 Priyanka Maridhi Nanjireddy1,2
Priyanka Maridhi Nanjireddy1,2 Nataliya Prokopenko Buxbaum
Nataliya Prokopenko Buxbaum- 1Department of Pediatric Oncology, Pediatric Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, United States
- 2Immunology Department, Roswell Park Comprehensive Cancer Center, Buffalo, NY, United States
- 3Department of Pediatrics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, United States
Genetically engineered chimeric antigen receptor (CAR) T cells can cure patients with cancers that are refractory to standard therapeutic approaches. To date, adoptive cell therapies have been less effective against solid tumors, largely due to impaired homing and function of immune cells within the immunosuppressive tumor microenvironment (TME). Cellular metabolism plays a key role in T cell function and survival and is amenable to manipulation. This manuscript provides an overview of known aspects of CAR T metabolism and describes potential approaches to manipulate metabolic features of CAR T to yield better anti-tumor responses. Distinct T cell phenotypes that are linked to cellular metabolism profiles are associated with improved anti-tumor responses. Several steps within the CAR T manufacture process are amenable to interventions that can generate and maintain favorable intracellular metabolism phenotypes. For example, co-stimulatory signaling is executed through metabolic rewiring. Use of metabolic regulators during CAR T expansion or systemically in the patient following adoptive transfer are described as potential approaches to generate and maintain metabolic states that can confer improved in vivo T cell function and persistence. Cytokine and nutrient selection during the expansion process can be tailored to yield CAR T products with more favorable metabolic features. In summary, improved understanding of CAR T cellular metabolism and its manipulations have the potential to guide the development of more effective adoptive cell therapies.
Introduction
Surgery, chemotherapy, and radiation remain the cornerstones of cancer treatment. However, many patients are not cured by these approaches and those that are cured may incur significant morbidities, demonstrating an urgent need for the development of novel therapeutic options. Cancer cells subvert normal metabolic pathways to favor their growth and evasion of the immune system. Altered metabolism of the tumor microenvironment (TME) plays a critical role in tumorigenesis by creating an immunosuppressive milieu (1–4). Several therapeutic approaches to modify and restore the immune system are currently being developed and applied. Genetically engineered T cells constitute a powerful new therapeutic approach in the treatment of cancer. Chimeric Antigen Receptors (CARs) are synthetic receptors that graft a defined specificity onto an immune effector cell, typically a T cell, and augment T cell function. Once infused into the patient they expand and kill tumor cells. They also prevent tumor recurrence by promoting immune surveillance in conjunction with tumor infiltrating lymphocytes or by their own persistence (5–7). While significant strides have been made in CAR T cell therapy for hematologic malignancies leading to FDA approval of multiple products, durable responses in solid tumors remain limited (8, 9). Strategies to improve CAR T cell function are actively being sought. Metabolic manipulation represents a potential approach for improving the former given that T cell function is closely tied to cellular metabolism. Energetic demands and consequences of T cell activation, cytokine production, proliferation, and survival are facilitated by metabolic rewiring (10, 11). Furthermore, each stage within the CAR T manufacture process can influence the eventual metabolic profile of the infusion product, while metabolic features of the latter are directly linked to in vivo efficacy and persistence (8). The goal of this manuscript is to review known aspects of T cell metabolism, in the context of CAR T therapy, and present potential metabolic interventions that can be undertaken at each step of the manufacture process, including CAR design, priming, and expansion to leverage metabolic fitness of CAR T cells to augment therapeutic outcomes. In general, CAR T products containing memory like cells with enhanced mitochondrial fitness and high reliance on oxidative phosphorylation (OXPHOS) along with fatty acid oxidation (FAO) have been shown to have superior in vivo anti-tumor efficacy and long-term persistence. While effector function is tied to glycolysis that is engaged upon antigen-driven activation, a high proportion of memory cells that can be maintained in diminishing levels of antigen are associated with sustained anti-tumor responses. Thus, strategies that favor mitochondrial biogenesis along the manufacture process of the CAR T cells and maintenance of this metabolic phenotype after infusion are discussed in this review.
Brief overview of T cell metabolism
T cells are specific effectors of the adaptive immune system, which continuously survey and eliminate pathogen infected cells as well as tumors. To elicit a robust immune response, T cells differentiate into diverse functional subsets, such as effector T cells (Teff) and memory T cells (Tmem), which can further differentiate into more diverse subsets based on cytokine milieu (12, 13). Such subsets have different functional and metabolic requirements. In the absence of antigen, naïve T cells are quiescent, rarely divide, have a low energetic demand, and continuously circulate through secondary lymphoid tissues (11, 14). Memory T cells are also mostly quiescent but display a greater mitochondrial mass that provides a bioenergetic advantage to support rapid recall responses upon antigen re-exposure. Both sets of T cells rely almost completely on the energy derived from mitochondrial oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) to maintain their basal energy level, cellular function, and viability (15–18). Following antigen exposure, T cell activation is orchestrated by TCR/peptide-MHC interaction providing the first signal forming an immune synapse. Further interaction at the synapse with costimulatory molecules provides the required second signal. Thus, a complete TCR-based activation of T cells requires two signals (15, 19, 20). Once activated, T cells predominantly engage in aerobic glycolysis, the pentose phosphate pathway (PPP), one-carbon metabolism, fatty acid oxidation (FAO), and glutaminolysis to facilitate proliferation and enable subsequent effector functions (17, 21–24), Figure 1A.
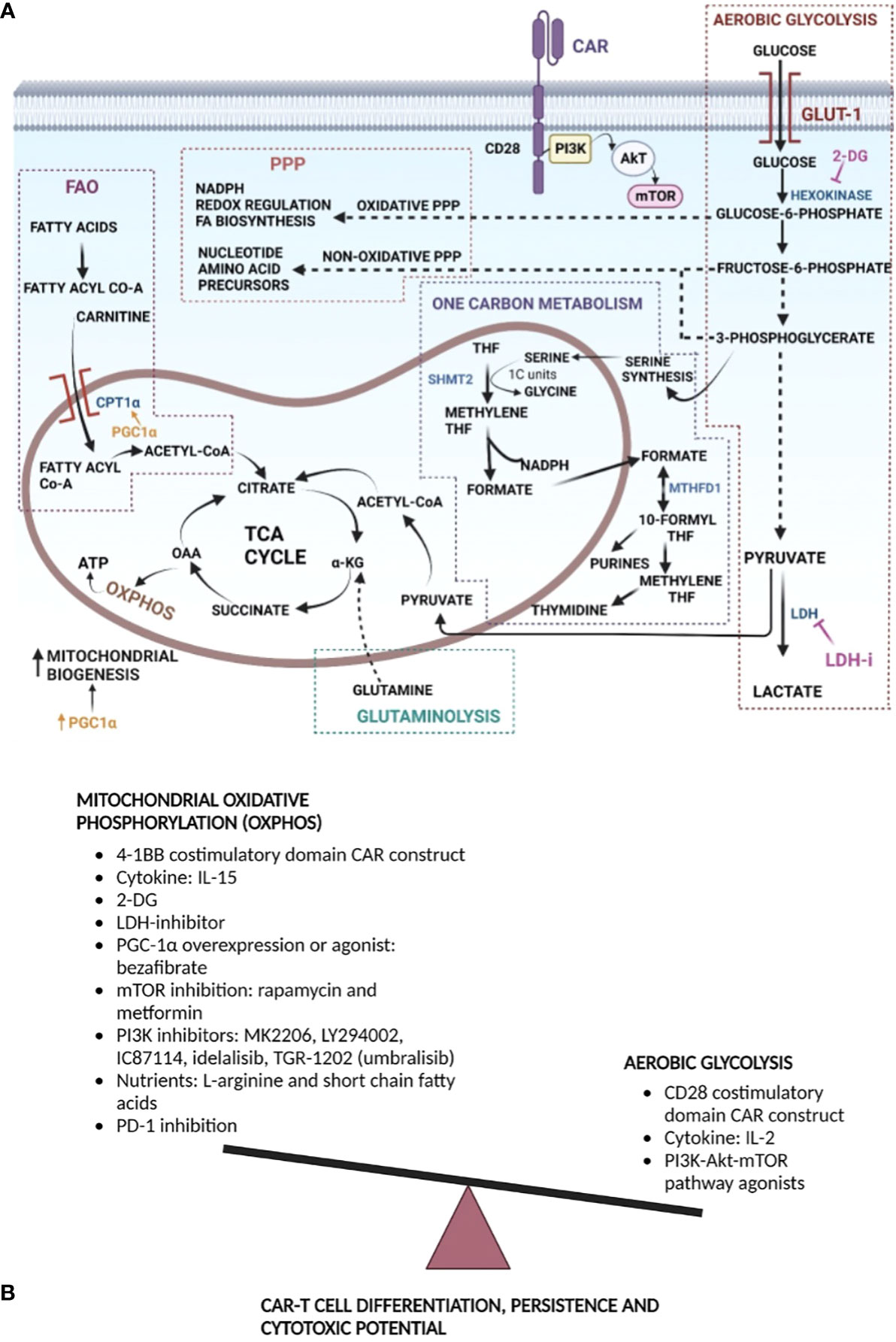
