 Renan Muniz-Santos1
Renan Muniz-Santos1 Giovanna Lucieri-Costa1
Giovanna Lucieri-Costa1 Matheus Augusto P. de Almeida2,3
Matheus Augusto P. de Almeida2,3 Isabelle Moraes-de-Souza1
Isabelle Moraes-de-Souza1 Maria Alice Dos Santos Mascarenhas Brito1
Maria Alice Dos Santos Mascarenhas Brito1 Adriana Ribeiro Silva3
Adriana Ribeiro Silva3 Cassiano Felippe Gonçalves-de-Albuquerque1,2,3*
Cassiano Felippe Gonçalves-de-Albuquerque1,2,3*- 1Laboratory of Immunopharmacology, Department of Physiology, Federal University of the State of Rio de Janeiro, Rio de Janeiro, Brazil
- 2Neuroscience Graduate Program, Federal Fluminense University, Niteroi, Brazil
- 3Laboratory of Immunopharmacology, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil
Sepsis is a life-threatening organ dysfunction caused by abnormal host response to infection. Millions of people are affected annually worldwide. Derangement of the inflammatory response is crucial in sepsis pathogenesis. However, metabolic, coagulation, and thermoregulatory alterations also occur in patients with sepsis. Fatty acid mobilization and oxidation changes may assume the role of a protagonist in sepsis pathogenesis. Lipid oxidation and free fatty acids (FFAs) are potentially valuable markers for sepsis diagnosis and prognosis. Herein, we discuss inflammatory and metabolic dysfunction during sepsis, focusing on fatty acid oxidation (FAO) alterations in the liver and muscle (skeletal and cardiac) and their implications in sepsis development.
1 Introduction
Sepsis is a Greek-derived term that initially meant “decomposition of animal or vegetable organic matter in the presence of bacteria.” This has been documented in Homer’s Iliad and Corpus Hippocraticum (1). Over the last 120 years, sepsis has received different modern definitions, most related to the interaction between pathogenic microorganisms and immune host defenses (2–5). Sepsis is defined “as life-threatening organ dysfunction caused by a dysregulated host response to infection” (6).
Sepsis is the most common cause of death by multiple organ dysfunction in critically ill patients, and, as an infectious condition, sepsis can be bacterial, viral, or fungal (7–9). Sepsis affects 1.7 million adults annually in the United States, and half of all sepsis cases are caused by gram-negative bacteria, making septic shock a common complication (10, 11). In addition, gram-positive bacteria cause septic shock. We also must consider that many studies use lipopolysaccharide (LPS), a component of gram-negative bacteria, which causes endotoxemia, but not sepsis, but can be biased considering gram-negative sepsis. More than 40 million annual cases are estimated to occur worldwide (12). Detecting the origin of sepsis is crucial in clinical practice because it presents different pathophysiologies, clinical patterns, and treatment approaches (13, 14).
To assess sepsis severity, the Sequential Organ Failure Assessment (SOFA) score evaluates six organ systems: the respiratory, cardiovascular, hepatic, coagulation, renal, and neurological systems (15, 16). Dysregulation of lipid oxidation plays a significant role in organ damage during sepsis, and studies have indicated a positive correlation between markers of lipid oxidation dysfunction and SOFA score. Serum malondialdehyde (MDA) and free fatty acids (FFA) levels also correlated with higher SOFA scores (17). For example, serum MDA is a marker of lipid peroxidation and oxidative stress that correlates with SOFA score and lactatemia (18). Recent research has also shown connections between MDA levels and SOFA score, as well as with APACHE-II and coagulation indices (19). In addition, a correlation between higher FFA levels and patient SOFA scores has been reported (20). Recent research has identified a correlation between SOFA score and various lipid metabolites involved in different pathways (21). Lipid metabolism dysfunction can cause muscle and liver damage but can also affect other systems evaluated by the SOFA score.
Inflammation is fundamental to sepsis pathogenesis but does not fully explain progressive organ dysfunction (22). In addition to inflammation, there are also reported impairments in metabolism, coagulation, and thermoregulation have also been reported (23). Mild alterations in metabolic pathways can be beneficial for regulating the immune response to infection and minimizing tissue damage (24). In sepsis, there is an interplay between inflammatory and metabolic changes, where the host response to infection plays a crucial role, potentially contributing to multiple organ dysfunction and, in severe cases, death (25). Recognizing the interdependency of inflammation and metabolism can be vital, as exploring metabolic alterations and inflammation may unveil new targets for sepsis treatment, which are currently primarily supportive rather than curative (26).
Mitochondrial or energetic dysfunction is among the most widely acknowledged metabolic impairments in sepsis and contributes to organ dysfunction and mortality (27, 28). As fatty acid oxidation (FAO) and the Krebs cycle are intramitochondrial events, a thorough exploration of lipid metabolism in sepsis has been conducted (29). As changes in fatty acid mobilization and oxidation are thought to play a role in the complex pathogenesis of sepsis, they hold significant promise for supporting the development of novel prognostic, therapeutic, and diagnostic tools (30).
Several investigations, including “-omics” and “multi-omics” analyses, have been performed to understand sepsis-induced FAO impairment, analyzing its consequences, predictive tools, and new therapeutic target compounds (31–36). Diverse metabolic adaptations occur in different organs during sepsis and some events can hamper these adaptations (22). For example, sepsis disturbances in lipid metabolism impair β-oxidation, which can lead to lipotoxicity, worsening the clinical status of these patients (37). To explore recent developments in our understanding of lipid metabolism in sepsis, we discuss the impairment of FFA metabolism induced by bacterial sepsis in the liver and muscles, including the skeletal and cardiac systems.
2 Sepsis inflammatory response
The latest international consensus (Sepsis-3) defines sepsis as life-threatening organ dysfunction caused by an abnormal host response to an infectious insult (6). Dysfunctional inflammatory and anti-inflammatory responses in sepsis may cause self-damage to the host organs (38). This is often observed in septic patients with acute respiratory failure, renal failure, or circulatory shock (39).
Sepsis can result from systemic inflammatory response syndrome (SIRS), which is caused by the release of pathogen-associated molecular patterns (PAMPs), such as LPS, peptidoglycans, viral ss/dsRNA, and bacterial DNA (40, 41). These molecules are recognized by pattern recognition receptors (PRRs) such as C-type lectin receptors (CLRs), NOD-like receptors (NLRs), and Toll-like receptors (TLRs) (42). To date, 13 TLRs have been described in mammals; ten are expressed in human cells. The activation of this receptor by its ligands triggers intracellular signaling by adaptor proteins; for instance, the MyD88-dependent pathway triggers several kinase proteins, such as p38 MAP kinase, JNK, ERK1, and ERK2, through phosphorylation. It culminates in activating transcription factors (e.g., NF-κB and AP-1) and the production of proinflammatory cytokines such as TNF-α, IL-6, IFN-γ, and enzymes such as cyclooxygenase-2 (COX-2) (43–45). Indeed, using the cecal ligation and puncture (CLP) sepsis model, TLR4 activation increased TNF-α serum levels as early as 4 h after surgery (46).
NOD-like receptors (NLRs) are important PRRs involved in sepsis caused by bacterial infections. They can detect PAMPs and DAMPs (41). NLR proteins form inflammasomes, which are multiprotein complexes (47, 48). Inflammasomes consist of NLR, the adaptor protein apoptosis-associated speck-like protein with a CARD domain (ASC), and pro-caspase-1, which activates caspase-1 and releases IL-1β and IL-18 (47).
The NLRP3 inflammasome modulates responses to sepsis by activation when cells are primed with cytokines or LPS and then stimulated with ATP, reactive oxygen species (ROS), mitochondrial dysfunction, or K+ efflux (49). Inhibition of the NLRP3 inflammasome during sepsis can improve survival and bacterial clearance (50), and alleviate acute lung injury. However, it may impair the immune response of monocytes, leading to higher mortality (51, 52). Higher levels of NLRP3, caspase-1, and IL-1 β in the bloodstream of patients with sepsis increase the risk of mortality (53–55).
PRRs activate cytokines that regulate the immune system and contribute to inflammatory responses (56). Although the expression of cytokines is beneficial in the inflammatory response and is necessary for pathogen clearance, excessive cytokine production during sepsis can cause harm, known as a cytokine storm (CS) or cytokine release syndrome (CRS). High levels can lead to organ failure, blood clotting, and even death. (57, 58). For example, in sepsis, cytokines such as IL-6, TNF-α, IL-1β, and CXCL8 increase antibody production, vascular permeability, and neutrophil recruitment (57, 59).
The altered state of endothelial cells, leukocytes, and platelets caused by cytokine production may cause sepsis-induced coagulopathy (SIC), which is linked to a poor outcome (60, 61). During sepsis, monocytes/macrophages express tissue factor and factor VII, activating the extrinsic coagulation pathway. Platelets are also activated, contributing to inflammation and thrombosis. Activated platelets can interact with leukocytes, promoting cytokine release and the neutrophils’ extracellular traps (NETs) (61–64).
Sepsis affects endothelial cells, causing an increase in vascular permeability and the expression of adhesion molecules such as E- and P-selectins, vascular adhesion molecule (VCAM)-1, and intracellular adhesion molecule (ICAM)-1, fostering leucocyte rolling and crawling. Damage to endothelial cells also impairs vascular tonus due to dysregulated NO production. These changes can lead to edema and impaired vascular tonus, compromising organ perfusion and metabolic function (65–69).
Under homeostatic conditions, the ATP demands of immune cells are regulated by the tricarboxylic acid (TCA) cycle (70). Sepsis rapidly increases ATP burning as immune cells prompt the fight against infection. Thus, these cells switch to aerobic glycolysis, quickly producing lactate and ATP but less efficiently. Interestingly, lactate is elevated in sepsis, showing a metabolic turnover in these patients (71), and has also been suggested to be a reliable prognostic tool that helps in the clinical management of patients (71). During sepsis, macrophages produce NO, which inhibits succinate dehydrogenase, a crucial enzyme in the TCA cycle (72). Also in sepsis, M2 macrophages increase lipid β-oxidation and upregulate IL-4 and peroxisome proliferator-activator receptor-γ (PPAR-γ) (72, 73).
3 Stressful state of sepsis
Sepsis is a sustained and extreme example of a stressful situation. Stress is “a state of homeostasis being challenged” (74). The stress system has two types of effectors: central and peripheral. The central system corresponds to the hypothalamic hormones arginine vasopressin (AVP), corticotropin-releasing hormone (CRH), proopiomelanocortin-derived peptides, and norepinephrine. The peripheral system includes glucocorticoids (the hypothalamic–pituitary–adrenal axis) and catecholamines (the sympathetic–adrenal–medullar axis). All these stress mediators reach the CNS and peripheral functions commanded by the gastrointestinal, cardiorespiratory, metabolic, and immune systems (75). PAMPs and DAMPs excessively activate PPRs, which activate proinflammatory signaling pathways, leading to the release of proinflammatory chemokines, cytokines, and the expression of endothelial adhesion molecules (76). Proinflammatory cytokines, such as TNF-α, IL-1-β, and IL-6, activate the hypothalamic–pituitary–adrenal (HPA) and sympathetic–adrenal–medullar axes (SAM) (77, 78). Activating these axes enhances the synthesis and release of glucocorticoids and catecholamines, thereby promoting metabolic alterations.
The HPA axis controls glucocorticoid and mineralocorticoid syntheses. Glucocorticoids (GCs) are steroid hormones that exert widespread hormone-physiological effects on homeostasis (79). Under internal and external signals, the hypothalamus secretes corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) (80). CRH is released into the hypophyseal portal circulation and stimulates the anterior pituitary to produce and secrete adrenocorticotropic hormone (ACTH). Consequently, glucocorticoids (cortisol in humans and corticosterone in rodents) are released from the zona fasciculata of the adrenal cortex (81). GCs are transported through the globulin-glucocorticoid protein in the blood. The glucocorticoid receptor (GR), which is ubiquitously expressed throughout the organism, mediates the glucocorticoid effects. The GC–GR complex translocates into the nucleus and modulates target gene expression through glucocorticoid-responsive elements (GREs) (82). Owing to hyperglucocorticoidemia, the HPA axis is controlled by negative feedback. During sepsis, immune cell-derived cytokines such as TNF-α, IL-IL-1-β, IL-6, and IL-10 activate the HPA axis (77). In experimental sepsis, adrenalectomized GRdim/dim mice treated with UX38, a GR blocker, showed poor outcomes (83). Thus, the disruption of the HPA axis plays a vital role in sepsis. Moreover, as in sepsis, higher vascular inflammation results in adrenal and HPA axis dysfunctions (84). Some studies showed that compared to a healthy state, plasma concentrations of total cortisol/corticosterone (CORT) rose in intensive care unit (ICU) septic patients and male mice and rats compared to a control group. However, they also observed reduced serum ACTH levels (85–88). This has been called “ACTH-cortisol disruption.” Likewise, Téblick et al. noticed that pro-opiomelanocortin gene expression was augmented more than usual. However, markers of processing POMC into ACTH and ACTH secretion were downregulated by glucocorticoid receptor–ligand binding. This could explain the low plasma ACTH and high POMC levels (88). Thus, impaired adrenal response during sepsis improves the circulation of FFA, triglycerides, glycerol, gluconeogenic amino acids, and glucose, guiding a starvation response (89, 90).
In conclusion, sepsis triggers a severe stress response that affects both central and peripheral systems, leading to hormonal and metabolic alterations. Disruption of the HPA axis and impaired adrenal response are crucial in sepsis pathophysiology and associated metabolic changes.
