 Yaqi Yang
Yaqi Yang- 1College of Chinese Medicine, Changchun University of Chinese Medicine, Changchun, Jilin, China
- 2Naniing Tongren Hospital, School of Medicine, Southeast University, Nanjing, China
- 3Ophthalmology Department, Affiliated Hospital of Changchun University of Traditional Chinese Medicine, Changchun, Jilin, China
Ferroptosis, a new type of programmed cell death proposed in recent years, is characterized mainly by reactive oxygen species and iron-mediated lipid peroxidation and differs from programmed cell death, such as apoptosis, necrosis, and autophagy. Ferroptosis is associated with a variety of physiological and pathophysiological processes. Recent studies have shown that ferroptosis can aggravate or reduce the occurrence and development of diseases by targeting metabolic pathways and signaling pathways in tumors, ischemic organ damage, and other degenerative diseases related to lipid peroxidation. Increasing evidence suggests that ferroptosis is closely linked to the onset and progression of various ophthalmic conditions, including corneal injury, glaucoma, age-related macular degeneration, diabetic retinopathy, retinal detachment, and retinoblastoma. Our review of the current research on ferroptosis in ophthalmic diseases reveals significant advancements in our understanding of the pathogenesis, aetiology, and treatment of these conditions.
1 Introduction
The pathology of ophthalmic diseases involves various mechanisms of cell death, including apoptosis, necrosis, and autophagy (1, 2). It is specifically regulated by iron levels and the antioxidant enzyme glutathione peroxidase 4 (GPX4), unlike apoptosis’s caspase-dependent mechanism, necrosis’s inflammatory cell rupture, and autophagy’s dual role in both cell survival and death (3). Each form of regulated cell death (RCD) exhibits distinct morphological features and is controlled by unique biochemical, genetic, and functional processes. Studies have shown that pharmaceutical agents modulate apoptosis, necrosis, and autophagy within ocular cells by targeting pathways such as inflammation, oxidative stress, and endoplasmic reticulum stress, leading to positive therapeutic effects (4).
The key membrane protein xCT related to ferroptosis was discovered as early as the 1980s (5). Subsequently, over the course of more than 20 years, the classic inducers of ferroptosis, erastin, which can kill RAS-mutated BeJLR cells, were also discovered in 2003 (6). However, it was not until 2012 that researchers such as Dixon defined this type of cell death as ferroptosis. Dixon and colleagues identified a form of programmed cell death (PCD) reliant on iron metabolism, particularly in cancer cells (7). The term “ferroptosis” was coined to describe a new form of non-apoptotic cell death activated by the small molecule erastin, which was found to induce cell death through iron-dependent lipid peroxidation.
This phenomenon is observed not only in mammals but also in plants, protozoa, and fungi, including Magnaporthe oryzae and Caenorhabditis elegans (8, 9). Emerging evidence suggests that ferroptosis significantly regulates the progression of numerous systemic diseases (Figure 1) (10–17).
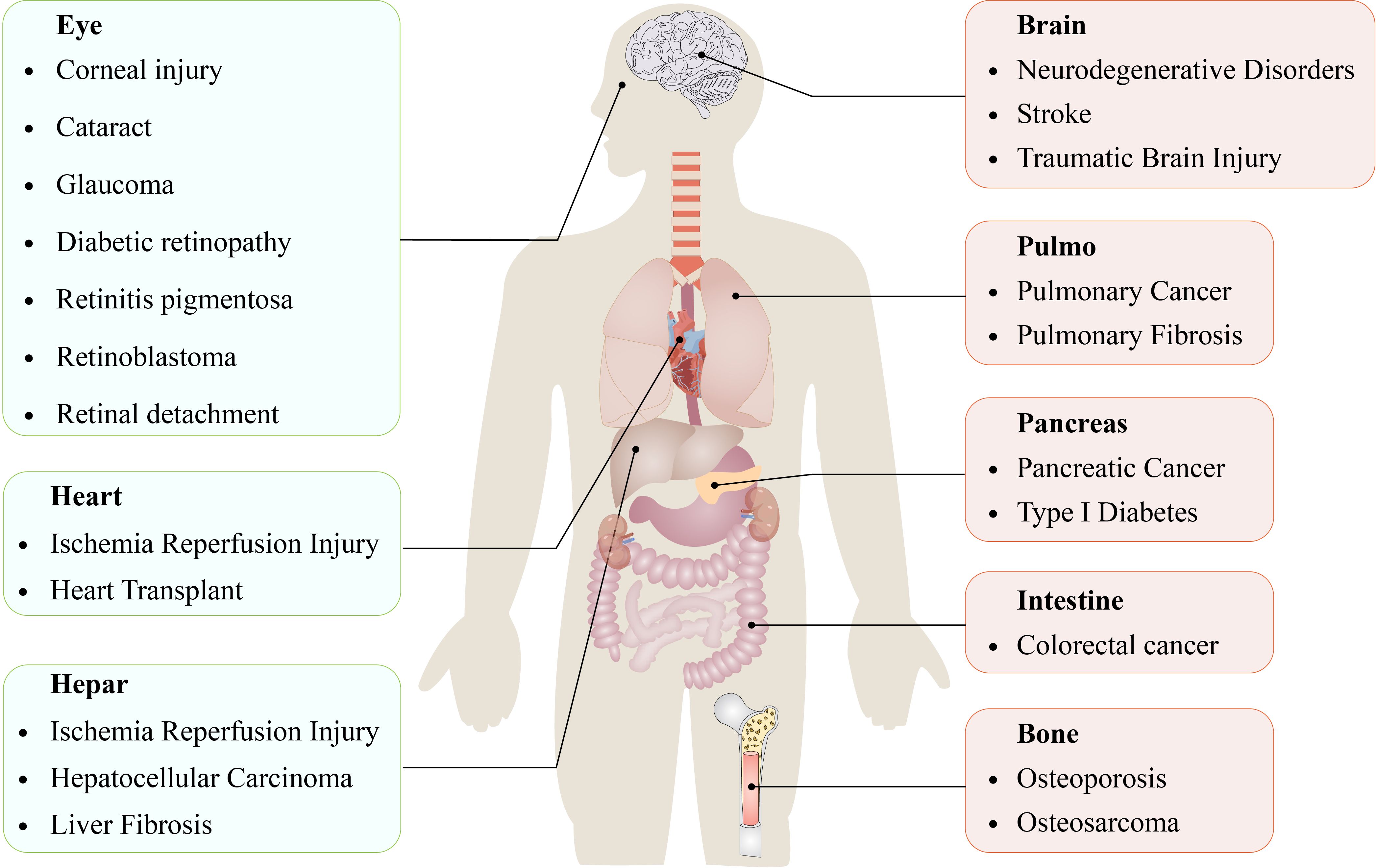
Figure 1 Ferroptosis is involved in the occurrence and development of various diseases in organs and tissues.
It is widely believed that ferroptosis primarily results from an overload of intracellular reactive oxygen species (ROS) dependent on iron, combined with a reduction in the detoxifying activity of GPX4, leading to an imbalance in ROS production and clearance (7). When the cell’s antioxidant defenses are overwhelmed and cannot neutralize the excess ROS, ferroptosis is triggered within the cell, with the subsequent peroxidation reactions being pivotal to its onset (18). Consequently, numerous molecules and signaling pathways involved in iron metabolism and peroxidation reactions are pivotal in the modulation of ferroptosis.
This review delves into the latest developments and future prospects in the research of ferroptosis in corneal injury, glaucoma, age-related macular degeneration, diabetic retinopathy, retinal detachment, and retinoblastoma. This study investigates the metabolic interplay between ferroptosis and ophthalmic pathology, aiming to establish a foundation for further exploration of the mechanisms and preventive strategies of ferroptosis in ophthalmic diseases.
2 Characteristics of ferroptosis
Iron ions play a pivotal role in metabolism in the ocular region, serving as essential micronutrients for a wide array of biological activities (19). They contribute significantly to the light conversion process in photoreceptor cells in the retina, participate in the electron transport chain within mitochondria, and are crucial for the activity of enzymes such as cytochrome oxidase (20). Excess iron ions produce ROS through the Fenton reaction, and iron overload and ROS accumulation are involved in the pathophysiological mechanisms of ferroptosis (21). Ferroptosis is a form of programmed cell death triggered by excessive iron accumulation and lipid peroxidation-induced damage. Signaling pathways and molecules involved in the regulatory mechanism of ferroptosis include iron metabolism, cysteine metabolism, GPX4 inactivation, polyunsaturated fatty acid (PUFA) synthesis, nuclear factor E2-related factor 2 (Nrf2), p53, heat shock proteins (HSPs), iron-regulated inhibitor-1 (FSP1), AMP-activated protein kinase (AMPK) activation, and nicotinamide adenine dinucleotide phosphate (NADPH), etc. We will classify and introduce ferroptosis-related molecules from signaling, execution, regulation and other functions.
2.1 Signaling
2.1.1 Iron metabolism
Iron metabolism is crucial in ferroptosis, influencing the accumulation of lipid peroxides (22). Iron uptake, iron transport across cell membranes, and iron storage profoundly influence the regulation of ferroptosis (23). Organisms carefully maintain a delicate balance to maintain iron homeostasis. The acquisition of iron through transferrin receptor 1 (TFR1) is crucial for ferroptosis; this receptor facilitates the transport of ferric ions (Fe3+) to endosomes, where they are converted to ferrous ions (Fe2+) (Figure 2). Divalent metal transporter 1 (DMT1) releases Fe2+ from endosomes into the labile intracellular iron pool. Excess iron is then sequestered within the cytoplasm in the form of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1) (24). Disruptions in the normal expression or function of these proteins related to iron metabolism can lead to an increase in intracellular iron levels, resulting from metabolic dysregulation (25). Under conditions of iron surplus, ferroportin 1 (FPN1) is notably expressed on the brush border membrane of renal proximal tubule cells, indicating its potential role in the excretion of excess iron into the urinary space (26). Hepcidin, which is predominantly produced in the liver, plays a crucial role in governing the release of iron by FPN1 in macrophages (27).
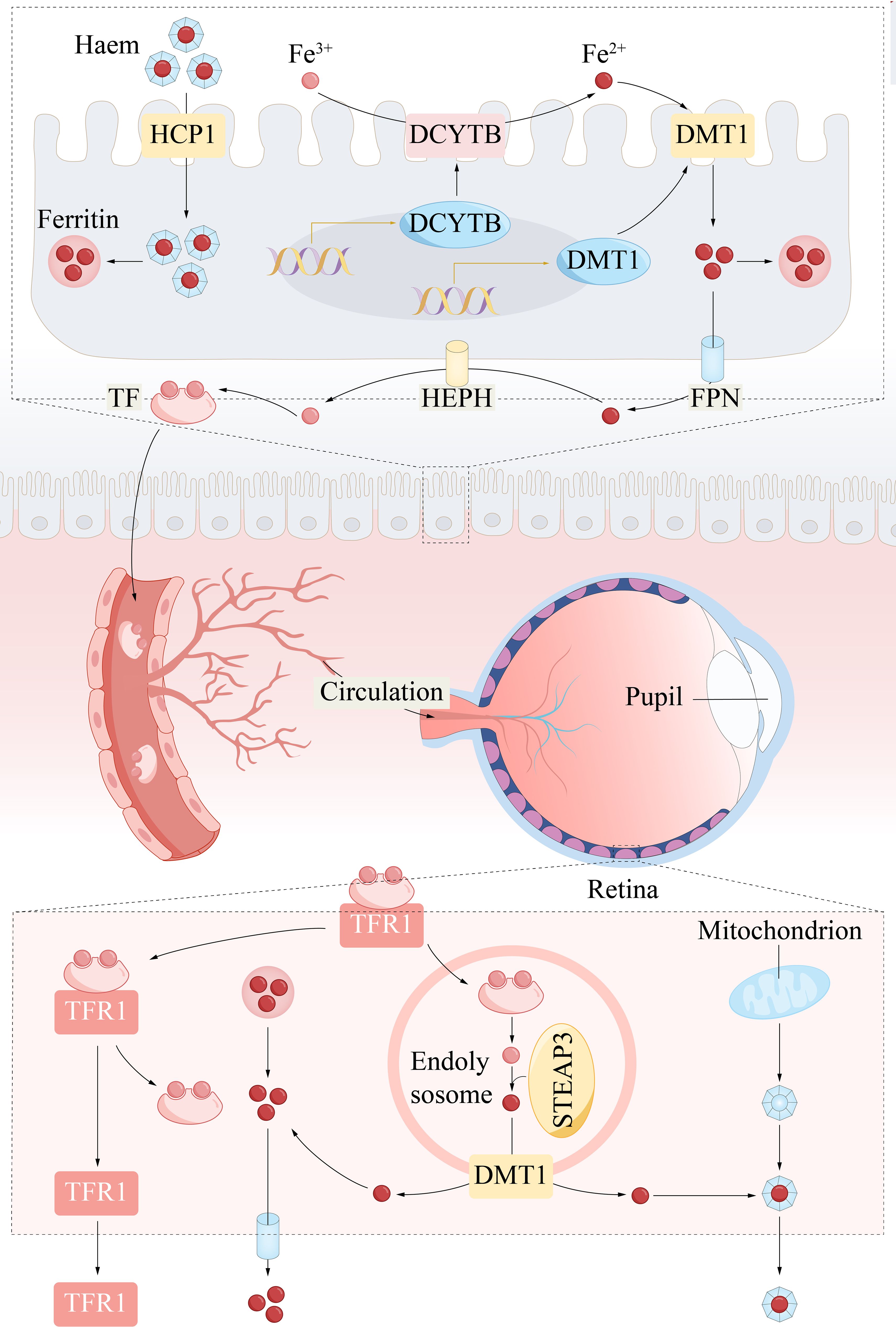
Figure 2 Systemic circulation of iron and iron metabolism. Iron ions are absorbed from the gut and reach the eyeball as blood circulates throughout the body. Specifically, Fe3+ is reduced to Fe2+ by DCYTB in the intestinal lumen. DMT1 then transports Fe2+ into enterocytes. HCP1 also incorporated heme iron into duodenal enterocytes. HIF2α (not shown, in the nucleus) binds to hypoxia response elements (HRE) in the regulatory regions of the DMT1 and DCYTB promoters and is therefore post-transcriptionally regulated. Intracellular iron storage is controlled by ferritin, whereas Fe2+ is exported into the blood via FPN and simultaneously oxidized to Fe3+ by hephaestin. Plasma transferrin captures and distributes Fe3+ to the retina. Cells take up iron via TFR1 -mediated endocytosis of holotransferrin. In retinal cells, DMT1 transports Fe2+ into the cytoplasm. Fe2+ can shuttle to mitochondria for heme biosynthesis. DCYTB, duodenal cytochrome B; DMT1, divalent metal transporter 1; FPN, ferroportin; HEPH, hephaestin; STEAP3, six transmembrane epithelial antigen of the prostate 3; TF, transferrin; TFR1, transferrin receptor 1.
Iron plays a crucial role in the visual process, particularly in phototransduction, where it acts as a cofactor for key enzymes involved in the regeneration of rhodopsin, converting light signals into neural signals. Additionally, iron is involved in maintaining the antioxidant balance of the retina, helping to eliminate free radicals and protect the retina from oxidative damage (28). However, disruptions in iron metabolism can lead to iron deposition and lipid peroxidation, which may have a negative impact in age-related macular degeneration (29). Therefore, the balance of iron ions is vital for retinal health, and excessive light exposure may cause iron metabolic disorders and harmful oxidative stress, damaging retinal cells.
2.2 Execution
2.2.1 GPX4 inactivation
The glutathione peroxidase (GPX) family comprises various isoforms, including GPX1 through GPX8, with GPX4 playing a particularly crucial role in the context of ferroptosis (Figure 3) (30). GPX4, a selenoprotein in mammals, is essential for the repair of lipid peroxidation within cells (31). It facilitates the conversion of the reduced form of glutathione (GSH) to its oxidized counterpart (GSSG) and transforms toxic lipid hydroperoxides (L-OOH) into nontoxic lipid alcohols (L-OH), thereby catalysing the breakdown of hydrogen peroxide (H2O2). This process is vital for safeguarding the cell membrane’s structure and function against oxidative stress and damage (32).
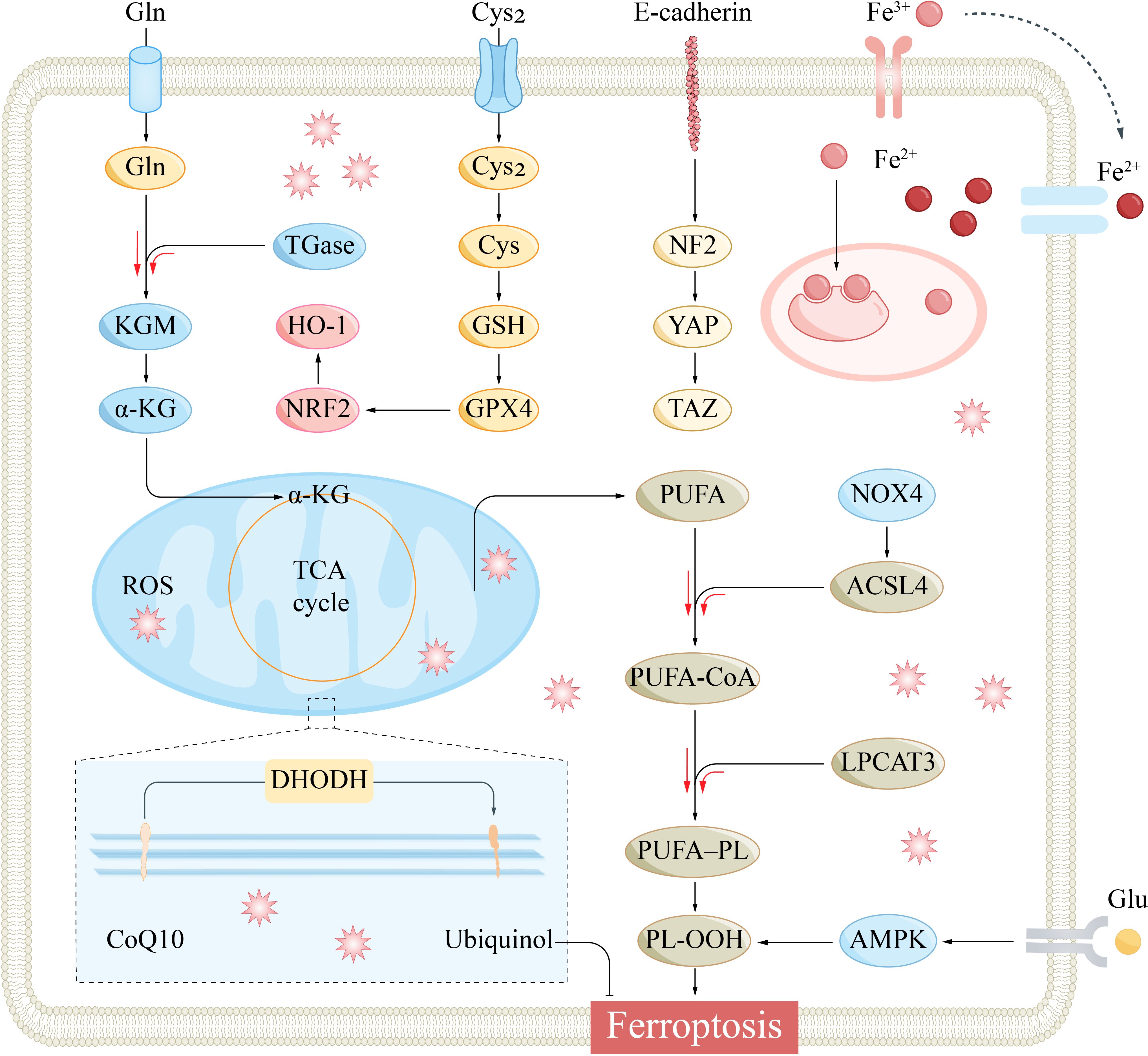
Figure 3 The main signaling pathways that regulate ferroptosis. Specifically, Cysteine metabolism, Iron metabolism, GPX4 inactivation, PUFA synthesis, Hippo pathway, AMPK, and NRF2 are involved in the regulatory mechanisms of ferroptosis. α-KG, α-ketoglutarate; ACSL4, acyl-CoA synthetase long-chain family member 4; AMPK, AMP-activated protein kinase; CoQ10, coenzyme Q10; Cys2, cystine; DHODH, dihydroorotate dehydrogenase; Gln, glutamine; Glu, glutamate; GPX4, glutathione peroxidase 4; GSH, glutathione; HO-1, heme oxygenase-1; KGM, konjac glucomannan; LPCAT3, lysophosphatidylcholine acyltransferase 3; NF2, Neurofibromin 2; NOX4, NADPH oxidase 4; Nrf2, nuclear factor erythroid 2-related factor 2; PL-OOH, phospholipid hydroperoxide; PUFA, polyunsaturated fatty acid; PUFA-PL, phospholipid containing polyunsaturated fatty acid chain; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; YAP, yes-associated protein.
