 Rida Rehman1,2
Rida Rehman1,2 Albrecht Froehlich1Florian olde Heuvel1Lobna Elsayed1
Albrecht Froehlich1Florian olde Heuvel1Lobna Elsayed1 Tobias Boeckers3,4
Tobias Boeckers3,4 Markus Huber-Lang5
Markus Huber-Lang5 Cristina Morganti-Kossmann6,7
Cristina Morganti-Kossmann6,7 Francesco Roselli1,3*†
Francesco Roselli1,3*†- 1Department of Neurology, Ulm University, Ulm, Germany
- 2Institute for Stem Cell Biology and Regenerative Medicine, Stanford University School of Medicine, Stanford, CA, United States
- 3German Center for Neurodegenerative Diseases (DZNE), Ulm, Germany
- 4Institute of Anatomy and Cell biology, Ulm University, Ulm, Germany
- 5Institute of Translational Trauma Immunology, Ulm University, Ulm, Germany
- 6Department of Child Health, Barrow Neurological Institute at Phoenix Children’s Hospital, Phoenix, AZ, United States
- 7University of Arizona College of Medicine, Phoenix, AZ, United States
Background: Traumatic brain injury (TBI) induces an acute reactive state of microglia, which contribute to secondary injury processes through phagocytic activity and release of cytokines. Several receptor tyrosine kinases (RTK) are activated in microglia upon TBI, and their blockade may reduce the acute inflammation and decrease the secondary loss of neurons; thus, RTKs are potential therapeutic targets. We have previously demonstrated that several members of the Fibroblast Growth Factor Receptor (FGFR) family are transiently phosporylated upon TBI; the availability for drug repurposing of FGFR inhibitors makes worthwhile the elucidation of the role of FGFR in the acute phases of the response to TBI and the effect of FGFR inhibition.
Methods: A closed, blunt, weight-drop mild TBI protocol was employed. The pan-FGFR inhibitor Rogaratinib was administered to mice 30min after the TBI and daily up to 7 days post injury. Phosphor-RTK Arrays and proteomic antibody arrays were used to determine target engagement and large-scale impact of the FGFR inhibitor. pFGFR1 and pFGFR3 immunostaining were employed for validation. As outcome parameters of the TBI injury immunostainings for NeuN, VGLUT1, VGAT at 7dpi were considered.
Results: Inhibition of FGFR during TBI restricted phosphorylation of FGFR1, FGFR3, FGFR4 and ErbB4. Phosphorylation of FGFR1 and FGFR3 during TBI was traced back to Iba1+ microglia. Rogaratinib substantially dowregulated the proteomic signature of the neuroimmunological response to trauma, including the expression of CD40L, CXCR3, CCL4, CCR4, ILR6, MMP3 and OPG. Prolonged Rogaratinib treatment reduced neuronal loss upon TBI and prevented the loss of excitatory (vGLUT+) synapses.
Conclusion: The FGFR family is involved in the early induction of reactive microglia in TBI. FGFR inhibition selectively prevented FGFR phosphorylation in the microglia, dampened the overall neuroimmunological response and enhanced the preservation of neuronal and synaptic integrity. Thus, FGFR inhibitors may be relevant targets for drug repurposing aimed at modulating microglial reactivity in TBI.
Introduction
Traumatic Brain Injury (TBI) is characterized by the dynamic interplay of multiple cellular actors, including neurons, astrocytes, microglia as well as vascular and immune cells, which may assume beneficial or detrimental roles, depending on time and space (1, 2). Microglial cells swiftly react to TBI by migrating to the site of injury (3), assuming an ameboid, chemotactic morphology (4) and diverse reactive functional states including ones characterized by increased interferon response (5) and by disease-associated-like microglial markers (6, 7). Unchecked acute microglial reactivity in TBI has been largely considered detrimental, leading to acidosis, oxidative stress, enhanced neuronal damage and synaptic loss (8–10) in the “secondary injury” phase.
However, early post-traumatic depletion of microglia by CSF1R inhibitor administration reduces the extent of neuronal apoptosis but does not affect the overall lesion size and actually increased the size of intracerebral haematoma (11). Actually, a number of microglia-associated responses, such as glial limitans repair and debris clearing may have neuroprotective outcomes (12–14) and may be carried out by specific subset or functional states of microglial cells [such as repopulating microglia; (15, 16)]. Thus, the goal of suppressing microglial reactivity should be substituted by the aim for a fine-tuning microglial reactivity and phenotype to maximize tissue preservation.
Receptor tyrosine kinases have emerged in the last 20 years as a class of drug targets, with more than 70 small-molecule kinase inhibitors approved for human use [mainly in oncology; (17, 18)] and therefore lend themselves to effective drug repurposing. Notably, RKT activation is a prominent response in TBI: a targeted phosphoproteomic screening of 39 RTK has revealed the significant increase in phosphorylation of multiple families of RTK including VEGFR1-3, EphB4, Met, MSPR, EGFR, ErbB3 and FGFR4 at 3h and 24h timepoints (19). In fact, Met and VEGFR, among others, were shown to be phosphorylated in microglial cells, contributing to their regulation. Proof of concept of the use of RTK as entry points for acute TBI treatment has been provided by the use of VEGFR and Met small-molecule inhibitors: both caused the substantial divergence in the phosphoproteomic profile after TBI (demonstrating target engagement) and resulted in improved motor performance (19). Furthermore, prolonged treatment with a Met inhibitor delivered persistent improvement in motor performace and enhanced neuronal preservation (19). These findings have opened up the possibility that multiple RTKs may be involved in the early induction of reactive microglial phenotype(s) and may lend themselves to therapeutic modulation. Among these, some FGFR family members displayed transient up-phosphorylation between 3h and 24h after trauma. Interestingly, the FGFR family does not only control proliferation and plasticity, but has been implicated also in the control of inflammation: blockade of FGFRs reduce the cytokine storm and macrophage proliferation in sepsis (20) and FGFR ligand FGF23 induces TNF-α in macrophages (21). Furthermore, FGFR inhibitor Infigratinib reduced microglial rand lymphocyte responses in a multiple sclerosis murine model (22) whereas a different FGFR inhibitor reduces the inflammatory response to B. burgdorferi antigens (23). FGFR inhibitors appear to reduce the availability of immunoproteasome subunits by inducing autophagy in immune cells (24). Taken together, this evidence supports the hypothesis that FGFR inhibitors may modulate inflammatory responses in TBI.
Since FGFR inhibitors have been recently introduced in clinical practice (25), we set out to explore the potential role of FGFR in the early stages of the neuroinflammatory response to TBI.
Results
Pan-FGFR inhibitor modifies the RTK phosphorylation landscape of acute TBI
We explored the effect of the pan-FGFR inhibitor BAY 1213802 [Rogaratinib-HCl; (26); henceforth BAY121], on the TBI-associated RTK phosphorylation landscape with the goal to demonstrate effective and specific target engagement. Mice were subjected to a blunt weight-drop mild TBI (or sham surgery), followed 30 min later by administration of BAY121 (or vehicle). NSS score ranged between 0 and 1 (coherently with the mild TBI protocol) at the 3h timepoints. At 3h post injury mice were sacrificed, samples from the injury site were obtained (Figure 1A) and processed for RTK phosphorylation screening using a nitrocellulose antibody arrays (RTK targets including FGFR 2, 3 and 4). Principal component analysis (PCA) demonstrated a substantial overlap among S-Veh, T-BAY and S-BAY samples, whereas T-Veh stood out (Figure 1B). Analysis of individual RTK phosphorylation revealed a significant upregulation of p-FGFR3 and p-FGFR4 (but not of pFGFR2) upon TBI which was negated by the BAY121 treatment (Figures 1C–E); of note, the antibody array did not include phosphoFGFR1 antibodies. In addition, TBI upregulated the phosphorylation of HGFR, cRet, ErbB4, EphA3 and downregulated phopsho-EphB2 (Figures 1F–J). Interestingly, BAY121 also prevented the phosphorylation of ErbB4 (Figure 1F) but did not affect the up-phosphorylation of cRet and EphA3 (Figures 1H, I; only statistical trends were detected for HGFR nd EphB2).
