 Xiaoying Zhang1,2
Xiaoying Zhang1,2 Yu’e Liu3
Yu’e Liu3 Tong Zhang1,2
Tong Zhang1,2 Yuying Tan1,2
Yuying Tan1,2 Xiangpeng Dai1,2*
Xiangpeng Dai1,2* Yong-Guang Yang1,2,4*
Yong-Guang Yang1,2,4* Xiaoling Zhang1,2*
Xiaoling Zhang1,2*- 1Key Laboratory of Organ Regeneration and Transplantation of Ministry of Education, First Hospital, Jilin University, Changchun, China
- 2National-Local Joint Engineering Laboratory of Animal Models for Human Disease, First Hospital, Jilin University, Changchun, China
- 3Tongji University Cancer Center, Shanghai Tenth People’s Hospital of Tongji University, School of Medicine, Tongji University, Shanghai, China
- 4International Center of Future Science, Jilin University, Changchun, China
Cullin-RING ligases (CRLs) are the largest class of E3 ubiquitin ligases regulating the stability and subsequent activity of a large number of important proteins responsible for the development and progression of various diseases, including autoimmune diseases (AIDs). However, the detailed mechanisms of the pathogenesis of AIDs are complicated and involve multiple signaling pathways. An in-depth understanding of the underlying regulatory mechanisms of the initiation and progression of AIDs will aid in the development of effective therapeutic strategies. CRLs play critical roles in regulating AIDs, partially by affecting the key inflammation-associated pathways such as NF-κB, JAK/STAT, and TGF-β. In this review, we summarize and discuss the potential roles of CRLs in the inflammatory signaling pathways and pathogenesis of AIDs. Furthermore, advances in the development of novel therapeutic strategies for AIDs through targeting CRLs are also highlighted.
1 Introduction
Autoimmune diseases (AIDs) refer to autoantibody and autoreactive immune cells attacking self-tissues and inducing severe inflammatory reactions, which damage a variety of host organs including the skin, kidneys, joints, bowel, and the nervous system, among others (1, 2). AIDs can be classified as either organ-specific or systemic AIDs based on whether a single organ or a system, respectively, is damaged. Organ-specific AIDs include Hashimoto’s thyroiditis (HT), pemphigus, insulin-dependent diabetes mellitus (IDDM), and ulcerative colitis (UC). Systemic AIDs include multiple sclerosis (MS), systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA) (3, 4). Environmental risk factors and genome instability are the common leading causes of AIDs (5). The immune system can recognize self from non-self in physiological conditions. However, in certain conditions, environmental triggers such as chemical toxicants, pollution, infection, or intrinsic genome changes can break the immune tolerance and generate excessive autoantibody and autoreactive lymphocytes, which will attack the host tissues and result in the onset of AIDs (3, 6, 7). However, the detailed mechanisms of the pathogenesis of AIDs remain largely unknown and need further in-depth investigation.
In recent years, a lot of studies have shown that the posttranslational modification (PTM) of proteins plays a pivotal role in the occurrence and progression of AIDs (8–10). Ubiquitination, one of the important PTM types, regulates the protein stability, activity, subcellular localization, and interactions in key signaling pathways, subsequently influencing the cell cycle, proliferation, apoptosis, autophagy, and inflammation (10–13). Dysregulation of the ubiquitination of critical proteins could induce excessive immune activation and AIDs (14, 15). The modification of proteins by ubiquitin is a reversible process controlled by ubiquitination and de-ubiquitination. De-ubiquitination modulates the ubiquitin removed from substrates, which is regulated by deubiquitinating enzymes (DUBs). On the other hand, ubiquitination is catalyzed by a cascade of reactions involving three types of key enzymes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) (11, 13). Ubiquitin is activated in an ATP-dependent manner by E1s and forms a thioester between the ubiquitin C-terminal carboxyl and the E1 active site sulfhydryl of cysteine. Subsequently, ubiquitin transfers from the E1 to the E2 active site cysteine and forms another thioester complex through the catalysis of E2s. In the final steps, E3s catalyze the ubiquitin from E2s to the lysine ϵ-amino group of substrates via isopeptide bonds (11, 16). The E3s play a pivotal role in the ubiquitination cascade because they are responsible for substrate specificity.
There are more than 600 E3s in humans, which comprise three families: the really interesting new gene (RING) family, the homology to E6AP C-terminus (HECT) family, and the RING-between-RING (RBR) family (16). The HECT and RBR E3s mediate the transfer of ubiquitin from E2 to the cysteine of the E3 active site and then transfer the ubiquitin from E3 to specific substrates. However, the RING E3s lack the cysteine of active sites and directly transfer the ubiquitin from the E2 ubiquitin intermedia to the substrates (17, 18). Cullin-RING ligases (CRLs), the largest subfamily of the RING E3 ligases, consist of 300 members distributed into different subclasses according to the Cullins. In humans, there are eight Cullin proteins—Cul1, Cul2, Cul3, Cul4A, Cul4B, Cul5, Cul7, and Cul9—that are organizers of the CRL complex (19). CRLs are responsible for the ubiquitination and subsequent degradation of approximately 20% of the proteins regulated by the ubiquitination proteasome system (UPS) in mammalian cells (20, 21). The CRLs play a crucial part in regulating autoimmunity and homeostasis in physiological and pathological conditions (22–24). Previous studies suggested that the F-box and WD repeat domain-containing 7 (FBW7), as a substrate receptor (SR) of SKP1–Cul1–F-box (SCF), was predominantly upregulated in the colon tissues of patients with inflammatory bowel disease (IBD) and was correlated with its severity (25, 26). In addition, the expression of DCAF2, which is a SR of CRL4, was significantly suppressed in biopsies from patients with psoriasis. DACF2 deficiency mediated the activation of the nuclear factor kappa B (NF-κB) signaling pathway and accelerated the severity of psoriasis (23). In this review, we summarize and discuss the potential roles of CRLs in AIDs and provide new insights for AIDs therapy via targeting CRLs.
2 Cullin-RING E3 ligases: Composition and mechanism
2.1 Composition and catalytic mechanism of CRLs
The multi-subunit CRL complex consists of the RBX1 or the RBX2 RING protein, the scaffold Cullin protein, and the adaptor and SR unit. The N-terminal domain of Cullins binds to the SRs with or without the adaptor protein SKP1 or Elongins B/C. The C-terminal domain of Cullins connects with the RING subunit (RBX1 for Cul1–Cul4 and RBX2 for Cul5) (19, 27, 28). The RING domains are responsible for the binding to the E2s and mediates the transfer of ubiquitin, while the SRs recognize and recruit specific substrates. The Cullins, as scaffold proteins, are essential for the assembly of the whole E3 complex and its functions (18, 19, 27).
2.2 The subfamily of CRLs and their distinct functions
The CRLs are divided into eight subfamilies according to the eight types of Cullins (Cul1, Cul2, Cul3, Cul4A, Cul4B, Cul5, Cul7, and Cul9). Importantly, each CRL has a distinct composition and structure. The first identified CRL is SCF E3 ligase, in which SKP1, as the adaptor protein, mediates the linkage between Cul1 and the F-box SR protein (27). There are 69 F-box proteins that determine the substrate diversity and specificity in humans. SCF ligases catalyze the mono- or poly-ubiquitination of substrates and affect various cellular processes such as the cell cycle, DNA damage and repair, and other signaling pathways (29). The adaptor proteins for Cul2- or Cul5-based RING E3 ubiquitin ligases are Elongins B and C, which mediate the linkage between the N-terminal of Cullin and the BC-box SRs (30). The first identified substrate of CRL2 is hypoxia-inducible factor 1α (HIF-1α), which regulates the hypoxia response (28, 31). CRL5 has similar components of adaptors and SRs to CRL2. However, the RING unit of CRL5 is RBX2. CRL5 is mainly responsible for signaling transduction, virus infection, and tumorigenesis (32, 33). For CRL3, the SRs directly interact with the Cul3 scaffold protein without the adaptor proteins Skp1 or Elongins B/C. The SRs of CRL3 are usually BTB/POZ (Broad-complex, Tramtrack, and Bric-a-brac/pox virus and zinc finger) domain proteins that share a fold with Skp1. The MATH and Kelch domains of the CRL3 SRs are generally associated with the BTB domain, which is responsible for substrate recognition and recruitment. CRL3 plays a crucial role in regulating the oxidative stress response, cellular homeostasis, tumorigenesis, and progression (28, 34). CRL4 includes two homologous Cullin proteins, i.e., Cul4A and Cul4B, that share the same adaptor protein and the SR proteins including damage-specific DNA binding protein 1 (DDB1) and DDB1/CUL4-associated factor (DCAF). CRL4s have great impact on the disorder of the nervous system and oncogenesis (35). CRL7 and CRL9 are two novel CRLs reported in recent years. Only two F-box proteins Fbxw8 and Fbxw11 have been identified as the SRs of CRL7. Moreover, Cul7, as a scaffold protein, interacts with the RBX1 RING protein and the Skp1 adaptor protein. Importantly, CRL7 could mediate proteolytic and non-proteolytic ubiquitination. There are approximately 10 substrates in total in the involvement of CRL7 in the regulation of cell proliferation, apoptosis, and DNA damage repair (36, 37). The exact components of CRL9 are largely unknown. CRL9 has been reported to mediate the ubiquitination and degradation of survivin and to maintain genome integrity. CRL9 also acts as an activator of p53 to inhibit cell proliferation and promote DNA damage repair. Therefore, CRL9 has been considered a tumor suppressor due to its function in the regulation of p53 and survivin (38–40).
2.3 Regulation of CRLs via NEDDylation, substrate adaptor exchange, and phosphorylation
The process of protein ubiquitination and the subsequent proteolytic or non-proteolytic functions that are controlled by CRLs can also be regulated by several mechanisms. The protein NEDD8 can covalently attach to a lysine moiety located in the WHB (winged-helix) domain of Cullins and enhance the activity of CRLs (41). Similar to ubiquitination, NEDDylation is a reversible process. It is catalyzed by an enzymatic cascade with the E1 NEDD8-activating enzyme (NAE), E2 NEDD8-conjugating enzyme, and E3 NEDD8 ligase. The COP9 signalosome (CSN) mediates the deNEDDylation of Cullins. The processes of NEDDylation/deNEDDylation regulate the activation of CRLs and influence the fate of the substrates of CRLs (20, 42).
Previous biochemical studies suggested that Cullin-associated and NEDDylation-dissociated protein 1 (CAND1) is a negative regulatory factor of SCF. CAND1 binds to the unNEDDylated Cul1 and inhibits the assembly of the SCF complex by blocking the combination of the adaptor protein Skp1 with Cul1 (43, 44). However, recent genetic studies have identified that CAND1 also promotes the activation of SCF; moreover, the CAND1 mutant showed a reduction of the SCF activity in Arabidopsis (45). A growing body of later studies demonstrated that CAND1, as an exchange factor, mediates the dynamic exchange of F-box–Skp1 substrate adaptors and regulates the substrate specificity of SCF (46–48).
The crosstalk between the phosphorylation and ubiquitination mediated by CRLs is prevalent in eukaryotic cells. The phosphorylation of substrates often inhibits or promotes their recognition and interaction with CRLs. It is well established that the SR of SCF, FBW7, recognizes and interacts with its substrates after the phosphorylation of special amino acids in their degron (49). In addition, as the SR of CRL3, the speckle-type POZ protein (SPOP) often recognizes the substrates with conserved Ser/Thr-rich motifs. In most cases, phosphorylation in the motif is essential for the interaction between substrates and SPOP (50). However, a previous study indicated that casein kinase 1 (CK1) mediated the phosphorylation of Ci at the Ser/Thr-rich degrons and inhibited the interaction between Ci and MATH and BTB domain-containing protein (HIB/SPOP), which is a SR of CRL3 (51). On the other hand, the phosphorylation of the adaptor or SR of CRLs also influences the substrates’ binding to CRLs and the subsequent ubiquitination and degradation (50). Targeting the CRL pathways might be a promising strategy for the treatment of related diseases through regulating the abundance of the key proteins in human diseases. Identification of more detailed mechanisms of the regulation of CRLs will help in the development of CRL-targeted therapy.
3 Critical signaling pathways of immunity and inflammation regulated by CRLs
3.1 NF-κB signaling pathway
NF-κB, as a transcription factor, regulates the expression of a series of genes involved in multiple cellular processes including inflammation, autoimmunity, and cell survival (52, 53). The NF-κB signaling pathway consists of canonical and non-canonical pathways. The transcription factor complexes assembled by p65 (RelA) and p50 can translocate into the nucleus and promote the transcription of a set of target genes, which leads to the activation of the canonical NK-κB signaling pathway. The p65/p50 complex is maintained in the cytoplasm by IkappaB (IκB), an inhibitor of the canonical NF-κB pathway. IκB can be phosphorylated by IκB kinase (IKK) and subsequently degraded by the 26S proteasome to release the p65/p50 complex from cytoplasmic retention (52, 54). RelB/p52 heterodimers are responsible for the transcriptional activation of the target genes in the non-canonical NF-κB signaling pathway. p100, the precursor of p52, acts as an inhibitor that blocks the translocation of RelB into the nucleus. The proteolysis of p100 results in the production of p52 and constitutes the RelB/p52 complex, which is important for the activation of the non-canonical NF-κB pathway (54, 55).