4 Alterations in lipid metabolism during sepsis
Lipids are the most significant energy reserves in the body, and FAO provides the most ATP total balance, becoming an essential energy source in high metabolic demand scenarios, such as sepsis. Lipids are usually conserved as triglycerides (TGs) in the cytoplasm of adipocytes. In order to be used as energy substrate, TGs suffer lipolysis to form fatty acids and glycerol, a reaction mediated by hormone-sensitive lipase (HSL). (91). Inflammatory stimuli trigger lipolysis via IRE1 kinase (92). Acyl-CoA synthase activates lipids and enters the mitochondria as carnitine derivatives through membrane transporters such as carnitine palmitoyltransferase 1 (CPT1), which is regulated by the nuclear receptors PGC-1α and PPAR-α (93). It has been reported that LPS suppresses FAO by reducing the expression of genes related to essential enzymes in lipid metabolism, including CPT-1, PGC-1α, and PPAR-α (94). Also, in an LPS-induced model, Li and coworkers 2021 (95) showed that CPT-1 and PGC-1α expression were reduced in cardiomyocytes and suggested a strong dependency of Nrf2 as a co-factor of PGC-1α. In addition, CPT1A inhibition is associated with a higher risk of infection and impairment of neutrophil response (96).
Elevated serum glucocorticoid levels promote endocrine and metabolic alterations, leading to a catabolic state, peripheral FAO, and ketogenesis (89, 97). Lpin1, Fabp4, Gpat4, Angptl4, Dgat1, Dgat2, PNPLA2, and LIPE are glucocorticoid-responsive genes that enhance adipose tissue lipolysis (98, 99). In patients with sepsis and sepsis animal models, FFA serum levels are high, and peripheral organs utilize these FFAs to produce energy. FFA also activates and upregulates the expression of PPAR-α, the central transcription factor responsible for inducing the β‐oxidation of FA and the production of ketone bodies (22). However, according to several studies, including human and animal models, PPAR-α levels are downregulated in sepsis, and β‐oxidation is compromised, causing FFA to accumulate in the liver, heart, kidney, and blood (20, 100–102).
Lipotoxicity occurs when lipids accumulate intra- or extracellularly, beyond physiological levels (103). In sepsis, mitochondrial oxidative phosphorylation is affected by the inflammatory state of the host (104). Therefore, this deficit in FFA breakdown during sepsis causes energy shortage, lipotoxicity, and mitochondrial damage due to FFA accumulation. Supplementation with oleic acid increases FAO and protects septic mice from organ dysfunction and mortality (105). On the other hand, FFA can trigger acute respiratory distress syndrome (ARDS) by inhibiting Na/K-ATPase in the lungs, causing tissue damage, edema, and inflammation (106–108).
Additionally, GR promotes the transcription of gluconeogenic genes such as Pck1, Pck2, Pcx, Pfkfb3, Mpc1, Mpc2, and G6Pc (99) by diminishing glucose uptake by suppressing GLUT4 translocation (109) and by reducing glucose utilization in skeletal muscle and white adipose tissue through the upregulation of PDK1 to PDK4 (22, 110). Hence, septic animal models and patients present with hyperglycemia during the acute phase of sepsis (111). Interestingly, patients with sepsis show insulin resistance, which causes an increase in lipid mobilization. In addition, inflammatory signaling elicited by LPS can induce lipolysis (92), further increasing FFA levels. Hyperglycemia, increased FFA levels, and insulin resistance are associated with worse outcomes in sepsis (112) (Figure 1).
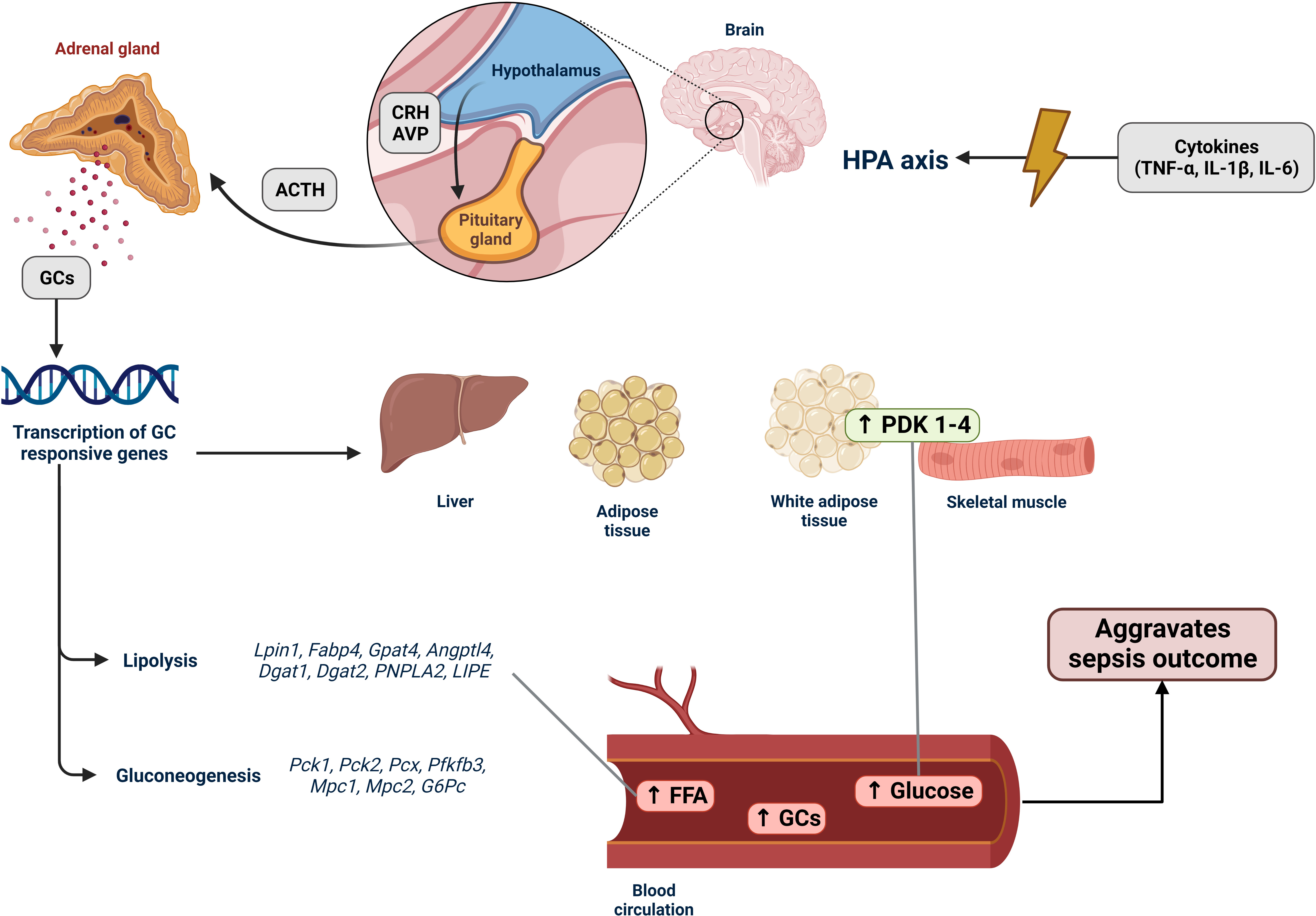
Figure 1 Activation of the HPA axis in sepsis and its effects on lipid metabolism. The hypothalamic–pituitary–adrenal (HPA) axis is an endocrine system that responds to stressful situations. Hormones produced by the HPA axis regulate cell metabolism and immune system functions. In sepsis, an imbalanced immune response leads to an increase in the production and release of various proinflammatory molecules, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6). These cytokines stimulate the HPA axis by activating the hypothalamus to secrete corticotropin-releasing hormone (CRH), which in turn stimulates the anterior pituitary gland to produce adrenocorticotropic hormone (ACTH) and arginine vasopressin (AVP). ACTH then promotes the production of glucocorticoids (GCs) in the zona fasciculata of the adrenal cortex. GCs are released into the bloodstream, reaching various tissues, particularly the liver, muscles, and adipose tissues. When coupled with glucocorticoid receptors (GRs), GCs induce transcription of GC-responsive genes. In sepsis, the expression of lipolysis-associated genes (Lpin1, Fabp4, Gpat4, Angptl4, Dgat1, Dgat2, PNPLA2, and LIPE) is upregulated. This upregulation increases the breakdown of fatty acids, resulting in the accumulation of free fatty acids (FFAs) in tissues and the bloodstream, indicating lipotoxicity during sepsis (Section 4). Genes associated with gluconeogenesis (Pck1, Pck2, Pcx, Pfkfb3, Mpc1, Mpc2, and G6Pc) were also upregulated, along with the increased expression of PDK1–4. These changes contribute to reduced glucose uptake and the hyperglycemia development. Hence, the HPA axis stress response system regulates the host response to internal and external signals, such as the intense inflammatory state observed in patients with sepsis. Consequently, it has a decisive impact on sepsis because hyperglycemia, accumulation of FFAs, and elevated GC levels are associated with poor patient outcomes.
Saturated fatty acids (SFA) activate the TLR4/MyD88/NF-kB pathway leading to endothelial inflammation in vitro upon FFA stimuli, with increased levels of CCL5, IL-6, IL-8, ROS, metalloproteinases (MMP-2 and MMP-9), and adhesion molecules (VCAM-1 and ICAM-1) (113). FFA uptake by cells is mediated by membrane receptors such as cluster of differentiation 36 (CD36), which is a transmembrane glycoprotein and a class B scavenger receptor (SR-B2) widely expressed throughout mammalian tissues (114). FFA can cause kidney lipotoxicity through the PPARγ/CD36 pathway in mice fed a high-fat diet. CD36 was found to be upregulated in the kidneys of obese mice alongside PPARγ, which is a crucial CD36 transcription factor, and inflammatory cytokines such as IL-6 and IL-18 (115). In addition, CD36 has been associated with metabolic diseases that present alterations in lipid metabolism (116) and is upregulated in septic patients (117) and sepsis animal models (118). Furthermore, CD36 deficiency improved sepsis outcomes in mice (119). CD36 plays a rate-limiting role in fatty acid uptake, making it a key regulator of cell lipid utilization.
Cell lipids can be used as substrates to generate oxylipins and other lipid mediators that act on immune cell signaling during inflammation. CPT1 also plays an essential role in transporting oxylipins to the mitochondria during infection, as evidenced by the accumulation of oxylipins in the peritoneal lavage of animals challenged with LPS and CPT1 inhibitors. Similarly, CPT1 controls COX/LOX-derived oxylipin secretion by increasing its uptake into the mitochondria. The authors suggested that CTP1 inhibition dampens the inflammatory response by increasing PGE2 and PGI2 levels (120).
In the face of sepsis, the body’s tissues confront a dire state of reduced oxygen supply, instigating a decline in FAO within the muscles. The liver tries to adapt swiftly by augmenting gluconeogenesis and FAO for energy production (22). However, intensified FAO by the liver can ultimately produce ketonic bodies (121). Moreover, the critical interplay between dwindling oxygen levels and the potential upsurge in FAO applies an unwavering pressure on the respiratory chain, culminating in a cascade of ROS generation (122, 123). Such intricate metabolic changes may have far-reaching implications for the pathogenesis of sepsis and clinical outcomes of affected patients.
Alterations in lipid metabolism are critical contributors to sepsis outcome. Despite being an important energy source, the accumulation of lipids in both the tissue and blood circulation can have harmful effects. In sepsis, the uptake of FFA by cells increases, but its oxidation by mitochondria is impaired, leading to lipotoxicity. FFA originating from augmented lipolysis activates inflammatory signaling pathways, thereby increasing the imbalanced host immune response. The endocrine system also plays a role in lipid metabolism by regulating the expression of lipid utilization genes through the activation of the HPA axis.
5 Heart and skeletal muscle metabolism during sepsis
Sepsis-induced myocardial injury is an acute type of cardiomyopathy that is a common complication of sepsis (124, 125). Bacterial exotoxins are primary triggers of the septic heart immune response (126). The pathogenesis of septic cardiomyopathy (SCM) is complex and involves both myocardial and peripheral vascular dysfunctions (124). Heart damage is associated with the left ventricle, particularly reducing systolic and diastolic function and ejection fraction (124, 125). Peripheral vascular injury is associated with endothelial dysfunction, resulting in leukocyte retention and ROS production (124).
The molecular mechanisms responsible for SCM include different pathways, most of which are related to inflammation. PAMP and DAMPs can activate cardiomyocyte TLR, fostering a septic inflammatory response (124, 125). TLRs play crucial roles in the pathophysiology of myocardial sepsis. For instance, a study using post-mortem myocardial cell lines found that TLR2, TLR4, and TLR5 stimulation increased inflammatory cytokines, especially IL-6 (127). In addition, an increased inflammatory response is related to worse ventricular function in a cell model (127). Furthermore, knockout of TLR4 results in decreased mortality and expression of IL-6 and TNF-α, with better global ventricular function in a sepsis animal model (128). Moreover, bacterial toxins are responsible for direct endothelial damage, causing microcirculation impairment (129, 130).
Once PAMPs prompt TLRs, they trigger intracellular signaling resulting in the release of cytokines, including TNF-α and interleukins, with a unique role in IL-1β and IL-18 (124, 125). A significant association between elevated plasma levels of interleukin-8 (IL-8) and cardiac dysfunction was observed in individuals with sepsis-induced myocardial dysfunction (131). TNF-α is also associated with depressed myocardial tissues (132). In addition, SFA can bind and activate TLR to promote tissue inflammation (133, 134).
Nitric oxide (NO) is also associated with cardiac dysfunction in sepsis. Initially, NO helps maintain hemostasis and hemodynamics. However, increased concentrations contribute to the deterioration of heart function, which may contribute to acute heart failure and dysfunction in sepsis (135, 136). Higher levels of NO induce decreased left ventricular contractility in animals treated with exotoxins (137). In addition, a previous study suggested that endothelial dysfunction could be responsible for leukocyte retention in the coronary vasculature (138). This increased leukocyte activity also increases inflammatory cytokine production and contributes to LV dysfunction.