GPX4 plays a crucial role in protecting retinal cells from oxidative stress and ferroptosis damage. It catalyzes the reduction of lipid peroxides by GSH, thereby protecting the cell membrane from oxidative damage, which is essential for maintaining retinal health (33). In ophthalmic diseases such as diabetic retinopathy, the downregulation of GPX4 may lead to increased oxidative stress, subsequently affecting the function and survival of retinal photoreceptor cells (34).
Agents that induce ferroptosis, such as RAS-selective lethal 3 (RSL3) and erastin, lead to an increase in lipid ROS levels (35). While erastin impacts GSH levels, RSL3 does not substantially alter GSH levels during ferroptosis (36). Subsequent investigations revealed that RSL3 specifically targets the GPX4 molecule, inhibiting its activity through covalent binding and resulting in the accumulation of lipid peroxides (37). Yang et al. noted that cells with diminished GPX4 expression are more susceptible to ferroptosis, whereas those with elevated GPX4 levels exhibit increased resistance (38). Consequently, GPX4 serves as a significant regulator of ferroptosis, and modulating its expression can either enhance or suppress the occurrence of ferroptosis (16).
2.2.2 PUFA synthesis
PUFAs play a significant physiological role in the ophthalmic environment, particularly in terms of retinal health and visual function. PUFAs are crucial components of the cell membrane, essential for maintaining the fluidity and integrity of the retinal cell membrane (39). They are also involved in regulating lipid metabolism in the retina, playing an important role in promoting growth and development, immune modulation, and antioxidation.
The molecular structure of PUFAs features vulnerable carbon−carbon double bonds, making them susceptible to a process known as lipid peroxidation, a key event in ferroptosis (40). Two pivotal enzymes in the lipid remodeling process are acyl-CoA synthase long-chain 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), which are crucial for the production of PUFAs during ferroptosis (Figure 3) (41). The deletion or silencing of these genes can curtail the synthesis of PUFAs, thereby impeding the progression of ferroptosis (42, 43). Conversely, an abundance of arachidonic acid and other PUFAs can exacerbate the ferroptosis response when triggered by an inducer, as it accelerates the formation of peroxide lipids within the cell (40). This result highlights the significance of PUFAs as targets for plasma membrane peroxidation during ferroptosis.
Additionally, the enzyme lipoxygenase (LOX) facilitates the peroxidation of PUFAs, and decreasing LOX expression has been shown to mitigate ferroptosis, particularly that induced by erastin (40, 42). Given that free PUFAs serve as substrates for lipid peroxidation, their concentration and cellular distribution can influence the extent of lipid peroxidation and, consequently, the intensity of ferroptosis (42). Understanding the dynamics of PUFA localization and concentration presents an alternative avenue for modulating ferroptosis.
Some PUFAs, such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), play a crucial role in visual development and retinal function. DHA is the most abundant omega-3 fatty acid in retinal photoreceptors and is essential for maintaining the structure and function of photoreceptor cells (44). EPA helps to reduce inflammatory responses and decrease the risk of age-related macular degeneration and other eye diseases. In addition, omega-3 PUFAs may also mitigate oxidative damage to ocular tissues through antioxidant mechanisms, protecting the retina from light-induced damage (45).
Future research may explore the possibility of modulating enzymes involved in the synthesis of phospholipids containing PUFAs as a strategy to either initiate or prevent ferroptosis. This approach could reveal novel regulatory mechanisms and potential therapeutic targets within the complex interplay of lipid metabolism and cell death pathways.
2.3 Regulation
2.3.1 Cysteine metabolism
Cysteine metabolism plays a pivotal role in ferroptosis, serving as a critical regulatory hub for the synthesis of GSH, the primary antioxidant defense against the peroxidation of lipids within the cell membrane. The cystine/glutamate antiporter System Xc- is a complex composed of two subunits, the light chain (SLC7A11) and the heavy chain (SLC3A2), which are linked by disulfide bonds (46). This transporter plays a vital role in cellular antioxidant defense mechanisms. It operates by taking up cystine while exporting glutamate in an equimolar exchange (47). Once internalized, cystine is converted to cysteine, which is a critical component in the assembly of GSH. GSH, under the influence of GPX4, facilitates the conversion of harmful lipid peroxides into harmless fatty alcohols, thereby acting as a pivotal cellular antioxidant (Figure 3) (48).
In this biochemical cascade, GSH acts as a reducing agent, with GPX4 being the central enzyme that catalyses the reduction of lipid peroxides and thereby suppresses ferroptosis (49). Nonetheless, the rate of GSH production is contingent upon the availability of its precursor, cysteine, making System Xc- a crucial regulatory element. Compounds such as erastin, which are known to induce ferroptosis, impede GSH synthesis by directly targeting System Xc- and curbing cysteine uptake. A subsequent deficiency in GSH can result in the accumulation of detrimental peroxides, leading to protein and cell membrane damage and ultimately triggering ferroptosis (7).
Cysteine metabolism plays a crucial role in the ophthalmic environment, particularly in maintaining the health and function of the retina. Cysteine is the rate-limiting precursor for the biosynthesis of GSH, which is one of the primary antioxidants within cells and essential for protecting retinal cells from oxidative stress and ferroptosis. In the retinal pigment epithelial cells and photoreceptor cells, GSH helps to neutralize light-induced ROS, which are continuous byproducts of the visual cycle (50). One of the hallmarks of ferroptosis is the accumulation of lipid peroxides, and GSH, in its reduced form, aids in maintaining the integrity of the cell membrane, preventing lipid peroxidation.
2.3.2 Nrf2
The Nrf2 pathway is integral to the cellular response against ferroptosis, acting as a master regulator of antioxidant and detoxification processes. In a 2016 study, Sun and colleagues showed that the transcription factor Nrf2 can modulate ferroptosis through the P62-KEAP1-NRF2 signaling axis (51) (Figure 3). Upon activation, Nrf2 enhances the sequestration of iron and reduces its cellular uptake, thereby curtailing ROS generation and exerting an inhibitory effect on ferroptosis (52). Furthermore, Nrf2 suppresses ferroptosis by upregulating the expression of genes pivotal for the metabolism of iron and ROS, including quinone oxidoreductase 1 (NQO1) and haem oxygenase-1 (HO-1) (53). Additionally, stimulation of the p62-KEAP1-NRF2 cascade can lead to the upregulation of System Xc-, which in turn augments the transport of cystine and glutamate. This accelerated transport aids in the elimination of accumulated lipid peroxides (54), contributing to the overall antioxidant response and cellular defense against ferroptosis.
Nrf2 mitigates oxidative stress-induced damage to ocular tissues by activating antioxidant enzymes that eliminate harmful reactive oxygen species and free radicals. It suppresses the production of inflammatory mediators, reducing ocular inflammatory responses, while also promoting the repair of damaged cells and the regeneration of retinal tissue, maintaining the integrity of ocular structures (55). Nrf2 also regulates cellular metabolic pathways, affecting the synthesis and breakdown of fatty acids, influencing the progression of metabolic diseases, and providing protective effects on retinal neurons, reducing neurodegeneration, and preserving visual function (56). Furthermore, it inhibits pathological angiogenesis, safeguarding vision, and is involved in the regulation of ferroptosis, the clearance of damaged organelles and misfolded proteins, thereby maintaining the stability of the intracellular environment (57). These integrated functions underscore the essential role of Nrf2 in maintaining ocular health and visual function.
2.3.3 P53
The tumor suppressor p53, which is activated by various stress signals, has been implicated in regulating ferroptosis (58). By repressing the SLC7A11 gene at the transcriptional level, p53 contributes to the onset of ferroptosis by hindering the uptake of cysteine, an essential amino acid for GSH synthesis, thereby promoting ferroptosis (54). The role of p53 in inducing ferroptosis is associated with a specific mutational mechanism involving the substitution of three lysine residues with arginine. These mutations created an acetylated mutant form of p53 known as p533KR, which significantly suppresses the expression of SLC7A11 without affecting other recognized p53 target genes, such as those linked to cell cycle regulation, apoptosis, or ageing (59).
System Xc- is a crucial transporter for cystine and glutamate, forming a heterodimeric complex with the subunits SLC7A11 and SLC3A2 (60). Consequently, p53 impedes cystine uptake by System Xc- through the downregulation of SLC7A11. This reduction in cystine uptake leads to a decrease in cystine-dependent glutathione peroxidase (GPX) activity, which weakens the cell’s antioxidant defenses (61). As a result, lipid ROS levels are increased, triggering ferroptosis within the cell (62). This regulatory pathway underscores the importance of p53 in modulating the cellular susceptibility to ferroptosis and provides a potential therapeutic target for interventions aimed at preventing or treating conditions associated with this form of cell death.
P53 plays a critical protective role in ophthalmic diseases by monitoring cellular DNA damage and stress signals. When retinal cells undergo photic damage, hypoxia, or metabolic stress, P53 is swiftly activated to induce cell cycle arrest, thereby allowing time for DNA repair (63). If the damage is too severe, P53 initiates the programmed cell death process to prevent the proliferation of damaged cells and potential tumor formation.
Moreover, P53 is involved in regulating cellular metabolism, influencing the progression of eye diseases such as diabetic retinopathy by affecting blood sugar balance and fatty acid oxidation (64). The activity of P53 is also linked with neuroprotection in age-related macular degeneration and glaucoma (65). In these conditions, it fosters the expression of stress-resistant molecules to shield retinal neurons from degeneration.
2.3.4 HSPs
HSPs are critical in modulating the cellular response to ferroptosis, serving as a network of molecular chaperones that assist in the preservation of cellular integrity under oxidative stress. HSPs form an evolutionarily conserved family of molecular chaperones that fulfil a variety of immunological functions (66). These functions include mitigating cellular stress, exhibiting antioxidant activity, modulating immune responses, and facilitating antigen presentation. Through these functions, HSPs confer cellular resilience to various modes of cell death, including ferroptosis (67). One member of this family, heat shock protein B1 (HSPB1), also known as HSP25 or HSP27, safeguards the integrity of the actin cytoskeleton. This function is achieved by mitigating ferroptosis through a reduction in iron uptake, thereby curtailing subsequent oxidative damage (68).
HSPB1 also regulates the expression of TFR1 (69). Furthermore, the protein kinase C-mediated phosphorylation of HSPB1 results in the upregulation of FTL and FTH1 expression (68). This increase in ferritin expression reduces the concentration of intracellular ferrous ions and the production of lipid ROS (70).
The HSPB1 pathway, shown to counteract erastin-induced ferroptosis in cancers like cervical, prostate, and osteosarcoma, reduces iron and ROS levels (68, 71). Additionally, another member of the HSP family, heat shock protein A5 (HSPA5), also known as binding immunoglobulin protein (BIP) or 78 kDa glucose-regulated protein (GRP78), is localized to the endoplasmic reticulum. HSPA5 can bind to GPX4, thereby significantly bolstering the cell’s antioxidant defenses (72).
HSPs play the role of protectors in ophthalmic diseases, participating in the maintenance of cellular proteostasis, especially when faced with damaging conditions such as oxidative stress, inflammation, heat shock, ultraviolet radiation, and metabolic stress. HSPs like HSP27, HSP70, and HSP90 can prevent protein aggregation and cellular dysfunction by promoting the proper folding of damaged proteins, repairing, or clearing misfolded proteins. In retinal diseases, HSPs help protect photoreceptor cells from light damage and slow the progression of age-related macular degeneration (73). In glaucoma and neurodegenerative eye diseases, the neuroprotective role of HSPs is particularly important, as they can protect retinal ganglion cells from mechanical stress or disease-related neurotoxic damage (74, 75). This intricate interplay between HSPs and cellular antioxidant systems presents a promising avenue for therapeutic intervention in conditions where ferroptosis contributes to pathology.
2.3.5 FSP1
A comprehensive genome-wide screening study revealed the role of FSP1 in regulating ferroptosis. Interestingly, the FSP1-CoQ10-NAD(P)H pathway operates as a significant intracellular antioxidant mechanism (76). This complements the traditional GSH-GPX4 pathway to collaboratively suppress phospholipid peroxidation and inhibit ferroptosis.
Specifically, FSP1 facilitates the reduction of CoQ10 to Ubiquinol using NAD(P)H (77). This reaction subsequently captures lipid peroxyl radicals, thereby halting the propagation of phospholipid peroxidation. In parallel, the GPX4 and glutathione systems aid in neutralizing phospholipid hydroperoxides and free radicals, further dampening peroxidative processes. In instances of GPX4 deficiency, the FSP1-CoQ10-NAD(P)H pathway steps up as the primary mechanism for combating ferroptosis.
In 2019, research conducted by Bersuker and colleagues indicated that the deletion of FSP1 resulted in tumor cells losing their resistance to the iron regulatory compound RSL3, concomitant with a marked increase in intracellular iron levels (78). FSP1, known for its myristoylation, contributes to the composition of numerous cellular membranes, and any modification of its myristoylated sites can impair its ferroptosis-inhibiting capabilities (79).
Mechanistically, FSP1 exerts its effects through the quinone oxidoreductase activity associated with NADH, converting ubiquinone to ubiquinol, which in turn inhibits lipid peroxidation and ferroptosis (80). Ubiquinol can directly neutralize lipid-derived free radicals, thereby halting the process of lipid autoxidation. Alternatively, it can indirectly cease lipid autoxidation through the regenerative oxidation of α-tocopherol free radicals (78).
FSP1 plays an integral role in the repair processes of the cornea and retina, facilitating the healing of damaged tissues by promoting cell adhesion and migration (81). Following a corneal injury, the deposition of FSP1 can provide a temporary substrate for epithelial cell regeneration, thereby expediting wound closure and repair.
Moreover, FSP1 significantly contributes to the functionality of RPE cells. It assists in maintaining the polarity and integrity of RPE cells, which are vital for the health and function of photoreceptor cells. In vascular diseases such as diabetic retinopathy, FSP1 may play a role in regulating the growth of abnormal blood vessels and scar formation (82). It influences the remodeling of the extracellular matrix and angiogenesis processes through interactions with growth factors and cell surface receptors. FSP1 may also have a role in age-related macular degeneration, affecting the formation of neovascularization and scar tissue beneath the retina (81).
This intricate regulatory network underscores the multifaceted role of FSP1 in modulating the cellular susceptibility to ferroptosis, providing novel insights into the complex interplay between cellular metabolism and iron homeostasis in the context of oxidative stress and cell death.
2.3.6 AMPK
AMPK serves as a pivotal sensor within the cell, monitoring energy metabolism and orchestrating a variety of metabolic pathways to maintain energy homeostasis (83). The activation of AMPK has been implicated in both the promotion and suppression of ferroptosis (84). The seemingly contradictory effects of AMPK on ferroptosis may stem from the intricate interplay of environmental factors and the degree of AMPK phosphorylation at specific sites (85–87).
Moreover, AMPK is recognized for its role in governing mitochondrial stability, which is integral to cellular health and the regulation of cell death pathways (88). The activation of AMPK triggers the degradation of ferritin, an iron-storage protein, leading to increased ROS levels and the initiation of ferroptosis (89, 90). A study identified AMPK as an upstream modulator of ferroptosis, and the depletion of AMPK sensitizes cells to this form of death (91). Conversely, another study reported that AMPK becomes activated under glucose-deprived conditions and that the AMPK signaling cascade curbs lipogenesis, thereby reducing the propensity for ferroptosis (Figure 3) (92).
In addition, AMPK is also involved in the regulation of mitochondrial biogenesis and inflammatory responses, both of which are significant factors in the development of eye diseases. For instance, the activation of AMPK can promote the process of mitochondrial ferroptosis, assisting in the replacement of dysfunctional mitochondria, which is crucial for maintaining the health of ocular cells (93). At the same time, AMPK can also inhibit the mTORC1 pathway, a key regulatory factor for cell growth and proliferation, and its inhibitory effect helps to control the progression of eye diseases (94). Therefore, the regulatory role of AMPK has potential application value in ophthalmic treatment and can serve as a new strategy for treating eye diseases.
2.3.7 NADPH
NADPH is a vital coenzyme in cellular redox homeostasis and plays a crucial role in ferroptosis by functioning as the primary reducing equivalent to counteract oxidative stress. It functions as a coenzyme for glutathione reductase, is integral to maintaining intracellular GSH levels (95). The influence of well-established ferroptosis initiators, including erastin, RSL3, and FIN56, has been explored across diverse cell lineages, notably those derived from human osteosarcoma (96). These studies consistently reported a significant decrease in the levels of intracellular NAD(H) and NADP(H), alongside the detection of ROS formation (97).
Current research shows NADPH donates electrons to cytochrome P450 via P450 oxidoreductase (POR), supplying them with electrons (98). This electron transfer may precipitate lipid peroxidation through several proposed mechanisms, either by facilitating the dehydrogenation of PUFAs or by impeding the reduction of Fe3+ to Fe2+ (99, 100).
These findings suggest that NADPH not only is central to the cellular redox balance but also plays a critical role in regulating ferroptosis, potentially by influencing iron metabolism and lipid peroxidation (101). Understanding the nuances of the role of NADPH in these processes could provide novel perspectives on the modulation of ferroptosis and its implications in various pathophysiological conditions.
Besides, NADPH is a key coenzyme for the synthesis of lipids and cholesterol within cells, which is crucial for the membrane structure and function of retinal cells. In ophthalmic diseases, the synthesis and utilization of NADPH may be affected, leading to dysfunction of retinal cells (102). Therefore, maintaining the levels and functionality of NADPH is essential for protecting vision and treating ophthalmic diseases (103). Supplementing NADPH or enhancing its synthetic pathways may help improve retinal health and delay the progression of ophthalmic diseases.
2.3.8 GCH1
A laboratory investigation revealed that GCH1, an essential enzyme in the biosynthesis of folates, can prevent ferroptosis (104). This protective effect is believed to be mediated by its metabolic products, tetrahydrobiopterin (BH4) and dihydrobiopterin (BH2), which are crucial for cellular redox homeostasis and the maintenance of nitric oxide synthase function (105).
Complementary lipidomic profiling revealed that cells with elevated GCH1 levels contain two PUFA chains that seem to shield phosphatidylcholine phospholipids from peroxidative damage. These PUFA chains may contribute to the cell’s defenses against oxidative stress by stabilizing the cell membrane and preventing the propagation of ROS. However, the precise biochemical pathways and molecular interactions that underlie this protective mechanism are not yet fully understood (106).
In the retina, the activity of GCH1 is particularly important for protecting photoreceptor cells from light damage (107). Photoreceptor cells are particularly susceptible to oxidative damage due to their continuous exposure to light. the function of GCH1 is potentially important for maintaining retinal health and preventing certain ophthalmic diseases, such as age-related macular degeneration and certain types of retinal degenerative diseases.
Further research is needed to elucidate the detailed mechanisms by which GCH1 and its metabolites modulate lipid peroxidation and ferroptosis. Understanding these processes could provide valuable insights into the development of novel therapeutic strategies for diseases in which ferroptosis plays a significant role, such as neurodegenerative disorders, ischemic injury, and certain types of cancer.
3 Ferroptosis and other forms of cell death
Ferroptosis is a unique form of regulated cell death that relies on iron and sets itself apart from other forms such as apoptosis, programmed necrosis, and pyroptosis (108–110).
From a morphological perspective, ferroptosis is characterized by specific features, including the atrophy of mitochondria, a significant reduction and shriveling of mitochondrial cristae, an increase in membrane density, and instances of membrane rupture (111, 112). Notably, the nucleus often appears normal, and an increase in cell membrane density occurs. In contrast, apoptosis is characterized by a decrease in cell volume, chromatin condensation, nuclear fragmentation, the preservation of a complete membrane structure, and the formation of apoptotic bodies (113). Programmed necrosis is identified by an increase in cell size, simultaneous rupture of the plasma membrane, the generation of substantial cell debris, and, frequently, the formation of necrosome structures (114, 115). Pyroptosis involves the swelling of cells, the formation of pores on the plasma membrane, and the release of inflammatory mediators (116).
