 Merijn Braams
Merijn Braams Karin Pike-Overzet
Karin Pike-Overzet Frank J. T. Staal
Frank J. T. Staal
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 11 July 2023
Sec. Primary Immunodeficiencies
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1210818
The mature lymphocyte population of a healthy individual has the remarkable ability to recognise an immense variety of antigens. Instead of encoding a unique gene for each potential antigen receptor, evolution has used gene rearrangements, also known as variable, diversity, and joining gene segment (V(D)J) recombination. This process is critical for lymphocyte development and relies on recombination-activating genes-1 (RAG1) and RAG2, here collectively referred to as RAG. RAG serves as powerful genome editing tools for lymphocytes and is strictly regulated to prevent dysregulation. However, in the case of dysregulation, RAG has been implicated in cases of cancer, autoimmunity and severe combined immunodeficiency (SCID). This review examines functional protein domains and motifs of RAG, describes advances in our understanding of the function and (dys)regulation of RAG, discuss new therapeutic options, such as gene therapy, for RAG deficiencies, and explore in vitro and in vivo methods for determining RAG activity and target specificity.
Throughout an individual’s lifetime, the immune system is exposed to numerous foreign antigens that must be promptly cleared before they can inflict substantial damage. While the innate immune response is often capable of handling such intruders without requiring additional support, there are instances where the adaptive immune response must coordinate a more elaborate defence strategy. To accomplish this, T and B lymphocytes from the adaptive immune response must be equipped to launch a response against any possible foreign agent that may invade the body.
Rather than encoding separate genes for each possible antigen receptor, the immune system has “invented” a mechanism of DNA rearrangement, known as V(D)J recombination. This mechanism allows for the antigen recognition gene segments of lymphocytes to be modified, creating one of the largest biological information banks in the world, capable of generating a vast repertoire of trillions of possible combinations (1). At the heart of this process lie RAG1 and RAG2, collectively referred to as RAG.
The RAG complex is a unique endonuclease that is responsible for inducing intentional DNA double-strand breaks (DSBs). RAG does so specifically around certain nonamer and heptamer sequences referred to as recombination signal sequences (RSSs) that flank the V, D and J segments in the genome (1, 2). By excising different variants of V(D)J segments, RAG (along with DNA repair mechanisms) creates coding joints that code for specific B-cell receptor (BCR) or T-cell receptor (TCR) gene segments. Thereby, generating clonal diversity (see Figure 1) from a relatively short piece of DNA when taking into perspective the number of different receptor possibilities (1, 2). As a result of its critical role in the V(D)J recombination process, RAG is indispensable for lymphocyte development. Loss-of-function mutations in RAG can completely block lymphocyte development at an early stage, leading to SCID (3, 4). On the other hand, dysregulation of RAG has been associated with autoimmunity and RAG-mediated oncogenic fusion genes that promote blood-borne cancer formations such as acute lymphoblastic leukaemia (ALL) (5). Therefore, the regulation of RAG is of the utmost importance to prevent dysregulation and adverse outcomes.
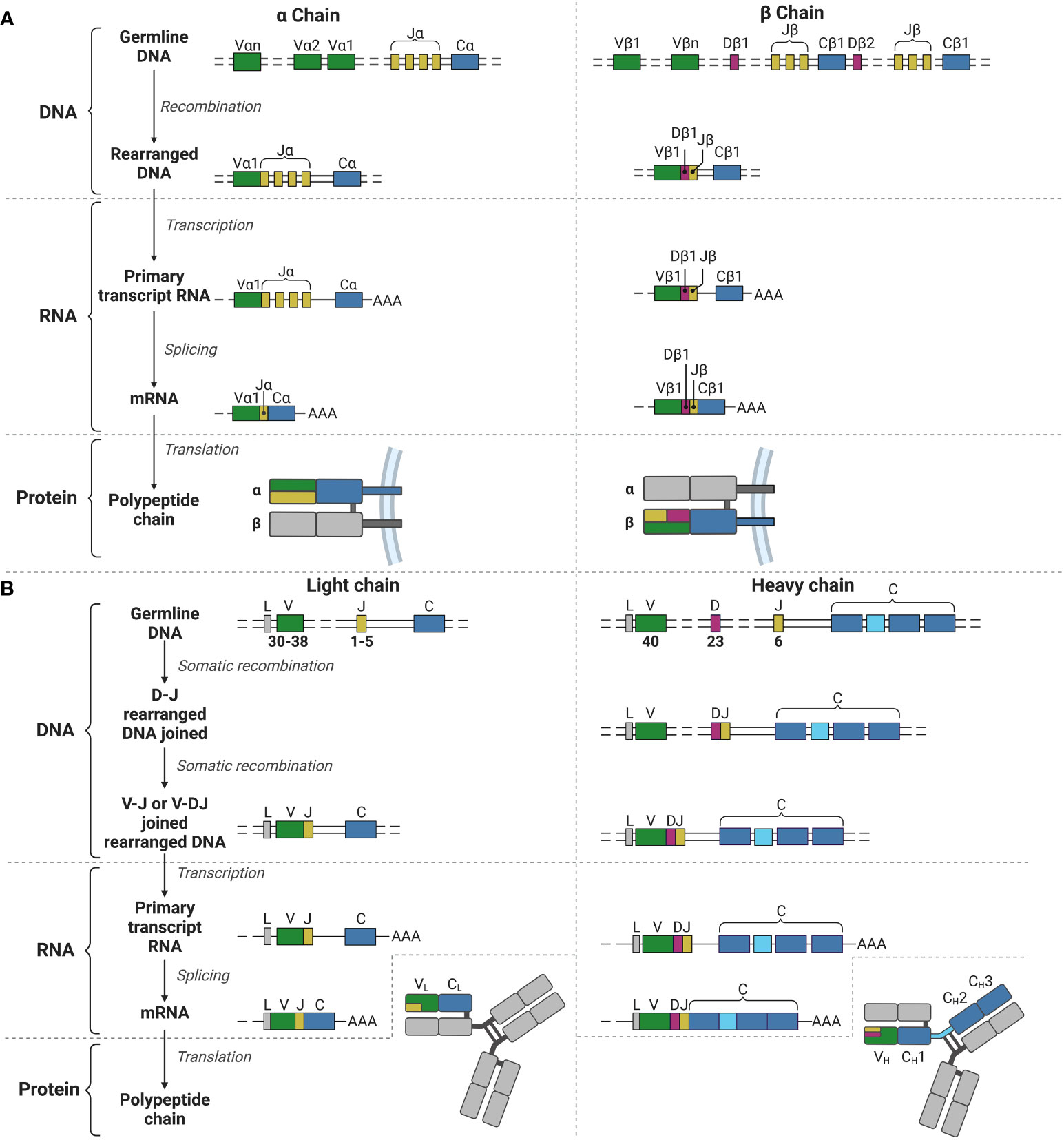
Figure 1 V(D)J recombination for the TCR α and β chains and the BCR light and heavy chains. (A) V(D)J recombination for the α (left) and β (right) chains for the TCR in T lymphocyte development. (B) V(D)J recombination for the light (left) and heavy (right) chains for the BCR (and immunoglobulins) in B lymphocyte development. V, Variable; J, joining; C, constant; D, diversity; L, leader; AAA, poly-A-tail. Created with BioRender.com.
The RAG1 gene is situated on chromosome 11p13 of the human genome and encodes the RAG1 protein which comprises 1.043 amino acids (aa) (6–8). The RAG1 protein can be subdivided into the N-terminal non-core region (aa 1-384), the core region (aa 384-1.008), and a short C-terminal non-core region (aa 1.008-1.040) (see Figure 2A) (2, 7, 8). The N-terminal non-core region contains nucleolar export and import domains, as well as a zinc dimerization and RING domain (2, 8–10). The nucleolar export and import domains regulate RAG1 protein levels as it moves in and out of the nucleus, where it exerts its function on the genome (11). The zinc dimerization domain (ZDD) comprises zinc-binding motifs in the form of zinc finger sequences that allow for homodimer formation (2, 12). The RING domain has a function in histone H3 monoubiquitylation and plays a role in V(D)J recombination activity (2, 10). Additionally, the N-terminal region is essential for full RAG1 activity with recombination enhancing domains and is shown to interact with several more proteins, including transcription and nuclear localisation factors, as well as non-homologous end-joining (NHEJ) components (2, 11, 13, 14).
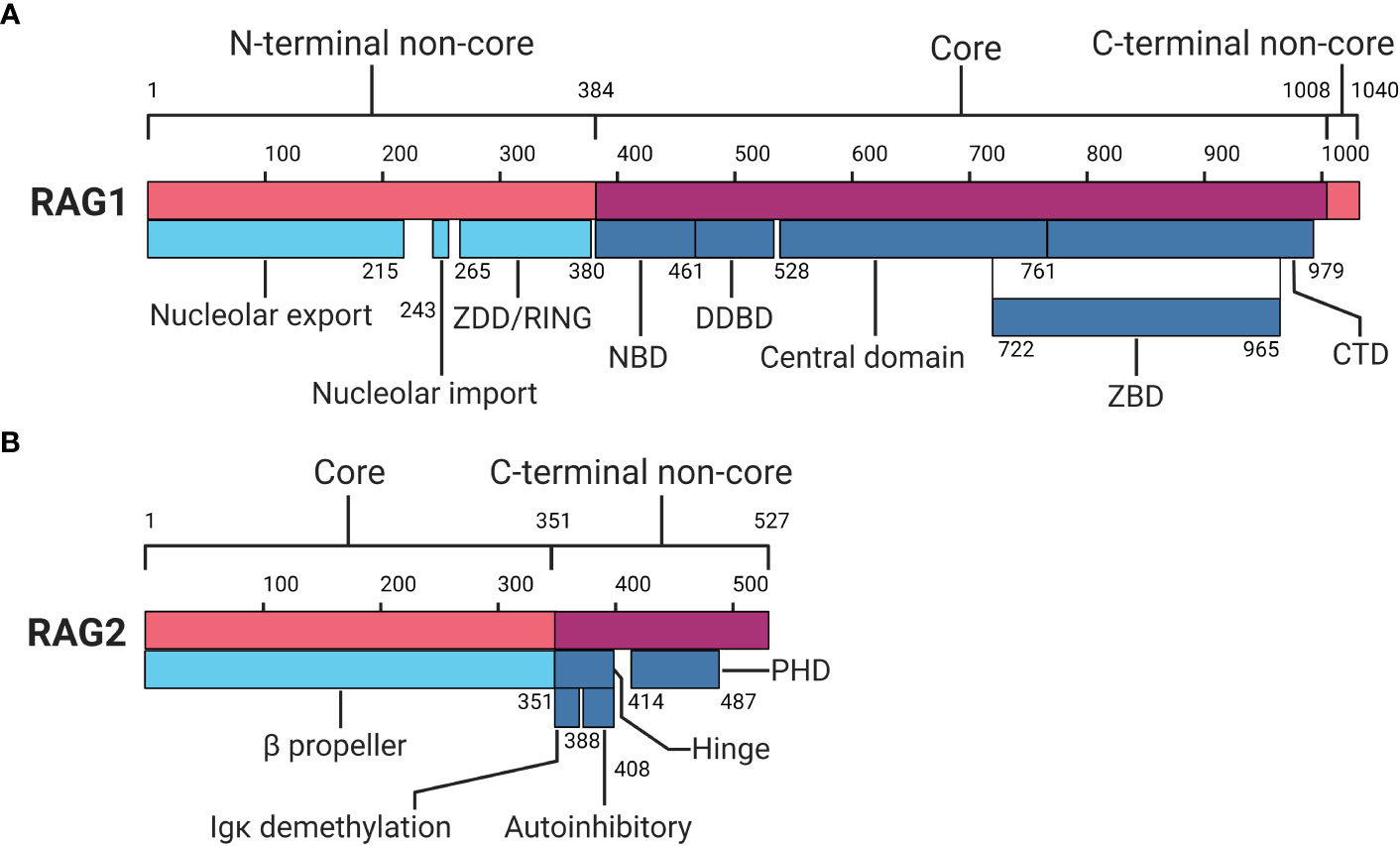
Figure 2 Protein domain map of RAG1 and RAG2. (A) RAG1 protein domain map divided in an N-terminal non-core, core and C-terminal non-core region. (B) RAG2 protein domain map divided in a core and C-terminal non-core region. RAG1, Recombination-activating gene-1;ZDD, zinc dimerization domain; NBD, nonamer binding domain; DDBD, dimerization and DNA binding domain; ZBD, zinc binding domain; CTD, C-terminal domain; RAG2, Recombination-activating gene-2; PHD, plant homeodomain. Numbers indicate amino acid positions. Inspired by Schatz and Swanson (2) and Christie et al (8). Created with BioRender.com.