In SCM, myocardial metabolism is impaired. Under physiological conditions, FFA is the preferential energy source of the myocardium. FAO generates approximately 70% of the heart’s adenosine triphosphate (ATP) (139), followed by carbohydrates (140). Cells can also generate ATP from amino acids and ketone bodies (140). However, the ability of heart cells to store high-energy phosphate is poor. Thus, mitochondria play a fundamental role in cellular and energy homeostasis (140).
In sepsis, cardiac metabolism is more dependent on glycolysis and lactate metabolism, and FFA and ketone bodies are reduced (141, 142). Under healthy conditions, FAO is the primary energy source for cardiomyocytes (142). However, a septic heart downregulates FAO and relies on other energy sources (143). The FAO is reduced for the following reasons as follows. FFA uptake by CD36 is impaired because IL-1β downregulates the expression of very-low-density proteins (VLDLs) receptors in cardiomyocytes (142). In addition, TLR-mediated inflammation is responsible for reducing the expression of FFA-binding proteins, acyl-CoA reductase, and FAO-associated transcription factors (142). Among the transcription factors, PPAR and its coactivator are the most essential. In sepsis, reduced expression of PPAR-γ in cardiomyocytes and adipose tissue was associated with lower levels of FAO and ATP production, as well as an increase in triglyceride accumulation, contributing to a general impairment in the heart’s metabolism (105, 139, 142, 144). Other molecular factors associated with decreased intracellular FAO include carnitine palmitoyl transferase-1 and acyl-CoA synthase (145).
Studies suggest that the oxygen supply to the heart is not generally affected during sepsis (142). Hence, it is hypothesized that metabolic changes, especially a reduction in FAO, are related to the genetic background (145). In inflammation, lipoprotein lipase (LpL) activity is markedly reduced, possibly due to the increased expression of Angptl4 (146), an LpL inhibitor. The expression of VLDL and CD36 receptors also decreases (142), compromising lipid uptake by the heart. Even though this causes a negative metabolic response, it is possible that the increased lipid clearance acts as a defense response since bacterial capsules are mainly composed of lipoproteins. The more these structures are removed from the organism, the less toxic the effects they exert (145).
Reduced PPAR expression in sepsis is associated with worse outcomes (147). A previous study by our group discussed the relationship between PPARγ and FAO (148). PPAR plays a significant role in modulating the immune response by regulating and controlling cytokine release (68). The higher the expression of PPARγ, the higher the immune response and its negative impacts. This is true not only for the myocardium but also for other organs. The effects of reduced FAO go further. Decreased oxidation leads to increased levels of non-esterified fatty acids (NEFA), which are also associated with a poorer prognosis in sepsis (105). In addition, increased circulating NEFA levels are associated with hypoalbuminemia and liver failure, which occurs during sepsis (134).
Metabolic changes in SCM also result from mitochondrial dysfunction because the heart is highly dependent on oxygen production (149). After endothelial injury, leukocytes are chemoattracted, increasing ROS production and causing organelle injury, including the mitochondria (150). Harmed mitochondria activate cAMP/protein kinase A signaling and produce more ROS by inhibiting the oxidative phosphorylation complex and lowering oxygen consumption (149). Consequently, this pathway triggers oxidative stress and apoptosis (124). In addition, mitochondrial DN (mtDNA) is essential for regulating immune responses. Studies have shown that individuals with an increased inflammatory response may present with increased mtDNA in their cytoplasm, related to mtDNA release from the organelle (149). Cardiolipin is one of the most critical targets of ROS damage in mitochondria. Cardiolipin causes structural damage to the mitochondria and impairs oxidative phosphorylation (151). Another possible target of ROS damage is poly(ADP-ribose) polymerase (PARP) enzyme, which is involved in DNA repair. This enzyme is upregulated in sepsis and may be associated with mitochondrial structural damage (151).
The Complement System is also linked to ROS damage in SCM, specifically C5a pathway (152, 153). Complement C5a can be activated early in sepsis and contributes to a robust inflammatory response (154). An imbalance in calcium homeostasis, caused by complement C5a and mediated by ROS, can harm cardiomyocytes (155). The activation of MAPks and Akt, which contributes to heart dysfunction in bacterial sepsis, has been linked to complement C5a (153). In addition, the complement system affects coagulation hemostasis, contributing to disseminated intravascular coagulation (DIC) during sepsis (154, 156).
In addition to causing damage to the myocardial muscle, sepsis affects the skeletal muscle, causing fiber wasting and sarcopenia. A meta-analysis by Liu et al. (157) suggested that sarcopenia may predict mortality risk in patients with sepsis. Muscle atrophy is a consequence of increased degradation and decreased repair of skeletal muscle. In addition, the increased secretion of cytokines causes increased activation of proteolytic enzymes, resulting in muscle loss.
The muscle structure and function are based on thinned-regulated homeostasis. Sepsis disrupts skeletal fiber homeostasis. Muscular homeostasis is a complex balance between muscle inflammation, degeneration, fibrosis, repair, and regeneration (158). However, literature suggests that muscle injury promoting regeneration and growth involves a pathophysiological process other than inflammatory wasting (158). Muscle injury and its repair mechanisms are mainly mediated by neutrophils and macrophages (158). After injury, neutrophils migrate to the tissue, and there is an increase in the production of inflammatory cytokines, including TNF-α, IL-1, and INF-γ (159). Specifically, TNF-α activates NF-kB, which causes muscle protein destruction (160). Nitric oxide secretion mediates macrophage degradation (158).
One of the apoptopic mechanisms seen in septic muscle wasting is related to the overwhelming endoplasmic reticulum (ER). In skeletal fibers, the ER, also called the sarcoplasmic reticulum, balances intracellular calcium according to the cell’s needs. ER is also involved in a protein-folding regulation mechanism that activates apoptosis-triggering pathways (161). Overstressed ER accumulates excess folded proteins, which activate three key molecules that participate in the unfolding process: protein kinase R-like endoplasmic reticulum kinase (PERK), inositol requiring protein 1a (IRE1a), and activating transcription factor 6 (ATF6) (161). If this process fails or the cell remains under stress, apoptosis pathways are activated (161). This process activates the ubiquitin–proteasome system and autophagy–lysosomal pathway, triggering cell death via proteases and caspases (161).
During muscle atrophy, contractile proteins are degraded by the ubiquitin-proteasome system (UPS), autophagy–lysosomal pathway (ALP), and proteases such as calpains and caspases (149). Studies suggest that muscle protein degradation follows a two-step pathway in which calpains and caspases first disassemble skeletal fibers, and then, proteases and other proteins from the ubiquitin complex act on the degradation of actin fibers (162). UPS-mediated protein degradation is regulated by a specific group of ubiquitin ligases including atrogin-1 and MuRF1. These enzymes target muscular proteins for ubiquitin-mediated degradation (162). However, this process is controversial because it has not yet been demonstrated in vivo (162).
Irisin is an essential protein related to muscle waste. Irisin was first discovered in 2012, and studies have shown that the expression of irisin and its precursors is related to muscular tissues (163). Irisin levels may be depleted during sepsis because of a cytokine storm (164). This is true because irisin modulation chemicals (TLRs, MAPK, NF-kB) are increased in a pro-inflammatory response, such as sepsis or COVID-19. This could explain the role of muscle waste in inflammation-related diseases (164).
Overall, an increased inflammatory response, lower oxygen supply, loss of tissue homeostasis, and cell destruction in the muscle result in decreased FAO and an increase in FFA/NEFA circulating levels.
6 Hepatic dysfunction during sepsis
The liver is central to human metabolism in health and disease states. The liver is pivotal in orchestrating the acute phase response (APR) in critical illness and systemic inflammation. It releases proteins that participate in essential activities such as coagulation, transport, and immunological functions. (165, 166). In clinical practice, preventing bacterial translocation in the gut is crucial because it can lead to bacteremia and sepsis (167). During translocation, the bacteria enter the hepatic sinusoids through the portal vein. The liver acts as the second line of defense, constituting the gut–liver axis and promoting bacterial clearance (168, 169). Sepsis is a hypermetabolic state wherein the patient’s feeding is often limited and does not supply daily energy requirements; therefore, metabolism mimics a starvation state (170). Thus, the liver promotes metabolic adaptations to sepsis, including alterations in glucose and fatty acid metabolism (Figure 2).
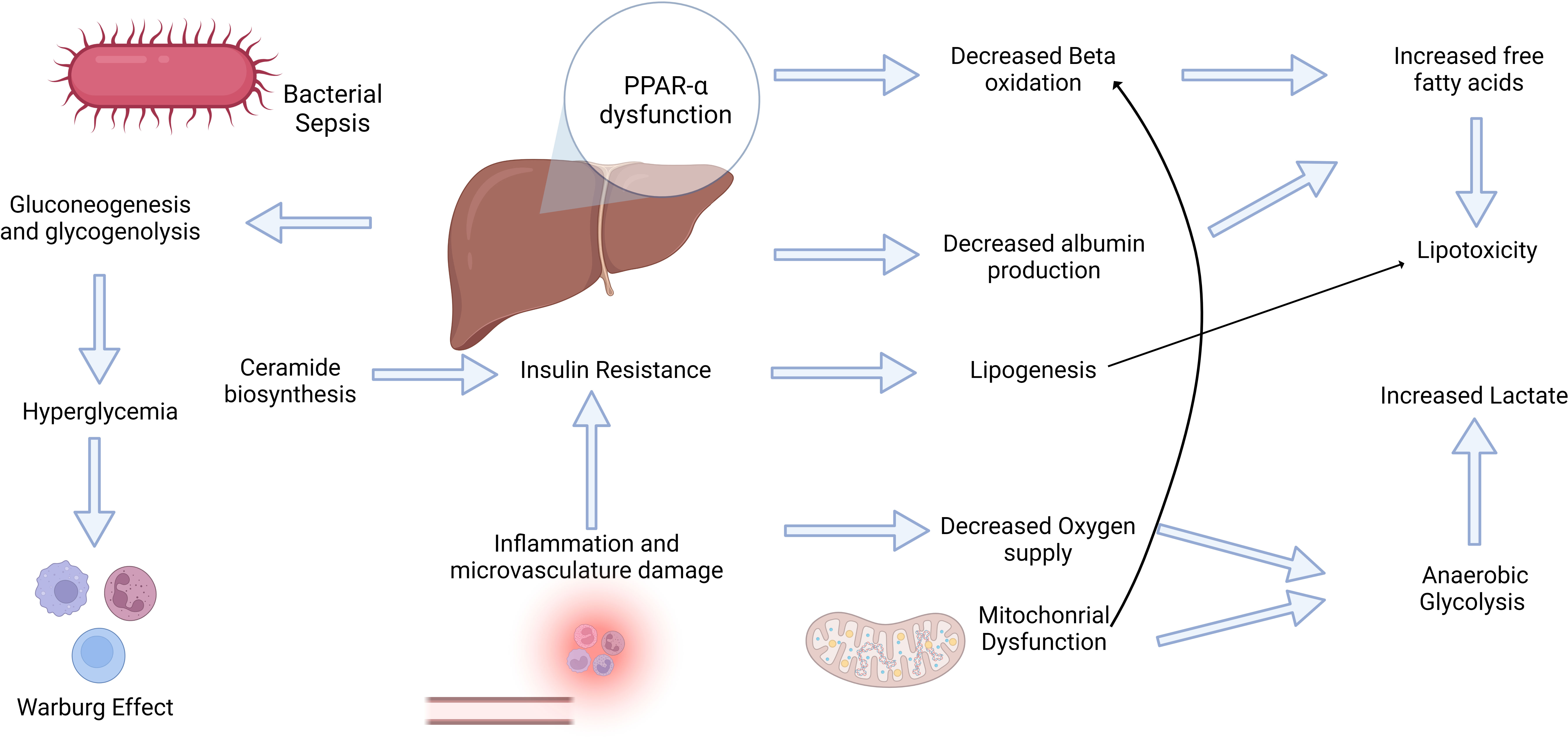
Figure 2 Sepsis induces a hypermetabolic state that triggers a complex interplay between inflammatory and metabolic changes. The bidirectional relationship between inflammation and metabolism plays a pivotal role in the development of sepsis. Inflammation triggers metabolic alterations and vice-versa. Inflammatory mediators, such as free fatty acids, can trigger insulin resistance, which is crucial for subsequent metabolic alterations. Conversely, hyperglycemia may elicit significant effects on immune cell activity, including the Warburg Effect. These multifaceted adaptations and dysfunctional PPAR-α levels contribute to increased oxidative stress, lipotoxicity, and elevated lactate levels, ultimately contributing to progressive organ failure.
