 Huikang Xu
Huikang Xu Shiying Sheng
Shiying Sheng Weiwei Luo
Weiwei Luo Xiaofang Xu
Xiaofang Xu Zhaocai Zhang
Zhaocai Zhang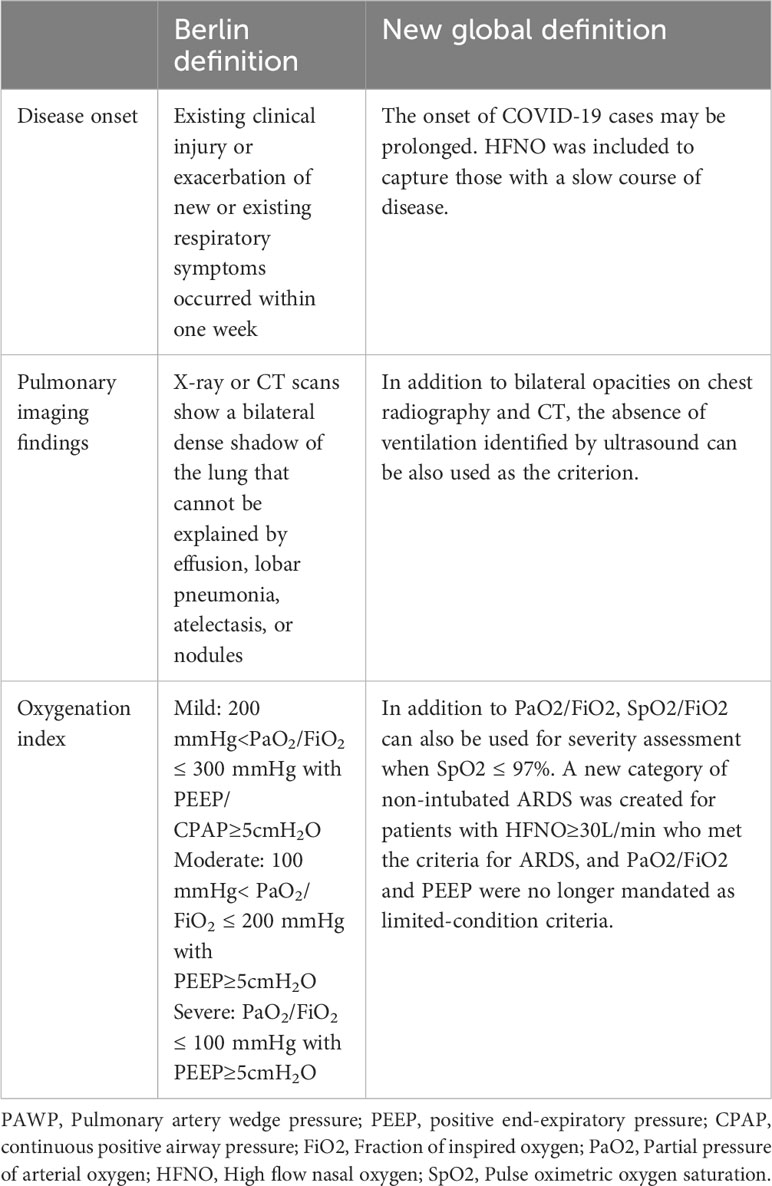

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 November 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1277161
This article is part of the Research Topic Characteristic clinical immune phenotypes and molecular mechanisms associated with inflammatory diseases View all 26 articles
Acute respiratory distress syndrome (ARDS) is an acute diffuse inflammatory lung injury characterized by the damage of alveolar epithelial cells and pulmonary capillary endothelial cells. It is mainly manifested by non-cardiogenic pulmonary edema, resulting from intrapulmonary and extrapulmonary risk factors. ARDS is often accompanied by immune system disturbance, both locally in the lungs and systemically. As a common heterogeneous disease in critical care medicine, researchers are often faced with the failure of clinical trials. Latent class analysis had been used to compensate for poor outcomes and found that targeted treatment after subgrouping contribute to ARDS therapy. The subphenotype of ARDS caused by sepsis has garnered attention due to its refractory nature and detrimental consequences. Sepsis stands as the most predominant extrapulmonary cause of ARDS, accounting for approximately 32% of ARDS cases. Studies indicate that sepsis-induced ARDS tends to be more severe than ARDS caused by other factors, leading to poorer prognosis and higher mortality rate. This comprehensive review delves into the immunological mechanisms of sepsis-ARDS, the heterogeneity of ARDS and existing research on targeted treatments, aiming to providing mechanism understanding and exploring ideas for accurate treatment of ARDS or sepsis-ARDS.
Acute respiratory distress syndrome (ARDS) is a diffuse lung injury caused by intrapulmonary and extrapulmonary factors in a short period, histologically characterized by diffuse alveolar damage, including pulmonary edema, hyaline membrane formation, alveolar hemorrhage and inflammation (1–3). ARDS is a heterogeneous syndrome, and its heterogeneity is mainly manifested in physiology, imaging findings, etiology, onset time, biomarker and gene differences, etc. Among the subtypes of ARDS, septic ARDS is a noteworthy and complex one. Sepsis is a major risk factor for ARDS, accounting for 32% of ARDS etiology (4). The prognosis of patients with sepsis-induced ARDS is worse than that of patients with sepsis or ARDS alone. This subgroup of ARDS usually faces a higher mortality rate and a lower success rate of extubation, which has received more attention in clinical practice (5, 6).
Sepsis is a systemic inflammatory reaction caused by a maladjusted response to infection induced by pathogenic microorganisms (7). The overwhelming inflammatory molecules, released due to infection in sepsis, not only flow along the systemic blood circulation, but also form a cascaded and amplified network with the participation of the nervous, endocrine and immune system (8). Sepsis is a common disease in intensive care units (ICU). Research had revealed that sepsis has an incidence of 437/100,000 on a global scale, with a case fatality rate of 17% (9). Another two-month prospective observational cohort study in China mainland showed that 54.8% of ICU patients would progress from severe sepsis to a comorbidity of sepsis-ARDS, and the proportion of non-surviving patients in sepsis-ARDS reached 71.0% (10). The cytokine storm caused by sepsis can bring about either direct damage to the lung epithelium or indirect damage to the lung endothelium (11, 12). Due to large individual differences, it is difficult to obtain reliable results in clinical trials of ARDS, and some treatments are only effective in a small portion of patients (13, 14). Therefore, it is crucial to explore the heterogeneity of ARDS and the immunological mechanism behind it for the effective therapy. This review mainly introduces the heterogeneity of ARDS, the related precision medicine and the immunological mechanism of sepsis-ARDS, hoping to provide some assistance and guidance for researchers in the study of the heterogeneity of ARDS and the treatment of sepsis-induced ARDS.
The clinical, physiological, biological and imaging manifestations of ARDS are highly heterogeneous. The etiological factors of ARDS can be divided into two types: intrapulmonary and extrapulmonary. The intrapulmonary factors, such as pneumonia and mechanical ventilation, primarily inflict damage on the alveolar epithelium, whereas extrapulmonary factors like sepsis and acute severe pancreatitis initially impair the endothelium and subsequently result in pulmonary edema (15). The 1994 American-European Consensus Conference(AECC) had suggested that this heterogeneity would greatly hinder the research on the treatment of ARDS (16). The Consensus Conference in 1994 laid the foundation for a better understanding of ARDS, and the Berlin Definition published in 2012 defined ARDS in more detail and had classified ARDS patients according to the PaO2/FiO2 level (17). With the proposal of the Berlin definition, some researchers conducted autopsies on 712 patients with ARDS according to the Berlin definition, which clarified the high sensitivity and low specificity of the Berlin definition, and researchers confirmed the pathological heterogeneity of ARDS as well (18). Recently, the new definition of ARDS has been proposed based on the original Berlin definition, which has been fine-tuned and modified. For example, the pulse oxygen saturation SpO2/FiO2 is allowed to replace PaO2/FiO2 in the classification criteria (3). These definitions reflect an evolving understanding of the heterogeneity of ARDS as a complex syndrome that needs to be treated differently, on a case-by-case basis, as compared with the previous defining as a whole. The specific changes in the new definition relative to the Berlin definition are shown in Table 1.
Many clinical trials that treated patients with ARDS as a unified group have failed (19). And latent class analysis of failed clinical trials briefly came into focus (20–22). Due to the failure of a large number of clinical trials, more individuals are paying attention to the subgrouping of ARDS to obtain the meaningful experimental results (Table 2). With this subgrouping, one hopes to harvest different and beneficial clinical trial results in the near future.
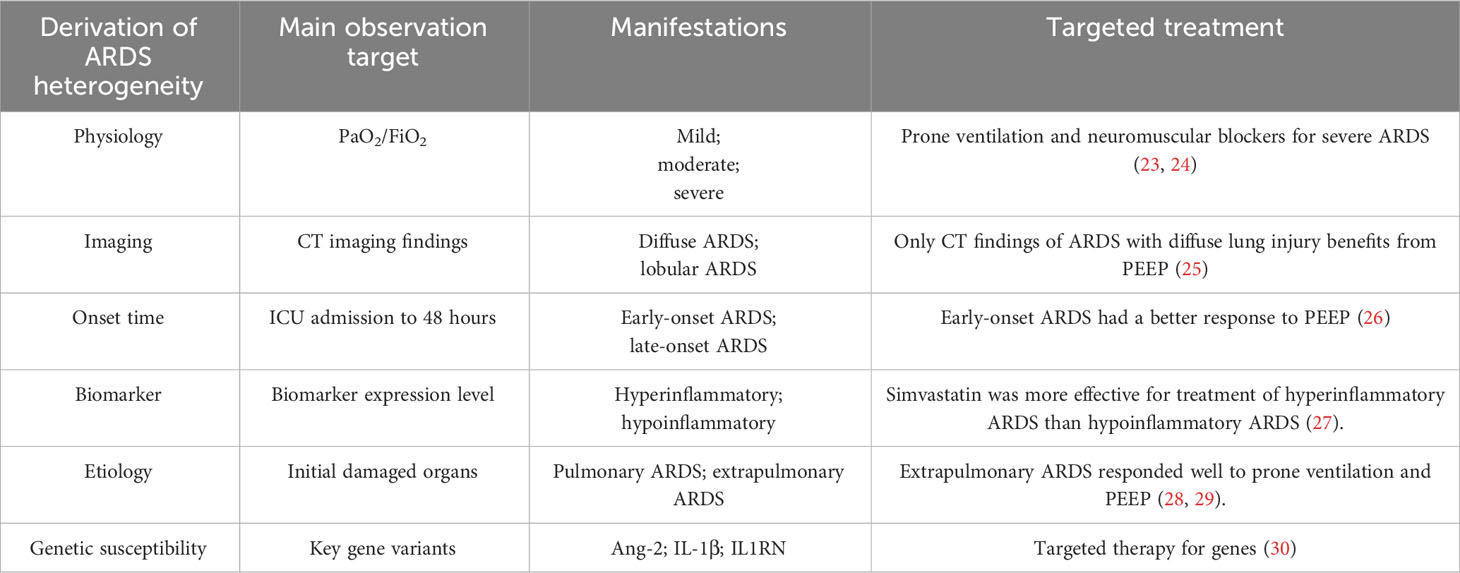
Table 2 ARDS heterogeneity summary.
ARDS is a multi-etiology disease, and different etiologies will bring physiological variability. The grouping of ARDS from a physiological perspective will be a key point to figure out its heterogeneity. According to the Berlin definition in 2012, ARDS was divided into mild, moderate and severe types based on PaO2/FiO2 ratio (17). Patients with severe ARDS (PaO2/FiO2 ≤ 150mmHg) were selected for randomized controlled trials of prone ventilation and neuromuscular blockers as a research object. Both trials shown that these two treatments can significantly improve 90-day survival (23, 24). The definition of severe ARDS here differs from the Berlin definition, in which PaO2/FiO2 ≤ 100mmHg, possibly because the two randomized trials were designed before the declaration of Berlin definition. In a multicenter prospective study, patients with increased lung dead-space tended to have a higher risk of mortality (31). The dead-space fraction is an independent risk factor for ARDS patients (31). Perhaps in future clinical trials, the lung dead-space fraction will be included in the grouping criteria.
In the progression of ARDS, the shadow range and lesion range in the imaging results of patients are varied, which brings a certain degree of hindrance to the diagnosis and treatment. Pulmonary ARDS is usually characterized by extensive ground-glass opacification and asymmetric lung consolidation, while extrapulmonary ARDS is characterized by symmetrical ground-glass opacification distributed in perihilar regions (32). Desai et al. tend to distinguish pulmonary ARDS and extrapulmonary ARDS by the typical or atypical CT manifestations. The typical CT manifestations are characterized by extensive and intense opacification of the dependent parenchyma, whereas the atypical CT manifestations involve more extensive and intense opacification of the non-dependent parenchyma. Researchers have observed that extrapulmonary ARDS often exhibit typical CT features (33). Some studies have divided the ARDS into diffuse ARDS and lobular ARDS by CT findings, and found that the two types have distinct responses to positive end expiratory pressure (PEEP) (25). In patients with diffuse ARDS, PEEP brought about significant alveolar dilation, while in patients with lobar ARDS, only mild alveolar dilation was induced by PEEP (25). These imaging findings have guiding implications for clinical management of ARDS. Radiological features are the most intuitive means to detect inflammatory infiltration in the lung, which can usually provide rapid guidance to clinicians when fighting against the illness.
The Berlin definition sets the onset of ARDS as within one week of the occurrence or exacerbation of the underlying injury (17). In 2009, Liao et al. took 48 hours as the cut-off line and divided patients admitted to ICU into early-onset(<48h after ICU admission) and late-onset ARDS(>48h after ICU admission), and they found that these two have different prognoses and mortality (34). Data from a multicenter observational study showed that patients with early-onset ARDS had higher scores in both Simplified Acute Physiology Score (SAPS) II and initial Sequential Organ Failure Assessment(SOFA), whereas those with late-onset ARDS had longer ICU and hospital stays (35). Therefore, the onset time of ARDS is also a very important heterogeneous factor affecting the outcome of patients. The classification of ARDS according to the onset time after admission can sometimes predict the possible clinical manifestations and thus make treatment more favorable. For example, studies have found that early-onset ARDS is associated with severe hemorrhagic shock, while late-onset ARDS is often characterized by pneumonia and multiple organ damage (26). Moreover, early-onset ARDS responds better to protective mechanical ventilation (26). The different onset time of ARDS can bring about protein biomarker diversity, such as receptor for advanced glycation end product (RAGE)s and Angiopoietin (Ang)-2, markers of alveolar capillary barrier injury, which are only elevated in early-onset ARDS (36). The variations in pathological manifestations, response to therapy, and levels of circulating proteins underscore the importance of classifying ARDS based on its onset time.
Biomarkers of ARDS can not only indicate the pathogenesis and progression of the disease, but also conduce to identify ARDS subgroups and provide targeted treatment (37–39). RAGE is mainly distributed in the basal surface of alveolar type 1(AT1) epithelial cells, and it engages in innate immunity and important alveolar inflammatory pathways (40, 41). Soluble RAGEs, a marker of type I alveolar epithelial cell injury, produced by cleavage of full-length RAGE, are increased only in the early stages of ARDS (39, 42). The randomized controlled trial of a single-center study found a significant difference in soluble RAGE level between direct ARDS and indirect ARDS (43). In addition to RAGE, the levels of Surfactant protein D (SP-D), IL-6, IL-8 and von Willebrand factor (vWF) were also significantly different between these two groups (43). In an observational study, patients with non-focal ARDS had higher plasma levels of soluble RAGE (44).
In ARDS, RAGE reflects alveolar epithelial damage, whereas Ang-2 is a marker of endothelial cell activation and increased capillary permeability. In a randomized controlled trial, plasma Ang-2 level was proved to predict the prognosis and mortality of patients with ARDS, and high levels of Ang-2 usually suggest poor prognosis (45). In critically ill patients, the early elevation of Ang-2 can predict the impending occurrence of acute lung injury (ALI) (46). Ang-2 in ARDS could reflect endothelial injury, which is common in extrapulmonary ARDS.
