 Olivia K. Favor1,2*Lichchavi D. Rajasinghe2,3Kathryn A. Wierenga2,4Krishna R. Maddipati5Kin Sing Stephen Lee1,2,6Andrew J. Olive7James J. Pestka2,3,7*
Olivia K. Favor1,2*Lichchavi D. Rajasinghe2,3Kathryn A. Wierenga2,4Krishna R. Maddipati5Kin Sing Stephen Lee1,2,6Andrew J. Olive7James J. Pestka2,3,7*- 1Department of Pharmacology and Toxicology, Michigan State University, East Lansing, MI, United States
- 2Institute for Integrative Toxicology, Michigan State University, East Lansing, MI, United States
- 3Department of Food Science and Human Nutrition, Michigan State University, East Lansing, MI, United States
- 4Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI, United States
- 5Department of Pathology, Wayne State University, Detroit, MI, United States
- 6Department of Chemistry, Michigan State University, East Lansing, MI, United States
- 7Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, United States
Introduction: Phagocytosis of inhaled crystalline silica (cSiO2) particles by tissue-resident alveolar macrophages (AMs) initiates generation of proinflammatory eicosanoids derived from the ω-6 polyunsaturated fatty acid (PUFA) arachidonic acid (ARA) that contribute to chronic inflammatory disease in the lung. While supplementation with the ω-3 PUFA docosahexaenoic acid (DHA) may influence injurious cSiO2-triggered oxylipin responses, in vitro investigation of this hypothesis in physiologically relevant AMs is challenging due to their short-lived nature and low recovery numbers from mouse lungs. To overcome these challenges, we employed fetal liver-derived alveolar-like macrophages (FLAMs), a self-renewing surrogate that is phenotypically representative of primary lung AMs, to discern how DHA influences cSiO2-induced eicosanoids.
Methods: We first compared how delivery of 25 µM DHA as ethanolic suspensions or as bovine serum albumin (BSA) complexes to C57BL/6 FLAMs impacts phospholipid fatty acid content. We subsequently treated FLAMs with 25 µM ethanolic DHA or ethanol vehicle (VEH) for 24 h, with or without LPS priming for 2 h, and with or without cSiO2 for 1.5 or 4 h and then measured oxylipin production by LC-MS lipidomics targeting for 156 oxylipins. Results were further related to concurrent proinflammatory cytokine production and cell death induction.
Results: DHA delivery as ethanolic suspensions or BSA complexes were similarly effective at increasing ω-3 PUFA content of phospholipids while decreasing the ω-6 PUFA arachidonic acid (ARA) and the ω-9 monounsaturated fatty acid oleic acid. cSiO2 time-dependently elicited myriad ARA-derived eicosanoids consisting of prostaglandins, leukotrienes, thromboxanes, and hydroxyeicosatetraenoic acids in unprimed and LPS-primed FLAMs. This cSiO2-induced eicosanoid storm was dramatically suppressed in DHA-supplemented FLAMs which instead produced potentially pro-resolving DHA-derived docosanoids. cSiO2 elicited marked IL-1α, IL-1β, and TNF-α release after 1.5 and 4 h of cSiO2 exposure in LPS-primed FLAMs which was significantly inhibited by DHA. DHA did not affect cSiO2-triggered death induction in unprimed FLAMs but modestly enhanced it in LPS-primed FLAMs.
Discussion: FLAMs are amenable to lipidome modulation by DHA which suppresses cSiO2-triggered production of ARA-derived eicosanoids and proinflammatory cytokines. FLAMs are a potential in vitro alternative to primary AMs for investigating interventions against early toxicant-triggered inflammation in the lung.
Introduction
Approximately 2.3 million American workers are exposed to cSiO2 levels that exceed the Occupational Health and Safety Administration’s (OSHA’s) Permissible Exposure Limits (1), with the highest exposure levels in construction, manufacturing, sandblasting, farming, ceramics, and dentistry work (2, 3). Occupational exposure to respirable crystalline silica (cSiO2) is etiologically linked to silicosis, lung cancer, and systemic autoimmune disease (4–7). When inhaled into the lung, cSiO2 particles travel to the alveoli where they are readily phagocytosed by alveolar macrophages (AMs) (8). Following phagocytosis, cSiO2 induces lysosomal membrane permeabilization (LMP), mitochondrial toxicity, reactive oxygen species (ROS) formation, NLRP3 inflammasome activation, oxylipin generation, release of proinflammatory proteins, and cell death (9, 10). The resultant bioactive oxylipins derived from the 20-carbon ω-6 polyunsaturated fatty acid (PUFA) arachidonic acid (ARA) (i.e., eicosanoids) and proinflammatory cytokines can act in a paracrine manner to induce expression and secretion of additional proinflammatory mediators by neighboring AMs and other cells. If not properly cleared, cSiO2 particles released from dead AMs can again be taken up by viable AMs or recruited macrophages, contributing to a perpetual cycle of inflammation, cell death, and tissue damage. Accordingly, tissue-resident AMs play a critical role in the initiation of persistent cSiO2-triggered lung inflammation and development of chronic disease.
Relative to eicosanoids, it is well-known that inflammatory stimuli in the lung can activate phospholipase A2 (PLA2) in macrophages which promotes release of ARA, one of the most abundant PUFAs in the sn-2 position of membrane phospholipids, into the cytosol. Free ARA is then converted into prostaglandins (PGs), leukotrienes (LTs), thromboxanes (TXs), and hydroxyeicosatetraenoic acids (HETEs) (11, 12). Some of these eicosanoids promote further immune cell infiltration and have been associated with damage to pulmonary tissue and increased symptom severity in many lung diseases including COVID-19, asthma, cystic fibrosis, chronic obstructive pulmonary disease (COPD), and extrinsic allergic alveolitis (13–17). Importantly, several previous studies have reported that exposing rat, bovine, and human AMs to cSiO2 drives biosynthesis of classical ARA-derived eicosanoids, such as PGE2, LTB4, TXB2, and 5-HETE (18–21), some of which have been recently described in human patients with silicosis and in a preclinical C57BL/6 mouse silicosis model (22). The possibility exists that many more oxylipins are elicited by AMs in response to the particle, perhaps tantamount to an “eicosanoid storm” (13–17), that contribute as a group to the potent inflammatory effects of the particle in the lung.
One potential intervention against cSiO2-induced ARA-derived eicosanoid generation and downstream lung inflammation is dietary supplementation with ω-3 polyunsaturated fatty acids (PUFAs) such as docosahexaenoic acid (C22:6, ω-3, DHA) (23, 24). ω-3 PUFAs can prevent excessive inflammatory responses and promote pro-resolving immune responses by displacing ω-6 PUFAs from the plasma membrane and i) competing with ω-6 PUFAs as substrates for cyclooxygenase (COX), lipoxygenase (LOX) and cytochrome P450 (CYP450) enzymes thereby reducing production of proinflammatory eicosoanoids while increasing potentially pro-resolving DHA-derived oxylipins (i.e., docosanoids), ii) altering lipid raft structures and attenuating downstream signal transduction, iii) preventing NF-κB-driven expression of proinflammatory mediators, and iv) enhancing efferocytosis of cell corpses by phagocytes (25–28). In both mice and humans, DHA consumption leads to decreased tissue and plasma levels of ARA and related eicosanoids concurrently with increased DHA and related docosanoids (24, 29–33). It is thus of great interest to understand how skewing AM membrane content from ω-6 PUFAs to ω-3 PUFAs influences the cSiO2-induced oxylipin profile, proinflammatory cytokine production, and cell death induction. As a first step toward that goal, we have demonstrated in RAW264.7 murine macrophages transfected with apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) that DHA displaces ω-6 ARA and ω-9 oleic acid (OA) from membrane phospholipids and suppresses cSiO2-induced NLRP3 inflammasome activation and IL-1 release (27). However, neither transformed macrophage cell lines (e.g. RAW264.7 or THP-1 cells) nor other widely used primary macrophages (e.g., bone marrow-derived macrophages [BMDMs] or peritoneal macrophages) adequately model the tissue-specific phenotype of primary AMs (34). Specifically, BMDMs originate from hematopoetic stem cells while peritoneal macrophages and AMs originate from erythromyeloid progenitor cells in the fetal liver (35). In addition, BMDMs express high CD115 and Ly6G (36) and peritoneal macrophages express high B7-H1 and CD11b (36, 37), whereas AMs express low CD11b and high CD11c, SiglecF, and Ly6G (38). Thus, there is a need to conduct these studies in primary AMs or a more physiologically relevant model.
Several barriers hinder mechanistic research efforts into primary AM function. Since only approximately 1-5 × 105 primary cells are typically recovered from one adult mouse by bronchoalveolar lavage, obtaining sufficient numbers of cells for rigorous in vitro investigations requires sacrificing large numbers of mice (39). Additionally, primary AMs may undergo phenotypic changes upon being cultured and therefore might not completely reflect in vivo AM function (40–42). Recently, our laboratory has overcome barriers to AM culture by developing an ex vivo fetal liver-derived alveolar-like macrophage (FLAM) model that is non-transformed and self-replicating in the presence of GM-CSF and TGF-β (43). Like primary AMs, FLAMs are characterized by high surface expression of SiglecF and CD11c, low surface expression of CD14, and stable expression of AM-specific genes such as Marco. In addition, the kinetics of cSiO2 phagocytosis, cell death, and IL-1 cytokine release in FLAMs mirrors that of primary AMs. Accordingly, FLAMs are convenient, phenotypically relevant AM surrogates for conducting thorough in vitro mechanistic studies involving cSiO2 exposure and the effects of ω-3 intervention.
Here, we hypothesized that DHA would inhibit proinflammatory eicosanoid production induced by cSiO2. To test our hypothesis, we employed FLAMs as an AM model and targeted LC-MS lipidomics to understand how DHA incorporation influences cSiO2-induced oxylipin generation within 4 h and further related these results to proinflammatory cytokine production and induction of cell death over the same time window. Our findings suggest FLAMs to be a promising in vitro alternative to primary AMs for future investigations of how modulation of their cellular lipidome influences eicosanoid production by cSiO2 and how these differential responses are potentially linked to cSiO2-induced inflammation in the lung.
Materials and methods
Key reagents
All key reagents used in this study and their corresponding catalog numbers are outlined in Supplementary Table 1.
Fetal liver-derived alveolar macrophage isolation and cell culture
Experimental protocols were approved by the Institutional Animal Care and Use Committee at Michigan State University (MSU) (Animal Use Form [AUF] #PROTO201800113) in accordance with guidelines established by the National Institutes of Health. Six- to 8-wk-old C57BL/6 mice (cat. #000664) were procured from the Jackson Laboratory (Bar Harbor, ME) and were given freely accessible food and water. Animal facilities were maintained with a 12 h light/dark cycle at consistent temperature (21-24°C) and humidity (40-55%). Mice were bred for FLAM isolation as previously described (43, 44). At 14-18 gestational days, pregnant dams (8-10 wk of age) were euthanized by CO2 asphyxiation for 10 min to ensure death to the neonatal mice (14-18 gestational days), which are resistant to anoxia. As a secondary method of euthanasia, cervical dislocation was performed on the dam and blood supply was cut off to the fetuses. Upon removing the fetuses from the dam, fetal livers were carefully removed and dissociated in ice-cold DPBS-/- (DPBS without calcium and magnesium) by gentle pipetting. Liver cells were pelleted by centrifugation at 220 x g for 5 min at 4°C, resuspended in complete FLAM medium (RPMI 1640 medium, 10% fetal bovine serum, 1% penicillin-streptomycin, 30 ng/ml recombinant mouse GM-CSF, and 20 ng/ml recombinant human TGF-β1), seeded in 100 mm tissue-culture treated dishes, and incubated at 37°C and 5% CO2. Medium was changed every 1-2 d and non-adherent dead cells discarded. Adherent fetal liver monocytes were lifted with DPBS-/- containing 10 mM EDTA and gentle scraping upon reaching 70-90% confluency. After 1-2 wk of culture, cells developed a round morphology resemblant of AMs and were frozen for future use. Mature, differentiated FLAMs were routinely confirmed to be CD14+ low, SiglecF+ high, and CD11c+ high by flow cytometry as previously described (43). Cells between passage 5 and 10 were used for this study.
Comparative effects of DHA delivery using ethanolic suspensions and bovine serum albumin complexes on phospholipid fatty acid profile in FLAMs
FLAMs were seeded in 6-well plates at a density of 4.50×105 cells/well in complete FLAM medium. Cells were incubated overnight to achieve 70-90% confluency before beginning treatments. The next day, cells were washed once with sterile DPBS-/-, and were then incubated for 24 h in fresh complete FLAM medium containing either 1) ethanolic DHA (25 µM) or ethanol (EtOH) vehicle (VEH), or 2) 25 µM DHA complexed to BSA at a 3:1 ratio as previously described (27, 28) or 8.33 µM BSA VEH. Following treatments, FLAMs were pelleted and stored in 100% methanol at -80°C before total fatty acids were measured at OmegaQuant Inc. using gas chromatography (GC) with flame ionization detection as previously described (45).
A standard mixture of fatty acids (GLC OQ-A; NuChek Prep, Elysian, MN) was used to generate calibration curves for individual fatty acids and identify fatty acids present in each sample. The following 24 fatty acids were identified (by class): i) saturated (14:0, 16:0, 18:0, 20:0, 22:0, 24:0), ii) cis monounsaturated (16:1, 18:1, 20:1, 24:1), iii) cis ω-6 polyunsaturated (18:2ω6, 18:3ω6, 20:2ω6, 20:3ω6, 20:4ω6, 22:4ω6, 22:5ω6), and iv) cis ω-3 polyunsaturated (18:3ω3, 20:5ω3, 22:5ω3, 22:6ω3). Proportions of individual fatty acids in each sample were expressed as a percentage of total identified fatty acids.
Two biomarkers of ω-3 tissue incorporation, percent ω-3 PUFAs and highly unsaturated fatty acid (HUFA) score, were calculated as previously described (23) using the following equations, with total fatty acids (FA) equaling the sum of all analyzed FA and total HUFA equaling the sum of dihomo-γ-linolenic acid (DGLA; 20:3ω6), ARA (20:4ω6), eicosatetraenoic acid (EPA; 20:5ω3), docosatetraenoic acid (DTA; 22:4ω6), ω-6 docosapentaenoic acid (DPA; 22:5ω6), ω-3 DPA (22:5ω3), and DHA (22:6ω3):
Determination of DHA’s effects on cSiO2-induced generation of oxylipins in FLAMs by targeted LC-MS lipidomics
For lipidomics analyses, FLAMs were seeded in 6-well plates at a density of 4.50×105 cells/well in complete FLAM medium. Cells were incubated overnight to achieve 70-90% confluency before beginning treatments. The next day, cells washed once with sterile DPBS-/-, fresh complete FLAM medium containing either 25 µM ethanolic DHA or EtOH VEH, then cells incubated for 24 h. Following DHA or VEH treatment, cells were washed once with sterile DPBS-/-, treated with either 20 ng/ml LPS in DPBS-/- or DPBS-/- VEH in DPBS+/+ (DPBS containing calcium and magnesium) for 2 h, then exposed to 12.5 µg/cm2 cSiO2 or DPBS-/- VEH for 1.5 or 4 h. These timepoints were selected for cSiO2 exposure to assess acute toxic effects of the particle on FLAMs instead of prolonged effects. LPS and cSiO2 exposures were done in DPBS+/+ to minimize interference from fatty acids present in cell culture medium during oxylipin analyses. Each treatment condition was tested using three biological replicates and one technical replicate.
Cell culture supernatants and cells were collected at 0, 1.5, and 4 h after cSiO2 treatment and pooled together prior to lipidomic analysis. Following treatments, ice-cold methanol was added to each well, resulting in a final sample volume of 3 ml (1 ml cell culture supernatant + 2 ml methanol). To each sample, 60 µl of antioxidant cocktail (0.2 mg/ml butylated hydroxytoluene, 0.2 mg/ml triphenylphosphine, 0.6 mg/ml EDTA) was added to achieve a total cocktail concentration of 5% (v/v) (46). After subjecting the cells and supernatant to centrifugation at 0°C and 10,000 x g to separate the cellular debris, the resulting supernatant was stored in tubes at -80°C under nitrogen gas. Cells and supernatants within each well were pooled together, then samples were frozen at -80°C until liquid chromatography-mass spectrometry (LC-MS) analysis. Targeted LC-MS lipidomics for 156 lipid metabolites was conducted at the Lipidomics Core Facility at Wayne State University as previously described (47–49). Briefly, 100 µl aliquots of cellular samples were thawed and spiked with a cocktail of deuterated internal standards (5 ng each of PGE1-d4, RvD2-d5, LTB4-d4, and 15[S]-HETE-d8; Cayman Chemical, Ann Arbor, MI) for quantification of oxylipins and recovery. Then, oxylipins were extracted by using C18 extraction columns that were washed with 15% (v/v) methanol and subsequently hexane, dried in a vacuum, eluted with methanol containing 0.1% (v/v) formic acid, dried under nitrogen gas, and dissolved in a 1:1 mixture of methanol:25 mM aqueous ammonium acetate. Extracted oxylipins were subjected to high-performance liquid chromatography (HPLC) using a Luna C18 (3 µm, 2.1×150 mm) column connected to a Prominence XR system (Shimadzu, Somerset, NJ) then analyzed with a QTrap5500 mass spectrometer (AB Sciex, Singapore) set to negative ion mode. Analyst 1.6 software (AB Sciex) and MultiQuant software (AB Sciex) were used to collect and quantify the data in units of ng, respectively. Lipid metabolite classifications are provided in Supplementary Table 2, and mass spectra of representative metabolites are given in reference (50).
Assessment of DHA’s effects on cSiO2-induced release of lysosomal cathepsins, LDH, and proinflammatory cytokines in FLAMs
FLAMs were seeded in 24-well plates at a density of 1.50×105 cells/well in complete FLAM medium and then cultured with DHA or VEH, LPS or VEH, and cSiO2 or VEH under the conditions described for the lipidomics study. LPS and cSiO2 exposures were done in DPBS+/+ to maintain consistent experimental conditions between oxylipin analyses and other analyses conducted in this study. Cell culture supernatants were collected by centrifugation at 200 x g at 0, 1.5, and 4 h post cSiO2 exposure and analyzed for proinflammatory cytokines, cathepsin B activity, and lactate dehydrogenase (LDH) release.
