 Carla Nel1
Carla Nel1 John Frater1,2*
John Frater1,2*- 1Peter Medawar Building for Pathogen Research, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- 2The National Institute for Health and Care Research (NIHR) Oxford Biomedical Research Centre, Oxford, United Kingdom
Although HIV infection can be managed with antiretroviral drugs, there is no cure and therapy has to be taken for life. Recent successes in animal models with HIV-specific broadly neutralising antibodies (bNAbs) have led to long-term virological remission and even possible cures in some cases. This has resulted in substantial investment in human studies to explore bNAbs as a curative intervention for HIV infection. Emerging data are encouraging, but suggest that combinations of bNAbs with other immunomodulatory agents may be needed to induce and sustain long-term viral control. As a result, a number of clinical trials are currently underway exploring these combinations. If successful, the impact for the millions of people living with HIV could be substantial. Here, we review the background to the use of bNAbs in the search for an HIV cure and how different adjunctive agents might be used together to enhance their efficacy.
1 Introduction
Human Immunodeficiency virus (HIV) infection continues to impact the lives of millions of people despite the availability of effective antiretroviral therapy (ART, 1). ART inhibits viral replication and maintains undetectable plasma viral loads in the majority of people with HIV (PWH, 2). Although ART has improved the length and quality of life for PWH there are unresolved issues (3, 4). Oral ART formulations need to be taken daily, can be associated with side effects and may interact with other medications (5). Furthermore, HIV has a rapid mutation rate causing the emergence of drug-resistant variants if the stringent regimen is not adhered to (6). These effects may impact children to a larger degree as they are still in an early immunological developmental phase, drug metabolism may be different (7, 8), and adherence can be problematic (9, 10). This highlights the need for therapies which are long-acting or have the potential to lead to drug-free virological remission and even cure, particularly in regions with the highest prevalence and incidence of new infections, such as some sub-Saharan African countries. Proposals for an optimal target product profile for such a curative treatment include to be safe and tolerable, to maintain plasma HIV RNA levels below that at which transmission may occur (200 copies HIV RNA/ml) for at least >3 years or indefinitely, and to be effective in >90% of individuals. Additionally, the treatment should result in minimal drug resistance for effective long-term results (11).
2 Difficulties in developing an HIV cure
HIV is difficult to eradicate as it establishes itself as a latent viral reservoir in CD4+ T cells (among other cells) early in infection (12). When HIV integrates into the human genome it can become a provirus that is transcriptionally silent – or ‘latent’ - preventing the presentation of HIV antigens by human leukocyte antigen (HLA) proteins (13). This prevents these cells from being identified by cytotoxic T and natural killer (NK) cells, and are therefore “hidden” from the immune system (14). ART successfully induces viral suppression but treatment cessation usually results in the return of plasma viraemia from the latent reservoir (15).
Additionally, some antiretroviral drugs may not fully penetrate certain anatomical locations known as ‘sanctuary sites’ (e.g. brain, genital tract, and gut-associated lymphoid tissues) resulting in suboptimal drug concentrations (16, 17). The varying ability of HIV drugs to penetrate certain sites depends on numerous factors such as their physicochemical properties, the rate of perfusion to the target site, protein-binding specificity and patient-specific polymorphisms for transporters and metabolic enzymes (17). Although there is little objective evidence to support on-going replication due to sub-optimal ART in these sanctuary sites, the potential adds to the complexity of eradicating the entirety of the HIV reservoir with ART alone.
Further problems are associated with the methods available for quantifying the true size of the replication-competent HIV reservoir. Current assays include, but are not limited to, the quantitative viral outgrowth assay (QVOA), proviral (total and integrated) DNA quantitation by the polymerase chain reaction (PCR) and primer/probe based digital methods [e.g. intact proviral DNA assay (IPDA, 18); Q4PCR (19, 20)]. There are drawbacks associated with each of these assays, and their variations, which prevents consistent determination of the replication-competent latent reservoir. The QVOA assay often underestimates the true reservoir size as not all cells with intact provirus are induced with a single round of activation thus the assay only gives an estimate of the minimum frequency of latently infected cells (21). Furthermore, this assay is laborious and costly. In contrast, proviral DNA PCRs greatly overestimate the replication-competent reservoir as they are unable to differentiate between defective and replication-competent provirus (22). The IPDA assay is a digital droplet PCR assay that is an improvement on the proviral DNA PCR as it is able to infer intact proviruses that lack common defects (90% of the defective proviral sequences can be identified and excluded) thereby greatly reducing the overestimation observed with the proviral DNA PCR (23). However, it has been shown that after many years on ART intact HIV proviruses can become permanently silenced which highlights the importance of developing an assay that accurately measures both provirus intactness as well as the inducibility of the reservoir in all cell types (20). The significant heterogeneity of sensitivity and specificity of currently available assays is further hindered by a lack of standardisation across laboratories (24). Additionally, these assays are generally applied to blood samples, when the bulk of the reservoir is likely to be found in tissue.
Additional difficulties in finding an HIV cure include the limitations associated with finding a translatable and ethically sound model to test potential treatments. Non-human primate models infected with Simian Immunodeficiency Virus (SIV) or Simian human immunodeficiency virus (SHIV) have been used extensively as models for human HIV infection, and proved valuable in studies using neutralising antibodies to prevent infection (25–27). In studies assessing the role of bNAbs as treatment, NHPs infected with SHIV have been seen to be more sensitive to achieving post-treatment virological control, but this has not yet been replicated in PWH (28, 29). Lessons may also be learned from vaccine prevention studies, for example in the STEP HIV clinical trial, the same Adenoviral 5 vector (containing HIV instead of SIV), that was highly effective in controlling viremia in non-human primate challenge studies (30–32), was seen to increase susceptibility to HIV when tested in healthy people at risk of developing HIV (33).
Additional complexities to studying viral cure include the lack of validated biomarkers to reliably predict HIV-infected replication-competent cells or immune correlates of protection to predict effective responses (34, 35). Therefore, to accurately test if investigational treatments are effective in preventing viral rebound and eliminating virus-infected cells for a prolonged period, PWH must undergo a period of antiretroviral treatment interruption (ATI, 36). Although there are associated risks, ATIs can be highly informative. In non-human primate preclinical trials, some animals control after ATI and some initially rebound but then undergo post-rebound viral suppression (37). Animals receiving an additional intervention, such as broadly neutralising antibodies (bnAbs) or immunomodulators (including therapeutic vaccination), have been seen to undergo post-rebound control more frequently than those receiving only ART (38).
In clinical ATI trials if viral rebound occurs, CD4+ T cell counts could drop to low levels resulting in the resurgence of opportunistic diseases (39). Thus, highly stringent trials are designed (involving regular testing for HIV viremia and CD4+ T cell count) to ensure people are placed back on ART as soon as viremia reaches a potentially dangerous level (40). There are various views on what these viral loads and CD4+ T cell count thresholds should be. Some groups only necessitate resumption of ART after one reading above 500 000 copies/mL is obtained (41, 42), whilst for others one reading above 1000 copies/mL triggers ART re-start (43–45); others require six readings >1000 copies/ml, each one week apart (46). The risks associated with prolonged viremia as a result of overly lenient restart criteria include HIV transmission to uninfected partners, expansion of the viral reservoir, immune exhaustion, and emergence of opportunistic infections (47). In contrast, setting a too stringent threshold may result in potential post-rebound controllers being missed. Thus, the optimal viral load threshold to restart ART must balance the risks associated with prolonged viremia as well as potential immune-mediated viral control that can arise after initial rebound (48). Restart criteria were defined in a collaborative meeting as a viral load >1000 copies/mL for over 4 weeks or any single reading of >100,000 copies/mL, and a CD4+ T cell count <350 cells/uL; this is due for further review in 2024 (35). These thresholds can, however, only act as guidelines, as there are few data available to define a gold standard for all ATI trials.
Participants undergoing ATI trials have been documented to have various views on the process. Some people react positively and enjoy the experience of aiding scientific research whilst others report feeling nervous and experiencing a sense of failure when viral loads increase. This highlights the importance of thorough education and psychological support during ATI trials to ensure participants fully understand the process and are aware that they have absolute authority to resume ART at any time point (49, 50).
Despite the limitations associated with the pre-clinical models and clinical trial designs mentioned above, they have provided valuable insight into HIV drug development. In this review, the current cure strategies for HIV will be discussed focusing on bNAbs used in combination with other treatments such as immune modulators, chromatin remodellers, and therapeutic vaccines.
3 HIV cure strategies – an overview
An effective, practical cure for HIV is yet to be discovered. Stem cell transplantation, with donor stem cells containing a CCR5 mutation (CCR5Δ32/Δ32) that prevents viral entry, has been the only successful strategy to date (51). Although this method is highly effective it has only been performed on patients requiring a transplant for another life-threatening disease such as leukaemia or lymphoma. It is too expensive and dangerous to use on every person with HIV and thus alternative strategies must be identified. Two different concepts for an HIV cure have been described: a ‘sterilizing’ cure in which HIV is eliminated from the body and a ‘functional’ cure where HIV is still present as a provirus but remains permanently suppressed without the need for any treatment. Although these terms are less widely used, different approaches might be considered if the goal is complete viral eradication versus long-term control. These strategies might be grouped into three broad categories: immunological, transcriptional regulation and genome editing. These strategies aim to either address the problem of the latent HIV reservoir directly, or enhance immune responses to eliminate circulating virus and infected cells, or both.
3.1 Immunological
Immunological strategies aim to achieve a functional cure by enhancing an individual’s immune responses to induce life-long protection against HIV. These interventions include therapeutic vaccines, bNAbs and immune modulators (such as toll-like receptor (TLR) agonists). Some of these strategies – such as passive infusions of bNAbs - have been successful in inducing long-term protection in some individuals. However, as of yet, any desired protective effect is temporary across most approaches, resulting in eventual viral rebound in the vast majority of trial participants.
3.2 Regulation of transcription
Two different HIV cure approaches utilise transcriptional regulation. The first, the “shock and kill” strategy, aims to achieve a sterilizing cure by reactivating latent HIV provirus using latency-reversing agents (LRA) such as immune modulators and chromatin remodellers and then killing newly reactive cells with enhanced immune responses brought about by increased antigen availability (52). This strategy is often combined with immunological strategies such as bNAbs, TLR agonists, Interleukin (IL) agonists, and vaccines in an attempt to enhance the natural immune responses required to eliminate the virus. Although this seems achievable in theory, so far researchers have been unable to reactivate a large enough proportion of the latent HIV reservoir to cause a significant effect (53, 54). Furthermore, it is still uncertain whether bNAbs and immune modulators are capable of enhancing long-term immune responses that are potent enough to neutralise all circulating virus and infected cells.
The other approach termed, the “Block and Lock” strategy, aims to achieve a functional cure by utilizing latency-promoting agents (LPA) to ‘permanently’ silence proviral genes and other treatments (e.g. ART/bNAbs) to neutralise residual circulating virus (55). LPAs must induce epigenetic silencing in all latently infected cells. This brings about a deep state of latency which would limit viral rebound after ATI and which is essential for this cure strategy to be effective (56). So far attempts to achieve a deep, permanent state of latency have been unsuccessful in clinical studies (57, 58). Regular dosing of LPAs has not yet been successful in silencing the entire latent reservoir thus it may be a challenge for permanent silencing to be achievable with this strategy alone.
3.3 “Genome editing”
A potential exciting approach for a genome editing strategy involves directly deleting proviral genomes from host cells using CRISPR/Cas9, Zinc finger nucleases or TALEN-based technologies and killing residual circulating virus with enhanced immune responses (59). In theory, this would make viral rebound impossible resulting in a sterilizing cure. Genome editing has not yet been perfected for use on humans due to potential off-target effects and has only been successfully achieved in vitro, in cell culture, and in vivo with mouse and rat models (59–61). A study investigating Zinc finger nucleases in vitro was able to successfully excise full-length proviruses in 30% of cells without any cytotoxicity (62). Even though this Zinc finger nuclease trial was relatively successful in vitro, ideal delivery vehicles still need to be developed that preferably only allow gene editing agents to enter infected cells, before this treatment can be tested on humans (55). Additional strategies employing CRISPR/Cas9 involve mutating integrated proviral HIV DNA to become replication incompetent, but with the risk of viral escape through non-homologous end joining repair of the DNA (63, 64).
An alternative approach is to use Zinc finger nucleases to induce the CCR5Δ32 mutation in CD4+ T cells, thereby altering the person’s own DNA, rendering them resistant to HIV. An example of such modified cells was made by Sangamo BioScience and have been used in human studies. In one trial the modified cells were seen to have an estimated half-life of 48 weeks and declined at a much slower rate (-1.81 cells/day) than unmodified cells (-7.25 cells/day) during ATI. Furthermore, blood HIV DNA levels declined in the majority of the participants with one person achieving undetectable HIV RNA levels (65). In a different trial most participants only experienced a slight delay in viral rebound after ATI (about 4 weeks) compared to historical controls receiving no treatment (about 2 weeks), however, 3/14 participants (of which 2 were naturally CCR5Δ32 heterozygous) experienced partial post rebound viral control (VL<1000 copies/mL) accompanied by the restoration of CD8+ T cell responses that was not seen with the other 11 participants (66). The experienced viral control was thus likely due to a combination of the modified cells preventing viral entry and CD8+ T cells killing already infected cells. Although genome editing appears to be an attractive HIV cure approach, many ethical issues first need to be addressed, especially when conducting these experiments on human embryos. In 2018, twins genetically modified using CRISPR-Cas9 (modification in CCR5 gene) to be resistant to HIV were born. This was performed unethically and illegally, and received serious backlash due to the full consequences of genetically modifying embryos (such as the modification being transferred through generations) still being unknown (67). Despite these challenges, genome editing remains a potent and promising treatment for a prospective sterilizing cure.
4 Broadly neutralising antibodies
Broadly neutralising antibodies (bNAbs) are antibodies targeting conserved regions of the HIV Envelope protein (Env) capable of neutralising multiple, diverse strains (68). These antibodies either target epitopes in the CD4 binding site (VRC01 and 3BNC17), V1/V2 loop (PG16), V3 loop (10-1074 and PGT121), membrane-proximal external region or gp120-gp41 proximal interface of Env (69). The first generation of bNAbs were produced using phage display and human hybridoma electrofusion (70). These bNAbs were not very effective in suppressing viremia and displayed inadequate potency and breadth causing the rapid emergence of drug-resistant strains (71). The newest generation of bNAbs were developed using single-cell antibody cloning methods (based on BCR sequencing and recombinant expression) and high throughput neutralisation assays (70, 72–75). These bNAbs showed significant increases in potency (10 – 100 fold) and breadth (2-fold) compared to the previous generation (70). In non-human primate trials, animals treated with these bNAb monotherapies showed rapid declines in plasma RNA and proviral DNA as well as significant increases in Gag-specific T-cell responses (76). Additionally, when bNAb cocktails were administered to non-human primates shortly after SHIV infection (24hrs) there was no evidence of viraemia or establishment of viral reservoirs, and although this may arguably represent highly effective post-exposure prophylaxis in this animal model (rather than treatment) it demonstrates the potential antiviral potency of bNAbs (77).