Another important plasma marker associated with ARDS severity is IL-1 receptor antagonist (IL-1RA), a naturally occurring substance secreted by monocytes, which can bind to the IL-1 receptor without initiating transcription and thus exert an anti-inflammatory effect (47). Due to the anti-inflammatory effect of IL-1RA, it is universally accepted that IL-1RA may play a protective role in ARDS and sepsis. In the 1994 randomized controlled trial, IL-1RA was found to have no statistically significant therapeutic effect in patients with sepsis (48). Nevertheless, in the post hoc analysis, IL-1RA improved survival in patients with severe sepsis who had organ failure or a high predicted mortality rate (>24%) (48). In another randomized controlled trial in sepsis (including septic ARDS), no clear therapeutic benefit of IL-1RA was found either (49). In the upgraded clinical trial, when sepsis patients (including ARDS patients) were grouped according to the plasma level of IL-1RA at enrollment, the high IL-1RA level group tended to obtain survival benefits (50). The ILRN variant rs315952C present in subjects of European ancestry increases plasma levels of ILR1A and patients with the variation had higher survival rate in patients with septic shock (50). Additionally, endothelin serves as a crucial biomarker for sepsis-induced ARDS, and its production actively contributes to the progression of organ dysfunction in sepsis, including ARDS (51). In the context of sepsis-induced ARDS, free fatty acid (FFA)s/non-esterified fatty acid (NEFA)s emerge as another crucial substances. Free fatty acid serum C18 can be used as an important predictor of ARDS development (52). In animal experiments, reducing plasma non-esterified fatty acid (NEFA) levels has been shown to decrease the incidence of lung injury (53).
Beyond that, other markers can also be used to observe the heterogeneity of ARDS including thrombomodulin, TNF-1 receptor and so on (54, 55). Latent class analysis after randomized controlled trials found that when ARDS patients were divided into hyperinflammatory and hypoinflammatory subgroups according to the expression levels of inflammatory cytokines, the clinical manifestations and prognoses of the two subgroups were widely different (56). In this subgrouping, ARDS patients were regrouped according to inflammatory biomarkers, and patients with hyperinflammatory ARDS had a higher incidence of shock and mortality rate, and they response better to PEEP (20). A suitable and stable biomarker not only enhance comprehension of the disease progression, pathogenesis and possible prognosis of ARDS, but also facilitate physicians to make appropriate treatment decisions for different subtypes of ARDS.
The etiology of ARDS is diverse and variable, including pneumonia, aspiration of stomach contents, sepsis, acute severe pancreatitis, extensive burn, etc. Pneumonia and non-pulmonary sepsis are the most common risk factors for ARDS (6). A retrospective cohort study of microbial-positive pneumonia found that bacterial infection was the leading cause of pneumonia, followed by fungal, viral, and mixed pathogen infections (57). However, the incidence of lung injury induced by bacterial pneumonia is lower than the latter (57). For patients with sepsis, the incidence of ALI caused by gram-positive and gram-negative bacterial infections was similar (57). For patients with viral infection, studies had pointed out that influenza A virus is the main viral cause of ARDS in adults (58). Compared with H1N1 influenza virus group, which mainly causes mild and moderate ARDS, H7N9 influenza virus infection has a high risk of severe ARDS (59, 60). In addition, the infection of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) could also lead to ARDS, namely the severe COVID‐19,which brings about significant lung injury by binding to the ACE2 receptor (61). Similar to sepsis induced ARDS, COVID-19 are characterized by coagulation dysfunction, which is prone to form pulmonary thrombosis and lung dysfunction (61). Vascular enlargement is a specific imaging finding of lung injury caused by COVID-19, differing from the etiology of other pathogens (61, 62).
Different etiology often leads to different clinical manifestations and mechanisms. As early as 1998, researchers had found that ARDS caused by pneumonia and extrapulmonary factors (trauma, peritonitis, shock, intestinal infection, etc.) has different pathologies and distinct responses to PEEP (63). Since then, ARDS had been divided into pulmonary and extrapulmonary subgroups according to etiology, and a series of studies have been conducted on ARDS according to this classification. Compared with indirect injury (extrapulmonary ARDS), patients with direct injury (pulmonary ARDS) would have higher lung compliance and less responsiveness to PEEP (64). Alveolar epithelial injury, alveolar collapse and fibrin deposition mostly occur in pulmonary ARDS, while extrapulmonary ARDS mainly causes vascular endothelial injury (15, 65). Wide differences in imaging manifestations and biomarkers between pulmonary ARDS and extrapulmonary ARDS arouse extensive concern (32, 43). Additionally, several studies had shown that the mortality of those two are not the same. The mortality rate of extrapulmonary ARDS was slightly higher than that of pulmonary ARDS, but the difference was not significant (66, 67). Various etiologies of ARDS often have different outcomes and pathophysiological mechanisms. Subgrouping of ARDS by etiologies can predict the likely progression of the disease and prevent the occurrence of serious conditions.
In clinical practice, the incidence of sepsis or ARDS is higher than that of sepsis-induced ARDS, but the prognosis of sepsis-induced ARDS is worse (68, 69). Sepsis-related ARDS has a lower PaO2/FiO2 ratio, more obvious dyspnea, longer recovery time, and lower success rate of extubation than non-sepsis-related ARDS (5, 6, 70). According to previous studies, sepsis is the main cause of ARDS, accounting for 31% of the etiology of ARDS, and ARDS is a serious and destructive complication of sepsis (4). Extreme hypoxia approximately account for 38.2% of mortality in ICU (5). Hence, sepsis-induced ARDS is a notable group, and further research on this subgroup may immensely reduce the mortality of respiratory causes in ICU.
When studying the genetic characteristics of ARDS, it is arduous to obtain a family pedigree of ARDS patients, because the disease is often secondary to other serious systemic diseases and most cases are sporadic (71). However, researchers can study the association between individual genetic variants about ARDS risk factors. Studies found that the single nucleotide polymorphism (SNP) rs1190286 in the POPDC3 gene was associated with a reduced risk of ARDS, and the -308A allele of TNF was associated with increased mortality of ARDS (72, 73). The SNP rs315952C of IL1RN can increase the plasma level of IL1RA and reduce the risk of ARDS (74). In the study of the causal relationship between genes and diseases, Mendelian randomization analysis is commonly used (75, 76). Through the analysis of haplotypes of IL-1β, it was found that plasma level of IL-1β can affect the 90-day mortality of sepsis (77). As a key marker of endothelial activation and permeability, Ang-2 gene variation is also crucial for the disease risk prediction of ARDS. Studies have found that gene variation in Ang-2 may lead to an increased risk of ARDS, such as the variant T allele of one tSNP (rs2515475) and haplotype TT in block 2 containing the T allele (78). The association between the development of ARDS and this SNP was found to be dependent on the linkage disequilibrium between rs2515475 and rs2959811, primarily occurring in subjects of extrapulmonary injury (78). The primary reason for the impact of Ang-2 gene variation on ARDS occurrence lies in its association with pulmonary vascular permeability, leading to an increased likelihood of pulmonary edema fluid accumulation when extrapulmonary injury affects the lungs (79). Furthermore, it has been demonstrated that genetic variations in the Ang-2 gene can alter its expression (80). The plasma level of Ang-2 can serve as a reliable prognostic and mortality predictor for patients with sepsis and ARDS (81). Genetic polymorphisms contributing to ARDS occurrence are generally relevant to innate immunity, surfactant function, oxidative stress, and capillary endothelium (71). Genetic variants correlated to the pathogenesis of ARDS are the root of the heterogeneity of ARDS. Candidate genes can be obtained by microarray gene expression analysis, genome-wide association analysis, RNA sequencing and so on. Studies of candidate genes can contribute to further identify the causal genes in ARDS pathogenesis.
Individuals with underlying diseases and diverse personal characteristics or living habits also exert a certain influence on the occurrence and progression of ARDS. Various studies have demonstrated that patients suffering from diabetes, severe obesity, hypertension and cardiovascular disease are more prone to experiencing unfavorable outcomes following infection with COVID-19 (82–84). Diabetic and obese individuals typically exhibit compromised innate and adaptive immunity, along with a persistent state of chronic low-grade inflammation (85). In a regression cohort study, advanced age was found to be significantly associated with an increased risk of mortality, and patients who developed ARDS as a complication of COVID-19 were observed to have a higher prevalence of hypertension and diabetes (86). Additionally, lifestyle behaviors such as smoking and drinking can affect ARDS progression and prognosis. A 15-year cohort study revealed a dose-response correlation between smoking and the development of ARDS, whereas no such association was observed among individuals who consumed alcohol (87). Studies found that exposure to cigarette smoke results in an increased influx of neutrophils and induces damage to the alveolar epithelium (88). However, the meta-analysis of observational studies conducted between 1985 and 2015 by Simon et al. revealed a significant association between alcohol consumption and an increased risk of ARDS (89). This may be attributed to the depletion of glutathione resulting from chronic alcohol exposure, which subsequently leads to dysfunction of alveolar epithelium and failure in clearing alveolar fluid (90). The presence of significant inter-individual variations and diverse risk factors among patients often exerts an impact on the onset and progression of ARDS, thereby posing challenges to clinical diagnosis and treatment.
Sepsis is the most common pathogenic factor of ARDS, which can cause ARDS in less than a week, and the prognosis of those patients is poor (91). ARDS, a common clinical critical disease, has a mortality rate as high as 40%, and septic ARDS has a worse prognosis and higher mortality (91). The underlying mechanism of sepsis-induced ARDS is very complex, and the general process may be as follows: under the action of injury factors (such as bacterial and viral attacks, traumas, peritonitis and so on), circulating immune cells are activated and inflammatory mediators are overwhelmingly released in the blood, which damages the systemic vascular endothelium, including the capillary endothelium of the lung. When the endothelium of pulmonary capillaries is damaged, it will cause the activation of immune cells in the lung. These cells aggravate the pulmonary inflammatory response and eventually leads to lung injury (Figure 1). Septic ARDS are mainly histologically manifested in five aspects: epithelial and endothelial injury, various cell death, oxidative stress, microcirculation disorder and pulmonary edema.
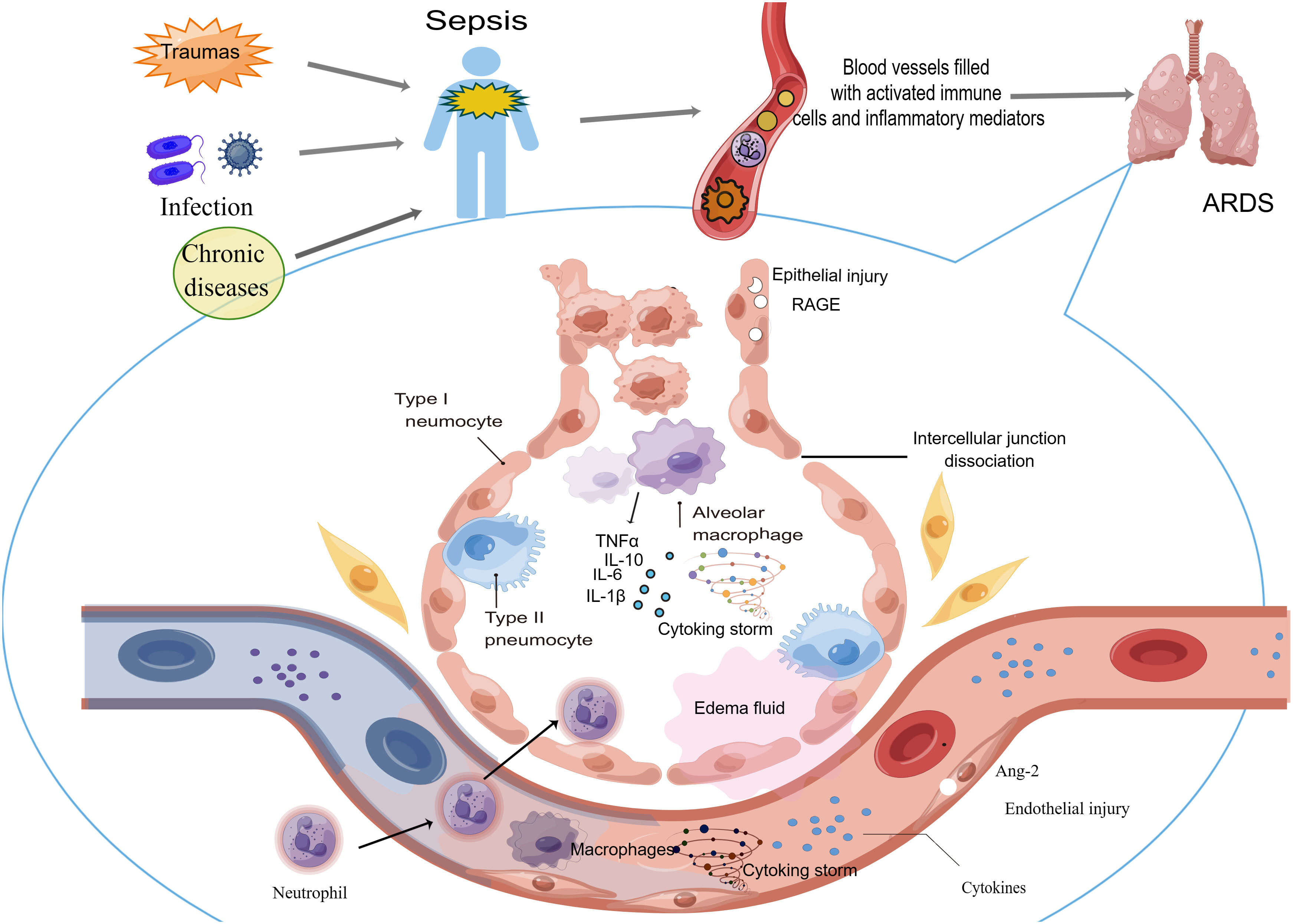
Figure 1 Sepsis can cause ARDS through circulating immune cells and cytokines. After the host undergoes the attack causing sepsis, circulating immune cells are activated and abundant inflammatory mediators are released in the blood. These cells and cytokines injure the vascular endothelium systemically, including the capillary endothelium of the lung. When the endothelium of pulmonary capillaries is damaged, activated immune cells migrating into the lung aggravate the pulmonary inflammatory response and eventually lead to ARDS occurrence.
The development of sepsis-induced ARDS can be attributed to both direct and indirect lung injury (11). The air-exchange function of the alveoli is maintained by the alveolar-capillary barrier, which is mainly composed of three parts, including the epithelial cell layer, the microvascular endothelial cell layer, and the interstitial space between the epithelium and the endothelium. The destruction of the barrier will disrupt the ventilation/perfusion ratio, leading to lung injury and dysfunction (92). Vascular endothelial integrity is synergistically established by vascular endothelial cadherin and the endothelial receptor kinase Tie2, and this process is regulated by vascular endothelial protein tyrosine phosphatase (VE-PTP) (93). Sepsis down-regulates the expression of Angpt-1, Tek, and KDR in systemic tissues at the transcriptional level, and Angpt-1, Tek, KDR corresponds to Ang-1, Tie2, and VEGFR2 or Flk-1.When these proteins are down-regulated, permeability of pulmonary capillary endothelial is increased and then lung injury develops (94).The alveolar epithelium, composed of alveolar type I and type II cells, is primarily responsible for gas exchange and alveolar fluid clearance in the lung. Epithelial cell death is the main feature of alveolar damage in ARDS, which can be caused by bacterial and viral infection, mechanical stretch, hypoxia and other reasons (95).
With the progress of scientific research, scientists have realized that the modes of cell death include but are not limited to necrosis and apoptosis necroptosis, pyroptosis, ferroptosis, lysosomal-dependent cell death, autophagy-dependent cell death and so on (96). Apoptosis is the main form of programmed cell death without producing inflammation (97). Excessive apoptosis of cells is critical in the progression of sepsis-induced ARDS. In sepsis-induced lung injury, apoptosis mainly appeared in endothelial rather than epithelial cells (98). Microvascular dysfunction induced by sepsis is closely related to vascular endothelial cell apoptosis, which is mediated by caspases and iNOS/NAPDH oxidase pathways. Besides, some studies had shown that in sepsis-induced ARDS, the degree of neutrophil apoptosis is inversely proportional to the disease severity, and the delay of neutrophil apoptosis will aggravate lung tissue injury (99).
In ARDS, inflammation and cell death interact such that both processes are gradually and automatically amplified, creating a vicious cycle. One study demonstrated that upregulation of IL-1R1promotes alveolar macrophage pyroptosis and worsens lung injury (100). In addition, inflammasomes released from pyroptotic cells can also induce IL-1β activation and enhance inflammatory responses in macrophages (101). Pulmonary endothelial cells also underwent caspase-11-mediated pyroptosis in sepsis-induced lung injury (102). Caspase-11, activated by LPS, can cleave Gasdermin D into polypeptides and form nanopores in the cell membrane, causing pyroptosis and the release of the cytokine IL-1β (103, 104). Extensive lung endothelial cell death, the release of lactate dehydrogenase (LDH) and IL-1β, and the destruction of endothelial cell barrier are important pathological mechanisms of sepsis-induced ARDS.
