 Chih-Chun Lee1†
Chih-Chun Lee1† Chia-Hsuan Tsai2,3†
Chia-Hsuan Tsai2,3† Chih-Hao Chen2,3
Chih-Hao Chen2,3 Yuan-Chieh Yeh4,5
Yuan-Chieh Yeh4,5 Wen-Hung Chung3,6,7,8,9,10,11,12,13,14,15,16
Wen-Hung Chung3,6,7,8,9,10,11,12,13,14,15,16 Chun-Bing Chen3,6,7,8,9,10,11,12,13,14,15,16,17*
Chun-Bing Chen3,6,7,8,9,10,11,12,13,14,15,16,17*- 11 Department of Medical Education, Chang Gung Memorial Hospital, Keelung, Taiwan
- 2Department of Plastic and Reconstructive Surgery, Chang Gung Memorial Hospital, Keelung, Taiwan
- 3College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 4Department of Traditional Chinese Medicine, Chang Gung Memorial Hospital, Keelung, Taiwan
- 5Program in Molecular Medicine, College of Life Sciences, National Yang Ming Chiao Tung University, Taipei, Taiwan
- 6Drug Hypersensitivity Clinical and Research Center, Department of Dermatology, Chang Gung Memorial Hospital, Linkou, Taiwan
- 7Drug Hypersensitivity Clinical and Research Center, Department of Dermatology, Chang Gung Memorial Hospital, Taipei, Taiwan
- 8Drug Hypersensitivity Clinical and Research Center, Department of Dermatology, Chang Gung Memorial Hospital, Keelung, Taiwan
- 9Cancer Vaccine and Immune Cell Therapy Core Laboratory, Chang Gung Memorial Hospital, Linkou, Taiwan
- 10Chang Gung Immunology Consortium, Chang Gung Memorial Hospital and Chang Gung University, Linkou, Taiwan
- 11Department of Dermatology, Xiamen Chang Gung Hospital, Xiamen, China
- 12Xiamen Chang Gung Allergology Consortium, Xiamen Chang Gung Hospital, Xiamen, China
- 13Whole-Genome Research Core Laboratory of Human Diseases, Chang Gung Memorial Hospital, Keelung, Taiwan
- 14Immune-Oncology Center of Excellence, Chang Gung Memorial Hospital, Linkou, Taiwan
- 15Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 16Genomic Medicine Core Laboratory, Chang Gung Memorial Hospital, Linkou, Taiwan
- 17School of Medicine, National Tsing Hua University, Hsinchu, Taiwan
Keloid is a type of disfiguring pathological scarring unique to human skin. The disorder is characterized by excessive collagen deposition. Immune cell infiltration is a hallmark of both normal and pathological tissue repair. However, the immunopathological mechanisms of keloid remain unclear. Recent studies have uncovered the pivotal role of both innate and adaptive immunity in modulating the aberrant behavior of keloid fibroblasts. Several novel therapeutics attempting to restore regulation of the immune microenvironment have shown variable efficacy. We review the current understanding of keloid immunopathogenesis and highlight the potential roles of immune pathway-specific therapeutics.
1 Introduction
Keloid is a type of pathological scarring unique to human skin. The disorder is characterized by dysregulated fibroproliferation with excessive production of extracellular matrix (ECM) and extension beyond the initial wound (1). Keloid scars are often disfiguring, profoundly impair the quality of life and cause immense physical and mental distress of affected individuals, especially in those with symptomatic (pruritic, painful) and/or hyperpigmented scars (2–4). Limited epidemiological data suggested a female predominance, and a higher prevalence among people of darker skin complexion, such as those of African and Asian descents (5, 6). The prevalence of excessive scarring in Black, Asians and Caucasians was recently reported at 2.4%, 1.1% and 0.4%, respectively (6). Association between excessive scarring and other systemic conditions including hypertension (7–9), vitamin D deficiency (10, 11), and atopic dermatitis (12, 13) has been suggested. A recent cohort of the UK biobank found atopic dermatitis significantly associated with excessive scarring across ethnic groups (6). Hypertension in Blacks and vitamin D deficiency in Asians also showed significant association with keloid formation (6).
In practice, several preventive and therapeutic therapies are used to manage keloids. Application of silicone gel sheets, topical corticosteroids, and intralesional corticosteroids are frequently utilized in individuals with a history of excessive scarring after trauma or surgeries (14). For established keloids, nonsurgical management commonly involves intralesional corticosteroids (e.g., triamcinolone acetonide) (5). Laser-assisted topical steroid application is a novel alternative with better reported aesthetic outcome (15). Intralesional injection of botulinum toxin A, 5-fluorouracil, verapamil, bleomycin, and interferon (IFN)-α2b are less common measures with varying efficacy (16, 17). Other methods include laser therapy (18), and intralesional cryosurgery (19). Monotherapy with radiation is less preferred due to the requirement of large radiation doses (14). Successful surgical management of keloids hinges on the ability to minimize dermal tension (20). Body site-specific techniques have been proposed (14). The high postsurgical recurrence rate can be ameliorated with adjunctive radiation and/or local corticosteroids (14). There are also anecdotal reports with tissue-engineered allografts (21) and platelet-rich plasma (22). The associated adverse effects of established therapies could be significant, especially with long-term or repeated treatment. Intralesional corticosteroids, one of the most frequent methods in both prophylactic and therapeutic management of keloids, is associated with skin hypo-/hyper-pigmentation, atrophy, and telangiectasia.
The pathogenesis of the exuberant scarring remains incompletely understood. No single determining pathway has been identified. Instead, roles of several transcription factors, growth factors, cytokines, ECM proteins, and their associated regulators/effectors have been implicated in experimental studies. The dysregulated molecular profile causes imbalance within and across stages of tissue repair. Wound healing consists of an overlapping sequence of hemostasis, inflammation, proliferation and re-epithelization, and remodelling (23, 24). At the inflammation stage, the innate immune system is activated in response to the damage-associate molecular patterns (DAMPs) and other danger signals (23, 24). Cell debris are removed via phagocytosis of neutrophils (23, 24). Macrophages are later recruited. In addition to phagocytosis, macrophages play an important role in the resolution of inflammation, setting the stage for proliferation (23, 24). The proliferation phase is characterized by migration of keratinocytes, angiogenesis, and formation of granulation tissue (23, 24). Remodelling ensues with replacement of collagen III with collagen I and regression of blood vessels (23, 24). Across the stages, there is a complex interplay between immune cells and fibroblasts. Moreover, the outcome of subsequent stages is closely associated with the integrity and functionality of prior events (23). Hence, excessive scarring could arise as primary dysfunction of the remodelling phase or secondary to an exaggerated inflammatory response (23, 25).
The etiology of keloids is likely multifactorial and hinges on a constellation of factors, including genetic predisposition (26–34), inflammation (35–39), mechanical stress (40–43), tissue hypoxia (44–48), delayed-type hypersensitivity (49), and metabolic dysfunction (50, 51). Familial cases of autosomal dominant inheritance with incomplete clinical penetrance and variable expression have been described (52–54). Several immune pathway-associated susceptible genotypes have been identified, including polymorphisms of interleukin (IL)-6 and transforming growth factor (TGF)-β receptors (26–28, 55–57). Moreover, immune cell infiltration is a hallmark of keloid tissue. Preferential recruitment of immune cells modulates the process of skin repair via interaction with keloid fibroblasts (58, 59). Since the 1970s, the immunological aspect of keloid formation has been proposed (60–63), and a potential role for autoimmunity was frequently evoked in early reports (60, 63). With the advent of novel technologies and laboratory methods, the interest in the immunological landscape of keloid formation has led to vigorous investigations over the past two decades.
The reticular dermis has been proposed as the main locale of chronic inflammation underscoring the formation of keloid scars with upregulation of various proinflammatory cytokines, including IL-1α, IL-1β, IL-6, and tumor necrosis factor (TNF)-α (35). Interestingly, there appeared to be a concomitant excess of regulatory cell types and cytokines (64, 65). Study of keloid histology demonstrated altered expression of ECM molecules with increased type I/III collagen ratio, and a hypercellular dermis with increased numbers of fibroblasts, mast cells and macrophages, as well as varying presence of lymphocytes (1, 36, 64, 66–75). Keloid tissue also harbored a higher percentage of mesenchymal stem cells, and the amount of which was found to be correlated with disease recurrence (76). The role of myofibroblasts is less defined. A recent report found that myofibroblasts, a key feature of cultured fibroblasts in several reports, are not characteristic of keloid lesion in vivo (77). The concept of keloid microenvironment has been frequently evoked to describe the complex cellular and molecular interplay that gives rise to and sustains keloidogenesis. Recent technologies, such as identification of differentially expressed genes via examination of RNA sequencing data sets (74, 75, 78–80), have led to more extensive analysis of keloid tissue. A skewed T helper (Th) 2 phenotype was recently characterized (81–85), along with a potential co-susceptibility of keloids and atopic dermatitis (6, 12, 86). Moreover, even in the absence of comorbid atopic dermatitis, both lesional and non-lesional skin of patients with chronic keloids exhibit Th2 predominance (81). These features of heightened immune response were validated in a recent transcriptomic study, in which a globally elevated expression of several immune pathways over the entire integument of keloid patients was seen, especially the Th2 and Janus kinase (JAK) 3 pathways. Increased expression of T cell, regulatory T cell (Treg), and dendritic cell (DC) markers was also observed, along with the expression of the innate, Th1- and Th17/Th22-signaling pathways (85). The change in cellular composition and function is accompanied by increased levels of IL-6, IL-10, IL-17, TGF-β, and TNF (38, 39, 48, 78, 85, 87–94). Increase in IL-4, IL-13, IL-18, granulocyte colony-stimulating factors, and granulocyte-monocyte colony-stimulating factors, were also observed (39, 82–85, 87, 88). On the other hand, reduced expression of potential anti-inflammatory mediators, such as IL-34 and IL-37, has been reported (85, 87, 95, 96). Single-cell RNA sequencing and spatial transcriptomics (72, 97–99), as well as epigenetics (34, 100) are emerging fields utilized to reveal potential pathophysiological features of keloids. One single-cell RNA sequencing study identified a distinct macrophage-centered communication regulatory network that may favor transition and proliferation of M2 macrophages (72). In addition to the local characteristics, corresponding abnormalities in the cytokine profile have been identified in the peripheral blood of keloid patients (39). Serum soluble human leukocyte antigen-E (sHLA-E) was recently identified as a potential biomarker of keloid occurrence and recurrence (101). Furthermore, aberrant immune cell composition and activity are increasingly recognized in the non-lesional skin of keloid patients (81, 82, 85). Reports on the involvement of humoral immunity were less consistent. Anti-hnRNPA2B1, an autoantibody against RNA-associated proteins, was found to be significantly elevated in the serum of keloid patients (73). The same study also showed deposition of immunoglobulins (IgA, IgM) and complements (IgA, IgM, C3 and C1q) via immunofluorescence in keloid skin tissue (73). These findings suggest a systemic pathological process underscoring the development of keloids, such that the risk-benefit of repeated local therapy for susceptible individuals is called into question. Further studies are required to elucidate the origin of the keloid-prone immunological signatures.
In this review, current understanding of keloid immunopathogenesis is discussed, with highlights of potential pathway-targeted therapeutics.
2 The roles of immune cells in keloid formation
2.1 Mast cells as profibrotic mediators
Mast cells cluster in tissue exposed to the external environment. In human skin, mature mast cells are abundant near the vasculature, lymphatics, nerves, and fibroblasts, and play a crucial role in wound healing by initiating inflammation, facilitating re-epithelialization, and inducing angiogenesis (68). It has been postulated that mast cells contribute to profibrotic chronic inflammation as well as to the common symptoms (pruritus and erythema) associated with keloid scars (Figure 1). Silicone gel sheeting has been shown to reduce mast cell infiltration in keloid lesions and thus provide symptomatic relief (102).
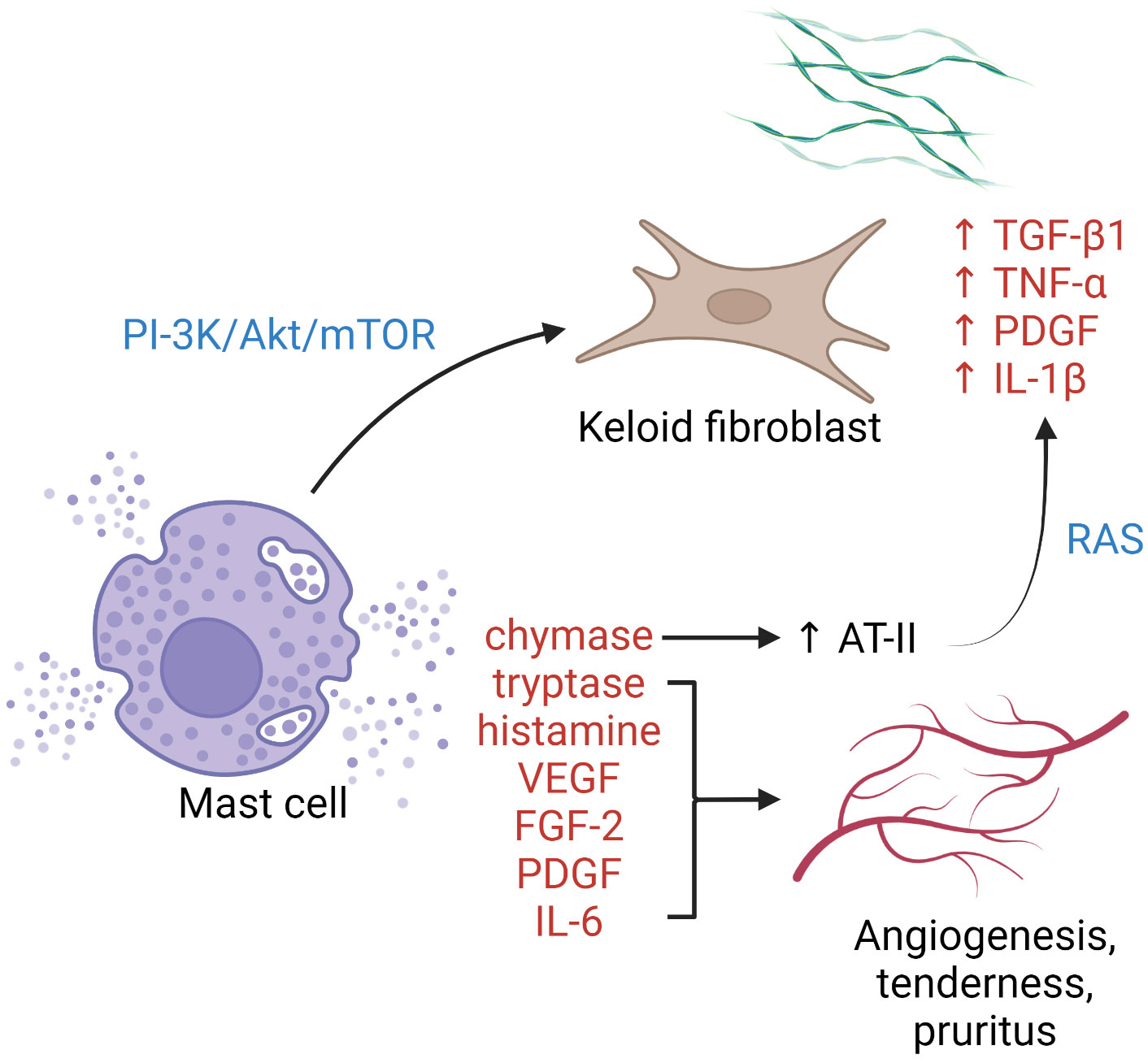
Figure 1 The role of mast cells in keloid pathogenesis. Degranulated mast cells crosstalk with activated keloid fibroblasts via the PI3K/Akt/mTOR pathway, leading to more collagen synthesis. Enzymes, growth factors, and cytokines released upon mast cell degranulation contribute to activation of the RAS, upregulation of keloid fibroblasts, angiogenesis, and cutaneous symptoms. AT-II, angiotensin II; FGF-2, fibroblast growth factor 2; IL, interleukin; PDGF, platelet-derived growth factor; PI3K/Akt/mTOR, phosphatidylinositol-3-kinase/Akt/mammalian target of rapamycin pathway; RAS, renin–angiotensin system; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor.
Increased intralesional and perilesional mast cells can be observed in keloid tissue, both perivascularly and within abnormal collagen bundles (36, 103). Degranulated mast cells are frequently seen in contact with active fibroblasts, indicating the presence of cell–cell interaction (67, 103). Attenuation of such cellular crosstalk has been achieved by blockading the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway using green tea extract (polyphenol EGCG), with a corresponding reduction in type I collagen production (104). Several pro-angiogenic factors are released by mast cells, including vascular endothelial growth factor (VEGF), fibroblast growth factor-2, platelet-derived growth factor, IL-6, tryptase, and chymase. Tryptase, a serine protease, is one of the most potent inducers of tissue angiogenesis. Tryptase-positive mast cell density and keloid angiogenesis are positively correlated (105). The use of transdermal tryptase inhibitors for hypertrophic scars and keloids has been described with symptomatic benefit (105). Mast cell chymase expression and activity are heightened in keloid tissue. The enzyme is profibrotic and stimulates fibroblast proliferation and collagen synthesis via the TGF-β1/Smad signaling pathway (106). Mast cell-derived chymase enhances angiotensin II expression, leading to local activation of the renin-angiotensin system and upregulation of TGF-β1, TNF-α, platelet-derived growth factor, and IL-1β in keloid fibroblasts (107). Chymase inhibitors have been shown to possess antifibrotic property in skin (108), cardiovascular system (109), and liver (110) in animal models. Other means of mast cell antagonization, e.g., with mast cell stabilizers (111) or tyrosine kinase inhibitors (112), have not been tested in keloids.
2.2 Macrophage polarization and chronic inflammation
M1 (classically activated, CD68-positive) and M2 (alternatively activated, CD163-positive) are two well-established macrophage subgroups. The two phenotypes possess opposing properties, with the former exerting a pro-inflammatory effect and the latter an anti-inflammatory effect (113). An imbalance between M1 and M2 macrophages has been described in several chronic inflammatory conditions such as rheumatoid arthritis (114). Normal wound healing is characterized by an orchestrated transition from M1-predominant early inflammatory stages to M2-predominant restitution (115). Dysregulation of this process leads to either prolonged inflammation with delayed wound closure or increased scarring. M2 macrophages are disproportionally elevated in keloid lesions (69–72, 74), in part due to local enrichment of Th2 cytokines. Although not yet verified, M2 dominance has also been linked to macrophage sensitivity to mechanical signals, including skin tension and stiffness (42). M2 macrophages initiate wound closure via secretion of TGF-β1, a potent inducer of both fibroblast proliferation and their differentiation into myofibroblasts (115). Moreover, M2 macrophages induce transcription factor forkhead box P3 (FOXP3) expression in circulating CD3+ T cells, contributing to the formation of Tregs (64) (Figure 2). Interestingly, while M2 predominance is clearly present, expression of both M1 (inducible nitric oxide synthase [iNOS], IL-12)- and M2 (IL-10, TGF-β)-associated genes and proteins is enhanced keloid lesions compared to normal skin (64).
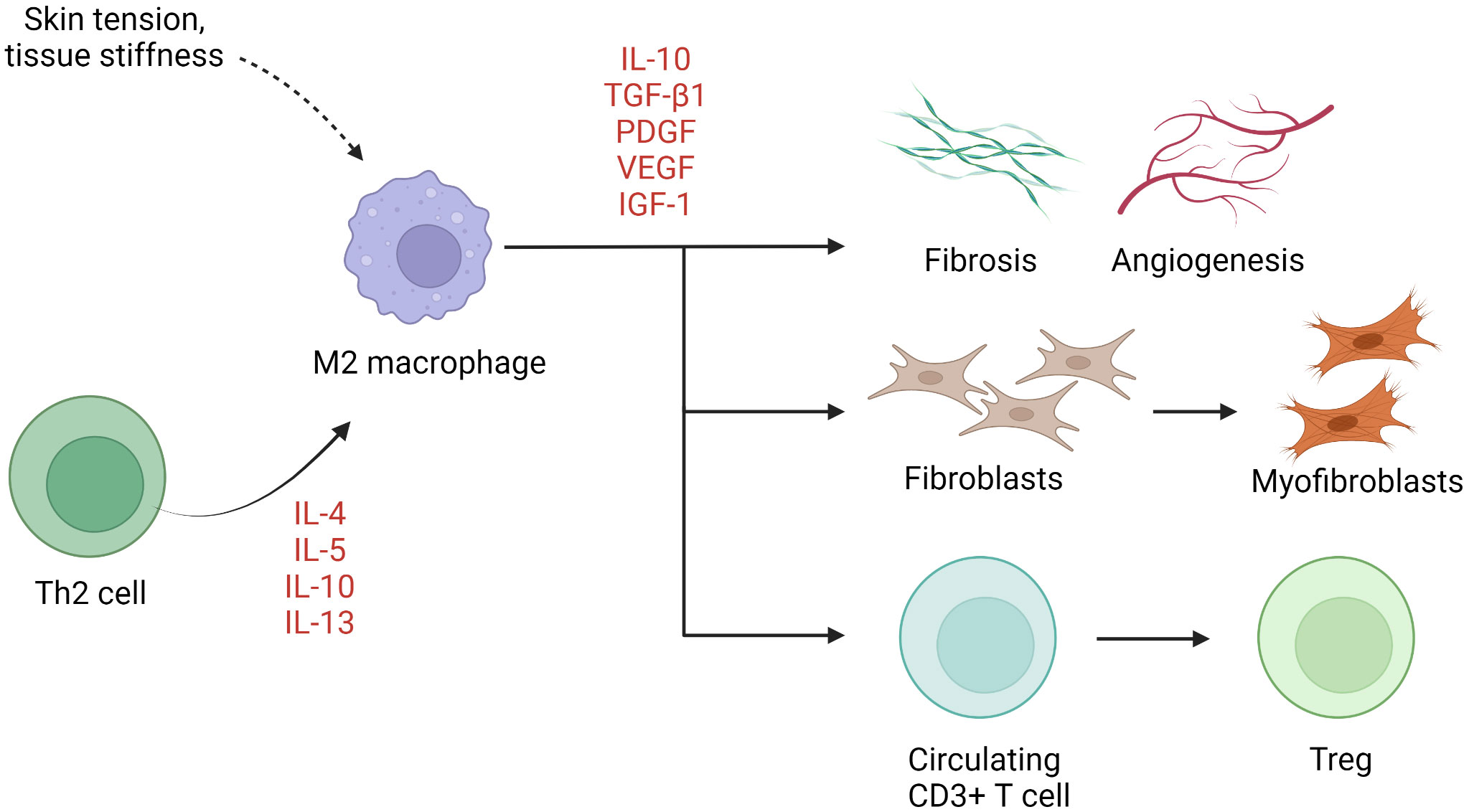
Figure 2 The role of macrophages in keloid pathogenesis. M2 macrophages predominate in keloids, resulting in activation of keloid fibroblasts, Treg differentiation, and fibrosis and angiogenesis. IGF-1, insulin-like growth factor 1; IL, interleukin; PDGF, platelet-derived growth factor; TGF-β1, transforming growth factor β1; Th, helper T cells; Treg, regulatory T cell; VEGF, vascular endothelial growth factor.
2.3 Tregs-derived TGF-β1 and collagen expression
The numbers of Tregs are increased in keloid lesions (64, 65). Tregs proliferate after cellular contact with dermal fibroblasts in the presence of IL-15 in chronically inflamed skin (116). In keloids, they promote preferential accumulation of collagen III in the presence of anti-CD3/CD28 (65). In patients with multiple keloid scars, the local infiltration of Tregs was found to be coupled with a reduction in circulating CD4+ CD25high FOXP3+ Tregs (117). Whether the apparent excess of local Tregs is pathogenic or merely represents a response to inflammation remains unclear. TGF-β1 and IL-10 are key cytokines secreted by Tregs and exert an autocrine effect (118, 119). The former mediates elaboration of matrix proteins and stimulates the production of IL-6 by mast cells (120), while the latter downregulates proinflammatory macrophages and promotes B cell activation and immunoglobulin secretion (121) (Figure 3). Interestingly, IL-10, rather than IFN-γ, antagonizes the TGF-β1 effect on keloid fibroblasts (93, 122). In muscle, Tregs are known to accumulate at injured sites and modulate the polarization of M1 macrophage to M2 macrophage (123). It is likely that they assume a similar coordinating role in wound healing. Further investigations are required to determine the extent to which Tregs alter the balance between M1 and M2 macrophages and contribute to keloidogenesis.
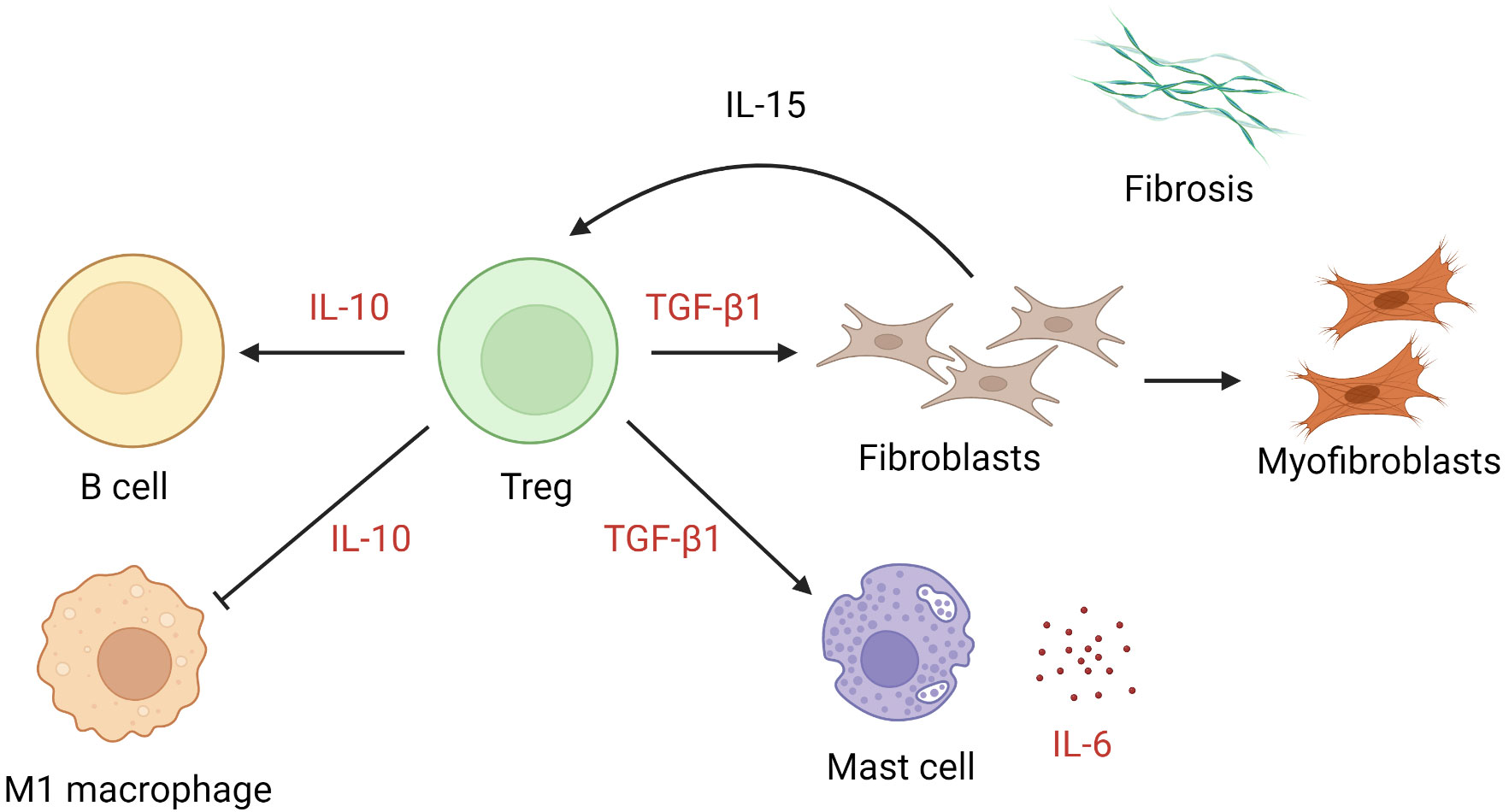
Figure 3 The role of regulatory T cells in keloid pathogenesis. Tregs exert effects through the action of IL-10 and TGF-β1, leading to suppression of M1 macrophages, activation of keloid fibroblasts, and mast cell production of IL-6. IL, interleukin; TGF-β1, transforming growth factor β1; Tregs, regulatory T cells.
2.4 Chronic stimulation and exhaustion of CD8+ T cells
In normal skin, the majority of T cells are CD45RO+ memory T cells. The same holds true in keloids, with a significantly higher proportion of effector memory CD8+ T cells (TEM) and CD103+CD8+ resident memory T cells (TRM) (117). TRM are known to trigger an exaggerated inflammatory response to stimuli (124). Keloid memory T cells are less adept at producing TNF-α and more prone to generating IFN-γ (117). FOXP3+ CD8- memory T cells are also defective with decreased IL-10 secretion, resulting in exuberant but dysregulated T cell responses in keloids (117). Further adding to the dysregulation, the expression of granzyme B+ CD8+ cytotoxic T cell is downregulated in keloids, a feature presumably related to the characteristic uncontrolled growth. A recent single-cell RNA study discovered that chronic antigenic stimulation in keloids result in enhanced surface NKG2A expression on CD8+ T cells and natural killer (NK) cells (125, 126), with resultant suppression of cytotoxic T cells via the NKG2A-soluble human leukocyte antigen-E (sHLA-E) axis (101). IL-15 (127) and TGF-β (128) were implicated in this process. The enhanced expression of the NKG2A/CD94 complex on CD8+ cytotoxic T cells is correlated with progression of keloids. The level of sHLA-E reflects clinical response to intralesional therapy (triamcinolone and 5-fluorouracil) and predicts recurrence risk (101). Furthermore, the degree of sHLA-E elevation could differentiate keloid scars from certain malignant mimics, with the former exhibiting significantly higher levels of sHLA-E (101). Monalizumab, a humanized anti-NKG2A IgG4 monoclonal antibody, exerts an antitumor effect by unleashing both cytotoxic T cells and NK cells (125). The agent has been tested in clinical trials as part of the immunotherapeutic regimens for advanced solid organ cancers, such as recurrent/metastatic squamous cell carcinoma of the head and neck (129), unresectable stage III non-small-cell lung cancer (130), and recurrent gynecologic malignancies (131). Further studies are required to determine the therapeutic potential of NKG2A/CD94 blockade for keloids.
2.5 Dendritic cells
Dermal infiltration of factor XIIIa (FXIIIa)-positive DCs is increased in keloid scars comparing to hypertrophic scars and mature scars (132, 133). These potent antigen-presenting cells are thought to take part in the pathogenic epidermal–dermal interactions in keloids (132), and DC-derived TGF-β could contribute to the differentiation of Tregs. RNA sequencing study confirmed increase of DC markers CD80 and CD86, as well as markers typical of atopic DCs (OX40L+, FCϵR1+) in both lesional and nonlesional skin of keloid patients (85). Unlike in atopic dermatitis, where DCs have been linked to mast cell activation and Th2, Th17 and Th22 differentiation (134, 135), the exact action of DCs in keloids is less clear.
2.6 Natural killer cells
Flow cytometric analyses of keloid single-cell suspensions have shown an unusually high number of NK cells (79). Although their role in keloidogenesis is less well described, NK cells express the surface NKG2A/CD94 complex and thus are implicated in the NKG2A-sHLA-E axis (101). Therefore, it is possible that NK cell activity is relatively suppressed in the TGF-β-rich, chronically inflamed keloid milieu, and that a phenomenon paralleling uncontrolled cancerous growth due to NK and cytotoxic T cell exhaustion is likely present.
3 Key cytokine pathways in keloid formation
Keloids are characterized by dysregulation of multiple signaling pathways and associated cytokines. The best described are IL-6/IL-17, IL-4/IL-13, canonical and non-canonical TGF-β1, and JAK/STAT signaling (Figure 4).
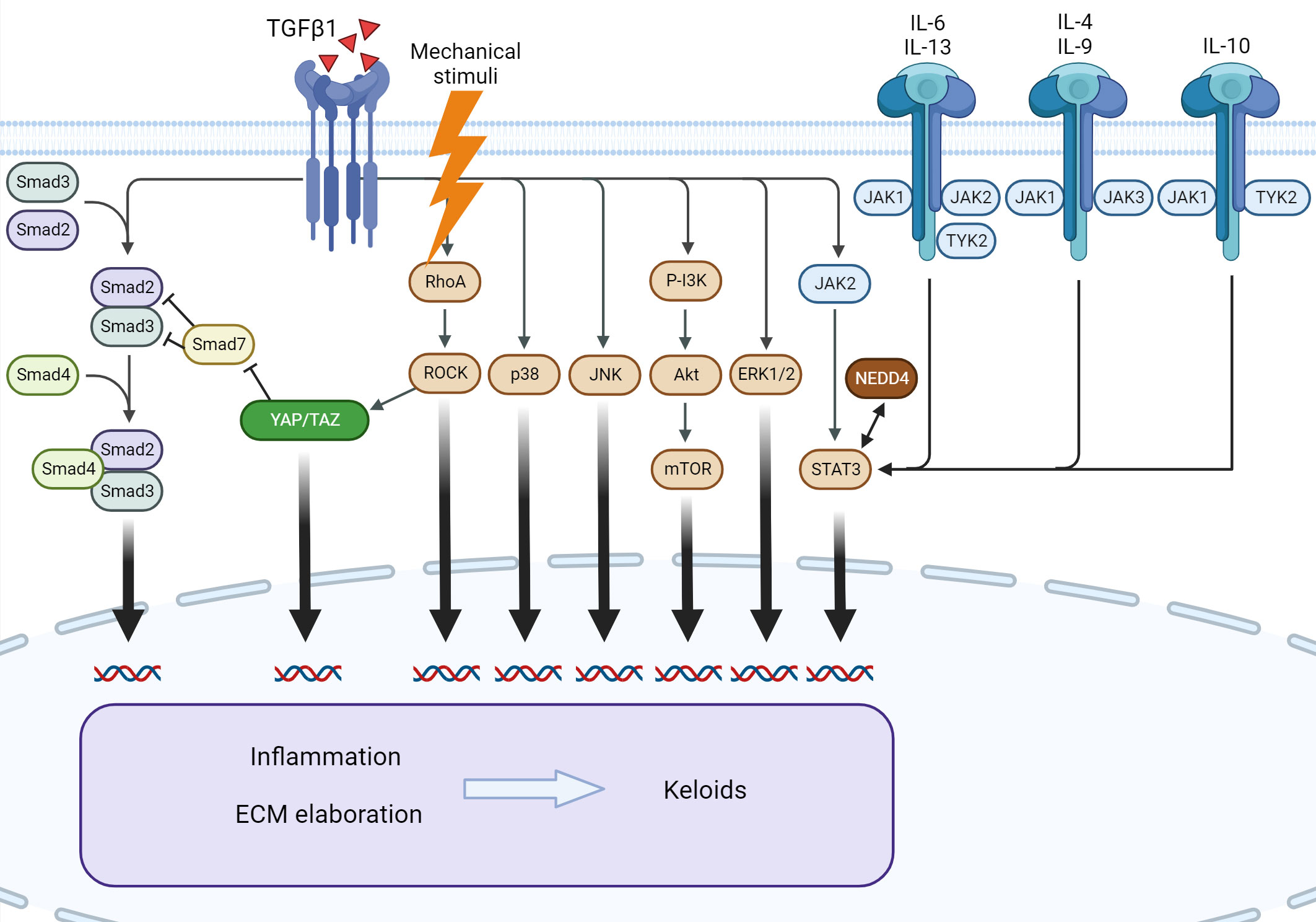
Figure 4 Signaling pathways involved in keloid formation. Several downstream pathways of TGF-β1 participate in keloid pathogenesis. Mechanical stimuli exert an effect through Rho/ROCK and YAP/TAZ, leading to the modulation of canonical TGF-β1 signaling. The interleukin family of cytokines and NEDD4 exert profibrotic action via STAT3, independent of TGF-β1. ERK, extracellular signal-regulated kinase; IL, interleukin; JAK, Janus kinase; JNK, c-Jun N-terminal kinase; mTOR, mammalian target of rapamycin; NEDD4, neural precursor cell expressed, developmentally downregulated 4; PI3K, phosphatidylinositol-3-kinase; ROCK, RhoA/Rho-associated protein kinase; STAT3, signal transducer and activator of transcription 3; TGF-β1, transforming growth factor β1; TYK2, tyrosine kinase 2; YAP/TAZ, Yes-associated protein/transcriptional coactivator with PDZ-binding motif.
3.1 The essential role of IL-6 in inflammation
IL-6 signals through the JAK1–signal transducer and activator of transcription (STAT) 3 pathway and the extracellular signal-regulated kinase (ERK) 1/2–mitogen-activated protein kinase (MAPK) pathway. Both pathways have been implicated in keloid ECM gene expression and collagen synthesis (90, 92). IL‐6 and the soluble IL‐6 receptor (sIL-6R) are essential for collagen production (136). Similar to patients with systemic sclerosis (137), patients with keloids show elevated IL-6 in the serum and skin (38, 55, 90). In addition, IL-6 polymorphisms have been associated with susceptibility to keloid formation across populations (55–57).
IL-6 is pivotal to the transition from acute to chronic inflammation via initiation of a profibrotic state (138–140). Specifically, the cytokine modulates the fibrogenic crosstalk between fibroblasts and keratinocytes by inducing proinflammatory cytokines (IL-1β and TNF-α) in monocytes via MAPK and nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) signaling (141). Keratinocyte growth factor production by fibroblasts is enhanced, and the activated keratinocytes in turn produce oncostatin M, triggering STAT3 signaling in dermal fibroblasts (141). IL-6 production is increased in response to enhanced TGF-β1 signaling as a downstream effector (via PI3K and p38-MAPK) (142), and in turn it enhances TGF-β1 production by macrophages (64), creating a positive feedback loop. IL-6 is also crucial to Th2 and M2 macrophage polarization by initiating IL-4 secretion by CD4+ T cells) and upregulating IL-4 receptors (IL-4R) on macrophages (141). Of note, several experimental therapies for keloids and other forms of cutaneous fibrosis directly or indirectly antagonize IL-6. Examples include corticosteroids, verapamil, angiotensin receptor blocker/angiotensin converting enzyme inhibitors, tocilizumab, pirfenidone, and ultraviolet A (87, 141). TNF-α-stimulated gene-6 (TSG-6), a protein suppressed in keloid fibroblasts, has been shown to attenuate IL-1β, IL-6, and TNF-α when intradermally injected into hypertrophic scars (143). The IL-17/IL-6 axis is crucial to sustaining a cytokine-rich, chronically inflamed niche, augmented by an autocrine loop with increased differentiation of Th17 and subsequent heightened secretion of IL-6 (38). IL-17-mediated enhancement of stromal cell-derived factor-1 (SDF-1) in keloid fibroblasts further reinforces Th17 differentiation via STAT3 mediation (89). This hyperinflammatory milieu is the most prominent perilesionally (89). Through upregulation of hypoxia-inducible factor-1α (HIF-1α) and STAT3, IL-17 impairs autophagy of both normal and keloid fibroblasts, resulting in increased necroptosis and fibrosis. Antagonization of IL-17 via HIF-1α or SDF-1α suppression has been demonstrated in vitro (48, 89).
3.2 The role of type 2 immunity: IL-4/IL-13
Several studies have investigated the association between keloids and other conditions characterized by the Th2 response. The results are variable, with some studies reporting a positive correlation with atopic dermatitis (6, 12, 13, 81). Th2 immunity is involved in normal wound healing as well as various fibrotic conditions (144). IL-4 and IL-13 are key Th2 cytokines that have wide-ranging influence across cell types as their receptors are commonly present (144). The binding of IL-4 and IL-13 to their cognate receptors activates the IL-4Rα/STAT6 signaling pathway, a TGF-β-independent profibrotic mechanism (145). Both IL-4 and IL-13 independently participate in normal and pathogenic healing. Topical IL-4 significantly accelerates the rate of fibrotic tissue formation, whereas IL-4 antisense oligonucleotides attenuate the healing process in animal models (146). In mouse models of systemic sclerosis, anti-IL-4 monoclonal antibodies prevent progression of cutaneous fibrosis by reducing dermal collagen deposition (147). On the other hand, IL-13 has been shown to directly contribute to fibroblast proliferation and differentiation. IL-13 enhances the expression of type I collagen, α-smooth muscle actin (α-SMA), and other essential proteins involved in fibrogenesis. Furthermore, tissue inhibitors of metalloproteinases are attenuated while matrix metalloproteinases are upregulated in keloid fibroblasts treated with IL-13 (83).
The expression of IL-4, IL-13 and their respective receptors is enhanced in keloid scars (81–83, 85, 144, 145). Regression of chronic keloids has been achieved with Th2-targeting therapy with dupilumab (an anti-IL-4Rα agent) in a case report (81). Others showed variable efficacy (148, 149). Molecular profiling of keloids with RNA sequencing demonstrated a significant increase in Th2 expression in both lesional and non-lesional skin of keloid patients (85). The relative dominance of the Th2 response has been attributed to the anti-apoptotic effect conferred to CD4+ T cells by IL-4 (39). IL−4− and IL−13−activated macrophages (M2 macrophages) are critical to resolving inflammation during wound repair (150, 151). In chronic inflammation, these cytokines have been shown to upregulate miR-142-5p and suppress miR-130a-3p in macrophages, leading to a sustained profibrogenic phenotype (152). Human dermal fibroblasts treated with IL-4 and IL-13 exhibit drastically elevated levels of periostin mRNA with enhanced secretion (82). Periostin is an important promoter of RhoA/Rho-associated protein kinase (ROCK) pathway-mediated TGF-β1 secretion impliacted in pathological scarring (82, 153). In systemic sclerosis, periostin is correlated with skin disease severity (154), and the serum level of IL-13 recflects severity of both skin fibrosis (155). The clinical utility of these biomarkers in keloids remains to be determined.
3.3 The pivotal role of TGF-β1
TGF-β1 is one of the most studied mediators of fibrosis. It is frequently implicated in keloid pathogenesis. TGF-β1 has a wide range of cellular sources, including fibroblasts, monocytes, T cells, and platelets (156). As a key regulator of fibrogenesis, this pleiotropic cytokine plays a pivotal role in various cutaneous and solid organ fibrotic disorders, as well as in tumorigenesis via induction of cancer-associated fibroblasts (156). Several monoclonal antibodies, small molecule inhibitors, small interfering RNAs (siRNAs), and antisense oligodeoxynucleotides targeting TGF-β1 signaling are currently under development (156).
The induction by TGF-β1 of various growth factors, including connective tissue growth factor (CTGF) and VEGF, is crucial to the maintenance of the ECM. Moreover, TGF-β1 exerts an autocrinal effect that downregulates dipeptidyl peptidase-4 (DPP4) expression, contributing to a chronically inflamed state with elevated levels of the extracellular C-X-C motif chemokine ligand 12 (CXCL12) (157). Fibroblasts in keloids are considerably sensitive to TGF-β compared to those in hypertrophic scars (158, 159). These abnormal fibroblasts are able to overcome Fas-mediated apoptosis when augmented by TGF-β1 (160). TGF-β1-induced smooth muscle actin (SMA) expression in keloid fibroblasts contributes to increased cell rigidity, a phenomenon common to both keloids and scleroderma (161). SMA expression is linked to wound contracture, and the process can be inhibited with treatment with recombinant human decorin, TNF-like weak inducer of apoptosis (TWEAK), and SB-431542, a novel specific inhibitor of TGF-β1 receptor kinase (162). TGF-β1 is also capable of upregulating C-MYC and its downstream splicing regulator polypyrimidine tract-binding protein—a key factor in tumorous growth—in keloid fibroblasts (163). Furthermore, altered interaction between TGF-β isoforms at the receptor level in keloid fibroblasts has been described (164–167). The ratio of these isoforms may cause a tendency to fibrosis (168). Accordingly, a novel truncated type II TGF-β receptor has been designed as an anti-scarring agent (169, 170). Both canonical and non-canonical TGF-β1 signaling are implicated in modulating the keloid keratinocytes to possess a metabolic profile similar to those undergoing epithelial–mesenchymal transition with increased invasiveness (45, 171–173).
3.3.1 Modulators of the TGF-β1/Smad pathway
The canonical TGF-β signaling is enhanced in keloids, and strategic targeting of TGF-β1/Smad has been shown to retard keloid fibroblasts in vitro or in animal models (Supplemental Table 1). Upstream modulators of the TGF-β1/Smad pathway, including activating transcription factor 3 (174), CR6-interacting factor 1 (175), NLR family CARD domain containing 5 (NLRC5) (176), and nuclear receptor subfamily 3, group C, member 1 (NR3C1), are overexpressed in keloid fibroblasts. HIF-1α and high temperature requirement factor A1 activate the TGF-β1/Smad pathway and promote keloid formation (46, 177). S100A4, a small, calcium-binding protein involved in skin and solid organ fibrosis, is upregulated in keloid fibroblasts and inhibited by calcimycin (178). Syndecan-1, a cell surface proteoglycan highly expressed in wounds, also enhances the pathway in keloids (179). Post-translational sumoylation amplifies TGF-β1/Smad signal transduction in keloids (47).
MicroRNAs are small regulatory RNAs capable of altering post-translational gene expression. In keloid fibroblasts, the anti-fibrotic regulators miR-200c (180), miR-92b (181), miR-1224-5p (182), and miR-133a-3p (183) are expressed at low levels and pro-fibrotic miR-21is overexpressed, altering the activity of the TGF-β1/Smad pathway (184). Peroxisome proliferator-activated receptor-γ agonists have been shown to induce miR-92b expression and thus lower TGF-β1 expression in keloids (181). MicroRNA expression is modulated by long-noncoding RNAs. In keloids, fibroblast behavior is altered in the presence of different long-noncoding RNAs (180, 185–188). For example, LINC01116 contributes to a pro-fibrotic state in keloid tissue via editing of miR-3141 (185). In addition, the BMP and activin membrane-bound inhibitor (189), Dickkopf-3 (190), and the receptor for activated C-kinase 1 (191) attenuate TGF-β1-induced fibrosis; all are downregulated in keloid fibroblasts. Smad-7 provides negative feedback to the TGF-β1/Smad system. The molecule is suppressed due to a marked increase in the level of TGF-β inducible early gene-1 in keloids (192). Downregulation of TRAF3IP2 in keloid fibroblasts by FOXO4 attenuates the growth of keloid scars (193). IL-37 is a broad inhibitor of innate inflammation and regulator of TGF-β (194, 195). Recent studies have uncovered its role in modulating several metabolic pathways and a potential role in reversing trained immunity (196). As seen in idiopathic pulmonary fibrosis (197), lower serum levels of IL-37 were found to indicate higher keloid severity (95).
3.3.2 Non-canonical TGF-β pathways
Several non-canonical TGF-β pathways are involved in keloid formation. These include the MAPK (94, 198, 199), ERK 1/2 (44, 200) phosphatidylinositol-3-kinase (P-I3K)/AKT (44, 104, 200–202), c-Jun amino-terminal kinase (94), p38 mitogen-activated protein kinase (p38/MAPK) (94, 203–205), and Rho-like (82) signaling pathways. The multi-kinase inhibitor sorafenib induces cell arrest of keloid fibroblasts by blockade of the intracellular TGF-β/Smad and MAPK/ERK pathways (206). JUN (an oncogene encoding the c-Jun protein) initiates fibrosis via CD36 in both human and murine hypertrophic scar fibroblasts, and the blockade of CD36 exerted an anti-scarring effect in the murine model (207).
3.3.3 Bridge to mechanical transduction: Reciprocal cross-regulation with the integrin and Yes-associated protein/transcriptional coactivator with PDZ-binding motif pathways
Crosstalk between TGF-β and mechanical transduction pathways is increasingly recognized. Among these pathways, the integrin pathway (79, 142) and the Hippo/Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) pathway (43, 208, 209) are the most recognized. In addition, TGF-β interacts with Wnt/β-catenin activity in dermal fibroblasts, upregulating ECM genes (210, 211). YAP/TAZ are important actors in cellular mechanical transduction. These transcriptional factors are regulated mostly by cell–cell adhesion and cell–ECM attachment via integrins (212). Conditions that cause stiffening of the ECM, such as inflammation, lead to a lower threshold of YAP/TAZ activation (212). IL-6 is also known to activate YAP through gp130 signaling (212). Activated YAP/TAZ translocate into keloid fibroblast nuclei, a step required for wound healing (208). In liver cirrhosis, YAP/TAZ contribute to tissue fibrosis via enhanced SMA expression, promoting the transformation of fibroblasts into myofibroblasts. YAP/TAZ are also implicated in the sustained profibrotic transcriptional profile of idiopathic pulmonary fibrosis (213). Targeted knockdown of YAP or TAZ has been shown to significantly inhibit the activity and induce apoptosis of keloid fibroblasts (208). Inhibition of Rho/Rho kinase signaling, a major upstream regulator of YAP/TAZ, also attenuates fibroblast activity (82, 214). Manipulation of the YAP/TAZ-associated pathways could potentially reduce keloid scarring. A recent study identified a subpopulation of dermal Engrailed-1 lineage-negative fibroblasts in cell transplantation and transgenic mouse models that could give rise to scar-forming Engrailed-1 lineage-positive fibroblasts during adult wound healing (215). The process is initiated by canonical mechanotransduction signaling and depends on YAP (215). Inhibition or knockout of YAP prohibits Engrailed-1 activation, favoring scarless (regenerative) wound healing via Engrailed-1 lineage-negative fibroblasts (215). Verteporfin, a small-molecule YAP inhibitor, has been proposed as a potential novel agent for promoting regenerative skin healing without compromising the healing process (216).
3.4 Janus kinase/signal transducers and activators of the transcription pathway
STAT3 is highly expressed and phosphorylated in keloid tissue with increased activation of JAK2 (217). Moreover, STAT3 activity is correlated with fibroblast proliferation and migration, as well as collagen deposition, mainly due to dysregulated secretion of cytokines resulting from altered epithelial–mesenchymal interactions (218). Attenuation of such activity can be achieved with JAK2/STAT3 inhibitors or STAT3 siRNA (217, 219). Cytokines enriched in the keloid microenvironment, especially IL-6 and OSM, are strong activators of the JAK/STAT system. Various Th2- and Th17-cytokines, including IL-4, IL-10, IL-13, and IL-17, also signal through JAK/STAT (85, 87). IL-6-specific hyperactivation of STAT3 has been shown to be profibrotic due to the induction of Gremlin (a bone morphogenetic protein [BMP] antagonist), which in turn sustains canonical TGF-β signaling (136). Recently, RNA sequencing analyses confirmed robust expression of JAK3 in keloid tissue (85), and positioned STAT3 in a feedforward loop regulating a myriad of downstream target genes involved in keloidogenesis (220). From a metabolic viewpoint, keloids exhibit accelerated glycolysis reminiscent of Warburg metabolism, a unique adaptive state presumably induced by JAK2/STAT3 (50, 221). Interestingly, an in vivo study demonstrated regulation of keloid fibroblast activity at the cost of a worsened hyperglycolytic state with JAK1/2 blockade (222). Epigallocatechin-3-gallate (EGCG), a green tea extract, has been found to possess chemopreventive properties, including suppression of STAT3 signaling, potentially inhibiting keloid growth (219). ASC-J9, an inhibitor of STAT3 phosphorylation, has shown efficacy in suppressing keloid fibroblasts (223). AG490, a selective JAK2/STAT3 inhibitor, and STAT3−specific decoy oligodeoxynucleotides are also beneficial in vitro (224). Oral small-molecule JAK inhibitors are effective in treating skin and pulmonary diseases of systemic sclerosis (225). In a case report, tofacitinib, a pan-JAK inhibitor, facilitated control of keloid scar (226).
STAT3 was recently discovered as a transcription factor for the neural precursor cell expressed, developmentally downregulated 4 (NEDD4) gene (227). NEDD4 encodes a ubiquitin ligase involved in protein degradation and has been associated with susceptibility to keloids (30, 32, 33, 228–231). NEDD4 transcript variant 3 is overexpressed in keloid skin and is responsible for heightened activation of NF-κB via interaction with receptor interacting protein, an adaptor protein (29). NF-kB is more prominent in keloids than in normal skin and contributes to impaired apoptosis of fibroblasts (37, 232). Aspirin may potentially prevent this effect (232). NEDD4 regulates cell contact inhibition and T cell factor/β-catenin transcriptional activity (231). It is also linked to fibronectin and type 1 collagen expression (231). A positive feedback loop between STAT3 and NEDD4 has been described (29), and silencing of NEDD4 also attenuates STAT3 (29, 227), making NEDD4 a potential therapeutic target in keloids.
4 Other potential therapeutics
Fibroblast activation protein (FAP), a membrane-bound enzyme with structural similarity to DPP4, is found almost exclusively on activated fibroblasts and myofibroblasts under pathological conditions (233), making it a potential target for selective inhibition. Similar to DPP4, FAP upregulates extracellular CXCL12 (234). In addition to its enzymatic activity against the ECM (and thus its association with lesion invasiveness (234, 235), the molecule is likely pluripotent with immunomodulatory properties (234). The FAP expression level is enhanced in keloid fibroblasts (234, 235) and FAP modulation has been shown to attenuate the invasiveness of scars (235). As a marker of pathological fibroblast activation, FAP is a novel subject of interest in solid tumors and connective tissue disorders. Previous studies have shown that FAP chimeric antigen receptor-T cell therapy may be limited by systemic toxicity as FAP is also expressed on multipotent bone marrow stromal cells (236, 237). On the other hand, FAP-inhibiting radiopharmaceuticals have shown theranostic promise in various malignancies and other disorders characterized by tissue fibrosis, such as systemic sclerosis (238), rheumatoid arthritis (239), and IgG4-related disease (240). Targeted photodynamic therapy with an anti-FAP photosensitizer exhibits a dose-dependent therapeutic effect on skin fibroblasts of patients with systemic sclerosis (238).
Additional pathway abnormalities, such as Notch and Toll-like receptor signaling pathways, have been implicated in keloid pathogenesis (51, 241, 242). Human adipose-, amnion-, bone marrow- and Wharton’s jelly-derived mesenchymal stem cells have been shown to inhibit proliferation, migration, and synthesis of keloid fibroblasts in vitro, presumably though paracrine effects (243–249). The TGF-β1/Smad and TGF−β2/Smad3 pathways, Notch-1, and cyclooxygenase-2/prostaglandin E2 cascade were all implicated (243–245). Further investigations are warranted to evaluate the in vivo effects of these pathways.
5 Current challenges and future direction in keloid research
Even with modern technologies, several factors complicate our understanding of keloidogenesis. The lack of an ideal animal model has impeded experimental investigations, and the examination of the nature of keloid scars is limited by sample size. Moreover, the lack of standardization of the site of tissue sampling complicates the interpretation of study results. We previously reported that the inflammatory activity within a keloid scar is most vigorous at the periphery, corresponding to the gradational change in skin tension (31). The gene signatures also varied at the leading edge, center and top of keloid lesions (250). Theoretically, anti-inflammatory measures would be most beneficial at the initial inflammatory stage of wound healing and at the periphery of the scar. Anti-fibrotic therapy, on the other hand, ameliorates the later stages and the more central part of the lesion before scar maturation. To allow for timely and appropriate (i.e., without compromising the healing process) modulation of immune pathways, the mechanisms regulating the transition and spatio-temporal overlap across stages need to be better understood. Studies focusing on explicating the cellular and molecular processes of wound healing, could be of immense value to our understanding and management of keloid disorder.
6 Conclusions
The keloid microenvironment is characterized by an exuberant inflammatory response to mechanical and non-mechanical stimuli, resulting in a complex interplay between various hyperactivated immune components with an ultimately profibrotic cytokine profile and signaling. Manipulation of isolated elements or pathways has shown variable efficacy, mostly in an experimental setting. Keloid is increasingly characterized by an inflammatory process, and local treatment might be insufficient for long-term control. Newer biologics and small molecule drugs allow for more specific and systemic targeting of immune pathways. For both approved and experimental drugs, a critical issue is the timing of intervention, as premature suppression of either inflammation or fibrosis could impair wound healing. Further investigations to disentangle the delicate process of wound healing are thus crucial for a more targeted management of keloids.
Author contributions
Conceptualization, C-CL, C-HT, and C-BC; methodology, C-CL, C-HT, and C-BC; resources, C-HT, W-HC, and C-BC; writing—original draft preparation, C-CL, C-HT, and C-BC; writing—review and editing, C-CL, C-HT, C-HC, Y-CY, W-HC, and C-BC; visualization, C-CL, C-HT, and C-BC; supervision, C-HT, W-HC, and C-BC; project administration, C-HT, C-HC, Y-CY, W-HC, and C-BC. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by research grants from the Ministry of Science and Technology, Taiwan (grant no. MOST 110-2314-B-182A-106-MY2 to C-HT) and Chang Gung Memorial Hospital, Taiwan (grant no. CMRPG2L0181 to C-HT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1117630/full#supplementary-material
References
1. Jumper N, Paus R, Bayat A. Functional histopathology of keloid disease. Histol Histopathol (2015) 30(9):1033–57. doi: 10.14670/hh-11-624
2. Sitaniya S, Subramani D, Jadhav A, Sharma YK, Deora MS, Gupta A. Quality-of-Life of people with keloids and its correlation with clinical severity and demographic profiles. Wound Repair Regener (2022) 30(3):409–16. doi: 10.1111/wrr.13015
3. Lu W, Chu H, Zheng X. Effects on quality of life and psychosocial wellbeing in chinese patients with keloids. Am J Transl Res (2021) 13(3):1636–42.
4. Bijlard E, Kouwenberg CA, Timman R, Hovius SE, Busschbach JJ, Mureau MA. Burden of keloid disease: A cross-sectional health-related quality of life assessment. Acta Derm Venereol (2017) 97(2):225–9. doi: 10.2340/00015555-2498
5. Davis SA, Feldman SR, McMichael AJ. Management of keloids in the united states, 1990-2009: An analysis of the national ambulatory medical care survey. Dermatol Surg (2013) 39(7):988–94. doi: 10.1111/dsu.12182
6. Ung CY, Warwick A, Onoufriadis A, Barker JN, Parsons M, McGrath JA, et al. Comorbidities of keloid and hypertrophic scars among participants in uk biobank. JAMA Dermatol (2023) . 159(2):172–81. doi: 10.1001/jamadermatol.2022.5607
7. Rutherford A, Glass DA 2nd. A case-control study analyzing the association of keloids with hypertension and obesity. Int J Dermatol (2017) 56(9):e187–e9. doi: 10.1111/ijd.13618
8. Adotama P, Rutherford A, Glass DA 2nd. Association of keloids with systemic medical conditions: A retrospective analysis. Int J Dermatol (2016) 55(1):e38–40. doi: 10.1111/ijd.12969
9. Snyder AL, Zmuda JM, Thompson PD. Keloid associated with hypertension. Lancet (1996) 347(8999):465–6. doi: 10.1016/s0140-6736(96)90042-2
10. El Hadidi HH, Sobhi RM, Nada AM, AbdelGhaffar MMM, Shaker OG, El-Kalioby M. Does vitamin d deficiency predispose to keloids Via dysregulation of koebnerisin (S100a15)? a case-control study. Wound Repair Regener (2021) 29(3):425–31. doi: 10.1111/wrr.12894
11. Yu D, Shang Y, Luo S, Hao L. The taqi gene polymorphisms of vdr and the circulating 1,25-dihydroxyvitamin d levels confer the risk for the keloid scarring in chinese cohorts. Cell Physiol Biochem (2013) 32(1):39–45. doi: 10.1159/000350121
12. Kwon HE, Ahn HJ, Jeong SJ, Shin MK. The increased prevalence of keloids in atopic dermatitis patients with allergic comorbidities: A nationwide retrospective cohort study. Sci Rep (2021) 11(1):23669. doi: 10.1038/s41598-021-03164-4
13. Lu YY, Lu CC, Yu WW, Zhang L, Wang QR, Zhang CL, et al. Keloid risk in patients with atopic dermatitis: A nationwide retrospective cohort study in taiwan. BMJ Open (2018) 8(7):e022865. doi: 10.1136/bmjopen-2018-022865
14. Ogawa R. The most current algorithms for the treatment and prevention of hypertrophic scars and keloids: A 2020 update of the algorithms published 10 years ago. Plast Reconstr Surg (2022) 149(1):79e–94e. doi: 10.1097/prs.0000000000008667
15. Abd El-Dayem DH, Nada HA, Hanafy NS, Elsaie ML. Laser-assisted topical steroid application versus steroid injection for treating keloids: A split side study. J Cosmet Dermatol (2021) 20(1):138–42. doi: 10.1111/jocd.13521
16. Wu W, Zhao Y, Chen Y, Zhong A. Comparing the efficacy of multiple drugs injection for the treatment of hypertrophic scars and keloid: A network meta-analysis. Aesthetic Plast Surg (2022). doi: 10.1007/s00266-022-03163-4
17. Lee JH, Kim SE, Lee AY. Effects of interferon-Alpha2b on keloid treatment with triamcinolone acetonide intralesional injection. Int J Dermatol (2008) 47(2):183–6. doi: 10.1111/j.1365-4632.2008.03426.x
18. Bouzari N, Davis SC, Nouri K. Laser treatment of keloids and hypertrophic scars. Int J Dermatol (2007) 46(1):80–8. doi: 10.1111/j.1365-4632.2007.03104.x
19. Har-Shai Y, Mettanes I, Zilberstein Y, Genin O, Spector I, Pines M. Keloid histopathology after intralesional cryosurgery treatment. J Eur Acad Dermatol Venereol (2011) 25(9):1027–36. doi: 10.1111/j.1468-3083.2010.03911.x
20. Ogawa R, Akaishi S, Huang C, Dohi T, Aoki M, Omori Y, et al. Clinical applications of basic research that shows reducing skin tension could prevent and treat abnormal scarring: The importance of Fascial/Subcutaneous tensile reduction sutures and flap surgery for keloid and hypertrophic scar reconstruction. J Nippon Med Sch (2011) 78(2):68–76. doi: 10.1272/jnms.78.68
21. Osswald SS, Elston DM, Vogel PS. Giant right plantar keloid treated with excision and tissue-engineered allograft. J Am Acad Dermatol (2003) 48(1):131–4. doi: 10.1067/mjd.2003.48
22. Jones ME, Hardy C, Ridgway J. Keloid management: A retrospective case review on a new approach using surgical excision, platelet-rich plasma, and in-office superficial photon x-ray radiation therapy. Adv Skin Wound Care (2016) 29(7):303–7. doi: 10.1097/01.Asw.0000482993.64811.74
23. Landén NX, Li D, Ståhle M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol Life Sci (2016) 73(20):3861–85. doi: 10.1007/s00018-016-2268-0
24. Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res (2012) 49(1):35–43. doi: 10.1159/000339613
25. Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: Molecular and cellular mechanisms. J Invest Dermatol (2007) 127(3):514–25. doi: 10.1038/sj.jid.5700701
26. Bayat A, Bock O, Mrowietz U, Ollier WE, Ferguson MW. Genetic susceptibility to keloid disease and transforming growth factor beta 2 polymorphisms. Br J Plast Surg (2002) 55(4):283–6. doi: 10.1054/bjps.2002.3853
27. Bayat A, Bock O, Mrowietz U, Ollier WE, Ferguson MW. Genetic susceptibility to keloid disease and hypertrophic scarring: Transforming growth factor Beta1 common polymorphisms and plasma levels. Plast Reconstr Surg (2003) 111(2):535–43. doi: 10.1097/01.Prs.0000041536.02524.A3
28. Bayat A, Bock O, Mrowietz U, Ollier WE, Ferguson MW. Genetic susceptibility to keloid disease: Transforming growth factor beta receptor gene polymorphisms are not associated with keloid disease. Exp Dermatol (2004) 13(2):120–4. doi: 10.1111/j.0906-6705.2004.00165.x
29. Fujita M, Yamamoto Y, Jiang JJ, Atsumi T, Tanaka Y, Ohki T, et al. Nedd4 is involved in inflammation development during keloid formation. J Invest Dermatol (2019) 139(2):333–41. doi: 10.1016/j.jid.2018.07.044
30. Nakashima M, Chung S, Takahashi A, Kamatani N, Kawaguchi T, Tsunoda T, et al. A genome-wide association study identifies four susceptibility loci for keloid in the japanese population. Nat Genet (2010) 42(9):768–71. doi: 10.1038/ng.645
31. Tsai CH, Ogawa R. Keloid research: Current status and future directions. Scars Burn Heal (2019) 5:2059513119868659. doi: 10.1177/2059513119868659
32. Velez Edwards DR, Tsosie KS, Williams SM, Edwards TL, Russell SB. Admixture mapping identifies a locus at 15q21.2-22.3 associated with keloid formation in african americans. Hum Genet (2014) 133(12):1513–23. doi: 10.1007/s00439-014-1490-9
33. Zhu F, Wu B, Li P, Wang J, Tang H, Liu Y, et al. Association study confirmed susceptibility loci with keloid in the chinese han population. PloS One (2013) 8(5):e62377. doi: 10.1371/journal.pone.0062377
34. Liu S, Yang H, Song J, Zhang Y, Abualhssain ATH, Yang B. Keloid: Genetic susceptibility and contributions of genetics and epigenetics to its pathogenesis. Exp Dermatol (2022) 31:1665–75. doi: 10.1111/exd.14671
35. Ogawa R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int J Mol Sci (2017) 18(3):606. doi: 10.3390/ijms18030606
36. Bagabir R, Byers RJ, Chaudhry IH, Müller W, Paus R, Bayat A. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol (2012) 167(5):1053–66. doi: 10.1111/j.1365-2133.2012.11190.x
37. Messadi DV, Doung HS, Zhang Q, Kelly AP, Tuan TL, Reichenberger E, et al. Activation of nfkappab signal pathways in keloid fibroblasts. Arch Dermatol Res (2004) 296(3):125–33. doi: 10.1007/s00403-004-0487-y
38. Zhang Q, Yamaza T, Kelly AP, Shi S, Wang S, Brown J, et al. Tumor-like stem cells derived from human keloid are governed by the inflammatory niche driven by il-17/Il-6 axis. PloS One (2009) 4(11):e7798. doi: 10.1371/journal.pone.0007798
39. Nangole FW, Ouyang K, Anzala O, Ogengo J, Agak GW. Multiple cytokines elevated in patients with keloids: Is it an indication of auto-inflammatory disease? J Inflammation Res (2021) 14:2465–70. doi: 10.2147/jir.S312091
40. Ogawa R, Okai K, Tokumura F, Mori K, Ohmori Y, Huang C, et al. The relationship between skin Stretching/Contraction and pathologic scarring: The important role of mechanical forces in keloid generation. Wound Repair Regener (2012) 20(2):149–57. doi: 10.1111/j.1524-475X.2012.00766.x
41. Song H, Liu T, Wang W, Pang H, Zhou Z, Lv Y, et al. Tension enhances cell proliferation and collagen synthesis by upregulating expressions of integrin αvβ3 in human keloid-derived mesenchymal stem cells. Life Sci (2019) 219:272–82. doi: 10.1016/j.lfs.2018.12.042
42. Zhou B, Gao Z, Liu W, Wu X, Wang W. Important role of mechanical microenvironment on macrophage dysfunction during keloid pathogenesis. Exp Dermatol (2022) 31(3):375–80. doi: 10.1111/exd.14473
43. Feng F, Liu M, Pan L, Wu J, Wang C, Yang L, et al. Biomechanical regulatory factors and therapeutic targets in keloid fibrosis. Front Pharmacol (2022) 13:906212. doi: 10.3389/fphar.2022.906212
44. Zhang Q, Oh CK, Messadi DV, Duong HS, Kelly AP, Soo C, et al. Hypoxia-induced hif-1 alpha accumulation is augmented in a co-culture of keloid fibroblasts and human mast cells: Involvement of Erk1/2 and pi-3k/Akt. Exp Cell Res (2006) 312(2):145–55. doi: 10.1016/j.yexcr.2005.10.006
45. Ma X, Chen J, Xu B, Long X, Qin H, Zhao RC, et al. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxic microenvironment in vitro. Int J Mol Med (2015) 35(5):1246–56. doi: 10.3892/ijmm.2015.2135
46. Lei R, Li J, Liu F, Li W, Zhang S, Wang Y, et al. Hif-1α promotes the keloid development through the activation of tgf-β/Smad and Tlr4/Myd88/Nf-κb pathways. Cell Cycle (2019) 18(23):3239–50. doi: 10.1080/15384101.2019.1670508
47. Lin X, Wang Y, Jiang Y, Xu M, Pang Q, Sun J, et al. Sumoylation enhances the activity of the tgf-β/Smad and hif-1 signaling pathways in keloids. Life Sci (2020) 255:117859. doi: 10.1016/j.lfs.2020.117859
48. Lee SY, Lee AR, Choi JW, Lee CR, Cho KH, Lee JH, et al. Il-17 induces autophagy dysfunction to promote inflammatory cell death and fibrosis in keloid fibroblasts Via the Stat3 and hif-1α dependent signaling pathways. Front Immunol (2022) 13:888719. doi: 10.3389/fimmu.2022.888719
49. Fong EP, Bay BH. Keloids – the sebum hypothesis revisited. Med Hypotheses (2002) 58(4):264–9. doi: 10.1054/mehy.2001.1426
50. Li Q, Qin Z, Nie F, Bi H, Zhao R, Pan B, et al. Metabolic reprogramming in keloid fibroblasts: Aerobic glycolysis and a novel therapeutic strategy. Biochem Biophys Res Commun (2018) 496(2):641–7. doi: 10.1016/j.bbrc.2018.01.068
51. Onoufriadis A, Hsu CK, Ainali C, Ung CY, Rashidghamat E, Yang HS, et al. Time series integrative analysis of rna sequencing and microrna expression data reveals key biologic wound healing pathways in keloid-prone individuals. J Invest Dermatol (2018) 138(12):2690–3. doi: 10.1016/j.jid.2018.05.017
52. Marneros AG, Norris JEC, Olsen BR, Reichenberger E. Clinical genetics of familial keloids. Arch Dermatol (2001) 137(11):1429–34. doi: 10.1001/archderm.137.11.1429
53. Clark JA, Turner ML, Howard L, Stanescu H, Kleta R, Kopp JB. Description of familial keloids in five pedigrees: Evidence for autosomal dominant inheritance and phenotypic heterogeneity. BMC Dermatol (2009) 9:8. doi: 10.1186/1471-5945-9-8
54. Chen Y, Gao JH, Liu XJ, Yan X, Song M. Characteristics of occurrence for han chinese familial keloids. Burns (2006) 32(8):1052–9. doi: 10.1016/j.burns.2006.04.014
55. Abdu Allah AMK, Mohammed KI, Farag AGA, Hagag MM, Essam M, Tayel NR. Interleukin-6 serum level and gene polymorphism in keloid patients. Cell Mol Biol (Noisy-le-grand) (2019) 65(5):43–8. doi: 10.14715/cmb/2019.65.5.7
56. Zhu XJ, Li WZ, Li H, Fu CQ, Liu J. Association of interleukin-6 gene polymorphisms and circulating levels with keloid scars in a chinese han population. Genet Mol Res (2017) 16(2):gmr16029110. doi: 10.4238/gmr16029110
57. Tosa M, Watanabe A, Ghazizadeh M. Il-6 polymorphism and susceptibility to keloid formation in a japanese population. J Invest Dermatol (2016) 136(5):1069–72. doi: 10.1016/j.jid.2016.01.019
58. Larouche J, Sheoran S, Maruyama K, Martino MM. Immune regulation of skin wound healing: Mechanisms and novel therapeutic targets. Adv Wound Care (New Rochelle) (2018) 7(7):209–31. doi: 10.1089/wound.2017.0761
59. Wilgus TA. Immune cells in the healing skin wound: Influential players at each stage of repair. Pharmacol Res (2008) 58(2):112–6. doi: 10.1016/j.phrs.2008.07.009
60. Yagi KI, Dafalla AA, Osman AA. Does an immune reaction to sebum in wounds cause keloid scars? beneficial effect of desensitisation. Br J Plast Surg (1979) 32(3):223–5. doi: 10.1016/s0007-1226(79)90037-7
61. Bloch EF, Hall MG Jr., Denson MJ, Slay-Solomon V. General immune reactivity in keloid patients. Plast Reconstr Surg (1984) 73(3):448–51. doi: 10.1097/00006534-198403000-00020
62. Smith CJ, Smith JC, Finn MC. The possible role of mast cells (Allergy) in the production of keloid and hypertrophic scarring. J Burn Care Rehabil (1987) 8(2):126–31. doi: 10.1097/00004630-198703000-00008
63. Kazeem AA. The immunological aspects of keloid tumor formation. J Surg Oncol (1988) 38(1):16–8. doi: 10.1002/jso.2930380106
64. Jin Q, Gui L, Niu F, Yu B, Lauda N, Liu J, et al. Macrophages in keloid are potent at promoting the differentiation and function of regulatory t cells. Exp Cell Res (2018) 362(2):472–6. doi: 10.1016/j.yexcr.2017.12.011
65. Chen Y, Jin Q, Fu X, Qiao J, Niu F. Connection between t regulatory cell enrichment and collagen deposition in keloid. Exp Cell Res (2019) 383(2):111549. doi: 10.1016/j.yexcr.2019.111549
66. Wilgus TA, Wulff BC. The importance of mast cells in dermal scarring. Adv Wound Care (New Rochelle) (2014) 3(4):356–65. doi: 10.1089/wound.2013.0457
67. Arbi S, Eksteen EC, Oberholzer HM, Taute H, Bester MJ. Premature collagen fibril formation, fibroblast-mast cell interactions and mast cell-mediated phagocytosis of collagen in keloids. Ultrastruct Pathol (2015) 39(2):95–103. doi: 10.3109/01913123.2014.981326
68. Ud-Din S, Wilgus TA, Bayat A. Mast cells in skin scarring: A review of animal and human research. Front Immunol (2020) 11:552205. doi: 10.3389/fimmu.2020.552205
69. Li X, Wang Y, Yuan B, Yang H, Qiao L. Status of M1 and M2 type macrophages in keloid. Int J Clin Exp Pathol (2017) 10(11):11098–105.
70. Seoudy WM, Mohy El Dien SM, Abdel Reheem TA, Elfangary MM, Erfan MA. Macrophages of the M1 and M2 types play a role in keloids pathogenesis. Int Wound J (2022) 20(1):38–45. doi: 10.1111/iwj.13834
71. Xu X, Gu S, Huang X, Ren J, Gu Y, Wei C, et al. The role of macrophages in the formation of hypertrophic scars and keloids. Burns Trauma (2020) 8:tkaa006. doi: 10.1093/burnst/tkaa006
72. Feng C, Shan M, Xia Y, Zheng Z, He K, Wei Y, et al. Single-cell rna sequencing reveals distinct immunology profiles in human keloid. Front Immunol (2022) 13:940645. doi: 10.3389/fimmu.2022.940645
73. Jiao H, Fan J, Cai J, Pan B, Yan L, Dong P, et al. Analysis of characteristics similar to autoimmune disease in keloid patients. Aesthetic Plast Surg (2015) 39(5):818–25. doi: 10.1007/s00266-015-0542-4
74. Xia Y, Wang Y, Xiao Y, Shan M, Hao Y, Zhang L. Identification of a diagnostic signature and immune cell infiltration characteristics in keloids. Front Mol Biosci (2022) 9:879461. doi: 10.3389/fmolb.2022.879461
75. Yin X, Bu W, Fang F, Ren K, Zhou B. Keloid biomarkers and their correlation with immune infiltration. Front Genet (2022) 13:784073. doi: 10.3389/fgene.2022.784073
76. Nang’ole WF, Omu A, Ogeng’o JA, Agak GW. Do mesenchymal stem cells influence keloid recurrence? Stem Cells Cloning: Adv Appl (2022) 15:77–84. doi: 10.2147/SCCAA.S373551
77. Hahn JM, McFarland KL, Combs KA, Powell HM, Supp DM. Myofibroblasts are not characteristic features of keloid lesions. Plast Reconstr Surg Glob Open (2022) 10(11):e4680. doi: 10.1097/gox.0000000000004680
78. Shan M, Liu H, Song K, Liu S, Hao Y, Wang Y. Immune-related gene expression in skin, inflamed and keloid tissue from patients with keloids. Oncol Lett (2022) 23(2):72. doi: 10.3892/ol.2022.13192
79. Li Y, Li M, Qu C, Li Y, Tang Z, Zhou Z, et al. The polygenic map of keloid fibroblasts reveals fibrosis-associated gene alterations in inflammation and immune responses. Front Immunol (2021) 12:810290. doi: 10.3389/fimmu.2021.810290
80. Yuan B, Miao L, Mei D, Li L, Hu Z. A signature of genes featuring Fgf11 revealed aberrant fibroblast activation and immune infiltration properties in keloid tissue. Emerg Med Int (2022) 2022:4452687. doi: 10.1155/2022/4452687
81. Diaz A, Tan K, He H, Xu H, Cueto I, Pavel AB, et al. Keloid lesions show increased il-4/Il-13 signaling and respond to Th2-targeting dupilumab therapy. J Eur Acad Dermatol Venereol (2020) 34(4):e161–e4. doi: 10.1111/jdv.16097
82. Maeda D, Kubo T, Kiya K, Kawai K, Matsuzaki S, Kobayashi D, et al. Periostin is induced by il-4/Il-13 in dermal fibroblasts and promotes Rhoa/Rock pathway-mediated tgf-β1 secretion in abnormal scar formation. J Plast Surg Handb Surg (2019) 53(5):288–94. doi: 10.1080/2000656x.2019.1612752
83. Oriente A, Fedarko NS, Pacocha SE, Huang SK, Lichtenstein LM, Essayan DM. Interleukin-13 modulates collagen homeostasis in human skin and keloid fibroblasts. J Pharmacol Exp Ther (2000) 292(3):988–94.
84. Shan M, Liu H, Hao Y, Meng T, Feng C, Song K, et al. Il-4 and Ccr7 play an important role in the development of keloids in patients with a family history. Am J Transl Res (2022) 14(5):3381–94. doi: 10.3390/ijms23168862
85. Wu J, Del Duca E, Espino M, Gontzes A, Cueto I, Zhang N, et al. Rna sequencing keloid transcriptome associates keloids with Th2, Th1, Th17/Th22, and Jak3-skewing. Front Immunol (2020) 11:597741. doi: 10.3389/fimmu.2020.597741
86. Hajdarbegovic E, Bloem A, Balak D, Thio B, Nijsten T. The association between atopic disorders and keloids: A case-control study. Indian J Dermatol (2015) 60(6):635. doi: 10.4103/0019-5154.169144
87. Zhang D, Li B, Zhao M. Therapeutic strategies by regulating interleukin family to suppress inflammation in hypertrophic scar and keloid. Front Pharmacol (2021) 12:667763. doi: 10.3389/fphar.2021.667763
88. Do DV, Ong CT, Khoo YT, Carbone A, Lim CP, Wang S, et al. Interleukin-18 system plays an important role in keloid pathogenesis Via epithelial-mesenchymal interactions. Br J Dermatol (2012) 166(6):1275–88. doi: 10.1111/j.1365-2133.2011.10721.x
89. Lee SY, Kim EK, Seo HB, Choi JW, Yoo JH, Jung KA, et al. Il-17 induced stromal cell-derived factor-1 and profibrotic factor in keloid-derived skin fibroblasts Via the Stat3 pathway. Inflammation (2020) 43(2):664–72. doi: 10.1007/s10753-019-01148-1
90. Ghazizadeh M, Tosa M, Shimizu H, Hyakusoku H, Kawanami O. Functional implications of the il-6 signaling pathway in keloid pathogenesis. J Invest Dermatol (2007) 127(1):98–105. doi: 10.1038/sj.jid.5700564
91. Uitto J. Il-6 signaling pathway in keloids: A target for pharmacologic intervention? J Invest Dermatol (2007) 127(1):6–8. doi: 10.1038/sj.jid.5700604
92. Tosa M, Ghazizadeh M, Shimizu H, Hirai T, Hyakusoku H, Kawanami O. Global gene expression analysis of keloid fibroblasts in response to electron beam irradiation reveals the involvement of interleukin-6 pathway. J Invest Dermatol (2005) 124(4):704–13. doi: 10.1111/j.0022-202X.2005.23592.x
93. Shi CK, Zhao YP, Ge P, Huang GB. Therapeutic effect of interleukin-10 in keloid fibroblasts by suppression of tgf-β/Smad pathway. Eur Rev Med Pharmacol Sci (2019) 23(20):9085–92. doi: 10.26355/eurrev_201910_19311
94. Li Q, Cheng F, Zhou K, Fang L, Wu J, Xia Q, et al. Increased sensitivity to tnf-α promotes keloid fibroblast hyperproliferation by activating the nf-κb, jnk and P38 mapk pathways. Exp Ther Med (2021) 21(5):502. doi: 10.3892/etm.2021.9933
95. Khattab FM, Samir MA. Correlation between serum il 37 levels with keloid severity. J Cosmet Dermatol (2020) 19(9):2428–31. doi: 10.1111/jocd.13290
96. Zhao Y, Shi J, Lyu L. Critical role and potential therapeutic efficacy of interleukin-37 in the pathogenesis of keloid scarring. J Cosmet Dermatol (2020) 19(7):1805–6. doi: 10.1111/jocd.13357
97. Liu X, Chen W, Zeng Q, Ma B, Li Z, Meng T, et al. Single-cell rna-sequencing reveals lineage-specific regulatory changes of fibroblasts and vascular endothelial cells in keloids. J Invest Dermatol (2022) 142(1):124–35.e11. doi: 10.1016/j.jid.2021.06.010
98. Shim J, Oh SJ, Yeo E, Park JH, Bae JH, Kim SH, et al. Integrated analysis of single-cell and spatial transcriptomics in keloids: Highlights on fibrovascular interactions in keloid pathogenesis. J Invest Dermatol (2022) 142(8):2128–39.e11. doi: 10.1016/j.jid.2022.01.017
99. Xie J, Chen L, Cao Y, Wu D, Xiong W, Zhang K, et al. Single-cell sequencing analysis and weighted co-expression network analysis based on public databases identified that tnc is a novel biomarker for keloid. Front Immunol (2021) 12:783907. doi: 10.3389/fimmu.2021.783907
100. Jones LR, Young W, Divine G, Datta I, Chen KM, Ozog D, et al. Genome-wide scan for methylation profiles in keloids. Dis Markers (2015) 2015:943176. doi: 10.1155/2015/943176
101. Xu H, Zhu Z, Hu J, Sun J, Wo Y, Wang X, et al. Downregulated cytotoxic Cd8(+) t-cell identifies with the Nkg2a-soluble hla-e axis as a predictive biomarker and potential therapeutic target in keloids. Cell Mol Immunol (2022) 19(4):527–39. doi: 10.1038/s41423-021-00834-1
102. Eishi K, Bae SJ, Ogawa F, Hamasaki Y, Shimizu K, Katayama I. Silicone gel sheets relieve pain and pruritus with clinical improvement of keloid: Possible target of mast cells. J Dermatolog Treat (2003) 14(4):248–52. doi: 10.1080/09546630310016808
103. Shaker SA, Ayuob NN, Hajrah NH. Cell talk: A phenomenon observed in the keloid scar by immunohistochemical study. Appl Immunohistochem Mol Morphol (2011) 19(2):153–9. doi: 10.1097/PAI.0b013e3181efa2ef
104. Zhang Q, Kelly AP, Wang L, French SW, Tang X, Duong HS, et al. Green tea extract and (-)-Epigallocatechin-3-Gallate inhibit mast cell-stimulated type i collagen expression in keloid fibroblasts Via blocking pi-3k/Akt signaling pathways. J Invest Dermatol (2006) 126(12):2607–13. doi: 10.1038/sj.jid.5700472
105. Ammendola M, Leporini C, Marech I, Gadaleta CD, Scognamillo G, Sacco R, et al. Targeting mast cells tryptase in tumor microenvironment: A potential antiangiogenetic strategy. BioMed Res Int (2014) 2014:154702. doi: 10.1155/2014/154702
106. Dong X, Zhang C, Ma S, Wen H. Mast cell chymase in keloid induces profibrotic response Via transforming growth factor-β1/Smad activation in keloid fibroblasts. Int J Clin Exp Pathol (2014) 7(7):3596–607.
107. Wang R, Chen J, Zhang Z, Cen Y. Role of chymase in the local renin-angiotensin system in keloids: Inhibition of chymase may be an effective therapeutic approach to treat keloids. Drug Des Devel Ther (2015) 9:4979–88. doi: 10.2147/dddt.S87842
108. Shiota N, Kakizoe E, Shimoura K, Tanaka T, Okunishi H. Effect of mast cell chymase inhibitor on the development of scleroderma in tight-skin mice. Br J Pharmacol (2005) 145(4):424–31. doi: 10.1038/sj.bjp.0706209
109. Takai S, Jin D. Improvement of cardiovascular remodelling by chymase inhibitor. Clin Exp Pharmacol Physiol (2016) 43(4):387–93. doi: 10.1111/1440-1681.12549
110. Takai S, Jin D. Chymase as a possible therapeutic target for amelioration of non-alcoholic steatohepatitis. Int J Mol Sci (2020) 21(20):7543. doi: 10.3390/ijms21207543
111. Gallant-Behm CL, Hildebrand KA, Hart DA. The mast cell stabilizer ketotifen prevents development of excessive skin wound contraction and fibrosis in red duroc pigs. Wound Repair Regener (2008) 16(2):226–33. doi: 10.1111/j.1524-475X.2008.00363.x
112. Mukhopadhyay A, Do DV, Ong CT, Khoo YT, Masilamani J, Chan SY, et al. The role of stem cell factor and c-kit in keloid pathogenesis: Do tyrosine kinase inhibitors have a potential therapeutic role? Br J Dermatol (2011) 164(2):372–86. doi: 10.1111/j.1365-2133.2010.10035.x
113. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
114. Fukui S, Iwamoto N, Takatani A, Igawa T, Shimizu T, Umeda M, et al. M1 and M2 monocytes in rheumatoid arthritis: A contribution of imbalance of M1/M2 monocytes to osteoclastogenesis. Front Immunol (2017) 8:1958. doi: 10.3389/fimmu.2017.01958
115. Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci (2017) 18(7):1545. doi: 10.3390/ijms18071545
116. Clark RA, Kupper TS. Il-15 and dermal fibroblasts induce proliferation of natural regulatory t cells isolated from human skin. Blood (2006) 109(1):194–202. doi: 10.1182/blood-2006-02-002873
117. Chen Z, Zhou L, Won T, Gao Z, Wu X, Lu L. Characterization of Cd45ro+ memory t lymphocytes in keloid disease. Br J Dermatol (2018) 178(4):940–50. doi: 10.1111/bjd.16173
118. Turner JA, Stephen-Victor E, Wang S, Rivas MN, Abdel-Gadir A, Harb H, et al. Regulatory t cell-derived tgf-β1 controls multiple checkpoints governing allergy and autoimmunity. Immunity (2020) 53(6):1202–14.e6. doi: 10.1016/j.immuni.2020.10.002
119. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by tgfbeta. Nat Rev Immunol (2010) 10(8):554–67. doi: 10.1038/nri2808
120. Ganeshan K, Bryce PJ. Regulatory t cells enhance mast cell production of il-6 Via surface-bound tgf-β. J Immunol (2012) 188(2):594–603. doi: 10.4049/jimmunol.1102389
121. Fickenscher H, Hör S, Küpers H, Knappe A, Wittmann S, Sticht H. The interleukin-10 family of cytokines. Trends Immunol (2002) 23(2):89–96. doi: 10.1016/S1471-4906(01)02149-4
122. Hasegawa T, Nakao A, Sumiyoshi K, Tsuboi R, Ogawa H. Ifn-gamma fails to antagonize fibrotic effect of tgf-beta on keloid-derived dermal fibroblasts. J Dermatol Sci (2003) 32(1):19–24. doi: 10.1016/s0923-1811(03)00044-6
123. Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, et al. Regulatory t cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med (2014) 6(258):258ra142. doi: 10.1126/scitranslmed.3009925
124. Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, et al. T cell memory. skin-resident memory Cd8⁺ t cells trigger a state of tissue-wide pathogen alert. Science (2014) 346(6205):101–5. doi: 10.1126/science.1254803
125. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-Nkg2a mab is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both t and nk cells. Cell (2018) 175(7):1731–43.e13. doi: 10.1016/j.cell.2018.10.014
126. McMahon CW, Zajac AJ, Jamieson AM, Corral L, Hammer GE, Ahmed R, et al. Viral and bacterial infections induce expression of multiple nk cell receptors in responding Cd8(+) t cells. J Immunol (2002) 169(3):1444–52. doi: 10.4049/jimmunol.169.3.1444
127. Mingari MC, Ponte M, Bertone S, Schiavetti F, Vitale C, Bellomo R, et al. Hla class i-specific inhibitory receptors in human t lymphocytes: Interleukin 15-induced expression of Cd94/Nkg2a in superantigen- or alloantigen-activated Cd8+ t cells. Proc Natl Acad Sci U.S.A. (1998) 95(3):1172–7. doi: 10.1073/pnas.95.3.1172
128. Bertone S, Schiavetti F, Bellomo R, Vitale C, Ponte M, Moretta L, et al. Transforming growth factor-Beta-Induced expression of Cd94/Nkg2a inhibitory receptors in human t lymphocytes. Eur J Immunol (1999) 29(1):23–9. doi: 10.1002/(sici)1521-4141(199901)29:01<23::Aid-immu23>3.0.Co;2-y
129. Galot R, Le Tourneau C, Saada-Bouzid E, Daste A, Even C, Debruyne P, et al. A phase ii study of monalizumab in patients with Recurrent/Metastatic squamous cell carcinoma of the head and neck: The I1 cohort of the eortc-Hncg-1559 upstream trial. Eur J Cancer (2021) 158:17–26. doi: 10.1016/j.ejca.2021.09.003
130. Herbst RS, Majem M, Barlesi F, Carcereny E, Chu Q, Monnet I, et al. Coast: An open-label, phase ii, multidrug platform study of durvalumab alone or in combination with oleclumab or monalizumab in patients with unresectable, stage iii non-Small-Cell lung cancer. J Clin Oncol (2022) 40(29):Jco2200227. doi: 10.1200/jco.22.00227
131. Tinker AV, Hirte HW, Provencher D, Butler M, Ritter H, Tu D, et al. Dose-ranging and cohort-expansion study of monalizumab (Iph2201) in patients with advanced gynecologic malignancies: A trial of the canadian cancer trials group (Cctg): Ind221. Clin Cancer Res (2019) 25(20):6052–60. doi: 10.1158/1078-0432.Ccr-19-0298
132. Onodera M, Ueno M, Ito O, Suzuki S, Igawa HH, Sakamoto H. Factor xiiia-positive dermal dendritic cells in keloids and hypertrophic and mature scars. Pathol Int (2007) 57(6):337–42. doi: 10.1111/j.1440-1827.2007.02105.x
133. Altman DA, Nickoloff BJ, Fivenson DP. Differential expression of factor xiiia and Cd34 in cutaneous mesenchymal tumors. J Cutan Pathol (1993) 20(2):154–8. doi: 10.1111/j.1600-0560.1993.tb00233.x
134. Luo X, Chen J, Yang H, Hu X, Alphonse MP, Shen Y, et al. Dendritic cell immunoreceptor drives atopic dermatitis by modulating oxidized camkii-involved mast cell activation. JCI Insight (2022) 7(5):e152559. doi: 10.1172/jci.insight.152559
135. Yoon J, Leyva-Castillo JM, Wang G, Galand C, Oyoshi MK, Kumar L, et al. Il-23 induced in keratinocytes by endogenous Tlr4 ligands polarizes dendritic cells to drive il-22 responses to skin immunization. J Exp Med (2016) 213(10):2147–66. doi: 10.1084/jem.20150376
136. O'Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (Il-6) trans signaling drives a Stat3-dependent pathway that leads to hyperactive transforming growth factor-β (Tgf-β) signaling promoting Smad3 activation and fibrosis Via gremlin protein. J Biol Chem (2014) 289(14):9952–60. doi: 10.1074/jbc.M113.545822
137. Sato S, Hasegawa M, Takehara K. Serum levels of interleukin-6 and interleukin-10 correlate with total skin thickness score in patients with systemic sclerosis. J Dermatol Sci (2001) 27(2):140–6. doi: 10.1016/s0923-1811(01)00128-1
138. Jones SA. Directing transition from innate to acquired immunity: Defining a role for il-6. J Immunol (2005) 175(6):3463–8. doi: 10.4049/jimmunol.175.6.3463
139. Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, et al. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity (2001) 14(6):705–14. doi: 10.1016/s1074-7613(01)00151-0
140. Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity (2014) 40(1):40–50. doi: 10.1016/j.immuni.2013.10.022
141. Johnson BZ, Stevenson AW, Prêle CM, Fear MW, Wood FM. The role of il-6 in skin fibrosis and cutaneous wound healing. Biomedicines (2020) 8(5):101. doi: 10.3390/biomedicines8050101
142. Tzavlaki K, Moustakas A. Tgf-β signaling. Biomolecules (2020) 10(3):487. doi: 10.3390/biom10030487
143. Wang H, Chen Z, Li XJ, Ma L, Tang YL. Anti-inflammatory cytokine tsg-6 inhibits hypertrophic scar formation in a rabbit ear model. Eur J Pharmacol (2015) 751:42–9. doi: 10.1016/j.ejphar.2015.01.040
144. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18(1):62–76. doi: 10.1038/nri.2017.90
145. Nguyen JK, Austin E, Huang A, Mamalis A, Jagdeo J. The il-4/Il-13 axis in skin fibrosis and scarring: Mechanistic concepts and therapeutic targets. Arch Dermatol Res (2020) 312(2):81–92. doi: 10.1007/s00403-019-01972-3
146. Salmon-Ehr V, Ramont L, Godeau G, Birembaut P, Guenounou M, Bernard P, et al. Implication of interleukin-4 in wound healing. Lab Invest (2000) 80(8):1337–43. doi: 10.1038/labinvest.3780141
147. Ong C, Wong C, Roberts CR, Teh HS, Jirik FR. Anti-Il-4 treatment prevents dermal collagen deposition in the tight-skin mouse model of scleroderma. Eur J Immunol (1998) 28(9):2619–29. doi: 10.1002/(sici)1521-4141(199809)28:09<2619::Aid-immu2619>3.0.Co;2-m
148. Luk K, Fakhoury J, Ozog D. Nonresponse and progression of diffuse keloids to dupilumab therapy. J Drugs Dermatol (2022) 21(2):197–9. doi: 10.36849/jdd.6252
149. Wong AJS, Song EJ. Dupilumab as an adjuvant treatment for keloid-associated symptoms. JAAD Case Rep (2021) 13:73–4. doi: 10.1016/j.jdcr.2021.04.034
150. Minutti CM, Knipper JA, Allen JE, Zaiss DM. Tissue-specific contribution of macrophages to wound healing. Semin Cell Dev Biol (2017) 61:3–11. doi: 10.1016/j.semcdb.2016.08.006
151. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-Mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu Rev Immunol (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
152. Su S, Zhao Q, He C, Huang D, Liu J, Chen F, et al. Mir-142-5p and mir-130a-3p are regulated by il-4 and il-13 and control profibrogenic macrophage program. Nat Commun (2015) 6:8523. doi: 10.1038/ncomms9523
153. Crawford J, Nygard K, Gan BS, O'Gorman DB. Periostin induces fibroblast proliferation and myofibroblast persistence in hypertrophic scarring. Exp Dermatol (2015) 24(2):120–6. doi: 10.1111/exd.12601
154. Yamaguchi Y, Ono J, Masuoka M, Ohta S, Izuhara K, Ikezawa Z, et al. Serum periostin levels are correlated with progressive skin sclerosis in patients with systemic sclerosis. Br J Dermatol (2013) 168(4):717–25. doi: 10.1111/bjd.12117
155. Fuschiotti P, Medsger TA Jr., Morel PA. Effector Cd8+ t cells in systemic sclerosis patients produce abnormally high levels of interleukin-13 associated with increased skin fibrosis. Arthritis Rheum (2009) 60(4):1119–28. doi: 10.1002/art.24432
156. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting tgf-β signal transduction for fibrosis and cancer therapy. Mol Cancer (2022) 21(1):104. doi: 10.1186/s12943-022-01569-x
157. Chen Z, Gao Z, Xia L, Wang X, Lu L, Wu X. Dysregulation of Dpp4-Cxcl12 balance by tgf-β1/Smad pathway promotes Cxcr4(+) inflammatory cell infiltration in keloid scars. J Inflammation Res (2021) 14:4169–80. doi: 10.2147/jir.S326385
158. Younai S, Nichter LS, Wellisz T, Reinisch J, Nimni ME, Tuan TL. Modulation of collagen synthesis by transforming growth factor-beta in keloid and hypertrophic scar fibroblasts. Ann Plast Surg (1994) 33(2):148–51. doi: 10.1097/00000637-199408000-00005
159. Babu M, Diegelmann R, Oliver N. Keloid fibroblasts exhibit an altered response to tgf-beta. J Invest Dermatol (1992) 99(5):650–5. doi: 10.1111/1523-1747.ep12668146
160. Chodon T, Sugihara T, Igawa HH, Funayama E, Furukawa H. Keloid-derived fibroblasts are refractory to fas-mediated apoptosis and neutralization of autocrine transforming growth factor-Beta1 can abrogate this resistance. Am J Pathol (2000) 157(5):1661–9. doi: 10.1016/s0002-9440(10)64803-1
161. Lee CH, Hong CH, Chen YT, Chen YC, Shen MR. Tgf-Beta1 increases cell rigidity by enhancing expression of smooth muscle actin: Keloid-derived fibroblasts as a model for cellular mechanics. J Dermatol Sci (2012) 67(3):173–80. doi: 10.1016/j.jdermsci.2012.06.004
162. Hasegawa T, Nakao A, Sumiyoshi K, Tsuchihashi H, Ogawa H. Sb-431542 inhibits tgf-Beta-Induced contraction of collagen gel by normal and keloid fibroblasts. J Dermatol Sci (2005) 39(1):33–8. doi: 10.1016/j.jdermsci.2005.01.013
163. Jiao H, Dong P, Yan L, Yang Z, Lv X, Li Q, et al. Tgf-β1 induces polypyrimidine tract-binding protein to alter fibroblasts proliferation and fibronectin deposition in keloid. Sci Rep (2016) 6:38033. doi: 10.1038/srep38033
164. Bran GM, Goessler UR, Schardt C, Hormann K, Riedel F, Sadick H. Effect of the abrogation of tgf-Beta1 by antisense oligonucleotides on the expression of tgf-Beta-Isoforms and their receptors i and ii in isolated fibroblasts from keloid scars. Int J Mol Med (2010) 25(6):915–21. doi: 10.3892/ijmm_00000422
165. Sadick H, Herberger A, Riedel K, Bran G, Goessler U, Hoermann K, et al. Tgf-Beta1 antisense therapy modulates expression of matrix metalloproteinases in keloid-derived fibroblasts. Int J Mol Med (2008) 22(1):55–60. doi: 10.3892/ijmm.22.1.55
166. Chin GS, Liu W, Peled Z, Lee TY, Steinbrech DS, Hsu M, et al. Differential expression of transforming growth factor-beta receptors i and ii and activation of smad 3 in keloid fibroblasts. Plast Reconstr Surg (2001) 108(2):423–9. doi: 10.1097/00006534-200108000-00022
167. Lee TY, Chin GS, Kim WJ, Chau D, Gittes GK, Longaker MT. Expression of transforming growth factor beta 1, 2, and 3 proteins in keloids. Ann Plast Surg (1999) 43(2):179–84.
168. Bock O, Yu H, Zitron S, Bayat A, Ferguson MW, Mrowietz U. Studies of transforming growth factors beta 1-3 and their receptors i and ii in fibroblast of keloids and hypertrophic scars. Acta Derm Venereol (2005) 85(3):216–20. doi: 10.1080/00015550410025453
169. Chu Y, Guo F, Li Y, Li X, Zhou T, Guo Y. A novel truncated tgf-beta receptor ii downregulates collagen synthesis and tgf-beta i secretion of keloid fibroblasts. Connect Tissue Res (2008) 49(2):92–8. doi: 10.1080/03008200801913924
170. Liu W, Cai Z, Wang D, Wu X, Cui L, Shang Q, et al. Blocking transforming growth factor-beta receptor signaling down-regulates transforming growth factor-Beta1 autoproduction in keloid fibroblasts. Chin J Traumatol (2002) 5(2):77–81.
171. Hahn JM, McFarland KL, Combs KA. Supp DM. partial epithelial-mesenchymal transition in keloid scars: Regulation of keloid keratinocyte gene expression by transforming growth factor-β1. Burns Trauma (2016) 4(1):30. doi: 10.1186/s41038-016-0055-7
172. Yan L, Cao R, Wang L, Liu Y, Pan B, Yin Y, et al. Epithelial-mesenchymal transition in keloid tissues and tgf-β1-Induced hair follicle outer root sheath keratinocytes. Wound Repair Regener (2015) 23(4):601–10. doi: 10.1111/wrr.12320
173. Xia W, Phan TT, Lim IJ, Longaker MT, Yang GP. Complex epithelial-mesenchymal interactions modulate transforming growth factor-beta expression in keloid-derived cells. Wound Repair Regener (2004) 12(5):546–56. doi: 10.1111/j.1067-1927.2004.012507.x
174. Wang XM, Liu XM, Wang Y, Chen ZY. Activating transcription factor 3 (Atf3) regulates cell growth, apoptosis, invasion and collagen synthesis in keloid fibroblast through transforming growth factor beta (Tgf-Beta)/Smad signaling pathway. Bioengineered (2021) 12(1):117–26. doi: 10.1080/21655979.2020.1860491
175. Nagar H, Kim S, Lee I, Kim S, Choi SJ, Piao S, et al. Downregulation of Cr6-interacting factor 1 suppresses keloid fibroblast growth Via the tgf-β/Smad signaling pathway. Sci Rep (2021) 11(1):500. doi: 10.1038/s41598-020-79785-y
176. Ma HL, Zhao XF, Chen GZ, Fang RH, Zhang FR. Silencing Nlrc5 inhibits extracellular matrix expression in keloid fibroblasts Via inhibition of transforming growth factor-β1/Smad signaling pathway. BioMed Pharmacother (2016) 83:1016–21. doi: 10.1016/j.biopha.2016.08.012
177. Jin SF, Wang YX, Xu N, Sun Q, Wang CC, Lv MZ, et al. High temperature requirement factor A1 (Htra1) regulates the activation of latent tgf-β1 in keloid fibroblasts. Cell Mol Biol (Noisy-le-grand) (2018) 64(1):107–10. doi: 10.14715/cmb/2018.64.2.19
178. Zhao YX, Ho CK, Xie Y, Chen YH, Li HZ, Zhang GY, et al. Calcimycin suppresses S100a4 expression and inhibits the stimulatory effect of transforming growth factor β1 on keloid fibroblasts. Ann Plast Surg (2018) 81(2):163–9. doi: 10.1097/sap.0000000000001502
179. Cui J, Jin S, Jin C, Jin Z. Syndecan-1 regulates extracellular matrix expression in keloid fibroblasts Via tgf-β1/Smad and mapk signaling pathways. Life Sci (2020) 254:117326. doi: 10.1016/j.lfs.2020.117326
180. Zhu HY, Bai WD, Li C, Zheng Z, Guan H, Liu JQ, et al. Knockdown of lncrna-atb suppresses autocrine secretion of tgf-β2 by targeting Znf217 Via mir-200c in keloid fibroblasts. Sci Rep (2016) 6:24728. doi: 10.1038/srep24728
181. Zhu HY, Bai WD, Li J, Tao K, Wang HT, Yang XK, et al. Peroxisome proliferator-activated receptor-Γ agonist troglitazone suppresses transforming growth factor-β1 signalling through mir-92b upregulation-inhibited axl expression in human keloid fibroblasts in vitro. Am J Transl Res (2016) 8(8):3460–70.
182. Yao X, Cui X, Wu X, Xu P, Zhu W, Chen X, et al. Tumor suppressive role of mir-1224-5p in keloid proliferation, apoptosis and invasion Via the tgf-β1/Smad3 signaling pathway. Biochem Biophys Res Commun (2018) 495(1):713–20. doi: 10.1016/j.bbrc.2017.10.070
183. Huang Y, Wang Y, Lin L, Wang P, Jiang L, Liu J, et al. Overexpression of mir-133a-3p inhibits fibrosis and proliferation of keloid fibroblasts by regulating Irf5 to inhibit the tgf-β/Smad2 pathway. Mol Cell Probes (2020) 52:101563. doi: 10.1016/j.mcp.2020.101563
184. Wu J, Fang L, Cen Y, Qing Y, Chen J, Li Z. Mir-21 regulates keloid formation by downregulating Smad7 Via the tgf-β/Smad signaling pathway. J Burn Care Res (2019) 40(6):809–17. doi: 10.1093/jbcr/irz089
185. Wu D, Zhou J, Tan M, Zhou Y. Linc01116 regulates proliferation, migration, and apoptosis of keloid fibroblasts by the tgf-β1/Smad3 signaling Via targeting mir-3141. Anal Biochem (2021) 627:114249. doi: 10.1016/j.ab.2021.114249
186. Huang H, Fu S, Liu D. Detection and analysis of the hedgehog signaling pathway-related long non-coding rna (Lncrna) expression profiles in keloid. Med Sci Monit (2018) 24:9032–44. doi: 10.12659/msm.911159
187. Guo L, Xu K, Yan H, Feng H, Chai L, Xu G. Expression profile of long noncoding rnas in human earlobe keloids: A microarray analysis. BioMed Res Int (2016) 2016:5893481. doi: 10.1155/2016/5893481
188. Liang X, Ma L, Long X, Wang X. Lncrna expression profiles and validation in keloid and normal skin tissue. Int J Oncol (2015) 47(5):1829–38. doi: 10.3892/ijo.2015.3177
189. Lin L, Wang Y, Liu W, Huang Y. Bambi inhibits skin fibrosis in keloid through suppressing tgf-β1-Induced hypernomic fibroblast cell proliferation and excessive accumulation of collagen i. Int J Clin Exp Med (2015) 8(8):13227–34.
190. Li Y, Liu H, Liang Y, Peng P, Ma X, Zhang X. Dkk3 regulates cell proliferation, apoptosis and collagen synthesis in keloid fibroblasts Via tgf-β1/Smad signaling pathway. BioMed Pharmacother (2017) 91:174–80. doi: 10.1016/j.biopha.2017.03.044
191. Zhou P, Shi L, Li Q, Lu D. Overexpression of Rack1 inhibits collagen synthesis in keloid fibroblasts Via inhibition of transforming growth factor-β1/Smad signaling pathway. Int J Clin Exp Med (2015) 8(9):15262–8.
192. Hu ZC, Shi F, Liu P, Zhang J, Guo D, Cao XL, et al. Tieg1 represses Smad7-mediated activation of tgf-β1/Smad signaling in keloid pathogenesis. J Invest Dermatol (2017) 137(5):1051–9. doi: 10.1016/j.jid.2016.12.019
193. Yan Q, Li B. Traf3ip2 regulated by Foxo4 affects fibroblast proliferation, migration, and extracellular matrix deposition in keloid through the tgf-β1/Smad pathway. J Cosmet Dermatol (2022) 21:5148–55. doi: 10.1111/jocd.15009
194. Cavalli G, Dinarello CA. Suppression of inflammation and acquired immunity by il-37. Immunol Rev (2018) 281(1):179–90. doi: 10.1111/imr.12605
195. Bulau AM, Nold MF, Li S, Nold-Petry CA, Fink M, Mansell A, et al. Role of caspase-1 in nuclear translocation of il-37, release of the cytokine, and il-37 inhibition of innate immune responses. Proc Natl Acad Sci U.S.A. (2014) 111(7):2650–5. doi: 10.1073/pnas.1324140111
196. Cavalli G, Tengesdal IW, Gresnigt M, Nemkov T, Arts RJW, Domínguez-Andrés J, et al. The anti-inflammatory cytokine interleukin-37 is an inhibitor of trained immunity. Cell Rep (2021) 35(1):108955. doi: 10.1016/j.celrep.2021.108955
197. Kim MS, Baek AR, Lee JH, Jang AS, Kim DJ, Chin SS, et al. Il-37 attenuates lung fibrosis by inducing autophagy and regulating tgf-β1 production in mice. J Immunol (2019) 203(8):2265–75. doi: 10.4049/jimmunol.1801515
198. Kuo YR, Wu WS, Wang FS. Flashlamp pulsed-dye laser suppressed tgf-Beta1 expression and proliferation in cultured keloid fibroblasts is mediated by mapk pathway. Lasers Surg Med (2007) 39(4):358–64. doi: 10.1002/lsm.20489
199. He S, Liu X, Yang Y, Huang W, Xu S, Yang S, et al. Mechanisms of transforming growth factor Beta(1)/Smad signalling mediated by mitogen-activated protein kinase pathways in keloid fibroblasts. Br J Dermatol (2010) 162(3):538–46. doi: 10.1111/j.1365-2133.2009.09511.x
200. Cui J, Li Z, Jin C, Jin Z. Knockdown of fibronectin extra domain b suppresses tgf-β1-Mediated cell proliferation and collagen deposition in keloid fibroblasts Via Akt/Erk signaling pathway. Biochem Biophys Res Commun (2020) 526(4):1131–7. doi: 10.1016/j.bbrc.2020.04.021
201. Zhang Q, Qian D, Tang DD, Liu J, Wang LY, Chen W, et al. Glabridin from glycyrrhiza glabra possesses a therapeutic role against keloid Via attenuating Pi3k/Akt and transforming growth factor-β1/Smad signaling pathways. J Agric Food Chem (2022) 70(35):10782–93. doi: 10.1021/acs.jafc.2c02045
202. Song R, Li G, Li S. Aspidin pb, a novel natural anti-fibrotic compound, inhibited fibrogenesis in tgf-β1-Stimulated keloid fibroblasts Via pi-3k/Akt and smad signaling pathways. Chem Biol Interact (2015) 238:66–73. doi: 10.1016/j.cbi.2015.06.005
203. Liang CJ, Yen YH, Hung LY, Wang SH, Pu CM, Chien HF, et al. Thalidomide inhibits fibronectin production in tgf-β1-Treated normal and keloid fibroblasts Via inhibition of the P38/Smad3 pathway. Biochem Pharmacol (2013) 85(11):1594–602. doi: 10.1016/j.bcp.2013.02.038
204. Xia W, Longaker MT, Yang GP. P38 map kinase mediates transforming growth factor-Beta2 transcription in human keloid fibroblasts. Am J Physiol Regul Integr Comp Physiol (2006) 290(3):R501–8. doi: 10.1152/ajpregu.00472.2005
205. Daian T, Ohtsuru A, Rogounovitch T, Ishihara H, Hirano A, Akiyama-Uchida Y, et al. Insulin-like growth factor-i enhances transforming growth factor-Beta-Induced extracellular matrix protein production through the P38/Activating transcription factor-2 signaling pathway in keloid fibroblasts. J Invest Dermatol (2003) 120(6):956–62. doi: 10.1046/j.1523-1747.2003.12143.x
206. Wang W, Qu M, Xu L, Wu X, Gao Z, Gu T, et al. Sorafenib exerts an anti-keloid activity by antagonizing tgf-β/Smad and Mapk/Erk signaling pathways. J Mol Med (Berl) (2016) 94(10):1181–94. doi: 10.1007/s00109-016-1430-3
207. Griffin MF, Borrelli MR, Garcia JT, Januszyk M, King M, Lerbs T, et al. Jun promotes hypertrophic skin scarring Via Cd36 in preclinical in vitro and in vivo models. Sci Transl Med (2021) 13(609):eabb3312. doi: 10.1126/scitranslmed.abb3312
208. Gao N, Lu L, Ma X, Liu Z, Yang S, Han G. Targeted inhibition of Yap/Taz alters the biological behaviours of keloid fibroblasts. Exp Dermatol (2022) 31(3):320–9. doi: 10.1111/exd.14466
209. Zhao MJ, Chen SY, Qu XY, Abdul-Fattah B, Lai T, Xie M, et al. Increased Cthrc1 activates normal fibroblasts and suppresses keloid fibroblasts by inhibiting tgf-β/Smad signal pathway and modulating yap subcellular location. Curr Med Sci (2018) 38(5):894–902. doi: 10.1007/s11596-018-1959-1
210. Hamburg-Shields E, DiNuoscio GJ, Mullin NK, Lafyatis R, Atit RP. Sustained β-catenin activity in dermal fibroblasts promotes fibrosis by up-regulating expression of extracellular matrix protein-coding genes. J Pathol (2015) 235(5):686–97. doi: 10.1002/path.4481
211. Sato M. Upregulation of the Wnt/Beta-catenin pathway induced by transforming growth factor-beta in hypertrophic scars and keloids. Acta Derm Venereol (2006) 86(4):300–7. doi: 10.2340/00015555-0101
212. Zanconato F, Cordenonsi M, Piccolo S. Yap and taz: A signalling hub of the tumour microenvironment. Nat Rev Cancer (2019) 19(8):454–64. doi: 10.1038/s41568-019-0168-y
213. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of yap and taz in physiology and disease. Nat Rev Mol Cell Biol (2017) 18(12):758–70. doi: 10.1038/nrm.2017.87
214. Kiya K, Kubo T, Kawai K, Matsuzaki S, Maeda D, Fujiwara T, et al. Endothelial cell-derived endothelin-1 is involved in abnormal scar formation by dermal fibroblasts through Rhoa/Rho-kinase pathway. Exp Dermatol (2017) 26(8):705–12. doi: 10.1111/exd.13264
215. Mascharak S, desJardins-Park HE, Davitt MF, Griffin M, Borrelli MR, Moore AL, et al. Preventing engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science (2021) 372(6540):eaba2374. doi: 10.1126/science.aba2374
216. Clark RAF. To scar or not to scar. N Engl J Med (2021) 385(5):469–71. doi: 10.1056/NEJMcibr2107204
217. Lim CP, Phan TT, Lim IJ, Cao X. Stat3 contributes to keloid pathogenesis Via promoting collagen production, cell proliferation and migration. Oncogene (2006) 25(39):5416–25. doi: 10.1038/sj.onc.1209531
218. Lim CP, Phan TT, Lim IJ, Cao X. Cytokine profiling and Stat3 phosphorylation in epithelial-mesenchymal interactions between keloid keratinocytes and fibroblasts. J Invest Dermatol (2009) 129(4):851–61. doi: 10.1038/jid.2008.337
219. Park G, Yoon BS, Moon JH, Kim B, Jun EK, Oh S, et al. Green tea polyphenol epigallocatechin-3-Gallate suppresses collagen production and proliferation in keloid fibroblasts Via inhibition of the Stat3-signaling pathway. J Invest Dermatol (2008) 128(10):2429–41. doi: 10.1038/jid.2008.103
220. Lee YS, Liang YC, Wu P, Kulber DA, Tanabe K, Chuong CM, et al. Stat3 signalling pathway is implicated in keloid pathogenesis by preliminary transcriptome and open chromatin analyses. Exp Dermatol (2019) 28(4):480–4. doi: 10.1111/exd.13923
221. Vinaik R, Barayan D, Auger C, Abdullahi A, Jeschke MG. Regulation of glycolysis and the warburg effect in wound healing. JCI Insight (2020) 5(17):e138949. doi: 10.1172/jci.insight.138949
222. McCann KJ, Yadav M, Alishahedani ME, Freeman AF, Myles IA. Differential responses to folic acid in an established keloid fibroblast cell line are mediated by Jak1/2 and Stat3. PloS One (2021) 16(3):e0248011. doi: 10.1371/journal.pone.0248011
223. Hong YK, Wu CH, Lin YC, Huang YL, Hung KS, Pai TP, et al. Asc-J9 blocks cell proliferation and extracellular matrix production of keloid fibroblasts through inhibiting Stat3 signaling. Int J Mol Sci (2022) 23(10):5549. doi: 10.3390/ijms23105549
224. Zhou Y, Sun Y, Hou W, Ma L, Tao Y, Li D, et al. The Jak2/Stat3 pathway inhibitor, Ag490, suppresses the abnormal behavior of keloid fibroblasts. In vitro. Int J Mol Med (2020) 46(1):191–200. doi: 10.3892/ijmm.2020.4592
225. Moriana C, Moulinet T, Jaussaud R, Decker P. Jak inhibitors and systemic sclerosis: A systematic review of the literature. Autoimmun Rev (2022) 21(10):103168. doi: 10.1016/j.autrev.2022.103168
226. Kazemi T, Farah R, Hordinsky M. Oral janus kinase inhibitors may have a beneficial role in the treatment of keloidal scars: A case report. In press. (2020).
227. Wang Y, Yuan B, Qiao L, Yang H, Li X. Stat3 operates as a novel transcription factor that regulates Nedd4 in keloid. Biochem Biophys Res Commun (2019) 518(4):638–43. doi: 10.1016/j.bbrc.2019.08.110
228. Farag AGA, Khaled HN, Hammam MA, Elshaib ME, Tayel NR, Hommos SEI, et al. Neuronal precursor cell expressed developmentally down regulated 4 (Nedd4) gene polymorphism contributes to keloid development in egyptian population. Clin Cosmet Investig Dermatol (2020) 13:649–56. doi: 10.2147/ccid.S253603
229. Zhao Y, Liu SL, Xie J, Ding MQ, Lu MZ, Zhang LF, et al. Nedd4 single nucleotide polymorphism Rs2271289 is associated with keloids in chinese han population. Am J Transl Res (2016) 8(2):544–55.
230. Ogawa R, Watanabe A, Than Naing B, Sasaki M, Fujita A, Akaishi S, et al. Associations between keloid severity and single-nucleotide polymorphisms: Importance of Rs8032158 as a biomarker of keloid severity. J Invest Dermatol (2014) 134(7):2041–3. doi: 10.1038/jid.2014.71
231. Chung S, Nakashima M, Zembutsu H, Nakamura Y. Possible involvement of Nedd4 in keloid formation; its critical role in fibroblast proliferation and collagen production. Proc Jpn Acad Ser B Phys Biol Sci (2011) 87(8):563–73. doi: 10.2183/pjab.87.563
232. Zhu G, Cai J, Zhang J, Zhao Y, Xu B. Abnormal nuclear factor (Nf)-kappab signal pathway and aspirin inhibits tumor necrosis factor alpha-induced nf-kappab activation in keloid fibroblasts. Dermatol Surg (2007) 33(6):697–708. doi: 10.1111/j.1524-4725.2007.33146.x
233. Niedermeyer J, Kriz M, Hilberg F, Garin-Chesa P, Bamberger U, Lenter MC, et al. Targeted disruption of mouse fibroblast activation protein. Mol Cell Biol (2000) 20(3):1089–94. doi: 10.1128/mcb.20.3.1089-1094.2000
234. Fitzgerald AA, Weiner LM. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev (2020) 39(3):783–803. doi: 10.1007/s10555-020-09909-3
235. Dienus K, Bayat A, Gilmore BF, Seifert O. Increased expression of fibroblast activation protein-alpha in keloid fibroblasts: Implications for development of a novel treatment option. Arch Dermatol Res (2010) 302(10):725–31. doi: 10.1007/s00403-010-1084-x
236. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210(6):1125–35. doi: 10.1084/jem.20130110
237. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med (2013) 210(6):1137–51. doi: 10.1084/jem.20122344
238. Dorst DN, van Caam APM, Vitters EL, Walgreen B, Helsen MMA, Klein C, et al. Fibroblast activation protein targeted photodynamic therapy selectively kills activated skin fibroblasts from systemic sclerosis patients and prevents tissue contraction. Int J Mol Sci (2021) 22(23):12681. doi: 10.3390/ijms222312681
239. Mousavi MJ, Farhadi E, Vodjgani M, Karami J, Tahmasebi MN, Sharafat Vaziri A, et al. Role of fibroblast activation protein alpha in fibroblast-like synoviocytes of rheumatoid arthritis. Iran J Allergy Asthma Immunol (2021) 20(3):338–49. doi: 10.18502/ijaai.v20i3.6335
240. Schmidkonz C, Rauber S, Atzinger A, Agarwal R, Götz TI, Soare A, et al. Disentangling inflammatory from fibrotic disease activity by fibroblast activation protein imaging. Ann Rheum Dis (2020) 79(11):1485–91. doi: 10.1136/annrheumdis-2020-217408
241. Chen J, Zeng B, Yao H, Xu J. The effect of Tlr4/7 on the tgf-β-Induced smad signal transduction pathway in human keloid. Burns (2013) 39(3):465–72. doi: 10.1016/j.burns.2012.07.019
242. Bagabir RA, Syed F, Rautemaa R, McGrouther DA, Paus R, Bayat A. Upregulation of toll-like receptors (Tlrs) 6, 7, and 8 in keloid scars. J Invest Dermatol (2011) 131(10):2128–30. doi: 10.1038/jid.2011.163
243. Li J, Li Z, Wang S, Bi J, Huo R. Exosomes from human adipose-derived mesenchymal stem cells inhibit production of extracellular matrix in keloid fibroblasts Via downregulating transforming growth factor-β2 and notch-1 expression. Bioengineered (2022) 13(4):8515–25. doi: 10.1080/21655979.2022.2051838
244. Yuan R, Dai X, Li Y, Li C, Liu L. Exosomes from mir-29a-Modified adipose-derived mesenchymal stem cells reduce excessive scar formation by inhibiting tgf-β2/Smad3 signaling. Mol Med Rep (2021) 24(5):758. doi: 10.3892/mmr.2021.12398
245. Yang J, Li S, He L, Chen M. Adipose-derived stem cells inhibit dermal fibroblast growth and induce apoptosis in keloids through the arachidonic acid-derived cyclooxygenase-2/Prostaglandin E2 cascade by paracrine. Burns Trauma (2021) 9:tkab020. doi: 10.1093/burnst/tkab020
246. Liu J, Ren J, Su L, Cheng S, Zhou J, Ye X, et al. Human adipose tissue-derived stem cells inhibit the activity of keloid fibroblasts and fibrosis in a keloid model by paracrine signaling. Burns (2018) 44(2):370–85. doi: 10.1016/j.burns.2017.08.017
247. Sato C, Yamamoto Y, Funayama E, Furukawa H, Oyama A, Murao N, et al. Conditioned medium obtained from amnion-derived mesenchymal stem cell culture prevents activation of keloid fibroblasts. Plast Reconstr Surg (2018) 141(2):390–8. doi: 10.1097/prs.0000000000004068
248. Fang F, Huang RL, Zheng Y, Liu M, Huo R. Bone marrow derived mesenchymal stem cells inhibit the proliferative and profibrotic phenotype of hypertrophic scar fibroblasts and keloid fibroblasts through paracrine signaling. J Dermatol Sci (2016) 83(2):95–105. doi: 10.1016/j.jdermsci.2016.03.003
249. Arno AI, Amini-Nik S, Blit PH, Al-Shehab M, Belo C, Herer E, et al. Effect of human wharton's jelly mesenchymal stem cell paracrine signaling on keloid fibroblasts. Stem Cells Transl Med (2014) 3(3):299–307. doi: 10.5966/sctm.2013-0120
Keywords: keloid, scar, immunity, macrophages, T lymphocytes, cytokines, signal transduction
Citation: Lee C-C, Tsai C-H, Chen C-H, Yeh Y-C, Chung W-H and Chen C-B (2023) An updated review of the immunological mechanisms of keloid scars. Front. Immunol. 14:1117630. doi: 10.3389/fimmu.2023.1117630
Received: 06 December 2022; Accepted: 07 March 2023;
Published: 22 March 2023.
Edited by:
Leonardo M.R. Ferreira, Medical University of South Carolina, United StatesReviewed by:
Xuanzhe Liu, Shanghai Jiao Tong University, ChinaHongtao Wang, Fourth Military Medical University, China
Copyright © 2023 Lee, Tsai, Chen, Yeh, Chung and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Bing Chen, Y2h1bmJpbmcuY2hlbkBnbWFpbC5jb20=; YjkyMDIwNTVAY2dtaC5vcmcudHc=
†These authors have contributed equally to this work and share first authorship