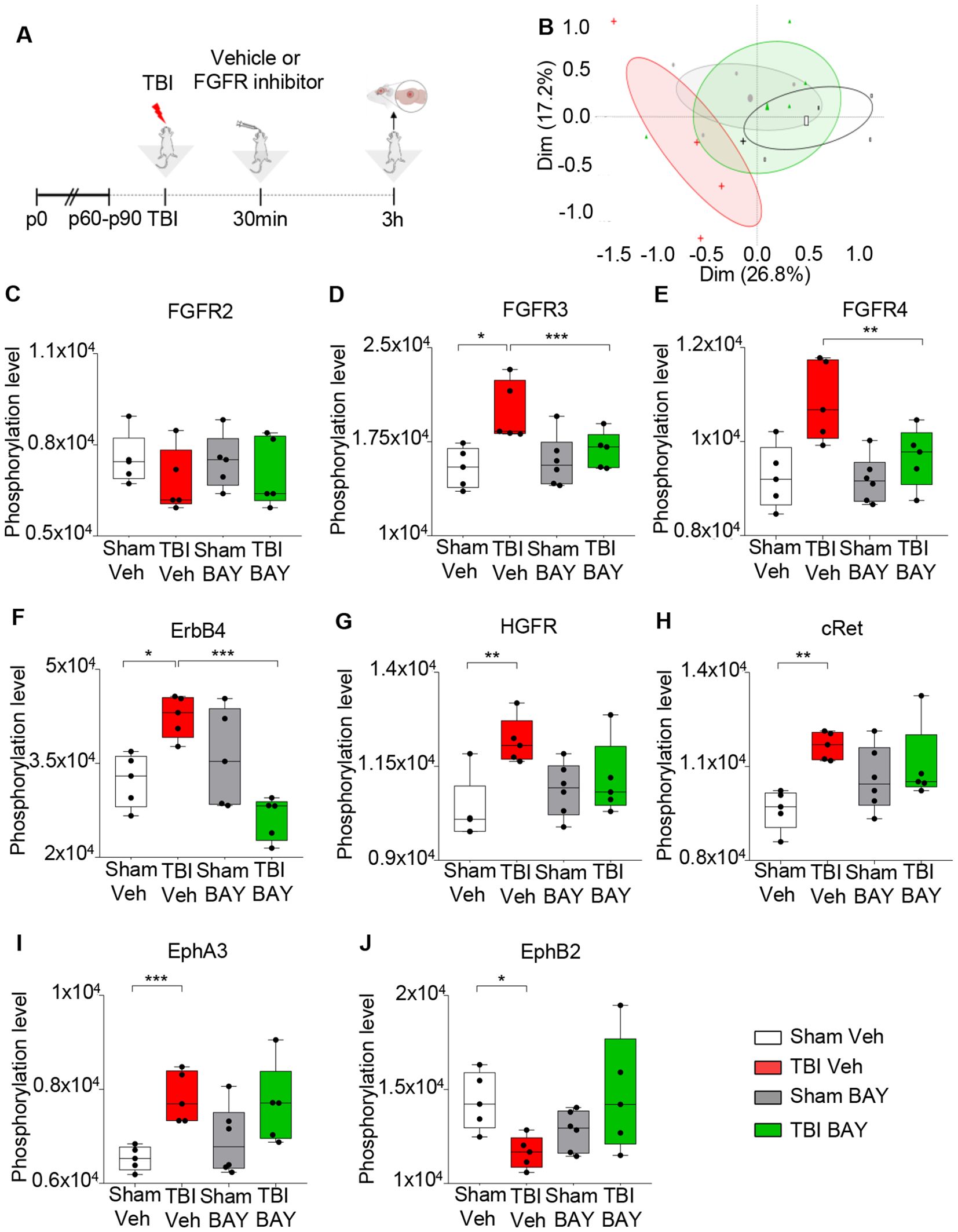
Figure 1. FGFR Inhibitor selectively alters RTK phosphorylation pattern at 3h post injury. (A) Outline of the experimental design. BAY121 (Rogaratinib) was administered by oral gavage at the dose of 25 mg/kg 30 min after TBI and samples were obtained 3h after TBI. (B) Principal component analysis (PCA) plot displayed group wise distribution of samples, highlighting the separation between TBI-Veh samples (red) and the BAY121-treated samples, which overlap with Sham-Veh samples. The group specific ellipses indicate 95% confidence interval. (C-J) Antibody phospho-array focused proteomic analysis of RTK phosphorylation patterns upon TBI with or withouth BAY121 treatment. Differential phosphorylation analysis showed the inhibitor significantly decreased the phosphorylated levels of (D) pFGFR3, (E), pFGFR4 (the array does not include pFGFR1) and (F) ErbB4 after TBI. However, the inhibitor showed no effect on phosphorylation levels of (C) FGFR2, (G) HGFR, (H) cRET, (I) EphA3, and (J) EphB2. Significance for differentially phosphorylated proteins was set at p<0.05 (FDR adjusted). [(B-J): n=5-6/group; *p<0.05, **p<0.01, ***p<0.001].
Taken together, these findings demonstrate that TBI upregulates the phosphorylation of FGFR and BAY121 successfully negates this event; BAY121 (confirming target engagement) does not appear to block the phosphorylation of other RKTs with the exception of ErbB4 (supporting the selectivity of BAY121).
FGFR inhibitor prevents FGFR1 and FGFR3 phophorylation in microglia upon trauma
We investigated the cellular sources and the spatial distribution of FGFR phosphorylation using immunohistological approaches. Only antibodies against FGFR1(pY654) and FGFR3(pY724) proved suitable for immunolabeling of brain sections, whereas no antibody against phospho-FGFR2 and phospho-FGFR4, suitable for immunohistochemistry, was available from commercial sources; therefore, we limited our immunohistochemical study to phospho-FGFR1 and phospho-FGFR3. Animals were subject to TBI and injected 30 min later with either vehicle or BAY121 and sacrificed 3h after the injury (2.5h after treatment; Figure 2A). In we could detect a significant fraction of Iba1+ cells displaying pFGFR1 immunoreactivity already in S-Veh samples. Moreover, immunoreactivity for both pFGFR1 and pFGFR3 was significantly upregulated in the site of injury (“core”, located in cortical layer II/III region) in T-Veh but not in T-BAY samples (Figures 2B, E). Interestingly, the immunoreactivity pattern of pFGFR1 and 3 highlighted large number of small cells of ramified morphology, resembling microglia. In fact, co-immunostaining with Iba1 demonstrated that 70-90% of Iba1+ cells in the site of injury displayed immunoreactivity for pFGFR1 and >50% of Iba1+ cells displayed immunoreactivity for FGFR3. In T-Veh samples, both the immunofluorescence intensity in Iba1+ cells (Figures 2C, F) and the fraction of Iba1+ immunopositive for phospho FGFR1 or phospho FGFR3 was significantly increased compared to S-Veh (Figures 2C, D, F, G). Notably, in T-BAY samples, both the immunofluorescence intensity and the number of Iba1+ cells immunopositive for phosphoFGFR1 and FGFR3 were strongly decreased (no change in the total number of Iba1+ cells was noted).
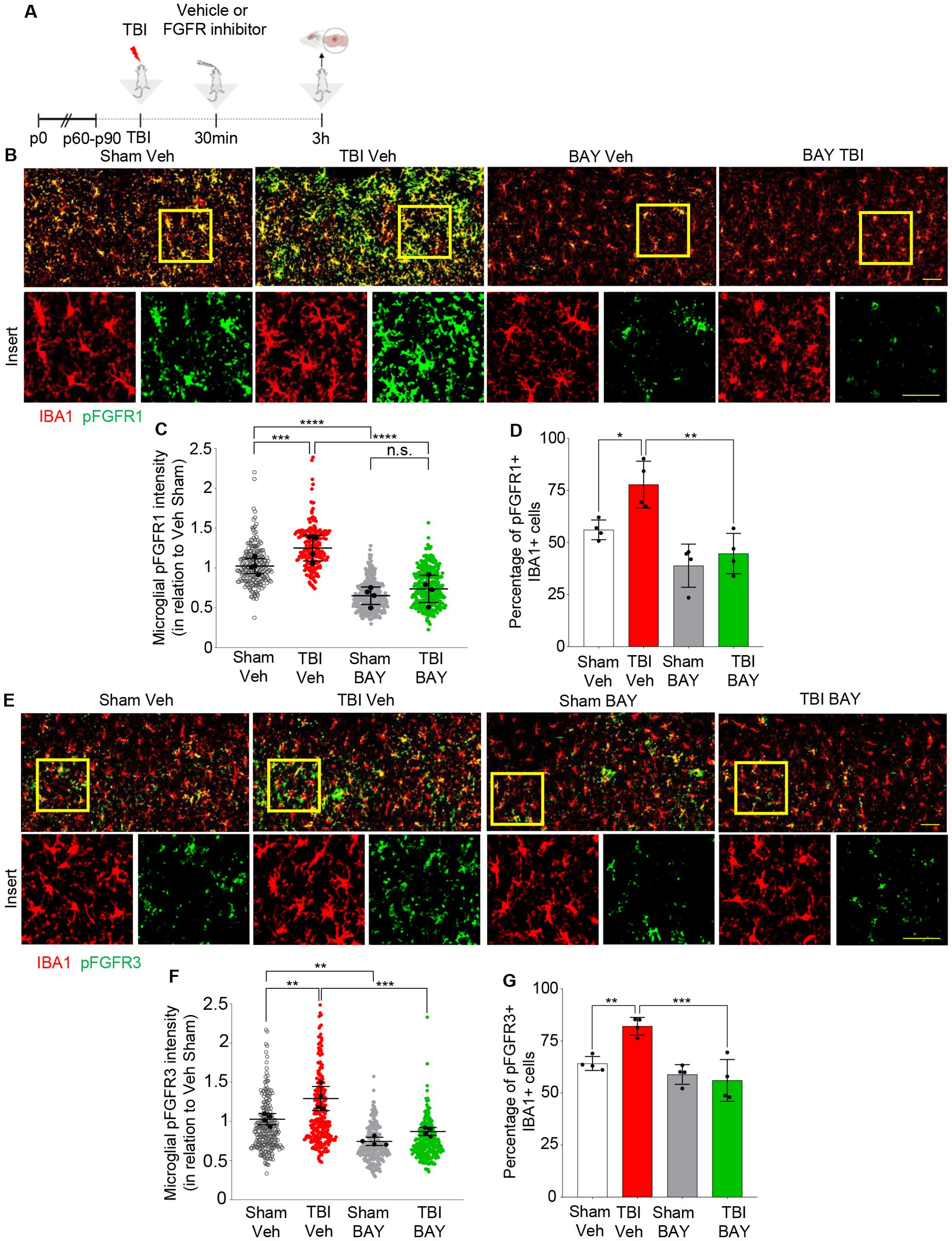
Figure 2. Upregulation of pFGFR1 and pFGFR3 in microglia 3h post injury. (A) Outline of the experimental design. BAY121 (Rogaratinib) was administered by oral gavage at the dose of 25 mg/kg 30 min after TBI and samples for immunohistology were obtained at 3h after TBI. (B-D) Immunostainings for pFGFR1 (green) and Iba1 (red) for Sham- Veh, TBI Veh, Sham- BAY121 and TBI BAY121 treated mice. Quantification of pFGFR1 immunostaining intensity in Iba1+ cells (C) and fraction of pFGFR1+ cells (D) display a significant increase upon TBI, which is negated by the treatment with BAY121. (D-F) Immunostaining for pFGFR3 (green) and Iba1 (red) shows the upregulation of pFGFR immunoreactivity in Iba1+ cells and the increase in the fraction of pFGFR3 upon TBI. Both indexes are decreased by treatment with BAY121. n=4/group; >300 cells per animal n.s., not significant; **p<0.01; ***p<0.001, ****p<0.0001. Overview Scale bar: 50µm. Inset scale bar: 20µm.
