 Qin Zhang1†
Qin Zhang1† Yujia Huo2†
Yujia Huo2† Qinggang Sun1Nan Liu1Hongchuan Shi1Minghui Wang1Jinming Xiao1Hanzi Yuan3
Qinggang Sun1Nan Liu1Hongchuan Shi1Minghui Wang1Jinming Xiao1Hanzi Yuan3 Xiangfeng Tang4*
Xiangfeng Tang4*- 1Department of Hematology, No.971 Hospital of People's Liberation Army Navy, Qingdao, Shandong, China
- 2Department of Traditional Chinese Medicine, Qingdao Special Service Sanatorium of People's Liberation Army Navy, Qingdao, Shandong, China
- 3Department of Pediatrics, the Sixth Medical Center of People's Liberation Army General Hospital, Beijing, China
- 4Department of Pediatrics, the Seventh Medical Center of People's Liberation Army General Hospital, Beijing, China
Unstable hemoglobinopathies are a rare, heterogeneous group of diseases that disrupt the stability of hemoglobin (Hb), leading to chronic hemolysis and anemia. Patients with severe phenotypes often require regular blood transfusions and iron chelation therapy. Although rare, studies have reported that hematopoietic stem cell transplantation (HSCT) seems to be an available curative approach in transfusion-dependent patients with unstable hemoglobinopathies. Here, we describe successful haploidentical HSCT for the treatment of an unstable Hb variant, Hb Bristol-Alesha, in a 6-year-old boy with severe anemia since early childhood. Two years after transplantation, he had a nearly normal hemoglobin level without evidence of hemolysis. DNA analysis showed complete chimerism of the donor cell origin, confirming full engraftment with normal erythropoiesis.
1 Introduction
Unstable hemoglobinopathies are a rare, heterogeneous group of hemolytic anemias caused by gene mutations in globin chains, leading to alterations in the solubility and stability of hemoglobin (Hb). Gene mutations result in the substitution or deletion of amino acids in the globin subunits. The general mechanisms that destabilize Hb include the weakening or altering of heme-globin interactions, interference with the secondary or tertiary structure of the Hb subunits, or interference with subunit interactions (1). Structure-altered Hbs have an increased tendency to denature and autoxidize to form methemoglobin, which results in the production of hemichromes. Hemichromes induce the clustering of band 3 at their membrane sites; these globular intracellular aggregates are called Heinz bodies (2, 3). The formation of Heinz bodies decreases membrane pliability and increases membrane permeability. Oxidative damage from free heme iron may further compromise the membrane. As a result, red blood cells (RBCs) containing Heinz bodies show reduced deformability and increased fragility (4, 5). The impairment of deformability causes RBCs to be preferentially trapped in the spleen. The spleen removes damaged cells or just a section of the membrane and Heinz body inclusions. Erythrocyte membrane loss reduces the lifespan of affected cells and eventually leads to their removal from circulation. Some Hbs are so unstable that they are difficult to detect in hemolysates because of their rapid denaturation and degradation. These Hbs are called hyperunstable Hb variants. The clinical presentations of the disease vary, ranging from mild to severe hemolytic anemia, depending on the instability of the Hb variants. Mildly unstable Hbs may not cause clinical symptoms; however, severely affected patients require chronic transfusion therapy from infancy or early childhood. Inheritance is autosomal dominant in most cases; however, de novo mutations have also been described (1, 6).
Hb Bristol-Alesha results from a G>A mutation at codon 67 of the β-globin gene, with a single amino acid substitution from normal β67 valine (Val) to β67 methionine (Met) or β67 aspartate (Asp). This substitution introduces a highly charged polar residue into the hydrophobic heme pocket, breaking the nonpolar bond between Val and the heme group. This results in the disruption of the molecular stability of Hb, which finally causes severe hemolytic anemia (7, 8). To date, there are no curative options for unstable Hb variants, except hematopoietic stem cell transplantation (HSCT). Although HSCT has been widely described as a therapeutic strategy for hemoglobinopathies such as thalassemia major (TM) and sickle cell disease (SCD), its application is rarely reported for the treatment of unstable hemoglobinopathies. Herein, we report successful haploidentical HSCT (haplo-HSCT) in a pediatric patient with Hb Bristol-Alesha.
2 Case presentation
A 3-month-old boy presented with dark urine, pallor, and mild jaundice. Moderate anemia (Hb 85 g/L) with reticulocytosis (13.4%) was detected in the peripheral blood cell count. Biochemical analysis revealed elevated serum lactate dehydrogenase and hyperbilirubinemia. Coombs and autoantibody test results were negative. DNA test results for thalassemia were normal. The patient was diagnosed with hemolytic anemia. The Hb level decreased to 58 g/L when he was 10 months. Physical examination revealed generalized icterus accompanied by marked splenomegaly (3 cm below the costal margin). From then on, he was treated conservatively with multiple blood transfusions of an average of 2.5 units every month to maintain a Hb level of 90 g/L. He was re-evaluated at 2 years of age. Malnutrition or growth retardation was not observed. A blood smear examination revealed erythrocyte abnormalities with marked anisocytosis, poikilocytosis, and basophilic stippling. Specific enzyme activity assays for glucose-6-phosphate dehydrogenase, phosphate isomerase, pyruvate kinase, and pyrimidine 5’-nucleotidase showed normal findings. Hb electrophoresis revealed elevated fetal Hb (21%) without anomalous Hb. Both heat stability and isopropanol tests showed positive results. DNA sequencing of the globin genes were subsequently performed and identified a heterozygous mutation in Hbβ codon 67, variant c.202G>A Val-Met (Figure 1A, GenBank accession No. OQ718455). The boy was definitively diagnosed with unstable hemoglobinopathy and Hb Bristol-Alesha. He had no family history of hemolytic anemia, and his parents were found to be normal based on DNA sequencing of globin chains. Deferasirox was administered at 4 years of age because of iron overload, with a serum ferritin level >2000 ng/mL (Supplementary Figure 1). However, the serum ferritin level did not decrease below 1000 ng/mL on periodical tests. Moreover, conventional hepatic magnetic resonance imaging at 6 years of age revealed a diffuse abnormal signal, indicating iron deposition in the liver.

