 Shuai Peng1,2†Lei Shen1,2†Xiaoyun Yu3Li Zhang4Ke Xu4Yuan Xia1,2Lanlan Zha1,2Jing Wu1,2
Shuai Peng1,2†Lei Shen1,2†Xiaoyun Yu3Li Zhang4Ke Xu4Yuan Xia1,2Lanlan Zha1,2Jing Wu1,2 Hesheng Luo1,2*
Hesheng Luo1,2*- 1Department of Gastroenterology, Renmin Hospital of Wuhan University, Wuhan, China
- 2Hubei Key Laboratory of Digestive Diseases, Wuhan, China
- 3Department of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 4Department of Neurology, Renmin Hospital of Wuhan University, Wuhan, China
Ulcerative colitis (UC) is a chronic inflammatory bowel disease involving mainly the colorectal mucosa and submucosa, the incidence of which has been on the rise in recent years. Nuclear factor erythroid 2-related factor 2 (Nrf2), known for its key function as a transcription factor, is pivotal in inducing antioxidant stress and regulating inflammatory responses. Numerous investigations have demonstrated the involvement of the Nrf2 pathway in maintaining the development and normal function of the intestine, the development of UC, and UC-related intestinal fibrosis and carcinogenesis; meanwhile, therapeutic agents targeting the Nrf2 pathway have been widely investigated. This paper reviews the research progress of the Nrf2 signaling pathway in UC.
1 Introduction
Ulcerative colitis (UC) is a nonspecific, chronic, relapsing inflammatory disorder mainly involving the mucosa and submucosa of the colorectum. The occurrence and frequency of UC have witnessed a continuous escalation in the progressive timeline of recent years (1). The disease has a long course and a wide range of lesions. Furthermore, it is essential to note that the principal clinical indications linked to this ailment encompass frequent diarrhea, excruciating abdominal pains, viscid mucus discharge, excrement that is conspicuously blood-stained, and even additional debilitating symptoms that cause a tremendous amount of distress and utterly corrode the quality of an individual’s life (2). It is also paramount to comprehend that this unfortunate medical predicament might lead to a sequence of menacing and detestable complications, such as damage to the intestinal tract resulting in the development of intestinal fibrosis and ultimately culminating in the malignant and life-threatening ailment commonly known as colorectal cancer (1).
The underlying causes of ulcerative colitis are murky and complicated. Yet the evidence is mounting that oxidative stress and inflammation are also closely related, except in genetics, intestinal flora, host immune system, and environmental factors (2). An accumulating corpus of empirical data has demonstrated the significant contribution of oxidative stress in inciting the inflammatory response that precipitates the onset of UC. Its multifaceted effects have impinged upon the disorder’s progression (3). Dysregulation of the immune system and pronounced inflammatory response events elevate reactive oxygen species (ROS) levels within the organism, thus perturbing redox balance and precipitating oxidative stress. The ensuing upsurge in ROS levels and consequential enhancement of oxidative stress indices result in deleterious cumulative damage to the fundamental biomolecules. Additionally, the imbalance between oxidative and antioxidant systems facilitates the activation of oxidative stress-associated pathways, which mediate cellular senescence, apoptosis, and necrosis (4). Nuclear factor erythroid 2-related factor 2 (Nrf2), a crucial transcriptional regulator involved in redox homeostasis, exerts a pivotal role in facilitating antioxidant responses within the organism (5). The Keap1/Nrf2 signaling pathway is constituted by the principal modulator Nrf2 and its counteractive inhibitor Kelch-like ECH-associated protein 1 (Keap1). This signaling pathway has been confirmed to exert a safeguarding influence on animal models and individuals with ulcerative colitis. In this regard, the Keap1/Nrf2 signaling pathway is vital as an antioxidant defense mechanism (3).
This paper aims to provide a comprehensive overview of Nrf2, including its physiological configuration and function, its involvement in intestinal maintenance and development, and its research progress in addressing ulcerative colitis and related complications. Among multiple aspects of Nrf2, we also focus on the therapeutic applications of modulating the Keap1/Nrf2 pathway in ulcerative colitis.
2 The physiological structure and function of Nrf2
2.1 The physiological structure of Nrf2
Nrf2 was initially cloned from the human leukemia cell line (K562) and identified as a Cap-n-collar (CNC) alkaline leucine zipper transcription factor family member (6). Nrf2 consists of seven Neh domains (Nrf2-ECH homology), each with a different function. The Neh1 domain is characterized by a remarkably conserved basic region-leucine zipper (bZIP) architecture. The Neh2 region mediates interactions with Keap1 through DLG and ETGE motifs. The carboxyl terminus of Neh3 has been found to facilitate the regulation of the antioxidant response element (ARE)-mediated transcription through its association with the chromo ATPase/helicase DNA binding protein (CHD6) (7). Neh4 and Neh5 regions, on the other hand, are instrumental in initiating downstream gene transcription, which is crucial for the transactivation of Nrf2. Notably, the regulatory region of Neh6 is characterized by the prevalence of serine residues and is responsible for the regulation of Nrf2 degradation via a mechanism independent of KEAP1. Furthermore, retinoic acid X receptor alpha (RXRα) has been reported to reduce the cytoprotective effect of Nrf2 by directly binding to the Neh7 domain (8).
Keap1 acts as a substrate adaptor protein for the E3 ubiquitin ligase complex, which comprises Cullin3 (Cul3) and Rbx1 to form a functional E3 ubiquitin ligase complex (Keap1-Cul3-E3). This complex plays a crucial role in regulating the activity of Nrf2 (9). Keap1 contains five domains, namely N-terminal region (NTR), intervention region (IVR), Broad complex, Tramtrack and Bric-à-Brac region (BTB), diglycine repeat region (DGR), and C-terminal domain C (CTR). The DGR region, the Kelch region, is the Neh2 junction region of Keap1 and Nrf2 (10) (Figure 1).
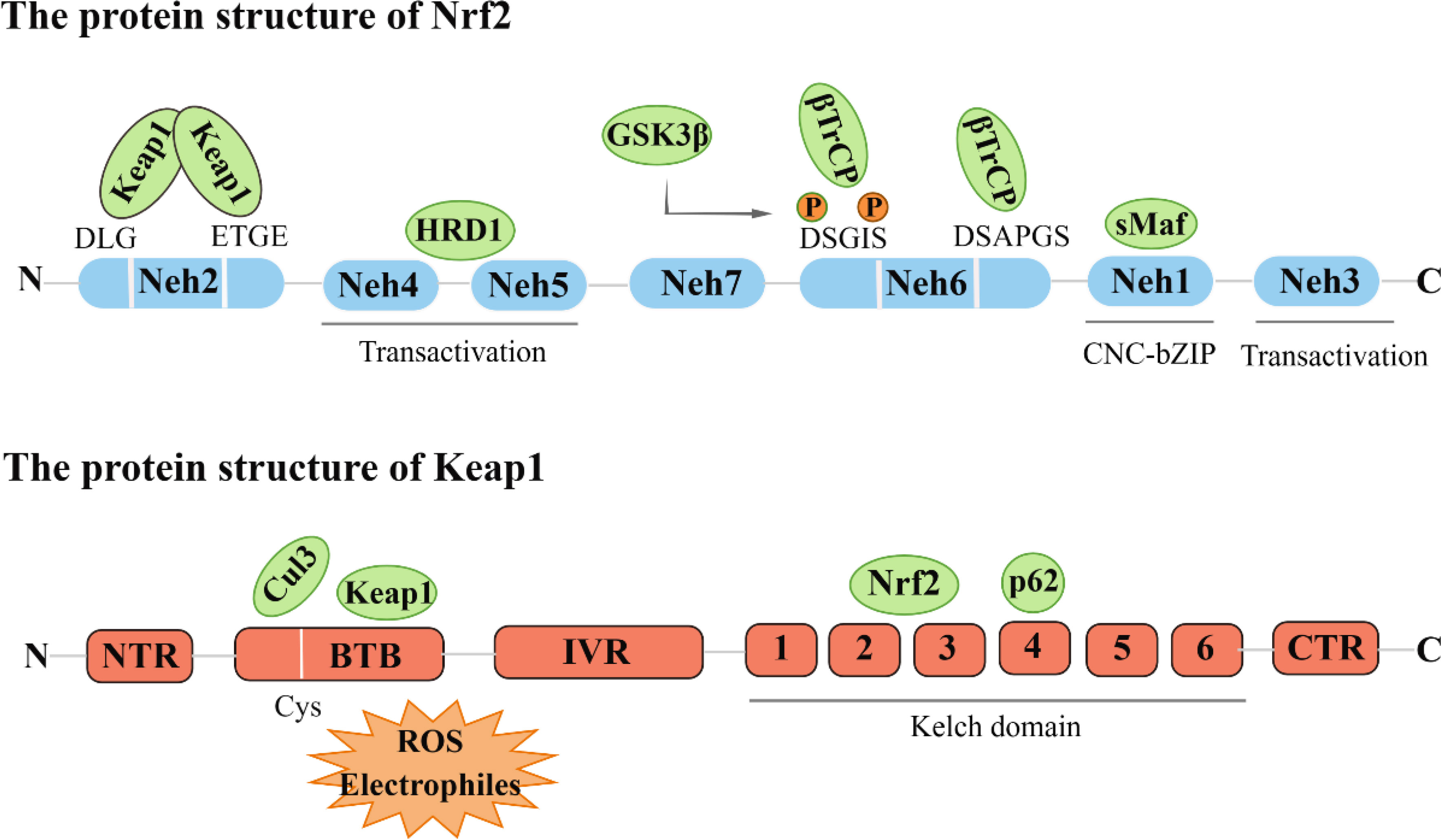
Figure 1 The protein structure of Nrf2 and Keap1.
2.2 Triggers for activation of the Nrf2 signaling pathway
2.2.1 Keap1-dependent activation of Nrf2 signaling pathway
Upon Nrf2 entering the nucleus, bZIP cooperates with small Maf proteins to form a heterodimer, enabling Nrf2 to recognize, bind to antioxidant response element (ARE), and initiate downstream related gene transcription (11). Under typical physiological circumstances, the BTB domain within the Keap1 protein interacts with the Cul3 protein. In contrast, the DGR domain targets various lysine residues in the Neh2 domain of Nrf2, thereby facilitating its ubiquitination. Consequently, the ubiquitinated Nrf2 undergoes degradation via the proteasome pathway (12). However, upon exposure to oxidative stress, particular cysteine residues within Keap1 undergo modification, which results in a structural alteration of the Keap1-Cul3-E3 ubiquitin ligase complex. This alteration disrupts the ubiquitination process of Nrf2, enabling its translocation into the nucleus. Subsequently, the nuclear Nrf2 forms a complex with sMaf proteins and binds to the ARE region. This binding event initiates a coordinated activation program that induces the expression of multiple cytoprotective genes, ultimately enhancing cellular defense mechanisms (13).
In addition to the Keap1/Nrf2 pathway, an atypical mechanism of Nrf2 activation, the autophagy-lysosome pathway, is driven by autophagy dysfunction, which also plays a crucial role in mediating oxidative stress (14, 15). The SQSTM1/p62 is a typical receptor for selective autophagy that degrades ubiquitinated substrates and plays a crucial role in regulating various signaling cascades, encompassing the Keap1/Nrf2 pathway (16). Dysregulation of autophagy processes culminates in the build-up of the autophagy-associated adaptor molecule SQSTM1/p62, consequently leading to the entrapment and subsequent functional impairment of various interacting proteins, including Keap1 (17). It has been reported that SQSTM1/p62 competes with Nrf2 for binding to Keap1, and this interaction can sequester Keap1 into autophagosomes, thereby preventing Keap1-mediated Nrf2 degradation and leading to Nrf2 pathway activation (18). Notably, the activation of Nrf2 must be tightly controlled. Over-accumulation or over-expression of Nrf2 has been reported to be detrimental and sometimes fatal (Figure 2).
