
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 October 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1477160
 Lan Li1,2Qingyuan Tan1,2
Lan Li1,2Qingyuan Tan1,2 Xueying Wu1Xiaowen Mou1,2Ziqi Lin1
Xueying Wu1Xiaowen Mou1,2Ziqi Lin1 Tingting Liu1
Tingting Liu1 Wei Huang1,3Lihui Deng1*
Wei Huang1,3Lihui Deng1* Tao Jin1,2*
Tao Jin1,2* Qing Xia1,2
Qing Xia1,2Coagulopathy is a critical pathophysiological mechanism of acute pancreatitis (AP), arising from the complex interplay between innate immune, endothelial cells and platelets. Although initially beneficial for the host, uncontrolled and systemic activation of coagulation cascade in AP can lead to thrombotic and hemorrhagic complications, ranging from subclinical abnormalities in coagulation tests to severe clinical manifestations, such as disseminated intravascular coagulation. Initiation of coagulation activation and consequent thrombin generation is caused by expression of tissue factor on activated monocytes and is ineffectually offset by tissue factor pathway inhibitor. At the same time, endothelial-associated anticoagulant pathways, in particular the protein C system, is impaired by pro-inflammatory cytokines. Also, fibrin removal is severely obstructed by inactivation of the endogenous fibrinolytic system, mainly as a result of upregulation of its principal inhibitor, plasminogen activator inhibitor type 1. Finally, increased fibrin generation and impaired break down lead to deposition of (micro) vascular clots, which may contribute to tissue ischemia and ensuing organ dysfunction. Despite the high burden of coagulopathy that have a negative impact on AP patients’ prognosis, there is no effective treatment yet. Although a variety of anticoagulants drugs have been evaluated in clinical trials, their beneficial effects are inconsistent, and they are also characterized by hemorrhagic complications. Future studies are called to unravel the pathophysiologic mechanisms involved in coagulopathy in AP, and to test novel therapeutics block coagulopathy in AP.
The activation of coagulation by inflammation cascade are essential reactions for host defense during inflammatory diseases (1). Pathogen-associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs) are recognized by pattern-recognition receptors on the cells of the innate immune system, which triggers the release of pro-inflammatory mediators (2). Pro-inflammatory mediators then activate the coagulation cascade, downregulate crucial endogenous anticoagulant mechanisms, and dysregulate fibrinolytic mechanisms. In turn coagulation disorders also markedly influences inflammatory response (2). The primitive response represents an effective strategy to slow inflammatory storm spread and maintain hemostasis, while this may come at the cost of immune-driven pathological thrombus formation, which is now commonly termed ‘immunothrombosis’ (3).
In acute pancreatitis (AP), one of the early events is the pancreas autodigestion due to premature trypsinogen activation (4). Injured acinar cells release cytokines, chemokines, and adhesion molecules into the circulatory system, which recruit the infiltration of immune cells to the site of injuries and initiate coagulation (5). Histologic evaluation of AP indeed shows inflammatory cell infiltration, elevated circulating tissue factor (TF), platelet aggregation, intravascular microthrombi, fibrin deposits (6–8) and microcirculation hypoperfusion of extrapancreatic organs (9, 10). From a clinical perspective, coagulation disorders are common in patients with severe AP, with severity ranging from clinically less apparent microvascular clot formation to devastating thrombotic and hemorrhagic complications (11–14).
Despite recognizing the potential deleterious of coagulopathy on the outcome in severe AP patients, effective treatments specifically aiming to block the devastating complications while maintaining its beneficial effects for the host, do not yet exist. Although a variety of anticoagulants drugs have been evaluated in clinical trials, their beneficial effects are inconsistent, and they are also characterized by a high rate of hemorrhage complication. Severe AP patients with coagulopathy, particular with disseminated intravascular coagulation (DIC) are at a higher risk for persistent organ failure and pancreatitis-associated death (15), hemorrhage complication may bring these patients into life threatening situation. International guidelines therefore discourage anticoagulant therapies in severe AP cases (16). Nowadays, there is still ongoing research assessing the effect of new molecules on thrombosis in severe AP, with agents targeting intracellular inflammatory pathways, P-selectin and neutrophil extracellular traps (NETs) formation demonstrating promising results. A better understanding of the underlying mechanisms and cellular interactions in AP-related immunothrombosis and coagulopathy is crucial to identifying new therapeutic targets. Our study summarizes the current literature regarding the role of innate immune cells, endothelial cells and platelet in coagulopathy in AP and summary clinical evidence on drugs targeting the critical pathological process.
Monocytes and macrophages have been found to play a vital role in inflammatory diseases-induced immunothrombosis (Figure 1). Upon stimulation by PAMPs, DAMPs or proinflammatory mediators (17, 18), monocytes are the main source of circulating TF (19, 20). TF is deemed critical for survival, as deletion in mice leads to universal embryonic death (21), and defects in TF gene expression are associated with differing clinical outcomes in patients with sepsis (22). The binding of lipopolysaccharide to transmembrane receptors in monocytes induces TF mRNA expression via NF-κB activation (23). The interaction of pathogen components either with TLRs or directly with intracellular pathways in monocytes result in inflammasome activation and subsequent TF release via pyroptosis (18, 23, 24). Moreover, pore formation on the cell membrane also induce calcium influx, which triggers phosphatidylserine exposure on the membrane, followed by TF activation (25). Sphingomyelin, another membrane lipid, is also involved in the activation of TF to its procoagulant form (26). Additionally, monocyte activation by PAMPs and DAMPs is followed by increased P-selectin glycoprotein ligand 1 (PSGL-1) expression and the release of TF- and PSGL-1-bearing microparticle (MPs). These MPs can fuse in vitro with platelets, leading to increased TF activity (19, 27). Pancreatic disruption leads to direct exposure of TF to the blood (28).
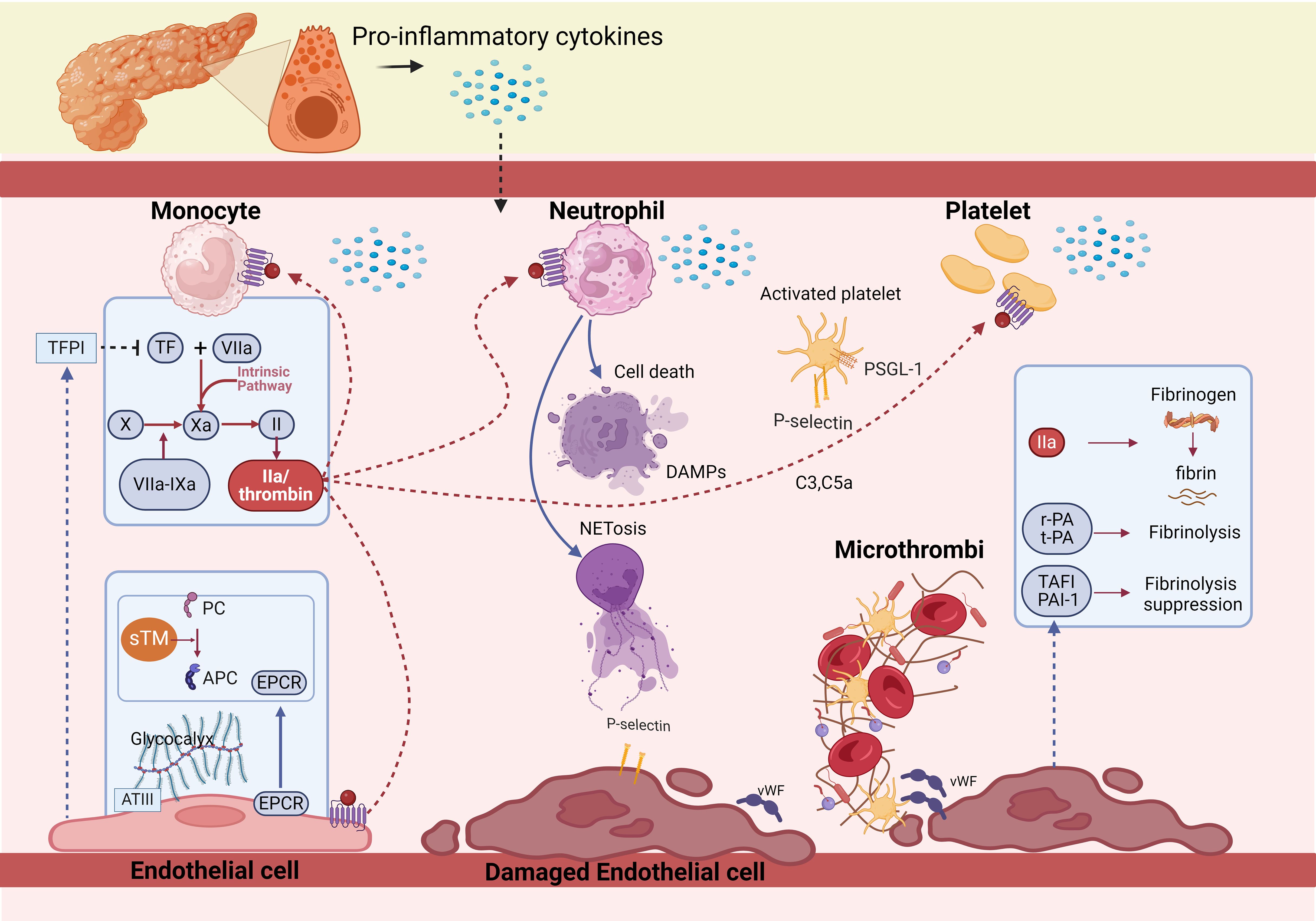
Figure 1. Pathophysiology of coagulopathy in acute pancreatitis. The activation of coagulation by inflammation cascade are essential reactions for host defense during acute pancreatitis. Initiation of coagulation activation is caused by expression of tissue factor on activated monocytes and is ineffectually offset by tissue factor pathway inhibitor. TF expression and release triggers the extrinsic coagulation pathway by binding factor VII/factor VIIa to form TF-FVIIa complex, converting factor X to factor Xa. FXa, as the prothrombinase then, thrombin is formed. At the same time, endothelial-associated anticoagulant pathways, in particular the protein C system, which includes PC, Thrombomodulin and endothelial cell protein C receptor, is impaired by pro-inflammatory cytokines. Also, fibrin removal is severely obstructed by inactivation of the endogenous fibrinolytic system, mainly as a result of upregulation of its principal inhibitors, plasminogen activator inhibitor type 1 and thrombin activated fibrinolytic inhibitor. Finally, increased fibrin generation and impaired break down lead to deposition of (micro) vascular clots.
