 Qiuyun Yi
Qiuyun Yi Jinxian Yang
Jinxian Yang Ying Wu
Ying Wu Ying Wang
Ying Wang Qiqi Cao
Qiqi Cao Wen Wen
Wen Wen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 July 2023
Sec. Cytokines and Soluble Mediators in Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1204524
This article is part of the Research Topic Community Series in Hepatic Immune Response underlying Liver Cirrhosis and Portal Hypertension, volume II View all 14 articles
Cirrhosis is a progressive and diffuse liver disease characterized by liver tissue fibrosis and impaired liver function. This condition is brought about by several factors, including chronic hepatitis, hepatic steatosis, alcohol abuse, and other immunological injuries. The pathogenesis of liver cirrhosis is a complex process that involves the interaction of various immune cells and cytokines, which work together to create the hepatic homeostasis imbalance in the liver. Some studies have indicated that alterations in the immune microenvironment of liver cirrhosis are closely linked to the development and prognosis of the disease. The noteworthy function of mesenchymal stem cells and their paracrine secretion lies in their ability to promote the production of cytokines, which in turn enhance the self-repairing capabilities of tissues. The objective of this review is to provide a summary of the alterations in liver homeostasis and to discuss intercellular communication within the organ. Recent research on MSCs is yielding a blueprint for cell typing and biomarker immunoregulation. Hopefully, as MSCs researches continue to progress, novel therapeutic approaches will emerge to address cirrhosis.
According to epidemiological data (1), approximately 1.5 billion people worldwide suffer from chronic liver disease (CLD), with about 20,000 deaths occurring annually, of which 10,000 are caused by liver cirrhosis. The global mortality for liver cirrhosis has risen by 47.15% in recent years (2, 3). Viral hepatitis, alcoholic liver disease, and non-alcoholic steatohepatitis are the leading causes of liver cirrhosis (4). Moreover, a wide range of other factors also can lead to cirrhosis, including genetic factors, autoimmune diseases, cholestatic diseases, iron or copper overload (5). Hepatitis B virus (HBV) and hepatitis C virus (HCV) are responsible for more than 60% of cirrhotic cases worldwide (6). The number of hospitalized patients with HCV-related cirrhosis is anticipated to decrease significantly by 2025 (7). Only a small number of patients infected with the hepatitis D virus (HDV) will develop liver cirrhosis (8). In pregnant women, low immune function often plays a role as a prerequisite for liver cirrhosis when infected with the hepatitis E virus (HEV), with up to 30% of pregnant patients dying from HEV infection (9). Alcohol-related cirrhosis (AC) has been shown to be a significant cause of hospitalization in the United States, with the number of hospitalized patients increasing rapidly (10). A survey of middle-aged women in the UK found that the higher the amount of alcohol consumed, the greater the incidence of liver cirrhosis (11). Due to the development of hepatitis virus vaccines and effective antiviral therapy, the incidence and prevalence of end-stage liver cirrhosis in non-alcoholic fatty liver disease (NAFLD) change to has risen sharply (12). The prevalence rate of NAFLD-related end-stage liver cirrhosis in China is growing at an alarming rate with the accelerating urbanization process. It is estimated that the number of NAFLD patients in China will reach 314.58 million by 2030 (13). We concluded the epidemiology and risk factors for liver cirrhosis (Figure 1).
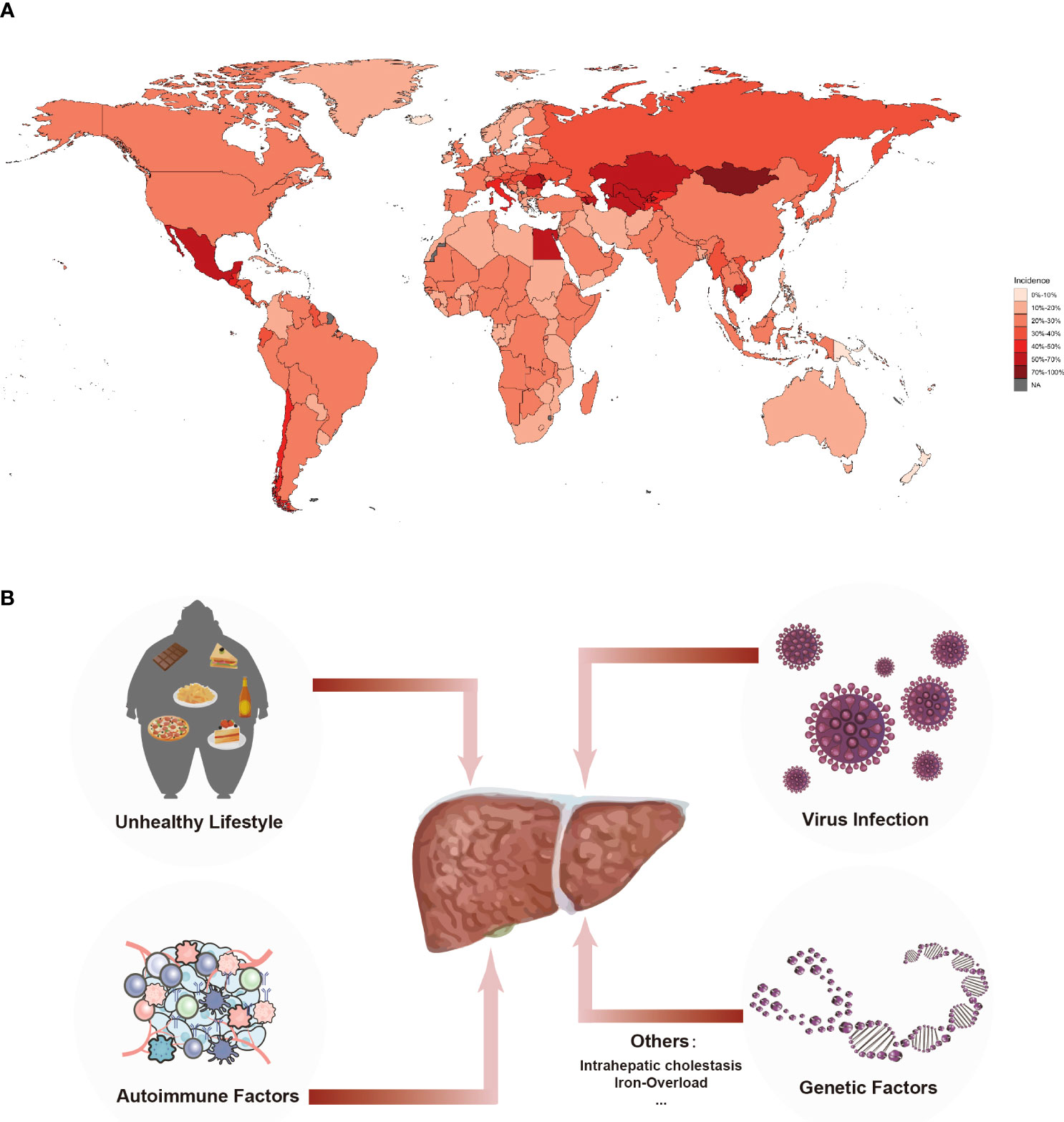
Figure 1 Incidence and Etiology of Liver Cirrhosis. (A) Incidence of liver cirrhosis in chronic liver diseases worldwide. Around 1.5 billion people worldwide suffer from chronic liver diseases that eventually progresses into fibrosis and cirrhosis, result in 10 thousand deaths globally. The data were extracted from the GHDx database (https://ghdx.healthdata.org). (B) The etiology of cirrhosis. Leading etiologies of cirrhosis were viral, alcoholic, unhealthy lifestyle and genetic factors.
Cirrhosis is an end-stage pathological process caused by a variety of chronic liver diseases that will result in persistent chronic liver injury (14). Cirrhosis, characterized by chronic inflammatory necrosis and dynamic fibrosis, is considered to be a diffuse pathological state with a transformation from normal liver tissue structure to abnormal nodular hyperplasia, which in turn progresses from compensated cirrhosis (asymptomatic stage) to decompensated cirrhosis (symptomatic stage) (5), eventually leading to hepatocellular carcinoma (15). However, studies have found that this process can be prevented, bringing about reversible liver fibrosis and the reversal of cirrhosis (15, 16). Regardless of the complexity and prevalence of the etiology of cirrhosis, liver fibrosis is a mandatory part of cirrhosis. Chronic inflammatory liver injury and liver fibrosis continue to increase, leading to dysregulated crosstalk between immune cells in the liver microenvironment, which drives the progression of cirrhosis (17–19).
In addition to its role in metabolism, nutrient storage, and detoxification, the liver is the body's most functionally complex immune organ. It has a profound impact on immune function (20). The liver is rich in blood circulation and the circulation system collects blood from the portal vein and the hepatic artery, which contains a large number of microbial-associated molecular patterns (MAMP), pathogen-associated molecular patterns (PAMP), damage-associated molecular patterns (DAMP), and various toxin and antigen molecules from the intestine (19, 21). Herein, the liver must simultaneously recognize antigenic components from the systemic circulation and the gastrointestinal tract. These antigens stimulate the liver through a series of pattern recognition receptors (PRR), such as Toll-like receptors (TLR) and nucleotide-binding oligomeric domain-like receptors (NOD-like receptors or NLR), which trigger unique immune responses to induce immune activation and immunomodulatory cytokine production. TLR is expressed on various hepatic cells, like Kupffer cells (KCs), dendritic cells, hepatic stellate cells, endothelial cells and hepatocytes (22). The hepatic immune microenvironment contains a variety of immune cells and molecules performing unique roles based on the association with non-immune cells, thus developing a complex and dynamic network system. Although the irreplaceable metabolic functions of the liver often obscure the perception of its role as an immune organ, hepatic metabolic functions create a microenvironment in which parenchymal and non-parenchymal cells communicate; in other words, the metabolic environment can alter the immune response in the liver (23).
The liver microenvironment consists of multiple components, including KCs, hepatic sinusoidal endothelial cells (HSECs), HSCs, immune cells, extracellular matrix (ECM), cytokines, and various growth factors (24, 25). Along with the liver’s inherent immune dysfunction, viral infections, alcohol abuse, metabolic disorders, and autoimmune abnormalities can indirectly inflict liver injury, inflammation, fibrosis, and cirrhosis. Changes to the immune microenvironment in liver cirrhosis involve a decrease in CD8+ T cells and natural killer (NK) cells and an increase in CD4+ memory T cell infiltration (26) (Figure 2).
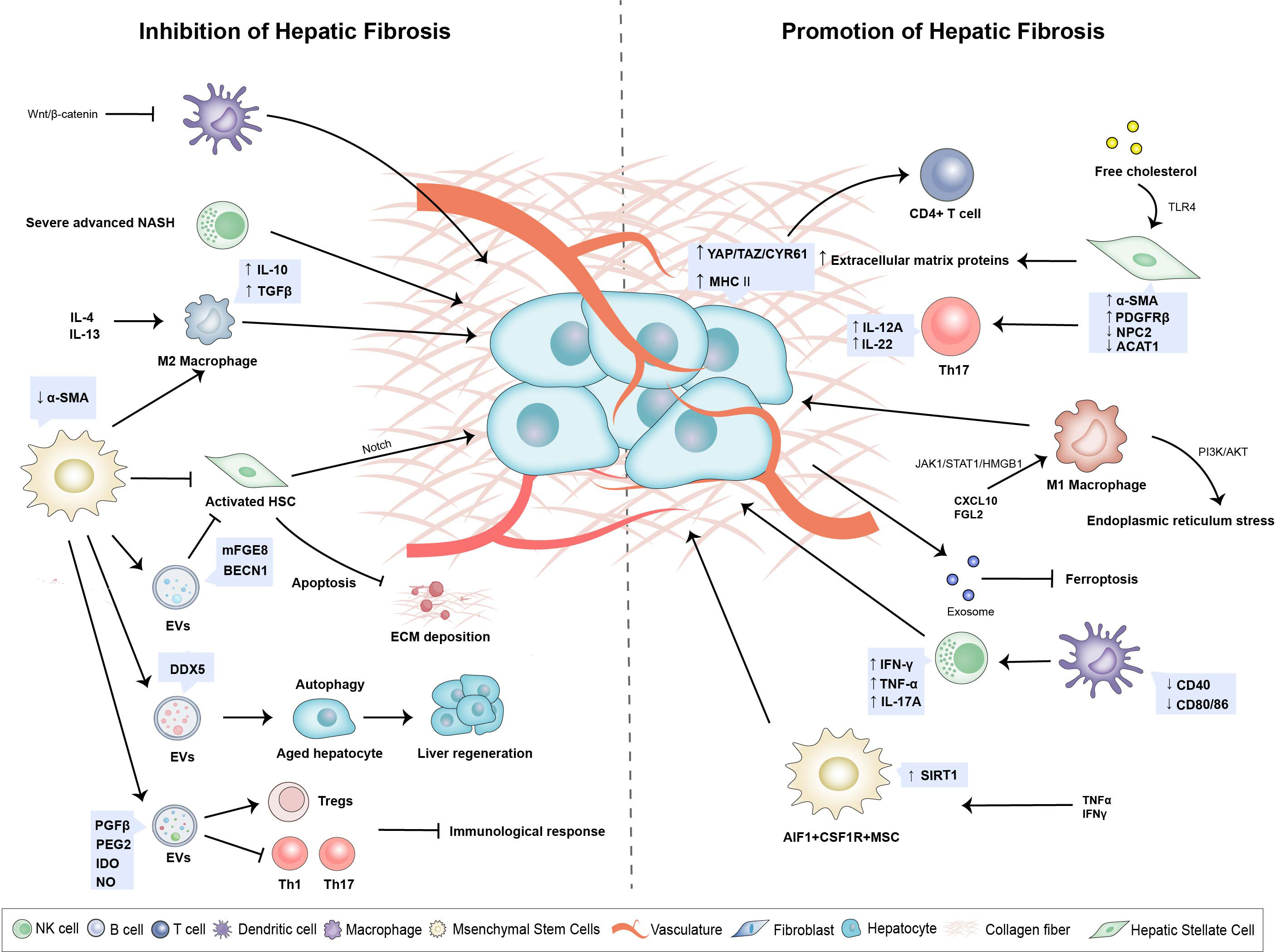
Figure 2 Changes of hepatic immune microenvironment play a pivotal role in hepatic fibrogenesis. A variety of immune cells and non-immune cells constitute a complex and dynamic network system. Upregulation of YAP/TAZ/CYR61 in activated Hepatocytes activating monocyte differentiation into pro-inflammatory macrophages. Activated HSCs promote lipid droplet loss and α-smooth muscle actin increase as well as secretion of extracellular matrix proteins and accelerate the development of liver fibrosis. Hepatic Macrophages promote autophagy and activation of HSC by secreting prostaglandin E2 and binding to receptor EP4, which leads to the development of liver fibrosis and cirrhosis. MSCs can inhibit the inflammation and immune response, inhibit the excessive ECM deposition, and promote the hepatocyte regeneration during liver fibrosis.