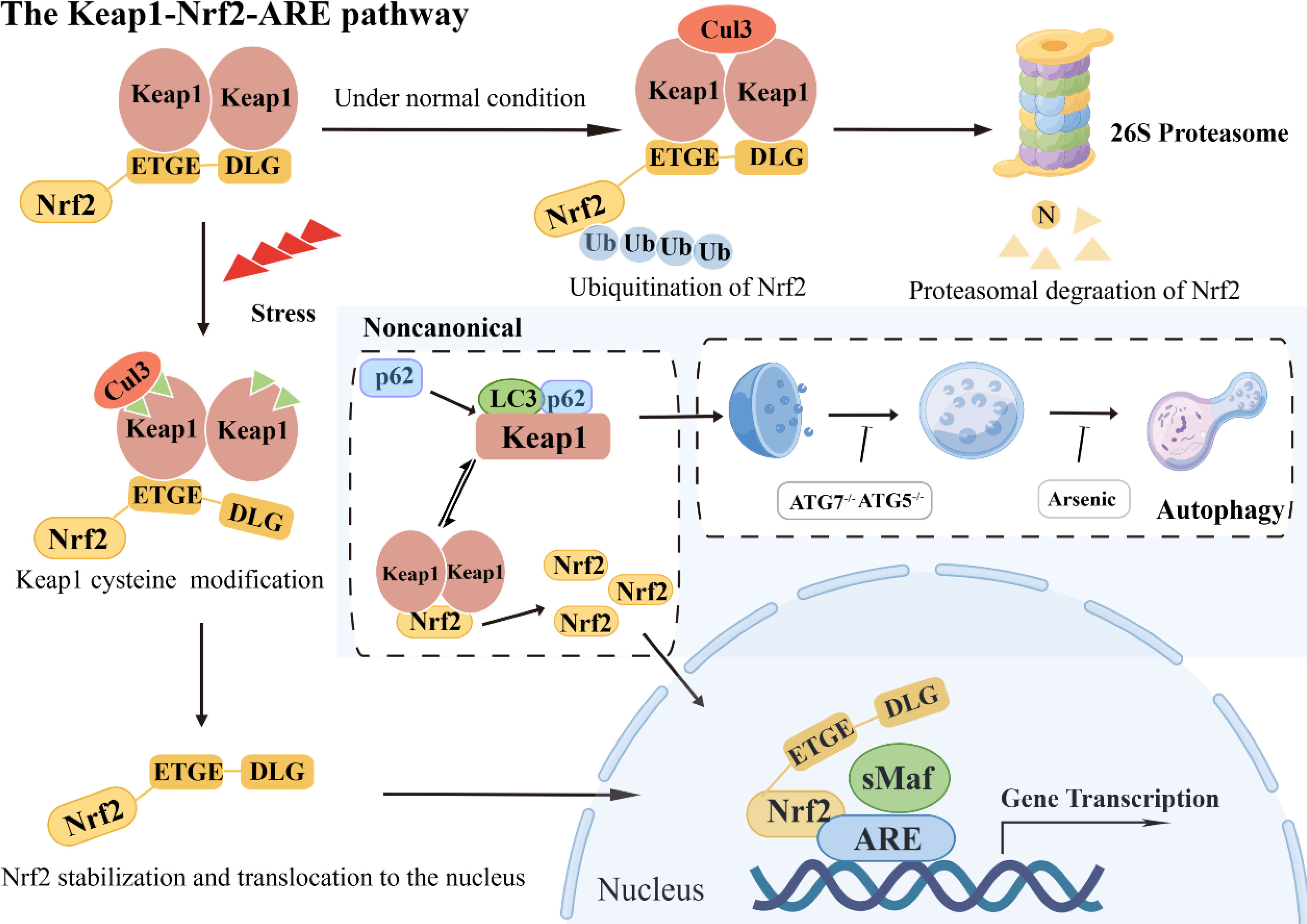
Figure 2 The Keap1-Nrf2-ARE pathway: classical and noncanonical pathways.
2.2.2 Keap1-independent activation of Nrf2 signaling pathway
Protein kinases are important regulators of many cellular processes through the phosphorylation of specific proteins. Recent studies have highlighted the crucial role of protein kinases in the modulation of Nrf2 activity. Among them, protein kinase C (PKC) has been shown to play a significant role in the activation and expression of Nrf2. Salvianolic acid B has been demonstrated to induce the expression of Nrf2, HO-1, and GCLC by activating the PI3K and PKC pathways, thus providing protection against APAP-induced liver injury (19). Similarly, protocatechualdehyde has been found to protect the liver from APAP-induced injury through the protein kinase Cϵ/Nrf2/HO-1 pathway and to alleviate cerebral ischemia-reperfusion-induced oxidative injury (20). Further investigations have revealed that the phosphorylation of Nrf2 at Ser-40 by PKC represents a key signaling event leading to the activation of ARE-mediated cellular antioxidant responses (21). Different isoforms of PKC may also play a role in mediating the phosphorylation of Nrf2 (22). Specifically, Chen et al. identified PKC-δ as the major PKC isoform responsible for the phosphorylation of Nrf2 Ser40, whereas PKC α and β activate Nrf2 at different time points, namely early and late stages, respectively (23).
Recent studies have highlighted the beneficial impact of AMP-activated protein kinase (AMPK) on the activation of Nrf2 and the subsequent induction of antioxidant enzymes in response to oxidative stress (24). AMPK has been found to trigger the expression of SOD and HO-1 via the Nrf2/ARE signaling pathway, leading to enhanced cellular antioxidant capacity and improved cell survival (25). Furthermore, AMPK has been shown to stimulate the nuclear accumulation of Nrf2 through phosphorylation at serine 550 (26). However, the interplay between AMPK and Nrf2 transcription warrants further investigation to better understand the underlying mechanisms of this signaling pathway.
Moreover, three Mitogen-activated protein kinases (MAPKs) have been identified as playing a role in the modulation of Nrf2 activity. Extracellular-signal-regulated kinases 1 and 2 (ERK1/2) are involved in both cell proliferation and defense mechanisms. Senkyunolide I has been found to protect the rat brain against focal cerebral ischemia-reperfusion injury by upregulating p-Erk1/2, Nrf2/HO-1, and inhibiting caspase 3 (27). Similarly, Gastrodin has been shown to safeguard the midbrain from oxidative stress in mice by blocking the ERK1/2-Nrf2 pathway (28). In addition, c-Jun N-terminal kinases (JNKs) and p38 MAPK are crucial mediators of oxidative stress reduction. Research indicates that Protocatechuic acid induces the expression of antioxidant/detoxification enzymes via JNK-mediated Nrf2 activation in mouse macrophages (29). Moreover, Ma et al. have identified p38 MAPK/Nrf2 signaling as a critical molecular network responsible for the development of temozolomide resistance in gliomas (30). Furthermore, Diallyl trisulfide has been shown to inhibit tumor growth by suppressing Nrf2/Akt activity and activating p38/JNK signaling (31). Meanwhile, Qi et al. have demonstrated that the activation and interplay between PI3K/Akt and Nrf2/HO-1 signaling pathways may play a role in regulating the hormesis of Z-ligustilide in PC12 cells subjected to oxygen and glucose deprivation (32). Similarly, reactive oxygen species and PI3K/Akt signaling have been shown to be key factors driving Nrf2-mediated heme oxygenase-1 expression in sulforaphane-treated human mesothelioma MSTO-211H cells (33). In conclusion, further investigation is necessary to gain a deeper understanding of the Keap1-independent Nrf2 pathway and its activation.
3 The role of Nrf2 in intestine development
The transcriptional activity of Nrf2 is essential for maintaining normal intestinal architecture in mice (34). The mouse intestine begins to establish around 9.5 days after conception. During the 9.5-14.5 days of embryonic development, pure epithelial cells transform into endodermal tubes, and the length and circumference of the intestine gradually increase (35). Subsequently, differentiated cells of absorptive and secretory lineages appeared from day 14.5, and epithelial remodeling and transient villi appeared in the gut on day 15. During 14.5-21 days of embryonic development, intestinal cells proliferate, and crypts and stem cell nests gradually form (36). Most colonic structures and cellular aggregates in adults exist prenatally, with fully developed colonic crypts manifested within 12-15 days following birth (35).
Intestinal development is mainly driven by the coordination between Notch and Wnt signaling pathways, and it is worth noting that Nrf2 can affect the activation of these signaling pathways (37). Studies have validated the significance of intercommunication between the Nrf2 and Notch pathways in regulating gastrointestinal tract maturation (38). Research in this area has shown that the proximal region of the promoter of the Notch downstream effector Math1 gene in mice has a functional ARE sequence, and the activation of Nrf2 signaling in the intestinal epithelium can lead to intestinal elongation and extension through negative transcriptional regulation of the Notch downstream effector Math1 (39). Meanwhile, Nrf2 knockout mouse embryonic fibroblasts decreased the expression of Notch-1 and its related gene signaling (40). There is a delicate balance between the Wnt and Nrf2 signaling pathways. A study confirmed that β-catenin in the Wnt pathway could activate the Nrf2 pathway to a certain extent, and Nrf2 could strongly inhibit β-catenin. Moreover, β-TrCP1 binds to β-catenin to weaken the inhibition effect of Nrf2 on β-catenin (41).
Twenty-five years ago, Chen et al. showed that the Nrf2 gene is highly expressed on the luminal side of the intestine during pregnancy in mice. In addition, they also observed substantial variations in Nrf2 mRNA levels across different organs and gestational days (42). A recent study has shown that Nrf2 levels continue to rise in the hindgut from 14.5 to 18.5 days of embryonic development, while Nrf2 levels decrease in other tissues, such as the lung or heart suggesting that Nrf2 plays a crucial role in intestine development (34). Nrf2 transcriptional deletion resulted in marked elongation of the colon, altered crypt distribution, enlarged goblet cells, and markedly elevated mucin levels (43). Thus, Nrf2 transcriptional activity constitutes an integral regulatory mechanism in the formation of the gastrointestinal tract, modulating the proliferation and differentiation of hindgut cells throughout varied embryonic stages.
4 The role of Nrf2 in ulcerative colitis
4.1 Nrf2 attenuates intestinal inflammation and damage by controlling oxidative stress
Oxidative stress results from dissonance amidst the production and clearance of ROS and reactive nitrogen species (RNS) following the body’s exposure to diverse injurious stimuli, which causes the body’s oxidation and oxidation antioxidant system imbalance. The antioxidant defense function is weakened, resulting in various pathological changes (44). Under physiological conditions, oxidation and anti-oxidation maintain a dynamic balance in the body. However, when inflammation occurs in the body, this balance is broken. Oxygen free radicals attack their tissues, participate in and generate inflammatory mediators through lipid peroxidation, and activate the inflammatory response (45).
Impairment of the antioxidant defense machinery in the gastrointestinal tract has been implicated in UC etiology. Infiltration of inflammatory cells exacerbates oxidative stress via the upregulation of ROS synthesis in the immune cells (46). Insurmountable ROS release, coupled with the persistent accumulation of oxidative stress, frequently culminates in DNA damage, protein oxidation, and lipid peroxidation, ultimately causing intestinal tissue damage, debilitating the immune system, and precipitating an array of severe pathologies, including UC (47). When UC occurs due to the inflammatory response, the activity of gut-derived vasoconstrictors is enhanced, resulting in intestinal ischemia. At the same time, inflammatory cells in the intestinal mucosa, such as neutrophils and macrophages, enter the intestinal tract from the blood circulation (48). In the damaged part, under the action of cell membrane reduced coenzyme II and NADPH oxidase, a large number of reactive oxygen radicals such as superoxide anion (H:0), hydrogen peroxide (H2O2), hydroxyl radical (H01), NO free radicals and lipid peroxides (LPO), thereby aggravating intestinal mucosal damage (47, 49).
Cells have evolved a complex protective system to defend against the damage mentioned above, and the Keap1/Nrf2 pathway is the foremost defense mechanism for counteracting oxidative stress (13). The initial research investigating the alleged involvement of Nrf2 in UC was reported first by Arisawa et al. in 2008. They identified that the -686*-684 genotype of the Nrf2 gene was significantly correlated with UC incidence among the Japanese population and closely associated with the chronic persistent phenotype (50). Recent research has established that the expression level of Nrf2 in individuals with UC is lower than in healthy cohorts. Nonetheless, some studies have also revealed a marked increase in the expression level of Nrf2 in the mucosa of the inflammatory intestinal tract among UC patients compared to controls (51, 52). Moreover, Milad’s research demonstrated that the phosphorylated form of Nrf2 was expressed at a significantly higher level in individuals diagnosed with moderate and severe UC than in healthy controls. Conversely, the expression level of non-phosphorylated Nrf2 was diminished in moderate to severe UC patients compared to healthy cohorts. These observations suggest that the altered levels of Nrf2 in UC patients may be influenced by the disease’s progression or the form of Nrf2 expression (51).
Over the years, related studies on the involvement of the Keap1/Nrf2 axis in the process of UC have found that mice lacking Nrf2 exhibit heightened vulnerability to dextran sodium sulfate (DSS)-induced colitis and an increased susceptibility to colorectal cancer (53–55). Furthermore, compared with wild-type mice, the levels of pro-inflammatory cytokines and lipid peroxidation in the colon of Nrf2 knockout mice treated with DSS were significantly increased, and the expression levels of antioxidant enzymes were decreased (56). Moreover, multiple genetic mutations on Nrf2 have been linked to increased susceptibility and progression of DSS-induced colitis in mice (57, 58). Considering these discoveries, modulation of the Keap1/Nrf2 signaling pathway could represent a promising avenue for managing UC.
In UC, the Keap1/Nrf2 pathway can reduce intestinal inflammation and injury by controlling oxidative stress and play an essential role in protecting intestinal integrity, mainly by regulating inflammatory mediators and inducing the production of antioxidant enzymes (59, 60). Oxidative stress provokes Nrf2 to translocate to the nucleus, where it mediates the transcription of a diverse suite of antioxidant genes, thereby conferring cell protection against the damage induced by oxidative stress (61). In addition, various antioxidant enzymes in the body, such as superoxide dismutase (SOD), glutathione (GSH), and other activations, establish an endogenous defense system against intestinal oxidative stress, protecting the intestinal mucosa from harmful stimuli (62). Currently, most studies focus on regulating inflammatory mediators, inducing the production of antioxidant enzymes, and regulating autophagy by activating Nrf2, thereby reducing oxidative stress damage caused by aggravated ROS and alleviating pathological inflammatory responses (63–66).