Investigating the interplay between metabolic and inflammatory alterations is crucial for understanding the inflammatory conditions (171). The role of inflammation in insulin resistance has been widely accepted in critically ill patients (172) (Figure 2). Cytokine signaling may stimulate the inhibitory phosphorylation of insulin receptor substrate 1 (IRS-1) by stress kinases such as JNK1 and IKKb (173). Cytokines may also enhance hepatocyte ceramide production, thereby increasing their production in sepsis (174, 175). Ceramide biosynthesis appears to be involved in insulin resistance (Figure 2), and TLR4 stimulation by SFA increase enzymes in this pathway (176–178). Supported mainly by the insulin resistance response, sympathetic stimulation, and limited oral starvation, the liver increases glucose production through gluconeogenesis and glycogenolysis during sepsis. However, glucocorticoid resistance and iron‐driven oxidative inhibition of glucose‐6‐phosphatase may hinder gluconeogenesis in the liver (179, 180). It usually leads to stress hyperglycemia during the acute phase of sepsis and in other critically ill patients, even in non-diabetic patients (181). Extremely high glycemic levels in these patients are associated with poor outcomes and an increased risk of developing type II diabetes (182, 183). However, hyperglycemia may be an adaptive mechanism to sustain immune cell activity because these cells shift their metabolism based primarily on glycolysis with lactate formation, even in an aerobic environment. This phenomenon is known as the Warburg effect, and it was first described in cancer cells; however, it similarly occurs in monocytes, dendritic cells, and T cells during their activation, mediated by increased succinate release into the cytoplasm and Akt-mTOR-HIF-1α (184–186). This metabolic shift may support the elimination of ROS and development of trained immunity (187, 188). However, in tissue ischemia, this phenomenon leads to an increased release of lactate. Lactate levels are correlated with adverse outcomes in patients with sepsis. They can activate TLR4, promoting the activation of the NF-kB pathway and release of inflammatory mediators (189). In addition, the increased accumulation of succinate may trigger inflammation via ROS generation mediated by succinate dehydrogenase (190, 191).
In addition, adipose tissue provides energy storage in mammals, releasing fatty acids that can be oxidized to produce ATP and maintain cell activities. It has been reported that lipolysis is upregulated during sepsis, increasing plasma levels of fatty acids and triglycerides (192). LPS challenge induces the enzymatic activation of hormone-sensitive lipase (HSL) by phosphorylation at Ser650 and an increase in perilipin-1 phosphorylation by protein kinase A (193). Insulin resistance in these patients also supports lipolysis and an increase in catabolic hormones, such as adrenaline, GH, and glucagon (194–196). Depressed lipoprotein lipase activity has also been postulated to cause hypertriglyceridemia, occurring not necessarily in sepsis but in endotoxemic and bacteremic states (197). It is worth highlighting that the shift in lipid metabolism can support even the conceptualization of new prognostic tools. Modified hepatic levels of selected phospholipids have been linked to impaired energy metabolism and the progression of sepsis (198). A recent metabolomic study has suggested differences in plasma lipid concentrations between survivors and non-survivors, medium/short-chain hydroxy acyl-CoA dehydrogenase (elevated in survivors), and crotonase (elevated in non-survivors). Hence, there is a future perspective for monitoring cellular energy beyond the currently established methods (199).
Unfavorable outcomes in sepsis are related to impaired metabolic adaptation in the liver (200). PPAR-α (encoded by the NR1C1 gene) is a crucial transcription factor that regulates gene transcription (e.g., CPT1A and FABP1) of proteins involved in β-oxidation (201, 202). During sepsis and other inflammatory conditions, PPAR-α expression is downregulated in the liver, thereby diminishing lipid oxidation in this organ (20). Malnourishment, which is common in patients with sepsis, is also associated with decreased PPAR-α levels (203). In addition, the entry of fatty acids into mitochondria is partially hindered by the increased concentration of malonyl-CoA generated from glucose, especially in the heart, muscles, and liver (204). In this context, if unable to capture and oxidize lipids, the liver does not shift to a starvation response properly, resulting in adverse outcomes for the patients. A recent transcriptomic study found that PPAR-α-deficient mice presented more severe glycemic disturbances and increased steatosis than control mice (205). Ketogenesis may also be impaired, which can be fatal after LPS-induced endotoxemia (206). Ketone bodies can protect against ROS and provide energy to extrahepatic tissues (207). Ultimately, downregulation of PPAR-α-dependent genes impairs liver metabolic adaptation in sepsis (205). Conversely, PPAR-α agonists may benefit septic patients (20, 208, 209).
Potential therapeutic approaches for dysfunctional lipid metabolism during sepsis, agonists of peroxisome proliferator-activated receptors α and γ, and omega-3 fatty acid supplementation have shown promise (210, 211). Saroglitazar (SAR), a dual PPAR-α/γ agonist, has anti-inflammatory and antioxidant effects that mitigate LPS-induced liver and kidney injury during sepsis (212). PPAR-α activation counters lipogenesis, endotoxemia, and dysbiosis, ameliorating the intestinal barrier structure and reducing TLR4 expression in the livers of high-fructose-fed C57BL/6 mice (213). In addition, PPAR activation by fenofibrate showed protective effects against cardiac damage in septic mice by reducing troponin-T, ROS, and IL-6 TNF-α (214). Rosiglitazone, another PPARγ agonist, mitigated apoptosis, and pro-inflammatory responses in LPS-stimulated myocardial cells (215). PPAR agonists improve sepsis-related conditions beyond the muscles and liver (216). A meta-analysis conducted in 2020 with 1,514 patients suggested that omega-3 fatty acid, a PPAR agonist, might be associated with decreased mortality in individuals with sepsis (217). Likewise, omega-3 fatty acids in severe COVID-19 patients with sepsis showed potential benefits, including reduced procalcitonin and IL-6 levels (218). Finally, intravenous omega-3 fatty acid lipid emulsion reduces systemic inflammation, endotoxemia, and sepsis in patients with acute liver failure (219).
In addition, liver metabolic adaptation is hindered by microcirculatory failure. Liver sinusoid endothelial capillaries (LSECs) regulate the immune response and blood flow (220). However, LSEC may be damaged during sepsis, contributing to liver failure. The architecture of LSECs is susceptible to LPS injection, leading to reduced flow velocity, increased heterogeneity, and blood perfusion deficits (221). In mice, toxin levels have been shown to alter the LSEC fenestrae diameter, and the direct effects of LPS and TNF-α can induce hepatocyte damage (222). Different substances have been linked to impaired liver microcirculation, including endothelin-1, carbon monoxide, and nitric oxide (223). In addition, hyperactivation of Kupffer cells (liver-resident macrophages) during sepsis may play a role in hepatic dysfunction and LSEC damage. Kupffer cells can intensely release ROS, TNFα, and IL-1β, leading to oxidative stress and damage to cellular structures, while recruiting other immune cells to the liver (224). Additionally, Kupffer cells can stimulate coagulation, forming microthrombi that further exacerbate LSEC damage (225). A mouse model has revealed that Kupffer cells can contribute to the onset of intrasinusoidal thrombosis, leading to acute liver failure (226). Therefore, oxygen supply mismatch in hepatocytes can impair aerobic energy production, leading to cell damage or death.
As described, sepsis patients present with increased lipid levels in the plasma and tissues due to stimulated lipolysis, impaired beta-oxidation, and hampered transport into the mitochondria. In addition, LPS and cytokines such as IL-6 and TNF-α may induce de novo synthesis of lipids, regulated by sterol regulatory element-binding proteins (SREBP) (227). High levels of insulinemia seem to increase SREBP expression in the hepatocytes. A recent transcriptomic investigation found upregulation of ERLIN1 (gene regulating SREBP) during sepsis (228, 229). In addition, LPS administration in murine livers upregulates SREBP-1 (230). Because of the production of toxic metabolites, mainly through lipid peroxidation, increased lipid levels can harm patients, causing cell damage and apoptosis, a phenomenon known as lipotoxicity (37). The double bonds of polyunsaturated fatty acids are major substrates for oxidization by free radicals. Sepsis induces the formation of many peroxidation products such as malondialdehyde (MDA) and 4‐hydroxynonenal (4‐HNE), which lead to toxicity by interacting with amino acids and nucleosides (231, 232). Additionally, diacylglycerol, ceramide, palmitate, and other SFA can be upregulated in sepsis, resulting in lipotoxic effects (233–236). SFA can promote inflammation mediated by TLR-4, while TLR-4 knockout mice may not suffer from SFA signaling (237). Possible therapeutic targets to prevent lipid peroxidation and lipotoxic effects in patients have been investigated. Administration of C75, a fatty acid synthase inhibitor, reduces inflammation and organ injury in sepsis (238). In addition, mitochondrial uncoupling proteins, such as UCP3, have been postulated to function in the defense against lipid-induced oxidative muscle damage (239, 240). Moreover, propofol, a common drug used to manage critically ill ICU patients, may protect against hepatic lipid peroxidation, oxidative stress, and inflammation (241–243).
In addition, albumin synthesis in the liver is decreased by sepsis-related liver failure and general inflammation, making albumin a negative acute-phase protein (244, 245). Serum albumin levels are decreased in most patients with sepsis, although it remains unclear whether this is due to suppressed albumin production or increased albumin clearance (246). Albumin is a crucial fatty acid transporter in systemic circulation. This decrease also favors increased plasma levels of fatty acids (247, 248). In addition, NEFA can activate toll-like receptors and inhibit Na+/K+-ATPase, causing lung injury and edema (249, 250). Our group showed a relationship between the oleic acid/albumin molar ratio and clinical outcomes in critically ill patients, with a higher ratio indicating higher mortality in these patients (251). Omega-9 treatment increases CPT1A mRNA in the livers of septic mice, reduces plasma NEFA levels, and improves survival and clinical status (105). CPT1A plays a crucial role in importing fatty acids into the mitochondrion, delivering them to their final destination oxidation, thus pointing out a possible therapeutic target related to ß-oxidation.
7 Study limitations
Sepsis can originate from different microbes, with infections originating in different organs. Here, we focused on bacterial infections and multiple foci of infection. Sepsis severity can influence lipid metabolism. Sex, age, metabolic status, pathological background, and alimentary status may influence lipid metabolism and sepsis outcomes. However, altered lipid metabolism in patients with sepsis has been described, regardless of different conditions. FFA will affect inflammatory and metabolic conditions, which may lead to poor outcomes.
8 Concluding remarks
Sepsis is a complex syndrome with a high morbidity and mortality rate. Sepsis pathogenesis disrupts metabolic and inflammatory systems. Derangement of the inflammatory response and metabolic alterations have been identified as targets for the diagnosis and treatment of patients with sepsis. We suggest a new protagonist in sepsis physiopathology, fatty acid oxidation, because FAO in the tissues is reduced and fatty acid blood levels are high. In addition, increased FFA levels are associated with poor prognosis in patients with sepsis. Different mechanisms account for alterations in lipid metabolism during sepsis, such as increased gluconeogenesis and lipolysis, lower oxygen supply to the tissues, increased inflammation, and worsening lipid oxidation. Hence, FAO and FFA can be potentially valuable markers for sepsis diagnosis and prognosis because alterations in their circulating levels and metabolism have a life-threatening impact on critically ill patients.
Author contributions
CG-d-A conceived and supervised the confection of the whole manuscript. RM-S, GA, MA, IS, MB, and CG-d-A wrote and revised the manuscript. CA and AS critically revised and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Universidade Federal Fluminense (PROPPI/UFF), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Grant (001), Programa de Biotecnologia da Universidade Federal Fluminense (UFF), Programa de Pós Graduação em Biologia Molecular Celular (UNIRIO), Universidade Federal do Estado do Rio de Janeiro (UNIRIO), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) Grants (E-26/010.000983/2019, E-26/203.290/2017, and E-26/2010.592/2019, E-26/201.448/2021), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Instituto Oswaldo Cruz, FIOCRUZ. We also acknowledge the financial support from the European Community’s Seventh Framework Programme (FP7-2007-2013) under a grant agreement (HEALTH-F4-2011-282095) (TARKINAID).
