 Eleonora Ponterio
Eleonora Ponterio Tobias Longin Haas
Tobias Longin Haas Ruggero De Maria
Ruggero De Maria- 1Department of Oncology and Molecular Medicine, Istituto Superiore di Sanità, Rome, Italy
- 2Dipartimento di Medicina e Chirurgia Traslazionali, Sezione di Patologia Generale, Università Cattolica del Sacro Cuore, Rome, Italy
- 3IIGM - Italian Institute for Genomic Medicine, Candiolo, TO, Italy
- 4Fondazione Policlinico Universitario “A. Gemelli” - I.R.C.C.S., Rome, Italy
Adoptive immunotherapy with T cells, genetically modified to express a tumor-reactive chimeric antigen receptor (CAR), is an innovative and rapidly developing life-saving treatment for cancer patients without other therapeutic opportunities. CAR-T cell therapy has proven effective only in hematological malignancies. However, although by now only a few clinical trials had promising outcomes, we predict that CAR-T therapy will eventually become an established treatment for several solid tumors. Oncolytic viruses (OVs) can selectively replicate in and kill cancer cells without harming healthy cells. They can stimulate an immune response against the tumor, because OVs potentially stimulate adaptive immunity and innate components of the host immune system. Using CAR-T cells along with oncolytic viruses may enhance the efficacy of CAR-T cell therapy in destroying solid tumors by increasing the tumor penetrance of T cells and reducing the immune suppression by the tumor microenvironment. This review describes recent advances in the design of oncolytic viruses and CAR-T cells while providing an overview of the potential combination of oncolytic virotherapy with CAR-T cells for solid cancers. In this review, we will focus on the host-virus interaction in the tumor microenvironment to reverse local immunosuppression and to develop CAR-T cell effector function.
1 From conventional therapies to CAR-T cell immunotherapy with OVs
Cancer treatment has undergone a radical transformation thanks to groundbreaking advancements in research and drug development. While chemotherapy once stood as the primary treatment, targeting rapidly dividing cancer cells, it also affected proliferating healthy cells, such as those of the epithelial and hematopoietic compartments. However, the advent of molecular profiling and understanding of cancer cell mutations has revolutionized treatment approaches. Random screening has been replaced by targeted therapies that specifically target molecular abnormalities in cancer cells (1).
Immunotherapy has rapidly ascended as a cutting-edge and highly promising domain within oncological treatments. While the response to immunotherapies varies among patients, those who do respond can achieve substantial improvements. A leading innovation in this field is Chimeric antigen receptor (CAR) T-cell therapy (2). CAR-T cell therapy involves the clinical use of adoptive cell therapies to combat cancer (3), introducing new possibilities alongside conventional treatments like surgery, radiotherapy, and chemotherapy. In this approach, a patient’s immune system is supported with infused T cells that have been genetically modified to specifically target tumor cells. This approach holds great promise and has demonstrated clinical efficacy in hematologic malignancies (4). These T cells have been modified to express a CAR. The CAR is an artificial molecule engineered to induce cytolytic T cell reactions in tumors. The CAR combines the extracellular single chain variable fragment (scFv) portion with the ability to recognize tumor-associated epitopes and the intracellular signalling domains that are required for T cell activation (3). At present, the clinical efficacy of CAR-T cell therapy is mainly limited to hematologic malignancies. Some successes were reported in preclinical studies on solid tumors but most of the CAR-T cell therapies targeting these types of cancers are yet not ready for FDA approval.
In this review we describe some of the drawbacks of CAR-T cells in solid tumors and how a combination of this therapy with oncolytic viruses (OVs) could potentially function in a synergistic manner, offering complementary and additive benefits.
2 CAR-T cells show limited efficacy in solid tumors, underscoring the need for new solutions
The therapy based on CAR-T cells faces challenges in targeting solid tumors, with issues related to identification of the optimal antigen, effective trafficking, infiltration, and persistence within the immunosuppressive tumor (5–8).
These issues often lead to reduced efficacy and potential toxicity (9). To address these limitations, researchers are developing advanced CAR designs that incorporate multiple costimulatory molecules, ligands, and soluble cytokines (10). Additionally, combining CAR-T cell therapy with checkpoint blockade and targeting inhibitory factors in the tumor microenvironment has shown promise in reducing T-cell exhaustion (11). Ongoing research aims to enhance CAR-T cell proliferation and tumor destruction capabilities in solid tumors by addressing mechanisms of CAR-T cell dysfunction (11). Strategies to improve CAR-T cell efficacy include enhancing tumor infiltration, persistence, and overall function (12).
2.1 Trafficking
After infusion, CAR-T cells need to accumulate at the tumor site. This process is favoured by interventions targeting the tumor vasculature and by promoting the expression of chemokine receptors that promote T cell infiltration (13). For instance, CAR-T cells engineered to co-express CXCR2/CCR2b (receptors for CXCL1) have shown enhanced targeting towards mesothelioma tumor cells expressing CXCL1. Similar results were obtained with αvβ3- or αvβ6-targeting CAR-T cells, which express integrins found on tumor vascular endothelium (14–18). Blocking endothelin B receptor has also demonstrated enhanced T cell infiltration into tumor lesions (19).
2.2 Infiltration
CAR-T cell therapy has been effective for hematological malignancies but struggles with solid tumors due to issues with infiltration and expansion (8). To overcome these challenges, researchers are exploring several strategies. One approach involves engineering CAR-T cells to express heparanase, an enzyme that breaks down extracellular matrix components, improving tumor infiltration (15). Combining CAR-T cells with vascular disrupting agents like combretastatin A-4 phosphate (CA4P) has also shown improved infiltration and efficacy in preclinical models (20). Additionally, Tian et al. (21) engineered CAR-T cells to express CXCL9, enhancing their cytokine secretion, cytotoxicity, and ability to recruit T cells while inhibiting tumor angiogenesis. Engineered CAR-T cells to express IL-8 receptors for improved migration and persistence in the tumor microenvironment (22). These innovative strategies aim to enhance CAR-T cell therapy’s efficacy in solid tumors by improving infiltration, antitumor activity, and survival outcomes in preclinical studies.