In addition, activation of Nrf2 can balance cellular homeostasis, activating a series of signaling pathways targeting inflammation, such as the NF-κB pathway. Numerous studies have shown an interaction between the Keap1/Nrf2 and NF-κB pathways (67, 68). First, sMaf (MafK) can positively regulate NF-κB activity by enhancing the Nrf2 transcriptional coactivator CBP-mediated acetylation of NF-κB p65, suggesting that Nrf2 may indirectly regulate NF-κB activity by inhibiting MafK. Second, Keap1 can inhibit the activation of NF-ĸB by inhibiting the ubiquitination degradation of IKKβ. Third, the inflammatory response can inhibit NF-κB activity by inducing inflammatory mediators and subsequently reacting with Keap1 to activate the expression of Nrf2 (69, 70). In conclusion, activation of Nrf2 in the gut can inhibit inflammatory pathways or reduce the overreaction of oxidative stress, thereby alleviating intestinal damage and inflammation.
However, we still need to control the expression of Nrf2 strictly. Recently, in a study of transgenic mice constitutively expressing active Nrf2 (caNrf2 mice), Gerstgrer et al. found that symptoms of acute colitis induced by DSS were exacerbated after constitutive Nrf2 expression but not worsened chronic colonic mucosal inflammation (71). The above phenomenon suggests that the redox balance in the body needs to be strictly regulated. Otherwise, the double-edged sword effect is prone to occur. Meanwhile, further extensive studies are therefore imperative for an enhanced understanding of the complex interplay between the Keap1/Nrf2 signaling network and oxidative stress as well as inflammation in the intestine.
4.2 Nrf2 facilitates the maintenance of the intestinal epithelial barrier
The intestine is an important digestive organ of the human body and the largest immune organ of the body. The intestinal epithelium forms a tightly regulated intestinal barrier between the external environment and the body, which can prevent the invasion of harmful substances such as pathogenic bacteria and toxins. It is indispensable in maintaining homeostasis and body health (72, 73).
The intestinal mucosal barrier is a multifaceted structure comprising the surface mucous layer, epithelial cell layer, and mucosal basal layer, alongside the biological barrier facilitated by the resident microflora of the gut. Furthermore, it encompasses a chemical barrier consisting of a range of digestive enzymatic secretions, lysozymes, mucopolysaccharides, glycoproteins, and glycolipids produced by the intestine, an immune barrier that is formed by intestinal associated lymphoid tissue (GALT) and secretory immunoglobulin A (sIgA), and a mechanical barrier consisting of intact intestinal mucosal epithelial cells and intercellular junctions (74–76).
The pivotal role of the intestinal mucosal barrier is attributed to its mechanical barrier, in which epithelial cells and their intercellular junctions are the structural basis for maintaining intestinal epithelial selective permeability and barrier function. It is the key to resisting the invasion of extraintestinal harmful substances or pathogens into the intestinal mucosa (77, 78). The junction between intestinal epithelial cells is the core part of the mechanical barrier. It is controlled by the myosin light chain (MIC), including tight junctions, gap junctions, adhesion junctions, and desmosome junctions, especially tight junctions, mainly composed of occlusal junctions. The constituents of this barrier include members of the tight junction protein group, such as occludin, claudin, and cadherin, as well as the Zonula Occludens (ZO) family (79, 80). The tight junctions between intact intestinal epithelial cells can prevent intestinal bacteria, toxins, and antigens from entering the lamina propria, prevent the activation of lamina propria immune cells, and induce abnormal intestinal immune responses (81, 82).
A key pathological event in UC is intestinal barrier dysfunction. The destruction of the intestinal barrier will increase intestinal permeability, and intestinal pathogenic bacteria and pathogens will further invade the intestinal mucosa, thereby exacerbating inflammatory cell infiltration and damage, forming a vicious circle, which eventually leads to damage to the intestinal mucosa and ulcer formation (83, 84). Current research confirms the indispensable involvement of Nrf2 in preventing UC. Activation of Nrf2 in animal models of UC exerts a regulatory effect on the expression of tight junction proteins located in the intestinal epithelium, specifically Zonula Occludens-1 (ZO-1) and claudin, thus safeguarding the integrity of the gut barrier (85–87). In DSS-induced mouse colitis models, tight junction protein expression was significantly lower than in the control group, resulting in increased intestinal permeability (88, 89). In the LPS-induced intestinal barrier damage model, the mitochondria-targeted antioxidant MitoQ can prevent intestinal barrier damage by upregulating the expression of Nrf2 downstream regulatory genes. The mechanism may involve activating the Keap1/Nrf2/ARE signaling pathway and inhibiting oxidative stress (90). Concurrently, the study revealed that Nrf2 confers a safeguarding effect in a traumatic brain injury-induced intestinal mucosal injury model. In contrast to wild-type mice, Nrf2-deficient mice demonstrated enhanced susceptibility to traumatic brain injury-induced intestinal inflammation, characterized by elevated intestinal permeability and augmented plasma endotoxin levels, exacerbating the decline in intestinal barrier function (91). Another study in the same model confirmed that ERK/Nrf2/HO-1-mediated stimulation of mitophagy ameliorates intestinal mucosal impairment and barrier dysfunction (92). Furthermore, Nrf2 activation is purported to enhance the protection of tight junction proteins by negating apoptosis of intestinal epithelial cells while concurrently instigating autophagy. In addition, in a study of reflux esophagitis, Nrf2 was found to bind to the promoter of claudin-4 and increase its expression but not to claudin-1. Nrf2 deficiency leads to mitochondrial dysfunction, downregulating claudin-4 expression and ultimately leading to tight junction damage in the esophageal epithelium (93). Further research has demonstrated that the inducement of the Keap1/Nrf2 pathway impacts the regulation of tight junction proteins and governs the regenerative processes of intestinal stem cells, thereby promoting intestinal homeostasis (94, 95).
The intestinal mucus barrier also plays an integral role in maintaining gut health (96). Intestinal mucus is a high-molecular glycoprotein mucus layer secreted by goblet cells, which forms a functional barrier between intestinal microbes and the intestinal epithelium, providing defense by hindering the direct contact of bacteria and harmful substances to the intestinal epithelium (97). Research has documented that diminished levels of intestinal mucus can hinder nutrient absorption through the intestinal mucosa while simultaneously triggering secretion of water and electrolytes within the intestinal lumen. At the same time, plasma-like fluid penetrates further into the lumen, increasing the volume of fluid and the permeation load in the lumen and causing diarrhea (98). Moreover, the study found that Low molecular Seleno-amino polysaccharide (LSA) can protect the intestinal mucosal barrier in rats by activating the Nrf2 pathway and mitigating the anomalous alterations of MUC2 (99). In addition, Singh and his team have found that gut microbial metabolism enhances the integrity of the intestinal barrier through the Nrf2 pathway (59). To conclude, the activation of Nrf2 aids in preserving the integrity of the intestinal epithelial barrier, while further investigations are required to elucidate the underlying mechanism involved.
4.3 Nrf2 is the regulator of intestinal immunity
The abnormal intestinal mucosal immune system is one of the main reasons for the development of UC. Meanwhile, initiating intestinal mucosal immunity and ensuing inflammation hinges upon activating effector T cells. In response to a plethora of stimuli, an array of effector T cell subsets emerge from precursor naive T cells, including Th1, Th2, Th17, and Treg cells (100, 101).
In recent years, in addition to the anti-inflammatory mentioned above and oxidative stress control effects of Nrf2, numerous research endeavors have additionally evidenced that Nrf2 activation holds the potential to modulate the Th1/Th2 equilibrium selectively. In 2012, Rockwell and his team’s research found that the induction of Nrf2 can preferentially direct CD4+ T cells toward Th2 differentiation. Specifically, the activation of Nrf2 by tBHQ, a food preservative, has been observed to hinder the production of the Th1 cytokine IFN-γ and simultaneously encourage the generation of Th2 cytokines such as IL-4, IL-5, and IL-13 (102). Three years later, the team found that the Nrf2 activator tBHQ inhibited the production of IL-2 and IFN-γ in activated CD4+ T cells (103). Five years later, the team continued to investigate the effects of Nrf2 activation on the initial events that follow T-cell activation. The results showed that different Nrf2 activators (tBHQ and CDDO-IM) had different effects on early T cell differentiation, and the activation of some cytokines did not depend on the Nrf2 pathway. Although Nrf2 inhibited the expression of early TNF-α and IFN-γ after activation, it was observed to facilitate the generation of IL-2. It displayed no discernible effect on the induction of CD25 and CD69. IL-2 serves as a growth factor for T cells that promotes the development of Th2 cells and is critical for the differentiation and function of Treg cells (104). Other recent studies on tBHQ also confirmed that tBHQ attenuated 5-fluorouracil-induced intestinal epithelial cell injury by activating Nrf2 (105). Moreover, tBHQ induced an Nrf2-dependent increase in IgM secretion by LPS-stimulated B cells (106). Meanwhile, studies have shown that Nrf2 can regulate the IL-22 response in CD4+ T cells through the AhR pathway (107). This suggests that activation of Nrf2 has a relevant role in regulating both T and B cells.
Another study focused on dimethyl fumarate (DMF). This Nrf2 activator demonstrated that DMF could reduce the inflammatory response in experimental colitis, mainly due to the activation of Nrf2 and its downstream antioxidant genes expression after administration of DMF and simultaneously inhibit the NF-κB signaling (108). A research investigation exploring acute graft-versus-host disease (AGVHD) unveiled that activation of Nrf2 by DMF promoted the development of donor Treg cells and reduced the deleterious response of allogeneic T cells (109). Although the activation of Nrf2 modulates intestinal immunity to varying degrees, the exact mechanism has not been fully elucidated (Figure 3).
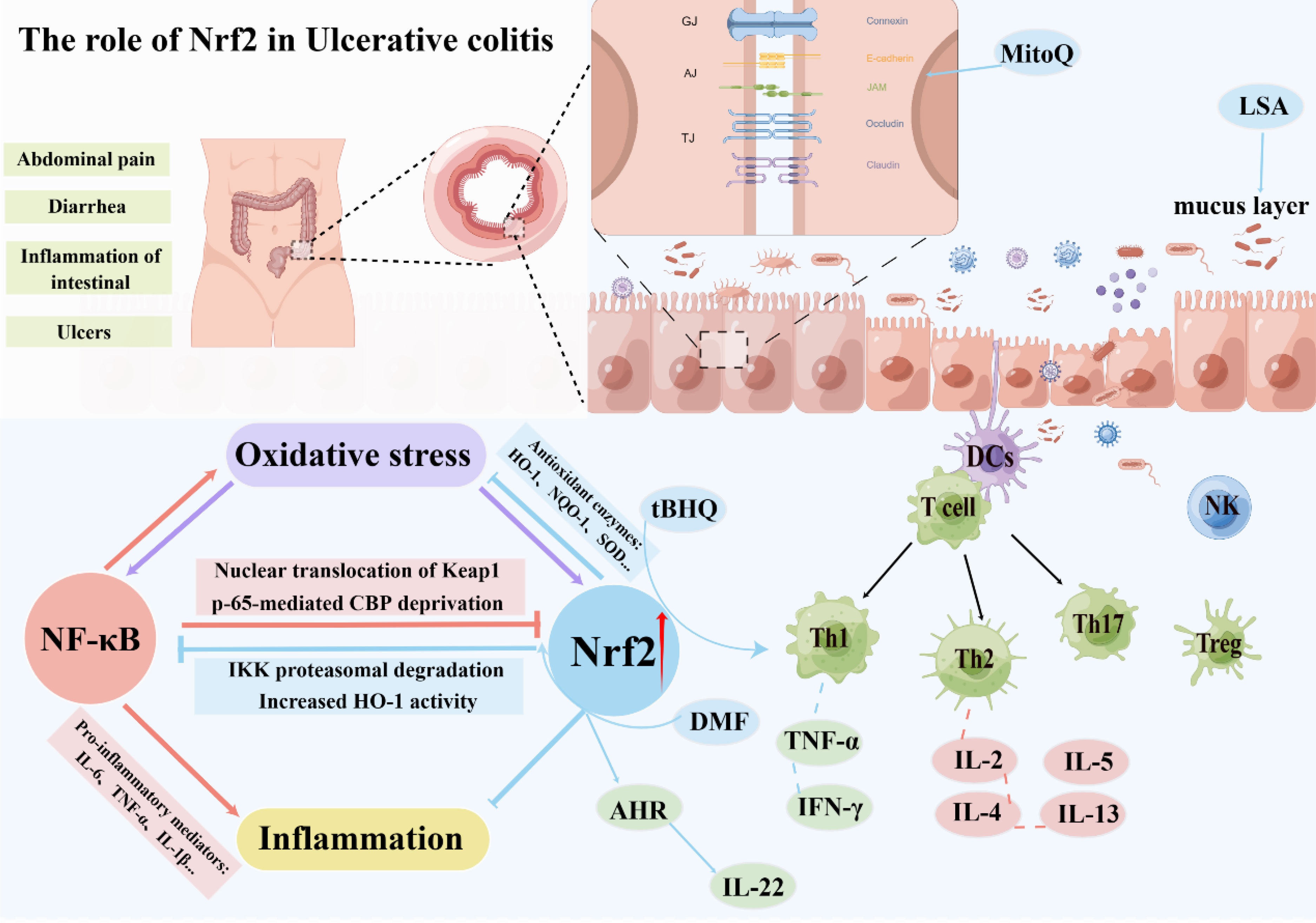
Figure 3 The role of Nrf2 in ulcerative colitis.