Figure 1 β-globin gene sequencing showing the point mutation at c.202G>A before HSCT (A) and a normal sequence after HSCT (B). The arrow shows the position of the mutation.
In view of serious transfusion dependence and the possibility of end-organ injury due to iron overload, HSCT was performed at the age of 6 years. As the patient did not have matched siblings or unrelated donors, he underwent haplo-HSCT with a myeloablative conditioning regimen (Figure 2). The donor was his father who has a human leukocyte antigen (HLA) 5/10 allele match. Donor-specific anti-HLA antibodies (DSAs) of the recipient before HSCT were negative; therefore, desensitization strategies were not conducted. The conditioning regimen consisted of cyclophosphamide (Cy) 55 mg/kg per day from days −10 to −9, busulfan (Bu) 4.8 mg/kg per day from days −8 to −6, fludarabine (Flu) 40 mg/m2 per day from days −8 to −4, and thiotepa (TT) 10 mg/kg on day −5. Anti-thymoglobulin (ATG) was also added to the regimen at a total dose of 9 mg/kg from days −4 to −2. Graft-versus-host disease (GVHD) prophylaxis included cyclosporine A (CsA), mycophenolate mofetil (MMF), and methotrexate (MTX). CsA was intravenously injected from day −1 to +25 and then orally administered to maintain its blood concentration at 200–250 ng/mL, and MMF was administered from day +1 to day +30 at a dose of 20–30 mg/kg per day. Short-term MTX was administered on days +1, +3, +6, and +11 at 15 mg/m2, 10 mg/m2, 10 mg/m2, and 10 mg/m2, respectively. Considering the different characteristics of hematopoietic stem cells from the bone marrow (BM) and peripheral blood stem cells (PBSCs), the patient received an infusion of a combination of BM and PBSCs mobilized by granulocyte colony-stimulating factor (G-CSF) on days 01 and 02, respectively, to improve engraftment and reduce the occurrence of GVHD. The cell dose of infusion was 21.44×108/kg of nucleated cells, 7.64×106/kg of CD34+ cells, and 2.14×106/kg of CD3+ cells. The time to neutrophil and platelet engraftment was 15 and 17 days, respectively. The patient developed cytomegalovirus reactivation and BKV+ hemorrhagic cystitis are on days 24 and 32, respectively, which were controlled using antiviral drugs and intravenous immunoglobulin. Grade II acute GVHD (skin type) was also detected on day 25 post-transplantation and was controlled by the infusion of CD52 monoclonal antibody. The patient showed complete chimerism on day 30, which was sustained at 3, 6, 9, 12, 18, and 24 months post-HSCT. Furthermore, the former mutation of Hbβ was not detected using DNA resequencing (Figure 1B, GenBank accession No. OQ718456). During a period of 2 years follow-up, Hb was maintained at a high level of >110 g/L. The serum ferritin level gradually decreased to 1100ng/mL and no chronic GVHD was observed. To date, he has been transfusion-free.
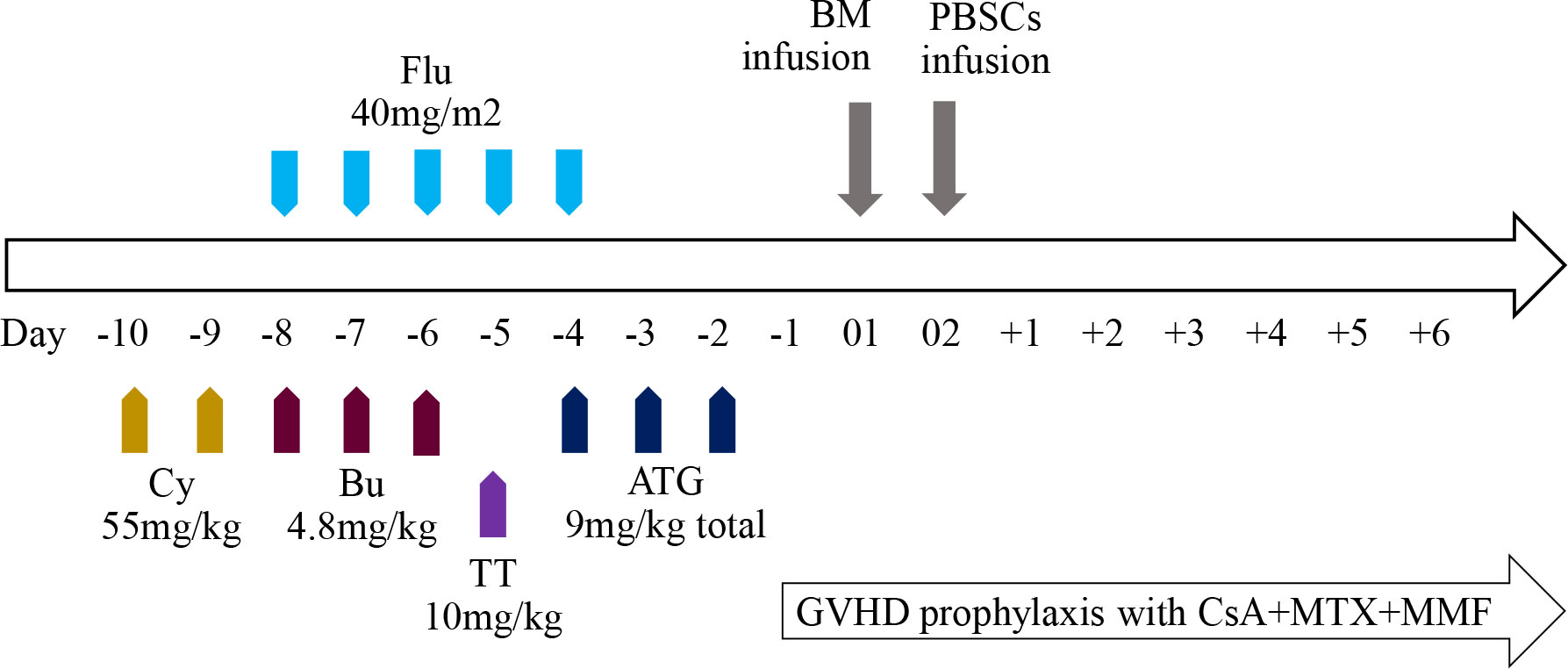
Figure 2 Overview of the haplo-HSCT protocol.