2.3 Tumor microenvironmental immune suppression
Tumors employ various mechanisms, including the recruitment of tumor-associated macrophages, myeloid-derived suppressor cells, and activated regulatory T cells, to create an immune-tolerant microenvironment (23). Understanding the tumor microenvironment (TME) in solid tumors reveals a complex interplay of cells and signals sustaining tumor growth and immune evasion. Strategies to counteract the immunosuppressive TME include immune checkpoint inhibitors and genetically engineered CAR-T cells to secrete immune-modulating compounds. Incorporating cytokines like IL-12, IL-18, or IL-36γ into CAR-T cells enhances bystander T cell activation, and co-stimulatory ligands such as CD40L boost nearby T cell effector functions. (24–28). Moreover, CAR-T cells expressing dendritic cell growth factors like Flt3L activate host T cells and promote an effective immune response (28). Addressing antigenic heterogeneity, researchers have developed EGFRvIII CAR-T BiTEs (29). While these approaches show promise, further research is needed to optimize CAR-T therapy in solid tumors, considering specific CAR constructs and patients’ clinical histories. Combining CAR-T therapy with checkpoint blockade holds potential (30), but requires careful consideration of efficacy and potential toxicity (31).
However, positive preclinical studies fostered new efforts that mesoCAR-T cells expressing anti-PD-1 antibodies are being evaluated in a phase II trial for treating mesothelin-positive advanced solid tumors (32).
Cell death within tumors is complex, and true “immunogenic” cell death, capable of triggering an anti-tumor immune response, is rare (33–35). Dying normal cells tend to be tolerogenic, and dying tumor cells often activate suppressive pathways, such as IDO, TGFβ, and Treg activation, hindering the immune response (36–38). Activated Tregs play a crucial role in establishing tolerance to dying tumor cells and can inhibit CAR-T cell activation through various mechanisms (39). The immunosuppressive properties of activated Tregs can limit CAR-T cell therapy effectiveness. Strategies to modulate Treg function or reduce their presence within the tumor microenvironment may enhance CAR-T cell efficacy. Combining therapies targeting both Tregs and tumor cells simultaneously could improve CAR-T cell therapy outcomes, particularly in the context of immune tolerance. Additionally, the enzyme IDO, which inhibits T-cell function, can be expressed in tumors and hinder CAR-T therapy, but targeting IDO with certain drugs may enhance the success of CAR-T treatments (40). Indeed, ongoing research, bolstered by technological advancements, offers the promise of maximizing the efficacy of immunotherapy. This entails also synergistically merging CAR-T therapy with oncolytic viruses (OVs) to address solid tumors. This evolving landscape holds significant promise for the future of cancer treatment.
2.4 Oncolytic viruses
OVs are a promising class of viruses that show the potential to selectively target and destroy cancer cells, leaving healthy cells largely unharmed. (41). Once inside the host cells, OVs replicate and may cause lysis of the cancer cells, or produce their primary therapeutic benefit by delivering immunostimulatory transgenes that trigger or augment an immune response against the tumor. OVs exert various effects that contribute to their anti-cancer activity, including: (i) the release of tumor antigens, (ii) the release of PAMP-like molecules, which trigger the innate immune system to recognize and eliminate infected cells, including cancer cells, (iii) the stimulation of the innate immune responses, (iv) the release of proinflammatory cytokines.
OVs induce oncolysis and immunogenic killing of tumor cells. OVs modulate specific cell death pathways such as apoptosis, autophagic cell death, necrosis, and necroptosis; many of these cell death pathways are by themself immunogenic (Figure 1). Moreover, OVs can be engineered to carry genes that produce immune-stimulating molecules, further boosting the anti-tumor response. These biological properties suggest that the combination of OVs with another immunotherapy agent may have considerable anti-tumor effects (42). Beyond OVs, CAR-T cells themselves can be engineered to express and deliver therapeutic factors. For instance, mesoCAR-T cells expressing anti-PD-1 antibodies are being evaluated in a phase II trial for treating mesothelin-positive advanced solid tumors (32). Additionally, IL-12-secreting Muc16-directed CAR-T cells have shown success in overcoming the immunosuppressive tumor microenvironment (TME) (43). Furthermore, OVs can be modified to express and deliver specific CAR antigens to the tumor surface, enhancing the targeting of corresponding CAR-T cells (44). In a study, a recombinant oncolytic vaccinia virus (OVV) was engineered to produce hyaluronidase (Hyal1) to break down hyaluronic acid in the tumor microenvironment. This degradation enhances drug delivery and immune cell infiltration, significantly improving antitumor effects both alone and in combination with other treatments (45). Recent research has found that combining oncolytic viruses with CAR-T cells enhances anti-tumor effects. However, delivering treatment to distant or metastatic tumors is challenging. To address this, scientists used CAR-T cells infected with HSV-1, which can systemically deliver the virus to solid tumors. These HSV-carrying CAR-T cells remained functional and, in mouse models, successfully delivered the virus to tumors, improving T-cell infiltration and significantly prolonging survival. The study demonstrates the potential of using CAR-T cells to target and treat distant tumors effectively (46). To improve CAR-T therapy for solid tumors, researchers used the A56 antigen, which is upregulated on tumor cells after administering an oncolytic vaccinia virus (OVV). A56-targeted CAR-T cells showed enhanced effectiveness in killing cancer cells in vitro. In mouse models, combining A56 CAR-T cells with OVV and hydroxyurea significantly reduced tumor size and delayed progression. This approach minimizes CAR-T effects on normal cells while improving targeting and treatment of various solid tumors. (47). Combining OVs with CAR-T cell therapy strategies could potentially function in a synergistic manner, offering complementary and additive benefits, that will be discussed in this review.