Acknowledgments
We acknowledge Patricia Burth for her help and discussion during this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Geroulanos S, Douka ET. Historical perspective of the word "sepsis". Intensive Care Med (2006) 32:2077. doi: 10.1007/s00134-006-0392-2
2. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest (1992) 101:1644–55. doi: 10.1378/chest.101.6.1644
3. Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Crit Care Med (2003) 31:1250–6. doi: 10.1097/01.CCM.0000050454.01978.3B
4. Kempker JA, Martin GS. The changing epidemiology and definitions of sepsis. Clin Chest Med (2016) 37:165–79. doi: 10.1016/j.ccm.2016.01.002
5. Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, et al. Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama (2016) 315:762–74. doi: 10.1001/jama.2016.0288
6. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama (2016) 315:801–10. doi: 10.1001/jama.2016.0287
7. Delaloye J, Calandra T. Invasive candidiasis as a cause of sepsis in the critically ill patient. Virulence (2014) 5:161–9. doi: 10.4161/viru.26187
8. Gu X, Zhou F, Wang Y, Fan G, Cao B. Respiratory viral sepsis: epidemiology, pathophysiology, diagnosis and treatment. Eur Respir Rev (2020) 29:1–12. doi: 10.1183/16000617.0038-2020
9. Potruch A, Schwartz A, Ilan Y. The role of bacterial translocation in sepsis: a new target for therapy. Therap Adv Gastroenterol (2022) 15:17562848221094214. doi: 10.1177/17562848221094214
10. Rangel-Frausto MS. The epidemiology of bacterial sepsis. Infect Dis Clin North Am (1999) 3:299–312. doi: 10.1016/S0891-5520(05)70076-3
11. Rhee C, Jones TM, Hamad Y, Pande A, Varon J, O’brien C, et al. Prevalence, underlying causes, and preventability of sepsis-associated mortality in US acute care hospitals. JAMA Network Open (2019) 2:e187571–e187571. doi: 10.1001/jamanetworkopen.2018.7571
12. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality 1990-2017: analysis for the Global Burden of Disease Study. Lancet (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
13. Dolin HH, Papadimos TJ, Chen X, Pan ZK. Characterization of pathogenic sepsis etiologies and patient profiles: A novel approach to triage and treatment. Microbiol Insights (2019) 12:1178636118825081. doi: 10.1177/1178636118825081
14. Minasyan H. Sepsis: mechanisms of bacterial injury to the patient. Scand J Trauma Resusc Emerg Med (2019) 27:19. doi: 10.1186/s13049-019-0596-4
15. Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med (1996) 22:707–10. doi: 10.1007/BF01709751
16. Lambden S, Laterre PF, Levy MM, Francois B. The SOFA score-development, utility and challenges of accurate assessment in clinical trials. Crit Care (2019) 23:374. doi: 10.1186/s13054-019-2663-7
17. Lorente L, Martín MM, Abreu-González P, Domínguez-Rodriguez A, Labarta L, Díaz C, et al. Sustained high serum malondialdehyde levels are associated with severity and mortality in septic patients. Crit Care (2013) 17:R290. doi: 10.1186/cc13155
18. Gobe GC, Coombes JS, Fassett RG, Endre ZH. Biomarkers of drug-induced acute kidney injury in the adult. Expert Opin Drug Metab Toxicol (2015) 11:1683–94. doi: 10.1517/17425255.2015.1083011
19. Lorente L, Martín MM, Abreu-González P, Domínguez-Rodríguez A, Labarta L, Díaz C, et al. Prognostic value of malondialdehyde serum levels in severe sepsis: a multicenter study. PloS One (2013) 8:e53741. doi: 10.1371/journal.pone.0053741
20. van Wyngene L, Vanderhaeghen T, Timmermans S, Vandewalle J, Van Looveren K, Souffriau J, et al. Hepatic PPARα function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol Med (2020) 12:e11319. doi: 10.15252/emmm.201911319
21. Mosevoll KA, Hansen BA, Gundersen IM, Reikvam H, Bruserud Ø., Wendelbo Ø. Systemic metabolomic profiles in adult patients with bacterial sepsis: characterization of patient heterogeneity at the time of diagnosis. Biomolecules (2023) 13:1–21. doi: 10.3390/biom13020223
22. van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med (2018) 10:1–18. doi: 10.15252/emmm.201708712
23. Cohen J, Vincent JL, Adhikari NK, Machado FR, Angus DC, Calandra T, et al. Sepsis: a roadmap for future research. Lancet Infect Dis (2015) 15:581–614. doi: 10.1016/S1473-3099(15)70112-X
24. Soares MP, Gozzelino R, Weis S. Tissue damage control in disease tolerance. Trends Immunol (2014) 35:483–94. doi: 10.1016/j.it.2014.08.001
25. Zhu XX, Zhang WW, Wu CH, Wang SS, Smith FG, Jin SW, et al. The novel role of metabolism-associated molecular patterns in sepsis. Front Cell Infect Microbiol (2022) 12:915099. doi: 10.3389/fcimb.2022.915099
26. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers (2016) 2:16045. doi: 10.1038/nrdp.2016.45
27. Rahmel T, Marko B, Nowak H, Bergmann L, Thon P, Rump K, et al. Mitochondrial dysfunction in sepsis is associated with diminished intramitochondrial TFAM despite its increased cellular expression. Sci Rep (2020) 10:21029. doi: 10.1038/s41598-020-78195-4
28. Preau S, Vodovar D, Jung B, Lancel S, Zafrani L, Flatres A, et al. Energetic dysfunction in sepsis: a narrative review. Ann Intensive Care (2021) 11:104. doi: 10.1186/s13613-021-00893-7
29. Ma Y, Wang W, Devarakonda T, Zhou H, Wang XY, Salloum FN, et al. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci Rep (2020) 10:1450. doi: 10.1038/s41598-020-58334-7
30. Langley RJ, Tsalik EL, Van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med (2013) 5:195ra195. doi: 10.1126/scitranslmed.3005893
31. Mecatti GC, Messias MCF, De Oliveira Carvalho P. Lipidomic profile and candidate biomarkers in septic patients. Lipids Health Dis (2020) 19:68. doi: 10.1186/s12944-020-01246-2
32. Mecatti GC, Sánchez-Vinces S, Fernandes A, Messias MCF, De Santis GKD, Porcari AM, et al. Potential lipid signatures for diagnosis and prognosis of sepsis and systemic inflammatory response syndrome. Metabolites (2020) 10:1–17. doi: 10.3390/metabo10090359
33. McReynolds CB, Cortes-Puch I, Ravindran R, Khan IH, Hammock BG, Shih PB, et al. Plasma linoleate diols are potential biomarkers for severe COVID-19 infections. Front Physiol (2021) 12:663869. doi: 10.3389/fphys.2021.663869
34. Li R, Li X, Zhao J, Meng F, Yao C, Bao E, et al. Mitochondrial STAT3 exacerbates LPS-induced sepsis by driving CPT1a-mediated fatty acid oxidation. Theranostics (2022) 12:976–98. doi: 10.7150/thno.63751
35. Montague B, Summers A, Bhawal R, Anderson ET, Kraus-Malett S, Zhang S, et al. Identifying potential biomarkers and therapeutic targets for dogs with sepsis using metabolomics and lipidomics analyses. PloS One (2022) 17:e0271137. doi: 10.1371/journal.pone.0271137
36. Wang S, Liang J, Shi H, Xia Y, Li J, Wu W, et al. [Nontargeted lipidomic analysis of sera from sepsis patients based on ultra-high performance liquid chromatography-mass spectrometry/mass spectrometry]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue (2022) 34:346–51. doi: 10.3760/cma.j.cn121430-20210612-00875
37. Lipke K, Kubis-Kubiak A, Piwowar A. Molecular mechanism of lipotoxicity as an interesting aspect in the development of pathological states-current view of knowledge. Cells (2022) 11:1–34. doi: 10.3390/cells11050844
38. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol (2018) 14:121–37. doi: 10.1038/nrneph.2017.165
39. Nishibori M. Novel aspects of sepsis pathophysiology: NETs, plasma glycoproteins, endotheliopathy and COVID-19. J Pharmacol Sci (2022) 150:9–20. doi: 10.1016/j.jphs.2022.06.001
40. Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence (2014) 5:36–44. doi: 10.4161/viru.25436
41. Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta Mol Basis Dis (2017) 1863:2564–73. doi: 10.1016/j.bbadis.2017.01.013
42. Dias ML, O'connor KM, Dempsey EM, O'halloran KD, Mcdonald FB. Targeting the Toll-like receptor pathway as a therapeutic strategy for neonatal infection. Am J Physiol Regul Integr Comp Physiol (2021) 321:R879–r902. doi: 10.1152/ajpregu.00307.2020
43. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11:373–84. doi: 10.1038/ni.1863
44. Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol (2013) 5:a011247. doi: 10.1101/cshperspect.a011247
45. Sasai M, Yamamoto M. Pathogen recognition receptors: ligands and signaling pathways by Toll-like receptors. Int Rev Immunol (2013) 32:116–33. doi: 10.3109/08830185.2013.774391
46. Lee EP, Lin MJ, Wu HP. Time-serial expression of toll-like receptor 4 signaling during polymicrobial sepsis in rats. Int J Immunopathol Pharmacol (2022) 36:3946320221090021. doi: 10.1177/03946320221090021
47. Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
48. Wang L, Hauenstein AV. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol Aspects Med (2020) 76:100889. doi: 10.1016/j.mam.2020.100889
49. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20:1–24. doi: 10.3390/ijms20133328
50. Jin L, Batra S, Jeyaseelan S. Deletion of nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol (2017) 198:1253–62. doi: 10.4049/jimmunol.1601745
51. Martínez-García JJ, Martínez-Banaclocha H, Angosto-Bazarra D, De Torre-Minguela C, Baroja-Mazo A, Alarcón-Vila C, et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat Commun (2019) 10:2711. doi: 10.1038/s41467-019-10626-x
52. Hu H, Jiang H, Zhang K, Zhang Z, Wang Y, Yi P, et al. Memantine nitrate MN-08 suppresses NLRP3 inflammasome activation to protect against sepsis-induced acute lung injury in mice. BioMed Pharmacother (2022) 156:113804. doi: 10.1016/j.biopha.2022.113804
53. Garnacho-Montero J, Palacios-García I, Díaz-Martín A, Gutiérrez-Pizarraya A, López-Sánchez JM, Gómez EA, et al. Sequential changes of NLRP3 inflammasome activation in sepsis and its relationship with death. Shock (2020) 54:294–300. doi: 10.1097/SHK.0000000000001521
54. Beltrán-García J, Osca-Verdegal R, Pérez-Cremades D, Novella S, Hermenegildo C, Pallardó FV, et al. Extracellular histones activate endothelial NLRP3 inflammasome and are associated with a severe sepsis phenotype. J Inflammation Res (2022) 15:4217–38. doi: 10.2147/JIR.S363693
55. Huang W, Wang X, Xie F, Zhang H, Liu D. Serum NLRP3: A biomarker for identifying high-risk septic patients. Cytokine (2022) 149:155725. doi: 10.1016/j.cyto.2021.155725
56. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol (2017) 39:517–28. doi: 10.1007/s00281-017-0639-8
57. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
58. Shapiro L, Scherger S, Franco-Paredes C, Gharamti AA, Fraulino D, Henao-Martinez AF. Chasing the ghost: hyperinflammation does not cause sepsis. Front Pharmacol (2022) 13:910516. doi: 10.3389/fphar.2022.910516
59. Jarczak D, Nierhaus A. Cytokine storm-definition, causes, and implications. Int J Mol Sci (2022) 23:1–30. doi: 10.3390/ijms231911740
60. Sygitowicz G, Sitkiewicz D. Molecular mechanisms of organ damage in sepsis: an overview. Braz J Infect Dis (2020) 24:552–60. doi: 10.1016/j.bjid.2020.09.004
61. Iba T, Levi M, Levy JH. Intracellular communication and immunothrombosis in sepsis. J Thromb Haemost (2022) 20:2475–84. doi: 10.1111/jth.15852
62. Dib PRB, Quirino-Teixeira AC, Merij LB, Pinheiro MBM, Rozini SV, Andrade FB, et al. Innate immune receptors in platelets and platelet-leukocyte interactions. J Leukoc Biol (2020) 108:1157–82. doi: 10.1002/JLB.4MR0620-701R
63. Iba T, Levy JH. Sepsis-induced coagulopathy and disseminated intravascular coagulation. Anesthesiology (2020) 132:1238–45. doi: 10.1097/ALN.0000000000003122
64. Iba T, Umemura Y, Wada H, Levy JH. Roles of coagulation abnormalities and microthrombosis in sepsis: pathophysiology, diagnosis, and treatment. Arch Med Res (2021) 52:788–97. doi: 10.1016/j.arcmed.2021.07.003
65. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun (2015) 21:827–46. doi: 10.1177/1753425915606525
66. Lipinska-Gediga M. Sepsis and septic shock-is a microcirculation a main player? Anaesthesiol Intensive Ther (2016) 48:261–5. doi: 10.5603/AIT.a2016.0037
67. Gonçalves-De-Albuquerque CF, Rohwedder I, Silva AR, Ferreira AS, Kurz ARM, Cougoule C, et al. The yin and yang of tyrosine kinase inhibition during experimental polymicrobial sepsis. Front Immunol (2018) 9:901. doi: 10.3389/fimmu.2018.00901
68. Medeiros-de-Moraes IM, Gonçalves-De-Albuquerque CF, Kurz ARM, Oliveira FMJ, De Abreu VHP, Torres RC, et al. Omega-9 oleic acid, the main compound of olive oil, mitigates inflammation during experimental sepsis. Oxid Med Cell Longev (2018) 2018:6053492. doi: 10.1155/2018/6053492
69. Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med (2020) 202:361–70. doi: 10.1164/rccm.201910-1911TR
70. Hortova-Kohoutkova M, Laznickova P, Fric J. How immune-cell fate and function are determined by metabolic pathway choice: The bioenergetics underlying the immune response. Bioessays (2021) 43:e2000067. doi: 10.1002/bies.202000067
71. Nolt B, Tu F, Wang X, Ha T, Winter R, Williams DL, et al. Lactate and immunosuppression in sepsis. Shock (2018) 49:120–5. doi: 10.1097/SHK.0000000000000958
72. Jung J, Zeng H, Horng T. Metabolism as a guiding force for immunity. Nat Cell Biol (2019) 21:85–93. doi: 10.1038/s41556-018-0217-x
73. Arts RJ, Gresnigt MS, Joosten LA, Netea MG. Cellular metabolism of myeloid cells in sepsis. J Leukoc Biol (2017) 101:151–64. doi: 10.1189/jlb.4MR0216-066R
74. Lu S, Wei F, Li G. The evolution of the concept of stress and the framework of the stress system. Cell Stress (2021) 5:76–85. doi: 10.15698/cst2021.06.250
75. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol (2009) 5:374–81. doi: 10.1038/nrendo.2009.106
76. Daniel M, Bedoui Y, Vagner D, Raffray L, Ah-Pine F, Doray B, et al. Pathophysiology of sepsis and genesis of septic shock: the critical role of mesenchymal stem cells (MSCs). Int J Mol Sci (2022) 23:1–34. doi: 10.3390/ijms23169274
77. Vardas K, Ilia S, Sertedaki A, Charmandari E, Briassouli E, Goukos D, et al. Increased glucocorticoid receptor expression in sepsis is related to heat shock proteins, cytokines, and cortisol and is associated with increased mortality. Intensive Care Med Exp (2017) 5:10. doi: 10.1186/s40635-017-0123-8
78. Kobbe P, Bläsius FM, Lichte P, Oberbeck R, Hildebrand F. Neuroendocrine modulation of the immune response after trauma and sepsis: does it influence outcome? J Clin Med (2020) 9:1–20. doi: 10.3390/jcm9072287
79. Spencer-Segal JL, Akil H. Glucocorticoids and resilience. Horm Behav (2019) 111:131–4. doi: 10.1016/j.yhbeh.2018.11.005
80. Gillies GE, Linton EA, Lowry PJ. Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature (1982) 299:355–7. doi: 10.1038/299355a0
81. Herman JP, Mcklveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, et al. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol (2016) 6:603–21. doi: 10.1002/cphy.c150015
82. Lee SJ, Shizu R, Negishi M. Glucocorticoid receptor dimerization in the cytoplasm might be essential for nuclear localization. Biochem Biophys Res Commun (2021) 553:154–9. doi: 10.1016/j.bbrc.2021.03.071
83. Kleiman A, Hübner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J (2012) 26:722–9. doi: 10.1096/fj.11-192112
84. Kanczkowski W, Alexaki VI, Tran N, Großklaus S, Zacharowski K, Martinez A, et al. Hypothalamo-pituitary and immune-dependent adrenal regulation during systemic inflammation. Proc Natl Acad Sci USA (2013) 110:14801–6. doi: 10.1073/pnas.1313945110
85. Carlson DE, Chiu WC, Scalea TM. Cecal ligation and puncture in rats interrupts the circadian rhythms of corticosterone and adrenocortical responsiveness to adrenocorticotrophic hormone. Crit Care Med (2006) 34:1178–84. doi: 10.1097/01.CCM.0000207340.24290.3C
86. Polito A, Sonneville R, Guidoux C, Barrett L, Viltart O, Mattot V, et al. Changes in CRH and ACTH synthesis during experimental and human septic shock. PloS One (2011) 6:e25905. doi: 10.1371/journal.pone.0025905
87. Boonen E, Vervenne H, Meersseman P, Andrew R, Mortier L, Declercq PE, et al. Reduced cortisol metabolism during critical illness. N Engl J Med (2013) 368:1477–88. doi: 10.1056/NEJMoa1214969
88. Téblick A, Vander Perre S, Pauwels L, Derde S, Van Oudenhove T, Langouche L, et al. The role of pro-opiomelanocortin in the ACTH-cortisol dissociation of sepsis. Crit Care (2021) 25:65. doi: 10.1186/s13054-021-03475-y
89. Goossens C, Weckx R, Derde S, Dufour T, Vander Perre S, Pauwels L, et al. Adipose tissue protects against sepsis-induced muscle weakness in mice: from lipolysis to ketones. Crit Care (2019) 23:236. doi: 10.1186/s13054-019-2506-6
90. Vandewalle J, Libert C. Sepsis: a failing starvation response. Trends Endocrinol Metab (2022) 33:292–304. doi: 10.1016/j.tem.2022.01.006
91. Wasyluk W, Zwolak A. Metabolic alterations in sepsis. J Clin Med (2021). 10(11):2412. doi: 10.3390/jcm10112412
92. Foley KP, Chen Y, Barra NG, Heal M, Kwok K, Tamrakar AK, et al. Inflammation promotes adipocyte lipolysis via IRE1 kinase. J Biol Chem (2021) 296:100440. doi: 10.1016/j.jbc.2021.100440
93. Schreurs M, Kuipers F, van der Leij FR. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes Rev (2010) 11:380–8. doi: 10.1111/j.1467-789X.2009.00642.x
94. Maitra U, Chang S, Singh N, Li L. Molecular mechanism underlying the suppression of lipid oxidation during endotoxemia. Mol Immunol (2009) 47:420–5. doi: 10.1016/j.molimm.2009.08.023
95. Li Y, Feng YF, Liu XT, Li YC, Zhu HM, Sun MR, et al. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol (2021) 38:101771. doi: 10.1016/j.redox.2020.101771
96. Pham L, Komalavilas P, Eddie AM, Thayer TE, Greenwood DL, Liu KH, et al. Neutrophil trafficking to the site of infection requires Cpt1a-dependent fatty acid β-oxidation. Commun Biol (2022) 5:1366. doi: 10.1038/s42003-022-04339-z
97. Dungan KM, Braithwaite SS, Preiser JC. Stress hyperglycaemia. Lancet (2009) 373:1798–807. doi: 10.1016/S0140-6736(09)60553-5
98. Koliwad SK, Kuo T, Shipp LE, Gray NE, Backhed F, So AY, et al. Angiopoietin-like 4 (ANGPTL4, fasting-induced adipose factor) is a direct glucocorticoid receptor target and participates in glucocorticoid-regulated triglyceride metabolism. J Biol Chem (2009) 284:25593–601. doi: 10.1074/jbc.M109.025452
99. Bose SK, Hutson I, Harris CA. Hepatic glucocorticoid receptor plays a greater role than adipose GR in metabolic syndrome despite renal compensation. Endocrinology (2016) 157:4943–60. doi: 10.1210/en.2016-1615
100. Drosatos K, Drosatos-Tampakaki Z, Khan R, Homma S, Schulze PC, Zannis VI, et al. Inhibition of c-Jun-N-terminal kinase increases cardiac peroxisome proliferator-activated receptor alpha expression and fatty acid oxidation and prevents lipopolysaccharide-induced heart dysfunction. J Biol Chem (2011) 286:36331–9. doi: 10.1074/jbc.M111.272146
101. Standage SW, Bennion BG, Knowles TO, Ledee DR, Portman MA, Mcguire JK, et al. PPARα augments heart function and cardiac fatty acid oxidation in early experimental polymicrobial sepsis. Am J Physiol Heart Circ Physiol (2017) 312:H239–49. doi: 10.1152/ajpheart.00457.2016
102. Iwaki T, Bennion BG, Stenson EK, Lynn JC, Otinga C, Djukovic D, et al. PPARα contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol Rep (2019) 7:e14078. doi: 10.14814/phy2.14078
103. Zhao X, Amevor FK, Cui Z, Wan Y, Xue X, Peng C, et al. Steatosis in metabolic diseases: A focus on lipolysis and lipophagy. BioMed Pharmacother (2023) 160:114311. doi: 10.1016/j.biopha.2023.114311
104. Lee I, Hüttemann M. Energy crisis: the role of oxidative phosphorylation in acute inflammation and sepsis. Biochim Biophys Acta (2014) 1842:1579–86. doi: 10.1016/j.bbadis.2014.05.031
105. Gonçalves-De-Albuquerque CF, Medeiros-De-Moraes IM, Oliveira FM, Burth P, Bozza PT, Castro Faria MV, et al. Omega-9 oleic acid induces fatty acid oxidation and decreases organ dysfunction and mortality in experimental sepsis. PloS One (2016) 11:e0153607. doi: 10.1371/journal.pone.0153607
106. Gonçalves-De-Albuquerque CF, Burth P, Silva AR, De Moraes IM, De Jesus Oliveira FM, Santelli RE, et al. Oleic acid inhibits lung Na/K-ATPase in mice and induces injury with lipid body formation in leukocytes and eicosanoid production. J Inflammation (Lond) (2013) 10:34. doi: 10.1186/1476-9255-10-34
107. Gonçalves-De-Albuquerque CF, Silva AR, Burth P, Castro-Faria MV, Castro-Faria-Neto HC. Acute respiratory distress syndrome: role of oleic acid-triggered lung injury and inflammation. Mediators Inflammation (2015) 2015:260465. doi: 10.1155/2015/260465
108. de Oliveira Rodrigues S, Patricio De Almeida MA, Castro-Faria-Neto HC, Silva AR, Felippe Gonçalves-De-Albuquerque C. Mouse model of oleic acid-induced acute respiratory distress syndrome. J Vis Exp (2022) 184:e63566. doi: 10.3791/63566
109. Wang X, Wang L, Zhu L, Pan Y, Xiao F, Liu W, et al. PAQR3 modulates insulin signaling by shunting phosphoinositide 3-kinase p110α to the Golgi apparatus. Diabetes (2013) 62:444–56. doi: 10.2337/db12-0244
110. Lu GP, Cui P, Cheng Y, Lu ZJ, Zhang LE, Kissoon N. Insulin control of blood glucose and GLUT4 expression in the skeletal muscle of septic rats. West Indian Med J (2015) 64:62–70. doi: 10.7727/wimj.2013.181
111. Fabbri A, Marchesini G, Benazzi B, Morelli A, Montesi D, Bini C, et al. Stress hyperglycemia and mortality in subjects with diabetes and sepsis. Crit Care Explor (2020) 2:e0152. doi: 10.1097/CCE.0000000000000152
112. Leonidou L, Michalaki M, Leonardou A, Polyzogopoulou E, Fouka K, Gerolymos M, et al. Stress-induced hyperglycemia in patients with severe sepsis: a compromising factor for survival. Am J Med Sci (2008) 336:467–71. doi: 10.1097/MAJ.0b013e318176abb4
113. Song M, Meng L, Liu X, Yang Y. Feprazone prevents free fatty acid (FFA)-induced endothelial inflammation by mitigating the activation of the TLR4/myD88/NF-κB pathway. ACS Omega (2021) 6:4850–6. doi: 10.1021/acsomega.0c05826
114. Brodie D, Slutsky AS, Combes A. Extracorporeal life support for adults with respiratory failure and related indications: A review. JAMA (2019) 322:557–68. doi: 10.1001/jama.2019.9302
115. Huang CC, Chou CA, Chen WY, Yang JL, Lee WC, Chen JB, et al. Empagliflozin ameliorates free fatty acid induced-lipotoxicity in renal proximal tubular cells via the PPARγ/CD36 pathway in obese mice. Int J Mol Sci (2021) 22:1–15. doi: 10.3390/ijms222212408
116. Glatz JFC, Luiken J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res (2018) 59:1084–93. doi: 10.1194/jlr.R082933
117. Boscolo A, Campello E, Bertini D, Spiezia L, Lucchetta V, Piasentini E, et al. Levels of circulating microparticles in septic shock and sepsis-related complications: a case-control study. Minerva Anestesiol (2019) 85:625–34. doi: 10.23736/S0375-9393.18.12782-9
118. Li J, Wang X, Ackerman WET, Batty AJ, Kirk SG, White WM, et al. Dysregulation of lipid metabolism in mkp-1 deficient mice during gram-negative sepsis. Int J Mol Sci (2018) 19:1–21. doi: 10.3390/ijms19123904
119. Leelahavanichkul A, Bocharov AV, Kurlander R, Baranova IN, Vishnyakova TG, Souza AC, et al. Class B scavenger receptor types I and II and CD36 targeting improves sepsis survival and acute outcomes in mice. J Immunol (2012) 188:2749–58. doi: 10.4049/jimmunol.1003445
120. Misheva M, Kotzamanis K, Davies LC, Tyrrell VJ, Rodrigues PRS, Benavides GA, et al. Oxylipin metabolism is controlled by mitochondrial β-oxidation during bacterial inflammation. Nat Commun (2022) 13:139. doi: 10.1038/s41467-021-27766-8
121. Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab (2017) 25:262–84. doi: 10.1016/j.cmet.2016.12.022
122. Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci (2008) 1147:37–52. doi: 10.1196/annals.1427.015
123. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
124. Walley KR. Sepsis-induced myocardial dysfunction. Curr Opin Crit Care (2018) 24:292–9. doi: 10.1097/MCC.0000000000000507
125. Lin H, Wang W, Lee M, Meng Q, Ren H. Current status of septic cardiomyopathy: basic science and clinical progress. Front Pharmacol (2020) 11:210. doi: 10.3389/fphar.2020.00210
126. Suffredini AF, Fromm RE, Parker MM, Brenner M, Kovacs JA, Wesley RA, et al. The cardiovascular response of normal humans to the administration of endotoxin. N Engl J Med (1989) 321:280–7. doi: 10.1056/NEJM198908033210503
127. Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res (2006) 72:384–93. doi: 10.1016/j.cardiores.2006.09.011
128. Zhou D, Zhu Y, Ouyang MZ, Zhang M, Tang K, Niu CC, et al. Knockout of Toll-like receptor 4 improves survival and cardiac function in a murine model of severe sepsis. Mol Med Rep (2018) 17:5368–75. doi: 10.3892/mmr.2018.8495
129. Argirò A, Olivotto I. The coronary microcirculation in sepsis: not of micro-importance. Glob Cardiol Sci Pract (2020) 2020:e202030. doi: 10.21542/gcsp.2020.30
130. McBride A, Chanh HQ, Fraser JF, Yacoub S, Obonyo NG. Microvascular dysfunction in septic and dengue shock: Pathophysiology and implications for clinical management. Glob Cardiol Sci Pract (2020) 2020:e202029. doi: 10.21542/gcsp.2020.29