CRLs regulate the activity of NF-κB both in the canonical and non-canonical signaling pathways (Figure 1, left). SCFβ-TrCP mediates the degradation of the NF-κB inhibitor IκB after its phosphorylation by IKK and releases the p65/p50 complex, which translocates into the nucleus and performs transcriptional activity in the NF-κB canonical pathway (56, 57). A recent study has reported that SCFFBW7 promoted the ubiquitination and proteolysis of IκB. The upregulation of FBW7 promoted the activation of the NF-κB pathway through the negative regulation of IκB and accelerated the intestinal inflammation in intestinal epithelial cells (IECs) (25). In addition, SCFβ-TrCP modifies p100 at the C-terminal domain, induces degradation, and generates the p52 mature subunit, constitutes the RelB/p52 complex then translocating into the nucleus and activating NF-κB in the non-canonical pathway (58, 59). SCFFBW7, another SCF E3 ligase, could recognize p100 based on the conserved degron in a GSK3-dependent manner. SCFFBW7 recognizes and destroys the p100 in the UPS after being phosphorylated by GSK3 at the Ser 707 and 711 sites, which subsequently contributes to the activation of the non-canonical NF-κB pathway (60–62). The Regulator of Cullins 1 (ROC1) mediates p-IκBα ubiquitination and the subsequent degradation induces the nuclear translocation of the P65 subunit and NF κB activation in bladder cancer. CRL5SPSB1 negatively regulates the activity of NF-κB through an undefined mechanism (63). Early studies have suggested that the NF-κB-inducing kinase (NIK) can be subjected to proteolysis in a TRAF3-dependent manner. TRAF3 mediates the ubiquitination and degradation of NIK by recruiting it to the cIAP1–cIAP2–TRAF2 ubiquitin ligase complex (64, 65). However, recent research has indicated that CRL4DCAF2 mediates the poly-ubiquitination and destruction of NIK in a TRAF3 pathway-independent manner. It was also found that CRL4DCAF2 inhibits the non-canonical activity of NF-κB in a NIK-dependent manner and ultimately reduces the production of interleukin 23 (IL-23), which can be a potential therapeutic target for psoriasis (23).
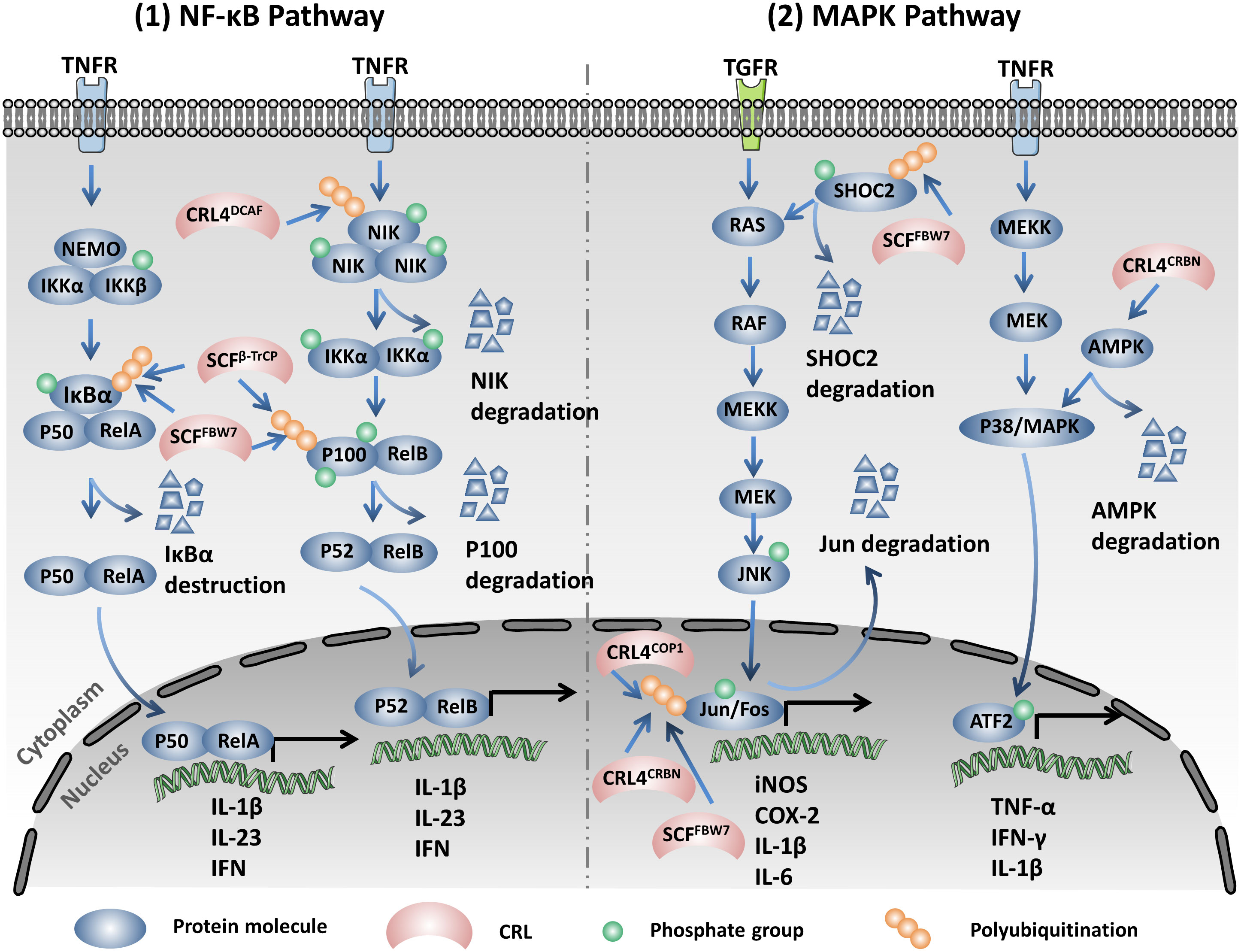
Figure 1 Cullin-RING ligases (CRLs) regulate the expression of multiple pro-inflammatory cytokines through modulating the nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways. Left: SCFβ-TrCP mediates the degradation of the NF-κB inhibitor IκB after phosphorylation by IκB kinase (IKK). CRL4DCAF2 mediates the poly-ubiquitination and destruction of the NF-κB-inducing kinase (NIK). SCFβ-TrCP modifies p100 at the C-terminal domain and induces degradation, generating the p52 mature subunit. SCFFBW7 recognizes and destroys the IκB and p100 in the ubiquitination proteasome system (UPS). These CRLs modulate the NF-κB pathway through regulating the stability of the key components. Right: SCFFBW7 mediates the ubiquitination and proteolysis of SHOC2 after being phosphorylated by MAPK at Thr507 and blocks the RAS-MAPK pathway. CRL4CRBN promotes the K48 linkage ubiquitination and the subsequent degradation of c-Jun and restrains the activity of the AP-1 complex. COP1, another Cul4-based E3 ubiquitin ligase, also regulates the protein stability of c-Jun. SCFFBW7 mediates the ubiquitination and destruction of c-Jun by proteasome. CRL4CRBN promotes a non-K48 linkage ubiquitination and the degradation of AMPKa.
3.2 MAPK signaling pathway
It is well known that the mitogen-activated protein kinase (MAPK) signaling pathway plays a central role in cell proliferation, apoptosis, and inflammation. Three main subgroups namely, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 build up the MAPK signaling pathway. The ERK pathway mainly regulates cell proliferation and differentiation. However, the JNK and p38 pathways generally participate in the response to oxidative stress and inflammation (66–68). A previous study showed that the RAS activator SHOC2 is a substrate of SCFFBW7. SCFFBW7 mediates the ubiquitination and proteolysis of SHOC2 after being phosphorylated by MAPK at Thr507 and blocks the RAS–MAPK pathway (69). CRL4CRBN promotes the K48 linkage ubiquitination and the subsequent degradation of c-Jun and restrains the activity of the AP-1 complex, which leads to the down-expression of the pro-inflammatory factors inducible nitric oxide synthase (iNOS) and COX-2 (70). COP1, another Cul4-based E3 ubiquitin ligase, also regulates the protein stability of c-Jun. In mice, COP1 deficiency induced tumorigenesis and tumor progression depending on the upregulation of c-Jun (71). In addition, SCFFBW7 recognizes c-Jun after being phosphorylated by GSK3 and mediates the ubiquitination and the subsequent degradation of c-Jun by proteasome (72). CRL4CRBN promotes a non-K48 linkage ubiquitination and degradation of the AMP-activated protein kinase alpha subunit (AMPKα). Notably, cereblon (CRBN) knockout decreases allergic responses in an AMPKα-dependent manner (73). CRBN plays an important role in the senescence process, and the depletion of CRBN activates p38/MAPK and downstream p53/p21 signaling and upregulates the senescence-associated markers SAHF (senescence-associated heterochromatic foci) and SA-β-Gal (74) (Figure 1, right).
3.3 JAK/STAT signaling pathway
The Janus kinase/signal transduction and activator of transcription (JAK/STAT) signaling pathway responds to a variety of inflammatory factors including cytokines, colony-stimulating factors, and growth factors. It also plays a central role in the pathogenesis of carcinoma and AIDs (75–77). Interleukins (ILs), interferons (IFNs), hormones, and colony-stimulating factors interact with specific type I/II cytokine receptors and induce the receptor dimerization and transphosphorylation of JAKs. As a step further, STATs are recruited and phosphorylated by JAKs, and the activated STATs dissociate from the receptors and form homodimers or heterodimers. The dimers then translocate into the nucleus and promote the transcription of associated genes (77, 78). The JAK family includes JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). Notably, in mammalian cells, STATs have seven subclasses: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 (78, 79). Targeting JAK signaling is thought to be a promising therapeutic strategy, and a series of JAK inhibitors (Jakinibs) have been approved by the US Food and Drug Administration (FDA) for the treatment of AIDs and lymphoma (76, 77).
CRL5SOCS3 promotes the ubiquitination and degradation of JAKs and STATs by the 26S proteasome (77, 80). The function of the negative regulation of the JAK/STAT signaling pathway makes suppressor of cytokine signaling (SOCS) proteins potential therapeutic targets for the treatment of JAK/STAT-associated diseases (81). Moreover, the Notch signaling pathway could transcriptionally activate Asb2, which is a SOCS-box-containing protein and is a SR of CRL5. Asb2 could replace SOCS in CRL5 and mediate the assembly of CRL5 and SCFSkp2 in a non-canonical E3 super complex, consequently promoting the degradation of JAK2 and E2A (82). SCFHOS recognizes and interacts with the type I IFN receptor IFNAR1 in a phosphorylation-dependent manner upon the stimulation of IFN-α and subsequently promotes its degradation, which influences the function of the JAK/STAK signaling pathway in cells (83).
The HIV virus protein Vif (viral infectivity factor) mediates the host STAT1 and STAT3 ubiquitination and degradation via the Elongin–Cullin–SOCS-box binding motif and subsequently reduces the production of the antiviral ISG15 induced by IFN-α (84). The Epstein–Barr virus (EBV) tegument protein BGLF2 utilizes the host SCF to promote the ubiquitination and degradation of STAT2 in the K48 linkage type by the 26S proteasome, which leads to the reduction of the expression of interferon-stimulating genes (ISGs) upon stimulation of IFN (85) (Figure 2, left).
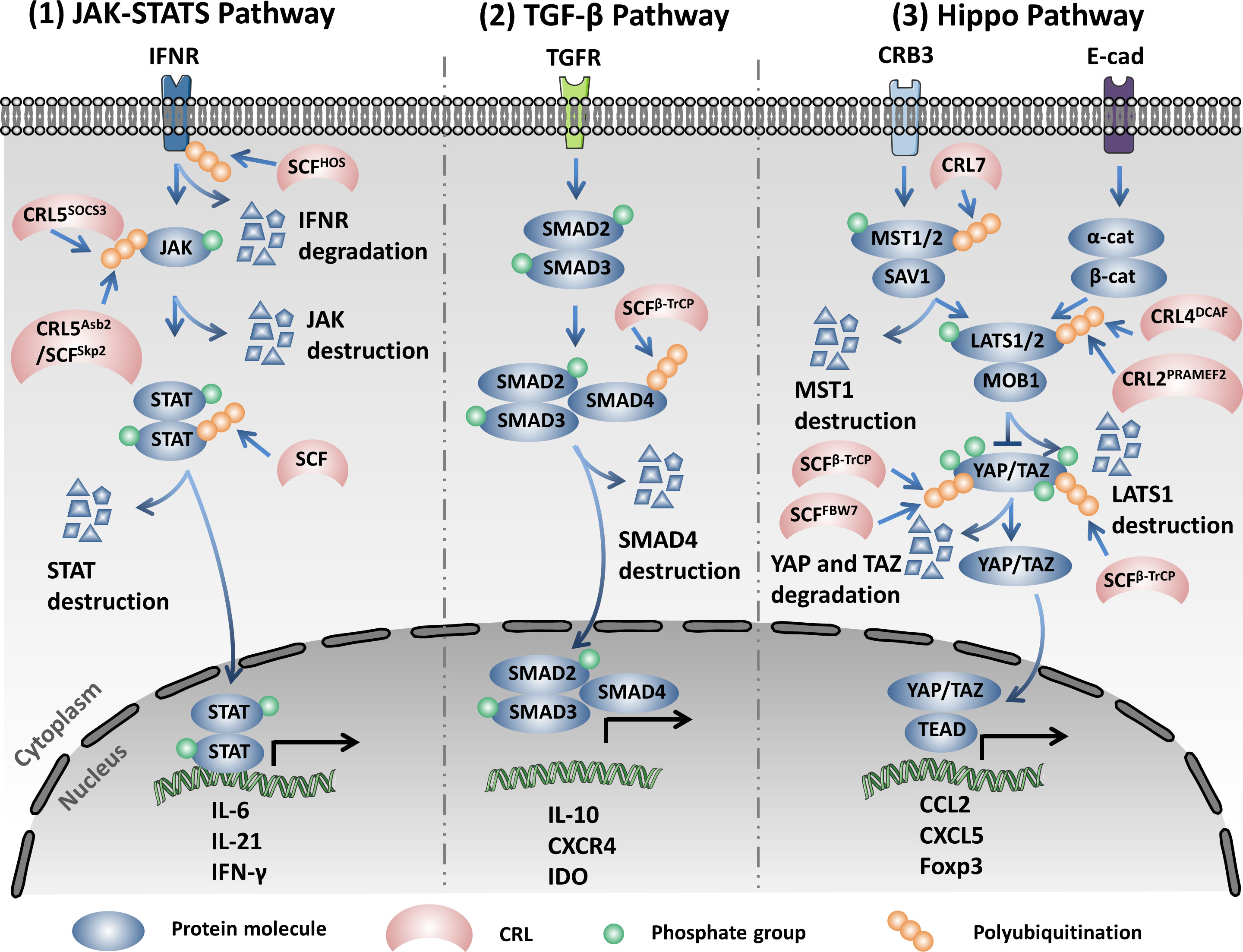
Figure 2 Cullin-RING ligases (CRLs) regulate the expression of multiple pro-inflammatory cytokines via modifying the Janus kinase/signal transduction and activator of transcription (JAK/STAT), TGF-β, and Hippo signaling pathways through the ubiquitination proteasome system (UPS). Left: SCFHOS recognizes and interacts with the type I interferon (IFN) receptor IFNR1 in a phosphorylation-dependent manner upon stimulation of IFN-α, subsequently promoting the degradation of IFNR1. CRL5SOCS3 promotes the ubiquitination and degradation of JAK and STAT by the 26S proteasome. Asb2 mediates the assembly of CRL5 and SCFSkp2 in the non-canonical E3 super complex and promotes the degradation of JAK2. The HIV protein Vif (viral infectivity factor) mediates the host STAT1 and STAT3 ubiquitination and degradation via the Elongin–Cullin–SOCS-box binding motif. The Epstein–Barr virus (EBV) utilizes the host SCF (SKP1–Cul1–F-box) to promote the ubiquitination and degradation of STAT2 in the K48 linkage type. Middle: SCFβ-TrCP1 specifically binds and destructs Smad4 to inhibit the expression of the genes in the TGF-β signaling pathway. Right: Cullin7 promotes the ubiquitination and destruction of Mst1. PRAMEF2, a substrate receptor (SR) of the Cul2-based ubiquitin ligase and CRL4DCAF promotes the degradation of LATS1. SCFβ-TrCP and SCFFBW7 promote the degradation of the phosphorylated YAP through UPS. Moreover, SCFβ-TrCP mediates the ubiquitination and degradation of TAZ (transcriptional coactivator with PDZ-binding motif) in a phosphorylation-dependent manner.