Figure 1 Metabolism manipulation strategies to improve CAR T cell efficacy and persistence: (A) T cells undergo metabolic rewiring upon encountering antigen. Aerobic glycolysis is upregulated upon T cell activation. Glycolysis provides energy for cell effector functions, as well as intermediates for the PPP that generates NADPH for anabolic processes and fuels nucleotide and amino acid biosynthesis. Proliferating T cells also rely on serine-glycine, folate, and methionine metabolism to generate one carbon units for de novo nucleotide synthesis, as well as NADPH production. Pyruvate produced at the end aerobic glycolysis can enter mitochondria for conversion to Acetyl CoA that feeds into the TCA cycle. Jointly, within the mitochondria, the TCA cycle, fatty acid oxidation, and glutaminolysis fuel T cell proliferation and differentiation. T cell activation is facilitated by increased mitochondrial biogenesis, generation of energy, OXPHOS and generation of reactive oxygen species, with NADPH providing reducing power for the latter. Potential metabolic points of stimulation or inhibition for enhancing CAR T cell function are provided. 2-DG inhibits hexokinase, re-directing metabolites toward PPP and OXPHOS. Inhibition of LDH drives pyruvate toward TCA and OXPHOS. PGC1α stimulates mitochondrial biogenesis, which promotes OXPHOS, and can stimulate FAO via CPT1α. (B) Generally, a memory like CAR T phenotype has been shown to improve in vivo persistence and anti-tumor function. Memory cells primarily rely on OXPHOS; hence, strategies that favor the former during CAR design, CAR T manufacture, expansion, or following infusion are provided. Mitochondrial OXPHOS can be increased directly or indirectly by decreasing aerobic glycolysis. Acronyms used: GLUT-1, glucose transporter-1; 2-DG, 2-deoxyglycose; LDH, lactate dehydrogenase; LDHi, lactate dehydrogenase inhibitor; α-KG, α-ketoglutarate; OAA- oxaloacetic acid; TCA Cycle, tricarboxylic acid cycle; THF, tetrahydrofolate; 1C, one carbon; SHMT2, serine hydroxymethyl transferase 2; MTHFD1, methylene tetrahydrofolate dehydrogenase 1; PPP, pentose phosphate pathway; FA, fatty acid; CPT1α, carnitine palmitoyl transferase 1α; PGC1α, peroxisome proliferator-activated receptor-gamma co-activator 1α; NADPH, reduced nicotinamide adenine dinucleotide phosphate; 4-1BB, distinct costimulatory molecule; CAR, chimeric antigen receptor; CD, cluster of differentiation; IL, interleukin; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase.
Glycolysis
Naïve T cells and non-proliferating cells generate ATP via OXPHOS. Once T cells are activated, they engage glycolysis, where pyruvate is fermented to lactate in the cytoplasm in the presence of sufficient oxygen, a process termed the Warburg effect (24–27). The process of glycolysis begins with the uptake of extracellular glucose, mediated by cell surface transporter Glut1 and ends in conversion to pyruvate, through a series of enzymatic reactions generating metabolites that can enter other pathways (21, 28, 29). Pyruvate produced under aerobic conditions can be converted in the mitochondria into acetyl Co-A, which then enters the tricarboxylic acid (TCA) cycle. Alternatively, under anaerobic conditions, it can be converted in the cytoplasm into lactate, which is then excreted from the cell (30), Figure 1A. While T cells require mitochondrial ATP from OXPHOS for activation, continued proliferation of the activated T cells relies on either aerobic glycolysis or OXPHOS (29). Chang et al. showed that OXPHOS and aerobic glycolysis can be used interchangeably as fuel for T cell proliferation and survival, but glycolysis is essential for T cell effector function (29). Glycolysis while relatively inefficient for energy production compared to OXPHOS, which produces 10 times higher energy yield, may nonetheless be preferred by rapidly proliferating cells secondary to the concurrent generation of biosynthetic precursor molecules that the cell needs (18, 24, 31–34). Following T cell activation there is also a reduction of ATP output from the mitochondria, which is instrumental to maintaining a low ATP : ADP ratio, promoting a high glycolytic rate (18, 21, 32, 35). Glycolysis is critical for effector differentiation as it is required for the post-transcriptional regulation of interferon (IFN)-γ production (29). However, persistently heightened glycolysis limits the Teff capacity to establish immunological memory making them short lived, while moderately dampened glycolysis supports generation of long-lived memory CD8+ T cells (36). T cells activated in limited concentrations of glucose fail to upregulate cytotoxic molecules, such as perforin and granzyme (16). The PI3K-AKT pathway also regulates glycolysis. AKT activity augments glycolysis by inducing Glut1 trafficking to the cell surface, increasing the activity of key glycolytic enzymes and more importantly, activating the kinase, mammalian target of rapamycin (mTOR), that favors cell growth, protein synthesis and proliferation (16, 21, 31–33).
Pentose phosphate pathway
Glycolysis is not the sole metabolic fate of glucose. The pentose phosphate pathway (PPP) starts from glucose-6-phosphate, an intermediate product of glycolysis, Figure 1A, and diverts it through several paths (37). The non-oxidative PPP branch shunts intermediates of glycolysis towards production of nucleotide and amino acid precursors that are needed for T cell growth and proliferation. Meanwhile, the oxidative PPP branch generates NADPH that is then used to generate reactive oxygen species (ROS) required for modulating redox balance and fatty acid biosynthesis (17, 37, 38).
Glutaminolysis
Glutaminolysis is a pathway of incomplete glutamine oxidation that occurs in immune cells (39). The rate of glutamine utilization is high in both resting and activated lymphocytes. Glutaminolysis is critical for T cell function and as an energy producing pathway. Glutamine is a major anaplerotic fuel required for maintaining the TCA cycle (40) and reductive carboxylation in effector T cells (41, 42). Glutamine can enter the TCA cycle via conversion to α-ketoglutarate, Figure 1A, which can be processed to oxaloacetate, and subsequently citrate (40, 43). The latter is then excreted into the cytosol where it can be converted to acetyl CoA, the backbone of lipid synthesis. Glutaminolysis also provides metabolites for other biosynthetic processes by increasing availability of intracellular glutamine, aspartate, and ammonia that are necessary for purine and pyrimidine synthesis (44). A catabolic pathway involving glutamine, in the presence of NADP+ dependent malate dehydrogenase, following a series of steps, yields pyruvate and large amounts of NADPH. NADPH is required for protein, DNA, and RNA synthesis (44, 45). In contrast to glycolytic energy production, glutaminolytic energy production requires mitochondrial OXPHOS (34, 39, 46).
One-carbon metabolism
While early studies in immune cells demonstrated glycolysis to be the predominant metabolic pathway (43, 47), more recent studies have shown a role for mitochondria in metabolic reprograming, respiration, and amino acid metabolism to support cellular proliferation (48, 49). Analysis of the mitochondrial proteome demonstrated that T cell activation induces mitochondrial proliferation and proteome remodeling, which then generates specialized mitochondria. The most striking change noted in these activated T cell mitochondria was the massive induction of enzymes involved in folate-mediated one carbon metabolism (50). One carbon metabolism is a key metabolic node in proliferating cells (51). It consists of serine-glycine metabolism, folate cycle and methionine cycle and is essential to processes such as de novo purine synthesis, methyl donor generation, as well as NADPH production that are critical for cell survival and function (52–54). Folate intermediates, such as, tetrahydrofolate (THF) are active carriers of one carbon units for de novo nucleotide synthesis (55). Ron-Harel et al. provided evidence for the former by showing a significant increase in the intracellular levels of precursors for these pathways during T cell activation, as well as 13C2-serine labeling of the media used to stimulate naïve T cells with tracing through the pathway to the yield of 13C labeled purines. This confirmed that metabolic changes in one carbon metabolism occur upon T cell activation and that T cells, upon activation, engage in both de novo biosynthesis and purine salvage pathways (50).
