 Jian-Yong Zhao1
Jian-Yong Zhao1 Hui Feng
Hui Feng- 1Hospital of Integrated Traditional Chinese and Western Medicine, Hebei University of Chinese Medicine, Cangzhou, Hebei, China
- 2School of Medicine, Chongqing University, Chongqing, China
- 3Department of Diseases and Infection, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
Phospholipase A and acyltransferase (PLAAT) 4 is a class II tumor suppressor with phospholipid metabolizing abilities. It was characterized in late 2000s, and has since been referred to as ‘tazarotene-induced gene 3’ (TIG3) or ‘retinoic acid receptor responder 3’ (RARRES3) as a key downstream effector of retinoic acid signaling. Two decades of research have revealed the complexity of its function and regulatory roles in suppressing tumorigenesis. However, more recent findings have also identified PLAAT4 as a key anti-microbial effector enzyme acting downstream of interferon regulatory factor 1 (IRF1) and interferons (IFNs), favoring protection from virus and parasite infections. Unveiling the molecular mechanisms underlying its action may thus open new therapeutic avenues for the treatment of both cancer and infectious diseases. Herein, we aim to summarize a brief history of PLAAT4 discovery, its transcriptional regulation, and the potential mechanisms in tumor prevention and anti-pathogen defense, and discuss potential future directions of PLAAT4 research toward the development of therapeutic approaches targeting this enzyme with pleiotropic functions.
Introduction
Vitamin A (retinol), an essential fat-soluble nutrient widely present in the diet, is vital for human health and embryonic development (1, 2). The metabolism and biological functions of vitamin A have long been studied, and even its anti-carcinogenic effect in columnar mucous epithelium of the respiratory tract was reported over half a century ago (3, 4). This led to an outburst of research enthusiasm in understanding the anti-cancer activities of retinoic acid (RA, one of the biologically active metabolites of vitamin A) and its analogs (retinoids) in the 1970s and 1980s (5). Not surprisingly, numerous RA- and retinoid-inducible genes were identified and their functions have been characterized ever since, including tazarotene (a synthetic retinoid)-induced gene 1 (TIG1) and TIG2 (6–8).
TIG3, also referred to as RA receptor responder 3 (RARRES3), retinoid-inducible gene 1 (RIG1) or H-Ras-like suppressor 4 (HRASLS4), was first identified in 1998 from tazarotene-stimulated primary human keratinocytes (9). Given the long history of research aimed at uncovering the biological functions of vitamin A metabolites, it is not surprising to identify TIG3/RARRES3 as a class II tumor suppressor that mediates the anti-proliferative effects of retinoids soon after its discovery. Since then, numerous studies have reported the connection between reduced TIG3/RARRES3 expression and tumor progression (10–13). Notably, despite being identified as a tumor suppressor, later studies provided proof-of-principle evidence for the phospholipase A1/A2 and acyltransferase activities of TIG3/RARRES3 involved in phospholipid metabolism (14, 15). These findings led to a change in gene nomenclature from TIG3/RARRES3 to phospholipase A and acyltransferase 4 (PLAAT4) that reflects the enzymatic activities associated with the TIG3/RARRES3 protein. Although each of these alternative names reflects the RA-inducible nature of PLAAT4, they have caused confusions over its annotation. For instance, RIG1 was widely used to refer to DExD/H-box helicase 58 (DDX58, also known as RIG-I), the pattern recognition receptor (PRR) that senses pathogen-associated molecular patterns (PAMPs) and triggers the rapid activation of innate immune responses. Hence, we only use the official term PLAAT4 below for clarity.
Since its discovery, over two decades of research have unveiled the molecular mechanisms underlying PLAAT4-mediated restriction of tumor development and progression. More recently, the anti-pathogen properties of PLAAT4 have come to light, expanding its focus from cancer biology to anti-pathogen defense mechanisms (16, 17). In this review, we summarize recent progress in understanding how PLAAT4 promotes tumor suppression and pathogen restriction, focusing on its biochemical properties and transcriptional regulation, and discuss potential mechanisms in tumor prevention and anti-pathogen defense.
PLAAT4 as a PLAAT family member
In early studies, PLAAT4 was shown to share significant homology with another class II tumor suppressor − H-REV 107 (most well known as PLA2G16/HRASLS3/PLAAT3 among other names) (18, 19). These two members, together with HRASLS1, HRASLS2 and HRASLS5, which were all noted to be homologous to H-REV 107, are thus referred to as the H-REV 107 subfamily proteins or HRAS-like suppressors (14, 20, 21). Interestingly, frequent changes in nomenclature have similarly occurred to all these four latter members, leading to an incredibly large number of aliases in the literature (Figure 1A). This reflects struggles in understanding the biological nature of the H-REV 107 subfamily proteins.
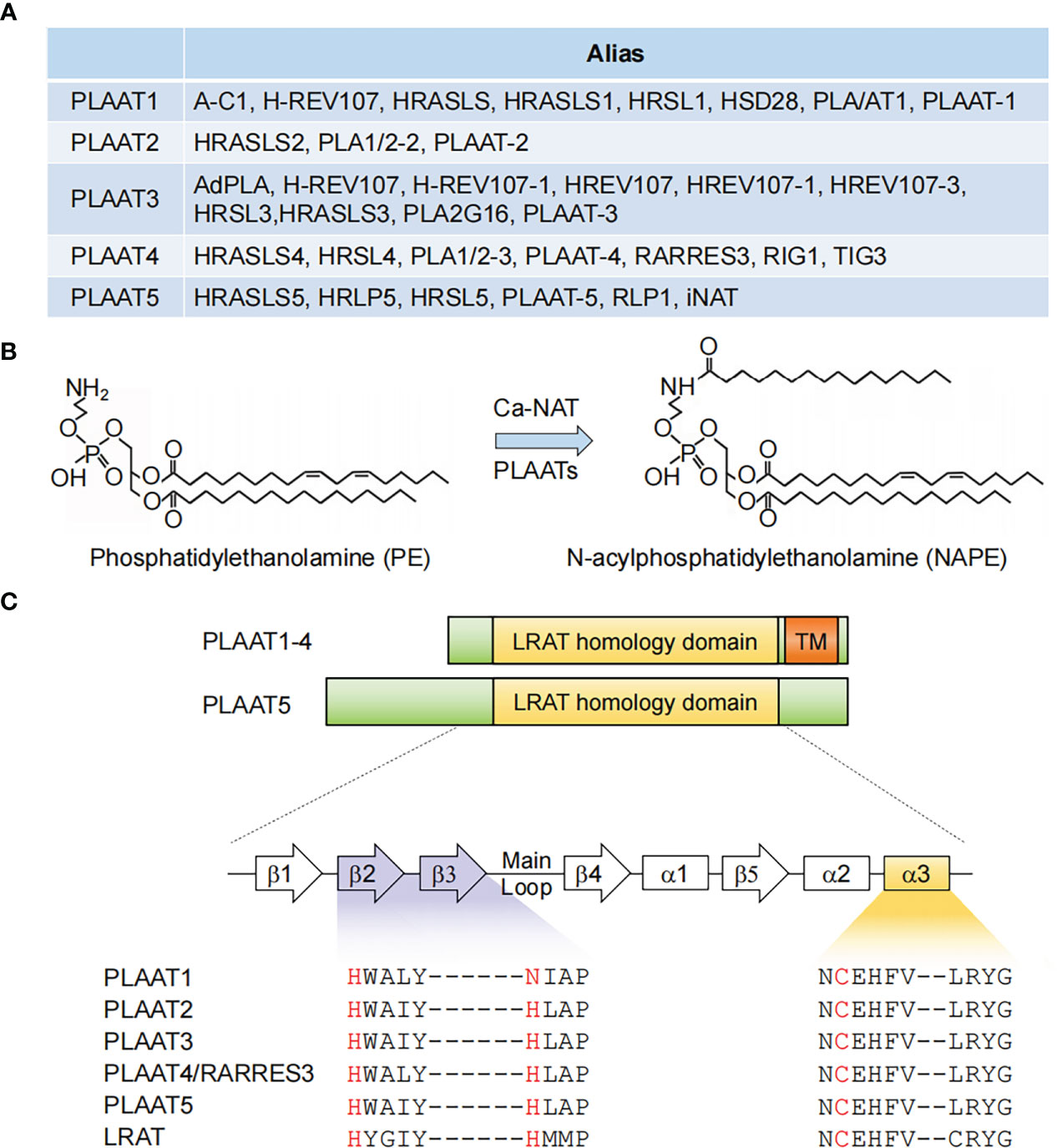
Figure 1 PLAAT family enzymes. (A) Gene names and aliases for all five PLAAT family proteins. (B) Illustration of an example of phospholipid metabolism reaction catalyzed by PLAAT family proteins. (C) Alignment of the structural domains of PLAAT family enzymes. The lecithin retinol acyltransferase (LRAT) homology and transmembrane (TM) domains are shown. The enzymatic activity of PLAAT proteins involves a cysteine-histidine-histidine triad, with the cysteine from a highly conserved ‘NCEHFV’ motif being the catalytically active site. α1−3, α helices 1-3; β1−5, β sheets 1-5.
In 2003, H-REV 107 was demonstrated to have originated from the lecithin retinol acyltransferase (LRAT) family, the representative member of vertebrate NlpC/P60 protein superfamily (22). Given that enzymes in this superfamily were known to be involved in phospholipid metabolism, the H-REV 107 subfamily proteins were subsequently identified to have in vitro Ca2+-independent phospholipase A1/2 and acyltransferase activities, and are thus capable of catalyzing several reactions in phospholipid metabolism (14, 21, 23, 24). For example, they have been shown to mediate the transfer of an acyl chain from glycerophospholipids, primarily phosphatidylcholine, to the amino group of the phosphatidylethanolamine, producing N-acylphosphatidylethanolamine (NAPE) that serves as the precursor for N-acylethanolamines (NAE) (Figure 1B). It should be noted, however, that this reaction was classically considered to be catalyzed by a Ca2+-dependent N-acyltransferase (Ca-NAT) (25–28). These seminal studies from the Ueda laboratory thus laid the foundation for renaming the H-REV 107 subfamily members as PLAAT1-5.
Functional domains of PLAAT4
The catalytic activity of PLAAT4 depends on a 134 amino-acid hydrophilic region at the N-terminus. It shares several common features with LRAT as well as all other PLAATs, among which a highly conserved ‘NCEHFV’ motif is particularly prominent, with the conserved cysteine being identified as the active site catalytic nucleophile (Figure 1C) (15). According to comparative structural analysis of the catalytic domains from PLAAT2-4, the cysteine active site is placed in close proximity to the conserved histidine from β2 strand, thereby forming a cysteine-histidine-histidine catalytic triad that requires the histidine from β3 strand (15, 29, 30). Notably, this latter histidine is replaced by an asparagine residue in PLAAT1 (Figure 1C). Although it might still be positioned in a similar manner, it is tempting to anticipate that this subtle change may affect the substrate specificity and/or balance between the phospholipase A and acyltransferase activities of PLAAT1. Importantly, similar catalytic triad (i.e. cysteine-histidine-a polar residue) was also identified in the NlpC/P60 superfamily, indicating an evolutionary conserved attribute of these enzymes (22). In addition to the catalytic triad residues, structural evidence suggested that the highly flexible loop region between β3 and β4 strands adds fundamental alterations in the enzymatic activities of different PLAATs (30, 31). This is consistent with the structural context of the cysteine active site embedded in the hydrophobic pocket formed by the extended loops between strands β1 and β2, β3 and β4, and the N-terminal α3 helix (15).