3 Discussion and conclusions
Unstable hemoglobinopathies are a group of rare congenital diseases that present with nonimmune hemolytic anemia of varying degrees. To date, more than 100 unstable Hb variants have been discovered using DNA sequencing (9). The incidence of this uncommon disorder is relatively low; therefore, only individual cases of each variant have been reported. Hb Bristol-Alesha is caused by a mutation of the β-globin gene, leading to the replacement of Val by Met. The Met residue is subsequently modified to Asp, probably via oxidative mechanisms (10). To the best of our knowledge, only 15 cases of this variant have been reported, all of which were dependent on blood transfusion (Table 1) (8, 11–23). According to this case and a previous analysis conducted on Hb Bristol-Alesha, it is always caused by de novo mutations. In addition, these cases have been reported in subjects of different origins, suggesting that this variant does not have a regional or racial propensity.
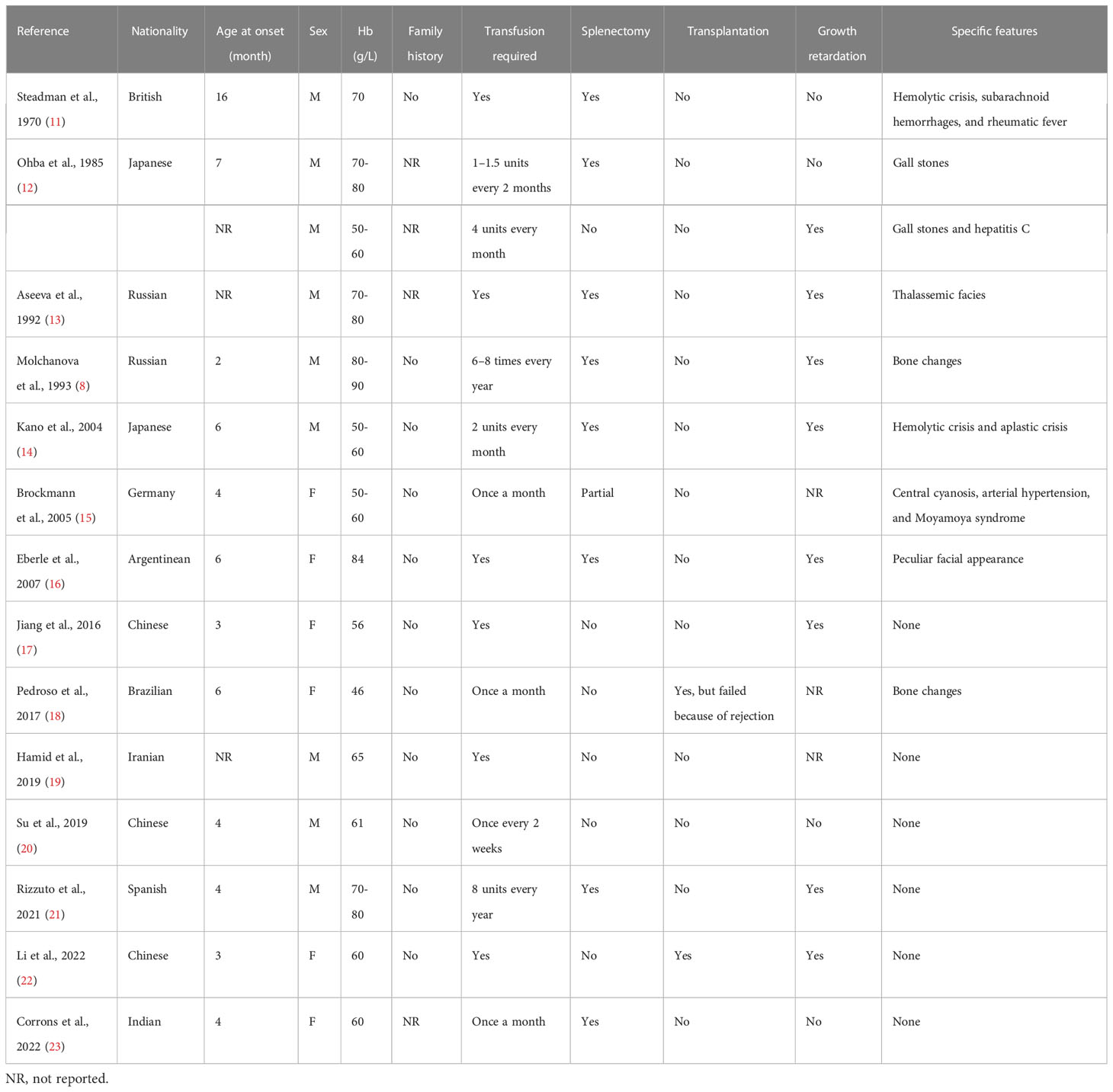
Table 1 Clinical features of published cases of Hb Bristol-Alesha.