Mixed success has been achieved in clinical trials with viraemic PWH when using short-acting bNAb monotherapy as resistant viral variants rapidly emerge (78, 79). The need for broad coverage was also an issue in two studies of bNAb monotherapy with VRC01 to prevent infection of healthy people at risk of HIV (the AMP study). The investigators concluded that there was no protection versus placebo for the cohort as a whole, but there was evidence of some protection against VRC01-sensitive strains (75% efficacy, 80, 81). Administering combinations of bNAbs has proven to be much more successful for treatment – especially in the context of concurrent ART - and reduces the frequency of viral escape mutations when both bNAbs are at therapeutic levels (82). bNAbs are being optimized to further increase their potency and breadth. One such optimization is the modification of two amino acids (M428L and N434S) in the Fc region of the antibody (83). This renders the antibodies LS variants with an extended half-life due to their increased binding affinity to the neonatal Fc receptor which promotes antibody recycling instead of degradation (70, 83). This modification has been shown to increase the period to post-treatment viral rebound, in many participants (84), and may have particular value for hard-to-reach populations.
bNAbs facilitate neutralisation of circulating virus through direct binding via the Fab fragment thereby preventing viral entry into cells. They also promote the activation of other components of the immune system using their Fc fragments by allowing for the opsonization of virus by antibody-dependent cellular phagocytosis, antibody-dependent cellular cytotoxicity, and activation of the complement cascade (Figure 1A, 85). Additionally, it has been postulated that bNAbs induce a ‘vaccinal effect’ (86). The exact mechanism underlying this ‘vaccinal effect’ is yet to be proven and it is still uncertain which immune cells are primarily responsible. However, it is suggested that the ‘vaccinal effect’ is a process whereby antibody-virus immune complexes form and enhance antigen processing, presentation, and subsequently proliferation/activation of immune cells (86, 87). A person displaying a vaccinal effect should experience viral control off ART when bNAb levels have waned to below therapeutic levels, along with the emergence of a potent, long-lasting immune response.
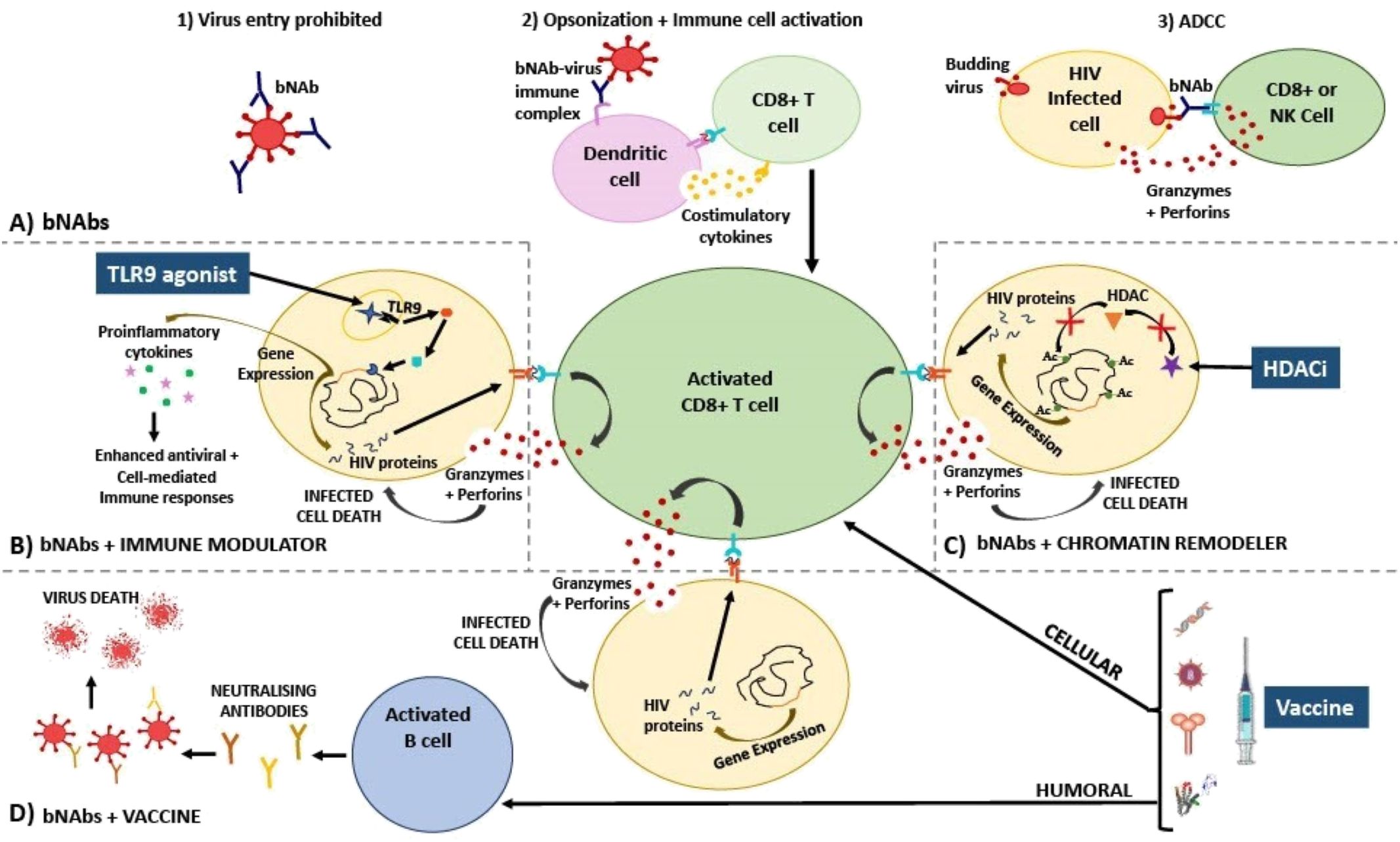
Figure 1. Diagram explaining the mechanism of action of HIV treatment strategies involving (A) bNAbs; used in combination with (B) immune modulators, (C) chromatin remodellers, and (D) therapeutic vaccines. (A) bNAbs mechanism of action has been described as inhibiting viral entry into cells via direct neutralisation (1), activating various immune cells via opsonization (2), and killing virus-infected cells via ADCC (3). (B) Immune modulators such as TLR9 agonists bind to receptors that activate signalling cascades in immune cells, resulting in proinflammatory cytokine gene expression, and as a result, HIV gene expression is also induced in once latently infected cells. Proinflammatory cytokines and bNAbs activate and enhance specific CD8+ T cell cytotoxic responses (release of perforin and granzyme) which are able to destroy newly activated CD4+ T cells that are presenting HIV peptides on HLA-I. (C) Chromatin remodellers such as HDAC inhibitors prevent the removal of acetyl groups from histone tails. As a result, nucleosomes are forced to remain further apart, keeping DNA in an open, transcriptionally active state. HIV gene expression is thus induced, reactivating the latent reservoir. Virus-infected cells are now recognized by CD8+ T cells that have been activated by bNAbs resulting in infected cell death. (D) HIV therapeutic vaccines should ideally amplify cellular immune responses, thereby additively enhancing CD8+ T cell activation along with bNAbs, as well as humoral (B cell) immune responses, initiating the production of neutralising antibodies towards a vast array of conserved epitopes. Additionally, these effective responses should be retained by memory T and B cells allowing sustained viral suppression when no therapy is taken, thereby bringing about a functional cure. Ac, acetyl group; ADCC, antibody-dependent cellular cytotoxicity; bNAbs, broadly neutralising antibodies; HDACi, histone deacetylase inhibitor; HIV, human immunodeficiency virus; NK, natural killer cell; TLR, toll-like receptor.
CD8+ T cells are potentially key to this ‘vaccinal effect’ as seen in studies of HIV ‘elite controllers’ and demonstrated in some non-human primate trials (88). The role of CD8+ T cells in viral control has been observed in macaques receiving only ART. During ATI CD8+ T cells were shown to be responsible for maintaining a lower and stable viral setpoint in untreated macaques compared to those treated with anti-CD8+ monoclonal antibodies (89). In humans, HIV ‘elite controllers’ are seen to have increased CD8+ HIV-specific T cell responses compared to non-controllers, thus, CD8+ T cells are suspected to play a critical role in prolonged viral suppression (88). Although CD8+ T cells are thought to be a major factor contributing to viral control it is likely many other mechanisms are at play. For example, viral relapse has also been linked to the loss of antibody induction when CD4+ T cells are depleted suggesting that autologous antibody responses and T cells combine forces to control HIV, with the former helping prevent CD8+ T cell escape (90).
In a study by Nishimura et al. a significant proportion (46%) of SHIV-infected macaques that received bNAb therapy were able to maintain post-treatment viral suppression (undetectable viral loads) during ATI and after bNAb levels had waned. When anti-CD8+ monoclonal antibodies were administered to deplete CD8+ T cells viral rebound rapidly occurred in all the macaques suggesting that CD8+ T cells were responsible for maintaining viral suppression during ATI (91). This highlights that bNAbs could be part of an effective long-term treatment or cure for HIV, through CD8+ T cell induction. However, the postulated ‘vaccinal effect’ has not yet been conclusively observed in bNAb clinical trials and more studies are needed (92, 93).
The majority of evidence for bNAbs as potential therapeutics comes from studies in which they are given as passive infusions. Alternative strategies that allow for long term expression of bNAbs in vivo have been trialled in non-human primates with promising results. The intramuscular administration of adeno-associated virus (AAV) expressing antibody like-neutralising proteins was able to protect against virulent SIV infection 4 weeks after AAV administration with 6/9 monkey remaining uninfected after challenge (94). In a different study a monkey that received a single dose of AAV coding for an anti-SIV antibody was able to express antibody continuously for 6 years, withstanding 6 different SIV challenges of increasing infectious dose (95). Although much success has been achieved with the long-term expression of bNAbs in non-human primates, translating these effects to humans is challenging. In clinical trials testing HIV-bNAb expressing AAVs, antibody levels are not maintained at high serum titers due to multiple reasons including anti-drug antibodies and preexisting immunity to the vector. However, in some participants high bNAb concentrations have been described in the serum for up to 3 years. None of these clinical trials have tested the treatments’ ability to suppress HIV off ART (96, 97). Whether the persistent bNAb expression using AAV delivery also drives a CD8+ T cell response or ‘vaccinal effect’ (as demonstrated following bNAb passive infusions) is not known, but raises potentially interesting questions for further study.
Limitations associated with bNAbs include the prevalence of pre-existing resistance, that they may only moderately reduce the size of the latent reservoir, and that resistant viral variants readily emerge (70, 98). As bNAbs are likely only effective in people harbouring antibody-sensitive viruses this may necessitate viral screening before administration. This – and geographical viral diversity – may impact the full value of bNAbs as therapy. To date, most studies have taken place in the U.S. and Europe targeting predominantly B clade HIV, and results may not translate to other clades and other regions. Although dual bNAb therapy has proven to be an extremely promising therapy for the treatment of HIV bNAb induced viral suppression is still temporary and short lived in some participants. Whether – as for ART – a three bNAb cocktail is needed for long-term efficacy is not clear. Concerningly, in one Phase 1 trial testing the bNAbs PGDM1400, PGT121 and VRC07-523LS on ART naïve PWH, plasma HIV RNA levels were noticeably decreased after administration, however, viral rebound occurred within a median of 20 days after nadir (99). How a triple bNAb regime would sustain control in PWH on ART and then undertaking a TI is an important follow-up research question. Other adjunctive treatments such as immune modulators, chromatin remodellers or vaccines may also enhance the efficacy of bNAbs, and are the focus of the rest of the review.
5 Enhancing bNAbs with immune modulation
Combining bNAbs with immune modulators may enhance their potential for viral suppression and this is being tested in clinical trials. Current immune modulators being assessed include signalling agonists (such as TLR, IL, and Interferon (IFN) agonists). These signalling agonists facilitate the transcription of type I IFN and proinflammatory cytokine genes via the nuclear factor-κB (NF-κB) pathway thereby enhancing antiviral/cell-mediated immune responses (such as phagocytosis, T cell proliferation, dendritic cell (DC) maturation, and NK cell activation) targeting HIV (100). Additionally, it has been shown that activation of the NF-κB pathway via TLR agonist-receptor binding also mediates activation of the latent integrated HIV provirus (101–104). This could result in the expression and presentation of HIV proteins on the HLA-I complex of once ‘hidden’ T cells allowing them to now be recognized and killed by the immune system (Figure 1B).
A humanized mouse trial testing cytotoxic T-lymphocyte associated protein 4 (CTLA4), I-BET151 (immune modulators) and Vorinostat (histone deacetylase inhibitor) with the bNAbs, 3BNC117, 10-1074 and PG16 in one treatment showed that a single inducer along with the bNAbs had no significant effect on viral rebound but that a combination of all the inducers with bNAbs significantly decreased the number of rebounding animals (105). This shows how the various inducers and bNAbs might work synergistically to reduce the chances of viral rebound and supports the use of combination therapies with bNAbs as a longer-term treatment for HIV.
Similarly, many non-human primate trials have tested combinations of immune modulators and bNAbs in SHIV-infected macaques. Two trials tested the efficacy of the TLR7 agonist, Vesatolimod and bNAb, PGT121. The first trial by Borducchi et al., initiated the ART and TLR7-bNAb combination treatment early, during acute SHIV infection (7 days post-SHIV infection, 106). At the end of the trial, 55% of the animals rebounded (at a median of 112 days vs 21 days in control) and no induction of CD8+ T cell responses were seen in any of the animals. The absence of CD8+ T cell responses was potentially due to low antigen availability because of early ART treatment intervention. In all animals that maintained viral suppression, SHIV RNA remained undetectable after CD8+ T cell depletion suggesting this combination potentially eliminated the viral reservoir (106). Although this trial yielded success in maintaining viral suppression in almost half of the animals, initiating treatment very early in infection limits the size and diversity of the viral reservoir and therefore does not resemble what is feasible in the majority of people living with HIV. The trial by Moldt et al. translates more readily into a clinical setting as ART was initiated during chronic infection (a year after SHIV infection, 107). In this study, the TLR7-bNAb combination treatment was initiated 2.5 years after initiation of ART. After ATI only 50% of the combination treatment animals rebounded compared to 100% in the placebo group. Similarly to Borducchi’s study, 71% of the animals that maintained viral suppression remained aviremic after CD8 + T and NK cell depletion, suggesting that the reservoir may have also been eradicated in these animals (107). These non-human primate trials show that the TLR7 agonist and PGT121 combination suppresses viral rebound in some SHIV-infected macaques but that viral suppression is not solely due to CD8+ T cells and the vaccinal effect, but also reservoir eradication.