PLAAT4 has a 30 amino-acid hydrophobic transmembrane domain following the N-terminal catalytic domain (Figure 1C). It encodes a single transmembrane-spanning segment, thereby directing PLAAT4 to cell membranes including plasma membrane, endoplasmic reticulum and Golgi apparatus (32, 33). Despite little sequence homology, this feature is shared by PLAAT1-3 but not PLAAT5, whose enzymatic activity is detected mainly in the cytosolic fraction (Figure 1C) (21, 23). Moreover, this transmembrane domain is not just an anchor tethering PLAAT4 to distinct membrane compartments, but itself is indispensable for an optimal biological function (34). For instance, it was demonstrated that the pro-apoptotic and anti-cancer activities of PLAAT4 are attributed primarily to the Golgi- rather than the endoplasmic reticulum-associated protein form (33). PLAAT4 at the plasma membrane, by contrast, was shown to facilitate the terminal stages in keratinocyte differentiation through interacting with type I transglutaminase (32, 35, 36).
Apart from the membrane system, PLAAT4 also distributes at the centrosome in skin cancer cells, leading to pericentrosomal organelle accumulation which in turn drives cancer cell apoptosis (24). A 24-amino acid segment (amino acids 102-125) that spans the β-sheet and α-helix immediately upstream of the hydrophobic tail was shown to be critical for the centrosome-targeting of PLAAT4 (37, 38). While the molecular mechanism underlying PLAAT4 translocation remains to be elucidated, a recent study showed that both zebrafish Plaat1 and murine PLAAT3 in the eye lens translocate from the cytosol to diverse organelles, which ultimately induces complete degradation of organelle membranes to achieve an optimal transparency of the lens (39). Likewise, cytosolic PLAAT3 in Hela cells can also translocate to endo-lysosomes following picornavirus infection, facilitating genome delivery into the cytoplasm from the micropores in endo-lysosomal membranes (40). Intriguingly, the translocation of Plaat1 and PLAAT3 both requires their C-terminal hydrophobic domain and is triggered by the membrane damage in organelle membranes. Therefore, it is highly plausible that both the hydrophobic tail itself and the upstream short-segment could serve as the recruitment cues, directing PLAAT4 towards target organelles where it acts as an acyltransferase and associates with other proteins for initiating signaling cascades.
Transcriptional regulation of PLAAT4
The PLAAT4 gene is expressed ubiquitously in human cells, implying a tissue-wide protective role of its product. In addition, PLAAT4 transcription is readily induced by retinoic acids and its natural and synthetic analogues, DNA-damaging stimuli, as well as interferons (IFNs), the secreted cytokines that orchestrate the induction of IFN-stimulated genes (ISGs) to establish an anti-pathogen state (discussed below) (16, 41, 42). Whether basally expressed or induced in response to different stimuli, the transcriptional activation of PLAAT4 involves a high degree of selectivity. It is tightly controlled by transcription factors that specifically recognize and bind to functional cis-elements in its promoter, and this is essential for understanding the biological importance and complexity of PLAAT4 during pathogenesis (Figure 2). Hence, we summarize how transcription of PLAAT4 is regulated under different biological contexts in the following subsections.
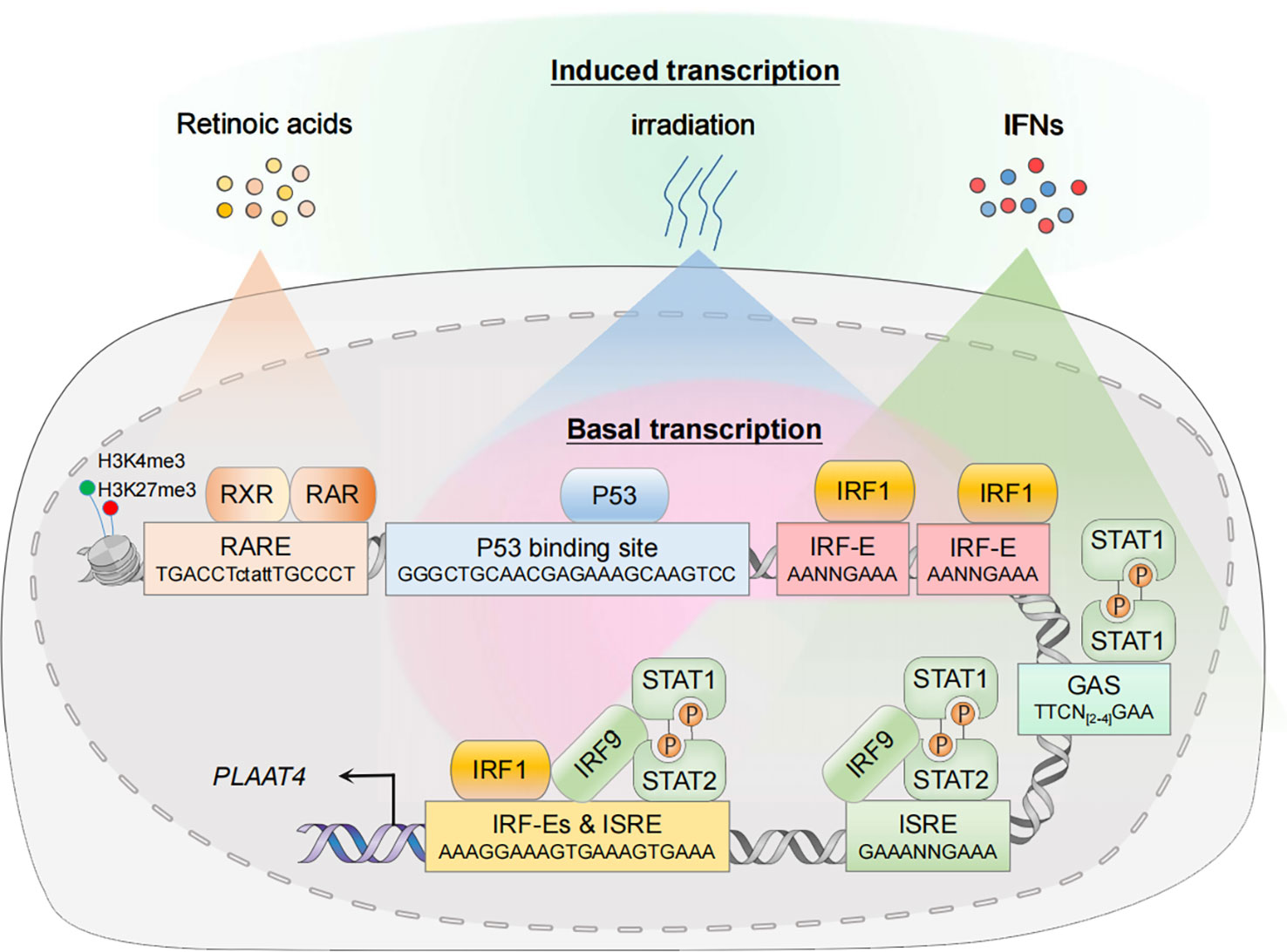
Figure 2 Transcriptional regulation of PLAAT4. In the resting state, nuclear transcription factors p53 and IRF1 mediate constitutive expression of PLAAT4. In the presence of agonists or acute stress, the corresponding signal transduction pathways are activated, thus leading to an enhanced PLAAT4 transcription. Epigenetic modifications such as histone methylation also contribute to dynamic regulation of chromatin states, affecting binding of the RXR-RAR heterodimer and other transcription factors. RXR, retinoid X receptor; RAR, retinoic acid receptor; RARE, retinoic acid response element; p53-RE, p53 response element; IRF-E, IRF-binding element; ISRE, IFN-stimulated response element; GAS, gamma-activated sequence.
Basal PLAAT4 expression and its repression during tumorigenesis
The PLAAT4 promoter contains the binding sites for transcription factors that constitutively activate PLAAT4 transcription to exert a protective role at the basal level (16, 43). For example, a functional p53 response element (p53RE, -5157 to -5134 relative to the translation start site) exists in the PLAAT4 promoter (44). Hence it is not surprising that p53, one of the best-characterized tumor suppressors expressed constitutively at low basal levels in unstressed cells, has the capacity to mediate PLAAT4 promoter activation. Apart from p53, interferon regulatory factor 1 (IRF1) is also present at low basal levels in the nucleus where it manifests anti-oncogenic and anti-pathogen activities through maintaining optimal constitutive expression of hundreds of target genes, including PLAAT4 (16, 45). Indeed, the PLAAT4 promoter contains several IRF-binding elements (IRF-E), the hexanucleotide units with a core ‘GAAA’ sequence which are recognized specifically by the N-terminal DNA-binding domains from all IRF family members (46–48). However, it remains to be determined which IRF-E(s) are critical for IRF1-mediated PLAAT4 transcriptional activation.
It has long been noted that at the heart of tumorigenesis and progression lies genetic mutations disrupting the harmonious checks and balances of normal cellular growth and development. In this context, TP53 (encoding wild-type p53) is among the most frequently mutated genes in cancer, and the majority of tumor-derived mutations occurs in the region encoding DNA binding domain of p53 (49). This results in a diminished or even completely abolished activity to transactivate p53 target genes, including class II tumor suppressors like CAV1, ING1b, SERPINB5, etc. (50–54). Likewise, it was shown that a common abnormality in human leukemia and myelodysplasia is an interstitial deletion mapped to chromosome 5q.31, and IRF1 is the only gene deleted consistently at one or both alleles within this region (55). Consistently, a decrease in PLAAT4 transcript levels has been widely noted in various types of tumors (11, 12, 56–58). It is noteworthy that the loss of IRF1 alleles per se does not lead to spontaneous tumor development (59). Rather, it favors a dramatic exacerbation of pre-existing genetic predispositions associated with other risk factors (e.g. TP53 nullizygosity). This does not necessarily suggest that p53 plays a dominant role in driving PLAAT4 transcription. In fact, PLAAT4 is abundantly expressed in T antigen-transformed cell lines in which p53 function is inactivated. Hence, while both IRF1 and p53 are key regulators of PLAAT4 transcription, p53 likely activates a much broader anti-tumor transcriptional spectrum than IRF1.
RA- and retinoid-induced PLAAT4 expression and its regulation
Expression of PLAAT4 is markedly induced by RA and its analogues, the biological effects of which are transduced through RA receptors (RARs) and retinoid X receptors (RXRs) as RAR-RXR heterodimers (60–63). They belong to the superfamily of nuclear receptors with ligand-dependent transcriptional activities, undergoing a shift from the repressive unliganded state towards its liganded active form that involves the exchange of co-repressors for co-activators, and could then drive the transcription of target genes via binding to a series of RA response elements (RAREs) that typically consist of hexameric direct repeats of (A/G)G(T/G)TCA with either a two (DR2) or five (DR5) nucleotide spacer (64–67). In agreement with the observation that PLAAT4 transcription is enhanced by both RAR- and RXR-selective agonists, a functional DR5-type RARE was identified in the PLAAT4 promoter (-5259 to -5243 relative to the translational start point) (10, 41). Interestingly, although this RARE is a non-canonical DR5-type element, it contains several conserved nucleotides which could form sequence-specific and water-mediated base contacts with residues Lys156 and Arg161 for RXR, and Lys109 for RAR (64, 68).