Our patient presented with a very unstable Hb phenotype, and abnormal Hb levels could not be detected on electrophoresis. Therefore, DNA sequencing of the globin genes was necessary for an accurate diagnosis. There is evidence that hyperunstable Hb variants cause symptoms clinically similar to those associated with TM (14). This patient developed severe hemolytic anemia and marked splenomegaly, requiring frequent transfusions and iron chelation to improve anemia and suppress ineffective erythropoiesis. It should be noted that transfusions are not risk-free. On the one hand, repetitive RBC transfusions expose patients to a high risk of developing alloantibodies, which can limit the availability of compatible RBCs for future transfusions and increase the risk of transfusion complications (24, 25). On the other hand, progressive iron loading resulting from repeated RBC transfusions and increased absorption of dietary iron is most likely to induce tissue-specific siderosis in the liver, myocardium, pancreas, and pituitary gland (26). As a result, patients receiving long-term transfusions tend to have organ dysfunction, which worsens with increasing age (27). Furthermore, the high economic burden related to lifelong transfusion therapy and iron chelation, together with the high usage of health care resources, provides more insights into possible curative therapies (28). Although splenectomy in later years seems to modestly improve the clinical condition in a few patients, it is not a curable option and may increase the risk of infections and thrombotic complications (29).
Currently, allogeneic HSCT offers an available curative approach for hemoglobinopathies. HSCT outcomes from matched identical donors have gradually improved over the past few decades as a result of optimized treatment protocols, leading to an overall survival (OS) rate exceeding 90% for TM and SCD (30–32). A retrospective follow-up study evaluated the long-term outcomes in 137 patients with TM who underwent allogeneic HSCT. The 39-year OS and disease-free survival rates were 81.4% and 74.5%, respectively, indicating a definitive cure for the majority of patients (33). For those lacking HLA-matched sibling donors or unrelated donors, HLA-haploidentical donors (in pediatrics, in most cases a parent) are increasingly considered and used (34). However, owing to the relatively low prevalence, there are currently no practical recommendations or treatment guidelines for patients with unstable hemoglobinopathies. Although allogeneic HSCT is the only curative option for severe cases, experience with this procedure is limited. To date, successful cases of HSCT have rarely been reported (21, 22, 35–38). These cases are summarized in Table 2.
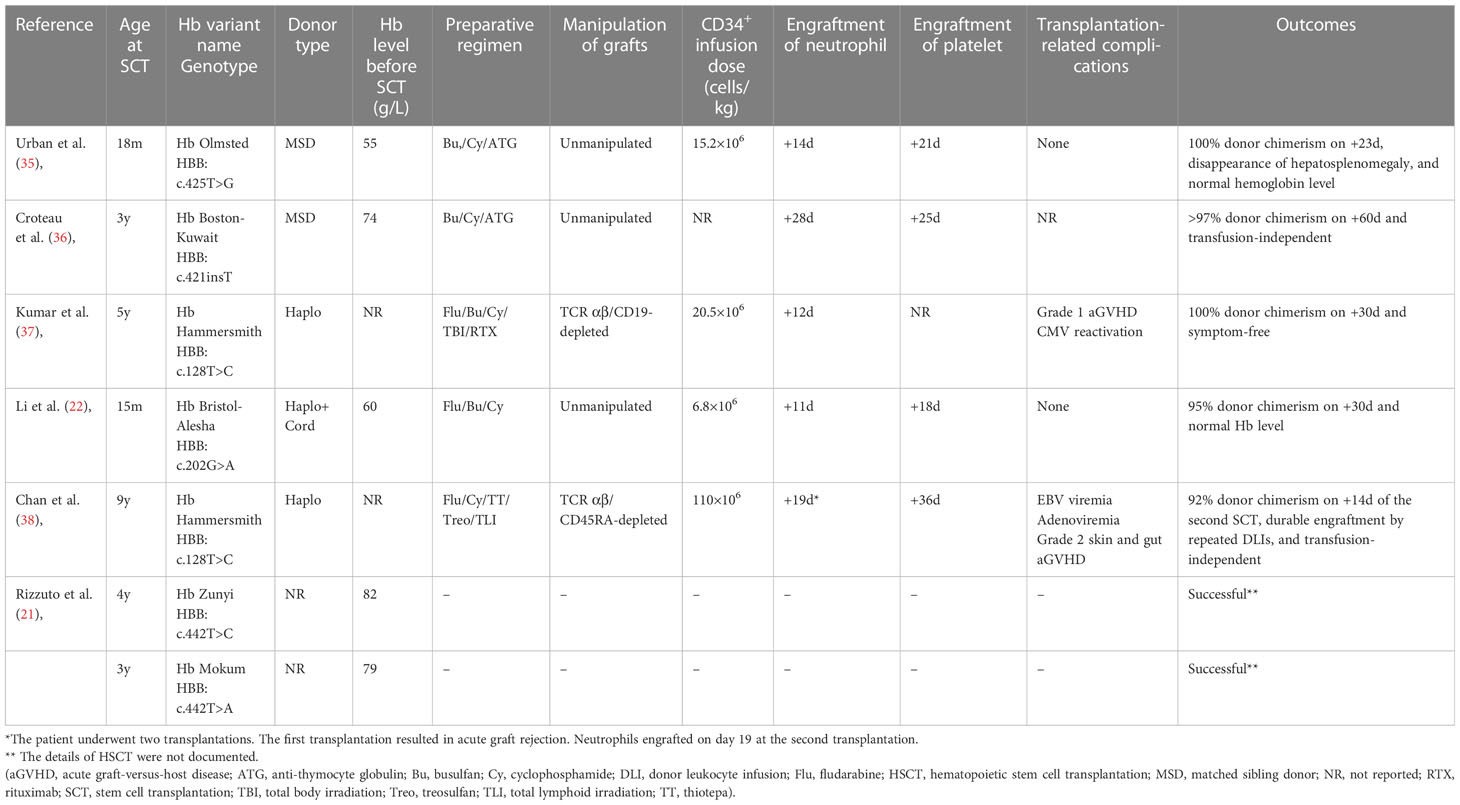
Table 2 Summary of allogeneic HSCT for unstable hemoglobinopathies.