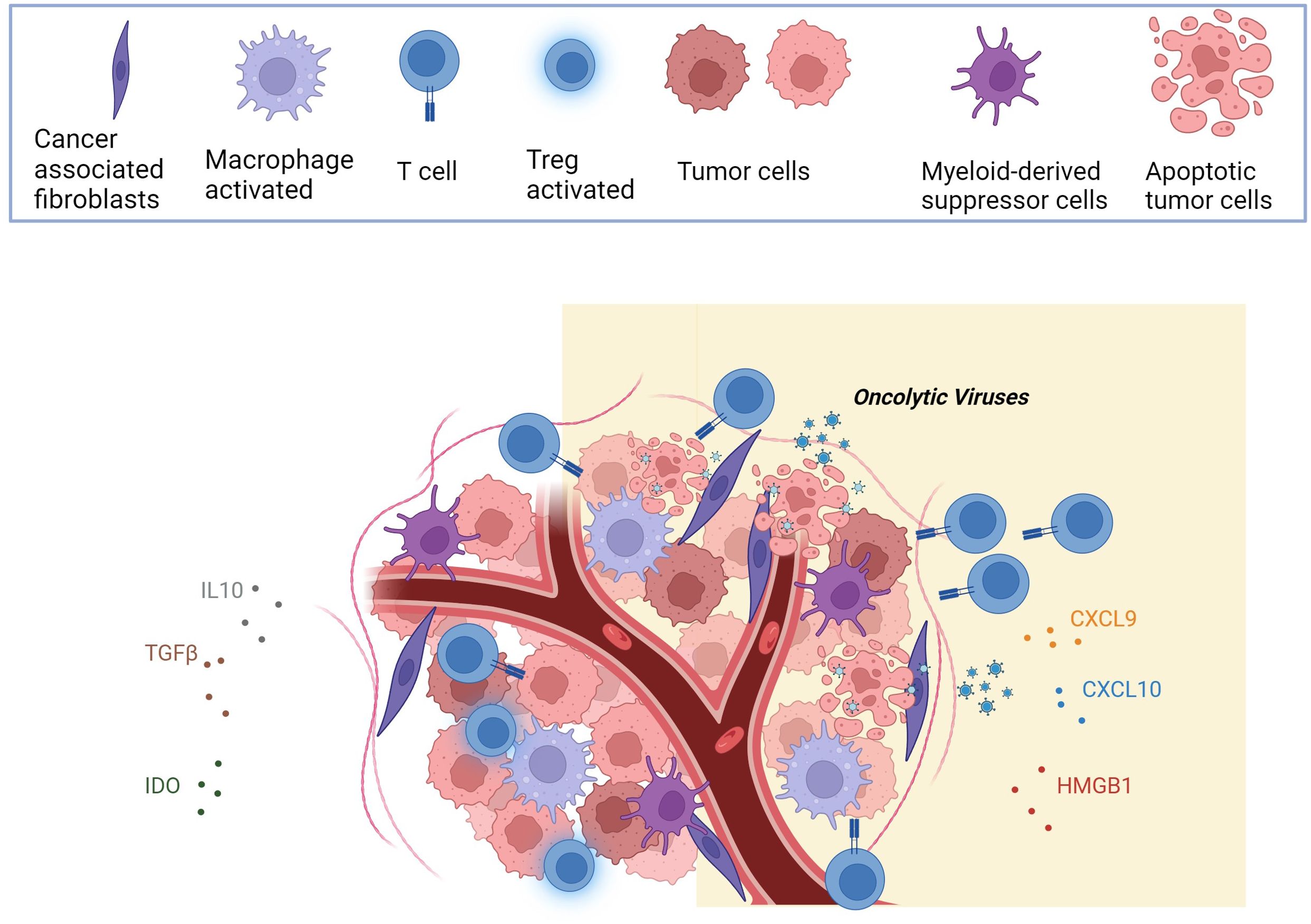
Figure 1. The figure illustrates that the heightened oncolytic immunogenicity is a distinctive feature of OVs. As OVs induce the lysis of tumor cells, they release a combination of viral progeny, tumor-specific antigens (TSAs), pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs), orchestrating immunogenic cell death (ICD). This intricate process not only activates innate immunity, with collaboration between immune cells for effective tumor clearance. This leads to the release of inflammatory factors and chemokines (CXCL9, CXCL10, HMGB1), ultimately reversing the immunosuppressive characteristics of the tumor microenvironment (TME). Created with BioRender.com.
3 Impact of Ovs on the tumor microenvironment
The use of OVs, has emerged as a potentially powerful approach for cancer treatment by harnessing the immune system’s ability to recognize and eliminate tumor cells (48), OVs exploit several molecular and cellular mechanisms to enhance antitumor immunity. Firstly, OVs can selectively infect and replicate within cancer cells due to their inherent tumor cell tropism, which is achieved through specific receptor interactions or alterations in the tumor microenvironment (49). Upon viral replication, cancer cells undergo cytolysis, resulting in the release of tumor-associated antigens (TAAs) and danger signals (50). These TAAs are presented by antigen-presenting cells, such as dendritic cells (DCs), to activate tumor-specific T cells and initiate an adaptive immune response against the tumor. In addition to direct tumor cell killing, OVs induce immunogenic cell death (ICD), a process characterized by the release of immunostimulatory molecules. During ICD, cancer cells release damage-associated molecular patterns (DAMPs) such as calreticulin, high-mobility group box 1 (HMGB1), and adenosine triphosphate (ATP) (51). Cellular damage or stress is induced by various triggers, such as tissue injury, radiation, drug treatment, infection, and ischemic shock, leading to the release of DAMPs (52, 53). These DAMPs serve as danger signals that alert the immune system to the presence of dying tumor cells. They activate innate immune cells, including DCs, macrophages, and natural killer (NK) cells, which process TAAs, leading to antigen presentation and activation of cytotoxic T lymphocytes (CTLs) and other effector cells (54). This immune activation promotes tumor-specific immune responses and long-term memory against the tumor. Furthermore, OVs can be genetically engineered to express therapeutic transgenes that enhance the antitumor immune response. These transgenes can encode cytokines (e.g., interferons, interleukins) (55, 56), chemokines (e.g., CXCL9, CXCL10), immune checkpoint inhibitors (e.g., anti-PD-1 antibodies (56), or co-stimulatory molecules (e.g., CD40 ligand, 4-1BB ligand) (57). By expressing these immune-stimulatory molecules, OVs can augment immune cell recruitment, activation, and effector functions within the tumor microenvironment. For example, the expression of cytokines can promote the infiltration of immune cells into the tumor, enhance antigen presentation, and activate cytotoxic immune responses. Immune checkpoint inhibitors expressed by OVs can block inhibitory signalling pathways, such as PD-1/PD-L1, leading to the reinvigoration of exhausted T cells and restoration of antitumor immune responses (56). The combination of direct oncolysis, induction of ICD, and expression of immune-stimulatory molecules by OVs creates a highly immunogenic tumor microenvironment, facilitating the generation of systemic antitumor immunity. This multimodal approach not only targets and eliminates primary tumors but also promotes immune memory and helps to control metastatic disease. Moreover, as described below, OVs can be used in combination with other immunotherapies, such as immune checkpoint inhibitors or adoptive cell therapies, to achieve synergistic effects and enhance treatment outcomes.
By harnessing these mechanisms, oncolytic viruses (OVs) show immense potential for enhancing cancer treatment and broadening the therapeutic arsenal available for patients, including CAR-T cell therapy. Numerous preclinical studies have been published demonstrating the efficacy of this approach. (Table 1).