Hepatocytes are involved in the innate immune response by undergoing organelle damage and releasing stress signals in response to injury and inflammatory stimulation, promoting the development of liver cirrhosis and cancer. This process occurs with complicated crosstalk between hepatocytes and immune cells in the liver microenvironment (27, 28). In mice, activated hepatocytes can induce monocytes into pro-inflammatory macrophages with increased YAP/TAZ/CYR61, stimulating liver inflammation and fibrosis (28, 29). YAP/TAZ is the vital effector in the Hippo pathway, which regulates TGF-β2–mediated fibrogenesis (30). MHC-II is highly expressed in hepatocytes of alcoholic hepatitis, and it can activate CD4-positive lymphocytes and trigger a pro-inflammatory response (31). Lipid deposition can increase the susceptibility of hepatocytes to apoptosis in patients with nonalcoholic steatohepatitis (NASH), which had demonstrated in high-fat diet (HFD) mice. Notably, lacking AMP-activated protein kinase (AMPK) can accelerate fibrosis in NASH (32). Virus-infected and hepatocyte-derived exocrine miR-222 promoted fibrosis by inhibiting TFRC and TFRC-induced ferroptosis (33).
The overexpression of transcription factor FoxM1 was dependent on Kupffer cells, and it triggered hepatocyte death and contributed to liver inflammation and injury (34). Hepatocyte autophagy is a steady-state process that protects against hepatocyte death (27, 35). In CCl4-induced mouse models and cirrhotic patients, hepatocyte autophagy was significantly inhibited by the miR-125a/VDR axis-dependent autophagy, which finally promoted liver fibrosis (36). Autophagy disorders were also observed in alcoholic liver disease (ALD) and NAFLD (37). While the situation was different in viral hepatitis. Hepatocyte autophagy could enhance HBV DNA replication (38), while autophagy disorders could inhibit HCV replication by enhancing intracellular immunity (39). Telomere shortening and the absence of telomerase in hepatocytes could lead to cell senescence, promoting virus replication and liver cirrhosis (40, 41).
HSCs reside in the Disse space between hepatic sinusoidal endothelial cells (LSECs) and hepatocytes. In their resting state, HSCs contain many retinol (vitamin A) lipid droplets (42). However, when the liver was subjected to inflammatory stimulation or hepatocyte death, HSCs received signals secreted by immune and non-immune cells in the liver microenvironment and underwent transdifferentiation into proliferative fibroblast myofibroblasts (MFs) (43). Activated HSCs lost lipid droplets and upregulated the expression of α-smooth muscle actin (α-SMA) (44), which led to the secretion of extracellular matrix proteins and the eventual development of liver fibrosis (17). The percentage of α-SMA positive hepatic stellate cells was significantly increased in patients with virus-associated cirrhosis (45). HSC activation was driven by the increased level of platelet-derived growth factor (PDGF) receptor β (46). Kupffer cells secreted PDGF, which could stimulate the production and deposition of collagen. Additionally, activation of the acid-sensing ion channel 1a (ASIC1a) via the PI3K/AKT pathway induced endoplasmic reticulum stress (ERS), thereby promoting the progression of liver fibrosis (47, 48). Apart from retinoids, cholesterol, triglycerides, phospholipids, and free fatty acids are also present in HSC lipid drops (49). The NPC2 protein expressed in resting HSC, as well as the ACAT1 isoenzyme, together bound directly to free cholesterol and played a critical role in cholesterol metabolic homeostasis. The accumulation of free cholesterol stimulated HSC through increasing TLR4 signaling and sensitizing HSC to transforming growth factor β (TGF-β) (50, 51). In a high-cholesterol diet mouse model of NASH, NPC2 and ACAT1 deficiency significantly boosted liver fibrosis progression (52, 53).
T helper cells (Th17s) collaborated with HSCs in a pro-inflammatory circumstance. Activated HSCs recruited more Th17 cells and provoked the secretion of IL-12A and IL-22 that contributed to cirrhosis in chronic hepatitis B (CHB) (54). Regulatory T cells (Tregs) possess anti-inflammatory properties. IL-8 produced by Foxp3+CD4+ Tregs activated HSCs and promoted liver fibrogenesis in chronic hepatitis C (43). 22-carbon hexanoic acid (DHA) plays a critical role in anti-fibrotic activity depending on peroxisome proliferator-activated receptor γ (PPARγ), while it is absent in liver cirrhosis patients, low level of DHA promotes NF-κB and TGF-β pathways in HSC and consecutively activates HSC (55, 56). Additionally, it was found that membrane-bound glycoprotein CD73 promoted activation and autophagy of HSCs by promoting AMPK/AKT/mTOR signaling pathway, which was conducive to alcohol-related liver fibrosis (57).
Cell-derived extracellular vesicles (EVs) have emerged as essential agents in the progression of liver injury and fibrosis (58). Delivering diverse cargo via EVs is a critical component of cell-to-cell communication (59). In the liver, EVs from injured hepatocytes and LSECs activate and migrate of HSCs (1, 58). Recent research shows that SHP2 in HSCs exerts its pro-fibrotic role by enhancing the release of fibrogenic EVs through inhibiting autophagy, REDD1, and activating the mTOR pathway (60).
Mesenchymal stromal cells (MSCs) are multipotent fibroblast-like cells that have the ability to differentiate into hepatocyte-like cells (HLCs) and immunomodulatory properties have received much attention in a wide range of medical and health fields (61).MSC was reported to express a specific set of surface markers, such as CD73, CD90 and CD105 (62).
Single-cell RNA sequencing analysis unveiled that different subsets of MSCs were functionally distinct, and even though CMKLR1+ MSCs had lower proliferative capacity than CMKLR1- MSCs, the former had superior immunomodulatory functions (63). In addition, Zong et al. also identified another isoform by using the high-throughput sequencing technology, AIF1+CSF1R+MSCs, with high expression of SIRT1 and induced by TNF-α, exerting pro-inflammatory and pro-tumorigenic effects (64). Similar to HSCs, MSCs are one of the sources of MFs in the liver and are highly differentiated (65). Nevertheless, MSCs could suppress HSC activation and protect hepatocytes from damage by inhibiting the Notch pathway, thus alleviating the progression of liver fibrosis to cirrhosis (66). It has been shown that MSCs only become immunosuppressive when exposed to sufficiently high levels of pro-inflammatory cytokines (67–69). Despite their higher pro-inflammatory potential, MSCs can exhibit pro-inflammatory phenotypes when exposed to low levels of IFN-γ and TNF-α. Through the production of chemokines, they enhance T cell response by bringing lymphocytes to areas of inflammation (67).
MSCs have been shown to exert significant therapeutic effects utilizing their soluble products, such as extracellular vesicles, cytokines, trophic factors, and chemokines. Research shows that the EVs generated by MSCs, such as exosomes, could make great contributions to the therapeutic potential in tissue repair, angiogenesis and immunomodulation by facilitating cell–cell interactions, and delivering paracrine factors (70). MicroRNA-618, the exosome of MSCs, acted as an instrumental player targeting Smad4 to reverse the progression of fibrosis to cirrhosis (71). Exosome-derived Bone marrow stromal cell-derived exosomes (BMSC-Exos) attenuated collagen deposition and liver impairment, enhanced hepatocyte proliferation and ultimately alleviated liver fibrosis in a rabbit cirrhosis model (72). BMSC-Exos suppressed hepatocyte pyroptosis by downregulating pyroptosis-related proteins which included NLRP3, caspase-1, and IL-1β, thereby remitting liver cirrhosis (73).
Moreover, the exosomes secreted by MSC have similar physiological functions as MSCs and play a major role in cellular communication (74, 75). They can also induce anti-inflammatory M2 polarization and facilitate the production of anti-inflammatory mediators such as IL-10 and TGF-β (76). HSC ferroptosis can also be mediated by MSC-exosome (MSC-Exo) to mitigate liver fibrosis (77).
It has been shown that MSCs regulate the innate and adaptive immune response through intercellular contacts or paracrine mechanisms (67). As an illustration, MSCs can produce HGF and IL-6, which inhibit monocyte differentiation into dendritic cells, lowering inflammation, decreasing the secretion of IL-12 and IFN-γ, and increasing the production of IL-10, continually weakening the activation of T cells (78). MSCs inhibit the Kupffer cell activity, reducing the production of the pro-inflammatory cytokine TNF. Furthermore, MSCs secrete PGE2 to transform pro-inflammatory M1 macrophages into anti-inflammatory M2 macrophages (79). MSCs suppress CD8+ T lymphocyte proliferation and enhance CD4+ T lymphocyte conversion from T-helper 1 to T-helper 2 phenotype by producing IDO and heme oxygenase 1 (80).
Furthermore, recent research suggests that autophagy and senescence are mechanisms through which MSCs acquire their antifibrotic properties. As a vital cellular process, autophagy prevents nutritional, metabolic, and infection-mediated stress while maintaining homeostasis (81). The efficacy of MSCs as a therapeutic intervention is contingent upon the maintenance of optimal levels of autophagy, which in turn can ameliorate the fibrotic cascade. Despite this, aging-related autophagic damage is associated with a decline in MSC number and function, which are crucial to liver fibrosis (82).
LSECs are non-substantial hepatic endothelial cells lacking basement membranes and rich in open window pores. These LSECs window pores serve as a parclose to protect hepatocytes from various damages and facilitate substance exchange by producing nitric oxide (NO) for stimulating vascular endothelial growth factor (VEGF) and reversing activated HSC to a resting state (83–85). The alcohol-metabolizing enzyme Cytochrome P4502E1 (CYP2E1) was expressed in alcohol-induced LSEC, leading to increased acetylation of mitochondrial heat shock protein 90 (Hsp90). This acetylation reduced the interaction between Hsp90 and nitric oxide synthase (eNOS), resulting in decreased NO production and increased alcohol-induced liver injury (86). Similarly, Notch signaling was activated in LSECs of NASH mice and exacerbated NASH progression in an eNOS-dependent mechanism (87). LSECs possessed endocytic and clearance abilities and vital immune functions, impacting the homeostasis of the liver microenvironment (88, 89). During the early stage of liver cirrhosis, LSECs exhibited anti-inflammatory effects (90). Upon microbial infection, LSECs triggered local activation of effector CD8 T cells that exerted the immune surveillance capacity of the liver (88). Immunoproteasome LMP7 levels in LSECs were elevated in cirrhosis patients and liver fibrosis mice models, and LSECs presented MCH-II antigen to CD4 T cells after liver injury stimulation (91).
Hepatocyte death can lead to LSECs capillarization, immune cell interactions, and HSCs activation (92). Hepatic sinusoidal capillarization is the underlying pathological change of liver cirrhosis (93). This transformation was deleterious in NASH to form a basement membrane on LESCs’ surface, which inhibited the release of very low-density lipoprotein (VLDL) from hepatocytes into the Disse cavity and finally promoted hepatic steatosis. Capillarization also invoked hedgehog (Hh) signaling and exacerbated liver cirrhosis development (94, 95). Adipocyte fatty acid binding protein (A-FABP) regulated lipid metabolism, and elevated expression of A-FABP was observed in cirrhosis-associated NAFLD. Meanwhile, A-FABP stimulated Hh signaling and promoted LSECs vascularization, which led to HSC activation to enhance TGF-β1 activity, resulting in more severe liver fibrosis (96). In the progression of liver fibrosis, CXCR4 and CXCR7 exerted opposite effects on LSECs. With the increase of HIF-1α, CXCR4 upregulated to promote the isoform PDGF-BB secretion by LSEC and binding to its receptors, forming an intercellular crosstalk that activated HSCs and aggravated fibrosis, promoting the development of cirrhosis. While CXCR7 downregulation facilitated the capillarization of LSEC to promote hepatic cirrhosis (97).