TF expression and release triggers the extrinsic coagulation pathway by binding factor VII/factor VIIa (FVII/FVIIa) to form TF-FVIIa complex, converting factor X (FX) to factor Xa (FXa). Then, FXa is incorporated into FXa-factor Va-Ca2+-phospholipids (FXa-FVa-Ca2+-PLs) complex known as the prothrombinase (29). Thrombin is formed, leading to fibrin clots (Figure 1). The process seems to be the most potent pathway leading to coagulation cascade activation and DIC (30–34). Studies of experimental or human AP have demonstrated a central role of the TF/FVIIa system in the initiation of thrombin generation (8, 35, 36). In the early stage of severe AP, TF is highly upregulated (8, 37–39), and it is a favorable predictive marker of severe AP (40, 41). Abrogation of the TF/FVIIa pathway by specific interventions aimed at TF or factor VIIa activity resulted in a complete abrogation of thrombin generation in experimental settings (42). As the initiator of the coagulation cascades, TF might play a large part in the development of severe AP, there needs to be a more basic experimental to explore their relationship.
The endothelium lines the lumen of the entire circulatory system, separating blood and subendothelial, and maintaining vascular health by exerting anticoagulant action via tissue factor pathway inhibitor (TFPI), Protein C (PC) system, Antithrombin (ATIII) and fibrinolysis (43, 44).
TFPI is the inhibitor of TF-mediated coagulation, primarily synthesized by endothelial cells (ECs, Figure 1), which binds to ECs via proteoglycans/glycosaminoglycans, inactivates TF-FVIIa-FXa complex and prothrombinase in the early phase of the coagulation process (45, 46). The deficiency of TFPI increases susceptibility to the development of DIC and thrombosis (37). However, in AP patients, the plasma TFPI levels were significantly increased, which might be compensatory to the rise of TF, and can be released from fibrin deposits after thrombosis. Elevation of TFPI delayed TF-initiated thrombin generation, the imbalance of TF/TFPI were markedly related to pancreatic necrosis and organ failure (OF) (38, 47).
The PC system, as main natural anticoagulants, harbors PC and Thrombomodulin (TM), which along with endothelial cell protein C receptor (EPCR) catalyzes the thrombin-mediated PC activation (Figure 1). Activated PC (APC) exerts potent anticoagulation by inactivating FVa and FVIIIa (48). TM is expressed on the endothelial surface, which switches the thrombotic activity of thrombin to antithrombotic through activating PC (49, 50). It is known that soluble TM (sTM), fragments of the extracellular region of membrane-bound TM cleaved by leukocyte-derived proteases or metal loproteases, are released into the circulation in inflammatory diseases (50). Multiple studies have reported the usefulness of measuring sTM to evaluate the severity of DIC. EPCR, a transmembrane glycoprotein present on the surface of ECs, increases the efficiency of APC generation by presenting PC zymogen to thrombin/TM complex (51). However, the PC system is damaged in AP patients characterized by low levels of PC and APC (52, 53), and significantly increased levels of plasma sTM and EPCR (54).
ATIII, as a serine protease inhibitor, which inactivates TF-FVIIa-FXa Complex, FIXa, FXIa, thrombin, and is the most abundant and most important physiological anticoagulant (55). ATIII makes complexes not only with thrombin but also bind to heparan sulfate of the glycocalyx at the ECs surface (56) (Figure 1). ATIII deficiency can result in severe venous thromboembolism, plasma levels of ATIII activity is positively correlated with the severity of DIC (57). In AP patients, the level of ATIII decreases as severity increases, which is rather pronounced in cases of biliary AP (52). This phenomenon could be ascribed to a combination of impaired synthesis because of the negative acute phase response, degradation by elastase, and consumption because of thrombin generation (37, 58).
Tissue-type plasminogen activator (t-PA) and urokinase-type PA (u-PA) released by ECs are the main activators in the fibrinolysis, which transform plasminogen into plasmin, and then catalyze clot dissolution and fibrinolysis. PA inhibitor, type 1 (PAI-1) and thrombin activated fibrinolytic inhibitor (TAFI) are the regulators of the fibrinolysis, of which PAI-1 is the principal inhibitor (59) (Figure 1). It has been shown that the production of PAI-1 is affected by proinflammatory, anti-inflammatory cytokines and the elevated levels sustain longer (60). In healthy volunteers, endotoxin induces a rapid activation in the coagulation system with a concurrent rise in tPA. This temporal activation in fibrinolysis is subsequently counteracted by a greater and sustained rise in PAI-1 (61), The marked increase in PAI-1 level causes fibrinolysis shutdown, subsequently failing to counteract the systemic deposition of fibrin clots during system inflammatory reaction syndrome, leading to thrombosis and DIC (59). Patients with OF have significantly higher plasma levels of PAI-1, and non-survivors demonstrate more potent suppression of fibrinolysis than survivors (36). In severe AP, the level of TAFI also rises at the onset of the disease (58), inhibits fibrinolysis by separating carboxyterminal lysine residues and preventing binding to plasminogen (62).
Cytokines (63) and thrombin (64–67) activate platelets by DAMP receptors (68) (Figure 1), myeloid differentiation factor 88 (MyD88) and cGMP-dependent protein kinase intracellular pathways (67), as well as protease associated receptors (64, 65). Upon platelet activation, dense and α-granules fuse with the cell membrane (65), dense granules are rich in adenosine diphosphate, which further stimulates and amplifies platelet activation via receptors P2Y1 and P2Y12, whilst α-granules contain P-selectin that mediates activation of leukocytes via binding to PSGL-1, chemokines, and pro-coagulant factors. Glycoproteins IIb/IIIa (GPIIb/IIIa) and Iba (GPIba), expressed on the surface of activated platelets (69), bridged by fibrinogen or von Willebrand Factor (vWF), which constitute another receptor category that promotes platelet degranulation and aggregation (69, 70). VWF, from α-granules, facilitate platelet adhesion to the endothelium (71) (Figure 1). FcgRIIa triggers an intracellular pathway for platelet activation by phosphorylating the tyrosine kinases Src, Syk, and phospholipase c gamma 2 (PLCg2) (70). Mechanical interactions are potentiated by change from discoid to stellate shape (72). Activated platelets have also been found to release polyphosphate (PolyP), an inorganic polymer that exerts procoagulant activity. In vitro, PolyP initiates the contact pathway by FXII activation (73). Further, activated platelets aggregate with leucocytes to form platelet-leucocyte aggregates (PLA) (74), PLA in turn cause release of platelets-activating neutrophil extracellular traps (NETs), which form a vicious cycle. Platelets are essential cellular components of the coagulation system in AP animal models (8). In AP, thrombocytopenia is associated with increased disease severity and an ominous prognosis (75).