Taken together these results confirm the elevation in FGFRs activation upon TBI and demonstrated the successful target engagement for BAY121 on microglial cells.
FGFR inhibition significantly suppresses immune responses in the site of injury
Next we explored if prolonged BAY121 administration could not only affect the acute reactive microglial phenotype, but generate a long-lasting, broad alteration of the proteomic neuroimmunological landscape associated with brain injury. Mice subjected to trauma were administered with BAY121 (or vehicle) 30 mins after trauma (or sham surgery) and continued daily for 3 days (Figure 3A). A targeted proteomic profile of the injury site, involving >1300 individual protein was obtained by antibody arrays. PCA plot showed a substantial separation between S-Veh and TBI-Veh samples on one side and S-BAY and T-BAY on the other (Figure 3B). When compared to S-Veh samples, TBI-Veh samples displayed the upregulation of 16 proteins and the downregulation of 3 proteins (Figure 3C). The upregulated proteins involved multiple mediators of inflammatory responses, including KC (murine homologue of the chemoattractant IL-8), the microglial regulator Axl, the chemotactic receptor CCR10 and the immune regulator CD40 and its ligand CD40L. On the other hand, BAY121 resulted in a substantial change in the proteome when administered after trauma: 51/1308 proteins were downregulated and 98/1308 were upregulated in TBI-BAY121 vs TBI-Veh samples (Figures 3D, E, Supplementary Figure S1A). The gene ontology analysis revealed that BAY121 treatment resulted in the downregulation of proteins involved in immune function and cytokine response (top 5 GO categories, Supplementary Figures S1B, C). Strikingly, Axl and CD40L (upregulated in TBI-Veh vs Sham-Veh) were among the top strongly downregulated along with microglia polarization indicator CD80 and, proinflammatory signaling and chemotaxis markers such as CXCR3, CCL4, CCR4, ILR6, MMP3 and Osteoprotegerin [recently involved in microglial reactivity; (7)] (Figure 3D). The upregulated proteins display an enrichment in the GO categories of cellular signaling, metabolism and protein synthesis, including the mitochondrial protein TRAP and the ion channel KCNAb3.
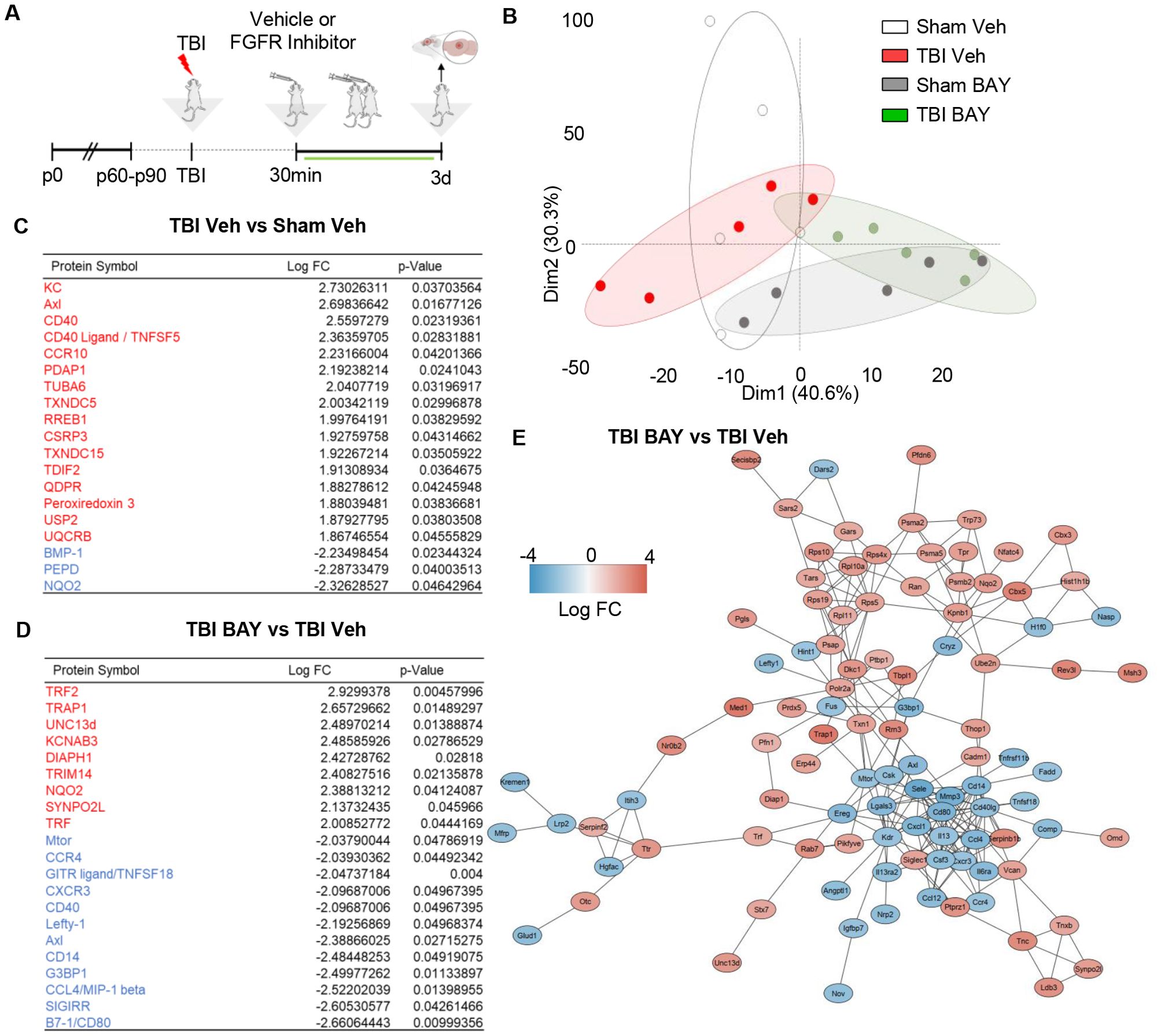
Figure 3. FGFR inhibitor suppresses immune-related proteome signature at 3d post injury. (A) Outline of the experimental design; BAY121 was administered 30 mins after trauma and continued for 3d (25 mg/Kg once daily by oral gavage, vehicle alone as control). Samples were collected 3d post injury. (B) PCA plot of proteomic data shows the separation of TBI-Veh from BAY121-treated samples. (C, D) After modified differential protein expression analysis (FDR <0.05), subsets of upregulated (Red) and downregulated (Blue) proteins with log fold change and individual significance for (D) TBI-Veh compared to Sham–Veh and (D) TBI-BAY121 compared to TBI-Veh (n=5/group). (E) Protein-protein-interaction analysis revealed distinct clustering of downregulated and upregulated proteins in TBI-BAY121 vs TBI Veh; the cluster of downregulated proteins is enriched with immune- and inflammation-related proteins.
We further characterized the distinct signature imposed by BAY121 treatment upon TBI by constructing a protein-protein interaction (PPI) network for the proteins up- or down-regulated by the treatment. We employed the String algorithm and visualized the network using the Cytoscape software. After preprocessing of the dataset, PPI network displayed 137 nodes and 243 edges (Figure 3E). Most notably, proteins up- and down-regulated by BAY121 were enriched in two distinct clusters, with most of the down-regulated proteins related to the immune response forming a tight cluster. Taken together, the proteomic data suggest that prolonged administration of the FGFR inhibitor BAY121 profoundly reduces the sub-acute neuroinflammatory response to TBI.
Prolonged FGFR inhibitor alters both neuron-specific and immune-specific proteins 7d post injury
We further investigated the proteomic signature of prolonged FGFR inhibition in TBI by taking into consideration samples obtained at 7 dpi. As before, mice subjected to trauma were administered with BAY121 (or vehicle) 30 mins after trauma. The treatment was continued for 7 days (1 dose/day) for 7 days (Figure 4A). PCA plot based on the targeted proteomics (1308 targets), revealed that, while the S-Veh and T-Veh samples largely clustered together, S-BAY121 and TBI-BAY121 minimally overlapped the Veh groups (Figure 4B), indicating a persistent and profound effect of BAY121 treatment.