In inflammation, necrosis of macrophages can be regulated by a receptor-interacting protein kinase (RIPK)1-RIPK3 complex-dependent pathway, namely necroptosis. Necroptosis is driven by necrosome and it alerts the immune system after overwhelming damage to the host, involving cell swelling, membrane rupture, and release of cytoplasmic contents (105). LPS-TLR4 signaling pathway can promote the occurrence of alveolar macrophage necroptosis (106). The recognition of TLRs towards bacteria is closely associated with bacterial cell wall. TLR2 primarily recognizes the cell wall components of Gram-positive bacteria and peptidoglycan from Staphylococcus aureus, while TLR4 plays a pivotal role in LPS signaling transduction from Gram-negative bacteria (107). Necroptosis of endothelial cells and epithelial cells can also exist in sepsis-induced ARDS (108–110). In a mouse model of sepsis, RIPK3 deficiency significantly prolonged the survival of mice, and pretreatment with necrostatin-1 can reduce mortality rate in systemic inflammatory response syndrome (111, 112).
Ferroptosis, as a form of cell death dependent on the regulation of iron and ROS, is also involved in the disease progression of ARDS caused by sepsis. Studies have shown that neutrophil extracellular traps (NET)s can regulate sepsis-induced ARDS by activating ferroptosis in alveolar epithelial cells (113). Ferroptosis exists not only in alveolar epithelial cells, but also in alveolar capillary endothelial cells and macrophages (114–117). Researchers are inclined to investigate targeted drugs that can effectively inhibit the process of ferroptosis, thereby potentially impeding the progression of sepsis-induced ARDS (117–120). In addition, autophagy also plays a role in the process of sepsis-induced lung injury. Autophagy is a cytoprotective process that promotes the degradation and recycling of cellular components, and its activation can alleviate lung injury and inflammation caused by sepsis (121). Dysregulation of autophagy can lead to the aggravation of lung injury, pulmonary fibrosis, chronic obstructive pulmonary disease and other lung diseases (122–125).
Oxidative stress generally refers to a state of imbalance between oxidation and antioxidant defense system (126). Oxidation is basically caused by highly oxidizing substances such as ROS and reactive nitrogen radicals (RNS).The production of these substances can not only fight against invading pathogens, but also oxidize or damage DNA, proteins and lipids, inducing gene mutations, protein denaturation and lipid peroxidation (127). Under physiological conditions, the oxidation and reduction of the human body can reach a state of equilibrium. In the pathological situation of ARDS caused by sepsis, plentiful neutrophils flood into lung tissue and alveolar space, and neutrophils contain NADPH oxidase complexes, which are the primary source of ROS (128). Excessive ROS can bring about lung epithelial and endothelial injury. A study showed that instillation of enzymes producing oxygen metabolites in the rat lung alone can cause non-neutrophil-dependent ALI (129). Moreover, oxidative stress can also cause pulmonary capillary endothelial injury by impairing vasodilation, increasing the adhesion of leukocytes and platelets to the blood vessel wall, and improving capillary permeability, which eventually leads to lung injury (130). Nowadays, molecules associated with oxidative stress has been considered a vital and new target for the treatment of septic ARDS disease, and antioxidant therapy is an essential part of the therapy of sepsis (131, 132).
Sepsis is a systemic disease that can affect the microcirculation of the lung through hemodynamic changes and vascular endothelial damage. In sepsis patients, the deformability of erythrocytes reduced and the cell aggregation increased, resulting in microvascular circulation disorders (133, 134). Sepsis can cause the damage of the endothelial and the release of the inflammatory cytokines, leading to the adhesion and aggregation of the immune cells and platelets on endothelial cells, which possibly results in microvascular thrombosis and the occurrence of ARDS. During sepsis, cytokine storms can lead to the overexpression of nitric oxide (NO) synthase on endothelial cells and the synthesis of NO, resulting in vasodilation and adrenergic hyporeactivity, which affects microcirculatory hemodynamics (135). It has been reported that intestinal ischemia can induce a massive accumulation of neutrophils, leading to extensive occlusion of pulmonary arteries, veins, and microvessels (136). Park et al. used customized video-rate laser scanning confocal microscopy and lung imaging windows to show that of in the sepsis-induced ALI, microvascular perfusion in the lung was greatly affected and regulated by Mac-1 in neutrophils (137). The diameter of neutrophils is larger than that of pulmonary capillaries, and neutrophils must deform to enter capillaries, which is known as neutrophil sequestration (138, 139). Neutrophils will form clusters that block capillaries and arterioles, leading to the disorder of microcirculation in the lung and the formation of dead space with ventilation-perfusion mismatch (137). Besides, NETs formed by neutrophils could block capillaries in the lungs (140). The occurrence of pulmonary microcirculation disorders can result in the accumulation of metabolic waste within the lung, leading to the development of pulmonary edema, inflammation, and hypoxia.
The pathological feature of ARDS includes non-cardiogenic pulmonary edema (141), primarily resulting from both alveolar-capillary barrier damage and impaired clearance of edema fluid (142). Moreover, the ability of clearing edematous fluid in ARDS patients was associated with shorter length of ICU stays and reduced mortality (143). The edema fluid of ARDS can be transported from the alveolar cavity to the alveolar interstitium and eliminated by lymphatic drainage, while the alveolar fluid can be transferred to the vasculature for equilibrium via blood circulation (142). During the clearance process, the transport of alveolar fluid is mainly driven by active Na+ transport located in the alveolar epithelium. Na+/K+-ATPase drives Na+ channels to transport Na+ to achieve the clearance of alveolar fluid (142). In ARDS patients, the impaired ability of lung to clear edema may be related to the dysfunction of Na+/K+-ATPase located in alveolar epithelium (144). Studies have found that improving the function of Na+/K+-ATPase and/or epithelial Na+ channels, such as using beta-adrenergic agonists and increasing sodium transport, can improve alveolar fluid clearance (145–147). Adenovirus-mediated transfer of Na+/K+-ATPase β1 subunit genes can also increase lung fluid clearance (148). When this enzyme is inhibited or the associated ion channels are inhibited, alveolar edematous fluid will be poorly absorbed (149). Hence, exploring the mechanism of edema clearance and targeting the NA+/K+-ATPase holds significant guiding implications for the treatment of ARDS.
When pathogens invade the host, the innate immune system responds immediately (150). The pattern recognition receptors (PRRs) can be expressed by both innate immune cells and epithelial cells of the lung (151). PRR can recognize invading pathogens and endogenous molecules through pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP). In this process, immune responses are activated and amplified, which forms cytokine storms. The secretion and release of numerous pro-inflammatory substances can result in vascular endothelial dysfunction, thereby further enhancing pulmonary microvascular permeability, which ultimately facilitates the occurrence and progression of ARDS (152, 153). Following lung injury, innate immune cells such as neutrophils and monocytes are recruited to the alveolar space, leading to subsequent damage of the alveolar epithelium and endothelium. Consequently, a significant accumulation of edema fluid occurs in both the alveoli and interstitium (95, 154). During the pathogenetic process of sepsis-induced ARDS, the host is not always in a state of hyperimmunity. Due to the complex negative feedback of the immune system and immune exhaustion, it will also show a state of immune paralysis in the later stage of the disease (155). The immune system exhibits diminished or absent responsiveness to infection at this juncture, resulting in an exceedingly high fatality rate among these sepsis patients. The signaling pathways, inflammatory cytokines, recruited immunocytes, immune paralysis and complement system mentioned below are all closely involved in the cascade of inflammatory reactions in sepsis and its associated lung injury (Figure 2).
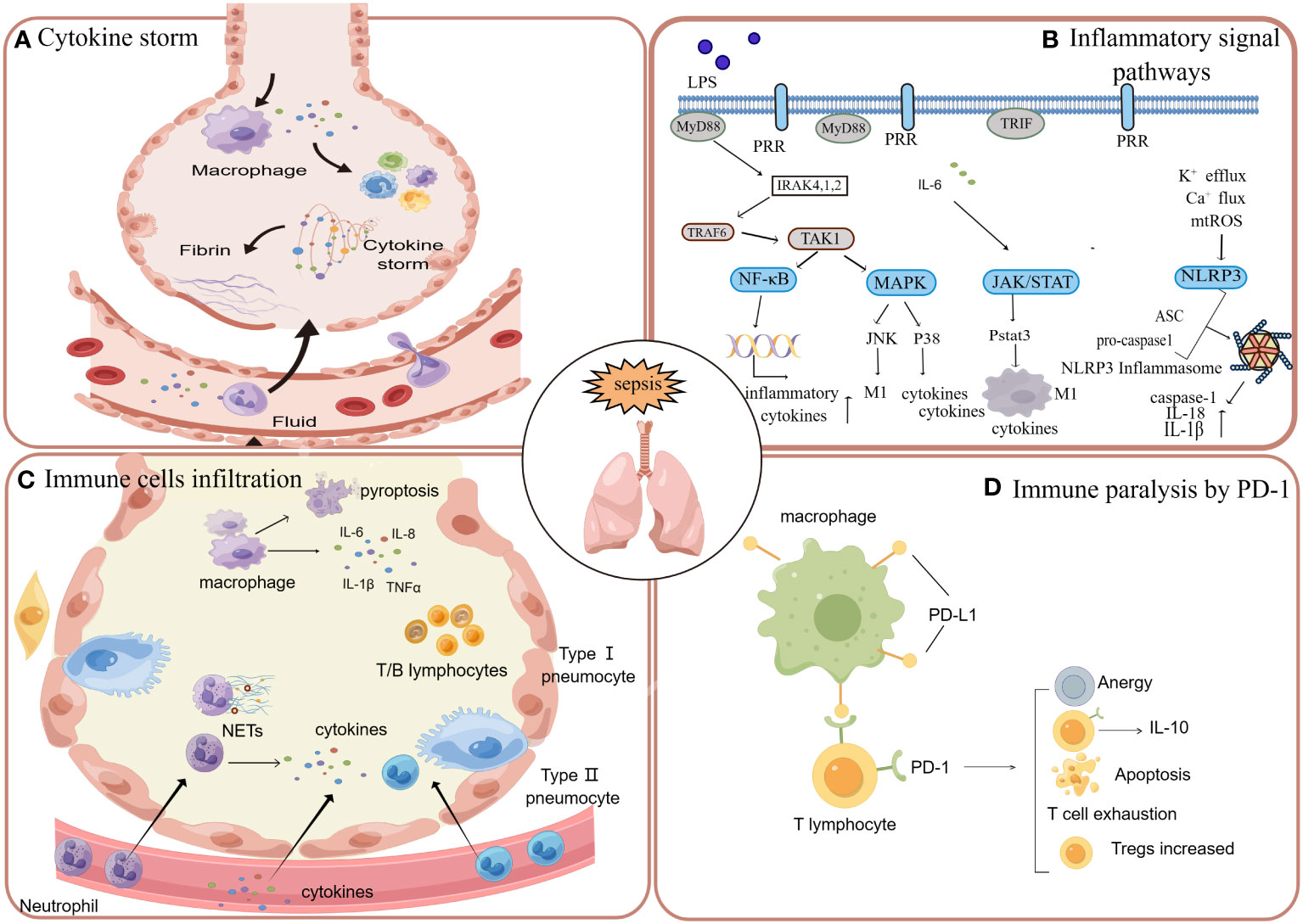
Figure 2 When the systemic system is in a state of sepsis, the inflammation in the circulation will affect various tissues and organs, in which the lung is often affected. (A) When the cytokine storm occurs in the circulation, cytokines can enter the lung through the broken endothelium and cause damage to the lung tissue. (B) Sepsis induces lung injury through inflammatory signaling pathways, including nuclear factor kappa B (NF-κB), JAK/STAT, and mitogen-activated protein kinase (MAPK). (C) In addition to the activation of resident macrophages in the alveoli, neutrophils, macrophages, a few lymphocytes and monocytes can also enter the alveoli through the damaged endothelium, causing inflammation and tissue damage. (D) With the massive activation of inflammation caused by sepsis, there will be apoptosis and depletion of immune cells, which is manifested as immune paralysis. The apoptosis, dysfunction and exhaustion of T cells are mainly mediated by the PD-1 pathway.
The progression of sepsis is significantly influenced by the involvement of crucial cytokines such as IL-1β, IL-18, IL-6, IL-12, IL-17, and others. Il-1β is a member of the IL-1 family and is produced by inflammasome activation. Studies have shown that the level of IL-1β in non-survivors is higher than that in survivors, indicating that the high level of IL-1β is probably related to the adverse outcome of sepsis (156). The key pathological alteration contributing to the progression of sepsis to septic ARDS is the progressive increase in pulmonary vascular permeability. IL-1β has been found to increase endothelial permeability by inhibiting the transcription of VE-cadherin (157), suggesting that IL-1β is an important cytokine in the pathogenesis of ARDS induced by sepsis. IL-18 in patients with septic ARDS were significantly higher than patients with sepsis alone, demonstrating that IL-18 in sepsis are key cytokines causing lung injury (158). In a randomized controlled trial, plasma IL-18 levels were also discovered to be associated with the mortality in sepsis-induced ARDS (159). In studies of cytokines, IL-6 has also been suggested to correlate with the severity of sepsis (160, 161). IL-6 was frequently found in the blood and lung of patients with sepsis, but the exact role of IL-6 in sepsis is still under investigation, which may be related to the activation of the complement pathway and capillary leakage (162, 163). In addition, IL-12, IL-17, IFN-γ, and TNF-α all play important pro-inflammatory roles during sepsis. They interact and promote each other, eventually inducing inevitable organ damage. For example, during sepsis, dendritic cells, macrophages, and lymphocytes could secrete IL-12, one cytokine able to promote T cell differentiation. The production of TH1 and TH17 cells promote the secretion of IFN-γ and IL-17 (164). As the core cytokine in the cytokine storm, IL-17 can stimulate endothelial cells, epithelial cells, fibroblasts or macrophages to synthesize and secrete GM-CSF, IL-1β, IL-6, TNF-α and other cytokines and chemokines (165). The findings suggest that inflammatory cytokines play a crucial role as mediators in the development of lung injury induced by sepsis, and they synergistically interact to facilitate the progression of inflammation.
PRR can interact with PAMP and DAMP, activating the myeloid differentiation primary response protein 88 (MyD88) dependent pathway or Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF) dependent pathway (166). Then, through this signal transduction, downstream nuclear factor κB (NF-κB) (167, 168), JAK/STAT (169, 170) mitogen-activated protein kinase (MAPK) (171–173) and other signaling pathways (174) are activated. These pathways upregulate the transcription of genes associated with inflammation, leading to the synthesis and release of various inflammatory molecules.
Some signal pathways in sepsis can cause lung injury and thus affect the disease process of ARDS. The NF-κB signaling pathway and its subsequent activation of NLRP3 inflammasome may be an important immune mechanism for ARDS induced by sepsis. In a sepsis induced ARDS rat mode, after the use of anti-TLR4 monoclonal antibodies, down-regulating expression of TLR4, MyD88, and NF-κB in macrophages and its alleviated pulmonary inflammatory injury were observed (168). NF-κB signaling pathway can further contribute to inflammasome production by activating the transcription of inflammasome components, such as the NLRP3 and the pro forms of cytokines (IL-18, IL-1β) (175). Blocking the activation of NLRP3 inflammasome and inhibiting the signaling pathway of NLRP3 can alleviate sepsis-induced lung injury (176).
The mitogen-activated protein kinases (MAPKs) pathway owns three major subfamilies, including the extracellular signal-regulated protein kinase (ERK) cascade, c-jun NH2 terminal kinase/stress-activated protein kinase (JNK/SAPK) cascade and p38-MAPK cascade (177). Multiple studies have found that ALI in sepsis patients can be significantly alleviated by inhibiting signaling pathways such as JNK and p38 MAPK (172, 178, 179). Pre-injection of specific inhibitors of JNK and p38 MAPK could decrease postoperative lung permeability and alleviate systemic inflammation after CLP inducing sepsis (172). One study on sepsis-induced ARDS also found that by inhibiting the activation of ERK1/2, p38 MAPK and p65, inflammatory infiltration and wet/dry ratio of lung tissue were all significantly decreased (171). Above all, pathways of ERK1/2, p38MAPK and JNK are probably involved in the lung injury induced by sepsis.
The activation of JAK/STAT signaling pathway has a dual effect on ARDS induced by sepsis. On the one hand, methotrexate (MTX), one inhibitor of the JAK/STAT signaling pathway, was shown to significantly reduce the production of pro-inflammatory cytokines and improve pulmonary inflammatory infiltration (169). Hence, inhibiting JAK/STAT signaling pathway could alleviate inflammatory lung injury in sepsis. On the other hand, several researchers have performed genome-wide analyses associated with ALI, the data suggest that the activation of STAT3 in type II alveolar pneumocytes (AT2) plays a dominant role in epithelial repair (180). Due to the bidirectional nature of this pathway, when targeting this pathway for anti-inflammation, it may be necessary to closely monitor the impact on epithelial repair when targeting this pathway for anti-inflammatory purposes.