PLAAT4 expression in response to cellular DNA damage
In addition to being induced by RA and its analogs, it was shown that the expression of PLAAT4 transcript and its protein product in human hepatoma HepG2 cells is robustly stimulated by DNA-damaging agents including 5-fluorouracil and UV irradiation (44). This is not surprising given that p53 and IRF1, besides acting at low basal levels, have been known as central hubs whose expression (i.e. transcription-coupled translation) is induced rapidly by various types of cellular stressors, resulting in either the repair or elimination of damaged cells by activating the expression of target genes (69, 70). DNA damage-induced PLAAT4 response is also consistent with the finding that tumor suppressor genes are activated prior to apoptosis effector genes as the levels of these two transcriptional factors increase (71). Interestingly, however, the PLAAT4 expression in HepG2 cells is not triggered by 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), a persistent and ubiquitous environmental contaminant that induces oxidative stress and DNA damage (44, 72). This may be due to the counteraction effect of TCDD on p53 response via enhancing the expression of many other genes (73, 74). For example, TCDD was shown to enhance the protein levels of murine double minute-2 (MDM2), a multifunctional E3 ubiquitin ligase which specifically counteracts p53 function by binding to its transcriptional activation domain, as well as by targeting p53 for ubiquitination and subsequent proteasome-dependent degradation (75–78). In fact, a notable property of p53 and IRF1 is their flexibility to act coordinately with a variety of other co-factors (p63, RELA, etc.), allowing for a dynamic fine-tuning of target gene expression (79–82). Importantly, although p53 and IRF1 tend to act cooperatively in driving the expression of common downstream targets under such circumstance (e.g. CDKN1A, a.k.a. P21), it should be noted that p53 plays an absolutely dominant role in activating PLAAT4 transcription in HepG2 cells treated with DNA damage agents (44, 83, 84).
PLAAT4 expression during host anti-pathogen responses
Being originally identified as a tumor suppressor, it is somewhat surprising that PLAAT4 is also one of the common downstream targets of IRF1 and IFN signals (16, 17, 85). IFN-dependent innate immunity is critical for host defense against invading pathogens. Cells engaged in such immune responses undergo sophisticated signal transduction that originates at the recognition of PAMPs by PRRs, leading to a rapid transcriptional activation of different types of IFNs and inflammatory cytokines. Secreted IFNs then initiate downstream Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling via their cognate receptors, resulting in a second wave of transcriptional induction of hundreds of IFN-stimulated genes (ISGs) that depend mainly on the specific binding of IFN-stimulated gene factor 3 (ISGF3) heterotrimeric complex and STAT1 homodimer (known as IFN gamma activating factor; i.e. GAF) to IFN-stimulated response element (ISRE) and gamma-activated sequence (GAS), respectively (86–88). In agreement with the findings that PLAAT4 functions as an ISG, both of these cis-elements exist in the PLAAT4 promoter (Figure 2) (17). Notably, IRF1 was initially identified from crude nuclear extracts of Newcastle disease virus-infected mouse L929 cells, where it strongly activates transcription of the genes encoding type I IFNs (89–92). Moreover, IRF1 itself is also highly responsive to IFNs, especially type II IFN (IFNγ) (42). In this regard, IRF1 can induce much more robust and long-lasting expression of PLAAT4 (IFN-independent and IFNγ-dependent) than any other types of IFN during host innate immunity. Given that IRF1 can act in concert with STAT1 to induce ISG expression in IFN-stimulated cells, it is also reasonable to anticipate an enhancement of IFN-dependent PLAAT4 expression by additional IRF1 binding (93, 94).
In addition to the classical role in cellular stress response, it is now widely appreciated that p53 plays essential roles during pathogen infections, in either an IFN-dependent or independent manner (95–97). Accordingly, pathogens have evolved sophisticated strategies to manipulate the p53 checkpoint for their own advantage. For instance, inhibition of p53 has been widely observed in the realm of viruses, bacteria as well as parasites (96, 98–101). While many studies are conducted in a p53-deficient background using the T antigen-immortalized and cancer cell lines, it is plausible to infer that PLAA4 transcription may also be affected indirectly via p53 inhibition during pathogen infection.
Mechanisms of PLAAT4 as a tumor suppressor
As a class II tumor suppressor, PLAAT4 that is found abundantly in normal tissues has been noted to favor differentiation and apoptosis but inhibit cell proliferation and attenuates tumor growth (9, 32, 33, 102–104). As our understanding of the tumorigenic process has grown significantly, many mechanistic aspects of the tumor-suppressing activity of PLAAT4 have become clear during the past two decades (Figure 3A). In normal keratinocyte, for example, PLAAT4 interacts with type I transglutaminase at the plasma membrane, resulting in a substantial increase in the proportion of sub-G1 cells as well as nuclear shrinkage, hence forming unique structures that resemble the cornified envelope close to the cell surface (32, 35, 107). It then suppresses cell proliferation and induces a shift toward terminal keratinocyte differentiation, the ultimate outcome of which is cell death (108). Follow-up studies with epidermal squamous cancer cells suggested that PLAAT4 is also important for limiting cancer cell proliferation. Notably, PLAAT4 in these cells is largely localized near the centrosome, and thus could inhibit centrosome separation during mitosis, leading to the induction of cancer cell apoptosis (37, 109). These observations indicate that the expression levels of PLAAT4 are the determinant for modulating the balance between cell proliferation and survival/death. However, it should be noted that, while PLAAT4 promotes normal keratinocyte death via enhancing transglutaminase activity, the pericentrosomal localization of PLAAT4 in cancer cells drives organelle accumulation which in turn triggers caspase-dependent apoptosis (32, 109).
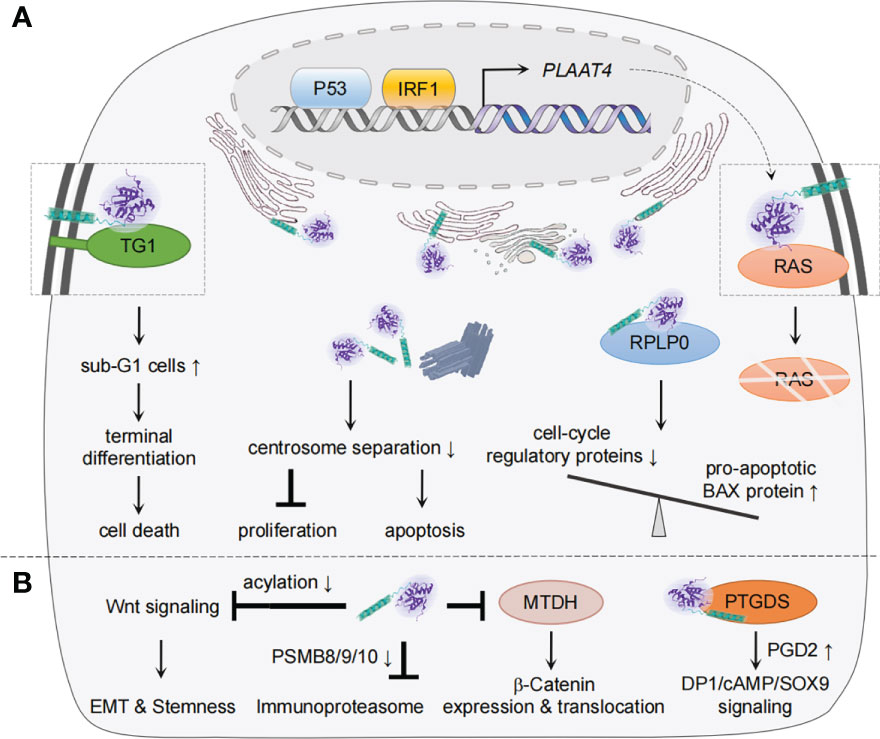
Figure 3 Mechanisms of PLAAT4 action during tumorigenesis and metastasis. (A) Nuclear p53 and IRF1 drive the basal expression of PLAAT4, which acts to regulate differentiation, proliferation, apoptosis, etc. to suppress tumorigenesis. The subcellular locations of PLAAT4 are shown, with a PLAAT4 structure predicted with alphaFold (105, 106). (B) PLAAT4 contributes to inhibition of epithelial-mesenchymal transition (EMT), cancer cell stemness, immunoproteasome, and β-Catenin expression and translocation, but induces prostaglandin D2 (PGD2)-mediated activation of DP1/cAMP/SOX9 signaling to suppress metastasis. For protein interactions where the subcellular location has not been identified, the illustration only depicts a physical interaction between PLAAT4 and target proteins.
Interestingly, PLAAT4 appears to inhibit proliferation and promote apoptosis via different mechanisms in other types of cancer cells. For instance, it was suggested that PLAAT4 could form a protein complex with the ribosomal protein P0 (RPLP0) in HtTA cervical cancer cells (110). This is associated with a decreased RPLP0 level, resulting in a reduction in cell-cycle regulatory proteins but an enhancement in the pro-apoptotic BAX. PLAAT4 in HtTA cervical cancer cells was also shown to interact with the RAS, a plasma membrane-associated GTPase that regulates proliferation, differentiation and apoptosis by serving as binary switches (33, 111–113). The association between PLAAT4 and RAS not just alters the subcellular distribution of RAS, but also facilitates its degradation, thus leading to a decreased activation of RAS and downstream signaling. A recent study performed in a hepatocellular carcinoma model further supported this notion (58).
Besides being identified as a tumor suppressor, there is growing evidence suggesting that PLAAT4 also plays crucial roles in restricting tumor metastasis, the main cause of cancer-related death (Figure 3B) (13, 114–116). In this regard, PLAAT4 is among the metastasis-associated gene signatures whose expression levels in primary breast tumors inversely correlate with the frequency of lung metastasis (117). Follow-up studies not only confirmed this observation, but unveiled the functional role of the catalytic activity of PLAAT4 in suppressing the lung metastasis (13, 118). Interestingly, while PLAAT4 dampens initial steps in the lung colonization through enforcing the retention of a phospholipase A1/2 activity-dependent differentiation features, it could also serve as an acyl protein thioesterase that hydrolyzes the acyl chains of Wnt proteins and a co-receptor in the canonical Wnt signaling to induce low density lipoprotein receptor protein 6 (LRP6). This contributes to the blockade of Wnt/β-catenin signaling, whereby suppressing epithelial-mesenchymal transition (EMT) and stem cell properties of tumor cells (114, 118). It is also noteworthy that PLAAT4 is expressed at significantly lower levels in steroid hormone receptors-positive (estrogen receptor-, progesterone receptor- and estrogen/progesterone receptors-positive (ER+, PR+ and ER+/PR+)) tissues than in ER-, PR- and ER/PR-negative tissues, hence providing a biomarker to identify a subgroup of patients with higher susceptibility to lung metastasis (103). In addition, a study reported the inhibitory effect of endogenous PLAAT4 on expression of the immunoproteasome subunits (PSMB8/9/10) in breast cancer cells (116). Although immunoproteasomes are most well-known for its role in antigen presentation, knocking down expression of its core subunit PSMB8 profoundly inhibits the migration and invasion of tumor cells (119–121). Hence, PLAAT4 could also indirectly suppress the distant metastasis of breast tumor cells by down-regulating PSMB8/9/10 expression (116).
In addition to the role in inhibiting breast tumor metastasis, PLAAT4 was shown to be capable of sequestering the oncoprotein metadherin (MTDH, a.k.a. AEG-1) that is involved in the development of various types of tumors, preventing MTDH from activating the cytoplasm-nuclear translocation of β-Catenin, whereby leading to suppression of the metastasis of colorectal cancer (115, 122, 123). The role of PLAAT4 in mediating cell migration and invasion was similarly demonstrated in NT2/D1 testicular cancer cells (124). Mechanistically, PLAAT4 physically interacts with glycoprotein prostaglandin D2 synthase (PTGDS, a.k.a. L-PGDS), a member of the lipocalin superfamily that has been shown to be involved in the tumorigenesis of solid tumors (125). It then promotes prostaglandin D2 (PGD2) production, resulting in the activation of PGD2 DP1 receptor/cAMP/SOX9 signaling. Notably, although an intact hydrophobic domain of PLAAT4 is critical for its interaction with PTGDS, the exact location for PLAAT4-PTGDS interaction remains unknown. It is interesting to note that H-REV107/PLA2G16/PLAAT3, a representative member of the PLAAT family, also inhibits migration and invasion of NT2/D1 testicular cancer cells by targeting at PTGDS (126).