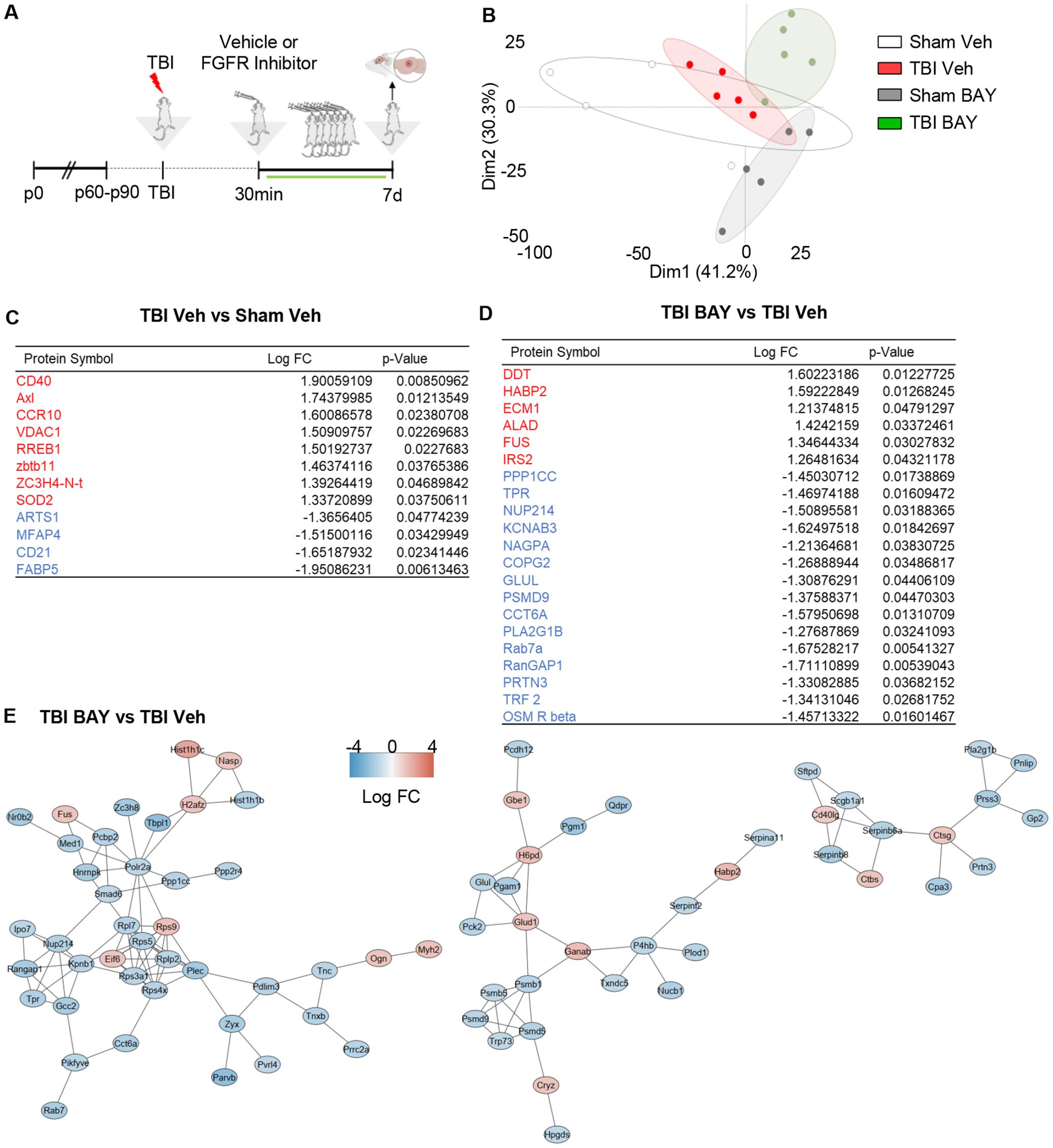
Figure 4. Prolonged FGFR inhibitor treatment significantly alters the TBI-related proteome profiles at 7d post injury. (A) Outline of the experimental design; BAY121 was administered 30 mins after trauma and continued for 7d (25 mg/Kg/day). Samples were collected 7d post injury. (B) PCA plot shows the substantial separation between TBI groups and groups treated with BAY121. (C, D) After modified differential protein expression analysis (FDR <0.05), subset of significantly upregulated (Red) and downregulated (Blue) proteins with log fold change and individual significance for (C) TBI-Veh vs Sham–Veh and (D) TBI-BAY121 vs TBI-Veh. (E) Protein-protein interaction analysis by STRING reveals three distinct networks affected by prolonged BAY121 treatment, related to protein synthesis proteasomal degradation and inflammation (n=5/group).
The analysis of differentially expressed proteins revealed the increased expression in TBI-Veh (vs Sham-Veh) of the proteins involved in inflammation such as Axl, CD40, CCR10 and SOD2, whereas the B-cell marker CD21 was downregulated (Figure 4C). Notably, the comparison of BAY121-treated TBI vs vehicle-treated TBI samples revealed a substantial divergence in the proteome: 123/1308 proteins were downregulated and 38/1308 proteins were upregulated (Figures 4D, E). The gene ontology analysis of the downregulated proteins revealed a substantial involvement of proteasome regulatory proteins and nucleo-cytoplasmic trafficking, pointing toward an impaired protein degradation and cellular stress (Supplementary Figure S2B). The comparatively small number of upregulated proteins did not lend itself to a reliable GO analysis.
Interestingly, when we mapped the PPI of proteins altered by BAY121 treatment (TBI-BAY121 vs TBI-Veh) using STRING/Cytoscape, three distinct sub-networks (cumulatively displaying 112 nodes and 135 edges; Figure 5E) were identified. The smallest of the network (13 proteins) involved downregulated proteins related to the inflammatory/phagocytic function (notably Catepsins and Serpins) and immune regulation (such as CD40L). The two larger networks (65 proteins) included many subunits of the proteasome system, chaperones and trafficking proteins, the largest majority of which were downregulated (Figure 4E, Supplementary Figure S2A). Based on these findings, the proteomic analysis at 7dpi suggested that blockade of FGFR signaling produced a persistend impact on the neuroinflammatory cascade but also substantially impacted the tissue protein homeostasis.
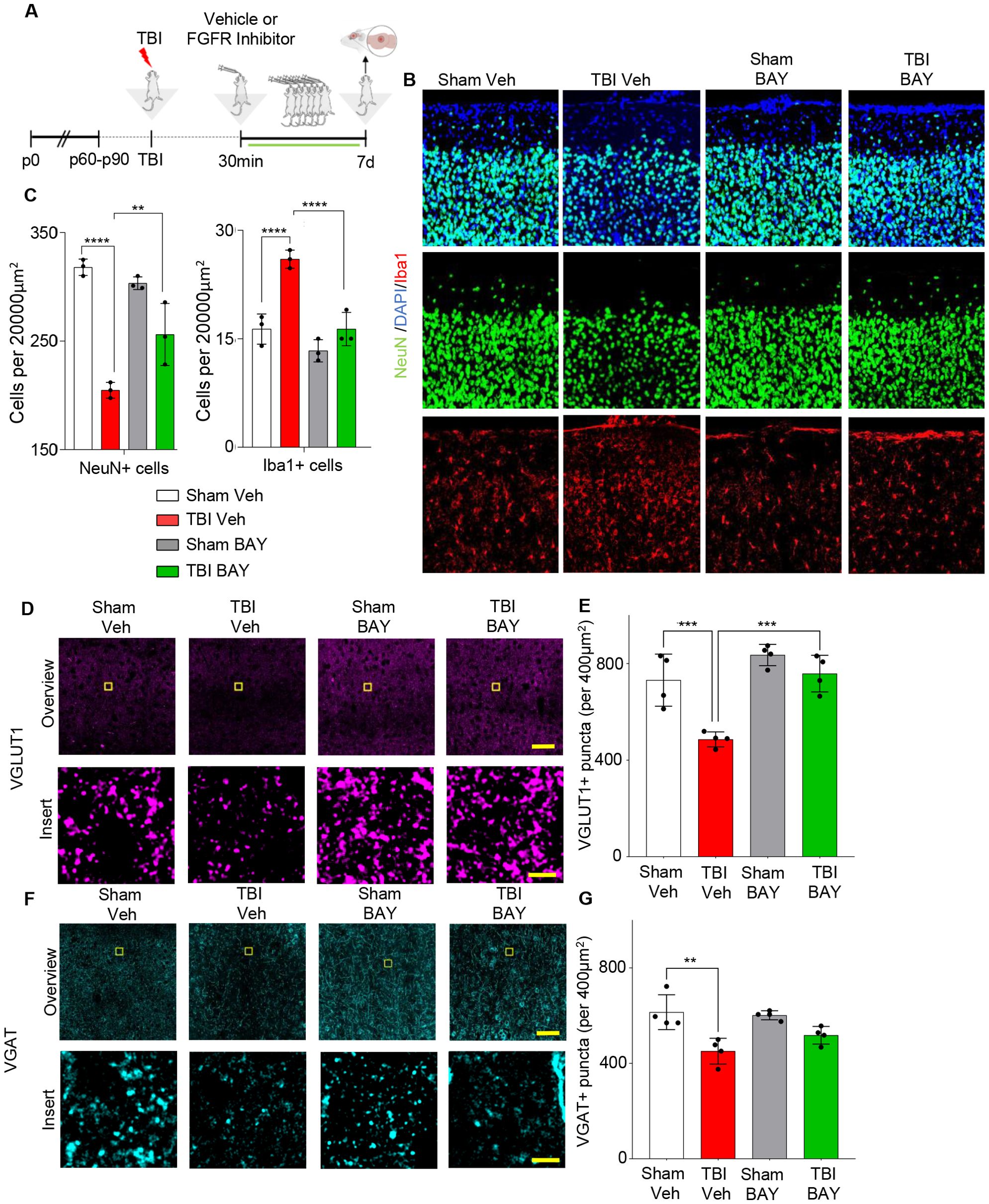
Figure 5. Prolonged FGFR inhibitor administration preserves neuronal density at 7 dpi. (A) Outline of the experimental design; BAY121 was administered 30 mins after trauma and continued for 7d (25 mg/Kg daily by oral gavage). Samples were collected 7d post injury. (B, C) Immunostaining for NeuN showed a significantly decreased number of neurons in the ijury site of TBI-Veh group compared to Sham–Veh; a significantly higher number of neurons was seen in the TBI-BAY121 group. Note that microglial density was also increased in TBI-Veh but not in TBI-BAY121. (n=4/group; **p<0.01, ***p<0.001, ****p<0.0001; Scalebar 70µm) (D, E) Loss of VGLUT1 density after TBI is dependent on FGFR signaling 7d after TBI. (n=4/group; ***p<0.001; Scale bar: overview = 70μm; insert = 5μm) (F, G) VGAT density in the TBI core is significantly reduced at 7dpi. (n=4/group; **p<0.01; Scale bar: overview = 70μm; insert = 5μm).