Cathepsin B activity was determined using a fluorescent assay as previously described (51). Briefly, 50 µl of cell culture supernatant and 2 µg Z-LR-AMC were combined in 96-well plates and adjusted to a final volume of 150 µl/well, then all samples were incubated for 1 h at 37°C. Sample fluorescence was then measured using a FilterMax F3 Multimode plate reader (Molecular Devices, San Jose, CA) set to 380 nm excitation and 460 nm emission. Cathepsin B activity in each well was calculated in units of relative fluorescence units (RFU) by the following equation:
LDH activity was measured as previously described (27, 28). Separate wells of untreated FLAMs were included and designated as max-kill (MK) samples and incubated with 0.2% Triton-X (Millipore Sigma) for 5 min. After all treatments, supernatants were collected from all wells and 50 µl was transferred to non-treated, flat-bottom 96-well plates in duplicate. DPBS+/+ was used as a sample blank, and DPBS+/+ containing 0.2% (v/v) Triton-X was used as a MK blank. 100 µl of LDH assay reagent (15 µM 1-methoxyphenazine methosulfate [PMS], 2 mM iodonitrotetrazolium [INT], 3.2 mM β-nicotinamide adenine dinucleotide [NAD] sodium salt, and 160 mM lithium lactate in 0.2 M Tris-HCl, pH 8.2) was added to each well, and assay plates were incubated at room temperature for 15 min in the dark. Sample absorbance was then measured using a FilterMax F3 Multimode plate reader (Molecular Devices, San Jose, CA) set to a wavelength of 492 nm. Percent cell death in each well was calculated by the following equation:
Concentrations of proinflammatory cytokines (i.e., IL-1α, IL-1β, TNF-α) were quantified by enzyme-linked immunosorbent assay (ELISA) using corresponding mouse R&D Systems DuoSet kits according to the manufacturer’s instructions.
Determination of DHA effects on cSiO2-induced lysosomal membrane permeabilization, mitochondrial depolarization, and cell death in FLAMs using live-cell microscopic imaging
FLAMs were seeded in 48-well plates 0.625×105 cells/well in complete FLAM medium to achieve 50% confluency after overnight incubation. The next day, cells were washed once with sterile DPBS-/- then treated with either 25 µM ethanolic DHA or EtOH VEH as a control in complete FLAM medium. After 24 h, cells were washed once with sterile DPBS-/- and primed with 20 ng/ml LPS or PBS VEH for 1.5 h. Following LPS priming, cells were washed with sterile DPBS+/+ then subsequently incubated for 30 min at 37°C in the dark with 50 nM LysoTracker Red DND-99 in DPBS+/+ to label lysosomes, 25 nM MitoTracker Red CMXRos to label mitochondria, or 200 nM SYTOX Green to detect cell death. All fluorescent dyes were diluted in DPBS+/+ prior to addition to the cells, and 20 ng/ml LPS was simultaneously added to appropriate wells to allow 2 h of total LPS priming prior to cSiO2 treatment. After 30 min of cell staining, a freshly prepared cSiO2 stock suspension was added dropwise to a final concentration of 0 or 12.5 µg/cm2. LPS and cSiO2 exposures were done in DPBS+/+ to maintain consistent experimental conditions between oxylipin analyses and other analyses conducted in this study.
Live-cell fluorescence microscopy began immediately after adding cSiO2 to the cells. Images of live cells were taken at 0, 1.5, and 4 h post cSiO2 treatment using an EVOS FL Auto 2 Cell Imaging System (ThermoFisher Scientific) with an onstage, temperature-controlled incubator. Each experimental condition was tested using 3 biological replicates. For each well, 2-4 images were acquired using fields of view defined before the beginning of the experiment. LysoTracker Red and MitoTracker Red were detected using the Texas Red light cube (Ex: 585/29 nm, Em: 628/32 nm), and SYTOX Green was detected using the GFP light cube (Ex: 482/25 nm, Em: 524/24 nm).
Acquired images were analyzed for lysosomal integrity, mitochondrial integrity, and cell death using CellProfiler 4.2.1 as previously described (43, 52). Briefly, lysosomal integrity, mitochondrial integrity, and cell death were assessed by quantifying the number of LysoTracker Red+, MitoTracker Red+, and SYTOX Green+ cells, respectively. LysoTracker Red+ and MitoTracker Red+ puncta were omitted if fluorescent intensity fell below preset minimum thresholds, which were chosen to omit false positives quantified from background fluorescence. Raw counts of LysoTracker Red+, MitoTracker Red+, and SYTOX Green+ cells were further analyzed using RStudio 2022.07.1 + 554 (Posit, Boston, MA).
Data analysis and statistics
For oxylipin data, MetaboAnalyst Version 5.0 (Xia Lab, Quebec, Canada, www.metaboanalyst.ca/) (53) was used to conduct statistical analyses. First, raw ng values were converted to corresponding pmol values in Microsoft Excel (50). Then, in MetaboAnalyst, the one factor statistical analysis module was chosen, and data were uploaded as a comma separated values (.csv) file with samples in unpaired columns and features (i.e., metabolites) in rows. Features with >70% missing data were removed from the dataset, and remaining missing values were estimated by replacing with the corresponding limits of detection (LODs; 1/5 of the minimum positive value of each variable). After the data was cleaned, the data was normalized by auto scaling only, then the data editor option was used to select experimental groups of interest to compare. For comparisons between experimental groups, one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test was used, with FDR q<0.05 considered statistically significant. Asterisks (*) indicate statistically significant differences (FDR q<0.05) for cSiO2-treated groups and their corresponding non-cSiO2-treated controls. Hashes (#) indicate statistically significant differences (FDR q<0.05) for DHA-treated groups and their corresponding non-DHA-treated controls. Crosses (†) indicate statistically significant differences (FDR q<0.05) for LPS-treated groups and their corresponding non-LPS-treated controls.
For all other endpoints, GraphPad Prism Version 9 (GraphPad Software, San Diego, CA, www.graphpad.com) was used to conduct statistical analyses. The ROUT outlier test (Q = 1%) and the Shapiro-Wilk test (p<0.01) were used to identify outliers and assess normality in the data, respectively. For comparisons between two experimental groups, non-normal and semiquantitative data were analyzed by the Mann-Whitney nonparametric test. The F test (p<0.05) was used to test the assumption of equal variances across both groups. Normal data with unequal variances were analyzed using an unpaired t test with Welch’s correction. Normal data that met the assumption of equal variance were analyzed using an unpaired t test. For comparisons between more than two experimental groups, non-normal and semi-quantitative data were analyzed by the Kruskal-Wallis nonparametric test followed by Dunn’s post-hoc test. The Brown-Forsythe test (p<0.01) was used to test the assumption of equal variances across treatment groups. Normal data with unequal variances were analyzed using the Brown-Forsythe/Welch analysis of variance (ANOVA) followed by Dunnett’s T3 post-hoc test. Normal data that met the assumption of equal variance were analyzed by standard one-way ANOVA followed by Tukey’s post-hoc test. Data are presented as mean ± standard error of the mean (SEM), with a p-value < 0.05 considered statistically significant.
Results
DHA displaces the ω-9 monounsaturated fatty acid oleic acid and the ω-6 polyunsaturated fatty acid arachidonic acid from membrane phospholipids in FLAMs
Two major methods for introducing ω-3 PUFAs to cell cultures, ethanolic suspensions and BSA complexes (54), were evaluated for their suitability for incorporating DHA into FLAMs as shown in Figure 1A. Ethanolic DHA-treated FLAMs had significantly greater DHA content (19.3% total fatty acids) compared to EtOH VEH-treated FLAMs (4.4% total fatty acids) (Figure 1B). Corresponding with these findings, the ω-9 monounsaturated fatty acid (MUFA) oleic acid (OA), the main fatty acid found in fetal bovine serum (55), and the ω-6 PUFA arachidonic acid (ARA), the major precursor PUFA for eicosanoid biosynthesis, were significantly decreased in DHA-treated FLAMs (16.5% and 7.2% total fatty acids, respectively) compared to VEH-treated FLAMs (26.5% and 10.6% total fatty acids, respectively). No notable changes were found for other saturated and unsaturated fatty acids that were analyzed. FLAMs incubated with DHA-BSA complexes or BSA VEH displayed similar DHA membrane incorporation at the expense of OA and ARA (Figure 1C) to that seen for ethanolic DHA (Figure 1B).
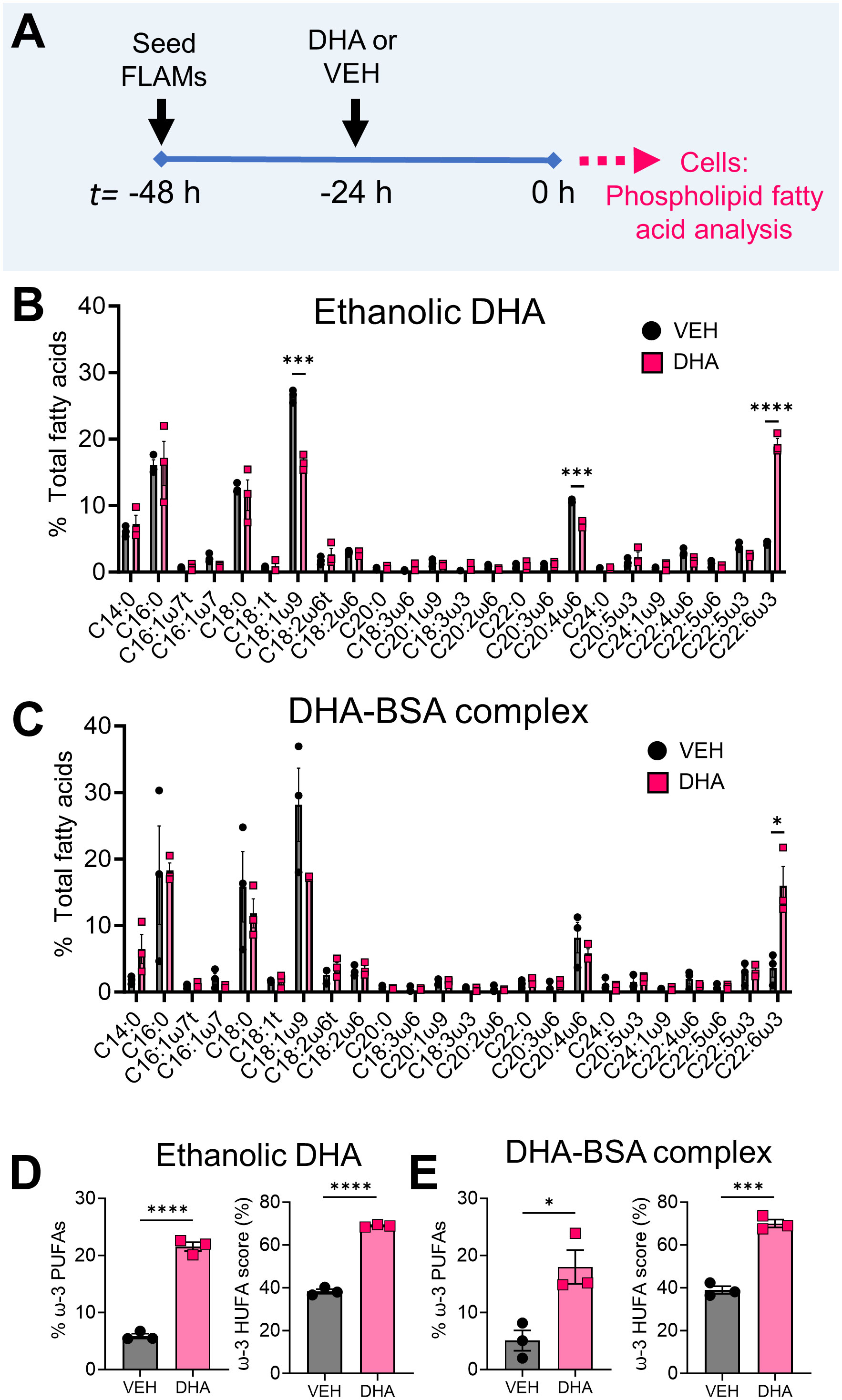
Figure 1 Supplementation of FLAMs with DHA significantly decreases oleic acid (OA) and arachidonic acid (ARA) content in FLAMs. (A) FLAMs were treated with i) 25 µM DHA as an ethanolic suspension or ethanol vehicle (VEH) or ii) 25 µM DHA as a BSA complex or 8.33 µM BSA VEH. After 24 h, cells were collected for membrane phospholipid fatty acid analysis by gas-chromatography (GC). (B, C) Following treatment with (B) ethanolic DHA and (C) DHA-BSA complexes, DHA (22:6ω3) displaces ω-9 OA (18:1ω9) and ω-6 ARA (20:4ω6) from FLAMs. (D, E) Percent ω-3 PUFAs (i.e., sum of EPA and DHA as a percentage of total fatty acids) and ω-3 highly unsaturated fatty acid (HUFA) score (i.e., sum of EPA, ω-3 DPA, and DHA as a percentage of the sum of 20:3ω6, 20:4ω6, 20:5ω3, 22:4ω6, 22:5ω6, 22:5ω3, and 22:6ω3) are elevated in FLAMs treated with (D) ethanolic DHA and (E) DHA-BSA complexes. Data are shown as mean ± SEM. *p<0.05, ***p<0.001, ****p<0.0001: Statistically significant differences between VEH-treated FLAMs and DHA-treated FLAMs.
Total fatty acid findings were related to two biomarkers: i) percent ω-3 fatty acids, which is the sum of eicosapentaenoic acid (EPA) and DHA as a percentage of all measured FA, and ii) ω-3 HUFA score, which equals the sum of ω-3 HUFAs (i.e., EPA, ω-3 DPA, and DHA) as a percentage of all measured ω-3/6 HUFAs (i.e., 20:3ω6, 20:4ω6, 20:5ω3, 22:4ω6, 22:5ω6, 22:5ω3, and 22:6ω3) (23). In FLAMs treated with ethanolic DHA, percent ω-3 fatty acids was 22%, while in VEH-treated FLAMs it was 6% (Figure 1D). FLAMs treated with ethanolic DHA also demonstrated a comparatively higher ω-3 HUFA score (69%) compared to VEH-treated FLAMs (38%). Similarly, FLAMs treated with DHA-BSA complexes exhibited significant increases in percent ω-3 fatty acids (18%) and ω-3 HUFA score (70%) compared to VEH-treated cells (5% and 39%, respectively) (Figure 1E). Based on its relative simplicity, ethanolic DHA delivery was used for all subsequent studies.
cSiO2 and DHA differentially impact production of ω-6 ARA-derived oxylipins and ω-3 DHA-/EPA-derived oxylipins in unprimed and LPS-primed FLAMs
The effects of cSiO2 on the combined intracellular and extracellular lipidome in VEH- and DHA-treated FLAMs was compared in unprimed and LPS-primed FLAMs (Figure 2A) and are illustrated in Figure 2B and Supplementary Figure 1. In addition, summarized oxylipin quantities (Supplementary Tables 3-5), and individual oxylipin quantities (50) were compared between experimental groups at each designated timepoint. Oxylipin profile shifts were highly pronounced following cSiO2 exposure (Figure 2B); therefore, we focused our analysis on the 1.5 h and 4 h post cSiO2 timepoints. cSiO2 elevated total levels of ARA-derived oxylipins and DHA-/EPA-derived oxylipins produced from VEH- and DHA-treated FLAMs, respectively (Figure 3A, Supplementary Tables 4, 5). In DHA-treated FLAMs, cSiO2-induced levels of ARA-derived oxylipins were significantly decreased, and, correspondingly, cSiO2-induced levels of DHA-/EPA-derived oxylipins were significantly elevated. cSiO2-induced levels of EPA-derived oxylipins also increased modestly in VEH-treated FLAMs. LPS priming elicited a marked increase in cSiO2-triggered ARA-derived oxylipins production in VEH-treated FLAMs and a marked decrease in cSiO2-triggered EPA-derived oxylipins levels in DHA-treated FLAMs. Findings at 4 h post cSiO2 reflected those found at 1.5 h with higher overall quantities of ARA-, EPA-, and DHA-derived metabolites.