The catalytic centre of RAG1 is located in the core region, which comprises the nonamer binding domain (NBD), the dimerization and DNA binding domain (DDBD), and the central domain holding the catalytic core. The central domain includes the heptamer binding region and shares a zinc binding domain (ZBD) with a C-terminal domain (CTD) within the core region (2, 7, 8). The NBD recognises and interacts with the nonamer sequence of the RSS, providing part of the specificity for RAG activity (2, 15–17). The DDBD has a role in binding DNA and provides another homodimerization domain for RAG1 (7, 8). The central domain holds motifs responsible for nicking ssDNA and recognising and interacting with the heptamer RSS (2, 8, 18, 19). The CTD functions as a dsDNA binding domain, although it does so non-specifically, relying on other domains and motifs for specificity (2, 8, 18, 20). Shared between the central domain and CTD, a ZBD is located which holds two zinc binding regions that interact with RAG2 (2, 8, 21, 22). Finally, the C-terminal non-core region, though small, has been reported to inhibit hairpin formation and modulate binding and cleavage activity together with the C terminus of RAG2 (2, 8, 23).
The RAG2 gene is also located on chromosome 11p13, encoding the RAG2 protein comprising 527 aa (2, 7, 8, 24). Similar to RAG1, RAG2 can be divided into two distinct regions: the core region (aa 1-351) and the C-terminal non-core region (aa 352-527) (see Figure 2B) (2, 7, 8). The core region of RAG2 contains a six bladed β-propeller shaped by six Kelch-like motifs, which enables efficient DNA cleavage and establishes a connection with RAG1 (2, 7, 8, 21). The non-core domain comprises a hinge and a plant homeodomain (PHD) (7, 25, 26). The hinge domain provides RAG2 with a flexible connection between the core region and the PHD. This flexibility is essential for RAG’s recombination activity, as research indicates that neutralisation of the hinge region, which is relatively acidic, increases genomic instability (8, 26). Within the hinge domain reside two functional regions important in recombination: the immunoglobulin κ (Igκ) demethylation motif and autoinhibitory motif (8, 27, 28). Demethylation of the Igκ locus by RAG2 may contribute to allelic exclusion, limiting additional local recombination (8). The autoinhibition motif is regulated by binding of histone H3 on lysine 4 containing a tri-methylation (H3K4me3) (28). Relief of autoinhibition by H3K4me3 triggers DNA cleavage and recombination by the PHD in RAG2 (8, 28–30). At the C-terminal non-core region of RAG2, degradation of RAG2 is promoted by phosphorylation, and additionally by ubiquitination by Skp2 ubiquitin ligase in a cell-cycle dependent manner (2, 8, 31, 32).
In the absence of DNA, RAG1 is predominantly present in a homodimer form, while RAG2 can exist as a monomer, dimer or even unresolved larger forms (33). Together, RAG1 and RAG2 form a “Y” shaped heterotetrametric structure comprising two heterodimer arms of RAG1 and RAG2 (25, 33). RAG1 forms the base of the “Y” shape, with RAG2 located on the upper tips, and the catalytic centre situated in the middle of the “Y” joint (25, 33). For a complete detailed and in-depth view of the exact (crystal) structure of RAG we refer the reader to seminal work by Kim et al., 2015 (25).
The recombination process begins with recognition of a suitable cleavage site. As one can imagine, the recombination process is an impactful genome editing mechanism that may cause serious problems if left unchecked. Thus, it only occurs at its intended sites, the V(D)J regions in the genome, and specifically in developing T and B lymphocytes. RAG exerts its function on the genome at sites where it detects signs of transcriptionally active chromatin and recognises the specific RSSs nonamer and heptamers (30, 34, 35). As noted above, RAG2 requires histone modification H3K4me3 in order to lift the autoinhibition of its PHD to catalyse RAGs function (28, 29). Additionally, H3K27 acetylation (H3K27ac) has been found to correlate with RAG binding to DNA (2, 8, 36). Interaction with H3K27ac was found to be RAG2 independent and seemed more dependent on N-terminal regions of RAG1, however, no clear evidence has proved this point yet (8, 36). Small sections within the V(D)J coding segments show highly active chromatin, promoting the recruitment of RAG to these sites, likely via the aforementioned histone interactions (2).
Furthermore, RAG cleavage activity is greatly dependent on nonamer and heptamer RSS recognition and binding by RAG1 NBD and the heptamer binding region in the central domain (2, 8). The NBD recognises the nonamer sequence ACAAAAACC, which binds strongly to this region with several highly conserved base pairs (bp) (8, 37, 38). The relatively weaker binding site of the heptamer CACAGTG only has the first 3 bp which are highly conserved and essential for DNA cleavage by RAG (8, 37, 38). Between the RSS nonamer and heptamer lies the spacer, which is always 12 or 23 bp long. RAG may only bind efficiently to one 12RSS and one 23RSS, not two 12RSSs or two 23RSSs. This is referred to as the 12/23 rule (1, 2, 8). It has been debated between multiple publications whether the 12RSS or the 23RSS pair is bound first or second by RAG prior to DNA cleavage (35, 39–41). Perhaps a more appealing model of 12/23 binding is one found from chromatin accessibility studies with histone modifications greatly influencing RAG binding capacities. This model suggests that the binding order of 12/23 has to do with the accessibility of the chromatin the sequence is located in (2, 35, 42). However, other factors are likely involved, such as the exact nonamer and heptamer sequences, which may vary apart from the highly conserved nucleotides. Even variations in the spacer sequence have been reported to influence RAG binding affinity (2, 8, 37, 38, 43–45).
However, not all RSS are located solely between the V(D)J segments. The so-called cryptic RSS (cRSS) are commonly present throughout the rest of the genome (8, 46). RAG’s variation in recognising RSSs can lead to different DSBs throughout the genome, causing genome instability. One example is a RAG-mediated DNA break in the c-Myc gene where only the 3 conserved bp (CAC) of the heptamer are enough for RAG to bind the sequence there with the heptamer binding region (8, 46). These off-target effects of RAG may lead to seriously threatening fusion genes with the risk of leukaemia’s as a result (5, 47–49).
The recombination process of DNA can be roughly divided into two phases, the DNA cleavage and joining phase. The first phase is initiated after RAG has gained access to the DNA via transcriptionally active histone modifications. RAG binds the first 12- or 23RSS heptamer and nonamer, forming a single RSS complex and subsequently recruits the second RSS (the 12RSS if the first was a 23RSS and vice versa) forming a paired complex (50). The heptamer sequences besides the 12 and 23 spacers flank the 3’ of the V segment and the 5’ of the J segment (when coding for a light chain, not containing the D segment, see example in Figure 3), and the nonamer sequences are positioned in between the heptamer sequences (see Figure 3). Once firmly in place, RAG induces a conformational change in the bound 12/23RSSs together with high mobility group box 1/2 (HMGB1/2), nicking the DNA on the 5’ single strand near the heptamer sequences, promoting an efficient double stranded cleavage initiated by RAG (see Figure 3) (2, 8). This cleavage creates two covalently sealed (hairpin) coding ends (at the V and J segments sites) and two blunt signal ends (at the heptamer ends).
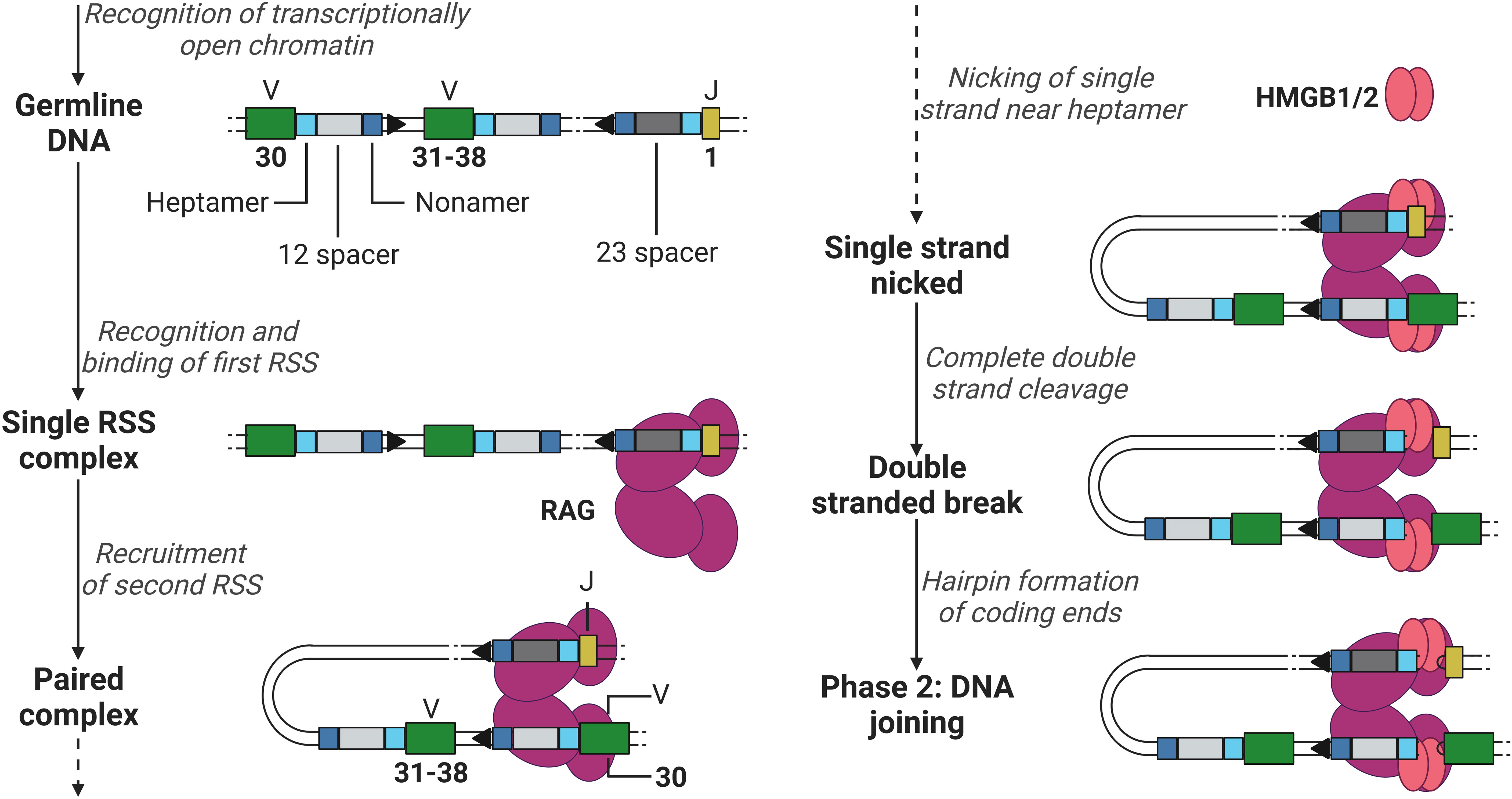
Figure 3 Phase 1 of recombination, DNA cleavage. Recombination example between V (green) segment 30 and J (yellow) segment 1 in a light chain, removing V segments 31 to 38. RAG (purple) binds to the first heptamer (cyan)/nonamer (blue) 23RSS (dark grey). Hereafter, binds the 12RSS (light grey) and initiates the first steps of DNA cleavage utilising HMGB1/2 (red). V, Variable; J, joining; RAG, recombination-activating gene (complex); HMGB1/2, high mobility group box 1/2. Created with BioRender.com.
After RAG has cleaved the DNA between the heptamer sequences and the V and J segments, the second recombination phase is initiated, the joining phase. Here, RAG dissociates from the coding ends, whilst holding on to the signal ends, giving space for NHEJ repair enzymes. Firstly, Ku70 and Ku80 heterodimers recognize the DSB site and bind to the lose DNA ends (51, 52). Once bound, Ku70/80 recruits DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which forms a holoenzyme together with Ku70/80 (51, 53, 54). Initially, the Ku/DNA-PKcs complex blocks any other factor from binding to the damaged DNA site. However, DNA-PKcs in the presence of DNA damage will be trans-phosphorylated by ataxia-telangiectasia mutated (ATM) and further be auto-phosphorylated, providing access to other factors to the DSB site (51, 55, 56).
Factors such as Artemis, a 5’ to 3’ endonuclease, now have access to the DSB site and are phosphorylated by DNA-PKcs (51, 57). Artemis opens the hairpin by introducing a single strand break in the DNA behind the first to third nucleotide of the coding ends of the V and J segments (see Figure 4A) (51, 58). This break generates one to three palindromic nucleotides (P nucleotides) as two strands which were first complementary are now in the same strand (1, 59). Hereafter, terminal deoxynucleotide transferase (TdT), a unique enzyme only present in developing lymphoid cells, attaches two to five (on average) random nucleotides which are not originally in the germline DNA on each coding end on the P-nucleotides, generating additional diversity to the junction (1, 60, 61). These nucleotides are called N nucleotides, because they were not encoded in the germline DNA. Complementary nucleotides at the ends of the coding ends pair and unpaired nucleotides are removed by exonucleases (51). Artemis is considered to be involved in the removal of unpaired nucleotides as it possesses exonuclease activity besides endonuclease activity (51, 58). However, its role in the removal of these unpaired nucleotides is still unclear (51, 58).