FGFR inhibitor reduces neuronal loss and synaptic loss 7d post trauma
Finally, we sought to determine if the effects of FGFR blockade on the neuroinflammatory response to TBI were associated with reduced neuronal vulnerability and synaptic integrity. As before, mice were subjected to trauma and treated with either vehicle or the FGFR inhibitor 30 mins after trauma for 7 days (1 dose/day) (Figure 5A). As expected, density of microglia was still increased in the injury site (“core”, located in layed II/III of cortex, on the axis of the site of injury) of TBI -Veh mice, but not in TBI-BAY121 group (Figures 5B, C). Conversely, the density of NeuN+ cells was significantly reduced in the site of injury in TBI Veh mice compared to Sham mice (Figures 5B, C). Notably, BAY121 treatment significant increased the number of surviving neurons in the injury site.
We further explored the preservation of synaptic structures in the injury site upon BAY121 treatment. We assessed the density (number of synaptic puncta per 400µm2) of excitatory synapses in the form of pre-synaptic VGLUT1 as well as of inhibitory synapses in the form of pre-synaptic VGAT. VGLUT1 showed a significant loss in synaptic density after TBI, which was not observable any more after BAY121 treatment 7dpi (Figures 5D, E). VGAT+ terminals were also significantly decreased in the lesion area at 7 dpi (Figures 5F, G) with a trend toward better preservation in T-BAY samples.
Discussion
Our data show that FGFR family is prominently activated in the early phases of mild TBI in particular in microglial cells. Inhibition of the FGFR family by BAY121 results in the suppression of early microglial reactivity and reduces the neuroinflammatory footprint at later stages, which ultimately leads to an improved preservation of neuronal and synaptic integrity in the site of injury.
TBI results in the simultaneous activation of multiple RTK families, often with distinct temporal dinamics (19): the activation of HGFR/Met has been shown to lead to a reactive, phagocytic microglial state with detrimental consequences (19), whereas the activation of the ErbB family in inhibitory interneurons controls synaptic plasticity and circuit activity after TBI (27). Likewise, activation of the VEGF-C/VEGFR3 contributes to drive microglial polarization after TBI (28) and multiple members of the Tyro-Axl-Mer RTK family regulate microglial reactivity and phagocytic activity in TBI (29) and other disease conditions (30). Our focused screening, together with the immunohistochemical confirmation, reproduce the activation of Met, ErbB4, EphB2 and ErbB4 previously identified (19, 27) and identify a significant phosphorylation of several members of the FGFR family. The pan FGFR inhibitor BAY121 effectively suppresses FGFR1, FGFR3 and FGFR4, with limited effect on the trauma-related activation of other RTK. A notable exception is the full blockade of the induction of ErbB4 phosphorylation; since ErbB4 is highly involved in synaptic plasticity and stability (31, 32) and it is activated by TBI (27), it is possible that this effect of Rogaratinib, possibly mediated by microglia modulation, may contribute to the maintained of circuit integrity and excitation/inhibition balance. The combined screening and immunohistochemistry data demonstrate that BAY121 is effective and relatively selective in preventing FGFR family receptors activation in microglia when administered 30 min after trauma, i.e. in a therapeutic window.
Interestingly, some degree of FGFR phosphorylation was detected also in sham samples; this suggests that a baseline FGFR activation in microglia may control the physiology of these cells in homeostatic conditions. Multiple FGFR ligands are normally expressed in the brain by neurons, astrocytes and other cells (33) and therefore are posited to control microglia as well. Microglial phagocytosis is involved not only in debris clearing upon injury but also in synaptic pruning (34), and FGF/FGFR may contribute to the regulation of this process as well.
The blockade of FGFR signaling by BAY121, both in acute (single dose) or protracted (3 to 7 days) administration, consistently results in the dampening of the neuroinflammatory response to TBI at the level of microglial reactivity as well as in terms of broad inflammatory proteomic footprint. In particular, whereas TBI elevates the protein level of CD40 and Axl both at 3d and 7d, Rogaratinib downregulates CD40 and Axl at 3dpi. Axl and CD40 expression corresponds to the induction of a reactive microglial state with upregulated phagocytosis, coherent with the tissue debris clearance occurring after injury (35–37) and the suppressed upregulation by Rogaratinib at 3d is compatible with the reduced reactivity of microglia (and better synaptic preservation at later stages). Although CD40 and Axl are still upregulated by TBI at 7dpi, they do not appear in the proteome subset downregulated by Rogaratinib. While the negative effect at 7dpi cannot be attributed only to biological effects without further investigations (heterogeneity of TBI mice increases at 7dpi -see (19) potentially increasing the number of false negatives), it could be possible to speculate that an escape from Rogaratinib effect, either in microglia or in infiltrating immune cells, could take place at 7dpi and account for the lack of downregulation in CD40 and Axl.
Interestingly, FGFR family members are expressed not only on microglia but also on a number of immune cells, suggesting their broad contribution to immunity and inflammation: FGFR2 mediates chemotaxis in neutrophils (38), and FGFR1 is expressed in T-cells (39, 40) as well as on macrophages and lymphocytes in lupus nephritis (41). At brain level, FGFR signaling contributes to microglial reactivity to bacterial products by inducing pro-inflammatory cytokines (42) and a small-molecule FGFR inhibitor reduces the infiltration of lymphocytes and activated macrophages, as well as the production of pro-inflammatory cytokines, in the EAE model (22). Thus, our data are coherent with a role of the FGFR family in modulating the overall neuroinflammatory cascade and cytokine response to injury, including microglial reactivity. Interestingly, FGFR expressed on oligodendrocytes also contribute to the regulation of neuroinflammatory cascades in EAE: conditional deletion of FGFR1 or FGFR2 from oligodendrocytes results in decreased microglial reactivity and lymphocytes infiltration in EAE as well as in reduced levels of pro-inflammatory cytokines (43, 44); thus, FGFR blockade may immunomodulatory effects through additional cell types. Although in our model blockade of FGFR resulted in reduced neuroinflammation and enhanced synaptic and neuronal preservation in our model, anti-inflammatory effects of FGFR have been also reported: systemic administration of FGF21 reduces the inflammatory response in a stroke model (45) and intranasal administration of FGF20 reduced blood-brain-barrier impairment in severe TBI (46). Thus, the net effect of FGFR activation may depend on the complex interplay of the inflammatory context and of the mix of FGFR ligands available (42). To this respect, multiple RTK are activated simultaneously in mild TBI [Figure 1, (19)]; blockade of FGFR does not affect other RTK also involved in regulation of microglial reactivity (such as HGFR/Met), although a similar anti-inflammatory effect is observed. This may imply a functional redundance of RTK regulation of reactive microglia, or may suggest that the ultimate phenotype is dependent on the combinatorial activation or one or more RTK, which would enable the fine-tuning of the response to the specific conditions. Hence, FGFR signaling may be ultimately pro- or anti-inflammatory depending on the ensemble of RTK activation and the context conditions.
Histological readouts at 7 dpi demonstrate that protracted BAY121 administration results in a reduced loss of excitatory and inhibitory synapses and in the overall preservation of neuronal survival. The proteomic analysis highlights the lack of inflammatory mediators in BAY121-treated samples (still present in the T-Veh samples) and the downregulation of two clusters of proteins involved in proteasome regulation, Golgi function, ribosome biosynthesis and a third cluster of proteases and protease inhibitors. The reduced microglial reactivity (reduced DAM-like phenotype and decreased CD68 expression) may contribute to the preservation of synaptic integrity, since microglia mediates synaptic elimination in several disease settings (47, 48); the downregulation in multiple proteases and protease inhibitors enriched in phagocytes, such as Cathepsins and Serpins, is compatible with this model. In addition, the downregulation of proteasome-regulators and in trafficking proteins may point toward a more limited synaptic de-stabilization and reduced degradation of synaptic components [many of which, are proteasome-dependent: (49–51)]. It must be stressed that FGF/FGFR themselves are regulator of synaptogenesis and synaptic stability (52, 53) and, although we observed a positive impact on synaptic preservation, this outcome may be the net consequence of effects on microglial as well as on other cells types.
A few limitations of the present work are worth addressing. BAY121 is a small-molecule pan FGFR inhibitor; however, the possibility of small-molecule tyrosine kinase inhibitors having additional targets (26, 54), possibly contributing to their biological effect, cannot be fully discounted; to date, our array RTK screening does not demonstrate substantial inhibition besides the FGFR family. In particular, BAY121 (Rogaratinib) is reported to have IC50 in the low-nanomolar range for FGFRs (26) but an IC50 in the sub-micromolar range for CSF1R. Although the IC50 is more than 100-fold larger for CSF1R than for FGFRs, given the relevance of CSF1R in microglia physiology and pathophysiology, the relative contribution of this low-affinity target to the overall Rogaratib efficacy remains to be fully elucidated.
Furthermore, the effect of systemic administration of a pan-FGFR inhibitor may be not restricted only to microglia but may involve additional players in the CNS [e.g., neurons, oligodendrocytes, immune cells; (33, 53, 55)] although at least in the acute phase, phosphorylation of FGFR1 and 3 is largely restricted to Iba1+ cells. Finally, FGFR are endowed with highly pleiotropic functions in synaptic stability (53), axonal guidance (56) and myelination (57) and the full spectrum of the beneficial and detrimental consequences of FGFR inhibition in acute TBI are not fully elucidated.