P-selectin stored in granular structures of ECs and platelets can be quickly mobilized towards the cell surface upon stimulation (76). P-selectin and its ligand, PSGL-1 linking is the first step for platelet adhesion (56) (Figure 1). PSGL-1 expressed on platelets, monocytes, and neutrophils mediate leukocyte and platelet rolling on the vascular wall as well as platelet-neutrophil and platelet–platelet aggregations to link inflammatory infiltration and thrombus formation (77). The expression of P-selectin on the platelet membrane not only mediates the adherence of platelets to leukocytes and endothelial cells but also enhances the expression of TF on monocytes (78). Notably, monocyte-derived, TF containing MPs fail to incorporate in thrombi when infused into P-selectin null mice, indicating that the accumulation of leukocyte-derived TF in growing thrombi is mediated by PSGL-1 on the MPs (79).
The levels of P-selectin are related to the development and course of AP, it’s value on admission may play a pivotal role as indicators of overall prognosis (80). The elevated level of P-selectin markedly strengthens the leukocyte–endothelium interaction and the thrombosis (81). Suppressing P-selectin inhibits leukocyte and platelet rolling in postcapillary venules of the inflamed pancreas (82), protecting against thrombosis (83) and improving pancreatic microcirculation and histopathology of acinar necrosis without causing any bleeding complications (84). Escopy et al. reviewed both preclinical and clinical trials that have evaluated therapeutic potential of biologic and small-molecule inhibitors as well as antibodies of P-selectin in a variety of diseases linked to immunothrombosis and coagulopathy (85). Wherein, crizanlizumab, a monoclonal antibody of P-selectin, has been evaluated for the treatment of vaso-occlusive crises with sickle cell disease (Food and Drug Administration approval in 2019) and COVID-19 vasculopathy (NCT04435184 and NCT04505774). Inclaclumab, a newly developed monoclonal antibody of P-selectin, was noted to reduce myocardial damage of non-ST-elevation in patients with myocardial infarction in a phase 2 clinical trial (NCT01327183). Currently, a multicenter phase 3 trial is in progress to determine whether inclaclumab could reduce the frequency of vaso-occlusive crises (Thrive-131; NCT04935879) and to evaluate its long-term safety (Thrive-133 open-label extension; NCT05348915). Except for these anti-P-selectin antibodies, PSI-697, PSI-421, as small-molecule inhibitors of P-selectin, have also been widely studied (85, 86). P-selectin and P-selectin glycoprotein ligand-1 play a fundamental role in aggravating pancreatic inflammation and their antibodies alleviate inflammatory responses in experimental severe AP (82). Therefore, it is believed that further research on the therapeutic potential of these inhibitors of P-selectin and related pathways in severe AP maybe promising.
DAMPs also activate neutrophils, which are typically the first responders to AP. Activated neutrophils exert their antimicrobial activity mainly through three processes: phagocytosis, degranulation, and the release of NETs (87). Neutrophil activation and release of NETs are considered as the initial and indispensable event in thrombus formation (88) (Figure 1). NETs as a meshwork of DNA fibers, comprise histones, antimicrobial proteins, and high-mobility group box 1 (89), which promote endothelial dysfunction (90), increase platelet activation, adhesion, aggregation (89–91), in turn contribute to thrombin-mediated fibrin generation (92, 93). NETs also propagate thrombosis by capturing TF and TF-positive extracellular vesicles from the circulation, further driving coagulation (94). Wherein, thrombin formation consists of NETs-induced platelet-dependent mechanisms and platelet-poor plasma via activation of the intrinsic coagulation pathway (95). Interaction of NETs with membrane-derived MPs released by activated neutrophils further enhanced NET-mediated intrinsic coagulation pathway activation (96). However, the role of NET components, or intact NETs on thrombosis is debatable, and merit further investigation. Except for prothrombotic role, NETs were also shown to interfere with the endogenous anticoagulant mechanisms. More specifically, extracellular nucleosomes within NETs facilitated TFPI degradation by neutrophil elastase on the surface of activated neutrophils (97), neutrophil elastase bound to DNA complexes was also shown to cleave plasminogen into fragments, cell-free DNA was capable of binding to plasmin and fibrin at the same time, resulting in decreased plasmin production and impaired fibrinolysis (98). H3 and H4, could also interact with TM and PC, leading to the inhibition of APC generation (99).
In AP patients, the plasma levels of NET components increase significantly compared to the controls (100). Platelets regulate the formation of NETs and NET-MPs aggregations (100–102) and in turn, NETs recruit platelets and neutrophils, reinforcing each other and injuring the endothelium within pancreatic microvasculature (100, 103). Exosomes adhere to NETs in vitro where they have a dose-dependent pro-coagulant effect. NETs also activate the intrinsic coagulation pathway via autoactivation of Factor XII (102). NETs are complex structures composed of DNA and cytotoxic granule proteins, including myeloperoxidase and neutrophil elastase. NETs targeting, either preventing their formation or degrading the NETs that have already formed, has been proved to prevent tissue damage and reduce risk of thrombus formation in the context of infections (104). Preventing NETs formation by inhibition protein-arginine deaminase 4, neutrophil elastase, or gasdermin D, has been proved to be effective in several preclinical inflammatory disease models. While whether inhibition of NETs formation has a detrimental effect on host defense mechanisms has not been determined (105). Another strategy could be to interfere with NETs that have already formed: a recombinant human deoxyribonuclease I (rhDNase) is already used for the treatment of cystic fibrosis with safety confirmed, making it a very viable option for other diseases. A phase Ib study for patients with systemic lupus erythematosus (SLE) showed that DNase was well tolerated without severe adverse effects. Significant recent developments in the field of rhDNase targeting the NET have led to testing of new NET-targeted drugs in clinical trials of patients with COVID-19 (NCT04409925, NCT04541979 and NCT05139901) (105). Heparins, as a class of anticoagulant drugs, has been proposed to destabilize NETs by dissociating histones from the chromatin backbone of the extracellular traps as well as to prevent phorbol myristate acetate-induced NET formation (106). Colchicine, destabilization of actin cytoskeleton in NETs has been tested in Gout. N-acetyl cysteine, a ROS scavenger, improved the condition of patients with SLE and acute liver failure. Anti-TNF monoclonal antibodies have been used for the treatment of rheumatoid arthritis (RA) and inflammatory bowel disease, and anti-IL-17 antibodies have also shown some efficacy for the treatment of RA (107). The involvement of NETs has been well established in the pathobiology in experimental models and patients of severe AP (100, 108, 109). All the aforementioned therapeutic agents targeting NETs are expected to have significant potential to mitigate severe AP in transitional research (105).
The complement system shares a common origin with the coagulation system and influences each other. It is activated through proteolytic cascades (110), leading to the formation of membrane attack complexes, ultimately polymerizing and inducing lysis of the cellular target (111). Recent studies have shown specific crosstalk between complement and coagulation in AP patients (112). First, in addition to activation by serine proteases, granzyme B and trypsin also cleave the central complement components, generating C3a and C5a (113). Second, C3a and C5a release TF from monocytes and ECs and promote platelet activation, leading to thrombogenesis (110, 111). Moreover, C3 is essential for the recruitment of neutrophils into the pancreas and NET formation (114). After C3a and C5a complement activation, the direct products also stimulate the platelets and promote coagulation by stimulating phosphatidylserine exposure (111) (Figure 1), enhancing the activation of platelets, granulocytes, and ECs, increasing the microcirculation thrombosis and pancreatic injury.