Biochemically and immunologically, ferroptosis is characterized by a significant reduction in the granular membrane potential, elevated levels of intracellular iron ions, a substantial increase in ROS levels, and intensified lipid peroxidation (117). It is also characterized by the release of damage-associated molecular patterns (DAMPs) and the promotion of an inflammatory response (118, 119). In apoptotic cells, DNA is cleaved into 180–200 base pair fragments, the mitochondrial membrane potential decreases significantly, phosphatidylserine flips from the inner to the outer membrane, and no endolysosomal release or inflammatory response occurs (119).
Programmed necrosis is characterized by the release of endolysates and DAMPs, which contribute to inflammation (120). In pyroptotic cells, inflammatory bodies such as the NLRP3 inflammasome can be observed (121). During pyroptosis, inflammatory caspases activate and cleave GSDMD, releasing its N-terminal fragment (GSDMD-N), which forms pores in bacterial membranes leading to their death without damaging the host cell (119).
These distinct characteristics of each form of cell death underscore the complexity of cellular demise and the importance of accurate identification for both diagnostic and therapeutic purposes. Understanding the biochemical and immunological signatures of ferroptosis, in particular, may provide novel avenues for intervention in diseases where this form of cell death plays a significant role.
3.1 Ferroptosis and apoptosis
Apoptosis, a form of PCD, is intricately governed by genetic mechanisms (122). It plays a vital role in maintaining tissue homeostasis, orchestrating immune and defense functions, and managing cellular damage associated with tumorigenesis (123).
A growing body of research indicates a close relationship between ferroptosis and apoptosis (124, 125). Under specific conditions, cells can transition from apoptosis to ferroptosis (126). This interplay can amplify the cellular susceptibility to apoptotic signals (127). Furthermore, the tumor suppressor protein P53, known for its role in halting the cell cycle and inducing apoptosis in tumor cells, can also trigger ferroptosis under certain circumstances (128). Both in vivo and in vitro investigations have shown that cells treated with metal organic network encapsulated with p53 plasmid (MON-p53) exhibit pronounced signs of ferroptosis and apoptosis. This dual action not only curbs tumor proliferation but also extends the lifespan of mice bearing tumors (129). Consequently, the convergence of ferroptosis and apoptosis pathways represents a promising avenue for therapeutic intervention in cancer treatment.
This emerging paradigm underscores the complexity of cell death mechanisms and highlights the potential for novel strategies that harness the interplay between ferroptosis and apoptosis (130). Targeting this combined pathway could represent a more comprehensive approach to managing tumor growth and potentially other diseases in which these cell death mechanisms are implicated. Future research is likely to explore the nuances of this interaction and develop targeted therapies that leverage the synergistic effects of ferroptosis and apoptosis.
3.2 Ferroptosis and programmed necrosis
Programmed necrosis is characterized by a cellular morphology resembling necrosis and involves a signaling pathway akin to apoptosis (131). Moreover, ferroptosis has been observed to coexist with programmed necrosis, particularly in the demise of neurons following hemorrhagic stroke (132). Studies have reported an increase in the mRNA levels of both ferroptosis and programmed necrosis markers during haem chloride-induced cell death (133).
In 2017, research conducted by Müller et al. (134) proposed that ferroptosis and programmed necrosis are complementary mechanisms of cell death. Within this framework, ACSL4, a key enzyme in lipid metabolism, was utilized as an indicator of ferroptosis, while mixed lineage kinase-like protein (MLKL), a protein involved in the execution of necrosis, served as a marker for programmed necrosis (135, 136). These findings indicated that a deficiency in ACSL4 resulted in the upregulation of MLKL, which sensitized cells to ferroptosis (137). This result suggests a compensatory relationship between the two pathways, where the suppression of one cell death mechanism can lead to the activation of the other.
Wulf Tonnus et al. (138) reported that NADPH, a critical coenzyme in cellular redox reactions, can diffuse freely between cells, potentially influencing both ferroptosis and programmed necrosis. The hypothesis posits that the consumption of NADPH during programmed necrosis may sensitize neighboring cells to ferroptosis (139). When cells undergo either programmed necrosis or ferroptosis, intracellular NADPH levels are reduced, potentially rendering adjacent cells more vulnerable to alternative cell death pathways (138). This perspective provides a more integrated view of how ferroptosis relates to and intersects with other PCD mechanisms, highlighting the complexity and interconnectivity of cell death processes.
3.3 Ferroptosis and autophagy
Autophagy, a process that relies on the lysosome for the degradation of cellular components, has been implicated in a spectrum of pathological conditions, including infectious diseases, immunological disorders, metabolic syndromes, and malignancies (140–142).
Recent investigations have revealed a novel role for autophagy in cancer cell death, showing that autophagy can initiate the breakdown of ferritin, consequently triggering ferroptosis (143). In fibroblasts and cancer cells, this form of iron-mediated autophagy has been shown to increase intracellular iron levels through the degradation of ferritin, further promoting ferroptosis (144).
Moreover, the deletion or inhibition of genes such as Atg5 and Atg7, which are essential for autophagy, has been reported to curtail ferroptosis induced by erastin (145, 146). This process is achieved by reducing both intracellular iron levels and the extent of lipid peroxidation (147). These discoveries shed light on the intricate relationships and cross-regulatory mechanisms between ferroptosis and autophagy, providing new insights into the complex network of cell death pathways and their interdependence (144).
4 Ferroptosis and ophthalmic disorders
Ferroptosis has been implicated in the progression of numerous diseases. Ferroptosis is particularly prevalent in ophthalmology and may be associated with corneal disease in the anterior segment, uveal disease in the middle segment, and retinal disease in the posterior segment of the eye (Figure 4). By exploring the regulatory mechanisms of ferroptosis, including the metabolism of iron ions, the production of reactive oxygen species, and the balance of antioxidant systems, we can obtain a better understanding of its role in ophthalmic diseases (Table 1).
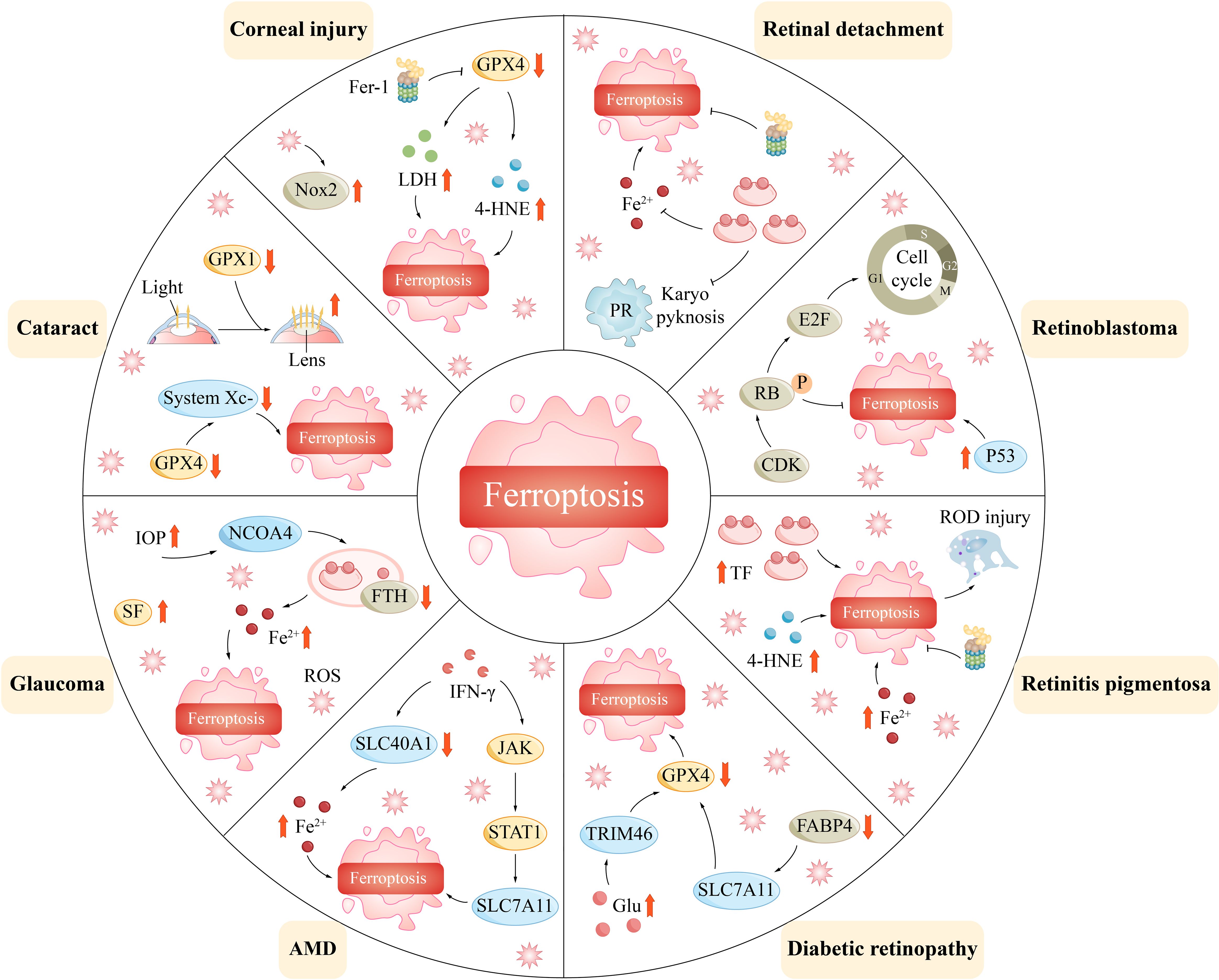
Figure 4 Ferroptosis mediates a variety of ophthalmic disorders.
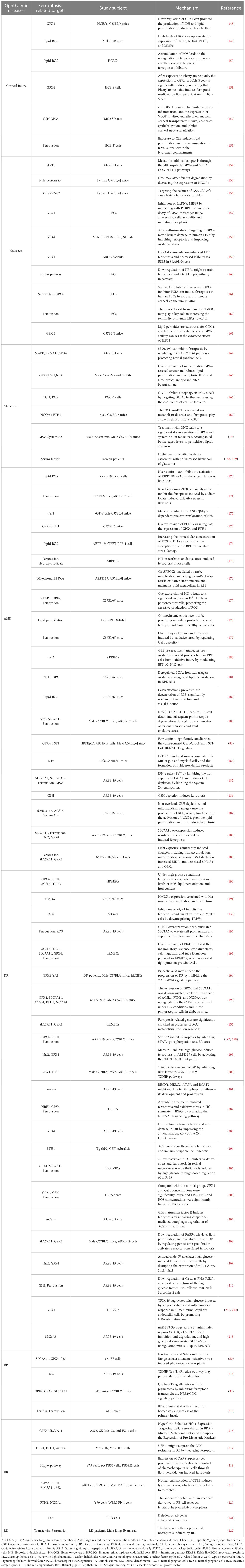
Table 1 The mechanism of ferroptosis-related targets in ophthalmic diseases.
4.1 Ferroptosis and corneal injury
Corneal injuries are medical conditions characterized by the disruption of the protective functions and structural integrity of the corneal epithelium due to various factors (223). This disruption can result in partial or complete loss of corneal epithelial cells. Clinical manifestations may include widespread punctate defects or erosions on the corneal surface, along with continuous detachment and damage to the epithelium (224). Additionally, varying levels of inflammation affecting the ocular surface may be present (225). In severe instances, such injuries can lead to pathological alterations in the corneal stroma, potentially impacting visual acuity (226).
Ferroptosis research, triggered by corneal injury, is attracting medical research interest. Particularly in cases of corneal trauma, damage to the corneal epithelial layer is notably prevalent. When the cornea sustains an injury, it initiates a series of intricate biochemical reactions, with oxidative stress being a particularly prominent factor (151, 227).
GPX4, an essential player in ferroptosis, is integral to maintaining the oxidative balance, ensuring cell survival, and facilitating the wound healing process in corneal epithelial cells (228). Research conducted by Sakai et al. (148) reported that a reduction in GPX4 levels, achieved through the transfection of GPX4 siRNA into the human corneal epithelial cell line HCEC, could lead to an increase in lipid peroxidation products such as lactate dehydrogenase (LDH) and 4-hydroxynonenal (4-HNE), thus triggering ferroptosis. Furthermore, the application of Fer-1, an inhibitor of ferroptosis, was found to mitigate cellular damage associated with the knockout of the GPX4 gene (229). Additional studies have indicated that mice with partial knockout of GPX4 exhibit slower corneal wound healing after epithelial injury than their wild-type counterparts, underscoring the significance of GPX4 in the repair process following corneal injury.
Corneal alkali burns represent an exceedingly severe form of corneal chemical injury that inflicts substantial damage on the ocular surface, with a particular emphasis on the cornea. Addressing this type of injury is complex, and the prognosis is often unfavorable, frequently resulting in a significant visual impairment or even blindness. During corneal alkali burn injury, ROS production is substantially increased, which can increase the expression of genes such as Nox2, Nox4, VEGF, and MMP. This upregulation contributes to further corneal damage and encourages the development of new blood vessels (149). Wang et al. (150) reported that ferroptosis, induced by lipid peroxidation, plays a pivotal role in the injury caused by alkali burns, along with ROS. The pathological changes linked to ferroptosis can be partially reversed by treatment with Fer-1. However, due to the hydrophobic nature of Fer-1, its direct application in clinical settings is not feasible. Consequently, Wang et al. (150) have engineered a liposomal delivery system for Fer-1 (Fer-1-NPs) to enhance its bioavailability and augment its therapeutic effects on corneal cloudiness and neovascularization.
In addition to the presence of ferroptosis during the process of corneal alkali burn, excessive ROS and reactive nitrogen species (RNS) may also be produced in corneal inflammation or corneal injury caused by trauma (230). These substances can promote the release of iron ions and lipid peroxidation, thereby triggering ferroptosis (231). Furthermore, corneal surgery or infection may also induce similar oxidative stress reactions, which can subsequently impact the metabolism of iron ions and the cellular redox state.
The collective findings from these studies highlight the critical role of ferroptosis in corneal injury and suggest that its inhibition could be beneficial for corneal preservation (152, 232). The development of pharmaceuticals based on the understanding of ferroptosis offers promising avenues for treating corneal injuries (233). Moreover, these investigations provide a substantial theoretical framework and empirical data that can inform the creation of novel therapeutic approaches for treating corneal injuries. Overall, an in-depth understanding of the ferroptosis mechanism is anticipated to pave the way for more efficacious treatment strategies for corneal injuries, providing renewed hope to those suffering from keratopathy.
In summary, the study of ferroptosis in the context of corneal injury represents a multifaceted and significant area of research. A thorough investigation of the roles played by key elements, such as GPX4, ROS, and lipid peroxidation, in the ferroptosis process, coupled with the development of potent ferroptosis inhibitors, can yield innovative strategies for treating corneal injuries. These research outcomes are also likely to serve as valuable references for addressing other related medical conditions. As scientific and technological advancements continue to progress, further breakthroughs and advancements in the field of corneal injury treatment will occur.
4.2 Ferroptosis and cataract
Cataract, a prevalent eye disorder, is identified by clouding of the ocular lens, which impairs visual acuity (234). This ailment can impact either a single eye or both simultaneously. Affected individuals often report a range of symptoms, including but not limited to, hazy vision, heightened nearsightedness, the occurrence of double vision, the appearance of light halos, photophobia, and diminished visual capacity in low-light conditions (235). Such symptoms can significantly impact daily activities, such as driving and reading, and can also hinder the ability to discern faces. Patients with critically impaired vision have an elevated risk of falls and an increased propensity for depressive symptoms (236). Cataract accounts for half of the global blindness cases and is responsible for 33% of visual impairments worldwide (237).
Recent scientific investigations have revealed that the ageing process can disrupt the redox equilibrium of the lens, leading to a decrease in the activity of enzymes linked to the glutathione synthesis pathway (238). This change results in a reduction in GPX activity, an increase in the accumulation of ROS, and an increase in lipid peroxidation byproducts (239). These discoveries point towards the potential involvement of ferroptosis in the genesis of cataracts.
In a study by Reddy and colleagues (163), a noted increase in the scattering of light within the nuclei of lenses from mice that were genetically deficient in GPX1 was documented. Over time, these mice exhibited signs of age-related cataracts, a phenomenon not observed in the wild-type control group. Wei et al. (161) reported that lens epithelial cells are susceptible to ferroptosis when exposed to inducers of this process. The level of GSH was identified as a pivotal determinant of cell vulnerability. Additionally, Wei et al. (161) observed that the sensitivity of lens epithelial cells to ferroptosis inducers increases with the extent of cellular aging.
Hydroxyl radicals are highly reactive and can attack PUFAs in the cell membrane, initiating lipid peroxidation (240). This process leads to damage and dysfunction of the cell membrane structure, impacting the normal physiological activities of lens cells. Apart from lipids, hydroxyl radicals can also target proteins and nucleic acids, resulting in protein cross-linking and DNA damage (241). These damages can impair the cell’s repair mechanisms and the stability of genetic information, ultimately influencing cell survival. Damage to lens cells can cause the accumulation and precipitation of proteins within the cells, diminishing the transparency of the lens and contributing to the development of cataracts.
Further analysis of the transcriptome revealed that in older cells, GPX4 expression was downregulated, System Xc- function was suppressed, and genes involved in iron ion uptake were upregulated, while genes responsible for iron ion export were downregulated. These findings indicate that ferroptosis could be instrumental in the progression of cataracts (154). Elucidating the relationship between ferroptosis and cataract development may pave the way for innovative preventative strategies and therapeutic approaches for cataract treatment.
4.3 Ferroptosis and glaucoma
Glaucoma encompasses a collection of eye disorders linked to abnormal levels of intraocular pressure (242). This condition has the potential to inflict damage on the optic nerve, leading to a reduction in the visual field and a loss of vision (243). The clinical manifestations of glaucoma can include a range of symptoms such as a decrease in visual acuity, discomfort or pain in the eyes, moderate enlargement of the pupils, redness and swelling of the ocular area, and nausea (244). Typically, glaucoma first affects peripheral vision; without timely intervention, it may progress to the central visual field, potentially resulting in irreversible blindness (245).
Recent scientific findings suggest that iron ions could be instrumental in injury to retinal ganglion cells (RGCs). Lin et al. (168) documented a positive correlation between increased levels of serum ferritin and the incidence of glaucoma. This correlation implies that disruptions in iron metabolism might be intertwined with the pathogenesis of the disease. Building on this information, Yao et al. (167) showed that pathological increases in intraocular pressure (IOP) can disrupt iron homeostasis, leading to an excessive accumulation of iron ions (Fe2+) in the retina, particularly affecting the RGC layer. This overabundance of iron ions compromises the retinal redox balance, triggering ferroptosis in RGCs. The administration of deferiprone, an iron ion chelator, has been found to significantly reduce iron ion levels in the retina, effectively inhibiting RGC death.
Moreover, comprehensive studies have indicated that pathologically elevated IOP might also expedite the degradation of ferritin by enhancing NCOA4-mediated ferritin autophagy (246). This process leads to the release of a substantial amount of previously bound iron ions into the cell. Once ferritin is degraded, the iron ions it had sequestered are released, intensifying the oxidative stress response and causing harm to RGCs. Knockdown of the NCOA4 gene can effectively prevent ferritin degradation and curtail the release of iron ions, thereby significantly diminishing iron ion levels and mitigating RGC damage (247).
The aforementioned research suggests that ferroptosis can contribute to the demise of RGCs through diverse pathways, thereby influencing the advancement of glaucoma. Recent studies have revealed that the reduction of GPX4 expression triggers ferroptosis in nerve cells, indicating that ferroptosis may further aggravate the progression of glaucoma by targeting optic nerve damage (248).
These insights not only illuminate the potential role of iron ions in the development of glaucoma but also present a novel perspective for therapeutic approaches (249). Regulating iron homeostasis, especially by reducing iron ion accumulation due to pathological IOP, could represent a groundbreaking strategy for glaucoma management (250). Future research may explore the regulatory mechanisms of iron metabolism and investigate how these pathways can be targeted by pharmaceuticals or other treatments. These investigations could lead to the development of more effective treatment plans for glaucoma patients. Collectively, these studies may pave the way for innovative preventative measures and therapeutic interventions for glaucoma with the potential to slow or even prevent the progression of this debilitating eye disease.