Considering the severe hemolytic phenotype of our patient and the unavailability of matched donors, haplo-HSCT was planned. Similar to TM and SCD, disease-specific features, including hyperplastic BM and allosensitization due to multiple transfusions, render haplo-HSCT a high risk for graft failure (GF) (39, 40), and this may be the main obstacle. Patients receiving transfusions have a higher prevalence of anti-HLA antibodies, which are defined as DSAs when their specificity corresponds to a mismatched donor antigen (41). DSAs are a major cause of primary GF, including graft rejection (GR) and poor graft function (PGF) in patients receiving haplo-HSCT (42, 43). The rejection rate is much higher in the DSA-positive group than in the DSA-negative group, and DSA-positive patients have worse OS and inferior progression-free survival (44, 45). Therefore, anti-HLA antibodies should be evaluated in all haplo-HSCT recipients, especially in those receiving multiple transfusions (46). Desensitization could be applied in recipients with pretransplant DSAs if alternative donor is unavailable (47). We performed DSA detection in our patient before haplo-HSCT and obtained a negative result. Consequently, neither GR nor PGF was found in the subsequent transplantation process.
The conditioning regimen also plays a vital role in stem cell engraftment, and the results from thalassemia could provide a valuable reference. Myeloablative preconditioning regimens with more advanced immunosuppression concepts and application of T-cell depletion strategies, either ex vivo T-cell depletion (CD3+/CD19+ or TCRαβ+/CD19+ depletion) or in vivo T-cell depletion (post-transplant Cy or G-CSF primed peripheral blood graft with ATG), have significantly improved the outcomes of haplo-HSCT (48–52). A previous study that adopted a novel NF-08-TM conditioning regimen reported high long-term OS and thalassemia-free survival rates in China (53, 54). More recently, additional research comparing HLA fully matched and mismatched HSCT for patients with TM demonstrated similar survival outcomes and incidences of complications (except for acute GVHD) based on the modified NF-08-TM regimen, adjusting the ATG dose according to HLA compatibility (55). Similar to that used in patients with thalassemia, a myeloablative regimen including Bu, Cy, Flu, TT, and ATG (at a total dose of 9 mg/kg) was administered in our patient. The boy achieved full donor chimerism and has remained transfusion-free to date. Notably, he developed CMV and BKV infection after transplantation. It has been speculated that viral infection complications are associated with delayed immune reconstitution caused by the addition of ATG (56–58).
In conclusion, unstable hemoglobinopathy is a rare disease, and a subset of patients present with severe hemolytic anemia. Based on our experience from this case, haplo-HSCT may be a curative option for patients with a transfusion-dependent phenotype of unstable hemoglobinopathy when matched donors are not available. Further clinical studies are required before haplo-HSCT can be widely applied in clinical practice.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: OQ718455, OQ718456 (NCBI GenBank database).
Ethics statement
The studies involving human participants were reviewed and approved by the Sixth Medical Center of PLA General Hospital, China. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
QZ and YH were responsible for data curation and drafting of the manuscript. QS, NL, HS, MW, and JX contributed to patient care and follow-up. HY collected the data. XT conceived and designed the study and guided the article revision. All the authors contributed to the manuscript and approved the submitted version.
Funding
This study is supported by the Capital Characteristic Clinic Project of China (Grant No. Z181100001718032) and Beijing E-Town Cooperation & Development Foundation (Grant No. YCXJ-JZ-2022-007).
Acknowledgments
This manuscript was edited for English Language by Charlesworth Author Services (www.cwauthors.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1188058/full#supplementary-material
References
1. Gallagher PG. Diagnosis and management of rare congenital nonimmune hemolytic disease. Hematol Am Soc Hematol Educ Program (2015) 2015:392–9. doi: 10.1182/asheducation-2015.1.392
2. Waugh SM, Low PS. Hemichrome binding to band 3: nucleation of Heinz bodies on the erythrocyte membrane. Biochemistry (1985) 24:34–9. doi: 10.1021/bi00322a006
3. Williamson D. The unstable haemoglobins. Blood Rev (1993) 7:146–63. doi: 10.1016/0268-960x(93)90002-l
4. Hasegawa S, Rodgers GP, Shio H, Schechter AN, Uyesaka N. Impaired deformability of Heinz body-forming red cells. Biorheology (1993) 30:275–86. doi: 10.3233/bir-1993-303-412
5. Chiu D, Lubin B. Oxidative hemoglobin denaturation and RBC destruction: the effect of heme on red cell membranes. Semin Hematol (1989) 26:128–35.