3.4 TGF-β signaling pathway
Abnormal activation of the transforming growth factor beta (TGF-β) signaling pathway is one of the main causes of inflammatory diseases and cancers. TGF-β signaling plays a key role in remodeling the tumor microenvironment and in promoting immune tolerance and tumor evasion (86–88). The active TGF-β interacts with TGF-β type I and II receptors in the cell membrane. TGF-β type I and II receptors are activated through interacting with TGF-β, subsequently activating Smad2 and Smad3 via phosphorylation. The activated Smad2 and Smad3 then bind to Smad4 to form trimeric complexes and translocate into the nucleus to regulate the expression of associated genes (86, 87). SCFβ-TrCP1 specifically binds to Smad4 instead of Smad2 and Smad3 to facilitate the ubiquitination and degradation of Smad4 and inhibits the expression of the genes in the TGF-β signaling pathway (89). Moreover, SCFFBXL15 promotes the ubiquitination and protein destruction of Smad ubiquitination regulatory factor 1 (Smurf1) by the 26S proteasome, which further regulates the bone morphogenetic protein (BMP) signaling pathway, thus affecting embryonic development and adult bone formation (90) (Figure 2, middle).
3.5 Hippo signaling pathway
The Hippo signaling pathway is an evolutionarily conserved pathway in mammalian cells that regulates a variety of biological processes including cell growth, tissue repair, organ regeneration, inflammation, and immunity. Dysregulation of the Hippo pathway leads to various human diseases such as cancer, AIDs, and abnormal development (91–93). The Hippo pathway is also involved in a series of kinase cascades in which MST1/2 interacts with SAV1 and phosphorylates SAV1, MOB1, and LATS1/2. Furthermore, the activated LATS1/2 subsequently mediates the phosphorylation of YAP/TAZ (yes-associated protein/transcriptional coactivator with PDZ-binding motif) at multiple sites and prevents them from translocating into the nucleus, ultimately inhibiting the transcription of the genes correlated with cell proliferation and survival. The activated Hippo signaling pathway plays a tumor suppressor role, while inactivated Hippo signaling promotes tumor progression (91, 93).
Overexpression of Cul4A is prevalent in human colon cancer (CC) cells. Cul4A induces the downregulation of MST1, LAST1, and p-YAP and promotes tumor progression by inactivating the Hippo pathway (94). Furthermore, a study on hepatocellular carcinoma (HCC) showed that the long non-coding RNA (lncRNA) uc.134 can inhibit the Cul4A-mediated ubiquitination and degradation of LATS1 and promote the phosphorylation of LATS1; moreover, it activates the Hippo pathway to suppress the cell proliferation of HCC (95). CRL4Mahj promotes the ubiquitination and degradation of Wts, the ortholog of LATS1/2, therefore inactivating the Hippo pathway and contributing to the reactivation of neural stem cells (NSCs) in Drosophila (96). In other ways, PRAMEF2, a SR of Cul2-based ubiquitin ligases, promotes the degradation of LATS1 and subsequently induces the nuclear translocation of YAP. Subsequently, the nucleus-localized YAP transcriptionally activates the pro-proliferation genes to facilitate tumor progression (97). NEDD8-mediated NEDDylation of Cul7 promotes the ubiquitination and destruction of Mst1 and enables the translocation of YAP into the nucleus. Therefore, cell proliferation-related genes have been activated to promote cardiomyocyte proliferation and ventricular chamber maturation (98). Large tumor suppressor kinase (LATS) phosphorylates YAP at Ser127 and Ser381 and promotes the binding of YAP with 14-3-3 and cytoplasmic retention. The phosphorylated YAP will then recruit the E3 ubiquitin ligase SCFβ-TrCP and be destroyed in a proteasome-dependent manner. In addition, the NAE inhibitor MLN4924 could inhibit the activity of CRL4DCAF to inhibit the degradation of LATS by CRL4DCAF, also promoting the phosphorylation and inactivation of YAP. MLN4924 in combination with the mammalian target of rapamycin–phosphatidylinositol-3-kinase (mTOR/PI3K) inhibitor GDC-0980 significantly suppresses the proliferation of NF2-mutant malignant pleural mesothelioma (MPM) cells (99). Moreover, previous studies indicated that FBW7 promoted the ubiquitination and destruction of YAP in HCC and KrasG12D-driven pancreatic cancer. YAP silencing inhibited the tumorigenesis induced by FBW7 depletion in KrasG12D-dependent pancreatic cancer (100, 101). Therefore, both the cytoplasmic location and the degradation of YAP by UPS inhibit its oncogenic functions in the Hippo pathway (102). The apical polarity protein Crumbs (Crb) promotes the phosphorylation and the subsequent degradation of the Moesin domain protein Expanded (Ex) by SCFSlimb/β-TrCP, which inactivates the Hippo pathway through inhibiting the phosphorylation of the Hpo–Wts–Yki cascade (103). TAZ, another transcription coactivator, could also be phosphorylated by LATS, which primes it for further phosphorylation by CKI at the phosphorylation degron and recruits the SCFβ-TrCP E3 ligase for ubiquitination and degradation (104). Similarly, the PI3K/AKT signaling pathway modulates the protein abundance of TAZ through inhibiting the activity of GSK3, which catalyzes the phosphorylation of the N-terminal domain of TAZ and promotes its degradation by SCFβ-TrCP (105) (Figure 2, right).
3.6 Autophagy signaling pathway
Autophagy is a fundamental catalytic process of mammalian cells that contributes to the elimination of dysfunctional organelles, pathological proteins, and invading microbes upon stimulation by hypoxia, oxidative stress, and infection (106–108). Autophagy plays an important part in the pathogenesis of neurodegenerative disorders, AIDs, and carcinomas. The process of autophagy involves a series of autophagy-related proteins (ATGs), the autophagosome infusion with lysosomes, and the hydrolyzed cargos (106, 107, 109).
CRL4Ambra1, as an E3 ligase, catalyzes the K63 linkage ubiquitination of Beclin1 under stimulation of starvation and promotes the interaction between Beclin1 and vacuolar protein sorting 34 (VPS34), which serves as a central component in the initiation of autophagy. However, WASH (Wiskott–Aldrich syndrome protein) competes with Ambra1 to bind to Beclin, reduces the K63 linkage of ubiquitination of Beclin1, induces the dissociation between Beclin1 and VPS34, and finally inhibits autophagy (110). In addition to being an E3 ligase, Ambra1, as a regulator, influences the activation and termination of autophagy by switching the interaction with Cul4 or Cul5. In detail, Cul4 binds with and reduces the abundance of the Ambral1 protein and inhibits the initiation of autophagy. Upon stimulation of autophagy, Ambral1 dissociates from Cul4 and binds with and inhibits Cul5, subsequently leading to the accumulation of the mTOR inhibitor DEPTOR and inactivating autophagy (111). However, SCFβ-TrCP, another CRL, influences the initiation of autophagy through the negative regulation of the protein abundance of DEPTOR. SCFβ-TrCP mediates ubiquitination and the subsequent degradation of DEPTOR after phosphorylation in its conserved degron in an mTOR- and CK1-dependent manner (112–114). Furthermore, DNA damage triggers the activation of cyclin-dependent kinase (CDK). The activated CDK further promotes the phosphorylation of VPS34, which facilitates the ubiquitination and degradation of VPS34 by SCFFBXL20, finally inhibiting autophagy. Interestingly, DNA damage could also trigger the p53-mediated transcriptional activation of FBXL20 and regulate the initiation of autophagy (115). CRL3ZBTB16 could especially promote the ubiquitination and degradation of ATG14L, which plays a key role in the formation of the phagophore nucleation PI3KC3 complex I and promotes the initiation of autophagy. The antagonist of G-protein-coupled receptors (GPCRs) activates GSK3β, which decreases ZBTB16 and elevates the protein abundance of ATG14L, which then promotes autophagy and benefits treatment outcomes of neurodegeneration (116). CRL3KLHL16 promotes the K48 linkage ubiquitination and degradation of ATG16L and influences the elongation of the autophagosome (117). Moreover, both the CRL4A- and CRL4B-based E3 ligase could induce the destruction of WIPI2 through the 26S proteasome to inhibit autophagosome biogenesis during mitosis (118). SCFFBXO27, a glycoprotein-specific E3 ligase, regulates the ubiquitination of the lysosomal glycoproteins LAMP1/2, GSN, PSAP, and TMEM192 and the SNARE (SNAP receptor) proteins VAMP3 and VAMP7 upon lysosomal damage. After ubiquitin modification, the lysosomal proteins will recruit the autophagic machinery to launch lysophagy (119). The ubiquitination mediated by CRLs plays a central role in the onset and elongation of autophagy and influences selective autophagy, such as mitophagy and lysophagy (13, 120) (Figure 3).
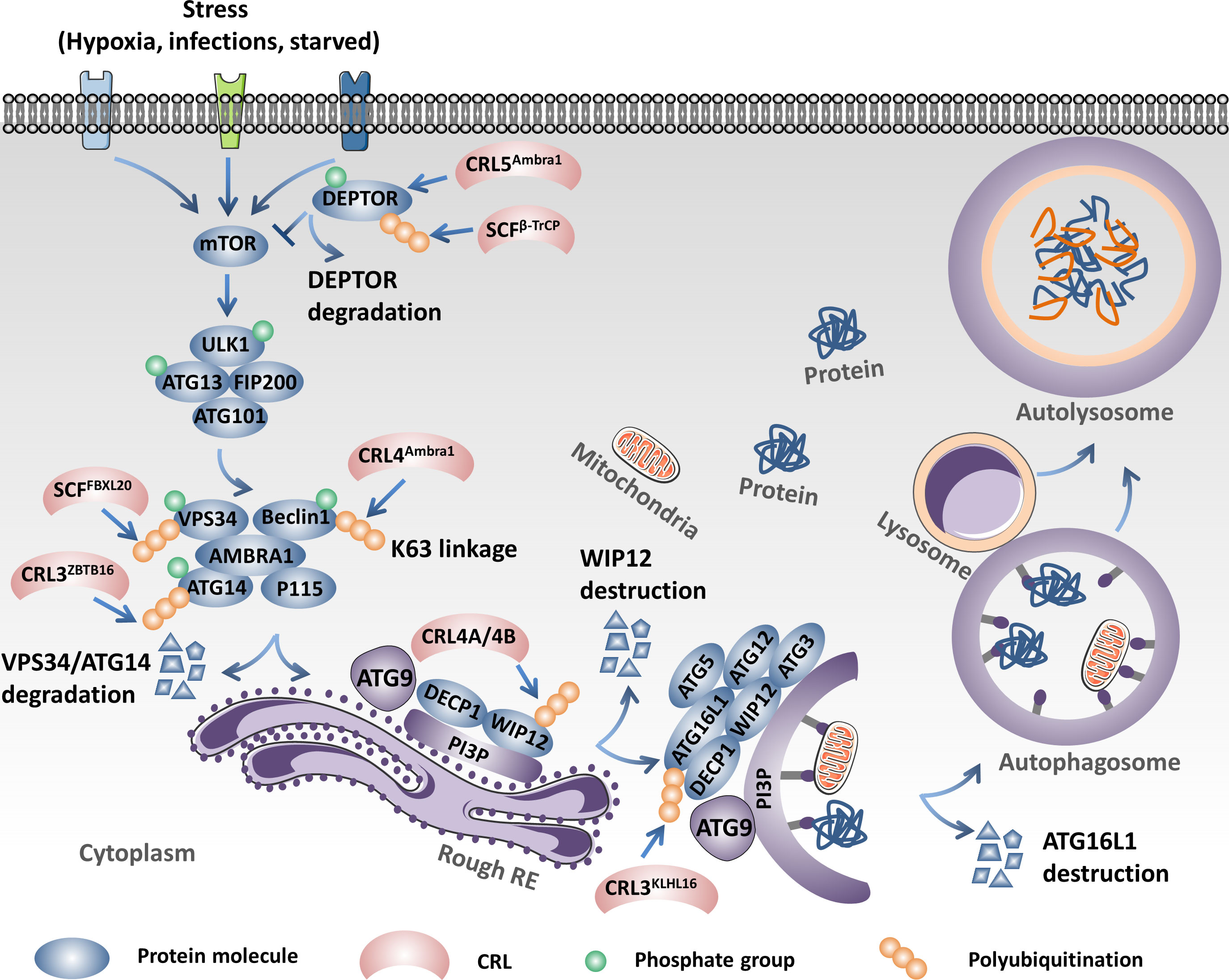
Figure 3 Cullin-RING ligases (CRLs) regulate the initiation and elongation of autophagy. Ambral1 binds with and inhibits Cul5, which leads to the accumulation of the mammalian target of rapamycin (mTOR) inhibitor DEPTOR, inactivating autophagy. However, SCFβ-TrCP mediates the ubiquitination and proteolysis of DEPTOR and activates autophagy. SCFFBXL20 facilitates the ubiquitination and degradation of VPS34 depending on the phosphorylation catalyzed by cyclin-dependent kinase (CDK). CRL4Ambra1 catalyzes the K63 linkage ubiquitination of Beclin1 under stimulation of starvation and promotes the interaction between Beclin1 and VPS34. CRL3ZBTB16 specifically promotes the ubiquitination and degradation of ATG14L. CRL4A- and CRL4B-based E3 ligases could induce the destruction of WIPI2 through the 26S proteasome to inhibit autophagosome biogenesis. CRL3KLHL16 promotes the K48 linkage ubiquitination and degradation of ATG16L1 and influences the elongation of the autophagosome.