4.4 Ferroptosis and age-related macular degeneration
Age-related macular degeneration (AMD) is a complex ocular disorder that occurs with ageing and is characterized by a deterioration in the macula structure and function (251). The macula, which is situated at the retina’s core, is pivotal for sharp central vision (252).
Recent in-depth examinations of retinal pigment epithelial (RPE) cells revealed that interferon-γ (IFN-γ) can trigger ferroptosis in these cells, contributing to the progression of AMD (185). The process is intricate and includes several stages: it starts with the suppression of the iron transporter SLC40A1, which results in an increase in intracellular iron (Fe2+) levels; this step is followed by the obstruction of System Xc−, which leads to a depletion of GSH within the cell. This process also involves the upregulation of acyl-CoA synthetase long-chain family member A (ACSLA) and 5-lipoxygenase (5-LOX), which promote lipid peroxidation (253). Finally, the downregulation of SLC7A11, GPX4, and GSH occurs through the activation of the JAK1–2/STAT1/SLC7A11 signaling pathways, stimulating ferroptosis (185).
In addition to IFN-γ, the pigment N-retinol-N-retinoethanolamine (A2E), a distinctive fluorophore within RPE cells, has also been implicated in ferroptosis (254). Through comprehensive transcriptome profiling, Scimone and colleagues noted the upregulation of A2E expression and the concomitant downregulation of SLC7A11 in H-RPE cells upon blue light exposure. This change curtailed the biosynthesis of GSH, further propagating ferroptosis in RPE cells.
Ferroptosis impacts not only RPE cells but also traditional AMD models (255). Liu and associates (256) analyzed sodium iodate (SI)-induced AMD cellular models and discovered that SI, while not influencing GPX4 activity, could deplete intracellular GSH, liberate Fe2+ from liposomes, increase intracellular ROS production, and increase lipid aggregation, consequently precipitating ferroptosis in ARPE-19 cells. Lee et al. (257) also detected that SI could provoke the generation of mitochondrial ROS, promoting ferroptosis in RPE cells.
Tang et al. (183) reported that ferroptosis induced by SI in RPE cells is linked to the Nrf2/SLC7A11/HO-1 pathway, with heightened HO-1 expression precipitating the upregulation of the TFR and the downregulation of SLC40A1, culminating in iron ion aggregation. The liposomal accumulation of Fe2+ can then increase HO-1 expression, perpetuating ferroptosis. The suppression of HO-1 or the application of the HO-1 inhibitor ZnPP has been shown to ameliorate the detrimental effects of ferroptosis on the morphology and functionality of RPE cells and photoreceptors (258).
Ferroptosis in photoreceptors also contributes significantly to the aetiology of AMD, particularly the dry form (50). Within the visual cycle, the regeneration of 11-cis-retinoic acid retinol (atRAL) between photoreceptors and the RPE is crucial for sustaining visual acuity (259). ATP-binding cassette transporter A4 (ABCA4) facilitates the transport of atRAL from the outer segment discs of photoreceptors to the cytoplasm, while all-trans retinol dehydrogenase 8 (RDH8) catalyses the conversion of atRAL back to all-trans retinol (260). In mouse models deficient in the Abca4 and Rdh8 genes, a deficiency in the clearance of photoreceptor outer segment discs, thinning of the retinal photoreceptor layer postlight exposure, the accumulation of atRAL in the retina, diminished GSH levels, aberrant expression of the lipid metabolism-related proteins COX2 and ACSLA, and heightened lipid peroxidation—indicators of ferroptosis—are observed (187). Subsequent studies have validated that atRAL can incite ferroptosis in the cone cell line 661W by impeding System Xc−, increasing Fe2+ levels, and amplifying mitochondrial ROS production.
Tang et al. (189) confirmed these findings, showing that light exposure can precipitate the degeneration of retinal photoreceptor cells through ferroptosis in 661W cells and male Sprague−Dawley rats. Illumination significantly diminishes the viability of photoreceptor cells and induces pro-ferroptosis alterations, such as iron buildup, mitochondrial condensation, GSH depletion, increased malondialdehyde (MDA) levels, and reduced expression of the SLC7A11 and GPX4 proteins.
The decrease in glutathione and GPX4 may result in the retinal cells’ ineffectiveness at efficiently eliminating lipid peroxides, thereby impairing the visual function of these cells and worsening AMD (81). The abnormal build-up of ROS and other oxidative stress molecules inflicts damage on the retinal cells, influencing the transmission and processing of visual signals (261). Moreover, as AMD is a neurodegenerative disease, ferroptosis could potentially hasten the progression of AMD by advocating the demise of nerve cells.
Ferristatin-1 has been shown to attenuate the impact of light on ferroptosis, ameliorate photoreceptor atrophy and ferroptosis, curb neuroinflammation, and protect the retinal structure and function from the effects of light (51, 189). Collectively, these insights provide innovative avenues for leveraging the mechanisms of ferroptosis in the prevention and treatment of retinal degeneration diseases, including AMD.
4.5 Ferroptosis and diabetic retinopathy
Diabetes mellitus is widely acknowledged as the primary cause of diabetic retinopathy (DR) (262). Persistently high blood sugar levels inflict damage on retinal vascular endothelial cells, resulting in a spectrum of retinal lesions, such as microaneurysms, hard exudates, cotton-wool spots, neovascularization, vitreous proliferation, macular oedema, and, in extreme cases, retinal detachment (263).
Recent research has revealed the crucial role of ferroptosis in the progression of DR, where it contributes to the disease by impairing the integrity of capillary endothelial cells. Uric acid, a significant biomarker for type 2 diabetes, influences serum uric acid levels through the TRIM46 gene, which exhibits increased expression in diabetic individuals (264). Under high-glucose conditions, the expression of TRIM46 in human retinal capillary endothelial cells (HRCECs) increases and correlates with the extent of cell death (212). Studies have indicated that TRIM46 overexpression facilitates the ubiquitination and subsequent degradation of GPX4 in HRCECs, thereby promoting ferroptosis.
Ferroptosis affects not only the microvasculature but also RPE cells. Research by Liu and colleagues has shown that in diabetic rat models, hyperglycemia induces the secretion of GMFB by Müller cells, and elevated levels of GMFB can impair retinal function by altering the quantity and connectivity of RPE cells (207). GMFB is capable of hindering lysosomal assembly and increasing alkalinity, which diminishes lysosomal functionality. This reduction in function impedes the autophagic degradation of ACSLA, leading to its accumulation and exacerbating lipid peroxidation in ARPE19 cells. This process results in a decrease in mitochondrial activity and the eventual induction of ferroptosis.
In addition to GMFB, fatty acid binding protein 4 (FABP4) has been shown to alleviate lipid peroxidation and oxidative stress associated with DR by modulating PPARγ-mediated ferroptosis (208). FABP4 is instrumental in sustaining the glucose and lipid balance and serves as an independent prognostic marker for patients with type 2 diabetic retinopathy.
PPARγ, a crucial modulator of fatty acid storage and glucose metabolism, regulates the expression of the FABP4 gene Conversely, FABP4 can initiate the ubiquitination and subsequent proteasomal degradation of PPARγ. In a streptomycin-induced diabetic mouse model, a marked increase in the expression of both the FABP4 and PPARγ proteins was observed in retinal tissue. Suppressing FABP4 has been shown to reverse the downregulation of the SLC7A11 and GPX4 proteins, thereby mitigating ferroptosis in RPE cells (208).
Furthermore, Tang and colleagues confirmed that RPE cells subjected to high-glucose conditions undergo ferroptosis (209). Persistent hyperglycemia intensifies oxidative stress, precipitating structural and functional alterations in retinal neurons (265). This degeneration characterizes the early neural damage in DR, potentially resulting in vision loss and other visual impairments. Oxidative stress induced by iron overload may also contribute to the manifestation of macrovascular complications of diabetes, including atherosclerosis and cardiovascular diseases (266). Within the context of DR, these changes could cause damage to and dysfunction of the retinal microvasculature. Therefore, inhibiting ferroptosis to curtail oxidative damage in the retina is a novel therapeutic approach for the management of diabetic retinopathy.
In summary, the pathogenesis of diabetic retinopathy involves a complex interplay of factors, including ferroptosis, uric acid metabolism, GMFB, and FABP4. These findings provide new insights into the intricate mechanisms underlying DR and present potential targets for future therapeutic interventions.
4.6 Ferroptosis and retinitis pigmentosa
Retinitis pigmentosa (RP), an inherited disorder, results in progressive degeneration of the retina, leading to night blindness, retinal atrophy, pigment deposition, and constriction of the visual field, potentially culminating in total blindness (267). The advancement of RP significantly diminishes the quality of life for those affected, underscoring the importance of investigating its underlying pathological processes to identify effective therapeutic approaches (268).
In the study of RP, researchers have focused on the expression levels of genes involved in iron metabolism, as well as the levels of iron and oxidative damage within the retina. In the rd10 mouse model, which exhibits rapid progression of retinal degeneration, a notable increase in the expression of several key proteins related to iron transport and storage, such as transferrin, ceruloplasmin, ferritin, and the transferrin receptor, was observed (215). Concurrently, an increase in the total iron content and the amount of iron bound to ferritin within the retina was observed. Moreover, the level of 4-hydroxy-2-nonenal (4-HNE), an indicator of lipid peroxidation, was significantly elevated, suggesting a strong correlation between excess iron and retinal damage (215).
Further research has established a connection between ferroptosis—the form of regulated cell death associated with iron metabolism and oxidative stress—and cell death induced by sodium iodate in ARPE-19 cells, highlighting the relevance of ferroptosis to the progression of RP (269). This discovery provides a novel perspective on the pathogenesis and potential treatment of this disease.
Mitochondrial dysfunction can result in the leakage of iron ions from the mitochondria into the cytoplasm. These iron ions react with ROS to generate hydroxyl radicals via the Fenton reaction (270). The accumulation of iron ions coupled with the production of hydroxyl radicals escalates the level of oxidative stress (271). Notably, oxidative stress is a pivotal factor in the onset of RP, which can inflict damage on photoreceptor cells, leading to cellular dysfunction and death (272). Moreover, the oxidative stress and cellular damage brought about during the process of ferroptosis can trigger inflammatory responses. The subsequent release of inflammatory mediators further impairs retinal cells, exacerbating the pathologic progression of RP (273).
Subsequent studies using RP models have revealed the efficacy of various iron homeostasis regulators in mitigating ferroptosis in photoreceptor cells and in protecting against photoreceptor degeneration. Notably, agents, such as zinc deferoxamine (274), the iron-chelating drugs (275) VK28 and VAR10303, the iron chelator deferiprone (276), and the ferroptosis inhibitors deferoxamine and hepstatin-1 (183), have all been proven to be effective in slowing ferroptosis. These findings offer a fresh scientific rationale for targeting iron homeostasis in therapeutic strategies for RP and propose innovative avenues for its management.
In conclusion, maintaining iron homeostasis is crucial in the pathogenesis of RP, with alterations in iron metabolism-related genes and iron levels being intricately linked to retinal damage. The utilization of iron chelators and inhibitors of ferroptosis represents innovative therapeutic avenues for RP and is anticipated to emerge as a promising treatment strategy in the future. These insights not only enhance our understanding of the pathological mechanisms of RP but also pave the way for the development of novel therapeutics, providing hope for more efficacious treatment solutions for individuals with RP.
4.7 Ferroptosis and retinoblastoma
Retinoblastoma (Rb), a malignant neoplasm originating from the precursor cells of the retinal photoreceptors, predominantly affects children under the age of three (277). This tumor presents with a variety of clinical signs and symptoms, which may affect one or both eyes and include conjunctival hyperemia, ocular oedema, corneal swelling, vitreous body turbidity, increased intraocular pressure, and ocular misalignment (278).
Delving into the pathogenesis of Rb, genetic alterations, particularly mutations in the p53 gene and the loss of the RB1 gene, have been pinpointed as pivotal for disease development. The p53 gene, known for its tumor-suppressive properties, modulates the phosphorylation of the RB protein by influencing cyclin-dependent kinases (CDKs), thereby regulating the E2F family of transcription factors, which are integral to the progression of the cell cycle. Disruptions in this regulatory system due to p53 mutations or RB1 gene deletions can lead to the abnormal activation of genes that drive the cell cycle and heightened vulnerability to ferroptosis (221).
Ferroptosis, an emerging form of regulated cell death, is closely linked to disruptions in iron metabolism and increased oxidative stress (279, 280). A significant challenge in Rb treatment is the potential for tumor cells to develop resistance to chemotherapy drugs. However, recent research has indicated that inducing autophagy-dependent ferroptosis can effectively counteract drug resistance in retinoblastoma cells. Itaconic acid derivatives have been shown to enhance ferroptosis by stimulating autophagy, thereby diminishing the activity of caspases, key mediators of apoptosis, and reversing the efficacy of chemotherapeutic agents such as carboplatin, etoposide, and vincristine (220).
By inducing selective ferritinophagy, a process that specifically degrades ferritin within the cell, the reserves of intracellular iron ions can be diminished, thereby heightening the cell’s sensitivity to ferroptosis. 4-Octyl itaconate (4-OI) is a metabolite capable of inducing ferritinophagy-dependent ferroptosis (281). Future studies could explore the possibility of 4-OI inducing ferritinophagy-dependent ferroptosis in Rb.
Furthermore, the therapeutic potential of ferroptosis has been noted in the treatment of other malignancies. In the context of advanced hepatocellular carcinoma (HCC), the RB protein has been shown to facilitate ferroptosis when exposed to sorafenib, thereby hastening the oxidative necrosis of cancer cells (282).
By synthesizing these findings, modulating ferroptosis is evidently a novel therapeutic strategy in oncology. By meticulously controlling the onset of ferroptosis, tumor cell resistance to chemotherapeutic agents can be significantly diminished and the death of cancer cells can be expedited. This approach is particularly pertinent to Rb, given that it is commonly diagnosed in early childhood, necessitating treatments with heightened sensitivity and responsiveness.
Consequently, the modulation of ferroptosis introduces a novel therapeutic perspective for Rb and may also offer novel insights into the treatment of other types of cancer. A deeper understanding of the molecular underpinnings of ferroptosis, coupled with the development of targeted modulators, holds promise for the delivery of more efficacious and targeted treatment regimens for patients afflicted with Rb. This area represents an auspicious frontier with the potential to bring about transformative advances in cancer therapy.
4.8 Ferroptosis and retinal detachment
Retinal detachment (RD) is a significant ophthalmological condition characterized by the separation of the retina from the underlying RPE and choroid (283). The formation of retinal tears or holes is a prevalent cause of this detachment, which, if neglected, can result in permanent vision impairment (284).
Contemporary medical research has detected an increase in iron concentrations and total iron-binding capacity within the vitreous and subretinal fluid as the duration of retinal detachment in patients increases (285). An examination of retinal tissue sections from individuals with RD revealed substantial iron accumulation within the RPE. This accumulation implies a potential correlation between aberrant iron ion levels and the pathogenesis of RD.
According to more in-depth experimental investigations, the administration of transferrin can efficiently mitigate the accumulation of iron ions in the RPE of rodent models of RD (285). This intervention also prevents the nuclear condensation of photoreceptor (PR) cells, thereby providing substantial protection against PR cell damage and reducing the incidence of ferroptosis (222). These findings indicate that iron ions may be partially responsible for PR cell damage during the onset of RD.
The discovery of the role of iron in RD opens the possibility of employing iron chelators as a novel therapeutic approach to diminish the damage associated with RD. The strategic use of iron chelators post-RD could reduce PR cell damage and enhance postoperative visual rehabilitation in patients. This approach introduces a novel avenue of research for the management of the RD prognosis.
The RPE cell layer provides support and nourishment for photoreceptor cells, and the integrity of its tight junctions is crucial for the structure and function of the retina (286). Ferroptosis could disrupt these tight junctions, leading to fluid permeation between the retinal layers, which could compromise the stability of the retina (287). RPE cells are responsible for phagocytosing and digesting the membrane discs of the outer segments of photoreceptor cells, a critical component of the visual cycle. Ferroptosis could possibly affect the phagocytic capability of RPE cells, disrupting the normal renewal of photoreceptor cells. This could potentially hinder the reattachment process and impede visual recovery (170).
In conclusion, iron ions are pivotal contributors to the pathophysiology of RD. The use of iron chelators is a promising therapeutic strategy for RD management. Subsequent studies may further elucidate the interplay between iron metabolism and the development of RD, as well as the prospective therapeutic utility of iron chelators in treatment protocols. This line of research could not only enhance the therapeutic outcomes for RD patients but also offer innovative treatment paradigms for a spectrum of ophthalmic conditions.
5 Therapeutic approaches targeting ferroptosis in ophthalmic disorders
Recently, significant progress has been achieved in the study of ferroptosis, revealing a diverse array of drugs associated with this process. These drugs include compounds derived from natural sources to synthetic small-molecule inhibitors and inducers. These discoveries provide fresh insights for mechanistic studies and potential therapeutic strategies for ferroptosis treatment.
In corneal injury, reducing the levels of GPX4, an antioxidant enzyme in corneal epithelial cells, leads to increased lipid peroxidation and ferroptosis. Fer-1, which inhibits ferroptosis, has been shown to decrease cell damage due to reduced GPX4 levels, suggesting its utility in corneal recovery. Fer-1 has been shown to partially ameliorate the associated pathology, especially in severe corneal injuries such as alkali burns, where ferroptosis significantly impacts tissue damage, as reported by Wang et al. (150). Given the hydrophobic properties of Fer-1, which can hinder its clinical use, researchers have developed a liposomal formulation (Fer-1-NPs) to improve its therapeutic efficacy. This development implies that drugs targeting ferroptosis could offer novel treatment options for corneal injuries, potentially enhancing treatment outcomes for keratopathy and similar conditions.
Cataract treatment can benefit significantly from understanding the association with ferroptosis, which affects the lens’s redox balance and the activity of enzymes in the glutathione pathway (239). Consequently, these changes lead to decreased GPX activity along with the accumulation of ROS and lipid peroxidation byproducts. In a study, mice deficient in GPX1 exhibited signs of age-related cataracts attributed to heightened light scattering within the lens nucleus (163). Additionally, lens epithelial cells are susceptible to ferroptosis, and the level of GSH plays a crucial role in determining their vulnerability (161). Gaining insights into this connection could pave the way for innovative preventative strategies and therapeutic interventions for cataract management.
In glaucoma, the physiological function of RGCs is influenced by iron ions and ferritin. Elevated serum ferritin levels are associated with the risk of glaucoma, and increased IOP can lead to the excessive accumulation of iron in the retina, triggering ferroptosis in RGCs (167, 168). Deferiprone, an iron chelator, has been reported to reduce iron levels and protect RGCs. Moreover, high IOP may induce ferritin degradation, resulting in the release of iron ions and exacerbating RGC injury. However, the inhibition of ferritin degradation by knocking down the NCOA4 gene attenuates RGC damage caused by high IOP (167). These findings indicate that targeting ferroptosis could serve as a novel therapeutic strategy for glaucoma and may offer innovative options for slowing disease progression.
Tang and colleagues developed an innovative iron chelator known as CaPB to address the issue of elevated iron levels in RPE cells and Bruch’s membrane in patients with AMD (182). This compound is characterized by its high biocompatibility and minimal cytotoxic effects, enabling it to selectively target and reduce intracellular bivalent iron concentrations. Additionally, CaPB mitigates oxidative stress by suppressing the expression of specific genes, thus potently inhibiting ferroptosis. This discovery suggests that the modulation of ferroptosis could emerge as a novel therapeutic approach for the treatment and prevention of AMD, providing a promising avenue for the development of new pharmaceutical interventions.
Ferroptosis plays a significant role in the progression of DR by impairing retinal endothelial cells and is associated with the upregulation of the TRIM46 gene in diabetic patients (212, 264). Inhibiting the effect of this gene on GPX4 degradation could alleviate this condition. Furthermore, interventions targeting FABP4 in PPARγ-mediated ferroptosis may decrease the oxidative stress and lipid peroxidation linked to DR (208, 209). These findings indicate that therapies concentrating on ferroptosis pathways could represent novel treatment options for diabetic retinopathy, underscoring the potential of molecular interventions in combating diabetic complications.