Serine is a major donor of one-carbon units during T cell activation and is essential for T cell effector responses (51). Other sources of one-carbon moieties for cytoplasmic one-carbon metabolism include formate, histidine, and purines (54, 56, 57). One-carbon units are generated in parallel pathways in the mitochondria and cytoplasm (50, 52, 54, 56, 57), and both sets of enzymes were found to be highly induced following T cell activation (50). However, the majority of one carbon units are generated within mitochondria in activated T cells. The key enzyme for mitochondrial one carbon metabolism and T cell survival is serine hydroxyl-methyltransferase-2 (SHMT2), Figure 1A. Evidence for the latter was generated in the SHMT2 knockdown (KD) model where a decrease in the one carbon unit pool, as well as an accumulation of metabolites upstream the de novo purine synthesis was observed (50). T cells from the SHMT2 KD displayed a 2- to 3-fold increase in cell death compared to wildtype. The latter was a result of increased cell death due a nucleotide imbalance, with an observed 50% reduction in purine levels but unaffected levels of pyrimidines leading to the inhibition of de novo purine synthesis and increased DNA damage. In addition, SHMT2 plays a critical role in glutathione synthesis; hence, the shortened T cell half-life was also attributable to increased oxidative stress promoting cell death under hypoxia in SHMT2 KD T cells (50). SHMT2 KD T cells could be rescued completely with the combination of formate, a product of mitochondrial one carbon metabolism, and N-acetyl cysteine (NAC), a glutathione precursor, reaffirming the important role of mitochondrial one-carbon metabolism in promoting T cell survival (50, 58, 59). Another critical regulator of CD4+ T cell proliferation and differentiation is methylenetetrahydrofolate dehydrogenase 2 (MTHFD2). Suguira et al. showed that MTHFD2 is selectively required for Teff cells. A deficiency of MTHFD2 alters de novo purine synthesis, resulting in insufficient generation of nucleotides (60). To summarize, one-carbon metabolism plays a major role in T cell proliferation and survival.
Fatty acid oxidation
Fatty acid oxidation (FAO) is a pathway that converts fatty acids to acetyl-CoA, NADH, FADH2 which are then used by cells for energy production (38). This pathway takes place in the mitochondria and can produce tremendous amounts of ATP. Starting in the cytoplasm with activation of fatty acids to a fatty acid acyl-CoA, short chain fatty acids diffuse passively into the mitochondria, while medium and long chain fatty acids are conjugated to carnitine and are consequently shuttled into the mitochondria. Once inside the mitochondria, carnitine conjugated fatty acids are converted back to fatty acid acyl-CoA that undergoes β-oxidation generating large amounts of acetyl-CoA, NADH and FADH2, which are used in the TCA cycle and electron transport chain (ETC) to produce ATP (38).
When in circulation, naïve T cells engage in FAO for ATP production or use OXPHOS to maintain low levels of glycolysis. Once activated, they switch from FAO to fatty acid synthesis (FAS) due an increased demand for lipids (11, 35). FAO promotes memory T cell production that is necessary for a long-lived immune response. CD8+ memory T cells are dependent on FAO for their development, persistence, and immediate response to stimulation (20, 23, 61). This is enabled by their greater mitochondrial mass and spare respiratory capacity compared to naïve and effector counterparts (20, 61). Therefore, lipid metabolism plays an important role in T cell activation and formation of memory phenotypes.
Brief overview of CAR T cell manufacture process and the associated cellular metabolism consequences
CARs are synthetic receptors that redirect T cells against a defined target in a major histocompatibility complex (MHC)-independent fashion. CAR T cell therapy aims to eliminate specific tumor cells in a sustained manner. The first step of the CAR T manufacturing process is the collection of PBMCs through leukapheresis, either patient’s own or from a donor (62, 63), followed by isolation of T cells. T cells are then activated with anti-CD3/CD28 magnetic beads to promote proliferation and differentiation. At this point, T cells transition from a naïve or quiescent state to an activated state, with a concurrent metabolic switch from FAO to glycolysis (20, 26, 29), whereby T cells differentiate into either high glucose requiring Teff cells and low glucose requiring Tmem cells (11, 36). The next step in the manufacturing process is viral transduction, where T cells are incubated with a lentiviral vector encoding the CAR construct. The final step in manufacture includes expansion within cytokine enriched media and represents yet another opportunity for T cells to differentiate into distinct functional phenotypes, i.e., Teff, Tem, Tscm, Tcm, based on culture conditions. CAR T products containing a high proportion Tcm or Tscm subsets have been shown to have an enhanced subsequent metabolic adaptability, mediated mainly through mitochondrial metabolism, and are able to maintain a long-term anti-tumor response in vivo (8, 10, 14, 64–67). Optimization of CAR T cell metabolism for the maintenance of early memory phenotypes, Tscm and Tcm, to improve CAR persistence and cytotoxic function can potentially be carried out at several stages, as described hereafter, Figures 1A, B.
CAR constructs: The first T cell activating receptors were CD3ζ chain fusions, which also elucidated the role of the ζ chain (68, 69). These early studies showed that T cell activation signaling, and initiation of cytotoxicity were possible by cross-linking the fusion receptors. Eshhar et al. then incorporated the immunoglobulin-derived single chain variable fragment (scFv) onto these receptors to direct and lyse hapten coated cells (5, 70), generating the first-generation CAR T. Since then, multiple generations of CAR T cells have been engineered. The current FDA approved CAR T cell receptor has four parts: 1) an extracellular target antigen-binding domain composed of a single chain variable fragment (scFv) of an antibody targeting the specific tumor antigen; 2) a hinge region; 3) a transmembrane domain; and 4) the intracellular domain, i.e., T cell receptor (TCR) signaling domain comprised of CD3ζ and costimulatory domains. The CD3ζ domain in the CAR structure serves as signal 1 and a costimulatory domain provides signal 2 (15). Second and third generation CARs have multiple costimulatory domains, such as immunoglobulin (Ig) superfamily members, CD28 (B7.1/B7.2-CD28) and inducible T cell costimulatory (ICOS, B7RP-1-ICOS) (71), and tumor necrosis factor receptor (TNFR) superfamily members 4-1BB, OX40 and CD27 (10, 15). Depending on the costimulatory domains incorporated into the CAR construct, different downstream signaling pathways are activated that impact in vivo persistence, susceptibility to exhaustion, generation of memory, and anti-tumor potency (10). CD28 and 4-1BB signaling domains are the most widely used and studied. Hence, their effects on CAR T cell metabolism are described below.
Metabolic phenotypes linked to CAR constructs: Kawalekar et al. found that 4-1BB based (BBζ) CAR T cells had higher proliferative capacity and persistence than CD28 based (28ζ) CAR T cells. Both sets of CARs started with a uniformly increased expression of CD69, an activation marker, on Day 1. Subsequently, the BBζ CAR T cells proliferated and persisted in culture for over 4 weeks, while the 28ζ CAR T cells had done so for 2 weeks (72). Even more striking persistence differences were observed in vivo, where CD28-based CAR T cells were detected for 30 days (73–75), while 4-1BB based CAR T cells persisted for years (76, 77). The increased persistence of BBζ CAR T cells was attributed to their differentiation into CD45RO+CCR7+ Tcm cells, and this phenotype was maintained through the culture process. In contrast, 28ζ T cell expansion resulted in a higher proportion of CD45RO+CCR7- Tem cells following stimulation through the CAR (20, 72, 78).
To characterize cellular metabolism changes upon CAR signaling, Kawalekar et al. measured the oxygen consumption rate (OCR), a surrogate measure of OXPHOS, before activation, as well as at 7- and 21-days post antigen stimulation; including following serial addition of an inhibitor of ATP synthesis, an uncoupling ionophore, and blocking agents of the ETC. The OCR profiles were similar in both groups on Day 0. On days 7- and 21-post antigenic stimulation an ~10-fold increase in OCR was observed in both groups. Maximal respiratory capacity of BBζ CAR T cells showed a robust increase following decoupling of the mitochondrial membrane. The extracellular acidification rate (ECAR), a surrogate of lactic acid production during glycolysis, was elevated in 28ζ CAR T cells. 28ζ CARs were found to be rapidly consuming glucose and generating lactate, consistent with observed high ECAR levels. Additionally, Glut1, PDK1 (26), G6PD, and phosphoglycerate kinase (PGK), and have also been shown to be elevated in 28ζ cells. These findings support the conclusion that 28ζ CAR T cells rely on glycolysis, Figure 1B, for their energy needs (26, 72), a characteristic of Teff cells (72, 78).