There are many clinical trials testing combinations of bNAbs with immune modulators in the pipeline. Many are still ongoing (NCT05281510, NCT05245292, NCT04340596, 108–110), whilst one has presented provisional results at a conference (NCT03588715 - BEAT-2, 111) and the other is completed and results have been published (NCT03837756 – TITAN Trial, 112). In the TITAN trial, the TLR9 agonist, Lefitolimod, and bNAbs (10-1074 and 3BNC117) were administered in combination and individually to PWH during ATI (28). The time to viral rebound was 0.5, 12.5, and 9.5 weeks longer than the placebo in the Lefitolimod only, bNAbs only, and Lefitolimod plus bNAbs groups, respectively. In general, HIV-specific CD8+ T cells were significantly increased in patients with higher viral loads but non-significantly increased in patients with lower viral loads suggesting that the increase was not due to the vaccinal effect but rather an increase in antigen availability. Furthermore, there was no change seen in Gag-specific CD8+ T cell responses or Gag-induced cytokine release. The authors proposed that the combination treatments’ inability to broadly stimulate cellular immunity was due to low amounts of antigen available at bNAb administration as the interventions were administered before ATI was induced. This suggests that administering immunostimulatory molecules in the same location and time where antigen is available may be important. This study showed that the addition of Lefitolimod added no clinical or immunological benefit to participants during ATI, and that the co-administration of Lefitolimod with bNAbs acted antagonistically leading to a significantly faster decline in bNAb serum concentrations compared to bNAbs alone (28). In the BEAT-2 trial where the immune modulator peginterferon alfa-2b and bNAbs (3BNC117 and 10-1074) were co-administered provisional results show that 86% of the participants maintained viral suppression for at least 26 weeks with 40% of these individuals not meeting ART restart criteria (and were still suppressing viremia) after the 38-week ATI (113). Despite the success of the treatment in inducing viral suppression, the combination did not decrease the reservoir size and there was no overall increase in HIV-specific T-cell responses.
6 Enhancing bNAbs with latency reversing agents
Chromatin structure plays a role in inhibiting HIV gene expression in latently infected cells. HIV proteins alter chromatin remodelling complexes as part of their natural cycle thereby controlling HIV latency and reactivation (114). Chromatin remodelling drugs such as histone deacetylase inhibitors (HDACi) can activate genes by altering the positioning of nucleosomes (115). HDACi prevent the removal of acetyl functional groups on histone lysine residues forcing the nucleosomes further apart thereby allowing DNA to remain in an open, transcriptionally active state (Figure 1C, 116). Additionally, the ‘Smac mimetic’ AZD5582 (a mimetic of the second mitochondrial-derived activator of caspases that antagonises apoptosis inhibitor proteins), can activate the non-canonical NF-κB signalling pathway and as a result induce HIV or SIV expression in humanized mice or non-human primates, respectively, thereby acting as a potent LRA in these models (117). AZD5582 has been tested in combination with four SHIV-specific rhesus monoclonal antibodies and the IL-15 superagonist, N-803 in SHIV-infected, ART supressed rhesus macaques (118). The trial showed that using AZD5582 in this combination resulted in a significant reduction of total and replication competent SIV DNA in lymph node derived CD4+ T cells compared to either of the treatments alone, indicating this combination successfully reduced the viral reservoir (118). In this study the bNAb-LRA combination worked additively to yield a greater effect than each intervention in isolation. Despite AZD5582 success in isolation and in combination with bNAbs in non-human primates, the drug has not yet been optimised for use in humans (119). As of yet, HDAC inhibitors are the only LRAs that have been used in combination with bNAbs in clinical trials. The HDAC inhibitors used in these clinical trials include Vorinostat and Romidepsin. Three clinical trials testing the combination treatment of bNAbs and HDAC inhibitors have been published to date.
Two of these trials tested the efficacy of the bNAb, 3BNC117, and HDACi, Romidepsin, in PWH during ATI. The trials differed in that one administered the treatment along with ARVs to people who had never initiated ARV therapy before (NCT03041012 – eCLEAR, 120, 121), whereas the other administered the treatment with ARVs to people who were virologically suppressed and taking ARVs for at least 18 months (NCT02850016 – ROADMAP, 92, 122). The eCLEAR trial reported increased elimination of plasma viruses and infected cells after treatment. Additionally, 80% of participants with pre-ART 3BNC117 sensitive viruses exhibited ART-free virological control for at least 12 weeks. The combination treatment also enhanced HIV-Gag-specific CD8+ T cell immunity (measured by the AIM assay) compared to ART alone. However, the combination of Romidepsin and 3BNC117 did not enhance clearance of the viral reservoir and maintain viral control more than 3BNC117 on its own. Therefore, Romidepsin exhibited no immunostimulatory or latency-reversing additive effect. In contrast, the ROADMAP trial yielded disappointing results with the same combination treatment. The combination treatment (Romidepsin and 3BNC117) did not significantly reduce HIV DNA or delay viral rebound when compared to Romidepsin alone (median time to viral rebound was 18 days and 28 days in the combination and Romidepsin-only groups, respectively). Additionally, no change in the size of the latent reservoir and no enhancement in HIV-specific cellular immunity was observed in either of the groups. The difference in success observed between the two trials may be due to the eCLEAR trial containing participants who were ART naïve at trial commencement. The immune responses induced by initial antigen availability could have been enhanced during treatment and throughout ATI as opposed to participants in ROADMAP where antigen, and as a result, likely immune responses, had been absent for over a year. Nevertheless, regardless of when ART was initiated, both trials showed that the combination of Romidepsin and 3BNC117 was no more effective in preventing viral rebound, decreasing the latent viral reservoir, and increasing HIV-specific cellular immunity than bNAbs alone.
The other clinical trial (NCT03803605 - VOR-07, 123) tested the efficacy of the bNAb, VRC07-523LS, and the HDACi, Vorinostat in stimulating expression of proviral HIV from resting CD4+ T cells whilst participants remained on uninterrupted ART (124). The combination therapy was unable to reduce low-level viremia or the latent HIV reservoir suggesting that the T cells induced by the combination treatment may be incapable of eliminating small populations of persistently infected cells when viral replication is inhibited by ART. Once again, these studies question the role of HIV antigen availability in stimulating strong, HIV-specific CD8 + T cell immune responses (88, 125).
7 Background to therapeutic HIV vaccines as a potential
The immune responses elicited by HIV ‘elite controllers’ are key to maintaining viral suppression off ART (88) and are a model that aims to be replicated by therapeutic vaccines (126). However, vaccine development is challenging due to HIV’s vast genetic diversity and high mutation rate (127). It is extremely difficult to find immunogens that are capable of eliciting broad and robust immune responses to all HIV strains and their possible escape mutants (128, 129). Nevertheless, many HIV therapeutic vaccines have been able to induce broad and effective cellular and humoral immune responses but none have been able to reduce the viral reservoir and prevent viral rebound (93, 130–132), challenging our understanding of the key correlates of protection.
In attempts to develop a functional cure, many researchers have designed various types of vaccines against HIV. These include, but are not limited to, DNA vaccines, viral vectored vaccines, RNA vaccines, inactivated vaccines and subunit vaccines (133).
7.1 DNA vaccines
DNA vaccines are recombinant bacterial plasmids containing a target gene encoding the vaccine antigen. In theory, DNA vaccines should be advantageous over some other vaccine types as they can induce both cellular (T cell) and humoral (antibody) immune responses without inducing anti-vector immunity and are easier to design and manufacture (134). However, in reality, most of these vaccines are only modestly immunogenic. In attempts to enhance the immunogenicity of DNA vaccines, adjuvants such as IL-12 and IL-15 have been added to vaccine formulations and methods have been developed to improve vaccine delivery e.g. through intradermal routes or electroporation (135, 136). These enhancements have had varying effects on the immunogenicity of DNA vaccines. In a study conducted by Jacobson et al. a multi-antigen DNA therapeutic vaccine was tested that encodes the HIV genes gag, pol, nef, tat, vif and env (MAG) alone and with a plasmid encoding the IL-12 gene administered at different concentrations (50ug, 250ug, 1000ug) via intramuscular injection combined with electroporation (135). The only significant response (measured by ICS) compared to placebo was the increase in IL-2-producing CD4+ T cells from baseline to week 14 in the MAG+IL-12(50ug) group, but no significant CD8+ T cell responses were observed (135). Seeing as the greatest response was induced by the lowest concentration of IL-12, higher IL-12 concentrations may induce negative-regulatory pathways that suppress T-cell responses. Although the MAG+IL-12(50ug) vaccine combination appeared to perform better than the MAG vaccine alone, the addition of IL-12 did not increase CD8+ T cell responses.
Some success has been achieved in the HVTN 070 and 080 studies testing the DNA vaccine PENNVAX-B (PV) alone, with IL-15 (0.8mg or 2mg) or with IL-12 (1.5mg or 1mg) administered via intramuscular injection (6mg) or intramuscular injection combined with electroporation (3mg, 136). The results showed that electroporation significantly increased the number of CD4+ and CD8+ T cell vaccine responders compared to intramuscular injection, despite containing only half the dose. In the electroporation group, the addition of IL-12 insignificantly increased the number of responders however in the intramuscular group both the addition of IL-12 and IL-15 resulted in a similar number of responders compared to PV alone. This study highlights electroporation’s superior ability to enhance immunogenicity and reduce the dose of vaccine required and demonstrates IL-12s potential to further increase immunogenicity if administered via electroporation.
Another study testing a multi-HIV antigen DNA vaccine, encoding Subtype B Rev, Nef, Tat, sections of Gag and an array of Pol and Env coded CTL epitopes, in ART naïve PWH who were infected with Subtype C viruses. Despite the discrepancy between the subtype of the vaccine and virus with which the participants were infected, significantly enhanced HIV-specific CD4+ and CD8+ T cell immune responses and a slight decrease in viral load were observed in the test group compared to placebo (137). In other DNA vaccine trials, although immune responses are sometimes enhanced, effects on viral load or viral replication are rarely seen (134).
7.2 Viral vectored vaccines
Viral vectored vaccines are recombinant viruses, containing a target gene encoding the vaccine antigen, rendered non-pathogenic/attenuated through the removal of genes involved in virulence. Most viral vectored vaccines are replication incompetent (genes involved with replication are removed) to improve their safety profile and to prevent off-target effects. Common viral vectors used for HIV vaccines include Poxviruses (e.g. the vaccinia viral vectors, New York Vaccinia (NYVAC) and Modified Vaccinia Virus Ankara (MVA), canarypox (ALVAC) and fowlpox), Adenoviruses (e.g. Chimpanzee Adenovirus (ChAd) and Human Adenoviruses (HAd) serotypes 5, 26 and 35) and Rhabdoviruses (e.g. Vesicular Stomatitis Virus (VSV), 138). Viral vectored vaccines are advantageous in that they elicit both potent antibody responses as well as cell-mediated immune responses. They are highly immunogenic on their own (don’t require an adjuvant) and are known to induce long-lasting immune responses that often only require a single dose (139). A key disadvantage associated with these vaccines is pre-existing immunity towards the viral vector. Pre-existing immunity can greatly decrease the efficacy of the vaccine by accelerating its elimination from the body or can cause severe side effects as a result of the immune reaction to the vector (140). Pre-existing immunity is only a problem when using viral vectors that normally infect humans such as the human adenoviruses or MVA (often in older people who were vaccinated against smallpox). There is also conflicting data that suggests pre-existing immunity may arise when the same viral vector is repeatedly administered to people as part of the same or different vaccine regimen thereby potentially neutralising the effect of other essential vaccines build on the same vector platform, but this is yet to be fully proven (141, 142).
Pre-existing immunity was seen to be a major problem during the STEP HIV prophylactic vaccine trial where a Human Adenovirus type 5 vaccine was used (143). In this trial, the vaccine group was associated with increased susceptibility to HIV compared to placebo (144). As a result, when designing viral vectored vaccines for HIV, one option is to use non-human adenoviral vectors boosted with a different vector or completely different vaccine. However, non-human adenoviral vectors or vectors with a very low seroprevalence in humans are not without fault. A rare side effect (Thrombosis with Thrombocytopenia Syndrome) was observed after vaccination with AstraZeneca’s SARS-CoV-2 vaccine (ChAdOx1-S) resulting in its withdrawal from the market in the European Union (145, 146). This vaccine utilised a Chimpanzee adenoviral vector and thus caution should be taken when developing HIV vaccines with this vector in the future. Furthermore, the use of vectors with low pre-existing immunity does not necessarily make for an effective vaccine as seen with the Phase 3 Mosaico HIV trial (HPX3002/HVTN706) where a low sero-prevalent vector, Adenovirus 26, was utilized (Ad26.Mos4.HIV, 147). The study was discontinued as the vaccine was seen to be ineffective at preventing the acquisition of HIV compared to placebo (148, 149).
More recently cytomegalovirus vectors (RhCMV) expressing SIV have been designed and tested in Rhesus macaques. The first generation of these vectors was effective in eliciting long lasting, broad cellular immune responses capable of protecting against pathogenic SIV mucosal challenge (150, 151). The macaques became infected with SIV after challenge, however, over time viral replication and spread was completely arrested, resulting in progressive viral clearance until challenged protected macaques were indistinguishable from control macaques – this method of control was termed “control and clear” (150, 152). Despite the RhCMV vectored vaccines great success, these vaccines were replication competent making their safety questionable for use in humans (153). This led to the development of an attenuated SIV RhCMV vectored vaccine with improved safety and retained ability to induce effective cellular immune responses capable of protecting against intravaginal SIV challenge using the “control and clear” concept. This vaccine was able to clear SIV infection in 59% of infected macaques and of these macaques 75% were able to control a second SHIV challenge three years after the last vaccination (153). Human CMV vectored vaccines harbouring HIV have not yet been developed and tested in humans but are awaited with interest (154).