RP studies have highlighted the significance of iron metabolism-related genes and oxidative damage in the retina. Therapies targeting iron homeostasis, including zinc deferoxamine, VK28, VAR10303, deferiprone, and ferroptosis inhibitors such as deferoxamine or hepstatin-1 (183, 274–276), have shown promise in slowing photoreceptor cell degeneration in individuals with RP. These findings underscore the importance of iron management in treating RP and suggest innovative approaches to address this degenerative eye disease, potentially leading to more effective treatments for affected individuals.
Targeting ferroptosis in Rb represents a pioneering therapeutic approach. Central to Rb are genetic mutations in the p53 and RB1 genes, which heighten cell susceptibility to ferroptosis (221). Inducing ferroptosis through autophagy has the potential to overcome drug resistance in Rb cells, and itaconic acid derivatives can enhance chemosensitivity to agents such as carboplatin and etoposide (220). The role of the RB protein in promoting ferroptosis has also been observed in HCC, indicating the broader applicability of this strategy.
Elevated iron levels in the vitreous and subretinal fluid of RD patients suggest a link to RD pathogenesis. In rodent models, transferrin administration reduced iron accumulation in RPE cells, prevented photoreceptor cell damage, and decreased ferroptosis, indicating the role of iron in cell damage during RD (222). The potential use of iron chelators post-RD could protect photoreceptor cells and enhance visual rehabilitation, suggesting a novel therapeutic strategy.
Targeting ferroptosis for the treatment of ophthalmic diseases has emerged as a significant area of research. However, current targeted therapies face several challenges. For instance, the cornea’s structure complicates drug delivery, and traditional eye drops have low bioavailability due to factors such as tear dilution and blinking. Nanomedicine drug delivery systems offer an effective strategy for addressing the challenges of ocular drug delivery. By enhancing corneal penetration, achieving sustained drug release, and improving bioavailability, nanomedicine could potentially enhance the therapeutic effect of treatments for eye diseases, thereby improving patients’ quality of life.
The eye’s immune system is distinct from that of other body parts and possesses a unique immune privilege. This means that the eye can avoid triggering a robust immune response, which is crucial for protecting the eye from damage. However, it also implies that therapeutic drugs need to be specially designed (combining targeted drugs with immunosuppressants) to prevent unnecessary inflammation or immune reactions.
Currently, there is limited clinical research on targeting ferroptosis for the treatment of ophthalmic diseases, with most studies focusing on animal experiments. Given the differences between disease models and human biology, understanding the absorption, distribution, metabolism, and excretion of drugs in the human eye is vital for effective treatment.
Finally, as scientists deepen their research on the mechanism of ferroptosis, we anticipate a more comprehensive understanding of ferroptosis as research progresses. This will likely result in more therapeutic targets for patients to choose from in clinical settings.
6 Conclusion
Since ferroptosis was identified in 2012, researchers have expressed a burgeoning interest in understanding its association with various ophthalmic conditions. This manuscript delves into the regulatory mechanisms of ferroptosis and their potential impacts on eye diseases. However, numerous aspects of how ferroptosis is implicated in the regulation of ophthalmic diseases remain largely unknown. Further research is essential to explore the mechanisms and roles of ferroptosis in conditions such as corneal injury, glaucoma, cataract, age-related macular degeneration, diabetic retinopathy, retinal detachment, and retinoblastoma.
The role of cell type and sensitivity in ferroptosis is intricate and tissue-specific. In the context of ocular diseases, this implies that ocular cells may be more prone to ferroptosis, which deviates from its manifestation in other tissues such as tumor cells and neurons. For instance, the retina, the light-sensitive tissue of the eye, contains a large number of neurons and glial cells. In conditions like AMD and certain types of retinal degenerative diseases, retinal cells are more susceptible to the effects of ferroptosis. Ferroptosis drives the progression of these diseases by affecting these cells’ redox balance.
Due to its high energy consumption and highly oxidative physiological characteristics, the retina forms an environment particularly susceptible to oxidative stress. Photoreceptor cells require a substantial amount of energy to maintain the transmission of visual signals. Mitochondria are extremely active in these cells, generating energy while also producing reactive oxygen species (ROS), which heighten oxidative stress.
Moreover, the eyes are directly exposed to light, especially high-energy visible light and ultraviolet rays. Such exposure can promote the generation of ROS and cause oxidative damage to retinal cells. Given that ferroptosis is a form of cell death closely linked to oxidative stress, its significance is even more pronounced in the eyes.
The focus of future research on ferroptosis in ophthalmic diseases involves uncovering its molecular mechanisms, including the regulation of iron ion metabolism and lipid peroxidation. Understanding these mechanisms is critical for identifying key factors in the development of ophthalmic diseases. This knowledge can aid in discovering new therapeutic targets and biomarkers, thereby enabling early diagnosis and monitoring of disease progression.
As our understanding of the molecular mechanisms of ferroptosis deepens, researchers will endeavor to develop new drugs that can target these pathways. These include small molecule inhibitors and inducers, as well as exploring the application of gene editing technology and stem cell therapy in the treatment of ophthalmic diseases. These strategies may offer new approaches to treatment, particularly when conventional treatments prove ineffective.
Furthermore, the advancement of non-invasive diagnostic technology and interdisciplinary collaboration will be pivotal to advancing research on ferroptosis. Non-invasive techniques can empower doctors to intervene at an early stage of the disease, while interdisciplinary collaboration can harness the expertise of professionals from diverse fields to collectively address complex issues in the treatment of ophthalmic diseases. This collaboration can optimize treatment strategies and ultimately deliver more effective treatment plans to patients.
The current manuscript does have certain limitations. For instance, our exploration of ophthalmic diseases is restricted to eight specific types: corneal injury, cataract, glaucoma, AMD, DR, RP, RB, and RD. This does not encompass all ophthalmic diseases potentially related to ferroptosis, such as Graves’ ophthalmopathy (288) and myopia (289). Our literature search was confined to English-language studies, thus excluding research in other languages, including Chinese. The review may inevitably incorporate lower-quality studies, potentially impacting the reliability of the conclusions. There may also be publication bias in the conclusions, as studies with negative or non-significant results may not have been published, possibly skewing the review towards supporting the role of ferroptosis in ophthalmic diseases. Given these limitations, future research should address a broader range of diseases, adopt more rigorous study designs, and conduct more comprehensive investigation into mechanisms.
In summary, ferroptosis is considered a significant form of cell death in ophthalmology. A more comprehensive understanding of ferroptosis may reveal new targets for the diagnosis and treatment of ophthalmic diseases, offering promising avenues for future therapeutic advancements.
Author contributions
YY: Writing – original draft. YL: Writing – original draft. ZH: Writing – original draft, Writing – review & editing. BW: Writing – original draft. WZ: Writing – review & editing. LW: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yuan H, Chen S, Duncan MR, de Rivero Vaccari JP, Keane RW, Dalton Dietrich W, et al. Ic100, a humanized therapeutic monoclonal anti-asc antibody alleviates oxygen-induced retinopathy in mice. Angiogenesis. (2024). doi: 10.1007/s10456-024-09917-9
2. Shi X, Zhou XZ, Chen G, Luo WF, Zhou C, He TJ, et al. Targeting the postsynaptic scaffolding protein Psd-95 enhances bdnf signaling to mitigate depression-like behaviors in mice. Sci Signal. (2024) 17:eadn4556. doi: 10.1126/scisignal.adn4556
3. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. (2021) 6:49. doi: 10.1038/s41392-020-00428-9
4. You W, Knoops K, Berendschot T, Benedikter BJ, Webers CAB, Reutelingsperger CPM, et al. Pgc-1a mediated mitochondrial biogenesis promotes recovery and survival of neuronal cells from cellular degeneration. Cell Death Discov. (2024) 10:180. doi: 10.1038/s41420-024-01953-0
5. Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. (1980) 255:2372–6. doi: 10.1016/S0021-9258(19)85901-X
6. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
7. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
8. Shen Q, Liang M, Yang F, Deng YZ, Naqvi NI. Ferroptosis contributes to developmental cell death in rice blast. New Phytol. (2020) 227:1831–46. doi: 10.1111/nph.16636
9. Jenkins NL, James SA, Salim A, Sumardy F, Speed TP, Conrad M, et al. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging caenorhabditis elegans. Elife. (2020) 9:e56580. doi: 10.7554/eLife.56580
10. Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. (2019) 10:449. doi: 10.1038/s41419-019-1678-y
11. Xu S, He Y, Lin L, Chen P, Chen M, Zhang S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. (2021) 12:289. doi: 10.1038/s41419-021-03559-1
12. Zhang Z, Yang Z, Wang S, Wang X, Mao J. Decoding ferroptosis: revealing the hidden assassin behind cardiovascular diseases. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 176:116761. doi: 10.1016/j.biopha.2024.116761
13. Li Z, Zhang Y, Ji M, Wu C, Zhang Y, Ji S. Targeting ferroptosis in neuroimmune and neurodegenerative disorders for the development of novel therapeutics. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 176:116777. doi: 10.1016/j.biopha.2024.116777
14. Zhang F, Xiang Y, Ma Q, Guo E, Zeng X. A deep insight into ferroptosis in lung disease: facts and perspectives. Front Oncol. (2024) 14:1354859. doi: 10.3389/fonc.2024.1354859
15. Miao R, Fang X, Zhang Y, Wei J, Zhang Y, Tian J. Iron metabolism and ferroptosis in type 2 diabetes mellitus and complications: mechanisms and therapeutic opportunities. Cell Death Dis. (2023) 14:186. doi: 10.1038/s41419-023-05708-0
16. Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, et al. Targeting ferroptosis in bone-related diseases: facts and perspectives. J Inflamm Res. (2023) 16:4661–77. doi: 10.2147/JIR.S432111
17. Han Z, Luo Y, Chen H, Zhang G, You L, Zhang M, et al. A deep insight into ferroptosis in renal disease: facts and perspectives. Kidney Dis (Basel Switzerland). (2024) 10:224–36. doi: 10.1159/000538106
18. Chen Y, Yang X, Li H, Wu X, Wu W, Chen J, et al. Self-assembled fe-phenolic acid network synergizes with ferroptosis to enhance tumor nanotherapy. Small (Weinheim an der Bergstrasse Germany). (2024), e2402073. doi: 10.1002/smll.202402073
19. Guo M, Zhu Y, Shi Y, Meng X, Dong X, Zhang H, et al. Inhibition of ferroptosis promotes retina ganglion cell survival in experimental optic neuropathies. Redox Biol. (2022) 58:102541. doi: 10.1016/j.redox.2022.102541
20. Shu W, Baumann BH, Song Y, Liu Y, Wu X, Dunaief JL. Ferrous but not ferric iron sulfate kills photoreceptors and induces photoreceptor-dependent rpe autofluorescence. Redox Biol. (2020) 34:101469. doi: 10.1016/j.redox.2020.101469
21. Zhuo Cai J, Lan Yu Y, Biao Yang Z, Xun Xu X, Chun Lv G, Lian Xu C, et al. Synergistic improvement of humus formation in compost residue by fenton-like and effective microorganism composite agents. Bioresour Technol. (2024) 400:130703. doi: 10.1016/j.biortech.2024.130703
22. Li J, Li Y, Wang D, Liao R, Wu Z. Plag1 interacts with gpx4 to conquer vulnerability to sorafenib induced ferroptosis through a Pvt1/Mir-195-5p axis-dependent manner in hepatocellular carcinoma. J Exp Clin Cancer research: CR. (2024) 43:143. doi: 10.1186/s13046-024-03061-4
23. Li H, Cao Z, Liu C, Wang Y, Wang L, Tang Y, et al. Quercetin inhibits neuronal pyroptosis and ferroptosis by modulating microglial M1/M2 polarization in atherosclerosis. J Agric Food Chem. (2024) 72:12156–70. doi: 10.1021/acs.jafc.4c01134
24. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. (2013) 13:342–55. doi: 10.1038/nrc3495
25. Pandey P, Elsori D, Kumar R, Lakhanpal S, Rautela I, Alqahtani TM, et al. Ferroptosis targeting natural compounds as a promising approach for developing potent liver cancer agents. Front Pharmacol. (2024) 15:1399677. doi: 10.3389/fphar.2024.1399677
26. Wolff NA, Liu W, Fenton RA, Lee W-K, Thévenod F, Smith CP. Ferroportin 1 is expressed basolaterally in rat kidney proximal tubule cells and iron excess increases its membrane trafficking. J Cell Mol Med. (2011) 15:209–19. doi: 10.1111/jcmm.2011.15.issue-2
27. Asshoff M, Petzer V, Warr MR, Haschka D, Tymoszuk P, Demetz E, et al. Momelotinib inhibits Acvr1/Alk2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. (2017) 129:1823–30. doi: 10.1182/blood-2016-09-740092
28. Zhao T, Guo X, Sun Y. Iron accumulation and lipid peroxidation in the aging retina: implication of ferroptosis in age-related macular degeneration. Aging Dis. (2021) 12:529–51. doi: 10.14336/AD.2020.0912
29. Liu D, Liu Z, Liao H, Chen ZS, Qin B. Ferroptosis as a potential therapeutic target for age-related macular degeneration. Drug Discov Today. (2024) 29:103920. doi: 10.1016/j.drudis.2024.103920
30. Mao C, Lei G, Horbath A, Wang M, Lu Z, Yan Y, et al. Unraveling etc complex I function in ferroptosis reveals a potential ferroptosis-inducing therapeutic strategy for lkb1-deficient cancers. Mol Cell. (2024) 84:1964–79.e6. doi: 10.1016/j.molcel.2024.04.009
31. Huang X, Yang X, Zhang M, Li T, Zhu K, Dong Y, et al. Selenoi functions as a key modulator of ferroptosis pathway in colitis and colorectal cancer. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2024), e2404073. doi: 10.1002/advs.202404073
32. Maiorino M, Conrad M, Ursini F. Gpx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. (2018) 29:61–74. doi: 10.1089/ars.2017.7115
33. Xiong M, Ou C, Yu C, Qiu J, Lu J, Fu C, et al. Qi-shen-Tang alleviates retinitis pigmentosa by inhibiting ferroptotic features via the Nrf2/Gpx4 signaling pathway. Heliyon. (2023) 9:e22443. doi: 10.1016/j.heliyon.2023.e22443
34. Niu T, Shi X, Liu X, Wang H, Liu K, Xu Y. Porous Se@Sio(2) nanospheres alleviate diabetic retinopathy by inhibiting excess lipid peroxidation and inflammation. Mol Med (Cambridge Mass). (2024) 30:24. doi: 10.1186/s10020-024-00785-z
35. Renner N, Schöb F, Pape R, Suciu I, Spreng AS, Ückert AK, et al. Modeling ferroptosis in human dopaminergic neurons: pitfalls and opportunities for neurodegeneration research. Redox Biol. (2024) 73:103165. doi: 10.1016/j.redox.2024.103165
36. Han Z, Luo Y, Chen H, Zhang G, You L, Zhang M, et al. A deep insight into ferroptosis in renal disease: facts and perspectives. Kidney Dis. (2024) 10:1–13. doi: 10.1159/000538106%JKidneyDiseases
37. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by Gpx4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
38. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-ras-harboring cancer cells. Chem Biol. (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
39. Zhang J, Ruiz M, Bergh PO, Henricsson M, Stojanović N, Devkota R, et al. Regulation of meiotic telomere dynamics through membrane fluidity promoted by Adipor2-Elovl2. Nat Commun. (2024) 15:2315. doi: 10.1038/s41467-024-46718-6
40. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. (2016) 113:E4966–E75. doi: 10.1073/pnas.1603244113
41. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
42. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic pes navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
43. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. Acsl4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
44. Lapaquette P, Terrat S, Proukhnitzky L, Martine L, Grégoire S, Buteau B, et al. Long-term intake of lactobacillus helveticus enhances bioavailability of omega-3 fatty acids in the mouse retina. NPJ biofilms microbiomes. (2024) 10:4. doi: 10.1038/s41522-023-00474-5
45. Perus M, Courtaut F, Pais de Barros JP, Aires V, Hermetet F, Delmas D. Vegf-R2/Cav-1 interaction induced by resveratrol/eicosapentaenoic acid/docosahexaenoic acid-enriched formulation through functional detergent-resistant membranes is associated with decreased vegf-a release in Arpe-19 cells. Mol Nutr Food Res. (2024), e2300893. doi: 10.1002/mnfr.202300893
46. Xue P, Zhuang H, Bai T, Zeng X, Deng J, Shao S, et al. Iron (Ii)-based metal-organic framework nanozyme for boosting tumor ferroptosis through inhibiting DNA damage repair and system Xc(). J Nanobiotechnol. (2024) 22:228. doi: 10.1186/s12951-024-02508-2
47. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. (1999) 274:11455–8. doi: 10.1074/jbc.274.17.11455
48. Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. (1982) 710:197–211. doi: 10.1016/0005-2760(82)90150-3
49. He Q, Lin Y, Chen B, Chen C, Zeng J, Dou X, et al. Vitamin K2 ameliorates osteoarthritis by suppressing ferroptosis and extracellular matrix degradation through activation Gpx4's dual functions. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 175:116697. doi: 10.1016/j.biopha.2024.116697
50. Yang Y, Wang Y, Deng Y, Lu J, Xiao L, Li J, et al. Fructus lycii and salvia miltiorrhiza bunge extract attenuate oxidative stress-induced photoreceptor ferroptosis in retinitis pigmentosa. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 167:115547. doi: 10.1016/j.biopha.2023.115547
51. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the P62-keap1-Nrf2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
52. Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. (2019) 23:4900–12. doi: 10.1111/jcmm.14511
53. Yao D, Bao L, Wang S, Tan M, Xu Y, Wu T, et al. Isoliquiritigenin alleviates myocardial ischemia-reperfusion injury by regulating the Nrf2/Ho-1/Slc7a11/Gpx4 axis in mice. Free Radical Biol Med. (2024) 221:1–12. doi: 10.1016/j.freeradbiomed.2024.05.012
54. Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, et al. Ferroptosis as a P53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
55. Alsabaani NA, Amawi K, Eleawa SM, Nabeel Ibrahim W, Aldhaban W, Alaraj AM, et al. Nrf-2-dependent antioxidant and anti-inflammatory effects underlie the protective effect of esculeoside a against retinal damage in streptozotocin-induced diabetic rats. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 173:116461. doi: 10.1016/j.biopha.2024.116461
56. Chen Y, Tong J, Liu C, He C, Xiang J, Yao G, et al. Msc-derived small extracellular vesicles mitigate diabetic retinopathy by stabilizing Nrf2 through mir-143-3p-mediated inhibition of neddylation. Free Radical Biol Med. (2024) 219:76–87. doi: 10.1016/j.freeradbiomed.2024.04.216
57. Feng J, Ji K, Pan Y, Huang P, He T, Xing Y. Resveratrol ameliorates retinal ischemia-reperfusion injury by modulating the Nlrp3 inflammasome and Keap1/Nrf2/Ho-1 signaling pathway. Mol Neurobiol. (2024). doi: 10.1007/s12035-024-04105-8
58. Zhang ZY, Yang ZH, Wang S, Feng SL, Wang XL, Mao JY. Regulation of optimized new shengmai powder on cardiomyocyte apoptosis and ferroptosis in ischemic heart failure rats: the mediating role of phosphatidylinositol-3-kinase/protein kinase B/tumor protein 53 signaling pathway. J Ethnopharmacol. (2024) 330:118264. doi: 10.1016/j.jep.2024.118264
59. Wang S-J, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for P53-mediated ferroptosis and tumor suppression. Cell Rep. (2016) 17:366–73. doi: 10.1016/j.celrep.2016.09.022
60. Lara O, Janssen P, Mambretti M, De Pauw L, Ates G, Mackens L, et al. Compartmentalized role of xct in supporting pancreatic tumor growth, inflammation and mood disturbance in mice. Brain behavior Immun. (2024) 118:275–86. doi: 10.1016/j.bbi.2024.03.001
61. Deng J, Lin X, Qin J, Li Q, Zhang Y, Zhang Q, et al. Sptbn2 suppresses ferroptosis in nsclc cells by facilitating Slc7a11 membrane trafficking and localization. Redox Biol. (2024) 70:103039. doi: 10.1016/j.redox.2024.103039
62. Liu W, Liu C, Xiao J, Qian C, Chen Z, Lin W, et al. Htra1 interacts with slc7a11 to modulate colorectal cancer chemosensitivity by inhibiting ferroptosis. Cell Death Discov. (2024) 10:228. doi: 10.1038/s41420-024-01993-6
63. Wang S, Zhao Y, Yao F, Wei P, Ma L, Zhang S. An anti-Gd2 aptamer-based bifunctional spherical nucleic acid nanoplatform for synergistic therapy targeting mdm2 for retinoblastoma. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 174:116437. doi: 10.1016/j.biopha.2024.116437