Conclusion
Our findings provide a proof-of-concept of the translational value of targeting the FGFR in acute TBI for the modulation of early microglial reactivity and enhanced preservation of neuronal and synaptic integrity. Recently, rogaratinib-HCl entered clinical trials (58, 59) and three FGFR inhibitors (pemigatinib, futibatinib, and infigratinib) have been approved for human use in the therapy of several gastrointestinal, urologic or haematopoietic neoplasms (25, 60). Although their chronic administration is not devoid of side effects (60), acute or short-term administration may maximize their immunomodulatory and anti-inflammatory effects without interfering with tissue regeneration. In this context, one may envision the administration of FGFR inhibitors in patients with severe neurotrauma and evidence of intense neuroinflammatory responses or synaptic damage (e.g., using synaptic damage biomarkers; 61), in order to limit mcroglia-driven damage. Our findings support the investigation of the repurposing of FGFR inhibitors in this direction. Preliminary to in-human applications, the long-term impact of acute or subacute FGFR inhibitors should be studied using behavioural readouts (e.g., motor and cognitive readouts), together with their impact on astrocyte prolieration, scar formation and oligodendrocyte survival. Given the multiple cellular subpopulations affected by FGFR in the brain, the use of a panel of peripheral biomarkers may contribute to disentangle the protective effects of FGFR inhibitors.
Materials and methods
Animals
All experimental procedures were performed in compliance with animal protocols approved by the local veterinary and animal experimentation committee at University Ulm and by the Regierungspraesidium Tubingen under the license no. 1370. B6SJL male mice aged between p60–p90 days were used throughout the study.
Pharmacological treatment
Rogaratinib (BAY 1213802) was obtained by Bayer Pharma and was administered by oral gavage (200µl) dissolved into the following vehicle: 10% ethanol, 40% Solutol® HS 15, 50% water (26); vehicle alone was administered as control. The dose of 25mg/kg (26) was used throughout this study.
Traumatic brain injury procedure
Modified closed, blunt weight drop model Traumatic Brain Injury (TBI) was performed as previously reported (19). For all procedures, mice were anesthetized with sevoflurane (2-4% in 96% O2) and were subcutaneously injected with buprenorphine (0.1mg/kg; 1 dose/day) as a pre- and postoperative analgesic. The scalp was shaved and eye ointment was applied preoperatively to protect the cornea. Scalp skin was then incised on the midline to expose the skull and the animals were positioned in the weight-drop apparatus in which the head was secured to a holding frame. Using the 3-axis mobile platform in the apparatus, the impactor was positioned to the coordinates of the injection site (From bregma ≈ x = +3.0mm, y = − 2.0mm, z = 0.0mm). TBI was delivered by dropping a weight of 120g from a height of 45 cm. A mechanical stop prevented a skull displacement (by the impactor) larger than 2.5 mm, in order to keep the brain damage comparable. Apnea time was monitored after injury. The Neurological Severity Score (NSS) was assessed after 3h, 1 dpi and at 7 dpi and never exceeded the score 1 for any mouse. As such, no animal met the criteria for early sacrifice. Mice were checked every 2 hours on the day of trauma. Effort was made to minimize the suffering of animals and reduce the number of animals used. The contralateral hemisphere was used as control samples throughout the study.
Neurological severity score measurement
Throughout all animal experiments the NSS (62) was measured at 3h, 1h and 7d, depending on sacrificial time of the individual mouse. The NSS is comprised of a total of 10 individual tasks mice were subjugated to each timepoint the NSS was measured with a 10 to 30 seconds break inbetween each individual test. The tests include an arena escape within 3min, mono-/hemiparesis, straight walking, search behaviour, startle reflex, balancing on a) a 7mm wide angular beam and b) a 5mm wide round beam and finally a beam walk test with a lenght of 30cm and a with of a) 3cm, b) 2cm or c) 1cm. Points were awarded when mice could not fulfill an individual task, which then were summed up into the total NSS score. The total NSS score for all animals sacrificed as part of the publication are reported in Supplementary Table S1.
Immunohistochemistry
Brain samples were processed as previously described (7, 19). Briefly, mice were sacrificed by trans-cardial perfusion with 4% PFA in PBS, and brains were dissected and postfixed in 4% PFA overnight. Brains were then transferred to 30% Sucrose for 2 days, after which the samples were embedded in OCT (Tissue Tek, Sakura, Germany). 40-micron sections were cut with a cryostat (Leica CM 1950 AG Protect cryostat). Sections spanning the injury site were selected and blocked (3% BSA, 0.3% Triton in 1x PBS) for 2h at room temperature, followed by incubation for 48h (Iba1, NeuN, VGAT, VGLUT1) or 72h (Iba1, phosphoFGFR1, phosphoFGFR3), at 4°C with primary antibodies diluted in blocking buffer. Identifiers of the antibody used and dilutions are reported in Supplementary Table S2. Sections were washed 3x 30 min with PBS and incubated for 2h at RT with secondary antibodies diluted in blocking buffer. The sections were washed with PBS and mounted using Prolong Gold Antifade Mounting Medium (Invitrogen, Germany). A list of the antibodies used in this study can be found in Supplementary Table S2.
Image acquisition and analysis
Confocal images were acquired in 1024 x 1024 pixel and 12-bit format, with a Leica DMi8 inverted microscope, equipped with an ACS APO 40x oil objective. Parameters were set to obtain the optimal signals from the stained antibody or mRNA and at the same time avoiding saturation. All fluorescent channels were acquired independently, to avoid cross-bleed. 3-4 sections spanning the core and perilesional area of the impact site were imaged of each mouse. For each image a tile scan was set up consisting of x by x tiles spanning the injury location.
For image analysis, stacks were collapsed in maximum intensity projection pictures and mean gray value or cell density per fixed region of interest (ROI) was measured. For quantification, we considered a 200µm x 200µm ROI centered on the axis of the injury site.
Synaptic density was detected after producing a mosaic image corresponding to 6 x 6 single optical sections (acquired with a 63x oil objective) with 1 μm optical section thickness. Each cortical section was imaged at a fixed depth 5-10µm inside the section, the composite image was positioned so that an uninterrupted coverage of the impact site with perilesional area was acquired. Quantification of density of synapses was performed using the IMARIS software (Bitplane AG, Zurich, CH) as previously described (63). For the quantification of the histological parameters, we considered a 450µm x 450µm (microglial imaging) or 200 µm × 200 µm (synapses count) ROI centered on the axis of the injury site (“core”) into the cerebral cortex (layer II/III). To measure the intensity of immunolabeling in microglia, we first subtracted the background intensity (Rolling Ball algorithm, ball diameter = 30), then individual cells were traced using the Iba1 immunolabeling as mask and the intensity of individual cell was logged. To determine the percentage of positive cells, for FGFR1 we considered all cells with average intensity higher than 400 arbitrary units (after background subtraction) and for FGFR3 we considered all cells with intensity higher than 100 arbitrary units (after background subtraction) counted every mean intensity above 400 (after subtracting the background mean value) as a positive. The number of positive cells was then divided by the number of total Iba1+ cells in the ROI under consideration.
Phospho RTK array processing
Proteome Profiler Mouse Phospho-RTK Array Kit (R&D Systems, Minneapolis) was used to determine the Phospho RTK activation pattern. The nitrocellulose membrane arrays provided in the kit were based on sandwich immunoassay and processed according to manufacturer’s instructions. Briefly, membranes spotted with the anti-RTK antibody were blocked in Array buffer 1 for 1h at RT. 160 ug of protein extracted from cortical samples was diluted in 1.5mL Array buffer 1 overnight at 4°C. After washing, arrays were incubated for 2 hours at RT with Anti-Phospho-Tyrosine-HRP Detection Antibody, diluted to 1:5000 in 1X Array Buffer 2. After final washing steps, HRP detection was performed by adding 1 ml Clarity Max™ Western ECL Blotting Substrates from Bio-Rad. Arrays were imaged using BioRad X-ray imager and quantified using ImageJ. ROI was drawn on each antibody spot with a constant diameter and mean gray value was recorded. Further analysis was performed using R software.
Proteome array processing
200 ug of protein extracted from cortical samples was loaded to the arrays. The arrays were processed according to the manufacturer’s instructions. Briefly, the sample was biotinylated and was mixed with reaction stop reagent and incubated for 30 mins at RT. The glass arrays were incubated in a blocking solution for 45 minutes and washed. After adding a biotinylated sample to the coupling solution, arrays were incubated for 2 hours at RT and washed. The arrays were incubated with a detection buffer with Cy5-streptavidin (ThermoFisher) at 1:1000 for 20 minutes at RT. The washing steps were repeated as described in the manufacturer’s instructions. After removing excess ddH20 from the slides, the arrays were dried. The arrays were imaged using a GenePix 4000B array scanner (Molecular Devices, LLC) and the image analysis was performed using GenePix Pro Software v7 (Molecular Devices, LLC). The settings for the analysis were kept constant in all cases. The GAL file was loaded in the software and the ROIs were adjusted on the protein spots. Each intensity on F635 was recorded and GPR files were saved. Further analysis was performed using R software.
Protein-protein interaction network and hub genes
We constructed PPI networks to analyze the functional interactions among differentially expressed proteins, using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING: http://www.stringdb.org) (64) and visualized the networks using Cytoscape (https://cytoscape.org/) (65).