7.3 RNA vaccines
mRNA vaccines contain a single-stranded piece of RNA encoding the target vaccine antigen of interest. Some nucleotides are modified in the RNA strand to extend half-life, decrease backbone immunogenicity and enhance translation (155). The RNA is encapsulated in delivery molecules, like lipid nanoparticles (LNPs), to allow for efficient uptake and expression by cells (156). mRNA vaccines pose many benefits such as being highly efficacious in inducing strong immune responses, having favourable safety profiles, and being easy to design and manufacture on a large scale (157). mRNA vaccine technology is still relatively new and thus clinical trials testing HIV mRNA vaccines are yet to be completed. However, rhesus macaque data is promising with strong humoral and cellular immune responses being elicited by mRNA vaccines at relatively low doses (158–160). Two phase 1 mRNA vaccine clinical trials currently underway in HIV-uninfected adults include the HVTN302 trial (NCT05217641, 161) testing 3 different mRNA vaccines (BG505 MD39.3 mRNA, BG505 MD39.3 gp151 mRNA or BG505 MD39.3 gp151 CD4KO mRNA) and the IAVIG002 trial (NCT05001373, 162) testing the eOD-GT8 60mer mRNA Vaccine (mRNA-1644) and Core-g28v2 60mer mRNA Vaccine (mRNA-1644v2-Core).
7.4 Inactivated vaccines
Inactivated vaccines are composed of whole virus that has been inactivated or killed via heat, chemicals or radiation - with formaldehyde and beta-propiolactone being the most common method for human vaccines (163). Very few inactivated vaccines have been designed for HIV to date. This is due to potential concerns of incomplete inactivation, technical challenges involved with vaccine manufacture and the known poorer immunogenicity associated with inactivated vaccines (164). However, inactivated vaccines are beneficial in their ability to present multiple antigens in their native conformation to the immune system which aids in the production of broadly neutralising antibodies (164). One intriguing inactivated HIV vaccine (SAV001) has been developed and tested in a clinical trial with PWH on ART (NCT01546818, 165, 166). SAV001 is a clade B HIV-1 virus genetically modified to be less virulent but more replication efficient (nef and vpu genes deleted) than the original virus, that is “killed” by inactivation with aldrithiol-2 and γ-irradiation. This vaccine was shown to be safe and effective in generating antibodies towards HIV p24, p17, gp120, and gp41 proteins as well as trimeric HIV Env glycoproteins on infected cells surfaces. Additionally, about 50% of participants showed an enhancement of broadly neutralising antibodies from baseline against HIV-1 B (tier I and II), D, and A subtypes using a luciferase-based assay to measure neutralisation activity. However, as ATI was not performed it is unknown whether the antibodies elicited by the vaccine are protective enough and capable of preventing viral replication.
7.5 Subunit vaccines
Subunit vaccines contain purified or recombinant immunogenic antigens of the virus that are responsible for inducing protective immune responses. Subunit vaccines only contain isolated parts of the virus making them a safe, although potentially less immunogenic, option. This makes adjuvants an essential component of the subunit vaccine formulation (167). Optimal recombinant immunogens that mimic the native conformation of the viral envelope spike of env are yet to be designed resulting mostly in a subpar elicitation of broadly neutralising antibodies by these vaccines (168–170). HIV-1 gp160 and gag p24 subunit vaccines have both been seen to induce HIV-specific immune responses and improve CD4+ T cell counts (171, 172). Most success has, however, been found when combining multiple HIV peptides into a singular recombinant subunit vaccine. An example of such a therapeutic vaccine is Vacc-4x, which contains four synthetic peptides of the HIV core protein, p24. Vacc-4x was seen to be safe, immunogenic and effective in reducing viral load (threefold reduction) when tested in a clinical trial, however, complete viral suppression was not induced (173).
7.6 Therapeutic vaccine combinations (clinical trials)
In attempts to maximize the benefits associated with the various vaccines discussed above, many trials use different vaccines in a combinatorial approach as part of a prime-boost vaccination strategy.
In the majority of HIV vaccine trials using different vaccine combinations, long-lasting viral suppression is not maintained during ATI. No significant effect on viral rebound was observed when using an HIVpDNA/IL-12 prime and rVSV gag boost vaccine (93). However, more promising results were seen in the AELIX-002 trial (NCT03204617, 174), which tested the efficacy of three different HIV therapeutic vaccines containing the HTI (HIVACAT) T-cell immunogen (175). The vaccine formulations tested in the trial included DNA.HTI, MVA.HTI and ChAdOx1.HTI. Of the vaccine recipients with no beneficial HLA class I controller alleles, 40% were able to continue ATI for 22 weeks as their plasma viral loads were below 10,000 copies/mL compared to only 8% of the placebo recipients. Additionally, the vaccine combination was able to induce robust, polyfunctional, and broad CD4+ and CD8+ T cell responses compared to the placebo group. Despite the vaccine immune responses being prominent and time to re-starting ART being delayed in the vaccine group, the criteria for re-start was a plasma viral load above 10,000 copies/mL, and a number of participants had detectable viral loads. This study demonstrates the promise combination vaccines might hold if administered in tandem with other treatments like LRAs and bNAbs. The majority of the vaccines described above have been shown to induce robust T-cell responses whilst LRAs [Romidepsin (120)]/immune modulators [Lefitolimod (28), peginterferon alfa-2b (113)], and dual bNAb therapies [10-1074-LS, 3BNC117-LS (82)] are thought to induce longer-term viral suppression off ART and sometimes cause reductions in the latent reservoir (69). Thus, the additive effects of these two treatments could prove to be invaluable in finding a cure or longer-term treatment for HIV.
8 Enhancing bNAbs with therapeutic vaccines
Neither bNAbs or vaccines alone have so far been able to consistently induce sustained viral control off ART in PWH. However, each has shown potential promise on their own. This raises the question of whether combining bNAbs with vaccination (and possibly other immune modulators) might be immunogenic enough to induce long-term viral remission. If an HIV vaccine combined with bNAbs is able to produce a strong, long-lasting, and specific cellular and humoral immune response against HIV, viral suppression could be mediated by the body’s own cells instead of ART (Figure 1D). This, in theory, would prevent viral rebound when no treatment is taken, bringing about a functional cure.
A non-human primate trial testing the TLR7 agonist vesatolimod, bNAb PGT121, and Ad26/MVA vaccination combination on SHIV-infected macaques supplied evidence for the potential additive effect of multiple therapeutic interventions administered together (38). Compared to the control group where 15/15 macaques rebounded, 8/12 (66%) rebounded in the PGT121 plus vesatolimod group (of which one subsequently controlled) and 6/10 (60%) rebounded in the triple combination group, of which three showed subsequent control. Overall, the triple combination treatment performed the best with 70% of the animals controlling at the end of the study compared to 41% and 33% in the Vesatolimod-bNAb and Vesatolimod-vaccine groups, respectively. This study supports the concept that vaccines and bNAbs may combine effectively to achieve viral suppression in the absence of ART.
Many clinical trials incorporating different combinations of vaccine types, immune modulators, chromatin remodellers, and bNAbs are in the pipeline. One trial has presented some provisional results at a conference (NCT04357821, 176, 177). This trial used a combination treatment comprising of the bNAbs, 10-1074 and VRC07-523LS, the TLR9 agonist, Lefitolimod, and the IL-12 adjuvanted Gag (p24) conserved element (CE)-targeted DNA prime and MVA/HIV62B boost vaccines. 70% of the combination therapy participants maintained at least partial virological control during ATI (with one participant maintaining complete viral control for over 18 months off ART) with an overall mean time to viral rebound of 15 weeks. This study shows promising results for the use of multiple combination therapies for HIV treatment, however, the study was single-armed so no results were obtained comparing the combination treatment to that of a placebo or one treatment individually. Thus, it is unknown whether the treatments induced a significant combinatorial effect or if one intervention was solely responsible for the outcomes.
The results from the ongoing clinical trials NCT04983030 (178), NCT05769569 (RV582, 179), NCT06071767 (A5374/HIV-CORE 009, 180), NCT03619278 (HIVACAR, 181), NCT06484335 (ACHIEV, 182) and AbVax are awaited with much anticipation as they are also combining multiple different LRA, bNAb, and vaccine strategies into one treatment intervention. The NCT04983030 trial will be testing the bNAbs PGT121, PGDM1400, and VRC07-523LS along with an Ad26.Mos4.HIV prime and MVA-BN-HIV boost vaccine. The RV582 trial will be testing three different vaccines (Ad26.Mos4.HIV; MVA-BN-HIV; A244d11 gp120 with a ALFQ Liposomal adjuvant) along with the bNAbs VRC07-523LS and PGDM1400LS and the superagonist IL-15 complex, N-803. The HIV-CORE 009 trial will be testing the TLR7 agonist, Vesatolimod, the bNAbs, 3BNC117-LS and 10-1074-LS and the vaccines, ChAdOx1.tHIVconsv1 + 6 and MVA.tHIVconsv3 + 4. The HIVACAR trial will be testing the efficacy of the combination treatment comprising of the LRA, Romidepsin, the bNAb, 10-1074, and a mRNA encoding HTI with TriMiXmRNAs prime and MVA.HTI boost vaccine. The ACHIEV trial will be testing the combination of the bNabs, VRC07-523LS, PGDM1400LS with a ChAdOx1.tHIVconsv1 and ChAdOx1.HIVconsv62 vaccine prime and MVA.tHIVconsv4 and A244d11 gp120/ALFQ vaccine boost. Lastly, the AbVax trial will test the ability of the combination of three vaccines (ChAdOx1.tHIVconsv1; ChAdOx1.HIVconsv62 and MVA.tHIVconsv4) and two bNAbs (10-1074-LS and 3BNC117-LS) to prevent viral rebound and elicit a vaccinal effect during ATI. The results from these trials will provide novel insights into whether combining many different HIV treatments acts synergistically to eliminate circulating and latently infected virus, antagonistically (e.g. as a result of overstimulation, immune exhaustion or drug interactions), or shows no beneficial additive effect.
9 Conclusion
Combinations of bNAbs with immune modulators, chromatin remodellers, and vaccines remain a promising strategy to achieve an effective functional or sterilizing cure for HIV. As seen by the clinical studies conducted above, many of these treatments enhance immune responses and maintain viral suppression off treatment for a reasonable period when administered alone, but their efficacy is not yet proven when administered together. Only a handful of clinical trials testing these combinations have been published with none yet available testing bNAb and vaccine combinations (Table 1). More research is currently on-going to determine which combinations will work synergistically or additively to amplify the responses achieved by each of the treatments alone (Table 2, Figure 2). It may very well be possible that a combination of bNAbs, immune modulators, chromatin remodellers, and vaccines will be the best strategy to achieve a cure, which is still desperately needed by many PWH. Many HIV combined bNAb-vaccine trials are currently in the pipeline or recruiting, and results from these trials over the next couple of years are greatly anticipated.
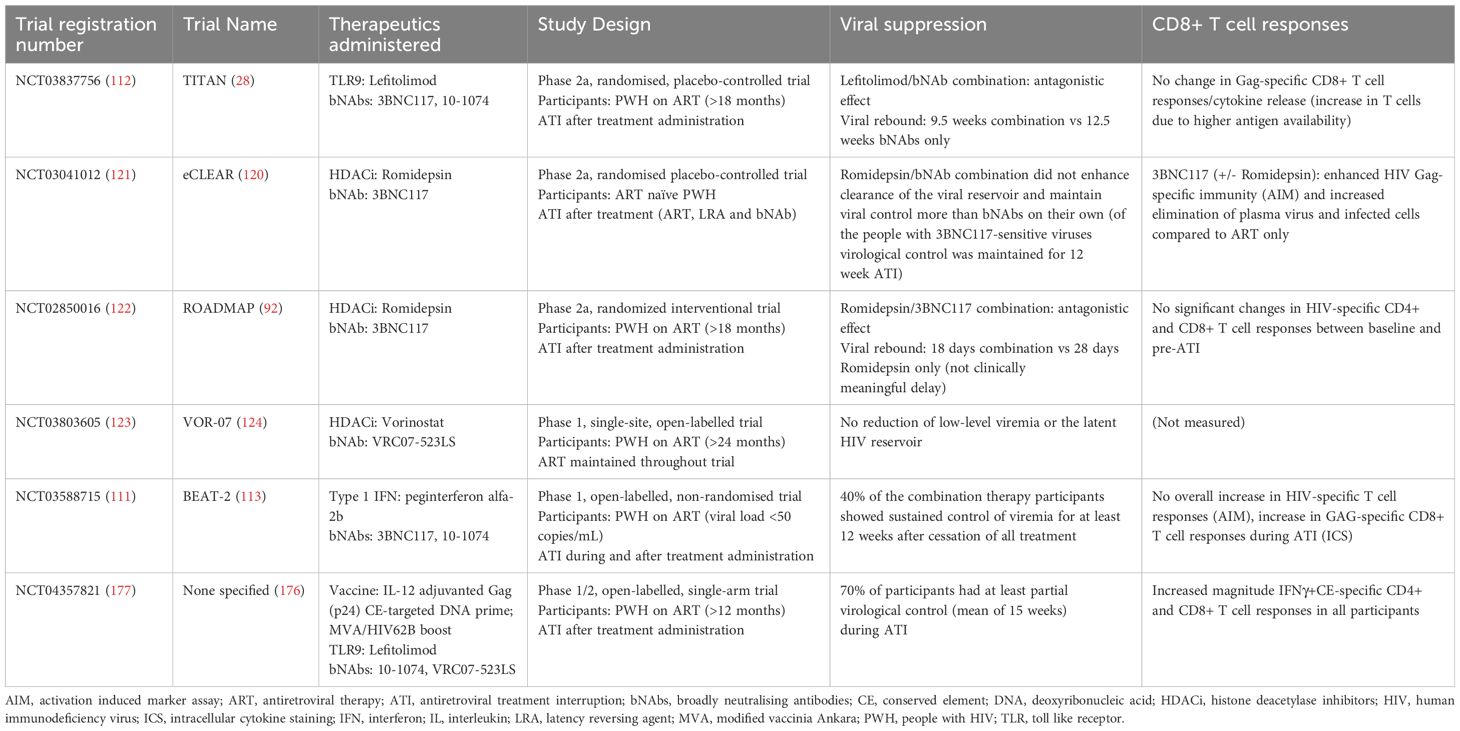
Table 1. Summary of the results of clinical trials involving bNAbs and other therapeutic interventions (immune modulators, chromatin remodellers and therapeutic vaccines).