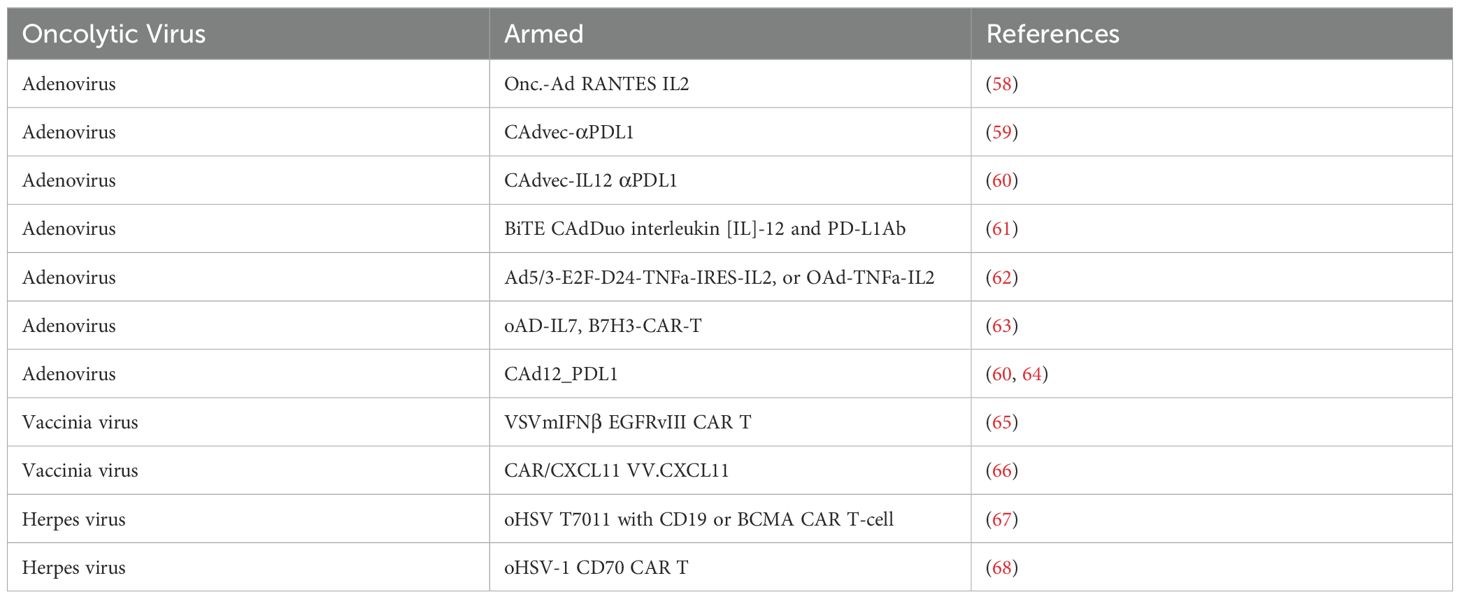
Table 1. Preclinical studies on oncolytic viruses and CAR-T cells.
4 Enhancement of CAR-T cell function by OVs
Setting up treatment strategies inducing complete therapeutic responses in patients with solid tumors presents significant challenges. However, OVs hold great promise as tools to overcome some of these obstacles and enhance the effectiveness of CAR-T cell therapy in solid tumors (Figure 2).
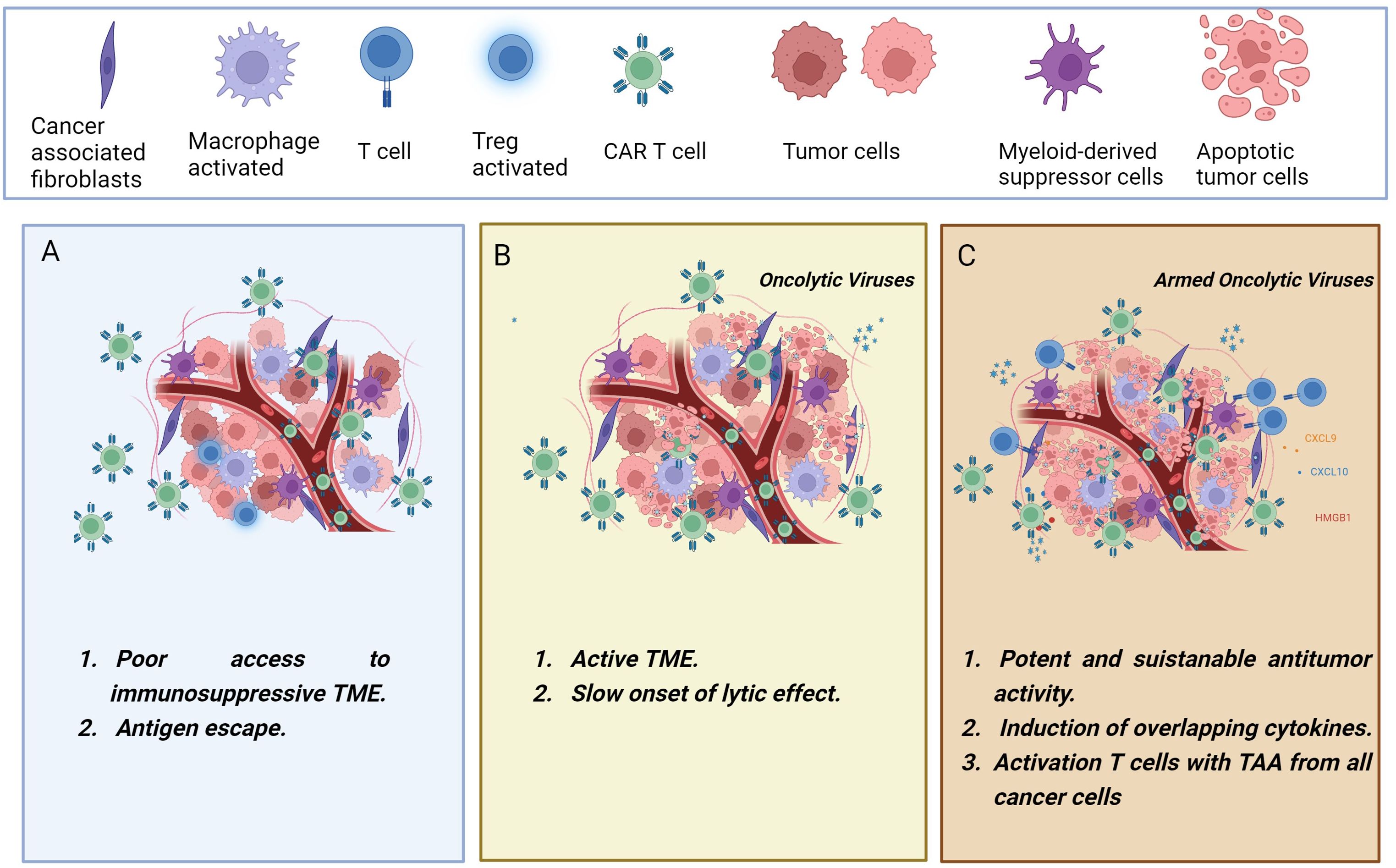
Figure 2. The figure illustrates the synergistic combination of CAR-T cells and oncolytic viruses. (A) CAR-T cells encounter various challenges in solid tumors, including an immunosuppressive environment that may lead to T cell dysfunction and treatment failure. (B) Administering oncolytic viruses for cancer treatment before CAR-T cell therapy leads to tumor debulking, immunogenic cell death, and a reversal of tumor immunosuppression. (C) In a collaborative effort, the engineered oncolytic viruses may transform the immunologically “cold” tumor into a “hot” tumor, exerting an upgraded and more powerful antitumor immunity. Oncolytic viruses can be genetically modified to deliver therapeutic transgenes into the tumor microenvironment, boosting T-cell effector functions. Combining CAR-T cells with oncolytic viruses armed with cytokines, chemokines, BiTEs, or immune checkpoint inhibitors has demonstrated enhanced therapeutic outcomes. Created with BioRender.com.