Although a significant number of studies mentioned above have demonstrated PLAAT4 as a tumor suppressor, it remains to be determined whether its action generally requires the catalytic activity of PLAAT4, and how its catalytic products regulate the cellular events. Moreover, results from a recent study highlighted a positive correlation between PLAAT4 expression and the glioma grade, indicating its potential as a prognostic marker for poor survival (127). Molecular mechanisms responsible for this paradox remain unexplained, but such a phenomenon was also observed within the tumor microenvironment (a major barrier to immunotherapy) that induces the production of RA (128, 129). Despite being long considered as an anti-cancer agent, RA in solid tumors was found to display tumorigenic capability via myeloid-mediated immune suppression. Mechanistically, it polarizes intratumoral monocyte differentiation away from immunostimulatory dendritic cells but toward tumor-associated macrophages through suppressing dendritic cell-promoting transcription factor IRF4 (129).
Mechanisms of PLAAT4 during anti-pathogen restriction
Despite the fact that IFN and IRF1 pathways have long been considered to be highly effective at resisting and controlling pathogen infections, their common target PLAAT4 has not been recognized as a restriction factor for any pathogen until recently (Figure 4) (45, 130–133). For example, PLAAT4 was identified as the most downregulated gene in IRF1-depleted human hepatocytes, and the most active one in limiting replication of hepatitis A virus (HAV), a notoriously stealthy picornavirus causing acute hepatitis in humans (16, 92, 134). Nevertheless, this robust antiviral activity of PLAAT4 seems virus-specific, as knocking down its expression only resulted in a modest (2- and 5-fold) enhancement in replication of dengue and Zika viruses (mosquito-borne flaviviruses causing dengue fever and Zika virus disease), and had no impact on replication of human rhinovirus (a picornavirus causing a variety of respiratory diseases) and hepatitis C virus (a hepatotropic flavivirus causing chronic hepatitis) (16). Likewise, PLAAT4 was also identified as an ISG that induces premature egress by reducing parasitic vacuole size, and thus actively restricts infection of the type III but not type I/II strains of Toxoplasma gondii, a parasite of warm-blooded animals that infects an estimated one-third of people worldwide (17, 135). Seemingly paradoxically, PLAAT4 as an effector of IRF1/IFNs does not affect or depend on the canonical innate immune signaling; rather, it directly restricts pathogen infections via the acyltransferase activity in both cases (16, 17). Considering the importance of innate immune responses in fighting against pathogen invasion, this may help to explain, at least partially, why anti-pathogen activity of PLAAT4 has long been neglected and why it is evident in a limited number of pathogens, especially those associated with mild or asymptomatic disease (e.g. HAV and type III T. gonidii) (134, 136). The anti-pathogen activity of PLAAT4 might also be related to degradation of specific host membranes through its phospholipase activity, a function that resembles PLAAT3 in the eye lens (39). Thus, identifying the selectivity of PLAAT4 would reveal the type of cellular organelle membranes specifically targeted by different pathogens for productive infection.
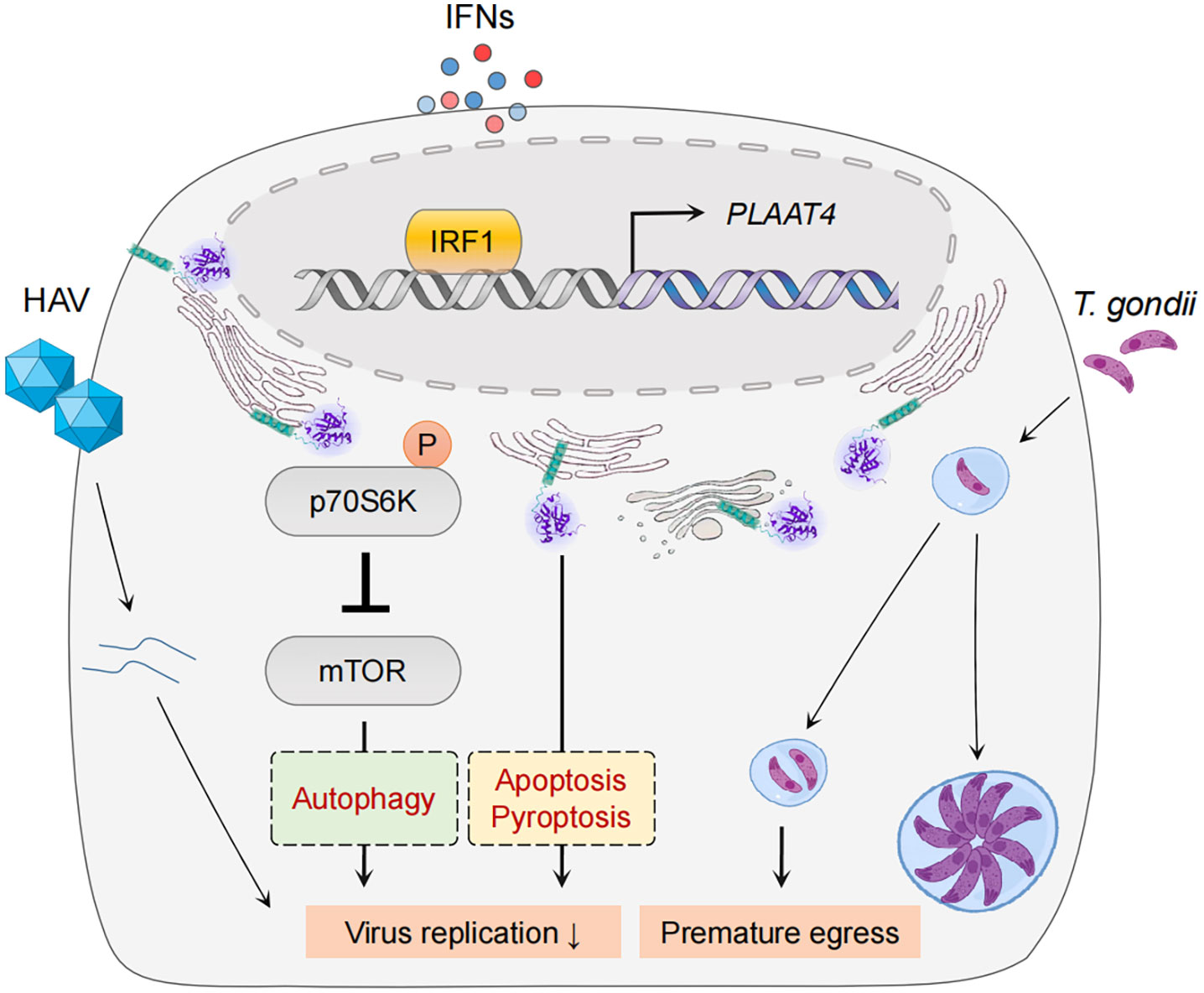
Figure 4 PLAAT4-mediated pathogen restriction. IRF1 and IFNs mediate expression of PLAAT4, which acts through in part by inactivating the mTOR signaling pathway to restrict hepatitis A virus replication (left), and induces premature egress to combat Toxoplasma gondii infection (right). PLAAT4-dependent resistance to viral replication might involve autophagy, pyroptosis and apoptosis (shown in dashed boxes).
It is currently unknown the detailed mechanisms underlying anti-pathogen activity of PLAAT4. Nevertheless, the catalytic activity of PLAAT4 mediates induction of p70 S6 kinase phosphorylation at Thr389 and subsequent phosphorylation of mechanistic/mammalian target of rapamycin (mTOR) at Ser2448 that inactivates mTOR activity to phosphorylate 4E-BP1 in immortalized human hepatocytes (16). The functional importance of mTOR inhibition in restricting HAV replication was further confirmed by the observation that mTOR inhibitors fully recapitulate the HAV-specific suppression by PLAAT4 (16). Since pharmacologic mTOR inhibition in various cell types induces autophagy, a major digestion process that removes damaged (in this case infected) substrates, it is possible that PLAAT4 suppresses HAV replication indirectly through autophagy (137–139). Interestingly, results from a recent study confirmed that activation of mTOR signaling and in turn a suppressed autophagy are required for Zika virus replication (140). Therefore, our observations regarding the role of PLAAT4 in restricting replication of Zika virus might also be due to its ability to induce mTOR-dependent autophagy. However, it is important to note that Zika virus NS4A and NS4B proteins cooperatively suppress the mTOR pathway (141). It thus is tempting to speculate that Zika virus might have evolved to overcome mTOR-dependent autophagy inhibition; and consequently, this might help explain the limited anti-pathogen activity of PLAAT4 (142). For instance, mTOR as one of the most important regulators plays a significant role in orchestrating several cell processes including apoptosis (143). Notably, pathogen infections could directly stimulate mTOR activation, regulating expression of apoptosis-related proteins (e.g. inducing anti-apoptotic Bcl2 but suppressing pro-apoptotic Bax proteins), and then leading to the survival of infected cells (144, 145). Hence, it is reasonable to anticipate that PLAAT4 is capable of counteracting such a process by inhibiting mTOR activation. In this context, further studies are needed to dissect whether PLAAT4, especially its catalytic activity, can regulate mTOR-dependent autophagy and/or apoptosis to restrict pathogen infections. Moreover, as we discussed earlier, PLAAT4 expression during pathogen infections is under the concerted control of p53, IRF1 and IFNs, which all display pleiotropic effects on diverse cellular pathways other than mTOR signaling. For example, they have been demonstrated to play an important role in inducing apoptosis and/or pyroptosis (146–152). While our study has shown that PLAAT4-mediated suppression of HAV does not involve cell death (16), it remains to be determined whether mTOR-independent signaling pathways (e.g. apoptosis and pyroptosis, etc) are involved in the restriction of other pathogens.
Intriguingly, it should be pointed out that PLAAT4 is not the only PLAAT family member that has been demonstrated to affect the virus life cycle. For example, two independent genome-wide screens have identified H-REV 107/PLA2G16/PLAAT3 as a pivotal host factor for picornaviruses including poliovirus, rhinovirus, enterovirus, encephalomyocarditis virus and Saffold virus (40, 153). Mechanistically, the catalytically active PLAAT3 functions at a post-entry step, competing with galectin-8 and thus facilitating the delivery of viral genome to the cytoplasm which enables viral protein synthesis and subsequent RNA replication. Otherwise the virions will proceed to a pore-activated autophagic degradation pathway, enabling virus clearance procedure (40, 154). Besides serving as the switch between viral genome delivery and clearance, PLAAT3 also confers a selective cytotoxicity effect of the host cells exposed to rhinovirus infection, and this function depends on its C-terminal domain that extends into the endosomal lumen (153). In sharp contrast, PLAAT3 restricts hepatitis A virus infection instead of preventing viral genome from autophagy degradation (155). It is currently uncertain how PLAAT3 elicits opposing roles on different picornaviruses. However, it is important to note that despite HAV belongs to the Picornaviridae family, it distinguishes itself from other mammalian picornaviruses in the capsid structure, genome organization and replication cycle (156–159).
Can PLAAT4 be an ideal therapeutic target?
A ubiquitous presence of PLAAT4 and its effector functions in tumor suppression and pathogen restriction suggest it might represent a self-protecting mechanism. In particular, PLAAT4 often declines in various types of tumor cells and tissues, but unlike the upstream transcriptional factors (e.g. p53 and IRF1), it is rarely mutated or deleted (9, 160). These characteristics of PLAAT4 make itself an attractive target for pharmacologic intervention to treat or palliate tumor-related symptoms, as well as to fight against pathogen infections.
In fact, the practical applications of vitamin A and its metabolites in anti-cancer therapy have been one of the most popular subjects (161–163). For example, all-trans retinoic acid (ATRA), the biologically active form of vitamin A and one of the first molecularly targeted drugs in oncology, has successfully eliminated fatal acute promyelocytic leukemia in cooperation with arsenic trioxide by targeting the prolyl isomerase Pin1 for degradation, and it is currently being evaluated for the feasibility in treating pancreatic cancer (164, 165). Notably, PLAAT4 exhibits a time- and concentration-dependent expression pattern in response to ATRA, similar to IFNs that also play functional roles in anti-cancer immunity (10, 57, 166–168). Hence, therapies that target PLAAT4 for increased gene expression (e.g. uptake of vitamin A and its metabolites) might favor both pathogen restriction and tumor suppression, particularly for patients with p53- and/or IRF1-deficient cancers.