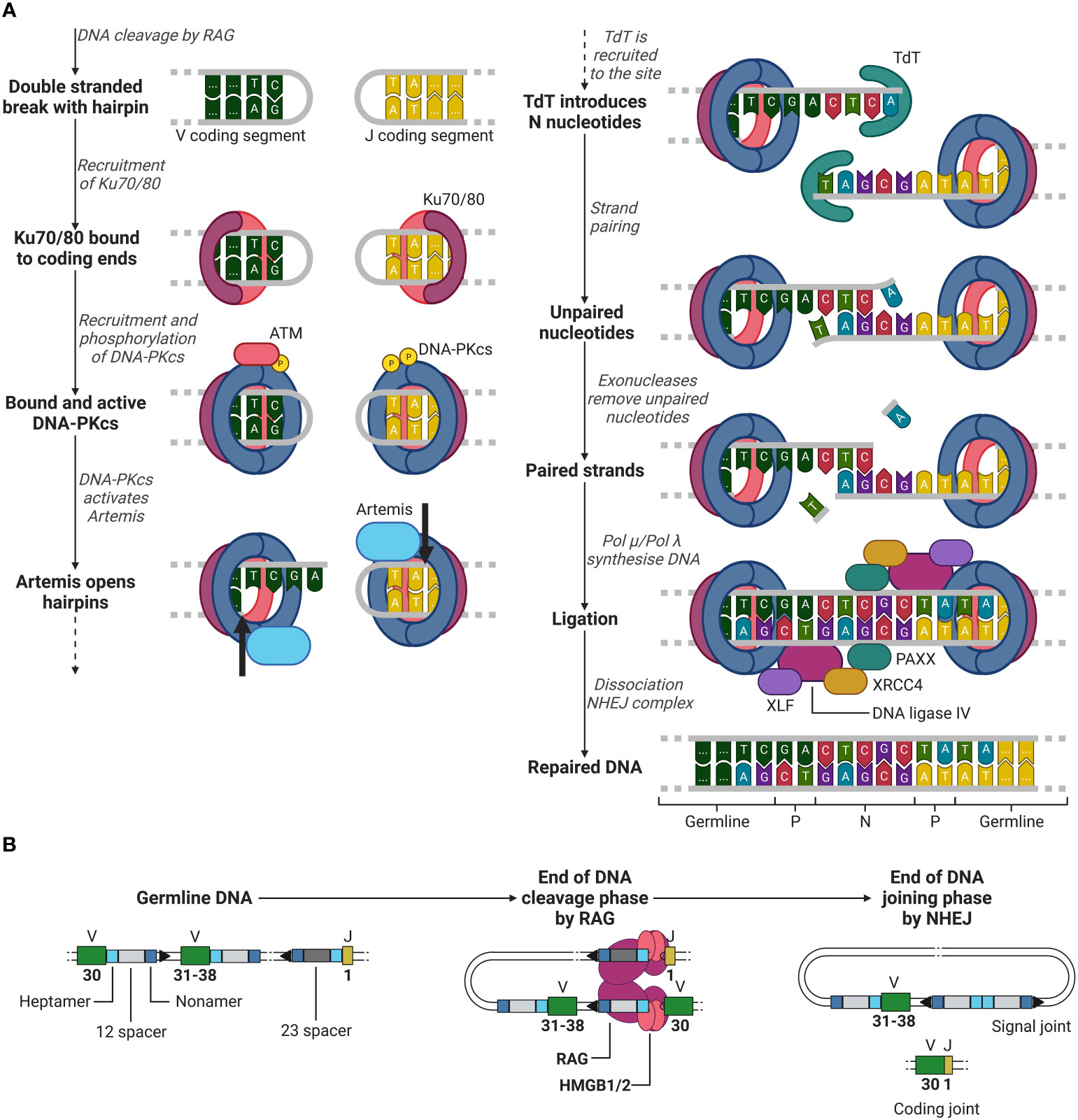
Figure 4 Phase 2 of recombination, DNA joining. (A) NHEJ example between a V (green) segment and J (yellow) segment after RAG mediated DNA cleavage. Ku70/80 (purple/red) is recruited to the double stranded break. Hereafter, DNA-PKcs (blue) is recruited, and trans/auto-phosphorylated. DNA-PKcs activates Artemis (light blue), which in turn opens the hairpins at both coding ends (cleavage location indicated by the black arrows) generating P nucleotides. TdT (cyan) then introduces N nucleotides to the P nucleotides. Strands are paired and unpaired nucleotides are removed by exonucleases. The DNA is then synthesised by either Pol µ or Pol λ and is then further ligated by DNA ligase IV (purple), supported by XRCC4 (yellow), XLF (dark purple) and PAXX (cyan). (B) Overview of the starting germline DNA to the end of the DNA cleavage and joining phases of recombination of a V and J segment. ATM, ataxia-telangiectasia mutated; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; TdT, terminal deoxynucleotide transferase; XRCC4, X-ray repair cross-complementing protein 4; XLF, XRCC4-like factor; RAG, recombination-activating gene(complex); HMGB1/2, high mobility group box 1/2. Created with BioRender.com.
Once unpaired nucleotides have been removed, DNA polymerase µ and λ (Pol µ and Pol λ) fill in the single strand DNA gaps by DNA synthesis (51). In the development of B lymphocytes, it has been shown that Pol µ is involved in the synthesis of the light chain junctions and Pol λ in the synthesis of the heavy chain junctions (61). It has yet to be investigated whether in T lymphocyte development the same distinction can be made for the use Pol µ or λ in the synthesis of the α and β (or γ and δ) TCR chain junctions. After all nucleotides are filled in, ligation is initiated by a complex of DNA ligase IV, X-ray repair cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF) and PAXX, completing the coding joint between the V and J segments (as by the example in Figures 3, 4) (1, 51, 62–65). Meanwhile, the signal ends of the two cut off heptamers are repaired in a similar manner with NHEJ and form the signal joint (see Figure 4B) (1).
Multiple tiers of regulatory elements orchestrate the regulation of RAG gene expression (66, 67). Extensive research conducted on the RAG locus has unveiled several cis-regulatory elements (68). However, the mere presence of these cis-elements falls short in elucidating the strictly controlled expression pattern of the RAG genes during lymphocyte development (68). Various studies have indicated that BCR signalling in immature B lymphocytes represses RAG gene transcription by means of phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt) (69–73). Diminished PI3K and Akt activity, mediated by B cell linker (BLNK) adaptor protein, leads to reduced levels of forkhead box O1 (FoxO1) and Fox3a phosphorylation (72, 73). In T lymphocytes, suppression of RAG gene expression has been observed to be indirectly regulated by linker for activation of T cells (LAT) and lymphocyte cytosolic protein 2 (LCP2) (74). Additionally, activation of Akt inhibits the nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1), thereby exerting a negative influence on RAG gene expression (75).
Further investigations have demonstrated that the RAG2 promotor exhibits greater specificity for lymphoid cells compared to the RAG1 promotor (68, 75, 76). Various lymphoid-specific transcription factors, including paired box 5 (PAX5), MYB, SP1, lymphoid enhancer binding factor 1 (LEF1), nuclear transcription factor Y (NF-Y), CCAAT enhancer binding protein (C/EBP), GATA binding protein 3 (GATA3), and NFATc1, have been identified to bind to the RAG2 promotor (68, 75, 76).
Moreover, it has been established that distant regulatory elements are essential for interacting with the RAG promotors to facilitate transcription (68). These regulatory elements vary between B and T lymphocytes (68). Upstream of the RAG2 promotor, approximately 23 kb away, lies the enhancer sequence Erag, which substantially enhances RAG expression in B lymphocytes (77). E2A, Foxp1, and FoxO1 are capable of binding to the Erag, thereby likely regulating enhancer activity (68, 77). Furthermore, two cis-elements situated 32 and 87 kb downstream of the RAG2 promotor have been found to govern RAG gene expression in T lymphocytes (78). Nestled between these cis-elements, an anti-silencing element (ASE) was found that counteracted silencing elements in the RAG locus at certain stages of T lymphocyte development (79). A common transcription factor in both B and T lymphocytes is Ikaros, which is thought to be an important regulator of RAG gene expression and function (80, 81).
An active regulator of the RAG complex is thought to be the transcription factor Ikaros (80). Ikaros has been described as a regulatory protein for TdT and is required for normal foetal T lymphocyte development, as well as in adult thymic development for pre-T lymphocyte TCR formation, and T lymphocyte differentiation choice to CD4 or CD8 (80, 82–86). Interaction of Ikaros with RAG was shown in murine B lymphocytes where hypomorphic Ikaros pro-B lymphocytes expressed lower levels of RAG1 and RAG2, additionally, Ikaros knockout lymphocytes even completely lacked RAG1 and RAG2 expression (80, 87, 88). Later studies found that Ikaros is not only required for RAG1 and RAG2 expression, retroviral expression of RAG1 and RAG2 in Ikaros knockout B lymphocytes were unable to form functional V-DJ heavy chain rearrangements (80, 88, 89). Further studies suggests that Ikaros may be involved in light chain rearrangement in B lymphocytes as well as in allelic exclusion (80, 90–92). These studies together suggest that Ikaros is required for the expression and function of RAG genes and protein as well as allelic exclusion in the development of B lymphocytes.
Multiple studies have shown that the recombination process is restricted to the G1 phase of the cell cycle in developing lymphocytes (31, 32, 93–96). RAG2 is the most studied of the two RAG proteins in this regard (97). It has been shown that RAG2 is undetectable throughout the S, G2 and M phase in dividing cells, however, during the G1 phase RAG2 accumulates and is degraded again before the S phase is initiated (32, 93). In the absence of RAG2, RAG1 aggregates in the nucleus. This aggregated form of RAG1 has an extremely low recombination activity (94). With the accumulation of RAG2 during the G1 phase, RAG1 is rescued from aggregation and forms functional RAG complexes with RAG2, allowing effective V(D)J recombination to take place (93, 94). Nearing the end of the G1 phase, degradation of RAG2 has been shown to be regulated post-transcriptionally through phosphorylation of Thr-490 by cyclin A/cyclin-dependent kinase 2 (cyclinA/Cdk2) (31, 93, 95, 96). A study further showed that in vitro addition of cyclin-dependent kinase inhibitor 1 (p21), an inhibitor for all cyclin-dependent kinases (CDKs) and a regulator of the G0 and 1 phase, prevented degradation of RAG2, further confirming the cyclin dependency of RAG2 (32, 98). Phosphorylation of RAG2 Thr-490 by cyclinA/Cdk2 is followed by ubiquitination by Skp2-SCF E3 ubiquitin ligase, a known regulator for G1-S transition (31, 32, 99).
Studies concerning regulation of RAG1 have demonstrated that DNA damage-binding protein 1 (DDB1) and cullin-4A (CUL4A) associated factor 1 (DCAF1) are necessary to control physiological levels of RAG1 in its protein form (97, 100, 101). RAG1 degradation is likely ubiquitin-dependent through proteasome degradation via CUL4A E3 ubiquitin ligase complexes (97). In mice, disruption of DCAF1 let to partial reduction of D-J recombination, whereas recombination of the V-DJ and V-J genes became severely impaired, resulting in a developmental block for B lymphocytes at the pro-B-to-pre-B lymphocyte stage (100). Similar to Skp2-SCF E3 ubiquitin ligase in regulation of RAG2, subunit CUL4A of the E3 ubiquitin ligase complex is a regulator of G1-S transition (102). All-in-all, these studies suggest that both RAG1 and RAG2 protein are regulated to function exclusively during the G1 phase and are marked for degradation through proteasomes during transition into the S phase of the cell cycle.
Using relatively simple assays like a DNase foot printing or electrophoretic mobility shift assay (EMSA) one can determine RAG binding to DNA or even specific (c)RSS sequences (103, 104). The DNase foot printing assay works on the principle that DNA bound by protein is protected from degradation by DNases (103). When RAG is bound to a given labelled DNA, specific sequences can be protected from DNase degradation. When these conditions are loaded onto a gel electrophoresis, the protected fragments can be observed (105). EMSA relies on a shift in molecular size when protein, in this case RAG, binds to a given DNA sequence (106). This shift in molecular size can be made visible on an electrophoresis gel, indicating an interaction (106).
However, DNase foot printing and EMSA only provide information on DNA binding of RAG. More advanced DNA-protein interaction measuring methods can be used, such as single-molecule fluorescence resonance energy transfer (smFRET) or single-molecule colocalization (smCL) assays. Besides information on specific DNA ((c)RSSs) binding, smFRET and smCL provide insight on the DNA bending kinetics of RAG (107). Using smFRET one can measure the bending of RAG target DNA by designing fluorescent donor and acceptor probes which recognise the 5’ and 3’ regions of the heptamer respectively (107). Interaction of RAG with the labelled region will induce a conformational chance in the DNA, bringing the donor and acceptor in closer proximity of each other (107). This proximity change of the donor and acceptor probes can be measured in FRET efficiency (107). With smCL RAG itself is fluorescently labelled besides a labelled sequence probe for the heptamer sequence (107). When RAG binds the labelled heptamer their colocalization and dwell time can be directly observed using total internal reflection fluorescence (TIRF) microscopy in real-time (107). All-in-all, DNase foot printing, EMSA, smFRET and smCL provide good information on DNA-RAG interaction concerning binding affinity to given DNA and DNA bending kinetics for the latter two methods.