First, OVs can be armed with therapeutic transgenes to boost the CAR-T activation. Second, OVs could be able to survive and maintain their cytotoxicity functions in a tumor microenvironment that is immunosuppressed. This may provide danger signals that can revert tumor immunosuppression. Third, the direct lytic effect of OV on cancer cells results in tumor cell death and thus the release of tumor-associated antigens (TAA) (69).
Wing et al. described that the combination of an adenovirus vector (OAd) and BiTE treatment-mediated oncolysis, improved CAR-T cell activation and proliferation. Furthermore, this co-treatment increased cytokine production and cytotoxicity and showed a favourable safety profile in vitro compared to the EGFR- targeting CAR-T cells alone (70). Another opportunity is the combination of CAR-T cells with an armed oncolytic virus that delivers chemokine such as RANTES and IL15 to enhance the trafficking and persistence of the CAR-T cells, resulting in antitumor effects (71). It was described that infecting tumor cells with an oncolytic vaccinia virus coding for CD19t produced de novo CD19 at the cell surface before virus-mediated tumor lysis. Co-culture with CD19 CAR-T cells with OV19t produced secretion of cytokines and exhibited potent cytolytic activity against infected tumors (72). In another study, the authors used a combination of DD7-IL7 and B7H3 CAR-T in vitro and showed increased proliferation and persistence of tumor-infiltrating B7H3 CAR-T for glioblastoma treatment (63). The oncolytic adenovirus LOAd703, encoding CD40L and 4-1BBL transgenes, demonstrates promise in B cell tumor therapy, activating both antigen-presenting cells and T cells through CD46 interaction (73). When combined with CAR-T cell therapy, it elicits robust anti-lymphoma immune responses, significantly enhancing the effectiveness of CAR-T treatment (73). Additionally, oncolytic vaccinia viruses engineered to produce CXCL11 or armed with IL21 have shown potential in bolstering adoptive T cell transfer and vaccine-based immunotherapy, offering promising avenues for treatment improvement (66).
Furthermore, OVs can be modified to express and deliver specific CAR antigens to the tumor surface, enhancing the targeting of corresponding CAR-T cells (44). Looking ahead, combining oncolytic virotherapy with established treatments and emerging strategies like immune checkpoint inhibitors and CAR-T therapy holds considerable potential for future cancer treatment paradigms for solid tumors (74).
5 Examples of oncolytic viruses combined with CAR-T cell
5.1 Adenovirus
Adenoviruses (Ads) are small, non-enveloped viruses with a double-stranded DNA genome. One advantage of adenoviral vectors is that their genome remains episomal, conferring a safer profile compared to integrating viral vectors. Adenovirus-based genetic vaccines have demonstrated the ability to drive robust and sustained T cell and B cell responses against the encoded transgenes. This may be attributed to the vector’s ability to persist in a transcriptionally active form at the site of inoculation and in lymphatic tissues (75) Adenovirus, holds significant potential in the realm of CAR-T cell therapy (CAR-T). Adenoviral vectors can be genetically modified to efficiently deliver CAR genes into T cells, enabling the production of CAR-T cells (63, 76, 77). The high infectivity and transduction efficiency of adenoviruses make them an appealing tool for CAR-T cell manufacturing (78). By employing adenoviral transduction, CAR expression can be effectively achieved on the surface of T cells, empowering them to target and eliminate cancer cells. Furthermore, adenoviral vectors can be engineered to enhance CAR-T cell persistence and augment their anti-tumor activity (79). However, it is essential to address challenges such as pre-existing immunity against adenoviruses in the human population and potential toxicity associated with viral vectors. Ongoing preclinical (Table 1) and clinical investigations aim to optimize adenoviral vectors for CAR-T cell generation, with the goal of improving the safety and efficacy of CAR-T cell therapy across diverse cancer types (70).
5.2 Vaccinia virus
Vaccinia virus (VV) is a large, enveloped, double-stranded DNA virus that belongs to the Poxviridae family. It has been extensively used as a smallpox vaccine and has a well-established safety profile in humans. By leveraging its inherent oncolytic properties, vaccinia virus can selectively infect and replicate within cancer cells, triggering their destruction and has shown promise in clinical trials for advanced solid cancers (80). When combined with CAR-T cells, vaccinia virus can enhance the anti-tumor immune response by promoting the release of tumor antigens and creating an inflammatory milieu that supports CAR-T cell activation and cytotoxicity (65). Ongoing preclinical investigations will be important to explore the synergistic potential of vaccinia virus and CAR-T cells aim to optimize this combination therapy for improved outcomes in cancer treatment (66).