Array bioinformatic analysis
Chemiluminescence signal for each spot was logged after microarray image analysis. The raw intensity values for each receptor/protein were recorded automatedly via image recorder software. The raw data files were loaded in R software and the dataset for each array was preliminary subjected to quality control assessment (QCA), outlier identification, data distribution, intra-array and inter-array normalization. Normalized data for each array was subjected to principal component analysis (PCA) to display group-based clustering. Confidence ellipses (assuming multivariate normal distribution) with the first two principal components were plotted to validate further analysis. Modified linear modeling-based analysis was then applied to the data to identify significant increase or decrease in phosphorylation or protein level. For protein array analyses, the code has been made publicly available on open-access GitHub repository PROTEAS (19).
Statistics
Statistical analysis was performed with the GraphPad Prism software suite. Normality was routinely checked using the Shapiro-Wilk test and equal variance was tested using the Brown-Forsythe test. Two-way ANOVA with Tukey correction was used for four group comparisons to examine statistical significance. Protein array analysis was performed using R software using the PROTEAS algorythm (previously published; 19), available on GitHub (https://github.com/Rida-Rehman/PROTEAS), with FDR correction at 0.05. Error bars represent standard deviation (SD), unless indicated otherwise. Statistical significance was set at P < 0.05.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by Animal Experimentation Commitee Regierungspraesidium-Tubingen. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
RR: Investigation, Writing – original draft. AF: Investigation, Writing – review & editing. Fo: Project administration, Resources, Writing – review & editing. LE: Investigation, Writing – review & editing. TB: Funding acquisition, Methodology, Resources, Writing – review & editing. MH-L: Conceptualization, Funding acquisition, Methodology, Resources, Writing – review & editing. CM-K: Conceptualization, Supervision, Writing – review & editing. FR: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The present work has been supported by the “Grant 4 Indication” program of Bayer Pharma. FR and RR are members of the MICRONET consortium funded by ERANET-NEURON (through BMBF: grant no. FKZ 01EW1705A). RR was also independently funded by the Hannelore Kohl Foundation Award. FR is also funded by DFG in the context of the SFB1149 (DFG No. 251293561) as well as through individual grants (DFG No. 443642953, 431995586 and 446067541).
Acknowledgments
RR and AF wish to thank the Hannelore Kohl Foundation for its support. All authors wish to thank Thomas Lenk and Gizem Yartas for their dedicated technical support and dr. Diana Wiesner and dr. Jelena Scekic-Zahirovic for discussions and technical suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1443940/full#supplementary-material
Supplementary Figure 1 | Comparison of TBI- Veh and TBI- BAY121 at 3d post injury. (A) Protein-protein interaction (PPI) of significantly upregulated (red) and down regulated (blue) proteins. Color distribution is based on fold change (FC). (B, C) Gene ontology (GO) analysis and for (B) down regulated proteins and (C) upregulated proteins.
Supplementary Figure 2 | Comparison of TBI- Veh and TBI- BAY121 at 7d post injury. (A) Protein-protein interaction (PPI) of significantly upregulated (red) and down regulated (blue) proteins. Color distribution is based on fold change (FC). (B) Gene ontology analysis for down regulated proteins.
Supplementary Table 1 | NSS scoring. This Excel file reports all the individual NSS scores calculated for each mouse sacrificed as part of this publication.
References
1. Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM. The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol. (2019) 137:731–55. doi: 10.1007/s00401-018-1944-6
2. Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron. (2017) 95:1246–65. doi: 10.1016/j.neuron.2017.07.010
3. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
4. Zanier ER, Fumagalli S, Perego C, Pischiutta F, De Simoni MG. Shape descriptors of the "never resting" microglia in three different acute brain injury models in mice. Intensive Care Med Exp. (2015) 3:39. doi: 10.1186/s40635-015-0039-0
5. Somebang K, Rudolph J, Imhof I, Li L, Niemi EC, Shigenaga J, et al. CCR2 deficiency alters activation of microglia subsets in traumatic brain injury. Cell Rep. (2021) 36:109727. doi: 10.1016/j.celrep.2021.109727
6. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–581.e9. doi: 10.1016/j.immuni.2017.08.008
7. Fröhlich A, Olde Heuvel F, Rehman R, Krishnamurthy SS, Li S, Li Z, et al. Neuronal nuclear calcium signaling suppression of microglial reactivity is mediated by osteoprotegerin after traumatic brain injury. J Neuroinflammation. (2022) 19:279. doi: 10.1186/s12974-022-02634-4
8. Ritzel RM, He J, Li Y, Cao T, Khan N, Shim B, et al. Proton extrusion during oxidative burst in microglia exacerbates pathological acidosis following traumatic brain injury. Glia. (2021) 69:746–64. doi: 10.1002/glia.23926
9. Wang J, Ma MW, Dhandapani KM, Brann DW. Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic Biol Med. (2017) 113:119–31. doi: 10.1016/j.freeradbiomed.2017.09.017
10. Jamjoom AAB, Rhodes J, Andrews PJD, Grant SGN. The synapse in traumatic brain injury. Brain. (2021) 144:18–31. doi: 10.1093/brain/awaa321
11. Wang Y, Wernersbach I, Strehle J, Li S, Appel D, Klein M, et al. Early posttraumatic CSF1R inhibition via PLX3397 leads to time- and sex-dependent effects on inflammation and neuronal maintenance after traumatic brain injury in mice. Brain Behav Immun. (2022) 106:49–66. doi: 10.1016/j.bbi.2022.07.164
12. Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. (2014) 505:223–8. doi: 10.1038/nature12808
13. Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. (2016) 353:783–5. doi: 10.1126/science.aaf6260
14. Loane DJ, Kumar A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol. (2016) 275 Pt 3:316–27. doi: 10.1016/j.expneurol.2015.08.018
15. Willis EF, MacDonald KPA, Nguyen QH, Garrido AL, Gillespie ER, Harley SBR, et al. Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell. (2020) 180:833–846.e16. doi: 10.1016/j.cell.2020.02.013
16. Ritzel RM, Li Y, Jiao Y, Lei Z, Doran SJ, He J, et al. Brain injury accelerates the onset of a reversible age-related microglial phenotype associated with inflammatory neurodegeneration. Sci Adv. (2023) 9:eadd1101. doi: 10.1126/sciadv.add1101
17. Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2023 update. Pharmacol Res. (2023) 187:106552. doi: 10.1016/j.phrs.2022.106552
18. Pottier C, Fresnais M, Gilon M, Jérusalem G, Longuespée R, Sounni NE. Tyrosine kinase inhibitors in cancer: breakthrough and challenges of targeted therapy. Cancers (Basel). (2020) 12:731. doi: 10.3390/cancers12030731
19. Rehman R, Miller M, Krishnamurthy SS, Kjell J, Elsayed L, Hauck SM, et al. Met/HGFR triggers detrimental reactive microglia in TBI. Cell Rep. (2022) 41:111867. doi: 10.1016/j.celrep.2022.111867
20. Huang Y, Wang F, Li H, Xu S, Xu W, Pan X, et al. Inhibition of fibroblast growth factor receptor by AZD4547 protects against inflammation in septic mice. Inflammation. (2019) 42:1957–67. doi: 10.1007/s10753-019-01056-4
21. Han X, Quarles LD. Multiple faces of fibroblast growth factor-23. Curr Opin Nephrol Hypertens. (2016) 25:333–42. doi: 10.1097/MNH.0000000000000240
22. Rajendran R, Rajendran V, Böttiger G, Stadelmann C, Shirvanchi K, von Au L, et al. The small molecule fibroblast growth factor receptor inhibitor infigratinib exerts anti-inflammatory effects and remyelination in a model of multiple sclerosis. Br J Pharmacol. (2023) 180(23):2989-3007. doi: 10.1111/bph.16186
23. Parthasarathy G. Fibroblast growth factor receptor inhibitors mitigate the neuropathogenicity of Borrelia burgdorferi or its remnants ex vivo. Front Immunol. (2024) 15:1327416. doi: 10.3389/fimmu.2024.1327416
24. Zhou J, Li C, Lu M, Jiang G, Chen S, Li H, et al. Pharmacological induction of autophagy reduces inflammation in macrophages by degrading immunoproteasome subunits. PloS Biol. (2024) 22:e3002537. doi: 10.1371/journal.pbio.3002537
25. Weaver A, Bossaer JB. Fibroblast growth factor receptor (FGFR) inhibitors: A review of a novel therapeutic class. J Oncol Pharm Pract. (2021) 27:702–10. doi: 10.1177/1078155220983425
26. Grünewald S, Politz O, Bender S, Héroult M, Lustig K, Thuss U, et al. Rogaratinib: A potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int J Cancer. (2019) 145:1346–57. doi: 10.1002/ijc.32224
27. Chandrasekar A, Olde Heuvel F, Wepler M, Rehman R, Palmer A, Catanese A, et al. The neuroprotective effect of ethanol intoxication in traumatic brain injury is associated with the suppression of erbB signaling in parvalbumin-positive interneurons. J Neurotrauma. (2018) 35:2718–35. doi: 10.1089/neu.2017.5270