3.7 Caspase signaling pathway
Caspases, members of the conserved cysteine protease family, play a critical roles in regulating cell apoptosis and inflammation (121, 122). The caspases involved in apoptosis signaling are caspases 3, 6, 7, 8, 9, and 10. Caspases 8, 9, and 10 are classified as initiator caspases, while caspases 3, 6, and 7 are executioner caspases. In humans, the caspases involved in inflammation are caspases 1, 3, 4, 5, and 12. Activation of the caspases in apoptosis or inflammation will induce programmed cell death or the release of inflammatory cytokines, including high-mobility group box (HMGB), IL-1β, and IL-18, which affect the development and progression of carcinomas and AIDs (121–123).
The NAE inhibitor MLN4924 inhibits the NEDDylation of Cullins and inactivates CRLs, resulting in the accumulation of activating transcription factor 4 (ATF4). ATF4 activates the transcription factor CHOP and then transcriptionally activates death receptor 5 (DR5) and caspase 8, which ultimately induces the extrinsic apoptosis of esophageal squamous cell carcinoma (ESCC) cells (124). It has been reported that the knockdown of CAND1 will activate caspase 8 and promote cell apoptosis in HCC via activating the CRLs (125). Interestingly, the Cul3-based ubiquitin ligase-mediated poly-ubiquitination and activation of caspase 8 is essential for the assembly of the death-inducing signaling complex (DISC) under the treatment of extrinsic apoptosis signaling. P62 associates with the DISC and promotes the aggregation of caspase 8 modified by Cul3 (126). SCFSkp2 promotes the ubiquitination and degradation of FLIP(L) and interrupts the interaction of p43-FLIP(L) and DISC. It also modulates the apoptosis mediated by TRAIL-R2 (DR5) (127). SCFβ-TrCP is the E3 ubiquitin ligase of pro-caspase 3 and mediates its degradation by the 26S proteasome, which protects cells from apoptosis (128) (Figure 4).
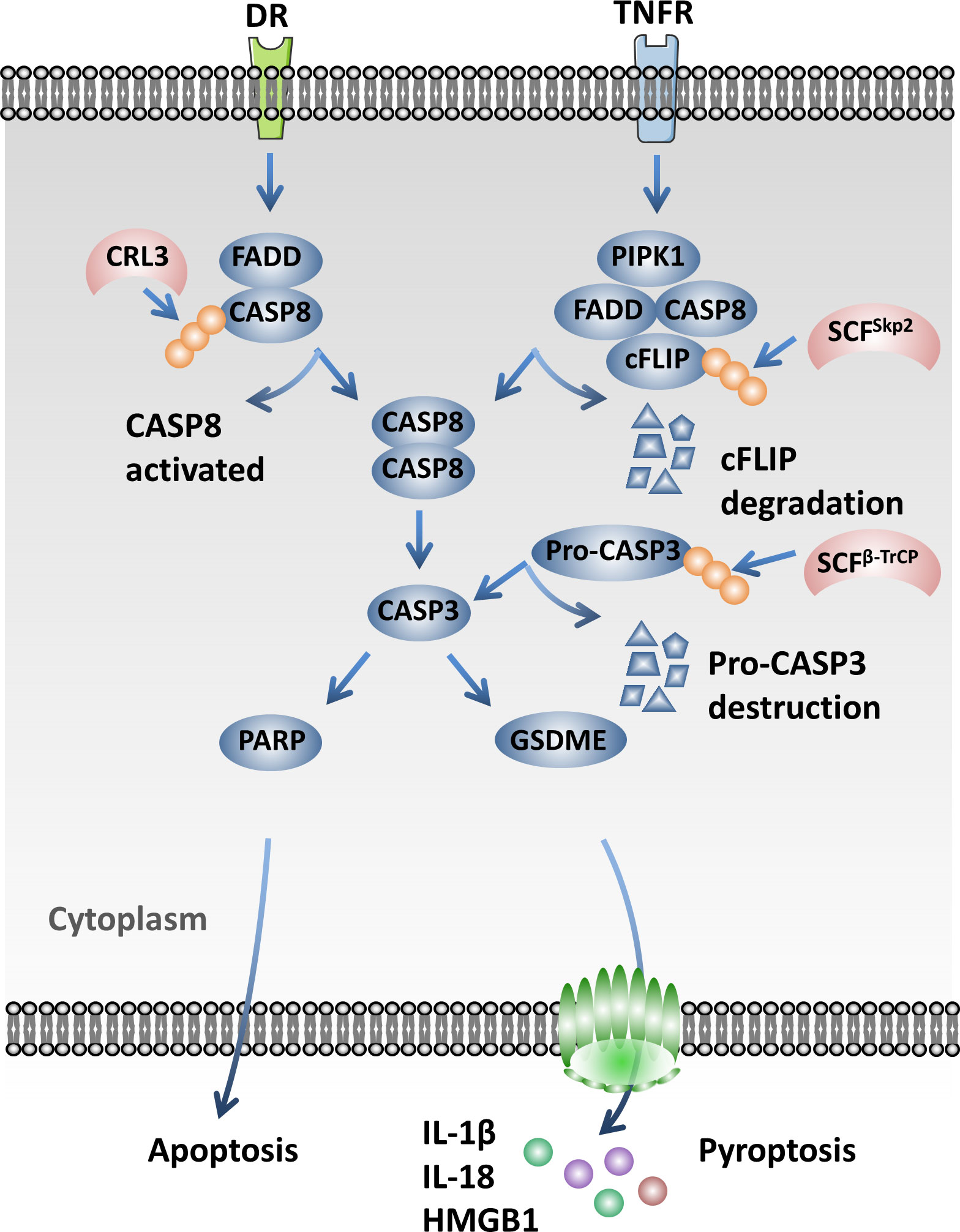
Figure 4 Cullin-RING ligases (CRLs) influence pyroptosis through regulating the caspase signaling pathway. Cul3-based ubiquitin ligases mediate the poly-ubiquitination and activation of caspase 8, which is essential for the assembly of the death-inducing signaling complex (DISC) under the treatment of extrinsic apoptosis signaling. SCFSkp2 promotes the ubiquitination and degradation of FLIP(L). SCFβ-TrCP mediates the degradation of pro-caspase 3 by the 26S proteasome and protects cells from apoptosis.
4 Cullin-RING ligases in autoimmune diseases
4.1 Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is termed based on the uncontrolled autoantibodies specific for the nuclear autoantigens, including double-strand DNA and the associated proteins induced to produce immune complex and tissue damage (129). SLE is a systemic AID that is triggered by genetic factors combined with a variety of environmental risk factors, such as exposure to ultraviolet radiation, smoking, infections, and environmental pollutants (129, 130). Autoreactive T and B cells produce diverse cytokines and autoantibodies that break the immune tolerance and induce immune dysfunction, promoting the occurrence of SLE. However, the pathological mechanisms of SLE remain largely unknown (129, 131).
Ubiquitination, as an important form of the PTM of proteins, regulates protein abundance, activity, subcellular localization, and their interactions, as well as a variety of signaling pathways. Ubiquitination plays a pivotal role in the pathogenesis of SLE (132, 133). A phase I clinical trial showed that the expression of Aiolos and Ikaros was significantly higher in patients with SLE than in healthy volunteers. CRLs, the largest family of E3 ubiquitin ligases, play a central role in the management of ubiquitin modification. CRBN, one of the SRs of CRL4, specifically promotes the transcriptional factors Ikaros (IKZF1) and Aiolos (IKZF3) for proteasomal degradation, which leads to the inactivation of T cells through downregulating the expression of IL-2. Lenalidomide and pomalidomide are immunomodulatory agents that activate T cells by promoting the ubiquitination and degradation of IKZF1 mediated by CRL4CRBN (134). Therefore, lenalidomide is used as an effective drug for the treatment of myeloma, which works by mediating the degradation of IKZF1 and IKZF3 in a CRL4CRBN-dependent manner (135).
Recent studies have shown that Iberdomide (CC-220), a new modulator of CRBN, interacts with CRBN at a higher affinity than lenalidomide or pomalidomide. It is used in the treatment of SLE. CC-220 promotes the binding of Aiolos and Ikaros to CRBN E3 ligase and the subsequent degradation by the 26S proteasome. CC-220 finally decreases the cell proliferation, plasmablast differentiation, and the immunoglobulin G (IgG) secretion of B cells stimulated by B-cell activating factor (BAFF) and CD40L, resulting in the attenuation of the progression of SLE (136, 137). CC-220 can help reduce the protein levels of Aiolos and Ikaros in B cells, T cells, and monocytes. In addition, CC-220 could markedly decrease the absolute population of CD19+ B cells and the expression of IL-1β and increase the production of IL-2 ex vivo (138). FBXW7 acts as a tumor suppressor by promoting the ubiquitination and degradation of various substrates including c-Myc, c-Jun, cyclin E, and MCL-1, which usually function as oncoproteins and promote tumor growth and survival (139, 140). SCFFBXW7 E3 ligase was also found to play a crucial role in the development of SLE. In tetramethylpentadecane (TMPD)-induced SLE, SCFFBXW7 induced cell apoptosis by promoting the K48 linkage ubiquitination and degradation of MCL-1. The apoptosis of peritoneal macrophages and neutrophils was lower in myeloid cell-specific Fbxw7-deficient (Lysm+Fbxw7f/f) C57BL/6 mice than in wild-type (WT) mice. In addition, the accumulation of immune complex, glomerulonephritis, the proliferation of glomerular mesangial cells, and the base membrane thickness decreased in the kidney of Lysm+Fbxw7f/f mice. Fewer anti-Sm/RNP and anti-ANA autoantibodies and a reduced expression of major histocompatibility complex (MHC) II in B cells were found in Lysm+Fbxw7f/f mice (141) (Table 1).
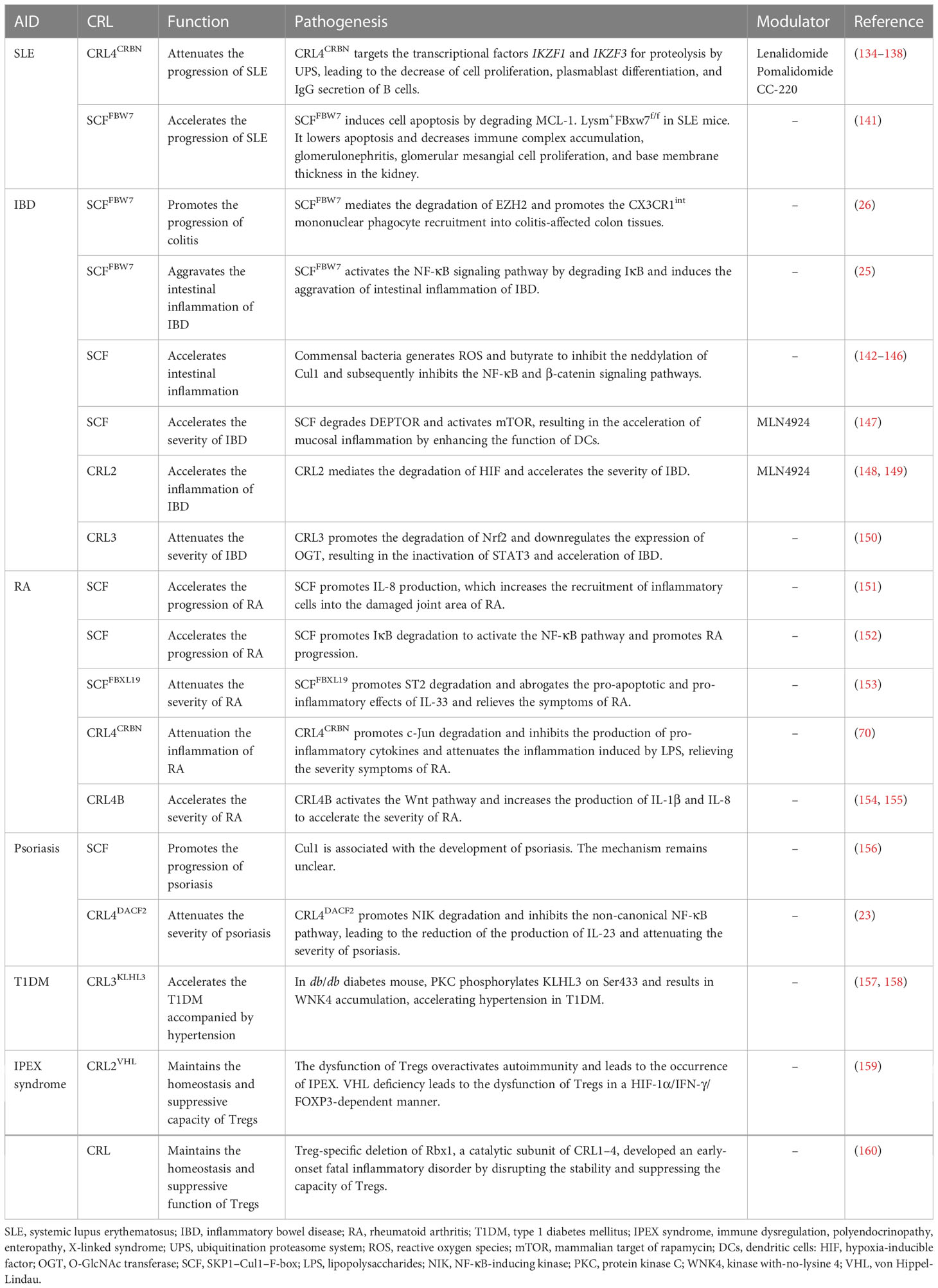
Table 1 Multiple functions of Cullin-RING ligases (CRLs) in autoimmune diseases (AIDs).