64. Zhang S, Wu J, Wang L, Mu L, Xu X, Li J, et al. Sirt1/P53 in retinal pigment epithelial cells in diabetic retinopathy: A gene co-expression analysis and He-Ying-Qing-Re formula treatment. Front Mol Biosci. (2024) 11:1366020. doi: 10.3389/fmolb.2024.1366020
65. Chang YY, Wang M, Yeh JH, Tsou SC, Chen TC, Hsu MY, et al. The Protective Effects of Beta-Mangostin against Sodium Iodate-Induced Retinal Ros-Mediated Apoptosis through Mek/Erk and P53 Signaling Pathways. Food Funct. (2023) 14:10896–909. doi: 10.1039/D3FO03568A
66. Zhang M, Bi X. Heat shock proteins and breast cancer. Int J Mol Sci. (2024) 25:876. doi: 10.3390/ijms25020876
67. Feng Y, Chen Q, Jin C, Ruan Y, Chen Q, Lin W, et al. Microwave-activated cu-doped zirconium metal-organic framework for a highly effective combination of microwave dynamic and thermal therapy. J Controlled release. (2023) 361:102–14. doi: 10.1016/j.jconrel.2023.07.046
68. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. Hspb1 as a novel regulator of ferroptotic cancer cell death. Oncogene. (2015) 34:5617–25. doi: 10.1038/onc.2015.32
69. Xu X, Wei Y, Hua H, Jing X, Zhu H, Xiao K, et al. Polyphenols sourced from ilex latifolia thunb. Relieve intestinal injury via modulating ferroptosis in weanling piglets under oxidative stress. Antioxidants (Basel Switzerland). (2022) 11:966. doi: 10.3390/antiox11050966
70. Zhu X, Zuo Q, Xie X, Chen Z, Wang L, Chang L, et al. Rocaglamide regulates iron homeostasis by suppressing hepcidin expression. Free Radical Biol Med. (2024) 219:153–62. doi: 10.1016/j.freeradbiomed.2024.04.232
71. Meng X, Peng X, Ouyang W, Li H, Na R, Zhou W, et al. Musashi-2 deficiency triggers colorectal cancer ferroptosis by downregulating the mapk signaling cascade to inhibit Hspb1 phosphorylation. Biol procedures Online. (2023) 25:32. doi: 10.1186/s12575-023-00222-1
72. Zhu S, Zhang Q, Sun X, Zeh HJ, Lotze MT, Kang R, et al. Hspa5 regulates ferroptotic cell death in cancer cells. Cancer Res. (2017) 77:2064–77. doi: 10.1158/0008-5472.CAN-16-1979
73. Jiang K, Fairless E, Kanda A, Gotoh N, Cogliati T, Li T, et al. Divergent effects of Hsp70 overexpression in photoreceptors during inherited retinal degeneration. Invest Ophthalmol Visual Sci. (2020) 61:25. doi: 10.1167/iovs.61.12.25
74. Geyer O, Levo Y. Glaucoma is an autoimmune disease. Autoimmun Rev. (2020) 19:102535. doi: 10.1016/j.autrev.2020.102535
75. Tsai T, Grotegut P, Reinehr S, Joachim SC. Role of heat shock proteins in glaucoma. Int J Mol Sci. (2019) 20:5160. doi: 10.3390/ijms20205160
76. Chen H, Wang C, Liu Z, He X, Tang W, He L, et al. Ferroptosis and its multifaceted role in cancer: mechanisms and therapeutic approach. Antioxidants (Basel Switzerland). (2022) 11:1504. doi: 10.3390/antiox11081504
77. Yang N, Pan X, Zhou X, Liu Z, Yang J, Zhang J, et al. Biomimetic nanoarchitectonics with chitosan nanogels for collaborative induction of ferroptosis and anticancer immunity for cancer therapy. Adv Healthc Mater. (2024) 13:e2302752. doi: 10.1002/adhm.202302752
78. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The coq oxidoreductase fsp1 acts parallel to Gpx4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
79. Nakamura T, Hipp C, Santos Dias Mourão A, Borggräfe J, Aldrovandi M, Henkelmann B, et al. Phase separation of Fsp1 promotes ferroptosis. Nature. (2023) 619:371–7. doi: 10.1038/s41586-023-06255-6
80. Elguindy MM, Nakamaru-Ogiso E. Apoptosis-inducing factor (Aif) and its family member protein, amid, are rotenone-sensitive Nadh : ubiquinone oxidoreductases (Ndh-2). J Biol Chem. (2015) 290:20815–26. doi: 10.1074/jbc.M115.641498
81. Yang M, Tsui MG, Tsang JKW, Goit RK, Yao KM, So KF, et al. Involvement of Fsp1-Coq(10)-Nadh and Gsh-Gpx-4 pathways in retinal pigment epithelium ferroptosis. Cell Death Dis. (2022) 13:468. doi: 10.1038/s41419-022-04924-4
82. Liu T, Zhao J, Lin C. Sprouty-related proteins with Evh1 domain (Spred2) prevents high-glucose induced endothelial-mesenchymal transition and endothelial injury by suppressing mapk activation. Bioengineered. (2022) 13:13882–92. doi: 10.1080/21655979.2022.2086351
83. Hardie DG. Amp-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. (2011) 25:1895–908. doi: 10.1101/gad.17420111
84. Wu Y, Jia C, Liu W, Zhan W, Chen Y, Lu J, et al. Sodium citrate targeting Ca(2+)/Camkk2 pathway exhibits anti-tumor activity through inducing apoptosis and ferroptosis in ovarian cancer. J Adv Res. (2024). doi: 10.1016/j.jare.2024.04.033
85. Han D, Jiang L, Gu X, Huang S, Pang J, Wu Y, et al. Sirt3 deficiency is resistant to autophagy-dependent ferroptosis by inhibiting the ampk/mtor pathway and promoting Gpx4 levels. J Cell Physiol. (2020) 235:8839–51. doi: 10.1002/jcp.29727
86. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. Ampk-mediated becn1 phosphorylation promotes ferroptosis by directly blocking system X activity. Curr Biol. (2018) 28:2388–99. doi: 10.1016/j.cub.2018.05.094
87. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z, et al. Hcar1/Mct1 regulates tumor ferroptosis through the lactate-mediated ampk-Scd1 activity and its therapeutic implications. Cell Rep. (2020) 33:108487. doi: 10.1016/j.celrep.2020.108487
88. Herzig S, Shaw RJ. Ampk: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
89. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. Dha inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radical Biol Med. (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
90. Zhu H-Y, Huang Z-X, Chen G-Q, Sheng F, Zheng Y-S. Typhaneoside prevents acute myeloid leukemia (Aml) through suppressing proliferation and inducing ferroptosis associated with autophagy. Biochem Biophys Res Commun. (2019) 516:1265–71. doi: 10.1016/j.bbrc.2019.06.070
91. Li C, Dong X, Du W, Shi X, Chen K, Zhang W, et al. Lkb1-ampk axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther. (2020) 5:187. doi: 10.1038/s41392-020-00297-2
92. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
93. Fomo KN, Perumal N, Manicam C, Pfeiffer N, Grus FH. Neuroretinal cell culture model as a tool for the development of new therapeutic approaches for oxidative stress-induced ocular diseases, with a focus on glaucoma. Cells. (2024) 13:775. doi: 10.3390/cells13090775
94. Dieguez HH, Romeo HE, Alaimo A, Bernal Aguirre NA, Calanni JS, Adán Aréan JS, et al. Mitochondrial quality control in non-exudative age-related macular degeneration: from molecular mechanisms to structural and functional recovery. Free Radical Biol Med. (2024) 219:17–30. doi: 10.1016/j.freeradbiomed.2024.03.024
95. Feng YQ, Liu X, Zuo N, Yu MB, Bian WM, Han BQ, et al. Nad(+) precursors promote the restoration of spermatogenesis in busulfan-treated mice through inhibiting Sirt2-regulated ferroptosis. Theranostics. (2024) 14:2622–36. doi: 10.7150/thno.92416
96. He L, Habibovic P, van Rijt S. Selenium-incorporated mesoporous silica nanoparticles for osteosarcoma therapy. Biomater Sci. (2023) 11:3828–39. doi: 10.1039/D2BM02102A
97. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify nadph as biomarker for ferroptosis sensitivity. Cell Chem Biol. (2016) 23:225–35. doi: 10.1016/j.chembiol.2015.11.016
98. Zhao X, Zhao Y, Zeng QY, Liu CJ. Cytochrome B5 diversity in green lineages preceded the evolution of syringyl lignin biosynthesis. Plant Cell. (2024). doi: 10.1093/plcell/koae120
99. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. (2020) 16:302–9. doi: 10.1038/s41589-020-0472-6
100. Ghosh MK, Mukhopadhyay M, Chatterjee IB. Nadph-initiated cytochrome P450-dependent free iron-independent microsomal lipid peroxidation: specific prevention by ascorbic acid. Mol Cell Biochem. (1997) 166:35–44. doi: 10.1023/A:1006841228483
101. Liu N, Wu WL, Wan XR, Wang J, Huang JN, Jiang YY, et al. Regulation of fsp1 myristoylation by nadph: A novel mechanism for ferroptosis inhibition. Redox Biol. (2024) 73:103176. doi: 10.1016/j.redox.2024.103176
102. Shi X, Li P, Herb M, Liu H, Wang M, Wang X, et al. Pathological high intraocular pressure induces glial cell reactive proliferation contributing to neuroinflammation of the blood-retinal barrier via the Nox2/Et-1 Axis-controlled Erk1/2 pathway. J Neuroinflamm. (2024) 21:105. doi: 10.1186/s12974-024-03075-x
103. Liao J, Lai Z, Huang G, Lin J, Huang W, Qin Y, et al. Setanaxib mitigates oxidative damage following retinal ischemia-reperfusion via Nox1 and Nox4 inhibition in retinal ganglion cells. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 170:116042. doi: 10.1016/j.biopha.2023.116042
104. Liu Z, Kang R, Yang N, Pan X, Yang J, Yu H, et al. Tetrahydrobiopterin inhibitor-based antioxidant metabolic strategy for enhanced cancer ferroptosis-immunotherapy. J colloid Interface Sci. (2024) 658:100–13. doi: 10.1016/j.jcis.2023.12.042
105. Jiang X, Shao Y, Liao Y, Zheng X, Peng M, Cai Y, et al. Mechanisms underlying the efficacy and limitation of dopa and tetrahydrobiopterin therapies for the deficiency of Gtp cyclohydrolase 1 revealed in a novel mouse model. Eur J Pharmacol. (2024) 967:176379. doi: 10.1016/j.ejphar.2024.176379
106. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. Gtp cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
107. Lohmann K, Redin C, Tönnies H, Bressman SB, Subero JIM, Wiegers K, et al. Complex and dynamic chromosomal rearrangements in a family with seemingly non-mendelian inheritance of dopa-responsive dystonia. JAMA Neurol. (2017) 74:806–12. doi: 10.1001/jamaneurol.2017.0666
108. Martin-Sanchez D, Fontecha-Barriuso M, Martinez-Moreno JM, Ramos AM, Sanchez-Niño MD, Guerrero-Hue M, et al. Ferroptosis and kidney disease. Nefrologia (Engl Ed). (2020) 40:384–94. doi: 10.1016/j.nefro.2020.03.005
109. Wang J, Liu Y, Wang Y, Sun L. The cross-link between ferroptosis and kidney diseases. Oxid Med Cell Longev. (2021) 2021:6654887. doi: 10.1155/2021/6654887
110. Tang S, Xiao X. Ferroptosis and kidney diseases. Int Urol Nephrol. (2020) 52:497–503. doi: 10.1007/s11255-019-02335-7
111. Radaelli E, Assenmacher CA, Verrelle J, Banerjee E, Manero F, Khiati S, et al. Mitochondrial defects caused by parl deficiency lead to arrested spermatogenesis and ferroptosis. eLife. (2023) 12:e84710. doi: 10.7554/eLife.84710
112. Li Y, Zhou Y, Liu D, Wang Z, Qiu J, Zhang J, et al. Glutathione peroxidase 3 induced mitochondria-mediated apoptosis via Ampk /Erk1/2 pathway and resisted autophagy-related ferroptosis via Ampk/Mtor pathway in hyperplastic prostate. J Trans Med. (2023) 21:575. doi: 10.1186/s12967-023-04432-9
113. Mazur O, Bałdysz S, Warowicka A, Nawrot R. Tap the sap - investigation of latex-bearing plants in the search of potential anticancer biopharmaceuticals. Front Plant Sci. (2022) 13:979678. doi: 10.3389/fpls.2022.979678
114. Lee RM, Choi H, Shin JS, Kim K, Yoo KH. Distinguishing between apoptosis and necrosis using a capacitance sensor. Biosensors bioelectronics. (2009) 24:2586–91. doi: 10.1016/j.bios.2009.01.028
115. Ohno Y, Yagi H, Nakamura M, Masuko K, Hashimoto Y, Masuko T. Simultaneous induction of apoptotic, autophagic, and necrosis-like cell death by monoclonal antibodies recognizing chicken transferrin receptor. Biochem Biophys Res Commun. (2008) 367:775–81. doi: 10.1016/j.bbrc.2008.01.030
116. den Hartigh AB, Loomis WP, Anderson MJ, Frølund B, Fink SL. Muscimol inhibits plasma membrane rupture and ninjurin-1 oligomerization during pyroptosis. Commun Biol. (2023) 6:1010. doi: 10.1038/s42003-023-05354-4
117. Wang J, Choi WG, Nguyen NK, Liu D, Kim SH, Lim D, et al. Cytoplasmic ca(2+) influx mediates iron- and reactive oxygen species-dependent ferroptotic cell death in rice immunity. Front Plant Sci. (2024) 15:1339559. doi: 10.3389/fpls.2024.1339559
118. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
119. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
120. Tojo K, Yamamoto N, Tamada N, Mihara T, Abe M, Nishii M, et al. Early alveolar epithelial cell necrosis is a potential driver of Covid-19-induced acute respiratory distress syndrome. iScience. (2023) 26:105748. doi: 10.1016/j.isci.2022.105748
121. Shadab A, Abbasi-Kolli M, Saharkhiz M, Ahadi SH, Shokouhi B, Nahand JS. The interplay between mitochondrial dysfunction and Nlrp3 inflammasome in multiple sclerosis: therapeutic implications and animal model studies. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 175:116673. doi: 10.1016/j.biopha.2024.116673
122. Ma J, Yuan H, Zhang J, Sun X, Yi L, Li W, et al. An ultrasound-activated nanoplatform remodels tumor microenvironment through diverse cell death induction for improved immunotherapy. J Controlled release. (2024) 370:501–15. doi: 10.1016/j.jconrel.2024.05.001
123. Yu M, Huo D, Yu K, Zhou K, Xu F, Meng Q, et al. Crosstalk of different cell-death patterns predicts prognosis and drug sensitivity in glioma. Comput Biol Med. (2024) 175:108532. doi: 10.1016/j.compbiomed.2024.108532
124. Chen F, Lin J, Kang R, Tang D, Liu J. Alkaliptosis induction counteracts paclitaxel-resistant ovarian cancer cells via Atp6v0d1-mediated Abcb1 inhibition. Mol carcinogenesis. (2024). doi: 10.1002/mc.23741
125. Chen B, Fu W, Jie C, Zhang G, Li Z, Liu Y, et al. Gpx7 reduces chondrocyte inflammation and extracellular matrix degradation triggered by il−1β, via a mechanism mediated by ferroptosis. Mol Med Rep. (2024) 30:118. doi: 10.3892/mmr.2024.13242
126. Li W, Shi J, Yu Z, Garcia-Gabilondo M, Held A, Huang L, et al. Slc22a17 as a cell death-linked regulator of tight junctions in cerebral ischemia. Stroke. (2024) 55:1650–9. doi: 10.1161/STROKEAHA.124.046736
127. He C, Gu X, Dong C, Xu Z, Liu L, Jiang B, et al. The association between ferroptosis-related patterns and tumor microenvironment in colorectal cancer. Int Immunopharmacol. (2024) 134:112258. doi: 10.1016/j.intimp.2024.112258
128. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. (2019) 12:34. doi: 10.1186/s13045-019-0720-y
129. Zheng D-W, Lei Q, Zhu J-Y, Fan J-X, Li C-X, Li C, et al. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. (2017) 17:284–91. doi: 10.1021/acs.nanolett.6b04060
130. Chen Z, Liu B, Zhou D, Lei M, Yang J, Hu Z, et al. Aqp4 regulates ferroptosis and oxidative stress of muller cells in diabetic retinopathy by regulating trpv4. Exp Cell Res. (2024) 439:114087. doi: 10.1016/j.yexcr.2024.114087
131. López-Gil JC, García-Silva S, Ruiz-Cañas L, Navarro D, Palencia-Campos A, Giráldez-Trujillo A, et al. The peptidoglycan recognition protein 1 confers immune evasive properties on pancreatic cancer stem cells. Gut. (2024). doi: 10.1136/gutjnl-2023-330995
132. Geng YQ, Qiu LN, Cheng YQ, Li JJ, Ma YL, Zhao CC, et al. Alleviating recombinant tissue plasminogen activator-induced hemorrhagic transformation in ischemic stroke via targeted delivery of a ferroptosis inhibitor. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2024), e2309517. doi: 10.1002/advs.202309517
133. Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. (2017) 48:1033–43. doi: 10.1161/STROKEAHA.116.015609
134. Müller T, Dewitz C, Schmitz J, Schröder AS, Bräsen JH, Stockwell BR, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. (2017) 74:3631–45. doi: 10.1007/s00018-017-2547-4
135. Ma L, Chen C, Zhao C, Li T, Ma L, Jiang J, et al. Targeting carnitine palmitoyl transferase 1a (Cpt1a) induces ferroptosis and synergizes with immunotherapy in lung cancer. Signal Transduct Target Ther. (2024) 9:64. doi: 10.1038/s41392-024-01772-w
136. Huang L, Zhang L, Zhang Z, Tan F, Ma Y, Zeng X, et al. Loss of nephric augmenter of liver regeneration facilitates acute kidney injury via Acsl4-mediated ferroptosis. J Cell Mol Med. (2024) 28:e18076. doi: 10.1111/jcmm.18076
137. Chen J, Jin Z, Zhang S, Zhang X, Li P, Yang H, et al. Arsenic trioxide elicits prophylactic and therapeutic immune responses against solid tumors by inducing necroptosis and ferroptosis. Cell Mol Immunol. (2023) 20:51–64. doi: 10.1038/s41423-022-00956-0
138. Tonnus W, Linkermann A. "Death is my heir"–ferroptosis connects cancer pharmacogenomics and ischemia-reperfusion injury. Cell Chem Biol. (2016) 23:202–3. doi: 10.1016/j.chembiol.2016.02.005
139. Meng X, Peng F, Yu S, Chi X, Wang W, Shao S. Knockdown of nadk promotes luad ferroptosis via Nadph/Fsp1 axis. J Cancer Res Clin Oncol. (2024) 150:228. doi: 10.1007/s00432-024-05752-z
140. Zhang Y, Liu P, Yang S, Lan J, Xu H, Jiang H, et al. Nogo-B promotes endoplasmic reticulum stress-mediated autophagy in endothelial cells of diabetic nephropathy. Antioxidants Redox Signaling. (2024). doi: 10.1089/ars.2023.0490
141. Li M, Lei P, Shuang S, Dong C, Zhang L. Visualization of polarity changes in endoplasmic reticulum (Er) autophagy and rheumatoid arthritis mice with near-infrared er-targeted fluorescent probe. Talanta. (2024) 275:126141. doi: 10.1016/j.talanta.2024.126141
142. Patergnani S, Bataillard MS, Danese A, Alves S, Cazevieille C, Valéro R, et al. The wolfram-like variant Wfs1(E864k) destabilizes mam and compromises autophagy and mitophagy in human and mice. Autophagy. (2024), 1–12. doi: 10.1080/15548627.2024.2341588
143. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies ncoa4 as the cargo receptor mediating ferritinophagy. Nature. (2014) 509:105–9. doi: 10.1038/nature13148
144. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
145. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
146. Liu J, Hu Z, Ma Q, Wang S, Liu D. Ferritin-dependent cellular autophagy pathway promotes ferroptosis in beef during cold storage. Food Chem. (2023) 412:135550. doi: 10.1016/j.foodchem.2023.135550
147. Zhong G, Qiao B, He Y, Liu H, Hong P, Rao G, et al. Co-exposure of arsenic and polystyrene-nanoplastics induced kidney injury by disrupting mitochondrial homeostasis and mtros-mediated ferritinophagy and ferroptosis. Pesticide Biochem Physiol. (2024) 201:105904. doi: 10.1016/j.pestbp.2024.105904
148. Sakai O, Uchida T, Imai H, Ueta T. Glutathione peroxidase 4 plays an important role in oxidative homeostasis and wound repair in corneal epithelial cells. FEBS Open Bio. (2016) 6:1238–47. doi: 10.1002/2211-5463.12141
149. Kubota M, Shimmura S, Kubota S, Miyashita H, Kato N, Noda K, et al. Hydrogen and N-acetyl-L-cysteine rescue oxidative stress-induced angiogenesis in a mouse corneal alkali-burn model. Invest Ophthalmol Visual Sci. (2011) 52:427–33. doi: 10.1167/iovs.10-6167
150. Wang K, Jiang L, Zhong Y, Zhang Y, Yin Q, Li S, et al. Ferrostatin-1-loaded liposome for treatment of corneal alkali burn via targeting ferroptosis. Bioengineering Trans Med. (2022) 7:e10276. doi: 10.1002/btm2.10276
151. Kandhari K, Kant R, Mishra N, Agarwal C, Agarwal R. Phenylarsine oxide induced corneal injury involves oxidative stress mediated unfolded protein response and ferroptotic cell death: amelioration by Nac. Free Radical Biol Med. (2023) 209:265–81. doi: 10.1016/j.freeradbiomed.2023.10.409
152. Xiong X, Jiang H, Liao Y, Du Y, Zhang Y, Wang Z, et al. Liposome-trimethyl chitosan nanoparticles codeliver insulin and sivegf to treat corneal alkali burns by inhibiting ferroptosis. Bioengineering Trans Med. (2023) 8:e10499. doi: 10.1002/btm2.10499
153. Otsu W, Ishida K, Chinen N, Nakamura S, Shimazawa M, Tsusaki H, et al. Cigarette smoke extract and heated tobacco products promote ferritin cleavage and iron accumulation in human corneal epithelial cells. Sci Rep. (2021) 11:18555. doi: 10.1038/s41598-021-97956-3
154. Mi Y, Wei C, Sun L, Liu H, Zhang J, Luo J, et al. Melatonin inhibits ferroptosis and delays age-related cataract by regulating Sirt6/P-Nrf2/Gpx4 and Sirt6/Ncoa4/Fth1 pathways. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 157:114048. doi: 10.1016/j.biopha.2022.114048
155. Wang L, Liu J, Ma D, Zhi X, Li L, Li S, et al. Glycine recalibrates iron homeostasis of lens epithelial cells by blocking lysosome-dependent ferritin degradation. Free Radical Biol Med. (2024) 210:258–70. doi: 10.1016/j.freeradbiomed.2023.11.020
156. Ma DY, Liu JX, Wang LD, Zhi XY, Luo L, Zhao JY, et al. Gsk-3β-dependent nrf2 antioxidant response modulates ferroptosis of lens epithelial cells in age-related cataract. Free Radical Biol Med. (2023) 204:161–76. doi: 10.1016/j.freeradbiomed.2023.04.022
157. Zhang X, Zheng C, Zhao J, Xu X, Yao J. Lncrna meg3 regulates ferroptosis of lens epithelial cells via Ptbp1/Gpx4 axis to participate in age-related cataract. J Cell Physiol. (2024). doi: 10.1002/jcp.31330
158. Kong D, Liu Y, Li L, Wang H, Li K, Zheng G. Astaxanthin ameliorates oxidative stress in lens epithelial cells by regulating Gpx4 and ferroptosis. Chemico-biological Interact. (2023) 383:110684. doi: 10.1016/j.cbi.2023.110684
159. Wang Y, Li P, Wang C, Bao S, Wang S, Zhang G, et al. Lens epithelium cell ferroptosis mediated by M(6)a-lncrna and gpx4 expression in lens tissue of age-related cataract. BMC Ophthalmol. (2023) 23:514. doi: 10.1186/s12886-023-03205-8
160. Jiang H, Yu Y, Yan Y. Kras depletion suppresses ferroptosis and affects hippo pathway in cataract. Gen Physiol Biophys. (2024) 43:243–53. doi: 10.4149/gpb_2024009
161. Wei Z, Hao C, Huangfu J, Srinivasagan R, Zhang X, Fan X. Aging lens epithelium is susceptible to ferroptosis. Free Radical Biol Med. (2021) 167:94–108. doi: 10.1016/j.freeradbiomed.2021.02.010
162. Liao S, Huang M, Liao Y, Yuan C. Hmox1 promotes ferroptosis induced by erastin in lens epithelial cell through modulates Fe(2+) production. Curr eye Res. (2023) 48:25–33. doi: 10.1080/02713683.2022.2138450
163. Reddy VN, Giblin FJ, Lin LR, Dang L, Unakar NJ, Musch DC, et al. Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice. Invest Ophthalmol Visual Sci. (2001) 42:3247–55.