28. Ju S, Xu C, Wang G, Zhang L. VEGF-C induces alternative activation of microglia to promote recovery from traumatic brain injury. J Alzheimers Dis. (2019) 68:1687–97. doi: 10.3233/JAD-190063
29. Wu H, Zheng J, Xu S, Fang Y, Wu Y, Zeng J, et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflammation. (2021) 18:2. doi: 10.1186/s12974-020-02041-7
30. Zhou S, Li Y, Zhang Z, Yuan Y. An insight into the TAM system in Alzheimer's disease. Int Immunopharmacol. (2023) 116:109791. doi: 10.1016/j.intimp.2023.109791
31. Bernard C, Exposito-Alonso D, Selten M, Sanalidou S, Hanusz-Godoy A, Aguilera A, et al. Cortical wiring by synapse type-specific control of local protein synthesis. Science. (2022) 378:eabm7466. doi: 10.1126/science.abm7466
32. Luo B, Liu Z, Lin D, Chen W, Ren D, Yu Z, et al. ErbB4 promotes inhibitory synapse formation by cell adhesion, independent of its kinase activity. Transl Psychiatry. (2021) 11:361. doi: 10.1038/s41398-021-01485-6
33. Klimaschewski L, Claus P. Fibroblast growth factor signalling in the diseased nervous system. Mol Neurobiol. (2021) 58:3884–902. doi: 10.1007/s12035-021-02367-0
34. Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao-Inoue H, et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell. (2020) 182:388–403.e15. doi: 10.1016/j.cell.2020.05.050
35. Huang Y, Happonen KE, Burrola PG, O'Connor C, Hah N, Huang L, et al. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat Immunol. (2021) 22:586–94. doi: 10.1038/s41590-021-00913-5
36. Fourgeaud L, Través PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, et al. TAM receptors regulate multiple features of microglial physiology. Nature. (2016) 532:240–4. doi: 10.1038/nature17630
37. Vinet J, Vainchtein ID, Spano C, Giordano C, Bordini D, Curia G, et al. Microglia are less pro-inflammatory than myeloid infiltrates in the hippocampus of mice exposed to status epilepticus. Glia. (2016) 64:1350–62. doi: 10.1002/glia.23008
38. Haddad LE, Khzam LB, Hajjar F, Merhi Y, Sirois MG. Characterization of FGF receptor expression in human neutrophils and their contribution to chemotaxis. Am J Physiol Cell Physiol. (2011) 301:C1036–45. doi: 10.1152/ajpcell.00215.2011
39. Byrd VM, Kilkenny DM, Dikov MM, Reich MB, Rocheleau JV, Armistead WJ, et al. Fibroblast growth factor receptor-1 interacts with the T-cell receptor signalling pathway. Immunol Cell Biol. (2003) 81:440–50. doi: 10.1046/j.1440-1711.2003.01199.x
40. Farahnak S, McGovern TK, Kim R, O'Sullivan M, Chen B, Lee M, et al. Basic fibroblast growth factor 2 is a determinant of CD4 T cell-airway smooth muscle cell communication through membrane conduits. J Immunol. (2017) 199:3086–93. doi: 10.4049/jimmunol.1700164
41. Rossini M, Cheunsuchon B, Donnert E, Ma LJ, Thomas JW, Neilson EG, et al. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1), and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. (2005) 68(6):2621–8. doi: 10.1111/j.1523-1755.2005.00734
42. Parthasarathy G, Pattison MB, Midkiff CC. The FGF/FGFR system in the microglial neuroinflammation with Borrelia burgdorferi: likely intersectionality with other neurological conditions. J Neuroinflammation. (2023) 20:10. doi: 10.1186/s12974-022-02681-x
43. Rajendran R, Giraldo-Velásquez M, Stadelmann C, Berghoff M. Oligodendroglial fibroblast growth factor receptor 1 gene targeting protects mice from experimental autoimmune encephalomyelitis through ERK/AKT phosphorylation. Brain Pathol. (2018) 28:212–24. doi: 10.1111/bpa.12487
44. Kamali S, Rajendran R, Stadelmann C, Karnati S, Rajendran V, Giraldo-Velasquez M, et al. Oligodendrocyte-specific deletion of FGFR2 ameliorates MOG35-55 -induced EAE through ERK and Akt signalling. Brain Pathol. (2021) 31:297–311. doi: 10.1111/bpa.12916
45. Wang D, Liu F, Zhu L, Lin P, Han F, Wang X, et al. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J Neuroinflammation. (2020) 17:257. doi: 10.1186/s12974-020-01921-2
46. Chen J, Wang X, Hu J, Du J, Dordoe C, Zhou Q, et al. FGF20 protected against BBB disruption after traumatic brain injury by upregulating junction protein expression and inhibiting the inflammatory response. Front Pharmacol. (2021) 11:590669. doi: 10.3389/fphar.2020.590669
47. Cangalaya C, Wegmann S, Sun W, Diez L, Gottfried A, Richter K, et al. Real-time mechanisms of exacerbated synaptic remodeling by microglia in acute models of systemic inflammation and tauopathy. Brain Behav Immun. (2023) 110:245–59. doi: 10.1016/j.bbi.2023.02.023
48. Tzioras M, Daniels MJD, Davies C, Baxter P, King D, McKay S, et al. Human astrocytes and microglia show augmented ingestion of synapses in Alzheimer's disease via MFG-E8. Cell Rep Med. (2023) 4:101175. doi: 10.1016/j.xcrm.2023.101175
49. Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. (2003) 6:231–42. doi: 10.1038/nn1013
50. Kerrisk Campbell M, Sheng M. USP8 deubiquitinates SHANK3 to control synapse density and SHANK3 activity-dependent protein levels. J Neurosci. (2018) 38:5289–301. doi: 10.1523/JNEUROSCI.3305-17.2018
51. Roselli F, Livrea P, Almeida OF. CDK5 is essential for soluble amyloid β-induced degradation of GKAP and remodeling of the synaptic actin cytoskeleton. PloS One. (2011) 6:e23097. doi: 10.1371/journal.pone.0023097
52. Li AJ, Suzuki S, Suzuki M, Mizukoshi E, Imamura T. Fibroblast growth factor-2 increases functional excitatory synapses on hippocampal neurons. Eur J Neurosci. (2002) 16:1313–24. doi: 10.1046/j.1460-9568.2002.02193.x
53. Dabrowski A, Terauchi A, Strong C, Umemori H. Distinct sets of FGF receptors sculpt excitatory and inhibitory synaptogenesis. Development. (2015) 142:1818–30. doi: 10.1242/dev.115568
54. Reinecke M, Brear P, Vornholz L, Berger BT, Seefried F, Wilhelm S, et al. Chemical proteomics reveals the target landscape of 1,000 kinase inhibitors. Nat Chem Biol. (2023) 20(5):577-85. doi: 10.1038/s41589-023-01459-3
55. Rajendran R, Böttiger G, Stadelmann C, Karnati S, Berghoff M. FGF/FGFR pathways in multiple sclerosis and in its disease models. Cells. (2021) 10:884. doi: 10.3390/cells10040884
56. Yang JJ, Bertolesi GE, Hehr CL, Johnston J, McFarlane S. Fibroblast growth factor receptor 1 signaling transcriptionally regulates the axon guidance cue slit1. Cell Mol Life Sci. (2018) 75:3649–61. doi: 10.1007/s00018-018-2824-x
57. Furusho M, Roulois AJ, Franklin RJ, Bansal R. Fibroblast growth factor signaling in oligodendrocyte-lineage cells facilitates recovery of chronically demyelinated lesions but is redundant in acute lesions. Glia. (2015) 63:1714–28. doi: 10.1002/glia.22838
58. Sternberg CN, Petrylak DP, Bellmunt J, Nishiyama H, Necchi A, Gurney H, et al. FORT-1: phase II/III study of rogaratinib versus chemotherapy in patients with locally advanced or metastatic urothelial carcinoma selected based on FGFR1/3 mRNA expression. J Clin Oncol. (2023) 41:629–39. doi: 10.1200/JCO.21.02303
59. Addeo A, Rothschild SI, Holer L, Schneider M, Waibel C, Haefliger S, et al. Fibroblast growth factor receptor (FGFR) inhibitor rogaratinib in patients with advanced pretreated squamous-cell non-small cell lung cancer over-expressing FGFR mRNA: The SAKK 19/18 phase II study. Lung Cancer. (2022) 172:154–9. doi: 10.1016/j.lungcan.2022.08.016
60. Subbiah V, Verstovsek S. Clinical development and management of adverse events associated with FGFR inhibitors. Cell Rep Med. (2023) 4:101204. doi: 10.1016/j.xcrm.2023.101204
61. Olde Heuvel F, Li Z, Riedel D, Halbgebauer S, Oeckl P, Mayer B, et al. Dynamics of synaptic damage in severe traumatic brain injury revealed by cerebrospinal fluid SNAP-25 and VILIP-1. J Neurol Neurosurg Psychiatry. (2024), 2:jnnp–2024-333413. doi: 10.1136/jnnp-2024-333413
62. Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. (2009) 4(9):1328–37. doi: 10.1038/nprot.2009.148
63. Olde Heuvel F, Holl S, Chandrasekar A, Li Z, Wang Y, Rehman R, et al. STAT6 mediates the effect of ethanol on neuroinflammatory response in TBI. Brain Behav Immun. (2019) 81:228–46. doi: 10.1016/j.bbi.2019.06.019
64. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. (2013) 41(Database issue):D808–15. doi: 10.1093/nar/gks1094
Keywords: reactive microglia, traumatic brain injury, receptor tyrosine kinase, proteomics, synapses
Citation: Rehman R, Froehlich A, olde Heuvel F, Elsayed L, Boeckers T, Huber-Lang M, Morganti-Kossmann C and Roselli F (2024) The FGFR inhibitor Rogaratinib reduces microglia reactivity and synaptic loss in TBI. Front. Immunol. 15:1443940. doi: 10.3389/fimmu.2024.1443940
Received: 04 June 2024; Accepted: 30 October 2024;
Published: 20 November 2024.
Edited by:
Tiziano Balzano, Centro Integral en Neurociencias A.C. HM CINAC, SpainReviewed by:
Hailong Song, University of Pennsylvania, United StatesWilma Friedman, Rutgers Univeristy, United States
Copyright © 2024 Rehman, Froehlich, olde Heuvel, Elsayed, Boeckers, Huber-Lang, Morganti-Kossmann and Roselli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Roselli, ZnJhbmNlc2NvLnJvc2VsbGlAdW5pLXVsbS5kZQ==
†ORCID: Francesco Roselli, orcid.org/0000-0001-9935-6899