4.2 Inflammatory bowel disease
Crohn’s disease (CD) and ulcerative colitis (UC) are two forms of IBD that comprise a type of chronic and relapsing intestinal inflammation disease (161, 162). The pathological mechanism of IBD is complicated, which includes alterations of genomic and environmental risk factors, destruction of the gut microbiome barrier, and immune dysfunction (161–163).
PTMs such as phosphorylation, acetylation, and ubiquitination play critical roles in the pathogenesis and progression of IBD by modulating a variety of signaling pathways involved in its regulation (14, 164). CRLs, the largest family of E3 ubiquitin ligases, have been reported to regulate the development of IBD. SCFFBW7, one of the most important CRLs involved in the regulation of the inflammation pathway, is significantly correlated with the severity of IBD. Notably, SCFFBW7 promotes the progression of colitis through mediating the ubiquitination and degradation of the histone-lysine-N-methyltransferase enhancer of zeste homolog 2 (EZH2). The degradation of EZH2 results in the inhibition of H3K27me3 modification and increases the expression of CCL2 and CCL7 in CXCR1hi macrophages, subsequently promoting the recruitment of CX3CR1int pro-inflammatory mononuclear phagocytes (MPhs) into colitis-affected colon tissues. Myeloid deficiency of FBW7 significantly alleviates the colitis induced either by dextran sodium sulfate (DSS) or 2,6,4-trinitrobenzene sulfonic acid (TNBS) in mouse models (26). On the other hand, SCFFBW7 activates the NF-κB signaling pathway by promoting the 26S proteasome-mediated IκB degradation and aggravates the intestinal inflammation in IBD. Interestingly, miR-129 could negatively regulate the expression of FBW7 through promoting the 3′-UTR for degradation. Therefore, the upregulation of miR-129 reduces the inflammation of colitis induced by TNBS in a FBW7-dependent manner (25).
In addition, commensal bacteria influence the host intestinal homeostasis and play a pivotal role in regulating mucosal immunity and inflammation. A pioneer work by Neish et al. reported that prokaryotic microflora attenuated the inflammation of IECs by inhibiting the activation of the NF-κB signaling pathway through the blockage of the ubiquitination and degradation of IκBα (142). The group further found that commensal bacteria generated reactive oxygen species (ROS) and butyrate to inhibit the NEDDylation of Cul1 by inactivating the NEDD8-conjugating enzyme Ubc12, which led to the consequent blockage of the NF-κB and β-catenin signaling pathways (143, 144). The probiotic bacteria Lactobacillus rhamnosus GG (LGG) can induce the production of ROS and consequently inactivate Ubc12 and inhibit the NF-κB signaling pathway through blocking the NEDDylation of Cul1, which contributes to preventing necrotizing enterocolitis (NEC) and relieving IBD in neonates (145). Inhibition of NEDDylation modification could reduce mucosal inflammation and alleviate the severity of IBD in mice because inhibition of NEDDylation blocks the degradation of the mTOR inhibitor DEPTOR by the Cul1-associated CRL. The inactivation of mTOR subsequently inhibits the function of dendritic cells (DCs) and induces their apoptosis in an mTOR pathway-dependent manner (147).
Human umbilical cord mesenchymal stem cell-derived exosomes (hucMSC-exosome) contain high levels of miR-326 that could attenuate the NEDDylation of Cul1 and consequently inhibit the NF-κB signaling pathway, contributing to the relief of IBD induced by DSS in mice (146). Adrenomedullin (ADM) downregulates the inflammation of IECs due to the stabilization of HIF mediated by the deNEDDylation of Cul2 (148). In addition, pharmacological inhibition of NEDDylation by MLN4924 could stabilize HIF through the inhibition of Cul2 NEDDylation, which potentially attenuates IBD. Furthermore, human deNEDDylase-1 (DEN-1) could reduce the inflammatory response by promoting the deNEDDylation of Cullins (149).
The JAK/STAT signaling pathway plays a pivotal role in regulating inflammation. STAT3 has been reported to be closely correlated with the pathogenesis of IBD. Deficiency of the myeloid-derived STAT3 promotes the development of chronic enterocolitis through activating Th1 cells (165, 166). Similarly, deficiency of the IEC-specific STAT3 accelerates mucosal inflammation (167). Cul3-based E3 ubiquitin ligase promotes the proteolysis of nuclear factor erythroid 2-related factor 2 (Nrf2) and subsequently downregulates the expression of O-GlcNAc transferase (OGT), which is responsible for the O-GlcNAcylation of STAT3. Li et al. reported that the O-GlcNAcylation of STAT3 on T717 inhibited its phosphorylation and consequently accelerated the intestinal chronic inflammation in Cul3-deficient myeloid cells (150) (Table 1).
4.3 Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic systemic AID. Joint pain and swelling are the prominent symptoms of RA. Anti-citrullinated protein antibodies (ACPAs), the RA-correlated autoantibody rheumatoid factor (RF), and C-reactive proteins (CRPs) are usually upregulated in patients with RA. The pathological mechanisms of RA include the change of the susceptibility genes and environmental risk factors. Of these, HLA-DRB1 is the most important genetic risk factor, while smoking is the main environmental risk factor (168, 169).
Importantly, Cul1 has been identified as one of the susceptibility genes of RA. Cul1 is often highly expressed in T and B lymphocytes. Suppression of the expression of Cul1 in T cells will reduce the production of IL-8. It has been reported that IL-8 plays a pivotal role in regulating the recruitment of inflammatory cells in the damaged joint area in RA (151). Another piece of research has also reported the close association of a promoter and two intronic polymorphisms of Cul1 with RA and the methotrexate response in patients with RA (170). Bmi1 regulates the stability of IκBα through binding with the SCF E3 ubiquitin complex via its N-terminus after the phosphorylation by IKKα/β. Consistently, Bmi1 deficiency inhibits the NF-κB pathway via the accumulation of IκBα and attenuates arthritis (152).
Cytokines regulate the progression of RA through influencing the function of multiple immune cells, including T and B lymphocytes and mast cells. The IL-33/ST2 axis is closely correlated with the severity of RA. Pro-inflammatory cytokines are secreted by mast cells upon the stimulation of IL-33. Moreover, IL-33 could induce macrophages to produce chemokines and recruit neutrophils in RA-affected tissues. ST2 is the receptor of IL-33, and ST2 deficiency can relieve the symptoms of RA. IL-33 is highly correlated with the response of patients with RA to tumor necrosis factor (TNF) inhibitors (171, 172). SCFFBXL19 was found to promote the ubiquitination and degradation of ST2 by the 26S proteasome after phosphorylation on Ser442 by GSK3β and to abrogate the pro-apoptotic and pro-inflammatory effects of IL-33 (153).
CRL4CRBN attenuates the inflammation induced by lipopolysaccharides (LPS) in a c-Jun-dependent manner and relieves the symptoms of inflammation-related diseases such as RA. CRL4CRBN promotes the K48 linkage ubiquitination and degradation of c-Jun, therefore inhibiting the production of pro-inflammatory cytokines such as COX-2, iNOS, IL-1β, and IL-6 (70). Cul4B was significantly upregulated in the synovium and fibroblast-like synoviocytes (FLS) of adjuvant-induced arthritis (AIA) rats, which is a RA rat model. Cul4B promotes the activation of the canonical Wnt signaling pathway and the production of the pro-inflammatory cytokines IL-1β and IL-8, accelerating the severity of AIA. Therefore, MiR-101-3p plays an important role in anti-inflammation in AIA by reducing the expression of Cul4B (154). The elevated expression of circ_0015756 in FLS and the synovium of RA upregulates the expression of Cul4B by inhibiting the expression of miR-942-5p, consequently promoting the progression of RA by activating the canonical Wnt signaling pathway (155) (Table 1).
4.4 Psoriasis
Psoriasis is a prevalent chronic inflammatory skin disease worldwide. The pathogenesis of psoriasis is complicated. Genetic susceptibility, depression, smoking, obesity, and streptococcal infection could induce the occurrence and development of psoriasis.
IL-17 and IL-23 are the key inducers of psoriasis. Targeting IL-17, IL-23, and TNF-α has been considered as the predominant therapeutic strategy for the treatment of psoriasis (173, 174). A study based on the microarray data of a cDNA library indicated that Cul1 is highly associated with the development of psoriasis (156). CRL4DCAF2 negatively regulates the production of IL-23 through the ubiquitination and degradation of NIK, which is associated with the non-canonical NF-κB pathway. Therefore, DCAF2 deficiency induced the accumulation of NIK, promoted the activation of the NF-κB non-canonical signaling pathway, and increased the production of IL-23. In a mouse model, MLN4924 treatment accelerated the severity of psoriasis through the inactivation of CRLs, including CRL4DCAF2. Furthermore, DCAF2 DC-conditional knockout mice showed increased susceptibility to AIDs (23) (Table 1).
4.5 Type 1 diabetes
Type 1 diabetes mellitus (T1DM) is IDDM and is an organ-specific AID and a severe metabolic disease. T1DM is frequently accompanied by the occurrence of hypertension. Autoreactive T cells and other components of the immune system attack pancreatic B cells to induce the occurrence of T1DM. Currently, multiple susceptibility genes and environment risk factors associated with the development of T1DM have been identified. Human leukocyte antigen (HLA) is one of the major susceptibility genes related to the autoantigen recognition and immune tolerance of T cells. Infections, commensal bacteria, and diet are the important environmental risk factors that influence the development of T1DM by modulating the functions of the immune system (175, 176).
Cul3KLHL3 is a SR of E3 ubiquitin ligase and plays an essential role in hypertension. Kinase with-no-lysine 4 (WNK4), a bona fide substrate of KLHL3, modulates the activation of the Na–Cl cotransporter (NCC). The phosphorylation of KLHL3 on Ser433 of the Kelch domain by protein kinase C (PKC) inhibited the interaction of WNK4 and KLHL3 (177). The phosphorylation of KLHL3 by PKC was also observed in the kidney of db/db mice (157). In streptozotocin-induced T1DM, the expression of KLHL3 was significantly decreased, therefore inducing the activation of the WNK–NCC cascade. Furthermore, KLHL3 plays a pivotal role in renal sodium reabsorption in conditions of T1DM (158) (Table 1).
4.6 Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked syndrome
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a multisystem AID with diverse clinical syndromes including enteropathy, skin manifestations, and endocrinopathy. Loss-of-function mutations in the transcription factor forkhead box P3 (FOXP3) are the dominant factors that induce the occurrence of IPEX (178–180). FOXP3 is mainly expressed on CD4+CD25+ regulatory T cells (Tregs) and modulates their development and functions. FOXP3 deficiency leads to the downregulation of a few of the core signature genes in Tregs and suppresses the activity of these cells. The dysfunction of Tregs overactivates autoimmunity and leads to the occurrence of IPEX (181).
A previous study indicated that the von Hippel-Lindau (VHL) E3 ubiquitin ligase played a pivotal role in regulating the functions of Tregs. VHL interacts with Elongins B and C, Cul2, and Rbx1 to form the CRL2 complex, which mediates the ubiquitination and subsequent degradation of HIF-1α. Conditional knockout of VHL in Tregs leads to the accumulation of HIF-1α, which induced the production of IFN-γ. VHL deficiency is accompanied with the downregulation of FOXP3, which can be reversed by IFN-γ deprivation. In conclusion, VHL modulates the stability and immunosuppressive functions of Tregs in a HIF-1α/IFN-γ/FOXP3-dependent manner (159). In addition, a recent study has suggested that Tregs with depletion of Rbx1, a catalytic subunit of CRL1–4, developed an early-onset fatal inflammatory disorder due to their disrupted stability and suppressive capacity. Moreover, deficiency of the Ube2m, but not the Ube2f, NEDDylation conjugation enzyme in Tregs presented similar but less severe phenotypes compared to Rbx1 deletion. Therefore, the Ube2m–Rbx1 axis plays a crucial role in regulating the homeostasis and function of Tregs (160) (Table 1).
5 Conclusion and outlook
AIDs are chronic inflammation-associated diseases induced by the dysfunction of the immune system. Overactivated autoreactive immune cells such as T cells, DCs, and macrophages produce pro-inflammatory cytokines, including IL-1β, IL-6, IL-17, IL-23, COX-2, and iNOS, or chemokines that promote the recruitment of inflammation-associated cells in the affected areas of AIDs. Ultimately, the tissue injury and the severity of AIDs are further accelerated (5). Hyper-activation of the inflammation-associated signaling pathways such as NF-κB, JAK/STAT, MAPK, and TGF-β is prevalent during the occurrence and progression of AIDs, including IBD, SA, and SLE.
Given that the activation of the inflammation-associated signaling pathways plays critical roles in the initiation and progression of AIDs, it is important to explore the underlying mechanisms of the abnormal activation of the key genes related to AIDs. These mechanisms include the transcriptional regulation of genes and the PTM of the correlated proteins. Ubiquitination, one of the PTMs of proteins, plays a pivotal role in regulating their stability, activation, and localization. The abnormal regulation of ubiquitination is closely related to the activation of the genes in the inflammatory pathways and the progression and recurrence of AIDs (10). CRLs comprise the largest class of E3 ubiquitin ligases and include more than 300 members that regulate the stability of about 20% proteins in a proteasome-dependent manner in mammalian cells (50). The results from clinical and animal model studies revealed that the dysregulation of CRLs usually functions as an inducer or an inhibitor in the development of inflammation and in the progression of AIDs. For instance, SCFFBW7 functions as a regulator of AIDs in the following aspects: 1) promoting the degradation of IκB, EZH2, and MCL-1; 2) accelerating the pro-inflammatory NF-κB signaling pathway; 3) promoting the recruitment of MPhs into colitis-affected colon tissues; 4) increasing the apoptosis of macrophages and neutrophils; and 5) leading to the accumulation of immune complex and inducing the aggravation of SLE (25, 26, 141). On the other hand, CRL4CRBN promotes the degradation of IKZF1, IKZF3, or c-Jun, which leads to the reduction of the secretion of IgG or the pro-inflammatory cytokines COX-2, iNOS, IL-1β, and IL-6, consequently attenuating SLE or RA (70, 138) (Table 1).