DCs, as the most important antigen-presenting cells (APC), serve as a bridge between innate and adaptive immunity. DCs recognized and ingested pathogenic antigens through phagocytosis with other immune cells and presenting MHC peptides to CD4 T cells and CD8 T cells to initiate the immune response for exogenous antigens (98). DCs include plasmacytoid DC (pDC) derived from common dendritic progenitor (CDP) cells and the conventional dendritic cell subtype generated by the entry of circulating cDC precursors into the peripheral environment (99).
Comprehensive single-cell RNA sequencing analysis revealed that cDCs were associated with NASH pathology. Elevated Xcr1cDC1 was observed in the NASH model to increase pro-inflammatory CD8T cells and exacerbated NASH to cirrhosis (100). The deficiency of Cbl-b and c-Cbl in DCs led to the excessive accumulation of cDC1 in the liver and promoted liver cirrhosis and premature death in mice (101). The Wnt/β-catenin pathway is crucial in liver homeostasis (102). Lack of Wnt/β-catenin signal should be triggered autoimmune hepatitis (AIH) and abnormal activation of hepatic dendritic cells (HDCs), promoting cholestatic liver injury and fibrosis (103). DCs induced NK cells to proliferate and produced IFN-γ, and DC-NK crosstalk severely impaired the ability of antiviral immune response in CHB patients (104). DCs were rapidly recruited to the liver of NASH mice model with elevated TNF-α, IL-6, and MCP-1 expression. DCs depleting delayed intrahepatic inflammation and fibrosis regression, thereby promoting NASH. Chronic alcohol consumption decreased the production of cytokines such as TNF-α, IFN-γ and IL-12 in DCs, and the number of peripheral blood DCs. It also decreased the expression of CD40, CD80, or CD86, which reduced the stimulatory function of DCs on T cells and led to immune deficiency in mice (105). CCL20, produced primarily by HSCs, is a chemoattractant for immature dendritic cells with inflammatory molecules mediating fibrosis. Interaction between immune cells resulted in the increased expression of CCL20 in NAFLD fibrosis patients (106). Indoleamine 2,3-dioxygenase 1 (IDO1), an immunomodulatory enzyme, was highly expressed in choledochotomy (BDL)-induced mice, inhibiting DCs maturation and T cell proliferation from recruiting immune cells and promoting hepatic fibrosis (107).
NK cells have a powerful killing function, whose amount and activity are always affected by the liver immune microenvironment. They serve as surveillance to monitor external infections, tumors, inflammatory stimuli, and autoimmunity and secrete cytokines and chemokines (108). NK cells are divided into two subgroups: CD56dim (> 90%) group and CD56bright group. The former has a more substantial cytotoxic effect and plays a role as an immunomodulator (109). NK cells are the first line of defense against viral hepatitis, exerting an antiviral immune response by directly clearing virally infected cells or activating antigen-specific T cells via the production of IFN-γ and TNF-α (110). The decrease of CD56dim NK cells, total NK cells, and their activated receptor NKG2D in peripheral blood monocytes (PBMC) of NAFLD patients can lead to NK cell dysfunction (111, 112). In particular, NK cell function was defective and inactivated in patients with CHB, and monocytes suppressed HBV-specific T cell immune responses, leading to chronic persistent HBV infection (113). The increase of CD56bright NK cells could be detected in patients with autoimmune-mediated liver disease, and elevated serum IFN-γ levels induced hepatocyte death by enhancing the cytotoxicity of NK cells, ultimately resulting in macrophage activation and the development of fibrosis (114).
NKT cells possess NK cell-like characteristics and express T cell receptors, which recognize lipid antigens from major MHC-1-associated protein CD1d (115, 116). Invariant natural killer cells (iNKTs) are the primary subtype of NKT cells (109). Patients with HBV-associated liver cirrhosis (HBV-LC) showed highly activated peripheral iNKT cells, which may lead to overhealing caused by extracellular matrix deposition and the progression from fibrosis to cirrhosis (117). Liver injury induced by concanavalin A (ConA) intravenous administration was considered as an experimental model of T cell-mediated AIH in mice, in which iNKTs were specifically activated to kill hepatocytes and accumulated in the mouse liver, increasing activated immune cells cytokine through upregulation of Fas/FasL in the liver, resulting in more severe immune damage (118). With the prevalence of obesity, excessive cholesterol uptake directly destroys the function of NKT cells through lipid oxidation during the progression of NAFLD disease to liver cirrhosis. Interestingly, NKT cell depletion occurred in the early stage of mild NASH. For severe advanced NASH, NKT cells were protective against disease progression and played an anti-fibrotic role (119, 120). Compared with healthy people, primary biliary cholangitis patients had more iNKT cells, which produced high levels of IL-17A and promoted the progression of PBC-related fibrosis (121).
For the complexity of the liver microenvironment and immune function, macrophages show great plasticity and heterogeneity (122, 123). Macrophages can polarize into M1 cells and M2 cells. The classical M1 subtype is activated by TLR ligand and IFN-γ and secretes pro-inflammatory cytokines. On the contrary, the alternative M2 subtype secretes anti-inflammatory cytokines, which are stimulated by IL-4 or IL-13 (124). In the progression of NAFLD, it was found that hepatic macrophages polarized toward M2 and promoted HSC autophagy and activation by secreting prostaglandin E2 (PGE2) and then binding with EP4, which in turn favored the development of liver fibrosis and cirrhosis (125). It was reported that fibrinogen-like 2 (Fgl2) mediated mitochondrial damage, disrupted mitochondrial HSP90-Akt interactions. Moreover, Fgl2 induced M1 Polarization to secrete pro-inflammatory factors in hepatitis B (126). CXCL10 promoted M1 polarization, resulting in the activation of the JAK/STAT1 pathway (127). The connection between macrophages and HSCs can facilitate liver fibrogenesis. Subtype M2C-like polarized macrophages activated tyrosine kinase receptors (MerTK) on their surface influencing the profibrogenic HSCs (128).
M1 Polarization of macrophages is critical in the liver. Xu et al. found that osteopontin promoted M1 Polarization in NAFLD by activating JAK1/STAT1/HMGB1 signaling, which aggravated liver injury and cirrhosis (129). Studies in a humanized mouse model of HBV infection revealed that HBV could induce the differentiation of human monocytes/macrophages into M2 macrophages, which then expressed IL-10 and other inhibitory cytokines (113, 130). In addition, soluble CD206 (sCD206) expressed by macrophages could promote T cell activity and inhibit the antiviral effect of CD8T cells. High expression of sCD206 accelerated the progression of cirrhosis in patients with hepatitis B virus-related decompensated cirrhosis (HBV-DeCi) (131).
Treatment of the etiology is the cornerstone of cirrhosis treatment. The means of treating the primary cause include alcohol abstinence for alcoholic cirrhosis, antiviral drugs for HBV and HCV, immunosuppressants for autoimmune hepatitis, and ursodeoxycholic acid for primary biliary cholangitis. Several studies have shown that etiologic treatment can effectively restrain the progression of cirrhosis and even reverse patients with decompensated cirrhosis to compensated cirrhosis (i.e., recompensated cirrhosis), thus reducing the rate of death and improving the quality of life. Therefore, better studies of the altered cirrhosis immune microenvironment would help to develop more effective targeted therapeutic regimens.
Currently approved antiviral therapies for HBV include pegylated interferon alpha (PEG-IFN-α) with immunomodulatory activity and nucleoside (acid) analogs (NAs) that inhibit HBV polymerase. Still, neither achieves the functional cure of HBV (i.e., scavenging HBsAg) (132). The conditions for the usage of the two drugs are different. NAs can prevent severe viral hepatitis relapse, especially in patients with liver cirrhosis. While PEG-IFN-α is contraindicated in cirrhosis patients, for it can cause more severe liver damage (133). In a randomized open phase II trial, treatment with elbasvir/grazoprevir (EBR/GZR) + sofosbuvir (SOF) for 12 weeks was highly effective for HCV patients treated either with or without peginterferon (PEG-IFN-α-2a) or for cirrhosis patients (134).
The most advanced approach in clinical development to date is the competitive inhibitor myrcludex-B (MyrB) based on the PreS1 peptide now called the hepatocyte entry inhibitor bulevirtide (BLV), which has successfully blocked HBV and HDV entry (135). In HDV-associated cirrhosis patients, for whom interferon is contraindicated, treatment with BLV alone results in a sustained virologic response (136), but the optimal duration remains determined (137). The combination of BLV and tenofovir disoproxil fumarate (TDF) has a favorable safety and efficacy profile for treating HDV-related compensated cirrhosis (138). Lonafarnib (LNF) and ritonavir (RTV) are promising therapies for treating HDV, and the combination of PEG-IFN-α may increase the efficacy. A phase III clinical trial of LNF is currently underway (139).
The risk of HCV progression to cirrhosis and HCC continues to increase after treatment with direct-acting antiviral agents (DAAs). Dysfunction of CD4+ and CD8+ T cells has been identified in patients with hepatitis C, making liver immunotherapy urgent (140). Compared with an oral agonist of TLR 7 (GS-9620), the agonist of TLR 8 (GS-9688) stimulated the expression of IFN-γ and TNF-α in NK cells while all increased the antiviral capacity of CD8+ T cells (141). The immunotherapy by GS-9688 achieved sustained efficacy in murine models of HBV (142). The safety and tolerability of oral selgantolimod (TLR 8 agonist) was evaluated in CHB patients in one phase Ib study, and the recent phase II study further supported the development of this immunomodulator (143). NASVAC, a vaccine formulation containing both hepatitis B surface antigen (HBsAg) and hepatitis B core antigen (HBcAg), targeted a lower proportion of patients who developed cirrhosis in phase III clinical trials compared with PEG-IFN (144). HCV vaccine in phase I-II clinical trials found that 78% of HCV-infected patients showed a specific response to T cells, reducing the peak of HCV RNA level, providing a basis for future immunotherapy (145). In a proof-of-concept clinical trial, combination therapy of entecavir (NA) plus PEG-IFN-α-2a followed by HBV vaccination developed a “blueprint” for serum clearance of HBsAg, suggesting that the combination of drugs and immunotherapy provides therapeutic interventions for functional cure of viral infections (146).
Campaigns for vaccination, screening, and antiviral treatment of hepatitis B and C have reduced the burden of chronic disease. However, concurrent increases in drug injection, alcohol abuse, and metabolic syndrome threaten these trends (1). A large randomized clinical trial discovered that long-term administration of albumin could improve survival in patients with decompensated cirrhosis (147). Whereas, in an open-label multicenter trial ATTIRE, increasing albumin infusion in patients with decompensated cirrhosis showed no more benefit due to most of the patients suffering alcohol-related liver disease (148). These results show that breaking down the etiology of cirrhosis is crucial for subsequent treatment.
Corticosteroids are currently recommended for the treatment of severe alcoholic hepatitis (SAH), but about 25% of SAH did not respond to prednisone treatment (149). Granulocyte colony-stimulating factor (G-CSF) can prolong the survival of alcoholic hepatitis (AH) patients, and the combination of N-acetylcysteine (NAC) with standard drug therapy (pentoxifylline) may also reduce AH liver injury and prolong survival (150). The immunomodulatory effect of G-CSF in the AH mouse model had shown to increase the number of immune cells entering the liver and promoting the polarization of macrophages toward M2, which facilitated liver repair (151). Macrophage and neutrophil infiltration diminished in AH mice treated with intraperitoneal MSC infusion, and skeletal muscle satellite cell-derived MSC counteracted ethanol-induced inflammation by secreting PEG2 and HGF, thus making MSC promising as an effective therapy for patients with alcoholic hepatitis (152, 153). In SAH patients, the fecal microbiome transplantation (FMT) treatment for 90 days could cause a reduction in the ratio of mucosa-associated invariant T and Th17 cells and a decrease in IL-17 and IFN-γ production. Besides, FMT could attenuate the hepatic inflammatory response and finally improve survival in SAH patients, which suggests that FMT may be an alternative to prednisone treatment (154). In the Defeat Alcoholic Steatohepatitis trial (DASH), the combination of IL-1β-receptor antagonist (anakinra) with pentoxifylline plus zinc supplementation offered the opportunity for prolonged survival in patients with AH (155). Significantly elevated markers of immune cell activation stimulated by DAMP and PAMP, including macrophages and neutrophils were found in AH subjects studied in four clinical centers in the United States and correlated with the severity of AH (156). Silybin, a kind of herbal plant, normalized alcohol-induced immune regulation of the liver and induced activation of T cells and downregulation of cytokines such as TNF (157). These immune cells play an irreplaceable role in the intricate liver immune microenvironment, which guides the direction for AH to discover immune targets and provides future treatment strategies to prevent disease progression, but further prospective clinical studies are needed to confirm this good desire.