In contrast, BBζ CAR T cells rely on mitochondrial oxidative phosphorylation (72). When glucose uptake and fatty acid utilization rates were evaluated by measuring residual glucose, lactate, and heavy-carbon-labeled long chain fatty acid, palmitic acid, in the media at different time points, BBζ cells showed high utilization of palmitic acid, measured by heavy-carbon-labeled acetyl CoA levels. As β-oxidation of fatty acids generates acetyl CoA, an increase in heavy-carbon-labeled acetyl-Co-A pool indicated that BBζ CAR T cells use FAO to fuel their bioenergetic needs. Also, carnitine palmitoyl transferase (CPT1A), which controls a rate limiting step in mitochondrial FAO and promotes mitochondrial biogenesis (20), was significantly elevated in BBζ cells. Additionally, fatty acid binding protein (FABP5) that is involved in fatty acid uptake, transport, and metabolism was also elevated in these cells. Together, these findings indicate that BBζ CAR T cells use fatty acids for their energy needs (23, 72). Finally, BBζ CAR T cells have a survival advantage due to their ability to generate increased mitochondrial mass (61, 72). BBζ CAR T cells consistently demonstrated high spare respiratory capacity (SRC), which is a characteristic of natural CD8+ T cell memory and supports T cell function in the hostile tumor environment (20, 72, 79–81).
Selection of CAR costimulation systems based on associated metabolic re-wiring: As discussed, the costimulatory signals in the CAR constructs that are necessary for T cell activation, expansion, cytokine secretion, cytotoxic function, memory formation, and survival are mediated through metabolic reprogramming. Depending on the costimulatory domain incorporated into the construct, different signaling pathways are triggered upon antigen activation (82–84). As already discussed, the CD28 domain in CARs leads to the activation of PI3K/Akt pathway, Figure 1A and aerobic glycolysis as the predominant metabolic program. In contrast, T cells with CAR constructs comprising of 4-1BB domain activate the NF-κB, MAPK, and ERK pathways. These CAR T cells exhibit enhanced OXPHOS and SRC derived from fatty acid oxidation. Mitochondrial biogenesis and oxidative metabolism associated with Tcm phenotype are preferred in CAR T therapy given enhanced in vivo persistence and function (72), Figure 1B.
Another costimulatory domain, ICOS, has been shown to lead to higher PI3K/Akt pathway activation compared to CD28, and increased secretion of IL-21, IL-17, and INF-gamma (85). OX40, a member of the TNFR family which is upregulated with T cell activation via the OX40L has a broad effect on T cell activation, proliferation, differentiation, and survival. It stimulates glycolysis and OXPHOS via PI3K/Akt, MAPK and NF-κB pathways, and induces antiapoptotic genes, Bcl-1and Bcl-xl, to promote T cell expansion and survival, respectively (86–88). To summarize, selection of co-stimulatory domains has a significant impact on persistence and antitumor function of CAR T therapies.
Improving CAR T cell efficacy using CAR systems encoding cytokines and/or chemokines: Cytokine- and chemokine- encoding genes can be added into the CAR construct, which may lead to distinct cellular metabolism features. While the metabolic consequences of such modifications have not been fully explored, they have been implemented to improve CAR T entry into and function within the TME. While first-generation CARs had a single CD3ζ signaling domain they were ultimately not very effective (89–91). Second and third generation CAR T cells have incorporated one or two costimulatory signals, respectively. Genetic modifications to CARs for co-expression of cytokine (92–98), chemokine (99–101), or both factors (102–104) have been successfully used to enhance therapeutic efficacy CAR T cells.
Use of cytokines in expansion media to promote CAR T cell metabolic rewiring: Cytokine composition in culture media impacts efficacy of the CAR T cell product. Cytokines that have been broadly investigated to date are IL-2, IL-7, IL-15, and IL-21 (105). IL-2 is a T cell growth factor that promotes effector differentiation and glycolysis in CD8 T cells (22, 106, 107). When CAR T cells are expanded using IL-2, they differentiate to effector CD8+ cells via Akt-m TOR pathway. However, aryl hydrocarbon receptor activation was also observed under such expansion conditions, suggesting that IL2 signaling contributes to CAR T cell exhaustion (105, 108). IL-7 and IL-15 can cause differentiation to memory T cells (20, 109, 110). IL-7 can induce glycerol channel expression and triglyceride (TAG) synthesis that results in a product with a high CD8 memory proportion. It also promotes Glut-1 cell surface expression thereby increasing glucose uptake and promoting cell survival (111). The combination of IL-2 and IL-7 in the ex vivo culture media during CAR T cell expansion enhances glycolysis and differentiation of T cells towards the effector phenotype necessary for cytotoxicity (112). Another cytokine used for CAR T expansion is IL-15. It downregulates mTORC1 activity and expression of several glycolysis enzymes, thereby improving mitochondrial fitness and maintenance of the Tscm phenotype (105, 113). Consequently, recent trials have used CAR T cells expanded in IL-7 and IL-15 and have demonstrated superior anti-tumor efficacy. Notably, CAR T cells expanded in IL-2 can show phenotypic features similar to those expanded in IL-15, when mTORC1 inhibition is provided by concurrent exposure to rapamycin (24).
Optimizing nutrients within expansion media: Optimization of expansion media, aside from cytokine milieu, can also impact on T cell differentiation and subsequent function. For example, L-arginine is consumed rapidly in activated T cells; hence, when exogenous L-arginine was supplemented, a shift from glycolysis to OXPHOS was observed in cultured cells (34, 114). A potential explanation for this switch is that with increased L-arginine leads to an upregulation of the serine biosynthesis pathway, fueling the TCA cycle and enhancing OXPHOS (114, 115). The decrease in glycolysis combined with increased L-arginine levels potentiates the generation of the Tcm subset, promoting anti-tumor activity in vivo (29, 36, 114). Another feature of T cells cultured in excess L-arginine is prolongation of survival (114). Other nutrients in the media that could be altered are fatty acids, especially short chain fatty acids (SCFA), such as butyrate, propionate, and acetate (116), which diffuse passively into the mitochondria. Depending on the concentration, the SCFAs have been shown to favor a memory-like T cell phenotype (116–120).
Following infusion, CAR T have to traffic to tumor sites, penetrate the TME, and persist in the patient to generate sustained anti-tumor activity. Strategies aimed at improving trafficking and persistence of CAR T, including nanoparticle RNA vaccines as well as oncolytic viruses have been described, albeit so far in pre-clinical studies, with clinical testing currently ongoing for the latter. While these approaches have the potential to enhance CAR T in vivo performance, they are not metabolic in nature and are reviewed elsewhere (121).
The impact of TME metabolism on endogenous and adaptively transferred T cells
Cancer involves abnormal cell growth, diminished apoptosis, and evasion of the normal host defenses that facilitate local invasion and potential distant metastasis (122). The high replication rate of tumor cells requires a continuous source of energy necessitating modification of normal metabolism. Cancer cells engage aerobic glycolysis to meet their metabolic requirements and ATP production (1, 123). Increased aerobic glycolysis by the tumor cells creates a glucose deprived TME, impairing effector T cell function within the endogenous anti-tumor immunity and adaptive cell therapy (1, 123). Tumor derived lactate accumulation via aerobic glycolysis leads to an acidified TME. This in turn impedes monocarboxylate transporter-1 mediated, gradient-dependent export of lactate from CD8+ T cells (1, 124, 125). Malignant cells within a solid tumor rapidly proliferate and organize, which can lead to poor vascularization that exacerbates TME hypoxia. In addition to the decreased oxygen delivery, TME hypoxia can be further enhanced by high tumor oxygen consumption. Hypoxia inhibits differentiation, proliferation, and cytokine production of cytotoxic T lymphocytes (CTL) and T helper (Th1) cells (10, 125, 126). Under hypoxic conditions, tumor cells respond with rapid induction of transcription factor hypoxia-inducible factor 1 alpha (HIF1α) and NFκB (1, 127). Both factors are involved in the regulation of genes implicated in inflammation and adaptation to hypoxia. These pathways control mitochondrial dynamics and mitophagy, promote TME acidification, and regulate the expression of cytokines and angiogenic factors (1, 10, 128–131). These transcription factors regulate the expression of a plethora of cytokines and angiogenic factors, including, IL-6, IL-10, and vascular endothelial growth factor (VEGF) (10, 127). These molecules then recruit cells with immunosuppressive function, including myeloid-derived suppressor cells (MDSCs), T regulatory (Treg) cells, innate lymphoid cells (ILCs), tumor associated macrophages (TAMs), and cancer associated fibroblasts (CAFs). This establishes an immunosuppressive TME and suppresses antigen processing and presentation (10, 132), leading to immune evasion. Furthermore, HIFα controls the expression of checkpoint molecule programmed death-ligand 1 (PD-L1) on the surface of cancer cells and PD-1 expression on T cells, which negatively impacts T cell survival and effector functions via the PD1/PD-L1 axis (10, 125). Additionally, hypoxia dampens activity of adenosine kinase and adenosine deaminase leading to adenosine accumulation in the TME that favors immunosuppressive cells and inhibits anti-tumor T cell function (125). As described, the TME plays an important role in augmenting endogenous T cell and CAR T cell function. Several excellent reviews on TME metabolism as a barrier to immunotherapy and metabolic strategies to manipulate the former have been recently published (3, 4, 133). The focus of this review is the manipulation of CAR T metabolism to improve in vivo function and persistence, which is described hereafter.