Being shared by anti-tumor and anti-pathogen responses, pathogen-triggered PLAAT4 expression may also offer potential to suppress tumorigenesis and development. This is reminiscent of applying microbial agents to treat malignant disease (169, 170). However, it is noteworthy that pathogenic microbial agents themselves are complicated, with some of them being well known to increase cancer incidence (e.g. human T lymphotropic virus type-1 oncovirus) (171, 172). Furthermore, although pathogen infections in general are expected to induce specific anti-tumor responses, a significant hurdle is that viral antigens often cause immunosuppressive effects that promote escape from the host immunity surveillance, thereby adding another layer of risk to the pathogen-triggered anti-tumor strategy (81, 92, 159, 171, 173). To bypass these impediments, live-attenuated or inactivated pathogens might be a suitable option for pathogen-initiated anti-tumor therapy (174–176).
It is important to note that PLAAT4 is just one of the hundreds of target genes induced by biological stimuli, and only a relatively small proportion of the gene products have been explored for their biological functions (133, 177–179). Although most of these effectors might not be potent enough to elicit tumor suppression and/or pathogen restriction when they act individually, they could mount an effective defense against a much wider spectrum of disease when induced as a combination with other effectors. This is exemplified by our findings that the antiviral effect of PLAAT4 is effectively amplified by contributions from other IRF1-effector gene products (e.g. NMI, MX1, ERAP2) with different mechanisms of action. However, the strategy to enhance the immune response is a double-edged sword that could potentially pose serious threats to the host if excessive and uncontrolled activation led to autoimmune-like disease. Thus, it would be necessary to identify other molecular targets that could act in synergy with PLAAT4 without adverse events associated with uncontrolled immune responses.
An alternative option would be to target specific expression of PLAAT4 or augment its phospholipase activity using small compounds, even though this sort of approaches may only partially restore the functions of its upstream agonists or transcriptional factors (16, 17, 57). Nonetheless, by identifying agonist-activated effectors that could target specific pathogens and cancers, host-directed therapeutic approaches that pinpoint such molecules would provide solid basis for more effective and safer therapies.
Closing marks
As a member of the class II tumor suppressor gene family, PLAAT4 exhibits phospholipase A1/2 and acyltransferase activities with pleiotropic effects that profoundly affect host immunity against tumors and pathogens. As summarized in this review, substantial advances have been made in understanding the regulation of its expression, functional consequences and the mechanisms underlying its inhibitory effects on tumorigenesis and pathogens. It remains to be determined how PLAAT4 responses affect regulatory circuits with multiple feedback loops to control upstream signals involving RA metabolism and tumor suppressor (p53 and IRF1) and anti-pathogen (IFNs and IRF1) functions, thereby reinforcing both stress response and immunity. Given the numerous interacting proteins identified with PLAAT4, it could serve as a potential multiplex switch, fine-tuning key pathways to ensure an adequate response, or otherwise triggering signaling processes that favor apoptotic cell death.
Since it has just begun to appreciate the roles for PLAAT4 in the restriction of tumors and pathogen infections, several important gaps have remained in terms of mechanistic understanding of its function. For example, are there any other contexts where a PLAAT4 transcription program is activated? What signals stimulate translocation of the membrane-localized PLAAT4 to other subcellular sites (e.g. centrosome) to transduce functional signals? What are the specific functions of endoplasmic reticulum-localized PLAAT4? Whether PLAAT4 regulates mTOR-dependent and independent autophagy and/or apoptosis in pathogen-infected cells? Understanding a comprehensive picture of PLAAT4 would open new avenues of PLAAT4 regulome that could impact biological events other than protection against cancers and pathogens. In addition to PLAAT4 itself, it would be important to clarify how its metabolite NAPE and changes in the host lipidome regulate the protein-protein interactions. It also remains to be determined whether PLAAT family proteins with similar biochemical features play redundant roles in protection from both invading pathogens and tumorigenesis. This is particularly important in understanding what cellular mechanisms may compensate for the lack of PLAAT4 orthologs in rodents.
In summary, PLAAT4 research, while still in its infancy, has just begun to be emerged as an important field with relevance to host immunity to infection and disease progression. Although more investigations are required to answer above questions, further understanding of the molecular details of the PLAAT4 action would contribute to the development of more effective therapeutic approaches to treat tumors and pathogen infections.
Author contributions
All authors contributed to conception of the topic. HF and J-YZ wrote the original paper and prepared the figures. DY provided intellectual input and wrote the paper. X-KY, R-ZL, L-XW and WG provided intellectual input. All authors contributed to the article and approved the submitted version.
Funding
Chongqing University (02270011080002); Japan Society for the Promotion of Science (JP21H02746); National Natural Science Foundation of China (81772061).
Acknowledgments
We thank alphaFold for predicted PLAAT4 structure.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Niederreither K, Subbarayan V, Dolle P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet (1999) 21:444–8. doi: 10.1038/7788
2. Carazo A, Macakova K, Matousova K, Krcmova LK, Protti M, Mladenka P. Vitamin a update: Forms, sources, kinetics, detection, function, deficiency, therapeutic use and toxicity. Nutrients (2021) 13. doi: 10.3390/nu13051703
3. Wolbach SB, Howe PR. Tissue changes following deprivation of fat-soluble a vitamin. J Exp Med (1925) 42:753–77. doi: 10.1084/jem.42.6.753
4. Saffiotti U, Montesano R, Sellakumar AR, Borg SA. Experimental cancer of the lung. inhibition by vitamin a of the induction of tracheobronchial squamous metaplasia and squamous cell tumors. Cancer (1967) 20:857–64. doi: 10.1002/1097-0142(1967)20:5<857::AID-CNCR2820200545>3.0.CO;2-3
5. Wolf G. A history of vitamin a and retinoids. FASEB J (1996) 10:1102–7. doi: 10.1096/fasebj.10.9.8801174
6. Nagpal S, Patel S, Asano AT, Johnson AT, Duvic M, Chandraratna RA. Tazarotene-induced gene 1 (TIG1), a novel retinoic acid receptor-responsive gene in skin. J Invest Dermatol (1996) 106:269–74. doi: 10.1111/1523-1747.ep12340668
7. Nagpal S, Patel S, Jacobe H, Disepio D, Ghosn C, Malhotra M, et al. Tazarotene-induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. J Invest Dermatol (1997) 109:91–5. doi: 10.1111/1523-1747.ep12276660
8. Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res (2002) 43:1773–808. doi: 10.1194/jlr.R100015-JLR200
9. Disepio D, Ghosn C, Eckert RL, Deucher A, Robinson N, Duvic M, et al. Identification and characterization of a retinoid-induced class II tumor suppressor/growth regulatory gene. Proc Natl Acad Sci U.S.A. (1998) 95:14811–5. doi: 10.1073/pnas.95.25.14811
10. Huang SL, Shyu RY, Yeh MY, Jiang SY. Cloning and characterization of a novel retinoid-inducible gene 1(RIG1) deriving from human gastric cancer cells. Mol Cell Endocrinol (2000) 159:15–24. doi: 10.1016/S0303-7207(99)00207-5
11. Casanova B, de la Fuente MT, Garcia-Gila M, Sanz L, Silva A, Garcia-Marco JA, et al. The class II tumor-suppressor gene RARRES3 is expressed in b cell lymphocytic leukemias and down-regulated with disease progression. Leukemia (2001) 15:1521–6. doi: 10.1038/sj.leu.2402243
12. Jiang SY, Chou JM, Leu FJ, Hsu YY, Shih YL, Yu JC, et al. Decreased expression of type II tumor suppressor gene RARRES3 in tissues of hepatocellular carcinoma and cholangiocarcinoma. World J Gastroenterol (2005) 11:948–53. doi: 10.3748/wjg.v11.i7.948
13. Morales M, Arenas EJ, Urosevic J, Guiu M, Fernandez E, Planet E, et al. RARRES3 suppresses breast cancer lung metastasis by regulating adhesion and differentiation. EMBO Mol Med (2014) 6:865–81. doi: 10.15252/emmm.201303675
14. Uyama T, Jin XH, Tsuboi K, Tonai T, Ueda N. Characterization of the human tumor suppressors TIG3 and HRASLS2 as phospholipid-metabolizing enzymes. Biochim Biophys Acta (2009) 1791:1114–24. doi: 10.1016/j.bbalip.2009.07.001
15. Golczak M, Kiser PD, Sears AE, Lodowski DT, Blaner WS, Palczewski K. Structural basis for the acyltransferase activity of lecithin:retinol acyltransferase-like proteins. J Biol Chem (2012) 287:23790–807. doi: 10.1074/jbc.M112.361550
16. Yamane D, Feng H, Rivera-Serrano EE, Selitsky SR, Hirai-Yuki A, Das A, et al. Basal expression of interferon regulatory factor 1 drives intrinsic hepatocyte resistance to multiple RNA viruses. Nat Microbiol (2019) 4:1096–104. doi: 10.1038/s41564-019-0425-6
17. Rinkenberger N, Abrams ME, Matta SK, Schoggins JW, Alto NM, Sibley LD. Overexpression screen of interferon-stimulated genes identifies RARRES3 as a restrictor of toxoplasma gondii infection. Elife (2021) 10. doi: 10.7554/eLife.73137.sa2
18. Hajnal A, Klemenz R, Schafer R. Subtraction cloning of h-rev107, a gene specifically expressed in h-ras resistant fibroblasts. Oncogene (1994) 9:479–90.