The methods mentioned above are all in vitro cell-free methods, which may not always provide all the necessary information. Measuring RAG activity in cells adds crucial cellular context of other interacting proteins that can provide valuable additional insights. However, direct measurements of RAG in cells presents challenges. RAG is so evolutionary conserved that it is difficult to obtain antibodies that can specifically mark RAG for flow cytometry or cytometry by time flight (CyTOF). Moreover, RAG is primarily located in the nucleus, and intranuclear staining, along with surface staining using flow cytometry, can be quite challenging. An alternative approach is to use PrimeFlow™, an in situ hybridization assay, to demonstrate RAG gene expression in flow cytometry. However, this approach does not provide information on the RAG protein, only RNA.
Indirect effects of recombination are easier to detect, such as lymphocyte development. Using flow cytometry, one can follow the developmental stages of lymphocytes and observe hindrances or a complete arrest of development when RAG is mutated or disrupted (108, 109). Recombination activity can be indirectly measured by (q)PCR of the signal joints from in vitro cultures. Furthermore, cells can be transduced with an inverted reporter gene (such as green fluorescent protein (GFP)) flanked by 12- and 23RSSs, along with RAG1 and RAG2, if the cells do not express RAG endogenously (94, 110, 111). Active RAG can perform recombination at the 12- and 23RSS flanking the reporter gene, generating a readable signal from the now-active reporter gene in the signal joint (94, 110). Moreover, this method can determine the recombination efficiency of different (c)RSSs by modifying the flanking RSSs of the inverted reporter gene (94, 110).
In an in vivo setting, the closest approximation to a real recombination scenario is achieved. However, measuring RAG activity directly in vivo is very challenging and has yet to be accomplished. Indirect measuring of the recombination activity is available in the form of the TCR excision circle (TREC) assay, following lymphocyte development in the bone marrow, thymus and peripheral blood through flow cytometry, repertoire analysis, and serum Ig quantification (108). These methods can be applied, for example, on mice, where RAG-deficient mice are available and serve as good negative controls and targets for RAG gene therapy (108, 112).
Clinical outcome for SCID has been greatly enhanced with early diagnosis and treatment (113). At birth, SCID affected infants often appear in good health, as a consequence the condition is diagnosed later, often when the infant already has multiple infections and presents with secondary organ damage (113–115). In 2010 the Department of Health and Human Services (HHS) in the USA recommended screening for SCID to be included in the new-born screening, and from 2018 and forward, all new-borns are screened for SCID throughout all states of the USA (116). Since the advice of the HHS, multiple countries like Taiwan (2012), Israel (2015), Iceland, New Zealand, Norway (2017), The Netherlands (2018), Switzerland, Sweden, Germany (2019) and Denmark (2020) have implemented SCID in their new-born screening programs (117, 118).
SCID is diagnosed using the TREC assay, a simple but effective PCR of the signal joint (113, 119, 120). TRECs are the signal joints that form after recombination (see Figure 4B) and therefore are abundantly present in recently formed T lymphocytes. Because TRECs are not replicated during cell division, they are divided over one of two daughter cells after division, and therefore only present in a fraction of the peripheral T lymphocytes. A TREC analysis, provides quantitative information on the replication rate of these cells (119). In healthy new-borns, TRECs represent about 10% of the peripheral T lymphocytes, and they in turn reflect thymic formation of naïve T lymphocytes (119, 121).
Dysregulated recombination has been shown to cause genomic instability in lymphoid cells (5, 47–49). Some translocations, amplifications and deletions in ALL could be traced back to recombination, as (c)RSS could be detected near breakpoints in the DNA (5, 122). In comparison to other non-lymphoid malignancies like breast, prostate and pancreatic cancers, these (c)RSS were not observed near translocations, amplifications or deletions present in their representative malignant cells (5, 122). However, these genomic alterations may not be the actual cause of malignancy in lymphoid cells, but a result of previous oncogenic alterations leading to further instability of the cell through RAG dysregulation (122).
Mutations in RAG itself may alter its activity. Failure to mark RAG2 for degradation by cyclinA/Cdk2 through mutation of its substrate, Thr-490, has shown to disconnect recombination activity from the G1 phase of the cell cycle, inducing lymphoid malignancies in p53 deficient mice (31). Furthermore, mutations in the autoinhibitory region of RAG2 may lift the histone recognition signal for activation, triggering increased off-target activity of RAG (8, 28). Besides dysregulations in RAG itself, dysregulation of NHEJ after DSB by RAG may result in genomic instability. In vivo murine studies have shown that RAG activity in p53 and NHEJ deficient mice resulted in genomic instability and leukemogenesis, often presenting with aneuploidy, amplification and chromosomal translocations (123–126). However, potentially oncogenic translocations caused in mice deficient for different components of NHEJ were not equal (123). Deficiencies in Prkdc (DNA-PKcs) did not result in a translocation between Ig heavy chain on chromosome 12 with c-Myc on chromosome 15, which were recurrent in mice deficient in p53 and KU80, LIG4 or XRCC4 (126, 127). All-in-all, dysregulation of RAG and/or NHEJ increases the likelihood of oncogenic genomic alterations, however dysregulation of recombination may not always be the cause for lymphoid malignancies, but a result of already preleukemic conditions in a cell (122).
Immunodeficiencies with partial recombination activity due to hypomorphic RAG mutations have been shown to carry risk for generating autoantibodies (128–131). When recombination is not completely impaired, functional lymphocytes can still develop, although restricted and not always in normal quantities (128–130, 132, 133). And just like during normal lymphocyte development, auto-reactive receptors are formed. In normal development these receptors are either edited, deleted or the cell becomes anergic (1). In the case of receptor editing, recombination is directly involved by trying other combinations of V(D)J segments if they are still possible to make (134). With partial recombination activity, this process may be impaired as well, resulting in an impaired tolerance selection if elimination or anergy fails (130). Studies have shown that partial recombination activity influences B lymphocyte tolerance in the periphery through generating a severely restricted B lymphocyte repertoire. These restricted B lymphocytes can become stimulated in an inflammatory environment which is often present in the form of infections for recombination impaired individuals, increasing the risk for autoimmunity (130, 135).
A complete or greatly deficient recombination machinery severely hampers lymphocyte development at an early stage, resulting in SCID (3, 4, 128, 136). SCID represents a heterogenous group of immunological disorders characterised by abnormalities in the function and development of T lymphocytes (and B lymphocytes, however not in all forms of SCID) (128, 137). Mutations in RAG make up a large part of SCID cases in humans (3). The most severe phenotypes of RAG deficiencies include T- and B-lymphocyte negative SCID, Omenn syndrome (OS), γδ T+ SCID and atypical SCID (128). Other RAG deficient immunodeficiencies with different clinical phenotypes cover: combined immunodeficiency associated with granulomas and/or autoimmunity (CID-G/AI), idiopathic CD4+ T cell lymphopenia, common variable immunodeficiency, IgA deficiency, selective deficiency of polysaccharide-specific antibody responses, hyper-IgM syndrome, and sterile chronic multifocal osteomyelitis (138–144). Specific mutations in RAG determine the phenotype of the disease, for a detailed list of known disease causing mutations in RAG we would refer the reader to an excellent review by Notarangelo et al., 2016 (128).
Amorphic mutations in RAG cause a complete loss-of-function in recombination, causing T and B lymphocyte negative SCID. T and B lymphocyte negative SCID can have two distinct phenotypes, one associated with a RAG deficiency, and one with a NHEJ deficiency. In the case of NHEJ deficiency, besides patients being severely immunocompromised, they also are radiosensitive due to the lack of NHEJ in DNA damage repair from ultraviolet light or ionizing radiation sources (128, 145).
OS is caused by hypomorphic mutations in RAG and still shows some recombination activity (146). OS is characterised by generalised erythroderma, lymphadenopathy, hepatosplenomegaly, eosinophilia, severe hypogammaglobulinemia with elevated levels of IgE, and multiple organs infiltrated with activated T lymphocytes (128, 147, 148). It is speculated that T lymphocytes in these patients are skewed towards T helper 2 lymphocytes, causing elevated levels of serum interleukin-5, it is however unclear how this skewing is initiated (128). Furthermore, distinct restrictions in TCRs may induce autoimmunity, causing T lymphocyte infiltrations in multiple organs (128, 148, 149). One explanation may be that specific mutations may alter V(D)J recombination in a particular way, favouring certain RSS above others (20, 128). In some conditions of autologous T lymphocytes without typical OS phenotype, the disease is referred to as atypical SCID (3, 150–152).
Some cases of hypomorphic RAG show relatively normal numbers of γδ T lymphocytes despite the patients having αβ T lymphocyte lymphopenia (153, 154). This so called γδ T+ SCID phenotype has been associated with cytomegalovirus (CMV) infection and does not present itself with the characteristic features of OS (128, 153, 154). It has been suggested that the expansion of γδ T lymphocytes is antigen-driven by the CMV infection, causing peripheral clonal expansion of these cells (153, 154). Patients with γδ T+ SCID phenotype often develop autoimmune reactions despite having minimal levels of B lymphocytes, however they do respond to some vaccines and infections (154). All-in-all RAG deficiencies can present themself in many different phenotypes depending on the nature of the mutation in RAG or NHEJ components, or the environmental exposure to pathogens, resulting in life-long and life threatening immunodeficiencies, often combined with autoimmunity (128).
For SCID, the curative treatment exists in the form of haematopoietic stem cell (HSC) transplantation, however, a well matched donor is not always available. Proper HLA matching decreases the chances of Graft versus Host Disease (GvHD) as mismatched HLA in an allograft transplant often results in Graft versus Host Disease and other comorbidities (155). An autologous bone marrow transplant with corrected genes, also known as autologous gene therapy, would be the ideal cure for these diseases. Ex vivo gene therapy treatment allows for control and selection before transplanting the treated cells back to the patient. This form of gene therapy may be achieved in different ways, through retro- or lentiviral vector transduction with corrected genes (gene addition) or by gene editing in which the affected locus is corrected through the use of nucleases, nowadays most frequently Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas.
Upon infection of a host cell, retroviruses integrate their genetic information more or less permanently in the host genome (156). Retroviruses have thus been seen as a natural gene delivery system and were actually used in the first recorded case of successful human gene therapy in the 1990s (157, 158). Besides a few of the viral DNA domains and long terminal repeat (LTR) elements, a large part of the viral genetic information can be replaced by exogenous DNA, up to 8 Kbp (159). The virus can be further modified by pseudotyping, replacing the envelope gene encoding for envelope glycoproteins with envelope genes of other viruses, changing the infectable host cells (159). Upon infection, integration is only possible during cell division, one of the downsides of a gamma-retroviral vector. Due to their specific integration proteins, integration by gamma-retroviral vectors is most likely to take place in euchromatin rather than heterochromatin, risking insertional mutagenesis which has led to cases of acute leukaemia in gamma-retroviral vector treated patients (159–165). However, since the first use of gamma-retroviral vectors, safety has been improved with insulator sequences that modulate promotor activity and the development of self-inactivating (SIN) vectors (166–169).
Close relatives to the gamma-retroviruses are lentiviruses and they too can be considered a good tool for gene delivery in gene therapy (and other applications) (159). Lentiviruses are considered to be more complex than gamma-retroviruses and thus are more complex to modify for specific gene delivery strategies. However, they come with more advantages as well. Lentiviruses come with additional regulatory genes that allow for integration in non-dividing cells (170, 171). Appropriate pseudotyping and modification of lentiviruses has resulted in space for up to 9 Kbp of exogenous DNA and the ability to effectively target and transduce difficult-to-transduce cells such as HCS, lymphoid cells, some myeloid cells, neurons and others (159, 172–175). Additionally, lentiviruses have low LTRs inducible promotor activity due to SIN modifications, and tend to integrate further away from start sites of cellular promotors, lowering the risk of insertional mutagenesis and oncogenicity compared to gamma-retroviruses (159, 160, 176).
In the ideal situation however, one should actually repair the mutated gene instead of inserting an additional one. Gene editing by CRISPR has great promise in this regard. Using CRISPER/Cas9, a DSB can be introduced in the mutated gene with a single guide RNA (sgRNA). This break can then be repaired through the cells homology-directed repair (HDR) mechanisms using donor DNA with homologous arms to the DSB site. This mechanism of gene editing can transport up to 4,7 Kbp of exogenous DNA and can be delivered via adeno-associated virus (AAV), which can be modified to target HSCs effectively (177–181). Because this method of gene editing relies on HDR, it can only be executed in dividing cells (182). Furthermore, CRISPR/Cas9 is not error-free, although great advances are being made to eliminate off-target activity (183–186). However, some studies using ex vivo gene editing with CRISPR/Cas9 transported in AAV with a donor sequence, triggered immune responses, negatively influencing the stemness of HSCs and reducing their long-term seeding after transplantation (178, 179, 185, 187–189). In addition, CRISPR also shows on target side effects, due to its imprecise nature and can generate indels at distances up to several hundred bps around the desired site, possibly disrupting normal gene regulation (190, 191). Especially for a locus that is subject to strict negative control, such as the RAG locus, these safety considerations are of utmost importance.
Besides gene editing through HDR, a modified Cas9 has been engineered with base editing enzymes, reverse transcriptase’s and prime editing enzymes (177, 192, 193). These CRISPR/Cas9 fusion mechanisms can edit single nucleotides, add insertions up to 44 bp, and delete up to 80 bp at 5 to 50 bp away from the target site (193). All-in-all, CRISPR-based gene editing caries great potential for gene therapy applications, however, further improvements in safety regarding off-target editing and delivery of the machinery need to be made in order for these methods to be safely implemented in clinical gene therapy.