164. Feng L, Wang C, Zhang C, Zhang W, Zhu W, He Y, et al. P38 mapk inhibitor sb202190 suppresses ferroptosis in the glutamate-induced retinal excitotoxicity glaucoma model. Neural Regen Res. (2024) 19:2299–309. doi: 10.4103/1673-5374.391193
165. Liu J, Pan Z, Tong B, Wang C, Yang J, Zou J, et al. Artesunate protects against ocular fibrosis by suppressing fibroblast activation and inducing mitochondria-dependent ferroptosis. FASEB J. (2023) 37:e22954. doi: 10.1096/fj.202201867R
166. Xu G, Wang J, Zhang Y, Chen Z, Deng R. Ggt1 suppresses the development of ferroptosis and autophagy in mouse retinal ganglion cell through targeting gclc. Eye Brain. (2023) 15:139–51. doi: 10.2147/EB.S434280
167. Yao F, Peng J, Zhang E, Ji D, Gao Z, Tang Y, et al. Pathologically high intraocular pressure disturbs normal iron homeostasis and leads to retinal ganglion cell ferroptosis in glaucoma. Cell Death Differ. (2023) 30:69–81. doi: 10.1038/s41418-022-01046-4
168. Lin SC, Wang SY, Yoo C, Singh K, Lin SC. Association between serum ferritin and glaucoma in the South Korean population. JAMA Ophthalmol. (2014) 132:1414–20. doi: 10.1001/jamaophthalmol.2014.2876
169. Gye HJ, Kim JM, Yoo C, Shim SH, Won YS, Sung KC, et al. Relationship between high serum ferritin level and glaucoma in a South Korean population: the kangbuk samsung health study. Br J Ophthalmol. (2016) 100:1703–7. doi: 10.1136/bjophthalmol-2015-307678
170. Tong Y, Wu Y, Ma J, Ikeda M, Ide T, Griffin CT, et al. Comparative mechanistic study of rpe cell death induced by different oxidative stresses. Redox Biol. (2023) 65:102840. doi: 10.1016/j.redox.2023.102840
171. Liu Z, Huang J, Li D, Zhang C, Wan H, Zeng B, et al. Targeting zip8 mediated ferroptosis as a novel strategy to protect against the retinal pigment epithelial degeneration. Free Radical Biol Med. (2024) 214:42–53. doi: 10.1016/j.freeradbiomed.2024.01.053
172. Zhi X, Lu H, Ma D, Liu J, Luo L, Wang L, et al. Melatonin protects photoreceptor cells against ferroptosis in dry amd disorder by inhibiting Gsk-3b/Fyn-Dependent Nrf2 nuclear translocation. Biochim Biophys Acta Mol basis Dis. (2024) 1870:166969. doi: 10.1016/j.bbadis.2023.166969
173. Xiang W, Li L, Zhao Q, Zeng Y, Shi J, Chen Z, et al. Pedf protects retinal pigment epithelium from ferroptosis and ameliorates dry amd-like pathology in a murine model. GeroScience. (2024) 46:2697–714. doi: 10.1007/s11357-023-01038-3
174. Neiteler A, Palakkan AA, Gallagher KM, Ross JA. Oxidative stress and docosahexaenoic acid injury lead to increased necroptosis and ferroptosis in retinal pigment epithelium. Sci Rep. (2023) 13:21143. doi: 10.1038/s41598-023-47721-5
175. Henning Y, Blind US, Larafa S, Matschke J, Fandrey J. Hypoxia aggravates ferroptosis in rpe cells by promoting the fenton reaction. Cell Death Dis. (2022) 13:662. doi: 10.1038/s41419-022-05121-z
176. Chen X, Wang Y, Wang JN, Cao QC, Sun RX, Zhu HJ, et al. M(6)a modification of circspecc1 suppresses rpe oxidative damage and maintains retinal homeostasis. Cell Rep. (2022) 41:111671. doi: 10.1016/j.celrep.2022.111671
177. Chen C, Yang K, He D, Yang B, Tao L, Chen J, et al. Induction of ferroptosis by ho-1 contributes to retinal degeneration in mice with defective clearance of all-trans-retinal. Free Radical Biol Med. (2023) 194:245–54. doi: 10.1016/j.freeradbiomed.2022.12.008
178. Lewis LLM, Dörschmann P, Seeba C, Thalenhorst T, Roider J, Iloki Assanga SB, et al. Properties of cephalopod skin ommochromes to inhibit free radicals, and the maillard reaction and retino-protective mechanisms in cellular models concerning oxidative stress, angiogenesis, and inflammation. Antioxidants (Basel Switzerland). (2022) 11:1574. doi: 10.3390/antiox11081574
179. Liu Y, Wu D, Fu Q, Hao S, Gu Y, Zhao W, et al. Chac1 as a novel contributor of ferroptosis in retinal pigment epithelial cells with oxidative damage. Int J Mol Sci. (2023) 24:1582. doi: 10.3390/ijms24021582
180. Li Y, Zhu X, Wang K, Zhu L, Murray M, Zhou F. Ginkgo biloba extracts (Gbe) protect human rpe cells from T-Bhp-induced oxidative stress and necrosis by activating the Nrf2-mediated antioxidant defence. J Pharm Pharmacol. (2023) 75:105–16. doi: 10.1093/jpp/rgac069
181. Gupta U, Ghosh S, Wallace CT, Shang P, Xin Y, Nair AP, et al. Increased Lcn2 (Lipocalin 2) in the rpe decreases autophagy and activates inflammasome-ferroptosis processes in a mouse model of dry amd. Autophagy. (2023) 19:92–111. doi: 10.1080/15548627.2022.2062887
182. Tang Z, Huo M, Ju Y, Dai X, Ni N, Liu Y, et al. Nanoprotection against retinal pigment epithelium degeneration via ferroptosis inhibition. Small Methods. (2021) 5:e2100848. doi: 10.1002/smtd.202100848
183. Tang Z, Ju Y, Dai X, Ni N, Liu Y, Zhang D, et al. Ho-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. (2021) 43:101971. doi: 10.1016/j.redox.2021.101971
184. Liu Y, Bell BA, Song Y, Kim HJ, Sterling JK, Kim BJ, et al. Intraocular iron injection induces oxidative stress followed by elements of geographic atrophy and sympathetic ophthalmia. Aging Cell. (2021) 20:e13490. doi: 10.1111/acel.13490
185. Wei TT, Zhang MY, Zheng XH, Xie TH, Wang W, Zou J, et al. Interferon-Γ Induces retinal pigment epithelial cell ferroptosis by a Jak1-2/Stat1/Slc7a11 signaling pathway in age-related macular degeneration. FEBS J. (2022) 289:1968–83. doi: 10.1111/febs.16272
186. Sun Y, Zheng Y, Wang C, Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. (2018) 9:753. doi: 10.1038/s41419-018-0794-4
187. Chen C, Chen J, Wang Y, Liu Z, Wu Y. Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance. J Biol Chem. (2021) 296:100187. doi: 10.1074/jbc.RA120.015779
188. Zhao X, Gao M, Liang J, Chen Y, Wang Y, Wang Y, et al. Slc7a11 reduces laser-induced choroidal neovascularization by inhibiting rpe ferroptosis and vegf production. Front Cell Dev Biol. (2021) 9:639851. doi: 10.3389/fcell.2021.639851
189. Tang W, Guo J, Liu W, Ma J, Xu G. Ferrostatin-1 attenuates ferroptosis and protects the retina against light-induced retinal degeneration. Biochem Biophys Res Commun. (2021) 548:27–34. doi: 10.1016/j.bbrc.2021.02.055
190. Liu Q, Liu CQ, Yi WZ, Ouyang PW, Yang BF, Liu Q, et al. Ferroptosis contributes to microvascular dysfunction in diabetic retinopathy. Am J Pathol. (2024) 194:1078–89. doi: 10.1016/j.ajpath.2024.01.019
191. Zhou H, Zhang L, Ding C, Zhou Y, Li Y. Upregulation of hmox1 associated with M2 macrophage infiltration and ferroptosis in proliferative diabetic retinopathy. Int Immunopharmacol. (2024) 134:112231. doi: 10.1016/j.intimp.2024.112231
192. Zhang G, Yu J, Wan Y. Usp48 deubiquitination stabilizes slc1a5 to inhibit retinal pigment epithelium cell inflammation, oxidative stress and ferroptosis in the progression of diabetic retinopathy. J bioenergetics biomembranes. (2024) 56:311–21. doi: 10.1007/s10863-024-10008-z
193. Xie HB, Guo JH, Yang MM, Wang JT. Kinase pim1 governs ferroptosis to reduce retinal microvascular endothelial cell dysfunction triggered by high glucose. In Vitro Cell Dev Biol Anim. (2024) 60:278–86. doi: 10.1007/s11626-024-00882-7
194. Luo L, Cai Y, Jiang Y, Gong Y, Cai C, Lai D, et al. Pipecolic acid mitigates ferroptosis in diabetic retinopathy by regulating Gpx4-Yap signaling. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 169:115895. doi: 10.1016/j.biopha.2023.115895
195. Gao S, Gao S, Wang Y, Li N, Yang Z, Yao H, et al. Inhibition of ferroptosis ameliorates photoreceptor degeneration in experimental diabetic mice. Int J Mol Sci. (2023) 24:16946. doi: 10.3390/ijms242316946
196. Lu C, Lan Q, Song Q, Yu X. Identification and validation of ferroptosis-related genes for diabetic retinopathy. Cell Signal. (2024) 113:110955. doi: 10.1016/j.cellsig.2023.110955
197. Xi X, Chen Q, Ma J, Wang X, Zhang J, Li Y. Sestrin2 ameliorates diabetic retinopathy by regulating autophagy and ferroptosis. J Mol Histol. (2024) 55:169–84. doi: 10.1007/s10735-023-10180-3
198. Xi X, Chen Q, Ma J, Wang X, Zhang J, Li Y. Correction: sestrin2 ameliorates diabetic retinopathy by regulating autophagy and ferroptosis. J Mol Histol. (2024) 55:185. doi: 10.1007/s10735-024-10181-w
199. Li Y, Liu J, Ma X, Bai X. Maresin-1 inhibits high glucose induced ferroptosis in Arpe-19 cells by activating the Nrf2/Ho-1/Gpx4 pathway. BMC Ophthalmol. (2023) 23:368. doi: 10.1186/s12886-023-03115-9
200. Liu Z, Gan S, Fu L, Xu Y, Wang S, Zhang G, et al. 1,8-cineole ameliorates diabetic retinopathy by inhibiting retinal pigment epithelium ferroptosis via Ppar-Γ/Txnip pathways. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 164:114978. doi: 10.1016/j.biopha.2023.114978
201. Yu F, Wang C, Su Y, Chen T, Zhu W, Dong X, et al. Comprehensive analysis of ferritinophagy-related genes and immune infiltration landscape in diabetic retinopathy. Front Endocrinol. (2023) 14:1177488. doi: 10.3389/fendo.2023.1177488
202. Li S, Lu S, Wang L, Liu S, Zhang L, Du J, et al. Effects of amygdalin on ferroptosis and oxidative stress in diabetic retinopathy progression via the Nrf2/are signaling pathway. Exp eye Res. (2023) 234:109569. doi: 10.1016/j.exer.2023.109569
203. Shao J, Bai Z, Zhang L, Zhang F. Ferrostatin-1 alleviates tissue and cell damage in diabetic retinopathy by improving the antioxidant capacity of the Xc(-)-Gpx4 system. Cell Death Discov. (2022) 8:426. doi: 10.1038/s41420-022-01141-y
204. Qi H, Kan K, Sticht C, Bennewitz K, Li S, Qian X, et al. Acrolein-inducing ferroptosis contributes to impaired peripheral neurogenesis in zebrafish. Front Neurosci. (2022) 16:1044213. doi: 10.3389/fnins.2022.1044213
205. Zhan D, Zhao J, Shi Q, Lou J, Wang W. 25-hydroxyvitamin D3 inhibits oxidative stress and ferroptosis in retinal microvascular endothelial cells induced by high glucose through down-regulation of Mir-93. BMC Ophthalmol. (2023) 23:22. doi: 10.1186/s12886-022-02762-8
206. Mu L, Wang D, Dong Z, Wu J, Wu X, Su J, et al. Abnormal levels of serum ferroptosis-related biomarkers in diabetic retinopathy. J Ophthalmol. (2022) 2022:3353740. doi: 10.1155/2022/3353740
207. Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang J, et al. Glia maturation factor-B Induces ferroptosis by impairing chaperone-mediated autophagic degradation of Acsl4 in early diabetic retinopathy. Redox Biol. (2022) 52:102292. doi: 10.1016/j.redox.2022.102292
208. Fan X, Xu M, Ren Q, Fan Y, Liu B, Chen J, et al. Downregulation of fatty acid binding protein 4 alleviates lipid peroxidation and oxidative stress in diabetic retinopathy by regulating peroxisome proliferator-activated receptor Γ-mediated ferroptosis. Bioengineered. (2022) 13:10540–51. doi: 10.1080/21655979.2022.2062533
209. Tang X, Li X, Zhang D, Han W. Astragaloside-iv alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of Mir-138-5p/Sirt1/Nrf2. Bioengineered. (2022) 13:8240–54. doi: 10.1080/21655979.2022.2049471
210. Zhu Z, Duan P, Song H, Zhou R, Chen T. Downregulation of circular rna psen1 ameliorates ferroptosis of the high glucose treated retinal pigment epithelial cells via Mir-200b-3p/Cofilin-2 axis. Bioengineered. (2021) 12:12555–67. doi: 10.1080/21655979.2021.2010369
211. Shen H, Gong Q, Zhang J, Wang H, Qiu Q, Zhang J, et al. Trim46 aggravated high glucose-induced hyper permeability and inflammatory response in human retinal capillary endothelial cells by promoting Iκbα Ubiquitination. Eye Vision (London England). (2022) 9:35. doi: 10.1186/s40662-022-00305-2
212. Zhang J, Qiu Q, Wang H, Chen C, Luo D. Trim46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating Gpx4 ubiquitination. Exp Cell Res. (2021) 407:112800. doi: 10.1016/j.yexcr.2021.112800
213. Zhou J, Sun C, Dong X, Wang H. A novel mir-338-3p/slc1a5 axis reprograms retinal pigment epithelium to increases its resistance to high glucose-induced cell ferroptosis. J Mol Histol. (2022) 53:561–71. doi: 10.1007/s10735-022-10070-0
214. Yumnamcha T, Devi TS, Singh LP. Auranofin mediates mitochondrial dysregulation and inflammatory cell death in human retinal pigment epithelial cells: implications of retinal neurodegenerative diseases. Front Neurosci. (2019) 13:1065. doi: 10.3389/fnins.2019.01065
215. Deleon E, Lederman M, Berenstein E, Meir T, Chevion M, Chowers I. Alteration in iron metabolism during retinal degeneration in rd10 mouse. Invest Ophthalmol Visual Sci. (2009) 50:1360–5. doi: 10.1167/iovs.08-1856
216. Cardile A, Passarini C, Zanrè V, Fiore A, Menegazzi M. Hyperforin enhances heme oxygenase-1 expression triggering lipid peroxidation in braf-mutated melanoma cells and hampers the expression of pro-metastatic markers. Antioxidants (Basel Switzerland). (2023) 12:1369. doi: 10.3390/antiox12071369
217. Liu H, Gan Q, Lai Y, Pan Z, Jin Q, Li J, et al. Usp14 increases the sensitivity of retinoblastoma to cisplatin by mediating the ferroptosis. Naunyn-Schmiedeberg's Arch Pharmacol. (2024) 13:1065. doi: 10.1007/s00210-024-03174-9
218. Zhong L, Meng X, Huang J, Hao W, Zuo Y. Expression of yap suppresses cell proliferation and elevates the sensitivity of chemotherapy in retinoblastoma cells through lipid-peroxidation induced ferroptosis. Chin Clin Oncol. (2023) 12:52. doi: 10.21037/cco-23-97
219. Li C, Sun S, Zhuang Y, Luo Z, Ji G, Liu Z. Ctsb nuclear translocation facilitates DNA damage and lysosomal stress to promote retinoblastoma cell death. Mol Biotechnol. (2023). doi: 10.1007/s12033-023-01042-0
220. Liu K, Huang J, Liu J, Klionsky DJ, Kang R, Tang D. Induction of autophagy-dependent ferroptosis to eliminate drug-tolerant human retinoblastoma cells. Cell Death Dis. (2022) 13:521. doi: 10.1038/s41419-022-04974-8
221. Kuganesan N, Dlamini S, Tillekeratne LMV, Taylor WR. Tumor suppressor P53 promotes ferroptosis in oxidative stress conditions independent of modulation of ferroptosis by P21, Cdks, Rb, and E2f. J Biol Chem. (2021) 297:101365. doi: 10.1016/j.jbc.2021.101365
222. Daruich A, Le Rouzic Q, Jonet L, Naud MC, Kowalczuk L, Pournaras JA, et al. Iron is neurotoxic in retinal detachment and transferrin confers neuroprotection. Sci Adv. (2019) 5:eaau9940. doi: 10.1126/sciadv.aau9940
223. Ma R, Li Y, Dong X, Zhang Y, Chen X, Zhang Y, et al. Pax6/cxcl14 regulatory axis promotes the repair of corneal injury by enhancing corneal epithelial cell proliferation. J Trans Med. (2024) 22:458. doi: 10.1186/s12967-024-05270-z
224. Agrawal P, Tiwari A, Chowdhury SK, Vohra M, Gour A, Waghmare N, et al. Kuragel: A biomimetic hydrogel scaffold designed to promote corneal regeneration. iScience. (2024) 27:109641. doi: 10.1016/j.isci.2024.109641
225. Sun R, Zhang J, Chen X, Deng Y, Gou J, Yin T, et al. An adaptive drug-releasing contact lens for personalized treatment of ocular infections and injuries. J Controlled release. (2024) 369:114–27. doi: 10.1016/j.jconrel.2024.03.040
226. Liu Z, Liu K, Shi S, Chen X, Gu X, Wang W, et al. Alkali injury-induced pathological lymphangiogenesis in the iris facilitates the infiltration of T cells and ocular inflammation. JCI Insight. (2024) 9:e175479. doi: 10.1172/jci.insight.175479