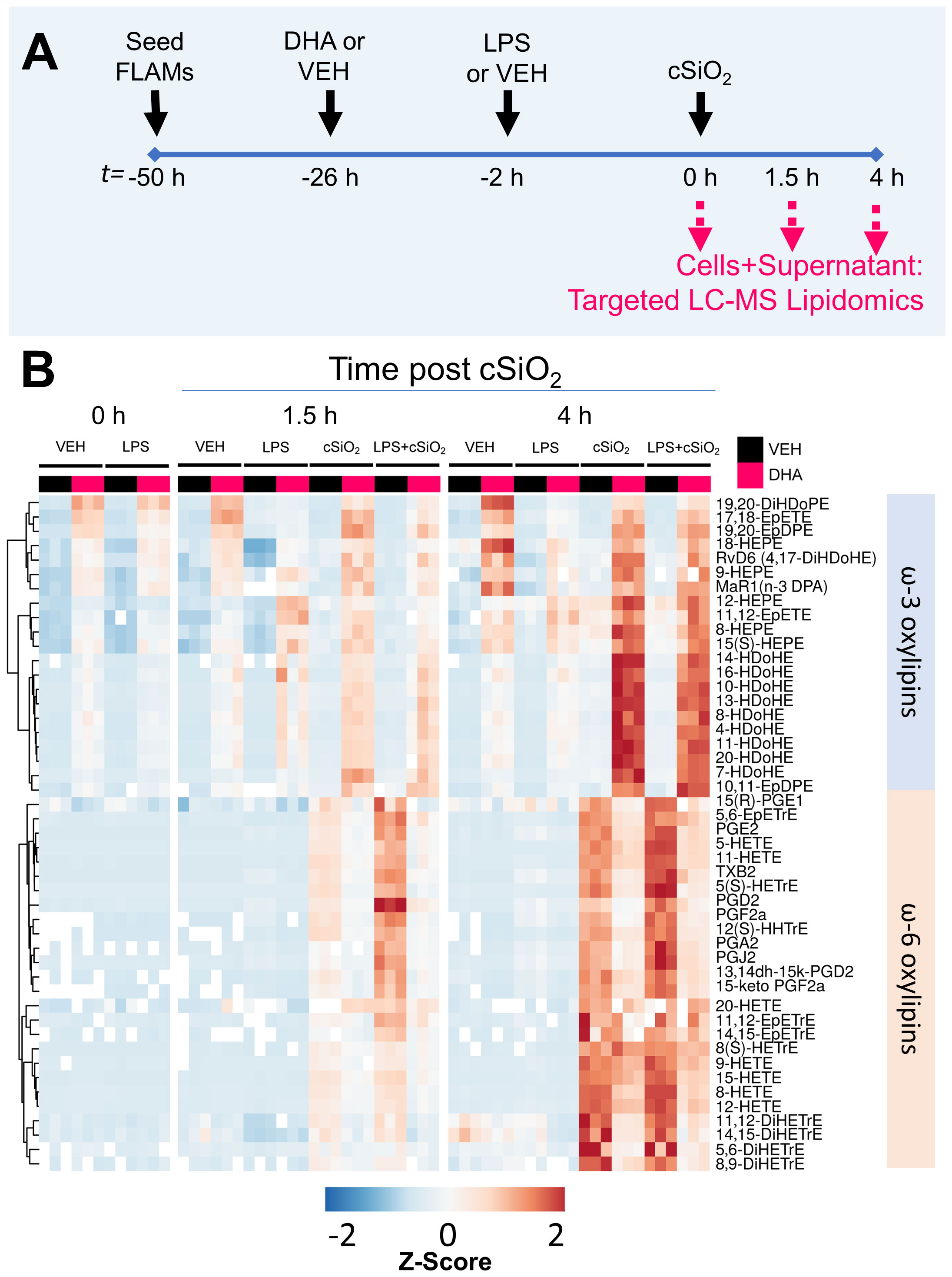
Figure 2 LPS, cSiO2, and DHA differentially impact generation of ω-3 and ω-6 oxylipins from FLAMs. (A) FLAMs were treated with ethanolic DHA (25 µM) or ethanol vehicle (VEH) for 24 h, primed with LPS (20 ng/ml), and/or exposed to cSiO2 (12.5 µg/cm2). Cultured FLAMs and supernatants were pooled at t = 0 h, 1.5 h, and 4 h post cSiO2 and 156 oxylipins profiled by targeted LC-MS. Treatment conditions were tested using three biological replicates, and oxylipins were measured using one technical replicate per sample. (B) Heat maps depicting the concentration of scaled ω-3 and ω-6 oxylipins, using unsupervised clustering with the Euclidean distance method. As an exception, 5(S)-HETrE is derived from ω-9 mead acid (C20:3ω9).
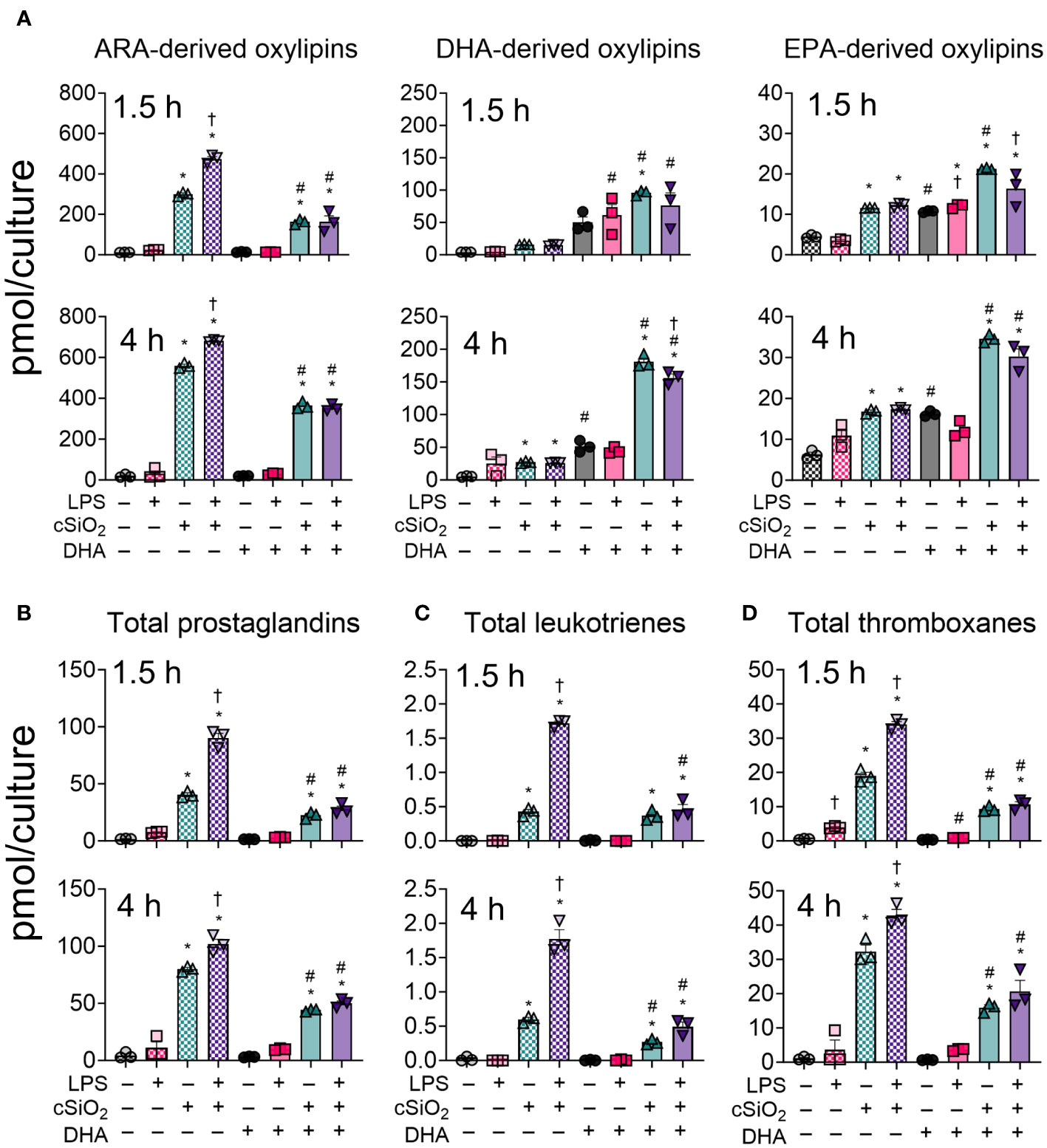
Figure 3 LPS, cSiO2, and DHA differentially impact generation of ARA-, DHA-, and EPA-derived oxylipins from FLAMs. FLAMs were treated with ethanolic DHA (25 µM) or ethanol vehicle (VEH) for 24 h, primed with LPS (20 ng/ml), and/or exposed to cSiO2 (12.5 µg/cm2). (A) Total ARA-, DHA-, and EPA-derived oxylipins were quantified for all experimental groups at 1.5 h and 4 h post cSiO2. (B) Total prostaglandins, (C) leukotrienes, and (D) thromboxanes were quantified for all experimental groups at 1.5 h and 4 h post cSiO2. Data are shown as mean ± SEM. MetaboAnalyst Version 5.0 was used for data normalization and statistically significant differences were determined by one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test. *FDR q<0.05 for cSiO2 vs. controls; #FDR q<0.05 for DHA vs. controls; †FDR q<0.05 for LPS vs. controls.
DHA suppresses cSiO2-induced production of ARA-derived prostaglandins, leukotrienes, and thromboxanes in unprimed and LPS-primed FLAMs
At 1.5 h and 4 h, cSiO2 induced robust increases in total prostaglandins (Figure 3B), leukotrienes (Figure 3C) and thromboxanes (Figure 3D) compared to VEH-treated FLAMs (Supplementary Tables 4, 5). Levels of representative oxylipins from each metabolite class including PGE2, PGJ2, PGD2, PGF2α, and PGA2 (Figure 4A), LTB4 (Figure 4B), and TXB2 (Figure 4C) were increased in like manner in the presence of cSiO2 (50).
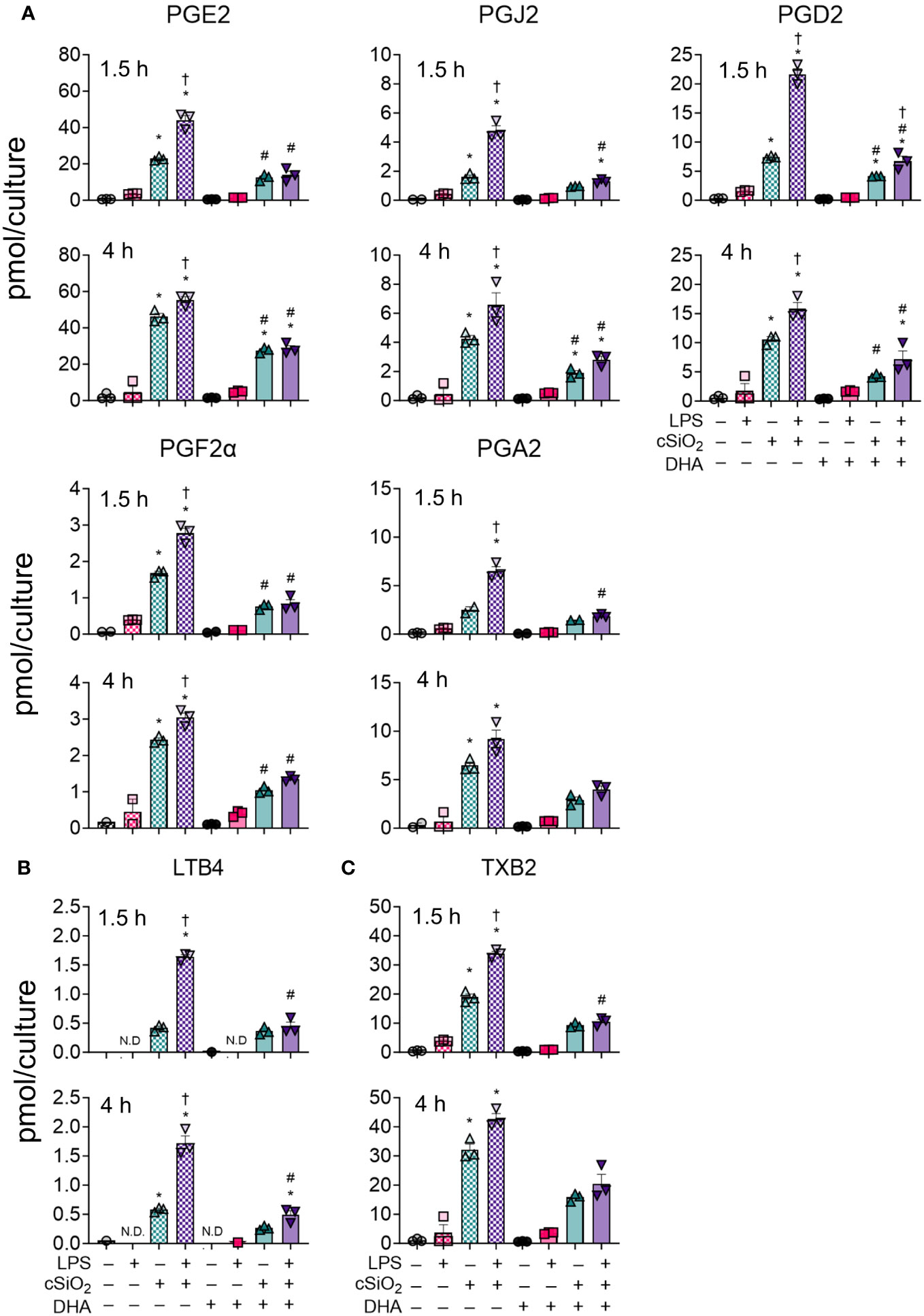
Figure 4 DHA dampens cSiO2-induced production of ARA-derived prostaglandins, LTB4, and TXB2 in FLAMs. (A) PGE2, PGJ2, PGD2, PGF2α, and PGA2, (B) LTB4, and (C) TXB2 were quantified by LC-MS for all experimental groups at 1.5 h and 4 h post cSiO2. Treatment conditions were tested using three biological replicates, and oxylipins were measured using one technical replicate per sample. Data are shown as mean ± SEM. MetaboAnalyst Version 5.0 was used for data normalization and statistical analysis by one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test. *FDR q<0.05 for cSiO2 vs. controls; #FDR q<0.05 for DHA vs. controls; †FDR q<0.05 for LPS vs. controls.
LPS priming augmented cSiO2-triggered production of total and individual prostaglandins, leukotrienes, and thromboxanes (Figures 3B-D, 4). DHA significantly reduced cSiO2-induced prostaglandin, leukotriene, and thromboxane production, yet induction of these metabolites was still significant compared to baseline levels in DHA-treated FLAMs. Interestingly, LPS priming did not significantly impact cSiO2-induced levels of prostaglandins, leukotrienes, and thromboxanes in DHA-treated FLAMs.
DHA broadly skews cSiO2-induced hydroxy fatty acids in FLAMS from being ω-6 PUFA-derived to being ω-3 PUFA-derived
Total hydroxy fatty acid (HFA) metabolites were significantly increased by cSiO2 exposure in both VEH-treated and DHA-treated FLAMs at 1.5 h and 4 h (Figure 5A, Supplementary Tables 4, 5). DHA supplementation significantly reduced ω-6 HFAs and increased ω-3 HFAs at both timepoints (Figures 5B, C). The suppressive effect of DHA on ω-6 HFA levels was more marked at 1.5 h than at 4 h, while ω-3 HFA levels steadily increased in DHA-treated FLAMs over time. LPS priming further increased levels of cSiO2-induced ω-6 HFAs at 1.5 h and decreased levels of cSiO2-induced ω-3 HFAs at 4 h.
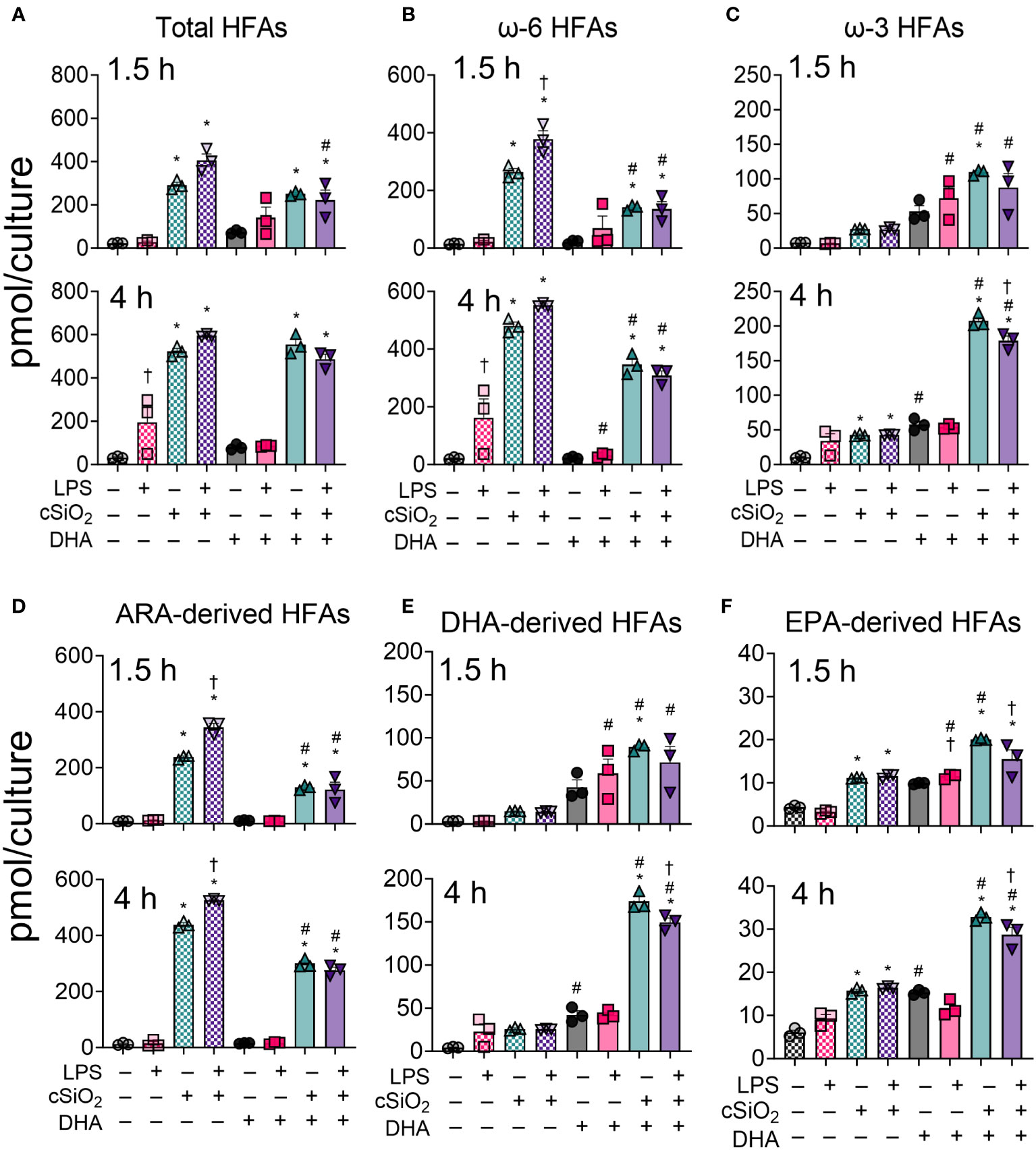
Figure 5 DHA skews cSiO2-induced hydroxy fatty acid (HFA) metabolites from being ω-6 PUFA-derived and toward being ω-3 PUFA-derived. (A) Total hydroxy fatty acids (HFAs), (B) ω-6 HFAs, (C) ω-3 HFAs, (D) ARA-derived HFAs, (E) DHA-derived HFAs, and (F) EPA-derived HFAs were quantified by LC-MS for all experimental groups at 1.5 h and 4 h post cSiO2. Treatment conditions were tested using three biological replicates, and oxylipins were measured using one technical replicate per sample. Data are shown as mean ± SEM. MetaboAnalyst Version 5.0 was used for data normalization and statistically significant differences were determined by one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test. *FDR q<0.05 for cSiO2 vs. controls; #FDR q<0.05 for DHA vs. controls; †FDR q<0.05 for LPS vs. controls.
Observed changes in ARA-derived HFAs reflected those in total ω-6 HFAs (Figures 5B, D, Supplementary Tables 4, 5). cSiO2 triggered significant increases in ARA-derived HFA levels in both VEH-treated and DHA-treated FLAMs (Figure 5D). DHA significantly reduced metabolite levels, and LPS priming further potentiated cSiO2-induced metabolite production at 1.5 h and 4 h. Likewise, DHA- and EPA-derived HFA levels reflected total levels of ω-3 HFAs, as these significantly increased in DHA-treated cells exposed to cSiO2 starting at 1.5 h and continuing at 4 h (Figures 5C, E, Supplementary Tables 4, 5). In both VEH-treated FLAMs and DHA-treated FLAMs, quantities of ARA-, EPA-, and DHA-derived HFAs were ranked as follows for both timepoints: ARA > DHA > EPA (Figures 5D-F). Furthermore, ARA-derived HFAs accounted for the majority of total measured ω-6 HFAs, whereas EPA and DHA both accounted for the majority of total measured ω-3 HFAs.
DHA suppresses cSiO2-induced production of ARA-derived HETEs and induces production of DHA-derived HDoHEs and EPA-derived HEPEs
cSiO2-induced ω-6 HFAs in VEH-treated FLAMs at 1.5 h and 4 h consisted primarily of ARA-derived 5-HETE, 8-HETE, 9-HETE, 11-HETE, 12-HETE, and 15-HETE (Figure 6) (50). Relative abundance of selected HETEs was: 5-HETE > 11-HETE > 15-HETE > 8-HETE > 12-HETE > 9-HETE. HETE levels also increased in DHA-treated FLAMs exposed to cSiO2 but were found to be significant only for 5-HETE, 8-HETE, and 9-HETE at 4 h and 15-HETE at both timepoints. In line with total ARA-derived HFA levels (Figure 5D), DHA significantly suppressed 8-HETE at 4 h and 9-HETE at both timepoints (Figure 6). Levels of other cSiO2-induced HETEs (e.g., 5-HETE, 11-HETE, 12-HETE, 15-HETE) also were reduced in DHA-treated FLAMs, but the findings were not statistically significant. LPS priming elicited a significant increase in cSiO2-induced 8-HETE and a non-significant trend toward increased levels of other HETEs in VEH-treated FLAMs at both timepoints.
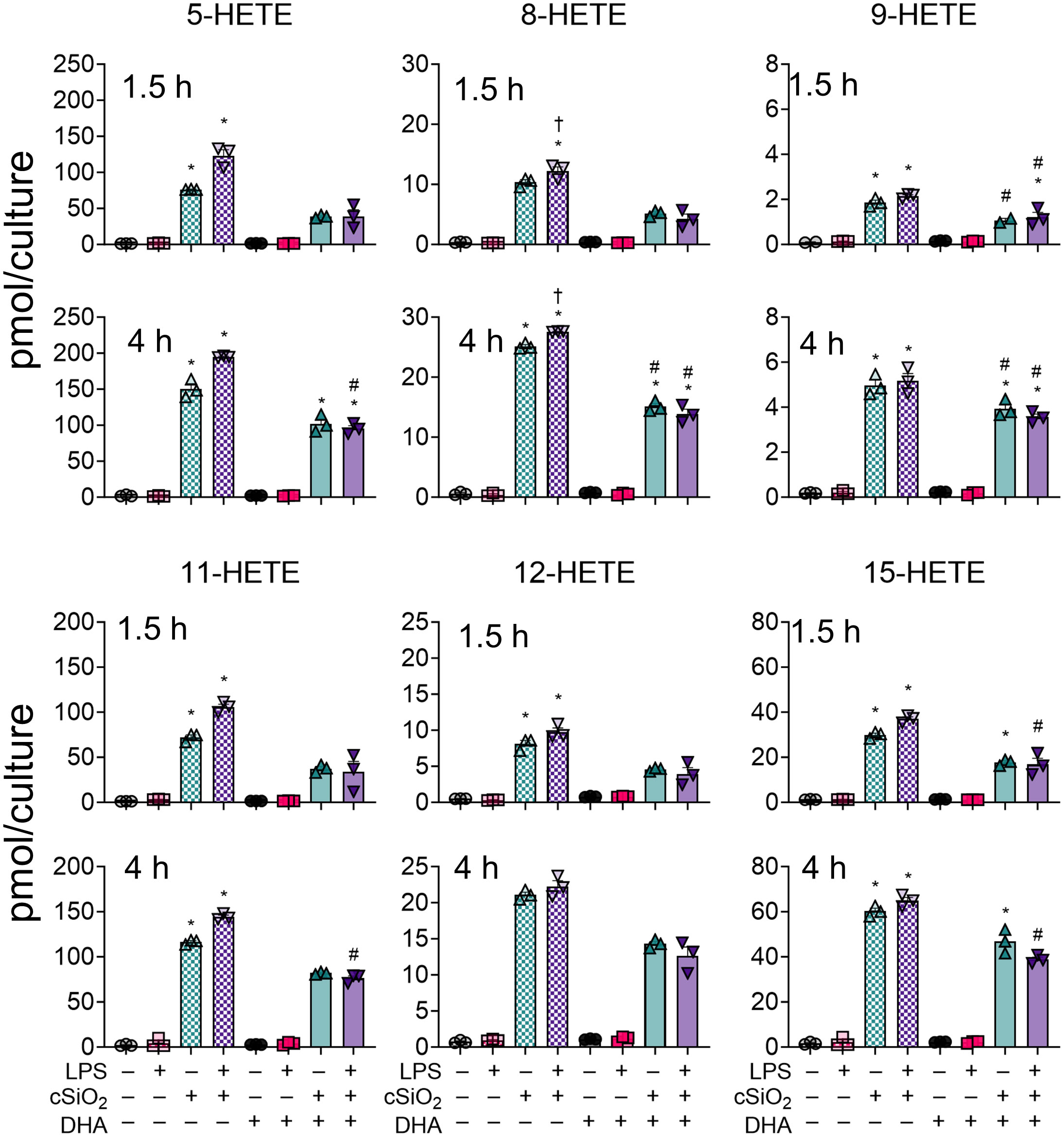
Figure 6 cSiO2-induced production of ARA-derived HFAs is diminished with DHA supplementation. 5-HETE, 8-HETE, 9-HETE, 11-HETE, 12-HETE, and 15-HETE were quantified by LC-MS for all experimental groups at 1.5 h and 4 h post cSiO2. Treatment conditions were tested using three biological replicates, and oxylipins were measured using one technical replicate per sample. Data are shown as mean ± SEM. MetaboAnalyst Version 5.0 was used for data normalization and statistically significant differences were determined by one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test. *FDR q<0.05 for cSiO2 vs. controls; #FDR q<0.05 for DHA vs. controls; †FDR q<0.05 for LPS vs. controls.
In DHA-treated FLAMs, cSiO2 induced significant increases in several DHA-derived HDoHEs (i.e., 4-HDoHE, 7-HDoHE, 8-HDoHE, 10-HDoHE, 11-HDoHE, 14-HDoHE, 16-HDoHE, 17-HDoHE, 20-HDoHE) (Figure 7) and in several EPA-derived HEPEs (i.e., 5-HEPE, 8-HEPE, 9-HEPE, 11-HEPE, 12-HEPE, 15[S]-HEPE) at both timepoints (Supplementary Figure 2) (50). Relative abundance of selected HDoHEs was: 4-HDoHE ≈ 20-HDoHE > 16-HDoHE > 7-HDoHE ≈ 8-HDoHE ≈ 10-HDoHE ≈ 11-HDoHE > 14-HDoHE > 17-HDoHE and relative abundance of selected HEPEs was: 5-HEPE > 8-HEPE ≈ 11-HEPE > 15(S)-HEPE > 12-HEPE > 9-HEPE. While cSiO2 exposure led to significant increases in EPA-derived 5-HEPE, 11-HEPE, and 12-HEPE in VEH-treated cells during the time-course, DHA-derived HDoHEs did not undergo significant increases in VEH-treated cells exposed to cSiO2. The effects of LPS priming on cSiO2-induced HEPEs and HDoHEs were minimal at 1.5 h and 4 h.
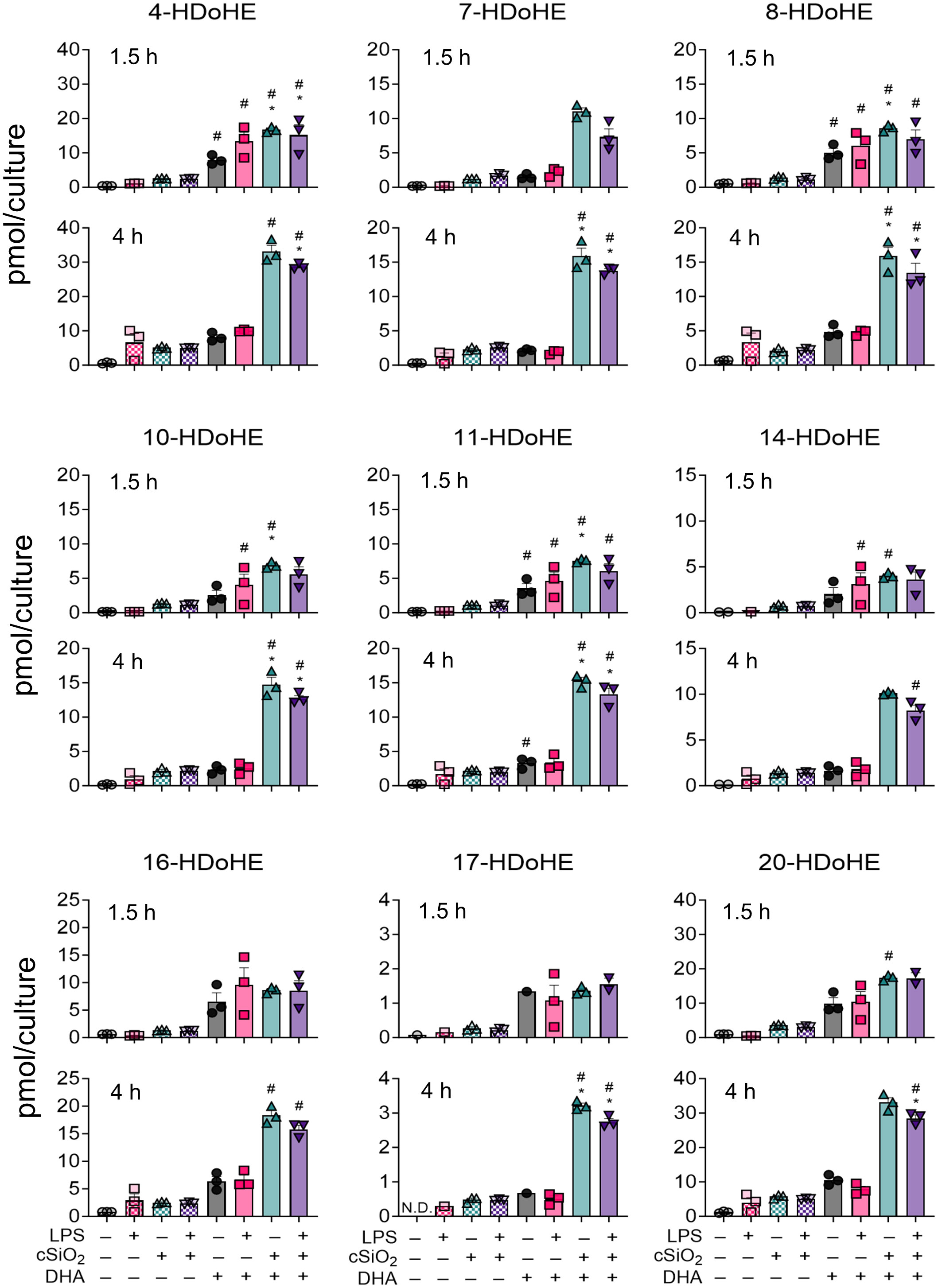
Figure 7 cSiO2 exposure of DHA-supplemented FLAMs triggers increased production of DHA-derived hydroxy fatty acids (HFAs). 4-HDoHE, 7-HDoHE, 8-HDoHE, 10-HDoHE, 11-HDoHE, 14-HDoHE, 16-HDoHE, 17-HDoHE, and 20-HDoHE were quantified by LC-MS for all experimental groups at 1.5 h and 4 h post cSiO2. Treatment conditions were tested using three biological replicates, and oxylipins were measured using one technical replicate per sample. Data are shown as mean ± SEM. MetaboAnalyst Version 5.0 was used for data normalization and statistically significant differences were determined by one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test. *FDR q<0.05 for cSiO2 vs. controls; #FDR q<0.05 for DHA vs. controls; †FDR q<0.05 for LPS vs. controls.
DHA modestly influences production of the specialized pro-resolving lipid mediators RvD6 (4,17-DiHDoPE) and MaR1ω-3 DPA in FLAMs
Specialized pro-resolving mediators (SPMs) are a class of oxylipins comprised of resolvins, maresins, protectins, and lipoxins derived from ARA, EPA, ω-3 DPA, and DHA that limit proinflammatory cytokine release and promote dead cell clearance by macrophages (56–58). Most SPMs assessed in our LC-MS oxylipin panel were not detected at any timepoint (50). DHA supplementation did, however, cause a modest increase in RvD6 (4,17-DiHDoPE) and MaR1ω-3 DPA at 1.5 h and 4 h (Figure 8) (50). In DHA-treated FLAMs, cSiO2 exposure did not significantly influence production of RvD6 at either timepoint (Figure 8A) but significantly increased MaR1ω-3 DPA at 1.5 h and decreased MaR1ω-3 DPA at 4 h (Figure 8B). LPS priming significantly suppressed MaR1ω-3 DPA production in DHA-treated FLAMs at 4 h, modestly inhibited MaR1ω-3 DPA at 1.5 h, and modestly decreased RvD6 production at both 1.5 h and 4 h.
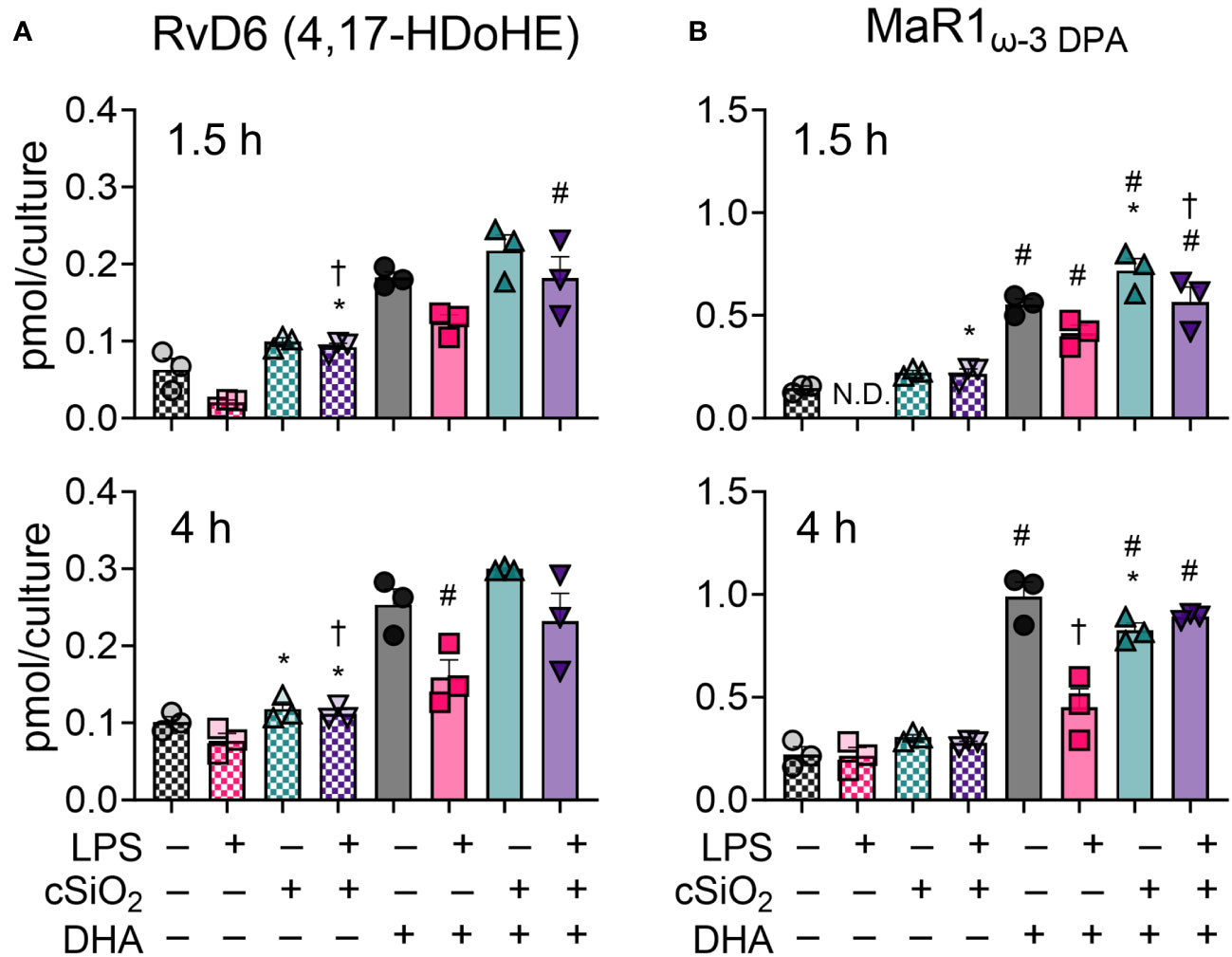
Figure 8 DHA supplementation contributes to modest production of specialized pro-resolving mediators RvD6 (4,17-DiHDoPE) and MaR1ω-3 DPA in FLAMs. (A) RvD6 (4,17-DiHDoPE) and (B) MaR1ω-3 DPA were quantified by LC-MS for all experimental groups at 1.5 h and 4 h post cSiO2. Treatment conditions were tested using three biological replicates, and oxylipins were measured using one technical replicate per sample. Data are shown as mean ± SEM. MetaboAnalyst Version 5.0 was used for data normalization and statistical analysis by one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test. *FDR q<0.05 for cSiO2 vs. controls; #FDR q<0.05 for DHA vs. controls; †FDR q<0.05 for LPS vs. controls.
DHA modestly influences production of EpFAs and DiHFAs in cSiO2-exposed FLAMs
Total epoxy fatty acids (EpFAs) and CYP450-derived dihydroxy fatty acids (DiHFAs) were quantified from VEH-treated and DHA-treated FLAMs (Figure 9A, Supplementary Tables 4, 5). In VEH-treated FLAMs, cSiO2 modestly induced production of EpFA metabolites at 1.5 h and 4 h and did not significantly impact production of DiHFA metabolites. Conversely, cSiO2 triggered significant increases in total EpFAs and DiHFAs in DHA-treated FLAMs at both timepoints. Interestingly, LPS priming significantly reduced total DiHFA metabolite levels in the absence of cSiO2 in DHA-treated FLAMs. Separate and simultaneous LPS priming and cSiO2 exposure elicited modest increases in EpFA : DiHFA ratios in both VEH-treated and DHA-treated FLAMs during the time-course.
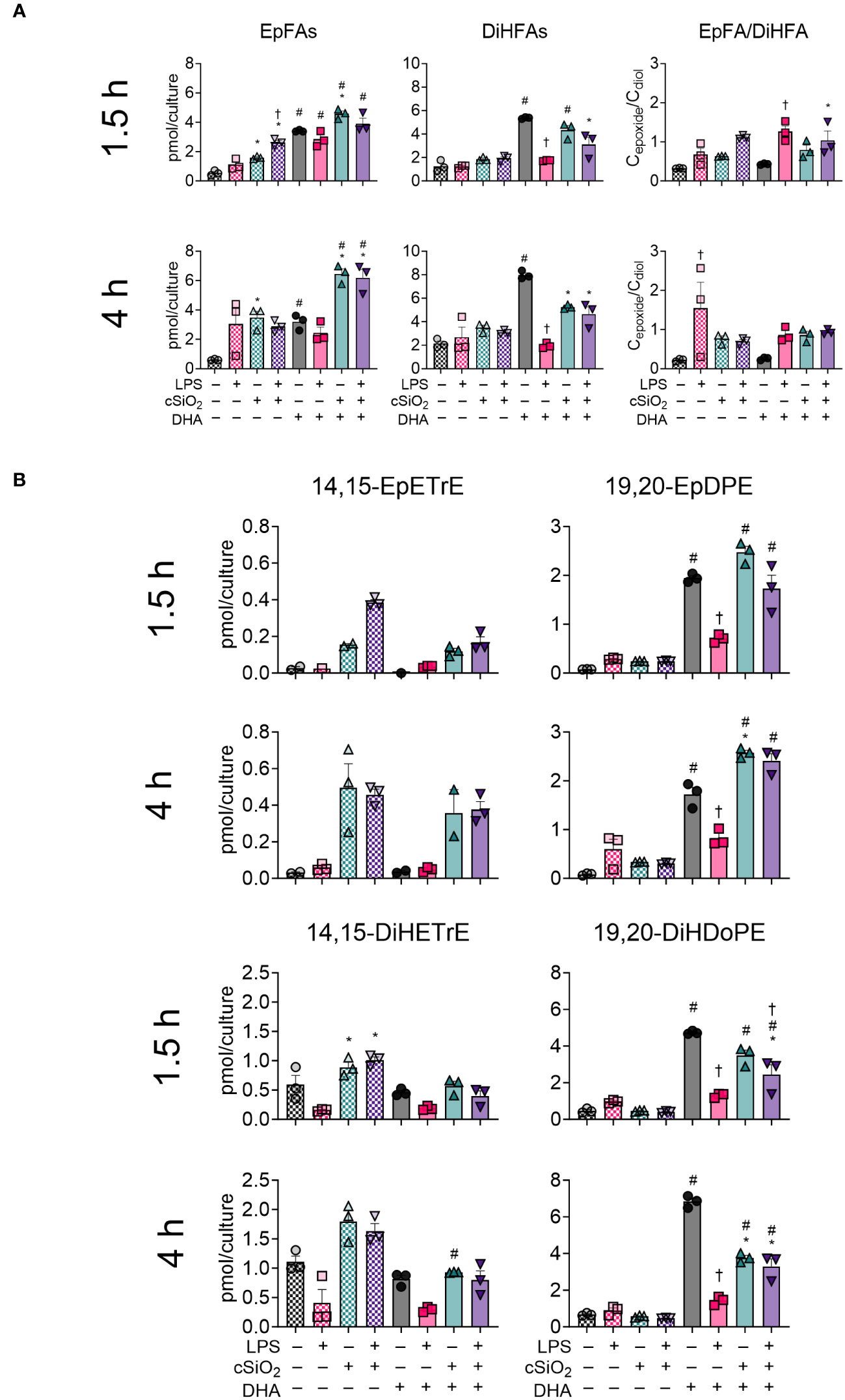
Figure 9 cSiO2 exposure and DHA supplementation contribute to increased production of epoxy fatty acids (EpFAs) and dihydroxy fatty acids (DiHFAs). (A) Total EpFAs, total DiHFAs, and EpFA : DiHFA ratios (Cepoxide/Cdiol) were quantified by LC-MS for all experimental groups at 1.5 h and 4 h post cSiO2. (B) 14,15-EpETrE, 19,20-EpDPE, 14,15-DiHETrE, and 19,20-DiHDoPE were quantified for all experimental groups at 1.5 h and 4 h post cSiO2. Treatment conditions were tested using three biological replicates, and oxylipins were measured using one technical replicate per sample. Data are shown as mean ± SEM. MetaboAnalyst Version 5.0 was used for data normalization and statistical analysis by one-way analysis of variance (ANOVA) (FDR = 0.05) followed by Tukey’s honestly significant difference (HSD) post-hoc test. *FDR q<0.05 for cSiO2 vs. controls; #FDR q<0.05 for DHA vs. controls; †FDR q<0.05 for LPS vs. controls.
Effects of cSiO2 and DHA were also analyzed for selected CYP450 oxylipin products of ARA (i.e., 14,15-EpETrE, 14,15-DiHETrE) and DHA (i.e., 19,20-EpDPE, 19,20-DiHDoPE) (Figure 9B) (50). cSiO2 evoked production of 14,15-EpETrE and 14,15-DiHETrE starting at 1.5 h and continuing through 4 h in VEH-treated FLAMs and, to a lesser degree, in DHA-treated FLAMs. LPS priming also modestly increased cSiO2-triggered production of 14,15-EpETrE in VEH-treated FLAMs. Changes in 14,15-EpETrE levels were not significant, and cSiO2-induced production of 14,15-DiHETrE was significant only at 1.5 h. In contrast, DHA treatment promoted robust production of 19,20-EpDPE and 19,20-DiHDoPE at both timepoints. Exposure to cSiO2 resulted in a subtle, yet non-significant, increase in 19,20-EpDPE and corresponding decrease in 19,20-DiHDoPE at both timepoints. Intriguingly, LPS priming alone significantly decreased levels of 19,20-EpDPE and 19,20-DiHDoPE during the experiment. Overall, levels of 19,20-EpDPE and 19,20-DiHDoPE were found to be higher than levels of 14,15-EpETrE and 14,15-DiHETrE.
DHA suppresses cSiO2-induced release of proinflammatory cytokines from LPS-primed FLAMs
The concurrent impacts of DHA on proinflammatory cytokine release from unprimed and LPS-primed FLAMs were evaluated at 1.5 h and 4 h post cSiO2-treatment (Figure 10A). At both timepoints, unprimed FLAMs treated with either VEH or cSiO2 alone secreted negligible amounts of IL-1α, IL-1β, and TNF-α (Figures 10B–D). In contrast, LPS-primed FLAMs released robust amounts of IL-1α, IL-1β, and TNF-α at both timepoints following cSiO2 exposure, with much higher cytokine levels observed at 4 h compared to 1.5 h post cSiO2. DHA pretreatment in LPS-primed FLAMs markedly reduced cSiO2-induced release of IL-1α, IL-1β, and TNF-α release at both timepoints. No notable DHA effects on proinflammatory cytokine release were evident in FLAMs treated with VEH or cSiO2 alone.
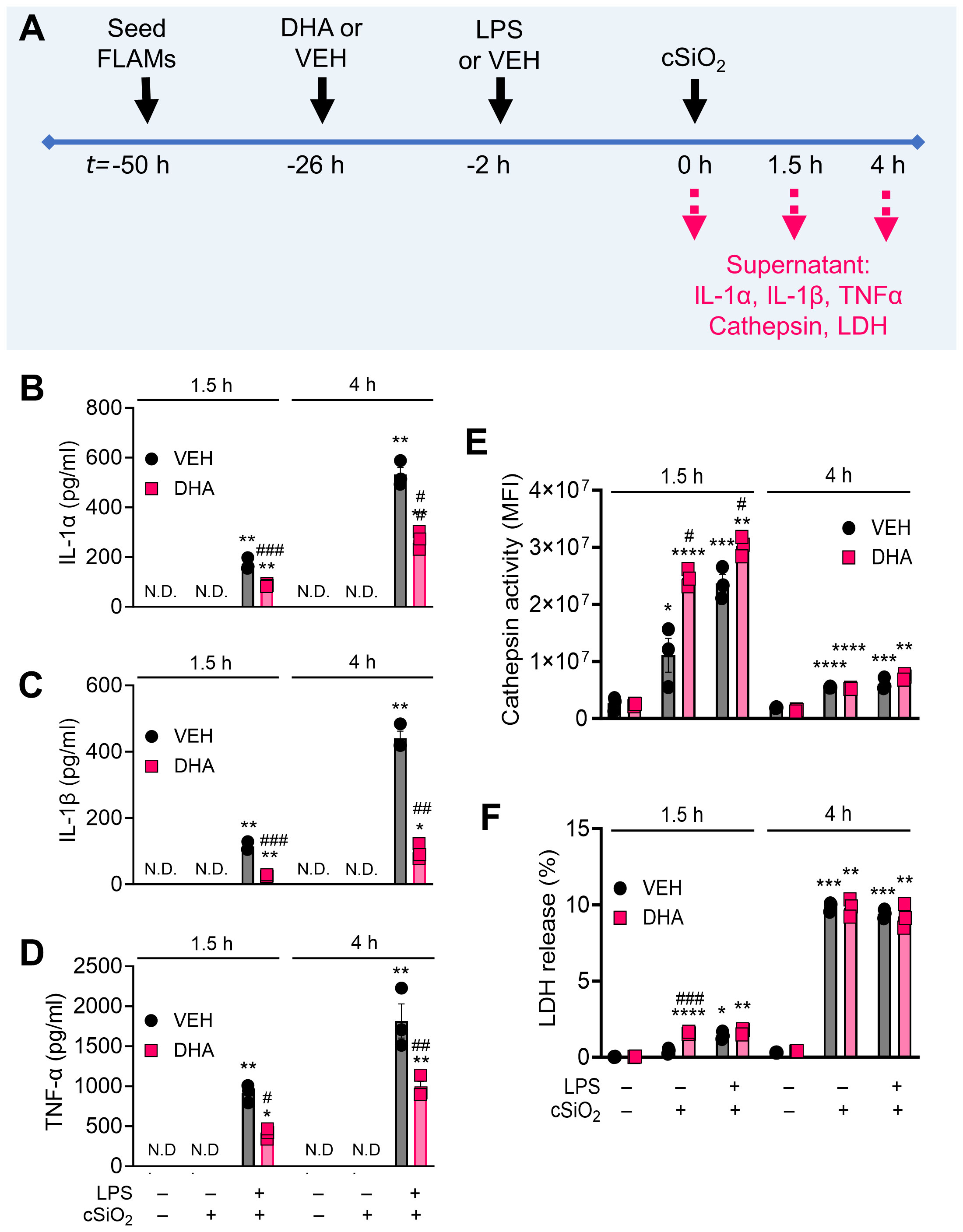
Figure 10 DHA suppresses cSiO2-induced release of proinflammatory cytokines in LPS-stimulated FLAMs. (A) FLAMs were treated with ethanolic DHA (25 µM) or ethanol vehicle (VEH) for 24 h, primed with LPS (20 ng/ml), then exposed to cSiO2 (12.5 µg/cm2). Cell culture supernatants were collected at t = 1.5 h and 4 h post cSiO2, and (B) IL-1α, (C) IL-1β, and (D) TNF-α were quantified by ELISA. (E) Lysosomal cathepsin activity (expressed in units of mean fluorescence intensity [MFI]) was quantified as a metric for lysosomal membrane permeabilization, and (F) percent LDH release was quantified as a metric for cell death. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001: Statistically significant differences between cSiO2 and its corresponding control. #p<0.05, ##p<0.01, ###p<0.001: Statistically significant differences between DHA and its corresponding control. N.D., not determined.
DHA does not suppress cSiO2-induced cathepsin or LDH release in FLAMs
In addition to analyzing the impacts of DHA, LPS, and cSiO2 on the cellular lipidome, we also measured lysosomal cathepsin and LDH activities in collected supernatants (Figure 10A). Cathepsin activity in VEH-treated cells exposed to cSiO2 alone was higher at 1.5 h (1.1×107 MFI) than at 4 h (5.5×106 MFI) post cSiO2 treatment (Figure 10E). Interestingly, DHA caused a moderate increase in cathepsin activity in FLAMs exposed to cSiO2 alone or to both LPS and cSiO2 at 1.5 h but not at 4 h. LDH release in VEH-treated FLAMs treated with cSiO2 alone was higher at 4 h (9.9%) than at 1.5 h (0.4%) (Figure 10F). LPS priming of FLAMs increased cSiO2 -induced extracellular lysosomal cathepsin and LDH response at 1.5 h post cSiO2 in both VEH- and DHA-treated FLAMs but not at 4 h post cSiO2 (Figures 10E, F). In line with cathepsin activity analyses, DHA caused a slight increase in LDH release in FLAMs exposed to cSiO2 alone at 1.5 h and did not significantly impact LDH release at 4 h.
DHA does not influence cSiO2-induced lysosomal membrane permeabilization, mitochondrial toxicity, or death in FLAMs
Live-cell imaging using LysoTracker Red (LTR), MitoTracker Red (MTR), and SYTOX Green (SG) was employed to further assess the impacts of DHA on cSiO2-induced lysosomal membrane permeabilization (LMP), mitochondrial depolarization, and cell death, respectively (Figure 11A, Supplementary Figure 3A). In a preliminary experiment, we found that LPS priming slightly expedited cSiO2-induced loss of LTR+ cells (Supplementary Figure 3B). LPS did not significantly impact cSiO2-induced development of SG+ cells (Supplementary Figure 3D). Intriguingly, LPS priming perpetuated cSiO2-triggered loss of MTR+ cells (Supplementary Figure 3C). In unprimed FLAMs, the proportions of LTR+ cells (Figure 10B), MTR+ cells (Figure 11C), and SG+ cells (Figure 11D) at time of cSiO2 addition were nearly 100%, 100%, and 0%, respectively, relative to total cells. Loss of lysosomal integrity occurred at very similar rates up to 4 h post cSiO2 in VEH- and DHA-treated unprimed FLAMs exposed to cSiO2 (Figure 11B). Mitochondrial depolarization progressed at approximately the same rate in VEH- and DHA-treated unprimed FLAMs from 0 to 1.5 h, and DHA slightly protected FLAMs from further mitochondrial depolarization from 1.5 to 4 h (Figure 11C). Minimal cell death was observed from 0 to 1.5 h for both VEH- and DHA-treated cells, and DHA slightly, albeit insignificantly, suppressed cell death from 1.5 to 4 h (Figure 11D). Interestingly, DHA pretreatment modestly enhanced cSiO2-induced loss of LTR+ (Figure 11B) and MTR+ cells (Figure 10C) while increasing SG+ cells (Figure 11D). Altogether, DHA had negligible effects on cSiO2-induced lysosomal membrane permeabilization, mitochondrial toxicity, and death in FLAMs.