Immunometabolic interventions to improve CAR T in vivo efficacy and persistence
Increased persistence and survival are characteristics of an effective CAR T anti-tumor response (134). Presence of specific T cell subsets, such as those with minimal differentiation and increased self-renewal capacity, can enhance CAR T cell anti-tumor efficacy (135). As already described, metabolism plays a major role in this process, with effector T cells depending more on glycolysis and memory T cells relying on mitochondrial oxidation and FAO for their bioenergetic needs (136). Pre-clinical studies have shown that an infusion of a high proportion of naïve T cells (Tn), Tscm, or Tcm leads to superior anti-tumor efficacy compared to products with higher Tem or Teff content (137, 138). However, during the ex vivo expansion process, a large proportion of Tem and Teff are generated due to a high rate of glycolysis induced during activation and expansion that drives T cells towards terminally differentiated phenotypes (36). The cytokine milieu during ex vivo expansion, antigen activation, and costimulation, contribute to this process, as previously described. Given that the functional phenotype, i.e., Tscm, and favorable metabolic features are linked, maintenance of this phenotype represents a promising strategy for generating effective adoptive T cell therapies, Figure 1B.
Inhibition of glycolysis: 2-deoxyglucose (2-DG) is a prototypical inhibitor of the glycolytic pathway that blocks hexokinase (139), Figure 1A. Fraietta et al. showed inhibition of glycolysis with 2-DG decreased T cell effector and promoted memory T cell generation (140). Additionally, the same group demonstrated that a glycolytic gene signature characterized CAR T cells obtained from patients with partial or no response to therapy. The former also displayed a higher uptake of a glucose analog than CAR T cells isolated from patients with complete responses (140). Thus, employing strategies to interfere with glycolysis is a potential solution to improve downstream in vivo CAR T cell efficacy. At high concentrations, however, 2-DG may interfere with T cell proliferation and cytolytic function. Cham et al. showed that 2-DG at 10-50 mM concentrations in culture interfered with T cell proliferation and almost completely abolished cytolytic ability of CD8+ T cells (123). Shi et al. showed that inhibition of glycolysis by 2-DG at a concentration of 1mM had minimal inhibitory effects on cell proliferation (139). Similarly, Sukumar et al. used 2 mM concentrations of 2-DG, which sufficiently inhibited glycolysis without interfering with cell proliferation and successfully induced OXPHOS. They also demonstrated that T cells primed in the presence of 2-DG accumulated at higher numbers in tumors (36). In conclusion, inducing a memory T cell phenotype by inhibiting glycolysis with relatively low levels of 2-DG during CAR T cell manufacture is a potential strategy to improve CAR T cell efficacy, Figure 1B.
Improving mitochondrial function: As already discussed, mitochondria play a key role in the regulation of T cell metabolism, biosynthesis, migration, cell fate and programmed cell death. Regulating mitochondrial OXPHOS is one approach to improve CD8+ T cell function. Lactate dehydrogenase (LDH) is an enzyme just distal to glycolysis that converts pyruvate into lactate. Hermans et al. demonstrated that a small molecule LDH inhibitor, at a concentration of 1 uM led to metabolic rewiring, blocking generation of lactate and promoting pyruvate entry into the TCA cycle, and ultimately enhancing OXPHOS (51), Figure 1A. The latter can inhibit terminal effector differentiation and exhaustion. The same group also demonstrated that LDH inhibition in combination with IL-21 exposure increased the formation of Tscm cells leading to an improved anti-tumor response and persistence. Interleukin-21 (IL-21) is a cytokine that uses the common cytokine receptor γ chain (γc) as a receptor component (141). It primarily activates STAT3 (142), along with IL-7 and IL-15 expands CD8+ T cells (143). Thus, transient inhibition of LDH in combination with IL-21 supplementation during expansion phase generated a more effective cell therapy product (51). Another strategy for improving mitochondrial function is through the peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α that belongs to a family of transcription coactivators (144). PGC-1α overexpression in CD8 T cells has been found to boost mitochondrial biogenesis and memory phenotype, enhancing anti-tumor immunity (121, 145, 146), Figure 1A. Additionally, overexpression of PGC-1α in exhausted T cells improved their mitochondrial function, restoring functionality (121, 147). CD8+ T cells with PGC-1α overexpression secondary to exposure to bezafibrate, a PGC-1α agonist, in the presence of PD-1 blockade, upregulated mitochondrial OXPHOS and increased FAO, which enhanced their survival (148). Bezafibrate is a drug already in clinical use for hypercholesterolemia and could potentially be deployed in the setting of adoptive immunotherapy, following CAR T infusion.
An exhausted T cell (Tex) phenotype has been described extensively in the setting of chronic infection but has also been recognized in tumor-resident endogenous T cells and in the setting of adoptive T cell therapy (105, 149). Tex vulnerability is at least in part attributable to tonic TCR stimulation, which leads to metabolic rewiring and epigenetic changes that can enforce terminal exhaustion (105). CAR T cells are thought to be especially susceptible to this process given continuous antigenic stimulation that occurs during their ex vivo expansion, ahead of exposure to tumor antigen in the TME. Enhancing mitochondrial fitness and the linked Tscm phenotype during priming and expansion appear to counteract this susceptibility (105). Tex display inhibitory receptors, diminished effector ability, and decreased proliferative capacity. Metabolically, these cells are characterized to mitochondrial dysfunction and decreased glycolysis utilization (105, 149). Interestingly, Tex functionality can be restored with metabolic manipulation, as described above, through PGC-1α overexpression (121, 147), further supporting the notion that ability to maintain OXPHOS and FAO metabolism are necessary to avoid Tex phenotype. Additionally, PD-1 blockade in T cells has been shown to drive increased FAO, enhancing their survival and memory phenotype (121, 150). However, terminally exhausted T cells may only partially respond to PD-1 blockade, presumably due to fixed epigenic modifications (149). Acetate supplementation, which can restore acetyl-CoA required for histone acetylation, can improve chromatic accessibility and restore functionality in CD8 T cells (149, 151). Other epigenetic modifiers may lower susceptibility or restore functionality in Tex and are explored in greater depth in other reviews (105, 149).
Sustained activation of the PI3K-Akt-mTOR pathway by activation beads, IL-2, or tonic signaling drives T cells towards terminal differentiation and inefficient tumor killing (152–154). mTOR is a main regulator of CD8 T cell differentiation. Inhibition of mTORC1 by rapamycin or metformin (AMPK activator) has been shown to enhance OXPHOS by increasing FAO, and ultimately promoting the CD8+ T memory phenotype (154, 155). In the setting of CAR T, use of rapamycin during priming and expansion promoted the memory phenotype and increased FAO metabolism (121, 152, 156, 157). The addition of PI3K inhibitors, including, MK2206, LY294002, IC87114, idelalisib, and TGR-1202, during expansion has been shown to maintain CAR T cells in a less differentiated state leading to increased downstream anti-tumor efficacy and persistence of CAR T cells in vivo (105, 121, 158).
To summarize, induction of mitochondrial biogenesis and promotion of T cell differentiation towards a memory phenotype is a promising strategy for improving anti-tumor efficacy and persistence of CAR T cells.