Early diagnosis and monitoring of coagulopathy is sometimes not straightforward and complicated in daily clinical practice. Among the items of the International Society on Thrombosis and Hemostasis (ISTH) score, fibrinogen concentrations and platelet counts might be increased in the early phase of AP because of inflammation (3), thrombocytopenia may also be due to other conditions, such as immune thrombocytopenia, heparin-induced thrombocytopenia, thrombotic microangiopathies, or medication-induced bone marrow depression (115), and poor sensitivity of the ISTH criteria for the diagnosis of DIC has been reported (116). Vitamin K deficit and liver insufficiency may also be present at the same time with AP associated coagulopathy, this differentiation is not always easy (117). Plasma D-dimer alone could predict coagulopathy and severity in patients with AP (118), while it might also be increased because of inflammation (119). It would be interesting to see whether new diagnostic criteria for DIC from the Japanese Society on Thrombosis and Hemostasis, which takes into account the underlying diseases (116). Thrombelastography is a viscoelastic assay that measures clotting of whole blood over time measured using a spinning wire probe, and is increasingly employed in severe AP patients with a hypercoagulable state (120). In total, early diagnosis and monitoring of coagulopathy in severe AP is still a challenge (15, 121), sequential assessment of fibrinogen (122), point-of-care tests (123) and biomarkers base on immunothrombosis might be more helpful and yield diagnostic insight (124). Further elucidation of the mechanisms of coagulopathy as well as the proper diagnostic criteria, and potential biomarkers would contribute to the improved management of prognosis of this intractable disease.
The efficacy of Food and Drug Administration (FDA)-approved drugs commonly used in clinical practice, such as heparin, has been fully tested AP patients by several small-scale clinical investigations (Table 1). In addition to FDA approved drugs, a variety of currently non-FDA-approved agents, including APC and rTM, have also been evaluated in AP (Table 1). According to the international guidelines, anticoagulant medications are not recommended for AP or severe AP, or AP patients coexisting DIC and splanchnic vein thrombosis (SVT) (146). Although the PC pathway defects are associated with the progress of multiple OF (53), the coagulation disorder in severe AP with APC treatment is not improved in patients (125), and the recovery from coagulopathy is slower than the placebo group (126). Studies assessing the efficacy of TM have shown more promising results, as rTM administration resulted in decreased mortality and was not associated with increased bleeding events in AP-induced coagulopathy (127–129). A recent study also showed that rTM effectively prevented the development of walled-off necrosis (127, 129). Similarly, low molecular weight heparin (LMWH) is not recommended in the initial managements of moderately severe AP and severe AP patients, although some studies have found a beneficial effect on OF, local complication, mortality, length of stay (LOS) without increase the risk of bleeding complications112-116. As for anticoagulation therapy in the AP patients complicated with SVT in the later stage, heparin or LMWH followed by warfarin or novel oral anticoagulant are not approved for clinical use in AP due to both inconclusive results for their efficacy (139, 140, 142–145, 147, 148) and an increased risk for bleeding side effects (139, 142). Carrying out larger, multicenter clinical trials designed to evaluate the potential treatment benefit of LMWH and rTM replacement in AP is encouraged.

Table 1. Selected clinically approved and pre-clinical therapeutics that may block coagulopathy in AP.
There are still many potential drug targets that are only being used in research, and seldom in clinical practice. In an animal study, ATIII (500 μg/kg) was injected intravenously 30 min before or after the induction of severe AP in rats, which in turn ameliorate SAP-induced kidney injury by inhibiting inflammation, oxidative stress, and apoptosis (149). Inhibitors of P-selectin has been proved to have the therapeutic potential on diseases linked to immunothrombosis and coagulopathy (85), NETs targeting might also reduce risk of thrombus formation in the context of infections (104, 105). Inhibition of P-selection or NET formation do attenuate OF and neutrophil recruitment in the inflamed pancreas (150) and improve survival by improving pancreatic microcirculation (82, 84, 151), thereby necessitating the translation of these findings into clinical trials of severe AP patients.
Taken together, pro-inflammatory cytokines activate the coagulation cascade, downregulate crucial endogenous anticoagulant mechanisms, and dysregulate fibrinolytic mechanisms (Figure 2). The coagulopathy has been ascribed a critical pathophysiological role in AP, arising from the complex interplay between innate immune, endothelial cells and platelets. Thus, a single-target therapy may be insufficient, requiring novel drugs. Large-scale clinical trials are needed to identify the appropriate drug and the adequate dose under various clinical situations.

Figure 2. Imbalance of coagulation system in acute pancreatitis. Pro-inflammatory cytokines activate the coagulation cascade by neutrophil extracellular traps, tissue factor and platelets, downregulate crucial endogenous anticoagulant mechanisms through tissue factor pathway inhibitor, protein C system and antithrombin, concurrently dysregulate fibrinolytic mechanisms with plasminogen activator inhibitor type 1 and thrombin activated fibrinolytic inhibitor.
LL: Writing – review & editing, Writing – original draft. QT: Writing – original draft. XW: Writing – original draft. XM: Writing – original draft. ZL: Writing – original draft. TL: Writing – original draft. WH: Writing – review & editing. LD: Writing – review & editing. TJ: Writing – review & editing, Writing – original draft. QX: Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by Sichuan Provincial Administration of Traditional Chinese Medicine, Innovative Team for Breakthrough in Traditional Chinese Medicine Efficacy in Severe Acute pancreatitis (No. 2023ZD04 to QX); Special Project for Scientific and Technological Research of Sichuan Provincial Administration of Traditional Chinese Medicine (No. 2023MS353 to TJ); the Program of Science and Technology Department of Sichuan Province (No. 2024NSFSC1826 to LL).
LD and TJ are correspondence authors with equal contributions. These authors thank all the staff from West China Center of Excellence for Pancreatitis, Institute of Integrated Traditional Chinese and Western Medicine, and Department of Integrated Traditional Chinese and Western Medicine, West China Tianfu Hospital, Sichuan University for their continuous support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Iba T, Levi M, Levy JH. Sepsis-induced coagulopathy and disseminated intravascular coagulation. Semin Thromb Hemostasis. (2019) 46:089–95. doi: 10.1002/ams2.2019.6.issue-3
2. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. NEJM. (2003) 348:138–50. doi: 10.1056/NEJMra021333
3. Dumnicka P, Kuśnierz-Cabala B, Sporek M, Mazur-Laskowska M, Gil K, Kuźniewski M, et al. Serum Concentrations of Angiopoietin-2 and Soluble fms-Like Tyrosine Kinase 1 (sFlt-1) Are Associated with Coagulopathy among Patients with Acute Pancreatitis. Int J Mol Sci. (2017) 18:753. doi: 10.3390/ijms18040753