6. Risinger M, Emberesh M, Kalfa TA. Rare hereditary hemolytic anemias: diagnostic approach and considerations in management. Hematol Oncol Clin North Am (2019) 33:373–92. doi: 10.1016/j.hoc.2019.01.002
7. Thom CS, Dickson CF, Gell DA, Weiss MJ. Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harb Perspect Med (2013) 3:a011858. doi: 10.1101/cshperspect.a011858
8. Molchanova TP, Postnikov Yu V, Pobedimskaya DD, Smetanina NS, Moschan AA, Kazanetz EG, et al. Hb alesha or alpha 2 beta (2)67(e11)Val→Met: a new unstable hemoglobin variant identified through sequencing of amplified DNA. Hemoglobin (1993) 17:217–25. doi: 10.3109/03630269308998896
9. Giardine BM, Joly P, Pissard S, Wajcman H, K Chui DH, Hardison RC, et al. Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res (2021) 49:D1192–d6. doi: 10.1093/nar/gkaa959
10. Strader MB, Hicks WA, Kassa T, Singleton E, Soman J, Olson JS, et al. Post-translational transformation of methionine to aspartate is catalyzed by heme iron and driven by peroxide: a novel subunit-specific mechanism in hemoglobin. J Biol Chem (2014) 289:22342–57. doi: 10.1074/jbc.M114.568980
11. Steadman JH, Yates A, Huehns ER. Idiopathic heinz’ body anaemia: hb-Bristol (beta67 (E11) Val to asp). Br J Haematol (1970) 18:435–46. doi: 10.1111/j.1365-2141.1970.tb01457.x
12. Ohba Y, Matsuoka M, Miyaji T, Shibuya T, Sakuragawa M. Hemoglobin Bristol or beta 67(E11) Val→Asp in Japan. Hemoglobin (1985) 9:79–85. doi: 10.3109/03630268508996986
13. Aseeva EA, Lutsenko IN, Pivnik AV, Spivak VA, Beliaeva LS, Zhestkova NM. Congenital hemolytic anemia caused by the carriage of hemoglobin Bristol beta 67 Bal→Asp. Ter Arkh (1992) 64:78–81.
14. Kano G, Morimoto A, Hibi S, Tokuda C, Todo S, Sugimoto T, et al. Hb Bristol-alesha presenting thalassemia-type hyperunstable hemoglobinopathy. Int J Hematol (2004) 80:410–5. doi: 10.1532/ijh97.04048
15. Brockmann K, Stolpe S, Fels C, Khan N, Kulozik AE, Pekrun A. Moyamoya syndrome associated with hemolytic anemia due to hb alesha. J Pediatr Hematol Oncol (2005) 27:436–40. doi: 10.1097/01.mph.0000175409.21342.ea
16. Eberle SE, Noguera NI, Sciuccati G, Bonduel M, Díaz L, Staciuk R, et al. Hb alesha [beta67(E11)Val→Met, GTG→ATG] in an Argentinian girl. Hemoglobin (2007) 31:379–82. doi: 10.1080/03630260701459408
17. Jiang H, Yan JM, Zhou JY, Li DZ. Hb alesha [β67(E11)Val→Met (GTG>ATG); HBB: c.202G > a] found in a Chinese girl. Hemoglobin (2016) 40:420–1. doi: 10.1080/03630269.2016.1273233
18. Pedroso GA, Kimura EM, Santos MNN, Albuquerque DM, Ferruzzi JLH, Jorge SE, et al. Coinheritance of hb Bristol-alesha [β67(E11)Val→Met; HBB: c.202G>A] and the α212 patchwork allele in a Brazilian child with severe congenital hemolytic anemia. Hemoglobin (2017) 41:203–8. doi: 10.1080/03630269.2017.1340305
19. Hamid M, Zargan Nezhad E, Galehdari H, Saberi A, Shariati G, Sedaghat A. A first report of hb alesha [β67(E11)Val>Met, GTG>ATG] in an Iranian patient. Iran BioMed J (2019) 23:429–31. doi: 10.29252/ibj.23.6.429
20. Su Q, Chen S, Wu L, Tian R, Yang X, Huang X, et al. Severe thalassemia caused by hb zunyi [β147(HC3)Stop→Gln; Hbb: c.442T>C)] on the β-globin gene. Hemoglobin (2019) 43(1):7–11. doi: 10.1080/03630269.2019.1582430
21. Rizzuto V, Koopmann TT, Blanco-Álvarez A, Tazón-Vega B, Idrizovic A, Díaz de Heredia C, et al. Usefulness of NGS for diagnosis of dominant beta-thalassemia and unstable hemoglobinopathies in five clinical cases. Front Physiol (2021) 12:628236. doi: 10.3389/fphys.2021.628236
22. Li S, Chen K, Huang C, Zhang N, Jiang H, Jiang S. First report of successful treatment for hemoglobin Bristol-alesha by hemopoietic stem cell transplantation. Ann Hematol (2022) 101:617–9. doi: 10.1007/s00277-021-04721-7
23. Corrons JV, Bain BJ. Haemoglobin Bristol-alesha in a child with non-spherocytic severe haemolytic anaemia and marked anisochromic poikilocytosis with basophilic stippling and amorphous intracellular content. Blood Cells Mol Dis (2022) 94:102652. doi: 10.1016/j.bcmd.2022.102652
24. Garraud O, Chiaroni J. An overview of red blood cell and platelet alloimmunisation in transfusion. Transfus Clin Biol (2022) 29:297–306. doi: 10.1016/j.tracli.2022.08.140
25. Tormey CA, Hendrickson JE. Transfusion-related red blood cell alloantibodies: induction and consequences. Blood (2019) 133:1821–30. doi: 10.1182/blood-2018-08-833962