As introduced above, SCID is a prime candidate to be treated by autologous stem cell based gene therapy. Indeed, the first successful gene therapy efforts were done with retroviral vectors for treating X-linked-SCID which is caused by mutations in the IL2RG gene (191). Based on the initial success, a similar approach was proposed to treat RAG1-SCID. While this kind of RAG1 gene therapy showed good efficacy in the mouse models, the occurrence of leukaemia due to insertional mutagenesis, forced the field to move to SIN LV vectors (164, 194). For some types of SCID, these vectors became quickly available, however for RAG1-SCID this proved to be a formidable challenge due to the high expression of RAG1 in very strictly defined lymphoid progenitor populations (195, 196). We first reported successful LV preclinical work with the spleen focus-forming virus (SFFV) promotor, which was later replaced by the more clinically acceptable myeloproliferative sarcoma virus enhancer, negative control region deleted, dl587rev primer-binding site substituted (MND) promotor, which has been used before in clinical trials for gene therapy (109, 112, 197). Since then, preclinical studies on lentiviral-based gene therapy for RAG1-SCID has advanced to a phase I/II clinical trial (NCT04797260) in 2021 (108, 109, 112, 198). These preclinical murine studies used RAG1 deficient mice and performed gene therapy with a lentiviral SIN vector containing codon optimised RAG1 (coRAG1). The treatment showed successful reconstruction of T and B lymphocytes in peripheral blood and developing lymphocytes seeded central lymphoid organs (109). And when challenged with foreign antigens, treated mice showed an antigen-specific immune response (109). Importantly, a relatively high level of coRAG1 expression was essential for successful recombination, as low levels of coRAG1 due to inefficient transduction resulted in OS-like T lymphocyte phenotypes (198, 199). After passing regulation standards, the lentiviral coRAG1 vector moved on to a phase I/II clinical trial, and as the clinical trial progresses, we eagerly await the results for the human in vivo setting. Thus far, two patients have been included and a third one will be transplanted with corrected autologous stem cells soon.
Recent work using HSC based gene therapy for cerebral adreno- leukodystrophy (CALD) using a similar MND promoter has showed 3 cases of myelodysplastic syndrome out of 70 patients treated showing the risk of insertional mutagenesis that is associated with this kind of therapy (197). For RAG1 LV gene therapy we tested 7 different promoters as high RAG1 expression is required to restore T cell development (108, 112, 198, 200). In safety assays, the MND-coRAG1 vector has not shown signs of insertional mutagenesis (112, 200). We have also opted for transduction efficiencies with approximately one integration per target cell, instead of VCNs of 5 to 10 that were aimed for in the CALD trial, where indeed 3-5 viral integration sites per dysplastic clone were found (197). Further follow up will show whether the perceived lower change of xeno-toxicity events in the RAG1 SCID trial holds up.
For RAG2-SCID, we have developed SIN LV vectors that correct RAG2-deficiency in mouse models (200). The vector is currently being produced good manufacturing practices (GMP) grade to facilitate clean room testing leading up to a phase I/II clinical trial anticipated early 2024 (an overview of other current developments in RAG gene therapy is provided in Table 1).
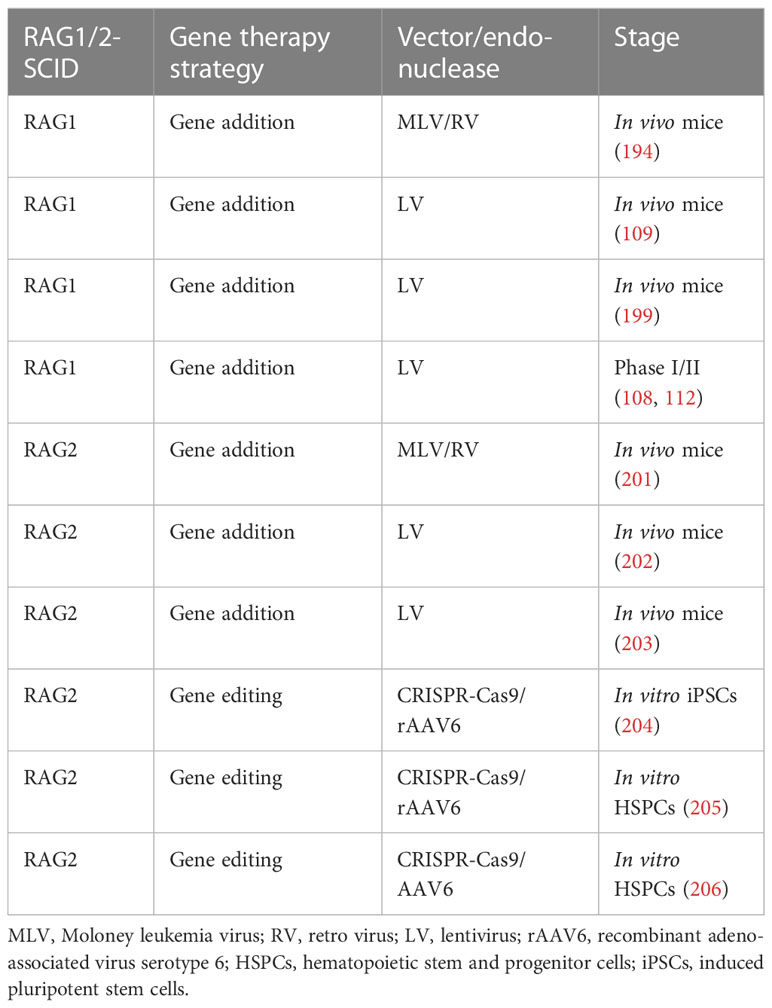
Table 1 Overview of the current developments on RAG gene therapy.
In summary, RAG plays a crucial role in the intricate process of recombination and lymphocyte development, leading to the generation of immune diversity. Thanks to extensive research, we have gained a more comprehensive understanding of RAG’s functional domains and motifs, and how it initiates the recombination process (2, 8). Furthermore, investigations into RAG have uncovered regulatory links to chromatin features, RSS, transcription factor Ikaros, the G1 phase of the cell cycle, and developing lymphocytes (2, 8, 28, 31, 80). Dysregulation of these components has been linked to diseases like cancer, autoimmunity, and SCID (2, 8, 128). However, with the emergence of precision medicine in the form of gene therapy, the possibility of treating diseases like SCID is now becoming a reality (108, 112). The current innovative methods for detecting and measuring recombination activity by RAG hold promise for advancing research on RAG deficiencies, gene therapy and related fields. However, the development of direct intranuclear measuring of RAG protein would be a significant addition to these methods, enabling even more precise and accurate measurements of recombination activity. Furthermore, the continued development of error-free CRISPR gene editing, efficient transduction delivery systems, and safe viral vectors will further enhance the potency of gene therapy, providing hope for patients suffering from a wide range of genetic disorders. As these technologies continue to evolve, it is likely that the future of gene therapy will be characterised by even greater precision and efficacy, paving the way for a new era of personalised medicine.
The first gene therapy trial for RAG1-SCID (NCT04797260) is currently underway, and the outcome is eagerly anticipated (108, 112). With existing literature providing strong support for further development of gene therapies for forms of SCID and other genetic disorders, advanced culture systems such as artificial thymic organoids (ATOs) can further facilitate this research by allowing for the use of patient material in culture and reducing the need for animal experimentation. However, creating effective and safe gene therapy for SCID poses significant challenges, given the powerful genome editing tools involved. As such, meticulous regulation is necessary to ensure the successful implementation of gene therapy for RAG in SCID. By carefully balancing innovation with regulatory oversight, we can continue to make strides towards safe and effective gene therapies that have the potential to transform the lives of patients with genetic disorders.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The research referred to is supported in part by ZonMW E-RARE grant (40 419000-98-020), NWO-NWA grant CURE4LIFE and EU H2020 grant RECOMB (755170-2) and has received funding from the European Union Horizon 2020 research and innovation program as well as from reNEW the Novo Nordisk Foundation for Stem Cell Research.
The authors would like to thank members of the Staal laboratory for helpful discussions and inspiration.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Parham P. Chapter 4: antibody structure and the generation of b-cell diversity, and chapter 5: antigen recognition by T lymphocytes. In: The immune system Garland science;, 4th ed (2015). p. :81–147.
2. Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet (2011) 45:167–202. doi: 10.1146/annurev-genet-110410-132552
3. Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human b cell-negative SCID. Science (1996) 274(5284):97–9doi: 10.1126/science.274.5284.97
4. Gennery A. Recent advances in understanding RAG deficiencies. F1000Res. (2019) 8. doi: 10.12688/f1000research.17056.1
5. Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet (2014) 46(2):116–25. doi: 10.1038/ng.2874
6. Oettinger MA, Stanger B, Schatz DG, Glaser T, Call K, Housman D, et al. The recombination activating genes, RAG 1 and RAG 2, are on chromosome 11p in humans and chromosome 2p in mice. Immunogenetics. (1992) 35(2):97–101. doi: 10.1007/BF00189518
7. Villa A, Notarangelo LD. RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev (2019) 287(1):73–90. doi: 10.1111/imr.12713
8. Christie SM, Fijen C, Rothenberg E. V(D)J recombination: recent insights in formation of the recombinase complex and recruitment of DNA repair machinery. Front Cell Dev Biol (2022) 10:886718. doi: 10.3389/fcell.2022.886718
9. Brecht RM, Liu CC, Beilinson HA, Khitun A, Slavoff SA, Schatz DG. Nucleolar localization of RAG1 modulates V(D)J recombination activity. Proc Natl Acad Sci U S A. (2020) 117(8):4300–9. doi: 10.1073/pnas.1920021117
10. Grazini U, Zanardi F, Citterio E, Casola S, Goding CR, McBlane F. The RING domain of RAG1 ubiquitylates histone H3: a novel activity in chromatin-mediated regulation of V(D)J joining. Mol Cell (2010) 37(2):282–93. doi: 10.1016/j.molcel.2009.12.035
11. McMahan CJ, Difilippantonio MJ, Rao N, Spanopoulou E, Schatz DG. A basic motif in the n-terminal region of RAG1 enhances V(D)J recombination activity. Mol Cell Biol (1997) 17(8):4544–52. doi: 10.1128/MCB.17.8.4544
12. Rodgers KK, Bu Z, Fleming KG, Schatz DG, Engelman DM, Coleman JE. A zinc-binding domain involved in the dimerization of RAG1. . J Mol Biol (1996) 260(1):70–84. doi: 10.1006/jmbi.1996.0382
13. Raval P, Kriatchko AN, Kumar S, Swanson PC. Evidence for Ku70/Ku80 association with full-length RAG1. Nucleic Acids Res (2008) 36(6):2060–72. doi: 10.1093/nar/gkn049
14. Maitra R, Sadofsky MJ. A WW-like module in the RAG1 n-terminal domain contributes to previously unidentified protein-protein interactions. Nucleic Acids Res (2009) 37(10):3301–9. doi: 10.1093/nar/gkp192
15. De P, Rodgers KK. Putting the pieces together: identification and characterization of structural domains in the V(D)J recombination protein RAG1. Immunol Rev (2004) 200:70–82. doi: 10.1111/j.0105-2896.2004.00154.x
16. Difilippantonio MJ, McMahan CJ, Eastman QM, Spanopoulou E, Schatz DG. RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell Oct 18 (1996) 87(2):253–62. doi: 10.1016/s0092-8674(00)81343-4
17. Spanopoulou E, Zaitseva F, Wang FH, Santagata S, Baltimore D, Panayotou G. The homeodomain region of rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell. (1996) 87(2):263–76. doi: 10.1016/s0092-8674(00)81344-6
18. Arbuckle JL, Fauss LA, Simpson R, Ptaszek LM, Rodgers KK. Identification of two topologically independent domains in RAG1 and their role in macromolecular interactions relevant to V(D)J recombination. J Biol Chem (2001) 276(40):37093–101. doi: 10.1074/jbc.M105988200
19. Qiu JX, Kale SB, Yarnell Schultz H, Roth DB. Separation-of-function mutants reveal critical roles for RAG2 in both the cleavage and joining steps of V(D)J recombination. Mol Cell (2001) 7(1):77–87. doi: 10.1016/s1097-2765(01)00156-3
20. Mo X, Bailin T, Sadofsky MJ. A c-terminal region of RAG1 contacts the coding DNA during V(D)J recombination. Mol Cell Biol Mar (2001) 21(6):2038–47. doi: 10.1128/MCB.21.6.2038-2047.2001
21. Aidinis V, Dias DC, Gomez CA, Bhattacharyya D, Spanopoulou E, Santagata S. Definition of minimal domains of interaction within the recombination-activating genes 1 and 2 recombinase complex. J Immunol (2000) 164(11):5826–32. doi: 10.4049/jimmunol.164.11.5826
22. Gwyn LM, Peak MM, De P, Rahman NS, Rodgers KK. A zinc site in the c-terminal domain of RAG1 is essential for DNA cleavage activity. J Mol Biol (2009) 390(5):863–78. doi: 10.1016/j.jmb.2009.05.076
23. Grundy GJ, Yang W, Gellert M. Autoinhibition of DNA cleavage mediated by RAG1 and RAG2 is overcome by an epigenetic signal in V(D)J recombination. Proc Natl Acad Sci U S A. (2010) 107(52):22487–92. doi: 10.1073/pnas.1014958107
24. Sherrington PD, Forster A, Seawright A, van Heyningen V, Rabbitts TH. Human RAG2, like RAG1, is on chromosome 11 band p13 and therefore not linked to ataxia telangiectasia complementation groups. Genes Chromosomes Cancer. (1992) 5(4):404–6. doi: 10.1002/gcc.2870050417
25. Kim MS, Lapkouski M, Yang W, Gellert M. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature. (2015) 518(7540):507–11. doi: 10.1038/nature14174
26. Coussens MA, Wendland RL, Deriano L, Lindsay CR, Arnal SM, Roth DB. RAG2’s acidic hinge restricts repair-pathway choice and promotes genomic stability. Cell Rep (2013) 4(5):870–8. doi: 10.1016/j.celrep.2013.07.041
27. Wu C, Dong Y, Zhao X, Zhang P, Zheng M, Zhang H, et al. RAG2 involves the igkappa locus demethylation during b cell development. Mol Immunol (2017) 88:125–34. doi: 10.1016/j.molimm.2017.06.026
28. Lu C, Ward A, Bettridge J, Liu Y, Desiderio S. An autoregulatory mechanism imposes allosteric control on the V(D)J recombinase by histone H3 methylation. Cell Rep (2015) 10(1):29–38. doi: 10.1016/j.celrep.2014.12.001
29. Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. (2007) 450(7172):1106–10. doi: 10.1038/nature06431
30. Teng G, Maman Y, Resch W, Kim M, Yamane A, Qian J, et al. RAG represents a widespread threat to the lymphocyte genome. Cell. (2015) 162(4):751–65. doi: 10.1016/j.cell.2015.07.009
31. Zhang L, Reynolds TL, Shan X, Desiderio S. Coupling of V(D)J recombination to the cell cycle suppresses genomic instability and lymphoid tumorigenesis. Immunity. (2011) 34(2):163–74. doi: 10.1016/j.immuni.2011.02.003
32. Jiang H, Chang FC, Ross AE, Lee J, Nakayama K, Nakayama K, et al. Ubiquitylation of RAG-2 by Skp2-SCF links destruction of the V(D)J recombinase to the cell cycle. Mol Cell (2005) 18(6):699–709. doi: 10.1016/j.molcel.2005.05.011
33. Bailin T, Mo X, Sadofsky MJ. A RAG1 and RAG2 tetramer complex is active in cleavage in V(D)J recombination. . Mol Cell Biol (1999) 19(7):4664–71. doi: 10.1128/MCB.19.7.4664
34. Lovely GA, Braikia F-Z, Singh A, et al. Direct observation of RAG recombinase recruitment to chromatin and the IgH locus in live pro-b cells. BioRxiv preprint. (2020). doi: 10.1101/2020.09.07.286484
35. Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. (2010) 141(3):419–31. doi: 10.1016/j.cell.2010.03.010
36. Maman Y, Teng G, Seth R, Kleinstein SH, Schatz DG. RAG1 targeting in the genome is dominated by chromatin interactions mediated by the non-core regions of RAG1 and RAG2. Nucleic Acids Res (2016) 44(20):9624–37. doi: 10.1093/nar/gkw633
37. Hirokawa S, Chure G, Belliveau NM, Lovely GA, Anaya M, Schatz DG, et al. Sequence-dependent dynamics of synthetic and endogenous RSSs in V(D)J recombination. Nucleic Acids Res (2020) 48(12):6726–39. doi: 10.1093/nar/gkaa418
38. Lee AI, Fugmann SD, Cowell LG, Ptaszek LM, Kelsoe G, Schatz DG. A functional analysis of the spacer of V(D)J recombination signal sequences. PloS Biol (2003) 1(1):E1. doi: 10.1371/journal.pbio.0000001
39. Jones JM, Gellert M. Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J (2002) 21(15):4162–71. doi: 10.1093/emboj/cdf394
40. Curry JD, Geier JK, Schlissel MS. Single-strand recombination signal sequence nicks in vivo: evidence for a capture model of synapsis. Nat Immunol (2005) 6(12):1272–9. doi: 10.1038/ni1270
41. Franchini DM, Benoukraf T, Jaeger S, Ferrier P, Payet-Bornet D. Initiation of V(D)J recombination by dbeta-associated recombination signal sequences: a critical control point in TCRbeta gene assembly. PloS One (2009) 4(2):e4575. doi: 10.1371/journal.pone.0004575
42. Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol (2011) 11(4):251–63. doi: 10.1038/nri2941
43. Feeney AJ, Goebel P, Espinoza CR. Many levels of control of V gene rearrangement frequency. Immunol Rev (2004) 200:44–56. doi: 10.1111/j.0105-2896.2004.00163.x
44. Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol (2000) 18:495–527. doi: 10.1146/annurev.immunol.18.1.495
45. Swanson PC. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol Rev (2004) 200:90–114. doi: 10.1111/j.0105-2896.2004.00159.x
46. Hu J, Zhang Y, Zhao L, Frock RL, Du Z, Meyers RM, et al. Chromosomal loop domains direct the recombination of antigen receptor genes. Cell. (2015) 163(4):947–59. doi: 10.1016/j.cell.2015.10.016
47. Thomson DW, Shahrin NH, Wang PPS, Wadham C, Shanmuganathan N, Scott HS, et al. Aberrant RAG-mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia. (2020) 34(8):2051–63. doi: 10.1038/s41375-020-0751-y
48. Paranjape AM, Desai SS, Nishana M, Roy U, Nilavar NM, Mondal A, et al. Nonamer dependent RAG cleavage at CpGs can explain mechanism of chromosomal translocations associated to lymphoid cancers. PloS Genet (2022) 18(10):e1010421. doi: 10.1371/journal.pgen.1010421
49. Kirkham CM, Scott JNF, Wang X, Smith AL, Kupinski AP, Ford AM, et al. Cut-and-Run: a distinct mechanism by which V(D)J recombination causes genome instability. Mol Cell (2019) 74(3):584–59.e9. doi: 10.1016/j.molcel.2019.02.025
50. Eastman QM, Leu TM, Schatz DG. Initiation of V(D)J recombination in vitro obeying the 12/23 rule. Nature. (1996) 380(6569):85–8. doi: 10.1038/380085a0
51. Rahimian E, Amini A, Alikarami F, Pezeshki SMS, Saki N, Safa M. DNA Repair pathways as guardians of the genome: therapeutic potential and possible prognostic role in hematologic neoplasms. DNA Repair (Amst). (2020) 96:102951. doi: 10.1016/j.dnarep.2020.102951
52. Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. (2001) 412(6847):607–14. doi: 10.1038/35088000
53. Shao Z, Davis AJ, Fattah KR, So S, Sun J, Lee K-J, et al. Persistently bound Ku at DNA ends attenuates DNA end resection and homologous recombination. DNA Repair (Amst). (2012) 11(3):310–6. doi: 10.1016/j.dnarep.2011.12.007
54. Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, et al. Ku And DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem Jan 8 (2010) 285(2):1414–23. doi: 10.1074/jbc.M109.065615
55. Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol (2007) 177(2):219–29. doi: 10.1083/jcb.200608077
56. DeFazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J (2002) 21(12):3192–200. doi: 10.1093/emboj/cdf299
57. Soubeyrand S, Pope L, De Chasseval R, Gosselin D, Dong F, Villartay J-P, et al. Artemis Phosphorylated by DNA-dependent protein kinase associates preferentially with discrete regions of chromatin. J Mol Biol (2006) 358(5):1200–11. doi: 10.1016/j.jmb.2006.02.061
58. Menon V, Povirk LF. End-processing nucleases and phosphodiesterases: an elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair (Amst). (2016) 43:57–68. doi: 10.1016/j.dnarep.2016.05.011
59. Srivastava SK, Robins HS. Palindromic nucleotide analysis in human T cell receptor rearrangements. PloS One (2012) 7(12):e52250. doi: 10.1371/journal.pone.0052250
60. Motea EA, Berdis AJ. Terminal deoxynucleotidyl transferase: the story of a misguided DNA polymerase. Biochim Biophys Acta May (2010) 1804(5):1151–66. doi: 10.1016/j.bbapap.2009.06.030
61. Bertocci B, De Smet A, Weill JC, Reynaud CA. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. (2006) 25(1):31–41. doi: 10.1016/j.immuni.2006.04.013
62. Lees-Miller SP, Meek K. Repair of DNA double strand breaks by non-homologous end joining. Biochimie. (2003) 85(11):1161–73. doi: 10.1016/j.biochi.2003.10.011
63. Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 27 (2006) 124(2):301–13. doi: 10.1016/j.cell.2005.12.031
64. Craxton A, Munnur D, Jukes-Jones R, Skalka G, Langlais C, Cain K, et al. PAXX and its paralogs synergistically direct DNA polymerase lambda activity in DNA repair. Nat Commun (2018) 9(1):3877. doi: 10.1038/s41467-018-06127-y
65. Tadi SK, Tellier-Lebegue C, Nemoz C, Drevet P, Audebert S, Roy S, et al. PAXX is an accessory c-NHEJ factor that associates with Ku70 and has overlapping functions with XLF. Cell Rep (2016) 17(2):541–55. doi: 10.1016/j.celrep.2016.09.026
66. Wilson A, Held W, MacDonald HR. Two waves of recombinase gene expression in developing thymocytes. J Exp Med (1994) 179(4):1355–60. doi: 10.1084/jem.179.4.1355
67. Grawunder U, Leu TM, Schatz DG, Werner A, Rolink AG, Melchers F, et al. Down-regulation of RAG1 and RAG2 gene expression in preB cells after functional immunoglobulin heavy chain rearrangement. Immunity. (1995) 3(5):601–8. doi: 10.1016/1074-7613(95)90131-0
68. Kuo TC, Schlissel MS. Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol (2009) 21(2):173–8. doi: 10.1016/j.coi.2009.03.008
69. Verkoczy L, Duong B, Skog P, Aït-Azzouzene D, Puri K, Vela JL, et al. Basal b cell receptor-directed phosphatidylinositol 3-kinase signaling turns off RAGs and promotes b cell-positive selection. J Immunol (2007) 178(10):6332–41. doi: 10.4049/jimmunol.178.10.6332
70. Schram BR, Tze LE, Ramsey LB, Liu J, Najera L, Vegoe AL, et al. B cell receptor basal signaling regulates antigen-induced ig light chain rearrangements. J Immunol (2008) 180(7):4728–41. doi: 10.4049/jimmunol.180.7.4728
71. Llorian M, Stamataki Z, Hill S, Turner M, Martensson IL. The PI3K p110delta is required for down-regulation of RAG expression in immature b cells. J Immunol (2007) 178(4):1981–5. doi: 10.4049/jimmunol.178.4.1981
72. Herzog S, Hug E, Meixlsperger S, Paik J-H, DePinho RA, Reth M, et al. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol (2008) 9(6):623–31. doi: 10.1038/ni.1616
73. Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during b cell development. Nat Immunol (2008) 9(6):613–22. doi: 10.1038/ni.1612
74. Roose JP, Diehn M, Tomlinson MG, Lin J, Alizadeh AA, Botstein D, et al. T Cell receptor-independent basal signaling via erk and abl kinases suppresses RAG gene expression. PloS Biol (2003) 1(2):E53. doi: 10.1371/journal.pbio.0000053
75. Patra AK, Drewes T, Engelmann S, Chuvpilo S, Kishi H, Hünig T, et al. PKB rescues calcineurin/NFAT-induced arrest of rag expression and pre-T cell differentiation. J Immunol (2006) 177(7):4567–76. doi: 10.4049/jimmunol.177.7.4567
76. Schlissel MS. Regulating antigen-receptor gene assembly. Nat Rev Immunol (2003) 3(11):890–9. doi: 10.1038/nri1225
77. Hsu LY, Lauring J, Liang HE, Greenbaum S, Cado D, Zhuang Y, et al. A conserved transcriptional enhancer regulates RAG gene expression in developing b cells. Immunity. (2003) 19(1):105–17. doi: 10.1016/s1074-7613(03)00181-x
78. Yu W, Misulovin Z, Suh H, Hardy RR, Jankovic M, Yannoutsos N, et al. Coordinate regulation of RAG1 and RAG2 by cell type-specific DNA elements 5’ of RAG2. Science. (1999) 285(5430):1080–4. doi: 10.1126/science.285.5430.1080
79. Yannoutsos N, Barreto V, Misulovin Z, Gazumyan A, Yu W, Rajewsky N, et al. A cis element in the recombination activating gene locus regulates gene expression by counteracting a distant silencer. Nat Immunol (2004) 5(4):443–50. doi: 10.1038/ni1053
80. Sellars M, Kastner P, Chan S. Ikaros in b cell development and function. World J Biol Chem (2011) 2(6):132–9. doi: 10.4331/wjbc.v2.i6.132
81. Miyazaki K, Miyazaki M. The interplay between chromatin architecture and lineage-specific transcription factors and the regulation of rag gene expression. Front Immunol (2021) 12:659761. doi: 10.3389/fimmu.2021.659761
82. Urban JA, Winandy S. Ikaros null mice display defects in T cell selection and CD4 versus CD8 lineage decisions. J Immunol (2004) 173(7):4470–8. doi: 10.4049/jimmunol.173.7.4470
83. Winandy S, Wu L, Wang JH, Georgopoulos K. Pre-T cell receptor (TCR) and TCR-controlled checkpoints in T cell differentiation are set by ikaros. J Exp Med (1999) 190(8):1039–48. doi: 10.1084/jem.190.8.1039
84. Avitahl N, Winandy S, Friedrich C, Jones B, Ge Y, Georgopoulos K. Ikaros sets thresholds for T cell activation and regulates chromosome propagation. Immunity. (1999) 10(3):333–43. doi: 10.1016/s1074-7613(00)80033-3
85. Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, et al. The ikaros gene is required for the development of all lymphoid lineages. Cell. (1994) 79(1):143–56. doi: 10.1016/0092-8674(94)90407-3
86. Lo K, Landau NR, Smale ST. LyF-1, a transcriptional regulator that interacts with a novel class of promoters for lymphocyte-specific genes. Mol Cell Biol (1991) 11(10):5229–43. doi: 10.1128/mcb.11.10.5229-5243.1991
87. Kirstetter P, Thomas M, Dierich A, Kastner P, Chan S. Ikaros is critical for b cell differentiation and function. Eur J Immunol Mar (2002) 32(3):720–30. doi: 10.1002/1521-4141(200203)32:3<720::AID-IMMU720>3.0.CO;2-P
88. Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, et al. Regulation of b cell fate commitment and immunoglobulin heavy-chain gene rearrangements by ikaros. Nat Immunol Aug (2008) 9(8):927–36. doi: 10.1038/ni.1626
89. Fuxa M, Skok J, Souabni A, Salvagiotto G, Roldan E, Busslinger M. Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev (2004) 18(4):411–22. doi: 10.1101/gad.291504
90. Liu Z, Widlak P, Zou Y, Xiao F, Oh M, Li S, et al. A recombination silencer that specifies heterochromatin positioning and ikaros association in the immunoglobulin kappa locus. Immunity. (2006) 24(4):405–15. doi: 10.1016/j.immuni.2006.02.001
91. Goldmit M, Ji Y, Skok J, Roldan E, Jung S, Cedar H, et al. Epigenetic ontogeny of the igk locus during b cell development. Nat Immunol (2005) 6(2):198–203. doi: 10.1038/ni1154
92. Macias-Garcia A, Heizmann B, Sellars M, Marchal P, Dali H, Pasquali J-L, et al. Ikaros is a negative regulator of B1 cell development and function. J Biol Chem (2016) 291(17):9073–86. doi: 10.1074/jbc.M115.704239
93. Li Z, Dordai DI, Lee J, Desiderio S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity. (1996) 5(6):575–89. doi: 10.1016/s1074-7613(00)80272-1
94. Gan T, Wang Y, Liu Y, Schatz DG, Hu J. RAG2 abolishes RAG1 aggregation to facilitate V(D)J recombination. Cell Rep (2021) 37(2):109824. doi: 10.1016/j.celrep.2021.109824
95. Byrum JN, Hoolehan WE, Simpson DA, Rodgers W, Rodgers KK. Full length RAG2 expression enhances the DNA damage response in pre-b cells. Immunobiology. (2021) 226(3):152089. doi: 10.1016/j.imbio.2021.152089
96. Lin WC, Desiderio S. Regulation of V(D)J recombination activator protein RAG-2 by phosphorylation. Science. (1993) 260(5110):953–9. doi: 10.1126/science.8493533
97. Schabla NM, Swanson PC. The CRL4VPRBP(DCAF1) E3 ubiquitin ligase directs constitutive RAG1 degradation in a non-lymphoid cell line. PloS One (2021) 16(10):e0258683. doi: 10.1371/journal.pone.0258683
98. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. (1993) 366(6456):701–4. doi: 10.1038/366701a0
99. Ou Y, Rattner JB. The centrosome in higher organisms: structure, composition, and duplication. Int Rev Cytol. (2004) 238:119–82. doi: 10.1016/S0074-7696(04)38003-4
100. Kassmeier MD, Mondal K, Palmer VL, Raval P, Kumar S, Perry GA, et al. VprBP binds full-length RAG1 and is required for b-cell development and V(D)J recombination fidelity. EMBO J (2012) 31(4):945–58. doi: 10.1038/emboj.2011.455
101. Schabla NM, Mondal K, Swanson PC. DCAF1 (VprBP): emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. J Mol Cell Biol (2019) 11(9):725–35. doi: 10.1093/jmcb/mjy085
102. Jang SM, Redon CE, Thakur BL, Bahta MK, Aladjem MI. Regulation of cell cycle drivers by cullin-RING ubiquitin ligases. Exp Mol Med (2020) 52(10):1637–51. doi: 10.1038/s12276-020-00508-4
103. Galas DJ, Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res (1978) 5(9):3157–70. doi: 10.1093/nar/5.9.3157
104. Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the escherichia coli lactose operon regulatory system. Nucleic Acids Res (1981) 9(13):3047–60. doi: 10.1093/nar/9.13.3047
105. Nagawa F, Ishiguro K, Tsuboi A, Yoshida T, Ishikawa A, Takemori T, et al. Footprint analysis of the RAG protein recombination signal sequence complex for V(D)J type recombination. Mol Cell Biol (1998) 18(1):655–63. doi: 10.1128/MCB.18.1.655
106. Zhao S, Gwyn LM, De P, Rodgers KK. A non-sequence-specific DNA binding mode of RAG1 is inhibited by RAG2. J Mol Biol (2009) 387(3):744–58. doi: 10.1016/j.jmb.2009.02.020
107. Zagelbaum J, Shimazaki N, Esguerra ZA, Watanabe G, Lieber MR, Rothenberg E. Real-time analysis of RAG complex activity in V(D)J recombination. Proc Natl Acad Sci U S A. (2016) 113(42):11853–8. doi: 10.1073/pnas.1606721113
108. Garcia-Perez L, Ordas A, Cante-Barrett K, Meij P, Pike-Overzet K, Lankester A, et al. Preclinical development of autologous hematopoietic stem cell-based gene therapy for immune deficiencies: a journey from mouse cage to bed side. Pharmaceutics. (2020) 12(6). doi: 10.3390/pharmaceutics12060549
109. Pike-Overzet K, Rodijk M, Ng YY, Baert MRM, Lagresle-Peyrou C, Schambach A, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. (2011) 25(9):1471–83. doi: 10.1038/leu.2011.106
110. Trancoso I, Bonnet M, Gardner R, Carneiro J, Barreto VM, Demengeot J, et al. A novel quantitative fluorescent reporter assay for RAG targets and RAG activity. Front Immunol (2013) 4:110. doi: 10.3389/fimmu.2013.00110
111. Tirosh I, Yamazaki Y, Frugoni F, Ververs FA, Allenspach EJ, Zhang Y, et al. Recombination activity of human recombination-activating gene 2 (RAG2) mutations and correlation with clinical phenotype. J Allergy Clin Immunol (2019) 143(2):726–35. doi: 10.1016/j.jaci.2018.04.027
112. Garcia-Perez L, van Eggermond M, van Roon L, Vloemans SA, Cordes M, Schambach A, et al. Successful preclinical development of gene therapy for recombinase-activating gene-1-Deficient SCID. Mol Ther Methods Clin Dev (2020) 17:666–82. doi: 10.1016/j.omtm.2020.03.016
113. Biggs CM, Haddad E, Issekutz TB, Roifman CM, Turvey SE. Newborn screening for severe combined immunodeficiency: a primer for clinicians. CMAJ. (2017) 189(50):E1551–7. doi: 10.1503/cmaj.170561
114. Adeli MM, Buckley RH. Why newborn screening for severe combined immunodeficiency is essential: a case report. Pediatrics. (2010) 126(2):e465–9. doi: 10.1542/peds.2009-3659
115. Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. (2011) 117(11):3243–6. doi: 10.1182/blood-2010-08-300384
116. Immune Deficiency Foundation (IDF)IDF SCID newborn screening campaign. In: Immune deficiency foundation. Available at: https://primaryimmune.org/scid-compass/idf-scid-newborn-screening-campaign#:~:text=On%20May%2021%2C%202010%2C%20the,programs%20in%20all%2050%20states.
117. Newborn screening for SCID expands to more countries! international patient organisation for primary immunodeficiencies (2023). Available at: https://e-news.ipopi.org/newborn-screening-for-scid-expands-to-more-countries/.
118. Onderzoek naar SCID - vanaf april 2018. RIVM (2023). Available at: https://www.rivm.nl/nieuws/onderzoek-naar-scid-vanaf-april-2018.
119. Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol (2005) 115(2):391–8. doi: 10.1016/j.jaci.2004.10.012
120. Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol (2012) 129(3):597–604. doi: 10.1016/j.jaci.2011.12.964
121. Schonland SO, Zimmer JK, Lopez-Benitez CM, Widmann T, Ramin KD, Goronzy JJ, et al. Homeostatic control of T-cell generation in neonates. Blood. (2003) 102(4):1428–34. doi: 10.1182/blood-2002-11-3591
122. Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer. (2018) 18(8):471–84. doi: 10.1038/s41568-018-0015-6
123. Gladdy RA, Taylor MD, Williams CJ, Grandal I, Karaskova J, Squirec JA, et al. The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell (2003) 3(1):37–50. doi: 10.1016/s1535-6108(02)00236-2
124. Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, Danska JS. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev (1996) 10(16):2038–54. doi: 10.1101/gad.10.16.2038
125. Vanasse GJ, Halbrook J, Thomas S, Burgess A, Hoekstra MF, Disteche CM, et al. Genetic pathway to recurrent chromosome translocations in murine lymphoma involves V(D)J recombinase. J Clin Invest. (1999) 103(12):1669–75. doi: 10.1172/JCI6658
126. Difilippantonio MJ, Zhu J, Chen HT, et al. DNA Repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. (2000) 404(6777):510–4. doi: 10.1038/35006670
127. Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. (2000) 404(6780):897–900. doi: 10.1038/35009138
128. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol Apr (2016) 16(4):234–46. doi: 10.1038/nri.2016.28
129. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. (2015) 125(11):4135–48. doi: 10.1172/JCI80477
130. Csomos K, Ujhazi B, Blazso P, Herrera JL, Tipton CM, Kawai T, et al. Partial RAG deficiency in humans induces dysregulated peripheral lymphocyte development and humoral tolerance defect with accumulation of T-bet(+) b cells. Nat Immunol (2022) 23(8):1256–72. doi: 10.1038/s41590-022-01271-6
131. Cassani B, Poliani PL, Marrella V, Schena F, Sauer AV, Ravanini M, et al. Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of omenn syndrome. J Exp Med (2010) 207(7):1525–40. doi: 10.1084/jem.20091928
132. Lawless D, Geier CB, Farmer JR, Allen HL, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol (2018) 141(6):2303–6. doi: 10.1016/j.jaci.2018.02.007
133. Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol (2014) 133(4):1099–108. doi: 10.1016/j.jaci.2013.10.007
134. Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow b cells. J Exp Med (1993) 177(4):1009–20. doi: 10.1084/jem.177.4.1009
135. Chen K, Wu W, Mathew D, Zhang Y, Browne SK, Rosen LB, et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J Allergy Clin Immunol (2014) 133(3):880–2 e10. doi: 10.1016/j.jaci.2013.11.038
136. Greenberg-Kushnir N, Lee YN, Simon AJ, Lev A, Marcus N, Abuzaitoun O, et al. A Large cohort of RAG1/2-deficient SCID patients-clinical, immunological, and prognostic analysis. J Clin Immunol (2020) 40(1):211–22. doi: 10.1007/s10875-019-00717-1
137. Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers. (2015) 1:15061. doi: 10.1038/nrdp.2015.61
138. Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol Feb (2015) 35(2):119–24. doi: 10.1007/s10875-014-0121-5
139. Kuijpers TW, Ijspeert H, van Leeuwen EM, Jansen MH, Hazenberg MD, Weijer KC, et al. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood. (2011) 117(22):5892–6. doi: 10.1182/blood-2011-01-329052
140. Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, et al. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol (2014) 134(6):1375–80. doi: 10.1016/j.jaci.2014.04.042
141. Kato T, Crestani E, Kamae C, et al. RAG1 deficiency may present clinically as selective IgA deficiency. J Clin Immunol (2015) 35(3):280–8. doi: 10.1007/s10875-015-0146-4
142. Geier CB, Piller A, Linder A, Sauerwein KM, Eibl MM, Wolf HM. Leaky RAG deficiency in adult patients with impaired antibody production against bacterial polysaccharide antigens. PloS One (2015) 10(7):e0133220. doi: 10.1371/journal.pone.0133220
143. Chou J, Hanna-Wakim R, Tirosh I, Kane .J, Fraulino D, Lee YN, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: omenn syndrome and hyper-IgM syndrome. J Allergy Clin Immunol (2012) 130(6):1414–6. doi: 10.1016/j.jaci.2012.06.012
144. Reiff A, Bassuk AG, Church JA, Campbell E, Bing X, Ferguson PJ. Exome sequencing reveals RAG1 mutations in a child with autoimmunity and sterile chronic multifocal osteomyelitis evolving into disseminated granulomatous disease. J Clin Immunol (2013) 33(8):1289–92. doi: 10.1007/s10875-013-9953-7
145. Nicolas N, Moshous D, Cavazzana-Calvo M, Papadopoulo D, Chasseval R, Deist Le F, et al. A human severe combined immunodeficiency (SCID) condition with increased sensitivity to ionizing radiations and impaired V(D)J rearrangements defines a new DNA recombination/repair deficiency. J Exp Med (1998) 188(4):627–34. doi: 10.1084/jem.188.4.627
146. Villa A, Santagata S, Bozzi F, et al. Partial V(D)J recombination activity leads to omenn syndrome. Cell. (1998) 93(5):885–96. doi: 10.1016/s0092-8674(00)81448-8
147. de Saint-Basile G, Le Deist F, de Villartay JP, et al. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome). J Clin Invest. (1991) 87(4):1352–9. doi: 10.1172/JCI115139
148. Rieux-Laucat F, Bahadoran P, Brousse N, et al. Highly restricted human T cell repertoire in peripheral blood and tissue-infiltrating lymphocytes in omenn’s syndrome. J Clin Invest. (1998) 102(2):312–21. doi: 10.1172/JCI332
149. Signorini S, Imberti L, Pirovano S, et al. Intrathymic restriction and peripheral expansion of the T-cell repertoire in omenn syndrome. Blood. (1999) 94(10):3468–78. doi: 10.1182/blood.V94.10.3468.422k34_3468_3478
150. Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood. Jan 1 (2001) 97(1):81–8. doi: 10.1182/blood.v97.1.81
151. Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst). (2014) 16:84–96. doi: 10.1016/j.dnarep.2014.02.011
152. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and omenn syndrome: the primary immune deficiency treatment consortium experience. J Allergy Clin Immunol (2014) 133(4):1092–8. doi: 10.1016/j.jaci.2013.09.044
153. de Villartay JP, Lim A, Al-Mousa H, Dupont S, Déchanet-Merville J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. (2005) 115(11):3291–9. doi: 10.1172/JCI25178
154. Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kühr J, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest. (2005) 115(11):3140–8. doi: 10.1172/JCI25221
155. Haddad E, Hoenig M. Hematopoietic stem cell transplantation for severe combined immunodeficiency (SCID). Front Pediatr (2019) 7:481. doi: 10.3389/fped.2019.00481
156. Wu X, Burgess SM. Integration target site selection for retroviruses and transposable elements. Cell Mol Life Sci (2004) 61(19-20):2588–96. doi: 10.1007/s00018-004-4206-9
157. Anderson WF. September 14, 1990: the beginning. Hum Gene Ther Winter (1990) 1(4):371–2. doi: 10.1089/hum.1990.1.4-371
158. Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T Lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. (1995) 270(5235):475–80. doi: 10.1126/science.270.5235.475
159. Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M. Viral vectors: a look back and ahead on gene transfer technology. New Microbiol (2013) 36(1):1–22.
161. Engelman A. The roles of cellular factors in retroviral integration. Curr Top Microbiol Immunol (2003) 281:209–38. doi: 10.1007/978-3-642-19012-4_6
162. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. (2003) 302(5644):415–9. doi: 10.1126/science.1088547
163. Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med (2003) 348(3):255–6. doi: 10.1056/NEJM200301163480314
164. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. Sep (2008) 118(9):3132–42. doi: 10.1172/JCI35700
165. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. (2008) 118(9):3143–50. doi: 10.1172/JCI35798
166. D’Apolito D, Baiamonte E, Bagliesi M, Marzo Di R, Calzolari R, Ferro L, et al. The sea urchin sns5 insulator protects retroviral vectors from chromosomal position effects by maintaining active chromatin structure. Mol Ther (2009) 17(8):1434–41. doi: 10.1038/mt.2009.74
167. Manic G, Maurin-Marlin A, Galluzzi L, Subra F, Mouscadet JF, Bury-Mone S. 3’ self-inactivating long terminal repeat inserts for the modulation of transgene expression from lentiviral vectors. Hum Gene Ther Methods (2012) 23(2):84–97. doi: 10.1089/hgtb.2011.154
168. Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum Gene Ther (2005) 16(11):1241–6. doi: 10.1089/hum.2005.16.1241
169. Yi Y, Hahm SH, Lee KH. Retroviral gene therapy: safety issues and possible solutions. Curr Gene Ther (2005) 5(1):25–35. doi: 10.2174/1566523052997514
170. Bukrinsky MI, Haffar OK. HIV-1 nuclear import: in search of a leader. Front Biosci (1999) . doi: 10.2741/bukrinsky
171. Freed EO. HIV-1 replication. Somat Cell Mol Genet (2001) 26(1-6):13–33. doi: 10.1023/a:1021070512287
172. Dropulic B. Lentiviral vectors: their molecular design, safety, and use in laboratory and preclinical research. Hum Gene Ther (2011) 22(6):649–57. doi: 10.1089/hum.2011.058
173. Howarth JL, Lee YB, Uney JB. Using viral vectors as gene transfer tools (Cell biology and toxicology special issue: ETCS-UK 1 day meeting on genetic manipulation of cells). Cell Biol Toxicol (2010) 26(1):1–20. doi: 10.1007/s10565-009-9139-5
174. Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med (2001) 7(1):33–40. doi: 10.1038/83324
175. Matrai J, Chuah MK, VandenDriessche T. Recent advances in lentiviral vector development and applications. Mol Ther (2010) 18(3):477–90. doi: 10.1038/mt.2009.319
176. Ciuffi A. Mechanisms governing lentivirus integration site selection. Curr Gene Ther (2008) 8(6):419–29. doi: 10.2174/156652308786848021
177. Pavel-Dinu M, Borna S, Bacchetta R. Rare immune diseases paving the road for genome editing-based precision medicine. Front Genome Ed (2023) 5:1114996. doi: 10.3389/fgeed.2023.1114996
178. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv (2020) 6(19):eaaz0571. doi: 10.1126/sciadv.aaz0571
179. Cromer MK, Camarena J, Martin RM, Lesch BJ, Vakulskas CA, Bode NM, et al. Gene replacement of alpha-globin with beta-globin restores hemoglobin balance in beta-thalassemia-derived hematopoietic stem and progenitor cells. Nat Med (2021) 27(4):677–87. doi: 10.1038/s41591-021-01284-y
180. Sweeney CL, Pavel-Dinu M, Choi U, Brault J, Liu T, Koontz S, et al. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Ther (2021) 28(6):373–90. doi: 10.1038/s41434-021-00251-z
181. Iancu O, Allen D, Knop O, Zehavi Y, Breier D, Arbiv A, et al. Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol Ther Nucleic Acids (2023) 31:105–21. doi: 10.1016/j.omtn.2022.12.006
182. Xue C, Greene EC. DNA Repair pathway choices in CRISPR-Cas9-Mediated genome editing. Trends Genet (2021) 37(7):639–56. doi: 10.1016/j.tig.2021.02.008
183. Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med Aug (2018) 24(8):1216–24. doi: 10.1038/s41591-018-0137-0
184. Gomez-Ospina N, Scharenberg SG, Mostrel N, Bak RO, Mantri S, Quadros RM, et al. Human genome-edited hematopoietic stem cells phenotypically correct mucopolysaccharidosis type I. Nat Commun (2019) 10(1):4045. doi: 10.1038/s41467-019-11962-8
185. Pavel-Dinu M, Wiebking V, Dejene BT, Srifa W, Mantri S, Nicolas CE, et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun (2019) 10(1):1634. doi: 10.1038/s41467-019-09614-y
186. Wang Q, Liu J, Janssen JM, Goncalves M. Precise homology-directed installation of large genomic edits in human cells with cleaving and nicking high-specificity Cas9 variants. Nucleic Acids Res (2023) 51(7):3465–94. doi: 10.1093/nar/gkad165
187. Allen D, Weiss LE, Saguy A, Rosenberg M, Iancu O, Matalon O, et al. High-throughput imaging of CRISPR- and recombinant adeno-associated virus-induced DNA damage response in human hematopoietic stem and progenitor cells. CRISPR J Feb (2022) 5(1):80–94. doi: 10.1089/crispr.2021.0128
188. Ferrari S, Jacob A, Cesana D, Laugel M, Beretta S, Varesi A, et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells. Cell Stem Cell (2022) 29(10):1428–1444 e9. doi: 10.1016/j.stem.2022.09.001
189. Scharenberg SG, Poletto E, Lucot KL, Colella P, Sheikali A, Montine TJ, et al. Engineering monocyte/macrophage-specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat Commun (2020) 11(1):3327. doi: 10.1038/s41467-020-17148-x
190. Turchiano G, Andrieux G, Klermund J, Blattner G, Pennucci V, Gaz ME, et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-seq. Cell Stem Cell (2021) 28(6):1136–1147 e5. doi: 10.1016/j.stem.2021.02.002
191. Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, Villartay De J-P, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med (2002) 346(16):1185–93. doi: 10.1056/NEJMoa012616
192. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. (2016) 533(7603):420–4. doi: 10.1038/nature17946
193. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576(7785):149–57. doi: 10.1038/s41586-019-1711-4
194. Lagresle-Peyrou C, Yates F, Malassis-Seris M, Hue C, Morillon E, Garrigue A, et al. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity. Blood. (2006) 107(1):63–72. doi: 10.1182/blood-2005-05-2032
195. Cordes M, Cante-Barrett K, van den Akker EB, Moretti FA, Kiełbasa SM, Vloemans SA, et al. Single-cell immune profiling reveals thymus-seeding populations, T cell commitment, and multilineage development in the human thymus. Sci Immunol (2022) 7(77):eade0182. doi: 10.1126/sciimmunol.ade0182
196. Cordes M, Pike-Overzet K, Van den Akker EB, Staal FJ, Cante-Barrett K. Multi-omic analyses in immune cell development with lessons learned from T cell development. Front Cell Dev Biol (2023) 11. doi: 10.3389/fcell.2023.1163529
197. Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. (2009) 326(5954):818–23. doi: 10.1126/science.1171242
198. Pike-Overzet K, Baum C, Bredius RG, Cavazzana M, Driessen G-J, Fibbe WE, et al. Successful RAG1-SCID gene therapy depends on the level of RAG1 expression. J Allergy Clin Immunol (2014) 134(1):242–3. doi: 10.1016/j.jaci.2014.04.033
199. van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, Velden der van G, et al. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune omenn-like syndrome. J Allergy Clin Immunol (2014) 133(4):1116–23. doi: 10.1016/j.jaci.2013.10.009
200. Pike-Overzet K. Gene therapy for RAG-deficient severe combined immunodeficiency. Erasmus MC, editor. Erasmus University Rotterdam: University Medical Centre Rotterdam; (2007).
201. Yates F, Malassis-Seris M, Stockholm D, Bouneaud C, Larousserie F, Noguiez-Hellin P, et al. Gene therapy of RAG-2-/- mice: sustained correction of the immunodeficiency. Blood. (2002) 100(12):3942–9. doi: 10.1182/blood-2002-03-0782
202. van Til NP, de Boer H, Mashamba N, Wabik A, Huston M, Visser TP, et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther (2012) 20(10):1968–80. doi: 10.1038/mt.2012.110
203. Capo V, Castiello MC, Fontana E, Penna S, Bosticardo M, Draghici E, et al. Efficacy of lentivirus-mediated gene therapy in an omenn syndrome recombination-activating gene 2 mouse model is not hindered by inflammation and immune dysregulation. J Allergy Clin Immunol Sep (2018) 142(3):928–941.e8. doi: 10.1016/j.jaci.2017.11.015
204. Gardner CL, Pavel-Dinu M, Dobbs K, Bosticardo M, Reardon PK, Lack J, et al. Gene editing rescues In vitro T cell development of RAG2-deficient induced pluripotent stem cells in an artificial thymic organoid system. J Clin Immunol (2021) 41(5):852–62. doi: 10.1007/s10875-021-00989-6
205. Allen D, Knop O, Itkowitz B, Iancu O, Beider K, Lee YN, et al. CRISPR-Cas9 RAG2 correction via coding sequence replacement to preserve endogenous gene regulation and locus structure, 17 February 2023, PREPRINT (Version 1). (2023). doi: 10.21203/rs.3.rs-2565742/v1.
Keywords: BCR - B cell receptor, TCR - T cell receptor, rearrangements of immunoglobulin and T cell receptor genes, gene therapy (GT), thymus, bone marrow, Recombination activating genes
Citation: Braams M, Pike-Overzet K and Staal FJT (2023) The recombinase activating genes: architects of immune diversity during lymphocyte development. Front. Immunol. 14:1210818. doi: 10.3389/fimmu.2023.1210818
Received: 23 April 2023; Accepted: 19 June 2023;
Published: 11 July 2023.
Edited by:
Yu Nee Lee, Sheba Medical Center, IsraelReviewed by:
Ayal Hendel, Bar-Ilan University, IsraelCopyright © 2023 Braams, Pike-Overzet and Staal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank J. T. Staal, Zi5qLnQuc3RhYWxAbHVtYy5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.