Sepsis-related circulating inflammatory overload causes pulmonary capillary endothelial damage and lung inflammatory injury, which involves a variety of immune cells. In the early stage of sepsis-induced lung injury, the massive release of pro-inflammatory cytokines results in disruption of vascular endothelial structures and endothelial leakage. Neutrophils and monocytes migrate across the endothelial cells and infiltrate in the alveoli (166).
Neutrophils are important components of innate immunity. There are four main mechanisms facilitating neutrophils fighting against microbial infection: degranulation, phagocytosis, cytokine production and NET. Neutrophil activation in the alveolar space is a universal feature of ALI in humans and animal models. During an episode of sepsis, circulating neutrophils undergo deformation, leading to their entrapment in the small capillaries of the pulmonary microcirculation. Trapped neutrophils respond to local chemokine gradients and migrate into lung tissue to cause lung injury (181).Neutrophils infiltrated in lung tissue can play a bactericidal and phagocytic role, simultaneously, their own secretion of cytotoxic products and the production of NETs can also lead to alveolar epithelial and endothelial damage (182, 183). In sepsis, the network structure of NET can limit the transmission and proliferation of pathogens, and the important components of NET such as histone, neutrophil elastase, and MPO can play a bactericidal role (184). NET has a definite role in controlling infection, but increasingly more studies have shown that excessive production of NET can cause thrombosis and tissue damage (182, 183, 185). In sepsis- induced ALI, activated platelets can promote the formation of tissue factor-rich NETs, which in turn can release thrombotic signals (182). Moreover, data from critically ill patients with sepsis and lung injury suggest that higher levels of circulating NETs will have adverse clinical consequences and even cause organ dysfunction (186, 187). For the moment, researchers tend to explore effective targeted drugs to inhibit NETs, so as to reduce multiple organ function damage and lung injury caused by sepsis (188–190).
During sepsis, alveolar macrophages are activated and they are able to secrete inflammatory cytokines to affect inflammatory environment. Alveolar macrophages can promote the trans-endothelial migration of neutrophils through the Src kinase/NAPDH oxidase pathway (191). Alveolar macrophages are essential for the maintenance of inflammatory homeostasis in the lung, and the impairment of 11β-hydroxysteroid dehydrogenase type-1(HSD-1) in alveolar macrophages leads to the aggravation of inflammation and the increased mortality in sepsis-induced ARDS (192). IFN-β can improve the dysfunction of alveolar macrophages in sepsis and reduce the mortality of septic ARDS (193). The mode of macrophage death can also affect lung inflammation, such as, pyroptosis. The up-regulation of IL-1R1 signaling caused by IL-1β can promote the pyroptosis of alveolar macrophages and aggravate lung injury (100).
Apart from that, lymphocytes and monocytes may also perform a crucial role in sepsis-induced ARDS. Single-cell sequencing of pneumonia, sepsis, and sepsis with ARDS found that the monocyte cluster in patients with early ARDS caused by sepsis was characterized by down-regulation of SOCS3 expression and up-regulation of multiple type I IFN-induced genes (194). In a bioinformatics analysis of key genes in sepsis-induced ARDS, compared with patients with sepsis alone, patients with septic-ARDS had higher levels of activated memory CD4+T lymphocytes and naive B lymphocytes, and lower levels of CD8+ T lymphocytes in their lung tissues (195). The findings of these studies suggest that lymphocytes and monocytes may play a pivotal role in the pathogenesis of this disease, necessitating further research.
The development and coexistence of systemic inflammatory response syndrome and compensatory anti-inflammatory response syndrome in sepsis exert a profound influence on the systemic immune environment (196).Initially, basic and clinical research on sepsis mainly focused on its excessive inflammation. But in contrast to the beneficial results of animal experiments, the clinical trials often failed, in which the management of inflammatory control and immunosuppressive drugs had been used (197–199). Due to the continuous development of immune paralysis in the process of sepsis progression, the use of immunosuppressive therapy may lead to treatment failure or even condition aggravation. The main mechanisms of septic immune paralysis include abnormal immune cell death, lymphocyte exhaustion, inhibition of antigen presentation, and expansion and activation of regulatory immune cells (200). When the apoptosis of immune cells is inhibited, the mortality rate of sepsis will decrease (201). Immune paralysis may also play an important role in the development of septic ARDS, and severe immunosuppression is often associated with adverse clinical outcomes. In one study, emergent lung and spleen harvest from patients who had died of sepsis revealed an expanded population of immunosuppressive cells in both organs and expression of inhibitory receptor ligands PD-L1 on lung epithelial cells, with extensive depletion of splenic CD4+, CD8+T cells and HLA-DR cells (202). After patients with sepsis were cured and discharged, systemic organs were still in a state of immunosuppression for a long time, and such patients were more likely to have secondary severe pulmonary infection (203).The suppression of pulmonary innate immunity induced by sepsis is mediated by IRAK-M molecules. When IRAK-M is knockout, septic mice would have a higher survival rate and bacterial clearance rate in the lung and blood (204). Immune paralysis dominates the systemic immune system in the later stages of sepsis, which contributes to the failure of immunosuppressive therapy. This compromised immune response renders patients vulnerable to infections and leads to a dismal prognosis, necessitating careful attention during the late-stage treatment process of septic ARDS.
The dysregulated systemic inflammatory response to infection can result in organ dysfunction throughout the body, leading to sepsis. Researchers found experimentally that complement is overactivated in septic hosts with adverse consequences (205–207). Complement activation is an innate immune response to fight infection, but excessive activation ultimately enhances organ dysfunction, including the heart, lungs and kidney (208–212). In sepsis, activation of the complement system is caused by recognition of PAMPS by mannose-binding lectins and ficolin, among others (213). DAMPs, the endogenous molecules released by distressed or damaged cell, can also lead to complement activation (213). The imbalanced activation of complement in the whole body can give rise to thrombotic inflammation and eventually bring about organ dysfunction. In sepsis-induced lung injury, complement activation can lead to the release of C3a and C5a, which can up-regulate the expression of endothelial adhesion molecules and recruit neutrophils to the alveoli (214). The released inflammatory molecules will recruit more inflammatory cells, eventually resulting in tissue damage and lung dysfunction (214). A marked increase in circulating complement activation products has been found in sepsis (215), and complement activation product C3a is associated with enhanced alveolar capillaries permeability (216). In addition, sepsis patients with enhanced capillary permeability are more likely to develop ARDS (217). The terminal complement complex exhibited an average increase of 110% two days prior to the progression from sepsis to ARDS, as demonstrated by a study (218). The above indicate that complement is closely related to the progression of sepsis to ARDS. The targeting of complement as a therapeutic approach for sepsis-induced lung injury had been investigated in several studies, yielding favorable outcomes (219, 220).
And beyond that, the coagulation and complement systems are closely related, and activation of the coagulation system is considered as part of the initial immune response to invading pathogens. Inflammation and coagulation are key host responses to infection and injury and have been implicated in the pathogenesis of both sepsis and ARDS (221). There is evidence that thrombosis and coagulation dysfunction are the main causes of ALI and ARDS (222). Both sepsis and ARDS are in a procoagulant state, and the interaction between the inflammatory response and the coagulation system plays a crucial role in the pathogenesis of sepsis-induced ARDS (223, 224). Sepsis can cause the alveolar-capillary barrier injury and further lead to the continuous state of promoting coagulation and fibrinolysis inhibition systemically (221, 225). In this process, tissue factor (TF) is considered to be the primary procoagulant initiation factor following infection and inflammation (226). Endotoxins or bacteria can activate TF-dependent clotting pathways to form a procoagulant state in the blood vessels and alveolar spaces of the lung (224, 227). TF can activate clotting factor VII, which further activates the downstream clotting cascade to produce thrombin. While thrombin causes fibrin deposition in interstitial spaces, perivascular spaces and pulmonary alveolus, high levels of thrombin may up-regulate lung inflammation genes and cause alveolar-capillary barrier damage (223). When subjected to pro-inflammatory stimulation (such as cytokine stimulation), alveolar epithelial cells will release TF-bearing microparticles, and the increase of circulating TF and microparticles expression levels will promote the coagulation cascade, resulting in the formation of thrombin and extensive fibrin inundation (228). Extensive pulmonary microthrombosis will increase dead space ventilation, which leads to the mismatch of ventilation-perfusion and causes respiratory dysfunction. An animal study on sepsis-induced ALI demonstrated that inhibiting the exogenous clotting pathway through inactivation of active site of FVIIa caused reduced fibrin deposition, decreased systemic cytokine response, and improved lung inflammation (229, 230). Nebulization of recombinant human-activated protein C(rh-APC) or plasma-derived human antithrombin had been found to attenuate lung coagulation activation, stimulate pulmonary fibrinolysis and alleviate lung inflammation without affecting blood clotting systemically (231). Nebulization of heparin or danaparoid also decreased pulmonary coagulopathy; however, they possess the drawback of impacting systemic coagulation (231, 232).
Researchers had found that thrombin-activated platelets and TF-enriched NETs facilitates immune thrombosis in sepsis-related lung injury, in which the two-hit procedures of thrombosis formation triggered by activated platelets and NETs contribute to the progression of ARDS (182). Besides, the expression of TF in peripheral blood monocytes induces intravascular thrombosis during sepsis as well (233).
Precision medicine is a medical strategy requiring clinicians to take into account the individual characteristics of patients, in which the patients will be grouped according to their heterogeneity, and receive targeted prevention, diagnosis and treatment. Numerous therapeutic approaches have garnered clinical attention for the treatment of ARDS, encompassing drug therapy, respiratory management, ECMO, and cell therapy. The utilization of inhaled surfactant therapy was initially employed in the neonatal respiratory distress syndrome (NRDS) due to the incomplete development of type II alveolar cells, which resulted in insufficient production of functional surfactant (234). Given the similarities between adult ARDS and NRDS, certain studies had also been conducted on inhaled surfactants for adult ARDS (235, 236). Although a multicenter randomized controlled trial in 1996 showed that inhaled surfactant did not improve 30-day mortality in sepsis-associated ARDS (237), recent studies in the treatment of COVID-19 have shown that surfactant administration can combat pulmonary dysfunction and enhance the diffusion of other drugs along the airway epithelium, which sheds new light on the application of surfactants (238, 239).
The inhibition of neutrophil elastase (NE) is crucial in mitigating lung injury caused by neutrophils, thus making NE inhibitors indispensable for the treatment of ARDS. In the past two years, the COVID-19 pandemic has brought renewed attention to the therapeutic role of NE inhibitors, which are anticipated to mitigate extracellular matrix degradation caused by neutrophils and restrict viral dissemination by inhibiting proteolytic activation of the S protein (240, 241). Compared with healthy individuals, the plasma level of NE in ARDS patients is significantly higher, and studies have found that SIRS patients with NE content over 220ng/ml are prone to ARDS (242). Elevated NE activity was also observed in bronchoalveolar lavage fluid of ARDS patients (243). The inhalation of prostaglandins has the potential to induce pulmonary vasodilation, decrease pulmonary artery pressure, and enhance oxygenation; however, there is substantial evidence suggesting that prostaglandins may elicit severe adverse reactions, such as hypotension (244).
The antifungal agent ketoconazole exhibits anti-inflammatory properties. Previous clinical studies have demonstrated its ability to prevent the progression of severe patients to ARDS (245), while showing limited efficacy in treating early-stage ARDS (246). The administration of ibuprofen did not demonstrate efficacy in preventing the development of ARDS in critically ill patients with sepsis (247); however, its early utilization for COVID-19 treatment has exhibited a preventive effect on complications and an improvement in prognosis (248).
ECMO is a form of extracorporeal life support technology, serving as an alternative therapeutic approach in cases of critical cardiac or pulmonary dysfunction (249). The device extracts blood, facilitates gas exchange outside the body, and then reinfuses the oxygenated blood back into the circulatory system of patients. Since the 2009 influenza A (H1N1) pandemic, ECMO has been documented to significantly reduce mortality in severe ARDS cases, thus garnering considerable attention and advancement (250–252). The utilization of ECMO in adults with severe ARDS was shown a significant reduction in 60-day mortality when compared to conventional mechanical ventilation (253). But a randomized controlled study demonstrated that there is no survival benefit associated with the early use of ECMO in severe ARDS when compared to its use as a rescue therapy after conventional mechanical ventilation (254). The use of ECMO is indicated for the treatment of severe ARDS following failure of protective mechanical ventilation and prone position ventilation (249, 255). Additionally, it can be employed in cases where the patient’s condition is unstable and urgent transfer to a specialized ward (256). ECMO has been extensively employed during the COVID-19 pandemic and showed significant improvements in patient outcomes and reductions in mortality rates across multiple studies (257, 258). Although ECMO has demonstrated survival benefits in several studies of severe ARDS, it still entails numerous risks, including infection (259) and coagulation related complications (257, 260). Therefore, careful attention must be paid to its indications and contraindications during ECMO utilization to reduce mortality.
Mesenchymal stem cells and their extracellular vesicles have been shown to modulate immunity and improve endothelial barrier integrity in cellular and animal experimental studies of ARDS (261, 262). However, in the recent randomized controlled trials of severe ARDS from COVID-19, no benefit was found on survival and ventilator-free days (263). Whether mesenchymal stem cell-related cell therapy is suitable for ARDS, a rapidly progressive disease, needs to be further studied and explored.
A randomized controlled trial found that only those patients with severe ARDS (PaO2/FiO2 ≤ 150mmHg) had a good response to prone position ventilation. And prone position ventilation significantly reduced 28-day and 90-day mortality in these patients (24). In ARDS caused by COVID-19, the use of dexamethasone combined with respiratory support can reduce the mortality of patients (264). A randomized controlled trial reported in 2020 claimed that the early use of dexamethasone reduced the duration of mechanical ventilation and mortality in patients with moderate and severe ARDS (265). Therefore, dexamethasone may be a good choice for patients with moderate-to-severe ARDS or patients with COVID-19 who have extensive lung injuries. In addition, in a randomized controlled trial, there was no significant difference between the simvastatin treated and the placebo-treated in the trial of ARDS. But in a secondary analysis of this study, patients were grouped according to inflammatory degrees, and the therapeutic effect of simvastatin on the hyperinflammatory phenotype was observed (27). The common treatments for ARDS are lung-protective ventilation, prone position ventilation, and fluid therapy. According to the heterogeneity of the disease, clinicians would not blindly use these methods for all patients without subgrouping, but take the individual characteristics into consideration to implement targeted and effective treatment.
In view of the particularity and risk of sepsis-induced ARDS, advanced treatment methods are expected to be developed urgently. Inflammatory infiltration and high vascular permeability are the key factors of sepsis-induced ARDS (266), which are two important targets worth cutting into. For example, in the aspect of inflammation, the inhibition of protein expression in key inflammatory signaling pathways and anti-complement antibodies are used to control septic ARDS in animals (171, 267, 268). In terms of vascular endothelial permeability, protection by endothelial connexin is enhanced by inhibiting proteins that affect the endothelial barrier or natural endothelial-protective factors (266, 269). In the study of sepsis patients, the levels of plasma Ang-2 were found to be associated with ANGRT2 genetic variants, which would lead to an increased risk of ARDS (270). For patients with high plasma ANG-2 levels, lowering ANG-2 levels may be an appropriate strategy for the prevention and treatment of sepsis-induced ARDS.
ARDS is a heterogeneous syndrome, and different etiology, severity of symptoms, and individual genetic differences can bring about various clinical manifestations and responses to treatment. In view of the heterogeneity of ARDS, increasingly more researchers and clinical staff are inclined to classify ARDS, and further carry out personalized treatment to obtain beneficial results. For example, in studies using statins and prone ventilation to treat ARDS, patients with severe ARDS (PaO2/FiO2 < 150mmHg) were specifically selected for these two trials, and treatment-related survival benefits were obtained (24, 271). Given the heterogeneity of ARDS, categorizing patients with ARDS into pulmonary and extrapulmonary groups based on the etiology is another solution having received much attention. There are differences between pulmonary ARDS and extrapulmonary ARDS in pathological manifestations, imaging findings, respiratory mechanics, mortality, biomarkers and even genetics (32, 43, 63, 72, 272).
Sepsis is a systemic disease in which the immune system reacts out of control in response to infection or injury, leading to systemic organ dysfunction. When sepsis progresses to the advanced stage of shock, it is more likely to be accompanied by ARDS and higher mortality rate (273). Sepsis-induced lung injury primarily arises from vascular endothelial damage, leading to the transmission of circulating inflammation to the lungs. This condition predominantly manifests as damage to the lung capillary endothelium and alveolar epithelium, cell death, oxidative stress, microcirculatory disorders and pulmonary edema. Pathological manifestations of that disease include diffuse alveolar damage, destruction of alveolar capillaries, pulmonary edema, and potentially even atelectasis (91).