26. Wood JC. Estimating tissue iron burden: current status and future prospects. Br J Haematol (2015) 170:15–28. doi: 10.1111/bjh.13374
27. De Simone G, Quattrocchi A, Mancini B, di Masi A, Nervi C, Ascenzi P. Thalassemias: from gene to therapy. Mol Aspects Med (2022) 84:101028. doi: 10.1016/j.mam.2021.101028
28. Kattamis A, Kwiatkowski JL, Aydinok Y. Thalassaemia. Lancet (2022) 399:2310–24. doi: 10.1016/s0140-6736(22)00536-0
29. Taher AT, Musallam KM, Cappellini MD. β-thalassemias. N Engl J Med (2021) 384:727–43. doi: 10.1056/NEJMra2021838
30. Baronciani D, Angelucci E, Potschger U, Gaziev J, Yesilipek A, Zecca M, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European society for blood and bone marrow transplantation hemoglobinopathy registry, 2000-2010. Bone Marrow Transplant (2016) 51:536–41. doi: 10.1038/bmt.2015.293
31. Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood (2017) 129:1548–56. doi: 10.1182/blood-2016-10-745711
32. Yesilipek MA, Uygun V, Kupesiz A, Karasu G, Ozturk G, Ertem M, et al. Thalassemia-free and graft-versus-host-free survival: outcomes of hematopoietic stem cell transplantation for thalassemia major, Turkish experience. Bone Marrow Transplant (2022) 57:760–7. doi: 10.1038/s41409-022-01613-w
33. Santarone S, Angelini S, Natale A, Vaddinelli D, Spadano R, Casciani P, et al. Survival and late effects of hematopoietic cell transplantation in patients with thalassemia major. Bone Marrow Transplant (2022) 57:1689–97. doi: 10.1038/s41409-022-01786-4
34. Oikonomopoulou C, Goussetis E. HSCT remains the only cure for patients with transfusion-dependent thalassemia until gene therapy strategies are proven to be safe. Bone Marrow Transplant (2021) 56:2882–8. doi: 10.1038/s41409-021-01461-0
35. Urban C, Preisegger KH, Krugluger W, Hopmeier P, Schwinger W, Lackner H, et al. Allogeneic bone marrow transplantation in a child with hemoglobinopathy Olmsted. J Pediatr Hematol Oncol (2002) 24:417–9. doi: 10.1097/00043426-200206000-00020
36. Croteau SE, Luo HY, Lehmann LE, Chui DH, Neufeld EJ. Novel dominant β-thalassemia: hb Boston-Kuwait [codon 139/140(+T)]. Pediatr Blood Cancer (2013) 60:E131–4. doi: 10.1002/pbc.24611
37. Kumar K, Badiger S, Damodar S, Bhat S. Haploidentical stem cell transplantation with TCR alpha/beta and CD19 depletion in a case of unstable hemoglobin disease. Transplantation (2018) 102:e45–e6. doi: 10.1097/tp.0000000000001986
38. Chan WYK, Chan NCN, So JCC, Lee PPW, Cheuk DKL, Ha SY, et al. Successful haploidentical hematopoietic stem cell transplantation (HSCT) and durable engraftment by repeated donor lymphocyte infusions for a Chinese patient with transfusion-dependent hemoglobin (Hb) Hammersmith and massive splenomegaly. Pediatr Transplant (2022) 26:e14278. doi: 10.1111/petr.14278
39. Oevermann L, Schulte JH, Hundsdörfer P, Hakimeh D, Kogel F, Lang P, et al. HLA-haploidentical hematopoietic stem cell transplantation in pediatric patients with hemoglobinopathies: current practice and new approaches. Bone Marrow Transplant (2019) 54(Suppl. 2):743–8. doi: 10.1038/s41409-019-0598-x
40. Leonard A, Tisdale J, Abraham A. Curative options for sickle cell disease: haploidentical stem cell transplantation or gene therapy? Br J Haematol (2020) 189:408–23. doi: 10.1111/bjh.16437
41. Huo MR, Xu YJ, Zhai SZ, Lv M, Wang Y, Cao LQ, et al. Prevalence and risk factors of antibodies to human leukocyte antigens in haploidentical stem cell transplantation candidates: a multi-center study. Hum Immunol (2018) 79:672–7. doi: 10.1016/j.humimm.2018.06.003
42. Chang YJ, Zhao XY, Xu LP, Zhang XH, Wang Y, Han W, et al. Donor-specific anti-human leukocyte antigen antibodies were associated with primary graft failure after unmanipulated haploidentical blood and marrow transplantation: a prospective study with randomly assigned training and validation sets. J Hematol Oncol (2015) 8:84. doi: 10.1186/s13045-015-0182-9
43. Krummey SM, Gareau AJ. Donor specific HLA antibody in hematopoietic stem cell transplantation: implications for donor selection. Front Immunol (2022) 13:916200. doi: 10.3389/fimmu.2022.916200
44. File B, Huang Y, Peedin A, Gergis U. The impact of HLA donor-specific antibodies on engraftment and the evolving desensitization strategies. Bone Marrow Transplant (2022) 57:526–31. doi: 10.1038/s41409-022-01578-w