Anti-inflammatory and anti-fibrotic immunotherapy strategies play a therapeutic role for NAFLD, with no standard treatments change to currently approved (158). FXR is expressed in immune cells. Cilofexor, an FXR agonist, improved hepatic steatosis in a 24-week phase II study in NASH patients. However, Liver Fibrosis scores and liver stiffness were not observed when were used only in noncirrhotic NASH patients (159). Meanwhile, a modified and optimized FXR agonist (MET409) had the same effect in another 12-week study in patients with NASH, despite the side effect of headache. MET409 is intended to be used as first-line monotherapy for NASH, while combinations of MET409 with other agents are in development (160). More recently, results of a phase II trial showed that the FXR agonist tropifexor led to sustained reductions in ALT and liver fat, but the side effect of pruritus was unavoidable, and further investigation for antifibrotic effects in combination with other agents is needed (161). NAFLD is associated with hypertriglyceridemia. In patients receiving the FXR agonist cilofexor (CILO) and the acetyl-coenzyme A carboxylase (ACAC) inhibitor firsocostat, inhibition of triglycerides by fenofibrate was strengthened (162), providing strong evidence for the combinations of multiple drugs. Saroglitazar is a PPAR-α/γ agonist that is no less effective than fenofibrate (163). Whether it can replace fenofibrate in combination therapy for NAFLD remains to be proven.
The immunotherapy based on chimeric antigen receptor (CAR) T cells targeting and destroying myofibroblasts can be used to reduce extracellular matrix deposition in NASH mice (164). But more evidence is needed to confirm the efficacy. Cenicriviroc (CVC), as a dual CCR2 and CCR5 antagonist, had confirmed in a phase IIb (CENTAUR) study in anti-fibrosis effect in NAFLD patients (165). Unfortunately, the CVC clinical trial was prematurely interrupted in the anticipated phase III (AURORA) study (166). Oral OKT3 (anti-CD3 mouse monoclonal antibody) was administered to NASH patients with diabetes to induce Tregs activation for ameliorating insulin resistance and liver injury. Although the number of study subjects was small, the related parameters showed promising results (167). Fibroblast growth factor 19 (FGF19) analog aldafermin (also known as NGM282 or M70) reduced liver fat content and fibrosis levels by 7.7% and 1%, respectively, compared with the placebo group (168). Pegbelfermin is a pegylated FGF21 analog. FGF21 improves the condition of NAFLD and NASH by directly regulating lipid metabolism and reducing fat accumulation in an insulin-independent manner (169). Some clinical trials have shown that BMS-986036 has a good effect on improving the liver fat content, inflammation, and fibrosis of NAFLD and NASH, and is well tolerated (170, 171). As new immunotherapeutic drugs, the clinical research of FGF19 and FGF21 is in the third stage, and more studies are needed to determine their biological characteristics and therapeutic effects (172).
Given that none of the new drugs currently under evaluation shows improvements in major clinical endpoints, it is expected that a reasonable combination will be more effective in controlling or preventing further deterioration of NASH. Several studies have now shown that MSC-secreted exosomes supply natural drug delivery vectors and offer prospective strategies for the treatment of NAFLD. MSC-Exo miR-24-3p, miR-223-3p, and miR-627-5p attenuated lipid deposition and liver fibrosis in NAFLD mice, but more studies are needed to provide meaningful evidence for clinical treatment (173–176). MSC-Exo extracted from human umbilical cords stimulated M2 polarization, exhibited downregulation of pro-inflammatory factors such as TNF-α, IL-6 and IL-1β, and detected high expression of PPARα in liver tissue, thereby alleviating methionine-cholesterol deficient (MCD) diet-induced progression to NASH in NAFLD mice (177). Exosomes originated from human adipose mesenchymal stem cells (hADMSCs-Exo) were shown to inhibit HSCs activation and rectify choline metabolism disorders via PI3K/Akt/mTOR pathway to ameliorate liver fibrosis, especially caused by NAFLD (178). Deeper exploration of immune-related mechanisms and further clinical trials may be needed to cure NAFLD patients.
Primary biliary cholangitis (formerly called primary biliary cirrhosis) is characterized by cholestasis and biliary fibrosis, with autoimmune destruction leading to immune-mediated damage (179). Ursodeoxycholic acid (UDCA) is used as first-line therapy for PBC or as second-line therapy (obectocholic acid and benzofibrate) if the patient does not respond to UDCA (180). In a phase III clinical trial, patients who did not respond to UDCA could also be treated with budesonide, which improved liver function markers in serum but had little effect on liver histology (181).
It is well recognized that PPARs are nuclear receptors that regulate a variety of immune cell functions and play an important role in regulating innate and adaptive immunity (182). During the 52-week study, seladelpar, a selective PPARγ-δ agonist, was safe and well tolerated in patients with PBC, which overcame the side effects of skin itching and caused no serious adverse events or deaths (183). Fatigue is also one of the symptoms of PBC. RITPBC is believed to be the first randomized, controlled phase II clinical trial to investigate fatigue in PBC. Rituximab is a monoclonal antibody against CD20 on the surface of B cells to improve serum alkaline phosphatase (ALP) of refractory UDCA in PBC and decimate B cells (184). Rituximab is also considered a potential treatment for PBC fatigue (185). In addition, the anti-IL12/23 monoclonal antibody ustekinumab led to a slight decrease in PBC in a proof-of-concept study, but the efficacy and safety of its immunomodulatory effect remain to be verified (186).
Primary sclerosing cholangitis is a chronic cholestatic liver disease characterized by progressive inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts, leading to multifocal biliary stricture and progressive liver disease (e.g., cirrhosis) (187). Immunotherapy for PSC is complicated. Although a variety of immunomodulators have been tested for the treatment of PSC, the general treatment regimen has not been proven to benefit patients (188). 24-Norursodoxycholic acid (norUDCA), a homolog of UDCA, is a novel bile acid that reduces ALP in PSC patients in a dose-dependent way and significantly improves cholestasis (189). According to a meta-analysis, immunosuppressants (mycophenolate mofetil, methotrexate, and tacrolimus) significantly reduced ALP and AST and improved liver function, which seemed to be the most effective treatment with severe side effects (188).
Liver transplantation (LT) is the only life-prolonging and curable treatment for PSC. However, an international observational study found that morbidity and mortality of recurrent primary sclerosing cholangitis (rPSC) increased after liver transplantation in children. It was thought that rPSC elicited a more robust immune response than PSC (190). In addition, a 36-year-old woman who had both PSC and ulcerative colitis was diagnosed with autoimmune hepatitis after treatment with the mRNA vaccine COVID-19 (191). We could speculate whether PSC or a specific immune response after vaccination led to the conversion of immune disease, but the specific mechanism was unclear. Proinflammatory cytokines, such as TNF-α and IL-1β, were highly expressed in patients with PSC and AIH, while the function of T lymphocytes and NK cells in the liver were impaired (192), so anti-TNF therapy was also one treatment option. Exploring the immunological changes in the liver microenvironment may provide a solid basis to clinical immunotherapy.
Autoimmune hepatitis is characterized by elevated serum aminotransferase, immunoglobulin G (IgG) levels, and positive autoantibodies (193). The International Autoimmune Hepatitis (IAIHG) defined “complete biochemical response” (CBR) as the normalization of serum transaminases and IgG (194). To achieve this goal, corticosteroids and/or azathioprine (AZA) are the standard treatment for AIH, but some patients still respond poorly to standard treatment. The CBR rate of patients treated with mycophenolate mofetil (MMF) was significantly higher than that of patients treated with AZA, so MMF became an alternative therapy for initial treatment (195–197). For children with AIH, MMF was a “life-saving drug” for children (198). In most patients with conventionally treated refractory AIH, one in three patients treated with the immunosuppressant calcium phosphatase inhibitor tacrolimus developed CBR and had good renal function after the withdrawal of the drug (199). At the same disease stage, selective depletion of B cells by rituximab, an anti-CD20 monoclonal antibody, also lowered transaminase and IgG levels (200, 201). In a multicenter study, liver stiffness was reduced compared with that before tacrolimus treatment, but no statistical significance was found, possibly due to a small sample size. It is worth noting that only one patient discontinued treatment due to serious adverse events (202). Future studies need to enlarge the number of patients and investigate the effects of immunomodulation on disease.
Depletion of Tregs was one of the methods to establish the AIH mouse model. In conjunction with the reduction of Tregs by steroid treatment, enhanced intrahepatic Tregs immunotherapy would be the preferred option for AIH patients (203, 204). The low dose of IL-2 improves the selectivity and number of Tregs after treatment, thereby re-regulating the hepatic immune microenvironment for improve AIH (204, 205).
Wilson disease (WD) is an inherited disorder of copper metabolism caused by mutations in ATP7B (hepatomegaly protein) (194). It causes liver damage and neurological symptoms due to abnormal copper ion metabolism in the body, leading to copper accumulation in the liver, brain, and other tissues, which can lead to cirrhosis in the long term (195, 196). Although liver transplantation can cure WD, can also cause serious immunosuppression (197). In most WD patients, oral chelating agents such as D-penicillamine and trientine were effective (206). Trientine tetrahydrochloride (TETA4) was found to be superior to penicillamine in phase III clinical trials (199). Besides, these drugs can decrease the number of whole blood cells in patients, weakening the immune system and increasing the risk of infection (200). Therefore, targeted liver immunotherapy would provide a better cure for WD.
The present study shows that a number of factors are upregulated by MSCs, such as MMPs and VEGF, to promote liver fibrosis regression (207). Besides, the study indicates A combination of MSCs and macrophages was more effective in reducing fibrotic gene expression and procollagen synthesis than either of them individually. In line with this, it has been observed that localized MSCs improve liver fibrosis by reducing the activation of fibroblasts and the production of collagen (208).
Evidence is mounting that MSC-mediated immune regulation can alleviate liver fibrosis through programmed death mechanisms, such as apoptosis, autophagy, ferroptosis, and pyroptosis (209). Maintaining the characteristics of MSCs is dependent on basal autophagy levels. Aged MSCs can benefit from activated or increased autophagy to slow metabolism and strengthen their functions to resist aging. Conversely, Aged MSCs are more susceptible to toxic substance accumulation and mitochondrial damage, exacerbating inflammatory responses and cell damage, and ultimately this accelerates the aging process. Increasing evidence suggests that fibrotic microenvironment induced MSC autophagy by upregulating Becn1, while Becn1 knockdown inhibited T lymphocyte infiltration, HSC proliferation and suppressed the production of cytokines by increasing PTGS2/PGE2 secretion, thereby further enhancing MSC antifibrotic activity (210). Therefore, the augmentation of the antifibrotic potential of MSCs through the manipulation of their autophagic processes presents a viable approach towards the management of liver fibrosis.
Recently, Zhang et al have proposed the therapeutic potential of extracellular vesicles derived from mesenchymal stromal cells (MSC-Evs) in facilitating liver regeneration (211). MSC-EVs have been observed to stimulate the rejuvenation of aged hepatocytes and augment their proliferation by upregulating mitophagy. Subsequent to a more in-depth inquiry into the mechanistic intricacies of this process, it was discovered that DDX5, which is abundant in MSC-EVs, can be transferred to aged hepatocytes to stimulate EF1 nuclear translocation and consequent upregulation of Atg4B expression, ultimately leading to the induction of mitophagy. The validity of these findings was confirmed through in vivo and in vitro experiments involving DDX5 knockdown in MSC-EVs. Therefore, MSC-EVs present a promising therapeutic modality for liver fibrosis patients by reversing senescence and promoting the regeneration of senescent hepatocytes (211).
The disruption of hepatic homeostasis may lead to a persistent inflammatory response and a reduction in immune function, thereby emphasizing the critical role of the immune microenvironment in the development of liver cirrhosis. Remarkably, recent clinical trials have demonstrated that transplantation of mesenchymal stromal cells derived from human umbilical cord blood can potentially improve the long-term survival of patients with decompensated cirrhosis (202). Moreover, the paracrine intercellular communication of MSCs may confer the benefit of effectively eliciting physiological effects through minute concentrations of EV. The diversity of MSCs derived from distinct tissue sources and the distinctive attributes of MSC-EVs indicate a significant therapeutic potential of MSCs for their antifibrotic properties. Nevertheless, the capacity of MSCs to proliferate indefinitely is limited, and they may exhibit senescence after cell division, which can disrupt homeostasis in vivo through senescent cells and cell-cell interactions. Hence, investigating the mechanisms of cellular communication in the aging microenvironment is a pressing necessity and a promising therapeutic strategy.
QY searched the literature and drafted the manuscript, Ywa and QC collected and sorted out the literature. JY made the figures, QY and JY designed the article structure, and Ywu revised the manuscript. All authors contributed to the article and approved the submitted version.
This review was supported by the National Natural Science Foundation of China (82072600). The plan of the Shanghai Municipal Health Commission (2022XD036) also provided funding to conduct this project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Moon AM, Singal AG, Tapper EB. Contemporary epidemiology of chronic liver disease and cirrhosis. Clin Gastroenterol Hepatol (2020) 18:2650–66. doi: 10.1016/j.cgh.2019.07.060
2. Ye F, Zhai M, Long J, Gong Y, Ren C, Zhang D, et al. The burden of liver cirrhosis in mortality: results from the global burden of disease study. Front Public Health (2022) 10:909455. doi: 10.3389/fpubh.2022.909455
3. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol (2019) 70:151–71. doi: 10.1016/j.jhep.2018.09.014
4. Smith A, Baumgartner K, Bositis C. Cirrhosis: diagnosis and management. Am Fam Physician (2019) 100:759–70.
5. Gines P, Krag A, Abraldes JG, Sola E, Fabrellas N, Kamath PS. Liver cirrhosis. Lancet (2021) 398:1359–76. doi: 10.1016/S0140-6736(21)01374-X
6. Alberts CJ, Clifford GM, Georges D, Negro F, Lesi OA, Hutin YJ, et al. Worldwide prevalence of hepatitis B virus and hepatitis C virus among patients with cirrhosis at country, region, and global levels: a systematic review. Lancet Gastroenterol Hepatol (2022) 7:724–35. doi: 10.1016/S2468-1253(22)00050-4
7. Rodriguez-Tajes S, Pocurull A, Castillo J, Casanova G, Vega L, Lens S, et al. Hepatitis c-related cirrhosis will be a marginal cause of hospital admissions by 2025. J Hepatol (2020) 73:1360–7. doi: 10.1016/j.jhep.2020.07.018
8. Rizzetto M, Hamid S, Negro F. The changing context of hepatitis d. J Hepatol (2021) 74:1200–11. doi: 10.1016/j.jhep.2021.01.014
9. LeDesma R, Nimgaonkar I, Ploss A. Hepatitis e virus replication. Viruses (2019) 11:719. doi: 10.3390/v11080719
10. Shirazi F, Singal AK, Wong RJ. Alcohol-associated cirrhosis and alcoholic hepatitis hospitalization trends in the United States. J Clin Gastroenterol (2021) 55:174–9. doi: 10.1097/MCG.0000000000001378
11. Simpson RF, Hermon C, Liu B, Green J, Reeves GK, Beral V, et al. Alcohol drinking patterns and liver cirrhosis risk: analysis of the prospective uk million women study. Lancet Public Health (2019) 4:e41–8. doi: 10.1016/S2468-2667(18)30230-5
12. Zhou F, Zhou J, Wang W, Zhang XJ, Ji YX, Zhang P, et al. Unexpected rapid increase in the burden of nafld in China from 2008 to 2018: a systematic review and meta-analysis. Hepatology (2019) 70:1119–33. doi: 10.1002/hep.30702
13. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling nafld disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
14. Melato M, Mucli E. Something new in liver cirrhosis epidemiology. Lancet (1989) 2:395–6. doi: 10.1016/s0140-6736(89)90578-3
15. Romanelli RG, Stasi C. Recent advancements in diagnosis and therapy of liver cirrhosis. Curr Drug Targets (2016) 17:1804–17. doi: 10.2174/1389450117666160613101413
16. Aydin MM, Akcali KC. Liver fibrosis. Turk J Gastroenterol (2018) 29:14–21. doi: 10.5152/tjg.2018.17330
17. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol (2021) 18:151–66. doi: 10.1038/s41575-020-00372-7
18. Zhangdi HJ, Su SB, Wang F, Liang ZY, Yan YD, Qin SY, et al. Crosstalk network among multiple inflammatory mediators in liver fibrosis. World J Gastroenterol (2019) 25:4835–49. doi: 10.3748/wjg.v25.i33.4835
19. Berumen J, Baglieri J, Kisseleva T, Mekeel K. Liver fibrosis: pathophysiology and clinical implications. WIREs Mech Dis (2021) 13:e1499. doi: 10.1002/wsbm.1499
20. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13:267–76. doi: 10.1038/cmi.2016.3
21. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
22. Nakamoto N, Kanai T. Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol (2014) 5:221. doi: 10.3389/fimmu.2014.00221
23. Li X, Ramadori P, Pfister D, Seehawer M, Zender L, Heikenwalder M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat Rev Cancer (2021) 21:541–57. doi: 10.1038/s41568-021-00383-9
24. Wang L, Sun Y, Yi M, Zhao W, Yuan X. Ieo model: a novel concept describing the complete metastatic process in the liver microenvironment. Oncol Lett (2020) 19:3627–33. doi: 10.3892/ol.2020.11525
25. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology (2013) 144:512–27. doi: 10.1053/j.gastro.2013.01.002
26. Liu Y, Dong Y, Wu X, Wang X, Niu J. Identification of immune microenvironment changes and the expression of immune-related genes in liver cirrhosis. Front Immunol (2022) 13:918445. doi: 10.3389/fimmu.2022.918445
27. Gong J, Tu W, Liu J, Tian D. Hepatocytes: a key role in liver inflammation. Front Immunol (2022) 13:1083780. doi: 10.3389/fimmu.2022.1083780
28. Wree A, Holtmann TM, Inzaugarat ME, Feldstein AE. Novel drivers of the inflammatory response in liver injury and fibrosis. Semin Liver Dis (2019) 39:275–82. doi: 10.1055/s-0039-1685515
29. Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, et al. Hepatocyte stress increases expression of yes-associated protein and transcriptional coactivator with pdz-binding motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology (2020) 71:1813–30. doi: 10.1002/hep.30928
30. Futakuchi A, Inoue T, Wei FY, Inoue-Mochita M, Fujimoto T, Tomizawa K, et al. Yap/Taz are essential for tgf-Beta2-Mediated conjunctival fibrosis. Invest Ophthalmol Vis Sci (2018) 59:3069–78. doi: 10.1167/iovs.18-24258
31. Lu JG, Iyasu A, French B, Tillman B, French SW. Overexpression of mhcii by hepatocytes in alcoholic hepatitis (Ah) compared to non-alcoholic steatohepatitis (Nash) and normal controls. Alcohol (2020) 84:27–32. doi: 10.1016/j.alcohol.2019.08.008
32. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol (2022) 22:429–43. doi: 10.1038/s41577-021-00639-3
33. Zhang Q, Qu Y, Zhang Q, Li F, Li B, Li Z, et al. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via Mir-222/Tfrc axis. Cell Biol Toxicol (2022) 39:467–81. doi: 10.1007/s10565-021-09684-z
34. Filliol A, Schwabe RF. Foxm1 induces Ccl2 secretion from hepatocytes triggering hepatic inflammation, injury, fibrosis, and liver cancer. Cell Mol Gastroenterol Hepatol (2020) 9:555–6. doi: 10.1016/j.jcmgh.2020.01.002
35. Francis H, Wu N, Alpini G, Meng F. Hepatocyte autophagy: maintaining a toxic-free environment. Hepatology (2020) 72:371–4. doi: 10.1002/hep.31219
36. He W, Ni W, Zhao L, Wang X, Liu L, Fan Z. Microrna-125a/Vdr axis impaired autophagic flux and contributed to fibrosis in a Ccl4-induced mouse model and patients with liver cirrhosis. Life Sci (2021) 264:118666. doi: 10.1016/j.lfs.2020.118666
37. Kim YS, Kim SG. Endoplasmic reticulum stress and autophagy dysregulation in alcoholic and non-alcoholic liver diseases. Clin Mol Hepatol (2020) 26:715–27. doi: 10.3350/cmh.2020.0173
38. Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U.S.A. (2010) 107:4383–8. doi: 10.1073/pnas.0911373107
39. Shiode Y, Hikita H, Tanaka S, Shirai K, Doi A, Sakane S, et al. Hepatitis C virus enhances Rubicon expression, leading to autophagy inhibition and intracellular innate immune activation. Sci Rep (2020) 10:15290. doi: 10.1038/s41598-020-72294-y
40. Barnard A, Moch A, Saab S. Relationship between telomere maintenance and liver disease. Gut Liver (2019) 13:11–5. doi: 10.5009/gnl18081
41. Nault JC, Ningarhari M, Rebouissou S, Zucman-Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol (2019) 16:544–58. doi: 10.1038/s41575-019-0165-3
42. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev (2008) 88:125–72. doi: 10.1152/physrev.00013.2007
43. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Delivery Rev (2017) 121:27–42. doi: 10.1016/j.addr.2017.05.007
44. Lee TF, Mak KM, Rackovsky O, Lin YL, Kwong AJ, Loke JC, et al. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (Adrp). J Cell Physiol (2010) 223:648–57. doi: 10.1002/jcp.22063
45. Carpino G, Morini S, Ginanni Corradini S, Franchitto A, Merli M, Siciliano M, et al. Alpha-sma expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig Liver Dis (2005) 37:349–56. doi: 10.1016/j.dld.2004.11.009
46. Barry AE, Baldeosingh R, Lamm R, Patel K, Zhang K, Dominguez DA, et al. Hepatic stellate cells and hepatocarcinogenesis. Front Cell Dev Biol (2020) 8:709. doi: 10.3389/fcell.2020.00709
47. Zuo L, Zhu Y, Hu L, Liu Y, Wang Y, Hu Y, et al. Pi3-Kinase/Akt pathway-regulated membrane transportation of acid-sensing ion channel 1a/Calcium ion Influx/Endoplasmic reticulum stress activation on pdgf-induced hsc activation. J Cell Mol Med (2019) 23:3940–50. doi: 10.1111/jcmm.14275
48. Ali FE, Abd El-Aziz MK, Sharab EI, Bakr AG. Therapeutic interventions of acute and chronic liver disorders: a comprehensive review. World J Hepatol (2023) 15:19–40. doi: 10.4254/wjh.v15.i1.19
49. Blaner WS, O'Byrne SM, Wongsiriroj N, Kluwe J, D'Ambrosio DM, Jiang H, et al. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta (2009) 1791:467–73. doi: 10.1016/j.bbalip.2008.11.001
50. Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology (2014) 59:154–69. doi: 10.1002/hep.26604
51. Teratani T, Tomita K, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, et al. A high-cholesterol diet exacerbates liver fibrosis in mice Via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology (2012) 142:152–164 e110. doi: 10.1053/j.gastro.2011.09.049
52. Twu YC, Lee TS, Lin YL, Hsu SM, Wang YH, Liao CY, et al. Niemann-pick type C2 protein mediates hepatic stellate cells activation by regulating free cholesterol accumulation. Int J Mol Sci (2016) 17:1122. doi: 10.3390/ijms17071122
53. Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, et al. Acyl-Coa:Cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol (2014) 61:98–106. doi: 10.1016/j.jhep.2014.03.018
54. Zhang H, Yan X, Yang C, Zhan Q, Fu Y, Luo H, et al. Intrahepatic T helper 17 cells recruited by hepatitis B virus X antigen-activated hepatic stellate cells exacerbate the progression of chronic hepatitis B virus infection. J Viral Hepat (2020) 27:1138–49. doi: 10.1111/jvh.13352
55. He J, Hong B, Bian M, Jin H, Chen J, Shao J, et al. Docosahexaenoic acid inhibits hepatic stellate cell activation to attenuate liver fibrosis in a ppargamma-dependent manner. Int Immunopharmacol (2019) 75:105816. doi: 10.1016/j.intimp.2019.105816
56. Enguita M, Razquin N, Pamplona R, Quiroga J, Prieto J, Fortes P. The cirrhotic liver is depleted of docosahexaenoic acid (Dha), a key modulator of nf-kappab and tgfbeta pathways in hepatic stellate cells. Cell Death Dis (2019) 10:14. doi: 10.1038/s41419-018-1243-0
57. Wu X, Liu XQ, Liu ZN, Xia GQ, Zhu H, Zhang MD, et al. Cd73 aggravates alcohol-related liver fibrosis by promoting autophagy mediated activation of hepatic stellate cells through Ampk/Akt/Mtor signaling pathway. Int Immunopharmacol (2022) 113:109229. doi: 10.1016/j.intimp.2022.109229
58. Hirsova P, Ibrahim SH, Verma VK, Morton LA, Shah VH, LaRusso NF, et al. Extracellular vesicles in liver pathobiology: small particles with big impact. Hepatology (2016) 64:2219–33. doi: 10.1002/hep.28814
59. Eguchi A, Kostallari E, Feldstein AE, Shah VH. Extracellular vesicles, the liquid biopsy of the future. J Hepatol (2019) 70:1292–4. doi: 10.1016/j.jhep.2019.01.030
60. Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S, et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol (2020) 73:1144–54. doi: 10.1016/j.jhep.2020.04.044
61. Lee KD, Kuo TK, Whang-Peng J, Chung YF, Lin CT, Chou SH, et al. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology (2004) 40:1275–84. doi: 10.1002/hep.20469
62. Viswanathan S, Shi Y, Galipeau J, Krampera M, Leblanc K, Martin I, et al. Mesenchymal stem versus stromal cells: international society for cell & gene therapy (Isct(R)) mesenchymal stromal cell committee position statement on nomenclature. Cytotherapy (2019) 21:1019–24. doi: 10.1016/j.jcyt.2019.08.002
63. Xie Z, Yu W, Ye G, Li J, Zheng G, Liu W, et al. Single-cell rna sequencing analysis of human bone-Marrow-Derived mesenchymal stem cells and functional subpopulation identification. Exp Mol Med (2022) 54:483–92. doi: 10.1038/s12276-022-00749-5
64. Zong C, Meng Y, Ye F, Yang X, Li R, Jiang J, et al. Aif1(+) Csf1r(+) mscs, induced by tnf-α, act to generate an inflammatory microenvironment and promote hepatocarcinogenesis. Hepatology (2022) doi: 10.1002/hep.32738
65. Yang X, Li Q, Liu W, Zong C, Wei L, Shi Y, et al. Mesenchymal stromal cells in hepatic Fibrosis/Cirrhosis: from pathogenesis to treatment. Cell Mol Immunol (2023) 20:583–99. doi: 10.1038/s41423-023-00983-5
66. Zhou Q, Rong C, Gu T, Li H, Wu L, Zhuansun X, et al. Mesenchymal stem cells improve liver fibrosis and protect hepatocytes by promoting microrna-148a-5p-Mediated inhibition of notch signaling pathway. Stem Cell Res Ther (2022) 13:354. doi: 10.1186/s13287-022-03030-8
67. Li W, Ren G, Huang Y, Su J, Han Y, Li J, et al. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ (2012) 19:1505–13. doi: 10.1038/cdd.2012.26
68. Renner P, Eggenhofer E, Rosenauer A, Popp FC, Steinmann JF, Slowik P, et al. Mesenchymal stem cells require a sufficient, ongoing immune response to exert their immunosuppressive function. Transplant Proc (2009) 41:2607–11. doi: 10.1016/j.transproceed.2009.06.119
69. Lin T, Pajarinen J, Nabeshima A, Lu L, Nathan K, Jämsen E, et al. Preconditioning of murine mesenchymal stem cells synergistically enhanced immunomodulation and osteogenesis. Stem Cell Res Ther (2017) 8:277. doi: 10.1186/s13287-017-0730-z
70. Wen D, Peng Y, Liu D, Weizmann Y, Mahato RI. Mesenchymal stem cell and derived exosome as small rna carrier and immunomodulator to improve islet transplantation. J Control Release (2016) 238:166–75. doi: 10.1016/j.jconrel.2016.07.044
71. Sun C, Shi C, Duan X, Zhang Y, Wang B. Exosomal microrna-618 derived from mesenchymal stem cells attenuate the progression of hepatic fibrosis by targeting Smad4. Bioengineered (2022) 13:5915–27. doi: 10.1080/21655979.2021.2023799
73. Zhang Y, Zhangdi H, Nie X, Wang L, Wan Z, Jin H, et al. Exosomes derived from bmmscs mitigate the hepatic fibrosis Via anti-pyroptosis pathway in a cirrhosis model. Cells (2022) 11:4004. doi: 10.3390/cells11244004
74. Shen Z, Huang W, Liu J, Tian J, Wang S, Rui K. Effects of mesenchymal stem cell-derived exosomes on autoimmune diseases. Front Immunol (2021) 12:749192. doi: 10.3389/fimmu.2021.749192
75. Joo HS, Suh JH, Lee HJ, Bang ES, Lee JM. Current knowledge and future perspectives on mesenchymal stem cell-derived exosomes as a new therapeutic agent. Int J Mol Sci (2020) 21:727. doi: 10.3390/ijms21030727
76. Arabpour M, Saghazadeh A, Rezaei N. Anti-inflammatory and M2 macrophage polarization-promoting effect of mesenchymal stem cell-derived exosomes. Int Immunopharmacol (2021) 97:107823. doi: 10.1016/j.intimp.2021.107823
77. Tan Y, Huang Y, Mei R, Mao F, Yang D, Liu J, et al. Hucmsc-derived exosomes delivered Becn1 induces ferroptosis of hepatic stellate cells Via regulating the Xct/Gpx4 axis. Cell Death Dis (2022) 13:319. doi: 10.1038/s41419-022-04764-2
78. Deng Y, Zhang Y, Ye L, Zhang T, Cheng J, Chen G, et al. Umbilical cord-derived mesenchymal stem cells instruct monocytes towards an Il10-producing phenotype by secreting Il6 and hgf. Sci Rep (2016) 6:37566. doi: 10.1038/srep37566
79. Prockop DJ. Concise review: two negative feedback loops place mesenchymal Stem/Stromal cells at the center of early regulators of inflammation. Stem Cells (2013) 31:2042–6. doi: 10.1002/stem.1400
80. Ezquer F, Ezquer M, Contador D, Ricca M, Simon V, Conget P. The antidiabetic effect of mesenchymal stem cells is unrelated to their transdifferentiation potential but to their capability to restore Th1/Th2 balance and to modify the pancreatic microenvironment. Stem Cells (2012) 30:1664–74. doi: 10.1002/stem.1132
81. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol (2015) 16:461–72. doi: 10.1038/nrm4024
82. Tao H, Liu Q, Zeng A, Song L. Unlocking the potential of mesenchymal stem cells in liver fibrosis: Insights into the impact of autophagy and aging. Int Immunopharmacol (2023) 121:110497. doi: 10.1016/j.intimp.2023.110497
83. Li H. Intercellular crosstalk of liver sinusoidal endothelial cells in liver fibrosis, cirrhosis and hepatocellular carcinoma. Dig Liver Dis (2022) 54:598–613. doi: 10.1016/j.dld.2021.07.006
84. Sorensen KK, Simon-Santamaria J, McCuskey RS, Smedsrod B. Liver sinusoidal endothelial cells. Compr Physiol (2015) 5:1751–74. doi: 10.1002/cphy.c140078
85. Yang M, Zhang C. The role of liver sinusoidal endothelial cells in cancer liver metastasis. Am J Cancer Res (2021) 11:1845–60.
86. Yang Y, Sangwung P, Kondo R, Jung Y, McConnell MJ, Jeong J, et al. Alcohol-induced Hsp90 acetylation is a novel driver of liver sinusoidal endothelial dysfunction and alcohol-related liver disease. J Hepatol (2021) 75:377–86. doi: 10.1016/j.jhep.2021.02.028
87. Fang ZQ, Ruan B, Liu JJ, Duan JL, Yue ZS, Song P, et al. Notch-triggered maladaptation of liver sinusoidal endothelium aggravates nonalcoholic steatohepatitis through endothelial nitric oxide synthase. Hepatology (2022) 76:742–58. doi: 10.1002/hep.32332
88. Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol (2016) 13:347–53. doi: 10.1038/cmi.2016.5
89. Bhandari S, Larsen AK, McCourt P, Smedsrod B, Sorensen KK. The scavenger function of liver sinusoidal endothelial cells in health and disease. Front Physiol (2021) 12:757469. doi: 10.3389/fphys.2021.757469
90. Furuta K, Guo Q, Hirsova P, Ibrahim SH. Emerging roles of liver sinusoidal endothelial cells in nonalcoholic steatohepatitis. Biol (Basel) (2020) 9:395. doi: 10.3390/biology9110395
91. Zhang Y, Yang X, Bi T, Wu X, Wang L, Ren Y, et al. Targeted inhibition of the immunoproteasome blocks endothelial mhc class ii antigen presentation to Cd4(+) T cells in chronic liver injury. Int Immunopharmacol (2022) 107:108639. doi: 10.1016/j.intimp.2022.108639
92. Odagiri N, Matsubara T, Sato-Matsubara M, Fujii H, Enomoto M, Kawada N. Anti-fibrotic treatments for chronic liver diseases: the present and the future. Clin Mol Hepatol (2021) 27:413–24. doi: 10.3350/cmh.2020.0187
93. Couvelard A, Scoazec JY, Feldmann G. Expression of cell-cell and cell-matrix adhesion proteins by sinusoidal endothelial cells in the normal and cirrhotic human liver. Am J Pathol (1993) 143:738–52.
94. Xie G, Choi SS, Syn WK, Michelotti GA, Swiderska M, Karaca G, et al. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut (2013) 62:299–309. doi: 10.1136/gutjnl-2011-301494
95. Nasiri-Ansari N, Androutsakos T, Flessa CM, Kyrou I, Siasos G, Randeva HS, et al. Endothelial cell dysfunction and nonalcoholic fatty liver disease (Nafld): a concise review. Cells (2022) 11:2511. doi: 10.3390/cells11162511
96. Wu X, Shu L, Zhang Z, Li J, Zong J, Cheong LY, et al. Adipocyte fatty acid binding protein promotes the onset and progression of liver fibrosis Via mediating the crosstalk between liver sinusoidal endothelial cells and hepatic stellate cells. Adv Sci (Weinh) (2021) 8:e2003721. doi: 10.1002/advs.202003721
97. Fang J, Ji Q, Gao S, Xiao Z, Liu W, Hu Y, et al. Pdgf-bb is involved in hif-1alpha/Cxcr4/Cxcr7 axis promoting capillarization of hepatic sinusoidal endothelial cells. Heliyon (2023) 9:e12715. doi: 10.1016/j.heliyon.2022.e12715
98. Wang Y, Xiang Y, Xin VW, Wang XW, Peng XC, Liu XQ, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol (2020) 13:107. doi: 10.1186/s13045-020-00939-6
99. Gardner A, de Mingo Pulido A, Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol (2020) 11:924. doi: 10.3389/fimmu.2020.00924
100. Deczkowska A, David E, Ramadori P, Pfister D, Safran M, Li B, et al. Publisher correction: Xcr1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med (2022) 28:214. doi: 10.1038/s41591-021-01668-0
101. Xu F, Liu C, Dong Y, Wu W, Xu J, Yan Y, et al. Ablation of cbl-b and c-cbl in dendritic cells causes spontaneous liver cirrhosis Via altering multiple properties of Cd103(+) Cdc1s. Cell Death Discovery (2022) 8:142. doi: 10.1038/s41420-022-00953-2
102. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/Beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther (2022) 7:3. doi: 10.1038/s41392-021-00762-6
103. Tan K, Xie X, Shi W, Miao L, Dong X, Yang W, et al. Deficiency of canonical Wnt/Beta-catenin signalling in hepatic dendritic cells triggers autoimmune hepatitis. Liver Int (2020) 40:131–40. doi: 10.1111/liv.14246
104. De Pasquale C, Campana S, Barberi C, Sidoti Migliore G, Oliveri D, Lanza M, et al. Human hepatitis b virus negatively impacts the protective immune crosstalk between natural killer and dendritic cells. Hepatology (2021) 74:550–65. doi: 10.1002/hep.31725
105. Fan J, Edsen-Moore MR, Turner LE, Cook RT, Legge KL, Waldschmidt TJ, et al. Mechanisms by which chronic ethanol feeding limits the ability of dendritic cells to stimulate T-cell proliferation. Alcohol Clin Exp Res (2011) 35:47–59. doi: 10.1111/j.1530-0277.2010.01321.x
106. Chu X, Jin Q, Chen H, Wood GC, Petrick A, Strodel W, et al. Ccl20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J Transl Med (2018) 16:108. doi: 10.1186/s12967-018-1490-y
107. Mo C, Xie S, Liu B, Zhong W, Zeng T, Huang S, et al. Indoleamine 2,3-dioxygenase 1 limits hepatic inflammatory cells recruitment and promotes bile duct ligation-induced liver fibrosis. Cell Death Dis (2021) 12:16. doi: 10.1038/s41419-020-03277-0
108. Mikulak J, Bruni E, Oriolo F, Di Vito C, Mavilio D. Hepatic natural killer cells: organ-specific sentinels of liver immune homeostasis and physiopathology. Front Immunol (2019) 10:946. doi: 10.3389/fimmu.2019.00946
109. Gan J, Mao XR, Zheng SJ, Li JF. Invariant natural killer T cells: not to be ignored in liver disease. J Dig Dis (2021) 22:136–42. doi: 10.1111/1751-2980.12968
110. Sajid M, Liu L, Sun C. The dynamic role of nk cells in liver cancers: role in hcc and hbv associated hcc and its therapeutic implications. Front Immunol (2022) 13:887186. doi: 10.3389/fimmu.2022.887186
111. Diedrich T, Kummer S, Galante A, Drolz A, Schlicker V, Lohse AW, et al. Characterization of the immune cell landscape of patients with nafld. PloS One (2020) 15:e0230307. doi: 10.1371/journal.pone.0230307
112. Ma Q, Dong X, Liu S, Zhong T, Sun D, Zong L, et al. Hepatitis b e antigen induces Nkg2a(+) natural killer cell dysfunction Via regulatory T cell-derived interleukin 10 in chronic hepatitis b virus infection. Front Cell Dev Biol (2020) 8:421. doi: 10.3389/fcell.2020.00421
113. Li TY, Yang Y, Zhou G, Tu ZK. Immune suppression in chronic hepatitis b infection associated liver disease: a review. World J Gastroenterol (2019) 25:3527–37. doi: 10.3748/wjg.v25.i27.3527
114. Ravichandran G, Neumann K, Berkhout LK, Weidemann S, Langeneckert AE, Schwinge D, et al. Interferon-Gamma-Dependent immune responses contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol (2019) 71:773–82. doi: 10.1016/j.jhep.2019.05.023
115. Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta (2013) 1832:1061–9. doi: 10.1016/j.bbadis.2012.09.008
116. Wu L, Van Kaer L. Natural killer T cells in health and disease. Front Biosci (Schol Ed) (2011) 3:236–51. doi: 10.2741/s148
117. Wei X, Qian J, Yao W, Chen L, Guan H, Chen Y, et al. Hyperactivated peripheral invariant natural killer T cells correlate with the progression of hbv-relative liver cirrhosis. Scand J Immunol (2019) 90:e12775. doi: 10.1111/sji.12775
118. Guan J, Wang G, Yang Q, Chen C, Deng J, Gu X, et al. Natural killer T cells in various mouse models of hepatitis. BioMed Res Int (2021) 2021:1782765. doi: 10.1155/2021/1782765
119. Zheng S, Yang W, Yao D, Tang S, Hou J, Chang X. A comparative study on roles of natural killer T cells in two diet-induced non-alcoholic steatohepatitis-related fibrosis in mice. Ann Med (2022) 54:2233–45. doi: 10.1080/07853890.2022.2108894
120. Tang W, Zhou J, Yang W, Feng Y, Wu H, Mok MTS, et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol (2022) 19:834–47. doi: 10.1038/s41423-022-00872-3
121. Jia H, Chen J, Zhang X, Bi K, Zhou H, Liu T, et al. Il-17a produced by invariant natural killer T cells and Cd3(+) Cd56(+) alphagalcer-Cd1d tetramer(-) T cells promote liver fibrosis in patients with primary biliary cholangitis. J Leukoc Biol (2022) 112:1079–87. doi: 10.1002/JLB.2A0622-586RRRR
122. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi: 10.1038/ni.2705
123. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol (2013) 3:785–97. doi: 10.1002/cphy.c120026
124. Sica A, Mantovani A. Macrophage plasticity and polarization: In vivo veritas. J Clin Invest (2012) 122:787–95. doi: 10.1172/JCI59643
125. Cao Y, Mai W, Li R, Deng S, Li L, Zhou Y, et al. Macrophages evoke autophagy of hepatic stellate cells to promote liver fibrosis in nafld mice Via the Pge2/Ep4 pathway. Cell Mol Life Sci (2022) 79:303. doi: 10.1007/s00018-022-04319-w
126. Tao R, Han M, Yuan W, Xiao F, Huang J, Wang X, et al. Fibrinogen-like protein 2 promotes proinflammatory macrophage polarization and mitochondrial dysfunction in liver fibrosis. Int Immunopharmacol (2023) 117:109631. doi: 10.1016/j.intimp.2022.109631
127. Zhang J, Liu Y, Chen H, Yuan Q, Wang J, Niu M, et al. Myd88 in hepatic stellate cells enhances liver fibrosis Via promoting macrophage M1 polarization. Cell Death Dis (2022) 13:411. doi: 10.1038/s41419-022-04802-z
128. Pastore M, Caligiuri A, Raggi C, Navari N, Piombanti B, Di Maira G, et al. Macrophage mertk promotes profibrogenic cross-talk with hepatic stellate cells Via soluble mediators. JHEP Rep (2022) 4:100444. doi: 10.1016/j.jhepr.2022.100444
129. Xu Z, Xi F, Deng X, Ni Y, Pu C, Wang D, et al. Osteopontin promotes macrophage M1 polarization by activation of the Jak1/Stat1/Hmgb1 signaling pathway in nonalcoholic fatty liver disease. J Clin Transl Hepatol (2023) 11:273–83. doi: 10.14218/JCTH.2021.00474
130. Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, et al. Hepatitis b virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PloS Pathog (2014) 10:e1004032. doi: 10.1371/journal.ppat.1004032
131. Zhang Y, Xiao N, Liu Q, Nie Y, Zhu X. Increased serum levels of Scd206 are associated with adverse prognosis in patients with hbv-related decompensated cirrhosis. Dis Markers (2022) 2022:7881478. doi: 10.1155/2022/7881478
132. Yardeni D, Ghany MG. Review article: hepatitis B-current and emerging therapies. Aliment Pharmacol Ther (2022) 55:805–19. doi: 10.1111/apt.16828
133. Jeng WJ, Papatheodoridis GV, Lok ASF. Hepatitis B. Lancet (2023) 401:1039–52. doi: 10.1016/S0140-6736(22)01468-4
134. Foster GR, Agarwal K, Cramp ME, Moreea S, Barclay S, Collier J, et al. Elbasvir/Grazoprevir and sofosbuvir for hepatitis c virus genotype 3 infection with compensated cirrhosis: a randomized trial. Hepatology (2018) 67:2113–26. doi: 10.1002/hep.29852
135. Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex b: first results of a phase Ib/Iia study. J Hepatol (2016) 65:490–8. doi: 10.1016/j.jhep.2016.04.016
136. Loglio A, Ferenci P, Uceda Renteria SC, Tham CYL, Scholtes C, Holzmann H, et al. Safety and effectiveness of up to 3 years' bulevirtide monotherapy in patients with hdv-related cirrhosis. J Hepatol (2022) 76:464–9. doi: 10.1016/j.jhep.2021.10.012
137. Jachs M, Schwarz C, Panzer M, Binter T, Aberle SW, Hartl L, et al. Response-guided long-term treatment of chronic hepatitis d patients with bulevirtide-results of a "Real world" study. Aliment Pharmacol Ther (2022) 56:144–54. doi: 10.1111/apt.16945
138. Loglio A, Ferenci P, Uceda Renteria SC, Tham CYL, van Bommel F, Borghi M, et al. Excellent safety and effectiveness of high-dose myrcludex-b monotherapy administered for 48 Weeks in hdv-related compensated cirrhosis: a case report of 3 patients. J Hepatol (2019) 71:834–9. doi: 10.1016/j.jhep.2019.07.003
139. Yurdaydin C, Keskin O, Yurdcu E, Caliskan A, Onem S, Karakaya F, et al. A phase 2 dose-finding study of lonafarnib and ritonavir with or without interferon alpha for chronic delta hepatitis. Hepatology (2022) 75:1551–65. doi: 10.1002/hep.32259
140. Park SJ, Hahn YS. Hepatocytes infected with hepatitis C virus change immunological features in the liver microenvironment. Clin Mol Hepatol (2023) 29:65–76. doi: 10.3350/cmh.2022.0032
141. Amin OE, Colbeck EJ, Daffis S, Khan S, Ramakrishnan D, Pattabiraman D, et al. Therapeutic potential of Tlr8 agonist gs-9688 (Selgantolimod) in chronic hepatitis b: remodeling of antiviral and regulatory mediators. Hepatology (2021) 74:55–71. doi: 10.1002/hep.31695
142. Daffis S, Balsitis S, Chamberlain J, Zheng J, Santos R, Rowe W, et al. Toll-like receptor 8 agonist gs-9688 induces sustained efficacy in the woodchuck model of chronic hepatitis B. Hepatology (2021) 73:53–67. doi: 10.1002/hep.31255
143. Gane EJ, Kim HJ, Visvanathan K, Kim YJ, Nguyen AH, Wallin JJ, et al. Safety, pharmacokinetics, and pharmacodynamics of the oral Tlr8 agonist selgantolimod in chronic hepatitis B. Hepatology (2021) 74:1737–49. doi: 10.1002/hep.31795
144. Al Mahtab M, Akbar SMF, Aguilar JC, Guillen G, Penton E, Tuero A, et al. Treatment of chronic hepatitis b naive patients with a therapeutic vaccine containing hbs and hbc antigens (a randomized, open and treatment controlled phase iii clinical trial). PloS One (2018) 13:e0201236. doi: 10.1371/journal.pone.0201236
145. Page K, Melia MT, Veenhuis RT, Winter M, Rousseau KE, Massaccesi G, et al. Randomized trial of a vaccine regimen to prevent chronic hcv infection. N Engl J Med (2021) 384:541–9. doi: 10.1056/NEJMoa2023345
146. Lee JH, Lee YB, Cho EJ, Yu SJ, Yoon JH, Kim YJ. Entecavir plus pegylated interferon and sequential hepatitis B virus vaccination increases hepatitis b surface antigen seroclearance: a randomized controlled proof-of-Concept study. Clin Infect Dis (2021) 73:e3308–16. doi: 10.1093/cid/ciaa807
147. Caraceni P, Riggio O, Angeli P, Alessandria C, Neri S, Foschi FG, et al. Long-term albumin administration in decompensated cirrhosis (Answer): an open-label randomised trial. Lancet (2018) 391:2417–29. doi: 10.1016/S0140-6736(18)30840-7
148. China L, Freemantle N, Forrest E, Kallis Y, Ryder SD, Wright G, et al. A randomized trial of albumin infusions in hospitalized patients with cirrhosis. N Engl J Med (2021) 384:808–17. doi: 10.1056/NEJMoa2022166
149. Shasthry SM, Sharma MK, Shasthry V, Pande A, Sarin SK. Efficacy of granulocyte colony-stimulating factor in the management of steroid-nonresponsive severe alcoholic hepatitis: a double-blind randomized controlled trial. Hepatology (2019) 70:802–11. doi: 10.1002/hep.30516
150. Singh V, Keisham A, Bhalla A, Sharma N, Agarwal R, Sharma R, et al. Efficacy of granulocyte colony-stimulating factor and n-acetylcysteine therapies in patients with severe alcoholic hepatitis. Clin Gastroenterol Hepatol (2018) 16:1650–1656 e1652. doi: 10.1016/j.cgh.2018.01.040
151. Cho Y, Joshi R, Lowe P, Copeland C, Ribeiro M, Morel C, et al. Granulocyte colony-stimulating factor attenuates liver damage by M2 macrophage polarization and hepatocyte proliferation in alcoholic hepatitis in mice. Hepatol Commun (2022) 6:2322–39. doi: 10.1002/hep4.1925
152. Wan YM, Li ZQ, Liu C, He YF, Wang MJ, Wu XN, et al. Mesenchymal stem cells reduce alcoholic hepatitis in mice Via suppression of hepatic neutrophil and macrophage infiltration, and of oxidative stress. PloS One (2020) 15:e0228889. doi: 10.1371/journal.pone.0228889
153. Chung JS, Hwang S, Hong JE, Jo M, Rhee KJ, Kim S, et al. Skeletal muscle satellite cell-derived mesenchymal stem cells ameliorate acute alcohol-induced liver injury. Int J Med Sci (2022) 19:353–63. doi: 10.7150/ijms.68971
154. Pande A, Sharma S, Khillan V, Rastogi A, Arora V, Shasthry SM, et al. Fecal microbiota transplantation compared with prednisolone in severe alcoholic hepatitis patients: a randomized trial. Hepatol Int (2023) 17:249–61. doi: 10.1007/s12072-022-10438-0
155. Szabo G, Mitchell M, McClain CJ, Dasarathy S, Barton B, McCullough AJ, et al. Il-1 receptor antagonist plus pentoxifylline and zinc for severe alcohol-associated hepatitis. Hepatology (2022) 76:1058–68. doi: 10.1002/hep.32478
156. Saha B, Tornai D, Kodys K, Adejumo A, Lowe P, McClain C, et al. Biomarkers of macrophage activation and immune danger signals predict clinical outcomes in alcoholic hepatitis. Hepatology (2019) 70:1134–49. doi: 10.1002/hep.30617
157. Li S, Tan HY, Wang N, Feng Y, Wang X, Feng Y. Recent insights into the role of immune cells in alcoholic liver disease. Front Immunol (2019) 10:1328. doi: 10.3389/fimmu.2019.01328
158. Lee YA, Friedman SL. Inflammatory and fibrotic mechanisms in nafld-implications for new treatment strategies. J Intern Med (2022) 291:11–31. doi: 10.1111/joim.13380
159. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal fxr agonist, in patients with noncirrhotic Nash: a phase 2 randomized controlled trial. Hepatology (2020) 72:58–71. doi: 10.1002/hep.31205
160. Harrison SA, Bashir MR, Lee KJ, Shim-Lopez J, Lee J, Wagner B, et al. A structurally optimized fxr agonist, Met409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J Hepatol (2021) 75:25–33. doi: 10.1016/j.jhep.2021.01.047
161. Sanyal AJ, Lopez P, Lawitz EJ, Lucas KJ, Loeffler J, Kim W, et al. Tropifexor for nonalcoholic steatohepatitis: an adaptive, randomized, placebo-controlled phase 2a/B trial. Nat Med (2023) 29:392–400. doi: 10.1038/s41591-022-02200-8
162. Lawitz EJ, Bhandari BR, Ruane PJ, Kohli A, Harting E, Ding D, et al. Fenofibrate mitigates hypertriglyceridemia in nonalcoholic steatohepatitis patients treated with Cilofexor/Firsocostat. Clin Gastroenterol Hepatol (2023) 21:143–152.e143. doi: 10.1016/j.cgh.2021.12.044
163. Rodriguez-Gutierrez R, Gonzalez JG, Parmar D, Shaikh F, Cruz-Lopez P. Saroglitazar is noninferior to fenofibrate in reducing triglyceride levels in hypertriglyceridemic patients in a randomized clinical trial. J Lipid Res (2022) 63:100233. doi: 10.1016/j.jlr.2022.100233
164. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic car T cells reverse senescence-associated pathologies. Nature (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
165. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b centaur study. Hepatology (2020) 72:892–905. doi: 10.1002/hep.31108
166. Anstee QM, Neuschwander-Tetri BA, Wong VW, Abdelmalek MF, Younossi ZM, Yuan J, et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: aurora phase 3 study design. Contemp Clin Trials (2020) 89:105922. doi: 10.1016/j.cct.2019.105922
167. Lalazar G, Mizrahi M, Turgeman I, Adar T, Ben Ya'acov A, Shabat Y, et al. Oral administration of Okt3 mab to patients with Nash, promotes regulatory T-cell induction, and alleviates insulin resistance: results of a phase iia blinded placebo-controlled trial. J Clin Immunol (2015) 35:399–407. doi: 10.1007/s10875-015-0160-6
168. Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and safety of aldafermin, an engineered Fgf19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology (2021) 160:219–231.e211. doi: 10.1053/j.gastro.2020.08.004
169. Xiao F, Shi X, Huang P, Zeng X, Wang L, Zeng J, et al. Dose-response relationship between serum fibroblast growth factor 21 and liver fat content in non-alcoholic fatty liver disease. Diabetes Metab (2021) 47:101221. doi: 10.1016/j.diabet.2020.101221
170. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (Bms-986036), a pegylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet (2019) 392:2705–17. doi: 10.1016/S0140-6736(18)31785-9
171. Verzijl CRC, Van De Peppel IP, Struik D, Jonker JW. Pegbelfermin (Bms-986036): an investigational pegylated fibroblast growth factor 21 analogue for the treatment of nonalcoholic steatohepatitis. Expert Opin Investig Drugs (2020) 29:125–33. doi: 10.1080/13543784.2020.1708898
172. Talukdar S, Kharitonenkov A. Fgf19 and Fgf21: in Nash we trust. Mol Metab (2021) 46:101152. doi: 10.1016/j.molmet.2020.101152
173. Yang F, Wu Y, Chen Y, Xi J, Chu Y, Jin J, et al. Human umbilical cord mesenchymal stem cell-derived exosomes ameliorate liver steatosis by promoting fatty acid oxidation and reducing fatty acid synthesis. JHEP Rep (2023) 5:100746. doi: 10.1016/j.jhepr.2023.100746
174. Niu Q, Wang T, Wang Z, Wang F, Huang D, Sun H, et al. Adipose-derived mesenchymal stem cell-secreted extracellular vesicles alleviate non-alcoholic fatty liver disease Via delivering mir-223-3p. Adipocyte (2022) 11:572–87. doi: 10.1080/21623945.2022.2098583
175. Du X, Li H, Han X, Ma W. Mesenchymal stem cells-derived exosomal mir-24-3p ameliorates non-alcohol fatty liver disease by targeting keap-1. Biochem Biophys Res Commun (2022) 637:331–40. doi: 10.1016/j.bbrc.2022.11.012
176. Cheng L, Yu P, Li F, Jiang X, Jiao X, Shen Y, et al. Human umbilical cord-derived mesenchymal stem cell-exosomal mir-627-5p ameliorates non-alcoholic fatty liver disease by repressing fto expression. Hum Cell (2021) 34:1697–708. doi: 10.1007/s13577-021-00593-1
177. Shi Y, Yang X, Wang S, Wu Y, Zheng L, Tang Y, et al. Human umbilical cord mesenchymal stromal cell-derived exosomes protect against mcd-induced Nash in a mouse model. Stem Cell Res Ther (2022) 13:517. doi: 10.1186/s13287-022-03201-7
178. Zhang Z, Shang J, Yang Q, Dai Z, Liang Y, Lai C, et al. Exosomes derived from human adipose mesenchymal stem cells ameliorate hepatic fibrosis by inhibiting Pi3k/Akt/Mtor pathway and remodeling choline metabolism. J Nanobiotechnology (2023) 21:29. doi: 10.1186/s12951-023-01788-4
179. Lleo A, Wang GQ, Gershwin ME, Hirschfield GM. Primary biliary cholangitis. Lancet (2020) 396:1915–26. doi: 10.1016/S0140-6736(20)31607-X
180. Barron-Millar B, Ogle L, Mells G, Flack S, Badrock J, Sandford R, et al. The serum proteome and ursodeoxycholic acid response in primary biliary cholangitis. Hepatology (2021) 74:3269–83. doi: 10.1002/hep.32011
181. Hirschfield GM, Beuers U, Kupcinskas L, Ott P, Bergquist A, Farkkila M, et al. A placebo-controlled randomised trial of budesonide for pbc following an insufficient response to udca. J Hepatol (2021) 74:321–9. doi: 10.1016/j.jhep.2020.09.011
182. Wang C, Shi Y, Wang X, Ma H, Liu Q, Gao Y, et al. Peroxisome proliferator-activated receptors regulate hepatic immunity and assist in the treatment of primary biliary cholangitis. Front Immunol (2022) 13:940688. doi: 10.3389/fimmu.2022.940688
183. Bowlus CL, Galambos MR, Aspinall RJ, Hirschfield GM, Jones DEJ, Dorffel Y, et al. A phase ii, randomized, open-label, 52-week study of seladelpar in patients with primary biliary cholangitis. J Hepatol (2022) 77:353–64. doi: 10.1016/j.jhep.2022.02.033
184. Myers RP, Swain MG, Lee SS, Shaheen AA, Burak KW. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol (2013) 108:933–41. doi: 10.1038/ajg.2013.51
185. Jopson L, Newton JL, Palmer J, Floudas A, Isaacs J, Qian J, et al. Ritpbc: b-cell depleting therapy (Rituximab) as a treatment for fatigue in primary biliary cirrhosis: study protocol for a randomised controlled trial. BMJ Open (2015) 5:e007985. doi: 10.1136/bmjopen-2015-007985
186. Hirschfield GM, Gershwin ME, Strauss R, Mayo MJ, Levy C, Zou B, et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof-of-Concept study. Hepatology (2016) 64:189–99. doi: 10.1002/hep.28359
187. Dyson JK, Beuers U, Jones DEJ, Lohse AW, Hudson M. Primary sclerosing cholangitis. Lancet (2018) 391:2547–59. doi: 10.1016/S0140-6736(18)30300-3
188. Liu X, Wang H, Liu X, Bridle K, Crawford D, Liang X. Efficacy and safety of immune-modulating therapy for primary sclerosing cholangitis: a systematic review and meta-analysis. Pharmacol Ther (2022) 237:108163. doi: 10.1016/j.pharmthera.2022.108163
189. Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Farkkila M, et al. Norursodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol (2017) 67:549–58. doi: 10.1016/j.jhep.2017.05.009
190. Martinez M, Perito ER, Valentino P, Mack CL, Aumar M, Broderick A, et al. Recurrence of primary sclerosing cholangitis after liver transplant in children: an international observational study. Hepatology (2021) 74:2047–57. doi: 10.1002/hep.31911
191. Zhou T, Fronhoffs F, Dold L, Strassburg CP, Weismuller TJ. New-onset autoimmune hepatitis following mrna covid-19 vaccination in a 36-year-old woman with primary sclerosing cholangitis - should we be more vigilant? J Hepatol (2022) 76:218–20. doi: 10.1016/j.jhep.2021.08.006
192. Bo X, Broome U, Remberger M, Sumitran-Holgersson S. Tumour necrosis factor alpha impairs function of liver derived T lymphocytes and natural killer cells in patients with primary sclerosing cholangitis. Gut (2001) 49:131–41. doi: 10.1136/gut.49.1.131
193. Mack CL, Adams D, Assis DN, Kerkar N, Manns MP, Mayo MJ, et al. Diagnosis and management of autoimmune hepatitis in adults and children: 2019 practice guidance and guidelines from the American association for the study of liver diseases. Hepatology (2020) 72:671–722. doi: 10.1002/hep.31065
194. Pape S, Snijders R, Gevers TJG, Chazouilleres O, Dalekos GN, Hirschfield GM, et al. Systematic review of response criteria and endpoints in autoimmune hepatitis by the international autoimmune hepatitis group. J Hepatol (2022) 76:841–9. doi: 10.1016/j.jhep.2021.12.041
195. Johnson PJ, McFarlane IG, Williams R. Azathioprine for long-term maintenance of remission in autoimmune hepatitis. N Engl J Med (1995) 333:958–63. doi: 10.1056/NEJM199510123331502
196. Zachou K, Gatselis N, Papadamou G, Rigopoulou EI, Dalekos GN. Mycophenolate for the treatment of autoimmune hepatitis: prospective assessment of its efficacy and safety for induction and maintenance of remission in a Large cohort of treatment-naive patients. J Hepatol (2011) 55:636–46. doi: 10.1016/j.jhep.2010.12.032
197. Dalekos GN, Arvaniti P, Gatselis NK, Gabeta S, Samakidou A, Giannoulis G, et al. Long-term results of mycophenolate mofetil vs. azathioprine use in individuals with autoimmune hepatitis. JHEP Rep (2022) 4:100601. doi: 10.1016/j.jhepr.2022.100601
198. Aw MM, Dhawan A, Samyn M, Bargiota A, Mieli-Vergani G. Mycophenolate mofetil as rescue treatment for autoimmune liver disease in children: a 5-year follow-up. J Hepatol (2009) 51:156–60. doi: 10.1016/j.jhep.2009.02.024
199. Hanouneh M, Ritchie MM, Ascha M, Ascha MS, Chedid A, Sanguankeo A, et al. A review of the utility of tacrolimus in the management of adults with autoimmune hepatitis. Scand J Gastroenterol (2019) 54:76–80. doi: 10.1080/00365521.2018.1551498
200. Than NN, Hodson J, Schmidt-Martin D, Taubert R, Wawman RE, Botter M, et al. Efficacy of rituximab in difficult-to-Manage autoimmune hepatitis: results from the international autoimmune hepatitis group. JHEP Rep (2019) 1:437–45. doi: 10.1016/j.jhepr.2019.10.005
201. Burak KW, Swain MG, Santodomingo-Garzon T, Lee SS, Urbanski SJ, Aspinall AI, et al. Rituximab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can J Gastroenterol (2013) 27:273–80. doi: 10.1155/2013/512624
202. Ferre-Aracil C, Riveiro-Barciela M, Trapero-Marugan M, Rodriguez-Peralvarez M, Llovet LP, Tellez L, et al. Tacrolimus as an effective and durable second-line treatment for chronic autoimmune hepatitis: a multicentric study. Dig Dis Sci (2021) 66:2826–32. doi: 10.1007/s10620-020-06569-9
203. Wang H, Feng X, Yan W, Tian D. Regulatory T cells in autoimmune hepatitis: unveiling their roles in mouse models and patients. Front Immunol (2020) 11:575572. doi: 10.3389/fimmu.2020.575572
204. Buitrago-Molina LE, Pietrek J, Noyan F, Schlue J, Manns MP, Wedemeyer H, et al. Treg-specific il-2 therapy can reestablish intrahepatic immune regulation in autoimmune hepatitis. J Autoimmun (2021) 117:102591. doi: 10.1016/j.jaut.2020.102591
205. Longhi MS, Mieli-Vergani G, Vergani D. Regulatory T cells in autoimmune hepatitis: an updated overview. J Autoimmun (2021) 119:102619. doi: 10.1016/j.jaut.2021.102619
206. Li Z, Zhou X, Han L, Shi M, Xiao H, Lin M, et al. Human umbilical cord blood-derived mesenchymal stem cell transplantation for patients with decompensated liver cirrhosis. J Gastrointest Surg (2023) 27:926–31. doi: 10.1007/s11605-022-05528-1
207. Watanabe Y, Tsuchiya A, Seino S, Kawata Y, Kojima Y, Ikarashi S, et al. Mesenchymal stem cells and induced bone marrow-derived macrophages synergistically improve liver fibrosis in mice. Stem Cells Transl Med (2019) 8:271–84. doi: 10.1002/sctm.18-0105
208. van der Helm D, Barnhoorn MC, de Jonge-Muller ESM, Molendijk I, Hawinkels L, Coenraad MJ, et al. Local but not systemic administration of mesenchymal stromal cells ameliorates fibrogenesis in regenerating livers. J Cell Mol Med (2019) 23:6238–50. doi: 10.1111/jcmm.14508
209. Weiss ARR, Dahlke MH. Immunomodulation by mesenchymal stem cells (Mscs): Mechanisms of action of living, apoptotic, and dead mscs. Front Immunol (2019) 10:1191. doi: 10.3389/fimmu.2019.01191
210. Wang HY, Li C, Liu WH, Deng FM, Ma Y, Guo LN, et al. Autophagy inhibition Via Becn1 downregulation improves the mesenchymal stem cells antifibrotic potential in experimental liver fibrosis. J Cell Physiol (2020) 235:2722–37. doi: 10.1002/jcp.29176
Keywords: liver cirrhosis, liver immune microenvironment, mesenchymal stromal cells, nonalcoholic fatty lives disease, autoimmune liver disease, chronic liver disease, therapy
Citation: Yi Q, Yang J, Wu Y, Wang Y, Cao Q and Wen W (2023) Immune microenvironment changes of liver cirrhosis: emerging role of mesenchymal stromal cells. Front. Immunol. 14:1204524. doi: 10.3389/fimmu.2023.1204524
Received: 12 April 2023; Accepted: 21 June 2023;
Published: 19 July 2023.
Edited by:
Yongzhan Nie, Fourth Military Medical University, ChinaReviewed by:
Debanjali Dasgupta, Mayo Clinic, United StatesCopyright © 2023 Yi, Yang, Wu, Wang, Cao and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Wen, d2Vud2VuX3NtbXVAMTYzLmNvbQ==
†These authors contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.