The development of small molecules targeting CRLs will be a promising therapeutic approach in the clinical intervention of AIDs. In line with this notion, treatment with the NEDDylation inhibitor MLN4924 effectively decreases inflammation through inhibiting the activation of SCF E3 ligases in IBD (143, 144, 147). Immunomodulatory imide drugs (IMiDs) such as thalidomide and its derivatives lenalidomide, pomalidomide, and Iberdomide (CC-220) act as molecular glue degraders to promote the ubiquitination and degradation of IKZF1 and IKZF3 by CRL4CRBN, ultimately relieving the symptoms of SLE (134, 137, 138, 182) (Table 1). Proteolysis-targeting chimera (PROTAC) technology is another novel targeted protein degradation (TPD) method that has rapidly developed in recent years. As bifunctional small molecules, PROTACs induce the ubiquitination and proteolysis of target proteins by E3 ubiquitin ligases (183). In addition, small molecules targeting the interface between CAND1 and Cullins comprise a new pharmocological strategy based on UPS. A recent study has shown that the chemical probe C60 perturbs the normal interaction between CAND1 and Cul1, resulting in the accumulation of p53, thus inducing the reactivation of EBV from latency (184). Either small-molecule inhibitors or the emerging TPD technology involving molecular glue degraders and PROTACs could be effective thrapeutic strategies to benefit the intervention of inflammation and AIDs. In particular, the TPD method, a novel pharmacological strategy to degrade the protein of interest (POI) using small-molecule degraders via hijacking CRLs, could be promising for the treatment of AIDs in the near future.
Author contributions
XYZ and XD conceived the manuscript. XYZ wrote the manuscript with partial help from YL, TZ, and YT. XLZ, Y-GY, and XD edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partly supported by the National Key R&D Program of China (2021YFA1100700); a grant from the Department of Human Resource and Social Security of Jilin Province (2022DJ02) and Bethune Medical Department of Jilin University (2022JBGS01); the National Natural Science Foundation of China (No: 81972558 and No: 81802815); the “Startup Funding of First Hospital, JLU”; and the Natural Science Foundation of Jilin Province (No: 20200201367JC, No: 20210204165YY, and No: 20200201473JC).
Acknowledgments
We thank the other members of the Dai Laboratory for their critical reading of the manuscript and for useful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jiang J, Zhao M, Chang C, Wu H, Lu Q. Type I interferons in the pathogenesis and treatment of autoimmune diseases. Clin Rev Allergy Immunol (2020) 59(2):248–72. doi: 10.1007/s12016-020-08798-2
2. Yasunaga M. Antibody therapeutics and immunoregulation in cancer and autoimmune disease. Semin Cancer Biol (2020) 64:1–12. doi: 10.1016/j.semcancer.2019.06.001
3. Barnas JL, Looney RJ, Anolik JH. B cell targeted therapies in autoimmune disease. Curr Opin Immunol (2019) 61:92–9. doi: 10.1016/j.coi.2019.09.004
4. Goulabchand R, Hafidi A, Van de Perre P, Millet I, Maria ATJ, Morel J, et al. Mastitis in autoimmune diseases: Review of the literature, diagnostic pathway, and pathophysiological key players. J Clin Med (2020) 9(4):958. doi: 10.3390/jcm9040958
5. Vojdani A. A potential link between environmental triggers and autoimmunity. Autoimmune Dis (2014) 2014:437231. doi: 10.1155/2014/437231
6. Liu R, Du S, Zhao L, Jain S, Sahay K, Rizvanov A, et al. Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Front Immunol (2022) 13:996469. doi: 10.3389/fimmu.2022.996469
7. Khan U, Ghazanfar H. T Lymphocytes and autoimmunity. Int Rev Cell Mol Biol (2018) 341:125–68. doi: 10.1016/bs.ircmb.2018.05.008
8. Kuwabara T, Matsui Y, Ishikawa F, Kondo M. Regulation of T-cell signaling by post-translational modifications in autoimmune disease. Int J Mol Sci (2018) 19(3):819. doi: 10.3390/ijms19030819
9. Dehnavi S, Sadeghi M, Johnston TP, Barreto G, Shohan M, Sahebkar A. The role of protein SUMOylation in rheumatoid arthritis. J Autoimmun (2019) 102:1–7. doi: 10.1016/j.jaut.2019.05.006
10. Beck DB, Werner A, Kastner DL, Aksentijevich I. Disorders of ubiquitylation: unchained inflammation. Nat Rev Rheumatol (2022) 18(8):435–47. doi: 10.1038/s41584-022-00778-4
11. Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ (2021) 28(2):427–38. doi: 10.1038/s41418-020-00648-0
12. Song Y, Liu Y, Pan S, Xie S, Wang ZW, Zhu X. Role of the COP1 protein in cancer development and therapy. Semin Cancer Biol (2020) 67(Pt 2):43–52. doi: 10.1016/j.semcancer.2020.02.001
13. Chen RH, Chen YH, Huang TY. Ubiquitin-mediated regulation of autophagy. J BioMed Sci (2019) 26(1):80. doi: 10.1186/s12929-019-0569-y
14. Chen R, Pang X, Li L, Zeng Z, Chen M, Zhang S. Ubiquitin-specific proteases in inflammatory bowel disease-related signalling pathway regulation. Cell Death Dis (2022) 13(2):139. doi: 10.1038/s41419-022-04566-6
15. Lewis M, Vyse S, Shields A, Boeltz S, Gordon P, Spector T, et al. Effect of UBE2L3 genotype on regulation of the linear ubiquitin chain assembly complex in systemic lupus erythematosus. Lancet (2015) 385(Suppl 1):S9 doi: 10.1016/S0140-6736(15)60324-5
16. Zheng N, Shabek N. Ubiquitin ligases: Structure, function, and regulation. Annu Rev Biochem (2017) 86:129–57. doi: 10.1146/annurev-biochem-060815-014922
17. Scott DC, Rhee DY, Duda DM, Kelsall IR, Olszewski JL, Paulo JA, et al. Two distinct types of E3 ligases work in unison to regulate substrate ubiquitylation. Cell (2016) 166(5):1198–214 e24. doi: 10.1016/j.cell.2016.07.027
18. Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol (2014) 21(4):301–7. doi: 10.1038/nsmb.2780
19. Duda DM, Scott DC, Calabrese MF, Zimmerman ES, Zheng N, Schulman BA. Structural regulation of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol (2011) 21(2):257–64. doi: 10.1016/j.sbi.2011.01.003
20. Yu Q, Jiang Y, Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B (2020) 10(5):746–65. doi: 10.1016/j.apsb.2019.09.005
21. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature (2009) 458(7239):732–6. doi: 10.1038/nature07884
22. Fan Y, Huo X, Guo B, Zhang X, Yang Y, Lian J, et al. Cullin 4b-RING ubiquitin ligase targets IRGM1 to regulate wnt signaling and intestinal homeostasis. Cell Death Differ (2022) 29(9):1673–88. doi: 10.1038/s41418-022-00954-9
23. Huang T, Gao Z, Zhang Y, Fan K, Wang F, Li Y, et al. CRL4(DCAF2) negatively regulates IL-23 production in dendritic cells and limits the development of psoriasis. J Exp Med (2018) 215(8):1999–2017. doi: 10.1084/jem.20180210
24. Bulatov E, Valiullina A, Sayarova R, Rizvanov A. Promising new therapeutic targets for regulation of inflammation and immunity: RING-type E3 ubiquitin ligases. Immunol Lett (2018) 202:44–51. doi: 10.1016/j.imlet.2018.08.001
25. Meng Q, Wu W, Pei T, Xue J, Xiao P, Sun L, et al. miRNA-129/FBW7/NF-kappaB, a novel regulatory pathway in inflammatory bowel disease. Mol Ther Nucleic Acids (2020) 19:731–40. doi: 10.1016/j.omtn.2019.10.048
26. He J, Song Y, Li G, Xiao P, Liu Y, Xue Y, et al. Fbxw7 increases CCL2/7 in CX3CR1hi macrophages to promote intestinal inflammation. J Clin Invest (2019) 129(9):3877–93. doi: 10.1172/JCI123374
27. Harper JW, Schulman BA. Cullin-RING ubiquitin ligase regulatory circuits: A quarter century beyond the f-box hypothesis. Annu Rev Biochem (2021) 90:403–29. doi: 10.1146/annurev-biochem-090120-013613
28. Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol (2005) 6(1):9–20. doi: 10.1038/nrm1547
29. Thompson LL, Rutherford KA, Lepage CC, McManus KJ. The SCF complex is essential to maintain genome and chromosome stability. Int J Mol Sci (2021) 22(16):8544. doi: 10.3390/ijms22168544
30. Mahrour N, Redwine WB, Florens L, Swanson SK, Martin-Brown S, Bradford WD, et al. Characterization of cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to elongin BC-based ubiquitin ligases. J Biol Chem (2008) 283(12):8005–13. doi: 10.1074/jbc.M706987200
31. Kaelin WG, Iliopoulos O, Lonergan KM, Ohh M. Functions of the von hippel-lindau tumour suppressor protein. J Intern Med (1998) 243(6):535–9. doi: 10.1046/j.1365-2796.1998.00335.x
32. Xu T, Ma Q, Li Y, Yu Q, Pan P, Zheng Y, et al. A small molecule inhibitor of the UBE2F-CRL5 axis induces apoptosis and radiosensitization in lung cancer. Signal Transduct Target Ther (2022) 7(1):354. doi: 10.1038/s41392-022-01182-w
33. Zhao Y, Xiong X, Sun Y. Cullin-RING ligase 5: Functional characterization and its role in human cancers. Semin Cancer Biol (2020) 67(Pt 2):61–79. doi: 10.1016/j.semcancer.2020.04.003
34. Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J (2013) 32(17):2307–20. doi: 10.1038/emboj.2013.173
35. Cheng J, Guo J, North BJ, Tao K, Zhou P, Wei W. The emerging role for cullin 4 family of E3 ligases in tumorigenesis. Biochim Biophys Acta Rev Cancer (2019) 1871(1):138–59. doi: 10.1016/j.bbcan.2018.11.007
36. Shi L, Du D, Peng Y, Liu J, Long J. The functional analysis of cullin 7 E3 ubiquitin ligases in cancer. Oncogenesis (2020) 9(10):98. doi: 10.1038/s41389-020-00276-w
37. Pan ZQ. Cullin-RING E3 ubiquitin ligase 7 in growth control and cancer. Adv Exp Med Biol (2020) 1217:285–96. doi: 10.1007/978-981-15-1025-0_17
38. Li Z, Xiong Y. Cytoplasmic E3 ubiquitin ligase CUL9 controls cell proliferation, senescence, apoptosis and genome integrity through p53. Oncogene (2017) 36(36):5212–8. doi: 10.1038/onc.2017.141
39. Li Z, Pei XH, Yan J, Yan F, Cappell KM, Whitehurst AW, et al. CUL9 mediates the functions of the 3M complex and ubiquitylates survivin to maintain genome integrity. Mol Cell (2014) 54(5):805–19. doi: 10.1016/j.molcel.2014.03.046
40. Pei XH, Bai F, Li Z, Smith MD, Whitewolf G, Jin R, et al. Cytoplasmic CUL9/PARC ubiquitin ligase is a tumor suppressor and promotes p53-dependent apoptosis. Cancer Res (2011) 71(8):2969–77. doi: 10.1158/0008-5472.CAN-10-4300
41. Baek K, Krist DT, Prabu JR, Hill S, Klugel M, Neumaier LM, et al. NEDD8 nucleates a multivalent cullin-RING-UBE2D ubiquitin ligation assembly. Nature (2020) 578(7795):461–6. doi: 10.1038/s41586-020-2000-y
42. Lyapina GC S, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, et al. Promotion of NEDD8-CUL1 conjugate cleavage by COP9 signalosome. Science (2001) 292(5520):1382–5. doi: 10.1126/science.1059780
43. Jidong Liu MF, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell (2002) 10(6):1511–8. doi: 10.1016/S1097-2765(02)00783-9
44. Jianyu Zheng XY, Jennifer M M, Harrell SR, Shim E.H, Lykke-Andersen Karin, Wei N. CAND1 binds to unneddylated CUL1 short article and regulates the formation of SCF ubiquitin E3 ligase complex. Mol Cell (2002) 10(6):1519–26. doi: 10.1016/S1097-2765(02)00784-0
45. Wenjing Zhang HI, Quint M, Huang H, Noe LD, Gray WM. Genetic analysis of CAND1–CUL1 interactions in arabidopsis supports a role for CAND1-mediated cycling of the SCFTIR1 complex. Proc Natl Acad Sci U S A (2008) 105(24):8470–5. doi: 10.1073/pnas.0804144105
46. Liu X, Reitsma JM, Mamrosh JL, Zhang Y, Straube R, Deshaies RJ. Cand1-mediated adaptive exchange mechanism enables variation in f-box protein expression. Mol Cell (2018) 69(5):773–86 e6. doi: 10.1016/j.molcel.2018.01.038
47. Zemla A, Thomas Y, Kedziora S, Knebel A, Wood NT, Rabut G, et al. CSN- and CAND1-dependent remodelling of the budding yeast SCF complex. Nat Commun (2013) 4:1641. doi: 10.1038/ncomms2628
48. Pierce NW, Lee JE, Liu X, Sweredoski MJ, Graham RL, Larimore EA, et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of f box proteins. Cell (2013) 153(1):206–15. doi: 10.1016/j.cell.2013.02.024
49. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer (2008) 8(2):83–93. doi: 10.1038/nrc2290
50. Chen Y, Shao X, Cao J, Zhu H, Yang B, He Q, et al. Phosphorylation regulates cullin-based ubiquitination in tumorigenesis. Acta Pharm Sin B (2021) 11(2):309–21. doi: 10.1016/j.apsb.2020.09.007
51. Shi Q, Li S, Li S, Jiang A, Chen Y, Jiang J. Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc Natl Acad Sci U S A (2014) 111(52):E5651–60. doi: 10.1073/pnas.1416652111
52. Chen J, Chen ZJ. Regulation of NF-kappaB by ubiquitination. Curr Opin Immunol (2013) 25(1):4–12. doi: 10.1016/j.coi.2012.12.005
53. Won M, Byun HS, Park KA, Hur GM. Post-translational control of NF-kappaB signaling by ubiquitination. Arch Pharm Res (2016) 39(8):1075–84. doi: 10.1007/s12272-016-0772-2
54. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-kappaB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther (2020) 5(1):209. doi: 10.1038/s41392-020-00312-6
55. Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev (2012) 246(1):125–40. doi: 10.1111/j.1600-065X.2011.01088.x
56. Jeffrey T, Winston PS, Beer-Romero P, . Chu CY, . Elledge SJ, Wade Harper J. The SCFb-TRCP–ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkBa and b-catenin and stimulates IkBa ubiquitination in vitro. Genes Dev (1999) 13(3):270–83. doi: 10.1101/gad.13.3.270
57. Erika Spencer JJ, . Chen ZJ. Signal-induced ubiquitination of IkBa by the f-box protein slimb/b-TrCP. Genes Dev (1999) 13):284–94. doi: 10.1101/gad.13.3.284
58. Mude Shi HC, Inn K-S, Yang A, Zhao Z, Liang Q, . Versteeg GA, et al. Negative regulation of NF-κB activity by brain-specific TRIpartite motif protein 9. Nat Commun (2014) 5(5):4820. doi: 10.1038/ncomms5820
59. Amir RE, Haecker H, Karin M, Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene (2004) 23(14):2540–7. doi: 10.1038/sj.onc.1207366
60. Fukushima H, Matsumoto A, Inuzuka H, Zhai B, Lau AW, Wan L, et al. SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep (2012) 1(5):434–43. doi: 10.1016/j.celrep.2012.04.002
61. Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O’Connor O, et al. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol (2012) 14(4):375–85. doi: 10.1038/ncb2463
62. Arabi A, Ullah K, Branca RM, Johansson J, Bandarra D, Haneklaus M, et al. Proteomic screen reveals Fbw7 as a modulator of the NF-kappaB pathway. Nat Commun (2012) 3:976. doi: 10.1038/ncomms1975
63. Georgana I, Maluquer de Motes C. Cullin-5 adaptor SPSB1 controls NF-kappaB activation downstream of multiple signaling pathways. Front Immunol (2019) 10:3121. doi: 10.3389/fimmu.2019.03121
64. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol (2008) 9(12):1371–8. doi: 10.1038/ni.1676
65. Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol (2008) 9(12):1364–70. doi: 10.1038/ni.1678
66. Peluso I, Yarla NS, Ambra R, Pastore G, Perry G. MAPK signalling pathway in cancers: Olive products as cancer preventive and therapeutic agents. Semin Cancer Biol (2019) 56:185–95. doi: 10.1016/j.semcancer.2017.09.002
67. Hammouda MB, Ford AE, Liu Y, Zhang JY. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells (2020) 9(4):857. doi: 10.3390/cells9040857
68. Yong H-Y, Koh M-S, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs (2009) 18(12):1893–905. doi: 10.1517/13543780903321490
69. Xie CM, Tan M, Lin XT, Wu D, Jiang Y, Tan Y, et al. The FBXW7-SHOC2-Raptor axis controls the cross-talks between the RAS-ERK and mTORC1 signaling pathways. Cell Rep (2019) 26(11):3037–50 e4. doi: 10.1016/j.celrep.2019.02.052
70. Yang J, Huang M, Zhou L, He X, Jiang X, Zhang Y, et al. Cereblon suppresses the lipopolysaccharide-induced inflammatory response by promoting the ubiquitination and degradation of c-jun. J Biol Chem (2018) 293(26):10141–57. doi: 10.1074/jbc.RA118.002246
71. Migliorini D, Bogaerts S, Defever D, Vyas R, Denecker G, Radaelli E, et al. Cop1 constitutively regulates c-jun protein stability and functions as a tumor suppressor in mice. J Clin Invest (2011) 121(4):1329–43. doi: 10.1172/JCI45784
72. Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr. The v-jun point mutation allows c-jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell (2005) 8(1):25–33. doi: 10.1016/j.ccr.2005.06.005
73. Kwon E, Li X, Deng Y, Chang HW, Kim DY. AMPK is down-regulated by the CRL4A-CRBN axis through the polyubiquitination of AMPKalpha isoforms. FASEB J (2019) 33(5):6539–50. doi: 10.1096/fj.201801766RRR
74. Jeon S, Yoon Y-S, Kim HK, Han J, Lee KM, Eun Seol J, et al. Ablation of CRBN induces loss of type I collagen and SCH in mouse skin by fibroblast senescence via the p38 MAPK pathway. Aging (Albany NY) (2021) 13(5):6406–19. doi: 10.18632/aging.202744
75. O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity (2012) 36(4):542–50. doi: 10.1016/j.immuni.2012.03.014
76. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of jak-STAT signaling in the immune system. Nat Immunol (2017) 18(4):374–84. doi: 10.1038/ni.3691
77. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
78. Salas A, Hernandez-Rocha C, Duijvestein M, Faubion W, McGovern D, Vermeire S, et al. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol (2020) 17(6):323–37. doi: 10.1038/s41575-020-0273-0
79. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: Current and future prospects. Drugs (2017) 77(5):521–46. doi: 10.1007/s40265-017-0701-9
80. Edmond M, Nicholson SE. Kinase inhibition, competitive binding and proteasomal degradation: resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol Rev (2015) 266(1):123–33. doi: 10.1111/imr.12305
81. Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol Sci (2019) 40(5):298–308. doi: 10.1016/j.tips.2019.03.001
82. Nie L, Zhao Y, Wu W, Yang Y-Z, Wang H-C, Sun X-H. Notch-induced Asb2 expression promotes protein ubiquitination by forming non-canonical E3 ligase complexes. Cell Res (2010) 21(5):754–69. doi: 10.1038/cr.2010.165
83. Kumar KGS, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J (2003) 22(20):5480–90. doi: 10.1093/emboj/cdg524
84. Gargan S, Ahmed S, Mahony R, Bannan C, Napoletano S, O’Farrelly C, et al. HIV-1 promotes the degradation of components of the type 1 IFN JAK/STAT pathway and blocks anti-viral ISG induction. EBioMedicine (2018) 30:203–16. doi: 10.1016/j.ebiom.2018.03.006
85. Jangra S, Bharti A, Lui WY, Chaudhary V, Botelho MG, Yuen KS, et al. Suppression of JAK-STAT signaling by Epstein-Barr virus tegument protein BGLF2 through recruitment of SHP1 phosphatase and promotion of STAT2 degradation. J Virol (2021) 95(20):e0102721. doi: 10.1128/JVI.01027-21
86. Daniele VF, Tauriello ES. Eduard Batlle overcoming TGFβ-mediated immune evasion in cancer. Nat Rev Cancer (2022) 22(1):25–44. doi: 10.1038/s41568-021-00413-6
87. Derynck R, Turley SJ, Akhurst RJ. TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol (2021) 18(1):9–34. doi: 10.1038/s41571-020-0403-1
88. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
89. Wan M, Tang Y, Tytler EM, Lu C, Jin B, Vickers SM, et al. Smad4 protein stability is regulated by ubiquitin ligase SCF beta-TrCP1. J Biol Chem (2004) 279(15):14484–7. doi: 10.1074/jbc.C400005200
90. Cui Y, He S, Xing C, Lu K, Wang J, Xing G, et al. SCFFBXL(1)(5) regulates BMP signalling by directing the degradation of HECT-type ubiquitin ligase Smurf1. EMBO J (2011) 30(13):2675–89. doi: 10.1038/emboj.2011.155
91. Xie Z, Wang Y, Yang G, Han J, Zhu L, Li L, et al. The role of the hippo pathway in the pathogenesis of inflammatory bowel disease. Cell Death Dis (2021) 12(1):79. doi: 10.1038/s41419-021-03395-3
92. Zheng Y, Pan D. The hippo signaling pathway in development and disease. Dev Cell (2019) 50(3):264–82. doi: 10.1016/j.devcel.2019.06.003
93. Ma S, Meng Z, Chen R, Guan KL. The hippo pathway: Biology and pathophysiology. Annu Rev Biochem (2019) 88:577–604. doi: 10.1146/annurev-biochem-013118-111829
94. Yi L-JY L-J, Ding N, Ren J. CUL4A promotes proliferation and inhibits apoptosis of colon cancer cells via regulating hippo pathway. Eur Rev Med Pharmacol Sci (2020) 24(20):10518–25. doi: 10.26355/eurrev_202010_23404
95. Ni W, Zhang Y, Zhan Z, Ye F, Liang Y, Huang J, et al. A novel lncRNA uc.134 represses hepatocellular carcinoma progression by inhibiting CUL4A-mediated ubiquitination of LATS1. J Hematol Oncol (2017) 10(1):91. doi: 10.1186/s13045-017-0449-4
96. Ly PT, Tan YS, Koe CT, Zhang Y, Xie G, Endow S, et al. CRL4Mahj E3 ubiquitin ligase promotes neural stem cell reactivation. PloS Biol (2019) 17(6):e3000276. doi: 10.1371/journal.pbio.3000276
97. Ghosh M, Das S. PRAMEF2-mediated dynamic regulation of YAP signaling promotes tumorigenesis. Proc Natl Acad Sci U S A (2021) 118(40):e2105523118. doi: 10.1073/pnas.2105523118
98. Zou J, Ma W, Li J, Littlejohn R, Zhou H, Kim IM, et al. Neddylation mediates ventricular chamber maturation through repression of hippo signaling. Proc Natl Acad Sci U S A (2018) 115(17):E4101–E10. doi: 10.1073/pnas.1719309115
99. Cooper J, Xu Q, Zhou L, Pavlovic M, Ojeda V, Moulick K, et al. Combined inhibition of NEDD8-activating enzyme and mTOR suppresses NF2 loss-driven tumorigenesis. Mol Cancer Ther (2017) 16(8):1693–704. doi: 10.1158/1535-7163.MCT-16-0821
100. Zhang Q, Zhang Y, Parsels JD, Lohse I, Lawrence TS, Pasca di Magliano M, et al. Fbxw7 deletion accelerates Kras(G12D)-driven pancreatic tumorigenesis via yap accumulation. Neoplasia (2016) 18(11):666–73. doi: 10.1016/j.neo.2016.08.009
101. Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C, et al. Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer (2014) 13(110). doi: 10.1186/1476-4598-13-110
102. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev (2010) 24(1):72–85. doi: 10.1101/gad.1843810
103. Ribeiro P, Holder M, Frith D, Snijders AP, Tapon N. Crumbs promotes expanded recognition and degradation by the SCF(Slimb/beta-TrCP) ubiquitin ligase. Proc Natl Acad Sci U S A (2014) 111(19):E1980–9. doi: 10.1073/pnas.1315508111
104. Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem (2010) 285(48):37159–69. doi: 10.1074/jbc.M110.152942
105. Huang W, Lv X, Liu C, Zha Z, Zhang H, Jiang Y, et al. The n-terminal phosphodegron targets TAZ/WWTR1 protein for SCFbeta-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem (2012) 287(31):26245–53. doi: 10.1074/jbc.M112.382036
106. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity (2021) 54(3):437–53. doi: 10.1016/j.immuni.2021.01.018
107. Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell (2019) 177(7):1682–99. doi: 10.1016/j.cell.2019.05.026
108. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol (2018) 19(6):349–64. doi: 10.1038/s41580-018-0003-4
109. Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol (2017) 176:55–62. doi: 10.1016/j.clim.2017.01.007
110. Xia P, Wang S, Du Y, Zhao Z, Shi L, Sun L, et al. WASH inhibits autophagy through suppression of beclin 1 ubiquitination. EMBO J (2013) 32(20):2685–96. doi: 10.1038/emboj.2013.189
111. Antonioli M, Albiero F, Nazio F, Vescovo T, Perdomo AB, Corazzari M, et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell (2014) 31(6):734–46. doi: 10.1016/j.devcel.2014.11.013
112. Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell (2011) 44(2):304–16. doi: 10.1016/j.molcel.2011.08.029
113. Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell (2011) 44(2):290–303. doi: 10.1016/j.molcel.2011.08.030
114. Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, et al. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol Cell (2011) 44(2):317–24. doi: 10.1016/j.molcel.2011.09.005
115. Xiao J, Zhang T, Xu D, Wang H, Cai Y, Jin T, et al. FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes Dev (2015) 29(2):184–96. doi: 10.1101/gad.252528.114
116. Zhang T, Dong K, Liang W, Xu D, Xia H, Geng J, et al. G-Protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. Elife (2015) 4:e06734. doi: 10.7554/eLife.06734.017
117. Scrivo A, Codogno P, Bomont P. Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production. Nat Commun (2019) 10(1):780. doi: 10.1038/s41467-019-08331-w
118. Lu G, Yi J, Gubas A, Wang YT, Wu Y, Ren Y, et al. Suppression of autophagy during mitosis via CUL4-RING ubiquitin ligases-mediated WIPI2 polyubiquitination and proteasomal degradation. Autophagy (2019) 15(11):1917–34. doi: 10.1080/15548627.2019.1596484
119. Yoshida Y, Yasuda S, Fujita T, Hamasaki M, Murakami A, Kawawaki J, et al. Ubiquitination of exposed glycoproteins by SCF(FBXO27) directs damaged lysosomes for autophagy. Proc Natl Acad Sci U S A (2017) 114(32):8574–9. doi: 10.1073/pnas.1702615114
120. Lu G, Wang L, Zhou J, Liu W, Shen HM. A destiny for degradation: Interplay between cullin-RING E3 ligases and autophagy. Trends Cell Biol (2021) 31(6):432–44. doi: 10.1016/j.tcb.2021.01.005
121. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity (2019) 50(6):1352–64. doi: 10.1016/j.immuni.2019.05.020
122. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
123. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol (2013) 5(4):a008656. doi: 10.1101/cshperspect.a008656
124. Chen P, Hu T, Liang Y, Li P, Chen X, Zhang J, et al. Neddylation inhibition activates the extrinsic apoptosis pathway through ATF4-CHOP-DR5 axis in human esophageal cancer cells. Clin Cancer Res (2016) 22(16):4145–57. doi: 10.1158/1078-0432.CCR-15-2254
125. Che Z, Liu F, Zhang W, McGrath M, Hou D, Chen P, et al. Targeting CAND1 promotes caspase-8/RIP1-dependent apoptosis in liver cancer cells. Am J Transl Res (2018) 10(5):1357–72.
126. Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell (2009) 137(4):721–35. doi: 10.1016/j.cell.2009.03.015
127. Roberts JZ, Holohan C, Sessler T, Fox J, Crawford N, Riley JS, et al. The SCF(Skp2) ubiquitin ligase complex modulates TRAIL-R2-induced apoptosis by regulating FLIP(L). Cell Death Differ (2020) 27(9):2726–41. doi: 10.1038/s41418-020-0539-7
128. Tan M, Gallegos JR, Gu Q, Huang Y, Li J, Jin Y, et al. SAG/ROC-SCF beta-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia (2006) 8(12):1042–54. doi: 10.1593/neo.06568
129. Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus-an update. Curr Opin Immunol (2012) 24(6):651–7. doi: 10.1016/j.coi.2012.10.004
130. Laura Durcan TOD. Michelle Petri Management strategies and future directions for systemic lupus erythematosus in adults. Lancet (2019) 393(10188):2332–43. doi: 10.1016/S0140-6736(19)30237-5
131. Fava A, Petri M. Systemic lupus erythematosus: Diagnosis and clinical management. J Autoimmun (2019) 96:1–13. doi: 10.1016/j.jaut.2018.11.001
132. Yang F, Lin J, Chen W. Post-translational modifications in T cells in systemic erythematosus lupus. Rheumatol (Oxford) (2021) 60(6):2502–16. doi: 10.1093/rheumatology/keab095
133. Navarro Quiroz E, Chavez-Estrada V, Macias-Ochoa K, Ayala-Navarro MF, Flores-Aguilar AS, Morales-Navarrete F, et al. Epigenetic mechanisms and posttranslational modifications in systemic lupus erythematosus. Int J Mol Sci (2019) 20(22):5679. doi: 10.3390/ijms20225679
134. Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors ikaros and aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol (2014) 164(6):811–21. doi: 10.1111/bjh.12708
135. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science (2014) 343(6168):305–9. doi: 10.1126/science.1244917
136. Matyskiela ME, Zhang W, Man HW, Muller G, Khambatta G, Baculi F, et al. A cereblon modulator (CC-220) with improved degradation of ikaros and aiolos. J Med Chem (2018) 61(2):535–42. doi: 10.1021/acs.jmedchem.6b01921
137. Nakayama Y, Kosek J, Capone L, Hur EM, Schafer PH, Ringheim GE. Aiolos overexpression in systemic lupus erythematosus b cell subtypes and BAFF-induced memory b cell differentiation are reduced by CC-220 modulation of cereblon activity. J Immunol (2017) 199(7):2388–407. doi: 10.4049/jimmunol.1601725
138. Schafer PH, Ye Y, Wu L, Kosek J, Ringheim G, Yang Z, et al. Cereblon modulator iberdomide induces degradation of the transcription factors ikaros and aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann Rheum Dis (2018) 77(10):1516–23. doi: 10.1136/annrheumdis-2017-212916
139. Fan J, Bellon M, Ju M, Zhao L, Wei M, Fu L, et al. Clinical significance of FBXW7 loss of function in human cancers. Mol Cancer (2022) 21(1):87. doi: 10.1186/s12943-022-01548-2
140. Sailo BL, Banik K, Girisa S, Bordoloi D, Fan L, Halim CE, et al. FBXW7 in cancer: What has been unraveled thus far? Cancers (Basel) (2019) 11(2):246. doi: 10.3390/cancers11020246
141. Chong Z, Bao C, He J, Chen T, Zhong L, Li G, et al. E3 ligase FBXW7 aggravates TMPD-induced systemic lupus erythematosus by promoting cell apoptosis. Cell Mol Immunol (2018) 15(12):1057–70. doi: 10.1038/s41423-018-0167-z
142. Andrew S, Neish ATG, Zeng H, . Young AN, . Hobert ME, Karmali V, et al. Prokaryotic regulation of epithelial responses by inhibition of IKB-a ubiquitination. Science (2000) 289(5484):1560–3. doi: 10.1126/science.289.5484.1560
143. Kumar A, Wu H, . Collier-Hyams LS, Kwon Y-M, Hanson JM, . Neish AS. The bacterial fermentation product butyrate influences epithelial signaling via reactive oxygen species-mediated changes in cullin-1 neddylation. J Immunol (2009) 182(1):538–46. doi: 10.4049/jimmunol.182.1.538
144. Kumar A, Wu H, Collier-Hyams LS, Hansen JM, Li T, Yamoah K, et al. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J (2007) 26(21):4457–66. doi: 10.1038/sj.emboj.7601867
145. Lin PW, Myers LE, Ray L, Song SC, Nasr TR, Berardinelli AJ, et al. Lactobacillus rhamnosus blocks inflammatory signaling in vivo via reactive oxygen species generation. Free Radic Biol Med (2009) 47(8):1205–11. doi: 10.1016/j.freeradbiomed.2009.07.033
146. Wang G, Yuan J, Cai X, Xu Z, Wang J, Ocansey DKW, et al. HucMSC-exosomes carrying miR-326 inhibit neddylation to relieve inflammatory bowel disease in mice. Clin Transl Med (2020) 10(2):e113. doi: 10.1002/ctm2.113
147. Wang L, Zhou J, Zheng S, Zhang T, Lin Y, Zhang M, et al. Inhibition of neddylation regulates dendritic cell functions via deptor accumulation driven mTOR inactivation. Oncotarget (2016) 7(24):35643–54. doi: 10.18632/oncotarget.9543
148. MacManus CF, Campbell EL, Keely S, Burgess A, Kominsky DJ, Colgan SP. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J (2011) 25(6):1856–64. doi: 10.1096/fj.10-170316
149. Curtis VF, Ehrentraut SF, Campbell EL, Glover LE, Bayless A, Kelly CJ, et al. Stabilization of HIF through inhibition of cullin-2 neddylation is protective in mucosal inflammatory responses. FASEB J (2015) 29(1):208–15. doi: 10.1096/fj.14-259663
150. Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, et al. Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J Exp Med (2017) 214(4):1093–109. doi: 10.1084/jem.20161105
151. Kawaida R, Yamada R, Kobayashi K, Tokuhiro S, Suzuki A, Kochi Y, et al. CUL1, a component of E3 ubiquitin ligase, alters lymphocyte signal transduction with possible effect on rheumatoid arthritis. Genes Immun (2005) 6(3):194–202. doi: 10.1038/sj.gene.6364177
152. Okuyama Y, Tanaka Y, Jiang JJ, Kamimura D, Nakamura A, Ota M, et al. Bmi1 regulates IkappaBalpha degradation via association with the SCF complex. J Immunol (2018) 201(8):2264–72. doi: 10.4049/jimmunol.1701223
153. Zhao J, Wei J, Mialki RK, Mallampalli DF, Chen BB, Coon T, et al. F-box protein FBXL19-mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat Immunol (2012) 13(7):651–8. doi: 10.1038/ni.2341
154. Miao C, Chang J, Zhang G, Yu H, Zhou L, Zhou G, et al. CUL4B promotes the pathology of adjuvant-induced arthritis in rats through the canonical wnt signaling. J Mol Med (Berl) (2018) 96(6):495–511. doi: 10.1007/s00109-018-1635-8
155. Wang X, Chang J, Zhou G, Cheng C, Xiong Y, Dou J, et al. The traditional Chinese medicine compound huangqin qingre chubi capsule inhibits the pathogenesis of rheumatoid arthritis through the CUL4B/Wnt pathway. Front Pharmacol (2021) 12:750233. doi: 10.3389/fphar.2021.750233
156. Szlavicz E, Olah P, Szabo K, Pagani F, Bata-Csorgo Z, Kemeny L, et al. Analysis of psoriasis-relevant gene expression and exon usage alterations after silencing of SR-rich splicing regulators. Exp Dermatol (2018) 27(6):656–62. doi: 10.1111/exd.13530
157. Ishizawa K, Wang Q, Li J, Xu N, Nemoto Y, Morimoto C, et al. Inhibition of sodium glucose cotransporter 2 attenuates the dysregulation of kelch-like 3 and NaCl cotransporter in obese diabetic mice. J Am Soc Nephrol (2019) 30(5):782–94. doi: 10.1681/ASN.2018070703
158. Guo Q, Zhang Y, Jiang GR, Zhang C. Decreased KLHL3 expression is involved in the activation of WNK-OSR1/SPAK-NCC cascade in type 1 diabetic mice. Pflugers Arch (2021) 473(2):185–96. doi: 10.1007/s00424-020-02509-8
159. Lee JH, Elly C, Park Y, Liu YC. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity (2015) 42(6):1062–74. doi: 10.1016/j.immuni.2015.05.016
160. Wu D, Li H, Liu M, Qin J, Sun Y. The Ube2m-Rbx1 neddylation-Cullin-RING-Ligase proteins are essential for the maintenance of regulatory T cell fitness. Nat Commun (2022) 13(1):3021. doi: 10.1038/s41467-022-30707-8
161. Agrawal M, Allin KH, Petralia F, Colombel JF, Jess T. Multiomics to elucidate inflammatory bowel disease risk factors and pathways. Nat Rev Gastroenterol Hepatol (2022) 19(6):399–409. doi: 10.1038/s41575-022-00593-y
162. Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol (2014) 20(1):91–9. doi: 10.3748/wjg.v20.i1.91
163. Liu S, Zhao W, Lan P, Mou X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell (2021) 12(5):331–45. doi: 10.1007/s13238-020-00745-3
164. Xiao Y, Huang Q, Wu Z, Chen W. Roles of protein ubiquitination in inflammatory bowel disease. Immunobiology (2020) 225(6):152026. doi: 10.1016/j.imbio.2020.152026
165. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for crohn’s disease. Nat Genet (2008) 40(8):955–62. doi: 10.1038/ng.175
166. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity (1999) 10(1):39–49. doi: 10.1016/S1074-7613(00)80005-9
167. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell (2009) 15(2):91–102. doi: 10.1016/j.ccr.2009.01.002
168. Scherer HU, Haupl T, Burmester GR. The etiology of rheumatoid arthritis. J Autoimmun (2020) 110:102400. doi: 10.1016/j.jaut.2019.102400
169. Sparks JA. Rheumatoid arthritis. Ann Intern Med (2019) 170(1):ITC1–ITC16. doi: 10.7326/AITC201901010
170. Negi S, Kumar A, Thelma BK, Juyal RC. Association of Cullin1 haplotype variants with rheumatoid arthritis and response to methotrexate. Pharmacogenet Genomics (2011) 21(9):590–3. doi: 10.1097/FPC.0b013e3283492af7
171. Dong Y, Zhong J, Dong L. IL-33 in rheumatic diseases. Front Med (Lausanne) (2021) 8:739489. doi: 10.3389/fmed.2021.739489
172. Iwaszko M, Wielinska J, Swierkot J, Kolossa K, Sokolik R, Bugaj B, et al. IL-33 gene polymorphisms as potential biomarkers of disease susceptibility and response to TNF inhibitors in rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis patients. Front Immunol (2021) 12:631603. doi: 10.3389/fimmu.2021.631603
173. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet (2021) 397(10281):1301–15. doi: 10.1016/S0140-6736(20)32549-6
174. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: A review. JAMA (2020) 323(19):1945–60. doi: 10.1001/jama.2020.4006
175. Ilonen J, Lempainen J, Veijola R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat Rev Endocrinol (2019) 15(11):635–50. doi: 10.1038/s41574-019-0254-y
176. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet (2018) 391(10138):2449–62. doi: 10.1016/S0140-6736(18)31320-5
177. Shibata S, Arroyo JP, Castaneda-Bueno M, Puthumana J, Zhang J, Uchida S, et al. Angiotensin II signaling via protein kinase c phosphorylates kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci U S A (2014) 111(43):15556–61. doi: 10.1073/pnas.1418342111
178. Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, et al. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: A systematic review. Autoimmun Rev (2020) 19(6):102526. doi: 10.1016/j.autrev.2020.102526
179. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4(4):337–42. doi: 10.1038/ni909
180. Bennett JC CL, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, et al. Polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
181. Zemmour D, Charbonnier LM, Leon J, Six E, Keles S, Delville M, et al. Single-cell analysis of FOXP3 deficiencies in humans and mice unmasks intrinsic and extrinsic CD4(+) T cell perturbations. Nat Immunol (2021) 22(5):607–19. doi: 10.1038/s41590-021-00910-8
182. Ito T, Yamaguchi Y, Handa H. Exploiting ubiquitin ligase cereblon as a target for small-molecule compounds in medicine and chemical biology. Cell Chem Biol (2021) 28(7):987–99. doi: 10.1016/j.chembiol.2021.04.012
183. He M, Cao C, Ni Z, Liu Y, Song P, Hao S, et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct Target Ther (2022) 7(1):181. doi: 10.1038/s41392-022-00999-9
Keywords: Cullin-RING ligases, autoimmune diseases, E3 ligases, ubiquitination, inflammation
Citation: Zhang X, Liu Y, Zhang T, Tan Y, Dai X, Yang Y-G and Zhang X (2023) Advances in the potential roles of Cullin-RING ligases in regulating autoimmune diseases. Front. Immunol. 14:1125224. doi: 10.3389/fimmu.2023.1125224
Received: 16 December 2022; Accepted: 28 February 2023;
Published: 17 March 2023.
Edited by:
Abhigyan Satyam, Harvard Medical School, United StatesReviewed by:
Xiaodong Lu, Northwestern University, United StatesYi Sun, Zhejiang University, China
Kankan Wang, Purdue University, United States
Copyright © 2023 Zhang, Liu, Zhang, Tan, Dai, Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangpeng Dai, daixiangpeng@jlu.edu.cn; Yong-Guang Yang, yongg@jlu.edu.cn; Xiaoling Zhang, xiaolingzhang@jlu.edu.cn