4. Saluja A, Dudeja V, Dawra R, Sah RP. Early intra-acinar events in pathogenesis of pancreatitis. Gastroenterology. (2019) 156:1979–93. doi: 10.1053/j.gastro.2019.01.268
5. Lee PJ, Papachristou GI. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:479–96. doi: 10.1038/s41575-019-0158-2
6. Bockman DE, Buchler M, Beger HG. Ultrastructure of human acute pancreatitis. Int J Pancreatol. (1986) 1:141–53. doi: 10.1007/BF02788446
7. Cuthbertson CM, Christophi C. Disturbances of the microcirculation in acute pancreatitis. Br J Surg. (2006) 93:518–30. doi: 10.1002/bjs.5316
8. Rao P, Niemann B, Szeligo B, Ivey AD, Murthy P, Schmidt CR, et al. Acute pancreatitis induces a transient hypercoagulable state in murine models. Pancreatology. (2023) 23:306–13. doi: 10.1016/j.pan.2023.02.007
9. Zhu R, Wei S, Wu C, Li S, Gong J. Utility of clot formation and lysis assay to monitor global coagulation state of patients with severe acute pancreatitis. Digest Dis Sci. (2012) 57:1399–403. doi: 10.1007/s10620-012-2034-6
10. Foitzik T, Eibl G, Hotz B, Hotz H, Kahrau S, Kasten C, et al. Persistent multiple organ microcirculatory disorders in severe acute pancreatitis experimental findings and clinical implications. Dig Dis Sci. (2002) 47:130–8. doi: 10.1023/A:1013284008219
11. Mole DJ, Olabi B, Robinson V, Garden OJ, Parks RW. Incidence of individual organ dysfunction in fatal acute pancreatitis: analysis of 1024 death records. Hpb. (2009) 11:166–70. doi: 10.1111/j.1477-2574.2009.00038.x
12. Lasson A, Ohlsson K. Consumptive coagulopathy, fibrinolysis and protease-antiprotease interactions during acute human pancreatitis. Thromb Res. (1986) 41:167–83. doi: 10.1016/0049-3848(86)90227-6
13. Salomone T, Tosi P, Palareti G, Tomassetti P, Migliori M, Guariento A, et al. Coagulative disorders in human acute pancreatitis role for the D-dimer. Pancreas. (2003) 26:111–6. doi: 10.1097/00006676-200303000-00003
14. Maeda K, Hirota M, Ichihara A, Ohmuraya M, Hashimoto D, Sugita H, et al. Applicability of disseminated intravascular coagulation parameters in the assessment of the severity of acute pancreatitis. Pancreas. (2006) 32:87–92. doi: 10.1097/01.mpa.0000186248.89081.44
15. Hamada S, Masamune A, Kikuta K, Shimosegawa T. Disseminated intravascular coagulation on admission predicts complications and poor prognosis of acute pancreatitis: analysis of the nationwide epidemiological survey in Japan. Pancreas. (2017) 46:e15–e6. doi: 10.1097/MPA.0000000000000739
16. Jaber S, Garnier M, Asehnoune K, Bounes F, Buscail L, Chevaux J-B, et al. Guidelines for the management of patients with severe acute pancreatitis, 2021. Anaesthesia Crit Care Pain Med. (2022) 41:101060. doi: 10.1016/j.accpm.2022.101060
17. Egorina EM, Sovershaev MA, Bjørkøy G, Gruber FXE, Olsen JO, Parhami-Seren B, et al. Intracellular and surface distribution of monocyte tissue factor. Arteriosclerosis Thrombosis Vasc Biol. (2005) 25:1493–8. doi: 10.1161/01.ATV.0000168413.29874.d7
18. Ryan TAJ, O’Neill LAJ. Innate immune signaling and immunothrombosis: New insights and therapeutic opportunities. Eur J Immunol. (2022) 52:1024–34. doi: 10.1002/eji.202149410
19. Pawlinski R, Wang J-G, Owens AP, Williams J, Antoniak S, Tencati M, et al. Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood. (2010) 116:806–14. doi: 10.1182/blood-2009-12-259267
20. Macey MG, Wolf SI, Wheeler-Jones CPD, Lawson C. Expression of blood coagulation factors on monocytes after exposure to TNF-treated endothelium in a novel whole blood model of arterial flow. J Immunol Methods. (2009) 350:133–41. doi: 10.1016/j.jim.2009.08.007
21. Carmeliet P, Mackman N, Moons L, Luther T, Gressens P, Vlaenderen I, et al. Role of tissue factor in embryonic blood vessel development. Nature. (1996) 383:73–5. doi: 10.1038/383073a0
22. Shi D, Song Z, Yin J, Xue M, Yao C, Sun Z, et al. Genetic variation in the tissue factor gene is associated with clinical outcome in severe sepsis patients. Crit Care. (2014) 18:631. doi: 10.1186/s13054-014-0631-9
23. Ryan TAJ, Preston RJS, O’Neill LAJ. Immunothrombosis and the molecular control of tissue factor by pyroptosis: prospects for new anticoagulants. Biochem J. (2022) 479:731–50. doi: 10.1042/BCJ20210522
24. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. (2019) 50:1401–11.e4. doi: 10.1016/j.immuni.2019.04.003
25. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity. (2019) 51:983–96.e6. doi: 10.1016/j.immuni.2019.11.005
26. Wang J, Pendurthi UR, Rao LVM. Acid sphingomyelinase plays a critical role in LPS- and cytokine-induced tissue factor procoagulant activity. Blood. (2019) 134:645–55. doi: 10.1182/blood.2019001400
27. del Conde I, Shrimpton CN, Thiagarajan P, López JA. Tissue-factor–bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. (2005) 106:1604–11. doi: 10.1182/blood-2004-03-1095
28. Lisman T, Porte R. Activation and regulation of hemostasis in acute liver failure and acute pancreatitis. Semin Thromb Hemostasis. (2010) 36:437–43. doi: 10.1055/s-0030-1254052
29. Schmaier AH. Transferrin: a blood coagulation modifier. Cell Res. (2020) 30:101–2. doi: 10.1038/s41422-020-0275-z
30. Zhou H, Wolberg AS, Roubey RAS. Characterization of monocyte tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood. (2004) 104:2353–8. doi: 10.1182/blood-2004-01-0145
32. Pereda J, Sabater L, Aparisi L, Escobar J, Sandoval J, Viña J, et al. Interaction between cytokines and oxidative stress in acute pancreatitis. Curr Med Chem. (2006) 13:2775–87. doi: 10.2174/092986706778522011
33. Dugina T, Kiseleva E, Chistov I, Umarova B, Strukova S. Receptors of the PAR family as a link between blood coagulation and inflammation. Biochemistry. (2002) 67:65–74. doi: 10.1023/a:1013952114485
34. von Brühl M-L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
35. Liu C, Zhou X, Ling L, Chen S, Zhou J. Prediction of mortality and organ failure based on coagulation and fibrinolysis markers in patients with acute pancreatitis. Medicine. (2019) 98:e15648. doi: 10.1097/MD.0000000000015648
36. Radenkovic D, Bajec D, Ivancevic N, Milic N, Bumbasirevic V, Jeremic V, et al. D-dimer in acute pancreatitis a new approach for an early assessment of organ failure. Pancreas. (2009) 38:655–60. doi: 10.1097/MPA.0b013e3181a66860
37. Sungurlu S, Kuppy J, Balk RA. Role of antithrombin III and tissue factor pathway in the pathogenesis of sepsis. Crit Care Clin. (2020) 36:255–65. doi: 10.1016/j.ccc.2019.12.002
38. Lindström OK, Tukiainen EM, Kylänpää M-L, Mentula PJ, Puolakkainen PA, Wartiovaara-Kautto UMK, et al. Thrombin Generation in vitro and in vivo, and Disturbed Tissue Factor Regulation in Patients with Acute Pancreatitis. Pancreatology. (2011) 11:557–66. doi: 10.1159/000333481
39. Zelaya H, Rothmeier AS, Ruf W. Tissue factor at the crossroad of coagulation and cell signaling. J Thromb Haemostasis. (2018) 16:1941–52. doi: 10.1111/jth.14246
40. Andersson E. Tissue factor in predicted severe acute pancreatitis. World J Gastroenterol. (2010) 16:6128–34. doi: 10.3748/wjg.v16.i48.6128
41. Sawa H, Ueda T, Takeyama Y, Yasuda T, Matsumura N, Nakajima T, et al. Elevation of plasma tissue factor levels in patients with severe acute pancreatitis. J Gastroenterol. (2006) 41:575–81. doi: 10.1007/s00535-006-1806-1
42. Ou Z-B, Miao C-M, Ye M-X, Xing D-P, He K, Li P-Z, et al. Investigation for role of tissue factor and blood coagulation system in severe acute pancreatitis and associated liver injury. Biomed Pharmacother. (2017) 85:380–8. doi: 10.1016/j.biopha.2016.11.039
43. Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. (2019) 133:906–18. doi: 10.1182/blood-2018-11-882993
44. Eibl G, Buhr HJ, Foitzik T. Therapy of microcirculatory disorders in severe acute pancreatitis: what mediators should we block? Intensive Care Med. (2002) 28:139–46. doi: 10.1007/s00134-001-1194-1
45. Venkatalaxmi A, Padmavathi BS, Amaranath T. A general solution of unsteady Stokes equations. Fluid Dynamics Res. (2004) 35:229–36. doi: 10.1016/j.fluiddyn.2004.06.001
46. Mast AE. Tissue factor pathway inhibitor. Arteriosclerosis Thrombosis Vasc Biol. (2016) 36:9–14. doi: 10.1161/ATVBAHA.115.305996
47. Yasuda T, Ueda T, Kamei K, Shinzaki W, Sawa H, Shinzeki M, et al. Plasma tissue factor pathway inhibitor levels in patients with acute pancreatitis. J Gastroenterol. (2009) 44:1071–9. doi: 10.1007/s00535-009-0096-9
48. Pendurthi UR, Rao LVM. Endothelial cell protein C receptor-dependent signaling. Curr Opin Hematol. (2018) 25:219–26. doi: 10.1097/MOH.0000000000000416
49. Esmon NL, Owen WG, Esmon CT. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. (1982) 257:859–64. doi: 10.1016/S0021-9258(19)68276-1
50. Loghmani H, Conway E. Exploring traditional and nontraditional roles for thrombomodulin. Blood. (2018) 132:148–58. doi: 10.1182/blood-2017-12-768994
51. Dinarvand P, Moser KA. Protein C deficiency. Arch Pathol Lab Med. (2019) 143:1281–5. doi: 10.5858/arpa.2017-0403-RS
52. Yang N, Hao J, Zhang D. Antithrombin III and D-dimer levels as indicators of disease severity in patients with hyperlipidaemic or biliary acute pancreatitis. J Int Med Res. (2017) 45:147–58. doi: 10.1177/0300060516677929
53. Lindstrom O, Kylanpaa L, Mentula P, Puolakkainen P, Kemppainen E, Haapiainen R, et al. Activated protein C retards recovery from coagulopathy in severe acute pancreatitis. Crit Care. (2006) 10:10–6. doi: 10.3109/00365513.2015.1084041
54. Chen Y, Ke L, Meng L, Yang Q, Tong Z, Pan Y, et al. Endothelial markers are associated with pancreatic necrosis and overall prognosis in acute pancreatitis: A preliminary cohort study. Pancreatology. (2017) 17:45–50. doi: 10.1016/j.pan.2016.12.005
55. Levi M, Welsby IJ, Sniecinski RM, Levy JH. Antithrombin: anti-inflammatory properties and clinical applications. Thromb Haemostasis. (2017) 115:712–28. doi: 10.1160/TH15-08-0687
56. Iba T, Levy JH, Thachil J, Susen S, Levi M, Scarlatescu E. Communication from the Scientific Standardization Committees of the International Society on Thrombosis and Haemostasis on vascular endothelium-related biomarkers in disseminated intravascular coagulation. J Thromb Haemostasis. (2023) 21:691–9. doi: 10.1016/j.jtha.2022.11.032
57. Iba T, Saitoh D. Efficacy of antithrombin in preclinical and clinical applications for sepsis-associated disseminated intravascular coagulation. J Intensive Care. (2014) 2:66. doi: 10.1186/s40560-014-0051-6
58. Fidan S, Erkut M, Cosar AM, Yogun Y, Orem A, Sonmez M, et al. Higher thrombin-antithrombin III complex levels may indicate severe acute pancreatitis. Dig Dis. (2018) 36:244–51. doi: 10.1159/000485613
59. Madoiwa S. Recent advances in disseminated intravascular coagulation: endothelial cells and fibrinolysis in sepsis-induced DIC. J Intensive Care. (2015) 3:8. doi: 10.1186/s40560-015-0075-6
60. Healy AM, Gelehrter TD. Induction of plasminogen activator inhibitor-1 in HepG2 human hepatoma cells by mediators of the acute phase response. J Biol Chem. (1994) 269:19095–100. doi: 10.1016/S0021-9258(17)32279-2
61. van Deventer SJ, Buller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. (1990) 76:2520–6. doi: 10.1182/blood.V76.12.2520.2520
62. Bajzar L, Jain N, Wang P, Walker JB. Thrombin activatable fibrinolysis inhibitor: Not just an inhibitor of fibrinolysis. Crit Care Med. (2004) 32:S320–S4. doi: 10.1097/01.CCM.0000126361.00450.B1
63. Houck KL, Yuan H, Tian Y, Solomon M, Cramer D, Liu K, et al. Physical proximity and functional cooperation of glycoprotein 130 and glycoprotein VI in platelet membrane lipid rafts. J Thromb Haemostasis. (2019) 17:1500–10. doi: 10.1111/jth.14525
64. Hoppensteadt D, Tsuruta K, Cunanan J, Hirman J, Kaul I, Osawa Y, et al. Thrombin generation mediators and markers in sepsis-associated coagulopathy and their modulation by recombinant thrombomodulin. Clin Appl Thrombosis/Hemostasis. (2013) 20:129–35. doi: 10.1177/1076029613492875
65. Parker WAE, Storey RF. Antithrombotic therapy for patients with chronic coronary syndromes. Heart. (2021) 107:925–33. doi: 10.1136/heartjnl-2020-316914
66. Galgano L, Guidetti GF, Torti M, Canobbio I. The controversial role of LPS in platelet activation in vitro. Int J Mol Sci. (2022) 23:10900. doi: 10.3390/ijms231810900
67. Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the cGMP-Dependent Protein Kinase Pathway. J Immunol. (2009) 182:7997–8004. doi: 10.4049/jimmunol.0802884
68. Vallance TM, Zeuner M-T, Williams HF, Widera D, Vaiyapuri S. Toll-like receptor 4 signalling and its impact on platelet function, thrombosis, and haemostasis. Mediators Inflammation. (2017) 2017:1–13. doi: 10.1155/2017/9605894
69. Stellos K, Sauter R, Fahrleitner M, Grimm J, Stakos D, Emschermann F, et al. Binding of oxidized low-density lipoprotein on circulating platelets is increased in patients with acute coronary syndromes and induces platelet adhesion to vascular wall in vivo—Brief report. Arteriosclerosis Thrombosis Vasc Biol. (2012) 32:2017–20. doi: 10.1161/ATVBAHA.111.244707
70. Arman M, Krauel K, Tilley DO, Weber C, Cox D, Greinacher A, et al. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood. (2014) 123:3166–74. doi: 10.1182/blood-2013-11-540526
71. Dempfle C-E. Coagulopathy of sepsis. Thromb Haemostasis. (2017) 91:213–24. doi: 10.1160/TH03-03-0182
72. Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascón GA, et al. The endothelium in sepsis. Shock. (2016) 45:259–70. doi: 10.1097/SHK.0000000000000473
73. Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. (2009) 139:1143–56. doi: 10.1016/j.cell.2009.11.001
74. Gawaz M, Moghadam S, Pilz G, Gurland H, Werdan K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J Clin Invest. (1995) 25:843–51. doi: 10.1111/j.1365-2362.1995.tb01694.x
75. McDonald B, Dunbar M. Platelets and intravascular immunity: guardians of the vascular space during bloodstream infections and sepsis. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.02400
76. Zinellu A, Mangoni AA. Systematic review and meta-analysis of the effect of statins on circulating E-selectin, L-selectin, and P-selectin. Biomedicines. (2021) 9:1707. doi: 10.3390/biomedicines9111707
77. Zhang X, Zhu M, Jiang X, Liu X, Liu X, Liu P, et al. P-selectin glycoprotein ligand 1 deficiency prevents development of acute pancreatitis by attenuating leukocyte infiltration. World J Gastroenterol. (2020) 26:6361–77. doi: 10.3748/wjg.v26.i41.6361
78. Shebuski RJ, Kilgore K. Role of inflammatory mediators in thrombogenesis. J Pharmacol Exp Ther. (2002) 300:729–35. doi: 10.1124/jpet.300.3.729
79. Zwicker JI, Trenor CC, Furie BC, Furie B. Tissue factor–bearing microparticles and thrombus formation. Arteriosclerosis Thrombosis Vasc Biol. (2011) 31:728–33. doi: 10.1161/ATVBAHA.109.200964
80. Tsaroucha AK, Schizas D, Vailas MG, Rachmani E, Kanavidis P, Asimakopoulos V, et al. E and P selectins as potential markers in the assessment of the severity of acute pancreatitis. Pancreas. (2018) 47:406–11. doi: 10.1097/MPA.0000000000001009
81. Setiadi H, Yago T, Liu Z, McEver RP. Endothelial signaling by neutrophil-released oncostatin M enhances P-selectin–dependent inflammation and thrombosis. Blood Adv. (2019) 3:168–83. doi: 10.1182/bloodadvances.2018026294