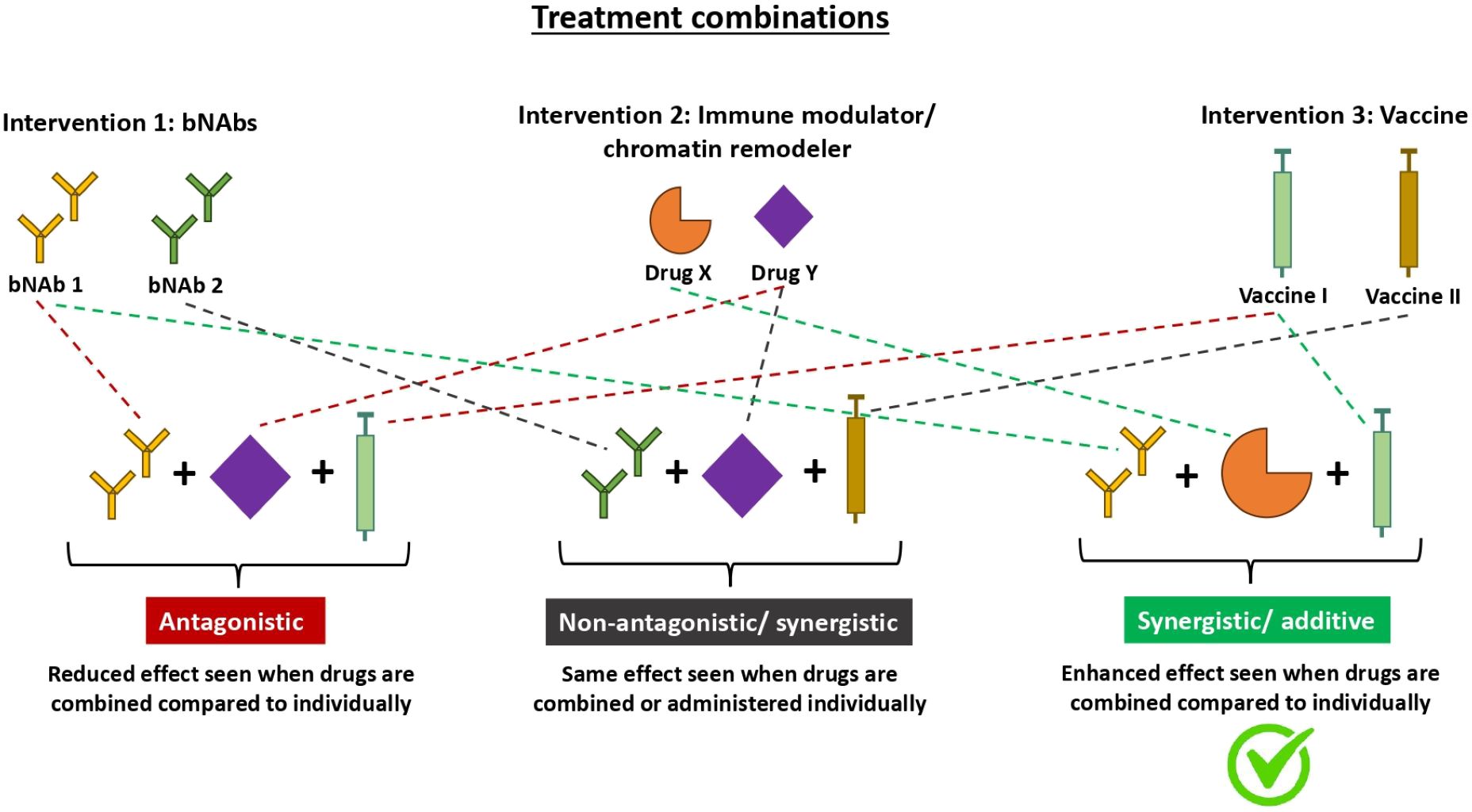
Figure 2. Diagram illustrating mechanisms in which different treatment combinations interact to yield various effects. Therapeutics do not always yield additive effects when used in combination. As seen with the many different types of HIV treatment combinations, the effect can either be antagonistic, additive, synergistic or neutral. Antagonistic treatment combinations (left) have a reduced effect when used in combination compared to individually. Non-antagonistic treatment combinations (middle) yield the same effect when used in combination vs individually. Whilst additive/synergistic treatment combinations (right) have an enhanced effect when used in combination compared to individually. Additive and synergistic both refer to an enhanced effect but they differ in the magnitude of enhancement. An additive interaction is when the effect of the combination treatment equals the sum of the effects of each treatment individually whereas a synergistic interaction is when the effect of the combination treatment is greater than the sum of the effects of each treatment individually. bNAbs, broadly neutralising antibodies.
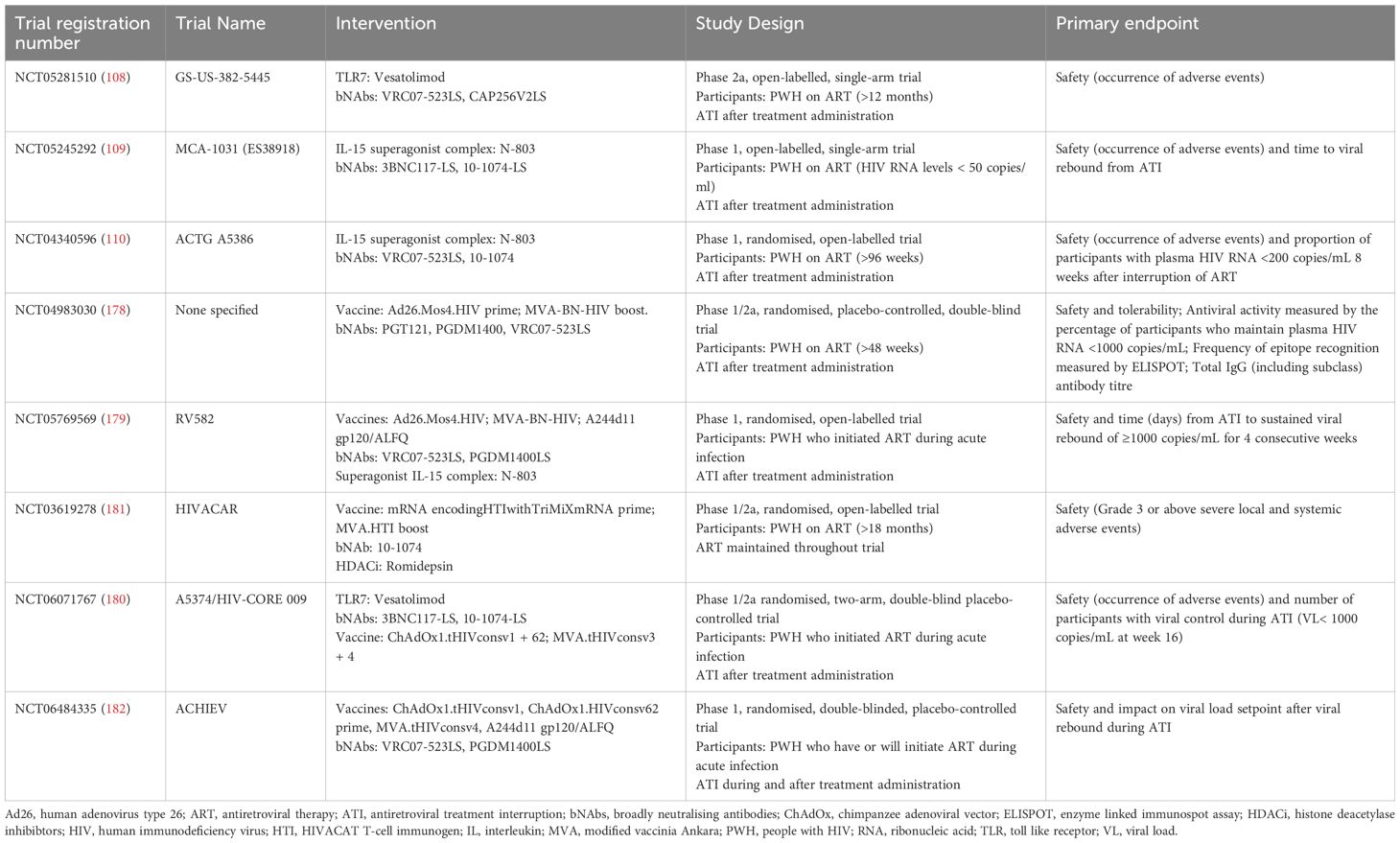
Table 2. Summary of the current and future clinical trials involving bNAbs and other therapeutic interventions registered on clinicaltrial.gov.
Author contributions
CN: Writing – original draft, Writing – review & editing. JF: Supervision, Writing – review & editing, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding was received from the Bill and Melinda Gates Foundation Grant (OPP1210792) to publish the article and conduct research on the use of bNAbs in people with HIV as well as the University of Oxford’s Nuffield Department of Medicine Studentship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tian X, Chen J, Wang X, Xie Y, Zhang X, Han D, et al. Global, regional, and national HIV/AIDS disease burden levels and trends in 1990–2019: A systematic analysis for the global burden of disease 2019 study. Front Public Health. (2023) 11:1068664. doi: 10.3389/fpubh.2023.1068664
2. HIV-CAUSAL Collaboration, Ray M, Logan R, Sterne JAC, Hernandez-Diaz S, Robins JM, et al. The effect of combined antiretroviral therapy on the overall mortality of HIV-infected individuals. AIDS. (2010) 24:123–37. doi: 10.1097/QAD.0b013e3283324283
3. Bhaskaran K, Hamouda O, Sannes M, Boufassa F, Johnson AM, Lambert PC, et al. Changes in the risk of death after HIV seroconversion compared with mortality in the general population. JAMA. (2008) 300:51–9. doi: 10.1001/jama.300.1.51
4. Lima VD, Hogg RS, Harrigan PR, Moore D, Yip B, Wood E, et al. Continued improvement in survival among HIV-infected individuals with newer forms of highly active antiretroviral therapy. AIDS. (2007) 21:685–92. doi: 10.1097/QAD.0b013e32802ef30c
5. Shubber Z, Mills EJ, Nachega JB, Vreeman R, Freitas M, Bock P, et al. Patient-reported barriers to adherence to antiretroviral therapy: A systematic review and meta-analysis. PloS Med. (2016) 13:e1002183. doi: 10.1371/journal.pmed.1002183
6. Ortego C, Huedo-Medina TB, Llorca J, Sevilla L, Santos P, Rodriguez E, et al. Adherence to highly active antiretroviral therapy (HAART): a meta-analysis. AIDS Behav. (2011) 15:1381–96. doi: 10.1007/s10461-011-9942-x
7. Palmer M, Chersich M, Moultrie H, Kuhn L, Fairlie L, Meyers T. Frequency of stavudine substitution due to toxicity in children receiving antiretroviral treatment in sub-Saharan Africa. AIDS. (2013) 27:781–5. doi: 10.1097/QAD.0b013e32835c54b8
8. Penazzato M, Gnanashanmugam D, Rojo P, Lallemant M, Lewis LL, Rocchi F, et al. Optimizing research to speed up availability of pediatric antiretroviral drugs and formulations. Clin Infect Dis. (2017) 64:1597–603. doi: 10.1093/cid/cix194
9. MacDonell K, Naar-King S, Huszti H, Belzer M. Barriers to medication adherence in behaviorally and perinatally infected youth living with HIV. AIDS Behav. (2013) 17:86–93. doi: 10.1007/s10461-012-0364-1
10. Haberer J, Mellins C. Pediatric adherence to HIV antiretroviral therapy. Curr HIV/AIDS Rep. (2009) 6:194–200. doi: 10.1007/s11904-009-0026-8
11. Lewin SR, Attoye T, Bansbach C, Doehle B, Dube K, Dybul M, et al. Multi-stakeholder consensus on a target product profile for an HIV cure. Lancet HIV. (2021) 8:e42–50. doi: 10.1016/S2352-3018(20)30234-4
12. Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. (2003) 9:727–8. doi: 10.1038/nm880
14. Kimata JT, Rice AP, Wang J. Challenges and strategies for the eradication of the HIV reservoir. Curr Opin Immunol. (2016) 42:65–70. doi: 10.1016/j.coi.2016.05.015
15. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U.S.A. (1997) 94:13193–7. doi: 10.1073/pnas.94.24.13193
16. Whyte-Allman S, Bendayan R. HIV-1 sanctuary sites-the role of membrane-associated drug transporters and drug metabolic enzymes. AAPS J. (2020) 22:118–1. doi: 10.1208/s12248-020-00498-1
17. Cory TJ, Schacker TW, Stevenson M, Fletcher CV. Overcoming pharmacologic sanctuaries. Curr Opin HIV AIDS. (2013) 8:190–5. doi: 10.1097/COH.0b013e32835fc68a
18. Bruner KM, Wang Z, Simonetti FR, Bender AM, Kwon KJ, Sengupta S, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature. (2019) 566:120–5. doi: 10.1038/s41586-019-0898-8
19. Gaebler C, Lorenzi JCC, Oliveira TY, Nogueira L, Ramos V, Lu C, et al. Combination of quadruplex qPCR and next-generation sequencing for qualitative and quantitative analysis of the HIV-1 latent reservoir. J Exp Med. (2019) 216:2253–64. doi: 10.1084/jem.20190896
20. Roux H, Chomont N. Measuring HIV reservoirs: Do we need to choose between quantity and quality? J Infect Dis. (2023) 229:635–43. doi: 10.1093/infdis/jiad381
21. Ho Y, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DIS, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. (2013) 155:540–51. doi: 10.1016/j.cell.2013.09.020
22. Eriksson S, Graf EH, Dahl V, Strain MC, Yukl SA, Lysenko ES, et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PloS Pathog. (2013) 9:e1003174. doi: 10.1371/journal.ppat.1003174
23. Bender AM, Simonetti FR, Kumar MR, Fray EJ, Bruner KM, Timmons AE, et al. The landscape of persistent viral genomes in ART-treated SIV, SHIV, and HIV-2 infections. Cell Host Microbe. (2019) 26:73–85.e4. doi: 10.1016/j.chom.2019.06.005
24. Lungu C, Banga R, Gruters RA, Procopio FA. Inducible HIV-1 reservoir quantification: clinical relevance, applications and advancements of TILDA. Front Microbiol. (2021) 12:686690. doi: 10.3389/fmicb.2021.686690
25. Mascola JR, Stiegler G, VanCott TC, Katinger H, Carpenter CB, Hanson CE, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med. (2000) 6:207–10. doi: 10.1038/72318
26. Ferrantelli F, Rasmussen RA, Buckley KA, Li P, Wang T, Montefiori DC, et al. Complete protection of neonatal rhesus macaques against oral exposure to pathogenic simian-human immunodeficiency virus by human anti-HIV monoclonal antibodies. J Infect Dis. (2004) 189:2167–73. doi: 10.1086/420833
27. Parren PW, Marx PA, Hessell AJ, Luckay A, Harouse J, Cheng-Mayer C, et al. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J Virol. (2001) 75:8340–7. doi: 10.1128/jvi.75.17.8340-8347.2001
28. Gunst JD, Hojen JF, Pahus MH, Rosas-Umbert M, Stiksrud B, McMahon JH, et al. Impact of a TLR9 agonist and broadly neutralizing antibodies on HIV-1 persistence: the randomized phase 2a TITAN trial. Nat Med. (2023) 29:2547–58. doi: 10.1038/s41591-023-02547-6
29. Hatziioannou T, Evans DT. Animal models for HIV/AIDS research. Nat Rev Microbiol. (2012) 10:852–67. doi: 10.1038/nrmicro2911
30. Liang X, Casimiro DR, Schleif WA, Wang F, Davies M, Zhang Z, et al. Vectored Gag and Env but not Tat show efficacy against simian-human immunodeficiency virus 89.6P challenge in Mamu-A*01-negative rhesus monkeys. J Virol. (2005) 79:12321–31. doi: 10.1128/JVI.79.19.12321-12331.2005
31. Casimiro DR, Wang F, Schleif WA, Liang X, Zhang Z, Tobery TW, et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with dna and recombinant adenoviral vaccine vectors expressing Gag. J Virol. (2005) 79:15547–55. doi: 10.1128/JVI.79.24.15547-15555.2005
32. Shiver JW, Fu T, Chen L, Casimiro DR, Davies M, Evans RK, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. (2002) 415:331–5. doi: 10.1038/415331a
33. Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. (2008) 372:1881–93. doi: 10.1016/S0140-6736(08)61591-3
34. Deeks SG, Archin N, Cannon P, Collins S, Jones RB, de Jong MAWP, et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat Med. (2021) 27:2085–98. doi: 10.1038/s41591-021-01590-5
35. Julg B, Dee L, Ananworanich J, Barouch DH, Bar K, Caskey M, et al. Recommendations for analytical antiretroviral treatment interruptions in HIV research trials-report of a consensus meeting. Lancet HIV. (2019) 6:e259–68. doi: 10.1016/S2352-3018(19)30052-9
36. Bailon L, Alarcon-Soto Y, Benet S. Challenges of HIV therapeutic vaccines clinical trials design. Curr Opin HIV AIDS. (2022) 17:345–51. doi: 10.1097/COH.0000000000000767
37. Lim SY, Osuna CE, Lee J, Silva-Ayala D, Vikhe P, Chen E, et al. Combination IL-15 therapy in a SHIV nhp model. In: Anonymous Conf Retroviruses Opportunistic Infections; 2020. (2020). Available at: https://www.croiconference.org/abstract/combination-il-15-therapy-in-a-shiv-nhp-model/ (Accessed December 20, 2023).