The immune mechanism of sepsis-induced ARDS is mostly related to the pathogenesis of sepsis itself, involving excessive activation of inflammatory signaling pathways and cytokines, activation of immune cells, immune paralysis and interaction of coagulation and complement. The cascade of cytokine activation in sepsis is able to cause systemic inflammation, including the lungs. Sepsis causes lung inflammation and injury by damaging the vascular endothelium, and the activation of endothelial cells give rise to the accumulation of inflammatory cells and inflammatory cytokines secretion. The aggregation of neutrophils can produce a large amount of myeloperoxidase and NETs, and then cause vascular thrombosis. Alveolar macrophages can regulate lung inflammation, and most of them can promote the amplification of inflammatory response and increase the concentration of cytokines. The death and apoptosis of alveolar macrophages can promote the accumulation of neutrophils, and their pyroptosis process also produces inflammatory cytokines. Obvious immune paralysis occurs in the later stage of sepsis, which often indicates poor prognosis and high mortality (274). The immunosuppression of sepsis is similar to that of tumors, which is related to the inhibition of T lymphocytes caused by PD-1 pathway. When immune paralysis is relieved, the prognosis of patients can be greatly improved. Perhaps, PD-1 and PD-L1 can also become important targets of septic-ARDS. After being discharged, surviving patients would undergo longer periods of immunosuppression and are more likely to lack of resistance against infection (202, 203).
At present, the solution to the heterogeneity of ARDS is to pay attention to the manifestations and biological subgroups of ARDS, such as PaO2/FiO2 level and inflammation degree, and give candidate therapeutic strategies according to the classification. The heterogeneity of ARDS affects the efficacy of various treatments, including drug therapy, respiratory management, ECMO (275). The discovery of the heterogeneity of ARDS had benefited from the latent class analysis of randomized controlled trials, which has made advances in the treatment of specific subgroups. There are still existing many unanswered questions regarding the advancement of precision medicine in ARDS, such as the biologic overlap between ARDS and sepsis, the impact on patient recruitment, the stability of ARDS subphenotypes and so on (276). The resolution of key issues can promote the progress of effective treatment of ARDS and usher in the progress of severe disease research.
According to a report from the Lancet Respiratory Medicine in 2022, precision medicine for ARDS is necessary, but there are still two major challenges (277). One is that the key nodes have limited information and cannot be determined. The pathogenesis of ARDS is complex, and the specific pathways involved will become deranged during the disease process. The other is rapid disease progression of ARDS, with a high mortality rate within 72 hours (278). At present, precision medicine is widely used in the field of cancer, but it still takes days to weeks to achieve staging and classification. The time requirements in the precision management of ARDS are deemed impractical, and it is crucial to rapidly determine our precise classification of the disease within a matter of hours.
HX: Writing – original draft, Writing – review & editing. SS: Writing – original draft, Writing – review & editing. WL: Conceptualization, Writing – review & editing. XX: Writing – review & editing. ZZ: Conceptualization, Funding acquisition, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National Key Research and Development Program of China, Grant No. 2021YFC2501800 (to ZZ) and the National Natural Science Foundation of China, Grant No. 82272182, 82072202 (to ZZ).
Figures are created using Figdraw (www.figdraw.com).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Banavasi H, Nguyen P, Osman H, Soubani AO. Management of ARDS - what works and what does not. Am J Med Sci (2021) 362(1):13–23. doi: 10.1016/j.amjms.2020.12.019
2. Katzenstein AL, Bloor CM, Leibow AA. Diffuse alveolar damage–the role of oxygen, shock, and related factors. A review. Am J Pathol (1976) 85(1):209–28.
3. Matthay MA, Arabi Y, Arroliga AC, Bernard G, Bersten AD, Brochard LJ, et al. A new global definition of acute respiratory distress syndrome. Am J Respir Crit Care Med (2023). doi: 10.1164/rccm.202303-0558WS
4. Bersten AD, Edibam C, Hunt T, Moran J. Incidence and mortality of acute lung injury and the acute respiratory distress syndrome in three Australian States. Am J Respir Crit Care Med (2002) 165(4):443–8. doi: 10.1164/ajrccm.165.4.2101124
5. Bice T, Cox CE, Carson SS. Cost and health care utilization in ARDS–different from other critical illness? Semin Respir Crit Care Med (2013) 34(4):529–36. doi: 10.1055/s-0033-1351125
6. Sheu CC, Gong MN, Zhai R, Chen F, Bajwa EK, Clardy PF, et al. Clinical characteristics and outcomes of sepsis-related vs non-sepsis-related ARDS. Chest (2010) 138(3):559–67. doi: 10.1378/chest.09-2933
7. Zhang YY, Ning BT. Signaling pathways and intervention therapies in sepsis. Signal Transduct Target Ther (2021) 6(1):407. doi: 10.1038/s41392-021-00816-9
8. Huang M, Cai S, Su J. The pathogenesis of sepsis and potential therapeutic targets. Int J Mol Sci (2019) 20(21):5376. doi: 10.3390/ijms20215376
9. Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med (2016) 193(3):259–72. doi: 10.1164/rccm.201504-0781OC
10. Zhou J, Qian C, Zhao M, Yu X, Kang Y, Ma X, et al. Epidemiology and outcome of severe sepsis and septic shock in intensive care units in mainland China. PloS One (2014) 9(9):e107181. doi: 10.1371/journal.pone.0107181
11. Englert JA, Bobba C, Baron RM. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight (2019) 4(2):e124061. doi: 10.1172/jci.insight.124061
12. Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med (2019) 40(1):31–9. doi: 10.1055/s-0039-1683996
13. Phua J, Stewart TE, Ferguson ND. Acute respiratory distress syndrome 40 years later: time to revisit its definition. Crit Care Med (2008) 36(10):2912–21. doi: 10.1097/CCM.0b013e31817d20bd
14. Subudhi S, Voutouri C, Hardin CC, Nikmaneshi MR, Patel AB, Verma A, et al. Strategies to minimize heterogeneity and optimize clinical trials in Acute Respiratory Distress Syndrome (ARDS): Insights from mathematical modelling. EBioMedicine (2022) 75:103809. doi: 10.1016/j.ebiom.2021.103809
15. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet (2021) 398(10300):622–37. doi: 10.1016/s0140-6736(21)00439-6
16. Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med (1994) 149(3 Pt 1):818–24. doi: 10.1164/ajrccm.149.3.7509706
17. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. Jama (2012) 307(23):2526–33. doi: 10.1001/jama.2012.5669
18. Thille AW, Esteban A, Fernández-Segoviano P, Rodriguez JM, Aramburu JA, Peñuelas O, et al. Comparison of the Berlin definition for acute respiratory distress syndrome with autopsy. Am J Respir Crit Care Med (2013) 187(7):761–7. doi: 10.1164/rccm.201211-1981OC
19. Frank AJ, Thompson BT. Pharmacological treatments for acute respiratory distress syndrome. Curr Opin Crit Care (2010) 16(1):62–8. doi: 10.1097/MCC.0b013e328334b151
20. Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med (2014) 2(8):611–20. doi: 10.1016/s2213-2600(14)70097-9
21. Dahmer MK, Yang G, Zhang M, Quasney MW, Sapru A, Weeks HM, et al. Identification of phenotypes in paediatric patients with acute respiratory distress syndrome: a latent class analysis. Lancet Respir Med (2022) 10(3):289–97. doi: 10.1016/s2213-2600(21)00382-9
22. Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, Calfee CS. Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med (2018) 44(11):1859–69. doi: 10.1007/s00134-018-5378-3
23. Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, Loundou A, et al. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med (2010) 363(12):1107–16. doi: 10.1056/NEJMoa1005372
24. Guérin C, Reignier J, Richard JC, Beuret P, Gacouin A, Boulain T, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med (2013) 368(23):2159–68. doi: 10.1056/NEJMoa1214103
25. Puybasset L, Gusman P, Muller JC, Cluzel P, Coriat P, Rouby JJ. Regional distribution of gas and tissue in acute respiratory distress syndrome. III. Consequences for the effects of positive end-expiratory pressure. CT Scan ARDS Study Group. Adult Respiratory Distress Syndrome. Intensive Care Med (2000) 26(9):1215–27. doi: 10.1007/s001340051340
26. Croce MA, Fabian TC, Davis KA, Gavin TJ. Early and late acute respiratory distress syndrome: two distinct clinical entities. J Trauma (1999) 46(3):361–6. doi: 10.1097/00005373-199903000-00001
27. Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med (2018) 6(9):691–8. doi: 10.1016/s2213-2600(18)30177-2
28. Lim CM, Jung H, Koh Y, Lee JS, Shim TS, Lee SD, et al. Effect of alveolar recruitment maneuver in early acute respiratory distress syndrome according to antiderecruitment strategy, etiological category of diffuse lung injury, and body position of the patient. Crit Care Med (2003) 31(2):411–8. doi: 10.1097/01.Ccm.0000048631.88155.39
29. Pelosi P, Brazzi L, Gattinoni L. Prone position in acute respiratory distress syndrome. Eur Respir J (2002) 20(4):1017–28. doi: 10.1183/09031936.02.00401702
30. Reilly JP, Christie JD, Meyer NJ. fifty years of research in ARDS. Genomic contributions and opportunities. . Am J Respir Crit Care Med (2017) 196(9):1113–21. doi: 10.1164/rccm.201702-0405CP
31. Nuckton TJ, Alonso JA, Kallet RH, Daniel BM, Pittet JF, Eisner MD, et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med (2002) 346(17):1281–6. doi: 10.1056/NEJMoa012835
32. Goodman LR, Fumagalli R, Tagliabue P, Tagliabue M, Ferrario M, Gattinoni L, et al. Adult respiratory distress syndrome due to pulmonary and extrapulmonary causes: CT, clinical, and functional correlations. Radiology (1999) 213(2):545–52. doi: 10.1148/radiology.213.2.r99nv42545
33. Desai SR, Wells AU, Suntharalingam G, Rubens MB, Evans TW, Hansell DM. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary injury: a comparative CT study. Radiology (2001) 218(3):689–93. doi: 10.1148/radiology.218.3.r01mr31689
34. Liao KM, Chen CW, Hsiue TR, Lin WC. Timing of acute respiratory distress syndrome onset is related to patient outcome. J Formos Med Assoc (2009) 108(9):694–703. doi: 10.1016/s0929-6646(09)60392-2
35. Vincent JL, Sakr Y, Groeneveld J, Zandstra DF, Hoste E, Malledant Y, et al. ARDS of early or late onset: does it make a difference? Chest (2010) 137(1):81–7. doi: 10.1378/chest.09-0714
36. Reilly JP, Bellamy S, Shashaty MG, Gallop R, Meyer NJ, Lanken PN, et al. Heterogeneous phenotypes of acute respiratory distress syndrome after major trauma. Ann Am Thorac Soc (2014) 11(5):728–36. doi: 10.1513/AnnalsATS.201308-280OC
37. Binnie A, Tsang JL, dos Santos CC. Biomarkers in acute respiratory distress syndrome. Curr Opin Crit Care (2014) 20(1):47–55. doi: 10.1097/mcc.0000000000000048
38. van der Zee P, Rietdijk W, Somhorst P, Endeman H, Gommers D. A systematic review of biomarkers multivariately associated with acute respiratory distress syndrome development and mortality. Crit Care (2020) 24(1):243. doi: 10.1186/s13054-020-02913-7
39. Blondonnet R, Constantin JM, Sapin V, Jabaudon M. A pathophysiologic approach to biomarkers in acute respiratory distress syndrome. Dis Markers 2016. (2016) p:3501373. doi: 10.1155/2016/3501373
40. Shirasawa M, Fujiwara N, Hirabayashi S, Ohno H, Iida J, Makita K, et al. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells (2004) 9(2):165–74. doi: 10.1111/j.1356-9597.2004.00712.x
41. Kuipers MT, van der Poll T, Schultz MJ, Wieland CW. Bench-to-bedside review: Damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care (2011) 15(6):235. doi: 10.1186/cc10437
42. Hanford LE, Enghild JJ, Valnickova Z, Petersen SV, Schaefer LM, Schaefer TM, et al. Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). J Biol Chem (2004) 279(48):50019–24. doi: 10.1074/jbc.M409782200
43. Calfee CS, Janz DR, Bernard GR, May AK, Kangelaris KN, Matthay MA, et al. Distinct molecular phenotypes of direct vs indirect ARDS in single-center and multicenter studies. Chest (2015) 147(6):1539–48. doi: 10.1378/chest.14-2454
44. Mrozek S, Jabaudon M, Jaber S, Paugam-Burtz C, Lefrant JY, Rouby JJ, et al. Elevated plasma levels of sRAGE are associated with nonfocal CT-based lung imaging in patients with ARDS: a prospective multicenter study. Chest (2016) 150(5):998–1007. doi: 10.1016/j.chest.2016.03.016
45. Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA. Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med (2012) 40(6):1731–7. doi: 10.1097/CCM.0b013e3182451c87
46. Agrawal A, Matthay MA, Kangelaris KN, Stein J, Chu JC, Imp BM, et al. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med (2013) 187(7):736–42. doi: 10.1164/rccm.201208-1460OC
47. Granowitz EV, Clark BD, Mancilla J, Dinarello CA. Interleukin-1 receptor antagonist competitively inhibits the binding of interleukin-1 to the type II interleukin-1 receptor. J Biol Chem (1991) 266(22):14147–50.
48. Fisher CJ Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. Jama (1994) 271(23):1836–43.
49. Opal SM, Fisher CJ Jr., Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. . Crit Care Med (1997) 25(7):1115–24. doi: 10.1097/00003246-199707000-00010
50. Meyer NJ, Ferguson JF, Feng R, Wang F, Patel PN, Li M, et al. A functional synonymous coding variant in the IL1RN gene is associated with survival in septic shock. Am J Respir Crit Care Med (2014) 190(6):656–64. doi: 10.1164/rccm.201403-0586OC
51. Sanai L, Haynes WG, MacKenzie A, Grant IS, Webb DJ. Endothelin production in sepsis and the adult respiratory distress syndrome. Intensive Care Med (1996) 22(1):52–6. doi: 10.1007/bf01728331
52. Bursten SL, Federighi DA, Parsons P, Harris WE, Abraham E, Moore EE Jr., et al. An increase in serum C18 unsaturated free fatty acids as a predictor of the development of acute respiratory distress syndrome. Crit Care Med (1996) 24(7):1129–36. doi: 10.1097/00003246-199607000-00011
53. Gonçalves de Albuquerque CF, Burth P, Younes Ibrahim M, Garcia DG, Bozza PT, Castro Faria Neto HC, et al. Reduced plasma nonesterified fatty acid levels and the advent of an acute lung injury in mice after intravenous or enteral oleic acid administration. Mediators Inflamm 2012. (2012) p:601032. doi: 10.1155/2012/601032
54. Sapru A, Calfee CS, Liu KD, Kangelaris K, Hansen H, Pawlikowska L, et al. Plasma soluble thrombomodulin levels are associated with mortality in the acute respiratory distress syndrome. Intensive Care Med (2015) 41(3):470–8. doi: 10.1007/s00134-015-3648-x
55. Proudfoot A, Bayliffe A, O'Kane CM, Wright T, Serone A, Bareille PJ, et al. Novel anti-tumour necrosis factor receptor-1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax (2018) 73(8):723–30. doi: 10.1136/thoraxjnl-2017-210305
56. Delucchi K, Famous KR, Ware LB, Parsons PE, Thompson BT, Calfee CS. Stability of ARDS subphenotypes over time in two randomised controlled trials. Thorax (2018) 73(5):439–45. doi: 10.1136/thoraxjnl-2017-211090
57. Kojicic M, Li G, Hanson AC, Lee KM, Thakur L, Vedre J, et al. Risk factors for the development of acute lung injury in patients with infectious pneumonia. Crit Care (2012) 16(2):R46. doi: 10.1186/cc11247
58. Kalil AC, Thomas PG. Influenza virus-related critical illness: pathophysiology and epidemiology. Crit Care (2019) 23(1):258. doi: 10.1186/s13054-019-2539-x
59. Li H, Weng H, Lan C, Zhang H, Wang X, Pan J, et al. Comparison of patients with avian influenza A (H7N9) and influenza A (H1N1) complicated by acute respiratory distress syndrome. Med (Baltimore) (2018) 97(12):e0194. doi: 10.1097/md.0000000000010194
60. Bonmarin I, Belchior E, Bergounioux J, Brun-Buisson C, Mégarbane B, Chappert JL, et al. Intensive care unit surveillance of influenza infection in France: the 2009/10 pandemic and the three subsequent seasons. Euro Surveill (2015) 20(46):2300044. doi: 10.2807/1560-7917.Es.2015.20.46.30066
61. Gibson PG, Qin L, Puah SH. COVID-19 acute respiratory distress syndrome (ARDS): clinical features and differences from typical pre-COVID-19 ARDS. Med J Aust (2020) 213(2):54–56.e1. doi: 10.5694/mja2.50674
62. Ye Z, Zhang Y, Wang Y, Huang Z, Song B. Chest CT manifestations of new coronavirus disease 2019 (COVID-19): a pictorial review. Eur Radiol (2020) 30(8):4381–9. doi: 10.1007/s00330-020-06801-0
63. Gattinoni L, Pelosi P, Suter PM, Pedoto A, Vercesi P, Lissoni A. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am J Respir Crit Care Med (1998) 158(1):3–11. doi: 10.1164/ajrccm.158.1.9708031
64. Albaiceta GM, Taboada F, Parra D, Blanco A, Escudero D, Otero J. Differences in the deflation limb of the pressure-volume curves in acute respiratory distress syndrome from pulmonary and extrapulmonary origin. Intensive Care Med (2003) 29(11):1943–9. doi: 10.1007/s00134-003-1965-y
65. Hoelz C, Negri EM, Lichtenfels AJ, Conceção GM, Barbas CS, Saldiva PH, et al. Morphometric differences in pulmonary lesions in primary and secondary ARDS. A preliminary study in autopsies. Pathol Res Pract (2001) 197(8):521–30.
66. Agarwal R, Aggarwal AN, Gupta D, Behera D, Jindal SK. Etiology and outcomes of pulmonary and extrapulmonary acute lung injury/ARDS in a respiratory ICU in North India. Chest (2006) 130(3):724–9. doi: 10.1378/chest.130.3.724
67. Monchi M, Bellenfant F, Cariou A, Joly LM, Thebert D, Laurent I, et al. Early predictive factors of survival in the acute respiratory distress syndrome. A multivariate analysis. Am J Respir Crit Care Med (1998) 158(4):1076–81. doi: 10.1164/ajrccm.158.4.9802009
68. Mikkelsen ME, Shah CV, Meyer NJ, Gaieski DF, Lyon S, Miltiades AN, et al. The epidemiology of acute respiratory distress syndrome in patients presenting to the emergency department with severe sepsis. Shock (2013) 40(5):375–81. doi: 10.1097/SHK.0b013e3182a64682
69. Kim WY, Hong SB. Sepsis and acute respiratory distress syndrome: recent update. Tuberc Respir Dis (Seoul) (2016) 79(2):53–7. doi: 10.4046/trd.2016.79.2.53
70. Dvorščak MB, Lupis T, Adanić M, Šarić JP. [Acute respiratory distress syndrome and other respiratory disorders in sepsis]. Acta Med Croatica (2015) 69(3):167–75.
71. Meyer NJ, Christie JD. Genetic heterogeneity and risk of acute respiratory distress syndrome. Semin Respir Crit Care Med (2013) 34(4):459–74. doi: 10.1055/s-0033-1351121
72. Tejera P, Meyer NJ, Chen F, Feng R, Zhao Y, O'Mahony DS, et al. Distinct and replicable genetic risk factors for acute respiratory distress syndrome of pulmonary or extrapulmonary origin. J Med Genet (2012) 49(11):671–80. doi: 10.1136/jmedgenet-2012-100972
73. Gong MN, Zhou W, Williams PL, Thompson BT, Pothier L, Boyce P, et al. -308GA and TNFB polymorphisms in acute respiratory distress syndrome. Eur Respir J (2005) 26(3):382–9. doi: 10.1183/09031936.05.00000505
74. Meyer NJ, Feng R, Li M, Zhao Y, Sheu CC, Tejera P, et al. IL1RN coding variant is associated with lower risk of acute respiratory distress syndrome and increased plasma IL-1 receptor antagonist. Am J Respir Crit Care Med (2013) 187(9):950–9. doi: 10.1164/rccm.201208-1501OC
75. Cohen JC, Boerwinkle E, Mosley TH Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med (2006) 354(12):1264–72. doi: 10.1056/NEJMoa054013
76. Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T, et al. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. Bmj (2011) 342:d548. doi: 10.1136/bmj.d548
77. Wang F, Meyer NJ, Walley KR, Russell JA, Feng R. Causal genetic inference using haplotypes as instrumental variables. Genet Epidemiol (2016) 40(1):35–44. doi: 10.1002/gepi.21940
78. Su L, Zhai R, Sheu CC, Gallagher DC, Gong MN, Tejera P, et al. Genetic variants in the angiopoietin-2 gene are associated with increased risk of ARDS. Intensive Care Med (2009) 35(6):1024–30. doi: 10.1007/s00134-009-1413-8
79. Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PloS Med (2006) 3(3):e46. doi: 10.1371/journal.pmed.0030046
80. Hegen A, Koidl S, Weindel K, Marmé D, Augustin HG, Fiedler U. Expression of angiopoietin-2 in endothelial cells is controlled by positive and negative regulatory promoter elements. Arterioscler Thromb Vasc Biol (2004) 24(10):1803–9. doi: 10.1161/01.ATV.0000140819.81839.0e
81. Rosenberger CM, Wick KD, Zhuo H, Wu N, Chen Y, Kapadia SB, et al. Early plasma angiopoietin-2 is prognostic for ARDS and mortality among critically ill patients with sepsis. Crit Care (2023) 27(1):234. doi: 10.1186/s13054-023-04525-3
82. Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. Jama (2020) 323(18):1775–6. doi: 10.1001/jama.2020.4683
83. Drucker DJ. Coronavirus infections and type 2 diabetes-shared pathways with therapeutic implications. Endocr Rev (2020) 41(3):bnaa011. doi: 10.1210/endrev/bnaa011
84. Abu-Farha M, Al-Mulla F, Thanaraj TA, Kavalakatt S, Ali H, Abdul Ghani M, et al. Impact of diabetes in patients diagnosed with COVID-19. Front Immunol (2020) 11:576818. doi: 10.3389/fimmu.2020.576818
85. Andersen CJ, Murphy KE, Fernandez ML. Impact of obesity and metabolic syndrome on immunity. Adv Nutr (2016) 7(1):66–75. doi: 10.3945/an.115.010207
86. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/s0140-6736(20)30566-3
87. Iribarren C, Jacobs DR Jr., Sidney S, Gross MD, Eisner MD. Cigarette smoking, alcohol consumption, and risk of ARDS: a 15-year cohort study in a managed care setting. Chest (2000) 117(1):163–8. doi: 10.1378/chest.117.1.163
88. Hirsch J, Chalkley RJ, Bentley T, Burlingame AL, Frank JA. Double impact of cigarette smoke and mechanical ventilation on the alveolar epithelial type II cell. Crit Care (2014) 18(2):R50. doi: 10.1186/cc13795
89. Simou E, Leonardi-Bee J, Britton J. The effect of alcohol consumption on the risk of ARDS: A systematic review and meta-analysis. Chest (2018) 154(1):58–68. doi: 10.1016/j.chest.2017.11.041
90. Fung C, Hyzy RC. The tipping point: alcohol as a risk factor for ARDS. Chest (2018) 154(1):6–7. doi: 10.1016/j.chest.2018.02.002
91. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. Jama (2016) 315(8):788–800. doi: 10.1001/jama.2016.0291
92. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med (2017) 377(6):562–72. doi: 10.1056/NEJMra1608077
93. Frye M, Dierkes M, Küppers V, Vockel M, Tomm J, Zeuschner D, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med (2015) 212(13):2267–87. doi: 10.1084/jem.20150718
94. Bomsztyk K, Mar D, An D, Sharifian R, Mikula M, Gharib SA, et al. Experimental acute lung injury induces multi-organ epigenetic modifications in key angiogenic genes implicated in sepsis-associated endothelial dysfunction. Crit Care (2015) 19(1):225. doi: 10.1186/s13054-015-0943-4
95. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers (2019) 5(1):18. doi: 10.1038/s41572-019-0069-0
96. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res (2019) 29(5):347–64. doi: 10.1038/s41422-019-0164-5
97. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol (2008) 9(3):231–41. doi: 10.1038/nrm2312
98. Pelosi P, Gattinoni L. Acute respiratory distress syndrome of pulmonary and extra-pulmonary origin: fancy or reality? Intensive Care Med (2001) 27(3):457–60. doi: 10.1007/s001340100879
99. Fialkow L, Fochesatto Filho L, Bozzetti MC, Milani AR, Rodrigues Filho EM, Ladniuk RM, et al. Neutrophil apoptosis: a marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit Care (2006) 10(6):R155. doi: 10.1186/cc5090
100. He X, Qian Y, Li Z, Fan EK, Li Y, Wu L, et al. TLR4-upregulated IL-1β and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep (2016) 6:31663. doi: 10.1038/srep31663
101. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation. Nat Immunol (2014) 15(8):727–37. doi: 10.1038/ni.2913
102. Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest (2017) 127(11):4124–35. doi: 10.1172/jci94495
103. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
104. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
105. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol (2014) 192(10):4709–17. doi: 10.4049/jimmunol.1302692
106. Li Z, Scott MJ, Fan EK, Li Y, Liu J, Xiao G, et al. Tissue damage negatively regulates LPS-induced macrophage necroptosis. Cell Death Differ (2016) 23(9):1428–47. doi: 10.1038/cdd.2016.21
107. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity (1999) 11(4):443–51. doi: 10.1016/s1074-7613(00)80119-3
108. Qing DY, Conegliano D, Shashaty MG, Seo J, Reilly JP, Worthen GS, et al. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med (2014) 190(11):1243–54. doi: 10.1164/rccm.201406-1095OC
109. Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC, et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ (2015) 22(8):1313–27. doi: 10.1038/cdd.2014.222
110. Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Peñaloza HF, et al. Necroptosis promotes staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep (2016) 16(8):2219–30. doi: 10.1016/j.celrep.2016.07.039
111. Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity (2011) 35(6):908–18. doi: 10.1016/j.immuni.2011.09.020
112. Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis (2012) 3(11):e437. doi: 10.1038/cddis.2012.176
113. Zhang H, Liu J, Zhou Y, Qu M, Wang Y, Guo K, et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Biol Sci (2022) 18(8):3337–57. doi: 10.7150/ijbs.69141
114. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discovery (2022) 8(1):43. doi: 10.1038/s41420-021-00807-3
115. Shen K, Wang X, Wang Y, Jia Y, Zhang Y, Wang K, et al. miR-125b-5p in adipose derived stem cells exosome alleviates pulmonary microvascular endothelial cells ferroptosis via Keap1/Nrf2/GPX4 in sepsis lung injury. Redox Biol (2023) 62:102655. doi: 10.1016/j.redox.2023.102655
116. Wang Y, Chen D, Xie H, Jia M, Sun X, Peng F, et al. AUF1 protects against ferroptosis to alleviate sepsis-induced acute lung injury by regulating NRF2 and ATF3. Cell Mol Life Sci (2022) 79(5):228. doi: 10.1007/s00018-022-04248-8
117. Zou X, Liu C, Huang Z, Xiang S, Li K, Yuan Y, et al. Inhibition of STEAP1 ameliorates inflammation and ferroptosis of acute lung injury caused by sepsis in LPS-induced human pulmonary microvascular endothelial cells. Mol Biol Rep (2023). 50(7): 5667–74. doi: 10.1007/s11033-023-08403-7
118. Lai K, Song C, Gao M, Deng Y, Lu Z, Li N, et al. Uridine alleviates sepsis-induced acute lung injury by inhibiting ferroptosis of macrophage. Int J Mol Sci (2023) 24(6):5093. doi: 10.3390/ijms24065093
119. Li J, Li M, Li L, Ma J, Yao C, Yao S. Hydrogen sulfide attenuates ferroptosis and stimulates autophagy by blocking mTOR signaling in sepsis-induced acute lung injury. Mol Immunol (2022) 141:318–27. doi: 10.1016/j.molimm.2021.12.003
120. Tang X, Liu J, Yao S, Zheng J, Gong X, Xiao B. Ferulic acid alleviates alveolar epithelial barrier dysfunction in sepsis-induced acute lung injury by activating the Nrf2/HO-1 pathway and inhibiting ferroptosis. Pharm Biol (2022) 60(1):2286–94. doi: 10.1080/13880209.2022.2147549
121. Zhao H, Chen H, Xiaoyin M, Yang G, Hu Y, Xie K, et al. Autophagy activation improves lung injury and inflammation in sepsis. Inflammation (2019) 42(2):426–39. doi: 10.1007/s10753-018-00952-5
122. Chang AL, Ulrich A, Suliman HB, Piantadosi CA. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med (2015) 78:179–89. doi: 10.1016/j.freeradbiomed.2014.10.582
123. Chu R, Wang J, Bi Y, Nan G. The kinetics of autophagy in the lung following acute spinal cord injury in rats. Spine J (2018) 18(5):845–56. doi: 10.1016/j.spinee.2018.01.001
124. Meng Y, Pan M, Zheng B, Chen Y, Li W, Yang Q, et al. Autophagy attenuates angiotensin II-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid Redox Signal (2019) 30(4):520–41. doi: 10.1089/ars.2017.7261
125. Li Y, Yu G, Yuan S, Tan C, Lian P, Fu L, et al. Cigarette smoke-induced pulmonary inflammation and autophagy are attenuated in ephx2-deficient mice. Inflammation (2017) 40(2):497–510. doi: 10.1007/s10753-016-0495-z
126. Gutteridge JM, Mitchell J. Redox imbalance in the critically ill. Br Med Bull (1999) 55(1):49–75. doi: 10.1258/0007142991902295
127. Huet O, Dupic L, Harrois A, Duranteau J. Oxidative stress and endothelial dysfunction during sepsis. Front Biosci (Landmark Ed) (2011) 16(5):1986–95. doi: 10.2741/3835
128. Yang SC, Tsai YF, Pan YL, Hwang TL. Understanding the role of neutrophils in acute respiratory distress syndrome. BioMed J (2021) 44(4):439–46. doi: 10.1016/j.bj.2020.09.001
129. Johnson KJ, . Fantone 3JC, Kaplan J, Ward PA. In vivo damage of rat lungs by oxygen metabolites. J Clin Invest (1981) 67(4):983–93. doi: 10.1172/jci110149
130. Joffre J, Hellman J. Oxidative stress and endothelial dysfunction in sepsis and acute inflammation. Antioxid Redox Signal (2021) 35(15):1291–307. doi: 10.1089/ars.2021.0027
131. Sang A, Wang Y, Wang S, Wang Q, Wang X, Li X, et al. Quercetin attenuates sepsis-induced acute lung injury via suppressing oxidative stress-mediated ER stress through activation of SIRT1/AMPK pathways. Cell Signal (2022) 96:110363. doi: 10.1016/j.cellsig.2022.110363
132. Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med (2007) 13(6):688–94. doi: 10.1038/nm1577
133. Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med (2008) 34(1):17–60. doi: 10.1007/s00134-007-0934-2
134. Piagnerelli M, Boudjeltia KZ, Vanhaeverbeek M, Vincent JL. Red blood cell rheology in sepsis. Intensive Care Med (2003) 29(7):1052–61. doi: 10.1007/s00134-003-1783-2
135. Colantuoni A, Martini R, Caprari P, Ballestri M, Capecchi PL, Gnasso A, et al. COVID-19 sepsis and microcirculation dysfunction. Front Physiol (2020) 11:747. doi: 10.3389/fphys.2020.00747
136. Yuan Y, Alwis I, Wu MCL, Kaplan Z, Ashworth K, Bark D Jr., et al. Neutrophil macroaggregates promote widespread pulmonary thrombosis after gut ischemia. Sci Transl Med (2017) 9(409):eaam5861. doi: 10.1126/scitranslmed.aam5861
137. Park I, Kim M, Choe K, Song E, Seo H, Hwang Y, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J (2019) 53(3):1800786. doi: 10.1183/13993003.00786-2018
138. Doerschuk CM. Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation (2001) 8(2):71–88.
139. Lien DC, Henson PM, Capen RL, Henson JE, Hanson WL, Wagner WW Jr., et al. Neutrophil kinetics in the pulmonary microcirculation during acute inflammation. Lab Invest (1991) 65(2):145–59.
140. Zhang H, Wang Y, Qu M, Li W, Wu D, Cata JP, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med (2023) 13(1):e1170. doi: 10.1002/ctm2.1170
141. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med (2000) 342(18):1334–49. doi: 10.1056/nejm200005043421806
142. Morty RE, Eickelberg O, Seeger W. Alveolar fluid clearance in acute lung injury: what have we learned from animal models and clinical studies? Intensive Care Med (2007) 33(7):1229–40. doi: 10.1007/s00134-007-0662-7
143. Sznajder JI. Alveolar edema must be cleared for the acute respiratory distress syndrome patient to survive. Am J Respir Crit Care Med (2001) 163(6):1293–4. doi: 10.1164/ajrccm.163.6.ed1801d
144. Sznajder JI, Factor P, Ingbar DH. Invited review: lung edema clearance: role of Na(+)-K(+)-ATPase. J Appl Physiol (1985) 93(5):1860–6. doi: 10.1152/japplphysiol.00022.2002
145. Sakuma T, Folkesson HG, Suzuki S, Okaniwa G, Fujimura S, Matthay MA. Beta-adrenergic agonist stimulated alveolar fluid clearance in ex vivo human and rat lungs. Am J Respir Crit Care Med (1997) 155(2):506–12. doi: 10.1164/ajrccm.155.2.9032186
146. Saldías FJ, Lecuona E, Comellas AP, Ridge KM, Rutschman DH, Sznajder JI. beta-adrenergic stimulation restores rat lung ability to clear edema in ventilator-associated lung injury. Am J Respir Crit Care Med (2000) 162(1):282–7. doi: 10.1164/ajrccm.162.1.9809058
147. Tibayan FA, Chesnutt AN, Folkesson HG, Eandi J, Matthay MA. Dobutamine increases alveolar liquid clearance in ventilated rats by beta-2 receptor stimulation. Am J Respir Crit Care Med (1997) 156(2 Pt 1):438–44. doi: 10.1164/ajrccm.156.2.9609141
148. Factor P, Saldias F, Ridge K, Dumasius V, Zabner J, Jaffe HA, et al. Augmentation of lung liquid clearance via adenovirus-mediated transfer of a Na,K-ATPase beta1 subunit gene. J Clin Invest (1998) 102(7):1421–30. doi: 10.1172/jci3214
149. Icard P, Saumon G. Alveolar sodium and liquid transport in mice. Am J Physiol (1999) 277(6):L1232–8. doi: 10.1152/ajplung.1999.277.6.L1232
150. Kumar V, Sharma A. Innate immunity in sepsis pathogenesis and its modulation: new immunomodulatory targets revealed. J Chemother (2008) 20(6):672–83. doi: 10.1179/joc.2008.20.6.672
151. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol (2004) 5(10):987–95. doi: 10.1038/ni1112
152. Chavez A, Smith M, Mehta D. New insights into the regulation of vascular permeability. Int Rev Cell Mol Biol (2011) 290:205–48. doi: 10.1016/b978-0-12-386037-8.00001-6
153. Sharp C, Millar AB, Medford AR. Advances in understanding of the pathogenesis of acute respiratory distress syndrome. Respiration (2015) 89(5):420–34. doi: 10.1159/000381102
154. Rosseau S, Hammerl P, Maus U, Walmrath HD, Schütte H, Grimminger F, et al. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol (2000) 279(1):L25–35. doi: 10.1152/ajplung.2000.279.1.L25
155. Yao RQ, Ren C, Zheng LY, Xia ZF, Yao YM. Advances in immune monitoring approaches for sepsis-induced immunosuppression. Front Immunol (2022) 13:891024. doi: 10.3389/fimmu.2022.891024
156. Mera S, Tatulescu D, Cismaru C, Bondor C, Slavcovici A, Zanc V, et al. Multiplex cytokine profiling in patients with sepsis. Apmis (2011) 119(2):155–63. doi: 10.1111/j.1600-0463.2010.02705.x
157. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, et al. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest (2020) 130(7):3684–98. doi: 10.1172/jci136908
158. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med (2012) 185(11):1225–34. doi: 10.1164/rccm.201201-0003OC
159. Rogers AJ, Guan J, Trtchounian A, Hunninghake GM, Kaimal R, Desai M, et al. Association of elevated plasma interleukin-18 level with increased mortality in a clinical trial of statin treatment for acute respiratory distress syndrome. Crit Care Med (2019) 47(8):1089–96. doi: 10.1097/ccm.0000000000003816
160. Wu HP, Chen CK, Chung K, Tseng JC, Hua CC, Liu YC, et al. Serial cytokine levels in patients with severe sepsis. Inflammation Res (2009) 58(7):385–93. doi: 10.1007/s00011-009-0003-0
161. Kellum JA, Kong L, Fink MP, Weissfeld LA, Yealy DM, Pinsky MR, et al. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch Intern Med (2007) 167(15):1655–63. doi: 10.1001/archinte.167.15.1655
162. Riedemann NC, Guo RF, Hollmann TJ, Gao H, Neff TA, Reuben JS, et al. Regulatory role of C5a in LPS-induced IL-6 production by neutrophils during sepsis. FASEB J (2004) 18(2):370–2. doi: 10.1096/fj.03-0708fje
163. Riedemann NC, Neff TA, Guo RF, Bernacki KD, Laudes IJ, Sarma JV, et al. Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J Immunol (2003) 170(1):503–7. doi: 10.4049/jimmunol.170.1.503
164. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (1993) 260(5107):547–9. doi: 10.1126/science.8097338
165. Reynolds JM, Angkasekwinai P, Dong C. IL-17 family member cytokines: regulation and function in innate immunity. Cytokine Growth Factor Rev (2010) 21(6):413–23. doi: 10.1016/j.cytogfr.2010.10.002
166. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol (2020) 11:1722. doi: 10.3389/fimmu.2020.01722
167. Chen G, Hou Y, Li X, Pan R, Zhao D. Sepsis-induced acute lung injury in young rats is relieved by calycosin through inactivating the HMGB1/MyD88/NF-κB pathway and NLRP3 inflammasome. Int Immunopharmacol (2021) 96:107623. doi: 10.1016/j.intimp.2021.107623
168. Zhou S, Wang G, Zhang W. Effect of TLR4/MyD88 signaling pathway on sepsis-associated acute respiratory distress syndrome in rats, via regulation of macrophage activation and inflammatory response. Exp Ther Med (2018) 15(4):3376–84. doi: 10.3892/etm.2018.5815
169. Bringué J, Guillamat-Prats R, Martinez ML, Torrents E, Camprubí-Rimblas M, Blanch L, et al. Methotrexate ameliorates systemic inflammation and septic associated-lung damage in a cecal ligation and puncture septic rat model. Int J Mol Sci (2021) 22(17):9612. doi: 10.3390/ijms22179612
170. Zhang J, Luo Y, Wang X, Zhu J, Li Q, Feng J, et al. Global transcriptional regulation of STAT3- and MYC-mediated sepsis-induced ARDS. Ther Adv Respir Dis (2019) 13:1753466619879840. doi: 10.1177/1753466619879840
171. Cong Z, Li D, Tao Y, Lv X, Zhu X. α(2A) -AR antagonism by BRL-44408 maleate attenuates acute lung injury in rats with downregulation of ERK1/2, p38MAPK, and p65 pathway. J Cell Physiol (2020) 235(10):6905–14. doi: 10.1002/jcp.29586
172. Fang W, Cai SX, Wang CL, Sun XX, Li K, Yan XW, et al. Modulation of mitogen−activated protein kinase attenuates sepsis−induced acute lung injury in acute respiratory distress syndrome rats. Mol Med Rep (2017) 16(6):9652–8. doi: 10.3892/mmr.2017.7811
173. Lou L, Hu D, Chen S, Wang S, Xu Y, Huang Y, et al. Protective role of JNK inhibitor SP600125 in sepsis-induced acute lung injury. Int J Clin Exp Pathol (2019) 12(2):528–38.
174. Li W, Li D, Chen Y, Abudou H, Wang H, Cai J, et al. Classic signaling pathways in alveolar injury and repair involved in sepsis-induced ALI/ARDS: new research progress and prospect. Dis Markers 2022. (2022) p:6362344. doi: 10.1155/2022/6362344
175. Lee S, Suh GY, Ryter SW, Choi AM. Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. Am J Respir Cell Mol Biol (2016) 54(2):151–60. doi: 10.1165/rcmb.2015-0231TR
176. Li J, Bai Y, Tang Y, Wang X, Cavagnaro MJ, Li L, et al. A 4-benzene-indol derivative alleviates LPS-induced acute lung injury through inhibiting the NLRP3 inflammasome. Front Immunol (2022) 13:812164. doi: 10.3389/fimmu.2022.812164
177. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science (2002) 298(5600):1911–2. doi: 10.1126/science.1072682
178. Zhao H, Zhao M, Wang Y, Li F, Zhang Z. Glycyrrhizic acid prevents sepsis-induced acute lung injury and mortality in rats. J Histochem Cytochem (2016) 64(2):125–37. doi: 10.1369/0022155415610168
179. Kim SJ, Baek KS, Park HJ, Jung YH, Lee SM. Compound 9a, a novel synthetic histone deacetylase inhibitor, protects against septic injury in mice by suppressing MAPK signalling. Br J Pharmacol (2016) 173(6):1045–57. doi: 10.1111/bph.13414
180. Paris AJ, Hayer KE, Oved JH, Avgousti DC, Toulmin SA, Zepp JA, et al. STAT3-BDNF-TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nat Cell Biol (2020) 22(10):1197–210. doi: 10.1038/s41556-020-0569-x
181. Goodman RB, Strieter RM, Frevert CW, Cummings CJ, Tekamp-Olson P, Kunkel SL, et al. Quantitative comparison of C-X-C chemokines produced by endotoxin-stimulated human alveolar macrophages. Am J Physiol (1998) 275(1):L87–95. doi: 10.1152/ajplung.1998.275.1.L87
182. Zhang H, Zhou Y, Qu M, Yu Y, Chen Z, Zhu S, et al. Tissue factor-enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis-induced lung injury. Front Cell Infect Microbiol (2021) 11:677902. doi: 10.3389/fcimb.2021.677902
183. Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep (2016) 6:37252. doi: 10.1038/srep37252
184. Kumar S, Payal N, Srivastava VK, Kaushik S, Saxena J, Jyoti A. Neutrophil extracellular traps and organ dysfunction in sepsis. Clin Chim Acta (2021) 523:152–62. doi: 10.1016/j.cca.2021.09.012
185. Yang S, Qi H, Kan K, Chen J, Xie H, Guo X, et al. Neutrophil extracellular traps promote hypercoagulability in patients with sepsis. Shock (2017) 47(2):132–9. doi: 10.1097/shk.0000000000000741
186. Margraf S, Lögters T, Reipen J, Altrichter J, Scholz M, Windolf J. Neutrophil-derived circulating free DNA (cf-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock (2008) 30(4):352–8. doi: 10.1097/SHK.0b013e31816a6bb1
187. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight (2018) 3(3):e98178. doi: 10.1172/jci.insight.98178
188. Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood (2021) 138(25):2702–13. doi: 10.1182/blood.2021011525
189. Hawez A, Taha D, Algaber A, Madhi R, Rahman M, Thorlacius H. MiR-155 regulates neutrophil extracellular trap formation and lung injury in abdominal sepsis. J Leukoc Biol (2022) 111(2):391–400. doi: 10.1002/jlb.3a1220-789rr
190. Qiao X, Kashiouris MG, L'Heureux M, Fisher BJ, Leichtle SW, Truwit JD, et al. Biological effects of intravenous vitamin C on neutrophil extracellular traps and the endothelial glycocalyx in patients with sepsis-induced ARDS. Nutrients (2022) 14(20):4415. doi: 10.3390/nu14204415
191. Wang Z, Rui T, Yang M, Valiyeva F, Kvietys PR. Alveolar macrophages from septic mice promote polymorphonuclear leukocyte transendothelial migration via an endothelial cell Src kinase/NADPH oxidase pathway. J Immunol (2008) 181(12):8735–44. doi: 10.4049/jimmunol.181.12.8735
192. Mahida RY, Lax S, Bassford CR, Scott A, Parekh D, Hardy RS, et al. Impaired alveolar macrophage 11β-hydroxysteroid dehydrogenase type 1 reductase activity contributes to increased pulmonary inflammation and mortality in sepsis-related ARDS. Front Immunol (2023) 14:1159831. doi: 10.3389/fimmu.2023.1159831
193. Hiruma T, Tsuyuzaki H, Uchida K, Trapnell BC, Yamamura Y, Kusakabe Y, et al. IFN-β Improves sepsis-related alveolar macrophage dysfunction and postseptic acute respiratory distress syndrome-related mortality. Am J Respir Cell Mol Biol (2018) 59(1):45–55. doi: 10.1165/rcmb.2017-0261OC
194. Jiang Y, Rosborough BR, Chen J, Das S, Kitsios GD, McVerry BJ, et al. Single cell RNA sequencing identifies an early monocyte gene signature in acute respiratory distress syndrome. JCI Insight (2020) 5(13):e135678. doi: 10.1172/jci.insight.135678
195. Chen Y, Qiu C, Cai W. Identification of key immune genes for sepsis-induced ARDS based on bioinformatics analysis. Bioengineered (2022) 13(1):697–708. doi: 10.1080/21655979.2021.2012621
196. Bone RC, Newton SI. sepsis, SIRS, and CARS. Crit Care Med (1996) 24(7):1125–8. doi: 10.1097/00003246-199607000-00010
197. Venkatesh B, Finfer S, Cohen J, Rajbhandari D, Arabi Y, Bellomo R, et al. Adjunctive glucocorticoid therapy in patients with septic shock. N Engl J Med (2018) 378(9):797–808. doi: 10.1056/NEJMoa1705835
198. Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature (1987) 330(6149):662–4. doi: 10.1038/330662a0
199. Fisher,. CJ Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med (1996) 334(26):1697–702. doi: 10.1056/nejm199606273342603
200. Nakamori Y, Park EJ, Shimaoka M. Immune deregulation in sepsis and septic shock: reversing immune paralysis by targeting PD-1/PD-L1 pathway. Front Immunol (2020) 11:624279. doi: 10.3389/fimmu.2020.624279
201. Lamkanfi M, Moreira LO, Makena P, Spierings DC, Boyd K, Murray PJ, et al. Caspase-7 deficiency protects from endotoxin-induced lymphocyte apoptosis and improves survival. Blood (2009) 113(12):2742–5. doi: 10.1182/blood-2008-09-178038
202. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. Jama (2011) 306(23):2594–605. doi: 10.1001/jama.2011.1829
203. Roquilly A, McWilliam HEG, Jacqueline C, Tian Z, Cinotti R, Rimbert M, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity (2017) 47(1):135–147.e5. doi: 10.1016/j.immuni.2017.06.021
204. Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest (2006) 116(9):2532–42. doi: 10.1172/jci28054
205. Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol (2004) 4(2):133–42. doi: 10.1038/nri1269
206. Gerard C. Complement C5a in the sepsis syndrome–too much of a good thing? N Engl J Med (2003) 348(2):167–9. doi: 10.1056/NEJMcibr022995
207. Schreiber H, Rittirsch D, Flierl M, Brueckner U, Schneider M, Weiss M, et al. Complement activation during sepsis in humans. Adv Exp Med Biol (2006) 586:217–26. doi: 10.1007/0-387-34134-x_15
208. Fattahi F, Kalbitz M, Malan EA, Abe E, Jajou L, Huber-Lang MS, et al. Complement-induced activation of MAPKs and Akt during sepsis: role in cardiac dysfunction. FASEB J (2017) 31(9):4129–39. doi: 10.1096/fj.201700140R
209. Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol (2012) 946:147–59. doi: 10.1007/978-1-4614-0106-3_9
210. Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol (2005) 23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835
211. Jongerius I, Porcelijn L, van Beek AE, Semple JW, van der Schoot CE, Vlaar APJ, et al. The role of complement in transfusion-related acute lung injury. Transfus Med Rev (2019) 33(4):236–42. doi: 10.1016/j.tmrv.2019.09.002
212. Poppelaars F, Thurman JM. Complement-mediated kidney diseases. Mol Immunol (2020) 128:175–87. doi: 10.1016/j.molimm.2020.10.015
213. Mannes M, Schmidt CQ, Nilsson B, Ekdahl KN, Huber-Lang M. Complement as driver of systemic inflammation and organ failure in trauma, burn, and sepsis. Semin Immunopathol (2021) 43(6):773–88. doi: 10.1007/s00281-021-00872-x
214. Bergh K, Olsen PO, Halgunset J, Iversen OJ. Complement activation and pulmonary dysfunction in experimental Escherichia coli septicaemia. Acta Anaesthesiol Scand (1991) 35(3):267–74. doi: 10.1111/j.1399-6576.1991.tb03286.x
215. Stöve S, Welte T, Wagner TO, Kola A, Klos A, Bautsch W, et al. Circulating complement proteins in patients with sepsis or systemic inflammatory response syndrome. Clin Diagn Lab Immunol (1996) 3(2):175–83. doi: 10.1128/cdli.3.2.175-183.1996
216. Hällgren R, Samuelsson T, Modig J. Complement activation and increased alveolar-capillary permeability after major surgery and in adult respiratory distress syndrome. Crit Care Med (1987) 15(3):189–93. doi: 10.1097/00003246-198703000-00001
217. Lange M, Hamahata A, Enkhbaatar P, Esechie A, Connelly R, Nakano Y, et al. Assessment of vascular permeability in an ovine model of acute lung injury and pneumonia-induced Pseudomonas aeruginosa sepsis. Crit Care Med (2008) 36(4):1284–9. doi: 10.1097/CCM.0b013e318169ef74
218. Langlois PF, Gawryl MS. Accentuated formation of the terminal C5b-9 complement complex in patient plasma precedes development of the adult respiratory distress syndrome. Am Rev Respir Dis (1988) 138(2):368–75. doi: 10.1164/ajrccm/138.2.368
219. Stevens JH, O'Hanley P, Shapiro JM, Mihm FG, Satoh PS, Collins JA, et al. Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J Clin Invest (1986) 77(6):1812–6. doi: 10.1172/jci112506
220. Li ZF, Wang YC, Feng QR, Zhang YS, Zhuang YF, Xie ZX, et al. Inhibition of the C3a receptor attenuates sepsis-induced acute lung injury by suppressing pyroptosis of the pulmonary vascular endothelial cells. Free Radic Biol Med (2022) 184:208–17. doi: 10.1016/j.freeradbiomed.2022.02.032
221. Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial damage in acute respiratory distress syndrome. Int J Mol Sci (2020) 21(22):8793. doi: 10.3390/ijms21228793
222. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med (2020) 46(6):1089–98. doi: 10.1007/s00134-020-06062-x
223. Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in acute respiratory distress syndrome: cross talks between inflammation and coagulation. Respiration (2017) 93(3):212–25. doi: 10.1159/000453002
224. Welty-Wolf KE, Carraway MS, Ortel TL, Piantadosi CA. Coagulation and inflammation in acute lung injury. Thromb Haemost (2002) 88(1):17–25.
225. Fourrier F, Chopin C, Goudemand J, Hendrycx S, Caron C, Rime A, et al. Septic shock, multiple organ failure, and disseminated intravascular coagulation. Compared patterns of antithrombin III, protein C, and protein S deficiencies. Chest (1992) 101(3):816–23. doi: 10.1378/chest.101.3.816
226. van der Poll T. Tissue factor as an initiator of coagulation and inflammation in the lung. Crit Care (2008) 12 Suppl 6(Suppl 6):S3. doi: 10.1186/cc7026
227. Gando S, Nanzaki S, Morimoto Y, Kobayashi S, Kemmotsu O. Systemic activation of tissue-factor dependent coagulation pathway in evolving acute respiratory distress syndrome in patients with trauma and sepsis. J Trauma (1999) 47(4):719–23. doi: 10.1097/00005373-199910000-00017
228. Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol (2009) 297(6):L1035–41. doi: 10.1152/ajplung.00214.2009
229. Welty-Wolf KE, Carraway MS, Idell S, Ortel TL, Ezban M, Piantadosi CA. Tissue factor in experimental acute lung injury. Semin Hematol (2001) 38(4 Suppl 12):35–8. doi: 10.1053/shem.2001.29505
230. Welty-Wolf KE, Carraway MS, Miller DL, Ortel TL, Ezban M, Ghio AJ, et al. Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med (2001) 164(10 Pt 1):1988–96. doi: 10.1164/ajrccm.164.10.2105027
231. Hofstra JJ, Cornet AD, de Rooy BF, Vlaar AP, van der Poll T, Levi M, et al. Nebulized antithrombin limits bacterial outgrowth and lung injury in Streptococcus pneumoniae pneumonia in rats. Crit Care (2009) 13(5):R145. doi: 10.1186/cc8040
232. Tuinman PR, Dixon B, Levi M, Juffermans NP, Schultz MJ. Nebulized anticoagulants for acute lung injury - a systematic review of preclinical and clinical investigations. Crit Care (2012) 16(2):R70. doi: 10.1186/cc11325
233. Oeth P, Parry GC, Mackman N. Regulation of the tissue factor gene in human monocytic cells. Role of AP-1, NF-kappa B/Rel, and Sp1 proteins in uninduced and lipopolysaccharide-induced expression. Arterioscler Thromb Vasc Biol (1997) 17(2):365–74. doi: 10.1161/01.atv.17.2.365
234. Serna D, Brenner M, Chen JC. A subject of ongoing investigation: can exogenous surfactant be used to treat ARDS? Crit Care Med (1998) 26(8):1307–8. doi: 10.1097/00003246-199808000-00007
235. Petty TL, Silvers GW, Paul GW, Stanford RE. Abnormalities in lung elastic properties and surfactant function in adult respiratory distress syndrome. Chest (1979) 75(5):571–4. doi: 10.1378/chest.75.5.571
236. Gregory TJ, Longmore WJ, Moxley MA, Whitsett JA, Reed CR, Fowler AA, et al. Surfactant chemical composition and biophysical activity in acute respiratory distress syndrome. J Clin Invest (1991) 88(6):1976–81. doi: 10.1172/jci115523
237. Anzueto A, Baughman RP, Guntupalli KK, Weg JG, Wiedemann HP, Raventós AA, et al. Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. Exosurf Acute Respiratory Distress Syndrome Sepsis Study Group. N Engl J Med (1996) 334(22):1417–21. doi: 10.1056/nejm199605303342201
238. Hidalgo A, Garcia-Mouton C, Autilio C, Carravilla P, Orellana G, Islam MN, et al. Pulmonary surfactant and drug delivery: Vehiculization, release and targeting of surfactant/tacrolimus formulations. J Control Release (2021) 329:205–22. doi: 10.1016/j.jconrel.2020.11.042
239. Hidalgo A, Cruz A, Pérez-Gil J. Barrier or carrier? Pulmonary surfactant and drug delivery. Eur J Pharm Biopharm (2015) 95(Pt A):117–27. doi: 10.1016/j.ejpb.2015.02.014
240. Silva V, Radic M. COVID-19 pathology sheds further light on balance between neutrophil proteases and their inhibitors. Biomolecules (2022) 13(1):82. doi: 10.3390/biom13010082
241. Mohamed MMA, El-Shimy IA, Hadi MA. Neutrophil Elastase Inhibitors: A potential prophylactic treatment option for SARS-CoV-2-induced respiratory complications? Crit Care (2020) 24(1):311. doi: 10.1186/s13054-020-03023-0
242. Kodama T, Yukioka H, Kato T, Kato N, Hato F, Kitagawa S. Neutrophil elastase as a predicting factor for development of acute lung injury. Intern Med (2007) 46(11):699–704. doi: 10.2169/internalmedicine.46.6182
243. McGuire WW, Spragg RG, Cohen AB, Cochrane CG. Studies on the pathogenesis of the adult respiratory distress syndrome. J Clin Invest (1982) 69(3):543–53. doi: 10.1172/jci110480
244. Fuller BM, Mohr NM, Skrupky L, Fowler S, Kollef MH, Carpenter CR. The use of inhaled prostaglandins in patients with ARDS: a systematic review and meta-analysis. Chest (2015) 147(6):1510–22. doi: 10.1378/chest.14-3161
245. Slotman GJ, Burchard KW, D'Arezzo A, Gann DS. Ketoconazole prevents acute respiratory failure in critically ill surgical patients. J Trauma (1988) 28(5):648–54. doi: 10.1097/00005373-198805000-00015
246. Ketoconazole for early treatment of acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. The ARDS Network. Jama (2000) 283(15):1995–2002. doi: 10.1001/jama.283.15.1995
247. Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. Ibuprofen Sepsis Study Group N Engl J Med (1997) 336(13):912–8. doi: 10.1056/nejm199703273361303
248. Kelleni MT. Early use of non-steroidal anti-inflammatory drugs in COVID-19 might reverse pathogenesis, prevent complications and improve clinical outcomes. BioMed Pharmacother (2021) 133:110982. doi: 10.1016/j.biopha.2020.110982
249. Combes A, Schmidt M, Hodgson CL, Fan E, Ferguson ND, Fraser JF, et al. Extracorporeal life support for adults with acute respiratory distress syndrome. Intensive Care Med (2020) 46(12):2464–76. doi: 10.1007/s00134-020-06290-1
250. Pham T, Combes A, Rozé H, Chevret S, Mercat A, Roch A, et al. Extracorporeal membrane oxygenation for pandemic influenza A(H1N1)-induced acute respiratory distress syndrome: a cohort study and propensity-matched analysis. Am J Respir Crit Care Med (2013) 187(3):276–85. doi: 10.1164/rccm.201205-0815OC
251. Patroniti N, Zangrillo A, Pappalardo F, Peris A, Cianchi G, Braschi A, et al. The Italian ECMO network experience during the 2009 influenza A(H1N1) pandemic: preparation for severe respiratory emergency outbreaks. Intensive Care Med (2011) 37(9):1447–57. doi: 10.1007/s00134-011-2301-6
252. Noah MA, Peek GJ, Finney SJ, Griffiths MJ, Harrison DA, Grieve R, et al. Referral to an extracorporeal membrane oxygenation center and mortality among patients with severe 2009 influenza A(H1N1). Jama (2011) 306(15):1659–68. doi: 10.1001/jama.2011.1471
253. Munshi L, Walkey A, Goligher E, Pham T, Uleryk EM, Fan E. Venovenous extracorporeal membrane oxygenation for acute respiratory distress syndrome: a systematic review and meta-analysis. Lancet Respir Med (2019) 7(2):163–72. doi: 10.1016/s2213-2600(18)30452-1
254. Combes A, Hajage D, Capellier G, Demoule A, Lavoué S, Guervilly C, et al. Extracorporeal membrane oxygenation for severe acute respiratory distress syndrome. N Engl J Med (2018) 378(21):1965–75. doi: 10.1056/NEJMoa1800385
255. Bein T, Grasso S, Moerer O, Quintel M, Guerin C, Deja M, et al. The standard of care of patients with ARDS: ventilatory settings and rescue therapies for refractory hypoxemia. Intensive Care Med (2016) 42(5):699–711. doi: 10.1007/s00134-016-4325-4
256. Abrams D, Ferguson ND, Brochard L, Fan E, Mercat A, Combes A, et al. ECMO for ARDS: from salvage to standard of care? Lancet Respir Med (2019) 7(2):108–10. doi: 10.1016/s2213-2600(18)30506-x
257. Lebreton G, Schmidt M, Ponnaiah M, Folliguet T, Para M, Guihaire J, et al. Extracorporeal membrane oxygenation network organisation and clinical outcomes during the COVID-19 pandemic in Greater Paris, France: a multicentre cohort study. Lancet Respir Med (2021) 9(8):851–62. doi: 10.1016/s2213-2600(21)00096-5
258. Schmidt M, Hajage D, Lebreton G, Monsel A, Voiriot G, Levy D, et al. Extracorporeal membrane oxygenation for severe acute respiratory distress syndrome associated with COVID-19: a retrospective cohort study. Lancet Respir Med (2020) 8(11):1121–31. doi: 10.1016/s2213-2600(20)30328-3
259. Barbaro RP, MacLaren G, Boonstra PS, Combes A, Agerstrand C, Annich G, et al. Extracorporeal membrane oxygenation for COVID-19: evolving outcomes from the international Extracorporeal Life Support Organization Registry. Lancet (2021) 398(10307):1230–8. doi: 10.1016/s0140-6736(21)01960-7
260. Shaefi S, Brenner SK, Gupta S, O'Gara BP, Krajewski ML, Charytan DM, et al. Extracorporeal membrane oxygenation in patients with severe respiratory failure from COVID-19. Intensive Care Med (2021) 47(2):208–21. doi: 10.1007/s00134-020-06331-9
261. Su Y, Silva JD, Doherty D, Simpson DA, Weiss DJ, Rolandsson-Enes S, et al. Mesenchymal stromal cells-derived extracellular vesicles reprogramme macrophages in ARDS models through the miR-181a-5p-PTEN-pSTAT5-SOCS1 axis. Thorax (2023) 78(6):617–30. doi: 10.1136/thoraxjnl-2021-218194
262. Dutra Silva J, Su Y, Calfee CS, Delucchi KL, Weiss D, McAuley DF, et al. Mesenchymal stromal cell extracellular vesicles rescue mitochondrial dysfunction and improve barrier integrity in clinically relevant models of ARDS. Eur Respir J (2021) 58(1):2002978. doi: 10.1183/13993003.02978-2020
263. Bowdish ME, Barkauskas CE, Overbey JR, Gottlieb RL, Osman K, Duggal A, et al. A randomized trial of mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome from COVID-19. Am J Respir Crit Care Med (2023) 207(3):261–70. doi: 10.1164/rccm.202201-0157OC
264. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in hospitalized patients with covid-19. N Engl J Med (2021) 384(8):693–704. doi: 10.1056/NEJMoa2021436
265. Villar J, Ferrando C, Martínez D, Ambrós A, Muñoz T, Soler JA, et al. Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med (2020) 8(3):267–76. doi: 10.1016/s2213-2600(19)30417-5
266. Jiang J, Huang K, Xu S, Garcia JGN, Wang C, Cai H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol (2020) 36:101638. doi: 10.1016/j.redox.2020.101638
267. Olson LM, Moss GS, Baukus O, Das Gupta TK. The role of C5 in septic lung injury. Ann Surg (1985) 202(6):771–6. doi: 10.1097/00000658-198512000-00018
268. Huber-Lang MS, Sarma JV, McGuire SR, Lu KT, Guo RF, Padgaonkar VA, et al. Protective effects of anti-C5a peptide antibodies in experimental sepsis. FASEB J (2001) 15(3):568–70. doi: 10.1096/fj.00-0653fje
269. Li W, Long L, Yang X, Tong Z, Southwood M, King R, et al. Circulating BMP9 protects the pulmonary endothelium during inflammation-induced lung injury in mice. Am J Respir Crit Care Med (2021) 203(11):1419–30. doi: 10.1164/rccm.202005-1761OC
270. Reilly JP, Wang F, Jones TK, Palakshappa JA, Anderson BJ, Shashaty MGS, et al. Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive Care Med (2018) 44(11):1849–58. doi: 10.1007/s00134-018-5328-0
271. Nagendran M, McAuley DF, Kruger PS, Papazian L, Truwit JD, Laffey JG, et al. Statin therapy for acute respiratory distress syndrome: an individual patient data meta-analysis of randomised clinical trials. Intensive Care Med (2017) 43(5):663–71. doi: 10.1007/s00134-016-4649-0
272. Peres e Serra A, Parra ER, Eher E, Capelozzi VL. Nonhomogeneous immunostaining of hyaline membranes in different manifestations of diffuse alveolar damage. Clinics (Sao Paulo) (2006) 61(6):497–502. doi: 10.1590/s1807-59322006000600002
273. Gajic O, Dabbagh O, Park PK, Adesanya A, Chang SY, Hou P, et al. Early identification of patients at risk of acute lung injury: evaluation of lung injury prediction score in a multicenter cohort study. Am J Respir Crit Care Med (2011) 183(4):462–70. doi: 10.1164/rccm.201004-0549OC
274. Heidecke CD, Weighardt H, Hensler T, Bartels H, Holzmann B. [Immune paralysis of T-lymphocytes and monocytes in postoperative abdominal sepsis. Correlation of immune function with survival]. . Chirurg (2000) 71(2):159–65. doi: 10.1007/s001040050028
275. Khan YA, Fan E, Ferguson ND. Precision medicine and heterogeneity of treatment effect in therapies for ARDS. Chest (2021) 160(5):1729–38. doi: 10.1016/j.chest.2021.07.009
276. Beitler JR, Thompson BT, Baron RM, Bastarache JA, Denlinger LC, Esserman L, et al. Advancing precision medicine for acute respiratory distress syndrome. Lancet Respir Med (2022) 10(1):107–20. doi: 10.1016/s2213-2600(21)00157-0
277. Liu S, Li H, Wang Y, Li H, Du S, Zou X, et al. High expression of IL-36γ in influenza patients regulates interferon signaling pathway and causes programmed cell death during influenza virus infection. Front Immunol (2020) 11:552606. doi: 10.3389/fimmu.2020.552606
Keywords: acute respiratory distress syndrome (ARDS), sepsis, heterogeneity, immune system, precision therapy
Citation: Xu H, Sheng S, Luo W, Xu X and Zhang Z (2023) Acute respiratory distress syndrome heterogeneity and the septic ARDS subgroup. Front. Immunol. 14:1277161. doi: 10.3389/fimmu.2023.1277161
Received: 14 August 2023; Accepted: 30 October 2023;
Published: 14 November 2023.
Edited by:
Chengjin Gao, Shanghai Jiao Tong University, ChinaReviewed by:
Cassiano Felippe Gonçalves-de-Albuquerque, Rio de Janeiro State Federal University, BrazilCopyright © 2023 Xu, Sheng, Luo, Xu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaocai Zhang, MjMxMzAwM0B6anUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.