82. Hartman H, Abdulla A, Awla D, Lindkvist B, Jeppsson B, Thorlacius H, et al. P-selectin mediates neutrophil rolling and recruitment in acute pancreatitis. Br J Surg. (2012) 99:246–55. doi: 10.1002/bjs.7775
83. Jin H, Gebska MA, Blokhin IO, Wilson KM, Ketsawatsomkron P, Chauhan AK, et al. Endothelial PPAR-γ Protects against vascular thrombosis by downregulating P-selectin expression. Arteriosclerosis Thrombosis Vasc Biol. (2015) 35:838–44. doi: 10.1161/ATVBAHA.115.305378
84. Hackert T, Sperber R, Hartwig W, Fritz S, Schneider L, Gebhard M-M, et al. P-selectin inhibition reduces severity of acute experimental pancreatitis. Pancreatology. (2009) 9:369–74. doi: 10.1159/000212098
85. Escopy S, Chaikof EL. Targeting the P-selectin/PSGL-1 pathway: discovery of disease-modifying therapeutics for disorders of thromboinflammation. Blood Vessels Thromb Hemostasis. (2024) 1:1–13. doi: 10.1016/j.bvth.2024.100015
86. Smith BAH, Bertozzi CR. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nat Rev Drug Discov. (2021) 20:217–43. doi: 10.1038/s41573-020-00093-1
87. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2017) 18:134–47. doi: 10.1038/nri.2017.105
88. Darbousset R, Thomas GM, Mezouar S, Frère C, Bonier R, Mackman N, et al. Tissue factor–positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. (2012) 120:2133–43. doi: 10.1182/blood-2012-06-437772
89. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukocyte Biol. (2016) 99:153–62. doi: 10.1189/jlb.3A0415-161R
90. Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, et al. Neutrophil extracellular traps promote angiogenesis. Arteriosclerosis Thrombosis Vasc Biol. (2016) 36:2078–87. doi: 10.1161/ATVBAHA.116.307634
91. Zucoloto AZ, Jenne CN. Platelet-neutrophil interplay: insights into neutrophil extracellular trap (NET)-driven coagulation in infection. Front Cardiovasc Med. (2019) 6. doi: 10.3389/fcvm.2019.00085
92. Carminita E, Crescence L, Panicot-Dubois L, Dubois C. Role of neutrophils and NETs in animal models of thrombosis. Int J Mol Sci. (2022) 23:85. doi: 10.3390/ijms23031411
93. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
94. Zhang H, Zhou Y, Qu M, Yu Y, Chen Z, Zhu S, et al. Tissue factor-enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis-induced lung injury. Front Cell Infect Microbiol. (2021) 11. doi: 10.3389/fcimb.2021.677902
95. Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SHC, Weitz JI, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arteriosclerosis Thrombosis Vasc Biol. (2014) 34:1977–84. doi: 10.1161/ATVBAHA.114.304114
96. Wang Y, Luo L, Braun OÖ, Westman J, Madhi R, Herwald H, et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. (2018) 8:1977–84. doi: 10.1038/s41598-018-22156-5
97. Massberg S, Grahl L, von Bruehl M-L, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96. doi: 10.1038/nm.2184
98. Cruz D, Helms J, Aquino LR, Stiel L, Cougourdan L, Broussard C, et al. DNA-bound elastase of neutrophil extracellular traps degrades plasminogen, reduces plasmin formation, and decreases fibrinolysis: proof of concept in septic shock plasma. FASEB J. (2019) 33:14270–80. doi: 10.1096/fj.201901363RRR
99. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemostasis. (2011) 9:1795–803. doi: 10.1111/j.1538-7836.2011.04422.x
100. Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. (2015) 149:1920–31.e8. doi: 10.1053/j.gastro.2015.08.026
101. Madhi R, Rahman M, Taha D, Linders J, Merza M, Wang Y, et al. Platelet IP6K1 regulates neutrophil extracellular trap-microparticle complex formation in acute pancreatitis. JCI Insight. (2019) 149:1920–1931.e8. doi: 10.1172/jci.insight.129270
102. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. (2014) 123:2768–76. doi: 10.1182/blood-2013-10-463646
103. Hu J, Kang H, Chen H, Yao J, Yi X, Tang W, et al. Targeting neutrophil extracellular traps in severe acute pancreatitis treatment. Ther Adv Gastroenterol. (2020) 13:2768–76. doi: 10.1177/1756284820974913
104. Adrover JM, McDowell SAC, He X-Y, Quail DF, Egeblad M. NETworking with cancer: The bidirectional interplay between cancer and neutrophil extracellular traps. Cancer Cell. (2023) 41:505–26. doi: 10.1016/j.ccell.2023.02.001
105. Bonilha CS, Veras FP, de Queiroz Cunha F. NET-targeted therapy: effects, limitations, and potential strategies to enhance treatment efficacy. Trends Pharmacol Sci. (2023) 44:622–34. doi: 10.1016/j.tips.2023.06.007
106. Herre M, Cedervall J, Mackman N, Olsson A-K. Neutrophil extracellular traps in the pathology of cancer and other inflammatory diseases. Physiol Rev. (2023) 103:277–312. doi: 10.1152/physrev.00062.2021
107. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. (2018) 15:206–21. doi: 10.1038/nrgastro.2017.183
108. Liu T, Huang W, Szatmary P, Abrams ST, Alhamdi Y, Lin Z, et al. Accuracy of circulating histones in predicting persistent organ failure and mortality in patients with acute pancreatitis. Br J Surg. (2017) 104:1215–25. doi: 10.1002/bjs.10538
109. Szatmary P, Huang W, Criddle D, Tepikin A, Sutton R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell Mol Med. (2018) 22:4617–29. doi: 10.1111/jcmm.2018.22.issue-10
110. Dzik S. Complement and coagulation: cross talk through time. Transfusion Med Rev. (2019) 33:199–206. doi: 10.1016/j.tmrv.2019.08.004
111. Conway EM. Complement-coagulation connections. Blood Coagulation Fibrinolysis. (2018) 29:243–51. doi: 10.1097/MBC.0000000000000720
112. Zhang L, Qiao Z, Feng H, Shen J. The early predictive role of complement C3 and C4 in patients with acute pancreatitis. J Clin Lab Anal. (2020) 34:243–51. doi: 10.1002/jcla.23205
113. Bettac L, Denk S, Seufferlein T, Huber-Lang M. Complement in pancreatic disease—Perpetrator or savior? Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00015
114. Linders J, Madhi R, Mörgelin M, King Ben C, Blom Anna M, Rahman M. Complement component 3 is required for tissue damage, neutrophil infiltration, and ensuring NET formation in acute pancreatitis. Eur Surg Res. (2020) 61:163–76. doi: 10.1159/000513845
115. Baughman RR, Lower EE, Flessa HC, Tollerud DJ. Thrombocytopenia in the intensive care unit. Chest. (1993) 104:1243–7. doi: 10.1378/chest.104.4.1243
116. Asakura H, Takahashi H, Uchiyama T, Eguchi Y, Okamoto K, Kawasugi K, et al. Proposal for new diagnostic criteria for DIC from the Japanese Society on Thrombosis and Hemostasis. Thromb J. (2016) 14:753. doi: 10.1186/s12959-016-0117-x
117. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol. (2009) 145:24–33. doi: 10.1111/j.1365-2141.2009.07600.x
118. Wan J, Yang X, He W, Zhu Y, Zhu Y, Zeng H, et al. Serum D-dimer levels at admission for prediction of outcomes in acute pancreatitis. BMC Gastroenterol. (2019) 19:42. doi: 10.1186/s12876-019-0989-x
119. Longstaff C, Adcock D, Olson JD, Jennings I, Kitchen S, Mutch N, et al. Harmonisation of D-dimer — A call for action. Thromb Res. (2016) 137:219–20. doi: 10.1016/j.thromres.2015.11.031
120. Fan C, Song Y, Wang X, Mao C, Xiong Y. Identification of early derangements of coagulation, hematological and biochemical profiles in patients with acute pancreatitis. Clin Biochem. (2022) 109-110:37–43. doi: 10.1016/j.clinbiochem.2022.08.005
121. Masamune A, Hamada S, Kikuta K. Diagnosis of disseminated intravascular coagulation in acute pancreatitis is still a challenge. Pancreas. (2022) 51:e116–7. doi: 10.1097/MPA.0000000000002124
122. Levi M, Meijers JC. DIC: Which laboratory tests are most useful. Blood Rev. (2011) 25:33–7. doi: 10.1016/j.blre.2010.09.002
123. Levi M, Hunt BJ. A critical appraisal of point-of-care coagulation testing in critically ill patients. J Thromb Haemost. (2015) 13:1960–7. doi: 10.1111/jth.13126