5.3 Herpes virus
Oncolytic herpes simplex virus (oHSV) have proven to be very potent OVs as they can selectively lyse tumor cells without harming healthy cells, addressing a key challenge in CAR-T cell therapy for solid tumors. Through genetic manipulation of the viral genome, oHSVs can be armed with various payloads such as cytokines, chemokines, and immune checkpoint inhibitors to convert immunologically “cold” tumors into “hot” tumors. Talimogene laherparepvec (T-VEC), the first clinically approved OV, is a genetically modified oHSV that expresses granulocyte-macrophage colony-stimulating factor (GM-CSF) and is utilized for treating melanoma (81). Possible combination with CAR-T cells are discussed in different papers. For instance, the authors Zhu et al. demonstrated that oncolytic herpes simplex virus type 1 (oHSV-1) enhances the therapeutic effect of CD70-specific CAR-T cells by promoting intratumoral T cell infiltration and the release of interferon-gamma (IFN-γ). This finding provides support for the incorporation of CAR-T therapy into glioblastoma (GB) therapeutic strategies (68). Moreover, in vitro and in vivo studies have demonstrated synergistic anti-tumor responses through combined treatment with oHSV T7011 and CAR-TCD19 or CAR-TBCMA cells (67). The efficacy assessments further validate the significant synergistic anti-tumor effects achieved by combining T7011 with either CAR-TCD19 or CAR-TBCMA cells across various solid tumor models. These findings collectively suggest that the next-generation oHSV T7011 holds substantial promise as a combinatorial therapy alongside CD19 or BCMA-specific CAR-T cells, offering a potential for the treatment of solid tumors (67).
6 Conclusion
The compelling potential of Onco-Immunotherapy, driven by the intricate molecular and cellular mechanisms inherent in oncolytic virus strategies, as discussed in the preceding sections of this review, strongly suggests that the synergistic use of CAR-T cells and OVs could be pivotal in overcoming tumor resistance and significantly improving therapeutic outcomes. When used in tandem, OVs can serve as powerful adjuvants to CAR-T cell therapy (71). They can sensitize tumors to CAR-T cell recognition by upregulating the expression of tumor antigens and immune-stimulatory molecules. This synergistic approach can address the limitations of CAR-T cell therapy in solid tumors and potentiate a more robust and durable anti-tumor immune response (82). OVs are genetically modified viruses designed to selectively infect and replicate within cancer cells. They possess several characteristics that make them attractive candidates for combination therapy with CAR-T cells. By infecting tumor cells, OVs can induce immunogenic cell death, release tumor antigens, and create an inflammatory environment favourable for CAR-T cell activation (83). Additionally, some OVs can directly modulate the tumor microenvironment, reversing immunosuppression and promoting an anti-tumor immune response. The integration of OVs with CAR-T cells in solid tumor treatment represents a promising avenue for future research and clinical applications (44). Ongoing studies are focused on optimizing the delivery methods, improving the safety profiles, and developing novel oncolytic viruses that can selectively target tumor cells while sparing normal tissues. Furthermore, combining these therapies with immune checkpoint inhibitors or other immunomodulatory agents may further enhance their effectiveness against most types of solid cancer. The combination of CAR-T cells and oncolytic viruses offers a promising strategy to address this hurdle. By harnessing the complementary mechanisms of action of these two modalities, researchers are striving to unlock the full potential of immunotherapy in the treatment of cancer. With continued advancements and clinical investigations, this combined approach holds great promise for the future of cancer treatment.
Author contributions
EP: Writing – original draft, Writing – review & editing. TLH: Writing – review & editing. RDM: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Italian Association for Cancer Research (AIRC; IG 2019 ID 23190 to RDM), by Italian Ministry of Health (Alliance Against Cancer CAR T cells project ID RCR23ACC to RDM), by Traiettoria 4 “Biotecnologia, Bioinformatica e Sviluppo Farmaceutico” - Linea di azione 4.1 “Creazione di Hub delle Scienze della Vita” - Piano Sviluppo e Coesione Salute – ID T4-AN-01 to RDM.
Acknowledgments
The authors extend their appreciation to Prof. Malcolm Brenner, Center for Cell and Gene Therapy Baylor College of Medicine Houston, TX US, for his insightful suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. (2018) 18:359–76. doi: 10.1038/s41568-018-0006-7
2. Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. (2017) 17:286–301. doi: 10.1038/nrc.2017.17
3. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/nejmra1706169
4. Ponterio E, De Maria R, Haas TL. Identification of targets to redirect CAR T cells in glioblastoma and colorectal cancer: an arduous venture. Front Immunol. (2020) 11:565631. doi: 10.3389/fimmu.2020.565631
5. Labanieh L, Majzner RG, Mackall CL. Programming CAR-T cells to kill cancer. Nat BioMed Eng. (2018) 2:377–91. doi: 10.1038/s41551-018-0235-9
6. Schmidts A, Maus MV. Making CAR T cells a solid option for solid tumors. Front Immunol. (2018) 9:2593. doi: 10.3389/fimmu.2018.02593
7. Tokarew N, Ogonek J, Endres S, Von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
8. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
9. Xia AL, Wang XC, Lu YJ, Lu XJ, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. (2017) 8:90521–31. doi: 10.18632/oncotarget.19361
10. Yeku O, Li X, Brentjens RJ. Adoptive T-cell therapy for solid tumors. Am Soc Clin Oncol Educ Book. (2017) 37:193–204. doi: 10.14694/edbk_180328
11. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. (2021) 12:81. doi: 10.1186/s13287-020-02128-1
12. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
13. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers (Basel). (2019) 11:674. doi: 10.3390/cancers11050674
14. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. (2009) 113:6392–402. doi: 10.1182/blood-2009-03-209650
15. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
16. Whilding LM, Vallath S, Maher J. The integrin alphavbeta6: a novel target for CAR T-cell immunotherapy? Biochem Soc Trans. (2016) 44:349–55. doi: 10.1042/bst20150249
17. Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RMG, Kao YV, et al. Targeting of aberrant alphavbeta6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol Ther. (2017) 25:259–73. doi: 10.1016/j.ymthe.2016.10.012
18. Phanthaphol N, Somboonpatarakun C, Suwanchiwasiri K, Chieochansin T, Sujjitjoon J, Wongkham S, et al. Chimeric antigen receptor T cells targeting integrin alphavbeta6 expressed on cholangiocarcinoma cells. Front Oncol. (2021) 11:657868. doi: 10.3389/fonc.2021.657868
19. Kandalaft LE, Facciabene A, Buckanovich RJ, Coukos G. Endothelin B receptor, a new target in cancer immune therapy. Clin Cancer Res. (2009) 15:4521–8. doi: 10.1158/1078-0432.ccr-08-0543
20. Deng C, Zhao J, Zhou S, Dong J, Cao J, Gao J, et al. The vascular disrupting agent CA4P improves the antitumor efficacy of CAR-T cells in preclinical models of solid human tumors. Mol Ther. (2020) 28:75–88. doi: 10.1016/j.ymthe.2019.10.010
21. Tian Y, Wen C, Zhang Z, Liu Y, Li F, Zhao Q, et al. CXCL9-modified CAR T cells improve immune cell infiltration and antitumor efficacy. Cancer Immunol Immunother. (2022) 71:2663–75. doi: 10.1007/s00262-022-03193-6
22. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
23. Iglesias-Escudero M, Arias-Gonzalez N, Martinez-Caceres E. Regulatory cells and the effect of cancer immunotherapy. Mol Cancer. (2023) 22:26. doi: 10.1186/s12943-023-01714-0
24. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. (2011) 121:4746–57. doi: 10.1172/jci58814
25. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. (2011) 19:751–9.