227. Wu Y, Du L, Xu X, Hu Y, Liu J, Zhang J, et al. Nano self-assemblies of caffeic acid-fibronectin mimic a peptide conjugate for the treatment of corneal epithelial injury. Mol pharmaceutics. (2023) 20:5937–46. doi: 10.1021/acs.molpharmaceut.3c00861
228. Dutta T, Sangwan J, Mondal M, Vohra M, Nidhi V, Gour A, et al. Prolonged inflammation and infectious changes in the corneal epithelium are associated with persistent epithelial defect (Ped). Pathog (Basel Switzerland). (2023) 12:261. doi: 10.3390/pathogens12020261
229. Liang N, Song W, Li J. Bpa promotes lung fibrosis in mice by regulating autophagy-dependent ferroptosis in alveolar epithelial cells. Ecotoxicol Environ Saf. (2024) 278:116412. doi: 10.1016/j.ecoenv.2024.116412
230. Wang Z, Li H, Zhou W, Lee J, Liu Z, An Z, et al. Ferrous sulfate-loaded hydrogel cures staphylococcus aureus infection via facilitating a ferroptosis-like bacterial cell death in a mouse keratitis model. Biomaterials. (2022) 290:121842. doi: 10.1016/j.biomaterials.2022.121842
231. Teng X, Xiong X, Sha X, Lei Y, Diao Y, Liu J, et al. Identification of hub genes and pathways of ferroptosis in fusarium keratitis by bioinformatics methods. Front Cell Infect Microbiol. (2023) 13:1103471. doi: 10.3389/fcimb.2023.1103471
232. Balla A, Tran B, Valtari A, Steven P, Scarpellini C, Augustyns K, et al. A novel ferroptosis inhibitor uamc-3203, a potential treatment for corneal epithelial wound. Pharmaceutics. (2022) 15:118. doi: 10.3390/pharmaceutics15010118
233. Guo XX, Pu Q, Hu JJ, Chang XJ, Li AL, Li XY. The role of regulated necrosis in inflammation and ocular surface diseases. Exp eye Res. (2023) 233:109537. doi: 10.1016/j.exer.2023.109537
234. Mi Y, Zhu Q, Zheng X, Wan M. The protective role of water intake in age-related eye diseases: insights from a Mendelian randomization study. Food Funct. (2024) 15:5147–57. doi: 10.1039/D4FO01559B
235. Radwan L, El Jalbout JD, Trad K, Radwan L, El Zein L, Brearley A, et al. Outcomes of phacoemulsification with or without kahook dual blade goniotomy for glaucoma patients with cataract. J glaucoma. (2024). doi: 10.1097/IJG.0000000000002429
236. van den Berg RM, van den Berg A, Rocha KM, Fetrin de Barros M, Dodhia M, Shahid M, et al. Prediction of the small aperture intraocular lens on visual acuity in patients with keratoconus. J cataract refractive Surg. (2024). doi: 10.1097/j.jcrs.0000000000001480
237. Asbell PA, Dualan I, Mindel J, Brocks D, Ahmad M, Epstein S. Age-related cataract. Lancet (London England). (2005) 365:599–609. doi: 10.1016/S0140-6736(05)17911-2
239. Babizhayev MA, Deyev AI, Yermakova VN, Brikman IV, Bours J. Lipid peroxidation and cataracts: N-acetylcarnosine as a therapeutic tool to manage age-related cataracts in human and in canine eyes. Drugs R&D. (2004) 5:125–39. doi: 10.2165/00126839-200405030-00001
240. Cao H, Xiong SF, Dong LL, Dai ZT. Study on the mechanism of lipid peroxidation induced by carbonate radicals. Molecules (Basel Switzerland). (2024) 29:1125. doi: 10.3390/molecules29051125
241. Wilson SE, Ingham CJ, Hunter IS, Smith MC. Control of lytic development in the streptomyces temperate phage Phi C31. Mol Microbiol. (1995) 16:131–43. doi: 10.1111/j.1365-2958.1995.tb02398.x
242. Fan N, Yan L, Li M, Li S. Comparative efficacies of 13 surgical interventions for primary congenital glaucoma in children: A network meta-analysis of randomized clinical trials. Int J Surg (London England). (2024) 110:3081–2. doi: 10.1097/JS9.0000000000001046
243. Gupta V, Kong GXY, Singh A, Panigrahi A, Gupta S, Prea S, et al. Measuring visual fields in children with glaucoma using a portable tablet. Trans Vision Sci Technol. (2024) 13:10. doi: 10.1167/tvst.13.5.10
244. Parakkel RR, Wong D, Li C, Cheong J, Nongpiur ME, Chong RS, et al. Retinal nerve fiber layer damage assessment in glaucomatous eyes using retinal retardance measured by polarization-sensitive optical coherence tomography. Trans Vision Sci Technol. (2024) 13:9. doi: 10.1167/tvst.13.5.9
245. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: A review. Jama. (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
246. Dixon A, Shim MS, Nettesheim A, Coyne A, Su CC, Gong H, et al. Autophagy deficiency protects against ocular hypertension and neurodegeneration in experimental and spontanous glaucoma mouse models. Cell Death Dis. (2023) 14:554. doi: 10.1038/s41419-023-06086-3
247. Hoelzgen F, Nguyen TTP, Klukin E, Boumaiza M, Srivastava AK, Kim EY, et al. Structural basis for the intracellular regulation of ferritin degradation. Nat Commun. (2024) 15:3802. doi: 10.1038/s41467-024-48151-1
248. Song S, Su Z, Kon N, Chu B, Li H, Jiang X, et al. Alox5-mediated ferroptosis acts as a distinct cell death pathway upon oxidative stress in Huntington's disease. Genes Dev. (2023) 37:204–17. doi: 10.1101/gad.350211.122
249. Liu P, Zhang M, Shoeb M, Hogan D, Tang L, Syed MF, et al. Metal chelator combined with permeability enhancer ameliorates oxidative stress-associated neurodegeneration in rat eyes with elevated intraocular pressure. Free Radical Biol Med. (2014) 69:289–99. doi: 10.1016/j.freeradbiomed.2014.01.039
250. Lin Y, Epstein DL, Liton PB. Intralysosomal iron induces lysosomal membrane permeabilization and cathepsin D-mediated cell death in trabecular meshwork cells exposed to oxidative stress. Invest Ophthalmol Visual Sci. (2010) 51:6483–95. doi: 10.1167/iovs.10-5410
251. Sendecki A, Ledwoń D, Nycz J, Wąsowska A, Boguszewska-Chachulska A, Mitas AW, et al. A deep learning approach to explore the association of age-related macular degeneration polygenic risk score with retinal optical coherence tomography: A preliminary study. Acta ophthalmologica. (2024). doi: 10.1111/aos.16710
252. Woo KM, Mahrous MA, D'Amico DJ, Kiss S, Kovacs KD. Prevalence of age-related macular degeneration in patients with chronic exposure to P2x7r inhibitors. Graefe's Arch Clin Exp Ophthalmol = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. (2024). doi: 10.1007/s00417-024-06507-9
253. Zhong R, Cui D, Richardson EA, Phillips DR, Azadi P, Lu G, et al. Cytosolic acetyl-coa generated by Atp-citrate lyase is essential for acetylation of cell wall polysaccharides. Plant Cell Physiol. (2020) 61:64–75. doi: 10.1093/pcp/pcz178
254. Scimone C, Donato L, Alibrandi S, Vadalà M, Giglia G, Sidoti A, et al. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp eye Res. (2021) 209:108641. doi: 10.1016/j.exer.2021.108641
255. Hanus J, Anderson C, Sarraf D, Ma J, Wang S. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell Death Discov. (2016) 2:16054. doi: 10.1038/cddiscovery.2016.54
256. Liu B, Wang W, Shah A, Yu M, Liu Y, He L, et al. Sodium iodate induces ferroptosis in human retinal pigment epithelium Arpe-19 cells. Cell Death Dis. (2021) 12:230. doi: 10.1038/s41419-021-03520-2
257. Lee JJ, Chang-Chien GP, Lin S, Hsiao YT, Ke MC, Chen A, et al. 5-lipoxygenase inhibition protects retinal pigment epithelium from sodium iodate-induced ferroptosis and prevents retinal degeneration. Oxid Med Cell Longev. (2022) 2022:1792894. doi: 10.1155/2022/1792894
258. Wei H, Chen C, Di F, Sun C, Wang X, Sun M, et al. Pm(2.5)-induced ferroptosis by nrf2/hmox1 signaling pathway led to inflammation in microglia. Environ pollut (Barking Essex: 1987). (2024) 352:124130. doi: 10.1016/j.envpol.2024.124130
259. Sohns C, Zabel M. [Current role of amiodarone in antiarrhythmic therapy]. Herzschrittmachertherapie Elektrophysiologie. (2010) 21:239–43. doi: 10.1007/s00399-010-0091-0
260. Chen Y, Okano K, Maeda T, Chauhan V, Golczak M, Maeda A, et al. Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J Biol Chem. (2012) 287:5059–69. doi: 10.1074/jbc.M111.315432
261. Kwon YS, Munsoor J, Kaufmann M, Zheng M, Smirnov AI, Han Z. Polydopamine nanoparticles as mimicking rpe melanin for the protection of retinal cells against blue light-induced phototoxicity. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2024), e2400230. doi: 10.1002/advs.202400230
262. Li T, Qian Y, Li H, Wang T, Jiang Q, Wang Y, et al. Cellular communication network factor 1 promotes retinal leakage in diabetic retinopathy via inducing neutrophil stasis and neutrophil extracellular traps extrusion. Cell communication signaling: CCS. (2024) 22:275. doi: 10.1186/s12964-024-01653-3
263. Gue CCY, Rahim NDA, Rojas-Carabali W, Agrawal R, Rk P, Abisheganaden J, et al. Evaluating the openai's Gpt-3.5 Turbo's performance in extracting information from scientific articles on diabetic retinopathy. Syst Rev. (2024) 13:135. doi: 10.1186/s13643-024-02523-2
264. Bone RN, Oyebamiji O, Talware S, Selvaraj S, Krishnan P, Syed F, et al. A computational approach for defining a signature of B-cell Golgi stress in diabetes. Diabetes. (2020) 69:2364–76. doi: 10.2337/db20-0636
265. Giri B, Dey S, Das T, Sarkar M, Banerjee J, Dash SK. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: an update on glucose toxicity. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2018) 107:306–28. doi: 10.1016/j.biopha.2018.07.157
266. Saxena R, Madhu SV, Shukla R, Prabhu KM, Gambhir JK. Postprandial hypertriglyceridemia and oxidative stress in patients of type 2 diabetes mellitus with macrovascular complications. Clinica chimica Acta. (2005) 359:101–8. doi: 10.1016/j.cccn.2005.03.036
267. Xue Y, Zhou Y, Cepko CL. Txnip deletions and missense alleles prolong the survival of cones in a retinitis pigmentosa mouse model. eLife. (2024) 12:RP90749. doi: 10.7554/eLife.90749
268. Humphries A, Bowman L, Nguyen T, So J, Duff M, Grover S, et al. Quality of life analysis in patients with retinitis pigmentosa. Ophthalmic Res. (2024) 67:348–57. doi: 10.1159/000539116
269. Yang M, So KF, Lam WC, Lo ACY. Cell ferroptosis: new mechanism and new hope for retinitis pigmentosa. Cells. (2021) 10:2153. doi: 10.3390/cells10082153
270. Park EJ, Lee KM, Kim T, Lee D, Kim MS, Lee C. Trivalent copper ion-mediated dual oxidation in the copper-catalyzed fenton-like system in the presence of histidine. Environ Sci Technol. (2024) 58:10852–62. doi: 10.1021/acs.est.4c03689
271. Ashraf AA, Aljuhani M, Hubens CJ, Jeandriens J, Parkes HG, Geraki K, et al. Inflammation subsequent to mild iron excess differentially alters regional brain iron metabolism, oxidation and neuroinflammation status in mice. Front Aging Neurosci. (2024) 16:1393351. doi: 10.3389/fnagi.2024.1393351
272. Santhanam A, Shihabeddin E, Wei H, Wu J, O'Brien J. Molecular basis of retinal remodeling in a zebrafish model of retinitis pigmentosa. Cell Mol Life sciences: CMLS. (2023) 80:362. doi: 10.1007/s00018-023-05021-1
273. Yang H, Zhang H, Li X. Navigating the future of retinitis pigmentosa treatments: A comprehensive analysis of therapeutic approaches in Rd10 mice. Neurobiol Dis. (2024) 193:106436. doi: 10.1016/j.nbd.2024.106436
274. Obolensky A, Berenshtein E, Lederman M, Bulvik B, Alper-Pinus R, Yaul R, et al. Zinc-desferrioxamine attenuates retinal degeneration in the rd10 mouse model of retinitis pigmentosa. Free Radical Biol Med. (2011) 51:1482–91. doi: 10.1016/j.freeradbiomed.2011.07.014
275. Wang K, Peng B, Xiao J, Weinreb O, Youdim MBH, Lin B. Iron-chelating drugs enhance cone photoreceptor survival in a mouse model of retinitis pigmentosa. Invest Ophthalmol Visual Sci. (2017) 58:5287–97. doi: 10.1167/iovs.17-22096
276. Song D, Song Y, Hadziahmetovic M, Zhong Y, Dunaief JL. Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radical Biol Med. (2012) 53:64–71. doi: 10.1016/j.freeradbiomed.2012.04.020
277. Zeng Z, Gao ZL, Zhang ZP, Jiang HB, Yang CQ, Yang J, et al. Downregulation of cks1b restrains the proliferation, migration, invasion and angiogenesis of retinoblastoma cells through the mek/erk signaling pathway. Int J Mol Med. (2024) 54:103–14. doi: 10.3892/ijmm.2024.5382
278. Thongkumkoon P, Sangphukieo A, Tongjai S, Noisagul P, Sangkhathat S, Laochareonsuk W, et al. Establishment, characterization, and genetic profiling of patient-derived osteosarcoma cells from a patient with retinoblastoma. Sci Rep. (2024) 14:11056. doi: 10.1038/s41598-024-60628-z
279. Bell HN, Stockwell BR, Zou W. Ironing out the role of ferroptosis in immunity. Immunity. (2024) 57:941–56. doi: 10.1016/j.immuni.2024.03.019
280. Xiong L, Liu Y, Wang Y, Zhao H, Song X, Fan W, et al. The protective effect of lonicera japonica thunb. Against lipopolysaccharide-induced acute lung injury in mice: modulation of inflammation, oxidative stress, and ferroptosis. J Ethnopharmacol. (2024) 331:118333. doi: 10.1016/j.jep.2024.118333
281. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. (2022) 8:43. doi: 10.1038/s41420-021-00807-3
282. Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. (2015) 356:971–7. doi: 10.1016/j.canlet.2014.11.014
283. Li S, Li M, Wu J, Li Y, Han J, Song Y, et al. Developing and validating a clinlabomics-based machine-learning model for early detection of retinal detachment in patients with high myopia. J Trans Med. (2024) 22:405. doi: 10.1186/s12967-024-05131-9
284. Wenzel DA, Gassel CJ, Druchkiv V, Neubauer J, Bartz-Schmidt KU, Dimopoulos S. A comparative analysis of traumatic retinal detachment after open and closed globe injuries in children. Retina (Philadelphia Pa). (2024). doi: 10.1097/IAE.0000000000004120
285. Moos T, Bernth N, Courtois Y, Morgan EH. Developmental iron uptake and axonal transport in the retina of the rat. Mol Cell Neurosci. (2011) 46:607–13. doi: 10.1016/j.mcn.2010.12.013
286. Esh Z, Suresh S, Ortolan D, Farnoodian M, Bose D, Ryu J, et al. Lipidunet-Machine Learning-Based Method of Characterization and Quantification of Lipid Deposits Using Ipsc-Derived Retinal Pigment Epithelium. J Vis Exp. JoVE. (2023) 197. doi: 10.3791/65503
287. Li SY, Zhao N, Wei D, Pu N, Hao XN, Huang JM, et al. Ferroptosis in the ageing retina: A malevolent fire of diabetic retinopathy. Ageing Res Rev. (2024) 93:102142. doi: 10.1016/j.arr.2023.102142
288. Ma R, Gan L, Guo J, Peng Z, Wu J, Harrison AR, et al. Insights into ferroptosis: targeting glycolysis to treat Graves' Orbitopathy. J Clin Endocrinol Metab. (2022) 107:1994–2003. doi: 10.1210/clinem/dgac163
Keywords: ferroptosis, metabolic pathway, eye diseases, therapy, corneal injury
Citation: Yang Y, Lin Y, Han Z, Wang B, Zheng W and Wei L (2024) Ferroptosis: a novel mechanism of cell death in ophthalmic conditions. Front. Immunol. 15:1440309. doi: 10.3389/fimmu.2024.1440309
Received: 29 May 2024; Accepted: 14 June 2024;
Published: 27 June 2024.
Edited by:
Lian Xiang Luo, Guangdong Medical University, ChinaReviewed by:
Peng Wang, Coriell Institute For Medical Research, United StatesXiaodong Zou, The Chinese University of Hong Kong, China
Zhuodong Chai, Texas A and M University, United States
Yan Li, Tongji University, China
Copyright © 2024 Yang, Lin, Han, Wang, Zheng and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongyu Han, hzyczy1997@163.com; Wei Zheng, drzhengwei@163.com; Lijuan Wei, 21102570672@stu.ccucm.edu.cn
†These authors have contributed equally to this work