19. Wang CH, Shyu RY, Wu CC, Tsai TC, Wang LK, Chen ML, et al. Phospholipase A/Acyltransferase enzyme activity of h-rev107 inhibits the h-RAS signaling pathway. J BioMed Sci (2014) 21:36. doi: 10.1186/1423-0127-21-36
20. Akiyama H, Hiraki Y, Noda M, Shigeno C, Ito H, Nakamura T. Molecular cloning and biological activity of a novel ha-ras suppressor gene predominantly expressed in skeletal muscle, heart, brain, and bone marrow by differential display using clonal mouse EC cells, ATDC5. J Biol Chem (1999) 274:32192–7. doi: 10.1074/jbc.274.45.32192
21. Jin XH, Uyama T, Wang J, Okamoto Y, Tonai T, Ueda N. cDNA cloning and characterization of human and mouse Ca(2+)-independent phosphatidylethanolamine n-acyltransferases. Biochim Biophys Acta (2009) 1791:32–8. doi: 10.1016/j.bbalip.2008.09.006
22. Anantharaman V, Aravind L. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol (2003) 4:R11. doi: 10.1186/gb-2003-4-2-r11
23. Jin XH, Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Discovery and characterization of a Ca2+-independent phosphatidylethanolamine n-acyltransferase generating the anandamide precursor and its congeners. J Biol Chem (2007) 282:3614–23. doi: 10.1074/jbc.M606369200
24. Shinohara N, Uyama T, Jin XH, Tsuboi K, Tonai T, Houchi H, et al. Enzymological analysis of the tumor suppressor a-C1 reveals a novel group of phospholipid-metabolizing enzymes. J Lipid Res (2011) 52:1927–35. doi: 10.1194/jlr.M015081
25. Uyama T, Ikematsu N, Inoue M, Shinohara N, Jin XH, Tsuboi K, et al. Generation of n-acylphosphatidylethanolamine by members of the phospholipase a/acyltransferase (PLA/AT) family. J Biol Chem (2012) 287:31905–19. doi: 10.1074/jbc.M112.368712
26. Ueda N, Tsuboi K, Uyama T. Metabolism of endocannabinoids and related n-acylethanolamines: Canonical and alternative pathways. FEBS J (2013) 280:1874–94. doi: 10.1111/febs.12152
27. Rahman IA, Tsuboi K, Uyama T, Ueda N. New players in the fatty acyl ethanolamide metabolism. Pharmacol Res (2014) 86:1–10. doi: 10.1016/j.phrs.2014.04.001
28. Mardian EB, Bradley RM, Duncan RE. The HRASLS (PLA/AT) subfamily of enzymes. J BioMed Sci (2015) 22:99. doi: 10.1186/s12929-015-0210-7
29. Wang L, Yu W, Ren X, Lin J, Jin C, Xia B. 1H, 13C, and 15N resonance assignments of the n-terminal domain of human TIG3. Biomol NMR Assign (2012) 6:201–3. doi: 10.1007/s12104-012-9357-2
30. Wei H, Wang L, Ren X, Yu W, Lin J, Jin C, et al. Structural and functional characterization of tumor suppressors TIG3 and h-REV107. FEBS Lett (2015) 589:1179–86. doi: 10.1016/j.febslet.2015.04.002
31. Golczak M, Sears AE, Kiser PD, Palczewski K. LRAT-specific domain facilitates vitamin a metabolism by domain swapping in HRASLS3. Nat Chem Biol (2015) 11:26–32. doi: 10.1038/nchembio.1687
32. Sturniolo MT, Dashti SR, Deucher A, Rorke EA, Broome AM, Chandraratna RA, et al. A novel tumor suppressor protein promotes keratinocyte terminal differentiation via activation of type I transglutaminase. J Biol Chem (2003) 278:48066–73. doi: 10.1074/jbc.M307215200
33. Tsai FM, Shyu RY, Jiang SY. RIG1 suppresses ras activation and induces cellular apoptosis at the golgi apparatus. Cell Signal (2007) 19:989–99. doi: 10.1016/j.cellsig.2006.11.005
34. Deucher A, Nagpal S, Chandraratna RA, Di Sepio D, Robinson NA, Dashti SR, et al. The carboxy-terminal hydrophobic domain of TIG3, a class II tumor suppressor protein, is required for appropriate cellular localization and optimal biological activity. Int J Oncol (2000) 17:1195–203. doi: 10.3892/ijo.17.6.1195
35. Sturniolo MT, Chandraratna RA, Eckert RL. A novel transglutaminase activator forms a complex with type 1 transglutaminase. Oncogene (2005) 24:2963–72. doi: 10.1038/sj.onc.1208392
36. Jans R, Sturniolo MT, Eckert RL. Localization of the TIG3 transglutaminase interaction domain and demonstration that the amino-terminal region is required for TIG3 function as a keratinocyte differentiation regulator. J Invest Dermatol (2008) 128:517–29. doi: 10.1038/sj.jid.5701035
37. Scharadin TM, Eckert RL. TIG3: an important regulator of keratinocyte proliferation and survival. J Invest Dermatol (2014) 134:1811–6. doi: 10.1038/jid.2014.79
38. Scharadin TM, Adhikary G, Shaw K, Grun DJB, Xu W, Eckert RL. Pericentrosomal localization of the TIG3 tumor suppressor requires an n-terminal hydrophilic region motif. J Invest Dermatol (2014) 134:1220–9. doi: 10.1038/jid.2013.533
39. Morishita H, Eguchi T, Tsukamoto S, Sakamaki Y, Takahashi S, Saito C, et al. Organelle degradation in the lens by PLAAT phospholipases. Nature (2021) 592:634–8. doi: 10.1038/s41586-021-03439-w
40. Staring J, Von Castelmur E, Blomen VA, Van Den Hengel LG, Brockmann M, Baggen J, et al. PLA2G16 represents a switch between entry and clearance of picornaviridae. Nature (2017) 541:412–6. doi: 10.1038/nature21032
41. Jiang SY, Wu MS, Chen LM, Hung MW, Lin HE, Chang GG, et al. Identification and characterization of the retinoic acid response elements in the human RIG1 gene promoter. Biochem Biophys Res Commun (2005) 331:630–9. doi: 10.1016/j.bbrc.2005.03.214
42. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature (2011) 472:481–5. doi: 10.1038/nature09907
43. Pappas K, Xu J, Zairis S, Resnick-Silverman L, Abate F, Steinbach N, et al. p53 maintains baseline expression of multiple tumor suppressor genes. Mol Cancer Res (2017) 15:1051–62. doi: 10.1158/1541-7786.MCR-17-0089
44. Hsu TH, Chu CC, Jiang SY, Hung MW, Ni WC, Lin HE, et al. Expression of the class II tumor suppressor gene RIG1 is directly regulated by p53 tumor suppressor in cancer cell lines. FEBS Lett (2012) 586:1287–93. doi: 10.1016/j.febslet.2012.03.020
45. Panda D, Gjinaj E, Bachu M, Squire E, Novatt H, Ozato K, et al. IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front Immunol (2019) 10:1019. doi: 10.3389/fimmu.2019.01019
46. Tanaka N, Kawakami T, Taniguchi T. Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol Cell Biol (1993) 13:4531–8. doi: 10.1128/mcb.13.8.4531-4538.1993
47. Escalante CR, Yie J, Thanos D, Aggarwal AK. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature (1998) 391:103–6. doi: 10.1038/34224
48. Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, Guo AY. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res (2019) 47:D33–8. doi: 10.1093/nar/gky822
49. Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep (2019) 28:1370–1384 e1375. doi: 10.1016/j.celrep.2019.07.001
50. Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene (1998) 16:1391–7. doi: 10.1038/sj.onc.1201661
51. Tokunaga E, Maehara Y, Oki E, Kitamura K, Kakeji Y, Ohno S, et al. Diminished expression of ING1 mRNA and the correlation with p53 expression in breast cancers. Cancer Lett (2000) 152:15–22. doi: 10.1016/S0304-3835(99)00434-6
52. Zou Z, Gao C, Nagaich AK, Connell T, Saito S, Moul JW, et al. p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem (2000) 275:6051–4. doi: 10.1074/jbc.275.9.6051
53. Feng Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb Perspect Biol (2010) 2:a001057. doi: 10.1101/cshperspect.a001057
54. Ezerskyte M, Paredes JA, Malvezzi S, Burns JA, Margison GP, Olsson M, et al. O(6)-methylguanine-induced transcriptional mutagenesis reduces p53 tumor-suppressor function. Proc Natl Acad Sci U.S.A. (2018) 115:4731–6. doi: 10.1073/pnas.1721764115
55. Willman CL, Sever CE, Pallavicini MG, Harada H, Tanaka N, Slovak ML, et al. And et AlDeletion of IRF-1, mapping to chromosome 5q31.1, in human leukemia and preleukemic myelodysplasia. Science (1993) 259:968–71. doi: 10.1126/science.8438156
56. Duvic M, Helekar B, Schulz C, Cho M, Disepio D, Hager C, et al. Expression of a retinoid-inducible tumor suppressor, tazarotene-inducible gene-3, is decreased in psoriasis and skin cancer. Clin Cancer Res (2000) 6:3249–59.
57. Zirn B, Samans B, Spangenberg C, Graf N, Eilers M, Gessler M. All-trans retinoic acid treatment of wilms tumor cells reverses expression of genes associated with high risk and relapse in vivo. Oncogene (2005) 24:5246–51. doi: 10.1038/sj.onc.1208725
58. Xu Y, Chen T, Liao D, Wu X, Zhong Y, Liu S, et al. The antitumor effect of TIG3 in liver cancer cells is involved in ERK1/2 inhibition. Tumour Biol (2016) 37:11311–20. doi: 10.1007/s13277-016-4998-x
59. Nozawa H, Oda E, Nakao K, Ishihara M, Ueda S, Yokochi T, et al. Loss of transcription factor IRF-1 affects tumor susceptibility in mice carrying the ha-ras transgene or nullizygosity for p53. Genes Dev (1999) 13:1240–5. doi: 10.1101/gad.13.10.1240
60. Giguere V, Ong ES, Segui P, Evans RM. Identification of a receptor for the morphogen retinoic acid. Nature (1987) 330:624–9. doi: 10.1038/330624a0
61. Petkovich M, Brand NJ, Krust A, Chambon P. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature (1987) 330:444–50. doi: 10.1038/330444a0
62. Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu XP, Haefner P, et al. Retinoids selective for retinoid X receptor response pathways. Science (1992) 258:1944–6. doi: 10.1126/science.1335166
63. Ghyselinck NB, Duester G. Retinoic acid signaling pathways. Development (2019) 146. doi: 10.1242/dev.167502
64. Predki PF, Zamble D, Sarkar B, Giguere V. Ordered binding of retinoic acid and retinoid-X receptors to asymmetric response elements involves determinants adjacent to the DNA-binding domain. Mol Endocrinol (1994) 8:31–9. doi: 10.1210/mend.8.1.8152429
65. Rochel N, Ciesielski F, Godet J, Moman E, Roessle M, Peluso-Iltis C, et al. Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacings. Nat Struct Mol Biol (2011) 18:564–70. doi: 10.1038/nsmb.2054
66. Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol (2015) 16:110–23. doi: 10.1038/nrm3932
67. Cordeiro TN, Sibille N, Germain P, Barthe P, Boulahtouf A, Allemand F, et al. Interplay of protein disorder in retinoic acid receptor heterodimer and its corepressor regulates gene expression. Structure (2019) 27:1270–1285 e1276. doi: 10.1016/j.str.2019.05.001
68. Osz J, Mcewen AG, Bourguet M, Przybilla F, Peluso-Iltis C, Poussin-Courmontagne P, et al. Structural basis for DNA recognition and allosteric control of the retinoic acid receptors RAR-RXR. Nucleic Acids Res (2020) 48:9969–85. doi: 10.1093/nar/gkaa697
69. Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, et al. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature (1995) 376:596–9. doi: 10.1038/376596a0
70. Younger ST, Rinn JL. p53 regulates enhancer accessibility and activity in response to DNA damage. Nucleic Acids Res (2017) 45:9889–900. doi: 10.1093/nar/gkx577
71. Garcia M, Mauro JA, Ramsamooj M, Blanck G. Tumor suppressor genes are larger than apoptosis-effector genes and have more regions of active chromatin: Connection to a stochastic paradigm for sequential gene expression programs. Cell Cycle (2015) 14:2494–500. doi: 10.1080/15384101.2015.1044179
72. Das DN, Panda PK, Sinha N, Mukhopadhyay S, Naik PP, Bhutia SK. DNA Damage by 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced p53-mediated apoptosis through activation of cytochrome P450/aryl hydrocarbon receptor. Environ Toxicol Pharmacol (2017) 55:175–85. doi: 10.1016/j.etap.2017.08.012
73. Ambolet-Camoit A, Bui LC, Pierre S, Chevallier A, Marchand A, Coumoul X, et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin counteracts the p53 response to a genotoxicant by upregulating expression of the metastasis marker agr2 in the hepatocarcinoma cell line HepG2. Toxicol Sci (2010) 115:501–12. doi: 10.1093/toxsci/kfq082
74. Yamaguchi M, Hankinson O. 2,3,7,8Tetrachlorodibenzopdioxin suppresses the growth of human liver cancer HepG2 cells in vitro: Involvement of cell signaling factors. Int J Oncol (2018) 53:1657–66. doi: 10.3892/ijo.2018.4507
75. Chen J, Lin J, Levine AJ. Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Mol Med (1995) 1:142–52. doi: 10.1007/BF03401562
76. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature (1997) 387:296–9. doi: 10.1038/387296a0
77. Paajarvi G, Viluksela M, Pohjanvirta R, Stenius U, Hogberg J. TCDD activates Mdm2 and attenuates the p53 response to DNA damaging agents. Carcinogenesis (2005) 26:201–8. doi: 10.1093/carcin/bgh289
78. Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer (2020) 20:471–80. doi: 10.1038/s41568-020-0262-1
79. Karsli Uzunbas G, Ahmed F, Sammons MA. Control of p53-dependent transcription and enhancer activity by the p53 family member p63. J Biol Chem (2019) 294:10720–36. doi: 10.1074/jbc.RA119.007965
80. Catizone AN, Uzunbas GK, Celadova P, Kuang S, Bose D, Sammons MA. Locally acting transcription factors regulate p53-dependent cis-regulatory element activity. Nucleic Acids Res (2020) 48:4195–213. doi: 10.1093/nar/gkaa147
81. Feng H, Zhang YB, Gui JF, Lemon SM, Yamane D. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PloS Pathog (2021) 17:e1009220. doi: 10.1371/journal.ppat.1009220
82. Zander DY, Burkart SS, Wust S, Magalhaes VG, Binder M. Cooperative effects of RIG-i-like receptor signaling and IRF1 on DNA damage-induced cell death. Cell Death Dis (2022) 13:364. doi: 10.1038/s41419-022-04797-7
83. Tanaka N, Ishihara M, Lamphier MS, Nozawa H, Matsuyama T, Mak TW, et al. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature (1996) 382:816–8. doi: 10.1038/382816a0
84. Purbey PK, Scumpia PO, Kim PJ, Tong AJ, Iwamoto KS, Mcbride WH, et al. Defined sensing mechanisms and signaling pathways contribute to the global inflammatory gene expression output elicited by ionizing radiation. Immunity (2017) 47421-434 e423. doi: 10.1016/j.immuni.2017.08.017
85. Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, et al. The UCSC genome browser database: 2018 update. Nucleic Acids Res (2018) 46:D762–9. doi: 10.1093/nar/gkx1020
86. Kessler DS, Veals SA, Fu XY, Levy DE. Interferon-alpha regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes Dev (1990) 4:1753–65. doi: 10.1101/gad.4.10.1753
87. Decker T, Lew DJ, Mirkovitch J, Darnell ,JE Jr. Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. EMBO J (1991) 10:927–32. doi: 10.1002/j.1460-2075.1991.tb08026.x
88. Mesev EV, Ledesma RA, Ploss A. Decoding type I and III interferon signalling during viral infection. Nat Microbiol (2019) 4:914–24. doi: 10.1038/s41564-019-0421-x
89. Fujita T, Sakakibara J, Sudo Y, Miyamoto M, Kimura Y, Taniguchi T. Evidence for a nuclear factor(s), IRF-1, mediating induction and silencing properties to human IFN-beta gene regulatory elements. EMBO J (1988) 7:3397–405. doi: 10.1002/j.1460-2075.1988.tb03213.x
90. Fujita T, Kimura Y, Miyamoto M, Barsoumian EL, Taniguchi T. Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor, IRF-1. Nature (1989) 337:270–2. doi: 10.1038/337270a0
91. Feng H, Lenarcic EM, Yamane D, Wauthier E, Mo J, Guo H, et al. NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR. Nat Immunol (2017) 18:1299–309. doi: 10.1038/ni.3853
92. Feng H, Sander AL, Moreira-Soto A, Yamane D, Drexler JF, Lemon SM. Hepatovirus 3ABC proteases and evolution of mitochondrial antiviral signaling protein (MAVS). J Hepatol (2019) 71:25–34. doi: 10.1016/j.jhep.2019.02.020
93. Xie D, Boyle AP, Wu L, Zhai J, Kawli T, Snyder M. Dynamic trans-acting factor colocalization in human cells. Cell (2013) 155:713–24. doi: 10.1016/j.cell.2013.09.043
94. Abou El Hassan M, Huang K, Eswara MB, Xu Z, Yu T, Aubry A, et al. Properties of STAT1 and IRF1 enhancers and the influence of SNPs. BMC Mol Biol (2017) 18:6. doi: 10.1186/s12867-017-0084-1
95. Siegl C, Rudel T. Modulation of p53 during bacterial infections. Nat Rev Microbiol (2015) 13:741–8. doi: 10.1038/nrmicro3537
96. Borchsenius SN, Daks A, Fedorova O, Chernova O, Barlev NA. Effects of mycoplasma infection on the host organism response via p53/NF-kappaB signaling. J Cell Physiol (2018) 234:171–80. doi: 10.1002/jcp.26781
97. Hao Z, Fu F, Cao L, Guo L, Liu J, Xue M, et al. Tumor suppressor p53 inhibits porcine epidemic diarrhea virus infection via interferon-mediated antiviral immunity. Mol Immunol (2019) 108:68–74. doi: 10.1016/j.molimm.2019.02.005
98. Kaushansky A, Ye AS, Austin LS, Mikolajczak SA, Vaughan AM, Camargo N, et al. Suppression of host p53 is critical for plasmodium liver-stage infection. Cell Rep (2013) 3:630–7. doi: 10.1016/j.celrep.2013.02.010
99. Gonzalez E, Rother M, Kerr MC, Al-Zeer MA, Abu-Lubad M, Kessler M, et al. Chlamydia infection depends on a functional MDM2-p53 axis. Nat Commun (2014) 5:5201. doi: 10.1038/ncomms6201
100. Zaika AI, Wei J, Noto JM, Peek RM. Microbial regulation of p53 tumor suppressor. PloS Pathog (2015) 11:e1005099. doi: 10.1371/journal.ppat.1005099
101. Breton Y, Desrosiers V, Ouellet M, Deshiere A, Torresilla C, Cohen EA, et al. Expression of MDM2 in macrophages promotes the early postentry steps of HIV-1 infection through inhibition of p53. J Virol (2019) 93:e01871–18. doi: 10.1128/JVI.01871-18
102. Shyu RY, Jiang SY, Chou JM, Shih YL, Lee MS, Yu JC, et al. RARRES3 expression positively correlated to tumour differentiation in tissues of colorectal adenocarcinoma. Br J Cancer (2003) 89:146–51. doi: 10.1038/sj.bjc.6601049
103. Shyu RY, Chang SC, Yu JC, Hsu SJ, Chou JM, Lee MS, et al. Expression and regulation of retinoid-inducible gene 1 (RIG1) in breast cancer. Anticancer Res (2005) 25:2453–60.
104. Wu CC, Shyu RY, Chou JM, Jao SW, Chao PC, Kang JC, et al. RARRES1 expression is significantly related to tumour differentiation and staging in colorectal adenocarcinoma. Eur J Cancer (2006) 42:557–65. doi: 10.1016/j.ejca.2005.11.015
105. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
106. Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Zidek A, et al. Highly accurate protein structure prediction for the human proteome. Nature (2021) 596:590–6. doi: 10.1038/s41586-021-03828-1
107. Eckert RL, Sturniolo MT, Jans R, Kraft CA, Jiang H, Rorke EA. TIG3: a regulator of type I transglutaminase activity in epidermis. Amino Acids (2009) 36:739–46. doi: 10.1007/s00726-008-0123-9
108. Rice G, Rompolas P. Advances in resolving the heterogeneity and dynamics of keratinocyte differentiation. Curr Opin Cell Biol (2020) 67:92–8. doi: 10.1016/j.ceb.2020.09.004
109. Scharadin TM, Jiang H, Jans R, Rorke EA, Eckert RL. TIG3 tumor suppressor-dependent organelle redistribution and apoptosis in skin cancer cells. PloS One (2011) 6:e23230. doi: 10.1371/journal.pone.0023230
110. Wang CH, Wang LK, Wu CC, Chen ML, Lee MC, Lin YY, et al. The ribosomal protein RPLP0 mediates PLAAT4-induced cell cycle arrest and cell apoptosis. Cell Biochem Biophys (2019) 77:253–60. doi: 10.1007/s12013-019-00876-3
111. Tsai FM, Shyu RY, Jiang SY. RIG1 inhibits the ras/mitogen-activated protein kinase pathway by suppressing the activation of ras. Cell Signal (2006) 18:349–58. doi: 10.1016/j.cellsig.2005.05.005
112. Tsai FM, Shyu RY, Lin SC, Wu CC, Jiang SY. Induction of apoptosis by the retinoid inducible growth regulator RIG1 depends on the NC motif in HtTA cervical cancer cells. BMC Cell Biol (2009) 10:15. doi: 10.1186/1471-2121-10-15
113. Simanshu DK, Nissley DV, Mccormick F. RAS proteins and their regulators in human disease. Cell (2017) 170:17–33. doi: 10.1016/j.cell.2017.06.009
114. Hsu TH, Chang TC. RARRES3 regulates signal transduction through post-translational protein modifications. Mol Cell Oncol (2015) 2:e999512. doi: 10.1080/23723556.2014.999512
115. Wang Z, Wang L, Hu J, Fan R, Zhou J, Wang L, et al. RARRES3 suppressed metastasis through suppression of MTDH to regulate epithelial-mesenchymal transition in colorectal cancer. Am J Cancer Res (2015) 5:1988–99.
116. Anderson AM, Kalimutho M, Harten S, Nanayakkara DM, Khanna KK, Ragan MA. The metastasis suppressor RARRES3 as an endogenous inhibitor of the immunoproteasome expression in breast cancer cells. Sci Rep (2017) 7:39873. doi: 10.1038/srep39873
117. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature (2005) 436:518–24. doi: 10.1038/nature03799
118. Hsu TH, Jiang SY, Chang WL, Eckert RL, Scharadin TM, Chang TC. Involvement of RARRES3 in the regulation of wnt proteins acylation and signaling activities in human breast cancer cells. Cell Death Differ (2015) 22:801–14. doi: 10.1038/cdd.2014.175
119. Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol (2011) 13:129–35. doi: 10.1038/ni.2203
120. Murata S, Takahama Y, Kasahara M, Tanaka K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat Immunol (2018) 19:923–31. doi: 10.1038/s41590-018-0186-z
121. Li S, Dai X, Gong K, Song K, Tai F, Shi J. PA28alpha/beta promote breast cancer cell invasion and metastasis via down-regulation of CDK15. Front Oncol (2019) 9:1283. doi: 10.3389/fonc.2019.01283
122. Dhiman G, Srivastava N, Goyal M, Rakha E, Lothion-Roy J, Mongan NP, et al. Metadherin: A therapeutic target in multiple cancers. Front Oncol (2019) 9:349. doi: 10.3389/fonc.2019.00349
123. Shen M, Xie S, Rowicki M, Michel S, Wei Y, Hang X, et al. Therapeutic targeting of metadherin suppresses colorectal and lung cancer progression and metastasis. Cancer Res (2021) 81:1014–25. doi: 10.1158/0008-5472.CAN-20-1876
124. Wu CC, Shyu RY, Wang CH, Tsai TC, Wang LK, Chen ML, et al. Involvement of the prostaglandin D2 signal pathway in retinoid-inducible gene 1 (RIG1)-mediated suppression of cell invasion in testis cancer cells. Biochim Biophys Acta (2012) 1823:2227–36. doi: 10.1016/j.bbamcr.2012.08.013
125. Hu S, Ren S, Cai Y, Liu J, Han Y, Zhao Y, et al. Glycoprotein PTGDS promotes tumorigenesis of diffuse large b-cell lymphoma by MYH9-mediated regulation of wnt-beta-catenin-STAT3 signaling. Cell Death Differ (2022) 29:642–56. doi: 10.1038/s41418-021-00880-2
126. Shyu RY, Wu CC, Wang CH, Tsai TC, Wang LK, Chen ML, et al. H-rev107 regulates prostaglandin D2 synthase-mediated suppression of cellular invasion in testicular cancer cells. J BioMed Sci (2013) 20:30. doi: 10.1186/1423-0127-20-30
127. Wang H, Xu H, Xu T, Tan C, Jiang M, Chen Y, et al. High expression of TIG3 predicts poor survival in patients with primary glioblastoma. Tumour Biol (2017) 39:1010428317712135. doi: 10.1177/1010428317712135
128. Balkwill FR. A darker side to retinoic acid revealed by sarcomas. N Engl J Med (2020) 383:491–3. doi: 10.1056/NEJMcibr2005538
129. Devalaraja S, To TKJ, Folkert IW, Natesan R, Alam MZ, Li M, et al. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell (2020) 180:1098–1114 e1016. doi: 10.1016/j.cell.2020.02.042
130. Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol (2008) 9:378–87. doi: 10.1038/ni1576
131. Harikumar KB, Yester JW, Surace MJ, Oyeniran C, Price MM, Huang WC, et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat Immunol (2014) 15:231–8. doi: 10.1038/ni.2810
132. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
133. Schoggins JW. Interferon-stimulated genes: What do they all do? Annu Rev Virol (2019) 6:567–84. doi: 10.1146/annurev-virology-092818-015756
134. Lemon SM. Hepatitis a: Current view of an ancient disease. J Hepatol (2022) 77:243–4. doi: 10.1016/j.jhep.2021.09.028
135. Sanchez-Arcila JC, Jensen KD. Eviction notice served on toxoplasma. Elife (2022) 11. doi: 10.7554/eLife.76246
136. Daher D, Shaghlil A, Sobh E, Hamie M, Hassan ME, Moumneh MB, et al. Comprehensive overview of toxoplasma gondii-induced and associated diseases. Pathogens (2021) 10. doi: 10.3390/pathogens10111351
137. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest (2015) 125:25–32. doi: 10.1172/JCI73939
138. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol (2020) 21:183–203. doi: 10.1038/s41580-019-0199-y
139. Sasaki-Tanaka R, Shibata T, Moriyama M, Okamoto H, Kogure H, Kanda T. Amantadine and rimantadine inhibit hepatitis a virus replication through the induction of autophagy. J Virol (2022) 96:e0064622. doi: 10.1128/jvi.00646-22
140. Sahoo BR, Pattnaik A, Annamalai AS, Franco R, Pattnaik AK. Mechanistic target of rapamycin signaling activation antagonizes autophagy to facilitate zika virus replication. J Virol (2020) 94:e01575-20. doi: 10.1128/JVI.01575-20
141. Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, et al. Zika virus NS4A and NS4B proteins deregulate akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell (2016) 19:663–71. doi: 10.1016/j.stem.2016.07.019
142. Lahon A, Arya RP, Banerjea AC. Dengue virus dysregulates master transcription factors and PI3K/AKT/mTOR signaling pathway in megakaryocytes. Front Cell Infect Microbiol (2021) 11:715208. doi: 10.3389/fcimb.2021.715208
143. Miricescu D, Totan A, Stanescu-Spinu Ii, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast cancer: From molecular landscape to clinical aspects. Int J Mol Sci (2020) 22. doi: 10.3390/ijms22010173
144. Aid M, Abbink P, Larocca RA, Boyd M, Nityanandam R, Nanayakkara O, et al. Zika virus persistence in the central nervous system and lymph nodes of rhesus monkeys. Cell (2017) 169:610–620 e614. doi: 10.1016/j.cell.2017.04.008
145. Meghil MM, Tawfik OK, Elashiry M, Rajendran M, Arce RM, Fulton DJ, et al. Disruption of immune homeostasis in human dendritic cells via regulation of autophagy and apoptosis by porphyromonas gingivalis. Front Immunol (2019) 10:2286. doi: 10.3389/fimmu.2019.02286
146. Sanceau J, Hiscott J, Delattre O, Wietzerbin J. IFN-beta induces serine phosphorylation of stat-1 in ewing's sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene (2000) 19:3372–83. doi: 10.1038/sj.onc.1203670
147. Dellacasagrande J, Ghigo E, Raoult D, Capo C, Mege JL. IFN-gamma-induced apoptosis and microbicidal activity in monocytes harboring the intracellular bacterium coxiella burnetii require membrane TNF and homotypic cell adherence. J Immunol (2002) 169:6309–15. doi: 10.4049/jimmunol.169.11.6309
148. Sun QH, Peng JP, Xia HF, Yang Y. IFN-gamma promotes apoptosis of the uterus and placenta in pregnant rat and human cytotrophoblast cells. J Interferon Cytokine Res (2007) 27:567–78. doi: 10.1089/jir.2006.0106
149. Bowie ML, Ibarra C, Seewalt VL. IRF-1 promotes apoptosis in p53-damaged basal-type human mammary epithelial cells: a model for early basal-type mammary carcinogenesis. Adv Exp Med Biol (2008) 617:367–74. doi: 10.1007/978-0-387-69080-3_35
150. Lee S, Hirohama M, Noguchi M, Nagata K, Kawaguchi A. Influenza a virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through the type I interferon signaling pathway in a mutually exclusive manner. J Virol (2018) 92:e00396-18. doi: 10.1128/JVI.00396-18
151. Yang L, Wang C, Shu J, Feng H, He Y, Chen J, et al. Porcine epidemic diarrhea virus induces vero cell apoptosis via the p53-PUMA signaling pathway. Viruses (2021) 13. doi: 10.3390/v13071218
152. Liu S, Luo L, Zuo F, Geng Y, Ou Y, Chen D, et al. Immunosuppression and apoptosis activation mediated by p53-Bcl2/Bax signaling pathway -the potential mechanism of goldfish (Carassius auratus Linnaeus) gill disease caused by myxobolus ampullicapsulatus. Front Immunol (2022) 13:998975. doi: 10.3389/fimmu.2022.998975
153. Elling U, Wimmer RA, Leibbrandt A, Burkard T, Michlits G, Leopoldi A, et al. A reversible haploid mouse embryonic stem cell biobank resource for functional genomics. Nature (2017) 550:114–8. doi: 10.1038/nature24027
154. McKnight KL, Lemon SM. Virology: Ins and outs of picornaviruses. Nature (2017) 541:299–300. doi: 10.1038/nature21116
155. Rivera-Serrano EE, Gonzalez-Lopez O, Das A, Lemon SM. Cellular entry and uncoating of naked and quasi-enveloped human hepatoviruses. Elife (2019) 8. doi: 10.7554/eLife.43983
156. Wang X, Ren J, Gao Q, Hu Z, Sun Y, Li X, et al. Hepatitis a virus and the origins of picornaviruses. Nature (2015) 517:85–8. doi: 10.1038/nature13806
157. McKnight KL, Lemon SM. Hepatitis a virus genome organization and replication strategy. Cold Spring Harb Perspect Med (2018) 8. doi: 10.1101/cshperspect.a033480
158. Stuart DI, Ren J, Wang X, Rao Z, Fry EE. Hepatitis a virus capsid structure. Cold Spring Harb Perspect Med (2019) 9. doi: 10.1101/cshperspect.a031807
159. Shirasaki T, Feng H, Duyvesteyn HME, Fusco WG, Mcknight KL, Xie L, et al. Nonlytic cellular release of hepatitis a virus requires dual capsid recruitment of the ESCRT-associated Bro1 domain proteins HD-PTP and ALIX. PloS Pathog (2022) 18:e1010543. doi: 10.1371/journal.ppat.1010543
160. Sager R. Tumor suppressor genes: the puzzle and the promise. Science (1989) 246:1406–12. doi: 10.1126/science.2574499
161. Heo SH, Kwak J, Jang KL. All-trans retinoic acid induces p53-depenent apoptosis in human hepatocytes by activating p14 expression via promoter hypomethylation. Cancer Lett (2015) 362:139–48. doi: 10.1016/j.canlet.2015.03.036
162. De The H. Differentiation therapy revisited. Nat Rev Cancer (2018) 18:117–27. doi: 10.1038/nrc.2017.103
163. Ni X, Hu G, Cai X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit Rev Food Sci Nutr (2019) 59:S71–80. doi: 10.1080/10408398.2018.1509201
164. Kozono S, Lin YM, Seo HS, Pinch B, Lian X, Qiu C, et al. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat Commun (2018) 9:3069. doi: 10.1038/s41467-018-05402-2
165. Kocher HM, Basu B, Froeling FEM, Sarker D, Slater S, Carlin D, et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat Commun (2020) 11:4841. doi: 10.1038/s41467-020-18636-w
166. Higuchi E, Chandraratna RA, Hong WK, Lotan R. Induction of TIG3, a putative class II tumor suppressor gene, by retinoic acid in head and neck and lung carcinoma cells and its association with suppression of the transformed phenotype. Oncogene (2003) 22:4627–35. doi: 10.1038/sj.onc.1206235
167. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15:405–14. doi: 10.1038/nri3845
168. Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
169. Kiselyov A, Bunimovich-Mendrazitsky S, Startsev V. Treatment of non-muscle invasive bladder cancer with bacillus calmette-guerin (BCG): Biological markers and simulation studies. BBA Clin (2015) 4:27–34. doi: 10.1016/j.bbacli.2015.06.002
170. Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, et al. Safety and survival with GVAX pancreas prime and listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol (2015) 33:1325–33. doi: 10.1200/JCO.2014.57.4244
171. Newman JH, Zloza A. Infection: A cause of and cure for cancer. Curr Pharmacol Rep (2017) 3:315–20. doi: 10.1007/s40495-017-0109-y
172. Ramezani S, Rezaee SA, Farjami Z, Ebrahimi N, Abdullabass HK, Ibrahim Jebur MI, et al. HTLV, a multi organ oncovirus. Microb Pathog (2022) 169:105622. doi: 10.1016/j.micpath.2022.105622
173. Zitvogel L, Ayyoub M, Routy B, Kroemer G. Microbiome and anticancer immunosurveillance. Cell (2016) 165:276–87. doi: 10.1016/j.cell.2016.03.001
174. Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther (2003) 10:292–303. doi: 10.1038/sj.gt.3301885
175. Zhang Q, Zhu H, Xu X, Li L, Tan H, Cai X. Inactivated Sendai virus induces apoptosis and autophagy via the PI3K/Akt/mTOR/p70S6K pathway in human non-small cell lung cancer cells. Biochem Biophys Res Commun (2015) 465:64–70. doi: 10.1016/j.bbrc.2015.07.130
176. Liang K, Zhang R, Luo H, Zhang J, Tian Z, Zhang X, et al. Optimized attenuated salmonella typhimurium suppressed tumor growth and improved survival in mice. Front Microbiol (2021) 12:774490. doi: 10.3389/fmicb.2021.774490
177. Hua S, Kittler R, White KP. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell (2009) 137:1259–71. doi: 10.1016/j.cell.2009.04.043
178. Kanki K, Akechi Y, Ueda C, Tsuchiya H, Shimizu H, Ishijima N, et al. Biological and clinical implications of retinoic acid-responsive genes in human hepatocellular carcinoma cells. J Hepatol (2013) 59:1037–44. doi: 10.1016/j.jhep.2013.06.024
Keywords: retinoic acid, class II tumor suppressor, lipid metabolizing enzyme, p53, IRF1, interferon, infection
Citation: Zhao J-Y, Yuan X-K, Luo R-Z, Wang L-X, Gu W, Yamane D and Feng H (2023) Phospholipase A and acyltransferase 4/retinoic acid receptor responder 3 at the intersection of tumor suppression and pathogen restriction. Front. Immunol. 14:1107239. doi: 10.3389/fimmu.2023.1107239
Received: 24 November 2022; Accepted: 22 March 2023;
Published: 31 March 2023.
Edited by:
Clett Erridge, Anglia Ruskin University, United KingdomReviewed by:
Bo Tang, Tianjin Medical University Cancer Institute and Hospital, ChinaJoseph Bird, Anglia Ruskin University, United Kingdom
Copyright © 2023 Zhao, Yuan, Luo, Wang, Gu, Yamane and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Feng, aHVpZmVuZzIwMjJAY3F1LmVkdS5jbg==; Daisuke Yamane, eWFtYW5lLWRzQGlnYWt1a2VuLm9yLmpw