124. Iba T, Levy JH. Sepsis-induced coagulopathy and disseminated intravascular coagulation. Anesthesiology. (2020) 132:1238–45. doi: 10.1097/ALN.0000000000003122
125. Pettilä V, Kyhälä L, Kylänpää M, Leppäniemi A, Tallgren M, Markkola A, et al. APCAP - activated protein C in acute pancreatitis a double-blind randomized human pilot trial. Crit Care. (2010) 14:R139. doi: 10.1016/j.blre.2010.09.002
126. Kyhälä L, Lindström O, Kylänpää L, Mustonen H, Puolakkainen P, Kemppainen E, et al. Activated protein C retards recovery from coagulopathy in severe acute pancreatitis. Scandinavian J Clin Lab Invest. (2015) 76:10–6. doi: 10.3109/00365513.2015.1084041
127. Eguchi T, Tsuji Y, Yamashita H, Fukuchi T, Kanamori A, Matsumoto K, et al. Efficacy of recombinant human soluble thrombomodulin in preventing walled-off necrosis in severe acute pancreatitis patients. Pancreatology. (2015) 15:485–90. doi: 10.1016/j.pan.2015.08.002
128. Yano T, Taniguchi M, Shirasaka T, Tsuneyoshi I. Effectiveness of soluble recombinant human thrombomodulin in patients with severe acute pancreatitis complicated by disseminated intravascular coagulation. Turkish J Anaesthesiol Reanimation. (2019) 47:320–6. doi: 10.5152/TJAR.2019.42709
129. Eguchi T, Tsuji Y, Okada A, Inoue D, Tokumasu H, Iwane K, et al. Reducing the risk of developing walled-off necrosis in patients with acute necrotic collection using recombinant human soluble thrombomodulin. J Hepato-Biliary-Pancreatic Sci. (2021) 28:788–97. doi: 10.1002/jhbp.v28.9
130. Lu XS, Qiu F, Li JQ, Fan QQ, Zhou RG, Ai YH, et al. Low molecular weight heparin in the treatment of severe acute pancreatitis: a multiple centre prospective clinical study. Asian J Surg. (2009) 32:89–94. doi: 10.1016/S1015-9584(09)60017-8
131. Du J-D, Zheng XI, Huang Z-Q, Cai S-W, Tan J-W, Li Z-L, et al. Effects of intensive insulin therapy combined with low molecular weight heparin anticoagulant therapy on severe pancreatitis. Exp Ther Med. (2014) 8:141–6. doi: 10.3892/etm.2014.1694
132. Tozlu M, Kayar Y, Ince AT, Baysal B, Senturk H. Low molecular weight heparin treatment of acute moderate and severe pancreatitis: A randomized, controlled,open-label study. Turk J Gastroenterol. (2019) 30:81–7. doi: 10.5152/tjg.2018.18583
133. Li H, Yang Z, Tian F. Clinical characteristics and risk factors for sinistral portal hypertension associated with moderate and severe acute pancreatitis: A seven-year single-center retrospective study. Med Sci Monitor. (2019) 25:5969–76. doi: 10.12659/MSM.916192
134. Kröner PT, Wallace MB, Raimondo M, Antwi SO, Ma Y, Li Z, et al. Systemic anticoagulation is associated with decreased mortality and morbidity in acute pancreatitis. Pancreatology. (2021) 21:1428–33. doi: 10.1016/j.pan.2021.09.003
135. Patil B, Meena LN, Sharma DC, Agarwal G, Dadhich Y, Gupta G. Impact of low-molecular-weight heparin in the treatment of moderately severe and severe acute pancreatitis; a randomized, single blind, phase 3 control trial. Int J Surg. (2022) 101:910–8. doi: 10.1016/j.ijsu.2022.106621
136. Gonzelez HJ, Sahay SJ, Samadi B, Davidson BR, Rahman SH. Splanchnic vein thrombosis in severe acute pancreatitis: a 2-year, single-institution experience. Hpb. (2011) 13:860–4. doi: 10.1111/j.1477-2574.2011.00392.x
137. Harris S, Nadkarni N, Naina H, Vege S. Splanchnic vein thrombosis in acute pancreatitis a single-center experience. Pancreas. (2013) 42:1251–4. doi: 10.1097/MPA.0b013e3182968ff5
138. Easler J, Muddana V, Furlan A, Dasyam A, Vipperla K, Slivka A, et al. Portosplenomesenteric venous thrombosis in patients with acute pancreatitis is associated with pancreatic necrosis and usually has a benign course. Clin Gastroenterol Hepatol. (2014) 12:854–62. doi: 10.1016/j.cgh.2013.09.068
139. Garret C, Péron M, Reignier J, Le Thuaut A, Lascarrou J-B, Douane F, et al. Risk factors and outcomes of infected pancreatic necrosis: Retrospective cohort of 148 patients admitted to the ICU for acute pancreatitis. United Eur Gastroenterol J. (2018) 6:910–8. doi: 10.1177/2050640618764049
140. Pagliari D, Cianci R, Brizi MG, Mancarella FA, Musso M, Cintoni M, et al. Anticoagulant therapy in the treatment of splanchnic vein thrombosis associated to acute pancreatitis: a 3-year single-centre experience. Internal Emergency Med. (2020) 15:1021–9. doi: 10.1007/s11739-019-02271-5
141. Junare PR, Udgirkar S, Nair S, Debnath P, Jain S, Modi A, et al. Splanchnic venous thrombosis in acute pancreatitis: does anticoagulation affect outcome? Gastroenterol Res. (2020) 13:25–31. doi: 10.14740/gr1223
142. Saleh S, Dalal S, Desai A, Thomas C, Chitsaz E. Outcomes of anticoagulation in patients with splanchnic vein thrombosis from acute pancreatitis A population-based nationwide retrospective cohort study. Pancreas. (2022) 51:e105–6. doi: 10.1097/MPA.0000000000002122
143. Thejasvin K, Chan S-J, Varghese C, Lim WB, Cheemungtoo GM, Akter N, et al. A selective anticoagulation policy for splanchnic vein thrombosis in acute pancreatitis is associated with favourable outcomes: experience from a UK tertiary referral centre. Hpb. (2022) 24:1937–43. doi: 10.1016/j.hpb.2022.06.003
144. Oyón D, Marra-López C, Bolado F, López-López S, Ibáñez-Beroiz B, Canaval-Zuleta HJ, et al. Determinants and impact of splanchnic vein thrombosis in acute pancreatitis. Digest Liver Dis. (2023) 55:1480–6. doi: 10.1016/j.dld.2023.04.026
145. Eltweri AM, Basamh M, Ting YY, Harris M, Garcea G, Kuan LL. A retrospective multicentre clinical study on management of isolated splenic vein thrombosis: risks and benefits of anticoagulation. Langenbeck’s Arch Surg. (2024) 409:89–94. doi: 10.1007/s00423-024-03295-y
146. Tenner S, Vege SS, Sheth SG, Sauer B, Yang A, Conwell DL, et al. American college of gastroenterology guidelines: management of acute pancreatitis. Am J Gastroenterol. (2024) 119:419–37. doi: 10.14309/ajg.0000000000002645
147. Toqué L, Hamy A, Hamel JF, Cesbron E, Hulo P, Robert S, et al. Predictive factors of splanchnic vein thrombosis in acute pancreatitis: A 6-year single-center experience. J Digest Dis. (2016) 16:734–40. doi: 10.5152/tjg.2018.18583
148. Wilson TM, Daneshmand A, Parys S, Watanabe Y. Splanchnic vein thrombosis in acute pancreatitis: a review of treatment indications, methods, and outcomes in a single institution. ANZ J Surg. (2023) 93:2487–91. doi: 10.1111/ans.v93.10
149. Kong Y, Yin J, Cheng D, Lu Z, Wang N, Wang F, et al. Antithrombin III attenuates AKI following acute severe pancreatitis. Shock. (2018) 49:572–9. doi: 10.1097/SHK.0000000000000946
150. Madhi R, Rahman M, Taha D, Mörgelin M, Thorlacius H. Targeting peptidylarginine deiminase reduces neutrophil extracellular trap formation and tissue injury in severe acute pancreatitis. J Cell Physiol. (2018) 234:11850–60. doi: 10.1002/jcp.v234.7
Keywords: coagulopathy, acute pancreatitis, immunothrombosis, pathophysiology, clinical treatment
Citation: Li L, Tan Q, Wu X, Mou X, Lin Z, Liu T, Huang W, Deng L, Jin T and Xia Q (2024) Coagulopathy and acute pancreatitis: pathophysiology and clinical treatment. Front. Immunol. 15:1477160. doi: 10.3389/fimmu.2024.1477160
Received: 07 August 2024; Accepted: 10 October 2024;
Published: 31 October 2024.
Edited by:
Brian A. Boone, West Virginia University, United StatesReviewed by:
Yanwen Chen, University of Pittsburgh, United StatesCopyright © 2024 Li, Tan, Wu, Mou, Lin, Liu, Huang, Deng, Jin and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lihui Deng, ZGVuZ2xpaHVpQHNjdS5lZHUuY24=; Tao Jin, amludGFvQHdjaHNjdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.