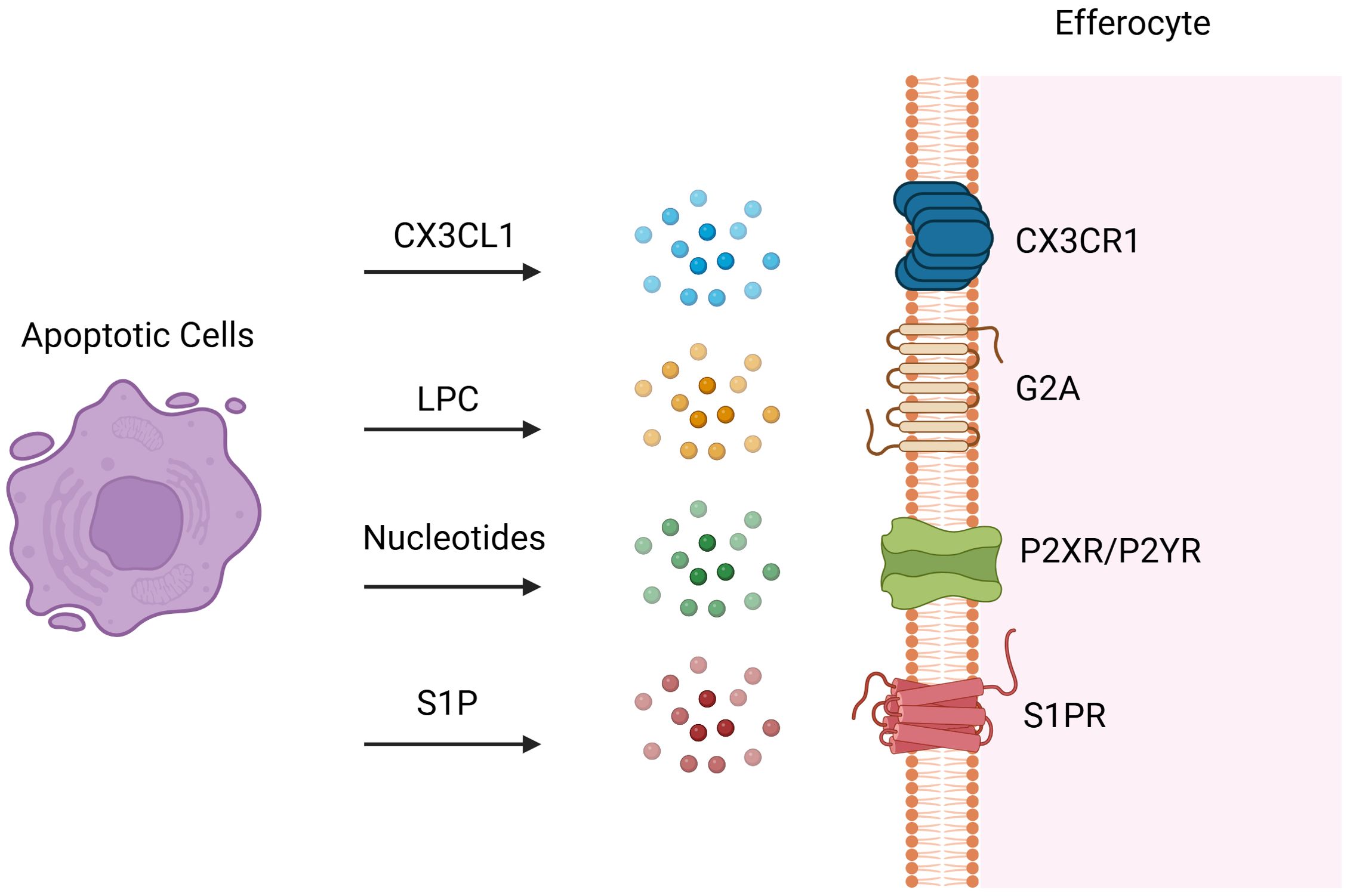
Figure 2. Find-me Signals. Following tissue injury, cells at the injury site undergo various programmed cell death pathways and dying cells release soluble find-me signals to attract efferocytes. These “find-me” signals encompass chemokines such as CX3C-chemokine ligand 1 (CX3CL1) also known as fractalkine, lipids such as sphingosine 1-phosphate (S1P) and lysophosphatidylcholine (LPC), and nucleotides such as ATP and UTP. Dying cells release CX3CL1, which binds to CX3CR1 on efferocytes and attract them to the injury site. LPC, an initial “find-me” signal, binds to G-protein-coupled receptor G2A to induce efferocytosis. S1P is synthesized in a caspase-dependent manner and abundantly released post-apoptosis and engages S1P receptors on efferocytes to induce efferocytosis. Nucleotides (ATP/UTP) also act as chemokines, promoting recruitment of efferocytes via binding to P2X or P2Y receptors. Created with BioRender.com.
Upon reaching the site of injury, phagocytes encounter “eat-me” signals displayed on the membranes of dying cells, initiating downstream efferocytosis either directly or indirectly through receptor interactions. These “eat-me” signals encompass phosphatidylserine (PtdSer), calreticulin, intercellular adhesion molecule 3 (ICAM3), and glycosylation moieties. In healthy cells, PtdSer is predominantly localized within the inner leaflet of plasma membranes, maintained by flippases. During apoptosis, caspase-3-mediated inactivation of flippases and activation of XKR8-independent scramblase activity induce rapid translocation of PtdSer from the inner to the outer leaflet, exposing it extracellularly for binding to phagocyte surface receptors directly (25). These receptors include stabilins (e.g., stabilin1 and stabilin2), T cell immunoglobulin mucin receptors (TIM family, such as TIM1, TIM3, and TIM4), adhesion G protein-coupled receptor B1 (ADGRB1, also known as BAI1), the receptor for advanced glycation end products (RAGE), and members of the CD300 family (CD300a and CD300b) (26, 27). Additionally, scavenger receptor CD36 interacts preferentially with oxidized PtdSer than non-oxidized PtdSer, enhancing phagocytic clearance of dying cells (28). Bridging ligands act as medium molecule between apoptotic cells and phagocytes, facilitating the binding of phagocytes to PtdSer indirectly. For instance, TAM receptor tyrosine kinases (TYRO3, AXL, and MerTK) recognize apoptotic cells via specific bridging ligands like growth arrest-specific protein 6 (GAS6) and Protein S. GAS6 interacts with all three TAM receptors to promote apoptotic cell binding, whereas Protein S only selectively binds to TYRO3 and MerTK (29). Integrins, another indirect receptor, require milk fat globule-EGF factor 8 (MFGE8) or its homologue developmental endothelial locus-1 (DEL-1) to bind PtdSer on dying cells (Figure 3) (30–32). Low-density lipoprotein receptor-related protein (LRP) can engage PtdSer with the aid of the bridging molecule beta-2-glycoprotein 1 (β2-GP1), facilitating engulfment by phagocytes (33). Other molecules acting as molecular bridges between apoptotic cargo and phagocytes include annexin A1 or lipocortin-1 interacting with PtdSer, and galectin-3 (Gal-3) interacting with MerTK (34, 35).
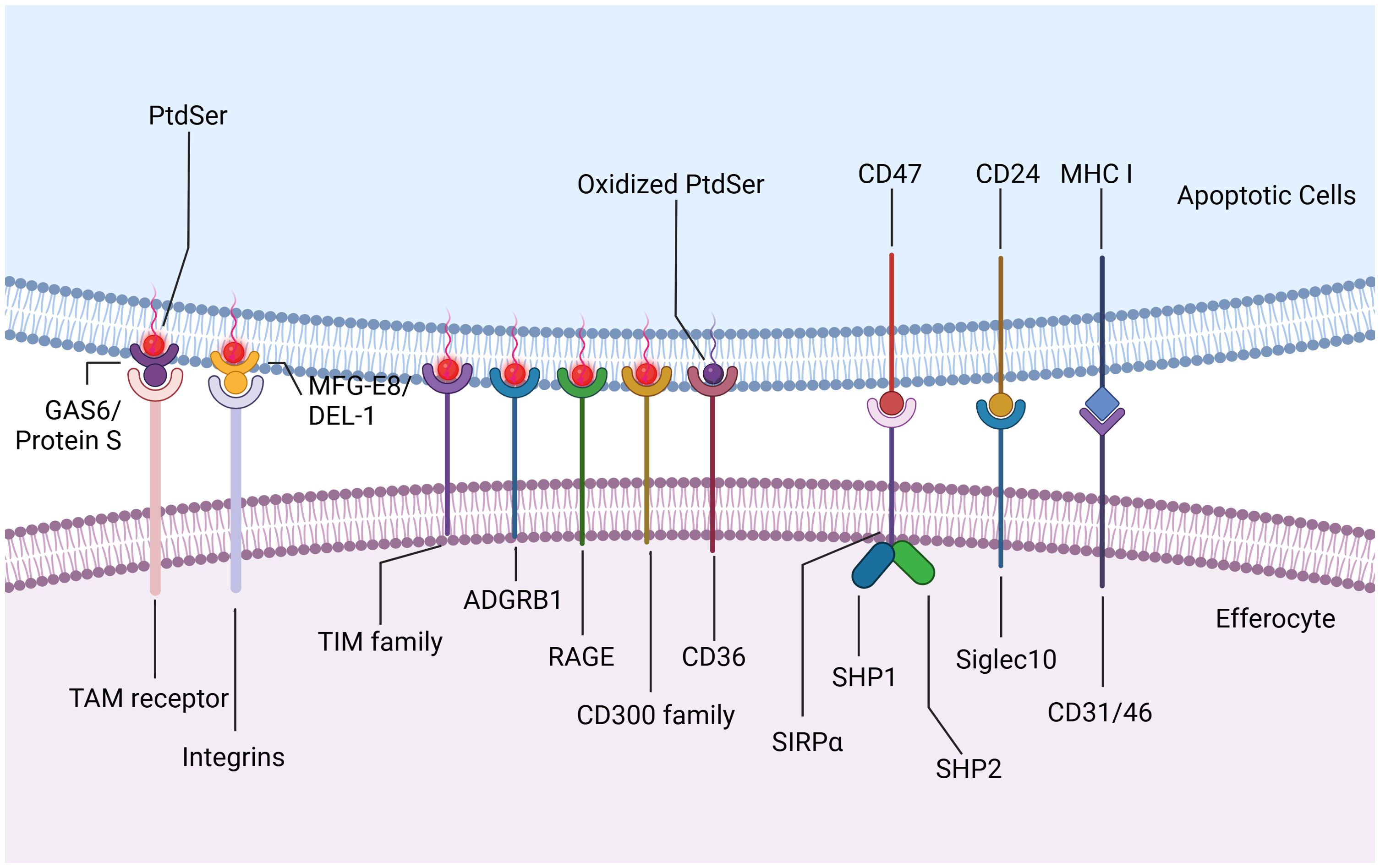
Figure 3. Eat-me Signals and Don’t eat-me Signals. Upon reaching the site of injury, efferocytes encounter “eat-me” signals displayed on the membranes of apoptotic cells, initiating downstream efferocytosis either directly or indirectly through receptor interactions. PtdSer translocates from the inner to the outer leaflet and exposes extracellularly for binding to efferocytes surface receptors directly. These receptors include stabilins such as stabilin1 and stabilin2, T cell immunoglobulin mucin receptors (TIM family) such as TIM1, TIM3, and TIM4, adhesion G protein-coupled receptor B1 (ADGRB1, also known as BAI1), the receptor for advanced glycation end products (RAGE), and members of the CD300 family such as CD300a and CD300b. Scavenger receptor CD36 interacts preferentially with oxidized PtdSer than non-oxidized PtdSer, enhancing phagocytic clearance of apoptotic cells. TAM receptor tyrosine kinases (TYRO3, AXL, and MerTK) recognize apoptotic cells via specific bridging ligands like growth arrest-specific protein 6 (GAS6) and Protein S. GAS6 interacts with all three TAM receptors to promote apoptotic cell binding, whereas Protein S only selectively binds to TYRO3 and MerTK. Integrins, another indirect receptor, require milk fat globule-EGF factor 8 (MFGE8) or its homologue developmental endothelial locus-1 (DEL-1) to bind PtdSer on apoptotic cells. In addition to “eat-me” signals, apoptotic cells also express “Don’t eat-me” signals to inhibit efferocytosis. CD47, typically expressed on healthy cells, interacts with SIRPα on phagocytes to prevent their engulfment. During apoptosis, CD47 on dying cells similarly engages with SIRPα, leading to the phosphorylation of SIRPα’s cytoplasmic domain and subsequent recruitment and activation of phosphatases SHP1/2. CD24 interacts with sialic acid binding immunoglobulin-like lectin 10 (Siglec10) on efferocytes, thereby evading efferocytosis. CD31, as well as CD46 with class I MHC molecules, contribute to preventing engulfment under healthy and apoptotic conditions. Created with BioRender.com.
In addition to “eat-me” signals, dying cells also express “Don’t eat-me” signals such as CD47 and CD24. CD47, typically expressed on healthy cells, interacts with SIRPα on phagocytes to prevent their engulfment. During apoptosis, CD47 on dying cells similarly engages with SIRPα, leading to the phosphorylation of SIRPα’s cytoplasmic domain and subsequent recruitment and activation of phosphatases SHP1/2. These phosphatases inhibit phagocytosis by suppressing non-muscle myosin IIA activity (36, 37). CD24, recently identified as highly expressed on tumor cells, interacts with sialic acid binding immunoglobulin-like lectin 10 (Siglec10) on tumor-associated macrophages, thereby evading efferocytosis (38). Additionally, homophilic interactions of CD31 on leukocytes and macrophages, as well as CD46 with class I MHC molecules, contribute to preventing engulfment under healthy and apoptotic conditions (Figure 3) (39, 40). Although targeting these anti-phagocytic receptors is an underexplored area, it holds promise for influencing cardiovascular and cerebrovascular inflammation pathways.
2.2 Engulfment of dying cells
Efferocytosis is a tightly regulated process that encompasses the recognition, engulfment, and subsequent degradation of dead and dying cells. Upon recognition of dying cells by phagocytes, the process of engulfment and subsequent degradation commences. This process involves the maturation of the phagosome, its transformation into a phagolysosome, and the subsequent breakdown and digestion of the engulfed material.
2.2.1 Uptake of dying cells
Upon recognizing dying cells, phagocytes initiate actin remodeling in the membrane and form phagosomes through alterations in membrane morphology, ultimately leading to engulfment (41). Actin remodeling involves two primary mechanisms converging on the RHO family small GTPase RAC1, a key regulator in this process. In one mechanism, LRP1 and the adapter protein GULP are implicated in RAC1 activation, although the specific mechanism remains unclear (42). In another mechanism, receptor ligation by dying cells and phagocytes activates the guanine nucleotide exchange factor (GEF) ‘TRIO’, which loads GTP onto the small GTPase RHOG. This activation leads to recruitment of the engulfment and cell motility protein (ELMO), facilitating interaction with the SH3 domain of DOCK180 (43). The resulting DOCK180-ELMO complex acts as another GEF for RAC1, thereby activating RAC1. Activated RAC1 then promotes localized actin polymerization, crucial for forming the actin nucleation core necessary to coat or grasp the cargo (Figure 4A) (44). This process involves nucleation-promoting factors from the WASP family, including SCAR and WAVE, which recruit the ARP2/3 complex to facilitate actin assembly (45).
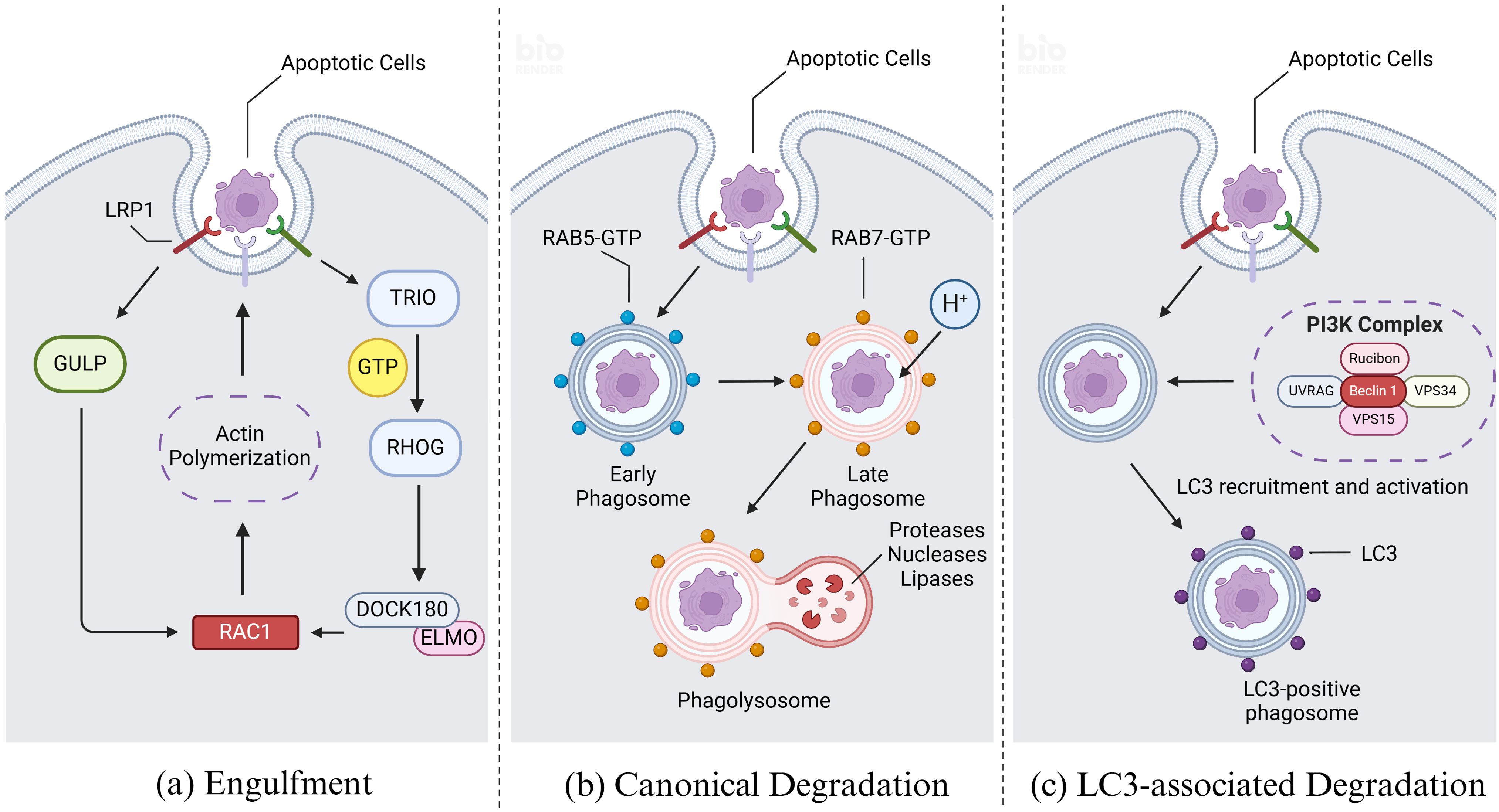
Figure 4. Engulfment and degradation of apoptotic cells. (A) Upon recognizing dying cells, efferocytes initiate actin remodeling and form phagosomes, ultimately leading to engulfment. Actin remodeling involves two primary mechanisms converging on the RHO family small GTPase RAC1. In the first mechanism, LRP1 and the adapter protein GULP are implicated in RAC1 activation, although the specific mechanism remains unclear. In the second mechanism, receptor ligation by apoptotic cells and efferocytes activates the guanine nucleotide exchange factor (GEF) ‘TRIO’, which loads GTP onto the small GTPase RHOG. This activation leads to recruitment of the engulfment and cell motility protein (ELMO), facilitating interaction with the SH3 domain of DOCK180. The resulting DOCK180-ELMO complex acts as another GEF for RAC1, thereby activating RAC1. Activated RAC1 then promotes the forming of the actin nucleation core which is necessary to coat or grasp the cargo. (B) Upon recognition of apoptotic cells by efferocytes, the membrane wraps around the apoptotic and enters the cell to form early phagosome. Afterwards the early phagosome matures and transforms into a late phagosome, distinguished by specific biochemical markers such as the acquisition of RAB7 and the concurrent loss of RAB5. Following the formation of the late phagosome, it undergoes fusion with lysosomes, which are rich in a diverse array of proteases, nucleases, and lipases responsible for the degradation of phagosomal cargo. Key to the regulation of these processes are the RAB GTPase family proteins, which cycle between an active, GTP-bound state and an inactive, GDP-bound state. RAB proteins interact with effector molecules that mediate various intracellular functions, including motor-driven vesicle trafficking, vesicle fusion events, and signaling pathways that regulate ‘RAB conversion’ and activation of downstream RAB GTPase family members. (C) Modification of phagosome maturation through LC3-associated phagocytosis (LAP) represents a noncanonical pathway involving autophagy proteins, specifically the microtubule-associated protein 1A/1B light chain 3 (LC3) family. Upon phagocytic engagement, a phosphatidylinositol 3-kinase (PI3K) complex assembles at the phagophore, comprising rubicon, UVRAG, beclin 1, VPS34, and VPS15. This complex phosphorylates phosphatidylinositides (PtdIns) to generate phosphatidylinositol 3-phosphate (PI3P), which is crucial for the activation of reactive oxygen species (ROS) production via NADPH oxidase-2 (NOX2). PI3P also facilitates the recruitment and activation of the LC3 conjugation machinery. Created with BioRender.com.
2.2.2 Degradation by lysosomes
Following the formation of the phagosome, it undergoes fusion with lysosomes, which are rich in a diverse array of proteases, nucleases, and lipases responsible for the degradation of phagosomal cargo (46). This fusion process is tightly regulated and influenced by multiple steps and biochemical changes at the phagosomal membrane. Key to the regulation of these processes are the RAB GTPase family proteins, which cycle between an active, GTP-bound state and an inactive, GDP-bound state. RAB proteins interact with effector molecules that mediate various intracellular functions, including motor-driven vesicle trafficking, vesicle fusion events, and signaling pathways that regulate ‘RAB conversion’ and activation of downstream RAB GTPase family members (Figure 4B) (47).
RAB5 plays a pivotal role in orchestrating phagosomes trafficking and the biogenesis of early phagosomes during maturation processes. It functions by recruiting and activating several critical effector proteins, including early endosomal antigen 1 (EEA1), the vacuolar fusion proteins MON1A and MON1B, and the class III phosphatidylinositol 3-kinase VPS34, all of which are essential for effective efferocytosis (48). Subsequently, VPS34 catalyzes the conversion of phosphatidylinositol into phosphatidylinositol 3-phosphate (PI3P), a key signaling lipid necessary for optimal phagosome maturation. This catalytic activity is further facilitated by the serine/threonine kinase VPS15, which forms a complex with VPS34 to activate RAB5 and promote its functions in phagosome biogenesis and trafficking (49, 50).
Early phagosomes undergo a transition to late phagosomes, distinguished by specific biochemical markers such as the acquisition of RAB7 and the concurrent loss of RAB5 (51). Late phagosomes further mature through the accumulation of two key RAB7 effector proteins: RAB7-interacting lysosomal protein (RILP) and oxysterol-binding protein-related protein 1 (ORP1 or ORPL1). These proteins facilitate interactions with the molecular motor dynein-dynactin, thereby coordinating microtubule-dependent vesicular trafficking of RAB7-positive late phagosomes (52). Concurrently, VAMP7 and syntaxin 7 are recruited to the phagosome membrane to form a Ca2+-dependent SNARE complex, promoting fusion between the phagosome and lysosome. Upon fusion, the resulting phagolysosome exhibits high acidity (pH 4.5-5.0), which is crucial for the efficient degradation of internalized cell corpse, facilitated by the presence of active cathepsins within the phagolysosomal lumen (53, 54).
2.3 Modification of phagosome maturation: LC3-associated phagocytosis
Modification of phagosome maturation through LC3-associated phagocytosis (LAP) represents a noncanonical pathway involving autophagy proteins, specifically the microtubule-associated protein 1A/1B light chain 3 (LC3) family (55). LC3 lipidation, critical for autophagosome trafficking and autophagosome/lysosome fusion, also plays a pivotal role in LAP, where the cargo primarily comprises dying cells and pathogens, distinct from the cellular organelles targeted in classical autophagy under intracellular stress conditions. LAP is particularly activated in response to injury or the phagocytosis of dying cells, in contrast to autophagy activation mechanism. Central to LAP is its regulatory function in innate immune cell activation subsequent to the recognition and engulfment of dead cell cargo (56). Upon phagocytic engagement, a phosphatidylinositol 3-kinase (PI3K) complex assembles at the phagophore, comprising rubicon, UVRAG, beclin 1, VPS34, and VPS15 (57). This complex phosphorylates phosphatidylinositides (PtdIns) to generate phosphatidylinositol 3-phosphate (PI3P), which is crucial for the activation of reactive oxygen species (ROS) production via NADPH oxidase-2 (NOX2). PI3P also facilitates the recruitment and activation of the LC3 conjugation machinery (Figure 4C). LC3 lipidation onto the phagosomal membrane promotes rapid phagosome maturation and subsequent phagosome-lysosome fusion, ensuring efficient clearance of dying cells and fostering an immune-silent environment (58).
3 Efferocytosis in cardiovascular disease
3.1 Atherosclerosis
Atherosclerotic cardiovascular diseases, such as atherosclerosis and myocardial infarction, result in more fatalities than all types of cancer combined. The principal pathophysiological mechanism involves the development of atherosclerotic plaques. These plaques originate from the accumulation of ApoB-containing lipoproteins within the subendothelial layer of arteries, initiating an inflammatory response that drives leukocyte influx into the vessel wall. Subsequently, these leukocytes undergo apoptosis and are cleared through efferocytosis. However, as the plaque advances, inefficient clearance of apoptotic cells leads to the accumulation of secondarily necrotic cells within the plaque, forming the necrotic core. These necrotic cores constitute the critical characteristic of plaque vulnerability, increasing the risk of rupture, luminal thrombosis, and thereby contributing significantly to the likelihood of myocardial infarction and stroke (2, 59). Hence, efferocytosis plays a pivotal role in resolving inflammation and mitigating atherosclerosis (Table 1).
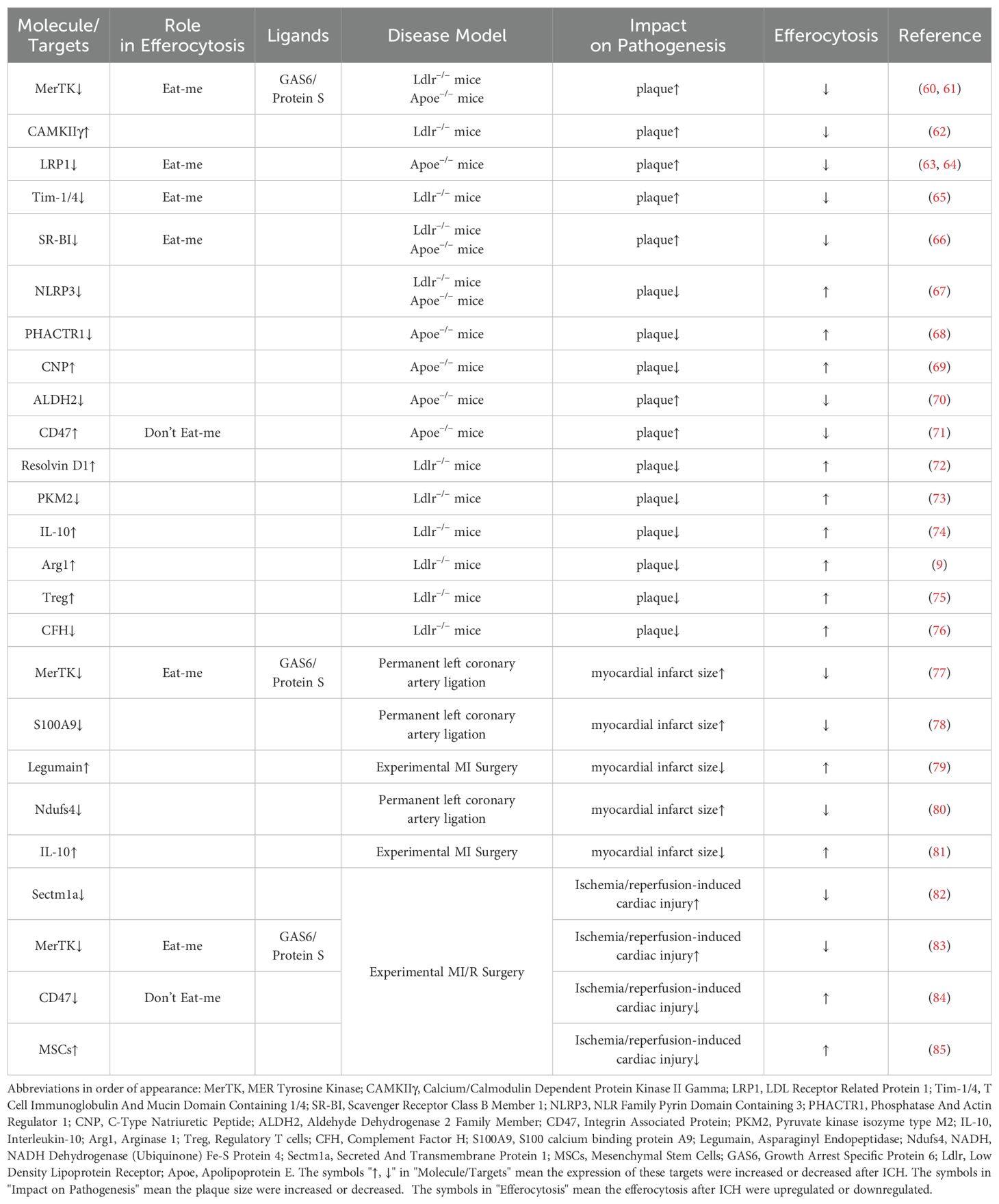
Table 1. Targets involved in efferocytosis in cardiovascular disease.
As atherosclerosis progresses, persistent inflammatory stimuli lead to the cleavage of MerTK from the cell surface. This cleaved MerTK competes with cell surface MerTK on phagocytes for binding with GAS6 and protein S, thereby impairing efferocytosis within the lesion. Genetically engineered mice expressing cleavage-resistant MerTK exhibit enhanced efferocytosis and reduced propensity for developing necrotic cores compared to control mice (60, 86). Conversely, mice lacking or expressing an inactive form of MerTK demonstrate increased plaque size and expanded necrotic core area (61, 87). Similarly, LRP1, progressively downregulated during atherosclerosis, can also be rendered inactive through ADAM17-mediated proteolytic cleavage akin to MerTK (86). Macrophages in advanced lesions in both human and murine models upregulate calcium/calmodulin-dependent protein kinase IIγ (CaMKIIγ), which suppresses the ATF6-LXRα-MERTK pathway. Consequently, Western diet-fed Ldlr−/− mice lacking myeloid CaMKIIγ exhibit elevated macrophage MerTK expression, improved efferocytosis, and reduced necrotic core area in atherosclerotic lesions (62). Loss of LRP1 in hematopoietic cells exacerbates atherosclerosis by impairing efferocytosis and increasing lesion area and necrotic core size in high-fat diet-fed atheroprone mice (63, 64). Additionally, blocking antibodies against TIM1 or TIM4, which serve as ‘Eat-me signals’, promote atherosclerosis progression by inhibiting efferocytosis within the lesion, leading to secondary accumulation of necrotic cells and heightened production of pro-inflammatory cytokines. Furthermore, TIM blockade induces substantial reductions in regulatory T cells in circulation, which otherwise stimulate lesional efferocytosis in an IL-10-dependent manner (65). Scavenger receptor class B member 1 (SRB1) facilitates efferocytosis via the SRC-PI3K-RAC1 pathway, and its deletion in macrophages results in defective efferocytosis, increased plaque size, larger necrotic core area, and heightened inflammation in atherosclerosis (66). Moreover, caspase 1 inhibitor VX765 could suppress NLRP3 inflammasome assembly and atherosclerosis progression by promoting mitophagy and efferocytosis (67).
CDKN2B, a key gene in atherosclerosis, exhibits reduced calreticulin levels in Cdkn2b−/−Apoe−/− mice compared to controls, contributing to increased plaque size and necrotic core area under Western diet conditions. Notably, atherosclerotic plaques from patients often exhibit genetic variations at the 9p21 locus, including the CDKN2B gene, which correlate with reduced calreticulin levels and compromised efferocytosis, thereby indicating poorer prognosis (68). C-type natriuretic peptide (CNP) negatively correlates with coronary atherosclerosis burden in patients by promoting an anti-inflammatory macrophage phenotype, enhancing efferocytosis, and reducing foam cell formation and necroptosis (69). Several genome-wide association studies have linked intronic variants in phosphatase and actin regulator 1 (PHACTR1) on chromosome 6p24 with coronary artery disease (CAD) risk. The rs9349379 risk allele G, which lowers PHACTR1 expression in macrophages, may increase CAD risk via impaired efferocytosis, as PHACTR1 prevents myosin light chain (MLC) dephosphorylation necessary for apoptotic cell engulfment (68). Clinical studies highlight ALDH2 rs671 mutation as a common risk factor for atherosclerotic cardiovascular diseases, where macrophage ALDH2 stabilizes Rac2 to facilitate efferocytosis and reduce atherosclerosis development. Wild-type ALDH2 interacts directly with Rac2 to inhibit its degradation, whereas the rs671 mutant enhances Rac2 instability through increased K48-linked polyubiquitination at lysine 123 (70).
In atherosclerotic plaques, CD47 expression is significantly upregulated on apoptotic and necroptotic cells, contributing to defective efferocytosis. Administration of CD47-blocking antibodies reverses this defect, normalizes vascular tissue clearance, and mitigates atherosclerosis in various mouse models (71). The long non-coding RNA myocardial infarction-associated transcript (MIAT), elevated in atherosclerosis patients, interferes with miR-149-5p post-translational processing, thereby increasing CD47 levels. Genetic targeting of MIAT in Apoe−/−mice decreases CD47 expression, improves efferocytosis, and reduces plaque necrosis (88).
Efferocytosis induces alterations in lipid mediator production that are pivotal for inflammation resolution. Macrophages exposed to apoptotic neutrophils or neutrophil-derived microparticles exhibit heightened expression of specialized pro-resolving mediators (SPMs), such as lipoxin A4, resolvins D1, D2, and E2, derived from long-chain fatty acids (72, 89). Concurrently, there is a reduction in pro-inflammatory prostaglandins and leukotriene B4. Advanced human atherosclerotic lesions demonstrate decreased levels of SPMs compared to early-stage lesions, correlating with impaired efferocytosis. Treatment of mice with advanced atherosclerosis using resolving mediators enhances lesional efferocytosis. Conversely, elevated levels of proinflammatory lipid mediators in advanced atherosclerosis can impair efferocytosis. For instance, 12(S)-hydroxyeicosatetraenoic acid, derived from arachidonic acid and elevated in progressing atherosclerotic lesions and serum of individuals with coronary artery disease, inhibits efferocytosis in human monocyte-derived macrophages by activating RHOA, thereby blocking phagosome internalization. This impairment can be rectified by co-incubation with statins, which inhibit RHOA through the blockade of isoprenylation, a critical RHOA-activating modification (90). Macrophage efferocytosis is intricately linked to energy metabolism. Elevated levels of an enzyme regulating energy metabolism have been observed in macrophages from patients with atherosclerotic coronary artery disease. Conditional deletion of PKM2 in myeloid cells of Ldlr-/- mice fed a high-fat Western diet for 14 weeks upregulated LRP-1 and enhanced macrophage efferocytosis, leading to regression of lesions in the entire aorta and aortic sinus. Depletion of PKM2 also suppressed MCP-1, IL-1β, and IL-12 expression, indicating inhibition of the macrophage proinflammatory phenotype (73).
Effective efferocytosis contributes to the synthesis of IL-10, TGF-β, and Arg-1 synthesis in atherosclerotic plaques. During efferocytosis, macrophage ornithine decarboxylase promotes the production of putrescine, which up-regulates IL-10 expression through the MerTK-MAPK-ERK1/2 pathway (74). IL-10, in turn, enhances efferocytosis in advanced atherosclerosis via the IL-10/Vav1/Rac1 pathway (91). Effective efferocytosis also induces the production of prostaglandin E2 (PGE2) via the CD36/ERK/Ptgs2/COX2 pathway, and PGE2 further stimulates TGF-β synthesis, which contributes to anti-inflammatory and pro-resolving properties (92). Additionally, effective efferocytosis up-regulates Arg-1 expression in macrophages, often associated with M2 polarization. Arg-1 metabolizes arginine and ornithine derived from apoptotic cells to produce putrescine, which enhances Dbl/Rac1-mediated engulfment of secondary apoptotic cells, thereby sustaining efferocytosis and promoting regression of atherosclerosis (9).
Adaptive immunity plays a pivotal role in the pathogenesis of atherosclerosis. Regulatory T cells (Tregs) are instrumental in attenuating proinflammatory responses and promoting resolution of inflammation. Single-cell RNA-sequencing of immune cells within atherosclerotic plaques has underscored the critical role of Tregs in enhancing efferocytosis and resolving atherosclerotic cardiovascular disease (75, 91). Cell-autonomous regulation of complement component C3 by complement factor H (CFH) modulates macrophage efferocytosis and impacts the progression of atherosclerosis. In a murine model of atherosclerosis, CFH deficiency has been shown to limit plaque necrosis in a manner dependent on complement C3. Specifically, deletion of CFH in monocyte-derived inflammatory macrophages leads to dysregulated cell-autonomous consumption of complement C3 without subsequent activation of downstream complement C5, thereby enhancing efferocytosis capacity (76).
3.2 Myocardial injury and repair
Heart failure subsequent to myocardial infarction (MI) continues to impose a significant burden of morbidity and mortality. Despite pharmacological advancements such as β-blockers and angiotensin-converting enzyme (ACE) inhibitors, which have effectively reduced mortality rates, the residual risk of developing heart failure post-MI remains considerable. Timely reperfusion through thrombolytic agents or primary percutaneous coronary intervention represents the most efficacious intervention for MI patients. However, reperfusion of the ischemic heart can induce additional damage, including extensive cardiomyocyte death and subsequent cardiac inflammation. This section will thus discuss recent research advances in efferocytosis in the context of MI (Table 1) (82, 93).
Following myocardial injury or ischemia/reperfusion injury, efferocytosis plays a critical role in the clearance of dying cells and damage-associated molecular patterns (DAMPs), thereby mitigating secondary cytokine storms and reducing inflammatory responses. Similar to its role in atherosclerosis, MerTK activation also facilitates efferocytosis in myocardial infarction. MerTK-mediated clearance of apoptotic cardiomyocytes accelerates inflammation resolution and enhances secretion of vascular endothelial growth factor A (VEGFA), thereby promoting cardiac repair post-injury (77). Maintaining elevated MerTK expression, potentially through extracellular vesicles secreted by cardiosphere-derived cells, could enhance efferocytosis efficacy and improve outcomes following myocardial infarction (94). Moreover, inhibiting the alarmin S100A9 may increase the population of reparative Ly6ClowMerTKhigh macrophages, thereby further enhancing efferocytosis and facilitating cardiac recovery (78).
As the principal phagocytes in the cardiovascular system, macrophages play a crucial role in efferocytosis following myocardial infarction. Cardiac resident macrophages, through the action of legumain, contribute to cardiac repair by facilitating the clearance and degradation of apoptotic cardiomyocytes post-myocardial infarction (79). Additionally, mitochondrial function in macrophages is pivotal in regulating cardiac repair processes after myocardial injury. Deletion of mitochondrial complex I protein Ndufs4 specifically in myeloid cells (mKO) recapitulates a proinflammatory metabolic phenotype in macrophages and exacerbates their response to lipopolysaccharide (LPS). Impaired efferocytosis in mKO macrophages results in reduced expression of anti-inflammatory cytokines and tissue repair factors, accompanied by heightened inflammatory responses. Mitochondria-targeted reactive oxygen species (ROS) scavenging ameliorates these impairments, enhances myofibroblast function, and reduces post-myocardial infarction mortality in mKO mice (80).
IL-10 is crucial in this context as an essential cytokine involved in MI. Co-incubation of apoptotic cells (ACs) with macrophages stimulates an increase in oxygen consumption rate and IL-10 production in macrophages, a process mediated by fatty acid oxidation. Fatty acids derived from apoptotic cells promote IL-10 synthesis through mitochondrial β-oxidation and regulation of electron transport, leading to an elevated NAD+/NADPH ratio. This heightened NAD+ level activates SIRT1, which in turn facilitates Pbx-1 binding to the apoptotic cell response element within the IL-10 promoter. Consequently, this cascade enhances IL-10 expression, thereby contributing to improved cardiac repair following ischemia/reperfusion injury (81).
Ischemia/reperfusion-induced cardiac injury represents a significant portion of myocardial damage. Macrophage-enriched Sectm1a plays a pivotal role in promoting effective efferocytosis to mitigate ischemia/reperfusion-induced cardiac injury and enhance cardiac function (82). Following ischemia/reperfusion-induced cardiac injury, MerTK undergoes cleavage similarly to the pattern observed in atherosclerosis in both humans and mice. The cleavage of MerTK on resident cardiac macrophages impairs efferocytosis and subsequent repair processes after myocardial ischemia/reperfusion injury (83). CD47 has been demonstrated to inhibit efferocytosis in the context of atherosclerosis. Consequently, genetically engineered macrophages co-loaded with CD47 inhibitors, such as PEP-20, could potentially synergistically restore efferocytosis and enhance cardiac remodeling following myocardial ischemia/reperfusion injury (84). Mesenchymal stem cells (MSCs) have shown considerable promise in the treatment of cardiovascular diseases, indicating their broad therapeutic potential. It has been established that MSC infusion can improve cardiac function in rats following myocardial ischemia/reperfusion, potentially through mechanisms that enhance M2 macrophage-mediated efferocytosis of apoptotic neutrophils (85).
4 Cerebrovascular disease
4.1 Intracerebral hemorrhage
Common cerebrovascular diseases in the population encompass ischemic stroke, intracerebral hemorrhage (ICH), and subarachnoid hemorrhage (SAH). Following the onset of these conditions, the efficient clearance of apoptotic cells is crucial for preserving central nervous system (CNS) homeostasis and facilitating recovery post-injury.
Intracerebral hemorrhage (ICH) is a severe condition characterized by hematoma-induced mass effect. Rupture of cerebral vessels leads to the accumulation of millions of red blood cells (RBCs) within the brain parenchyma, forming a hematoma. Surgical evacuation of hematoma is generally not recommended for most ICH cases due to uncertain clinical benefits and potential surgical complications. Hemolysis within the hematoma can generate toxic byproducts that contribute to significant secondary injuries and irreversible neurological deficits. Hence, promoting rapid hematoma clearance is crucial (95, 96).
Microglia and macrophages, acting as phagocytes, are swiftly recruited to the hemorrhage site to remove RBCs via erythrophagocytosis, a process vital for detoxifying hemolytic products and fostering neurological recovery post-ICH (Table 2) (120). Monocyte-derived macrophages (MDMs) exhibit heightened phagocytic activity and erythrophagocytosis within the ICH-afflicted brain. Recent research has discerned distinct roles of brain tissue-resident microglia and MDMs in the context of hemorrhagic brain injury. Initially, distinguishing between macrophages and microglia in vivo was challenging due to their shared origin and functional similarities. However, advancements in specific cell markers and multichannel flow cytometry have enabled researchers to differentiate these cell types. Consequently, researchers now refer to both microglia and macrophages collectively as Mφ (121).
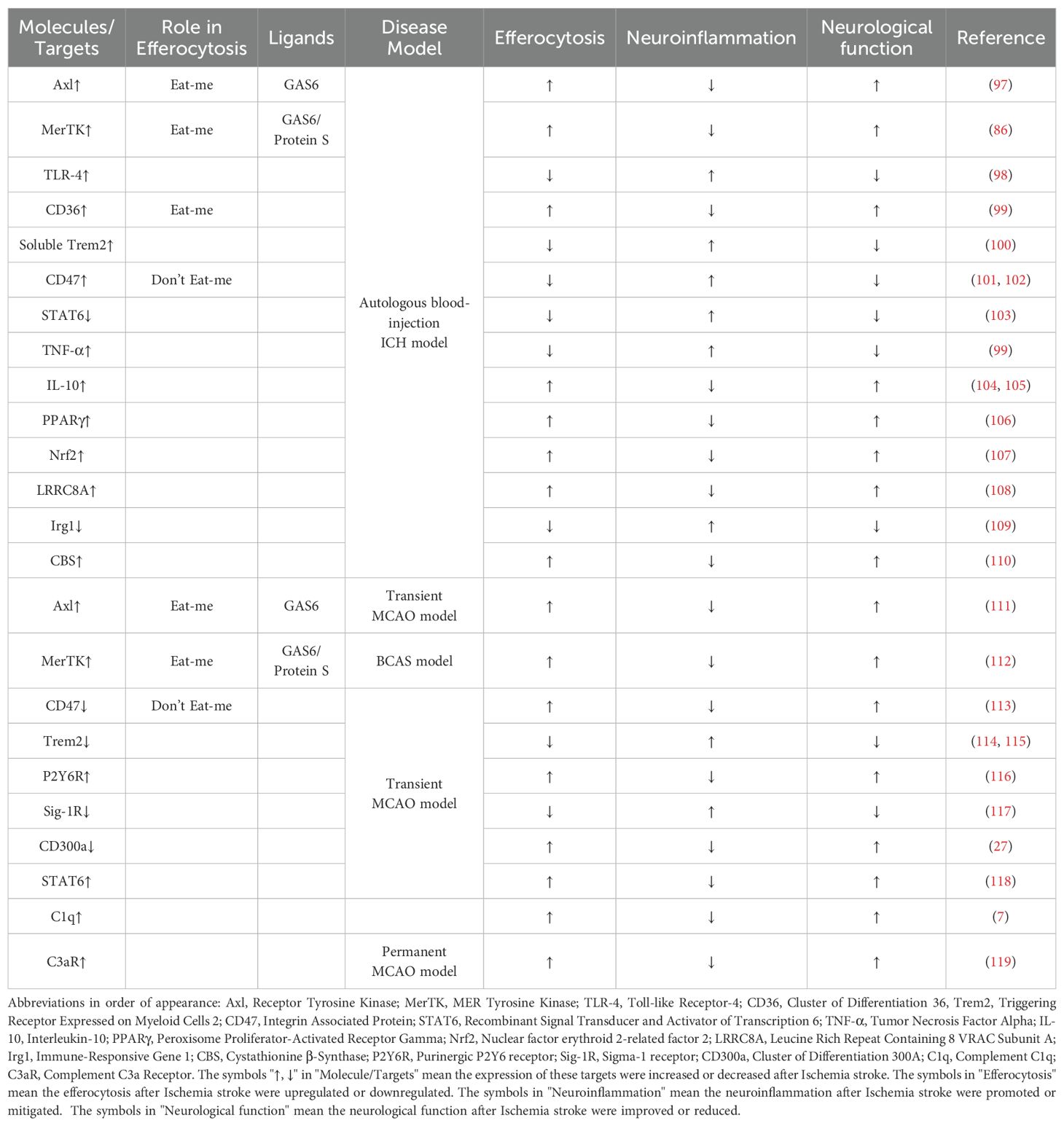
Table 2. Targets involved in efferocytosis in cardiovascular disease.
Tyro3, predominantly expressed on neurons rather than Mφ within the central nervous system (CNS), contrasts with Axl and MerTK, which are primarily expressed on Mφ. In a murine model, the transcriptional levels of Axl, MerTK, and their ligand Gas6 increase within 24 hours post-intracerebral hemorrhage (ICH), and deficiency of Axl/MerTK impairs macrophage-mediated erythrophagocytosis in ICH (97). Toll-like receptors (TLRs) induce a pro-inflammatory Mφ phenotype (M1), whereas Axl/MerTK activation suppresses TLR signaling through suppressors such as SOC1 and SOC3, promoting an anti-inflammatory Mφ phenotype (M2) (98, 122). Exogenous ligands, including recombinant Gas6, can target Axl/MerTK-mediated erythrophagocytosis akin to approaches used in atherosclerosis. Following ICH, Axl/Mertk undergo cleavage from the cell membrane, generating soluble but non-functional forms (sAxl/sMertk). These soluble forms competitively bind to endogenous ligands (Gas6 and Pros1), thereby depleting ligands crucial for maintaining homeostasis. Consequently, exogenous recombinant Gas6 enhances efferocytosis and resolves inflammation in an Axl-dependent manner following ICH (86, 97).
CD36, recognized as an ‘eat-me’ signal, exhibits elevated transcription in microglia cultures treated with erythrocytes and within the perihematomal region following intracerebral hemorrhage (ICH) (123, 124). Genetic deletion or antibody blockade of CD36 impedes erythrocyte phagocytosis by microglia. CD36 knockout mice demonstrate delayed hematoma resolution and exacerbated deficits compared to wild-type (WT) mice post-ICH. Patients lacking CD36 exhibit impaired hematoma resolution and poorer clinical outcomes. Upregulation of CD36 accelerates erythrophagocytosis and enhances hematoma resolution (99). Soluble Trem2 has been shown to negatively regulate erythrophagocytosis post-ICH via CD36 receptor recycling, mediated by reduced VPS35 (100). Similarly, CD47, ubiquitously expressed across various cell types including erythrocytes, serves as a ‘don’t eat me’ signal by binding to signal regulatory protein α (SIRPα) on macrophages (Mφ) to inhibit erythrophagocytosis. Perihematomal levels of CD47 initially rise within hours post-ICH but subsequently decline, coinciding with Mφ infiltration and erythrophagocytosis (101, 125). Intracranial injection of CD47 knockout blood accelerates hematoma resolution and reduces brain edema, effects attenuated by intracranial clodronate liposome administration, a specific Mφ depletion agent (102). Furthermore, CD47 blocking antibodies significantly enhance erythrophagocytosis and promote hematoma clearance following ICH (126, 127).
Interleukin-4 (IL-4) serves as a canonical activator of signal transducer and activator of transcription 6 (STAT6), and exogenous IL-4 administration has been shown to activate STAT6 and enhance erythrophagocytosis in intracerebral hemorrhage (ICH). STAT6 knockout (KO) mice demonstrate exacerbated outcomes compared to wild-type (WT) counterparts in ICH models, showing reduced responsiveness to IL-4 treatment and impaired phagocytic capacity of red blood cells by phagocytes (103). Transcriptomic analyses revealed diminished expression of IL-1 receptor-like 1 (ST2) in microglia/macrophages of STAT6 KO mice post-ICH, underscoring the significance of IL-4/STAT6/ST2 signaling in hematoma resolution and functional recovery after ICH (128). Intranasal IL-4 treatment warrants further investigation as a potential therapeutic strategy for ICH. Moreover, the IL-4/STAT6 axis has been observed to upregulate CD36, a scavenger receptor critical for initiating efferocytosis, potentially through direct binding of STAT6 to the promoter regions of the CD36 gene (129). IL-4/STAT6 signaling also transcriptionally upregulates anti-inflammatory cytokines such as Arg1, which are essential for STAT6-mediated pro-phagocytic activity in Mφ and sustained efferocytosis (118). Conversely, TNF-α inhibits apoptotic cell clearance and polarizes Mφ towards a proinflammatory phenotype, concurrently downregulating CD36 expression in microglia and impairing erythrophagocytosis, while also upregulating the ‘don’t eat-me’ signal CD47 in vascular smooth muscle cells, further hindering erythrophagocytosis (99). IL-10, known for its anti-inflammatory properties, has been reported to promote erythrophagocytosis by regulating CD36 expression (104). Additionally, IL-10 delivery via phosphatidylserine liposomes has shown promise in improving erythrophagocytosis and clinical outcomes in ICH (105).
Transcription factors within Mφ are pivotal in regulating erythrophagocytosis and recovery following intracerebral hemorrhage (ICH). PPARγ transcriptionally upregulates critical scavenger receptors such as Axl, MerTK, and CD36, which are essential for erythrophagocytosis (106, 124). Nrf2 serves as a principal transcription factor protecting cells against endogenous and exogenous stressors, and its activation has been demonstrated to enhance erythrophagocytosis by upregulating CD36 (107). Both PPAR γ and Nrf2 also modulate the NF-κB pathway, contributing to inflammation attenuation in ICH, thereby highlighting targeting PPAR γ and Nrf2 as promising strategies to augment hematoma resolution (130). Bexarotene, an FDA-approved selective RXR (retinoid X receptor) agonist used clinically in cutaneous T-cell lymphoma, enhances the expression of PPARγ-dependent genes through RXR heterodimerization, thereby improving erythrophagocytosis and recovery in ICH (131). Recent insights underscore the regulation of Mφ phagocytosis via the LRRC8A channel through the AMPK-Nrf2-CD36 pathway following ICH, suggesting LRRC8A as a potential therapeutic target for enhancing hematoma clearance (108). Itaconate, an intermediate of the tricarboxylic acid cycle, is produced from the decarboxylation of cis-aconitate by immune-responsive gene 1 (Irg1) within mitochondria. Depletion of Irg1 in macrophages/microglia diminishes erythrocyte phagocytosis, exacerbating outcomes in ICH. Administration of sodium itaconate or 4-octyl itaconate (4-OI) promotes macrophage phagocytosis via the Keap1-Nrf2-CD36 pathway (109). The Irg1/itaconate axis represents a potential therapeutic target for disorders characterized by phagocytosis deficiency, such as ICH.
In addition to the aforementioned findings, hydrogen sulfide (H2S), a gasotransmitter, acts as an endogenous regulator facilitating sustained phagocytosis following intracerebral hemorrhage (ICH). Expression of the H2S synthase cystathionine β-synthase (CBS) and CBS-derived H2S is upregulated in brain-resident phagocytic microglia post-ICH, thereby enhancing continuous erythrocyte phagocytosis via the CBS-H2S-complex I axis (110). Fan et al. have developed pH-responsive pro-efferocytic nanoparticles resembling neutrophils, designed to enhance neurological recovery by promoting erythrophagocytosis after ICH (132).
4.2 Ischemic stroke
Ischemic stroke represents a significant public health burden and remains the leading cause of mortality and disability worldwide. Apart from thrombolysis and thrombectomy during the acute stage, effective therapeutic strategies remain limited (133). The disruption of regional blood supply initiates an ischemic cascade that results in neuronal dysfunction and subsequent cell death. During the sub-acute phase, brain edema and inflammatory responses further contribute to secondary injury processes (134).Following ischemic stroke onset, microglia and macrophages undergo rapid activation and recruitment to the infarct site, promoting efferocytosis (Table 2). TAM receptors, including Axl and MerTK, play crucial roles in enhancing efferocytosis post-ischemic stroke (111, 112). Phagocytic processes are initiated by exposure of “eat-me” signals on target cells or debris, such as phosphatidylserine (PS) exposed on the membranes of dying cells, which interacts with MFG-E8, Axl, and MerTK to facilitate efferocytosis and mitigate injury. Additionally, the ligands Gas6 and Protein S are involved in mediating these processes after ischemic stroke (135). Conversely, ‘don’t eat-me’ signals can modulate microglia and macrophage efferocytosis during ischemic stroke. Research utilizing CD47 knockout mice indicates that deletion of CD47 reduces brain infarction and edema during the acute phase in the middle cerebral artery occlusion (MCAO) model by mitigating neuroinflammation (113).
Accumulating evidence underscores the significant impact of the Triggering receptor expressed on myeloid cells-2 (TREM2)-activating protein of 12kDa (DAP12) system in central nervous system CNS disorders such as neurodegenerative diseases and stroke (136). TREM2, initially expressed on macrophages and microglia, relies on DAP12 as an intracellular membrane adaptor (137). Microglial deficiency in TREM2 impedes the clearance of apoptotic neurons and exacerbates the production of inflammatory cytokines like TNF-α (138). Moreover, TREM2 facilitates efferocytosis of dying cells following experimental stroke. Deficiency in TREM2 exacerbates outcomes after ischemic stroke by diminishing efferocytosis of dying neurons and microglia, underscoring its pivotal role over circulating macrophages in ischemic conditions (114). Furthermore, a high-salt diet has been shown to reduce the efferocytic capacity of macrophages by downregulating TREM2 expression, thereby hindering the resolution of neuroinflammation post-ischemic stroke (139). Augmenting TREM2 signaling in monocytes/macrophages represents a promising therapeutic approach to enhance efferocytosis and promote inflammation resolution following stroke. Recent studies employing microscale thermophoresis (MST), surface plasmon resonance (SPR), and liquid chromatography-tandem mass spectrometry (LC-MS/MS) have revealed sphingosine-1-phosphate (S1P) as a novel ligand for TREM2, which targets the TREM2-DAP12 complex to promote microglial efferocytosis and protect against ischemic brain injury (115).
P2 purinoceptors play a significant role in the pathogenesis of ischemic stroke. These receptors are categorized into two families: ionotropic receptors (P2X) and metabotropic receptors (P2Y). The P2Y6 receptor expressed on microglia is activated by UDP released from dying neurons, initiating microglial efferocytosis to clear apoptotic cells. Upon activation by UDP, P2Y6 receptors induce actin cytoskeleton rearrangement, forming filopodia-like protrusions that facilitate the engulfment of cellular debris (140). Expression of P2Y6 receptors in microglia increases following transient middle cerebral artery occlusion (tMCAO), and inhibition of P2Y6 receptors with the antagonist MRS2578 suppresses microglial phagocytosis of cell debris, thereby exacerbating neurological deficits (116). In addition to the P2Y6/UDP signaling pathway, neuronal injury leads to ATP or ADP release, which recruits microglia to the site of injury through P2Y12 receptors (141). Post-ischemic stroke, P2Y12-mediated chemotaxis of microglia plays a critical role in maintaining blood-brain barrier (BBB) integrity (142). Sigma-1 receptor (Sig-1R) is a chaperone protein that modulates diverse cellular functions, including cell death, autophagy, apoptosis, neuronal differentiation, and neuroinflammation. Depletion of Sig-1R markedly impairs the phagocytic activity of macrophages and microglia, resulting in exacerbated brain damage and neurological deficits. Mechanistically, Sig-1R-mediated efferocytosis relies on Rac1 activation, and specific binding pockets responsible for Sig-1R interactions have been identified (117). CD300a, has a long cytoplasmic region that contains immunoreceptor tyrosine-based inhibitory motifs (ITIMs) akin to SIRPα, also enhances efferocytosis by infiltrating myeloid cells and ameliorates neuronal deficit after ischemic stroke via blocking by its neutralizing antibody (27).
Macrophages play a pivotal role in the phagocytic clearance of deceased neurons following ischemic stroke. Robust transcriptomic alterations occur in monocytes/macrophages infiltrating the post-stroke brain. These changes include significant upregulation of numerous efferocytosis-related genes within brain macrophages. Transcriptomic analyses further reveal that PPARγ and STAT6 act as potential upstream regulators that shape the pro-efferocytic and inflammation-resolving transcriptome of macrophages in the post-stroke brain, akin to their roles observed in intracerebral hemorrhage (ICH) (143). Members of the STAT family are critical in regulating the functional state of microglia/macrophages, with the STAT6/Arg1 axis identified as a key signaling mechanism for phenotypic modulation in the context of ischemic stroke. Activation of the STAT6/Arg1 axis promotes efferocytosis by microglia/macrophages and contributes to inflammation resolution in mouse models of stroke (118).
In addition to the previously discussed non-specific and standard receptors, adaptive immunity also plays a pivotal role in the pathogenesis of ischemia stroke. Complement components C1q and C3 play significant roles in inducing efferocytosis through their interaction with dying cell surfaces. C1q is widely distributed in the central nervous system (CNS), including neutrophils, microglia, and a subset of interneurons. Following ischemic stroke, activation of the complement system leads to increased C1q levels, which enhance microglial efferocytosis by binding to apoptotic cells and neuronal blebs, thereby protecting the CNS (7). C3 is activated and cleaved by C3 convertase into C3a, a small protein that recruits immunocytes and modulates immune responses through interaction with its receptor, C3aR (144). Intracortical administration of the complement C3 receptor antagonist trifluoroacetate has been shown to modulate microglial responses and attenuate neuronal death following brain injury (119). C3b, a product of C3 cleavage, facilitates the clearance of dying cells and modulates adaptive immune responses. Modification of glycoproteins or glycolipids represents a prominent strategy to regulate efferocytosis induced by C1q and C3. Desialylation of glycoproteins or glycolipids enhances recognition by C1q, thereby promoting efferocytosis. Conversely, sialic acid modification of glycoproteins or glycolipids acts as a ‘don’t eat-me’ signal by preventing binding of complement C1q and C3b, thereby modulating the clearance of dying cells (145).
4.3 Subarachnoid hemorrhage
Subarachnoid hemorrhage (SAH) is a relatively common cause of stroke, with an annual incidence of approximately 6-7 cases per 100,000 individuals, predominantly affecting those under 55 years of age. Despite accounting for only 5% of all stroke cases, SAH exhibits a high initial mortality rate, reaching up to 67% in the first few months. Recent studies have highlighted the crucial role of efferocytosis in early brain injury (EBI) following SAH (146). SAH triggers the upregulation of Axl and its ligand Gas6. Administration of recombinant Gas6 (rGas6) has been shown to enhance efferocytosis, reduce inflammation, and mitigate SAH-induced blood-brain barrier (BBB) breakdown and neurological deficits through the Axl/Rac1 signaling pathway (97). Although the influence of other efferocytosis-related molecules on EBI after SAH remains less explored, many of these molecules are known to regulate neuroinflammation, which can be alleviated through efferocytosis (147). Consequently, they represent potential targets for future therapeutic interventions.
5 Concluding remarks
Cardiovascular and cerebrovascular diseases impose significant burdens on both patients and society owing to their unfavorable prognostic outcomes. Inflammation assumes a pivotal role in these conditions, whereas efferocytosis represents a potent mechanism for resolving inflammation and maintaining homeostasis. In recent years, scholarly attention has been directed towards investigating the mechanisms and exploring drug discovery avenues related to efferocytosis in cardiovascular and cerebrovascular diseases. Despite considerable advancements, significant challenges persist in developing methods that substantially enhance the prognosis of patients afflicted with cardiovascular and cerebrovascular disease. Nevertheless, efferocytosis and its underlying mechanisms continue to constitute a pivotal area for future research, offering potential to markedly improve the prognosis of patients with cardiovascular and cerebrovascular disease.
Author contributions
BZ: Conceptualization, Data curation, Writing – original draft. YZ: Investigation, Writing – review & editing. ZY: Data curation, Writing – review & editing. KJ: Writing – review & editing. ZZ: Writing – review & editing. SC: Conceptualization, Writing – review & editing. XZ: Conceptualization, Writing – review & editing. QW: Conceptualization, Writing – review & editing. XZ: Conceptualization, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (82071328).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
2. Adkar SS, Leeper NJ. Efferocytosis in atherosclerosis. Nat Rev Cardiol. (2024) 21(11), 762–779. doi: 10.1038/s41569-024-01037-7
3. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. (2015) 16:907–17. doi: 10.1038/ni.3253
4. Jia J, Yang L, Chen Y, Zheng L, Chen Y, Xu Y, et al. The role of microglial phagocytosis in ischemic stroke. Front Immunol. (2021) 12:790201. doi: 10.3389/fimmu.2021.790201
5. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
6. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. (2014) 509:310–7. doi: 10.1038/nature13085
7. Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. (2010) 112:733–43. doi: 10.1111/j.1471-4159.2009.06494.x
8. Elliott MR, Koster KM, Murphy PS. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J Immunol. (2017) 198:1387–94. doi: 10.4049/jimmunol.1601520
9. Yurdagul A Jr., Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. (2020) 31:518–33 e10. doi: 10.1016/j.cmet.2020.01.001
10. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of apoptotic cells in resolution of inflammation. Front Immunol. (2020) 11:553. doi: 10.3389/fimmu.2020.00553
11. Tajbakhsh A, Bianconi V, Pirro M, Gheibi Hayat SM, Johnston TP, Sahebkar A. Efferocytosis and atherosclerosis: regulation of phagocyte function by microRNAs. Trends Endocrinol Metab. (2019) 30:672–83. doi: 10.1016/j.tem.2019.07.006
12. Mehrotra P, Ravichandran KS. Drugging the efferocytosis process: concepts and opportunities. Nat Rev Drug Discovery. (2022) 21:601–20. doi: 10.1038/s41573-022-00470-y
13. Morioka S, Maueroder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity. (2019) 50:1149–62. doi: 10.1016/j.immuni.2019.04.018
14. Szondy Z, Sarang Z, Kiss B, Garabuczi E, Koroskenyi K. Anti-inflammatory mechanisms triggered by apoptotic cells during their clearance. Front Immunol. (2017) 8:909. doi: 10.3389/fimmu.2017.00909
15. Kloditz K, Fadeel B. Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discovery. (2019) 5:65. doi: 10.1038/s41420-019-0146-x
16. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. (2008) 112:5026–36. doi: 10.1182/blood-2008-06-162404
17. Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. (2003) 113:717–30. doi: 10.1016/S0092-8674(03)00422-7
18. Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem. (2008) 283:5296–305. doi: 10.1074/jbc.M706586200
19. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. (2008) 22:2629–38. doi: 10.1096/fj.08-107169
20. Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, Brune B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood. (2006) 108:1635–42. doi: 10.1182/blood-2006-04-014852
21. Medina CB, Ravichandran KS. Do not let death do us part: ‘find-me’ signals in communication between dying cells and the phagocytes. Cell Death Differ. (2016) 23:979–89. doi: 10.1038/cdd.2016.13
22. Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. (2006) 16:189–97. doi: 10.1016/j.tcb.2006.02.003
23. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. (2010) 467:863–7. doi: 10.1038/nature09413
24. Huveneers S, Truong H, Danen HJ. Integrins: signaling, disease, and therapy. Int J Radiat Biol. (2007) 83:743–51. doi: 10.1080/09553000701481808
25. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. (2013) 341:403–6. doi: 10.1126/science.1236758
26. Tajbakhsh A, Rezaee M, Kovanen PT, Sahebkar A. Efferocytosis in atherosclerotic lesions: Malfunctioning regulatory pathways and control mechanisms. Pharmacol Ther. (2018) 188:12–25. doi: 10.1016/j.pharmthera.2018.02.003
27. Nakahashi-Oda C, Fujiyama S, Nakazawa Y, Kanemaru K, Wang Y, Lyu W, et al. CD300a blockade enhances efferocytosis by infiltrating myeloid cells and ameliorates neuronal deficit after ischemic stroke. Sci Immunol. (2021) 6:eabe7915. doi: 10.1126/sciimmunol.abe7915
28. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. (2006) 203:2613–25. doi: 10.1084/jem.20060370
29. Vago JP, Amaral FA, van de Loo FAJ. Resolving inflammation by TAM receptor activation. Pharmacol Ther. (2021) 227:107893. doi: 10.1016/j.pharmthera.2021.107893
30. Tajbakhsh A, Gheibi Hayat SM, Butler AE, Sahebkar A. Effect of soluble cleavage products of important receptors/ligands on efferocytosis: Their role in inflammatory, autoimmune and cardiovascular disease. Ageing Res Rev. (2019) 50:43–57. doi: 10.1016/j.arr.2019.01.007
31. Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, et al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol. (2019) 20:40–9. doi: 10.1038/s41590-018-0249-1
32. Geng K, Kumar S, Kimani SG, Kholodovych V, Kasikara C, Mizuno K, et al. Requirement of gamma-carboxyglutamic acid modification and phosphatidylserine binding for the activation of tyro3, axl, and mertk receptors by growth arrest-specific 6. Front Immunol. (2017) 8:1521. doi: 10.3389/fimmu.2017.01521
33. Maiti SN, Balasubramanian K, Ramoth JA, Schroit AJ. Beta-2-glycoprotein 1-dependent macrophage uptake of apoptotic cells. Binding to lipoprotein receptor-related protein receptor family members. J Biol Chem. (2008) 283:3761–6. doi: 10.1074/jbc.M704990200
34. Nomura K, Vilalta A, Allendorf DH, Hornik TC, Brown GC. Activated microglia desialylate and phagocytose cells via neuraminidase, galectin-3, and mer tyrosine kinase. J Immunol. (2017) 198:4792–801. doi: 10.4049/jimmunol.1502532
35. Tzelepis F, Verway M, Daoud J, Gillard J, Hassani-Ardakani K, Dunn J, et al. Annexin1 regulates DC efferocytosis and cross-presentation during Mycobacterium tuberculosis infection. J Clin Invest. (2015) 125:752–68. doi: 10.1172/JCI77014
36. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
37. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPalpha immune checkpoint. Immunity. (2020) 52:742–52. doi: 10.1016/j.immuni.2020.04.011
38. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
39. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. (2014) 14:166–80. doi: 10.1038/nri3607
40. Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, et al. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem. (2005) 280:36342–54. doi: 10.1074/jbc.M506579200
41. Richards DM, Endres RG. The mechanism of phagocytosis: two stages of engulfment. Biophys J. (2014) 107:1542–53. doi: 10.1016/j.bpj.2014.07.070
42. Ma Z, Thomas KS, Webb DJ, Moravec R, Salicioni AM, Mars WM, et al. Regulation of Rac1 activation by the low density lipoprotein receptor-related protein. J Cell Biol. (2002) 159:1061–70. doi: 10.1083/jcb.200207070
43. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. (2007) 450:430–4. doi: 10.1038/nature06329
44. Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. (2002) 4:574–82. doi: 10.1038/ncb824
45. Evans IR, Ghai PA, Urbancic V, Tan KL, Wood W. SCAR/WAVE-mediated processing of engulfed apoptotic corpses is essential for effective macrophage migration in Drosophila. Cell Death Differ. (2013) 20:709–20. doi: 10.1038/cdd.2012.166
46. Yin C, Heit B. Cellular responses to the efferocytosis of apoptotic cells. Front Immunol. (2021) 12:631714. doi: 10.3389/fimmu.2021.631714
47. Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. (2005) 122:735–49. doi: 10.1016/j.cell.2005.06.043
48. Rubino M, Miaczynska M, Lippe R, Zerial M. Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J Biol Chem. (2000) 275:3745–8. doi: 10.1074/jbc.275.6.3745
49. Vieira OV, Bucci C, Harrison RE, Trimble WS, Lanzetti L, Gruenberg J, et al. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol. (2003) 23:2501–14. doi: 10.1128/MCB.23.7.2501-2514.2003
50. Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. (2001) 155:19–25. doi: 10.1083/jcb.200107069
51. Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. (2003) 23:6494–506. doi: 10.1128/MCB.23.18.6494-6506.2003
52. Johansson M, Lehto M, Tanhuanpaa K, Cover TL, Olkkonen VM. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol Biol Cell. (2005) 16:5480–92. doi: 10.1091/mbc.e05-03-0189
53. Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. (2012) 33:397–405. doi: 10.1016/j.it.2012.03.003
54. Braun V, Fraisier V, Raposo G, Hurbain I, Sibarita JB, Chavrier P, et al. TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. EMBO J. (2004) 23:4166–76. doi: 10.1038/sj.emboj.7600427
55. Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. (2011) 108:17396–401. doi: 10.1073/pnas.1113421108
56. Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-associated phagocytosis and inflammation. J Mol Biol. (2017) 429:3561–76. doi: 10.1016/j.jmb.2017.08.012
57. Heckmann BL, Green DR. Correction: LC3-associated phagocytosis at a glance. J Cell Sci. (2019) 132. doi: 10.1242/jcs.222984
58. Wong SW, Sil P, Martinez J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J. (2018) 285:1379–88. doi: 10.1111/febs.2018.285.issue-8
59. Kojima Y, Weissman IL, Leeper NJ. The role of efferocytosis in atherosclerosis. Circulation. (2017) 135:476–89. doi: 10.1161/CIRCULATIONAHA.116.025684
60. Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B, et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. (2017) 127:564–8. doi: 10.1172/JCI90520
61. Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol. (2008) 28:1421–8. doi: 10.1161/ATVBAHA.108.167197
62. Doran AC, Ozcan L, Cai B, Zheng Z, Fredman G, Rymond CC, et al. CAMKIIgamma suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J Clin Invest. (2017) 127:4075–89. doi: 10.1172/JCI94735
63. Mueller PA, Zhu L, Tavori H, Huynh K, Giunzioni I, Stafford JM, et al. Deletion of macrophage low-density lipoprotein receptor-related protein 1 (LRP1) accelerates atherosclerosis regression and increases C-C chemokine receptor type 7 (CCR7) expression in plaque macrophages. Circulation. (2018) 138:1850–63. doi: 10.1161/CIRCULATIONAHA.117.031702
64. Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, et al. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler Thromb Vasc Biol. (2010) 30:787–95. doi: 10.1161/ATVBAHA.109.202051
65. Foks AC, Engelbertsen D, Kuperwaser F, Alberts-Grill N, Gonen A, Witztum JL, et al. Blockade of tim-1 and tim-4 enhances atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2016) 36:456–65. doi: 10.1161/ATVBAHA.115.306860
66. Tao H, Yancey PG, Babaev VR, Blakemore JL, Zhang Y, Ding L, et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res. (2015) 56:1449–60. doi: 10.1194/jlr.M056689
67. Jin Y, Liu Y, Xu L, Xu J, Xiong Y, Peng Y, et al. Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell Death Dis. (2022) 13:512. doi: 10.1038/s41419-022-04966-8
68. Kasikara C, Schilperoort M, Gerlach B, Xue C, Wang X, Zheng Z, et al. Deficiency of macrophage PHACTR1 impairs efferocytosis and promotes atherosclerotic plaque necrosis. J Clin Invest. (2021) 131. doi: 10.1172/JCI145275
69. Bao Q, Zhang B, Zhou L, Yang Q, Mu X, Liu X, et al. CNP ameliorates macrophage inflammatory response and atherosclerosis. Circ Res. (2024) 134:e72–91. doi: 10.1161/CIRCRESAHA.123.324086
70. Zhang J, Zhao X, Guo Y, Liu Z, Wei S, Yuan Q, et al. Macrophage ALDH2 (Aldehyde dehydrogenase 2) stabilizing rac2 is required for efferocytosis internalization and reduction of atherosclerosis development. Arterioscler Thromb Vasc Biol. (2022) 42:700–16. doi: 10.1161/ATVBAHA.121.317204
71. Jarr KU, Kojima Y, Weissman IL, Leeper NJ. 2021 Jeffrey M. Hoeg award lecture: defining the role of efferocytosis in cardiovascular disease: A focus on the CD47 (Cluster of differentiation 47) axis. Arterioscler Thromb Vasc Biol. (2022) 42:e145–e54. doi: 10.1161/atvbbaha.122.317049
72. Gerlach BD, Marinello M, Heinz J, Rymut N, Sansbury BE, Riley CO, et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. (2020) 27:525–39. doi: 10.1038/s41418-019-0370-1
73. Doddapattar P, Dev R, Ghatge M, Patel RB, Jain M, Dhanesha N, et al. Myeloid cell PKM2 deletion enhances efferocytosis and reduces atherosclerosis. Circ Res. (2022) 130:1289–305. doi: 10.1161/CIRCRESAHA.121.320704
74. Yurdagul A Jr., Kong N, Gerlach BD, Wang X, Ampomah P, Kuriakose G, et al. ODC (Ornithine decarboxylase)-dependent putrescine synthesis maintains merTK (MER tyrosine-protein kinase) expression to drive resolution. Arterioscler Thromb Vasc Biol. (2021) 41:e144–e59. doi: 10.1161/ATVBAHA.120.315622
75. Sharma M, Schlegel MP, Afonso MS, Brown EJ, Rahman K, Weinstock A, et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res. (2020) 127:335–53. doi: 10.1161/CIRCRESAHA.119.316461
76. Kiss MG, Papac-Milicevic N, Porsch F, Tsiantoulas D, Hendrikx T, Takaoka M, et al. Cell-autonomous regulation of complement C3 by factor H limits macrophage efferocytosis and exacerbates atherosclerosis. Immunity. (2023) 56:1809–24 e10. doi: 10.1016/j.immuni.2023.06.026
77. Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res. (2013) 113:1004–12. doi: 10.1161/CIRCRESAHA.113.301198
78. Marinkovic G, Koenis DS, de Camp L, Jablonowski R, Graber N, de Waard V, et al. S100A9 links inflammation and repair in myocardial infarction. Circ Res. (2020) 127:664–76. doi: 10.1161/CIRCRESAHA.120.315865
79. Jia D, Chen S, Bai P, Luo C, Liu J, Sun A, et al. Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation. (2022) 145:1542–56. doi: 10.1161/CIRCULATIONAHA.121.057549
80. Cai S, Zhao M, Zhou B, Yoshii A, Bugg D, Villet O, et al. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J Clin Invest. (2023) 133. doi: 10.1172/JCI159498
81. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–56 e5. doi: 10.1016/j.cmet.2018.12.004
82. Wang X, Du W, Li Y, Yang HH, Zhang Y, Akbar R, et al. Macrophage-enriched Sectm1a promotes efficient efferocytosis to attenuate ischemia/reperfusion-induced cardiac injury. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.173832
83. DeBerge M, Yeap XY, Dehn S, Zhang S, Grigoryeva L, Misener S, et al. MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ Res. (2017) 121:930–40. doi: 10.1161/CIRCRESAHA.117.311327
84. Tan H, Li W, Pang Z, Weng X, Gao J, Chen J, et al. Genetically engineered macrophages co-loaded with CD47 inhibitors synergistically reconstruct efferocytosis and improve cardiac remodeling post myocardial ischemia reperfusion injury. Adv Healthc Mater. (2024) 13:e2303267. doi: 10.1002/adhm.202303267
85. Zhang Z, Tian H, Yang C, Liu J, Zhang H, Wang J, et al. Mesenchymal stem cells promote the resolution of cardiac inflammation after ischemia reperfusion via enhancing efferocytosis of neutrophils. J Am Heart Assoc. (2020) 9:e014397. doi: 10.1161/JAHA.119.014397
86. Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A. (2016) 113:6526–31. doi: 10.1073/pnas.1524292113
87. Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. (2008) 28:1429–31. doi: 10.1161/ATVBAHA.108.169078
88. Ye ZM, Yang S, Xia YP, Hu RT, Chen S, Li BW, et al. LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. (2019) 10:138. doi: 10.1038/s41419-019-1409-4
89. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. (2000) 164:1663–7. doi: 10.4049/jimmunol.164.4.1663
90. Manega CM, Fiorelli S, Porro B, Turnu L, Cavalca V, Bonomi A, et al. 12(S)-Hydroxyeicosatetraenoic acid downregulates monocyte-derived macrophage efferocytosis: New insights in atherosclerosis. Pharmacol Res. (2019) 144:336–42. doi: 10.1016/j.phrs.2019.03.012
91. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–77 e6. doi: 10.1016/j.immuni.2018.07.015
92. Ampomah PB, Cai B, Sukka SR, Gerlach BD, Yurdagul A Jr., Wang X, et al. Macrophages use apoptotic cell-derived methionine and DNMT3A during efferocytosis to promote tissue resolution. Nat Metab. (2022) 4:444–57. doi: 10.1038/s42255-022-00551-7
93. Halade GV, Lee DH. Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine. (2022) 79:103992. doi: 10.1016/j.ebiom.2022.103992
94. de Couto G, Jaghatspanyan E, DeBerge M, Liu W, Luther K, Wang Y, et al. Mechanism of enhanced merTK-dependent macrophage efferocytosis by extracellular vesicles. Arterioscler Thromb Vasc Biol. (2019) 39:2082–96. doi: 10.1161/ATVBAHA.119.313115
95. Chang CF, Goods BA, Askenase MH, Hammond MD, Renfroe SC, Steinschneider AF, et al. Erythrocyte efferocytosis modulates macrophages towards recovery after intracerebral hemorrhage. J Clin Invest. (2018) 128:607–24. doi: 10.1172/JCI95612
96. Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. (2010) 92:463–77. doi: 10.1016/j.pneurobio.2010.08.001
97. Tong LS, Shao AW, Ou YB, Guo ZN, Manaenko A, Dixon BJ, et al. Recombinant Gas6 augments Axl and facilitates immune restoration in an intracerebral hemorrhage mouse model. J Cereb Blood Flow Metab. (2017) 37:1971–81. doi: 10.1177/0271678X16658490
98. Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol. (2011) 70:646–56. doi: 10.1002/ana.22528
99. Fang H, Chen J, Lin S, Wang P, Wang Y, Xiong X, et al. CD36-mediated hematoma absorption following intracerebral hemorrhage: negative regulation by TLR4 signaling. J Immunol. (2014) 192:5984–92. doi: 10.4049/jimmunol.1400054
100. Zhou H, Li J, Hu L, Yu J, Fu X, Liang F, et al. Soluble Trem2 is a negative regulator of erythrophagocytosis after intracerebral hemorrhage in a CD36 receptor recycling manner. J Adv Res. (2023) 44:185–99. doi: 10.1016/j.jare.2022.03.011
101. Zhou X, Xie Q, Xi G, Keep RF, Hua Y. Brain CD47 expression in a swine model of intracerebral hemorrhage. Brain Res. (2014) 1574:70–6. doi: 10.1016/j.brainres.2014.06.003
102. Ni W, Mao S, Xi G, Keep RF, Hua Y. Role of erythrocyte CD47 in intracerebral hematoma clearance. Stroke. (2016) 47:505–11. doi: 10.1161/STROKEAHA.115.010920
103. Xu J, Chen Z, Yu F, Liu H, Ma C, Xie D, et al. IL-4/STAT6 signaling facilitates innate hematoma resolution and neurological recovery after hemorrhagic stroke in mice. Proc Natl Acad Sci U S A. (2020) 117:32679–90. doi: 10.1073/pnas.2018497117
104. Li Q, Lan X, Han X, Durham F, Wan J, Weiland A, et al. Microglia-derived interleukin-10 accelerates post-intracerebral hemorrhage hematoma clearance by regulating CD36. Brain Behav Immun. (2021) 94:437–57. doi: 10.1016/j.bbi.2021.02.001
105. Han R, Lan X, Han Z, Ren H, Aafreen S, Wang W, et al. Improving outcomes in intracerebral hemorrhage through microglia/macrophage-targeted IL-10 delivery with phosphatidylserine liposomes. Biomaterials. (2023) 301:122277. doi: 10.1016/j.biomaterials.2023.122277
106. Zhuang J, Peng Y, Gu C, Chen H, Lin Z, Zhou H, et al. Wogonin accelerates hematoma clearance and improves neurological outcome via the PPAR-gamma pathway after intracerebral hemorrhage. Transl Stroke Res. (2021) 12:660–75. doi: 10.1007/s12975-020-00842-9
107. Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. (2004) 94:609–16. doi: 10.1161/01.RES.0000119171.44657.45
108. Liu J, Shen D, Wei C, Wu W, Luo Z, Hu L, et al. Inhibition of the LRRC8A channel promotes microglia/macrophage phagocytosis and improves outcomes after intracerebral hemorrhagic stroke. iScience. (2022) 25:105527. doi: 10.1016/j.isci.2022.105527
109. Luo Z, Sheng Z, Hu L, Shi L, Tian Y, Zhao X, et al. Targeted macrophage phagocytosis by Irg1/itaconate axis improves the prognosis of intracerebral hemorrhagic stroke and peritonitis. EBioMedicine. (2024) 101:104993. doi: 10.1016/j.ebiom.2024.104993
110. Yan X, He M, Huang H, Wang Q, Hu Y, Wang X, et al. Endogenous H(2)S targets mitochondria to promote continual phagocytosis of erythrocytes by microglia after intracerebral hemorrhage. Redox Biol. (2022) 56:102442. doi: 10.1016/j.redox.2022.102442
111. Wu G, McBride DW, Zhang JH. Axl activation attenuates neuroinflammation by inhibiting the TLR/TRAF/NF-kappaB pathway after MCAO in rats. Neurobiol Dis. (2018) 110:59–67. doi: 10.1016/j.nbd.2017.11.009
112. Zheng L, Jia J, Chen Y, Liu R, Cao R, Duan M, et al. Pentoxifylline alleviates ischemic white matter injury through up-regulating Mertk-mediated myelin clearance. J Neuroinflamm. (2022) 19:128. doi: 10.1186/s12974-022-02480-4
113. Jin G, Tsuji K, Xing C, Yang YG, Wang X, Lo EH. CD47 gene knockout protects against transient focal cerebral ischemia in mice. Exp Neurol. (2009) 217:165–70. doi: 10.1016/j.expneurol.2009.02.004
114. Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. (2015) 35:3384–96. doi: 10.1523/JNEUROSCI.2620-14.2015
115. Xue T, Ji J, Sun Y, Huang X, Cai Z, Yang J, et al. Sphingosine-1-phosphate, a novel TREM2 ligand, promotes microglial phagocytosis to protect against ischemic brain injury. Acta Pharm Sin B. (2022) 12:1885–98. doi: 10.1016/j.apsb.2021.10.012
116. Wen RX, Shen H, Huang SX, Wang LP, Li ZW, Peng P, et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci Ther. (2020) 26:416–29. doi: 10.1111/cns.13296
117. Zhang G, Li Q, Tao W, Qin P, Chen J, Yang H, et al. Sigma-1 receptor-regulated efferocytosis by infiltrating circulating macrophages/microglial cells protects against neuronal impairments and promotes functional recovery in cerebral ischemic stroke. Theranostics. (2023) 13:543–59. doi: 10.7150/thno.77088
118. Cai W, Dai X, Chen J, Zhao J, Xu M, Zhang L, et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.131355
119. Surugiu R, Catalin B, Dumbrava D, Gresita A, Olaru DG, Hermann DM, et al. Intracortical administration of the complement C3 receptor antagonist trifluoroacetate modulates microglia reaction after brain injury. Neural Plast. (2019) 2019:1071036. doi: 10.1155/2019/1071036
120. Trahtemberg U, Mevorach D. Apoptotic cells induced signaling for immune homeostasis in macrophages and dendritic cells. Front Immunol. (2017) 8:1356. doi: 10.3389/fimmu.2017.01356
121. Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. (2017) 13:420–33. doi: 10.1038/nrneurol.2017.69
122. Wu H, Zheng J, Xu S, Fang Y, Wu Y, Zeng J, et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflamm. (2021) 18:2. doi: 10.1186/s12974-020-02041-7
123. Roszer T. Transcriptional control of apoptotic cell clearance by macrophage nuclear receptors. Apoptosis. (2017) 22:284–94. doi: 10.1007/s10495-016-1310-x
124. Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. (2007) 61:352–62. doi: 10.1002/ana.21097
125. Cao S, Zheng M, Hua Y, Chen G, Keep RF, Xi G. Hematoma changes during clot resolution after experimental intracerebral hemorrhage. Stroke. (2016) 47:1626–31. doi: 10.1161/STROKEAHA.116.013146
126. Tao C, Keep RF, Xi G, Hua Y. CD47 blocking antibody accelerates hematoma clearance after intracerebral hemorrhage in aged rats. Transl Stroke Res. (2020) 11:541–51. doi: 10.1007/s12975-019-00745-4
127. Jing C, Bian L, Wang M, Keep RF, Xi G, Hua Y. Enhancement of hematoma clearance with CD47 blocking antibody in experimental intracerebral hemorrhage. Stroke. (2019) 50:1539–47. doi: 10.1161/STROKEAHA.118.024578
128. Buckley JM, Liu JH, Li CH, Blankson S, Wu QD, Jiang Y, et al. Increased susceptibility of ST2-deficient mice to polymicrobial sepsis is associated with an impaired bactericidal function. J Immunol. (2011) 187:4293–9. doi: 10.4049/jimmunol.1003872
129. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. (2010) 33:699–712. doi: 10.1016/j.immuni.2010.11.009
130. Zhao XR, Gonzales N, Aronowski J. Pleiotropic role of PPARgamma in intracerebral hemorrhage: an intricate system involving Nrf2, RXR, and NF-kappaB. CNS Neurosci Ther. (2015) 21:357–66. doi: 10.1111/cns.2015.21.issue-4
131. Chang CF, Massey J, Osherov A, Angenendt da Costa LH, Sansing LH. Bexarotene enhances macrophage erythrophagocytosis and hematoma clearance in experimental intracerebral hemorrhage. Stroke. (2020) 51:612–8. doi: 10.1161/STROKEAHA.119.027037
132. Fan L, Jin L, Tang T, Zheng Y, Chen Z, Lin H, et al. Neutrophil-like pH-responsive pro-efferocytic nanoparticles improve neurological recovery by promoting erythrophagocytosis after intracerebral hemorrhage. Theranostics. (2024) 14:283–303. doi: 10.7150/thno.90370
133. Cheng J, Wang W, Xia Y, Li Y, Jia J, Xiao G. Regulators of phagocytosis as pharmacologic targets for stroke treatment. Front Pharmacol. (2023) 14:1122527. doi: 10.3389/fphar.2023.1122527
134. Xie XD, Dong SS, Liu RJ, Shi LL, Zhu T. Mechanism of efferocytosis in determining ischaemic stroke resolution-diving into microglia/macrophage functions and therapeutic modality. Mol Neurobiol. (2024) 61(10), 7583–7602. doi: 10.1007/s12035-024-04060-4
135. Burstyn-Cohen T, Lew ED, Traves PG, Burrola PG, Hash JC, Lemke G. Genetic dissection of TAM receptor-ligand interaction in retinal pigment epithelial cell phagocytosis. Neuron. (2012) 76:1123–32. doi: 10.1016/j.neuron.2012.10.015
136. Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. (2020) 26:131–42. doi: 10.1038/s41591-019-0695-9
137. Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener. (2017) 12:56. doi: 10.1186/s13024-017-0197-5
138. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. (2005) 201:647–57. doi: 10.1084/jem.20041611
139. Hu M, Lin Y, Men X, Wang S, Sun X, Zhu Q, et al. High-salt diet downregulates TREM2 expression and blunts efferocytosis of macrophages after acute ischemic stroke. J Neuroinflamm. (2021) 18:90. doi: 10.1186/s12974-021-02144-9
140. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. (2007) 446:1091–5. doi: 10.1038/nature05704
141. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. (2006) 9:1512–9. doi: 10.1038/nn1805
142. Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A. (2016) 113:1074–9. doi: 10.1073/pnas.1520398113
143. Zhang W, Zhao J, Wang R, Jiang M, Ye Q, Smith AD, et al. Macrophages reprogram after ischemic stroke and promote efferocytosis and inflammation resolution in the mouse brain. CNS Neurosci Ther. (2019) 25:1329–42. doi: 10.1111/cns.v25.12
144. Ma Y, Liu Y, Zhang Z, Yang GY. Significance of complement system in ischemic stroke: A comprehensive review. Aging Dis. (2019) 10:429–62. doi: 10.14336/AD.2019.0119
145. Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci. (2012) 32:946–52. doi: 10.1523/JNEUROSCI.3830-11.2012
146. Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, et al. Aneurysmal subarachnoid hemorrhage and neuroinflammation: A comprehensive review. Int J Mol Sci. (2016) 17:497. doi: 10.3390/ijms17040497
Keywords: efferocytosis, phagocytosis, inflammation, cardiovascular disease, cerebrovascular disease
Citation: Zhang B, Zou Y, Yuan Z, Jiang K, Zhang Z, Chen S, Zhou X, Wu Q and Zhang X (2024) Efferocytosis: the resolution of inflammation in cardiovascular and cerebrovascular disease. Front. Immunol. 15:1485222. doi: 10.3389/fimmu.2024.1485222
Received: 23 August 2024; Accepted: 11 November 2024;
Published: 26 November 2024.
Edited by:
Yachen Shi, Wuxi People’s Hospital, ChinaReviewed by:
Zhenbao Li, The First Affiliated Hospital of Wannan Medical College, ChinaYunpeng Liu, Capital Medical University, China
Copyright © 2024 Zhang, Zou, Yuan, Jiang, Zhang, Chen, Zhou, Wu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Zhang, emhhbmd4c3BAMTYzLmNvbQ==