26. Zhao J, Zhao J, Perlman S. Differential effects of IL-12 on Tregs and non-Treg T cells: roles of IFN-gamma, IL-2 and IL-2R. PloS One. (2012) 7:e46241. doi: 10.1371/journal.pone.0046241
27. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol. (2018) 9:1977. doi: 10.3389/fimmu.2018.01977
28. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. (2020) 21:914–26. doi: 10.1038/s41590-020-0676-7
29. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
30. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.ccr-13-0458
31. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol Ther. (2016) 24:1135–49. doi: 10.1038/mt.2016.63
32. Adusumilli PS, Zauderer MG, Riviere I, Solomon SB, Rusch VW, O’cearbhaill RE, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with Malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discovery. (2021) 11:2748–63. doi: 10.1158/2159-8290.cd-21-0407
33. Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U.S.A. (2012) 109:3909–14. doi: 10.1073/pnas.1117736109
34. Bezu L, Gomes-De-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. (2015) 6:187. doi: 10.3389/fimmu.2015.00187
35. Yang J, Bae H. Drug conjugates for targeting regulatory T cells in the tumor microenvironment: guided missiles for cancer treatment. Exp Mol Med. (2023) 55:1996–2004. doi: 10.1038/s12276-023-01080-3
36. Hirata F, Ohnishi T, Hayaishi O. Indoleamine 2,3-dioxygenase. Characterization and properties of enzyme. O2- complex. J Biol Chem. (1977) 252:4637–42. doi: 10.1016/s0021-9258(17)40208-0
37. Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. (2002) 101:151–5. doi: 10.1002/ijc.10645
38. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. (2015) 125:3905–16. doi: 10.1182/blood-2015-01-621474
39. Hosseinalizadeh H, Rabiee F, Eghbalifard N, Rajabi H, Klionsky DJ, Rezaee A. Regulating the regulatory T cells as cell therapies in autoimmunity and cancer. Front Med (Lausanne). (2023) 10:1244298. doi: 10.3389/fmed.2023.1244298
40. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. (2016) 39:1–6. doi: 10.1016/j.coi.2015.10.009
41. Sakhawat A, Ma L, Muhammad T, Khan AA, Chen X, Huang Y. A tumor targeting oncolytic adenovirus can improve therapeutic outcomes in chemotherapy resistant metastatic human breast carcinoma. Sci Rep. (2019) 9:7504. doi: 10.1038/s41598-019-43668-8
42. Jafari M, Kadkhodazadeh M, Shapourabadi MB, Goradel NH, Shokrgozar MA, Arashkia A, et al. Immunovirotherapy: The role of antibody based therapeutics combination with oncolytic viruses. Front Immunol. (2022) 13:1012806. doi: 10.3389/fimmu.2022.1012806
43. Li T, Wang J. Therapeutic effect of dual CAR-T targeting PDL1 and MUC16 antigens on ovarian cancer cells in mice. BMC Cancer. (2020) 20:678. doi: 10.1186/s12885-020-07180-x
44. Mcgrath K, Dotti G. Combining oncolytic viruses with chimeric antigen receptor T cell therapy. Hum Gene Ther. (2021) 32:150–7. doi: 10.1089/hum.2020.278
45. Wang S, Li Y, Xu C, Dong J, Wei J. An oncolytic vaccinia virus encoding hyaluronidase reshapes the extracellular matrix to enhance cancer chemotherapy and immunotherapy. J Immunother Cancer. (2024) 7:e008431. doi: 10.1136/jitc-2023-008431
46. Zhang Z, Yang N, Xu L, Lu H, Chen Y, Wang Z, et al. Systemic delivery of oncolytic herpes virus using CAR-T cells enhances targeting of antitumor immuno-virotherapy. Cancer Immunol Immunother. (2024) 73:173. doi: 10.1007/s00262-024-03757-8
47. Cho E, An MH, Lee YS, Ryu EJ, Lee YR, Park SY, et al. Development of chimeric antigen receptor (CAR)-T cells targeting A56 viral protein implanted by oncolytic virus. iScience. (2024), 16.
48. Garmaroudi GA, Karimi F, Naeini LG, Kokabian P, Givtaj N. Therapeutic efficacy of oncolytic viruses in fighting cancer: recent advances and perspective. Oxid Med Cell Longev. (2022) 2022:3142306. doi: 10.1155/2022/3142306
49. Rahman MM, McFadden G. Oncolytic viruses: newest frontier for cancer immunotherapy. Cancers (Basel). (2021) 13:5452. doi: 10.3390/cancers13215452
50. Lin D, Shen Y, Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Sig Transduct Target Ther. (2023) 8:156. doi: 10.1038/s41392-023-01407-6
51. Mardi A, Shirokova AV, Mohammed RN, Keshavarz A, Zekiy AO, Thangavelu L, et al. Biological causes of immunogenic cancer cell death (ICD) and anti-tumor therapy; Combination of Oncolytic virus-based immunotherapy and CAR T-cell therapy for ICD induction. Cancer Cell Int. (2022) 22:168. doi: 10.1186/s12935-022-02585-z
52. Ma M, Jiang W, Zhou R. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity. (2024) 57:752–71. doi: 10.1016/j.immuni.2024.03.002
53. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
54. Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. (2014) 21:26–38. doi: 10.1038/cdd.2013.48
55. Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum Gene Ther Clin Dev. (2016) 27:69–78. doi: 10.1089/humc.2016.031
56. Chen CY, Wang PY, Hutzen B, Sprague L, Swain HM, Love JK, et al. Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci Rep. (2017) 7:2396. doi: 10.1038/s41598-017-02503-8
57. Huang JH, Zhang SN, Choi KJ, Choi IK, Kim JH, Lee MG, et al. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol Ther. (2010) 18:264–74. doi: 10.1038/mt.2009.205
58. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. (2014) 74:5195–205. doi: 10.1158/0008-5472.can-14-0697
59. Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, et al. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. (2017) 77:2040–51. doi: 10.1158/0008-5472.can-16-1577
60. Rosewell Shaw A, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. (2017) 25:2440–51. doi: 10.1016/j.ymthe.2017.09.010
61. Porter CE, Rosewell Shaw A, Jung Y, Yip T, Castro PD, Sandulache VC, et al. Oncolytic adenovirus armed with biTE, cytokine, and checkpoint inhibitor enables CAR T cells to control the growth of heterogeneous tumors. Mol Ther. (2020) 28:1251–62. doi: 10.1016/j.ymthe.2020.02.016
62. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. (2018) 3:e99573. doi: 10.1172/jci.insight.99573
63. Huang J, Zheng M, Zhang Z, Tang X, Chen Y, Peng A, et al. Interleukin-7-loaded oncolytic adenovirus improves CAR-T cell therapy for glioblastoma. Cancer Immunol Immunother. (2021) 70:2453–65. doi: 10.1007/s00262-021-02856-0
64. Mckenna MK, Englisch A, Brenner B, Smith T, Hoyos V, Suzuki M, et al. Mesenchymal stromal cell delivery of oncolytic immunotherapy improves CAR-T cell antitumor activity. Mol Ther. (2021) 29:1808–20. doi: 10.1016/j.ymthe.2021.10.007
65. Evgin L, Huff AL, Wongthida P, Thompson J, Kottke T, Tonne J, et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun. (2020) 11:3187. doi: 10.1038/s41467-020-17011-z
66. Moon EK, Wang LS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. (2018) 7:e1395997. doi: 10.1080/2162402x.2017.1395997
67. Liu Y, Zheng Y, Deng T, Huang Y, Liu Z, Zhan B, et al. Oncolytic herpes simplex virus delivery of dual CAR targets of CD19 and BCMA as well as immunomodulators to enhance therapeutic efficacy in solid tumors combined with CAR T cell therapy. Front Oncol. (2022) 12:1037934. doi: 10.3389/fonc.2022.1037934
68. Zhu G, Zhang J, Zhang Q, Jin G, Su X, Liu S, et al. Enhancement of CD70-specific CAR T treatment by IFN-gamma released from oHSV-1-infected glioblastoma. Cancer Immunol Immunother. (2022) 71:2433–48. doi: 10.1007/s00262-022-03172-x
69. Evgin L, Vile RG. Parking CAR T cells in tumours: oncolytic viruses as valets or vandals? Cancers (Basel). (2021) 13:1106. doi: 10.3390/cancers13051106
70. Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. (2018) 6:605–16. doi: 10.1158/2326-6066.cir-17-0314
71. Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. (2015) 4:e988098. doi: 10.4161/21505594.2014.988098
72. Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med. (2020) 12:e988098. doi: 10.1126/scitranslmed.aaz1863
73. Wenthe J, Naseri S, Labani-Motlagh A, Enblad G, Wikstrom KI, Eriksson E, et al. Boosting CAR T-cell responses in lymphoma by simultaneous targeting of CD40/4-1BB using oncolytic viral gene therapy. Cancer Immunol Immunother. (2021) 70:2851–65. doi: 10.1007/s00262-021-02895-7
74. Abd-Aziz N, Poh CL. Development of oncolytic viruses for cancer therapy. Transl Res. (2021) 237:98–123. doi: 10.1016/j.trsl.2021.04.008
75. Tatsis N, Fitzgerald JC, Reyes-Sandoval A, Harris-McCoy KC, Hensley SE, Zhou D, et al. Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines. Blood. (2007) 110:1916–23. doi: 10.1182/blood-2007-02-062117
76. Zhang C, Fang L, Wang X, Yuan S, Li W, Tian W, et al. Oncolytic adenovirus-mediated expression of decorin facilitates CAIX-targeting CAR-T therapy against renal cell carcinoma. Mol Ther Oncolytics. (2022) 24:14–25.
77. Wang G, Zhang Z, Zhong K, Wang Z, Yang N, Tang X, et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol Ther. (2023) 31:134–53. doi: 10.1016/j.ymthe.2022.08.021
78. Fang L, Tian W, Zhang C, Wang X, Li W, Zhang Q, et al. Oncolytic adenovirus-mediated expression of CCL5 and IL12 facilitates CA9-targeting CAR-T therapy against renal cell carcinoma. Pharmacol Res. (2023) 189:106701. doi: 10.1016/j.phrs.2023.106701
79. Tang X, Li Y, Ma J, Wang X, Zhao W, Hossain MA, et al. Adenovirus-mediated specific tumor tagging facilitates CAR-T therapy against antigen-mismatched solid tumors. Cancer Lett. (2020) 487:1–9. doi: 10.1016/j.canlet.2020.05.013
80. Kaynarcalidan O, Moreno Mascaraque S, Drexler I. Vaccinia virus: from crude smallpox vaccines to elaborate viral vector vaccine design. Biomedicines. (2021) 9:1780. doi: 10.3390/biomedicines9121780
81. Kaufman HL, Kim DW, Deraffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. (2010) 17:718–30. doi: 10.1245/s10434-009-0809-6
82. Rezaei R, Esmaeili Gouvarchin Ghaleh H, Farzanehpour M, Dorostkar R, Ranjbar R, Bolandian M, et al. Combination therapy with CAR T cells and oncolytic viruses: a new era in cancer immunotherapy. Cancer Gene Ther. (2022) 29:647–60. doi: 10.1038/s41417-021-00359-9
Keywords: CAR T cells, oncolytic viruses, cancer, immunotherapy, solid tumor
Citation: Ponterio E, Haas TL and De Maria R (2024) Oncolytic virus and CAR-T cell therapy in solid tumors. Front. Immunol. 15:1455163. doi: 10.3389/fimmu.2024.1455163
Received: 26 June 2024; Accepted: 08 October 2024;
Published: 30 October 2024.
Edited by:
Don J. Diamond, City of Hope National Medical Center, United StatesReviewed by:
Lance Hellman, Nevada State College, United StatesMasmudur Mohammed Rahman, Arizona State University, United States
Copyright © 2024 Ponterio, Haas and De Maria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleonora Ponterio, ZWxlb25vcmEucG9udGVyaW9AaXNzLml0