45. Huang Y, Luo C, Wu G, Huang X, Ding Y, Huang Z, et al. Effects of donor-specific antibodies on engraftment and long-term survival after allogeneic hematopoietic stem cell transplantation-a systematic review and meta-analysis. Bone Marrow Transplant (2023) 58:544–51. doi: 10.1038/s41409-023-01932-6
46. Ciurea SO, Cao K, Fernandez-Vina M, Kongtim P, Malki MA, Fuchs E, et al. The European society for blood and marrow transplantation (EBMT) consensus guidelines for the detection and treatment of donor-specific anti-HLA antibodies (DSA) in haploidentical hematopoietic cell transplantation. Bone Marrow Transplant (2018) 53:521–34. doi: 10.1038/s41409-017-0062-8
47. Zhang XH, Chen J, Han MZ, Huang H, Jiang EL, Jiang M, et al. The consensus from the Chinese society of hematology on indications, conditioning regimens and donor selection for allogeneic hematopoietic stem cell transplantation: 2021 update. J Hematol Oncol (2021) 14:145. doi: 10.1186/s13045-021-01159-2
48. Ciurea SO, Al Malki MM, Kongtim P, Fuchs EJ, Luznik L, Huang XJ, et al. The European society for blood and marrow transplantation (EBMT) consensus recommendations for donor selection in haploidentical hematopoietic cell transplantation. Bone Marrow Transplant (2020) 55:12–24. doi: 10.1038/s41409-019-0499-z
49. Hashem H, Najjar R, Abu-Shanap M, Khattab E, Rihani R, Tbakhi A, et al. Haploidentical hematopoietic cell transplantation using post-transplant cyclophosphamide for children with non-malignant diseases. J Clin Immunol (2021) 41:1754–61. doi: 10.1007/s10875-021-01113-4
50. Oostenbrink LVE, Pool ES, Jol-van der Zijde CM, Jansen-Hoogendijk AM, Vervat C, van Halteren AGS, et al. Successful mismatched hematopoietic stem cell transplantation for pediatric hemoglobinopathy by using ATG and post-transplant cyclophosphamide. Bone Marrow Transplant (2021) 56:2203–11. doi: 10.1038/s41409-021-01302-0
51. Gaziev J, Isgrò A, Sodani P, Paciaroni K, De Angelis G, Marziali M, et al. Haploidentical hsct for hemoglobinopathies: improved outcomes with TCRαβ(+)/CD19(+)-depleted grafts. Blood Adv (2018) 2:263–70. doi: 10.1182/bloodadvances.2017012005
52. Merli P, Pagliara D, Galaverna F, Li Pira G, Andreani M, Leone G, et al. TCRαβ/CD19 depleted HSCT from an HLA-haploidentical relative to treat children with different nonmalignant disorders. Blood Adv (2022) 6:281–92. doi: 10.1182/bloodadvances.2021005628
53. Li C, Wu X, Feng X, He Y, Liu H, Pei F, et al. A novel conditioning regimen improves outcomes in β-thalassemia major patients using unrelated donor peripheral blood stem cell transplantation. Blood (2012) 120:3875–81. doi: 10.1182/blood-2012-03-417998
54. He Y, Jiang H, Li C, Zhu Y, Wu X, Liu S, et al. Long-term results of the NF-08-TM protocol in stem cell transplant for patients with thalassemia major: a multi-center large-sample study. Am J Hematol (2020) 95:E297–e9. doi: 10.1002/ajh.25969
55. Huang C, Qu Y, Liu S, Nie S, Jiang H. Hematopoietic stem cell transplantation for thalassemia major using HLA fully-matched and mismatched donor grafts. Transl Pediatr (2021) 10:1552–65. doi: 10.21037/tp-20-415
56. Nishihori T, Al-Kadhimi Z, Hamadani M, Kharfan-Dabaja MA. Antithymocyte globulin in allogeneic hematopoietic cell transplantation: benefits and limitations. Immunotherapy (2016) 8:435–47. doi: 10.2217/imt.15.128
57. Qin F, Shi L, Li Q, Zhang Z, Liu L, Li J, et al. Immune recovery after in vivo T-cell depletion myeloablative conditioning hematopoietic stem cell transplantation in severe beta-thalassemia children. Eur J Haematol (2019) 103:342–50. doi: 10.1111/ejh.13289
Keywords: unstable hemoglobinopathy, hemolytic anemia, Hb Bristol-Alesha, hematopoietic stem cell transplantation, haploidentical
Citation: Zhang Q, Huo Y, Sun Q, Liu N, Shi H, Wang M, Xiao J, Yuan H and Tang X (2023) Case report: Curing a rare, unstable hemoglobin variant Hb Bristol-Alesha using haploidentical hematopoietic stem cell transplantation. Front. Immunol. 14:1188058. doi: 10.3389/fimmu.2023.1188058
Received: 16 March 2023; Accepted: 13 June 2023;
Published: 30 June 2023.
Edited by:
Ying-Jun Chang, Peking University People’s Hospital, ChinaReviewed by:
Isabel Badell, Sant Pau Institute for Biomedical Research, SpainErden Atilla, Fred Hutchinson Cancer Research Center, United States
Copyright © 2023 Zhang, Huo, Sun, Liu, Shi, Wang, Xiao, Yuan and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangfeng Tang, bTE4NjAwMzE3ODE2QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship