 Barney Viengkhou
Barney Viengkhou Markus J. Hofer
Markus J. Hofer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 February 2023
Sec. Multiple Sclerosis and Neuroimmunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1110593
This article is part of the Research TopicTrends in Neuroimmunology: cross-talk between brain-resident and peripheral immune cells in both health and diseaseView all 12 articles
Since their original discovery, type I interferons (IFN-Is) have been closely associated with antiviral immune responses. However, their biological functions go far beyond this role, with balanced IFN-I activity being critical to maintain cellular and tissue homeostasis. Recent findings have uncovered a darker side of IFN-Is whereby chronically elevated levels induce devastating neuroinflammatory and neurodegenerative pathologies. The underlying causes of these ‘interferonopathies’ are diverse and include monogenetic syndromes, autoimmune disorders, as well as chronic infections. The prominent involvement of the CNS in these disorders indicates a particular susceptibility of brain cells to IFN-I toxicity. Here we will discuss the current knowledge of how IFN-Is mediate neurotoxicity in the brain by analyzing the cell-type specific responses to IFN-Is in the CNS, and secondly, by exploring the spectrum of neurological disorders arising from increased IFN-Is. Understanding the nature of IFN-I neurotoxicity is a crucial and fundamental step towards development of new therapeutic strategies for interferonopathies.
Central nervous system (CNS) inflammation is involved in a wide range of neurological disorders and diseases, from pathogen-driven encephalitis and autoimmune disorders to trauma, aging, and neurodegeneration (1–4). The complex nature of inflammation is typically portrayed as either beneficial, such as pathogen elimination, or detrimental, like induction of cell death. Yet in many cases, these processes occur simultaneously and are driven by multiple mediators. The type I interferons (IFN-Is) are master regulators of inflammation. They include the IFN-α subtypes and IFN-β and were originally identified due to their ability to interfere with viral replication (5). However, a vast amount of research over the past 60 years has revealed that IFN-Is have a wide range of roles in addition to regulating inflammation and immunity.
There are three main mechanisms by which IFN-I production and signaling can be increased. Firstly, activation of innate immune sensors by pathogens or cellular danger signals triggers increased expression of IFN-I genes. For example, cytosolic dsDNA from viruses, damaged mitochondria, or improperly processed self-nucleic acids are recognized by cyclic GMP–AMP synthase (cGAS), which in turn activates the stimulator of interferon genes (STING) (6). Activated STING then triggers a signaling cascade resulting in the upregulation of IFN-I expression (6). In addition to STING, there are multiple other immune sensors that upregulate IFN-I expression in similar ways (7, 8). Secondly, genetic changes can result in increased IFN-I signaling such as in trisomy 21 due to an extra copy of IFN-I receptor 1 (IFNAR1) (9), or reduced negative regulation of the IFN-I pathway such as in patients with mutations in USP18 or ISG15 (10, 11). Thirdly, IFN-Is are used as treatment for a range of diseases including chronic viral infections (12), multiple sclerosis (MS), and several cancers and tumors (13–16).
All IFN-Is mediate their cellular effects through binding to a single heterodimeric cell surface receptor consisting of the IFNAR1 and IFNAR2 chains. Activation of the receptor complex triggers two distinct signaling phases (Figure 1). The first phase induces rapid and widespread changes to protein phosphorylation and affects multiple signaling pathways including mitogen-activated protein kinase, cyclin-dependent kinase, and AKT (17). While still not fully understood, it appears that this widespread change in protein phosphorylation prepares the cell for the second phase, which modulates the expression of several hundreds of IFN-regulated genes (IRGs). To make matters more complex, this transcriptional phase mediates its effects through several signaling pathways. Of these, the best understood is the activation of the interferon-stimulated gene factor 3 (ISGF3) complex, which consists of the transcription factors signal transducer and activator of transcription (STAT1) 1, STAT2, and interferon regulatory factor 9 (IRF9). The ISGF3 pathway is often also called the canonical IFN-I signaling pathway and is critical to activate the antiviral response. By contrast, all other pathways are termed ‘non-canonical’ and are thought to modulate the antiviral response in a cell- and stimulus-dependent context (18–22). Moreover, the signaling components in the IFN-I pathway and can be activated by other cytokines, which complicates defining the precise contribution of IFN-Is in inflammation and immunity in vivo. In particular, while IFN-IIIs bind to their unique cell surface receptor, they also mediate their effects through the ISGF3 complex. Recent findings suggest that IFN-IIIs, which consist of the IFN-λs, contribute to neuroinflammation, however, many aspects remain unclear. It appears that IFN-Is are more potent than IFN-IIIs (23, 24) and that the expression of the IFN-III receptor is restricted (25) with very low transcript levels in the brain (23, 24). Thus, while we will not discuss the role of IFN-IIIs in detail, it is important to keep in mind that synergism and antagonism of signaling pathways between IFN-Is and other cytokines influences the outcomes of IFN-I-induced cellular and tissue responses.
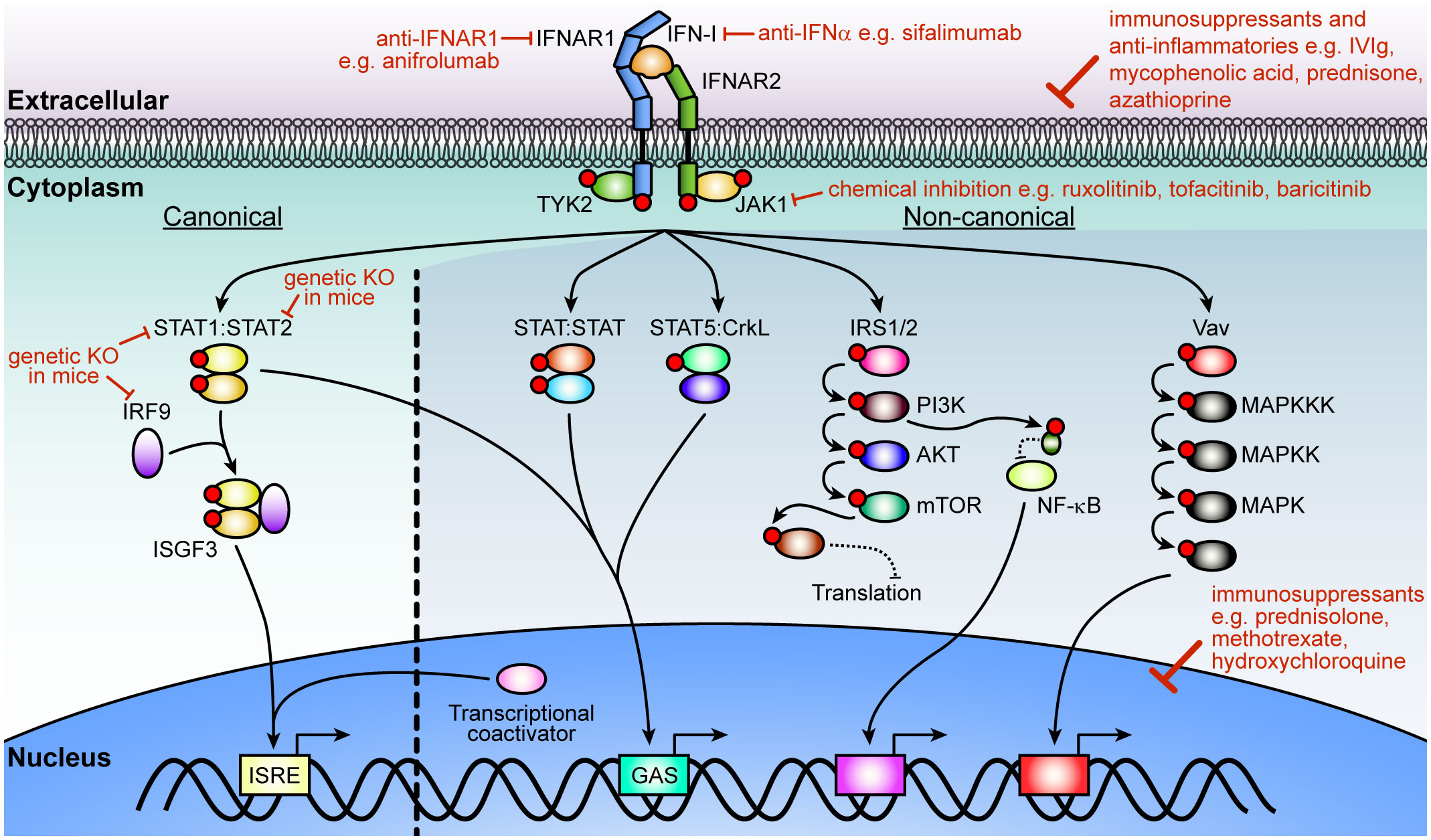
Figure 1 IFN-I signaling pathway and strategies of inhibition. After ligation of IFN-Is with its cognate receptor chains, IFNAR1 and IFNAR2, JAK1 and TYK2 transphosphorylate each other before phosphorylating the receptors. In the canonical pathway, STAT1 and STAT2 dock at the receptor to become phosphorylated by the JAKs. Phosphorylated STAT1 and STAT2 then form a trimolecular complex (ISGF3) with IRF9 and translocate into the nucleus to bind ISREs to regulate the expression of hundreds of interferon-regulated genes (IRGs). Non-canonical signaling involves homodimers or heterodimers of STATs, STAT5 binding to CrkL, or recruitment of transcriptional coactivators to regulate ISRE or GAS elements. Additional kinases are activated (PI3K, NF-κB and MAPK pathways) which modulate the cellular response to IFN-Is that includes translation of a subset of genes, regulation of transcription or a range of cellular functions. Multiple strategies have been employed to target IFN-I signaling including inhibition or elimination of proteins in the pathway or its overall effects with immunosuppressants and anti-inflammatories that act on the cell or affect the expression of genes associated with inflammation. Red circles indicate phosphorylation of a protein. IFN-I, type I interferon; IFNAR, IFN-α/β receptor; JAK1, Janus kinase 1; TYK, tyrosine kinase 2; STAT, signal transducer and activator of transcription; IRF9, interferon regulatory factor 9; ISFG3, interferon-stimulated gene factor; ISRE, interferon-stimulated response-elements; GAS, γ-activated sequence; CrkL, Crk like proto-oncogene, adaptor protein; IRS, insulin receptor substrate; PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor-κB; MAPK, mitogen-activated protein kinase.
Although IFN-Is are critical for the physiological regulation of inflammation, they are associated with a range of adverse effects. These adverse effects manifest often as neurological deficits and are commonly observed when IFN-Is are used as a drug or in patients with chronically elevated IFN-I production in the brain (26). Importantly, the cellular and molecular basis for this IFN-I neurotoxicity remains unclear and its study is complicated by the presence of multiple cell types in the CNS (e.g., neurons, glia, and vascular cells), each of which shows unique cell type-specific responses (17, 27–30). Accordingly, in this review, we dissect the complexity of IFN-I neurotoxicity at two levels: firstly, by analyzing the cell-type specific responses to IFN-I in the CNS, and secondly, exploring the spectrum of diseases and symptoms of neurological disorders with increased IFN-Is.
The existence of a homeostatic level of IFN-I signaling in the brain is demonstrated by the presence of IRG products in the healthy brain (31, 32) and reduced expression of IRGs in unstimulated IFNAR1-deficient mice (18) and cells lacking IFN-I signaling proteins (33). The role of homeostatic IFN-I signaling in the brain is diverse and ranges from priming cells for detection and response to pathogens to roles in learning and memory. For example, several studies have shown that neutralization of IFNAR1 results in synapse reduction and impaired synaptic plasticity (34) and ablation of IFN-β leads to defective neuronal autophagy (35). In addition to homeostatic production, IFN-I expression can be markedly increased in most if not all brain-resident cells in response to a range of stimuli. Recent progress in omic analyzes, particularly at the single-cell level, has demonstrated that within the diseased brain, a spectrum of cellular response states occurs simultaneously rather than a uniform response (36–42). Moreover, while all cell types in the CNS can respond to IFN-Is, each cell type mounts its specific response to IFN-Is. Consequently, the sum of the individual responses determines the local tissue response. In the following sections, we will summarize these cell-type specific responses.
Neurons require IFN-I signaling for normal development. Homeostatic IFN-β signaling in neurons is involved in the formation of dendritic spines, neurite branching, and neuronal autophagy, while loss of IFNAR1 signaling in neurons leads to formation of protein aggregates or Lewy bodies (35). However, IFN-β injected into the brain also causes a reduction of synapses (43), demonstrating the importance of balanced IFN-I signaling for neuronal function. In response to viral infections, neurons show limited production of IFN-Is (44, 45).
Importantly, while they mount a robust response to IFN-Is, neurons only regulate the expression of a limited set of IRGs (46, 47). This comparatively (to other CNS cell types - see below) narrow response provides antiviral protection and may serve to limit adverse or detrimental effects of IFN-I signaling in these delicate cells. The need to protect neurons from damage is also supported by the elevated basal expression of some IRGs like ISG15 in neurons compared with other cells contributing to an intrinsic antiviral resistance (48). IFN-I mediated neurotoxicity manifests in neurons after IFN-α treatment with fewer dendrites (49, 50), decreased neuronal neurogenesis (51), reduced neurotrophic signaling (52), and increased apoptosis of precursor cells (53). In addition, IFN-α alters glutamate-induced excitatory potentials in hippocampal neurons and inhibitory post synaptic potentials in pyramidal neurons (47, 54–56). This in turn may increase epileptiform discharges associated with seizures and inhibit long term potentiation, a process important in memory formation (47, 54–56). Moreover, antagonizing the glutamate receptor, N-methyl D-aspartate receptor (NMDAR), reduces the neurotoxicity of IFN-α, indicating a toxic role of IFNAR and NMDAR coactivation (50). IFN-β also modulates ion channels to increase the number of action potentials elicited after activation of protein kinase C (56) and is in line with IFN-β altering glutamatergic neurotransmission (57). In addition, increased cerebral IFN-α levels in transgenic mice with CNS-targeted overproduction of IFN-α (termed GFAP-IFN mice) results in a progressive loss of neurons (58), impaired learning (59), and changes in phosphoproteins that are associated with various neuronal functions (17). Thus, increased IFN-I signaling has detrimental effects on neuronal health and survival.
Astrocytes are the most abundant glia cell and tile the CNS. Similar to neurons, basal IFN-I signaling in astrocytes is required for a healthy brain. Astrocyte-specific deletion of IFNAR1 results in impaired learning, reduced synapse plasticity, and fewer synapses (34). Following infection with neurotropic viruses, astrocytes are the main producers of IFN-β in mice (44, 60). Their response to IFN-Is is required to limit pathogen replication (61) and to promote blood–brain barrier (BBB) integrity following virus infection (23). Astrocytes alter morphology in response to IFN-Is as observed in brains of patients with increased cerebral IFN-I production (62–64) and GFAP-IFN mice (17, 59). Treatment of astrocytes with IFN-α or IFN-β reduces astrocytic process complexity and domain range and also upregulates genes involved in antiviral responses, metabolism, apoptosis, and major histocompatibility complex (MHC) (17, 27, 39, 59, 62–64). Of note, increased levels of MHC on astrocytes negatively impact neuronal function, activate microglia, and are correlated with social and cognitive deficits in mice (65). Astrocytes can facilitate leukocyte infiltration by increasing chemokine expression after IFN-α treatment (66). In line with this, a subset of astrocytes located around outer cortical blood vessels, and thought to regulate leukocyte access, has been identified as being highly responsive to IFN-Is (39). This highly IFN-I-responsive subset has also been identified in mouse models of Alzheimer’s disease (AD), MS, and acute cortical trauma (39). Hypertrophic astrocytes and increased parenchymal leukocytes are also observed in brains of GFAP-IFN mice, supporting a role for astrocytes in mediating leukocyte infiltration (58, 59). While these findings suggest an inflammation-promoting role of IFN-Is on astrocytes, IFN-I signaling in astrocytes can also limit neuroinflammation through the production of the aryl hydrocarbon receptor and suppressor of cytokine signaling 2, dampening activation of proinflammatory signaling pathways (67). Specifically, mice with astrocyte-restricted Ifnar1-knockdown show exaggerated neuroinflammation in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS (67). In addition, IFN-α but not IFN-β treatment of human astrocytes reduces proliferation and glucose uptake (68) which impacts the metabolic heath of the CNS. Thus, while the contribution of astrocytes to IFN-I neurotoxicity is not clear, these findings suggest a complex role for astrocytes in modulating IFN-I responses, one that is of increasing interest.
Unlike neurons or astrocytes, microglia do not originate from the neuroectoderm. They are derived from the yolk sac and colonize the brain early during embryonic development (69). Microglia are highly plastic and sensitive to the local environment and are considered the key immunoresponsive cell type in the CNS. Microglia produce IFN-α and IFN-β in a wide range of neurological diseases ranging from viral infection to autoimmune disorders (44, 70, 71). Microglia show a more rapid and diverse response to IFN-α compared with astrocytes and neurons (17, 27, 46). Similar to astrocytes, microglia morphology has been used as an indicator of their functional state (72). However, rather than changing into an amoeboid morphology, which is typically observed of microglia in inflammatory situations, in response to IFN-Is, microglia become hyper-ramified with increased process complexity (73). This is also observed in AD and aging (74), indicating microglia are responding to IFN-Is in these conditions. In response to IFN-α, microglia upregulate expression of IRGs, cytokines and chemokines and increase antigen presentation (27), enabling them to act as antigen-presenting cells, propagate inflammation, and promote leukocyte infiltration. This transcriptomic response has been similarly identified in microglia in the aged brain, AD or demyelination in humans or mouse models (38, 40–42). Although most microglia upregulate IRGs, there is a small subset of microglia that are IFN-I-hyperresponsive as identified by single-cell sequencing of a large number of microglia (36, 38, 40, 41). It has been suggested that this hyperresponsive subset contributes to age-dependent cognitive decline and increased synaptic stripping (75–77). In support, minocycline inhibition of microglia activation reduced features of depression and impaired learning of fear extinction in mice injected with IFN-α (78) and use of anti-IFNAR1 treatment in a mouse model of AD demonstrated that IFN-Is promote microglial engulfment of synapses (79). Additionally, minocycline has been used in various neurodegenerative diseases with varied outcomes in animal and human studies (80). However, a recent study using GFAP-IFN mice has demonstrated that depletion of microglia exaggerated disease (81), suggesting that the role of these cells in IFN-I-driven disease may be both beneficial and detrimental.
Oligodendrocytes have limited responses to IFN-α and IFN-β. In viral infections, oligodendrocytes have low production of IFN-Is and show less expression of IRGs, compared with microglia (82). Additionally, IFN-α or IFN-β have no effect on oligodendrocyte proliferation or survival (31, 51, 83). This suggests on the one hand a partial refractory state of oligodendrocytes to IFN-Is, and on the other hand, that the loss of myelin in neurodegenerative diseases may be an indirect response due to actions from surrounding cells or other mediators rather directly through IFN-I signaling. In support of this, a study using single-cell transcriptomics in a mouse model for MS identified a subset of oligodendrocytes that actively recruit T cells, driving the loss of myelin (37). However, data on oligodendrocyte responses to IFN-Is remains limited and further studies are needed to provide a deeper understanding how IFN-Is affect these cells.
The BBB is critical for maintaining CNS homeostasis and brain function (84) and plays crucial roles in neuroinflammation by regulating the migration of leukocytes and diffusion of plasma proteins into the brain parenchyma (85). This separation between blood and brain tissue differs from most other vascular barriers, resulting in vascular cells of the BBB adopting a comparatively distinct phenotype (86). The vascular cells forming the BBB include endothelial cells, pericytes, and mural cells. In particular, cerebral endothelial cells may contribute more to IFN-I signaling in the murine CNS than other cell types as single-cell transcriptomics indicate expression of Ifnar1 and Ifnar2 is higher in these cells than in microglia, astrocytes, and neurons (87, 88). Similarly, in humans, IFNAR2 expression is higher in endothelial cells than glia and neurons (89). This responsiveness of the vasculature is also evident from reports of systemic vasculitis and loss of BBB integrity in patients receiving IFN-Is (55, 90, 91). This vasculopathy is amplified in patients with cerebral interferonopathies and in GFAP-IFN mice, where aneurysms and perivascular calcification are hallmarks of the disease (58, 62, 63, 91). However, the mechanisms leading to these pathologies are unclear, and studies suggest opposing actions of IFN-Is. IFN-α blocks angiogenesis and is toxic to endothelial progenitor cells, contributing to irregular vasculogenesis, abnormal repair and increased atherosclerosis (92). IFN-I therapy can also cause thrombotic microangiopathy and aneurysms (91). The response of endothelial cells in the BBB to IFN-β leads to the secretion of C-X-C motif chemokine 10 resulting in compromised neuronal function and sickness behavior (30). In vitro studies support the BBB-damaging effects of IFN-Is, showing that IFN-α and IFN-β enhance endothelial apoptosis and reduce angiogenesis (93–96). Yet, other studies found that IFN-α induces endothelial proliferation (97, 98) and that IFN-β signaling in endothelial cells has anti-inflammatory roles by inhibiting intracellular signaling of proinflammatory pathways and promoting BBB integrity in the host response to viruses and in MS (23, 99, 100). While the basis for these reported differences in endothelial responses to IFN-Is remains unclear, it points to the importance of the subtype of IFN-Is involved and also the context in which IFN-I signaling occurred. Nevertheless, the impact of IFN-Is on the cerebral vasculature has an active role in disease progression of patients with cerebral interferonopathies and in other neurodegenerative diseases. Accordingly, should further studies demonstrate a direct pathogenic role for the brain’s vasculature, this would open new therapeutic avenues as in contrast to the brain’s parenchyma, the vessels are easily targeted by peripheral drugs.
There is growing evidence that inflammatory processes and, in particular, IFN-Is, are involved in a wide range of neurological diseases (Table 1) (1–3). The symptomatic overlap between these diseases, as well as the reported adverse effects of IFN-I therapy, suggests a causal contribution of increased IFN-I signaling to their pathogenesis (Figure 2). However, the specific contribution of IFN-Is to the pathogenesis of these diseases is often not well understood.
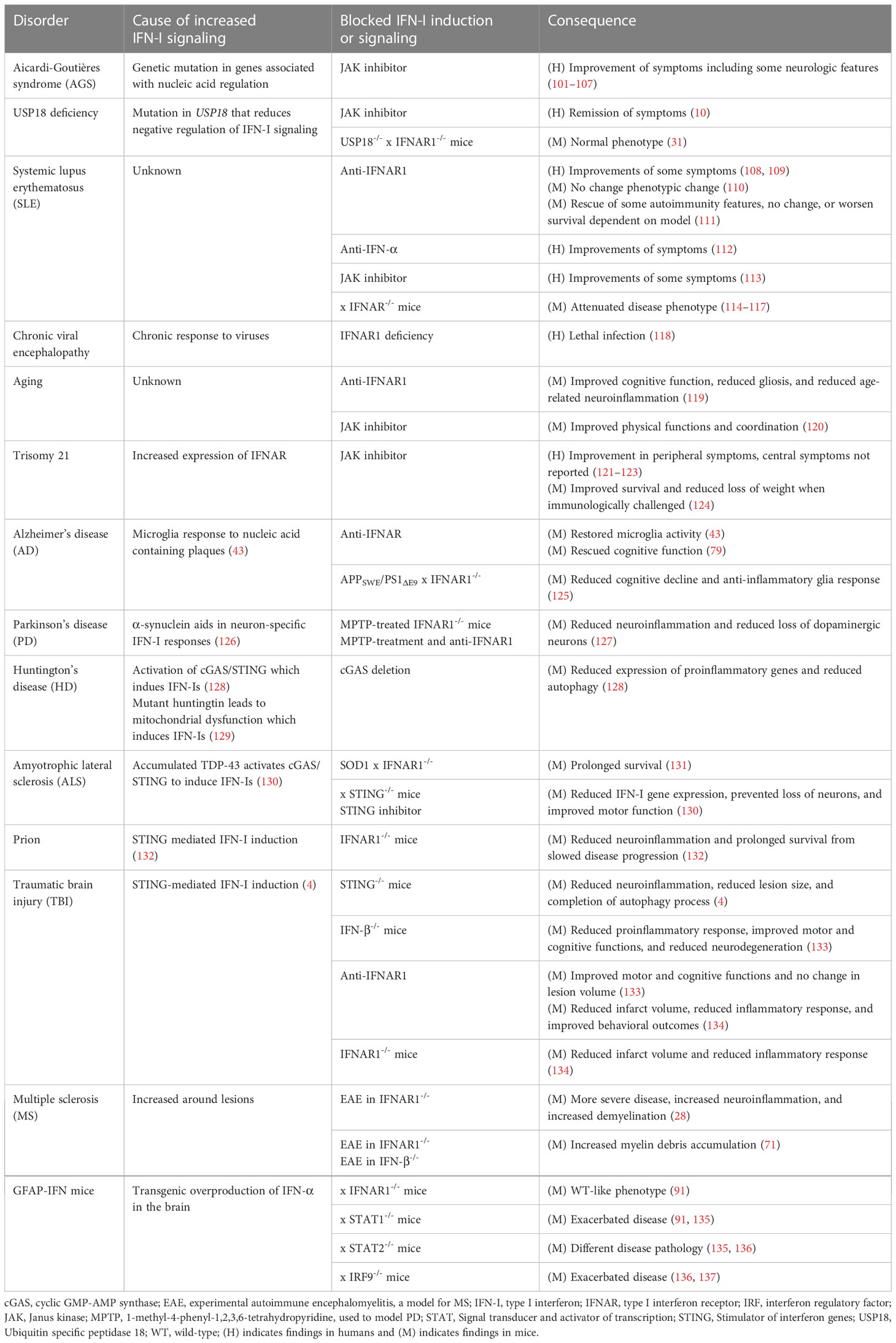
Table 1 IFN-I signaling and its inhibition in neurological disorders.
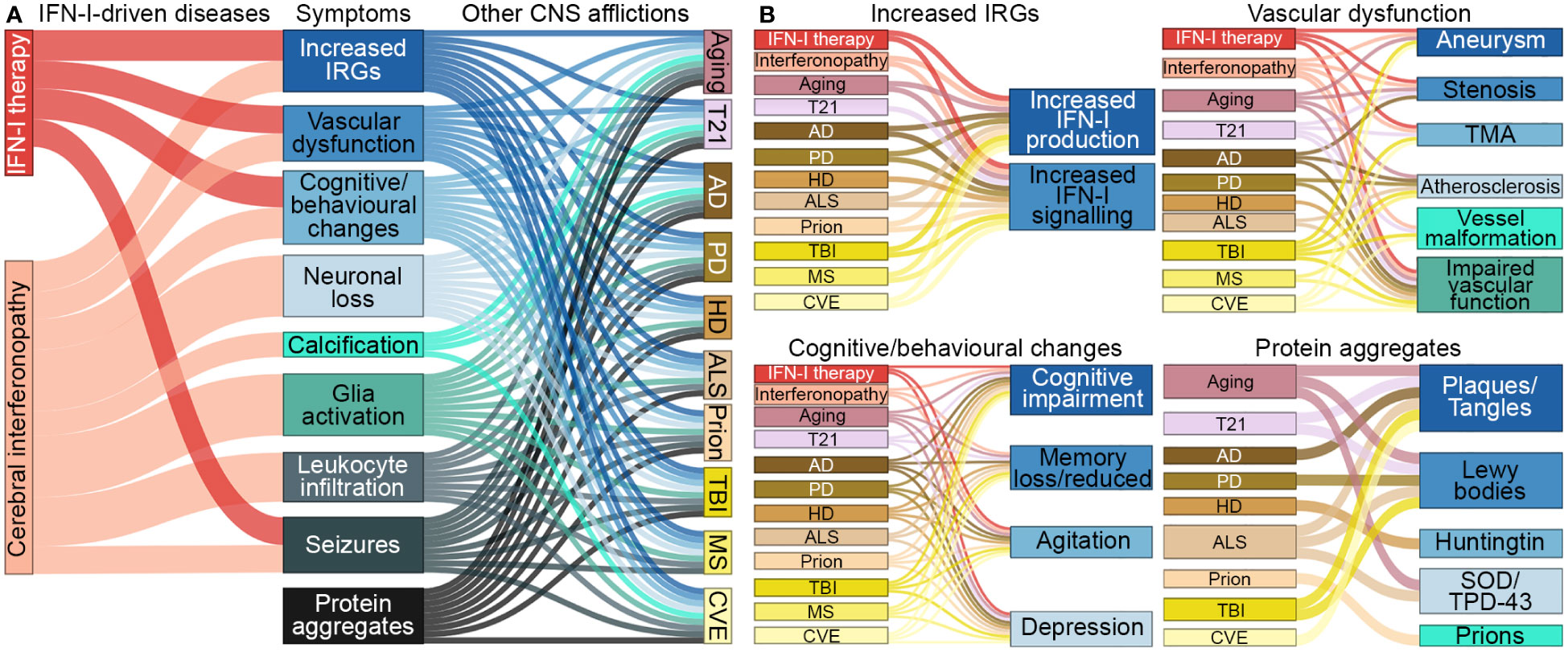
Figure 2 Symptomatic links between IFN-I-driven diseases and CNS afflictions. (A) Symptoms that arise in diseases driven by IFN-Is overlap with symptoms that occur in aging, trisomy 21 and several neurodegenerative diseases, trauma, autoimmune diseases and chronic viral infections, CNS afflictions found to have increased IFN-I signaling. (B) Further breakdown of symptoms linked to each of the CNS afflictions. Note, protein aggregates can lead to increased IFN-Is and expression of IRGs. CNS-centric symptoms were compared and linked if there was prevalence in several human cases. Size of nodes and links are arbitrary. T21, Trisomy 21; AD, Alzheimer’s disease; PD, Parkinson’s disease; HD, Huntington’s disease; ALS, amyotrophic lateral sclerosis; TBI, traumatic brain injury; MS, multiple sclerosis; CVE, chronic viral encephalopathy; TMA, thrombotic microangiopathy; SOD, superoxide dismutase; TPD-43, transactive response DNA binding protein 43 kDa.
The direct neurotoxic effects of IFN-Is are well documented due to their clinical use (53, 138–143). Common (>20%) adverse neurological reactions in patients include flu-like symptoms, fatigue, and depression. Less commonly (<5%) observed adverse events include personality changes, cognitive dysfunction, memory loss, mood disorders, psychomotor slowing, and rare (<1%) but severe reactions including psychosis, mania, and seizures. Nature and severity of adverse reactions is dose dependent and generally worsens over time. Fortunately, cessation of treatment leads to an eventual recovery in most cases (140), indicating that these reactions are mediated by IFN-Is rather than the underlying condition for which IFN-Is have been used as treatment. Importantly, the requirement of basal IFN-I signaling for normal brain development suggests a threshold above which IFN-Is become neurotoxic. This is further supported by findings in glioblastomas. In a subset of glioblastoma, stem cells that display elevated cell-intrinsic IFN-I signaling, which contributes to tumor growth, IFN-β treatment can induce cell death, but not in tumor stem cells that have lower cell-intrinsic IFN-I signaling (144, 145). Several mechanisms by which IFN-Is mediate neurotoxicity have been proposed. For example, IFN-α-induced neuropsychiatric symptoms have been associated with changes in glucose metabolism and neuronal circuitry activity in the basal ganglia and prefrontal cortex (146–148), decreased tryptophan availability with altered serotonergic signaling (149–152) and increased presence of proinflammatory cytokines (141, 149, 152–154). IFN-α treatment can also cause retinopathy (30–86% occurrence) (90, 155) and focal BBB leakage which potentially induces seizures in patients (55). Although rare, IFN-α and IFN-β can prompt extensive vascular changes including thrombotic microangiopathy which encompasses endothelial dysfunction, microvascular ischemia, and microangiopathic hemolytic anemia with vascular microaneurysms and stenoses (91).
Diseases associated with chronically elevated levels of IFN-I in the CNS are collectively termed ‘cerebral interferonopathies’. This diverse group of diseases may be genetic/hereditary (e.g., Aicardi-Goutières Syndrome (AGS), ISG15 deficiency, and USP18 deficiency), autoinflammatory [e.g., systemic lupus erythematosus (SLE) with neurological manifestation], caused by congenital and chronic viral infections (e.g., infections with Toxoplasma gondii, rubella virus, cytomegalovirus, herpes simplex virus, hepatitis B and C virus, and human immunodeficiency virus), or without known etiologies such as Degos disease (156–158). Given their many shared symptoms and pathological features, cerebral interferonopathies provide valuable insights into the long-term biological effects of increased IFN-I signaling in the CNS.
AGS is the commonly exemplified cerebral interferonopathy whereby mutations in genes involved in nucleic acid detection and metabolism lead to increased intrathecal IFN-α production (159, 160). So far, mutations in nine genes have been identified to cause AGS: TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, IFIH1, LSM11, and RNU7-1 (26, 161). It is proposed that loss-of-function mutations in TREX1, RNASEH2, and SAMHD1 lead to the accumulation of immunostimulatory nucleic acid species derived from endogenous retroviral element expression which activate sensors that induces the expression of IFN-Is (162). Similarly, loss of function in ADAR1 results in lack of posttranscriptional modification of endogenous retroviral element transcripts, resulting activation of MDA5, PKR, and ZBP1, which induces IFN-Is and cell death (163–165). Gain-of-function mutations in IFIH1 cause an overactive gene product, MDA5, and consequently abnormal induction of IFN-Is (166). In contrast to aberrant IFN-I induction through sensing or regulating endogenous retroviral elements, mutations in LSM11 and RNU7-1 result in disrupted histone packing of DNA leading to the activation of cGAS/STING to induce IFN-Is (161).
Clinically, AGS has an early onset that mimics transplacental-acquired infections and includes increased mortality before adulthood, irritability, slowed cognitive growth, abnormal movements that develop into ataxia, and epileptic seizures (26, 156, 162, 167, 168). Neuroimaging reveals features including microcephaly, white matter disease, intracranial calcification, necrosis, and vasculopathy with stenosis, moyamoya (small and inadequate vessels formed due to the narrowed cerebral artery), aneurysms, infarcts, and hemorrhage (26, 162, 167, 168). Neuropathological brain examinations show demyelination, perivascular calcification, T-cell infiltration, and apoptotic cells (62–64, 169, 170). Consequently, the clinical and neuropathological observations have led to the proposal of AGS being either a leukodystrophy (171, 172) or a microangiopathy (63, 173). Notably, while vessel disease is a common feature in brains from patients with AGS, whether it mediates pathology or is a consequence of disease has not been clarified. Further, immunohistochemistry has revealed that astrocytes are the main source of IFN-α in the CNS in patients with AGS (62–64) and AGS has thus also been classified as an astrocytopathy by some authors (174). Similar to IFN-I therapy, elevated IFN-α plasma and CSF levels correlate with clinical severity in patients with AGS (160). However, there is a lack of knowledge regarding which cell types and molecular mechanisms mediate disease pathology in AGS, a deficit that also extends to other cerebral interferonopathies. This lack of knowledge stems in large parts from the fact that mouse or zebrafish models that mimic the genetic mutations of patients with AGS, do not recapitulate the human disease (175). By contrast, transgenic mice with increased cerebral IFN-I production (GFAP-IFN mice) – recapitulating the one feature common to of all cerebral interferonopathies – develop closely overlapping clinical and pathological changes also present in patients (Figure 3) (58, 59).
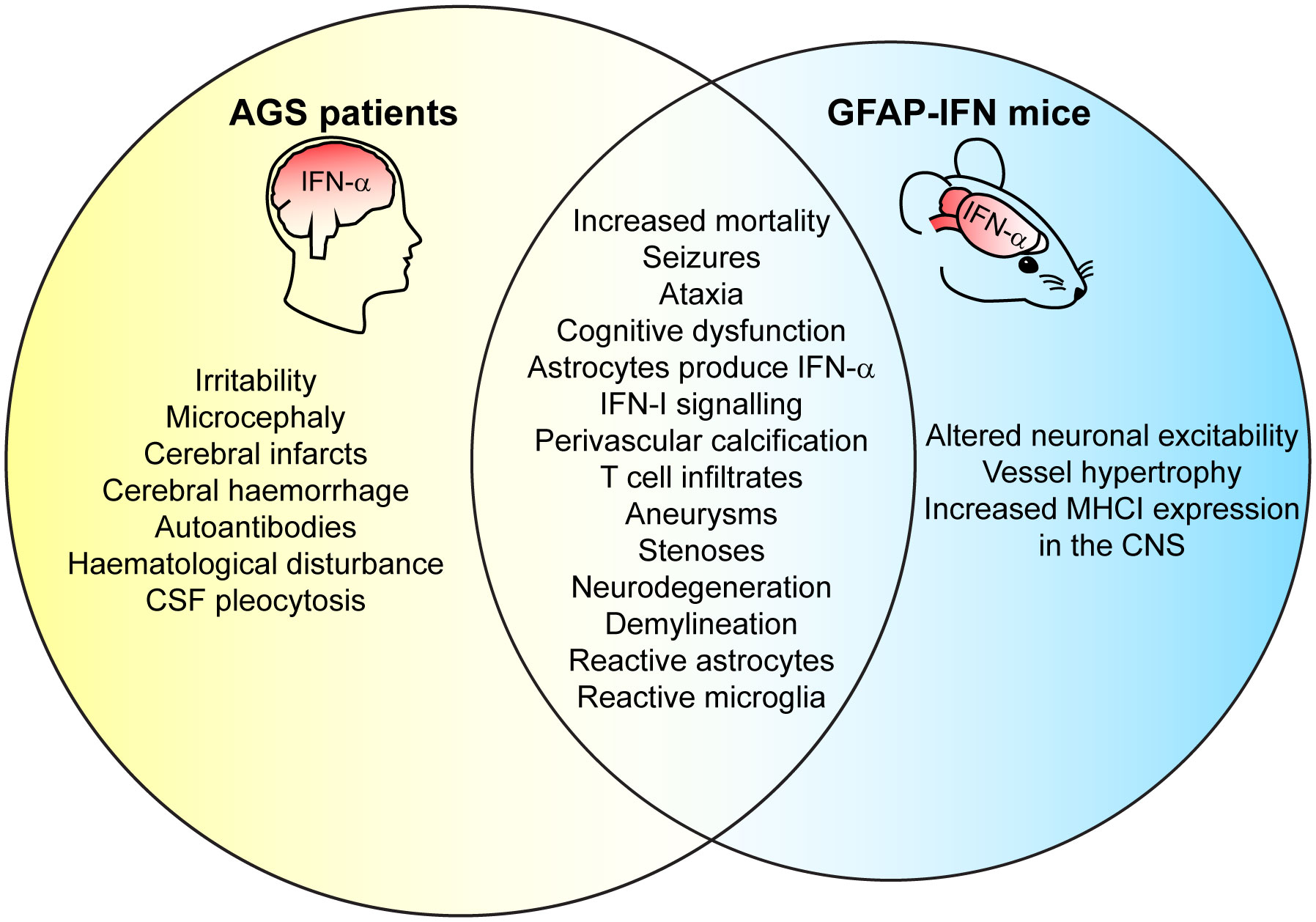
Figure 3 GFAP-IFN mice recapitulate clinical and pathological features of patients with AGS. Venn diagram showing overlap between clinical CNS symptoms and neuropathology observed in the GFAP-IFN mice and patients with AGS. Features that do not overlap and/or have yet to been shown in mice or in patients are also indicated.
A key feature of the host immune response to pathogens is the rapid production of IFN-Is that activate and regulate both the innate and adaptive immune response (176). The ultimate aim of this immune response is to limit damage to the host, eliminate the pathogen, and re-establish organismal homeostasis. However, in situations where pathogen elimination is not achieved, chronic production of IFN-Is occurs. This is evident in a range of congenital and chronic infections of the CNS including toxoplasmosis, syphilis, rubella, cytomegalovirus, Zika virus, herpes simplex virus and human immunodeficiency virus (177). Many of the clinical and neuropathological findings mirror those observed in patients with AGS (157) including cognitive and motor dysfunction, microcephaly, leukodystrophy, cerebral calcification, loss of neurons, and gliosis (178, 179) (Figure 2). Importantly, these changes are paralleled by elevated cerebral IFN-α levels (180). Further, increased IFN-α levels detected in patients with human immunodeficiency virus are linked to developing neurocognitive disorders (50, 181). Together, these findings indicate a direct association between increased chronic cerebral IFN-I and disease.
Aging of the brain concurrently occurs with cognitive decline, reduced neurogenesis, cerebral atrophy, waning of cerebral vascular function, and increased neuroinflammation (182, 183), symptoms which are also seen in patients with cerebral interferonopathies (Figure 2). The mechanisms of aging are not well understood and are made more complex by the presence of comorbidities like BBB breakdown (184, 185), dementia, cerebral small vessel disease and neurodegenerative disorders (182, 186). Notably, IFN-β protein and IFN-I signaling are increased in the choroid plexus in the aged CNS of humans and mice (75, 119). Antibody-mediated neutralization of IFNAR1 in mice reversed the aged transcriptomic phenotype while increased IFN-β expression in the choroid plexus of young mice resulted in a transcriptome that reflected that of aged mice (75, 119). Additionally, JAK inhibition reduced cellular senescence and improved physical functions in aged mice (120). Thus, aging and increased IFN-I signaling in the CNS appear to be interlinked, with implications for the further study of age-related cognitive decline.
One important and so far, understudied aspect of neurodegenerative diseases is the co-occurrence of inflammation and increased IFN-I signaling. To date, this has probably been best studied in AD. In brain tissue from patients with AD, expression of IFN-Is and IRGs is increased (32, 187, 188), which is supported by similar findings in mouse models of AD (43, 79, 188). A recent study demonstrated that the induction of IFN-I is due to nucleic acid contained in amyloid-beta plaques that stimulates IFN-β production and IFN-I signaling in microglia (43). A role for increased IFN-Is in AD pathogenesis (rather than just being a bystander effect) has been demonstrated in mouse models, where IFNAR1 deletion or neutralization resulted in downregulated expression of proinflammatory cytokines, attenuated microgliosis, increased complement-mediated synapse engulfment, enhanced astrogliosis, and partial improvement in learning (43, 79, 125). Likewise, patients with mild cognitive impairment had increased blood IFN-I signaling compared with healthy controls, which was further increased in those with AD (189). Of note, in a rat model of AD, IFN-β treatment improved memory and reduced inflammatory markers (190), and in humans with subtle cognitive decline, a preclinical feature of AD, reduced blood IFN-I signaling levels is linked to an increased risk of progression to mild cognitive impairment (189). Thus, IFN-Is display protective and damaging properties in AD.
In Parkinson’s disease (PD), increased IFN-Is and IRG products surround Lewy bodies (32, 127, 187), the disease-defining pathological hallmark of PD. Additionally, the protein α-synuclein, that form into Lewy bodies, enhances the signaling of IFN-Is in neurons (126). Ablation of IFNAR1 in a mouse model of PD reduced neuroinflammation and decreased dopaminergic neuronal death (127). The increased IFN-I signaling in the vicinity of protein aggregation, pathological hallmarks of AD and PD, indicates that protein aggregation facilitates localized IFN-I production in surrounding cells. This is supported by studies in mouse models on prion disease, which also involves abnormal protein aggregation. Here, robust IFN-I signaling is seen in microglia (132), and in mice lacking IFNAR1 or STING, disease pathology was delayed (132). Furthermore, increased IFN-I signaling is also observed in the CNS of mouse models of Huntington’s disease (HD) (128, 129, 191) and amyotrophic lateral sclerosis (ALS) (130, 131), other disorders with prominent protein aggregates. Together, these findings suggest that protein aggregates are strong inducers of IFN-I signaling and may contribute to disease progression (Table 1).
Unlike the previous CNS conditions, traumatic brain injury (TBI) involves external physical disruption to the CNS. Symptoms reflect both trauma severity and impact location and may include depression, memory problems, anxiety, agitation, and motor coordination problems (192, 193). The pathological features around the CNS injury site include necrosis, glial cell activation, BBB leakage, neuron degeneration, neuroinflammation, and leukocyte infiltrates (194), features that also occur in cerebral interferonopathies (Figure 2). In response to TBI, chronic local upregulation of IRGs occurs at the injury site, persisting for several months post-injury in both humans and mice (4, 133, 195, 196). Additional increase in IFN-β or IFN-I signaling, for example, in the case of traumatic infection or an aged brain, exacerbates disease outcomes in patients and mice, whilst loss of Ifnb and anti-IFNAR1 treatment in mice attenuates the damage from TBI (133, 134, 196–198), further demonstrating the neurotoxic capacity of IFN-Is.
An extra copy of chromosome 21 in humans (trisomy 21) results in diverse symptoms affecting many organs including the CNS. Although symptoms may not all manifest together (9), they include cognitive dysfunction, moyamoya, craniofacial abnormalities, autoimmunity, hematological disorders, intracranial calcification, and early-onset AD (9, 199–203). Some degree of resistance to the development of solid tumors has been observed (9, 199). IFNAR1 and IFNAR2 are located on chromosome 21 and their levels are elevated in trisomy 21 (9, 204–206), possibly rendering cells hyperresponsive to IFN-Is. In support, both transcriptomic and proteomic studies of various cell types from trisomy 21 patients show elevated IFN-I signaling and IRG products (9, 204). Notably, many CNS-associated symptoms mirror those observed in cerebral interferonopathies (Figure 2) indicating that increased cerebral IFN-Is may contribute to disability in these patients, and trisomy 21 has been suggested to be an interferonopathy by some authors (9, 204). This in turn opens new therapeutic options for patients with trisomy 21 and accordingly, JAK inhibitors, which block formation of the ISGF3 signaling complex, have been used with some success in case studies and mouse models showing improvements in disease (121–124) and is in a clinical trial (ClinicalTrials.gov Identifier: NCT04246372).
MS is a demyelinating disease with unclear etiology (207). Patients exhibit a diverse range of symptoms which are largely associated with the location of lesions that occur in the CNS (207). These lesions contain inflammatory leukocytes that presumably mediate oligodendrocyte damage, loss of myelin (208), and local disruption of the BBB (209). IFN-I serum and CSF levels in MS patients do not differ from healthy controls (210). However, there is a focal increase of IFN-I production and IRGs in brain lesions of MS patients and mouse models of MS (71, 211). This mirrors the increase in IFN-I around abnormal protein aggregates and TBI lesions described above, indicating that local production of IFN-I to cellular damage is a common response in the brain. Further, pathological overlaps with AGS/leukodystrophies (212) and MS (Figure 2) such as cerebral small vascular disease exist (213).
Although IFN-Is are produced locally in MS and some mouse models, overall, IFN-I signaling appears to be protective. Genetic ablation of IFNAR1 or IFN-β in mice, results in more severe EAE (28). IFN-β is highly effective for the treatment of MS (IFN-α, although effective, is less well tolerated due to adverse effects including increased occurrence of depression) (16, 214, 215). However, the mechanisms by which IFN-Is are beneficial in MS remain unclear and there is variability in the responses to IFN-β, with some MS patients showing improvement, while others having no change or worsening of disease (216, 217). It has been suggested that some MS patients with IFN-I-induced worsening of disease may have been misdiagnosed; MS and neuromyelitis optica spectrum disorder (NMOSD) can cause very similar symptoms, but in contrast to most MS patients, IFN-Is exacerbate disease in NMOSD (218). In addition, variations in responses to IFN-Is could be due to subnormal serum responses to IFN-Is (219, 220). Thus, it is possible that IFN-β treatment rebalances host IFN-I signaling activity in these patients, rather than being excessive or detrimental.
Currently, there is no cure for cerebral interferonopathies, such as AGS and SLE, and available treatments are primarily aimed at managing symptoms. Treatment is complicated by differences in etiologies, disease progression, severity, and symptoms and importantly by a lack of knowledge regarding the vulnerable and disease-mediating cell types (162). Anti-inflammatory and immunosuppressant drugs (Figure 1) such as corticosteroids or methotrexate are often given to dampen inflammation and reduce infiltrating immune cells, while antiepileptics are used to manage seizures (101, 158, 162, 221–223). Careful consideration is required when devising therapeutic strategies as inactivating canonical signaling factors STAT1, STAT2, or IFR9 in GFAP-IFN mice results in exacerbated disease (135–137), demonstrating that maintaining balanced IFN-I signaling is critical.
Recently, targeting the IFN-I signaling pathway has shown some promise. Treatments with anti-interferon, anti-IFNAR, or JAK inhibitors (Figure 1, Table 1) results in dramatic improvements in some patients with AGS, SLE, and even recovery of patients with peripheral interferonopathies (10, 101–106, 108–112, 221, 224–226). However, these treatments lack support from larger clinical trials, especially in regards to changes in neurological symptoms (162). Importantly, the ability of these treatments to bypass the BBB and improve CNS pathology is yet to be confirmed. Furthermore, the safety profiles of the therapies are noted to include an increased risk of opportunistic infections due to the generalized immunosuppression, as well as an increased risk of major adverse cardiovascular events (227–230). Currently, several clinical trials are underway for patients with AGS (ClinicalTrials.gov Identifier: NCT03921554, NCT04517253, and NCT01724580) and their outcomes will hopefully provide the necessary rationale for the wider use of these treatments. The therapeutic potential of IFN-I signaling inhibition is less clear in the other discussed neurological disorders, with evidence suggesting it may be beneficial in some cases and detrimental in others (Table 1).
IFN-Is are a double-edged sword in the CNS. While they are critical for normal brain function and antimicrobial immunity, chronically elevated levels of IFN-Is can be highly neurotoxic. In addition to both the level and signaling duration of IFN-Is, these opposing effects of IFN-Is are in part due to cell-type specific responses, disease-specific contexts, and biological differences between IFN-I subtypes. These parameters modulate the overall tissue response to IFN-Is in the brain. The detrimental effects of IFN-Is are most evident in cerebral interferonopathies which can serve as a paradigm of IFN-I neurotoxicity, providing valuable insight into a broad spectrum of neurological diseases. Recent advancements with single-cell technologies have provided us with a glimpse of the diversity of the IFN-I responses in the CNS. These studies have provided novel insights into the cell-type specificity of the responses to IFN-Is and demonstrated their variability within a single-cell type. Together, this evidence points to a complex coordination to IFN-Is resulting in a highly stimulus- and time-specific response of CNS-resident cells.
BV wrote the review with revisions by MH. All authors contributed to the article and approved the submitted version.
This review was supported by an Ideas Grant from the National Health and Medical Research Council (NHMRC), Australia, to MH (APP2001543).
We thank Claire Thompson for editorial help with the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schwartz M, Deczkowska A. Neurological disease as a failure of brain-immune crosstalk: The multiple faces of neuroinflammation. Trends Immunol (2016) 37(10):668–79. doi: 10.1016/j.it.2016.08.001
2. Gilhus NE, Deuschl G. Neuroinflammation - a common thread in neurological disorders. Nat Rev Neurol (2019) 15(8):429–30. doi: 10.1038/s41582-019-0227-8
3. Ní Chasaide C, Lynch MA. The role of the immune system in driving neuroinflammation. Brain Neurosci Adv (2020) 4:2398212819901082. doi: 10.1177/2398212819901082
4. Abdullah A, Zhang M, Frugier T, Bedoui S, Taylor JM, Crack PJ. STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J Neuroinflamm (2018) 15(1):323. doi: 10.1186/s12974-018-1354-7
5. Lindenmann J, Burke DC, Isaacs A. Studies on the production, mode of action and properties of interferon. Br J Exp Pathol (1957) 38(5):551–62.
6. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol (2020) 21(9):501–21. doi: 10.1038/s41580-020-0244-x
7. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
8. Okude H, Ori D, Kawai T. Signaling through nucleic acid sensors and their roles in inflammatory diseases. Front Immunol (2020) 11:625833. doi: 10.3389/fimmu.2020.625833
9. Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, et al. Trisomy 21 consistently activates the interferon response. Elife (2016) 5:e16220. doi: 10.7554/eLife.16220
10. Alsohime F, Martin-Fernandez M, Temsah MH, Alabdulhafid M, Le Voyer T, Alghamdi M, et al. JAK inhibitor therapy in a child with inherited USP18 deficiency. N Engl J Med (2020) 382(3):256–65. doi: 10.1056/NEJMoa1905633
11. Martin-Fernandez M, Bravo García-Morato M, Gruber C, Murias Loza S, Malik MNH, Alsohime F, et al. Systemic type I IFN inflammation in human ISG15 deficiency leads to necrotizing skin lesions. Cell Rep (2020) 31(6):107633. doi: 10.1016/j.celrep.2020.107633
12. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov (2007) 6(12):975–90. doi: 10.1038/nrd2422
13. Hauschild A. Adjuvant interferon alfa for melanoma: new evidence-based treatment recommendations? Curr Oncol (2009) 16(3):3–6. doi: 10.3747/co.v16i3.447
14. Shirani A, Zhao Y, Karim ME, Evans C, Kingwell E, van der Kop ML, et al. Association between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. Jama (2012) 308(3):247–56. doi: 10.1001/jama.2012.7625
15. Groves MD, Puduvalli VK, Gilbert MR, Levin VA, Conrad CA, Liu VH, et al. Two phase II trials of temozolomide with interferon-alpha2b (pegylated and non-pegylated) in patients with recurrent glioblastoma multiforme. Br J Cancer (2009) 101(4):615–20. doi: 10.1038/sj.bjc.6605189
16. Cohan SL, Hendin BA, Reder AT, Smoot K, Avila R, Mendoza JP, et al. Interferons and multiple sclerosis: Lessons from 25 years of clinical and real-world experience with intramuscular interferon beta-1a (Avonex). CNS Drugs (2021) 35(7):743–67. doi: 10.1007/s40263-021-00822-z
17. Viengkhou B, White MY, Cordwell SJ, Campbell IL, Hofer MJ. A novel phosphoproteomic landscape evoked in response to type I interferon in the brain and in glial cells. J Neuroinflamm (2021) 18(1):237. doi: 10.1186/s12974-021-02277-x
18. Mostafavi S, Yoshida H, Moodley D, LeBoité H, Rothamel K, Raj T, et al. Parsing the interferon transcriptional network and its disease associations. Cell (2016) 164(3):564–78. doi: 10.1016/j.cell.2015.12.032
19. de Weerd NA, Nguyen T. The interferons and their receptors–distribution and regulation. Immunol Cell Biol (2012) 90(5):483–91. doi: 10.1038/icb.2012.9
20. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
21. Platanias LC. Mechanisms of type-i- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5(5):375–86. doi: 10.1038/nri1604
22. Suprunenko T, Hofer MJ. The emerging role of interferon regulatory factor 9 in the antiviral host response and beyond. Cytokine Growth Fact Rev (2016) 29:35–43. doi: 10.1016/j.cytogfr.2016.03.002
23. Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, White JP, et al. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J Clin Invest (2017) 127(3):843–56. doi: 10.1172/jci88720
24. Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, et al. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med (2015) 7(284):284ra59. doi: 10.1126/scitranslmed.aaa4304
25. Broggi A, Granucci F, Zanoni I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J Exp Med (2020) 217(1):e20190295. doi: 10.1084/jem.20190295
26. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A (2015) 167a(2):296–312. doi: 10.1002/ajmg.a.36887
27. Li W, Viengkhou B, Denyer G, West PK, Campbell IL, Hofer MJ. Microglia have a more extensive and divergent response to interferon-alpha compared with astrocytes. Glia (2018) 66(10):2058–78. doi: 10.1002/glia.23460
28. Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity (2008) 28(5):675–86. doi: 10.1016/j.immuni.2008.03.011
29. Chhatbar C, Detje CN, Grabski E, Borst K, Spanier J, Ghita L, et al. Type I interferon receptor signaling of neurons and astrocytes regulates microglia activation during viral encephalitis. Cell Rep (2018) 25(1):118–29.e4. doi: 10.1016/j.celrep.2018.09.003
30. Blank T, Detje CN, Spiess A, Hagemeyer N, Brendecke SM, Wolfart J, et al. Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity (2016) 44(4):901–12. doi: 10.1016/j.immuni.2016.04.005
31. Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, et al. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J (2015) 34(12):1612–29. doi: 10.15252/embj.201490791
32. Yamada T, Horisberger MA, Kawaguchi N, Moroo I, Toyoda T. Immunohistochemistry using antibodies to alpha-interferon and its induced protein, MxA, in alzheimer's and parkinson's disease brain tissues. Neurosci Lett (1994) 181(1-2):61–4. doi: 10.1016/0304-3940(94)90560-6
33. Li W, Hofer MJ, Songkhunawej P, Jung SR, Hancock D, Denyer G, et al. Type I interferon-regulated gene expression and signaling in murine mixed glial cells lacking signal transducers and activators of transcription 1 or 2 or interferon regulatory factor 9. J Biol Chem (2017) 292(14):5845–59. doi: 10.1074/jbc.M116.756510
34. Hosseini S, Michaelsen-Preusse K, Grigoryan G, Chhatbar C, Kalinke U, Korte M. Type I interferon receptor signaling in astrocytes regulates hippocampal synaptic plasticity and cognitive function of the healthy CNS. Cell Rep (2020) 31(7):107666. doi: 10.1016/j.celrep.2020.107666
35. Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjørn M, Kuss M, et al. Lack of neuronal IFN-β-IFNAR causes lewy body- and parkinson's disease-like dementia. Cell (2015) 163(2):324–39. doi: 10.1016/j.cell.2015.08.069
36. Ellwanger DC, Wang S, Brioschi S, Shao Z, Green L, Case R, et al. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of alzheimer's disease. Proc Natl Acad Sci USA (2021) 118(3):e2017742118. doi: 10.1073/pnas.2017742118
37. Falcão AM, van Bruggen D, Marques S, Meijer M, Jäkel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med (2018) 24(12):1837–44. doi: 10.1038/s41591-018-0236-y
38. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity (2019) 50(1):253–71.e6. doi: 10.1016/j.immuni.2018.11.004
39. Hasel P, Rose IVL, Sadick JS, Kim RD, Liddelow SA. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci (2021) 24(10):1475–87. doi: 10.1038/s41593-021-00905-6
40. Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep (2017) 21(2):366–80. doi: 10.1016/j.celrep.2017.09.039
41. Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with alzheimer's disease. Nat Commun (2020) 11(1):6129. doi: 10.1038/s41467-020-19737-2
42. Plemel JR, Stratton JA, Michaels NJ, Rawji KS, Zhang E, Sinha S, et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci Adv (2020) 6(3):eaay6324. doi: 10.1126/sciadv.aay6324
43. Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest (2020) 130(4):1912–30. doi: 10.1172/jci133737
44. Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, Staeheli P. Visualizing production of beta interferon by astrocytes and microglia in brain of la crosse virus-infected mice. J Virol (2012) 86(20):11223–30. doi: 10.1128/jvi.01093-12
45. Roth-Cross JK, Bender SJ, Weiss SR. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol (2008) 82(20):9829–38. doi: 10.1128/jvi.01199-08
46. Wang J, Campbell IL. Innate STAT1-dependent genomic response of neurons to the antiviral cytokine alpha interferon. J Virol (2005) 79(13):8295–302. doi: 10.1128/jvi.79.13.8295-8302.2005
47. Wang J, Campbell IL, Zhang H. Systemic interferon-alpha regulates interferon-stimulated genes in the central nervous system. Mol Psychiatry (2008) 13(3):293–301. doi: 10.1038/sj.mp.4002013
48. Cavanaugh SE, Holmgren AM, Rall GF. Homeostatic interferon expression in neurons is sufficient for early control of viral infection. J Neuroimmunol (2015) 279:11–9. doi: 10.1016/j.jneuroim.2014.12.012
49. Kessing CF, Tyor WR. Interferon-alpha induces neurotoxicity through activation of the type I receptor and the GluN2A subunit of the NMDA receptor. J Interferon Cytokine Res (2015) 35(4):317–24. doi: 10.1089/jir.2014.0105
50. Sas AR, Bimonte-Nelson H, Smothers CT, Woodward J, Tyor WR. Interferon-alpha causes neuronal dysfunction in encephalitis. J Neurosci (2009) 29(12):3948–55. doi: 10.1523/jneurosci.5595-08.2009
51. Zheng LS, Hitoshi S, Kaneko N, Takao K, Miyakawa T, Tanaka Y, et al. Mechanisms for interferon-α-induced depression and neural stem cell dysfunction. Stem Cell Rep (2014) 3(1):73–84. doi: 10.1016/j.stemcr.2014.05.015
52. Dedoni S, Olianas MC, Ingianni A, Onali P. Type I interferons impair BDNF-induced cell signaling and neurotrophic activity in differentiated human SH-SY5Y neuroblastoma cells and mouse primary cortical neurons. J Neurochem (2012) 122(1):58–71. doi: 10.1111/j.1471-4159.2012.07766.x
53. Borsini A, Cattaneo A, Malpighi C, Thuret S, Harrison NA, Zunszain PA, et al. Interferon-alpha reduces human hippocampal neurogenesis and increases apoptosis via activation of distinct STAT1-dependent mechanisms. Int J Neuropsychopharmacol (2018) 21(2):187–200. doi: 10.1093/ijnp/pyx083
54. Mendoza-Fernández V, Andrew RD, Barajas-López C. Interferon-alpha inhibits long-term potentiation and unmasks a long-term depression in the rat hippocampus. Brain Res (2000) 885(1):14–24. doi: 10.1016/s0006-8993(00)02877-8
55. Pavlovsky L, Seiffert E, Heinemann U, Korn A, Golan H, Friedman A. Persistent BBB disruption may underlie alpha interferon-induced seizures. J Neurol (2005) 252(1):42–6. doi: 10.1007/s00415-005-0596-3
56. Reetz O, Stadler K, Strauss U. Protein kinase c activation mediates interferon-beta-induced neuronal excitability changes in neocortical pyramidal neurons. J Neuroinflamm (2014) 11:185. doi: 10.1186/s12974-014-0185-4
57. Di Filippo M, Tozzi A, Arcangeli S, de Iure A, Durante V, Di Gregorio M, et al. Interferon-β1a modulates glutamate neurotransmission in the CNS through CaMKII and GluN2A-containing NMDA receptors. Neuropharmacology (2016) 100:98–105. doi: 10.1016/j.neuropharm.2015.06.009
58. Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, et al. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol (1998) 161(9):5016–26. doi: 10.4049/jimmunol.161.9.5016
59. Campbell IL, Krucker T, Steffensen S, Akwa Y, Powell HC, Lane T, et al. Structural and functional neuropathology in transgenic mice with CNS expression of IFN-alpha. Brain Res (1999) 835(1):46–61. doi: 10.1016/S0006-8993(99)01328-1
60. Pfefferkorn C, Kallfass C, Lienenklaus S, Spanier J, Kalinke U, Rieder M, et al. Abortively infected astrocytes appear to represent the main source of interferon beta in the virus-infected brain. J Virol (2016) 90(4):2031–8. doi: 10.1128/jvi.02979-15
61. Hidano S, Randall LM, Dawson L, Dietrich HK, Konradt C, Klover PJ, et al. STAT1 signaling in astrocytes is essential for control of infection in the central nervous system. mBio (2016) 7(6):e01881-16. doi: 10.1128/mBio.01881-16
62. Cuadrado E, Jansen MH, Anink J, De Filippis L, Vescovi AL, Watts C, et al. Chronic exposure of astrocytes to interferon-alpha reveals molecular changes related to aicardi-goutieres syndrome. Brain (2013) 136(Pt 1):245–58. doi: 10.1093/brain/aws321
63. Klok MD, Bakels HS, Postma NL, van Spaendonk RM, van der Knaap MS, Bugiani M. Interferon-alpha and the calcifying microangiopathy in aicardi-goutieres syndrome. Ann Clin Transl Neurol (2015) 2(7):774–9. doi: 10.1002/acn3.213
64. van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW. Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in aicardi-goutieres syndrome. Glia (2008) 56(5):568–78. doi: 10.1002/glia.20639
65. Sobue A, Ito N, Nagai T, Shan W, Hada K, Nakajima A, et al. Astroglial major histocompatibility complex class I following immune activation leads to behavioral and neuropathological changes. Glia (2018) 66(5):1034–52. doi: 10.1002/glia.23299
66. Rizzo MD, Crawford RB, Bach A, Sermet S, Amalfitano A, Kaminski NE. Imiquimod and interferon-alpha augment monocyte-mediated astrocyte secretion of MCP-1, IL-6 and IP-10 in a human co-culture system. J Neuroimmunol (2019) 333:576969. doi: 10.1016/j.jneuroim.2019.576969
67. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22(6):586–97. doi: 10.1038/nm.4106
68. Wang T, Takikawa Y, Sawara K, Yoshida Y, Suzuki K. Negative regulation of human astrocytes by interferon (IFN) α in relation to growth inhibition and impaired glucose utilization. Neurochem Res (2012) 37(9):1898–905. doi: 10.1007/s11064-012-0806-1
69. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol (2018) 18(4):225–42. doi: 10.1038/nri.2017.125
70. Shiozawa S, Kuroki Y, Kim M, Hirohata S, Ogino T. Interferon-alpha in lupus psychosis. Arthritis Rheum (1992) 35(4):417–22. doi: 10.1002/art.1780350410
71. Kocur M, Schneider R, Pulm AK, Bauer J, Kropp S, Gliem M, et al. IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol Commun (2015) 3:20. doi: 10.1186/s40478-015-0192-4
72. Walker FR, Beynon SB, Jones KA, Zhao Z, Kongsui R, Cairns M, et al. Dynamic structural remodelling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain Behav Immun (2014) 37:1–14. doi: 10.1016/j.bbi.2013.12.010
73. West PK, McCorkindale AN, Guennewig B, Ashhurst TM, Viengkhou B, Hayashida E, et al. The cytokines interleukin-6 and interferon-α induce distinct microglia phenotypes. J Neuroinflamm (2022) 19(1):96. doi: 10.1186/s12974-022-02441-x
74. West PK, Viengkhou B, Campbell IL, Hofer MJ. Microglia responses to interleukin-6 and type I interferons in neuroinflammatory disease. Glia (2019) 67(10):1821–41. doi: 10.1002/glia.23634
75. Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-i-dependent manner. Nat Commun (2017) 8(1):717. doi: 10.1038/s41467-017-00769-0
76. Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci (2017) 20(8):1162–71. doi: 10.1038/nn.4597
77. Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, et al. A transcriptomic atlas of aged human microglia. Nat Commun (2018) 9(1):539. doi: 10.1038/s41467-018-02926-5
78. Zheng LS, Kaneko N, Sawamoto K. Minocycline treatment ameliorates interferon-alpha- induced neurogenic defects and depression-like behaviors in mice. Front Cell Neurosci (2015) 9:5. doi: 10.3389/fncel.2015.00005
79. Roy ER, Chiu G, Li S, Propson NE, Kanchi R, Wang B, et al. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity (2022) 55(5):879–94.e6. doi: 10.1016/j.immuni.2022.03.018
80. Romero-Miguel D, Lamanna-Rama N, Casquero-Veiga M, Gómez-Rangel V, Desco M, Soto-Montenegro ML. Minocycline in neurodegenerative and psychiatric diseases: An update. Eur J Neurol (2021) 28(3):1056–81. doi: 10.1111/ene.14642
81. West PK, Viengkhou B, Campbell IL, Hofer MJ. Microglia shield the murine brain from damage mediated by the cytokines IL-6 and IFN-α. Front Immunol (2022) 13:1036799. doi: 10.3389/fimmu.2022.1036799
82. Kapil P, Butchi NB, Stohlman SA, Bergmann CC. Oligodendroglia are limited in type I interferon induction and responsiveness in vivo. Glia (2012) 60(10):1555–66. doi: 10.1002/glia.22375
83. Heine S, Ebnet J, Maysami S, Stangel M. Effects of interferon-beta on oligodendroglial cells. J Neuroimmunol (2006) 177(1-2):173–80. doi: 10.1016/j.jneuroim.2006.04.016
84. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell (2015) 163(5):1064–78. doi: 10.1016/j.cell.2015.10.067
85. Takata F, Nakagawa S, Matsumoto J, Dohgu S. Blood-brain barrier dysfunction amplifies the development of neuroinflammation: Understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction. Front Cell Neurosci (2021) 15:661838. doi: 10.3389/fncel.2021.661838
86. Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell (2020) 180(4):764–79.e20. doi: 10.1016/j.cell.2020.01.015
87. Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell (2018) 174(4):1015–30.e16. doi: 10.1016/j.cell.2018.07.028
88. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci (2014) 34(36):11929–47. doi: 10.1523/jneurosci.1860-14.2014
89. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron (2016) 89(1):37–53. doi: 10.1016/j.neuron.2015.11.013
90. Al-Zahrani H, Gupta V, Minden MD, Messner HA, Lipton JH. Vascular events associated with alpha interferon therapy. Leuk Lymphoma (2003) 44(3):471–5. doi: 10.1080/1042819021000055066
91. Kavanagh D, McGlasson S, Jury A, Williams J, Scolding N, Bellamy C, et al. Type I interferon causes thrombotic microangiopathy by a dose-dependent toxic effect on the microvasculature. Blood (2016) 128(24):2824–33. doi: 10.1182/blood-2016-05-715987
92. Denny MF, Thacker S, Mehta H, Somers EC, Dodick T, Barrat FJ, et al. Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood (2007) 110(8):2907–15. doi: 10.1182/blood-2007-05-089086
93. Kaiser WJ, Kaufman JL, Offermann MK. IFN-alpha sensitizes human umbilical vein endothelial cells to apoptosis induced by double-stranded RNA. J Immunol (2004) 172(3):1699–710. doi: 10.4049/jimmunol.172.3.1699
94. Jia H, Thelwell C, Dilger P, Bird C, Daniels S, Wadhwa M. Endothelial cell functions impaired by interferon in vitro: Insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb Res (2018) 163:105–16. doi: 10.1016/j.thromres.2018.01.039
95. Minischetti M, Vacca A, Ribatti D, Iurlaro M, Ria R, Pellegrino A, et al. TNP-470 and recombinant human interferon-alpha2a inhibit angiogenesis synergistically. Br J Haematol (2000) 109(4):829–37. doi: 10.1046/j.1365-2141.2000.02087.x
96. Pammer J, Reinisch C, Birner P, Pogoda K, Sturzl M, Tschachler E. Interferon-alpha prevents apoptosis of endothelial cells after short-term exposure but induces replicative senescence after continuous stimulation. Lab Invest (2006) 86(10):997–1007. doi: 10.1038/labinvest.3700461
97. Cozzolino F, Torcia M, Lucibello M, Morbidelli L, Ziche M, Platt J, et al. Interferon-alpha and interleukin 2 synergistically enhance basic fibroblast growth factor synthesis and induce release, promoting endothelial cell growth. J Clin Invest (1993) 91(6):2504–12. doi: 10.1172/jci116486
98. Gomez D, Reich NC. Stimulation of primary human endothelial cell proliferation by IFN. J Immunol (2003) 170(11):5373–81. doi: 10.4049/jimmunol.170.11.5373
99. Axtell RC, Steinman L. Type 1 interferons cool the inflamed brain. Immunity (2008) 28(5):600–2. doi: 10.1016/j.immuni.2008.04.006
100. Benveniste EN, Qin H. Type I interferons as anti-inflammatory mediators. Sci STKE (2007) 2007(416):pe70. doi: 10.1126/stke.4162007pe70
101. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest (2018) 128(7):3041–52. doi: 10.1172/jci98814
102. Kothur K, Bandodkar S, Chu S, Wienholt L, Johnson A, Barclay P, et al. An open-label trial of JAK 1/2 blockade in progressive IFIH1-associated neuroinflammation. Neurology (2018) 90(6):289–91. doi: 10.1212/wnl.0000000000004921
103. McLellan KE, Martin N, Davidson JE, Cordeiro N, Oates BD, Neven B, et al. JAK 1/2 blockade in MDA5 gain-of-Function. J Clin Immunol (2018) 38(8):844–6. doi: 10.1007/s10875-018-0563-2
104. Vanderver A, Adang L, Gavazzi F, McDonald K, Helman G, Frank DB, et al. Janus kinase inhibition in the aicardi-goutières syndrome. N Engl J Med (2020) 383(10):986–9. doi: 10.1056/NEJMc2001362
105. Cattalini M, Galli J, Zunica F, Ferraro RM, Carpanelli M, Orcesi S, et al. Case report: The JAK-inhibitor ruxolitinib use in aicardi-goutieres syndrome due to ADAR1 mutation. Front Pediatr (2021) 9:725868. doi: 10.3389/fped.2021.725868
106. Li W, Wang W, Wang W, Zhong L, Gou L, Wang C, et al. Janus kinase inhibitors in the treatment of type I interferonopathies: A case series from a single center in China. Front Immunol (2022) 13:825367. doi: 10.3389/fimmu.2022.825367
107. Han VX, Mohammad SS, Jones HF, Bandodkar S, Crow YJ, Dale RC. Cerebrospinal fluid neopterin as a biomarker of treatment response to janus kinase inhibition in aicardi-goutières syndrome. Dev Med Child Neurol (2022) 64(2):266–71. doi: 10.1111/dmcn.15025
108. Kalunian KC, Furie R, Morand EF, Bruce IN, Manzi S, Tanaka Y, et al. A randomized, placebo-controlled phase III extension trial of the long-term safety and tolerability of anifrolumab in active systemic lupus erythematosus. Arthritis Rheumatol (2022). doi: 10.1002/art.42392
109. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med (2020) 382(3):211–21. doi: 10.1056/NEJMoa1912196
110. Huang MW, Stock AD, Mike EV, Herlitz L, Kolbeck R, Putterman C. Anti-IFNAR treatment does not reverse neuropsychiatric disease in MRL/lpr lupus mice. Lupus (2019) 28(13):1510–23. doi: 10.1177/0961203319872265
111. Baccala R, Gonzalez-Quintial R, Schreiber RD, Lawson BR, Kono DH, Theofilopoulos AN. Anti-IFN-α/β receptor antibody treatment ameliorates disease in lupus-predisposed mice. J Immunol (2012) 189(12):5976–84. doi: 10.4049/jimmunol.1201477
112. Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann Rheum Dis (2016) 75(11):1909–16. doi: 10.1136/annrheumdis-2015-208562
113. Richter P, Cardoneanu A, Burlui AM, Macovei LA, Bratoiu I, Buliga-Finis ON, et al. Why do we need JAK inhibitors in systemic lupus erythematosus? Int J Mol Sci (2022) 23(19):11788. doi: 10.3390/ijms231911788
114. Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, et al. Deficiency of type I IFN receptor in lupus-prone new Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol (2009) 183(9):6021–9. doi: 10.4049/jimmunol.0803872
115. Braun D, Geraldes P, Demengeot J. Type I interferon controls the onset and severity of autoimmune manifestations in lpr mice. J Autoimmun (2003) 20(1):15–25. doi: 10.1016/s0896-8411(02)00109-9
116. Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med (2003) 197(6):777–88. doi: 10.1084/jem.20021996
117. Hron JD, Peng SL. Type I IFN protects against murine lupus. J Immunol (2004) 173(3):2134–42. doi: 10.4049/jimmunol.173.3.2134
118. Bastard P, Manry J, Chen J, Rosain J, Seeleuthner Y, AbuZaitun O, et al. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J Clin Invest (2021) 131(1):e139980. doi: 10.1172/jci139980
119. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science (2014) 346(6205):89–93. doi: 10.1126/science.1252945
120. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA (2015) 112(46):E6301–10. doi: 10.1073/pnas.1515386112
121. Rachubinski AL, Estrada BE, Norris D, Dunnick CA, Boldrick JC, Espinosa JM. Janus kinase inhibition in down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Rep (2019) 5(4):365–7. doi: 10.1016/j.jdcr.2019.02.007
122. Jones JT. Treatment of down syndrome-associated arthritis with JAK inhibition. Case Rep Rheumatol (2022) 2022:4889102. doi: 10.1155/2022/4889102
123. Pham AT, Rachubinski AL, Enriquez-Estrada B, Worek K, Griffith M, Espinosa JM. JAK inhibition for treatment of psoriatic arthritis in down syndrome. Rheumatol (Oxf) (2021) 60(9):e309–e11. doi: 10.1093/rheumatology/keab203
124. Tuttle KD, Waugh KA, Araya P, Minter R, Orlicky DJ, Ludwig M, et al. JAK1 inhibition blocks lethal immune hypersensitivity in a mouse model of down syndrome. Cell Rep (2020) 33(7):108407. doi: 10.1016/j.celrep.2020.108407
125. Minter MR, Moore Z, Zhang M, Brody KM, Jones NC, Shultz SR, et al. Deletion of the type-1 interferon receptor in APPSWE/PS1ΔE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol Commun (2016) 4(1):72. doi: 10.1186/s40478-016-0341-4
126. Monogue B, Chen Y, Sparks H, Behbehani R, Chai A, Rajic AJ, et al. Alpha-synuclein supports type 1 interferon signalling in neurons and brain tissue. Brain (2022) 145(10):3622–36. doi: 10.1093/brain/awac192
127. Main BS, Zhang M, Brody KM, Ayton S, Frugier T, Steer D, et al. Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of parkinson's disease. Glia (2016) 64(9):1590–604. doi: 10.1002/glia.23028
128. Sharma M, Rajendrarao S, Shahani N, Ramírez-Jarquín UN, Subramaniam S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in huntington disease. Proc Natl Acad Sci USA (2020) 117(27):15989–99. doi: 10.1073/pnas.2002144117
129. Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, et al. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron (2020) 107(5):891–908.e8. doi: 10.1016/j.neuron.2020.06.021
130. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell (2020) 183(3):636–49.e18. doi: 10.1016/j.cell.2020.09.020
131. Wang R, Yang B, Zhang D. Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia (2011) 59(6):946–58. doi: 10.1002/glia.21167
132. Nazmi A, Field RH, Griffin EW, Haugh O, Hennessy E, Cox D, et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia (2019) 67(7):1254–76. doi: 10.1002/glia.23592
133. Barrett JP, Henry RJ, Shirey KA, Doran SJ, Makarevich OD, Ritzel RM, et al. Interferon-β plays a detrimental role in experimental traumatic brain injury by enhancing neuroinflammation that drives chronic neurodegeneration. J Neurosci (2020) 40(11):2357–70. doi: 10.1523/jneurosci.2516-19.2020
134. Karve IP, Zhang M, Habgood M, Frugier T, Brody KM, Sashindranath M, et al. Ablation of type-1 IFN signaling in hematopoietic cells confers protection following traumatic brain injury. eNeuro (2016) 3(1):ENEURO.0128-15.2016. doi: 10.1523/eneuro.0128-15.2016
135. Wang J, Schreiber RD, Campbell IL. STAT1 deficiency unexpectedly and markedly exacerbates the pathophysiological actions of IFN-alpha in the central nervous system. Proc Natl Acad Sci USA (2002) 99(25):16209–14. doi: 10.1073/pnas.252454799
136. Wang J, Pham-Mitchell N, Schindler C, Campbell IL. Dysregulated sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-gamma in the brain. J Clin Invest (2003) 112(4):535–43. doi: 10.1172/jci18637
137. Hofer MJ, Li W, Lim SL, Campbell IL. The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9. J Neurosci (2010) 30(3):1149–57. doi: 10.1523/jneurosci.3711-09.2010
138. Su KP, Lai HC, Peng CY, Su WP, Chang JP, Pariante CM. Interferon-alpha-induced depression: Comparisons between early- and late-onset subgroups and with patients with major depressive disorder. Brain Behav Immun (2019) 80:512–8. doi: 10.1016/j.bbi.2019.04.032
139. Reyes-Vázquez C, Prieto-Gómez B, Dafny N. Interferon modulates central nervous system function. Brain Res (2012) 1442:76–89. doi: 10.1016/j.brainres.2011.09.061
140. Rubin KM, Vona K, Madden K, McGettigan S, Braun IM. Side effects in melanoma patients receiving adjuvant interferon alfa-2b therapy: a nurse's perspective. Support Care Cancer (2012) 20(8):1601–11. doi: 10.1007/s00520-012-1473-0
141. Hoyo-Becerra C, Schlaak JF, Hermann DM. Insights from interferon-alpha-related depression for the pathogenesis of depression associated with inflammation. Brain Behav Immun (2014) 42:222–31. doi: 10.1016/j.bbi.2014.06.200
142. Fritz-French C, Tyor W. Interferon-α (IFNα) neurotoxicity. Cytokine Growth Fact Rev (2012) 23(1-2):7–14. doi: 10.1016/j.cytogfr.2012.01.001
143. Hosoda S, Takimura H, Shibayama M, Kanamura H, Ikeda K, Kumada H. Psychiatric symptoms related to interferon therapy for chronic hepatitis c: clinical features and prognosis. Psychiatry Clin Neurosci (2000) 54(5):565–72. doi: 10.1046/j.1440-1819.2000.00754.x
144. Han XX, Jin S, Yu LM, Wang M, Hu XY, Hu DY, et al. Interferon-beta inhibits human glioma stem cell growth by modulating immune response and cell cycle related signaling pathways. Cell Regener (2022) 11(1):23. doi: 10.1186/s13619-022-00123-w
145. Khan S, Mahalingam R, Sen S, Martinez-Ledesma E, Khan A, Gandy K, et al. Intrinsic interferon signaling regulates the cell death and mesenchymal phenotype of glioblastoma stem cells. Cancers (Basel) (2021) 13(21):5284. doi: 10.3390/cancers13215284
146. Haroon E, Felger JC, Woolwine BJ, Chen X, Parekh S, Spivey JR, et al. Age-related increases in basal ganglia glutamate are associated with TNF, reduced motivation and decreased psychomotor speed during IFN-alpha treatment: Preliminary findings. Brain Behav Immun (2015) 46:17–22. doi: 10.1016/j.bbi.2014.12.004
147. Taylor MJ, Godlewska B, Near J, Christmas D, Potokar J, Collier J, et al. Effect of interferon-alpha on cortical glutamate in patients with hepatitis c: a proton magnetic resonance spectroscopy study. Psychol Med (2014) 44(4):789–95. doi: 10.1017/s0033291713001062
148. Abe K, Wada A, Oshima S, Kono S, Takahashi A, Kanno Y, et al. Reduced frontal activation during verbal fluency task in chronic hepatitis c patients with interferon-based therapy as measured by near-infrared spectroscopy. Hepatol Res (2017) 47(3):E55–e63. doi: 10.1111/hepr.12721
149. Lotrich FE, Ferrell RE, Rabinovitz M, Pollock BG. Risk for depression during interferon-alpha treatment is affected by the serotonin transporter polymorphism. Biol Psychiatry (2009) 65(4):344–8. doi: 10.1016/j.biopsych.2008.08.009
150. Murakami Y, Ishibashi T, Tomita E, Imamura Y, Tashiro T, Watcharanurak K, et al. Depressive symptoms as a side effect of interferon-α therapy induced by induction of indoleamine 2,3-dioxygenase 1. Sci Rep (2016) 6:29920. doi: 10.1038/srep29920
151. Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, et al. Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol Psychiatry (2003) 54(9):906–14. doi: 10.1016/s0006-3223(03)00173-2
152. Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry (2002) 7(5):468–73. doi: 10.1038/sj.mp.4000995
153. Lotrich FE. Inflammatory cytokine-associated depression. Brain Res (2015) 1617:113–25. doi: 10.1016/j.brainres.2014.06.032
154. Bonaccorso S, Marino V, Puzella A, Pasquini M, Biondi M, Artini M, et al. Increased depressive ratings in patients with hepatitis c receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J Clin Psychopharmacol (2002) 22(1):86–90. doi: 10.1097/00004714-200202000-00014
155. Abd El-Badie Mohamed M, Abd-El Azeem Eed K. Retinopathy associated with interferon therapy in patients with hepatitis c virus. Clin Ophthalmol (2012) 6:1341–5. doi: 10.2147/opth.S32469
156. McGlasson S, Jury A, Jackson A, Hunt D. Type I interferon dysregulation and neurological disease. Nat Rev Neurol (2015) 11(9):515–23. doi: 10.1038/nrneurol.2015.143
157. Hofer MJ, Campbell IL. Type I interferon in neurological disease-the devil from within. Cytokine Growth Fact Rev (2013) 24(3):257–67. doi: 10.1016/j.cytogfr.2013.03.006
158. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med (2016) 213(12):2527–38. doi: 10.1084/jem.20161596
159. Lodi L, Melki I, Bondet V, Seabra L, Rice GI, Carter E, et al. Differential expression of interferon-alpha protein provides clues to tissue specificity across type I interferonopathies. J Clin Immunol (2021) 41(3):603–9. doi: 10.1007/s10875-020-00952-x
160. Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med (2017) 214(5):1547–55. doi: 10.1084/jem.20161451
161. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet (2020) 52(12):1364–72. doi: 10.1038/s41588-020-00737-3
162. Crow YJ, Shetty J, Livingston JH. Treatments in aicardi-goutieres syndrome. Dev Med Child Neurol (2020) 62(1):42–7. doi: 10.1111/dmcn.14268
163. de Reuver R, Verdonck S, Dierick E, Nemegeer J, Hessmann E, Ahmad S, et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature (2022) 607(7920):784–9. doi: 10.1038/s41586-022-04974-w
164. Hubbard NW, Ames JM, Maurano M, Chu LH, Somfleth KY, Gokhale NS, et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature (2022) 607(7920):769–75. doi: 10.1038/s41586-022-04896-7
165. Jiao H, Wachsmuth L, Wolf S, Lohmann J, Nagata M, Kaya GG, et al. ADAR1 averts fatal type I interferon induction by ZBP1. Nature (2022) 607(7920):776–83. doi: 10.1038/s41586-022-04878-9
166. Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, et al. Aicardi-goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet (2014) 95(1):121–5. doi: 10.1016/j.ajhg.2014.06.007
167. La Piana R, Uggetti C, Roncarolo F, Vanderver A, Olivieri I, Tonduti D, et al. Neuroradiologic patterns and novel imaging findings in aicardi-goutieres syndrome. Neurology (2016) 86(1):28–35. doi: 10.1212/wnl.0000000000002228
168. Ramesh V, Bernardi B, Stafa A, Garone C, Franzoni E, Abinun M, et al. Intracerebral large artery disease in aicardi-goutieres syndrome implicates SAMHD1 in vascular homeostasis. Dev Med Child Neurol (2010) 52(8):725–32. doi: 10.1111/j.1469-8749.2010.03727.x
169. Livingston JH, Stivaros S, van der Knaap MS, Crow YJ. Recognizable phenotypes associated with intracranial calcification. Dev Med Child Neurol (2013) 55(1):46–57. doi: 10.1111/j.1469-8749.2012.04437.x
170. Livingston JH, Stivaros S, Warren D, Crow YJ. Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Dev Med Child Neurol (2014) 56(7):612–26. doi: 10.1111/dmcn.12359
171. Goutières F, Aicardi J, Barth PG, Lebon P. Aicardi-goutières syndrome: an update and results of interferon-alpha studies. Ann Neurol (1998) 44(6):900–7. doi: 10.1002/ana.410440608
172. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of aicardi-goutieres syndrome. Am J Hum Genet (2007) 81(4):713–25. doi: 10.1086/521373
173. Barth PG, Walter A, van Gelderen I. Aicardi-goutières syndrome: a genetic microangiopathy? Acta Neuropathol (1999) 98(2):212–6. doi: 10.1007/s004010051071
174. van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol (2017) 134(3):351–82. doi: 10.1007/s00401-017-1739-1
175. Rutherford HA, Kasher PR, Hamilton N. Dirty fish versus squeaky clean mice: Dissecting interspecies differences between animal models of interferonopathy. Front Immunol (2020) 11:623650. doi: 10.3389/fimmu.2020.623650
176. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
177. de Vries LS. Viral infections and the neonatal brain. Semin Pediatr Neurol (2019) 32:100769. doi: 10.1016/j.spen.2019.08.005
178. Gray F, Scaravilli F, Everall I, Chretien F, An S, Boche D, et al. Neuropathology of early HIV-1 infection. Brain Pathol (1996) 6(1):1–15. doi: 10.1111/j.1750-3639.1996.tb00775.x
179. Bale JF Jr. Fetal infections and brain development. Clin Perinatol (2009) 36(3):639–53. doi: 10.1016/j.clp.2009.06.005
180. Krivine A, Force G, Servan J, Cabée A, Rozenberg F, Dighiero L, et al. Measuring HIV-1 RNA and interferon-alpha in the cerebrospinal fluid of AIDS patients: insights into the pathogenesis of AIDS dementia complex. J Neurovirol (1999) 5(5):500–6. doi: 10.3109/13550289909045379
181. Anderson AM, Lennox JL, Mulligan MM, Loring DW, Zetterberg H, Blennow K, et al. Cerebrospinal fluid interferon alpha levels correlate with neurocognitive impairment in ambulatory HIV-infected individuals. J Neurovirol (2017) 23(1):106–12. doi: 10.1007/s13365-016-0466-z
182. Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature (2016) 539(7628):180–6. doi: 10.1038/nature20411
183. Lee J, Kim HJ. Normal aging induces changes in the brain and neurodegeneration progress: Review of the structural, biochemical, metabolic, cellular, and molecular changes. Front Aging Neurosci (2022) 14:931536. doi: 10.3389/fnagi.2022.931536
184. Bowman GL, Dayon L, Kirkland R, Wojcik J, Peyratout G, Severin IC, et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimers Dement (2018) 14(12):1640–50. doi: 10.1016/j.jalz.2018.06.2857
185. Senatorov VV Jr., Friedman AR, Milikovsky DZ, Ofer J, Saar-Ashkenazy R, Charbash A, et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci Transl Med (2019) 11(521):eaaw8283. doi: 10.1126/scitranslmed.aaw8283
186. Peters R. Ageing and the brain. Postgrad Med J (2006) 82(964):84–8. doi: 10.1136/pgmj.2005.036665
187. Akiyama H, Ikeda K, Katoh M, McGeer EG, McGeer PL. Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J Neuroimmunol (1994) 50(2):195–201. doi: 10.1016/0165-5728(94)90046-9
188. Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ. Type-1 interferon signaling mediates neuro-inflammatory events in models of alzheimer's disease. Neurobiol Aging (2014) 35(5):1012–23. doi: 10.1016/j.neurobiolaging.2013.10.089
189. Song L, Chen J, Lo CZ, Guo Q, Feng J, Zhao XM. Impaired type I interferon signaling activity implicated in the peripheral blood transcriptome of preclinical alzheimer's disease. EBioMedicine (2022) 82:104175. doi: 10.1016/j.ebiom.2022.104175
190. Chavoshinezhad S, Mohseni Kouchesfahani H, Salehi MS, Pandamooz S, Ahmadiani A, Dargahi L. Intranasal interferon beta improves memory and modulates inflammatory responses in a mutant APP-overexpressing rat model of alzheimer's disease. Brain Res Bull (2019) 150:297–306. doi: 10.1016/j.brainresbull.2019.06.015
191. Bhambri A, Pinto A, Pillai B. Interferon mediated neuroinflammation in polyglutamine disease is not caused by RNA toxicity. Cell Death Dis (2020) 11(1):3. doi: 10.1038/s41419-019-2193-x
192. Dikmen S, Machamer J, Fann JR, Temkin NR. Rates of symptom reporting following traumatic brain injury. J Int Neuropsychol Soc (2010) 16(3):401–11. doi: 10.1017/s1355617710000196
193. O'Connor C, Colantonio A, Polatajko H. Long term symptoms and limitations of activity of people with traumatic brain injury: a ten-year follow-up. Psychol Rep (2005) 97(1):169–79. doi: 10.2466/pr0.97.1.169-179
194. Ng SY, Lee AYW. Traumatic brain injuries: Pathophysiology and potential therapeutic targets. Front Cell Neurosci (2019) 13:528. doi: 10.3389/fncel.2019.00528
195. Todd BP, Chimenti MS, Luo Z, Ferguson PJ, Bassuk AG, Newell EA. Traumatic brain injury results in unique microglial and astrocyte transcriptomes enriched for type I interferon response. J Neuroinflamm (2021) 18(1):151. doi: 10.1186/s12974-021-02197-w
196. Moro F, Pischiutta F, Portet A, Needham EJ, Norton EJ, Ferdinand JR, et al. Ageing is associated with maladaptive immune response and worse outcome after traumatic brain injury. Brain Commun (2022) 4(2):fcac036. doi: 10.1093/braincomms/fcac036
197. Mastorakos P, Russo MV, Zhou T, Johnson K, McGavern DB. Antimicrobial immunity impedes CNS vascular repair following brain injury. Nat Immunol (2021) 22(10):1280–93. doi: 10.1038/s41590-021-01012-1
198. Kesinger MR, Kumar RG, Wagner AK, Puyana JC, Peitzman AP, Billiar TR, et al. Hospital-acquired pneumonia is an independent predictor of poor global outcome in severe traumatic brain injury up to 5 years after discharge. J Trauma Acute Care Surg (2015) 78(2):396–402. doi: 10.1097/ta.0000000000000526
199. Colvin KL, Yeager ME. What people with down syndrome can teach us about cardiopulmonary disease. Eur Respir Rev (2017) 26(143):160098. doi: 10.1183/16000617.0098-2016
200. Sadana KS, Goraya JS. Intracranial calcification in down syndrome. J Pediatr Neurosci (2018) 13(1):120–1. doi: 10.4103/jpn.Jpn_114_17
201. Mann DM. Calcification of the basal ganglia in down's syndrome and alzheimer's disease. Acta Neuropathol (1988) 76(6):595–8. doi: 10.1007/bf00689598
202. Cramer SC, Robertson RL, Dooling EC, Scott RM. Moyamoya and down syndrome. Clin Radiol Features Stroke (1996) 27(11):2131–5. doi: 10.1161/01.str.27.11.2131
203. Wilcock DM, Griffin WS. Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflamm (2013) 10:84. doi: 10.1186/1742-2094-10-84
204. Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci Rep (2017) 7(1):14818. doi: 10.1038/s41598-017-13858-3
205. Powers RK, Culp-Hill R, Ludwig MP, Smith KP, Waugh KA, Minter R, et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat Commun (2019) 10(1):4766. doi: 10.1038/s41467-019-12739-9
206. Waugh KA, Araya P, Pandey A, Jordan KR, Smith KP, Granrath RE, et al. Mass cytometry reveals global immune remodeling with multi-lineage hypersensitivity to type I interferon in down syndrome. Cell Rep (2019) 29(7):1893–908.e4. doi: 10.1016/j.celrep.2019.10.038
207. Dobson R, Giovannoni G. Multiple sclerosis - a review. Eur J Neurol (2019) 26(1):27–40. doi: 10.1111/ene.13819
208. Lassmann H. Pathology and disease mechanisms in different stages of multiple sclerosis. J Neurol Sci (2013) 333(1-2):1–4. doi: 10.1016/j.jns.2013.05.010
209. van Horssen J, Brink BP, de Vries HE, van der Valk P, Bø L. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol (2007) 66(4):321–8. doi: 10.1097/nen.0b013e318040b2de
210. Varley JA, Andersson M, Grant E, Berretta A, Zandi MS, Bondet V, et al. Absence of neuronal autoantibodies in neuropsychiatric systemic lupus erythematosus. Ann Neurol (2020) 88(6):1244–50. doi: 10.1002/ana.25908
211. Traugott U, Lebon P. Multiple sclerosis: involvement of interferons in lesion pathogenesis. Ann Neurol (1988) 24(2):243–51. doi: 10.1002/ana.410240211
212. van der Knaap MS, Schiffmann R, Mochel F, Wolf NI. Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol (2019) 18(10):962–72. doi: 10.1016/s1474-4422(19)30143-7
213. Filippi M, Preziosa P, Banwell BL, Barkhof F, Ciccarelli O, De Stefano N, et al. Assessment of lesions on magnetic resonance imaging in multiple sclerosis: practical guidelines. Brain (2019) 142(7):1858–75. doi: 10.1093/brain/awz144
214. Magyari M, Bach Søndergaard H, Sellebjerg F, Soelberg Sørensen P. Preserved in vivo response to interferon-alpha in multiple sclerosis patients with neutralising antibodies against interferon-beta (REPAIR study). Mult Scler Relat Disord (2013) 2(2):141–6. doi: 10.1016/j.msard.2012.10.001
215. Durelli L, Bongioanni MR, Ferrero B, Ferri R, Imperiale D, Bradac GB, et al. Interferon alpha-2a treatment of relapsing-remitting multiple sclerosis: disease activity resumes after stopping treatment. Neurology (1996) 47(1):123–9. doi: 10.1212/wnl.47.1.123
216. Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, et al. T Helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med (2010) 16(4):406–12. doi: 10.1038/nm.2110
217. Axtell RC, Raman C. Janus-like effects of type I interferon in autoimmune diseases. Immunol Rev (2012) 248(1):23–35. doi: 10.1111/j.1600-065X.2012.01131.x
218. Feng X, Reder NP, Yanamandala M, Hill A, Franek BS, Niewold TB, et al. Type I interferon signature is high in lupus and neuromyelitis optica but low in multiple sclerosis. J Neurol Sci (2012) 313(1-2):48–53. doi: 10.1016/j.jns.2011.09.032
219. Feng X, Petraglia AL, Chen M, Byskosh PV, Boos MD, Reder AT. Low expression of interferon-stimulated genes in active multiple sclerosis is linked to subnormal phosphorylation of STAT1. J Neuroimmunol (2002) 129(1-2):205–15. doi: 10.1016/s0165-5728(02)00182-0
220. Reder AT, Feng X. Aberrant type I interferon regulation in autoimmunity: Opposite directions in MS and SLE, shaped by evolution and body ecology. Front Immunol (2013) 4:281. doi: 10.3389/fimmu.2013.00281
221. Briand C, Fremond ML, Bessis D, Carbasse A, Rice GI, Bondet V, et al. Efficacy of JAK1/2 inhibition in the treatment of chilblain lupus due to TREX1 deficiency. Ann Rheum Dis (2019) 78(3):431–3. doi: 10.1136/annrheumdis-2018-214037
222. Stock AD, Wen J, Putterman C. Neuropsychiatric lupus, the blood brain barrier, and the TWEAK/Fn14 pathway. Front Immunol (2013) 4:484. doi: 10.3389/fimmu.2013.00484
223. Cattalini M, Galli J, Andreoli L, Olivieri I, Ariaudo G, Fredi M, et al. Exploring autoimmunity in a cohort of children with genetically confirmed aicardi-goutières syndrome. J Clin Immunol (2016) 36(7):693–9. doi: 10.1007/s10875-016-0325-y
224. Fremond ML, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, et al. Efficacy of the janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol (2016) 138(6):1752–5. doi: 10.1016/j.jaci.2016.07.015
225. Konig N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tungler V, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis (2017) 76(2):468–72. doi: 10.1136/annrheumdis-2016-209841
226. Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med (2018) 5(1):e000261. doi: 10.1136/lupus-2018-000261
227. Kerrigan SA, McInnes IB. JAK inhibitors in rheumatology: Implications for paediatric syndromes? Curr Rheumatol Rep (2018) 20(12):83. doi: 10.1007/s11926-018-0792-7
228. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O'Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discovery (2017) 16(12):843–62. doi: 10.1038/nrd.2017.201
229. Kontzias A, Kotlyar A, Laurence A, Changelian P, O'Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol (2012) 12(4):464–70. doi: 10.1016/j.coph.2012.06.008
Keywords: type I interferons, cerebral interferonopathies, neurotoxin, neurodegenerative diseases, aging, multiple sclerosis, Aicardi-Goutières syndrome, traumatic brain injury
Citation: Viengkhou B and Hofer MJ (2023) Breaking down the cellular responses to type I interferon neurotoxicity in the brain. Front. Immunol. 14:1110593. doi: 10.3389/fimmu.2023.1110593
Received: 29 November 2022; Accepted: 25 January 2023;
Published: 03 February 2023.
Edited by:
Sundararajan Jayaraman, University of Illinois, United StatesReviewed by:
Anthony T. Reder, The University of Chicago, United StatesCopyright © 2023 Viengkhou and Hofer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus J. Hofer, bWFya3VzLmhvZmVyQHN5ZG5leS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.