5 The role of Nrf2 in intestinal fibrosis in UC
The accumulation of excessive extracellular matrix caused by chronic inflammation is a characteristic feature of intestinal fibrosis, which is a prevalent complication found in patients with Crohn’s disease (CD) and has more recently been observed in those with UC (110). During UC, ECM accumulates in the mucosa and submucosa, leading to its thickening, which results in the shortening and stiffening of the colon (111). Furthermore, fibrosis in UC is notably intertwined with inflammation and the disruption of the epithelial layer, often attributed to damage inflicted on the tight junctions (112).
The abnormal accumulation of ECM characterized by fibrosis may be due to overproduction or reduced degradation of ECM. Despite recognizing the TGF-β1/SMADs signaling pathway as the chief driving force behind intestinal fibrosis, numerous pro- and anti-fibrotic endogenous factors, such as ROS and Nrf2, have been identified as interactors of this pathway (113, 114). Several studies have shown that Nrf2 exerts an anti-fibrotic effect in various organs and that this protective effect is closely related to the classical pathway of fibrosis, TGF-β1/SMADs (115–117).
TGF-β1 and its receptor are highly expressed in animal models of fibrous stenosis and intestinal fibrosis. They can signal to the downstream Smads family of proteins, promoting extracellular matrix protein deposition and fibroblast transformation, thereby accelerating fibrosis (118, 119). At the same time, TGF-β1 promotes the generation of ROS while concurrently obstructing the activity of antioxidant enzymes, thereby creating an asymmetry within the redox homeostasis system, while ROS is also an essential mediator in activating the TGF-β1/Smads pathway (120). Therefore, redox disorders caused by dysregulation of the Nrf2 pathway induce fibrosis through massive ROS production and activation of the TGF-β1/Smads pathway (113). Guan et al. showed that tBHQ reduced fibrosis in mice with chronic fibrous colitis and human intestinal fibroblasts by inhibiting the TGF-β1/Smads signaling pathway. At the same time, Nrf2-/- promoted TGF-β1-induced intestinal fibroblast differentiation by pretreating human intestinal fibroblasts with tBHQ or siNrf2. Nrf2-/- could promote TGF-β1-induced intestinal fibroblast differentiation (114). Thus, the inhibition of intestinal fibrosis by Nrf2 is accomplished through the ROS/TGF-β1/Smads pathway, which has been demonstrated in both in vitro and in vivo.
Parallel results were observed in CCD-18Co, a type of normal human colonic fibroblasts, following stimulation with TGF-β1. Suppression of Nrf2 amplified the expression of the TGF-β1/Smad signaling cascade in CCD-18Co fibroblasts (116, 121, 122). These findings highlight the potential of Nrf2 activation in constraining the TGF-β1/Smad signaling axis and its consequent alleviation of intestinal fibrosis. Among colon-derived CCD-18Co fibroblasts, Nrf2 equipped itself to tone down intestinal fibrosis by putting the brakes on the ROS-dependent TGF-β1 signaling pathway, eliciting ROS scavenging (114, 116, 123).
MMPs and TIMPs regulate the degradation of the extracellular matrix. An irregularity in the functioning of these specific enzymes results in the accumulation of extracellular matrix (ECM), thereby contributing to the development of fibrosis (124, 125). Among the myriad matrix metalloproteinases (MMPs), MMP7 is the most critical component within the intestinal context. Research has demonstrated that in human intestinal epithelial cells, the Nrf2/HO-1 axis effectively suppresses MMP7 activity. Reducing fibrosis by specifically inhibiting MMP7 through modulation of the Nrf2 signaling pathway may greatly benefit the treatment of IBD (113, 126). In addition, growing evidence elucidates the role of MMP-3 in IBD.MMP3 concentrations have been observed to increase in response to oxidative stress, and both its expression and activity exhibit augmentation in mice lacking Nrf2. Significantly, patients diagnosed with CD and UC exhibit elevated levels of MMP-3 (127, 128). Furthermore, research has highlighted that MMP3 concentrations are pivotal in determining the responsiveness to infliximab therapy among individuals diagnosed with IBD. Those who did not respond within one year had significantly higher serum MMP3 levels than controls (129). Furthermore, MMP3 possesses the discriminatory capacity for differentiating pediatric patients with UC from their healthy counterparts (130). The implications of MMP3 in regulating intestinal barrier functionality have been comprehensively examined and elucidated by Giuffrida et al. (131). The above findings reveal new therapeutic strategies to modulate Nrf2 signaling to re-establish cellular homeostasis in intestinal fibrosis.
6 Two-sidedness of Nrf2 in UC-associated colorectal cancer
Colorectal cancer (CRC) is a frequently occurring malignancy with a substantial global burden, characterized by elevated incidence and mortality rates. Part of it is caused by chronic colitis, called colitis-related colon cancer (2). Numerous epidemiological, experimental pathological, and clinical studies have shown that the prolonged presence of inflammatory bowel disease, especially chronic ulcerative colitis, can lead to malignant transformation into colon cancer and even promote the progression and early metastasis of colon cancer (132). Studies have reported that UC is a precancerous lesion of colorectal cancer and that the prevalence of CRC in patients with UC is two to six times higher than in the general population, with the incidence increasing with the number of years of diagnosis (132).
As a classical pathway for cellular defense and survival signaling, the role of Nrf2 in tumors has been of great interest. In recent studies, evidence has emerged indicating that Nrf2 plays a role in the carcinogenic transformation of UC. In patients with active ulcerative colitis, a complex ROS-rich microenvironment consisting of cytokines, chemokines, and inflammatory cells in the inflammatory state of the intestine is a major factor in promoting the cancerous transformation of UC (3, 126). It is thought that Nrf2 plays a dual and controversial role in developing and progressing colorectal cancer associated with colitis (Figure 4).
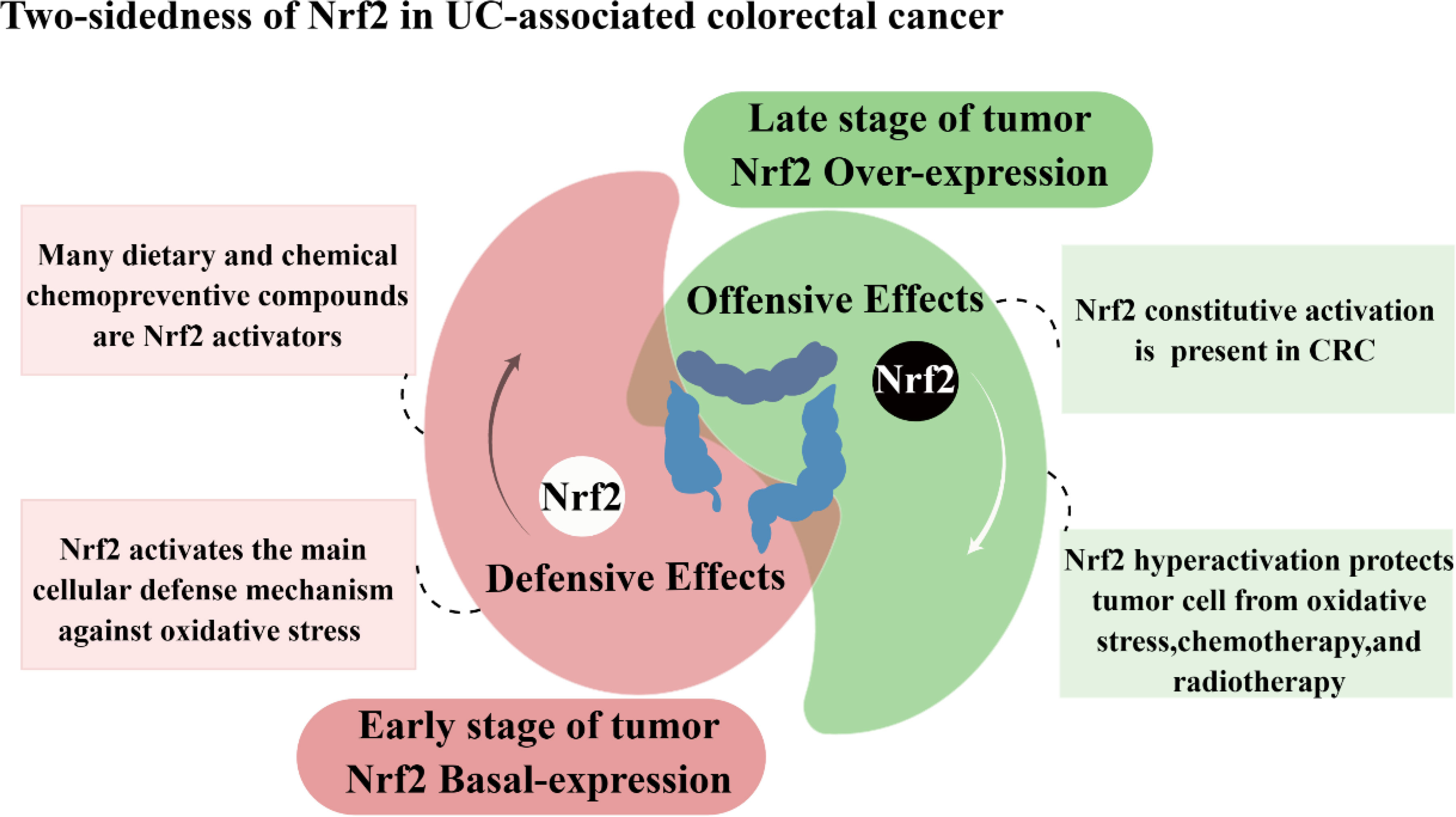
Figure 4 Two-sidedness of Nrf2 in UC-associated colorectal cancer.
Nrf2 has a protective role in the early stages of CRC development, and ensuring the appropriate basal regulation of Nrf2 is crucial for averting carcinogenesis in colon tissues. In colon cells, activation of Nrf2 protects colon cells from damage by reducing genotoxic damage produced by oxidative stress, thereby inhibiting colon cancer progression (133). Existing research suggests that when colon cells are subjected to dextran sodium sulfate, it leads to the induction of carcinogenesis in the colonic tissue. At the same time, the addition of CPUY192018, a Keap1-Nrf2-PPI signaling pathway inhibitor, activates Nrf2 to reduce the risk of conversion of ulcerative enteritis to colon cancer (134). In addition, digitalis flavonoids can activate Nrf2 via p38 MAPK, promote Nrf2 nuclear translocation, and stimulate the expression of downstream phase II detoxification enzymes. This process safeguards colonic cells from oxidative stress and subsequently decreases the occurrence of AOM-DSS-induced colorectal cancer (135). Conversely, the absence of Nrf2 regulation compromises the cellular capacity to withstand genotoxic and oxidative stress, resulting in a disrupted intestinal microenvironment, and normal colon cells become more susceptible to damage from oxidative stress and various genotoxic compounds. The study showed that Nrf2 knockout mice and mice normally expressing the Nrf2 gene was exposed to both the colitis-inducing agent DSS and the colon cancer-inducing agent AOM to observe the risk of colon cancer in both groups. The results showed that Nrf2 knockout mice exposed to DSS or AOM developed prolapse, rectal bleeding, and inflammation and increased the number of abnormal crypt foci, indicating that Nrf2 knockouts are more susceptible to colitis or colorectal cancer (53, 54, 136). In addition, the presence of single nucleotide polymorphisms (SNPs) within the promoter region of the Nrf2 gene is correlated with an elevated risk of neoplastic development, and Nrf2-silenced mice showed significantly higher levels of neoplastic damage and tumorigenicity than wild-type mice, as well as significantly higher levels of 8-hydroxydeoxyguanosine in the epithelial DNA of mice. This suggests that identifying molecules that maintain a constant state of Nrf2 could help prevent cancer development at an early stage (137).
Nrf2 has been implicated in the processes of cancer cell proliferation, metastasis, and resistance to radiotherapy during advanced tumor stages. Although Nrf2 was initially identified as a tumor suppressor owing to its protective function against exogenous and endogenous damage. However, mounting evidence suggests that the over-activation of the Nrf2 pathway facilitates tumor cell survival and protects them from oxidative stress and drug effects. Nrf2 is now thought to accelerate tumor progression, promote metastasis and participate in resistance to radiotherapy in advanced tumor stages.
Nrf2 overexpression can promote the development of colon cancer. One study found that colon cells were subjected to oxidative stress and produced excess ROS, which induced Nrf2 overexpression, leading to inflammation of colon tissue and promoting colon carcinogenesis (138). Arlt and colleagues discovered that heightened Nrf2 expression in colon cancer cells and augmented expression of proteasomal subunit proteins S5a/PSMD4 and α-5/PSMA5 increased proteasomal activity provided anti-apoptotic protection and adequate clearance of abnormal proteins in cancer cells and promoted colon carcinogenesis (139). Sebens et al. found that co-culture of M1-type macrophages with colonic epithelial cells activated Nrf2 expression and proteasome activity in colonic epithelial cells, rendering colon cells resistant to apoptosis, promoting inflammatory carcinogenesis, and increasing the risk of colon carcinogenesis (140, 141).
Nrf2 exhibits a strong association with tumor metastasis. Research has demonstrated that Nrf2 is potentially one of the indicative markers related to metastatic tumor processes. Nrf2 is highly expressed in highly invasive colorectal cancer tissues, and its expression is positively correlated with Duke’s stage and clinical prognosis, making it an important marker of colon cancer prognosis (142). Nrf2 regulates colon cancer metastasis by regulating vascular endothelial growth factor (VEGF) and its receptors. Neoangiogenesis is a critical factor contributing to the growth of colorectal cancer tissues. VEGF and its receptor display elevated activity levels during early and advanced (metastatic) stages of colon cancer. At the same time, VEGF family proteins and receptors trigger multiple signaling networks that cause endothelial cell survival, mitosis, and migration (143). One study found that inhibition of Nrf2 blocked the accumulation of HIF-1α in colon cancer cells under hypoxic conditions and inhibited the expression of VEGF and HIF-1α target genes while reducing the growth and angiogenesis of xenograft tumors in mice (144). Currently, anti-VEGF/VEGFR therapy is essential in treating metastatic colon cancer, improving progression-free survival (FPS) and overall survival (OS) in patients with colon cancer (145). Thus, the suppression of Nrf2 may potentially manifest inhibitory effects on the metastasis of colon cancer, thereby highlighting its prospective significance as a target for the treatment of metastatic colon cancer.
Nrf2 plays a vital role in colon cancer chemoresistance. Nrf2, an essential transcriptional regulator of oxidative stress, protects cells from oxidative stress and toxic damage from chemical drugs. However, many studies have reported that persistent overexpression of Nrf2 causes increased resistance of cancer cells to chemotherapeutic drugs, including adriamycin, etoposide, and cisplatin, suggesting that Nrf2 is an important transcription factor for tumor drug resistance (146). 5-Fluorouracil (5-FU) is the most commonly used chemotherapeutic agent in the treatment of colon cancer, but the development of resistance to 5-FU has dramatically reduced its clinical efficacy. The chemotherapeutic agent 5-FU exerts cytotoxic effects on colon cancer cells by inducing the generation of ROS, consequently causing oxidative damage and ensuing cell death. Nonetheless, tumor stem cells (CSCs), a subgroup of colon cancer cells, can counteract 5-FU-induced oxidative damage in colon cancer cells by producing an adaptive cellular response to ROS, closely related to Nrf2 activation, which causes upregulation of antioxidant enzymes and increases cancer cell resistance to 5-FU (147, 148). It was found that FoxO3 overexpression increased the sensitivity of colon cancer cells SW620 and HCT-8 to 5-FU and that the reversal of resistance of human colorectal cancer cells to 5-FU was demonstrated through the involvement of FoxO3 in the inhibition of the Nrf2/TR1 signaling pathway (149). In addition, Kang et al. found that the mechanism of 5-FU resistance in colon cancer was associated with epigenetic modifications such as DNA demethylation upregulating Nrf2 and HO-1 expression. By comparing the epigenetic changes associated with Nrf2 induction in the 5-FU-resistant colon cancer cell line SNUC5, it was concluded that Nrf2 expression, as well as its nuclear translocation and promoter binding, were markedly elevated in SNUC5/5-FUR cells compared to SNUC5 cells, and further Nrf2 or HO-1 knockdown mediated by siRNA considerably curtailed the proliferation of colon cancer cells both in vitro and in vivo, leading to heightened sensitivity to 5-FU (150). Cheng et al. demonstrated that cNrf2 exhibited resistance towards 5-FU and oxaliplatin, both in vitro, using the HCT116 cell line, and in vivo, employing the CRC animal model. This resistance was attributed to the PSMD4-mediated nuclear export of Nrf2, which ultimately activated the NF-κB/AKT/β-catenin cascade, further supporting these findings (151). In addition, cNrf2 and PSMD4-positive CRC patients had a higher rate of chemoresistance.
Nrf2 also plays a key role in promoting resistance to other chemotherapeutic agents in colon cancer. The significant abatement in SW480/Res cell migration and increased induction of apoptosis by oxaliplatin was observed upon inhibition of Nrf2 in colon cancer cells (152). In addition, Nrf2 reduced the sensitivity of NCM460 or Colo320 cells to TRAIL/etoposide by inducing proteasome activity, thereby reducing the apoptosis induced, and tissue immunostaining further confirmed the activation of Nrf2 in the colonic epithelium in the inflammatory region, as well as the increased proteasome expression (140)a. These studies suggest that Nrf2 has an important influence on the development of chemoresistance in colon cancer.
Nrf2 co-regulates CRC progression through interactions with other signaling pathways. In CRC, Nrf2 enhances NF-κB transcriptional activity, which is strongly associated with CRC cell invasion; positive and negative regulation of NF-κB and Nrf2 signaling pathways coexist, which may be closely linked to cell type and tissue microenvironment (153). At the same time, it has been found that Keap1 mutations lead to impairment of the Nrf2-Keap1-ARE signaling pathway, affecting its binding to Nrf2, causing a large accumulation of Nrf2, and increasing the resistance of tumor cells. In malignant tumors, the incidence of Keap1 loss of function is high. Keap1 mutations affect the inhibitory activity of Keap1 on Nrf2, and Keap1 loss of function enhances the survival of tumor cells (154). DeNicola et al. found that Nrf2 transcription was significantly increased in primary mouse cells following the expression of Kras, BRaf, and myc endogenous oncogenic alleles. The upregulation of Nrf2 target genes and the augmented stability of Nrf2 engendered by somatic mutations in both Nrf2 and Keap1 might serve as a mechanism for the increased expression of Nrf2 during tumorigenesis and progression (155). In addition, overexpression of Nrf2 in colon cancer cells could promote colon cancer progression through ERK and AKT signaling pathways (156).
7 The therapeutic potential of modulation of the Keap1-Nrf2 pathway in UC
Nrf2, a crucial transcription factor responsible for regulating cellular defense mechanisms, is intricately associated with the progression of UC, intestinal fibrosis, and CRC. Activation of Nrf2 activity is an effective therapeutic modality against oxidative stress-related diseases (8, 157). Recent studies have demonstrated both sides of Nrf2 in treating malignant tumors. It has been demonstrated through previous studies that the excessive activation of Nrf2 contributes significantly to malignant tumor transformation, treatment resistance, and unfavorable clinical outcomes. Inhibition of over-activated Nrf2 activity in tumor cells can exert anti-tumor effects by disrupting redox homeostasis, antagonizing tumor metabolism, and reversing drug resistance in various ways (15, 158, 159). Therefore, the study of the role of Nrf2 and the molecular mechanism of its activity is becoming a new hot topic.
The literature shows that many medicinal plants and phytochemicals, synthetic chemicals or inducers, and others, such as metformin and short-chain fatty acids, modulate the effects of Nrf2 on UC and UC-associated colorectal cancer by activating Nrf2-mediated antioxidant expression and attenuating NF-κB-associated inflammation (Table 1). Studies have shown that Nrf2 inhibits NF-κB, the most important mediator of UC inflammation, through several cellular and biochemical mechanisms (68, 160, 161). At its peak expression, Nrf2 attenuates the activity of NF-κB primarily by hampering the generation of ROS. The activation of Nrf2 reduces ROS levels, consequently inhibiting the production of NF-κB-dependent pro-inflammatory factors activation mediated by ROS (68). The maintenance of stable Nrf2 activation levels during the initial stages of UC is likely to enhance the intestinal environment, bolster the mucosal barrier, prevent the disruption of the colon, reduce ulceration and microbial metastasis, ultimately suppressing the disease activity index (DAI), restraining the progression of UC, and mitigating the likelihood of subsequent complications (162–164). Moreover, Nrf2 signaling modulates a multitude of genes implicated in redox regulation, protein degradation, DNA repair, xenobiotic metabolism, and apoptosis, which collectively impede the development of colorectal cancer associated with ulcerative colitis (55, 165, 166). Research indicates that in the early stages of various inflammatory disorders, maintaining constant levels of Nrf2 activators can inhibit progression, thereby preventing complications like fibrosis and cancer. However, in advanced cancers wherein Nrf2 expression is elevated, Nrf2 inhibitors may serve as efficacious therapeutic adjuvants that can significantly reduce radiotherapy resistance (3, 126). Nevertheless, further investigations are essential to illuminate the intricate role of Nrf2, along with developing novel drugs capable of modulating the Nrf2 pathway and potentiating its defensive effects.
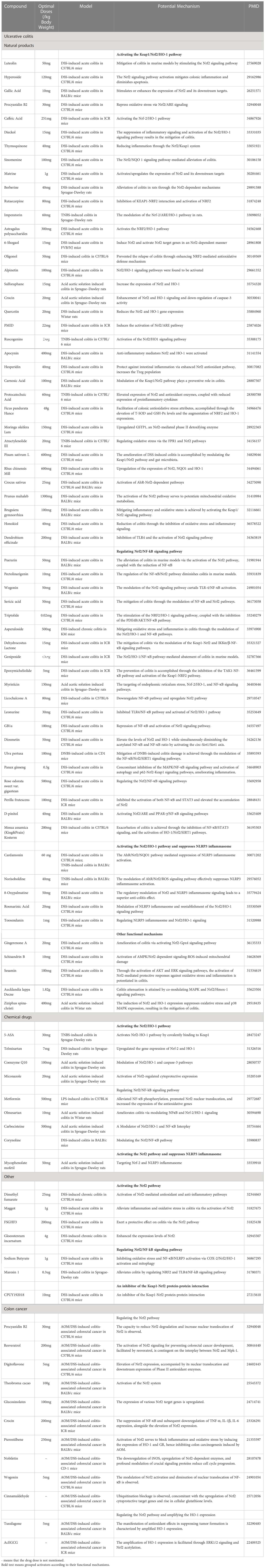
Table 1 Studies the therapeutic impact of controlling the Nrf2 signaling pathway using medication on UC.
8 Conclusions and perspective
The Keap1/Nrf2 axis significantly influences the healthy development and maintenance of the gastrointestinal tract’s normal function. Given its significant association with UC and its dire complications, the Keap1/Nrf2 axis is a potential therapeutic target for preventing such ailments. In this review, we describe the structure and function of Nrf2 and its role in intestinal development while we elucidate the critical role of Nrf2 in UC and its complications. First, Nrf2 can alleviate intestinal injury and inflammation by controlling oxidative stress. Secondly, Nrf2 activation modulates the expression of tight junction proteins, consequently supporting and fortifying the integrity of the intestinal epithelial barrier. Thirdly, Nrf2 activation regulates intestinal immunity to varying degrees and influences the development of UC. Finally, Nrf2 is closely associated with intestinal fibrosis and colorectal cancer that occur in UC. In summary, comprehending the intricacies underlying the structure and functionality of Nrf2, developing drugs that target the molecule effectively, and meticulously regulating its activity at different stages of the disease progression could offer new and innovative hypotheses to tackle ulcerative colitis management.
Author contributions
SP and LS wrote the draft of the manuscript. SP drew the figures. XY, LZ, KX, YX, LLZ, and JW contributed to the literature search and discussion. LS and HL designed and revised this manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
The Figures in our article were drawn by Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Krugliak CN, Torres J, Rubin DT. What does disease progression look like in ulcerative colitis, and how might it be prevented? Gastroenterology (2022) 162:1396–408. doi: 10.1053/j.gastro.2022.01.023
2. Kobayashi T, Siegmund B, Le Berre C, Wei SC, Ferrante M, Shen B, et al. Ulcerative colitis. Nat Rev Dis Primers (2020) 6:74. doi: 10.1038/s41572-020-0205-x
3. Piotrowska M, Swierczynski M, Fichna J, Piechota-Polanczyk A. The Nrf2 in the pathophysiology of the intestine: molecular mechanisms and therapeutic implications for inflammatory bowel diseases. Pharmacol Res (2021) 163:105243. doi: 10.1016/j.phrs.2020.105243
4. Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discovery (2021) 20:689–709. doi: 10.1038/s41573-021-00233-1
5. He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci (2020) 21(13):4777. doi: 10.3390/ijms21134777
6. Kopacz A, Kloska D, Forman HJ, Jozkowicz A, Grochot-Przeczek A. Beyond repression of Nrf2: an update on Keap1. Free Radic Biol Med (2020) 157:63–74. doi: 10.1016/j.freeradbiomed.2020.03.023
7. Crisman E, Duarte P, Dauden E, Cuadrado A, Rodriguez-Franco MI, Lopez MG, et al. KEAP1-NRF2 protein-protein interaction inhibitors: design, pharmacological properties and therapeutic potential. Med Res Rev (2023) 43:237–87. doi: 10.1002/med.21925
8. Kasai S, Shimizu S, Tatara Y, Mimura J, Itoh K. Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules (2020) 10(2):320. doi: 10.3390/biom10020320
9. Baird L, Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol (2020) 40(13):e00099-20. doi: 10.1128/MCB.00099-20
10. Yu C, Xiao JH. The Keap1-Nrf2 system: a mediator between oxidative stress and aging. Oxid Med Cell Longev (2021) 2021:6635460. doi: 10.1155/2021/6635460
11. Ulasov AV, Rosenkranz AA, Georgiev GP, Sobolev AS. Nrf2/Keap1/ARE signaling: towards specific regulation. Life Sci (2022) 291:120111. doi: 10.1016/j.lfs.2021.120111
12. Liu S, Pi J, Zhang Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol (2022) 54:102389. doi: 10.1016/j.redox.2022.102389
13. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res (2018) 1865:721–33. doi: 10.1016/j.bbamcr.2018.02.010
14. Komatsu M. p62 bodies: phase separation, NRF2 activation, and selective autophagic degradation. IUBMB Life (2022) 74:1200–08. doi: 10.1002/iub.2689
15. Jiang T, Harder B, Rojo DLVM, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med (2015) 88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014
16. Sanchez-Martin P, Saito T, Komatsu M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J (2019) 286:8–23. doi: 10.1111/febs.14712
17. Sanchez-Martin P, Komatsu M. p62/SQSTM1 - steering the cell through health and disease. J Cell Sci (2018) 131(21):jcs222836. doi: 10.1242/jcs.222836
18. Silva-Islas CA, Maldonado PD. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol Res (2018) 134:92–9. doi: 10.1016/j.phrs.2018.06.013
19. Lin M, Zhai X, Wang G, Tian X, Gao D, Shi L, et al. Salvianolic acid b protects against acetaminophen hepatotoxicity by inducing Nrf2 and phase II detoxification gene expression via activation of the PI3K and PKC signaling pathways. J Pharmacol Sci (2015) 127:203–10. doi: 10.1016/j.jphs.2014.12.010
20. Guo C, Wang S, Duan J, Jia N, Zhu Y, Ding Y, et al. Protocatechualdehyde protects against cerebral ischemia-Reperfusion-Induced oxidative injury via protein kinase Cepsilon/Nrf2/HO-1 pathway. Mol Neurobiol (2017) 54:833–45. doi: 10.1007/s12035-016-9690-z
21. Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at ser-40 by protein kinase c regulates antioxidant response element-mediated transcription. J Biol Chem (2002) 277:42769–74. doi: 10.1074/jbc.M206911200
22. Lee KM, Kang K, Lee SB, Nho CW. Nuclear factor-E2 (Nrf2) is regulated through the differential activation of ERK1/2 and PKC alpha/betaII by gymnasterkoreayne b. Cancer Lett (2013) 330:225–32. doi: 10.1016/j.canlet.2012.11.053
23. Chen L, Ran Q, Xiang Y, Xiang L, Chen L, Li F, et al. Co-Activation of PKC-delta by CRIF1 modulates oxidative stress in bone marrow multipotent mesenchymal stromal cells after irradiation by phosphorylating NRF2 Ser40. Theranostics (2017) 7:2634–48. doi: 10.7150/thno.17853
24. Zimmermann K, Baldinger J, Mayerhofer B, Atanasov AG, Dirsch VM, Heiss EH. Activated AMPK boosts the Nrf2/HO-1 signaling axis–a role for the unfolded protein response. Free Radic Biol Med (2015) 88:417–26. doi: 10.1016/j.freeradbiomed.2015.03.030
25. Liu XM, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. . doi: 10.1152/ajpheart.00749.2010
26. Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol (2016) 36:1931–42. doi: 10.1128/MCB.00118-16
27. Hu Y, Duan M, Liang S, Wang Y, Feng Y. Senkyunolide I protects rat brain against focal cerebral ischemia-reperfusion injury by up-regulating p-Erk1/2, Nrf2/HO-1 and inhibiting caspase 3. Brain Res (2015) 1605:39–48. doi: 10.1016/j.brainres.2015.02.015
28. Wang XL, Xing GH, Hong B, Li XM, Zou Y, Zhang XJ, et al. Gastrodin prevents motor deficits and oxidative stress in the MPTP mouse model of parkinson’s disease: involvement of ERK1/2-Nrf2 signaling pathway. Life Sci (2014) 114:77–85. doi: 10.1016/j.lfs.2014.08.004
29. Vari R, D’Archivio M, Filesi C, Carotenuto S, Scazzocchio B, Santangelo C, et al. Protocatechuic acid induces antioxidant/detoxifying enzyme expression through JNK-mediated Nrf2 activation in murine macrophages. J Nutr Biochem (2011) 22:409–17. doi: 10.1016/j.jnutbio.2010.03.008
30. Ma L, Liu J, Zhang X, Qi J, Yu W, Gu Y. p38 MAPK-dependent Nrf2 induction enhances the resistance of glioma cells against TMZ. Med Oncol (2015) 32:69. doi: 10.1007/s12032-015-0517-y
31. Jiang XY, Zhu XS, Xu HY, Zhao ZX, Li SY, Li SZ, et al. Diallyl trisulfide suppresses tumor growth through the attenuation of Nrf2/Akt and activation of p38/JNK and potentiates cisplatin efficacy in gastric cancer treatment. Acta Pharmacol Sin (2017) 38:1048–58. doi: 10.1038/aps.2016.176
32. Qi H, Han Y, Rong J. Potential roles of PI3K/Akt and Nrf2-Keap1 pathways in regulating hormesis of z-ligustilide in PC12 cells against oxygen and glucose deprivation. Neuropharmacology (2012) 62:1659–70. doi: 10.1016/j.neuropharm.2011.11.012
33. Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY, Shim JH, et al. Reactive oxygen species and PI3K/Akt signaling play key roles in the induction of Nrf2-driven heme oxygenase-1 expression in sulforaphane-treated human mesothelioma MSTO-211H cells. Food Chem Toxicol (2012) 50:116–23. doi: 10.1016/j.fct.2011.10.035
34. Kopacz A, Kloska D, Klimczyk D, Kopec M, Jozkowicz A, Piechota-Polanczyk A. Nrf2 transcriptional activity governs intestine development. Int J Mol Sci (2022) 23(11):6175. doi: 10.3390/ijms23116175
35. Noah TK, Donahue B, Shroyer NF. Intestinal development and differentiation. Exp Cell Res (2011) 317:2702–10. doi: 10.1016/j.yexcr.2011.09.006
36. Fazilaty H, Brugger MD, Valenta T, Szczerba BM, Berkova L, Doumpas N, et al. Tracing colonic embryonic transcriptional profiles and their reactivation upon intestinal damage. Cell Rep (2021) 36:109484. doi: 10.1016/j.celrep.2021.109484
37. Tian H, Biehs B, Chiu C, Siebel CW, Wu Y, Costa M, et al. Opposing activities of notch and wnt signaling regulate intestinal stem cells and gut homeostasis. Cell Rep (2015) 11:33–42. doi: 10.1016/j.celrep.2015.03.007
38. Koch S. Extrinsic control of wnt signaling in the intestine. Differentiation (2017) 97:1–08. doi: 10.1016/j.diff.2017.08.003
39. Yagishita Y, McCallum ML, Kensler TW, Wakabayashi N. Constitutive activation of Nrf2 in mice expands enterogenesis in small intestine through negative regulation of Math1. Cell Mol Gastroenterol Hepatol (2021) 11:503–24. doi: 10.1016/j.jcmgh.2020.08.013
40. Wakabayashi N, Chartoumpekis DV, Kensler TW. Crosstalk between Nrf2 and notch signaling. Free Radic Biol Med (2015) 88:158–67. doi: 10.1016/j.freeradbiomed.2015.05.017
41. Long MJ, Lin HY, Parvez S, Zhao Y, Poganik JR, Huang P, et al. Beta-TrCP1 is a vacillatory regulator of wnt signaling. Cell Chem Biol (2017) 24:944–57. doi: 10.1016/j.chembiol.2017.06.009
42. Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S (1996) 93:13943–48. doi: 10.1073/pnas.93.24.13943
43. Hou Q, Huang J, Ayansola H, Masatoshi H, Zhang B. Intestinal stem cells and immune cell relationships: potential therapeutic targets for inflammatory bowel diseases. Front Immunol (2020) 11:623691. doi: 10.3389/fimmu.2020.623691
44. Bourgonje AR, Kloska D, Grochot-Przeczek A, Feelisch M, Cuadrado A, van Goor H. Personalized redox medicine in inflammatory bowel diseases: an emerging role for HIF-1alpha and NRF2 as therapeutic targets. Redox Biol (2023) 60:102603. doi: 10.1016/j.redox.2023.102603
45. McGarry T, Biniecka M, Veale DJ, Fearon U. Hypoxia, oxidative stress and inflammation. Free Radic Biol Med (2018) 125:15–24. doi: 10.1016/j.freeradbiomed.2018.03.042
46. Dziabowska-Grabias K, Sztanke M, Zajac P, Celejewski M, Kurek K, Szkutnicki S, et al. Antioxidant therapy in inflammatory bowel diseases. Antioxidants (Basel) (2021) 10(3):412. doi: 10.3390/antiox10030412
47. Jena G, Trivedi PP, Sandala B. Oxidative stress in ulcerative colitis: an old concept but a new concern. Free Radic Res (2012) 46:1357–73. doi: 10.3109/10715762.2012.717692
48. Feuerstein JD, Moss AC, Farraye FA. Ulcerative Colitis. Mayo Clin Proc (2019) 94:1357–73. doi: 10.1016/j.mayocp.2019.01.018
49. Kim YJ, Kim EH, Hahm KB. Oxidative stress in inflammation-based gastrointestinal tract diseases: challenges and opportunities. J Gastroenterol Hepatol (2012) 27:1004–10. doi: 10.1111/j.1440-1746.2012.07108.x
50. Arisawa T, Tahara T, Shibata T, Nagasaka M, Nakamura M, Kamiya Y, et al. Nrf2 gene promoter polymorphism is associated with ulcerative colitis in a Japanese population. Hepatogastroenterology (2008) 55:394–97.
51. Sabzevary-Ghahfarokhi M, Shohan M, Shirzad H, Rahimian G, Soltani A, Ghatreh-Samani M, et al. The regulatory role of Nrf2 in antioxidants phase2 enzymes and IL-17A expression in patients with ulcerative colitis. Pathol Res Pract (2018) 214:1149–55. doi: 10.1016/j.prp.2018.06.001
52. Myers JN, Schaffer MW, Korolkova OY, Williams AD, Gangula PR, M’Koma AE. Implications of the colonic deposition of free hemoglobin-alpha chain: a previously unknown tissue by-product in inflammatory bowel disease. Inflammation Bowel Dis (2014) 20:1530–47. doi: 10.1097/MIB.0000000000000144
53. Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res (2006) 66:11580–84. doi: 10.1158/0008-5472.CAN-06-3562
54. Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila) (2008) 1:187–91. doi: 10.1158/1940-6207.CAPR-08-0028
55. Hammad A, Zheng ZH, Gao Y, Namani A, Shi HF, Tang X. Identification of novel Nrf2 target genes as prognostic biomarkers in colitis-associated colorectal cancer in Nrf2-deficient mice. Life Sci (2019) 238:116968. doi: 10.1016/j.lfs.2019.116968
56. Osburn WO, Karim B, Dolan PM, Liu G, Yamamoto M, Huso DL, et al. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer (2007) 121:1883–91. doi: 10.1002/ijc.22943
57. Zhang Y, Yan T, Sun D, Xie C, Wang T, Liu X, et al. Rutaecarpine inhibits KEAP1-NRF2 interaction to activate NRF2 and ameliorate dextran sulfate sodium-induced colitis. Free Radic Biol Med (2020) 148:33–41. doi: 10.1016/j.freeradbiomed.2019.12.012
58. Bai X, Gou X, Cai P, Xu C, Cao L, Zhao Z, et al. Sesamin enhances Nrf2-mediated protective defense against oxidative stress and inflammation in colitis via AKT and ERK activation. Oxid Med Cell Longev (2019) 2019:2432416. doi: 10.1155/2019/2432416
59. Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun (2019) 10:89. doi: 10.1038/s41467-018-07859-7
60. Sabzevary-Ghahfarokhi M, Soltani A, Luzza F, Larussa T, Rahimian G, Shirzad H, et al. The protective effects of resveratrol on ulcerative colitis via changing the profile of Nrf2 and IL-1beta protein. Mol Biol Rep (2020) 47:6941–47. doi: 10.1007/s11033-020-05753-4
61. Tonelli C, Chio I, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal (2018) 29:1727–45. doi: 10.1089/ars.2017.7342
62. Perez S, Talens-Visconti R, Rius-Perez S, Finamor I, Sastre J. Redox signaling in the gastrointestinal tract. Free Radic Biol Med (2017) 104:75–103. doi: 10.1016/j.freeradbiomed.2016.12.048
63. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ (2015) 22:377–88. doi: 10.1038/cdd.2014.150
64. Chen GH, Song CC, Pantopoulos K, Wei XL, Zheng H, Luo Z. Mitochondrial oxidative stress mediated fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol Med (2022) 180:95–107. doi: 10.1016/j.freeradbiomed.2022.01.012
65. Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med (2011) 32:234–46. doi: 10.1016/j.mam.2011.10.006
66. Sabzevary-Ghahfarokhi M, Shohan M, Shirzad H, Rahimian G, Bagheri N, Soltani A, et al. The expression analysis of fra-1 gene and IL-11 protein in Iranian patients with ulcerative colitis. BMC Immunol (2018) 19:17. doi: 10.1186/s12865-018-0257-9
67. Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis (2017) 1863:585–97. doi: 10.1016/j.bbadis.2016.11.005
68. Wardyn JD, Ponsford AH, Sanderson CM. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem Soc Trans (2015) 43:621–26. doi: 10.1042/BST20150014
69. Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta (2008) 1783:713–27. doi: 10.1016/j.bbamcr.2008.01.002
70. Hwang YJ, Lee EW, Song J, Kim HR, Jun YC, Hwang KA. MafK positively regulates NF-kappaB activity by enhancing CBP-mediated p65 acetylation. Sci Rep (2013) 3:3242. doi: 10.1038/srep03242
71. Gerstgrasser A, Melhem H, Leonardi I, Atrott K, Schafer M, Werner S, et al. Cell-specific activation of the Nrf2 antioxidant pathway increases mucosal inflammation in acute but not in chronic colitis. J Crohns Colitis (2017) 11:485–99. doi: 10.1093/ecco-jcc/jjw172
72. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med (2018) 50:1–09. doi: 10.1038/s12276-018-0126-x
73. Funk MC, Zhou J, Boutros M. Ageing, metabolism and the intestine. EMBO Rep (2020) 21:e50047. doi: 10.15252/embr.202050047
74. Breugelmans T, Oosterlinck B, Arras W, Ceuleers H, De Man J, Hold GL, et al. The role of mucins in gastrointestinal barrier function during health and disease. Lancet Gastroenterol Hepatol (2022) 7:455–71. doi: 10.1016/S2468-1253(21)00431-3
75. Di Tommaso N, Gasbarrini A, Ponziani FR. Intestinal barrier in human health and disease. Int J Environ Res Public Health (2021) 18(23):12836. doi: 10.3390/ijerph182312836
76. Martens EC, Neumann M, Desai MS. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat Rev Microbiol (2018) 16:457–70. doi: 10.1038/s41579-018-0036-x
77. Dokladny K, Zuhl MN, Moseley PL. Intestinal epithelial barrier function and tight junction proteins with heat and exercise. J Appl Physiol (1985) (2016) 120:692–701. doi: 10.1152/japplphysiol.00536.2015
78. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14:141–53. doi: 10.1038/nri3608
79. Buckley A, Turner JR. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb Perspect Biol (2018) 10(1):a029314. doi: 10.1101/cshperspect.a029314
80. Mehandru S, Colombel JF. The intestinal barrier, an arbitrator turned provocateur in IBD. Nat Rev Gastroenterol Hepatol (2021) 18:83–4. doi: 10.1038/s41575-020-00399-w
81. Chen Y, Cui W, Li X, Yang H. Interaction between commensal bacteria, immune response and the intestinal barrier in inflammatory bowel disease. Front Immunol (2021) 12:761981. doi: 10.3389/fimmu.2021.761981
82. Kayama H, Okumura R, Takeda K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu Rev Immunol (2020) 38:23–48. doi: 10.1146/annurev-immunol-070119-115104
83. Salim SY, Soderholm JD. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflammation Bowel Dis (2011) 17:362–81. doi: 10.1002/ibd.21403
84. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol (2017) 4:33–46. doi: 10.1016/j.jcmgh.2017.03.007
85. Wang L, Wang J. Honokiol ameliorates DSS-induced mouse colitis by inhibiting inflammation and oxidative stress and improving the intestinal barrier. Oxid Med Cell Longev (2022) 2022:1755608. doi: 10.1155/2022/1755608
86. Yuan Z, Yang L, Zhang X, Ji P, Hua Y, Wei Y. Huang-Lian-Jie-Du decoction ameliorates acute ulcerative colitis in mice via regulating NF-kappaB and Nrf2 signaling pathways and enhancing intestinal barrier function. Front Pharmacol (2019) 10:1354. doi: 10.3389/fphar.2019.01354
87. Wang YJ, Li QM, Zha XQ, Luo JP. Dendrobium fimbriatum hook polysaccharide ameliorates dextran-sodium-sulfate-induced colitis in mice via improving intestinal barrier function, modulating intestinal microbiota, and reducing oxidative stress and inflammatory responses. Food Funct (2022) 13:143–60. doi: 10.1039/d1fo03003e
88. Guo K, Ren J, Gu G, Wang G, Gong W, Wu X, et al. Hesperidin protects against intestinal inflammation by restoring intestinal barrier function and up-regulating treg cells. Mol Nutr Food Res (2019) 63:e1800975. doi: 10.1002/mnfr.201800975
89. Wu T, Wang X, Xiong H, Deng Z, Peng X, Xiao L, et al. Bioactives and their metabolites from tetrastigma hemsleyanum leaves ameliorate DSS-induced colitis via protecting the intestinal barrier, mitigating oxidative stress and regulating the gut microbiota. Food Funct (2021) 12:11760–76. doi: 10.1039/d1fo02588k
90. Zhang S, Zhou Q, Li Y, Zhang Y, Wu Y. MitoQ modulates lipopolysaccharide-induced intestinal barrier dysfunction via regulating Nrf2 signaling. Mediators Inflammation (2020) 2020:3276148. doi: 10.1155/2020/3276148
91. Jin W, Wang H, Ji Y, Hu Q, Yan W, Chen G, et al. Increased intestinal inflammatory response and gut barrier dysfunction in Nrf2-deficient mice after traumatic brain injury. Cytokine (2008) 44:135–40. doi: 10.1016/j.cyto.2008.07.005
92. Liu Y, Bao Z, Xu X, Chao H, Lin C, Li Z, et al. Extracellular signal-regulated Kinase/Nuclear factor-Erythroid2-like2/Heme oxygenase-1 pathway-mediated mitophagy alleviates traumatic brain injury-induced intestinal mucosa damage and epithelial barrier dysfunction. J Neurotrauma (2017) 34:2119–31. doi: 10.1089/neu.2016.4764
93. Chen H, Hu Y, Fang Y, Djukic Z, Yamamoto M, Shaheen NJ, et al. Nrf2 deficiency impairs the barrier function of mouse oesophageal epithelium. Gut (2014) 63:711–19. doi: 10.1136/gutjnl-2012-303731
94. Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in drosophila. Cell Stem Cell (2011) 8:188–99. doi: 10.1016/j.stem.2010.12.006
95. Chi X, Yao W, Xia H, Jin Y, Li X, Cai J, et al. Elevation of HO-1 expression mitigates intestinal ischemia-reperfusion injury and restores tight junction function in a rat liver transplantation model. Oxid Med Cell Longev (2015) 2015:986075. doi: 10.1155/2015/986075
96. Pelaseyed T, Bergstrom JH, Gustafsson JK, Ermund A, Birchenough GM, Schutte A, et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev (2014) 260:8–20. doi: 10.1111/imr.12182
97. Petersson J, Schreiber O, Hansson GC, Gendler SJ, Velcich A, Lundberg JO, et al. . doi: 10.1152/ajpgi.00422.2010
98. Lee D, Albenberg L, Compher C, Baldassano R, Piccoli D, Lewis JD, et al. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology (2015) 148:1087–106. doi: 10.1053/j.gastro.2015.01.007
99. Wen ZS, Du M, Tang Z, Zhou TY, Zhang ZS, Song HH, et al. Low molecular seleno-aminopolysaccharides protect the intestinal mucosal barrier of rats under weaning stress. Int J Mol Sci (2019) 20(22):5727. doi: 10.3390/ijms20225727
100. Saez A, Gomez-Bris R, Herrero-Fernandez B, Mingorance C, Rius C, Gonzalez-Granado JM. Innate lymphoid cells in intestinal homeostasis and inflammatory bowel disease. Int J Mol Sci (2021) 22(14):7618. doi: 10.3390/ijms22147618
101. Zou J, Liu C, Jiang S, Qian D, Duan J. Cross talk between gut microbiota and intestinal mucosal immunity in the development of ulcerative colitis. Infect Immun (2021) 89:e1421. doi: 10.1128/IAI.00014-21
102. Rockwell CE, Zhang M, Fields PE, Klaassen CD. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J Immunol (2012) 188:1630–37. doi: 10.4049/jimmunol.1101712
103. Turley AE, Zagorski JW, Rockwell CE. The Nrf2 activator tBHQ inhibits T cell activation of primary human CD4 T cells. Cytokine (2015) 71:289–95. doi: 10.1016/j.cyto.2014.11.006
104. Zagorski JW, Turley AE, Freeborn RA, VanDenBerg KR, Dover HE, Kardell BR, et al. Differential effects of the Nrf2 activators tBHQ and CDDO-im on the early events of T cell activation. Biochem Pharmacol (2018) 147:67–76. doi: 10.1016/j.bcp.2017.11.005
105. Deng S, Wu D, Li L, Li J, Xu Y. TBHQ attenuates ferroptosis against 5-fluorouracil-induced intestinal epithelial cell injury and intestinal mucositis via activation of Nrf2. Cell Mol Biol Lett (2021) 26:48. doi: 10.1186/s11658-021-00294-5
106. Bursley JK, Rockwell CE. Nrf2-dependent and -independent effects of tBHQ in activated murine b cells. Food Chem Toxicol (2020) 145:111595. doi: 10.1016/j.fct.2020.111595
107. Lin X, Tawch S, Wong HT, Roy S, Gaudino S, Castillo P, et al. Nrf2 through aryl hydrocarbon receptor regulates IL-22 response in CD4(+) T cells. J Immunol (2021) 206:1540–48. doi: 10.4049/jimmunol.1900656
108. Casili G, Cordaro M, Impellizzeri D, Bruschetta G, Paterniti I, Cuzzocrea S, et al. Dimethyl fumarate reduces inflammatory responses in experimental colitis. J Crohns Colitis (2016) 10:472–83. doi: 10.1093/ecco-jcc/jjv231
109. Han J, Ma S, Gong H, Liu S, Lei L, Hu B, et al. Inhibition of acute graft-versus-Host disease with retention of graft-versus-Tumor effects by dimethyl fumarate. Front Immunol (2017) 8:1605. doi: 10.3389/fimmu.2017.01605
110. Wang J, Lin S, Brown JM, van Wagoner D, Fiocchi C, Rieder F. Novel mechanisms and clinical trial endpoints in intestinal fibrosis. Immunol Rev (2021) 302:211–27. doi: 10.1111/imr.12974
111. Derkacz A, Olczyk P, Olczyk K, Komosinska-Vassev K. The role of extracellular matrix components in inflammatory bowel diseases. J Clin Med (2021) 10(5):1122. doi: 10.3390/jcm10051122
112. D’Alessio S, Ungaro F, Noviello D, Lovisa S, Peyrin-Biroulet L, Danese S. Revisiting fibrosis in inflammatory bowel disease: the gut thickens. Nat Rev Gastroenterol Hepatol (2022) 19:169–84. doi: 10.1038/s41575-021-00543-0
113. Latella G. Redox imbalance in intestinal fibrosis: beware of the TGFbeta-1, ROS, and Nrf2 connection. Dig Dis Sci (2018) 63:312–20. doi: 10.1007/s10620-017-4887-1
114. Guan Y, Tan Y, Liu W, Yang J, Wang D, Pan D, et al. NF-E2-Related factor 2 suppresses intestinal fibrosis by inhibiting reactive oxygen species-dependent TGF-beta1/SMADs pathway. Dig Dis Sci (2018) 63:366–80. doi: 10.1007/s10620-017-4710-z
115. Wang H, Wang B, Wei J, Zheng Z, Su J, Bian C, et al. Sulforaphane regulates Nrf2-mediated antioxidant activity and downregulates TGF-beta1/Smad pathways to prevent radiation-induced muscle fibrosis. Life Sci (2022) 311:121197. doi: 10.1016/j.lfs.2022.121197
116. Wang R, Wang D, Wang H, Wang T, Weng Y, Zhang Y, et al. Therapeutic targeting of Nrf2 signaling by maggot extracts ameliorates inflammation-associated intestinal fibrosis in chronic DSS-induced colitis. Front Immunol (2021) 12:670159. doi: 10.3389/fimmu.2021.670159
117. Cui Y, Xin H, Tao Y, Mei L, Wang Z. Arenaria kansuensis attenuates pulmonary fibrosis in mice via the activation of Nrf2 pathway and the inhibition of NF-kB/TGF-beta1/Smad2/3 pathway. Phytother Res (2021) 35:974–86. doi: 10.1002/ptr.6857
118. Sedda S, Marafini I, Dinallo V, Di Fusco D, Monteleone G. The TGF-beta/Smad system in IBD pathogenesis. Inflammation Bowel Dis (2015) 21:2921–25. doi: 10.1097/MIB.0000000000000542
119. Hu HH, Chen DQ, Wang YN, Feng YL, Cao G, Vaziri ND, et al. New insights into TGF-beta/Smad signaling in tissue fibrosis. Chem Biol Interact (2018) 292:76–83. doi: 10.1016/j.cbi.2018.07.008
120. Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol (2015) 6:565–77. doi: 10.1016/j.redox.2015.09.009
121. Wang Y, Wang Z, Yang H, Chen S, Zheng D, Liu X, et al. Metformin ameliorates chronic colitis-related intestinal fibrosis via inhibiting TGF-beta1/Smad3 signaling. Front Pharmacol (2022) 13:887497. doi: 10.3389/fphar.2022.887497
122. Laudadio I, Bastianelli A, Fulci V, Carissimi C, Colantoni E, Palone F, et al. ZNF281 promotes colon fibroblast activation in TGFbeta1-induced gut fibrosis. Int J Mol Sci (2022) 23(18):10261. doi: 10.3390/ijms231810261
123. Tan HH, Thomas NF, Inayat-Hussain SH, Chan KM. (E)-N-(2-(3, 5-dimethoxystyryl) phenyl) furan-2-carboxamide (BK3C231) induces cytoprotection in CCD18-Co human colon fibroblast cells through Nrf2/ARE pathway activation. Sci Rep (2021) 11:4773. doi: 10.1038/s41598-021-83163-7
124. Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech (2014) 7:193–203. doi: 10.1242/dmm.012062
125. Robert S, Gicquel T, Victoni T, Valenca S, Barreto E, Bailly-Maitre B, et al. Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci Rep (2016) 36(4):e00360. doi: 10.1042/BSR20160107
126. Pompili S, Sferra R, Gaudio E, Viscido A, Frieri G, Vetuschi A, et al. Can Nrf2 modulate the development of intestinal fibrosis and cancer in inflammatory bowel disease? Int J Mol Sci (2019) 20(16):4061. doi: 10.3390/ijms20164061
127. Silosi I, Boldeanu MV, Mogoanta SS, Ghilusi M, Cojocaru M, Biciusca V, et al. Matrix metalloproteinases (MMP-3 and MMP-9) implication in the pathogenesis of inflammatory bowel disease (IBD). Rom J Morphol Embryol (2014) 55:1317–24.
128. Biancheri P, Brezski RJ, Di Sabatino A, Greenplate AR, Soring KL, Corazza GR, et al. Proteolytic cleavage and loss of function of biologic agents that neutralize tumor necrosis factor in the mucosa of patients with inflammatory bowel disease. Gastroenterology (2015) 149:1564–74. doi: 10.1053/j.gastro.2015.07.002
129. Barberio B, D’Inca R, Facchin S, Dalla GM, Fohom TC, Cardin R, et al. Matrix metalloproteinase 3 predicts therapeutic response in inflammatory bowel disease patients treated with infliximab. Inflammation Bowel Dis (2020) 26:756–63. doi: 10.1093/ibd/izz195
130. Kofla-Dlubacz A, Matusiewicz M, Krzesiek E, Noga L, Iwanczak B. Metalloproteinase-3 and -9 as novel markers in the evaluation of ulcerative colitis activity in children. Adv Clin Exp Med (2014) 23:103–10. doi: 10.17219/acem/37031
131. Giuffrida P, Biancheri P, MacDonald TT. Proteases and small intestinal barrier function in health and disease. Curr Opin Gastroenterol (2014) 30:147–53. doi: 10.1097/MOG.0000000000000042
132. Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology (2022) 162:715–30. doi: 10.1053/j.gastro.2021.10.035
133. Trivedi PP, Jena GB, Tikoo KB, Kumar V. Melatonin modulated autophagy and Nrf2 signaling pathways in mice with colitis-associated colon carcinogenesis. Mol Carcinog (2016) 55:255–67. doi: 10.1002/mc.22274
134. Lu MC, Ji JA, Jiang YL, Chen ZY, Yuan ZW, You QD, et al. An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis. Sci Rep (2016) 6:26585. doi: 10.1038/srep26585
135. Yang Y, Cai X, Yang J, Sun X, Hu C, Yan Z, et al. Chemoprevention of dietary digitoflavone on colitis-associated colon tumorigenesis through inducing Nrf2 signaling pathway and inhibition of inflammation. Mol Cancer (2014) 13:48. doi: 10.1186/1476-4598-13-48
136. Cheung KL, Lee JH, Khor TO, Wu TY, Li GX, Chan J, et al. Nrf2 knockout enhances intestinal tumorigenesis in apc(min/+) mice due to attenuation of anti-oxidative stress pathway while potentiates inflammation. Mol Carcinog (2014) 53:77–84. doi: 10.1002/mc.21950
137. Yokoo Y, Kijima A, Ishii Y, Takasu S, Tsuchiya T, Umemura T. Effects of Nrf2 silencing on oxidative stress-associated intestinal carcinogenesis in mice. Cancer Med (2016) 5:1228–38. doi: 10.1002/cam4.672
138. Stachel I, Geismann C, Aden K, Deisinger F, Rosenstiel P, Schreiber S, et al. Modulation of nuclear factor E2-related factor-2 (Nrf2) activation by the stress response gene immediate early response-3 (IER3) in colonic epithelial cells: a novel mechanism of cellular adaption to inflammatory stress. J Biol Chem (2014) 289:1917–29. doi: 10.1074/jbc.M113.490920
139. Arlt A, Bauer I, Schafmayer C, Tepel J, Muerkoster SS, Brosch M, et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene (2009) 28:3983–96. doi: 10.1038/onc.2009.264
140. Kruse ML, Friedrich M, Arlt A, Rocken C, Egberts JH, Sebens S, et al. Colonic lamina propria inflammatory cells from patients with IBD induce the nuclear factor-E2 related factor-2 thereby leading to greater proteasome activity and apoptosis protection in human colonocytes. Inflammation Bowel Dis (2016) 22:2593–606. doi: 10.1097/MIB.0000000000000925
141. Sebens S, Bauer I, Geismann C, Grage-Griebenow E, Ehlers S, Kruse ML, et al. Inflammatory macrophages induce Nrf2 transcription factor-dependent proteasome activity in colonic NCM460 cells and thereby confer anti-apoptotic protection. J Biol Chem (2011) 286:40911–21. doi: 10.1074/jbc.M111.274902
142. Ji L, Wei Y, Jiang T, Wang S. Correlation of Nrf2, NQO1, MRP1, cmyc and p53 in colorectal cancer and their relationships to clinicopathologic features and survival. Int J Clin Exp Pathol (2014) 7:1124–31.
143. Lopez A, Harada K, Vasilakopoulou M, Shanbhag N, Ajani JA. Targeting angiogenesis in colorectal carcinoma. Drugs (2019) 79:63–74. doi: 10.1007/s40265-018-1037-9
144. Kim TH, Hur EG, Kang SJ, Kim JA, Thapa D, Lee YM, et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res (2011) 71:2260–75. doi: 10.1158/0008-5472.CAN-10-3007
145. Zhao M, Yu Z, Li Z, Tang J, Lai X, Liu L. Expression of angiogenic growth factors VEGF, bFGF and ANG1 in colon cancer after bevacizumab treatment in vitro: a potential self-regulating mechanism. Oncol Rep (2017) 37:601–07. doi: 10.3892/or.2016.5231
146. Mirzaei S, Zarrabi A, Hashemi F, Zabolian A, Saleki H, Azami N, et al. Nrf2 signaling pathway in chemoprotection and doxorubicin resistance: potential application in drug discovery. Antioxidants (Basel) (2021) 10(3):349. doi: 10.3390/antiox10030349
147. Cui Q, Wang JQ, Assaraf YG, Ren L, Gupta P, Wei L, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Update (2018) 41:1–25. doi: 10.1016/j.drup.2018.11.001
148. Blondy S, David V, Verdier M, Mathonnet M, Perraud A, Christou N. 5-fluorouracil resistance mechanisms in colorectal cancer: from classical pathways to promising processes. Cancer Sci (2020) 111:3142–54. doi: 10.1111/cas.14532
149. Liu C, Zhao Y, Wang J, Yang Y, Zhang Y, Qu X, et al. FoxO3 reverses 5-fluorouracil resistance in human colorectal cancer cells by inhibiting the Nrf2/TR1 signaling pathway. Cancer Lett (2020) 470:29–42. doi: 10.1016/j.canlet.2019.11.042
150. Kang KA, Piao MJ, Kim KC, Kang HK, Chang WY, Park IC, et al. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: involvement of TET-dependent DNA demethylation. Cell Death Dis (2014) 5:e1183. doi: 10.1038/cddis.2014.149
151. Cheng YM, Lin PL, Wu DW, Wang L, Huang CC, Lee H. PSMD4 is a novel therapeutic target in chemoresistant colorectal cancer activated by cytoplasmic localization of Nrf2. Oncotarget (2018) 9:26342–52. doi: 10.18632/oncotarget.25254
152. Payandeh Z, Pirpour TA, Mansoori B, Khaze V, Asadi M, Baradaran B, et al. The impact of Nrf2 silencing on Nrf2-PD-L1 axis to overcome oxaliplatin resistance and migration in colon cancer cells. Avicenna J Med Biotechnol (2021) 13:116–22. doi: 10.18502/ajmb.v13i3.6371
153. Lin PL, Chang JT, Wu DW, Huang CC, Lee H. Cytoplasmic localization of Nrf2 promotes colorectal cancer with more aggressive tumors via upregulation of PSMD4. Free Radic Biol Med (2016) 95:121–32. doi: 10.1016/j.freeradbiomed.2016.03.014
154. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev (2013) 27:2179–91. doi: 10.1101/gad.225680.113
155. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature (2011) 475:106–09. doi: 10.1038/nature10189
156. Lee YJ, Kim WI, Bae JH, Cho MK, Lee SH, Nam HS, et al. Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann Surg Treat Res (2020) 98:159–67. doi: 10.4174/astr.2020.98.4.159
157. Lu MC, Ji JA, Jiang ZY, You QD. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev (2016) 36:924–63. doi: 10.1002/med.21396
158. He F, Antonucci L, Karin M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis (2020) 41:405–16. doi: 10.1093/carcin/bgaa039
159. Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol (2013) 1:45–9. doi: 10.1016/j.redox.2012.10.001
160. He J, Liu L, Liu X, Chen H, Liu K, Huang N, et al. Epoxymicheliolide prevents dextran sulfate sodium-induced colitis in mice by inhibiting TAK1-NF-kappaB pathway and activating Keap1-NRF2 signaling in macrophages. Int Immunopharmacol (2022) 113:109404. doi: 10.1016/j.intimp.2022.109404
161. Qiu S, Li P, Zhao H, Li X. Maresin 1 alleviates dextran sulfate sodium-induced ulcerative colitis by regulating NRF2 and TLR4/NF-kB signaling pathway. Int Immunopharmacol (2020) 78:106018. doi: 10.1016/j.intimp.2019.106018
162. Liu H, Johnston LJ, Wang F, Ma X. Triggers for the Nrf2/ARE signaling pathway and its nutritional regulation: potential therapeutic applications of ulcerative colitis. Int J Mol Sci (2021) 22(21):11411. doi: 10.3390/ijms222111411
163. Dong S, Lu Y, Peng G, Li J, Li W, Li M, et al. Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2-Gpx4 signaling pathway. Dig Liver Dis (2021) 53:1276–85. doi: 10.1016/j.dld.2021.02.011
164. Wang R, Luo Y, Lu Y, Wang D, Wang T, Pu W, et al. Maggot extracts alleviate inflammation and oxidative stress in acute experimental colitis via the activation of Nrf2. Oxid Med Cell Longev (2019) 2019:4703253. doi: 10.1155/2019/4703253
165. Wang X, Saud SM, Wang F, He S, Zhang X, Hua B, et al. Protective effect of ShaoYao decoction on colitis-associated colorectal cancer by inducing Nrf2 signaling pathway. J Ethnopharmacol (2020) 252:112600. doi: 10.1016/j.jep.2020.112600
Keywords: Nrf2, ulcerative colitis, oxidative stress, intestinal fibrosis, colorectal cancer
Citation: Peng S, Shen L, Yu X, Zhang L, Xu K, Xia Y, Zha L, Wu J and Luo H (2023) The role of Nrf2 in the pathogenesis and treatment of ulcerative colitis. Front. Immunol. 14:1200111. doi: 10.3389/fimmu.2023.1200111
Received: 04 April 2023; Accepted: 22 May 2023;
Published: 05 June 2023.
Edited by:
Natividad Garrido Mesa, Kingston University, United KingdomReviewed by:
Longhuan Ma, University of Florida, United StatesNader Bagheri, Shahrekord University of Medical Sciences, Iran
Copyright © 2023 Peng, Shen, Yu, Zhang, Xu, Xia, Zha, Wu and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hesheng Luo, cGVuZ3BlbmdzcGFjZUB3aHUuZWR1LmNu
†These authors have contributed equally to this work