131. Chen X, Liu X, Dong R, Zhang D, Qin S. A retrospective observational study of the association between plasma levels of interleukin 8 in 42 patients with sepsis-induced myocardial dysfunction at a single center between 2017 and 2020. Med Sci Monit (2021) 27:e933065. doi: 10.12659/MSM.933065
132. Eichenholz PW, Eichacker PQ, Hoffman WD, Banks SM, Parrillo JE, Danner RL, et al. Tumor necrosis factor challenges in canines: patterns of cardiovascular dysfunction. Am J Physiol (1992) 263:H668–675. doi: 10.1152/ajpheart.1992.263.3.H668
133. Wang Y, Qian Y, Fang Q, Zhong P, Li W, Wang L, et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat Commun (2017) 8:13997. doi: 10.1038/ncomms13997
134. Goncalves-De-Albuquerque CF, Barnese MRC, Soares MA, Castro-Faria MV, Silva AR, De Castro-Faria-Neto HC, et al. Serum albumin saturation test based on non-esterified fatty acids imbalance for clinical employment. Clin Chim Acta (2019) 495:422–8. doi: 10.1016/j.cca.2019.05.003
135. van de Sandt AM, Windler R, Gödecke A, Ohlig J, Zander S, Reinartz M, et al. Endothelial NOS (NOS3) impairs myocardial function in developing sepsis. Basic Res Cardiol (2013) 108:330. doi: 10.1007/s00395-013-0330-8
136. Khalid N, Patel PD, Alghareeb R, Hussain A, Maheshwari MV. The effect of sepsis on myocardial function: A review of pathophysiology, diagnostic criteria, and treatment. Cureus (2022) 14:e26178. doi: 10.7759/cureus.26178
137. Herbertson MJ, Werner HA, Walley KR. Nitric oxide synthase inhibition partially prevents decreased LV contractility during endotoxemia. Am J Physiol (1996) 270:H1979–1984. doi: 10.1152/ajpheart.1996.270.6.H1979
138. Goddard CM, Allard MF, Hogg JC, Herbertson MJ, Walley KR. Prolonged leukocyte transit time in coronary microcirculation of endotoxemic pigs. Am J Physiol (1995) 269:H1389–1397. doi: 10.1152/ajpheart.1995.269.4.H1389
139. Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, et al. Cardiac myocyte KLF5 regulates ppara expression and cardiac function. Circ Res (2016) 118:241–53. doi: 10.1161/CIRCRESAHA.115.306383
140. Kolwicz SC Jr., Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res (2013) 113:603–16. doi: 10.1161/CIRCRESAHA.113.302095
141. Dhainaut JF, Huyghebaert MF, Monsallier JF, Lefevre G, Dall'ava-Santucci J, Brunet F, et al. Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation (1987) 75:533–41. doi: 10.1161/01.CIR.75.3.533
142. Lv X, Wang H. Pathophysiology of sepsis-induced myocardial dysfunction. Mil Med Res (2016) 3:30. doi: 10.1186/s40779-016-0099-9
143. Jia L, Takahashi M, Morimoto H, Takahashi S, Izawa A, Ise H, et al. Changes in cardiac lipid metabolism during sepsis: the essential role of very low-density lipoprotein receptors. Cardiovasc Res (2006) 69:545–55. doi: 10.1016/j.cardiores.2005.11.014
144. Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, et al. Peroxisome proliferator-activated receptor-γ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail (2013) 6:550–62. doi: 10.1161/CIRCHEARTFAILURE.112.000177
145. Drosatos K, Lymperopoulos A, Kennel PJ, Pollak N, Schulze PC, Goldberg IJ. Pathophysiology of sepsis-related cardiac dysfunction: driven by inflammation, energy mismanagement, or both? Curr Heart Fail Rep (2015) 12:130–40. doi: 10.1007/s11897-014-0247-z
146. Lu B, Moser A, Shigenaga JK, Grunfeld C, Feingold KR. The acute phase response stimulates the expression of angiopoietin like protein 4. Biochem Biophys Res Commun (2010) 391:1737–41. doi: 10.1016/j.bbrc.2009.12.145
147. Kaplan J, Nowell M, Chima R, Zingarelli B. Pioglitazone reduces inflammation through inhibition of NF-κB in polymicrobial sepsis. Innate Immun (2014) 20:519–28. doi: 10.1177/1753425913501565
148. Silva AR, Gonçalves-De-Albuquerque CF, Pérez AR, Carvalho VF. Immune-endocrine interactions related to a high risk of infections in chronic metabolic diseases: The role of PPAR gamma. Eur J Pharmacol (2019) 854:272–81. doi: 10.1016/j.ejphar.2019.04.008
149. Pan P, Wang X, Liu D. The potential mechanism of mitochondrial dysfunction in septic cardiomyopathy. J Int Med Res (2018) 46:2157–69. doi: 10.1177/0300060518765896
150. Cimolai MC, Alvarez S, Bode C, Bugger H. Mitochondrial mechanisms in septic cardiomyopathy. Int J Mol Sci (2015) 16:17763–78. doi: 10.3390/ijms160817763
151. Stanzani G, Duchen MR, Singer M. The role of mitochondria in sepsis-induced cardiomyopathy. Biochim Biophys Acta Mol Basis Dis (2019) 1865:759–73. doi: 10.1016/j.bbadis.2018.10.011
152. Fattahi F, Ward PA. Complement and sepsis-induced heart dysfunction. Mol Immunol (2017) 84:57–64. doi: 10.1016/j.molimm.2016.11.012
153. Fattahi F, Kalbitz M, Malan EA, Abe E, Jajou L, Huber-Lang MS, et al. Complement-induced activation of MAPKs and Akt during sepsis: role in cardiac dysfunction. FASEB J (2017) 31:4129–39. doi: 10.1096/fj.201700140R
154. Nong Y, Wei X, Yu D. Inflammatory mechanisms and intervention strategies for sepsis-induced myocardial dysfunction. Immun Inflammation Dis (2023) 11:e860. doi: 10.1002/iid3.860
155. Kalbitz M, Fattahi F, Herron TJ, Grailer JJ, Jajou L, Lu H, et al. Complement destabilizes cardiomyocyte function in vivo after polymicrobial sepsis and in vitro. J Immunol (2016) 197:2353–61. doi: 10.4049/jimmunol.1600091
156. Abe T, Kubo K, Izumoto S, Shimazu S, Goan A, Tanaka T, et al. Complement activation in human sepsis is related to sepsis-induced disseminated intravascular coagulation. Shock (2020) 54:198–204. doi: 10.1097/SHK.0000000000001504
157. Liu W, Hu C, Zhao S. Sarcopenia and mortality risk of patients with sepsis: A meta-analysis. Int J Clin Pract (2022) 2022:4974410. doi: 10.1155/2022/4974410
158. Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol (2005) 288:R345–353. doi: 10.1152/ajpregu.00454.2004
159. Costamagna D, Costelli P, Sampaolesi M, Penna F. Role of inflammation in muscle homeostasis and myogenesis. Mediators Inflammation (2015) 2015:805172. doi: 10.1155/2015/805172
160. Thoma A, Lightfoot AP. NF-kB and inflammatory cytokine signalling: role in skeletal muscle atrophy. Adv Exp Med Biol (2018) 1088:267–79. doi: 10.1007/978-981-13-1435-3_12
161. Kny M, Fielitz J. Hidden agenda - the involvement of endoplasmic reticulum stress and unfolded protein response in inflammation-induced muscle wasting. Front Immunol (2022) 13:878755. doi: 10.3389/fimmu.2022.878755
162. Haberecht-Müller S, Krüger E, Fielitz J. Out of control: the role of the ubiquitin proteasome system in skeletal muscle during inflammation. Biomolecules (2021) 11:1–42. doi: 10.3390/biom11091327
163. Huh JY, Panagiotou G, Mougios V, Brinkoetter M, Vamvini MT, Schneider BE, et al. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metabolism (2012) 61:1725–38. doi: 10.1016/j.metabol.2012.09.002
164. Alves HR, Lomba GSB, Gonçalves-De-Albuquerque CF, Burth P. Irisin, exercise, and COVID-19. Front Endocrinol (Lausanne) (2022) 13:879066. doi: 10.3389/fendo.2022.879066
165. Srivastava B, Gimson A. Hepatic changes in systemic infection. Best Pract Res Clin Gastroenterol (2013) 27:485–95. doi: 10.1016/j.bpg.2013.06.011
166. Yan J, Li S. The role of the liver in sepsis. Int Rev Immunol (2014) 33:498–510. doi: 10.3109/08830185.2014.889129
167. Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol (2012) 12:201–13. doi: 10.1038/nri3169
168. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology (2014) 146:1513–24. doi: 10.1053/j.gastro.2014.01.020
169. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol (2017) 14:55–66. doi: 10.1038/nrgastro.2016.168
170. Englert JA, Rogers AJ. Metabolism, metabolomics, and nutritional support of patients with sepsis. Clin Chest Med (2016) 37:321–31. doi: 10.1016/j.ccm.2016.01.011
171. Chimenti MS, Triggianese P, Conigliaro P, Candi E, Melino G, Perricone R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis (2015) 6:e1887. doi: 10.1038/cddis.2015.246
172. Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res (2020) 126:1549–64. doi: 10.1161/CIRCRESAHA.119.315896
173. Andreozzi F, Laratta E, Procopio C, Hribal ML, Sciacqua A, Perticone M, et al. Interleukin-6 impairs the insulin signaling pathway, promoting production of nitric oxide in human umbilical vein endothelial cells. Mol Cell Biol (2007) 27:2372–83. doi: 10.1128/MCB.01340-06
174. Podbielska M, Szulc ZM, Kurowska E, Hogan EL, Bielawski J, Bielawska A, et al. Cytokine-induced release of ceramide-enriched exosomes as a mediator of cell death signaling in an oligodendroglioma cell line. J Lipid Res (2016) 57:2028–39. doi: 10.1194/jlr.M070664
175. Mecatti GC, Fernandes Messias MC, Sant'anna Paiola RM, Figueiredo Angolini CF, Da Silva Cunha IB, Eberlin MN, et al. Lipidomic profiling of plasma and erythrocytes from septic patients reveals potential biomarker candidates. biomark Insights (2018) 13:1177271918765137. doi: 10.1177/1177271918765137
176. Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest (2011) 121:1858–70. doi: 10.1172/JCI43378
177. Chaurasia B, Talbot CL, Summers SA. Adipocyte ceramides-the nexus of inflammation and metabolic disease. Front Immunol (2020) 11:576347. doi: 10.3389/fimmu.2020.576347
178. Smith AB, Schill JP, Gordillo R, Gustafson GE, Rhoads TW, Burhans MS, et al. Ceramides are early responders in metabolic syndrome development in rhesus monkeys. Sci Rep (2022) 12:9960. doi: 10.1038/s41598-022-14083-3
179. Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, et al. Metabolic adaptation establishes disease tolerance to sepsis. Cell (2017) 169:1263–1275.e1214. doi: 10.1016/j.cell.2017.05.031
180. Vandewalle J, Timmermans S, Paakinaho V, Vancraeynest L, Dewyse L, Vanderhaeghen T, et al. Combined glucocorticoid resistance and hyperlactatemia contributes to lethal shock in sepsis. Cell Metab (2021) 33:1763–1776.e1765. doi: 10.1016/j.cmet.2021.07.002
181. Tao J, Hu Z, Lou F, Wu J, Wu Z, Yang S, et al. Higher stress hyperglycemia ratio is associated with a higher risk of stroke-associated pneumonia. Front Nutr (2022) 9:784114. doi: 10.3389/fnut.2022.784114
182. Gornik I, Vujaklija A, Lukić E, Madžarac G, Gašparović V. Hyperglycemia in sepsis is a risk factor for development of type II diabetes. J Crit Care (2010) 25:263–9. doi: 10.1016/j.jcrc.2009.10.002
183. Gunst J, Van den Berghe G. Blood glucose control in the ICU: don't throw out the baby with the bathwater! Intensive Care Med (2016) 42:1478–81. doi: 10.1007/s00134-016-4350-3
184. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345:1250684. doi: 10.1126/science.1250684
185. Tseng PL, Wu WH, Hu TH, Chen CW, Cheng HC, Li CF, et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci Rep (2018) 8:3081. doi: 10.1038/s41598-018-21361-6
186. Wang X, Wang Z, Tang D. Aerobic exercise improves LPS-induced sepsis via regulating the Warburg effect in mice. Sci Rep (2021) 11:17772. doi: 10.1038/s41598-021-97101-0
187. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
188. Gómez H, Kellum JA, Ronco C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat Rev Nephrol (2017) 13:143–51. doi: 10.1038/nrneph.2016.186
189. Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol (2009) 182:2476–84. doi: 10.4049/jimmunol.0802059
190. Tretter L, Patocs A, Chinopoulos C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta (BBA) - Bioenergetics (2016) 1857:1086–101. doi: 10.1016/j.bbabio.2016.03.012
191. Harber KJ, De Goede KE, Verberk SGS, Meinster E, De Vries HE, Van Weeghel M, et al. Succinate is an inflammation-induced immunoregulatory metabolite in macrophages. Metabolites (2020) 10:1–14. doi: 10.3390/metabo10090372
192. Nogueira AC, Kawabata V, Biselli P, Lins MH, Valeri C, Seckler M, et al. Changes in plasma free fatty acid levels in septic patients are associated with cardiac damage and reduction in heart rate variability. Shock (2008) 29:342–8. doi: 10.1097/SHK.0b013e31815abbc6
193. Rittig N, Bach E, Thomsen HH, Pedersen SB, Nielsen TS, Jørgensen JO, et al. Regulation of lipolysis and adipose tissue signaling during acute endotoxin-induced inflammation: A human randomized crossover trial. PloS One (2016) 11:e0162167. doi: 10.1371/journal.pone.0162167
194. Şimşek T, Şimşek HU, Cantürk NZ. Response to trauma and metabolic changes: posttraumatic metabolism. Ulus Cerrahi Derg (2014) 30:153–9 doi: 10.5152/UCD.2014.2653.
195. Hamzaoui O, Shi R. Early norepinephrine use in septic shock. J Thorac Dis (2020) 12:S72–7. doi: 10.21037/jtd.2019.12.50
196. Raje V, Ahern KW, Martinez BA, Howell NL, Oenarto V, Granade ME, et al. Adipocyte lipolysis drives acute stress-induced insulin resistance. Sci Rep (2020) 10:18166. doi: 10.1038/s41598-020-75321-0
197. Scholl RA, Lang CH, Bagby GJ. Hypertriglyceridemia and its relation to tissue lipoprotein lipase activity in endotoxemic, Escherichia coli bacteremic, and polymicrobial septic rats. J Surg Res (1984) 37:394–401. doi: 10.1016/0022-4804(84)90205-1
198. van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr (2017) 1859:1558–72. doi: 10.1016/j.bbamem.2017.04.006
199. Khaliq W, Großmann P, Neugebauer S, Kleyman A, Domizi R, Calcinaro S, et al. Lipid metabolic signatures deviate in sepsis survivors compared to non-survivors. Comput Struct Biotechnol J (2020) 18:3678–91. doi: 10.1016/j.csbj.2020.11.009
200. Smith RL, Soeters MR, Wüst RCI, Houtkooper RH. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr Rev (2018) 39:489–517. doi: 10.1210/er.2017-00211
201. Warden A, Truitt J, Merriman M, Ponomareva O, Jameson K, Ferguson LB, et al. Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep (2016) 6:27618. doi: 10.1038/srep27618
202. Sasaki Y, Asahiyama M, Tanaka T, Yamamoto S, Murakami K, Kamiya W, et al. Pemafibrate, a selective PPARα modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci Rep (2020) 10:7818. doi: 10.1038/s41598-020-64902-8
203. Suh JH, Kim KH, Conner ME, Moore DD, Preidis GA. Hepatic PPARα Is destabilized by SIRT1 deacetylase in undernourished male mice. Front Nutr (2022) 9:831879. doi: 10.3389/fnut.2022.831879
204. Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest (2012) 122:1958–9. doi: 10.1172/JCI63967
205. Paumelle R, Haas JT, Hennuyer N, Baugé E, Deleye Y, Mesotten D, et al. Hepatic PPARα is critical in the metabolic adaptation to sepsis. J Hepatol (2019) 70:963–73. doi: 10.1016/j.jhep.2018.12.037
206. Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell (2016) 166:1512–1525.e1512. doi: 10.1016/j.cell.2016.07.026
207. Gough SM, Casella A, Ortega KJ, Hackam AS. Neuroprotection by the ketogenic diet: evidence and controversies. Front Nutr (2021) 8:782657. doi: 10.3389/fnut.2021.782657
208. Standage SW, Caldwell CC, Zingarelli B, Wong HR. Reduced peroxisome proliferator-activated receptor α expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock (2012) 37:164–9. doi: 10.1097/SHK.0b013e31823f1a00
209. Yoo SH, Abdelmegeed MA, Song BJ. Activation of PPARα by Wy-14643 ameliorates systemic lipopolysaccharide-induced acute lung injury. Biochem Biophys Res Commun (2013) 436:366–71. doi: 10.1016/j.bbrc.2013.05.073
210. Hill MR, Clarke S, Rodgers K, Thornhill B, Peters JM, Gonzalez FJ, et al. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect Immun (1999) 67:3488–93. doi: 10.1128/IAI.67.7.3488-3493.1999
211. Zingarelli B, Cook JA. Peroxisome proliferator-activated receptor-gamma is a new therapeutic target in sepsis and inflammation. Shock (2005) 23:393–9. doi: 10.1097/01.shk.0000160521.91363.88
212. Francis MR, El-Sheakh AR, Suddek GM. Saroglitazar, a dual PPAR-α/γ agonist, alleviates LPS-induced hepatic and renal injury in rats. Int Immunopharmacol (2023) 115:109688. doi: 10.1016/j.intimp.2023.109688
213. Silva-Veiga FM, Miranda CS, Martins FF, Daleprane JB, Mandarim-De-Lacerda CA, Souza-Mello V. Gut-liver axis modulation in fructose-fed mice: a role for PPAR-alpha and linagliptin. J Endocrinol (2020) 247:11–24. doi: 10.1530/JOE-20-0139
214. Lv M, Xie D, Long X. The effect of fenofibrate, a peroxisome proliferator-activated receptor α agonist, on cardiac damage from sepsis in BALB/c mice. Cell Mol Biol (Noisy-le-grand) (2022) 67:260–6. doi: 10.14715/cmb/2021.67.6.34
215. Zhao H, Wang Y, Zhu X. Chrysophanol exerts a protective effect against sepsis-induced acute myocardial injury through modulating the microRNA-27b-3p/Peroxisomal proliferating-activated receptor gamma axis. Bioengineered (2022) 13:12673–90. doi: 10.1080/21655979.2022.2063560
216. Bai Y, Han Q, Dong B, Lin H, Jiang Y, Zhang X, et al. PPARα contributes to the therapeutic effect of hydrogen gas against sepsis-associated encephalopathy with the regulation to the CREB-BDNF signaling pathway and hippocampal neuron plasticity-related gene expression. Brain Res Bull (2022) 184:56–67. doi: 10.1016/j.brainresbull.2022.03.015
217. Wang C, Han D, Feng X, Wu J. Omega-3 fatty acid supplementation is associated with favorable outcomes in patients with sepsis: an updated meta-analysis. J Int Med Res (2020) 48:300060520953684. doi: 10.1177/0300060520953684
218. Erdem D, Segmen F, Uysal E, Kilicarslan G. Effect of omega-3 fatty acid use on sepsis and mortality in patients with Covıd-19. Niger. J Clin Pract (2023) 26:102–8. doi: 10.4103/njcp.njcp_415_22
219. Kulkarni AV, Anand L, Vyas AK, Premkumar M, Choudhury AK, Trehanpati N, et al. Omega-3 fatty acid lipid emulsions are safe and effective in reducing endotoxemia and sepsis in acute-on-chronic liver failure: An open-label randomized controlled trial. J Gastroenterol Hepatol (2021) 36:1953–61. doi: 10.1111/jgh.15400
220. Caparrós E, Juanola O, Gómez-Hurtado I, Puig-Kroger A, Piñero P, Zapater P, et al. Liver sinusoidal endothelial cells contribute to hepatic antigen-presenting cell function and th17 expansion in cirrhosis. Cells (2020) 9:1–19. doi: 10.3390/cells9051227
221. Ito Y, Abril ER, Bethea NW, Mccuskey MK, Cover C, Jaeschke H, et al. Mechanisms and pathophysiological implications of sinusoidal endothelial cell gap formation following treatment with galactosamine/endotoxin in mice. Am J Physiol Gastrointest Liver Physiol (2006) 291:G211–218. doi: 10.1152/ajpgi.00312.2005
222. Zapotoczny B, Szafranska K, Kus E, Braet F, Wisse E, Chlopicki S, et al. Tracking fenestrae dynamics in live murine liver sinusoidal endothelial cells. Hepatology (2019) 69:876–88. doi: 10.1002/hep.30232
223. Spapen H. Liver perfusion in sepsis, septic shock, and multiorgan failure. Anat Rec (Hoboken) (2008) 291:714–20. doi: 10.1002/ar.20646
224. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
225. Jackson MA, Patel SS, Yu F, Cottam MA, Glass EB, Hoogenboezem EN, et al. Kupffer cell release of platelet activating factor drives dose limiting toxicities of nucleic acid nanocarriers. Biomaterials (2021) 268:120528. doi: 10.1016/j.biomaterials.2020.120528
226. Tsutsui H, Nishiguchi S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int J Mol Sci (2014) 15:7711–30. doi: 10.3390/ijms15057711
227. Horton JD. Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis. Biochem Soc Trans (2002) 30:1091–5. doi: 10.1042/bst0301091
228. Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Lièpvre X, et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol (1999) 19:3760–8. doi: 10.1128/MCB.19.5.3760
229. Huang SSY, Toufiq M, Saraiva LR, Van Panhuys N, Chaussabel D, Garand M. Transcriptome and literature mining highlight the differential expression of ERLIN1 in immune cells during sepsis. Biol (Basel) (2021) 10:1–28. doi: 10.3390/biology10080755
230. Dembek A, Laggai S, Kessler SM, Czepukojc B, Simon Y, Kiemer AK, et al. Hepatic interleukin-6 production is maintained during endotoxin tolerance and facilitates lipid accumulation. Immunobiology (2017) 222:786–96. doi: 10.1016/j.imbio.2017.01.003
231. Gentile F, Arcaro A, Pizzimenti S, Daga M, Cetrangolo GP, Dianzani C, et al. DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genet (2017) 4:103–37. doi: 10.3934/genet.2017.2.103
232. Aranda-Rivera AK, Cruz-Gregorio A, Arancibia-Hernández YL, Hernández-Cruz EY, Pedraza-Chaverri J. RONS and oxidative stress: an overview of basic concepts. Oxygen (2022) 2:1–42. doi: 10.3390/oxygen2040030
233. Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci USA (2014) 111:9597–602. doi: 10.1073/pnas.1409229111
234. Ciregia F, Bugliani M, Ronci M, Giusti L, Boldrini C, Mazzoni MR, et al. Palmitate-induced lipotoxicity alters acetylation of multiple proteins in clonal β cells and human pancreatic islets. Sci Rep (2017) 7:13445. doi: 10.1038/s41598-017-13908-w
235. Ogawa Y, Imajo K, Honda Y, Kessoku T, Tomeno W, Kato S, et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci Rep (2018) 8:11365. doi: 10.1038/s41598-018-29735-6
236. Bandet CL, Tan-Chen S, Bourron O, Le Stunff H, Hajduch E. Sphingolipid metabolism: new insight into ceramide-induced lipotoxicity in muscle cells. Int J Mol Sci (2019) 20:1–26. doi: 10.3390/ijms20030479
237. Saberi M, Woods NB, De Luca C, Schenk S, Lu JC, Bandyopadhyay G, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab (2009) 10:419–29. doi: 10.1016/j.cmet.2009.09.006
238. Idrovo JP, Yang WL, Jacob A, Corbo L, Nicastro J, Coppa GF, et al. Inhibition of lipogenesis reduces inflammation and organ injury in sepsis. J Surg Res (2016) 200:242–9. doi: 10.1016/j.jss.2015.06.059
239. Nabben M, Hoeks J, Briedé JJ, Glatz JFC, Moonen-Kornips E, Hesselink MKC, et al. The effect of UCP3 overexpression on mitochondrial ROS production in skeletal muscle of young versus aged mice. FEBS Lett (2008) 582:4147–52. doi: 10.1016/j.febslet.2008.11.016
240. Čater M, Križančić Bombek L. Protective role of mitochondrial uncoupling proteins against age-related oxidative stress in type 2 diabetes mellitus. Antioxidants (Basel) (2022) 11:1–21. doi: 10.3390/antiox11081473
241. Corcoran TB, Engel A, Sakamoto H, O'shea A, O'callaghan-Enright S, Shorten GD. The effects of propofol on neutrophil function, lipid peroxidation and inflammatory response during elective coronary artery bypass grafting in patients with impaired ventricular function. Br J Anaesthesia (2006) 97:825–31. doi: 10.1093/bja/ael270
242. Liu Y-C, Chang AYW, Tsai Y-C, Chan JYH. Differential protection against oxidative stress and nitric oxide overproduction in cardiovascular and pulmonary systems by propofol during endotoxemia. J Biomed Sci (2009) 16:8. doi: 10.1186/1423-0127-16-8
243. Romuk E, Szczurek W, Nowak P, Kwiecień I, Stolecka D, Birkner E. Influence of propofol on oxidative-antioxidative system parameters in peripheral organs of rats with Parkinson disease. Postepy Hig Med Dosw (Online) (2015) 69:661–7. doi: 10.5604/17322693.1156935
244. Jain S, Gautam V, Naseem S. Acute-phase proteins: As diagnostic tool. J Pharm Bioallied Sci (2011) 3:118–27. doi: 10.4103/0975-7406.76489
245. Carvalho JR, Machado MV. New insights about albumin and liver disease. Ann Hepatol (2018) 17:547–60. doi: 10.5604/01.3001.0012.0916
246. Omiya K, Sato H, Sato T, Wykes L, Hong M, Hatzakorzian R, et al. Albumin and fibrinogen kinetics in sepsis: a prospective observational study. Crit Care (2021) 25:436. doi: 10.1186/s13054-021-03860-7
247. Burth P, Younes-Ibrahim M, Santos MC, Castro-Faria Neto HC, De Castro Faria MV. Role of nonesterified unsaturated fatty acids in the pathophysiological processes of leptospiral infection. J Infect Dis (2005) 191:51–7. doi: 10.1086/426455
248. Haeri HH, Schunk B, Tomaszewski J, Schimm H, Gelos MJ, Hinderberger D. Fatty acid binding to human serum albumin in blood serum characterized by EPR spectroscopy. ChemistryOpen (2019) 8:650–6. doi: 10.1002/open.201900113
249. Kelly RA, O'hara DS, Mitch WE, Smith TW. Identification of NaK-ATPase inhibitors in human plasma as nonesterified fatty acids and lysophospholipids. J Biol Chem (1986) 261:11704–11. doi: 10.1016/S0021-9258(18)67301-6
250. Goncalves-De-Albuquerque CF, Burth P, Silva AR, De Moraes IM, De Oliveira FM, Santelli RE, et al. Na/K-ATPase assay in the intact mice lung subjected to perfusion. BMC Res Notes (2014) 7:798. doi: 10.1186/1756-0500-7-798
Keywords: fatty acid oxidation (FAO), inflammation, metabolic dysfunction, free fatty acid (FFA), sepsis
Citation: Muniz-Santos R, Lucieri-Costa G, Almeida MAP, Moraes-de-Souza I, Brito MASM, Silva AR and Gonçalves-de-Albuquerque CF (2023) Lipid oxidation dysregulation: an emerging player in the pathophysiology of sepsis. Front. Immunol. 14:1224335. doi: 10.3389/fimmu.2023.1224335
Received: 17 May 2023; Accepted: 30 June 2023;
Published: 03 August 2023.
Edited by:
Praveen Papareddy, Lund University, SwedenReviewed by:
Jingbo Pang, University of Illinois Chicago, United StatesChristian Karl Braun, Ulm University Medical Center, Germany
Lynn Xiaoling Qiang, Northwell Health, United States
Copyright © 2023 Muniz-Santos, Lucieri-Costa, Almeida, Moraes-de-Souza, Brito, Silva and Gonçalves-de-Albuquerque. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cassiano Felippe Gonçalves-de-Albuquerque, Y2Fzc2lhbm8uYWxidXF1ZXJxdWVAdW5pcmlvLmJy