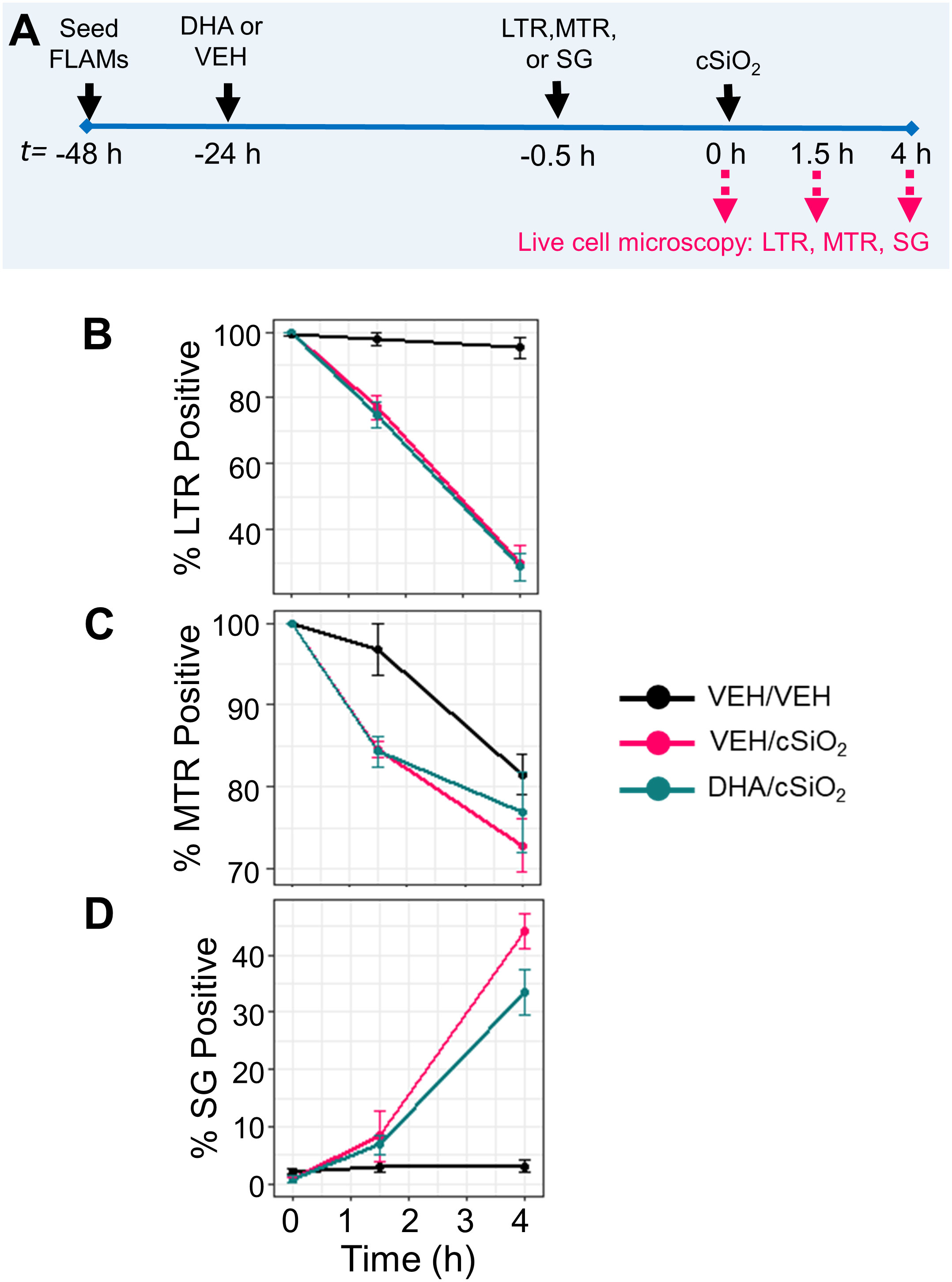
Figure 11 DHA does not affect early cSiO2-induced lysosomal membrane permeabilization, mitochondrial toxicity, and death in FLAMs. (A) FLAMs were treated with ethanolic DHA (25 µM) or ethanol vehicle (VEH) for 24 h. VEH-treated and DHA-treated FLAMs were then stained with LysoTracker Red (LTR; 50 nM), MitoTracker Red (MTR; 25 nM), or SYTOX Green (SG; 200 nM) in DPBS+/+ for 30 min. After 30 minutes to allow fluorescent dyes to equilibrate, cSiO2 was added dropwise at 0 or 12.5 μg/cm2. (B) Percent LTR+, (C) MTR+, and (D) SG+ cells from 0 to 4 h post cSiO2 in VEH/VEH, VEH/cSiO2, and DHA/cSiO2 treated cells quantified using CellProfiler 4.2.1 and RStudio Desktop. Data are shown as mean ± SEM.
Discussion
AMs comprise the first line of defense against cSiO2 and other inhaled particles (59). Preclinical studies have demonstrated that cSiO2 elicits robust proinflammatory mediators and death in AMs, which might be critical first steps for subsequent development of inflammation and autoimmunity observed in the lungs of animals exposed to the particle. Importantly, intervention by dietary DHA supplementation dramatically suppresses these responses (27, 60, 61). While bioactive oxylipins potentially play key roles in these responses, the effects of cSiO2 and DHA on oxylipin production in naïve and TLR-activated AMs have not yet been systematically addressed. Herein, we hypothesized that DHA would inhibit cSiO2-induced production of proinflammatory eicosanoids in FLAMs. This investigation is the first to comprehensively assess how LPS, cSiO2, and DHA time-dependently influence the oxylipin signature in an AM surrogate. Several novel findings were made. First, DHA delivery to FLAM cultures as either an ethanolic suspension or as BSA complexes were equally effective in displacing the ω-6 PUFA ARA and ω-9 PUFA OA from cellular phospholipids, resulting in increased percent ω-3 PUFAs and ω-3 HUFA scores. Second, cSiO2 exposure within 4 h elicited an ARA-derived eicosanoid storm consisting of prostaglandins, leukotrienes, thromboxanes, and HETEs coupled with less prominent changes in ω-6 EpFAs and DiHFAs. Third, supplementing FLAMs with DHA dramatically suppressed cSiO2-induced ARA-derived oxylipins while promoting production of DHA- and EPA-derived oxylipins, including HDoHEs and HEPEs. Fourth, LPS priming modestly enhanced cSiO2-induced ARA-derived oxylipin generation. Finally, within a concurrent time period, DHA suppressed cSiO2-triggered release of IL-1α, IL-1β, and TNF-α in LPS-primed FLAMs but modestly enhanced both cSiO2-induced decrements in lysosome and mitochondrial integrity concurrently and cell death.
Preclinical and clinical studies suggest that increased ω-3 PUFA intake—and consequently increased ω-3 PUFA tissue content—are associated with decreased symptom severity in chronic inflammatory conditions such as rheumatoid arthritis (62, 63), lupus (23, 64), and cardiovascular disease (65, 66). The pro-resolving impacts of the ω-3 PUFA DHA are multifaceted. At the cellular level, DHA 1) modulates membrane fluidity by displacing ω-6 PUFAs from the sn-2 position of membrane phospholipids, 2) suppresses expression and release of proinflammatory cytokines, 3) competes with ω-6 PUFAs as substrates for fatty acid metabolizing enzymes, and 4) undergoes conversion into several classes of highly pro-resolving oxylipins [reviewed in (25, 67, 68)]. In previously published studies, we have found in several macrophage models that DHA is readily incorporated into membrane phospholipids at the expense of ω-6 ARA and ω-9 OA, suppresses LPS-induced transcription and translation of proinflammatory genes, dampens cSiO2-induced proinflammatory cytokine release, and stimulates efferocytosis of cSiO2-killed cell corpses (27, 28, 45). Our prior investigations also suggest that DHA protects primary AMs, fetal liver-derived macrophages maintained with GM-CSF without TGF-β, and murine RAW264.7 macrophage-like cells from cSiO2-induced death (28), dissimilar to the present study.
Our findings that supplementation of FLAMs with DHA in the form of either an ethanolic suspension or BSA complexes were equivalent correspond with previous findings of Wiesenfeld and coworkers (54), who reported that delivery of DHA as ethanolic suspensions and BSA complexes resulted in roughly equal displacement of ARA by DHA in two different transformed macrophage cell lines. Here, we used a physiologically relevant dose of DHA that resulted in plasma membrane incorporation at levels comparable to erythrocyte DHA content observed in previous in vivo studies, where mice were fed a realistic human equivalent dose of 5 g/day (61, 69, 70). However, the cell culture conditions used here do not completely reflect other dietary components that could influence AM inflammatory responses from a translational perspective. For instance, the recommended ω-6/ω-3 ratio is 2-3:1 yet the ω-6/ω-3 ratio in the standard Western diet is approximately 20:1 (71), which may increase the risk of inflammatory ARA-derived oxylipin cascades (72). It would therefore be informative in future lipidomics investigations to treat FLAMs with various ratios of ω-6 PUFAs (e.g., LA, ARA) and ω-3 PUFAs (e.g., EPA, DHA) prior to cSiO2 exposure to more closely model dietary patterns in rodent and human studies.
Although our lipid metabolite panel may not account for all potential oxylipin species present in FLAMs, we found here that cSiO2 induced production of numerous bioactive oxylipins derived from ARA (e.g., PGE2, LTB4, TXB2, HETEs), DHA (e.g., HDoHEs), and EPA (e.g., HEPEs) in VEH-treated and DHA-treated FLAMs. Oxylipins derived from other less abundant ω-6 PUFAs (e.g., LA, DGLA) were also detected in our analysis, which may play roles in modulating cSiO2-triggered toxicity in FLAMs (73). These results suggest that cSiO2 may induce PLA2-mediated release of ω-6 PUFAs and ω-3 PUFAs from the sn-2 position of phospholipids in VEH-treated FLAMs and DHA-treated FLAMs, respectively (74), freeing these PUFAs for subsequent conversion into oxylipins inside the cell. A previously published study by Sager and coworkers (75) suggests that cSiO2 can induce expression of various PLA2 isozymes in the rat lungs, but the impacts of cSiO2 on PLA2 expression and activity remain unresearched at large. Additional studies in FLAMs involving genetic deletion or pharmacological inhibition of different PLA2 isozymes would help to clarify the role of PLA2 in cSiO2-induced oxylipin production.
LPS priming enhanced cSiO2-induced production of ARA-derived PGE2, LTB4, and TXB2 at both timepoints. Accordingly, our observations suggest that LPS priming may upregulate expression of COX and LOX in FLAMs, contributing to heightened production of classical proinflammatory eicosanoids after cSiO2 exposure. Previous studies have shown that LPS-triggered TLR4 activation in macrophages contributes to upregulation and activation of COX and LOX enzymes (76–80). In the context of inflammation and infection, classical eicosanoids are important modulators of the immune response. For instance, LTB4 in neutrophils promotes chemotaxis, enhances phagocytosis, triggers degranulation, and induces ROS production as antimicrobial defenses (81). Contrastingly, the immunoregulatory roles of PGE2 are more nuanced. While PGE2 has been previously shown to upregulate expression of proinflammatory cytokines (e.g., IL-1β, IL-6, IL-23) (82), PGE2 has also been shown to induce production of anti-inflammatory cytokines (e.g., IL-10, TGF-β) (83), suppress LPS-induced TNF-α production (84), inhibit bacterial phagocytosis (85), and stimulate M2-associated gene expression via transcription factor CREB (e.g., Arg1, Mrc1, Fizz1, Ym1) in macrophages (83, 86). Based on our study, LPS priming contributed to increased rates of lysosomal and mitochondrial integrity loss, but it is unclear whether augmented eicosanoid biosynthesis directly contributed to these observations, as proinflammatory cytokine production significantly increased after LPS priming.
In addition to promoting biosynthesis of classical proinflammatory eicosanoids, LPS priming also elicited release of the proinflammatory cytokines IL-1α, IL-1β, and TNF-α. It remains unclear whether LPS-stimulated proinflammatory cytokines (e.g., IL-1α, IL-1β, TNF-α) interact with their corresponding receptors (e.g., IL-1R, TNFR1) on neighboring FLAMs to stimulate production of ARA-derived oxylipins. Previous investigations suggest that certain proinflammatory cytokines, including IL-1β and TNF-α, can induce production of PGE2 and TXB2 in various contexts (87–89). In the future, it will be informative to either genetically knock out or pharmacologically inhibit proinflammatory cytokine receptors of interest to clarify the roles that cytokine-receptor signaling might play in influencing the cellular lipidome.
While numerous HFAs can be produced via the LOX or CYP450 enzymatic pathways (29, 90), HFAs can also be produced via non-enzymatic oxidation by reactive oxygen species (ROS) (91–93). Here, HFA levels were not significantly changed after LPS priming, which suggests these oxylipins may result from non-enzymatic conversion triggered by cSiO2 instead of conversion by LOX enzymes upregulated by LPS. Relatedly, cSiO2 caused steady declines in lysosomal and mitochondrial integrity that occurred at similar rates in VEH-treated and DHA-treated FLAMs and corresponded with increased HFA production. cSiO2 uptake by macrophages has been previously demonstrated to increase ROS levels in the cytoplasm and phagolysosome, resulting in LMP (94). Furthermore, mitochondrial depolarization has been shown to occur after cSiO2-induced LMP (9), and cSiO2 exposure has been linked to increased lysosomal, mitochondrial, and total cytosolic ROS production (95, 96). Although we did not directly measure ROS production in the present study, it is plausible that cSiO2-triggered HFA production in FLAMs is largely caused by non-enzymatic oxidation via ROS released from damaged lysosomes and mitochondria, as no subsets of HFAs were selectively produced in our oxylipin panel. Follow-up studies should aim to quantify total ROS and mitochondrial ROS produced from cSiO2-exposed FLAMs and utilize antioxidant agents (e.g., N-acetylcyteine, Trolox) to elucidate the impacts of ROS on HFA and total oxylipin production.
Interestingly, we found that DHA-treated FLAMs produced minimal SPMs, including RvD6 and MaR1ω-3 DPA, in response to LPS and cSiO2, which was unexpected because macrophages have been reported to biosynthesize a variety of SPMs in response to infectious agents and other inflammatory stimuli (97–99). We speculate that this occurred because the acute timepoints we selected may not adequately capture the kinetics of SPM biosynthesis. For instance, Dalli and coworkers demonstrated in zymosan-treated mice that MaR1ω-3 DPA was negligible at 4 h post-treatment but accumulated between 12-24 h post-treatment (100). In line with this conjecture, we also speculate that most of the administrated DHA was non-enzymatically converted to HDoHE metabolites following cSiO2 exposure, leaving insufficient quantities available for enzymatic conversion into various SPMs. It will therefore be important to extend the time-course in future experiments to better understand the kinetics of SPM production in our FLAM model.
We chose to focus our investigation on C57BL/6-derived FLAMs first because we recently characterized this model from a functional perspective and found that these cells are amenable to genetic modulation. This prompted us to assess whether this model was also amenable to lipidome modulation. Previously, we have demonstrated in female autoimmune-prone NZBWF1 mice that dietary DHA administered at human caloric equivalents of 2 or 5 g/d dose-dependently reduces perivascular leukocyte infiltration and expression of proinflammatory proteins in the lung (61, 70, 101). These changes correspond with increased levels of ω-3 PUFAs in erythrocytes and lungs; suppressed levels of cSiO2-induced inflammatory proteins and autoantibodies in bronchoalveolar lavage fluid (BALF) and plasma; and delayed onset of resultant glomerulonephritis and proteinuria. Our use of FLAMs from non-autoimmune C57BL/6 mice somewhat limits the translatability of the present study to other studies analyzing respirable cSiO2 as an autoimmune trigger in genetically-susceptible mice and humans. Nevertheless, developing a baseline oxylipin profile for C57BL/6 FLAMs will aid us in future investigations comparing effects of LPS, cSiO2, and DHA on the lipidome of FLAMs derived from autoimmune-prone mice (e.g., female NZBWF1 mice). Additional perspectives are needed to understand not only the influence of cSiO2 and DHA on the oxylipin signatures of non-autoimmune FLAMs and autoimmune-prone FLAMs but also on the lipidome of primary AMs and whole lung homogenates from non-autoimmune mice and autoimmune-prone mice.
Prior studies of protective effects of DHA against lysosomal toxicity, mitochondrial toxicity, and cell death might be highly dependent on cellular phenotype (27, 28, 102–107). As seen here, DHA did not inhibit cSiO2-induced LMP, mitochondrial toxicity, or cell death in FLAMs. DHA’s inability to protect against or even enhance cSiO2-triggered cell death in FLAMs suggests that these processes might not be critical targets in DHA-mediated lung protection. Nevertheless, cathepsin release and cell death may be vehicles by which cSiO2 drives production of pro-resolving DHA-derived oxylipins in FLAMs, as DHA-derived HDoHE levels rose at similar rates to ARA-derived HETE levels following cSiO2 exposure.
One limitation of our investigation is that intracellular and extracellular oxylipin content was pooled for all LC-MS analyses, making it impossible to discern quantities of secreted oxylipins from quantities of non-secreted oxylipins. By conducting LC-MS on separated cell cultures and supernatants, we would be able to better understand not only how cSiO2 impacts overall oxylipin production but also how cSiO2 impacts individual oxylipin release from FLAMs. Accordingly, prostaglandins, thromboxanes, leukotrienes, HFAs, and other subclasses of oxylipins elicit biological activity through transmembrane G protein-coupled receptors (GPCRs) and intracellular receptors such as PPARγ (108–112). While receptor-mediated biological effects have been reported—some proinflammatory and some pro-resolving—for numerous individual oxylipins (Supplementary Table 6), it is very likely that oxylipins elicit their biological activity as mixtures. To this end, it would be of interest to generate conditioned medium containing ARA-derived oxylipins and DHA-derived oxylipins from cSiO2-exposed VEH-treated FLAMs and cSiO2-exposed DHA-treated FLAMs, respectively, and then measure paracrine effects of the oxylipin mixtures on cSiO2-induced toxic responses in separate FLAM cultures.
FLAMs are not only useful for conducting broad genomic and lipidomic screens, but they may also be a useful AM surrogate for screening novel drug candidates in vitro. Accordingly, Hoffman and coworkers assessed the toxicity and efficacy of 13 known inhalable compounds using rat NR8383 and human U937-derived AM cell lines (113). This approach could easily be applied to our FLAM model to efficiently screen many drug candidates at once without needing to sacrifice large numbers of mice for primary AM isolation. Toxicity and efficacy results from preliminary drug screens in FLAMs could then be used to inform decisions regarding whether to discontinue or continue development of specific drugs in primary AMs, animal models, and ultimately human beings.
Conclusions
Figure 12 depicts potential mechanisms that underlie how DHA influences the cSiO2-induced eicosanoid storm, lysosomal permeability, and mitochondrial depolarization in FLAMs. To summarize, the results of the present study suggest that cSiO2 induces robust biosynthesis of ω-6 ARA-derived eicosanoids and DHA supplementation broadly skews the cSiO2-triggered lipidome from ω-6 eicosanoids to ω-3 DHA-derived docosanoids and ω-3 EPA-derived eicosanoids. cSiO2-triggered lipidome modulation corresponded with release of proinflammatory cytokines, loss of lysosomal/mitochondrial integrity, and cell death. DHA suppressed proinflammatory cytokine release but not LMP, mitochondrial toxicity, or cell death. LPS was required for proinflammatory cytokine release and modestly accelerated cSiO2-induced oxylipin production and loss of lysosomal/mitochondrial integrity. Together, these findings suggest that dietary ω-3 PUFAs may protect against cSiO2-triggered inflammatory lung disease by quelling proinflammatory ω-6 eicosanoid cascades and promoting biosynthesis of ω-3 oxylipins in lung AMs. Future investigations are necessary to characterize the lipidome in primary AMs and lung tissue from non-autoimmune and autoimmune-prone mice and relate oxylipin profiles to biomarkers of cSiO2-induced toxicity and inflammatory lung disease.
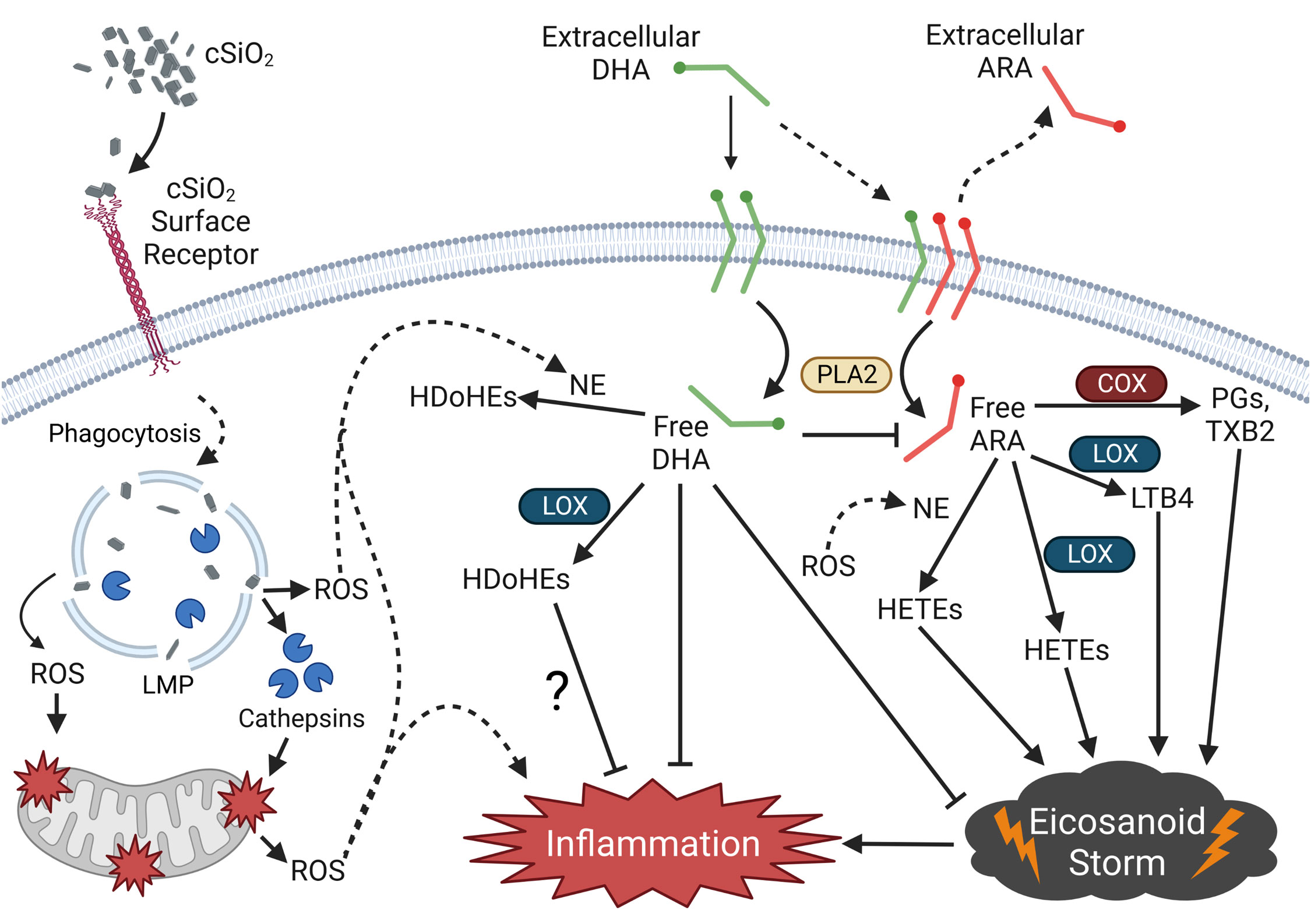
Figure 12 Putative model for the effects of DHA on the cSiO2-triggered proinflammatory eicosanoid storm in the FLAM. cSiO2 binds a cSiO2-specific surface receptor and is phagocytosed by the FLAM. Following phagocytosis, cSiO2 triggers lysosomal membrane permeabilization (LMP), causing release of lysosomal proteolytical cathepsins and reactive oxygen species (ROS). Lysosomal cathepsin release and ROS elicit mitochondrial membrane depolarization and further ROS release into the cytosol. cSiO2 also triggers phospholipase A2 (PLA2)-mediated release of the ω-6 polyunsaturated fatty acid (PUFA) arachidonic acid (ARA) from the plasma membrane, freeing it for enzymatic and non-enzymatic conversion to various proinflammatory eicosanoids, resulting in an “eicosanoid storm.” Pre-incubation of FLAMs with DHA causes displacement of ARA from the plasma membrane, thereby allowing PLA2-mediated release of DHA into the cytosol following cSiO2 exposure. Cytosolic DHA competes with ARA as a substrate of enzymatic and non-enzymatic oxylipin production, ultimately leading to generation of pro-resolving docosanoids, suppression of the eicosanoid storm, and reduced inflammation. Created with BioRender.com. cSiO2, crystalline silica; COX, cyclooxygenase; LOX, lipoxygenase; NE, non-enzymatic conversion; PGs, prostaglandins; TXB2, thromboxane B2; LTB4, leukotriene B4; HETEs, hydroxyeicosatetraenoic acids; HDoHEs, hydroxydocosahexaenoic acid.
Data availability statement
The data presented in the study are deposited in the Dryad repository. This data can be found here: https://doi.org/10.5061/dryad.w3r2280wn.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee at Michigan State University (MSU) (Animal Use Form [AUF] #PROTO201800113). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
OF: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review and editing. LR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review and editing. KW: Conceptualization, Investigation, Methodology, Validation, Writing – review and editing. KM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Writing – review and editing. KL: Methodology, Project administration, Resources, Writing – review and editing. AO: Conceptualization, Investigation, Methodology, Resources, Writing – review and editing. JP: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by NIH RO1ES027353 (JP), Lupus Foundation of America (JP), Dr. Robert and Carol Deibel Family Endowment (JP), NIH R03AG075465 (KSSL), MSU Discretionary Funding Initiative (KSSL), NIH T32GM142521 (OF), and in part by NIH/NCRR grant S10RR027926 (KM).
Acknowledgments
We would like to thank Adrianna Kirby at Michigan State University for her technical assistance with in vitro FLAM assays, the Lipidomics Core Facility at Wayne State University for their assistance with oxylipin and lipidomic analyses, and Dr. Charles Serhan at Harvard Medical School for providing experimental expertise on processing cellular samples for lipidomic analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1274147/full#supplementary-material
Abbreviations
AM, alveolar macrophage; ARA, arachidonic acid; COX, cyclooxygenase; cSiO2, crystalline silica; CYP450, cytochrome P450; DHA, docosahexaenoic acid; DiHFA, dihydroxy fatty acid; EPA, eicosapentaenoic acid; EpFA, epoxy fatty acid; FLAM, fetal liver-derived alveolar-like macrophage; HFA, hydroxy fatty acid; LOX, lipoxygenase; OA, oleic acid.
References
1. OSHA. Workers’ exposure to respirable crystalline silica: Final rule overview (2016). Available at: https://www.osha.gov/Publications/OSHA3683.pdf (Accessed 6/2/2020).
2. Finckh A, Cooper GS, Chibnik LB, Costenbader KH, Watts J, Pankey H, et al. Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheum (2006) 54(11):3648–54. doi: 10.1002/art.22210
3. Parks CG, De Roos AJ. Pesticides, chemical and industrial exposures in relation to systemic lupus erythematosus. Lupus (2014) 23(6):527–36. doi: 10.1177/0961203313511680
4. Farhat SC, Silva CA, Orione MA, Campos LM, Sallum AM, Braga AL. Air pollution in autoimmune rheumatic diseases: A review. Autoimmun Rev (2011) 11(1):14–21. doi: 10.1016/j.autrev.2011.06.008
5. OSHA. Silica, crystalline (2016). Available at: https://www.osha.gov/dsg/topics/silicacrystalline/index.html (Accessed 6/1/2020).
6. Hoy RF, Chambers DC. Silica-related diseases in the modern world. Allergy (2020) 75(11):2805–17. doi: 10.1111/all.14202
7. Boudigaard SH, Schlunssen V, Vestergaard JM, Sondergaard K, Toren K, Peters S, et al. Occupational exposure to respirable crystalline silica and risk of autoimmune rheumatic diseases: A nationwide cohort study. Int J Epidemiol (2021) 50(4):1213–26. doi: 10.1093/ije/dyaa287
8. Joshi N, Walter JM, Misharin AV. Alveolar macrophages. Cell Immunol (2018) 330:86–90. doi: 10.1016/j.cellimm.2018.01.005
9. Joshi GN, Knecht DA. Silica phagocytosis causes apoptosis and necrosis by different temporal and molecular pathways in alveolar macrophages. Apoptosis (2013) 18(3):271–85. doi: 10.1007/s10495-012-0798-y
10. Hindman B, Ma Q. Carbon nanotubes and crystalline silica stimulate robust ros production, inflammasome activation, and il-1beta secretion in macrophages to induce myofibroblast transformation. Arch Toxicol (2019) 93(4):887–907. doi: 10.1007/s00204-019-02411-y
11. Lundstrom SL, Balgoma D, Wheelock AM, Haeggstrom JZ, Dahlen SE, Wheelock CE. Lipid mediator profiling in pulmonary disease. Curr Pharm Biotechnol (2011) 12(7):1026–52. doi: 10.2174/138920111795909087
12. Wang B, Wu L, Chen J, Dong L, Chen C, Wen Z, et al. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct Target Ther (2021) 6(1):94. doi: 10.1038/s41392-020-00443-w
13. Claar D, Hartert TV, Peebles RS Jr. The role of prostaglandins in allergic lung inflammation and asthma. Expert Rev Respir Med (2015) 9(1):55–72. doi: 10.1586/17476348.2015.992783
14. Teopompi E, Rise P, Pisi R, Buccellati C, Aiello M, Pisi G, et al. Arachidonic acid and docosahexaenoic acid metabolites in the airways of adults with cystic fibrosis: Effect of docosahexaenoic acid supplementation. Front Pharmacol (2019) 10:938. doi: 10.3389/fphar.2019.00938
15. Hammock BD, Wang W, Gilligan MM, Panigrahy D. Eicosanoids: The overlooked storm in coronavirus disease 2019 (covid-19)? Am J Pathol (2020) 190(9):1782–8. doi: 10.1016/j.ajpath.2020.06.010
16. Mazidimoradi A, Alemzadeh E, Alemzadeh E, Salehiniya H. The effect of polyunsaturated fatty acids on the severity and mortality of covid patients: A systematic review. Life Sci (2022) 299. doi: 10.1016/j.lfs.2022.120489
17. Mohammed A, Kalle AM, Reddanna P. Managing sars-cov2 infections through resolution of inflammation by eicosanoids: A review. J Inflamm Res (2022) 15:4349–58. doi: 10.2147/jir.S355568
18. Dubois CM, Bissonnette E, Rola-Pleszczynski M. Asbestos fibers and silica particles stimulate rat alveolar macrophages to release tumor necrosis factor. Autoregulatory role of leukotriene b4. Am Rev Respir Dis (1989) 139(5):1257–64. doi: 10.1164/ajrccm/139.5.1257
19. Garcia JG, Griffith DE, Cohen AB, Callahan KS. Alveolar macrophages from patients with asbestos exposure release increased levels of leukotriene b4. Am Rev Respir Dis (1989) 139(6):1494–501. doi: 10.1164/ajrccm/139.6.1494
20. Koren HS, Joyce M, Devlin RB, Becker S, Driscoll K, Madden MC. Modulation of eicosanoid production by human alveolar macrophages exposed to silica in vitro. Environ Health Perspect (1992) 97:77–83. doi: 10.1289/ehp.929777
21. Mohr C, Davis GS, Graebner C, Hemenway DR, Gemsa D. Enhanced release of prostaglandin e2 from macrophages of rats with silicosis. Am J Respir Cell Mol Biol (1992) 6(4):390–6. doi: 10.1165/ajrcmb/6.4.390
22. Pang J, Qi X, Luo Y, Li X, Shu T, Li B, et al. Multi-omics study of silicosis reveals the potential therapeutic targets pgd(2) and txa(2). Theranostics (2021) 11(5):2381–94. doi: 10.7150/thno.47627
23. Wierenga KA, Strakovsky RS, Benninghoff AD, Rajasinghe LD, Lock AL, Harkema JR, et al. Requisite omega-3 hufa biomarker thresholds for preventing murine lupus flaring. Front Immunol (2020) 11:1796(1796). doi: 10.3389/fimmu.2020.01796
24. Favor OK, Pestka JJ, Bates MA, Lee KSS. Centrality of myeloid-lineage phagocytes in particle-triggered inflammation and autoimmunity. Front Toxicol (2021) 3:777768(51). doi: 10.3389/ftox.2021.777768
25. Calder PC. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem Soc Trans (2017) 45(5):1105–15. doi: 10.1042/BST20160474
26. Gutierrez S, Svahn SL, Johansson ME. Effects of omega-3 fatty acids on immune cells. Int J Mol Sci (2019) 20(20). doi: 10.3390/ijms20205028
27. Wierenga KA, Wee J, Gilley KN, Rajasinghe LD, Bates MA, Gavrilin MA, et al. Docosahexaenoic acid suppresses silica-induced inflammasome activation and il-1 cytokine release by interfering with priming signal. Front Immunol (2019) 10:2130. doi: 10.3389/fimmu.2019.02130
28. Rajasinghe LD, Chauhan PS, Wierenga KA, Evered AO, Harris SN, Bates MA, et al. Omega-3 docosahexaenoic acid (dha) impedes silica-induced macrophage corpse accumulation by attenuating cell death and potentiating efferocytosis. Front Immunol (2020) 11:2179. doi: 10.3389/fimmu.2020.02179
29. Kuda O. Bioactive metabolites of docosahexaenoic acid. Biochimie (2017) 136:12–20. doi: 10.1016/j.biochi.2017.01.002
30. Schuchardt JP, Ostermann AI, Stork L, Fritzsch S, Kohrs H, Greupner T, et al. Effect of dha supplementation on oxylipin levels in plasma and immune cell stimulated blood. Prostaglandins Leukot Essent Fatty Acids (2017) 121:76–87. doi: 10.1016/j.plefa.2017.06.007
31. Ostermann AI, West AL, Schoenfeld K, Browning LM, Walker CG, Jebb SA, et al. Plasma oxylipins respond in a linear dose-response manner with increased intake of epa and dha: Results from a randomized controlled trial in healthy humans. Am J Clin Nutr (2019) 109(5):1251–63. doi: 10.1093/ajcn/nqz016
32. Calder PC. N-3 pufa and inflammation: From membrane to nucleus and from bench to bedside. Proc Nutr Soc (2020) 79(4):1–13. doi: 10.1017/S0029665120007077
33. Hartling I, Cremonesi A, Osuna E, Lou PH, Lucchinetti E, Zaugg M, et al. Quantitative profiling of inflammatory and pro-resolving lipid mediators in human adolescents and mouse plasma using uhplc-ms/ms. Clin Chem Lab Med (2021) 59(11):1811–23. doi: 10.1515/cclm-2021-0644
34. Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol (2011) 89(4):557–63. doi: 10.1189/jlb.0710409
35. Lazarov T, Juarez-Carreno S, Cox N, Geissmann F. Physiology and diseases of tissue-resident macrophages. Nature (2023) 618(7966):698–707. doi: 10.1038/s41586-023-06002-x
36. Wang C, Yu X, Cao Q, Wang Y, Zheng G, Tan TK, et al. Characterization of murine macrophages from bone marrow, spleen and peritoneum. BMC Immunol (2013) 14(1):6. doi: 10.1186/1471-2172-14-6
37. Cassado Ados A, D'Imperio Lima MR, Bortoluci KR. Revisiting mouse peritoneal macrophages: Heterogeneity, development, and function. Front Immunol (2015) 6:225. doi: 10.3389/fimmu.2015.00225
38. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol (2013) 49(4):503–10. doi: 10.1165/rcmb.2013-0086MA
39. Busch CJ, Favret J, Geirsdóttir L, Molawi K, Sieweke MH. Isolation and long-term cultivation of mouse alveolar macrophages. Bio Protoc (2019) 9(14). doi: 10.21769/BioProtoc.3302
40. McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M, et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J Clin Invest (2021) 131(4). doi: 10.1172/JCI140299
41. Gorki AD, Symmank D, Zahalka S, Lakovits K, Hladik A, Langer B, et al. Murine ex vivo cultured alveolar macrophages provide a novel tool to study tissue-resident macrophage behavior and function. Am J Respir Cell Mol Biol (2022) 66(1):64–75. doi: 10.1165/rcmb.2021-0190OC
42. Subramanian S, Busch CJ-L, Molawi K, Geirsdottir L, Maurizio J, Vargas Aguilar S, et al. Long-term culture-expanded alveolar macrophages restore their full epigenetic identity after transfer in vivo. Nat Immunol (2022) 23(3):458–68. doi: 10.1038/s41590-022-01146-w
43. Thomas ST, Wierenga KA, Pestka JJ, Olive AJ. Fetal liver-derived alveolar-like macrophages: A self-replicating ex vivo model of alveolar macrophages for functional genetic studies. Immunohorizons (2022) 6(2):156–69. doi: 10.4049/immunohorizons.2200011
44. Fejer G, Wegner MD, Gyory I, Cohen I, Engelhard P, Voronov E, et al. Nontransformed, gm-csf-dependent macrophage lines are a unique model to study tissue macrophage functions. Proc Natl Acad Sci U.S.A. (2013) 110(24):E2191–2198. doi: 10.1073/pnas.1302877110
45. Wierenga KA, Riemers FM, Westendorp B, Harkema JR, Pestka JJ. Single cell analysis of docosahexaenoic acid suppression of sequential lps-induced proinflammatory and interferon-regulated gene expression in the macrophage. Front Immunol (2022) 13:993614. doi: 10.3389/fimmu.2022.993614
46. Rand AA, Helmer PO, Inceoglu B, Hammock BD, Morisseau C. Lc-ms/ms analysis of the epoxides and diols derived from the endocannabinoid arachidonoyl ethanolamide. Methods Mol Biol (2018) 1730:123–33. doi: 10.1007/978-1-4939-7592-1_10
47. Markworth JF, Kaur G, Miller EG, Larsen AE, Sinclair AJ, Maddipati KR, et al. Divergent shifts in lipid mediator profile following supplementation with n-3 docosapentaenoic acid and eicosapentaenoic acid. FASEB J (2016) 30(11):3714–25. doi: 10.1096/fj.201600360R
48. Lamon-Fava S, So J, Mischoulon D, Ziegler TR, Dunlop BW, Kinkead B, et al. Dose- and time-dependent increase in circulating anti-inflammatory and pro-resolving lipid mediators following eicosapentaenoic acid supplementation in patients with major depressive disorder and chronic inflammation. Prostaglandins Leukot Essent Fatty Acids (2021) 164:102219. doi: 10.1016/j.plefa.2020.102219
49. So J, Wu D, Lichtenstein AH, Tai AK, Matthan NR, Maddipati KR, et al. Epa and dha differentially modulate monocyte inflammatory response in subjects with chronic inflammation in part via plasma specialized pro-resolving lipid mediators: A randomized, double-blind, crossover study. Atherosclerosis (2021) 316:90–8. doi: 10.1016/j.atherosclerosis.2020.11.018
50. Favor OK, Wierenga KA, Rajasinghe LD, Maddipati KR, Olive A, Pestka JJ. Data from: Omega-3 docosahexaenoic acid suppresses silica-induced proinflammatory cytokine release and oxylipin mediator production in novel fetal liver-derived alveolar-like macrophages. Dryad (2023). doi: 10.5061/dryad.w3r2280wn
51. Sager TM, Wolfarth M, Leonard SS, Morris AM, Porter DW, Castranova V, et al. Role of engineered metal oxide nanoparticle agglomeration in reactive oxygen species generation and cathepsin b release in nlrp3 inflammasome activation and pulmonary toxicity. Inhal Toxicol (2016) 28(14):686–97. doi: 10.1080/08958378.2016.1257664
52. Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. Cellprofiler 4: Improvements in speed, utility and usability. BMC Bioinf (2021) 22(1):433. doi: 10.1186/s12859-021-04344-9
53. Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, et al. Using metaboanalyst 5.0 for lc-hrms spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc (2022) 17(8):1735–61. doi: 10.1038/s41596-022-00710-w
54. Wiesenfeld PW, Babu US, O'Donnell MW. Effect of long-chain fatty acids in the culture medium on fatty acid composition of wehi-3 and j774a.1 cells. Comp Biochem Physiol B Biochem Mol Biol (2001) 128(1):123–34. doi: 10.1016/s1096-4959(00)00305-5
55. Else PL. The highly unnatural fatty acid profile of cells in culture. Prog Lipid Res (2020) 77:101017. doi: 10.1016/j.plipres.2019.101017
56. Basil MC, Levy BD. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat Rev Immunol (2016) 16(1):51–67. doi: 10.1038/nri.2015.4
57. Chiang N, Serhan CN. Specialized pro-resolving mediator network: An update on production and actions. Essays Biochem (2020) 64(3):443–62. doi: 10.1042/EBC20200018
58. Cagnina RE, Duvall MG, Nijmeh J, Levy BD. Specialized pro-resolving mediators in respiratory diseases. Curr Opin Clin Nutr Metab Care (2022) 25(2):67–74. doi: 10.1097/MCO.0000000000000805
59. Woo YD, Jeong D, Chung DH. Development and functions of alveolar macrophages. Mol Cells (2021) 44(5):292–300. doi: 10.14348/molcells.2021.0058
60. Hamilton RF Jr, Thakur SA, Holian A. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med (2008) 44(7):1246–58. doi: 10.1016/j.freeradbiomed.2007.12.027
61. Bates MA, Brandenberger C, Langohr II, Kumagai K, Lock AL, Harkema JR, et al. Silica-triggered autoimmunity in lupus-prone mice blocked by docosahexaenoic acid consumption. PloS One (2016) 11(8):e0160622. doi: 10.1371/journal.pone.0160622
62. Morin C, Blier PU, Fortin S. Eicosapentaenoic acid and docosapentaenoic acid monoglycerides are more potent than docosahexaenoic acid monoglyceride to resolve inflammation in a rheumatoid arthritis model. Arthritis Res Ther (2015) 17(1):142. doi: 10.1186/s13075-015-0653-y
63. Dawczynski C, Dittrich M, Neumann T, Goetze K, Welzel A, Oelzner P, et al. Docosahexaenoic acid in the treatment of rheumatoid arthritis: A double-blind, placebo-controlled, randomized cross-over study with microalgae vs. Sunflower oil. Clin Nutr (2018) 37(2):494–504. doi: 10.1016/j.clnu.2017.02.021
64. Charoenwoodhipong P, Harlow SD, Marder W, Hassett AL, McCune WJ, Gordon C, et al. Dietary omega polyunsaturated fatty acid intake and patient-reported outcomes in systemic lupus erythematosus: The michigan lupus epidemiology and surveillance program. Arthritis Care Res (Hoboken) (2020) 72(7):874–81. doi: 10.1002/acr.23925
65. Khan RS, Chokshi A, Drosatos K, Jiang H, Yu S, Harris CR, et al. Fish oil selectively improves heart function in a mouse model of lipid-induced cardiomyopathy. J Cardiovasc Pharmacol (2013) 61(4):345–54. doi: 10.1097/FJC.0b013e318283d845
66. Kris-Etherton PM, Richter CK, Bowen KJ, Skulas-Ray AC, Jackson KH, Petersen KS, et al. Recent clinical trials shed new light on the cardiovascular benefits of omega-3 fatty acids. Methodist Debakey Cardiovasc J (2019) 15(3):171–8. doi: 10.14797/mdcj-15-3-171
67. Calder PC. Very long-chain n-3 fatty acids and human health: Fact, fiction and the future. Proc Nutr Soc (2018) 77(1):52–72. doi: 10.1017/S0029665117003950
68. Serhan CN, Levy BD. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J Clin Invest (2018) 128(7):2657–69. doi: 10.1172/JCI97943
69. Efsa Panel on Dietetic Products, N., and Allergies. Scientific opinion on the tolerable upper intake level of eicosapentaenoic acid (epa), docosahexaenoic acid (dha) and docosapentaenoic acid (dpa). EFSA J (2012) 10(7):2815. doi: 10.2903/j.efsa.2012.2815
70. Bates MA, Akbari P, Gilley KN, Wagner JG, Li N, Kopec AK, et al. Dietary docosahexaenoic acid prevents silica-induced development of pulmonary ectopic germinal centers and glomerulonephritis in the lupus-prone nzbwf1 mouse. Front Immunol (2018) 9:2002. doi: 10.3389/fimmu.2018.02002
71. Simopoulos AP. An increase in the omega-6/omega-3 fatty acid ratio increases the risk for obesity. Nutrients (2016) 8(3):128. doi: 10.3390/nu8030128
72. Lands B. Omega-3 pufas lower the propensity for arachidonic acid cascade overreactions. BioMed Res Int (2015) 2015:285135. doi: 10.1155/2015/285135
73. Sarparast M, Pourmand E, Hinman J, Vonarx D, Reason T, Zhang F, et al. Dihydroxy-metabolites of dihomo-gamma-linolenic acid drive ferroptosis-mediated neurodegeneration. bioRxiv (2023). doi: 10.1101/2023.01.05.522933
74. Mouchlis VD, Dennis EA. Phospholipase a(2) catalysis and lipid mediator lipidomics. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(6):766–71. doi: 10.1016/j.bbalip.2018.08.010
75. Sager TM, Umbright CM, Mustafa GM, Yanamala N, Leonard HD, McKinney WG, et al. Tobacco smoke exposure exacerbated crystalline silica-induced lung toxicity in rats. Toxicol Sci (2020) 178(2):375–90. doi: 10.1093/toxsci/kfaa146
76. Wilborn J, DeWitt DL, Peters-Golden M. Expression and role of cyclooxygenase isoforms in alveolar and peritoneal macrophages. Am J Physiol (1995) 268(2 Pt 1):L294–301. doi: 10.1152/ajplung.1995.268.2.L294
77. Wuest SJ, Crucet M, Gemperle C, Loretz C, Hersberger M. Expression and regulation of 12/15-lipoxygenases in human primary macrophages. Atherosclerosis (2012) 225(1):121–7. doi: 10.1016/j.atherosclerosis.2012.07.022
78. Abrial C, Grassin-Delyle S, Salvator H, Brollo M, Naline E, Devillier P. 15-lipoxygenases regulate the production of chemokines in human lung macrophages. Br J Pharmacol (2015) 172(17):4319–30. doi: 10.1111/bph.13210
79. Lee SJ, Seo KW, Kim CD. Lps increases 5-lo expression on monocytes via an activation of akt-sp1/nf-kappab pathways. Korean J Physiol Pharmacol (2015) 19(3):263–8. doi: 10.4196/kjpp.2015.19.3.263
80. Vijayan V, Srinu T, Karnati S, Garikapati V, Linke M, Kamalyan L, et al. A new immunomodulatory role for peroxisomes in macrophages activated by the tlr4 ligand lipopolysaccharide. J Immunol (2017) 198(6):2414–25. doi: 10.4049/jimmunol.1601596
81. He R, Chen Y, Cai Q. The role of the ltb4-blt1 axis in health and disease. Pharmacol Res (2020) 158:104857. doi: 10.1016/j.phrs.2020.104857
82. Vallerie SN, Kramer F, Barnhart S, Kanter JE, Breyer RM, Andreasson KI, et al. Myeloid cell prostaglandin e2 receptor ep4 modulates cytokine production but not atherogenesis in a mouse model of type 1 diabetes. PloS One (2016) 11(6):e0158316. doi: 10.1371/journal.pone.0158316
83. Burkett JB, Doran AC, Gannon M. Harnessing prostaglandin e(2) signaling to ameliorate autoimmunity. Trends Immunol (2023) 44(3):162–71. doi: 10.1016/j.it.2023.01.004
84. Miles EA, Allen E, Calder PC. In vitro effects of eicosanoids derived from different 20-carbon fatty acids on production of monocyte-derived cytokines in human whole blood cultures. Cytokine (2002) 20(5):215–23. doi: 10.1006/cyto.2002.2007
85. Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin e2 inhibits alveolar macrophage phagocytosis through an e-prostanoid 2 receptor-mediated increase in intracellular cyclic amp. J Immunol (2004) 173(1):559–65. doi: 10.4049/jimmunol.173.1.559
86. Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. Creb pathway links pge2 signaling with macrophage polarization. Proc Natl Acad Sci USA (2015) 112(51):15642–7. doi: 10.1073/pnas.1519644112
87. Conti P, Cifone MG, Alesse E, Reale M, Fieschi C, Dinarello CA. In vitro enhanced thromboxane b2 release by polymorphonuclear leukocytes and macrophages after treatment with human recombinant interleukin 1. Prostaglandins (1986) 32(1):111–5. doi: 10.1016/0090-6980(86)90151-6
88. Smith GS, Rieckenberg C, Longo WE, Kaminski DL, Mazuski JE, Deshpande Y, et al. The effect of an interleukin receptor antagonist (il-1ra) on colonocyte eicosanoid release. Mediators Inflammation (1996) 5(6):449–52. doi: 10.1155/S0962935196000622
89. Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of cox-2 and pge(2) biosynthesis by il-1beta is mediated by pkc and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol (2000) 131(1):152–9. doi: 10.1038/sj.bjp.0703557
90. Massey KA, Nicolaou A. Lipidomics of oxidized polyunsaturated fatty acids. Free Radic Biol Med (2013) 59(100):45–55. doi: 10.1016/j.freeradbiomed.2012.08.565
91. Derogis PB, Freitas FP, Marques AS, Cunha D, Appolinario PP, de Paula F, et al. The development of a specific and sensitive lc-ms-based method for the detection and quantification of hydroperoxy- and hydroxydocosahexaenoic acids as a tool for lipidomic analysis. PloS One (2013) 8(10):e77561. doi: 10.1371/journal.pone.0077561
92. Fischer R, Konkel A, Mehling H, Blossey K, Gapelyuk A, Wessel N, et al. Dietary omega-3 fatty acids modulate the eicosanoid profile in man primarily via the cyp-epoxygenase pathway. J Lipid Res (2014) 55(6):1150–64. doi: 10.1194/jlr.M047357
93. Mattmiller SA, Carlson BA, Gandy JC, Sordillo LM. Reduced macrophage selenoprotein expression alters oxidized lipid metabolite biosynthesis from arachidonic and linoleic acid. J Nutr Biochem (2014) 25(6):647–54. doi: 10.1016/j.jnutbio.2014.02.005
94. Joshi GN, Goetjen AM, Knecht DA. Silica particles cause nadph oxidase-independent ros generation and transient phagolysosomal leakage. Mol Biol Cell (2015) 26(18):3150–64. doi: 10.1091/mbc.E15-03-0126
95. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ros-induced ros release: An update and review. Biochim Biophys Acta (2006) 1757(5-6):509–17. doi: 10.1016/j.bbabio.2006.04.029
96. Fazzi F, Njah J, Di Giuseppe M, Winnica DE, Go K, Sala E, et al. Tnfr1/phox interaction and tnfr1 mitochondrial translocation thwart silica-induced pulmonary fibrosis. J Immunol (2014) 192(8):3837–46. doi: 10.4049/jimmunol.1103516
97. Thatcher TH, Woeller CF, McCarthy CE, Sime PJ. Quenching the fires: Pro-resolving mediators, air pollution, and smoking. Pharmacol Ther (2019) 197:212–24. doi: 10.1016/j.pharmthera.2019.02.001
98. Dyall SC, Balas L, Bazan NG, Brenna JT, Chiang N, da Costa Souza F, et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog Lipid Res (2022) 86:101165. doi: 10.1016/j.plipres.2022.101165
99. Jordan PM, Werz O. Specialized pro-resolving mediators: Biosynthesis and biological role in bacterial infections. FEBS J (2022) 289(14):4212–27. doi: 10.1111/febs.16266
100. Dalli J, Colas RA, Serhan CN. Novel n-3 immunoresolvents: Structures and actions. Sci Rep (2013) 3:1940. doi: 10.1038/srep01940
101. Benninghoff AD, Bates MA, Chauhan PS, Wierenga KA, Gilley KN, Holian A, et al. Docosahexaenoic acid consumption impedes early interferon- and chemokine-related gene expression while suppressing silica-triggered flaring of murine lupus. Front Immunol (2019) 10:2851(2851). doi: 10.3389/fimmu.2019.02851
102. Stillwell W, Jenski LJ, Crump FT, Ehringer W. Effect of docosahexaenoic acid on mouse mitochondrial membrane properties. Lipids (1997) 32(5):497–506. doi: 10.1007/s11745-997-0064-6
103. Ng Y, Barhoumi R, Tjalkens RB, Fan YY, Kolar S, Wang N, et al. The role of docosahexaenoic acid in mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis (2005) 26(11):1914–21. doi: 10.1093/carcin/bgi163
104. Pacheco FJ, Almaguel FG, Evans W, Rios-Colon L, Filippov V, Leoh LS, et al. Docosahexanoic acid antagonizes tnf-alpha-induced necroptosis by attenuating oxidative stress, ceramide production, lysosomal dysfunction, and autophagic features. Inflammation Res (2014) 63(10):859–71. doi: 10.1007/s00011-014-0760-2
105. Chen CY, Chen CY, Liu CC, Chen CP. Omega-3 polyunsaturated fatty acids reduce preterm labor by inhibiting trophoblast cathepsin s and inflammasome activation. Clin Sci (Lond) (2018) 132(20):2221–39. doi: 10.1042/CS20180796
106. Fletcher P, Hamilton RF Jr., Rhoderick JF, Pestka JJ, Holian A. Docosahexaenoic acid impacts macrophage phenotype subsets and phagolysosomal membrane permeability with particle exposure. J Toxicol Environ Health A (2021) 84(4):152–72. doi: 10.1080/15287394.2020.1842826
107. Li G, Li Y, Xiao B, Cui D, Lin Y, Zeng J, et al. Antioxidant activity of docosahexaenoic acid (dha) and its regulatory roles in mitochondria. J Agric Food Chem (2021) 69(5):1647–55. doi: 10.1021/acs.jafc.0c07751
108. Powell WS, Rokach J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (hetes) and oxoeicosatetraenoic acids (oxo-etes) derived from arachidonic acid. Biochim Biophys Acta (2015) 1851(4):340–55. doi: 10.1016/j.bbalip.2014.10.008
109. Milligan G, Shimpukade B, Ulven T, Hudson BD. Complex pharmacology of free fatty acid receptors. Chem Rev (2017) 117(1):67–110. doi: 10.1021/acs.chemrev.6b00056
110. Biringer RG. A review of prostanoid receptors: Expression, characterization, regulation, and mechanism of action. J Cell Commun Signal (2021) 15(2):155–84. doi: 10.1007/s12079-020-00585-0
111. Biringer RG. A review of non-prostanoid, eicosanoid receptors: Expression, characterization, regulation, and mechanism of action. J Cell Commun Signal (2022) 16(1):5–46. doi: 10.1007/s12079-021-00630-6
112. Liu J, Sahin C, Ahmad S, Magomedova L, Zhang M, Jia Z, et al. The omega-3 hydroxy fatty acid 7(s)-hdha is a high-affinity pparalpha ligand that regulates brain neuronal morphology. Sci Signal (2022) 15(741):eabo1857. doi: 10.1126/scisignal.abo1857
Keywords: crystalline silica, docosahexaenoic acid, alveolar macrophage, lipidome, oxylipin
Citation: Favor OK, Rajasinghe LD, Wierenga KA, Maddipati KR, Lee KSS, Olive AJ and Pestka JJ (2023) Crystalline silica-induced proinflammatory eicosanoid storm in novel alveolar macrophage model quelled by docosahexaenoic acid supplementation. Front. Immunol. 14:1274147. doi: 10.3389/fimmu.2023.1274147
Received: 07 August 2023; Accepted: 16 October 2023;
Published: 09 November 2023.
Edited by:
Philip Calder, University of Southampton, United KingdomReviewed by:
Andrij Holian, University of Montana, United StatesHong Yong Peh, Brigham and Women’s Hospital and Harvard Medical School, United States
Copyright © 2023 Favor, Rajasinghe, Wierenga, Maddipati, Lee, Olive and Pestka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivia K. Favor, favoroli@msu.edu; James J. Pestka, pestka@msu.edu