Conclusion
Aside from identifying tumor specific antigens and engineering appropriate CARs, several strategies to enhance in vivo CAR T function are currently being pursued. As greater understanding of T cell metabolism and immunosuppressive features of the TME is gained, this knowledge can be potentially leveraged to enhance anti-tumor responses. As described, manipulation of the T cell activation machinery that is linked to cellular metabolism reprogramming can enhance in vivo CAR T cell performance and persistence. Improved trafficking and function of CAR T cells in the TME have been observed with CAR T products containing distinct functional phenotypes that are linked to corresponding metabolic rewiring. These phenotypes can be generated via manipulations of the CAR construct itself, i.e., through selection of costimulatory elements, or during the expansion phase using cytokines and nutrients to skew CAR T products towards more favorable metabolic characteristics. Additionally, metabolic regulators can be used either during expansion or after infusion to generate and maintain metabolic phenotypes within CAR T cells, respectively. Metabolic rewiring of cellular therapies represents a promising clinically relevant approach to improve immunotherapy responses.
Author contributions
All authors contributed to the writing of this review after NB received the invitation to submit.
Acknowledgments
The authors are grateful to Dr. Meghan Higman and Mackenzie Pautler for assisting PMN with literature searches. We are thankful to Jill Duzen for editing the manuscript. Biorender program was used for figure generation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
2. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science (2011) 331(6024):1565–70. doi: 10.1126/science.1203486
3. Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell (2020) 78(6):1019–33. doi: 10.1016/j.molcel.2020.05.034
4. DePeaux K, Delgoffe GM. Metabolic barriers to cancer immunotherapy. Nat Rev Immunol (2021) 21(12):785–97. doi: 10.1038/s41577-021-00541-y
5. Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol (2009) 21(2):215–23. doi: 10.1016/j.coi.2009.02.009
6. Sadelain M, Riviere I, Riddell S. Therapeutic t cell engineering. Nature (2017) 545(7655):423–31. doi: 10.1038/nature22395
7. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor t cells. Sci Transl Med (2012) 4(132):132ra53. doi: 10.1126/scitranslmed.3003761
8. Zhang M, Jin X, Sun R, Xiong X, Wang J, Xie D, et al. Optimization of metabolism to improve efficacy during CAR-t cell manufacturing. J Transl Med (2021) 19(1):499. doi: 10.1186/s12967-021-03165-x
9. Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: Driving t cells to solid tumors. Cancer Discov (2016) 6(2):133–46. doi: 10.1158/2159-8290.CD-15-0583
10. Pellegrino M, Del Bufalo F, De Angelis B, Quintarelli C, Caruana I, de Billy E. Manipulating the metabolism to improve the efficacy of CAR t-cell immunotherapy. Cells (2020) 10(1). doi: 10.3390/cells10010014
11. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. doi: 10.1084/jem.20151159
12. Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, et al. A single naive CD8+ t cell precursor can develop into diverse effector and memory subsets. Immunity (2007) 27(6):985–97. doi: 10.1016/j.immuni.2007.10.012
13. Jameson SC, Masopust D. Understanding subset diversity in t cell memory. Immunity (2018) 48(2):214–26. doi: 10.1016/j.immuni.2018.02.010
14. O'Sullivan D, Pearce EL. Targeting t cell metabolism for therapy. Trends Immunol (2015) 36(2):71–80. doi: 10.1016/j.it.2014.12.004
15. Forcados C, Joaquina S, Casey NP, Caulier B, Walchli S. How CAR t cells breathe. Cells (2022) 11(9). doi: 10.3390/cells11091454
16. Lugli E, Gattinoni L. Harnessing stem cell-like memory t cells for adoptive cell transfer therapy of cancer. In Developments in T Cell Based Cancer Immunotherapies, (2015), Chapter 8, pp 183–209, (Springer). doi: 10.1007/978-3-319-21167-1_8 (book chapter
17. Xu X, Gnanaprakasam JNR, Sherman J, Wang R. A metabolism toolbox for CAR t therapy. Front Oncol (2019) 9:322. doi: 10.3389/fonc.2019.00322
18. Cammann C, Rath A, Reichl U, Lingel H, Brunner-Weinzierl M, Simeoni L, et al. Early changes in the metabolic profile of activated CD8(+) t cells. BMC Cell Biol (2016) 17(1):28. doi: 10.1186/s12860-016-0104-x
19. Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, et al. Memory CD8+ t cells require CD28 costimulation. J Immunol (2007) 179(10):6494–503. doi: 10.4049/jimmunol.179.10.6494
20. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ t cell memory development. Immunity (2012) 36(1):68–78. doi: 10.1016/j.immuni.2011.12.007
21. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in t cell activation and requires CD28-mediated akt-dependent and independent pathways. J Immunol (2008) 180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476
22. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ t cells. J Exp Med (2012) 209(13):2441–53. doi: 10.1084/jem.20112607
23. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 t-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
24. Gerriets VA, Rathmell JC. Metabolic pathways in t cell fate and function. Trends Immunol (2012) 33(4):168–73. doi: 10.1016/j.it.2012.01.010
25. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: Energy metabolism and the t-cell response. Nat Rev Immunol (2005) 5(11):844–52. doi: 10.1038/nri1710
26. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/s1074-7613(02)00323-0
27. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
28. Chang CH, Pearce EL. Emerging concepts of t cell metabolism as a target of immunotherapy. Nat Immunol (2016) 17(4):364–8. doi: 10.1038/ni.3415
29. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of t cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
30. Toriyama K, Kuwahara M, Kondoh H, Mikawa T, Takemori N, Konishi A, et al. T cell-specific deletion of Pgam1 reveals a critical role for glycolysis in t cell responses. Commun Biol (2020) 3(1):394. doi: 10.1038/s42003-020-01122-w
31. Dimeloe S, Burgener AV, Grahlert J, Hess C. T-cell metabolism governing activation, proliferation and differentiation; A modular view. Immunology (2017) 150(1):35–44. doi: 10.1111/imm.12655
32. Dugnani E, Pasquale V, Bordignon C, Canu A, Piemonti L, Monti P. Integrating t cell metabolism in cancer immunotherapy. Cancer Lett (2017) 411:12–8. doi: 10.1016/j.canlet.2017.09.039
33. Frauwirth KA, Thompson CB. Regulation of t lymphocyte metabolism. J Immunol (2004) 172(8):4661–5. doi: 10.4049/jimmunol.172.8.4661
34. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
35. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon t lymphocyte activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
36. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ t cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/JCI69589
37. Daneshmandi S, Cassel T, Higashi RM, Fan TW, Seth P. 6-phosphogluconate dehydrogenase (6PGD), a key checkpoint in reprogramming of regulatory t cells metabolism and function. Elife (2021) 10. doi: 10.7554/eLife.67476
38. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
39. Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol (1985) 70(4):473–89. doi: 10.1113/expphysiol.1985.sp002935
40. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA (2007) 104(49):19345–50. doi: 10.1073/pnas.0709747104
41. Sugiura A, Rathmell JC. Metabolic barriers to t cell function in tumors. J Immunol (2018) 200(2):400–7. doi: 10.4049/jimmunol.1701041
42. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
43. Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: Metabolism and tumor cell growth. Curr Opin Genet Dev (2008) 18(1):54–61. doi: 10.1016/j.gde.2008.02.003
44. Newsholme P. Why is l-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr (2001) 131(9 Suppl):2515S–22S; discussion 2523S-4S. doi: 10.1093/jn/131.9.2515S
45. Newsholme P, Costa Rosa LF, Newsholme EA, Curi R. The importance of fuel metabolism to macrophage function. Cell Biochem Funct (1996) 14(1):1–10. doi: 10.1002/cbf.644
46. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during t-cell differentiation and memory development. Immunol Rev (2012) 249(1):27–42. doi: 10.1111/j.1600-065X.2012.01150.x
47. Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol (2015) 35 Suppl:S129–50. doi: 10.1016/j.semcancer.2015.10.002
48. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481(7381):380–4. doi: 10.1038/nature10602
49. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162(3):552–63. doi: 10.1016/j.cell.2015.07.017
50. Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for t cell activation. Cell Metab (2016) 24(1):104–17. doi: 10.1016/j.cmet.2016.06.007
51. Clare CE, Brassington AH, Kwong WY, Sinclair KD. One-carbon metabolism: Linking nutritional biochemistry to epigenetic programming of long-term development. Annu Rev Anim Biosci (2019) 7:263–87. doi: 10.1146/annurev-animal-020518-115206
52. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature (2014) 510(7504):298–302. doi: 10.1038/nature13236
53. Fox JT, Stover PJ. Folate-mediated one-carbon metabolism. Vitam Horm (2008) 79:1–44. doi: 10.1016/S0083-6729(08)00401-9
54. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector t cell expansion. Cell Metab (2017) 25(2):482. doi: 10.1016/j.cmet.2017.01.014
55. Ron-Harel N, Notarangelo G, Ghergurovich JM, Paulo JA, Sage PT, Santos D, et al. Defective respiration and one-carbon metabolism contribute to impaired naive t cell activation in aged mice. Proc Natl Acad Sci USA (2018) 115(52):13347–52. doi: 10.1073/pnas.1804149115
56. Kurniawan H, Kobayashi T, Brenner D. The emerging role of one-carbon metabolism in t cells. Curr Opin Biotechnol (2021) 68:193–201. doi: 10.1016/j.copbio.2020.12.001
57. Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi SC, et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis (2013) 4(10):e877. doi: 10.1038/cddis.2013.393
58. Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov (2014) 4(12):1406–17. doi: 10.1158/2159-8290.CD-14-0250
59. Sugiura A, Andrejeva G, Voss K, Heintzman DR, Xu X, Madden MZ, et al. MTHFD2 is a metabolic checkpoint controlling effector and regulatory t cell fate and function. Immunity (2022) 55(1):65–81 e9. doi: 10.1016/j.immuni.2021.10.011
60. van der Windt GJ, O'Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory t cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA (2013) 110(35):14336–41. doi: 10.1073/pnas.1221740110
61. Benjamin R, Jain N, Maus MV, Boissel N, Graham C, Jozwik A, et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor t-cell therapy for adults with relapsed or refractory b-cell acute lymphoblastic leukaemia (CALM): a phase 1, dose-escalation trial. Lancet Haematol (2022) 9(11):e833–43. doi: 10.1016/S2352-3026(22)00245-9
62. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR t cells: development and challenges. Nat Rev Drug Discov (2020) 19(3):185–99. doi: 10.1038/s41573-019-0051-2
63. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human b-cell malignancies. Blood (2016) 128(4):519–28. doi: 10.1182/blood-2015-11-683847
64. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified t cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi: 10.1038/leu.2015.247
65. O'Sullivan D. The metabolic spectrum of memory t cells. Immunol Cell Biol (2019) 97(7):636–46. doi: 10.1111/imcb.12274
66. Rostamian H, Fallah-Mehrjardi K, Khakpoor-Koosheh M, Pawelek JM, Hadjati J, Brown CE, et al. A metabolic switch to memory CAR t cells: Implications for cancer treatment. Cancer Lett (2021) 500:107–18. doi: 10.1016/j.canlet.2020.12.004
67. Irving BA, Weiss A. The cytoplasmic domain of the t cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell (1991) 64(5):891–901. doi: 10.1016/0092-8674(91)90314-o
68. Romeo C, Amiot M, Seed B. Sequence requirements for induction of cytolysis by the t cell antigen/Fc receptor zeta chain. Cell (1992) 68(5):889–97. doi: 10.1016/0092-8674(92)90032-8
69. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and t-cell receptors. Proc Natl Acad Sci USA (1993) 90(2):720–4. doi: 10.1073/pnas.90.2.720
70. van Berkel ME, Oosterwegel MA. CD28 and ICOS: similar or separate costimulators of t cells? Immunol Lett (2006) 105(2):115–22. doi: 10.1016/j.imlet.2006.02.007
71. Kawalekar OU, O. CRS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR t cells. Immunity (2016) 44 712(3). doi: 10.1016/j.immuni.2016.02.023
72. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted t cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra38. doi: 10.1126/scitranslmed.3005930
73. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3
74. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted t cells in patients with relapsed or chemotherapy refractory b-cell leukemias. Blood (2011) 118(18):4817–28. doi: 10.1182/blood-2011-04-348540
75. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor t cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7(303):303ra139. doi: 10.1126/scitranslmed.aac5415
76. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates t cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
77. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of t lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
78. Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today (2008) 13(5-6):268–74. doi: 10.1016/j.drudis.2007.12.008
79. Nicholls DG. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans (2009) 37(Pt 6):1385–8. doi: 10.1042/BST0371385
80. Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex i with rotenone. J Neurosci (2007) 27(27):7310–7. doi: 10.1523/JNEUROSCI.0212-07.2007
81. Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted t cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res (2007) 13(18 Pt 1):5426–35. doi: 10.1158/1078-0432.CCR-07-0674
82. Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred t cells. Cancer Res (2006) 66(22):10995–1004. doi: 10.1158/0008-5472.CAN-06-0160
83. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of t cells and increased antileukemic efficacy in vivo. Mol Ther (2009) 17(8):1453–64. doi: 10.1038/mt.2009.83
84. Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci Transl Med (2010) 2(55):55ra78. doi: 10.1126/scitranslmed.3000448
85. Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem (1998) 273(10):5808–14. doi: 10.1074/jbc.273.10.5808
86. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes bcl-xL and bcl-2 expression and is essential for long-term survival of CD4 t cells. Immunity (2001) 15(3):445–55. doi: 10.1016/s1074-7613(01)00191-1
87. So T, Choi H, Croft M. OX40 complexes with phosphoinositide 3-kinase and protein kinase b (PKB) to augment TCR-dependent PKB signaling. J Immunol (2011) 186(6):3547–55. doi: 10.4049/jimmunol.1003156
88. Brocker T, Karjalainen K. Signals through t cell receptor-zeta chain alone are insufficient to prime resting t lymphocytes. J Exp Med (1995) 181(5):1653–9. doi: 10.1084/jem.181.5.1653
89. Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient t cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia (1999) 1(2):123–7. doi: 10.1038/sj.neo.7900018
90. Globerson Levin A, Riviere I, Eshhar Z, Sadelain M. CAR t cells: Building on the CD19 paradigm. Eur J Immunol (2021) 51(9):2151–63. doi: 10.1002/eji.202049064
91. Liu Y, An L, Huang R, Xiong J, Yang H, Wang X, et al. Strategies to enhance CAR-t persistence. biomark Res (2022) 10(1):86. doi: 10.1186/s40364-022-00434-9
92. Brentjens RJ, Curran KJ. Novel cellular therapies for leukemia: CAR-modified t cells targeted to the CD19 antigen. Hematol Am Soc Hematol Educ Program (2012) 2012:143–51. doi: 10.1182/asheducation-2012.1.143
93. Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary t lymphocytes. J Exp Med (1998) 188(4):619–26. doi: 10.1084/jem.188.4.619
94. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor t cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.CIR-13-0027
95. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR t cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics (2018) 8:41–51. doi: 10.1016/j.omto.2017.12.003
96. He C, Zhou Y, Li Z, Farooq MA, Ajmal I, Zhang H, et al. Co-expression of IL-7 improves NKG2D-based CAR t cell therapy on prostate cancer by enhancing the expansion and inhibiting the apoptosis and exhaustion. Cancers (Basel) (2020) 12(7). doi: 10.3390/cancers12071969
97. Hombach AA, Geumann U, Gunther C, Hermann FG, Abken H. IL7-IL12 engineered mesenchymal stem cells (MSCs) improve a CAR t cell attack against colorectal cancer cells. Cells (2020) 9(4). doi: 10.3390/cells9040873
98. Lesch S, Blumenberg V, Stoiber S, Gottschlich A, Ogonek J, Cadilha BL, et al. T cells armed with c-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat BioMed Eng (2021) 5(11):1246–60. doi: 10.1038/s41551-021-00737-6
99. Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor t cells targeting mesothelin in a non-small-cell lung carcinoma model. Front Immunol (2021) 12:628906. doi: 10.3389/fimmu.2021.628906
100. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a hodgkin tumor model. Blood (2009) 113(25):6392–402. doi: 10.1182/blood-2009-03-209650
101. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-t cells improves immune cell infiltration and CAR-t cell survival in the tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
102. Luo H, Su J, Sun R, Sun Y, Wang Y, Dong Y, et al. Coexpression of IL7 and CCL21 increases efficacy of CAR-t cells in solid tumors without requiring preconditioned lymphodepletion. Clin Cancer Res (2020) 26(20):5494–505. doi: 10.1158/1078-0432.CCR-20-0777
103. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR t cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun (2019) 10(1):4016. doi: 10.1038/s41467-019-11869-4
104. Huang Y, Si X, Shao M, Teng X, Xiao G, Huang H. Rewiring mitochondrial metabolism to counteract exhaustion of CAR-t cells. J Hematol Oncol (2022) 15(1):38. doi: 10.1186/s13045-022-01255-x
105. Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity (2013) 38(1):13–25. doi: 10.1016/j.immuni.2013.01.004
106. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic t cells. Immunity (2010) 32(1):79–90. doi: 10.1016/j.immuni.2009.11.012
107. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. IL-2 regulates tumor-reactive CD8(+) t cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol (2021) 22(3):358–69. doi: 10.1038/s41590-020-00850-9
108. Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory t cells by opposing cytokines. Science (2000) 288(5466):675–8. doi: 10.1126/science.288.5466.675
109. Schluns KS, Lefrancois L. Cytokine control of memory t-cell development and survival. Nat Rev Immunol (2003) 3(4):269–79. doi: 10.1038/nri1052
110. Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. IL-7-Induced glycerol transport and TAG synthesis promotes memory CD8+ t cell longevity. Cell (2015) 161(4):750–61. doi: 10.1016/j.cell.2015.03.021
111. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of akt to support t-cell survival. Blood (2008) 111(4):2101–11. doi: 10.1182/blood-2007-06-096297
112. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-t cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res (2019) 7(5):759–72. doi: 10.1158/2326-6066.CIR-18-0466
113. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates t cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–842e13. doi: 10.1016/j.cell.2016.09.031
114. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476(7360):346–50. doi: 10.1038/nature10350
115. Rial Saborido J, Volkl S, Aigner M, Mackensen A, Mougiakakos D. Role of CAR t cell metabolism for therapeutic efficacy. Cancers (Basel) (2022) 14(21). doi: 10.3390/cancers14215442
116. Bachem A, Makhlouf C, Binger KJ, de Souza DP, Tull D, Hochheiser K, et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8(+) t cells. Immunity (2019) 51(2):285–297e5. doi: 10.1016/j.immuni.2019.06.002
117. Balmer ML, Ma EH, Bantug GR, Grahlert J, Pfister S, Glatter T, et al. Memory CD8(+) t cells require increased concentrations of acetate induced by stress for optimal function. Immunity (2016) 44(6):1312–24. doi: 10.1016/j.immuni.2016.03.016
118. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory t cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
119. Kespohl M, Vachharajani N, Luu M, Harb H, Pautz S, Wolff S, et al. The microbial metabolite butyrate induces expression of Th1-associated factors in CD4(+) t cells. Front Immunol (2017) 8:1036. doi: 10.3389/fimmu.2017.01036
120. Lopez-Cantillo G, Uruena C, Camacho BA, Ramirez-Segura C. CAR-t cell performance: How to improve their persistence? Front Immunol (2022) 13:878209. doi: 10.3389/fimmu.2022.878209
121. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
122. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ t cells. Eur J Immunol (2008) 38(9):2438–50. doi: 10.1002/eji.200838289
123. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human t cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
124. Shen L, Xiao Y, Tian J, Lu Z. Remodeling metabolic fitness: Strategies for improving the efficacy of chimeric antigen receptor t cell therapy. Cancer Lett (2022) 529:139–52. doi: 10.1016/j.canlet.2022.01.006
125. Nakagawa Y, Negishi Y, Shimizu M, Takahashi M, Ichikawa M, Takahashi H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic t lymphocytes. Immunol Lett (2015) 167(2):72–86. doi: 10.1016/j.imlet.2015.07.003
126. Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood (2009) 114(4):844–59. doi: 10.1182/blood-2008-12-195941
127. Capece D, Verzella D, Di Francesco B, Alesse E, Franzoso G, Zazzeroni F. NF-kappaB and mitochondria cross paths in cancer: mitochondrial metabolism and beyond. Semin Cell Dev Biol (2020) 98:118–28. doi: 10.1016/j.semcdb.2019.05.021
128. D'Ignazio L, Batie M, Rocha S. Hypoxia and inflammation in cancer, focus on HIF and NF-kappaB. Biomedicines (2017) 5(2). doi: 10.3390/biomedicines5020021
129. Laforge M, Rodrigues V, Silvestre R, Gautier C, Weil R, Corti O, et al. NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ (2016) 23(1):89–98. doi: 10.1038/cdd.2015.42
130. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of c-MYC activity. Cancer Cell (2007) 11(5):407–20. doi: 10.1016/j.ccr.2007.04.001
131. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory t cells and natural killer t cells. Immunology (2013) 138(2):105–15. doi: 10.1111/imm.12036
132. Mao W. Overcoming current challenges to t-cell receptor therapy via metabolic targeting to increase antitumor efficacy, durability, and tolerability. Front Immunol (2022) 13:1056622. doi: 10.3389/fimmu.2022.1056622
133. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory t cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi: 10.1038/nm.2446
134. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector t cell differentiation and generates CD8+ memory stem cells. Nat Med (2009) 15(7):808–13. doi: 10.1038/nm.1982
135. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) t cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41(1):75–88. doi: 10.1016/j.immuni.2014.06.005
136. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ t cells. J Clin Invest (2005) 115(6):1616–26. doi: 10.1172/JCI24480
137. Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ t cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood (2011) 117(3):808–14. doi: 10.1182/blood-2010-05-286286
138. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
139. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) t cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
140. Hermans D, Gautam S, Garcia-Canaveras JC, Gromer D, Mitra S, Spolski R, et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8(+) t cell stemness and antitumor immunity. Proc Natl Acad Sci USA (2020) 117(11):6047–55. doi: 10.1073/pnas.1920413117
141. Leonard WJ, Lin JX, O'Shea JJ. The gamma(c) family of cytokines: Basic biology to therapeutic ramifications. Immunity (2019) 50(4):832–50. doi: 10.1016/j.immuni.2019.03.028
142. Spolski R, Leonard WJ. Interleukin-21: A double-edged sword with therapeutic potential. Nat Rev Drug Discovery (2014) 13(5):379–95. doi: 10.1038/nrd4296
143. Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21-mediated proliferation. Blood (2007) 109(10):4135–42. doi: 10.1182/blood-2006-10-054973
144. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J (2015) 282(4):647–72. doi: 10.1111/febs.13175
145. Li W, Zhang L. Rewiring mitochondrial metabolism for CD8(+) t cell memory formation and effective cancer immunotherapy. Front Immunol (2020) 11:1834. doi: 10.3389/fimmu.2020.01834
146. Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, et al. Enforced PGC-1alpha expression promotes CD8 t cell fitness, memory formation and antitumor immunity. Cell Mol Immunol (2021) 18(7):1761–71. doi: 10.1038/s41423-020-0365-3
147. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) t cell exhaustion. Immunity (2016) 45(2):358–73. doi: 10.1016/j.immuni.2016.07.008
148. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in t cells increases the number of tumor-reactive CD8(+) t cells and facilitates anti-PD-1 therapy. Cancer Immunol Res (2018) 6(11):1375–87. doi: 10.1158/2326-6066.CIR-18-0095
149. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of t-cell exhaustion. Nat Metab (2020) 2(10):1001–12. doi: 10.1038/s42255-020-00280-9
150. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters t-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
151. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes t cell effector function during glucose restriction. Cell Rep (2019) 27(7):2063–2074e5. doi: 10.1016/j.celrep.2019.04.022
152. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 t-cell differentiation. Nature (2009) 460(7251):108–12. doi: 10.1038/nature08155
153. Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, et al. Signal integration by akt regulates CD8 t cell effector and memory differentiation. J Immunol (2012) 188(9):4305–14. doi: 10.4049/jimmunol.1103568
154. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8(+) t cell differentiation. J Clin Invest (2015) 125(5):2090–108. doi: 10.1172/JCI77746
155. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res (2015) 75(2):296–305. doi: 10.1158/0008-5472.CAN-14-2277
156. Gattinoni L, Klebanoff CA, Restifo NP. Pharmacologic induction of CD8+ t cell memory: better living through chemistry. Sci Transl Med (2009) 1(11):11ps12. doi: 10.1126/scitranslmed.3000302
157. Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR signalling triggers the formation of stem cell-like memory t cells. EBioMedicine (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
Keywords: CAR T cell, adoptive cell therapy (ACT), cell metabolism, immunometabolism, tumor microenvironment
Citation: Nanjireddy PM, Olejniczak SH and Buxbaum NP (2023) Targeting of chimeric antigen receptor T cell metabolism to improve therapeutic outcomes. Front. Immunol. 14:1121565. doi: 10.3389/fimmu.2023.1121565
Received: 11 December 2022; Accepted: 17 February 2023;
Published: 14 March 2023.
Edited by:
Hildegard Theresia Greinix, Medical University of Graz, AustriaReviewed by:
Paramita Chakraborty, Medical University of South Carolina, United StatesLuciana D’Apice, National Research Council (CNR), Italy
Copyright © 2023 Nanjireddy, Olejniczak and Buxbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nataliya Prokopenko Buxbaum, TmF0YWxpeWEuQnV4YmF1bUBSb3N3ZWxsUGFyay5vcmc=