38. Walker-Sperling VEK, Mercado NB, Chandrashekar A, Borducchi EN, Liu J, Nkolola JP, et al. Therapeutic efficacy of combined active and passive immunization in ART-suppressed, SHIV-infected rhesus macaques. Nat Commun. (2022) 13:3463. doi: 10.1038/s41467-022-31196-5
39. Strategies for Management of Antiretroviral Therapy (SMART) Study Group, El-Sadr WM, Lundgren JD, Neaton JD, Gordin F, Abrams D, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. (2006) 355:2283–96. doi: 10.1056/NEJMoa062360
40. Zheng L, Tierney C, Bosch RJ. Analytical treatment interruption in HIV trials: statistical and study design considerations. Curr HIV/AIDS Rep. (2021) 18:475–82. doi: 10.1007/s11904-021-00569-8
41. Fagard C, Oxenius A, Gunthard H, Garcia F, Le Braz M, Mestre G, et al. A prospective trial of structured treatment interruptions in human immunodeficiency virus infection. Arch Intern Med. (2003) 163:1220–6. doi: 10.1001/archinte.163.10.1220
42. Papasavvas E, Grant RM, Sun J, Mackiewicz A, Pistilli M, Gallo C, et al. Lack of persistent drug-resistant mutations evaluated within and between treatment interruptions in chronically HIV-1-infected patients. AIDS. (2003) 17:2337–43. doi: 10.1097/00002030-200311070-00008
43. Bar KJ, Sneller MC, Harrison LJ, Justement JS, Overton ET, Petrone ME, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N Engl J Med. (2016) 375:2037–50. doi: 10.1056/NEJMoa1608243
44. Kroon E, Ananworanich J, An L, Intasan J, Benjapornpong K, Pinyakorn S, et al. 50LB Central nervous system impact of vorinostat, hydroxychloroquine and maraviroc combination therapy followed by treatment interruption in individuals treated during acute HIV infection. J Virus Eradication. (2016) 2:27. doi: 10.1016/S2055-6640(20)30995-X
45. Carcelain G, Tubiana R, Samri A, Calvez V, Delaugerre C, Agut H, et al. Transient mobilization of human immunodeficiency virus (HIV)-specific CD4 T-helper cells fails to control virus rebounds during intermittent antiretroviral therapy in chronic HIV type 1 infection. J Virol. (2001) 75:234–41. doi: 10.1128/JVI.75.1.234-241.2001
46. Lee MJ, Collins S, Babalis D, Johnson N, Falaschetti E, Prevost AT, et al. The RIO trial: rationale, design, and the role of community involvement in a randomised placebo-controlled trial of antiretroviral therapy plus dual long-acting HIV-specific broadly neutralising antibodies (bNAbs) in participants diagnosed with recent HIV infection—study protocol for a two-stage randomised phase II trial. Trials. (2022) 23:263. doi: 10.1186/s13063-022-06151-w
47. Leal L, Feher C, Richart V, Torres B, Garcia F. Antiretroviral therapy interruption (ATI) in HIV-1 infected patients participating in therapeutic vaccine trials: surrogate markers of virological response. Vaccines (Basel. (2020) 8:442. doi: 10.3390/vaccines8030442
48. Wen Y, Bar KJ, Li JZ. Lessons learned from HIV antiretroviral treatment interruption trials. Curr Opin HIV AIDS. (2018) 13:416–21. doi: 10.1097/COH.0000000000000484
49. Bilger A, Plenn E, Barg FK, Rendle KA, Carter WB, Lamour-Harrington A, et al. Participant experiences in HIV cure-directed trial with an extended analytical treatment interruption in Philadelphia, United States. HIV Res Clin Pract. (2023) 24:2267825. doi: 10.1080/25787489.2023.2267825
50. van Paassen P, Dijkstra M, Peay HL, Rokx C, Verbon A, Reiss P, et al. Perceptions of HIV cure and willingness to participate in HIV cure-related trials among people enrolled in the Netherlands cohort study on acute HIV infection. J Virus Erad. (2022) 8:100072. doi: 10.1016/j.jve.2022.100072
51. Jensen BO, Knops E, Cords L, Lubke N, Salgado M, Busman-Sahay K, et al. In-depth virological and immunological characterization of HIV-1 cure after CCR5Delta32/Delta32 allogeneic hematopoietic stem cell transplantation. Nat Med. (2023) 29:583–7. doi: 10.1038/s41591-023-02213-x
52. Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity. (2018) 48:872–95. doi: 10.1016/j.immuni.2018.04.030
53. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang H, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. (2012) 36:491–501. doi: 10.1016/j.immuni.2012.01.014
54. Fidler S, Stöhr W, Pace M, Dorrell L, Lever A, Pett S, et al. Antiretroviral therapy alone versus antiretroviral therapy with a kick and kill approach, on measures of the HIV reservoir in participants with recent HIV infection (the RIVER trial): a phase 2, randomised trial. Lancet. (2020) 395:888–98. doi: 10.1016/S0140-6736(19)32990-3
55. Sadowski I, Hashemi FB. Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs. Cell Mol Life Sci. (2019) 76:3583–600. doi: 10.1007/s00018-019-03156-8
56. Vansant G, Bruggemans A, Janssens J, Debyser Z. Block-and-lock strategies to cure HIV infection. Viruses. (2020) 12:84. doi: 10.3390/v12010084
57. Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, et al. In vivo suppression of HIV rebound by didehydro-cortistatin A, a "Block-and-lock" Strategy for HIV-1 treatment. Cell Rep. (2017) 21:600–11. doi: 10.1016/j.celrep.2017.09.080
58. Nguyen K, Dobrowolski C, Shukla M, Cho W, Luttge B, Karn J. Inhibition of the H3K27 demethylase UTX enhances the epigenetic silencing of HIV proviruses and induces HIV-1 DNA hypermethylation but fails to permanently block HIV reactivation. PloS Pathog. (2021) 17:e1010014. doi: 10.1371/journal.ppat.1010014
59. Wang G, Zhao N, Berkhout B, Das AT. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. (2018) 244:321–32. doi: 10.1016/j.virusres.2017.07.020
60. Qu X, Wang P, Ding D, Li L, Wang H, Ma L, et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. (2013) 41:7771–82. doi: 10.1093/nar/gkt571
61. Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther. (2017) 25:1168–86. doi: 10.1016/j.ymthe.2017.03.012
62. Qu X, Wang P, Ding D, Wang X, Zhang G, Zhou X, et al. Zinc finger nuclease: a new approach for excising HIV-1 proviral DNA from infected human T cells. Mol Biol Rep. (2014) 41:5819–27. doi: 10.1007/s11033-014-3456-3
63. Hussein M, Molina MA, Berkhout B, Herrera-Carrillo E. A CRISPR-cas cure for HIV/AIDS. Int J Mol Sci. (2023) 24:1563. doi: 10.3390/ijms24021563
64. Wang G, Zhao N, Berkhout B, Das AT. CRISPR-cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol Ther. (2016) 24:522–6. doi: 10.1038/mt.2016.24
65. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. (2014) 370:901–10. doi: 10.1056/NEJMoa1300662
66. Tebas P, Jadlowsky JK, Shaw PA, Tian L, Esparza E, Brennan AL, et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J Clin Invest. (2021) 131:e144486. doi: 10.1172/JCI144486
67. BBC News. China jails 'gene-edited babies' scientist for three years (2019). Available online at: https://www.bbc.co.uk/news/world-asia-China-50944461 (Accessed January 4, 2024).
68. Griffith SA, McCoy LE. To bnAb or Not to bnAb: Defining Broadly Neutralising Antibodies Against HIV-1. Front Immunol. (2021) 12:708227. doi: 10.3389/fimmu.2021.708227
69. Hsu DC, Mellors JW, Vasan S. Can broadly neutralizing HIV-1 antibodies help achieve an ART-free remission? Front Immunol. (2021) 12:710044. doi: 10.3389/fimmu.2021.710044
70. Liu Y, Cao W, Sun M, Li T. Broadly neutralizing antibodies for HIV-1: efficacies, challenges and opportunities. Emerg Microbes Infect. (2020) 9:194–206. doi: 10.1080/22221751.2020.1713707
71. West APJ, Scharf L, Horwitz J, Klein F, Nussenzweig MC, Bjorkman PJ. Computational analysis of anti-HIV-1 antibody neutralization panel data to identify potential functional epitope residues. Proc Natl Acad Sci U.S.A. (2013) 110:10598–603. doi: 10.1073/pnas.1309215110
72. Scheid JF, Mouquet H, Feldhahn N, Walker BD, Pereyra F, Cutrell E, et al. A method for identification of HIV gp140 binding memory B cells in human blood. J Immunol Methods. (2009) 343:65–7. doi: 10.1016/j.jim.2008.11.012
73. Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. Antibodies in HIV-1 vaccine development and therapy. Science. (2013) 341:1199–204. doi: 10.1126/science.1241144
74. Sok D, Burton DR. Recent progress in broadly neutralizing antibodies to HIV. Nat Immunol. (2018) 19:1179–88. doi: 10.1038/s41590-018-0235-7
75. Pedrioli A, Oxenius A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. (2021) 42:1143–58. doi: 10.1016/j.it.2021.10.008
76. Barouch DH, Whitney JB, Moldt B, Klein F, Oliveira TY, Liu J, et al. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature. (2013) 503:224–8. doi: 10.1038/nature12744
77. Hessell AJ, Jaworski JP, Epson E, Matsuda K, Pandey S, Kahl C, et al. Early short-term treatment with neutralizing human monoclonal antibodies halts SHIV infection in infant macaques. Nat Med. (2016) 22:362–8. doi: 10.1038/nm.4063
78. Caskey M, Klein F, Lorenzi JCC, Seaman MS, West APJ, Buckley N, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. (2015) 522:487–91. doi: 10.1038/nature14411
79. Caskey M, Schoofs T, Gruell H, Settler A, Karagounis T, Kreider EF, et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat Med. (2017) 23:185–91. doi: 10.1038/nm.4268
80. Miner MD, Corey L, Montefiori D. Broadly neutralizing monoclonal antibodies for HIV prevention. J Int AIDS Soc. (2021) 24 Suppl 7:e25829. doi: 10.1002/jia2.25829
81. Corey L, Gilbert PB, Juraska M, Montefiori DC, Morris L, Karuna ST, et al. Two randomized trials of neutralizing antibodies to prevent HIV-1 acquisition. N Engl J Med. (2021) 384:1003–14. doi: 10.1056/NEJMoa2031738
82. Mendoza P, Gruell H, Nogueira L, Pai JA, Butler AL, Millard K, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. (2018) 561:479–84. doi: 10.1038/s41586-018-0531-2
83. Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IWL, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. (2010) 28:157–9. doi: 10.1038/nbt.1601
84. Gaudinski MR, Coates EE, Houser KV, Chen GL, Yamshchikov G, Saunders JG, et al. Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PloS Med. (2018) 15:e1002493. doi: 10.1371/journal.pmed.1002493
85. Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. (2014) 158:1243–53. doi: 10.1016/j.cell.2014.08.023
86. Tipoe T, Fidler S, Frater J. An exploration of how broadly neutralizing antibodies might induce HIV remission: the 'vaccinal' effect. Curr Opin HIV AIDS. (2022) 17:162–70. doi: 10.1097/COH.0000000000000731
87. Lambour J, Naranjo-Gomez M, Piechaczyk M, Pelegrin M. Converting monoclonal antibody-based immunotherapies from passive to active: bringing immune complexes into play. Emerg Microbes Infect. (2016) 5:e92. doi: 10.1038/emi.2016.97
88. Rutishauser RL, Trautmann L. CD8 + T-cell responses in HIV controllers: potential implications for novel HIV remission strategies. Curr Opin HIV AIDS. (2022) 17:315–24. doi: 10.1097/COH.0000000000000748
89. Okoye AA, Duell DD, Fukazawa Y, Varco-Merth B, Marenco A, Behrens H, et al. CD8+ T cells fail to limit SIV reactivation following ART withdrawal until after viral amplification. J Clin Invest. (2021) 131:e141677. doi: 10.1172/JCI141677
90. Wikramaratna PS, Lourenco J, Klenerman P, Pybus OG, Gupta S. Effects of neutralizing antibodies on escape from CD8+ T-cell responses in HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140290. doi: 10.1098/rstb.2014.0290
91. Nishimura Y, Gautam R, Chun T, Sadjadpour R, Foulds KE, Shingai M, et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature. (2017) 543:559–63. doi: 10.1038/nature21435
92. Gruell H, Gunst JD, Cohen YZ, Pahus MH, Malin JJ, Platten M, et al. Effect of 3BNC117 and romidepsin on the HIV-1 reservoir in people taking suppressive antiretroviral therapy (ROADMAP): a randomised, open-label, phase 2A trial. Lancet Microbe. (2022) 3:e203. doi: 10.1016/s2666-5247(21)00239-1
93. Sneller MC, Justement JS, Gittens KR, Petrone ME, Clarridge KE, Proschan MA, et al. A randomized controlled safety/efficacy trial of therapeutic vaccination in HIV-infected individuals who initiated antiretroviral therapy early in infection. Sci Transl Med. (2017) 9:eaan8848. doi: 10.1126/scitranslmed.aan8848
94. Johnson PR, Schnepp BC, Zhang J, Connell MJ, Greene SM, Yuste E, et al. Vector-mediated gene transfer engenders long-lived neutralizing activity and protection against SIV infection in monkeys. Nat Med. (2009) 15:901–6. doi: 10.1038/nm.1967
95. Martinez-Navio JM, Fuchs SP, Mendes DE, Rakasz EG, Gao G, Lifson JD, et al. Long-term delivery of an anti-SIV monoclonal antibody with AAV. Front Immunol. (2020) 11:449. doi: 10.3389/fimmu.2020.00449
96. Priddy FH, Lewis DJM, Gelderblom HC, Hassanin H, Streatfield C, LaBranche C, et al. Adeno-associated virus vectored immunoprophylaxis to prevent HIV in healthy adults: a phase 1 randomised controlled trial. Lancet HIV. (2019) 6:e230–9. doi: 10.1016/S2352-3018(19)30003-7
97. Casazza JP, Cale EM, Narpala S, Yamshchikov GV, Coates EE, Hendel CS, et al. Safety and tolerability of AAV8 delivery of a broadly neutralizing antibody in adults living with HIV: a phase 1, dose-escalation trial. Nat Med. (2022) 28:1022–30. doi: 10.1038/s41591-022-01762-x
98. Zacharopoulou P, Ansari MA, Frater J. A calculated risk: Evaluating HIV resistance to the broadly neutralising antibodies10-1074 and 3BNC117. Curr Opin HIV AIDS. (2022) 17:352–8. doi: 10.1097/COH.0000000000000764
99. Julg B, Stephenson KE, Wagh K, Tan SC, Zash R, Walsh S, et al. Safety and antiviral activity of triple combination broadly neutralizing monoclonal antibody therapy against HIV-1: a phase 1 clinical trial. Nat Med. (2022) 28:1288–96. doi: 10.1038/s41591-022-01815-1
100. Adams S. Toll-like receptor agonists in cancer therapy. Immunotherapy. (2009) 1:949–64. doi: 10.2217/imt.09.70
101. Offersen R, Nissen SK, Rasmussen TA, Østergaard L, Denton PW, Søgaard OS, et al. A novel toll-like receptor 9 agonist, MGN1703, enhances HIV-1 transcription and NK cell-mediated inhibition of HIV-1-infected autologous CD4+ T cells. J Virol. (2016) 90:4441–53. doi: 10.1128/JVI.00222-16
102. Novis CL, Archin NM, Buzon MJ, Verdin E, Round JL, Lichterfeld M, et al. Reactivation of latent HIV-1 in central memory CD4+T cells through TLR-1/2 stimulation. Retrovirology. (2013) 10:119. doi: 10.1186/1742-4690-10-119
103. Thibault S, Imbeault M, Tardif MR, Tremblay MJ. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4+ T cells. Virology. (2009) 389:20–5. doi: 10.1016/j.virol.2009.04.019
104. Schlaepfer E, Speck RF. TLR8 activates HIV from latently infected cells of myeloid-monocytic origin directly via the MAPK pathway and from latently infected CD4+ T cells indirectly via TNF-α. J Immunol. (2011) 186:4314–24. doi: 10.4049/jimmunol.1003174
105. Halper-Stromberg A, Lu C, Klein F, Horwitz J, Bournazos S, Nogueira L, et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. (2014) 158:989–99. doi: 10.1016/j.cell.2014.07.043
106. Borducchi EN, Liu J, Nkolola JP, Cadena AM, Yu W, Fischinger S, et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature. (2018) 563:360. doi: 10.1038/s41586-018-0600-6
107. Moldt B, Chandrashekar A, Borducchi EN, Nkolola JP, Stephenson H, Nagel M, et al. HIV envelope antibodies and TLR7 agonist partially prevent viral rebound in chronically SHIV-infected monkeys. PloS Pathog. (2022) 18:e1010467. doi: 10.1371/journal.ppat.1010467
108. A Phase 2a Study to Evaluate the Safety and Tolerability of a Regimen of Dual Anti-HIV Envelope Antibodies, VRC07-523LS and CAP256V2LS, in a Sequential Regimen With a TLR7 Agonist, Vesatolimod, in Early Antiretroviral-Treated HIV-1 Clade C-Infected Women (2022). ClinicalTrials.gov identifier: NCT05281510. Available online at: https://clinicaltrials.gov/study/NCT05281510 (Accessed December 19, 2023)
109. An open label, single arm study of the safety and antiretroviral activity of the combination of 2 long-acting broadly neutralizing antibodies plus an IL-15 superagonist complex in ART-treated adults living with HIV during analytical treatment interruption (2022). ClinicalTrials.gov identifier: NCT05245292. Available online at: https://clinicaltrials.gov/study/NCT05245292 (Accessed December 19, 2023).
110. A Phase I Clinical Trial of the Safety, Tolerability, and Efficacy of IL-15 Superagonist (N-803) With and Without Combination Broadly Neutralizing Antibodies to Induce HIV-1 Control During Analytic Treatment Interruption (2021). ClinicalTrials.gov identifier: NCT04340596. Available online at: https://clinicaltrials.gov/study/NCT04340596 (Accessed December 19, 2023).
111. Pilot Study on Innate Activation and Viral Control in HIV-Infected Adults Undergoing an Analytical Treatment Interruption After Administration of Pegylated Interferon Alpha 2b With Broadly HIV-1 Neutralizing Antibodies (3BNC117, 10-1074) (2020). ClinicalTrials.gov identifier: NCT03588715. Available online at: https://clinicaltrials.gov/study/NCT03588715 (Accessed December 19, 2023).
112. Combining a TLR9 Agonist With Broadly Neutralizing Antibodies for Reservoir Reduction and Immunological Control of HIV Infection: An Investigator-initiated Randomized, Placebo-controlled, Phase IIa Trial (2019). ClinicalTrials.gov identifier: NCT03837756. Available online at: https://clinicaltrials.gov/study/NCT03837756 (Accessed December 19, 2023).
113. Tebas P, Azzoni L, Papasavvas E, Choi G, Ellenberg S, Caskey M, et al. (2023). BEAT2 PRIMARY TRIAL OUTCOMES: PEG-IFN-α2B + 3BNC117 & 10-1074 IN CHRONIC HIV INFECTION, in: Anonymous Conference on Retroviruses and Opportunistic Infections; 2023. Available online at: https://www.croiconference.org/wp-content/uploads/sites/2/posters/2023/Tebas_poster_CROI-2023_431_PT_LJM3-133207934336133464.pdf (Accessed December 21,2023).
114. Pumfery A, Deng L, Maddukuri A, de la Fuente C, Li H, Wade JD, et al. Chromatin remodeling and modification during HIV-1 Tat-activated transcription. Curr HIV Res. (2003) 1:343–62. doi: 10.2174/1570162033485186
115. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. (1997) 389:349–52. doi: 10.1038/38664
116. Chen HP, Zhao YT, Zhao TC. Histone deacetylases and mechanisms of regulation of gene expression. Crit Rev Oncog. (2015) 20:35–47. doi: 10.1615/critrevoncog.2015012997
117. Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, et al. Systemic HIV and SIV latency reversal via non-canonical NF-kappaB signalling in vivo. Nature. (2020) 578:160–5. doi: 10.1038/s41586-020-1951-3
118. Dashti A, Sukkestad S, Horner AM, Neja M, Siddiqi Z, Waller C, et al. AZD5582 plus SIV-specific antibodies reduce lymph node viral reservoirs in antiretroviral therapy-suppressed macaques. Nat Med. (2023) 29:2535–46. doi: 10.1038/s41591-023-02570-7
119. Lichterfeld M. Reactivation of latent HIV moves shock-and-kill treatments forward. Nature. (2020) 578:42–3. doi: 10.1038/d41586-020-00010-x
120. Gunst JD, Pahus MH, Rosás-Umbert M, Lu I, Benfield T, Nielsen H, et al. Early intervention with 3BNC117 and romidepsin at antiretroviral treatment initiation in people with HIV-1: a phase 1b/2a, randomized trial. Nat Med. (2022) 28:2424. doi: 10.1038/s41591-022-02023-7
121. Early Administration of Latency Reversing Therapy and Broadly Neutralizing Antibodies to Limit the Establishment of the HIV-1 Reservoir During Initiation of Antiretroviral Treatment - a Randomized Controlled Trial (2017). ClinicalTrials.gov identifier: NCT03041012. Available online at: https://clinicaltrials.gov/study/NCT03041012 (Accessed January 5, 2024).
122. A Phase 2a, Randomized Study of Romidepsin With or Without 3BNC117 to Evaluate the Effects on the HIV-1 Reservoir (ROADMAP) (2017). ClinicalTrials.gov identifier: NCT02850016. Available online at: https://clinicaltrials.gov/study/NCT02850016 (Accessed January 5, 2024).
123. IGHID 11802 - Combination Therapy With the Novel Clearance Modality (VRC07-523LS) and the Latency Reversal Agent (Vorinostat) to Reduce the Frequency of Latent, Resting CD4+ T Cell Infection (The VOR-07 Study) (2019). ClinicalTrials.gov identifier: NCT03803605. Available online at: https://clinicaltrials.gov/study/NCT03803605 (Accessed January 5, 2024).
124. Gay CL, James KS, Tuyishime M, Falcinelli SD, Joseph SB, Moeser MJ, et al. Stable latent HIV infection and low-level viremia despite treatment with the broadly neutralizing antibody VRC07-523LS and the latency reversal agent vorinostat. J Infect Dis. (2022) 225:856–61. doi: 10.1093/infdis/jiab487
125. Collins DR, Gaiha GD, Walker BD. CD8+ T cells in HIV control, cure and prevention. Nat Rev Immunol. (2020) 20:471–82. doi: 10.1038/s41577-020-0274-9
126. Sobieszczyk ME. Therapeutic HIV vaccines and broadly neutralizing antibodies. Top Antivir Med. (2020) 27:97–101.
127. Gao F, Weaver EA, Lu Z, Li Y, Liao H, Ma B, et al. Antigenicity and immunogenicity of a synthetic human immunodeficiency virus type 1 group m consensus envelope glycoprotein. J Virol. (2005) 79:1154–63. doi: 10.1128/JVI.79.2.1154-1163.2005
128. Magro G, Calistri A, Parolin C. How to break free: HIV-1 escapes from innovative therapeutic approaches. Front Virol (online. (2022) 2:933418. doi: 10.3389/fviro.2022.933418
129. Akbari E, Seyedinkhorasani M, Bolhassani A. Conserved multiepitope vaccine constructs: A potent HIV-1 therapeutic vaccine in clinical trials. Braz J Infect Dis. (2023) 27:102774. doi: 10.1016/j.bjid.2023.102774
130. Mothe B, Manzardo C, Sanchez-Bernabeu A, Coll P, Morón-López S, Puertas MC, et al. Therapeutic vaccination refocuses T-cell responses towards conserved regions of HIV-1 in early treated individuals (BCN 01 study). EClinicalMedicine. (2019) 11:65. doi: 10.1016/j.eclinm.2019.05.009
131. Colby DJ, Sarnecki M, Barouch DH, Tipsuk S, Stieh DJ, Kroon E, et al. Safety and immunogenicity of Ad26 and MVA vaccines in acutely treated HIV and effect on viral rebound after antiretroviral therapy interruption. Nat Med. (2020) 26:498–501. doi: 10.1038/s41591-020-0774-y
132. Mothe B, Rosas-Umbert M, Coll P, Manzardo C, Puertas MC, Moron-Lopez S, et al. HIVconsv vaccines and romidepsin in early-treated HIV-1-infected individuals: safety, immunogenicity and effect on the viral reservoir (Study BCN02). Front Immunol. (2020) 11:823. doi: 10.3389/fimmu.2020.00823
133. Chen Z, Julg B. Therapeutic vaccines for the treatment of HIV. Transl Res. (2020) 223:61–75. doi: 10.1016/j.trsl.2020.04.008
134. Munson P. Progress towards a therapeutic HIV DNA vaccine. Expert Rev Vaccines. (2022) 21:783–98. doi: 10.1080/14760584.2022.2059467
135. Jacobson JM, Zheng L, Wilson CC, Tebas P, Matining RM, Egan MA, et al. The safety and immunogenicity of an interleukin-12-enhanced multiantigen DNA vaccine delivered by electroporation for the treatment of HIV-1 infection. J Acquir Immune Defic Syndr. (2016) 71:163–71. doi: 10.1097/QAI.0000000000000830
136. Kalams SA, Parker SD, Elizaga M, Metch B, Edupuganti S, Hural J, et al. Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J Infect Dis. (2013) 208:818–29. doi: 10.1093/infdis/jit236
137. Vardas E, Stanescu I, Leinonen M, Ellefsen K, Pantaleo G, Valtavaara M, et al. Indicators of therapeutic effect in FIT-06, a Phase II trial of a DNA vaccine, GTU((R))-Multi-HIVB, in untreated HIV-1 infected subjects. Vaccine. (2012) 30:4046–54. doi: 10.1016/j.vaccine.2012.04.007
138. Johnson JA, Barouch DH, Baden LR. Nonreplicating vectors in HIV vaccines. Curr Opin HIV AIDS. (2013) 8:412–20. doi: 10.1097/COH.0b013e328363d3b7
139. Travieso T, Li J, Mahesh S, Mello JDFRE, Blasi M. The use of viral vectors in vaccine development. NPJ Vaccines. (2022) 7:1–10. doi: 10.1038/s41541-022-00503-y
140. Orlova OV, Glazkova DV, Bogoslovskaya EV, Shipulin GA, Yudin SM. Development of modified vaccinia virus ankara-based vaccines: advantages and applications. Vaccines (Basel. (2022) 10:1516. doi: 10.3390/vaccines10091516
141. Goepfert PA, Elizaga ML, Sato A, Qin L, Cardinali M, Hay CM, et al. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J Infect Dis. (2011) 203:610–9. doi: 10.1093/infdis/jiq105
142. Altenburg AF, van Trierum SE, de Bruin E, de Meulder D, van de Sandt CE, van der Klis FRM, et al. Effects of pre-existing orthopoxvirus-specific immunity on the performance of Modified Vaccinia virus Ankara-based influenza vaccines. Sci Rep. (2018) 8:6474–2. doi: 10.1038/s41598-018-24820-2
143. Gray G, Buchbinder S, Duerr A. Overview of STEP and Phambili trial results: two phase IIb test-of-concept studies investigating the efficacy of MRK adenovirus type 5 gag/pol/nef subtype B HIV vaccine. Curr Opin HIV AIDS. (2010) 5:357–61. doi: 10.1097/COH.0b013e32833d2d2b
144. Duerr A, Huang Y, Buchbinder S, Coombs RW, Sanchez J, del Rio C, et al. Extended follow-up confirms early vaccine-enhanced risk of HIV acquisition and demonstrates waning effect over time among participants in a randomized trial of recombinant adenovirus HIV vaccine (Step Study). J Infect Dis. (2012) 206:258–66. doi: 10.1093/infdis/jis342
145. World Health Organization. The Oxford/AstraZeneca (ChAdOx1-S [recombinant] vaccine) COVID-19 vaccine: what you need to know (2022). Available online at: https://www.who.int/news-room/feature-stories/detail/the-oxford-astrazeneca-covid-19-vaccine-what-you-need-to-know (Accessed April 10, 2024).
147. AIDS Vaccine Advocacy Coalition. HVTN 706/ HPX3002/ mosaico (2024). Available online at: https://avac.org/trial/hvtn-706-hpx3002-mosaico/ (Accessed June 1, 2024).
148. HIV Vaccine Trials Network. Phase 3 mosaic-based investigational HIV vaccine study discontinued following disappointing results of planned data review (2023). Available online at: https://www.hvtn.org/news/news-releases/2023/01/phase-3-mosaic-based-investigational-hiv-vaccine-study-discontinued-following-disappointing-results-planned-data-review.html (Accessed June 1, 2024).
149. Johnson&Johnson. Janssen and global partners to discontinue phase 3 mosaico HIV vaccine clinical trial (2023). Available online at: https://www.jnj.com/media-center/press-releases/janssen-and-global-partners-to-discontinue-phase-3-mosaico-hiv-vaccine-clinical-trial (Accessed June 1, 2024).
150. Hansen SG, Piatak MJ, Ventura AB, Hughes CM, Gilbride RM, Ford JC, et al. Immune clearance of highly pathogenic SIV infection. Nature. (2013) 502:100–4. doi: 10.1038/nature12519
151. Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. (2009) 15:293–9. doi: 10.1038/nm.1935
152. Okoye AA, Hansen SG, Vaidya M, Fukazawa Y, Park H, Duell DM, et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat Med. (2018) 24:1430–40. doi: 10.1038/s41591-018-0130-7
153. Hansen SG, Marshall EE, Malouli D, Ventura AB, Hughes CM, Ainslie E, et al. A live-attenuated RhCMV/SIV vaccine shows long-term efficacy against heterologous SIV challenge. Sci Transl Med. (2019) 11:eaaw2607. doi: 10.1126/scitranslmed.aaw2607
154. Picker LJ, Lifson JD, Gale MJ, Hansen SG, Fruh K. Programming cytomegalovirus as an HIV vaccine. Trends Immunol. (2023) 44:287–304. doi: 10.1016/j.it.2023.02.001
155. Granados-Riveron JT, Aquino-Jarquin G. Engineering of the current nucleoside-modified mRNA-LNP vaccines against SARS-CoV-2. BioMed Pharmacother. (2021) 142:111953. doi: 10.1016/j.biopha.2021.111953
156. Fortner A, Schumacher D. First COVID-19 vaccines receiving the US FDA and EMA emergency use authorization. Discoveries (Craiova. (2021) 9:e122. doi: 10.15190/d.2021.1
157. Fortner A, Bucur O. mRNA-based vaccine technology for HIV. Discoveries (Craiova. (2022) 10:e150. doi: 10.15190/d.2022.9
158. Pardi N, LaBranche CC, Ferrari G, Cain DW, Tombacz I, Parks RJ, et al. Characterization of HIV-1 nucleoside-modified mRNA vaccines in rabbits and rhesus macaques. Mol Ther Nucleic Acids. (2019) 15:36–47. doi: 10.1016/j.omtn.2019.03.003
159. Bogers WM, Oostermeijer H, Mooij P, Koopman G, Verschoor EJ, Davis D, et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J Infect Dis. (2015) 211:947–55. doi: 10.1093/infdis/jiu522
160. Zhang P, Narayanan E, Liu Q, Tsybovsky Y, Boswell K, Ding S, et al. A multiclade env-gag VLP mRNA vaccine elicits tier-2 HIV-1-neutralizing antibodies and reduces the risk of heterologous SHIV infection in macaques. Nat Med. (2021) 27:2234–45. doi: 10.1038/s41591-021-01574-5
161. A Phase 1, Randomized, Open-label Clinical Trial to Evaluate the Safety and Immunogenicity of BG505 MD39.3, BG505 MD39.3 gp151, and BG505 MD39.3 gp151 CD4KO HIV Trimer mRNA Vaccines in Healthy, HIV-uninfected Adult Participants (2022). ClinicalTrials.gov identifier: NCT05217641. Available online at: https://clinicaltrials.gov/study/NCT05217641 (Accessed December 19, 2023).
162. A Phase 1, Randomized, First-in-human, Open-label Study to Evaluate the Safety and Immunogenicity of eOD-GT8 60mer mRNA Vaccine (mRNA-1644) and Core-g28v2 60mer mRNA Vaccine (mRNA-1644v2-Core) in HIV-1 Uninfected Adults in Good General Health (2021). ClinicalTrials.gov identifier: NCT05001373. Available online at: https://clinicaltrials.gov/study/NCT05001373 (Accessed December 19, 2023).
163. Sanders B, Koldijk M, Schuitemaker H. Inactivated viral vaccines. Vaccine Analysis: Strategies Principles Control. (2014), 45–80. doi: 10.1007/978-3-662-45024-6_2
164. Kang CY, Gao Y. Killed whole-HIV vaccine; employing a well established strategy for antiviral vaccines. AIDS Res Ther. (2017) 14:47–5. doi: 10.1186/s12981-017-0176-5
165. Choi E, Michalski CJ, Choo SH, Kim GN, Banasikowska E, Lee S, et al. First Phase I human clinical trial of a killed whole-HIV-1 vaccine: demonstration of its safety and enhancement of anti-HIV antibody responses. Retrovirology. (2016) 13:82–2. doi: 10.1186/s12977-016-0317-2
166. A Phase I, Randomized, Observer-blinded, Placebo-controlled Clinical Study to Assess the Safety, Tolerability, and Immune Response of Killed-Whole HIV-1 Vaccine (SAV001-H) Administered Intramuscularly to Chronic HIV-1 Infected Patients Currently Under Treatment With Highly Active Antiretroviral Therapy (HAART) (2012). ClinicalTrials.gov identifier: NCT01546818. Available online at: https://clinicaltrials.gov/study/NCT01546818 (Accessed January 6, 2024).
168. Joyce JG, Hurni WM, Bogusky MJ, Garsky VM, Liang X, Citron MP, et al. Enhancement of alpha -helicity in the HIV-1 inhibitory peptide DP178 leads to an increased affinity for human monoclonal antibody 2F5 but does not elicit neutralizing responses in vitro. Implications for vaccine design. J Biol Chem. (2002) 277:45811–20. doi: 10.1074/jbc.M205862200
169. Ho J, Uger RA, Zwick MB, Luscher MA, Barber BH, MacDonald KS. Conformational constraints imposed on a pan-neutralizing HIV-1 antibody epitope result in increased antigenicity but not neutralizing response. Vaccine. (2005) 23:1559–73. doi: 10.1016/j.vaccine.2004.09.037
170. Schief WR, Ban YA, Stamatatos L. Challenges for structure-based HIV vaccine design. Curr Opin HIV AIDS. (2009) 4:431–40. doi: 10.1097/COH.0b013e32832e6184
171. Gudmundsdotter L, Bostrom A, Burton C, Rosignoli G, Sandstrom E, Hejdeman B, et al. Long-term increase of CD4+ central memory cells in HIV-1-infected individuals by therapeutic HIV-1 rgp160 immunization. Vaccine. (2008) 26:5107–10. doi: 10.1016/j.vaccine.2008.03.063
172. Harrer T, Plettenberg A, Arasteh K, Van Lunzen J, Fatkenheuer G, Jaeger H, et al. Safety and immunogenicity of an adjuvanted protein therapeutic HIV-1 vaccine in subjects with HIV-1 infection: a randomised placebo-controlled study. Vaccine. (2014) 32:2657–65. doi: 10.1016/j.vaccine.2013.10.030
173. Pollard RB, Rockstroh JK, Pantaleo G, Asmuth DM, Peters B, Lazzarin A, et al. Safety and efficacy of the peptide-based therapeutic vaccine for HIV-1, Vacc-4x: a phase 2 randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. (2014) 14:291–300. doi: 10.1016/S1473-3099(13)70343-8
174. A Phase I, Randomized, Double-Blind, Placebo-Controlled Safety, Tolerability and Immunogenicity Study of Candidate HIV-1 Vaccines DNA.HTI, MVA.HTI and ChAdOx1.HTI in Early Treated HIV-1 Positive Individuals (2017). ClinicalTrials.gov identifier: NCT03204617. Available online at: https://clinicaltrials.gov/study/NCT03204617 (Accessed January 5, 2024).
175. Bailón L, Llano A, Cedeño S, Escribà T, Rosás-Umbert M, Parera M, et al. Safety, immunogenicity and effect on viral rebound of HTI vaccines in early treated HIV-1 infection: a randomized, placebo-controlled phase 1 trial. Nat Med. (2022) 28:2611–21. doi: 10.1038/s41591-022-02060-2
176. Peluso M, Deitchman A, Magombedze G, Williams M, Hoh R, Kumar N, et al. (2023). Rebound Dynamics Following Immunotherapy with an HIV vaccine, TLR-9 agonist, and Broadly Neutralizing Antibodies, in: Anonymous Conference on Retroviruses and Opportunistic Infections, , Feb 19-22, 2023. Seattle, Washington.
177. Combinatorial Therapy With a Therapeutic Conserved Element DNA Vaccine, MVA Vaccine Boost, TLR9 Agonist and Broadly Neutralizing Antibodies: a Proof-of-concept Study Aimed at Inducing an HIV Remission (2020). ClinicalTrials.gov identifier: NCT04357821. Available online at: https://clinicaltrials.gov/study/NCT04357821 (Accessed January 5, 2024).
178. A Safety, Immunogenicity and Efficacy Phase 1/2a Study of a Heterologous Ad26.Mos4.HIV, MVA-BN-HIV Vaccine Regimen Plus Broadly Neutralizing Antibodies PGT121, PGDM1400, and VRC07-523LS in HIV-1-Infected Adults on Suppressive ART (2022). ClinicalTrials.gov identifier: NCT04983030. Available online at: https://clinicaltrials.gov/study/NCT04983030 (Accessed January 5, 2024).
179. Safety and Efficacy of Broadly Neutralizing Antibodies Followed by Innate Immune Stimulation and Therapeutic Vaccination for the Induction of HIV Remission (2023). ClinicalTrials.gov identifier: NCT05769569. Available online at: https://clinicaltrials.gov/study/NCT05769569 (Accessed January 10, 2024).
180. A Phase I/IIa Randomized, Placebo-Controlled Trial of Conserved-Mosaic T-cell Vaccine in a Regimen With Vesatolimod and Broadly Neutralizing Antibodies in Adults Initiated on Suppressive Antiretroviral Therapy During Acute HIV-1 (2024). ClinicalTrials.gov identifier: NCT06071767. Available online at: https://clinicaltrials.gov/study/NCT06071767 (Accessed June 3, 2024).
181. A Phase I/IIa, Randomised Study to Evaluate the Safety and the Effectiveness of a Combination of Therapeutic Vaccine, Broadly Neutralising Antibody (10-1074), and the Latency Reversing Agent Romidepsin to Achieve a Remission of HIV Infection in Chronically HIV-infected Participants Under Stable Combined Antiretroviral Therapy (2020). ClinicalTrials.gov identifier: NCT03619278. Available online at: https://clinicaltrials.gov/study/NCT03619278 (Accessed January 10, 2024).
182. Approach to Control HIV With Immune Enhancement and Vaccination (ACHIEV): Safety and Efficacy of Broadly Neutralizing Antibodies Combined With Therapeutic Vaccination for the Induction of HIV Remission (2024). ClinicalTrials.gov identifier: NCT06484335. Available online at: https://clinicaltrials.gov/study/NCT06484335 (Accessed January 10, 2024).
Keywords: HIV, therapeutics, broadly neutralising antibodies, latency-reversing agents, immune modulators, vaccines, cure
Citation: Nel C and Frater J (2024) Enhancing broadly neutralising antibody suppression of HIV by immune modulation and vaccination. Front. Immunol. 15:1478703. doi: 10.3389/fimmu.2024.1478703
Received: 10 August 2024; Accepted: 17 October 2024;
Published: 07 November 2024.
Edited by:
Heinz Kohler, Retired, Carlsbad, CA, United StatesReviewed by:
Thorsten Demberg, Baylor College of Medicine, United StatesNancy L. Haigwood, Oregon Health and Science University, United States
Copyright © 2024 Nel and Frater. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John Frater, Sm9obi5mcmF0ZXJAbmRtLm94LmFjLnVr